

Here, Boutin et al. investigated the role of oncogenic KRAS in colorectal cancer (CRC). They generated a mouse model of metastatic CRC that harbors an inducible oncogenic Kras allele (Krasmut) and conditional null alleles of Apc and Trp53 (iKAP), which provides genetic evidence that Krasmut drives CRC invasion and maintenance of metastases.
Keywords: colorectal cancer, metastasis, Kras, Apc, P53, invasion
Abstract
Human colorectal cancer (CRC) is a major cause of cancer mortality and frequently harbors activating mutations in the KRAS gene. To understand the role of oncogenic KRAS in CRC, we engineered a mouse model of metastatic CRC that harbors an inducible oncogenic Kras allele (Krasmut) and conditional null alleles of Apc and Trp53 (iKAP). The iKAP model recapitulates tumor progression from adenoma through metastases. Whole-exome sequencing revealed that the Krasmut allele was heterogenous in primary tumors yet homogenous in metastases, a pattern consistent with activated Krasmut signaling being a driver of progression to metastasis. System-level and functional analyses revealed the TGF-β pathway as a key mediator of Krasmut-driven invasiveness. Genetic extinction of Krasmut resulted in specific elimination of the Krasmut subpopulation in primary and metastatic tumors, leading to apoptotic elimination of advanced invasive and metastatic disease. This faithful CRC model provides genetic evidence that Krasmut drives CRC invasion and maintenance of metastases.
Human colorectal cancer (CRC) is a major killer worldwide, responsible for >52,000 deaths in the United States and ∼655,000 deaths globally each year (Siegel et al. 2016). Activating mutations in the KRAS oncogene (referred to here as KRASmut) and inactivating mutations/deletions in APC and TP53 tumor suppressor genes are classical genetic events associated with disease initiation and progression (Wood et al. 2007). The majority of KRAS mutations occurs at codons 12, 13, and 61 of the KRAS gene, with the most common being glycine (G)-to-aspartic acid (D) substitution in codon 12. The presence of KRASmut correlates with increased invasive stage and liver metastasis of human CRC (Li et al. 2011; Modest et al. 2011; Mannan and Hahn-Stromberg 2012). There is also high concordance between primary CRC tumors and their liver metastasis in regard to KRAS mutation, as, 96% of the time, both have the same mutation or neither does (Knijn et al. 2011). Furthermore, lung metastases are twofold more common in CRC cases harboring an oncogenic KRAS mutation (Pereira et al. 2015)—patterns suggesting a role for oncogenic KRAS in tumor progression.
Numerous therapeutic strategies targeting components of the KRAS signaling pathway have yielded limited clinical success. KRAS signaling is comprised of a core RAF–MEK–ERK cascade, which transmits signals from extracellular receptors to transcription factors. The meager clinical impact of targeting RAF–MEK–ERK components could be the result of a nonessential role for KRASmut in CRC maintenance, the plethora of pathways downstream from activated KRAS (Pylayeva-Gupta et al. 2011), the existence of alternative KRAS-independent proliferation/survival signaling pathways such as YAP (Kapoor et al. 2014), and/or the intratumoral genomic heterogeneity of the KRASmut allele across the cancer cell population. Indeed, KRASmut has been shown to be heterogenous in cancer cells of primary CRC tumors and selected for during EGFR inhibitor therapy (Diaz et al. 2012; Misale et al. 2012). Furthermore, intratumoral genomic heterogeneity increases resistance to therapy and confers a worse prognosis (Marusyk et al. 2012). These clinical observations suggest a role for oncogenic KRAS in disease progression and in governing therapeutic responses to targeted therapy. A study in CRC cell lines showed that many KRAS mutant lines are not strictly dependent on continued KRAS expression, and the subset that was dependent required TAK1 kinase (Singh et al. 2012). Whether oncogenic KRAS serves a role in the maintenance of CRC tumors in situ is not known.
CRC models engineered with key mutations and capturing genomic heterogeneity and disease evolution are needed to understand the role of Krasmut in disease progression, tumor maintenance, and therapeutic responses. The first intestinal cancer model harboring a germline Apc mutation (ApcMin/+) generated many adenomatous polyps located primarily in the small intestine rather than the large intestine, where the disease arises in familial adenomatous polyposis patients (Moser et al. 1990; Su et al. 1992; Fodde et al. 1994; Oshima et al. 1995; Sasai et al. 2000; Kuraguchi et al. 2006; Pollard et al. 2009). Refinement of this model has been achieved by restricting Apc loss to the colon using a conditional floxed version of the allele combined with either local administration of adenovirus-Cre (Shibata et al. 1997) or a colon-specific expressing Cre such as FabplCre (Robanus-Maandag et al. 2010), CAC-Cre (Xue et al. 2010), or CDX2-Cre (Hinoi et al. 2007). These colonic lesions are predominantly benign adenomas that rarely progress to invasive adenocarcinoma and fail to metastasize. More recently, CRC mouse models have been engineered with multiple mutations such as combined activation of oncogenic KRAS and loss of APC, which leads to a synergistic increase in CRC over APC alone (Janssen et al. 2006; Sansom et al. 2006; Phelps et al. 2009). Another CRC model uses conditional alleles of Apc and oncogenic Kras that are somatically activated in the colon via surgery and local adenovirus-Cre administration (Hung et al. 2010). Collectively, these studies indicate that mice engineered with CRC signature mutations can model important aspects of the human disease.
Hallmark features of human CRC include its intratumoral genomic heterogeneity and progressive nature (Taketo and Edelmann 2009). Thus, the ideal mouse model of CRC would be sporadic and limited to the colon; have high penetrance; show reasonable latency of a few months; generate one or a few tumors per mouse; progress to adenocarcinoma at high frequency; metastasize reliably to the lymph nodes, liver, and/or lungs; mimic the human genetics and pathobiology; allow manipulation of a single variable amid a defined set of complementing mutations; allow the identification of additional mutations; and allow tumors to be monitored in vivo (Jonkers and Berns 2002).
In this study, we sought to generate a genetically engineered mouse model (GEMM) of CRC possessing the most common mutations in human CRC (KRAS, APC, and TP53) and exhibiting genomic heterogeneity and disease progression from adenoma to invasive adenocarcinoma to lung and liver metastases. By engineering an inducible Krasmut allele with spatial and temporal control of expression, we examined the tumor biological and maintenance roles of oncogenic Kras in advanced CRC disease harboring deletion of Apc and Trp53 genes. This CRC model faithfully recapitulates human disease evolution, exhibits intratumoral heterogeneity of Krasmut cells, and affords analysis of oncogenic KRAS signaling in tumor maintenance. The experimental merits of this model have revealed the critical roles and underlying mechanisms for oncogenic KRAS signaling in disease progression to metastases and in the continued maintenance of invasive and metastatic disease yet have also shown KRAS as dispensable for noninvasive primary tumor growth.
