

Abstract
Natural products that contain functional groups with heteroatom-heteroatom linkages (X–X, where X = N, O, S, and P) are a small yet intriguing group of metabolites. The reactivity and diversity of these structural motifs has captured the interest of synthetic and biological chemists alike. Functional groups containing X–X bonds are found in all major classes of natural products and often impart significant biological activity. This review presents our current understanding of the biosynthetic logic and enzymatic chemistry involved in the construction of X–X bond containing functional groups within natural products. Elucidating and characterizing biosynthetic pathways that generate X–X bonds could both provide tools for biocatalysis and synthetic biology, as well as guide efforts to uncover new natural products containing these structural features.
Graphical Abstract

1. Introduction
Functional groups containing heteroatom-heteroatom bonds (X–X, where X = N, O, S, and P) have been identified in natural products isolated from a variety of sources (Figure 1). The diverse structures and reactivities of these motifs have captured the attention of chemists and chemical biologists. While many of these functional groups are rare, they have been found in all major families of secondary metabolites, including nonribosomal peptides, polyketides, ribosomally-synthesized and post-translationally modified peptides (RiPPs), terpenes, alkaloids, and nonproteinogenic amino acids.
Figure 1.

X–X bond containing functional groups covered in this review
This wide distribution suggests that these structural features have important biological roles. Indeed, many metabolites containing such linkages are used clinically, including the N-nitrosourea-containing chemotherapeutic streptozotocin (1), the isoxazolidinone-containing antibiotic D-cycloserine (2), and the nitro group-containing antibiotic chloramphenicol (3).1–3 X–X bond containing functional groups can be essential for the activity of natural products. For example, the N-nitrosourea substituent of streptozotocin is a precursor to diazomethane, which alkylates DNA. Similarly the presence of both diazo groups in lomaiviticin A (4) are required for efficient generation of DNA double strand breaks.4
In addition to their presence in reactive functional groups, X–X bonds play other important roles within natural products. The N-hydroxyl groups of siderophores e.g. aerobactin (5) are crucial for chelating insoluble ferric iron and the N–O glycosidic linkage in calicheamicin (6) is critical for optimal binding to DNA.5 The N–N bond of piperazic acid imparts increased conformational rigidity compared with L-proline, potentially impacting the activity of piperazic acid-containing nonribosomal peptides, such as kutzneride 1 (7).6–8 Generally, the lone pairs and polarized X–H bonds present within these functional groups facilitate binding to biological macromolecules via hydrogen bonding and polar interactions.
In addition to their presence in secondary metabolites, structural motifs with N–N and N–O bonds, including tetrazoles, pyrazoles, hydrazides, and hydroxamates, are abundant in synthetic screening libraries and were present in 10% of the 100 top selling drugs in 2013, further suggesting the privileged status of X–X bond containing molecules in imparting biological activity.9,10 Synthetic methods for accessing these structural motifs are available, however developing new strategies for constructing X–X linkages is an active area of investigation in synthetic organic chemistry.11–15 An underexplored but promising approach for accessing such scaffolds is the use of X–X bond forming enzymes in the context of biocatalysis and synthetic biology. Such applications could help to alleviate the relative lack of synthetic methods for X–X bond formation in comparison to C–C and C–X bond construction.
This review compiles and updates our current understanding of the enzymology of X–X bond construction in natural product biosynthesis. This topic is of general interest given the influence these structural motifs have on the activity of natural products, their presence in both natural product-based and synthetic drugs, and the potential applications of X–X bond forming enzymes and metabolic pathways in biocatalysis and synthetic biology. We organize our discussion according to the type of X–X linkage being assembled, the functional group into which it is incorporated, and the type of enzyme involved in its construction (Figure 2). We include a brief discussion of the synthetic methods available for accessing particular structural motifs at the beginning of several sections to provide a framework for discussing biosynthetic strategies. While the enzymes responsible for certain transformations, such as N–O bond formation, have been extensively studied, we are only starting to understand the enzymatic chemistry and biosynthetic logic used to construct many X–X bond containing functional groups. Although we primarily focus on pathways where a specific enzyme is known to catalyze X–X bond formation, we will also discuss examples in which genetic experiments and/or feeding studies have provided insights into the chemical logic underlying X–X bond construction. We expect these latter examples to stimulate future investigations. We will not extensively discuss X–X bond formation in primary metabolism, including nitrification, anammox, or nitric oxide synthase, as they have been reviewed elsewhere.16–18 Additionally, we will not cover the enzymatic chemistry used in phosphorylation of natural products or biosynthetic intermediates, as this likely parallels well-studied enzymes involved in primary metabolism and posttranslational modification.19,20
Figure 2.

Selected bioactive natural products containing an X–X bond
2. N–O bond forming enzymes
N-hydroxyl, isoxazolidine, oxime, nitrone, nitroso, and nitro functional groups comprise many of the N–O linkages found in natural product scaffolds. The predominant biosynthetic logic used to access these structural features involves successive oxidations of amines, with each oxidation step conferring unique reactivity to the intermediates and products. Many enzymes have evolved to catalyze these oxidative transformations, including flavin-dependent N-oxygenases, cytochrome P450s, and iron- and copper-containing oxygenases. Throughout this review, a general mechanism for an enzyme class will be presented, and existing reviews detailing in-depth discussions about their enzymology will be cited accordingly. A common feature of these enzymes is the use of various cofactors to access electrophilic or radical oxygen species from molecular oxygen. Furthermore, amine oxygenases can display exquisite selectivity, avoiding the problematic overoxidation often encountered in analogous non-enzymatic processes. This particular feature of enzymatic N-oxygenation has therefore fueled interest in applying these systems for biocatalysis. This section will discuss enzymes from natural product biosynthesis that catalyze N–O bond formation, including several pathways in which the N–O bond forming enzyme has not yet been characterized.
2.1. Hydroxylamine
Much of our knowledge of enzymatic N-oxidation has come from extensive research into the origins of hydroxylamines, which are key intermediates in siderophore biosynthesis. In addition to being precursors to the more highly oxidized nitroso and nitro functional groups, hydroxylamines are emerging as key building blocks in pathways that generate other types of X–X linkages. For example, the biosynthesis of azoxy compounds (Section 3.1) and piperazic acid (Section 3.3–3.4), which contain a N–N bond, proceeds through N-hydroxylated intermediates.21,22 The oxygen atoms of the carbonyl and the N–OH group in hydroxamic acids, often found in siderophores, impart metal chelating properties by serving as bidentate ligands after deprotonation of the hydroxyl group (pKa ~ 9).23 For some drugs, including the histone deacetylase inhibitors trichostatin A and vorinostat, the N-hydroxyl group is a key pharmacophore required for target binding.24,25
In all known examples, hydroxylamines are installed on natural product scaffolds via hydroxylation of the corresponding primary amine. Notably, this biosynthetic strategy differs substantially from methods used by organic chemists to access this functional group. The chemical synthesis of hydroxylamines by direct oxidation of amines is difficult to control due to the challenge of avoiding overoxidation and typically results in variable yields.26 To circumvent this problem, an amine is usually converted into a nitrone intermediate, which is incapable of further hydroxylation, and hydrolyzed to yield a hydroxylamine. Other non-enzymatic approaches to access hydroxylamines include reduction of the corresponding nitro or oxime groups.
The majority of N-hydroxylases discovered to date are either flavin-dependent N-monooxygenases (NMOs) or cytochrome P450 monooxygenase enzymes, and the following section will be organized based on these two enzyme families. Numerous reviews have been published on the enzymology of flavin-containing monooxygenases27–29 and cytochrome P450s.30–32 Interestingly, both families have evolved to install hydroxyl groups on amines despite the use of different cofactors and oxygen activation mechanisms. This convergent evolution between heme-based and flavin-dependent enzymes to catalyze the same reaction is not unique to N-oxygenation, as exemplified by epoxidation33 and chlorination reactions.34
2.1.1. Flavin-dependent
Flavin-dependent N-monooxygenases (NMOs) have been extensively studied in the context of siderophore biosynthesis. Iron is an essential micronutrient, and bacteria sequester insoluble ferric iron (FeIII) from the environment using small molecule metal chelators called siderophores.35 Hydroxamic acid is a common functional group in siderophores for coordinating metals and is installed by the sequential action of NMOs and acetyltransferases on a variety of amine substrates.35,36 The addition of an acetyl or formyl group not only confers additional chelating properties to siderophores, but also prevents non-enzymatic oxidation of hydroxylamines to the corresponding nitro groups.37
Many microbial pathogens, including Aspergillus fumigatus and Pseudomonas aeruginosa, secrete siderophores in the iron-limited environment of the human body.38–41 Thus, targeting pathogen virulence by disrupting siderophore biosynthesis has been actively investigated.42 Deletion of siderophore genes, including those encoding NMOs, adversely affects bacterial pathogenicity and growth in iron-deficient medium.39–41,43 This vital role of NMOs in conferring virulence has thus motivated many studies of NMO structures and mechanisms of catalysis. The insights gained from this work may inform the development of enzyme inhibitors that can target siderophore production by inactivating monooxygenases.
Hydroxamic acid precursors commonly incorporated into siderophores include the amino acids L-ornithine and L-lysine.44–58 (Figure 3, Table 1). In addition to amino acids, some siderophores, including rhizobactin (8) and alcaligin (9), contain hydroxylated aliphatic diamines like putrescine or cadaverine as their chelating functional groups.36,44,55 These building blocks are incorporated into the siderophores either by (1) nonribosomal peptide synthetase (NRPS) assembly lines, which activate, load, and elongate pre-assembled hydroxamate-containing amino acid building blocks, or (2) NRPS-independent siderophore synthetases that use ATP to form amide or ester bonds in a non-templated manner.36 Several published reviews present in-depth analysis of the enzymatic chemistry and biosynthetic logic used in siderophore assembly.59,60 A comprehensive list of more than 290 siderophores with references as of 2010 has been compiled in Appendix A of a review by Hider and Kong.23 Here we will focus on selected N-hydroxylases from siderophore biosynthesis that have been extensively characterized using a combination of biochemical, structural, and computational approaches.
Figure 3.

Structures of selected siderophores, highlighting key amino acid and amine building blocks and the presence of hydroxylamines
Table 1.
Selected siderophore N-monooxygenases
| NMO | Siderophore | Amine | References |
|---|---|---|---|
| AlcA | Alcaligin | Putrescene | 44 |
| CchBa | Coelichelin | Ornithine | 45 |
| BibB | Bisucaberin | Cadavarine | 46 |
| DesB | Desferroxamine | Cadaverine | 47 |
| EtcBa | Erythrochelin | Ornithine | 48 |
| FscE | Fuscachelin | Ornithine | 49 |
| IucDa | Aerobactin | Lysine | 50 |
| MbsGa | Mycobactin | Lysine | 51 |
| NbtGa | Nocobactin | Lysine | 52 |
| PsbA | Pseudobactin | Ornithine | 53 |
| PvdAa | Pyoverdine | Ornithine | 54 |
| RhbE | Rhizobactin | 1,3-diaminopropane | 55 |
| Rmoa | Rhodochelin | Ornithine | 56 |
| SidAa | Ferrocrocin | Ornithine | 57 |
| VcbOa | Vicibactin | Ornithine | 58 |
Biochemically characterized
N-monooxygenases (NMOs), like all enzymes that employ molecular oxygen as a substrate, evolved to address the challenge of engaging the inert ground state triplet molecular oxygen species (3O2).27–29 Organic cofactors that can form stabilized radicals (flavins and pterins), as well as redox active transition metals (Fe, Cu, Mn), are used to transfer single electrons to molecular oxygen, generating reactive species that can interact with a variety of different partner substrates. To date, all of the characterized N-hydroxylases in siderophore biosyntheses are flavin-dependent. These NMOs, along with Baeyer–Villiger (BVMO) and microsomal flavin-containing monoxygenases (mFMO), are classified as Class B flavoprotein monooxygenases based on similarities in their sequences, structures, and mechanistic features.28 In contrast to the two-component NMOs (discussed in Section 2.2.1 and Section 3.1.1), Class B NMOs do not require a separate flavin reductase partner to supply the reduced cofactor. Class B NMOs are all encoded by a single gene, preferentially utilize NADPH over NADH to generate reduced flavin, and bind oxidized NADP+ tightly throughout catalysis. These shared features of NMOs have implications for both substrate specificity and flavin intermediate stabilization.
IucD, SidA, and PvdA, NMOs from aerobactin (5), ferrocrocin (10), and pyoverdine (11) biosynthesis, have been extensively characterized biochemically, and crystal structures have been elucidated for SidA and PvdA.61,62 These enzymes share conserved Rossman dinucleotide binding domains that serve as binding pockets for FAD and NADPH. As with the general flavin monooxygenase mechanism, substrate oxygenation can be separated into reductive and oxidative half-reactions.29 Both of these half-reactions rely on the unique reactivity of the flavin isoalloxazine ring, which allows for both the one-electron reduced semiquinone radical species (15) and the two-electron reduced flavin (14) to be kinetically accessible.29 In the reductive half-reaction, hydride transfer from bound NADPH to flavin N5 generates 14 (Scheme 1). This initial reduction (13→14) occurs independently of substrate binding.63–65 The NMOs EtcB, PvdA, SidA, and CchB are specific for NADPH over NADH in this reduction reaction.45,48,54,66,67 Next, oxygen is proposed to diffuse from the bulk solvent into the enzyme active site housing the flavin cofactor through enzyme multichannels.68 A one-electron transfer reaction from 14 to molecular oxygen results in a radical pair consisting of flavin semiquinone (15) and superoxide anion.27 The exact mechanism of this electron transfer in NMOs is still being investigated, but the unstable pair of radicals rapidly recombines to form a C4a-hydroperoxyflavin species (16) that has been detected spectrophotometrically at 360–380nm in numerous NMOs (15 → 16, Scheme 1).63–65 The proton source for 16 is proposed to derive from the charged amino acid side chain, as evidenced by a spectroscopic shift from 361nm, which is indicative of a peroxyflavin intermediate, to the 380nm of 16.63,65,69 A hallmark of these class B NMOs is the stability of 16, which has a half-life of about 30 minutes.
Scheme 1.

General flavin-dependent monooxygenation mechanism
To prevent the unproductive generation of hydrogen peroxide from the electrophilic hydroperoxo group of 16, flavin-dependent monooxygenases employ different strategies to stabilize this intermediate. In many cases, both the nicotinamide ring and ribose moieties of NADP+ as well as active site residues of the enzyme interact favorably with 16 via hydrogen bonding, extending its half-life. 27,61,62,64,70–73 In the crystal structure of SidA, for example, the amide nitrogen of NADP+ forms a hydrogen bond with the N5 proton (Scheme 2).61 This interaction disrupts a hydrogen bond between the N5 proton and the proximal hydroperoxo oxygen, thereby preventing intramolecular proton transfer and the release of hydrogen peroxide.74 Once 16 is generated, the nucleophilic nitrogen atom of a bound amine substrate attacks the distal electrophilic hydroperoxo oxygen of this intermediate, breaking its O–O bond to produce the corresponding hydroxylamine and hydroxyflavin intermediate (17). The oxidized flavin cofactor is regenerated after eliminating water (17 → 13).65 This oxygenation mechanism has been proposed for numerous siderophore NMOs highlighted in Table 1.
Scheme 2.

C4a-hydroperoxyflavin intermediate stabilization by NADP+ prevents uncoupling and generation of H2O2
Siderophore NMOs generally exhibit narrow substrate specificity. In the presence of the appropriate substrate, a 5-fold and 80-fold increase in the formation of 16 by SidA and PvdA, respectively, were observed.63,64 Despite the structural similarities of L-ornithine and L-lysine, the lysine NMO IucD from aerobactin biosynthesis cannot hydroxylate L-ornithine75 and PvdA and SidA exclusively hydroxylate L-ornithine over L-lysine.57,63,66 In the crystal structures of PvdA and SidA in complex with L-ornithine, this substrate is positioned such that its charged α-amino and carboxylate groups interact favorably with conserved active site asparagine and lysine residues.61,62 The side chain amino group forms hydrogen bond interactions with the ribose moiety of NADP+ via a water molecule. Co-crystallization of SidA with L-lysine also reveals that while this amino acid can bind in a manner similar to ornithine, its side chain amino group is not optimally positioned near the C4a position for nucleophilic attack.61 Instead, the extra methylene group in lysine appears to orient the terminal amino group closer to the N5 proton, potentially facilitating the increase in H2O2 release observed in the presence of this amino acid.76
More recently, the first crystal structure of a lysine N-monooxgenase was reported with the structural characterization of NbtG from nocobactin biosynthesis in Nocardia farcinica.52 Unlike the canonical Class B NMOs, NbtG releases unusually high amounts of uncoupled hydrogen peroxide and superoxide. While all the key residues for binding ornithine in SidA are conserved in NbtG, its NADPH binding domain is significantly rotated compared to that of SidA. This domain rotation alters the active site geometry and relative positions of the substrate binding residues. More importantly, the nicotinamide binding domain is obstructed by an α-helix, which contributes to a 10–1000 fold increase in measured KD for NADP+ when compared to other Class B monooxygenases.52 NbtG’s weak binding interaction with NADP+ provides an explanation as to why its oxidation reaction is highly uncoupled, as NADP+ plays a crucial role in stabilizing 16. Similar deviations in reactivity were observed for lysine monooxygenase MbsG from Mycobacterium smegmatis, in which the enzyme shows no preference for NADH or NADPH and exhibits high uncoupling rates of 16 to form hydrogen peroxide.51 The evolutionary origin of substrate specificity among all the siderophore NMOs remains unclear. However, Sobrado et al. have suggested that NbtsG may have evolved as a dual NADPH oxidase.52 Peroxide production by this enzyme may also serve signaling roles in response to amino acid levels inside the cell.76
Functional groups containing more extensively oxidized nitrogen atoms can arise from successive oxidations by N-monooxygenases, which will be covered in later sections (Section 2.2–2.6). The mechanism by which siderophore NMOs avoid a second hydroxylation event is not well understood. Given the importance of the acyl group in protecting the hydroxylamine from overoxidation, acylation of the hydroxylamine may be tightly coupled to the oxidation event,. Moreover, pH-dependence studies indicate that the amine substrate is bound to the enzyme in the protonated state.76 While the mechanism by which substrate accelerates formation of 16 remains to be studied, the absence of an acidic proton in the hydroxylamine (pKa ~ 5)64 may play in role in preventing the formation of 16. Finally, given the strict substrate specificity of NMOs toward the substrate, the N-hydroxylated amino acid may not be in a position for nucleophilic attack of the distal oxygen of 16. The crystal structure of PvdA with N5-hydroxy-L-ornithine bound reveals that the oxygen atom of the substrate is taking the place of the distal peroxo oxygen of 16.76 Indeed, the hydroxylamine product was shown to be a competitive inhibitor of the L-ornithine binding to 16 without being catalyzed to form a dihydroxylated intermediate.64 Further structural comparisons between siderophore NMO and other oxime- and nitrone-forming flavin-dependent NMOs (Section 2.2 and 2.3) may reveal additional structural features or motifs that can account for these enzymes’ ability to avoid overoxidation.
In addition to siderophores, other naturally occurring N-hydroxylamines have been identified, including alanosine77 and dopastin.78 As mentioned in the introduction to this section, N-hydroxylated intermediates are involved in constructing the azoxy group of valanimycin and the piperazic acid building block use in kutzneride (7) biosynthesis.21,77,79 The ornithine hydroxylase KtzI from kutzeneride biosynthesis and the isobutylamine hydroxylase VlmH from valanimycin biosynthesis will be covered in Section 3.4.2 and Section 3.1.1, respectively. Flavin-dependent N-oxidation enzymes are also found in oxime, nitroso, nitrone, and nitro-group biosynthesis and will be discussed below in Sections 2.2–2.6.
2.1.2. P450-dependent
Cytochrome P450s are heme-dependent enzymes that catalyze a diverse set of chemical transformations in both primary metabolism and natural product biosynthesis.34,80 Motivation for studying the mechanisms of cytochrome P450s stems from their roles in oxidative degradation of xenobiotics and synthesis of key signaling metabolites such as nitric oxide.18 In addition to performing C-oxidations, including hydroxylation of unactivated carbon centers, epoxidation, and oxidative crosslinking, P450s also catalyze N-hydroxylation of amines. To date, only a handful of cytochrome P450s have been identified that perform N-oxidation in the context of natural product biosynthesis. As a result, most of the published work on heme-based N-hydroxylation mechanisms has involved the more ubiquitous arylamine hydroxylases81–83 and nitric oxide synthases (NOS)18,84–88. This section will first discuss the two different mechanisms proposed for N–O bond formation by arylamine hydroxylases and will then describe studies of CalE10, an N-hydroxylase from calicheamicin (6) biosynthesis.89 N-hydroxylation by NOS will be discussed briefly in the context of D-cycloserine (2) biosynthesis in Section 2.4.1. Similar P450-dependent monooxygenases are found in oxime biosynthesis, and they will be discussed in Section 2.2.
Hydroxylation of primary aromatic and heteroaromatic amines (ArNH2) by mammalian P450 enzymes, specifically CYP1A2, has received widespread interest, as the corresponding arylhydroxylamine products are precursors to nitrenes. These species are potent DNA alkylating agents and have been linked to development of colorectal, pancreatic, and prostate cancer in humans.90–94 This particular biotransformation has also received attention in the context of drug metabolism, in which hydroxylation of amine-containing drugs can generate electrophilic reactive metabolites that can alkylate proteins and DNAs, causing adverse drug reactions95. These considerations have fueled interest in understanding the mechanisms of P450-dependent hydroxylation.83 Although detailed biochemical and mechanistic analysis of P450-catalyzed arylamine N-hydroxylation have not been reported, insights into reaction mechanism have been gained from theroretical studies. Numerous computational models have been developed to investigate as many as five different potential mechanisms for N-oxidation of aromatic amines.81–83 While a consensus remains to be reached, computational analyses support two mechanisms: a hydrogen atom transfer (HAT) mechanism (Path A, Scheme 3) and a proton transfer (PT) mechanism (Path B, Scheme 3).
Scheme 3.