Results
Development of a GEMM of CRC with intratumoral genomic heterogeneity and metastatic propensity
We constructed a GEMM in which a colonic enema of 4-hydroxytamoxifen (4-OHT) deletes loxP conditional Apc and Trp53 tumor suppressor genes and reconstitutes a doxycycline (Dox)-responsive tet-O-LSL-KrasG12D transgene in a small number of colonic intestinal epithelial cells expressing the Cre-ERT2 transgene under the control of the intestine-specific Villin promoter (Fig. 1A). This GEMM with the full allelic series is designated here as iKAP, while mice possessing only conditional Apc and Trp53 alleles are designated iAP. We titrated 4-OHT dosage via enema using a Cre-activated membrane targeted tandem dimer Tomato (mT)/membrane targeted green fluorescent protein (mG) reporter strain and identified 1 mg/mL 4-OHT as a dose that produced one to three colonic tumors in iAP and iKAP mice (Supplemental Fig. S1A,B).
Figure 1.

iKAP GEMM of CRC. (A) GEMM design: Conditional alleles of Apc, Trp53, and tet-O-LSL-KrasG12D (Krasmut) were crossed with the tamoxifen-inducible VillinCreERT2, and 4-OHT tamoxifen was introduced into the adult colon via enema. (B) The model produces the full range of CRC progression, including macroscopic liver metastasis with Krasmut but not with Kraswt. (C) Median tumor-free survival is 6 mo and significantly longer without Krasmut. (D) Metastasis is dependent on Krasmut. (E) Endpoint tumors histologically staged by the TNM standard show that mutant Kras shifts the spectrum toward more invasive and malignant tumors. Bars: gross liver mets, 2 mm; histology, 500 µm; insets, 50 µm. (****) P < 0.0001 versus −C; (*) P < 0.05 versus −C; (NA) not applicable; (n.s.) not significant.
Following administration of 4-OHT and Dox at 8 wk of age, a weekly colonoscopy revealed an average age of tumor onset at 16 wk for iAP and 6 wk for iKAP (P = 0.009). The median tumor-free survival was 38 and 23 wk of age for iAP and iKAP mice, respectively (P = 0.008), where death in both models resulted primarily from intestinal obstruction by primary tumors (Fig. 1C; Moser et al. 1990).
Gross inspection and histopathological analyses of the tumors revealed a classical adenoma-to-carcinoma progression sequence as reported for the human disease (Fig. 1B; Fearon and Vogelstein 1990). In our model, the use of the colon-restricted 4-OHT enema produced tumors that arose primarily in the colon (85% of all gastorintestinal tumors) (Table 1), which is in contrast to the preponderance of small intestine tumors observed in most extant models (Moser et al. 1990; Johnson and Fleet 2013; Chanrion et al. 2014; Dow et al. 2015). Detailed histopathological staging was performed to document the occurrence of T1 (invasion through the muscularis mucosa), T2 (invasion into the muscularis propria), T3 (invasion into the subserosa), and T4 (invasion through the serosa and outer intestinal wall) stage tumors. The iKAP mice exhibited an overall higher incidence of invasive and metastatic disease relative to iAP mice: Twenty-three out of 60 (38%) iKAP tumors were classified as T2–T4 tumors with invasion into the colon muscle wall and beyond, whereas one out of 18 (6%) iAP tumors was T2, and zero out of 25 was T3–T4 (P = 0.0005, Fisher's exact test) (Fig. 1D,E). All T3/T4 adenocarcinomas appeared as flat invasive lesions, whereas the majority of adenomas had classical benign polypoid morphology.
Table 1.
Cancer outcomes by genotype
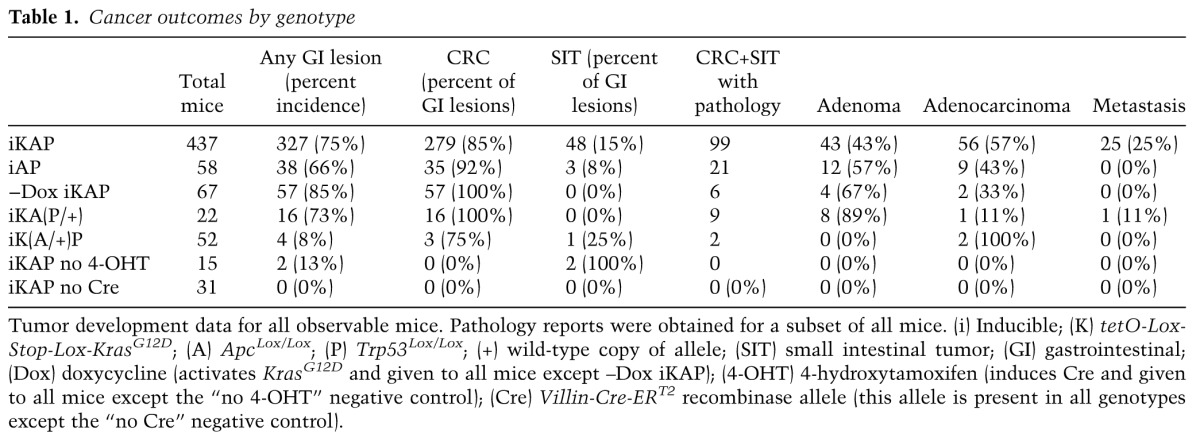
All intestinal adenocarcinomas exhibited moderate to poor differentiation and stained positively for nuclear β-catenin and the intestinal epithelial marker CK20 (Supplemental Fig. S2A). Thus, the iKAP model faithfully recapitulates human CRC evolution and reveals a role for oncogenic Kras in driving disease progression and metastases. Necropsy revealed that 17 out of 99 (17%) iKAP mice had lymph node metastasis, and an additional eight out of 99 (8%) had additional organ metastases. The CRC origin of these metastatic lesions was confirmed using markers of intestinal epithelium lineage such as CK20 (Supplemental Fig. S2B), WSS-CK, or CDX2 (data not shown) and nuclear β-catenin staining consistent with WNT activation (Supplemental Fig. S2B). In contrast, zero out of 21 (0%) iAP mice had lymph or organ metastasis (Table 1). It is worth noting that all iKAP mice with metastatic disease also possessed at least one T3–T4 adenocarcinoma, supporting the view that invasion beyond the muscularis mucosa is associated with increased risk of metastasis, as observed in the human disease.
Inefficiencies in Cre-mediated recombination have been exploited to create mosaic tissues with different allelic combinations (Wong et al. 2000). A notable feature of the iKAP GEMM is the mosaic expression of the Krasmut allele in most primary tumors, mimicking the intratumoral mutational heterogeneity of human CRC. Specifically, GFP expression patterns from the Cre-activated Rosa26-LSL-rtTA-IRES-GFP allele were often mosaic and regionally clustered in the same tumor (Fig. 2A–C). In the iKAP tumors, activation of Kras signaling (i.e., pERK) exhibited a near 1:1 concordance with GFP staining across 20 tumors examined (Supplemental Fig. S3A; data not shown). Correspondingly, GFP/pERK-positive cells had a strikingly different histology compared with adjacent GFP/Kras-negative cells within the same tumor, the former exhibiting larger oncocytic cytology with nuclear enlargement and hyperchromasia as well as dramatic reduction in adjacent interstitial stroma (Supplemental Fig. S3A). In contrast, WNT pathway signaling as indicated by nuclear β-catenin staining was seen in every cancer cell (Supplemental Fig. S3A,B). Together, these patterns are consistent with the role of APC loss as a gatekeeper event in adenoma initiation (Fearon and Vogelstein 1990) with contemporaneous or subsequent Cre-directed activation of the Krasmut allele in these Apc-deficient neoplasms.
Figure 2.