Proposed mechanisms for cytochrome P450-catalyzed N-hydroxylation
Several published reviews delineate the catalytic cycle of P450 monooxygenases.31,32,80 To summarize, the reaction begins with a six-coordinate low-spin ferric heme with water or hydroxide as the distal ligand (18). Substrate binding displaces the bound water molecule, generating a five-coordinate high-spin ferric heme (19). This change in the heme spin state increases the redox potential and lowers the energy barrier for electron transfer from a reduced electron transfer protein to the ferric heme (19 → 20). The electron transport proteins, termed P450-reductases, utilize NADH or NADPH as an electron source. The different types of P450-reductases and the electron transfer mechanism have been described in a separate review.96 Once reduced, 20 is primed to bind oxygen, forming a ferric-superoxide complex (21). An additional electron then reduces 21 to the ferric-peroxo species (22), and at this stage the two proposed mechanisms for N–O bond formation diverge.
In the HAT mechanism (Path A, Scheme 3), 22 is first protonated to form a hydroperoxo-heme intermediate (23). An additional protonation step results in O–O bond cleavage to generate water and an oxo-ferryl porphyrin radical cation species commonly referred to as Compound I (24). Hydrogen abstraction from the amine by 24 leads to the formation of ferryl-hydroxo (25) and a nitrogen-centered radical intermediate. Finally, oxygen rebound forges the key N–O bond. Computational modeling by Schüürmann et al. recently suggested that this pathway is the most feasible route for ArNH2 hydroxylation.83 Furthermore, radical-based mechanisms for N–O bond formation have been previously proposed, but not supported with mechanistic studies, for nitroso formation by the di-copper enzyme NspF in ferroverdin biosynthesis (Section 2.5.1)97 and nitro formation by the di-iron enzyme CmlI in chloramphenicol biosynthesis (Section 2.6.1.1).98 For NOS, extensive electron paramagnetic resonance (EPR) and electron nuclear double resonance (ENDOR) studies have provided evidence that N–O bond formation proceeds through 24 (Section 2.4.1).87,85
In the PT mechanism (Path B), the substrate amine is deprotonated by 22 to form 23 and an anionic intermediate. The pKa of the amine proton of the arylamines tested range from −1.3 to 4.5.82 Nucleophilic attack of the distal hydroperoxo oxygen of 23 by the nitrogen atom of this anionic intermediate affords the corresponding hydroxylamine. This alternative mechanism has also been previously proposed for N–O bond formation by NOS based on inconsistent experimental evidence for the involvement of 24 as the active oxygenating species99 and ArNH2 hydroxylase modeling studies by Shamovsky et al.82 As mentioned above, whether N-oxygenation proceeds via radical or anionic pathway is currently unresolved for P450-dependent ArNH2 hydroxylases. Nonetheless, these potential mechanisms should provide adequate background knowledge for discussing heme-based N-hydroxylation and oxime installation (Section 2.2) in natural product biosynthesis.
P450-catalyzed N-hydroxylation appears in the biosynthesis of the enediyne natural product calicheamicin (6, Figure 4A). Calicheamicin is produced by Micromonospora echinospora ssp. calichensisis and exhibits potent antimicrobial and antitumor activities.100 Other natural products containing the unique enediyne warhead include esperamicin from Actinomadura verrucosopora101,102, neocarzinostatin from Streptomyces carzinostaticus F-41103, maduropeptin, kedaricidin, and dynemicin.104 6 induces DNA double strand breaks via initiation of trisulfide cleavage, followed by rearrangement of the enediyne to form a highly reactive diradical phenylene. Hydrogen atom abstraction from DNA nucleotides by the biradical induces for formation of a DNA double strand break.104–108 The unusual N–O glycosidic linkage in the disaccharide moieties of esperamicin and calicheamicin plays a pivotal role in orienting the two sugar residues for optimal binding to the DNA helix minor groove.5,109–111 In both antibiotics, the two sugars linked are 4-hydroxyamino-6-deoxy-α-D-glucose and 2,4,6,-trideoxy-4-methylthio-α-D-ribo-hexopyranose.112
Figure 4.

A) Structure of calicheamicin (6) B) N-hydroxylation reaction catalyzed by CalE10 and substrate scope of CalE10
The N-hydroxylase that generates intermediate TDP-4-hydroxyamino-6-deoxy-α-D-glucose (27, Figure 4B) was identified in M. echinospora by comparing the contents of various enediyne biosynthetic gene clusters.89,113–115 After excluding genes involved in forming the aminosugar, orsenillic acid, and enediyne core, cytochrome P450 CalE10 was identified as a potential candidate for N-oxidation and was the first hydroxylamino sugar-forming enzyme to be characterized in vitro.89,116 UV-spectroscopy of carbon monoxide (CO) bound CalE10 revealed the characteristic absorbance at 450nm. Using NADPH and the spinach ferredoxin/ferredoxin reductase (Fd/Fr) system, which is often used in in vitro studies to supply the heme iron with electrons from NADPH, CalE10 is able to convert TDP-4-amino-6-deoxy-α-D-glucose (26) to 27 in vitro as analyzed by liquid chromatography-mass spectrometry(LC-MS) and infrared(IR) spectroscopy (Figure 4B). Conversion of the aminosugar analogue 29 to the hydroxylated product was also observed with lower efficiency. The oxygenation reaction is selective for the 4-amino group, and CalE10 is able to limit overoxidation of the hydroxylamine to the corresponding nitrosugar (28) (less than 20% relative to the production of 27). Monitoring UV absorption changes upon ligand binding revealed that (26) is preferably bound compared to various sugar analogues tested (29–31). Further structural studies may provide insights into the factors contributing to substrate specificity and the mechanism by which CalE10 prevents overoxidation. Once the N-hydroxyl group is installed, CLM glycosyltransferase CalG2 transfers 27 to the thiosugar, forming an N–O linked disaccharide.117,118
Given the nucleophilicity of the nitrogen atom, it is perhaps surprising that a cytochrome P450 enzyme has evolved to hydroxylate 26. The amino acids and diamines encountered in Section 2.1.1 are sufficiently nucleophilic to attack the electrophilic distal oxygen atom of the key C4a-hydroperoxyflavin intermediate 16, breaking its O–O bond to afford an N-hydroxylamine product. Moreover, aminosugar N-oxygenases that will be discussed in Section 2.6.1.4 are flavin-dependent. Metalloenzymes access more reactive, high valent oxoiron intermediates for oxygenation reactions.119 The nitrogen lone pairs of the arylamines are less reactive because of conjugation with an aromatic ring π-system, potentially explaining why P450s, di-iron, and di-copper enzymes have evolved to catalyze N-oxygenation of such substrates to form ferroverdin (83), chloramphenicol (3), and aureothin (Section 2.5.1, 2.6.1.1). P450s that catalyze oxime formation using non-arylamine substrates have been characterized and will be discussed further in Section 2.2.
2.1.3. Unknown: L-Canavanine
L-canavanine (32, Scheme 4) is an L-arginine analogue produced by various species of the leguminous plant Canavalia.120 The compound was originally isolated for its ability to release high levels of ammonia and urea decomposition products, which led to the speculation that it played a key role in nitrogen storage.121–123 Its structural resemblance to L-arginine allows 32 to function as an antimetabolite. Replacement of the δ-carbon of L-arginine with an oxygen atom neutralizes the basic guanidinium moiety, lowering its pKa from 12.5 to 7.124 Incorporation of 32 into proteins instead of L-arginine results in altered electrostatic interactions that can lead to protein malfunction and eventually cellular toxicity.125 This amino acid has also been shown recently at high concentrations to be a precursor of cyanamide, an insecticide and fungicide.126
Scheme 4.

Proposed biosynthetic pathway for L-canavanine (32)
While the enzyme responsible for the key N–O bond formation in L-canavanine biosynthesis has not been isolated or characterized, extensive in vivo feeding experiments and biochemical assays with crude enzyme extracts have revealed several important biosynthetic intermediates and laid the groundwork for understanding this key reaction.127,128 The steps in the proposed pathway parallel those of L-arginine biosynthesis (Scheme 4). L-homoserine (33) undergoes O-amination to form L-canaline (34), which is converted to O-ureido-L-homoserine (35) by a carbamoyltransferase. 35 subsequently condenses with aspartic acid to form L-canavaninosuccinic acid (36), which then undergoes an elimination reaction catalyzed by an argininosuccinic acid lyase-like enzyme, generating 32 and fumarate.128
Crude enzyme extracts from the jack bean plant were able to convert [14C]-carbamoylphosphate and 34 to labeled 35 in vitro, and both 35 and 36 were converted to 32 after incubation with the same extracts..127 The involvement of these intermediates was further corroborated by in vivo feeding experiments with [14C]-carbamoyl phosphate and [14C]-35.128 However, labeled [14C]-33 did not label 32, nor were 34, 35, and 36 isolated from plant extracts. The specific enzymes involved in this pathway have not yet been identified in Canavalia enuiformis. Deciphering the enzymatic chemistry involved in constructing 34 would not only advance the current state of knowledge regarding biological N–O bond formation but also provide valuable insights into how plants accomplish an intriguing biosynthetic process that has not yet been observed in the microbial world.
2.2. Oxime
Oxime-containing secondary metabolites are rare, but such compounds usually display potent bioactivities. In a similar manner to hydroxylamines, an oxime group can alter a natural product’s acid-base properties, solubility, and target binding.24 Furthermore, the E/Z-geometry of an oxime can influence these features.129 Early biosynthetic studies of oxime-containing natural products focused largely on plant glucosinolates, a family of N-hydroximinosulfate ester-containing metabolites which have anticancer activity.130 The labile sulfated oxime readily rearranges to form electrophilic isothiocyanates, which can influence expression of cytoprotective proteins in mammalian cells by direct conjugation with nucleophilic thiol residues and alter transcription factor binding.131 Aside from plants, oxime natural products are also made by fungi and microbes derived from soil and marine sources.
While oxime formation in synthetic chemistry usually involves the condensation of hydroxylamine with either an aldehyde or ketone, biological routes to oximes employ a distinct logic that involves oxygenation of primary amine (Figure 5).26 This alternative disconnection is possible because of oxime-forming N-oxygenases’s ability to control this four-electron oxidation and avoid the formation nitroso or nitro compounds. The four-electron oxidation required to generate an oxime could arise from successive monooxygenation of the amine to form an N,N-dihydroxylamine intermediate before dehydration to yield the oxime or could result from dehydrogenation of a hydroxylamine. To date, the former mechanism involving an N,N-dihydrointermediate has been supported by experimental work investigating glucosinolate biosynthesis (Section 2.2.2.) and nitrone biosynthesis (Section 2.3.1.)
Figure 5.