Kras clonal evolution. (A) Early stage iKAP tumors are heterogenous for Krasmut, but those cells predominate in late stage tumors and metastasis as visualized by native GFP fluorescence. (B) Immunohistochemistry (IHC) against GFP-tagged Krasmut cells showing Krasmut at the invasive front (red arrow) and increasing with tumor stage. (C) Quantification of GFP and pERK IHC signal across 251 tumors shows a dramatic selection for Krasmut cells at the invasive front of T1 adenocarcinomas and then throughout the bulk of T4 adenocarcinomas and metastases. Each tumor or invasive region was binned based on the percentage of GFP+ cells. (Ad) Adenoma; (AdC) adenocarcinoma; (Inv) invasive front; (Met) metastasis. Bars: 500 µm; insets, 50 µm.
Given this intratumoral heterogeneity, we sought to determine definitively whether the Krasmut heterogeneity arose from Cre-mediated activation in different parental cells of origin or one parental cell followed by several rounds of cell division in an early APC-deficient neoplasm. To that end, whole-exome sequencing cataloged somatic single-nucleotide variations (SNVs) in the GFP/pERK-positive (Krasmut) and GFP/pERK-negative (Kraswt) regions microdissected from the same tumor. In three out of five intratumor pairs, a small number of shared SNVs were detected in each member of the pair—a pattern consistent with a monoclonal tumor origin where one cell underwent Cre recombination of Apc, and a daughter cell subsequently recombined Kras (Supplemental Fig. S4B). The two clones then diverged significantly, as a vast majority of the SNVs was private to each clone. A lack of overlapping SNVs in the other two intratumor pairs suggested either a polyclonal origin of the clones or a monoclonal origin with lack of early shared mutations due to low mutation rate (Supplemental Figs. S4A, S5, S6). In the case of polyclonal origin, two distinct cells within either the same crypt or neighboring crypts had recombination events affecting Apc in one cell and Apc + Kras in the other. These neoplasms then merged to form the larger tumor. This coexistence of two clones within the same lesion is consistent with the proposed mechanism of adenoma growth by crypt fission (bottom-up histogenesis of CRC) (Preston et al. 2003; Humphries and Wright 2008). Polyclonal tumor origin has been documented previously in mouse models (Thliveris et al. 2005) and human cancers (Novelli et al. 1996; Parsons 2008). Finally, two pairs of neighboring but distinct tumors were sequenced as negative controls, and, as expected, no shared SNVs were found (Supplemental Fig. S5B).
This intratumoral mosaicism enables a side-by-side comparison of the behavior of cancer cells of the same tumor with and without Krasmut. We noted that all invasive fronts of T1–T4 tumors contained morphologically distinct cells that had further lost their columnar architecture and much of their basal/apical orientation, consistent with a less differentiated and more invasive phenotype. All 61 GFP/pERK mosaic invasive adenocarcinomas exhibited GFP and pERK staining at the morphologically distinct invasive front, while the GFP/pERK-negative plus β-catenin-positive tumor cells resided exclusively in the noninvasive bulk of the tumor mass (Fig. 2B,C; Supplemental Figs. S3A, S7, S8). Notably, this GFP/pERK pattern was evident even in early stage invasive T1 tumors, implying that Krasmut activation plays a critical role in the initial break through the submucosa (Supplemental Figs. S7, S8). In contrast, all nine iAP mice with T1 disease were GFP- and pERK-negative at the invasion front (Supplemental Fig. S3B). Furthermore, 57 out of 58 histologically identified iKAP metastases expressed GFP and pERK in every cell (Fig. 2B,C; Supplemental Fig. S8B). These data indicate that activated Krasmut subclones can significantly outcompete nonactivated Kraswt subclones in invasive and metastatic components within the same primary tumor. In line with these observations, examination of human CRC TCGA (The Cancer Genome Atlas) data showed a consistent trend toward higher allelic frequency of KRAS from stage I to stage IV (Supplemental Fig. S11B; Supplemental Table S2). Further emphasizing the role of Krasmut in driving invasion and metastasis, invasive iKAP tumors were consistently identified as early as 3–8 wk after tumor induction (Fig. 1D,E; Supplemental Fig. S7). In contrast, iAP mice never progressed beyond T2 stage disease even at >28 wk after tumor induction (Supplemental Fig. S7). We conclude that oncogenic KRAS signaling promotes CRC invasion and metastasis.
Oncogenic Krasmut is required for maintenance of advanced CRC disease yet dispensable for growth of APC-deficient cancer cells in the same primary tumor
Apc restoration has been shown to reverse the malignant CRC phenotype fully (Dow et al. 2015), prompting us to assess the role of Krasmut in tumor maintenance. First, standard two-dimensional (2D) and three-dimensional (3D)/organoid Matrigel cell culture systems were used to examine the growth of primary cultures derived from iKAP tumors. Both culture systems exhibited cytostasis upon extinction of KRASmut expression (withdrawal of Dox)=(Supplemental Fig. S9A,B). Xenografts implanted subcutaneously or intravenously into nude mice were also dependent on continued KRASmut expression (Supplemental Fig. S9C–E). Next, we examined the Krasmut dependency of autochthonous primary and metastatic tumors. To that end, 34 Dox-treated iKAP mice with endoscopically confirmed tumors were enlisted into a study in which 10 tumor-bearing mice were withdrawn from Dox, and the remaining 24 mice were maintained on Dox. All mice were followed by weekly endoscopy for change in tumor growth. Withdrawal of Dox induced transient tumor regression followed by resumption of tumor growth after 4 wk, consistent with the aforementioned mosaicism and continued growth of Kraswt subclones in the iKAP primary tumors (Fig. 3A–C).
Figure 3.

Kras withdrawal in situ. (A) Mouse endoscopy showing iKAP tumor growth of tumors remaining on Dox (+Dox/+Kras) and after Dox withdrawal (−Dox/−Kras) within the same mouse over time. (B,C) Tumor growth was significantly reduced in −Dox/−Kras (n = 16) (B) as well as final tumor diameter (C) compared with “on Dox” (n = 48). (D,E) TNM staging of CRC tumors after random assignment to either remain +Dox/+Kras (D) or be taken off (−Dox/−Kras) (E) for 3 wk. The −Dox/−Kras tumors were exclusively adenomas, while the mice left on Dox had the full range of progression to metastasis. (F) H&E and IHC of iKAP tumors show GFP- and pERK-positive regions +Dox/+Kras. Upon Dox withdrawal for 2 d, GFP cells remained but no longer had activated pERK (arrows), and, after 2 wk off Dox, GFP-positive cells were mostly gone (‡), leaving large pockets of necrosis (†). (G) Biopsies were taken from iKAP tumors +Dox/+Kras, and then Dox was withdrawn for 2 wk. The previously biopsied tumors revealed that the GFP+/pERK+ population of cells was gone, and cCASP3-positive pockets of apoptotic cells were present (†). Bars: F, 500 µm; F inset, 50 µm; G, 200 µm; G inset, 50 µm. (**) P < 0.01; (*) P < 0.05.
Next, the histopathological impact of Krasmut extinction on tumor stage was examined in a second cohort of 29 endoscopically confirmed tumor-bearing iKAP mice (19 Dox continued and 10 Dox withdrawn). Necropsy at 3 wk following Dox continuation/withdrawal revealed an incidence of 14 out of 19 (74%) invasive adenocarcinomas and five out of 19 (26%) metastases in the Dox-continued mice versus zero out of 10 (0%) adenocarcinoma or metastases in the Dox withdrawal cohort (Fig. 3D,E). Instead, the Dox withdrawal mice all harbored noninvasive adenomas, demonstrating that extinction of oncogenic KRAS signaling selectively prevents or eliminates invasive and metastatic cancer cells.