Logic of oxime installation in organic synthesis versus biosynthesis
Similar to hydroxylamine biosynthesis, multiple enzyme classes catalyze oxime formation, including flavin-dependent monooxygenases, cytochrome P450s, and more recently a di-iron enzyme. In plants, a large family of cytochrome P450s termed CYP79s install oxime groups onto glucosinolate biosynthetic intermediates.132 The first microbial oxime-forming cytochrome P450 was discovered in the pathway that produces the β-lactam antibiotic nocardicin A (37).133 In recent years, flavin-dependent enzymes have also been shown to synthesize this functional group in the bacterial pathways that produce the bipyridine natural product caerulomycin A and its analogues (38, 39).133,134 Finally, an oxime forming di-iron enzyme has been recently identified in althiomycin (41) biosynthesis.135 Novel oxime-containing natural products continue to be isolated, including the stachylines from the sponge-derived fungus Stachylidium, brevioxime from Penicillium brevicompactum, Aspergillusol A from Aspergillus aculeatus, and vibralactoxime from the Basidiomycete Boreostereum vibrans.136–139 The biosynthetic pathways generating this latter group of compounds have not yet been elucidated and will not be included in the review, but these metabolites may represent interesting targets for future studies. The organization of this section will follow Section 2.1, with each subsection grouped by the type of N-oxygenase.
2.2.1. Flavin-dependent
The first oxime-forming, two-component flavin-dependent monooxygenase to be characterized in vitro participates in caerulomycin A (38, Figure 6) biosynthesis.134 The caerulomycins (CRM) were originally discovered in Streptomyces caeruleus in 1959140–143 and reisolated from Actinoalloteichus cyanogriseus WH1-2216-6 along with six new analogues.144 These antibiotics, together with collismycin A (39), the cyanogrisides, and pyrisulfoxin A (40), share a unique 2,2-bipyridyl ring system.145–147 The caerulomycins exhibit general antibiotic and immunosuppressive activities.144
Figure 6.

Structures of caerulomycin A (38) and related analogues
A 45 kb caerulomycin A biosynthetic gene cluster was identified by searching the A. cyanogriseus WH1-2216-6 genome for pyridine (Pyr) biosynthetic genes. Heterologous expression of this gene cluster in S. coelicolor resulted in production of 38, confirming its link to caerulomycin biosynthesis.148 The most likely candidate for N-oxygenation in this pathway was CrmH, a predicted two-component flavin monooxgenase with 30% sequence similarity to the isobutylamine hydroxylase VlmH in valanimycin biosynthesis (Section 3.1.1)134,149 Gene inactivation of crmH resulted in accumulation of the corresponding amine (42, Scheme 5) in culture extracts, suggesting that CrmH catalyzed oxime formation via N-oxidation of an amine precursor (42 → 38).134
Scheme 5.

Proposed reaction leading to the formation of oxime 43 by CrmH
Two-component flavin monooxygenases like CrmH require a separately encoded flavin reductase enzyme to generate the reduced flavin intermediate required for reaction with molecular oxygen. Protein-protein interactions between the flavin reductase and the monooxygenase can facilitate efficient transfer of the reduced flavin, thus preventing non-enzymatic oxidation of this redox sensitive species.150 Unexpectedly, the flavin reductase partner for CrmH is not present in the crm gene cluster. Instead, CrmH was capable of accepting reduced flavin from six different flavin reductases found in A. cyanogriseus WH1-2216-6, as well as E. coli flavin reductases SsuE and Fre. When the amine substrate (42) was incubated with purified CrmH, FAD, NADH, and Fre, both the E- and Z-oxime products (43ab) were detected alongside the aldehyde product of oxime hydrolysis (44).134 The Z-isomer was found to be unstable and quickly equilibrated to the more stable E-isomer. CrmH accepts both FMNH2 and FADH2 cofactors when catalyzing oxime formation in vitro, and similar flexibility has been observed in other two-component flavin monooxygenase systems.28
The hydroxylamine 45 was also observed in the CrmH in vitro assay. However, it rapidly transformed into oximes 43, aldehyde 44, and nitrone 46 upon purification. Since the rate of non-enzymatic oxidation was slow, the authors hypothesized that another hydroxylation event occurs on the nitrogen to form an N,N-dihydroxylamine intermediate (47). 47 can subsequently dehydrate to form 43 directly by removal of a benzylic proton and elimination of water (Path A) or via a nitroso intermediate (48) which can tautomerize to form 43 (Path B). Key residues for substrate and flavin binding were identified from homology modeling with other two-component oxygenases, and site-directed mutagenesis confirmed the residues’ importance for catalysis. Further mechanistic studies will be required to understand how CrmH avoids oxidation to the nitro group.
The biosynthesis of collismycin A (39), a structural analog of 38, has been characterized in vivo. The collismycins were isolated from Streptomyces sp. CS40 and displays antibacterial, antifungal, cytotoxic, and neuroprotective activities 151,152 The collismycin A biosynthetic gene cluster was identified using degenerate primers targeting the lysine aminotransferase required for picolinic acid biosynthesis.146,152,153 A flavin-dependent monooxygenase ClmM has been implicated in late-stage oxime formation because a clmM deletion mutant accumulated the corresponding N-acetylamine shunt product.154 In vitro characterization of ClmM has not yet been reported, but based on the similarities of the collismycin and caerulomycin structures and biosynthetic gene custers, ClmM is expected to display reactivity similar to CrmH.
Lastly, oxime-forming flavin-dependent enzymes have been shown to participate in the biosynthesis of ribosomally synthesized and post-translationally modified peptides (RiPPs). The oxime-containing RiPP azolemycin (49, Figure 7) and related analogues were isolated from Streptomyces sp. FXJ1.264 and display antiproliferative activity against human cancer cell lines.155 The azolemycin (azm) biosynthetic gene cluster was identified by searching the genome of the producing organism for the core peptide sequence VVSTCTI. The azm gene cluster encodes an enzyme (AzmF) that shares 65% amino acid sequence similarity with ClmM and is hypothesized to install the oxime group by oxidizing the N-terminal valine residue of the peptide. When azmF was knocked out, the mutant accumulated the corresponding core peptide with an amine in lieu of the oxime group as well as two proteolysed shunt products in extracts. This was the first report of oxime formation as a RiPP post-translational modification.
Figure 7.

Structure of the RiPPs azolemycin (49) and ustiloxin B (50)
More recently, an oxime intermediate was discovered in the biosynthetic pathway of the fungal RiPP ustiloxin B (50, Figure 7).156 The ustiloxins are phytotoxins originally isolated from the plant pathogen Ustilaginoidea virens, and the ust biosynthetic gene cluster was identified in Aspergillus flavus. This gene cluster encodes a class B flavin monooxygenase UstF2 that resembles CrmH (24% amino acid identity). When ustF2 was inactivated, the amino congener ustiloxin H (51, Scheme 6) was identified in the culture broth.157 Incubating 51 with purified UstF2 and NADH resulted in the formation of 52 as an E/Z-mixture (Scheme 6). Similarly to other oxime-forming flavin monooxygenases, the proposed mechanism for UstF2 involves successive oxidation of 51 to form a N,N-dihydroxylamine intermediate 53. Decarboxylative dehydration may lead to the formation of oxime 52. Alternatively, dehydrogenation of the two-electron hydroxylamine intermediate to form a primary nitroso species could be followed by decarboxylation to give 52. After 52 hydrolyzes to an aldehyde intermediate (54), a PLP-dependent enzyme UstD ligates aspartic acid and 54 to afford 50.
Scheme 6.

Involvement of an oxime intermediate in ustiloxin B (50) biosynthesis
2.2.2. P450-dependent
Some of the first oxime-forming enzymes characterized in vitro were the plant cytochrome P450s involved in glucosinolate biosynthesis. Glucosinolates are plant natural products with anticancer and insecticidal properties. 130–132,158 All glucosinolates share a similar architecture consisting of a β-pyranose sugar, an amino acid-derived side chain, and a sulfate group. Myrosinases nonspecifically hydrolyze the sugar moiety from glucosinolates, forming an aglycone that spontaneously degrades into various thiocyanate- and nitrile-containing bioactive products (Scheme 7A). Glucosinolate biosynthesis has been thoroughly studied and is discussed in detail in several reviews.130–132 An amino acid is first oxidized and decarboxylated by a cytochrome P450 to the corresponding aldoxime (55–60, Scheme 7B). This intermediate is then oxidized to a reactive aci-nitro or nitrile oxide species and conjugated with a sulfur donor to form an alkylthiohydroximate (61) by another cytochrome P450 enzyme. Subsequent glycosylation and sulfonation reactions yield the final glucosinolate scaffold, which can be further diversified by methylation, hydroxylation, oxidation, and desaturation. The CYP79 family of cytochrome P450s that catalyze the oxidation of amino acids to their corresponding aldoximes (55–60) will be discussed here.
Scheme 7.

A) Mechanism of action for glucosinolates. B) Aldoxime formation catalyzed by CYP79s
Glucosinolates can be classified as aliphatic, aromatic, or indolic based on the amino acid precursors used, and the cytochrome P450s catalyzing aldoxime formation usually display strict specificity for a single amino acid substrate. Phylogenetic analysis using the amino acid sequences of the CYP79 family and other plant cytochrome P450s involved in secondary metabolism suggests that the CYP79 family forms a unique clade. Cytochrome P450s can be distinguished from one another based on sequences found in the heme binding domain, PERF motif, K-helix and I-helix regions.30 The CYP79 family contains unique residues in these motifs, but how these differences translate to substrate specificity and reactivity is poorly understood.159
The proposed mechanism for CYP79-mediated aldoxime formation involves two successive hydroxylations of the amino group of the amino acid, followed by a concerted decarboxylation and dehydration step.160 This proposal is based on early experiments that demonstrated two equivalents of molecular oxygen are consumed during the conversion of L-tyrosine to the corresponding oxime using an oxygen electrode, which would be inconsistent with a dehydrogenation mechanism.161 The resulting aldoxime is usually isolated as a mixture of the E/Z-isomers. For example, CYP79F1 from Arabidopsis thaliana catalyzes the conversion of di- and tri-homomethionine to the respective aldoximes in aliphatic glucosinolate (55, Scheme 7B) biosynthesis.162 Knockout of this gene in Arabidopsis resulted in the abolishment of short chain aliphatic glucosinolate production.163 When E. coli spheroplasts overexpressing CYP79F1 were incubated with substrate and NADPH:cytochrome P450 reductase, both E- and Z-5-methylthio-pentanaldoxime and 6-methylthiohexanaldoxime were formed (55). CYP79F1 is specific for homoelongated methionines and did not accept L-methionine. CYP79F1 and a related cytochrome P450 CYP79F2 from Arabidopsis have been subsequently purified and shown in vitro that CYP79F1 catalyzes oxime formation for short- and long-chain elongated methionines whereas CYP79F2 exclusively hydroxylates long-chain derivatives.164
Characterized cytochrome P450s involved in indolic and aromatic glucosinolate biosynthesis include CYP79A1 (Sorghum bicolor), CYP79A2 (A. thaliana), and CYP79E1 (Triglochin maritima). CYP79A1 and CYP79E1 oxidize L-tyrosine to (E/Z)-p-hydroxyphenylacetaldoxime (56) in dhurrin and taxiphillin biosynthesis.160,161,165,166 No activity was observed when L-DOPA and L-phenylalanine were used as substrates.166 While the E-oxime was found to be the initial product of the reaction, equilibration to a mixture of E/Z-oxime occurred rapidly. CYP79A2 is involved in benzylglucosinolate biosynthesis in A. thaliana, and has no oxidative activity towards L-methionine, L-tryptophan, and L-tyrosine.167 CYP79B2 and CYP79B3 convert L-tryptophan to indole-3-acetaldoxime (58) and did not oxidize L-phenylalanine and L-tyrosine.168,169 CYPD1 and CYPD2 from Cassava (Manihot esculenta Crantz) catalyze the conversion of L-valine and L-isoleucine to first steps in the biosynthesis of the glucosides linamarin and lotaustralin, in which the aldoximes 59 and 60 are derived from respectively.170 Information about the structures of CYP79s would provide valuable insights into the basis for the strict substrate specificity. Further mechanistic work and biophysical characterization of the CYP79s would also elucidate whether N–O bond formation occurs via the radical or anionic pathway discussed in Section Section 2.1.2.
Oxime formation in nocardicin A (37, Scheme 8) biosynthesis uses a cytochrome P450 (NocL), which was the first microbial oxime-forming enzyme to be characterized. This β-lactam antibiotic was discovered in culture extracts of Nocardia uniformis subsp. tsuyamanesis in 1976.171–173 Structural characterization of 37 revealed an oxime moiety.174 Although 37 exhibits comparatively weak antimicrobial activity in comparison to other β-lactams, it is less susceptibile to cleavage by β-lactamases.171,175–178 The noc biosynthetic gene cluster encodes an NRPS that builds a β-lactam-containing tripeptide scaffold.179,180 The biosynthesis of 37 has been studied extensively, and a unique role for the NRPS in promoting β-lactam formation has been elucidated.181–184
Scheme 8.