To understand the molecular consequences of Krasmut extinction in advanced CRC, serial iKAP tumor biopsies were obtained for histologic and expression analysis at baseline and at day 2 and week 2 following Dox withdrawal. On day 2, pERK signal was lost, and the remaining GFP+ cells were rounded up and detached, exhibited early signs of cell death (including increased cleaved Caspase-3), and showed decreased Ki-67 (Fig. 3F; Supplemental Fig. S10A,B). By week 2 off Dox, most GFP+ cells were eliminated, and cleaved Caspase-3 and Ki-67 returned back to baseline levels (Fig. 3F,G; Supplemental Fig. S10C–E). We conclude that the shift toward benign adenoma following Krasmut extinction results from the elimination and/or reversion of the malignant phenotype of Krasmut adenocarcinoma cells.
Activated Krasmut drives invasion at least partially through activation of TGF-β and induces a transcriptional profile enriched for an invasive human CRC signature
Given the prominent role of Krasmut in driving invasion, we sought to elucidate the pathways mediating the Krasmut-driven motile behavior of the cancer cells. We conducted RNA sequencing (RNA-seq) profiling of 39 iKAP tumors and 10 iAP tumors from 44 mice at various stages. The late stage Krasmut invasive and metastatic tumors clustered tightly together, suggesting that the transcriptional program is stable once T3/T4 invasion is achieved (Fig. 4A). The top 10 gene set enrichment analysis (GSEA) pathways up-regulated in the invasive and metastastic tumors compared with noninvasive tumors included gene sets related to invasion, epithelial-to-mesenchymal transition (EMT), stemness, and TGF-β signaling (Fig. 4B; Supplemental Table S1). Given the known role of TGF-β signaling in invasion (Siegel and Massague 2003), we selected it for further validation. Expression of TGFβ1, the principal ligand in TGF-β signaling, was increased in invasive primary iKAP tumors and metastases when compared with noninvasive primary tumors (Fig. 4C). TGFβ1 expression correlated very well with KRAS expression as well as a 20-gene RAS signature in vivo (Fig. 4D; Supplemental Fig. S11A).
Figure 4.

Invasive iKAP tumor cells activate Tgfβ1 in a Krasmut -dependent manner. (A) Unsupervised hierarchical clustering of all mouse CRC samples showed similarity of expression by Krasmut status, KrasG12D transcript expression, and tumor stage. Three mice with T3/4 tumors show 0%–3% KRAS expression; we note that this is because the noninvasive regions were intentionally profiled. (B) Selected GSEA enrichment plots from analysis of the full 17,179-gene list from the in vivo data set. One-thousand-five-hundred-fifty pathways were significant at a false discovery rate (FDR) of <0.1. (C) Tgfβ1 expression in vivo. (D) Kras expression in vivo. (****) P < 0.0001; (n.s.) not significant.
We next determined whether TGF-β pathway activation is associated with KRASmut signaling in vivo using immunohistochemistry (IHC) on mosaic iKAP tumors. Consistent with the bioinformatic analyses, TGFβ1 expression and pSMAD2 nuclear expression were prominently enhanced in invasive Krasmut tumor cells but not in Kraswt cells within the same tumor (Fig. 5D). Interestingly, noninvasive Krasmut tumor cells showed an intermediate or low level of TGFβ1 and pSMAD2 staining, suggesting that additional events are necessary to fully activate the TGF-β pathway and invasion (Oft et al. 1996; Fujimoto et al. 2001), which may include stromal interactions. Next, we functionally validated TGF-β signaling as a regulator of invasion in primary iKAP cells in vitro. Recombinant TGF-β strongly enhanced invasion in iKAP cells, and inhibition of TGF-β reduced invasion (Fig. 5A,B). Western blots for pSMAD2/3 validated the expected pathway modulation by the exogenous agents (Fig. 5C). Collectively, these data confirm that TGF-β activation is Krasmut and invasion-specific in vivo and that TGFβ1 modulation at least partially accounts for the invasive phenotype.
Figure 5.

TGF-β modulates invasion of iKAP cells. (A) Primary iKAP CRC cells cultured with Dox to induce Krasmut and with or without recombinant TGF-β and/or TGF-β inhibitor added to the medium. Crystal violet-stained invasion chambers showing recombinant TGF-β increases invasion when Krasmut is active. In Krasmut cells, invasion is increased further with additional TGF-β and decreased substantially by adding TGF-β inhibitor. (B) Invasion was quantified by counting migrated cells in three independent experiments. (C) Western blot on the same culture conditions as in A showing pSMAD2/3 as a readout of TGF-β pathway activation and pERK as a readout of RAS pathway activation. (D) IHC against GFP, TGFβ1, and pSMAD2 in invasive versus noninvasive tumors showing TGF-β activation in GFP+/Krasmut regions of tumors. Bars: 10×, 200 µm; 40×, 50 µm. (****) P < 0.0001.
To determine the human relevance of these findings, we undertook a comparison of the iKAP tumors with human CRC subtype transcriptomes. Using the recently designated consensus molecular subtypes (CMS1–4) (Guinney et al. 2015), we found that the EMT/TGF-β signature enriched in invasive and metastatic iKAP tumors had a strong overlap with the signature associated with the CMS4 subtype, which is defined as mesenchymal with stromal invasion and high TGF-β signaling (Fig. 6A,B). However, these assessments are relative, not absolute: iKAP/iAP tumor analyses compare adenocarcinomas with adenomas, while the human TCGA data include only adenocarcinomas. We therefore assessed this CMS4-related signature in two publicly available human data sets that include both adenomas and adenocarcinomas. These analyses demonstrated that human adenocarcinomas showed a significant enrichment of the metastasis-associated CMS4 signature and of TGFβ1 gene expression relative to adenomas (Fig. 6C–E; Supplemental Fig. S12). Moreover, TGFβ1 correlated well with the CMS4 gene signature across data sets (Supplemental Fig. S12). Altogether, these data suggest that TGFβ1 activation is a dominant feature of the adenoma-to-adenocarcinoma transition and at least partially underlies the cardinal features of invasion and metastasis in the iKAP model.
Figure 6.

iKAP tumors resemble human CRC subtype CMS4. (A) TCGA CRC expression data for the 416 genes in the CMS signature showing segmentation into the four distinct CMS subtypes across the bottom. Mutation status of KRAS, SMAD4, and SMAD2 in the TCGA samples is represented at the top. (B) iKAP mouse CRC expression data demonstrate that the invasive/metastatic iKAP tumors share the CMS4 signature, defined as mesenchymal with stromal invasion and high TGF-β signaling. (C) TGFβ1 expression in the human TCGA CRC data set, separated by CMS subtypes. (D) Quantification of TGFB1 expression comparing human adenocarcinomas (AdC) with adenomas in data set GSE41258. (E) Quantification of the CMS4 gene signature in the same data set.
Discussion
In this study, we report a mouse model of CRC engineered with the three most common human genetic alterations that recapitulates disease progression, including metastases. The inducible nature of the Krasmut allele, coupled with its intratumoral heterogeneity, enabled exploration of its role in CRC tumor biology and disease progression in the context of Apc and Trp53 deficiency. Consistent with the acquisition of KRAS mutation following APC loss in human disease, our studies provide clear experimental evidence that oncogenic KRAS signaling is dispensable for adenomatous growth yet essential for progression to invasive and metastatic growth in this model. Through differential expression analysis and in vitro experiments, we observed the TGF-β pathway to be one of the major downstream effectors. Notably, extinction of oncogenic KRAS signaling in fully advanced disease resulted in the specific elimination of invasive and metastatic disease while allowing for sustained primary tumor growth through the presence of intratumoral Kras genotype heterogeneity. This suggests that, on a tumor cell-intrinsic level, loss of oncogenic Kras is either cell-lethal or reverts advanced cells to a benign adenomatous morphology, whereas Apc reactivation in a CRC mouse model led to differentiation toward a normal colonic morphology (Dow et al. 2015). These differences underscore the distinctly different roles of APC loss in tumor initiation and KRAS oncogenic activation in tumor progression. This paradigm of a metastasis maintenance gene holds important therapeutic implications for treatment of advanced CRC or for interceptive strategies to impede disease progression.
The iKAP model mirrors many aspects of the human disease. The similarity of iKAP tumor gene signatures to those in the recently described CMS4 human subtype of CRC (Guinney et al. 2015) is consistent with TGF-β pathway activation playing a role in the adenoma-to-adenocarcinoma transition in both mice and humans. One feature of the iKAP model that does not track well with the human disease is the lack of the heavy mutational load typical of human CRC. Specifically, whole-exome sequencing of the iKAP tumors revealed few additional somatic mutations beyond the “baked-in” drivers (Supplemental Fig. S6). As dietary factors have been implicated in fueling mutation in human CRC, the iKAP model may serve as a platform for identifying dietary factors driving somatic mutation of the human CRC genome.
Intratumoral heterogeneity is a critical aspect of cancer development and therapeutic response but is poorly modeled. The iKAP model displays heterogeneity for Krasmut activation in early stage CRC, with clonal evolution driving Krasmut cell expansion during tumor progression. By sequencing Krasmut and Kraswt clones within a single tumor, we discovered that this heterogeneity may arise from both monoclonal and polyclonal origins as reported for human solid tumors (Parsons 2008). Genetic ablation of the Krasmut cells temporarily reduces tumor size, but the Kraswt clones remain and eventually regrow the tumor, leading to bowel obstruction. Further testing of drug combinations using this model could be useful in understanding how heterogeneity contributes to the drug resistance and recurrence seen in human CRC.
The importance of continued oncogene expression for tumor maintenance has been well established in animal models of melanoma (Chin et al. 1999), lung (Sotillo et al. 2010), and PDAC (Ying et al. 2012) cancers. These tumors were deemed “addicted” to their driver oncogenes, including Kras, as the cancer regressed completely upon extinction of the oncogene. In contrast, the mosaicism in the iKAP mouse faithfully models intratumoral heterogeneity and reveals that, despite addiction of individual cells to Krasmut signaling, the heterogeneity of the tumor subverts anti-Krasmut therapy from killing the entire tumor outright but converts invasive/metastatic tumors to more benign disease. However, KRASwt human CRC tumors are still capable of metastasizing, indicating that the positive prophylactic effects of anti-KRASmut therapy, although potentially highly beneficial, would be improved by further therapeutic intervention.
TGF-β signaling emerged as a top pathway enriched in invasive and metastatic tumors in the iKAP model. IHC, functional assays, and comparison with human expression data further underscored the central role of TGFβ1 in mediating invasion in the iKAP model and the adenoma-to-adenocarcinoma transition. Our findings are consistent with TGF-β increasing invasion in late stage tumors (Siegel and Massague 2003) and with RAS and TGF-β collaborating in invasion (Oft et al. 1996), including in the intestine (Fujimoto et al. 2001). Interestingly, in our Krasmut iKAP cells, which are already highly invasive, additional recombinant TGF-β increased invasion further, presumably through a dose-dependent mechanism as described by David et al. (2016).
In conclusion, Krasmut drives invasion and metastasis in our CRC GEMM at least in part through up-regulation of TGF-β signaling and maintains metastases. That the metastatic cells remain dependent on continued KRASmut expression represents a clear clinical opportunity for metastatic CRC patients. Upon genetic Krasmut extinction in mouse CRC, all invasive and metastatic tumors reverted back to adenomas. Due to heterogeneity within our GEM tumors, Kraswt cells persisted, resulting in only moderate reduction of tumor bulk. This will likely also be the case in the clinical setting, which may explain the modest results seen from MAPK inhibitors thus far, and will necessitate combination therapy, potentially with WNT pathway inhibitors.
Materials and methods
Generation of iKAP mice
The tamoxifen-inducible Villin-Cre-ERT2 allele (El Marjou et al. 2004) was crossed with the Dox-inducible Tet-Kras-G12D allele (Ying et al. 2012), the reverse tetracycline transactivator (rtTA)-GFP allele (Belteki et al. 2005), the Apc Lox allele (Kuraguchi et al. 2006), and the Tp53 Lox allele (Marino et al. 2000) and backcrossed to C57BL/6. The iKAP mice have no phenotype without tamoxifen administration. To activate KrasG12D, Dox was administered in the drinking water at 2 g of Dox (Sigma, D9891) and 40 g of sucrose (Sigma, S7903) per 1 L of autoclaved water. All manipulations were performed under Institutional Animal Care and Use Committee approval (protocol 00001123-RN0.) See Supplemental Table S3 for genotyping primers and protocols.
Tamoxifen enema
Cre expression driven by the Villin promoter was detected throughout the gastrointestinal tract. To limit Cre activity to the colon, we used the tamoxifen-inducible Villin-Cre-ERT2 and delivered tamoxifen directly to the colon with an enema. Different forms of tamoxifen and different concentrations were tested. To achieve specific spatial induction of Cre, we had to use the active metabolite of tamoxifen (4-OHT; Sigma, H7904) freshly solubilized in 100% EtOH for 10 min at 55°C and diluted to working concentrations of 10 mg/mL (26 mM), 1 mg/mL (2.6 mM), 0.1 mg/mL (260 µM), or 0.01 mg/mL (26 µM). We found that the 1 mg/mL dose gave the best response. The enema was administered via a small, flexible, sterile plastic catheter attached to a 1-mL syringe (Instech Solomon, FTP2038); the catheter tip was dipped in lubricating jelly before insertion (MediChoice, 4067063031). Eight-week-old to 12-wk-old mice were scruffed without anesthesia, 1 cm of the catheter was inserted into the anus, and 50 µL of solution was administered.
mT/mG Cre reporter
To quantify and calibrate the level of Cre activation by the tamoxifen enema, we used the mT/mG reporter strain described by Muzumdar et al. (2007). Briefly, mice were injected intraperitoneally with 1.0 mg of tamoxifen (Sigma) dissolved in corn oil or the 4-OHT enema described above. For whole-mount organ analysis, mice were sacrificed using CO2 plus cervical dislocation, and organs were isolated and imaged with a CCD camera mounted on a fluorescence microscope. For cryosection preparation, tissues were fixed for 24 h in cold 4% PFA at 4°C. Fixed tissues were cryoprotected in 30% sucrose overnight at 4°C and embedded in OCT. Ten-micrometer sections were cut using a cryostat. Slides were washed three times with PBS, treated with DAPI, washed again, mounted in gel/mount (Biomeda Corp.), and imaged as above or with confocal microscopy. Neither mT nor mG required immunostaining for visualization.