Structure of nocardicin A (37) and proposed mechanism of the oxidation catalyzed by NocL
Early feeding experiments using 15N-labeled p-hydroxylphenylglycine showed that the oxime derives from N-oxidation of this building block.185 Cytochrome P450 NocL was hypothesized to be involved in this transformation, and gene inactivation supported this proposal as the amine precursor nocardicin C (62) was detected in the extract of the nocL mutant.186 NocL was able to catalyze conversion of 62 to 37 in vitro in the presence of NADPH and the spinach Fd/Fr reductase system. Oxime formation in this system is proposed to parallel aldoxime formation in glucosinolate biosynthesis. A four-electron oxidation of 62 involving two sequential monooxygenation reactions by NocL would form N,N-dihydroxylamine intermediate 63. Elimination of water, followed by tautomerization, would produce the oxime group.133 No detailed mechanistic experiments were performed to support this proposal, although the authors also proposed that the dehydrogenation reaction of the hydroxylamine would be a feasible, alternative route to oxime formation. NocL was unable to accept nocardicin G, which lacks the homoserine substituent, indicating that this structural motif might be important for substrate recognition. NocL shows low amino acid sequence identity to both nitric oxide synthase (<10% identity) and the CYP79 enzymes involved in glucosinolate production (~30% identity).
2.2.3. Non-heme di-iron
Non-heme di-iron enzymes also catalyze N-oxidation reactions. As extensive mechanistic experiments have been performed on di-iron enzymes that form nitro groups, a general introduction to this enzyme family is included in Section 2.6. An oxime-catalyzing di-iron enzyme was discovered in the althiomycin biosynthetic pathway. Althiomycin (41, Scheme 9) was originally isolated from Streptomyces althioticus and displays broad spectrum antibiotic activity.187,188 Additionally, 41 is also produced by Serratia marcescens Db10 and Myxococcus xanthus DK897. The geometry of the aldoxime group has been shown to affect the bioactivity significantly, with the Z-isomer exhibiting > 4-fold decrease in minimum inhibitory concentration against various Streptococcus strains compared to the E-isomer. 129,189 The alm gene cluster was recently identified in Myxococcus xanthus DK897 using a genome mining approach guided by the hypothesis that 41 is NRPS-derived.135
Scheme 9.

Proposed biosynthetic pathway for althiomycin (41). A, adenylation domain. T, thiolation domain
AlmD, originally annotated as a hypothetical protein in the kerritin-like family, was identified as a putative N-oxygenase based on sequence similarity to AurF and NorF from aureothin and neoaureothin biosynthesis (Section 2.6). Inactivation of almD resulted in complete abolishment of althiomycin production.135 Aligning the amino acid sequence of AlmD with those of AurF and NorF revealed two EX2H motifs potentially involved in binding a di-iron cofactor. While no in vitro characterization of AlmD has been performed, it has been proposed that AlmD installs the oxime on glycine (64 → 65) while the amino acid is still tethered to the NRPS because the almD mutant did not accumulate althiomycin derivatives lacking the oxime group. The alm gene cluster was also discovered in Serratia marcescens via transposon mutagenesis and screening for reduced killing of B. subtilis and S. aureus.190 Gene inactivation of the almD homolog from this gene cluster (alb2) also supported its role in oxime formation.
Compared with other oxime-containing nonribosomal peptides discussed in this review (ustiloxin, azolemycin, and nocardicin), oxime installation in althiomycin biosynthesis appears to occur at an early stage. Late-stage installation of this group would prevent premature hydrolysis during the biosynthetic pathway and possibly avoid formation of shunt products. If the hypothesis that oxime installation occurs on the glycine residue is substantiated with in vitro studies, this could provide an opportunity to understand how the althiomycin NRPS prevents hydrolysis of the oxime group during assembly line elongations and modifications.
2.3. Nitrone
Compounds containing the nitrone functional group have been explored as potential therapeutics due to their ability to trap radical intermediates implicated in oxidative stress (Scheme 10A).191–193 For example, the antiproliferative activity of avrainvillamide (66, Scheme 10B), a nitrone-containing alkaloid isolated from a strain of Aspergillus, involves the formation of stable covalent adducts between cysteine thiols in proteins and an electrophilic, conjugated nitrone.194,195 Notwithstanding the scarcity of natural products bearing the nitrone group, the enzymatic synthesis of this functionality has recently been investigated in the context of prenylated indole alkaloids (PIAs) biosynthesis. Specifically, a flavin-dependent N-oxygenase (OxaD) responsible for installing the nitrone group in roquefortine L (67), an intermediate in oxaline (68, Scheme 11) biosynthesis, has been characterized in vitro.
Scheme 10.

A) Trapping of radical intermediates by nitrones. B) Mechanism of action proposed for avrainvillamide (66)
Scheme 11.

The proposed biosynthetic pathway of oxaline (68) and nitrone formation catalyzed by OxaD
2.3.1. Flavin-dependent
Roquefortine C (69, Scheme 11) is a neurotoxic PIA originally isolated from Penicillium roqueforti196 and subsequently obtained from various other Penicillium strains.197–199 Early feeding experiments with radiolabeled 69 revealed this metabolite is a precursor to oxaline (68, Scheme 11), a related antiproliferative alkaloid produced by Penicillium oxalicum.200,201 68 contains a unique triazaspirocyclic scaffold which arises from a rearrangement of the diketopiperazine core of 69.202 Recently, the biosynthetic gene cluster that produces glandicoline (70) and meleagrin (71), demethylated precursors to 68, was identified in Penicillium chrysogenum.203 Specifically, the authors searched the genome for cyclodipeptide synthetase and prenyltransferase genes that could afford the prenylated diketopiperazine core of 69. Gene inactivation experiments revealed that the rearrangement reaction that forms the triazaspirocyclic skeleton proceeds via a nitrone intermediate, named roquefortine L (67).204 Flavin-dependent monooxygenase RoqM was identified as the enzyme responsible for nitrone installation because ΔroqM mutants did not produce 70 and 71, and instead accumulated 69. Furthermore, gene inactivation of roqO, which encodes a predicted P450 monooxygenase, suggested that this enzyme could catalyze a requisite oxidation at C16 on 67 to promote the rearrangement reaction to the triazaspirocycle (72 → 70).205
The RoqM homologue involved in oxaline biosynthesis (OxaD) was identified in Penicillium oxalicum and has been characterized in vitro.205 OxaD was purified with a high incorporation of FAD. Incubation of the enzyme with 69 and either NADH or NADPH resulted in formation of the nitrone product (67) along with the hydroxylamine congener (73) and a hydrated product (74). The conversion of 69 to 67 was shown to consume two equivalents of NAD(P)H, which is consistent with a four-election oxidation of an amine to a nitrone. When only one equivalent of reduced nicotinamide was used with respect to 69, the reaction yielded a 1:2 ratio of 73 to 67, supporting the hypothesis that nitrone formation proceeds via a two-electron oxidized hydroxylamine intermediate. Increased amounts of the hydroxylamine product were observed when excess starting material was utilized, indicating that N-oxidation to the nitrone proceeds iteratively with 73 formed initially. Encouragingly, OxaD was able to accept several halogenated analogues of 67, rendering this enzyme an attractive potential biocatalyst for accessing diverse nitrone-containing alkaloid scaffolds via engineered pathways.
2.4. Isoxazolidine
The isoxazolidines are a class of natural products often found in organic extracts of marine sponges, and these compounds often display potent anticancer activity.206,207 Very few biosynthetic studies have been performed on this class of compounds. Sponge-associated metabolites present difficult challenges for biosynthetic studies because many of the strains are uncultivatable, precluding in vivo studies. With the notable exception of D-cycloserine (2), most reports exploring the biosynthesis of microbial- and plant-derived isoxazolidine compounds have employed feeding experiments and in vitro assays with cell extracts.
2.4.1. D-Cycloserine
D-Cycloserine (2, Scheme 12A) is produced by Streptomyces lavendulae and Streptomyces garyphalus208, and is currently used clinically as an antibiotic to treat Mycobacterium tuberculosis infections.209 This natural product is an antimetabolite to D-alanine and inhibits D-ala-D-ala ligase, a critical enzyme in bacterial cell wall biosynthesis.2,210 It has been postulated that the isoxazolidine oxygen may form hydrogen bonds with active site residues, leading to competitive inhibition of D-alanine binding.211 The D-cycloserine (dcs) biosynthetic gene cluster was identified in Streptomyces lavendulae by proximity to a previously characterized self-resistance gene in Streptoymces garyphalus.212,213 Heterologous expression of this gene cluster in S. lividans and E. coli resulted in the production of 2.213,214
Scheme 12.