IHC
Five-micrometer sections were deparaffinized, rehydrated, and treated in a pressure cooker with citrate buffer for antigen retrieval. Sections were incubated in primary antibodies (diluted 1:500 in blocking solution) overnight at 4°C. We used the following rabbit polyclonal antibodies: anti-GFP (Cell Signaling Technology, 2956), anti-phospho-ERK (Cell Signaling Technology, 4370), anti-β-catenin (Cell Signaling Technology, 9587), anti-cleaved Caspase 3 (Cell Signaling Technology, 9661), anti-cytokeratin 20 (Progen, GP-K20), anti-LYVE-1 (RELIATech, 103-PA50AG), anti-P53 (Vector Laboratories, CM5), anti-Ki67 (Vector Laboratories, VP-RM04), anti-TGFb1 (Proteintech, 18978-1-AP), and p-Smad2 (ThermoFisher, 44-244G). An anti-rabbit IgG HRP-linked secondary antibody (DAKO's Envision dual-link system, K4063) was used to visualize binding of primary antibody. Sections were developed with the DAB chromogen (Vector Laboratories), counterstained with Mayer's hematoxylin, dehydrated, and coverslipped with Permount.
Endoscopy
Normal chow was replaced with a nonsolid diet the night before endoscopy. Anesthesia was administered through the regulated flow of 1%–2% isoflurane vapor through a nose cone. The colon was flushed with 10 mL of warm PBS enema. The colon was carefully insufflated with air from a standard 10-gallon air pump, and endoscopic images were acquired with a Storz veterinary endoscope. Once tumors were established, we quantified their size in situ by photographing them and measuring the percent of the lumen occluded by tumor. Tumor size was measured in Adobe Photoshop relative to the luminal diameter and recorded as the percent tumor occlusion of the lumen. The change in percent tumor occlusion relative to the baseline measurement was calculated weekly and averaged within the two groups.
Care of immunocompromised mice
Nude mice were housed in pathogen-free conditions. They were observed daily for tumor formation and poor health. Mice were carefully monitored, and steps were taken to alleviate pain and stress. Moribund animals (animals that had visceral tumors; were not eating, drinking, or eliminating; were exhibiting cachexia; or presented tumor ulceration or inhibition of mobility) and animals with obvious tumors (≤1.5 cm) or obvious neurological symptoms or under obvious stress were sacrificed and necropsied, at which time tumor and normal tissue biopsies were taken.
Subcutaneous injection
Human or mouse cell lines were injected subcutaneously to study the ability of cells to grow and generate tumors/metastases in vivo. The cell suspensions were injected in the subcutaneous space of the loose skin between the shoulder blades. Injections were given without anesthesia. Prior to injection, the skin was cleaned with betadine. A disposable syringe and a 25- to 27-gauge 0.5- to 0.75-inch needle were used to enter the subcutaneous space at an acute angle with care not to extend beyond the subcutaneous space into the muscle beneath. Skin was tented with two fingers, and the needle was inserted through the skin and advanced 5–10 mm through the subcutaneous tissue to prevent leakage from the site. Once the needle entered the subcutaneous space, the bevel of the needle was pointed up to tent the skin. The cells (106) were injected in a volume of 0.5 mL.
Primary cell culture
Colon tumors were dissected out and washed in 1% bleach in PBS for 15 min. Tumors were then minced with a razor and dissociated to single cells following the MACS dissociation kit protocol (Miltenyi Biotec, 130-096-730). Primary growth medium consisted of DMEM Advanced (ThermoFisher, 12491-015), 2 mM L-glutamine (ThermoFisher, 25030-081), 1× penicillin/streptomycin (ThermoFisher, 15140-122), 2.5 µg/mL Fungizone (ThermoFisher, 15290-018), 1% Tet-free FBS (Clontech, 631106), and 1 µg/mL Dox (Sigma, D9891). Cells were incubated at 37°C in a 5% CO2 atmosphere. For 3D culture, cells were mixed with cold Matrigel (BD, 354230), and 50-µL droplets were plated on 24-well low-attachment plates with 500 µL of growth medium from above.
Invasion chambers
Invasion chambers were adapted from BD/Corning (354480) using BioCoat Matrigel invasion chambers with 8.0-µm PET membrane in 24-well plates. Primary cells were trypsinized, counted, and resuspended in serum-free DMEM Advanced. Next, 1 × 105 cells were added to the upper chamber in 200 µL of serum-free DMEM Advanced. In the lower chamber, 800 µL of DMEM Advanced plus 20% FBS was added as a chemoattractant. As an input control, 50 µL of cells (1/10 vol) was seeded into an empty well with 1 mL of 1% FBS DMEM Advanced. After 24 h of incubation in a cell culture incubator, the upper well membrane was carefully scrubbed with a cotton swab, and the lower membrane was fixed in 10% neutral buffered formalin for 10 min, rinsed in PBS, and stained in 0.5% crystal violet in 25% methanol for 10 min. The wells were rinsed several times in water. Whole chambers were scanned from the bottom of each well, and pictures were taken under a microscope (20×) for counting. Migration was calculated as the average number of cells per 20× field.
Western blotting
Primary cultured iKAP CRC cells were treated with 2 ng/mL recombinant TGFB1 protein or 10 µM TGFβR1 inhibitor galunisertib for 24 h and harvested. Western blotting was performed under standard methods using anti-phospho-SMAD2/3 (Cell Signaling Technology, 8828) and anti-phospho-ERK (Cell Signaling Technology, 4370).
RT-qPCR
RNA was harvested using the Qiagen RNeasy minikit (no. 74104), and 1 µg was reverse-transcribed to cDNA using the SuperScript III kit (ThermoFisher, 18080-051). qPCR was run on a ABI 7500 machine using the fast two-step program with dissociation curve and SYBR Green 2× master mix (ThermoFisher, 4309155). Transgenic and endogenous Kras expression was normalized to TBP. The primer sequences used were mKrasG12-1F (GCTTATCGATACCGTCGATCG), mKrasG12-1R (GGTCGTACTCATCCACAAAGTG), mKras-wt-2F (GAGCAAAGATGGGAAGAAGA), mKras-wt-2R (TTTGCACGAACAGAAGAAAAA), m TBP F (ACATCTCAGCAACCCACACA), and m TBP R (CAGCCAAGATTCACGGTAGA).