A) The biosynthetic pathway of D-cycloserine (2). B) Reaction catalyzed by nitric oxide synthase
Feeding experiments identified hydroxyurea (75) as a key biosynthetic intermediate.209 DcsB, annotated as an arginase, was demonstrated to hydrolyze N-hydroxyl-L-arginine (76) to generate 75 in vitro. 76 was originally proposed to be generated by nitric oxide synthase (NOS) because this compound is an intermediate generated by NOS en route to NO (Scheme 12B). However, gene inactivation of NOS in the producing organism did not disrupt production of 2. Surprisingly, disruption of dcsA, a gene encoding a protein of unknown function, completely abolished the biosynthesis of 2. Feeding 76 to ΔdcsA mutants restored production of D-cycloserine, supporting the involvement of 76 in biosynthesis and suggesting DcsA might be responsible for its synthesis. UV spectrophotometric studies on purified DcsA revealed that this enzyme contains a heme prosthetic group cofactor.215 Despite much effort, in vitro reconstitution of DcsA activity has not been successful. The authors hypothesized that the spinach Fd/Fr system they used to reduce the heme-iron was incompatible with the oxygenase. Therefore, the mechanism of N–O bond formation in the biosynthesis of 2 remains unclear.
As mentioned above, arginine N-hydroxylation to form 76 is not unique to DcsA. NOS, a heme-dependent enzyme found in both prokaryotic and eukaryotic cells, catalyzes the formation of nitric from arginine via a three-electron oxidation (Scheme 12B). The enzymology of NOS has been studied extensively because NO is an important signaling molecule in eukaryotes.216 The role of NO in microbes is still an active area of research, although recent research suggests that NO may play a role in protection against oxidative stress and providing a nitrogen oxide source for metabolite nitrations.84,217 In the first step of the reaction, NOS catalyzes the two-electron oxidation of arginine to form hydroxylarginine. A subsequent one-electron oxidation releases citrulline and NO. Oxygen activation by NOS occurs in a manner analogous to cytochrome P450s, and details for the overall nitric oxide forming mechanism have been highlighted in many published reviews.84,217 As mentioned in Section 2.1.2, N–O bond formation by NOS has been proposed to involve either radical or anionic mechanisms. Evidence for Compound I as the active oxygenating species from EPR and ENDOR studies, along with explanations of these biophysical methods, are presented in several manuscripts and review.85–87 Given that DscA and NOS both contain a heme-iron cofactor, it is conceivable that N-hydroxylation of L-arginine by DcsA may also proceed via one of these two mechanisms.
After 75 is generated by DcsB, a pyridoxal phosphate (PLP)-dependent enzyme (DcsD) catalyzes C–O bond formation between this intermediate and O-acetyl-L-serine to form the O-ureido-L-serine (L-isomer of 77).218 The PLP cofactor generates a key electrophilic aminoacrylate species that can accept a variety of nucleophiles via conjugate addition. DcsC then racemizes the L-isomer to form 77.214,219 Finally, DcsG catalyzes cyclization (77 → 2) to form the final isoxazolidine ring in an ATP-dependent manner while also hydrolyzing the urea moiety.220 The L-isomer of 77 was not accepted by DcsG. These enzyme activities have all been deomonstrated in vitro.
2.4.2. Acivicin, L-quisqualic acid, β-(isoxazolin-5-on-2-yl)-L-alanine
L-Acivicin (78, Figure 8A) is produced by Streptomyces sviceus, and as a result of its anticancer and antiparasitic properties it has received great interest as a therapeutic. Its target in cancer cells has been recently identified as ALDH4A1 using affinity chromatography with alkyne-modified 78.221 Extensive feeding experiments with isotopically enriched biosynthetic intermediates suggest that N-hydroxyl-L-ornithine is a key biosynthetic intermediate to the isoxazolidine ring, with the oxygen atom of this heterocycle deriving from molecular oxygen (Figure 8A).222,223 Two possible ring formation mechanisms have been proposed. The oxygen atom in the hydroxylamine may be acting as a nucleophile that attacks the β-carbon equipped with a leaving group. In this case, the nitrogen and oxygen atom would be incorporated intact to the final structure. On the other hand, functionalization of the amine with an oxygen atom can facilitate generation of an electrophilic intermediate, which could be attacked by a hydroxyl group installed on the β-carbon. This latter proposal would parallel the biosynthetic logic of N–N bond formation (Section 3.5). Further genetic and biochemical characterization will help resolve which strategy is used to construct the N–O bond in the isoxazolidine ring.
Figure 8.

A) Feeding experiments reveal the precursors to the N–O linkage found in L-acivicin (78). B): In vitro assays examining the biosynthesis of L-quisqualic acid (79) and BIA (76)
L-quisqualic acid (79) and the related metabolite p-(isoxazolin-5-on-2-yl)-L-alanine (BIA) (80) were isolated from Quisqualis indica and Latkyrus satiuus, respectively. These amino acids are known for their neuroexcitatory bioactivities.224,225 The biosyntheses of these compounds are largely unknown. Early characterization primarily focused on exploring the enzymatic synthesis of these molecules using cysteine synthase isoforms obtained from crude extracts of the producing plants. These cysteine synthase-like enzymes can ligate O-acetylserine to isoxazollin-5-one (81) to form 80 and 3,5-dioxo-1,2,4-oxadiazolidine (82) to form 79 (Figure 8B) in a manner similar to the formation of the O-ureido-L-serine intermediate mentioned in Section 2.4.1. However, further biochemical and genetic work will be needed to elucidate the origin of the isoxazolidine scaffolds and the biosynthetic enzymes involved in forming the key N–O bond.
2.5. Nitroso
Natural products containing a nitroso group are rare, as nitroso groups formed from primary and secondary amines often tautomerize to the corresponding oximes. In addition, overoxidation to the nitro group and dimerization to the azoxy compound often complicate efforts to synthesize and isolate nitroso compounds. As a result, C-nitroso groups in natural products are often attached to aromatic or tertiary carbon centers.226 For N-nitroso compounds (nitrosamines), the nitroso group is usually attached to a secondary amine as generation of primary nitrosamines leads to diazo formation.227 This section will focus on the assembly of C-nitroso compounds while the biosynthesis of N-nitroso natural products will be discussed in Section 3.6.
C-nitroso compounds function as metal chelators and nitric oxide donors. The ferroverdins, a class of aryl-nitroso compounds isolated from Streptomyces, are examples of the former. In the context of biosynthesis, C-nitroso compounds are also generated as intermediates in pathways that produce azoxy (Section 3.1) and nitro compounds (Section 2.6). While synthetic methods often construct C-nitrosamines via C–N bond formation using nitrosating agents, biological systems often employ the four-electron oxidation of amino groups.
2.5.1. Di-copper
The ferroverdins are a family of green pigments isolated from various Streptomyces species. For example, 4-hydroxy-3-nitrosobenzoate (deferroviridomycin) and the viridomycins are produced by Streptomyces viridans.228,229 These nitrosophenols are chelators used to acquire iron or cobalt by the bacteria, and some ferroverdins even exhibit weak antibiotic activities.230 Biosynthetic studies have been performed for 4-hydroxy-3-nitrosobenzamide (4,3-HNBAm, 83, Scheme 13A), a ferroverdin produced by Streptomyces murayamaensis.231 Degenerate primers targeting 3-amino-4-hydoxybenzoic acid (3,4-AHBA, 91) biosynthetic genes were used to identify a putative ferroverdin biosynthetic gene cluster. Heterologous expression in S. lividans linked the gene cluster to the production of 83. Within the ferroverdin gene cluster, NspF was identified as a putative N-oxygenase based on sequence homology to GriF (70% identity), a tyrosinase-like monooxygenase that oxidizes o-aminophenol (84) to o-quinone imine (85) in grixazone A (86) biosynthesis (Scheme 13B).232 The nsp gene cluster also contains a homolog to GriE (NspE), a transport protein that facilitates the incorporation of copper ions into the GriF monooxygenase active site.
Scheme 13.

A) Biosynthetic pathway of 4,3-HMBAm (83). B) Biosynthetic pathway of grixazone (86). C) Other substrates tested for NspF activity
Tyrosinases are di-copper monooxygenases that can catalyze the dehydrogenation of catechols to o-quinones (Scheme 14A) and hydroxylation of phenols to catechols. The ability of tyrosinases to perform both dehydrogenation and monooxygenation reactions are dictated by the amino acid residues in the active site environment, and detailed mechanistic insights are provided in published reviews.233–235 Six histidine residues are required for coordinating the binuclear copper ions, and these residues are conserved between NspF, GriF, and other characterized tyrosinases from Streptomyces.232 Similar to di-iron enzymes, a reduced copper site Cu2I/I is required for activating oxygen and generating the active oxygenating μ-η2:η2-peroxo-Cu2II/II complex (87 → 88, Scheme 14A).233 In the dehydrogenation mechanism, hydride transfer from the hydroxyl or amine group to the peroxo oxygen generates the quinone or quinone imine (88 → 89). While GriF displays the dehydrogenation reactivity of tyrosinases, the authors hypothesized that NspF may instead perform monooxygenation.
Scheme 14.