Whole-exome sequence
All sequence data have been deposited in NCBI under Sequence Read Archive (SRA) number SRP097890. DNA was harvested from fresh-frozen tissue using the Qiagen AllPrep DNA/RNA microkit (no. 80284) or from formalin-fixed paraffin-embedded tissue using the QIAamp DNA FFPE tissue kit (no. 56404). Exome capture was performed on 500 ng of genomic DNA per sample according to the manufacturer's instructions, and paired-end multiplex sequencing of samples was performed on the Illumina HiSeq 2000 sequencing platform. The average sequencing depth was 277× per sample (ranging from 138× to 437×, standard deviation ± 67). For variant calling, paired-end reads in FastQ format generated by the Illumina pipeline were aligned to the reference mouse genome mm10 (Ensembl GRCm38) using Burrows-Wheeler Aligner (BWA) on default settings except for using a seed length of 40, maximum edit distance of 3, and maximum edit distance in the seed of 2. GATK best practices of duplicate read removal, indel realignment, and base recalibration were used on aligned reads. Somatic single-nucleotide substitutions were detected by using MuTect. In addition to MuTect's built-in filters, the following filtering criteria were applied: (1) total read count in tumor DNA ≥ 100, (2) total read count in germline DNA ≥ 50, (3) variant present on both strands, (4) VAF in tumor DNA ≥ 1%, (5) minimum depth in tumor = 20, (6) minimum depth in matched normal = 10, and (7) variants in positions listed in dbSNP1.37 removed. Substitutions were annotated using ANNOVAR based on mouse University of California at Santa Cruz known genes. Tumors from the same mouse were compared using Venny 2.1 (http://bioinfogp.cnb.csic.es/tools/venny/index.html). Samtools (Li et al. 2009) was used to determine the read depth for the deleted exons in Apc and Trp53.
TCGA analysis
For the analysis of APC and KRAS mutations in stage I–IV CRC, somatic nonsilent mutations and corresponding allelic frequencies were obtained from the TCGA data portal. A total of 79 CRCs were identified to harbor mutations in both APC and KRAS. In those tumors where more than one somatic mutation in APC or KRAS was identified, only the allelic frequency of the dominant mutation was considered for calculations. KRAS allelic frequencies were corrected using APC allelic frequencies (TCGA data portal, https://tcga-data.nci.nih.gov).
RNA-seq
All sequence data have been deposited in NCBI under SRA number SRP097890. RNA was harvested from fresh-frozen tissue using the Qiagen AllPrep DNA/RNA microkit (no. 80284). Libraries were made, and samples were run on an Illumina HiSeq 2000 at The University of Texas M.D. Anderson Cancer Center. Transcriptome reads were mapped to the reference mouse genome mm10, normalized, and quantified as FPKM (fragments per kilobase per million mapped fragments) values using HTSeq-count (HTSeq) (Anders et al. 2015). The values were further transformed into Z-values for downstream statistical analysis. The gene selections for supervised hierarchical clustering were made by “limma” R package (Gentleman et al. 2004; Smyth 2004) using a P-value of <0.05 and a false discovery rate (FDR) P-value of <0.05. The hierarchical clustering was implemented by Gene Cluster version 3 (de Hoon et al. 2004) using centroid linkage as the distance measure.
GSEA
For the in vivo data, the R package FCROS (Dembele and Kastner 2014) was used to generate a unified statistic incorporating fold change and significance for input into the GSEA preranked algorithm.
Statistical analysis
Survival curves were calculated using GraphPad Prism 6 and compared using the log-rank (Mantel-Cox) test. Pairwise comparisons were performed using the unpaired two-tailed Student's t-test, and comparisons of three or more groups were done using one-way ANOVA with a Turkey correction for multiple testing, also done in GraphPad Prism 6. For all experiments with error bars, the standard error of the mean (SEM) was calculated and plotted.
Supplementary Material
Acknowledgments
We thank Raghu Kalluri for scientific feedback, Kathleen Quast for help with editing figures, Denise Spring for careful editing of the manuscript, members of the DePinho laboratory for discussion, Edward Chang for help with histology and slide scanning, David Menter and Deepavali Chakravarti for help with primary cell culture, and Guocan Wang for help with analyzing expression data. This research is supported by The Helen Hay Whitney Foundation Post-doctoral Fellowship (A.T.B), University of Texas Stars award (R.A.D.), and National Institutes of Health R01 CA084628 (R.A.D.).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.293449.116.
References
- Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belteki G, Haigh J, Kabacs N, Haigh K, Sison K, Costantini F, Whitsett J, Quaggin SE, Nagy A. 2005. Conditional and inducible transgene expression in mice through the combinatorial use of Cre-mediated recombination and tetracycline induction. Nucleic Acids Res 33: e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanrion M, Kuperstein I, Barriere C, El Marjou F, Cohen D, Vignjevic D, Stimmer L, Paul-Gilloteaux P, Bieche I, Tavares SD, et al. 2014. Concomitant Notch activation and p53 deletion trigger epithelial-to-mesenchymal transition and metastasis in mouse gut. Nat Commun 5: 5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, Shen Q, O'Hagan R, Pantginis J, Zhou H, et al. 1999. Essential role for oncogenic Ras in tumour maintenance. Nature 400: 468–472. [DOI] [PubMed] [Google Scholar]
- David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, Iacobuzio-Donahue CA, Massague J. 2016. TGF-β tumor suppression through a lethal EMT. Cell 164: 1015–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoon MJ, Imoto S, Nolan J, Miyano S. 2004. Open source clustering software. Bioinformatics 20: 1453–1454. [DOI] [PubMed] [Google Scholar]
- Dembele D, Kastner P. 2014. Fold change rank ordering statistics: a new method for detecting differentially expressed genes. BMC Bioinformatics 15: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, et al. 2012. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 486: 537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow LE, O'Rourke KP, Simon J, Tschaharganeh DF, van Es JH, Clevers H, Lowe SW. 2015. Apc restoration promotes cellular differentiation and reestablishes crypt homeostasis in colorectal cancer. Cell 161: 1539–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, Robine S. 2004. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39: 186–193. [DOI] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B. 1990. A genetic model for colorectal tumorigenesis. Cell 61: 759–767. [DOI] [PubMed] [Google Scholar]
- Fodde R, Edelmann W, Yang K, van Leeuwen C, Carlson C, Renault B, Breukel C, Alt E, Lipkin M, Khan PM, et al. 1994. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci 91: 8969–8973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto K, Sheng H, Shao J, Beauchamp RD. 2001. Transforming growth factor-β1 promotes invasiveness after cellular transformation with activated Ras in intestinal epithelial cells. Exp Cell Res 266: 239–249. [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, et al. 2015. The consensus molecular subtypes of colorectal cancer. Nat Med 21: 1350–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinoi T, Akyol A, Theisen BK, Ferguson DO, Greenson JK, Williams BO, Cho KR, Fearon ER. 2007. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res 67: 9721–9730. [DOI] [PubMed] [Google Scholar]
- Humphries A, Wright NA. 2008. Colonic crypt organization and tumorigenesis. Nat Rev Cancer 8: 415–424. [DOI] [PubMed] [Google Scholar]