A) Proposed mechanism for the catecholase activity of tyrosinases. B) Proposed mechanism of C-nitrosation from Ref. 97
NspF prepared with excess CuSO4 was able to catalyze nitroso formation in vitro using the amide substrate 90 and without the need for NspE (Scheme 13A).231 Sequence alignment with known di-copper-containing enzymes like GriF and characterized tyrosinases revealed the six conserved histidine ligands. Site-directed mutagenesis experiments confirmed that two of the six conserved histidine residues are essential for copper binding and catalysis. Little activity was observed when the carboxylic acid 91 was used as substrate. Further studies of substrate scope revealed that NspE can catalyze nitroso-forming reactions using a variety of o-aminophenols (84,92,93 → 94–96, Scheme 13C), albeit with lower conversion rates. For these substrates, NspF also formed the corresponding non-enzymatic dimerization phenoxazinone products (97–99). While additional mechanistic studies will be required to understand this N-oxidation chemistry which is unique among di-copper enzymes, Dorrestein et al. proposed a mechanism involving radical recombination between a peroxo-Cu2II/II intermediate and the aniline substrate to form the hydroxylamine intermediate, followed by an additional two-electron oxidation and water release to form 83 (Scheme 14B).97 Given the high sequence similarities between NspF and GriF, structural comparisons between the two enzymes could potentially provide new and exciting insights into how di-copper enzymes control N-oxidation, accomplishing either dehydrogenation to quinone imine or monooxygenation to form N-oxygenated compounds.
2.6. Nitro
The nitro functional group is an integral part of important therapeutics, however many compounds containing this structural feature are toxic and highly prevalent within the environment.236 Therefore, a large body of research exists pertaining to the biology and chemistry of nitro-containing compounds, which includes both synthetic- and naturally-derived molecules.236–244 Natural products containing a nitro group comprise a family of greater than 200 members and exhibit a wide range of structural diversity.238 These compounds display varied bioactivities including antibacterial, antifungal, and antiproliferative effects.
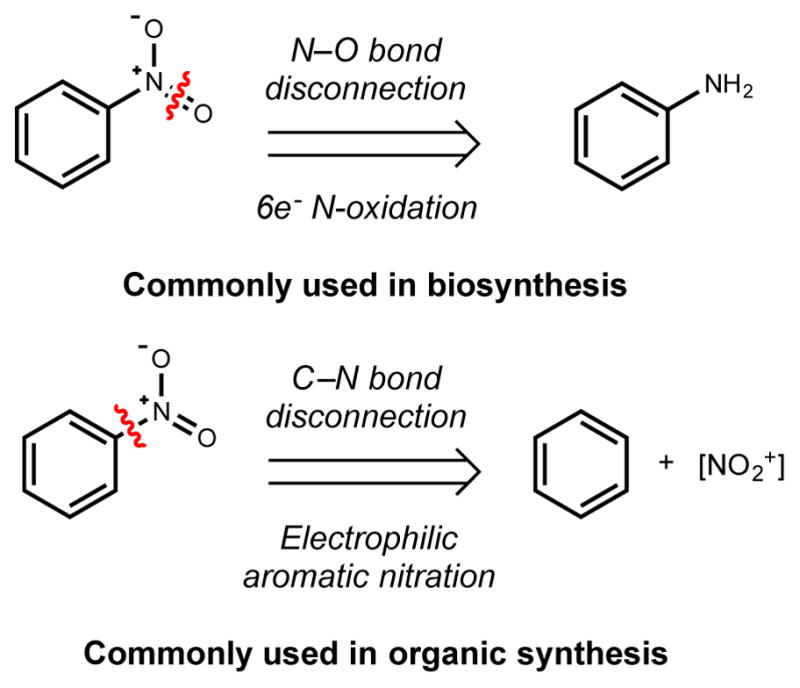
Synthetic nitro compounds derive from both the incomplete combustion of fossil fuels and numerous industrial processes, leading to their great abundance in the environment – over five metric tons of nitrobenzene and 1.1 metric tons of 2,4-dinitrotoluene were released into the soil in the United States in 2002 alone.243,245 Synthetic methods for installing nitro groups include the six-electron oxidation of amines and the direct nitration of aromatic compounds via electrophilic aromatic substitution, among other recently developed methods (Figure 9).237 Electrophilic aromatic nitration is by far the more common method, especially on industrial scale, as the generation of oxidized by-products is less problematic compared with the oxidation of amines. Unfortunately, direct nitration is a hazardous and environmentally-unfriendly reaction as it uses large excesses of nitric and sulfuric acids.237,246
Figure 9.
Logic of nitro group installation in organic synthesis versus biosynthesis
Due to their relevance to human health and the environment, the biosynthesis, degradation, and bioactivation of nitro compounds has been extensively studied.240,243,244,247,248 An understanding of the biosynthetic pathways that install nitro groups could provide enzymatic routes to this functional group that may be attractive alternatives to current synthetic methods. The biosynthesis of nitro-containing natural products has been the subject of several excellent reviews.110,238,239 Over the last decade, researchers have obtained deeper mechanistic insights into known nitro group biosynthetic enzymes and uncovered additional pathways for installing this functional group.
Here we provide an update of our current understanding of nitro group biosynthesis. Current experimental evidence reveals two main strategies for nitro group construction that parallel methods used in synthetic chemistry: oxidation of amines and direct nitration of aromatic scaffolds (Figure 9). While direct nitration involves C–N bond formation, generation of the active nitrating species involves N–O bond formation and thus will be discussed briefly here. As observed for the biosynthesis of other N–O bond containing functional groups, nitro group formation can be catalyzed by several distinct classes of enzymes. The following section is divided by the type of biosynthetic logic used in nitro group formation (N-oxygenation vs. electrophilic nitration) and further subdivided according to enzyme class.
2.6.1. N-oxygenation
Our current understanding of nitro group biosynthesis points toward the six-electron N-oxidation of amines as the most common strategy used for the construction of this functional group. Before nitro group-forming enzymes were discovered, the isolation of amine congeners alongside nitro group-containing products provided a strong clue that N-oxidation was involved. At present, non-heme di-iron oxygenases, Rieske non-heme mononuclear iron oxygenases, flavin-dependent monooxygenases, and cytochrome P450s are all known to oxidize amines to nitro groups. Additionally, an unknown N-oxygenase enzyme is believed to participate in 3-nitropropanoic acid biosynthesis. Access to multiple different enzyme classes capable of nitro group formation should prove useful in biocatalysis and synthetic biology applications.
2.6.1.1. Non-heme di-iron
Oxidoreductases containing a non-heme di-iron metallocofactor catalyze a diverse range of challenging oxidative reactions, including hydroxylation of unactivated carbon centers, desaturation of alkanes, N-oxygenation, and epoxidation of alkenes (Scheme 15).249,250 These enzymes utilize an oxygen-bridged di-iron cofactor, with each iron subsite ligated by histidine and acidic residues (aspartate and glutamate), to activate molecular oxygen and affect subsequent oxidation reactions. Oxygen activation occurs upon oxidative addition of O2 to the Fe2II/II center with the di-iron center delivering two electrons to O2 to generate a bridging, μ-(hydro)peroxo-Fe2III/III species. Interestingly, spectroscopic characterization of this intermediate in several systems has revealed structural differences that may be important for catalyzing these diverse oxidative transformations.250 In some cases, this μ-(hydro)peroxo-Fe2III/III species has been shown to be a competent oxidation catalyst, however, in other systems this intermediate is further transformed to high-valent iron-oxo species, including the Fe2IV/IV complex of soluble methane monooxygnease. Both types of intermediates subsequently catalyze oxidation of the substrate, completing a net four-electron reduction of O2 in which two electrons came from the substrate and two from the initial Fe2II/II center.
Scheme 15.

Proposed peroxo-Fe2III/III and bis-μ-oxo Fe2IV/IV species involved in diverse reactions catalyzed by non-heme di-iron enzymes. We show the peroxo-Fe2 III/III species as having a μ-1,2 bridging mode, although additional binding modes have been suggested
The reaction with substrate typically generates a stable Fe2III/III species. Thus, to complete the catalytic cyclic and regenerate the oxygen-reactive Fe2II/II species, two external electrons must be provided. The electron delivery system varies between enzymes, but generally uses reduced nicotinamide cofactors as the external electron source. Recently, characterization of the non-heme di-iron nitro-forming N-oxygenases AurF and CmlI (Section 2.6.1.1) has suggested two novel mechanisms that differ from typical di-iron oxidoreductases in terms of their reaction stoichiometry and requirement for external electrons. In depth biochemical, spectroscopic, and structural investigations of these enzymes have provided the most detailed insights of nitro group biosynthesis obtained to date.
The nitro-forming enzyme AurF, which participates in aureothin (100) biosynthesis, was the first non-heme di-iron N-oxygenase to be discovered.251,252 Aureothin is a polyketide natural product isolated in 1953 from Streptomyces thioluteus that exhibits antitumor, antifungal and insecticidal properties.253–255 Feeding studies confirmed its polyketide origin and suggested that the nitro group likely derived from the oxidation of p-aminobenzoic acid (PABA, 101) to p-nitrobenzoic acid (PNBA, 102) (Scheme 16).256–260 Subsequent discovery of the aur gene cluster revealed the presence of PABA synthase genes, but no candidate N-oxygenase was identified.261 Expression of the aureothin (aur) gene cluster in S. lividans ZX1 and systematic gene deletion experiments demonstrated that AurF was responsible for generating PNBA.252 Conversion of 101 to 102 could also be reconstituted in vivo in S. lividans ZX1 and E. coli when AurF was overexpressed, further supporting its role as the nitro-forming enzyme.252,262 This enzyme represented a novel type of nitro-forming N-oxygenase and was only the second enzyme after PrnD (Section 2.6.1.2) identified to catalyze nitro formation via amine oxidation.
Scheme 16.
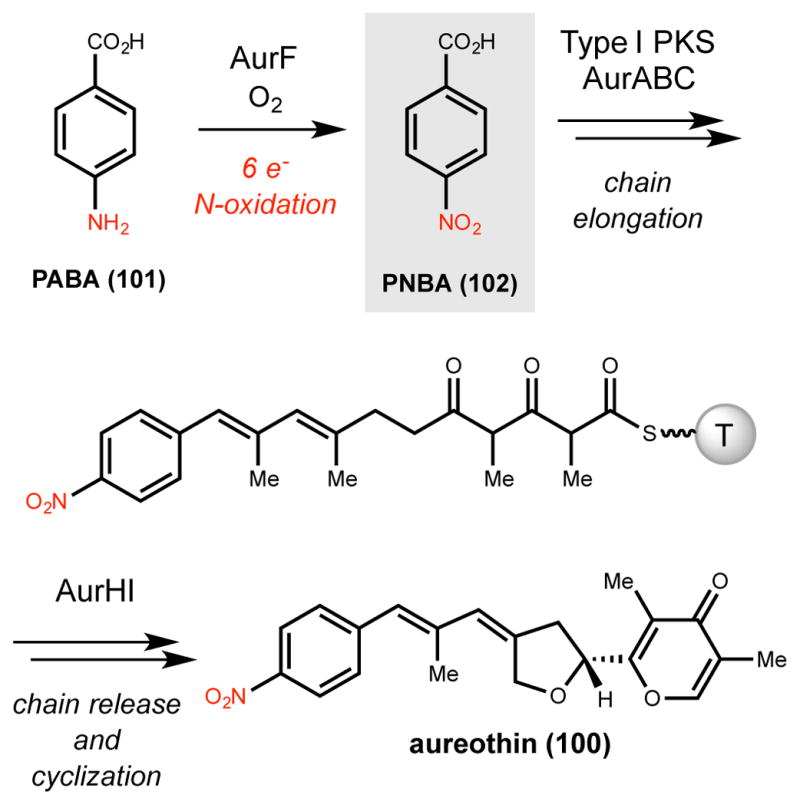
Oxidation of PABA to PNBA by AurF in the biosynthesis of aureothin. T = thiolation domain
After this initial discovery, many subsequent studies aimed at characterizing the mechanism and structure of AurF appeared, with contributions from the Hertweck, Schulz, Zhao, Krebs, and Bollinger groups.251,262–271 In particular, the nature of the binuclear metallocofactor and the mechanism of oxidation were debated. An initial analysis of the AurF sequence revealed two copies of an EX28–37 DEXXH motif that is conserved among several non-heme di-iron oxygenases.262 The first experimental evidence for a di-iron active site came from inductively coupled plasma mass spectrometry (ICP-MS) atomic emission spectrometric analysis of inactive AurF purified from E. coli.262 This analysis showed an average iron-to-enzyme ratio of 2.18, which is in close agreement with the expected value for a di-iron enzyme. Subsequently, additional proposals were put forth claiming AurF contained a di-manganese or an iron-manganese cofactor.263–265,269 More recently, several avenues of investigation conclusively demonstrated that AurF is a di-iron containing enzyme.266–268 When E. coli was supplemented with various ratios of iron and manganese, the relative in vitro activity of purified AurF decreased as the manganese content of its cofactor increased.266 Only 5.8% relative activity was seen with AurF preparations containing a 0.1:1.9 Fe:Mn ratio per AurF monomer compared with 100% relative activity for preparations containing a 2.2:0.0 Fe:Mn ratio per AurF monomer. Nitro-forming activity with di-iron AurF could be reconstituted in vitro using both a chemical reductant system consisting of ascorbate and phenazine methosulfate, as well as an enzyme-based system involving NADPH and a Fd/Fr reductase system from Anabaena sp. PCC 7119. The specific activity seen in vitro with di-iron AurF is 40–50× higher than that reported for dimanganese AurF.263,265,266
Crystal structures of AurF reconstituted both with a di-manganese and di-iron metallocofactor exhibit significant differences in the active site architectures that are consistent with the highly attenuated activity of Mn-reconstituted AurF.264,266 The active site of di-iron AurF is located within the core of a four helix bundle and is bound by four glutamate and three histidine residues (Figure 10). While global structure features are conserved between the two structures, di-manganese AurF contains a smaller, more collapsed active site that may prevent binding of 101, leading to the difference in observed activity.266
Figure 10.