- Hung KE, Maricevich MA, Richard LG, Chen WY, Richardson MP, Kunin A, Bronson RT, Mahmood U, Kucherlapati R. 2010. Development of a mouse model for sporadic and metastatic colon tumors and its use in assessing drug treatment. Proc Natl Acad Sci 107: 1565–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen KP, Alberici P, Fsihi H, Gaspar C, Breukel C, Franken P, Rosty C, Abal M, El Marjou F, Smits R, et al. 2006. APC and oncogenic KRAS are synergistic in enhancing Wnt signaling in intestinal tumor formation and progression. Gastroenterology 131: 1096–1109. [DOI] [PubMed] [Google Scholar]
- Johnson RL, Fleet JC. 2013. Animal models of colorectal cancer. Cancer Metastasis Rev 32: 39–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers J, Berns A. 2002. Conditional mouse models of sporadic cancer. Nat Rev Cancer 2: 251–265. [DOI] [PubMed] [Google Scholar]
- Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, Zhong Y, Wu CJ, Sadanandam A, Hu B, et al. 2014. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 158: 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knijn N, Mekenkamp LJ, Klomp M, Vink-Borger ME, Tol J, Teerenstra S, Meijer JW, Tebar M, Riemersma S, van Krieken JH, et al. 2011. KRAS mutation analysis: a comparison between primary tumours and matched liver metastases in 305 colorectal cancer patients. Br J Cancer 104: 1020–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraguchi M, Wang XP, Bronson RT, Rothenberg R, Ohene-Baah NY, Lund JJ, Kucherlapati M, Maas RL, Kucherlapati R. 2006. Adenomatous polyposis coli (APC) is required for normal development of skin and thymus. PLoS Genet 2: e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing Subgroup. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HT, Lu YY, An YX, Wang X, Zhao QC. 2011. KRAS, BRAF and PIK3CA mutations in human colorectal cancer: relationship with metastatic colorectal cancer. Oncol Rep 25: 1691–1697. [DOI] [PubMed] [Google Scholar]
- Mannan A, Hahn-Stromberg V. 2012. K-ras mutations are correlated to lymph node metastasis and tumor stage, but not to the growth pattern of colon carcinoma. APMIS 120: 459–468. [DOI] [PubMed] [Google Scholar]
- Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. 2000. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev 14: 994–1004. [PMC free article] [PubMed] [Google Scholar]
- Marusyk A, Almendro V, Polyak K. 2012. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer 12: 323–334. [DOI] [PubMed] [Google Scholar]
- Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, et al. 2012. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 486: 532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modest DP, Stintzing S, Laubender RP, Neumann J, Jung A, Giessen C, Haas M, Aubele P, Schulz C, Boeck S, et al. 2011. Clinical characterization of patients with metastatic colorectal cancer depending on the KRAS status. Anticancer Drugs 22: 913–918. [DOI] [PubMed] [Google Scholar]
- Moser AR, Pitot HC, Dove WF. 1990. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 247: 322–324. [DOI] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. 2007. A global double-fluorescent Cre reporter mouse. Genesis 45: 593–605. [DOI] [PubMed] [Google Scholar]
- Novelli MR, Williamson JA, Tomlinson IP, Elia G, Hodgson SV, Talbot IC, Bodmer WF, Wright NA. 1996. Polyclonal origin of colonic adenomas in an XO/XY patient with FAP. Science 272: 1187–1190. [DOI] [PubMed] [Google Scholar]
- Oft M, Peli J, Rudaz C, Schwarz H, Beug H, Reichmann E. 1996. TGF-β1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev 10: 2462–2477. [DOI] [PubMed] [Google Scholar]
- Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M. 1995. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci 92: 4482–4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons BL. 2008. Many different tumor types have polyclonal tumor origin: evidence and implications. Mutat Res 659: 232–247. [DOI] [PubMed] [Google Scholar]
- Pereira AA, Rego JF, Morris V, Overman MJ, Eng C, Garrett CR, Boutin AT, Ferrarotto R, Lee M, Jiang ZQ, et al. 2015. Association between KRAS mutation and lung metastasis in advanced colorectal cancer. Br J Cancer 112: 424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps RA, Chidester S, Dehghanizadeh S, Phelps J, Sandoval IT, Rai K, Broadbent T, Sarkar S, Burt RW, Jones DA. 2009. A two-step model for colon adenoma initiation and progression caused by APC loss. Cell 137: 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard P, Deheragoda M, Segditsas S, Lewis A, Rowan A, Howarth K, Willis L, Nye E, McCart A, Mandir N, et al. 2009. The Apc 1322T mouse develops severe polyposis associated with submaximal nuclear β-catenin expression. Gastroenterology 136: 2204–2213.e13. [DOI] [PubMed] [Google Scholar]
- Preston SL, Wong WM, Chan AO, Poulsom R, Jeffery R, Goodlad RA, Mandir N, Elia G, Novelli M, Bodmer WF, et al. 2003. Bottom-up histogenesis of colorectal adenomas: origin in the monocryptal adenoma and initial expansion by crypt fission. Cancer Res 63: 3819–3825. [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. 2011. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 11: 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robanus-Maandag EC, Koelink PJ, Breukel C, Salvatori DC, Jagmohan-Changur SC, Bosch CA, Verspaget HW, Devilee P, Fodde R, Smits R. 2010. A new conditional Apc-mutant mouse model for colorectal cancer. Carcinogenesis 31: 946–952. [DOI] [PubMed] [Google Scholar]
- Sansom OJ, Meniel V, Wilkins JA, Cole AM, Oien KA, Marsh V, Jamieson TJ, Guerra C, Ashton GH, Barbacid M, et al. 2006. Loss of Apc allows phenotypic manifestation of the transforming properties of an endogenous K-ras oncogene in vivo. Proc Natl Acad Sci 103: 14122–14127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai H, Masaki M, Wakitani K. 2000. Suppression of polypogenesis in a new mouse strain with a truncated Apc(Delta474) by a novel COX-2 inhibitor, JTE-522. Carcinogenesis 21: 953–958. [DOI] [PubMed] [Google Scholar]
- Shibata H, Toyama K, Shioya H, Ito M, Hirota M, Hasegawa S, Matsumoto H, Takano H, Akiyama T, Toyoshima K, et al. 1997. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science 278: 120–123. [DOI] [PubMed] [Google Scholar]
- Siegel PM, Massague J. 2003. Cytostatic and apoptotic actions of TGF-β in homeostasis and cancer. Nat Rev Cancer 3: 807–821. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. 2016. Cancer statistics, 2016. CA Cancer J Clin 66: 7–30. [DOI] [PubMed] [Google Scholar]
- Singh A, Sweeney MF, Yu M, Burger A, Greninger P, Benes C, Haber DA, Settleman J. 2012. TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell 148: 639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. 2004. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: 1–25. [DOI] [PubMed] [Google Scholar]
- Sotillo R, Schvartzman JM, Socci ND, Benezra R. 2010. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature 464: 436–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, Dove WF. 1992. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 256: 668–670. [DOI] [PubMed] [Google Scholar]
- Taketo MM, Edelmann W. 2009. Mouse models of colon cancer. Gastroenterology 136: 780–798. [DOI] [PubMed] [Google Scholar]
- Thliveris AT, Halberg RB, Clipson L, Dove WF, Sullivan R, Washington MK, Stanhope S, Newton MA. 2005. Polyclonality of familial murine adenomas: analyses of mouse chimeras with low tumor multiplicity suggest short-range interactions. Proc Natl Acad Sci 102: 6960–6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong MH, Saam JR, Stappenbeck TS, Rexer CH, Gordon JI. 2000. Genetic mosaic analysis based on Cre recombinase and navigated laser capture microdissection. Proc Natl Acad Sci 97: 12601–12606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. 2007. The genomic landscapes of human breast and colorectal cancers. Science 318: 1108–1113. [DOI] [PubMed] [Google Scholar]
- Xue Y, Johnson R, Desmet M, Snyder PW, Fleet JC. 2010. Generation of a transgenic mouse for colorectal cancer research with intestinal Cre expression limited to the large intestine. Mol Cancer Res 8: 1095–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. 2012. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149: 656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.