A) Proposed geometric arrangements of the peroxo-Fe2III/III species in several di-iron enzymes and AurF. B) Coo1 rdination sphere of the μ-oxo-Fe2III/III species from the crystal structure of AurF
Several possible mechanisms have been put forth for the AurF-catalyzed six-electron oxidation of 101 to 102 (Scheme 17). Formation of the two-electron oxidized product p-hydroxyaminobenzoic acid (103) has been detected in vitro and in vivo. While the four-electron oxidized product p-nitrosobenzoic acid (104) has been detected in vitro, the assertion that its formation is enzyme-catalyzed has been challenged.251,266,267 These compounds are potential intermediates in several of the proposed mechanisms. The first two possibilities described both invoke three sequential two-electron oxidations, but the transformations involved differ (Scheme 17A). The first proposed mechanism (Path A) employs three sequential monooxygenation reactions that each oxidize the substrate by two electrons and require an equivalent of molecular oxygen.251 PABA is first oxidized to 103 and then hydroxylated again to afford the N,N-dihydroxylated intermediate 105, which undergoes dehydration to generate 104. This nitroso-containing intermediate 104 is subsequently hydroxylated to give PNBA. A second mechanism (Path B) was later proposed based upon studies of an unrelated nitro-forming enzyme, the Rieske non-heme mononuclear iron oxygenase PrnD (Section 2.6.1.2).262 Path B differs from Path A in that it involves two monoxygenation reactions and one dehydrogenation. An initial monooxygenation would still convert PABA to 103, but dehydrogenation would provide 104 directly. A final hydroxylation step would then afford PNBA.
Scheme 17.

A) Proposed mechanisms and B) reaction stoichiometries of the AurF-catalyzed oxidation of PABA (101) to PNBA (102)
The results of 18O2 labeling experiments argued against Path A as the route for nitro group formation.262 When unlabeled 103 was added to AurF-expressing cells grown under an atmosphere of 18O2 the resulting PNBA showed incorporation of only one heavy atom of oxygen. These results suggested that only one monooxygenation reaction had occurred and supported a route involving a dehydrogenation step. If AurF oxidized 103 via Path A, the resulting PNBA would be expected to be a mixture containing one and two incorporations of 18O, unless AurF dehydrates dihydroxy intermediate 105 selectively to provide a single isotopologue of PNBA.
More recently, evidence from in vitro enzyme assays and spectroscopic studies has supported a different mechanism (Path C) involving a distinct stoichiometry (Scheme 17).267 The spectroscopic analyses also revealed an unusual peroxo di-iron species involved in N-oxygenation. 267,268 Analysis of a mixture containing reduced Fe2II/II AurF and O2 by UV/Vis and Mössbauer spectroscopy revealed an exceptionally long-lived intermediate (t1/2 ~ 7 min at 20 °C) proposed to be a peroxo-Fe2III/III species.268 Based on spectral comparisons the geometry of this peroxo-Fe2III/III species (106) likely differs from the more canonical cis-μ-1,2(μ-η1:η1)-peroxo arrangement proposed in several related enzymes, including ribonucleotide reductase subunit R2 and stearoyl acyl carrier protein Δ9 desaturase (Figure 10). It has been proposed that 106 adopts a μ-1,1-hydroperoxo arrangement, which may activate the distal oxygen atom for attack by PABA.268 Addition of PABA or PHABA to assay mixtures containing 106 and excess oxygen resulted in rapid decay to a proposed μ-oxo-Fe2III/III species (107) suggesting that peroxo-Fe2III/III AurF is involved in oxidizing these substrates.267,268 Interestingly, when ~ 0.3 equivalents of PABA was added relative to peroxo-Fe2III/III AurF in this experiment, greater than 80% conversion to PNBA was achieved, suggesting that the peroxo-Fe2III/III is competent for the full oxidation of PABA to PNBA.268
Further insights into the reaction stoichiometry and mechanism were gained from in vitro experiments in which the relative amounts of substrate, O2, and AurF were precisely controlled.267 When assay mixtures contained the intermediate 103, O2 and Fe2II/II AurF at a ratio of 1:2:0.03, respectively, greater than 95% conversion to PNBA was observed. This unexpected result demonstrated that AurF could catalyze the four-electron oxidation of 103 to PNBA without the input of any exogenous reducing equivalents, suggesting that 103 provides all four of the electrons required to reduce O2. Further evidence for this reaction stoichiometry was obtained when 103, O2 and Fe2II/II AurF were mixed in a ratio of 1:1:1 and analyzed by UV/Vis absorption over time. Initial formation of the oxidizing peroxo-Fe2III/III species (formed from Fe2II/II reaction with O2) was followed by rapid decay to regenerate the fully reduced Fe2II/II species, which is needed to initiate another catalytic cycle. This experiment suggests that only one molecule of O2 is required to oxidize 103 to PNBA.
Taken together these results support a mechanism for AurF that is very different than those proposed previously (Scheme 17).251,262,267 Oxidation of PABA to PNBA begins with the two-electron oxidation of PABA to 103, consistent with earlier proposals. This monooxygenation reaction is proposed to be catalyzed by the μ-1,1-peroxo-Fe2III/III species via nucleophilic attack of the aniline nitrogen of PABA onto the distal oxygen atom which generates the stable μ-oxo-Fe2III/III intermediate (Scheme 18A). Reduction of the μ-oxo-Fe2III/III species by two external electrons regenerates reduced Fe2II/II AurF. This reduced intermediate is proposed to react with a second equivalent of O2 to again afford the μ-1,1-peroxo-Fe2III/III species, which subsequently catalyzes the full four-electron oxidation of 103 to PNBA without requirement for any exogenous reducing equivalents (Scheme 18B). The conversion of 103 to PNBA is proposed to occur via initial nucleophilic attack of 103 onto the μ-1,1-peroxo-Fe2III/III species to afford 105, followed by a formal dehydrogenation to give PNBA and reduced Fe2II/II AurF. 267 Thus, this proposed mechanism requires only two molecules of O2 and two external electrons to oxidize PABA to PNBA (Scheme 17).251,262 Moreover, this proposed catalytic cycle sets AurF apart from other non-heme di-iron oxidases due to the ability of the substrate to provide all four electrons to reduce one of the equivalents of O2, thus regenerating reduced Fe2II/II rather than an oxidized Fe2III/III species (Scheme 19A).250 This potential mechanism is consistent with earlier 18O2 labeling studies in which only one 18O atom was incorporated in conversion of 103 to PNBA.262
Scheme 18.

A) Proposed mechanism for the AurF-mediated N-hydroxylation of PABA to 103. B) Proposed mechanism for the AurF-mediated four-electron of oxidation of 103 to PNBA
Scheme 19.

Proposed catalytic cycles for the six-electron N-oxidation of amines to nitro groups by A) AurF and B) CmlI
Since the initial characterization of AurF, a related non-heme di-iron N-oxygenase has been discovered to generate a nitro group via oxidation of an amine in chloramphenicol biosynthesis. Chloramphenicol (3) is a nonribosomal peptide antibiotic isolated from Streptomyces venezuelae ISP5230 that targets both Gram-positive and Gram-negative bacteria by inhibiting protein synthesis.272–276 Chloramphenicol biosynthesis has been extensively studied due to its important biological activity and unusual structural features.3,277–281 As with aureothin, feeding studies provided initial evidence that the nitro group was installed via N-oxidation, as 4-amino-L-phenylalanine (108) was shown to be an intermediate.277,278 Identification of the chloramphenicol (cml) gene cluster enabled the biochemical characterization of several biosynthetic enzymes and a proposed order of events (Scheme 20).282–287 This gene cluster encodes a homolog of AurF, CmlI (34% amino acid identity). Subsequent characterization in vitro and in vivo in a heterologous expression system demonstrated that CmlI could convert the amino precursor, NH2-Cam (109), to chloramphenicol, confirming its role as a nitro-forming N-oxygenase and suggesting that nitro group installation occurs as the last step in the biosynthetic pathway following product release from the NRPS assembly line.288 Notably, this proposed timing of nitro group formation differs substantially from that observed in aureothin biosynthesis, which uses a nitro group-containing building block to initiate a PKS assembly line.
Scheme 20.

Proposed pathway for chloramphenicol biosynthesis. A, adenylation domain; T, thiolation domain; R, reductase domain
Very recently, biochemical, spectroscopic, and structural investigations of CmlI have provided insights into the mechanism of N-oxygenation and revealed the involvement of a peroxo-Fe2III/III intermediate in catalysis.98,289,290 Optical, EPR, Mössbauer, and structural studies have demonstrated conclusively that CmlI, like AurF, contains a di-iron metallocofactor. Also similar to AurF, a combination of UV/Vis, Mössbauer, and resonance Raman spectroscopies have showed that the reaction of reduced Fe2II/II ClmI with O2 generates a peroxo-Fe2III/III species (110) that has spectral properties distinct from those previously characterized in other non-heme di-iron enzymes.98 Based on these unique spectral features it has been proposed that the peroxo-Fe2III/III intermediate of CmlI does not exhibit the canonical cis-μ-1,2(μ-η1:η1)-peroxo geometry usually found in this enzyme family nor the μ-1,1-peroxo arrangement recently proposed for AurF. Instead, it has been suggested that the intermediate adopts a novel μ-η1:η2-peroxo (110) arrangement (Figure 11A).267,268
Figure 11.

A) Proposed geometric arrangements of the peroxo-Fe2III/III species in several di-iron enzymes and CmlI. B) Coordination sphere of the μ-oxo-Fe2III/III species from the crystal structure of CmlI
Similar to the AurF peroxo-Fe2III/III intermediate, the CmlI peroxo-Fe2III/III is very long-lived (t1/2 = 3 h at 4 °C), and rapidly decays upon addition of substrate 109, demonstrating its competence as an N-oxygenating species. A series of detailed 18O2 in vitro labeling and spectroscopic experiments with CmlI have provided insights into the mechanism and reaction stoichiometry of the six-electron oxidation of 109 to chloramphenicol.290 Interestingly, while the overall reaction stoichiometry is the same as that of AurF, the proposed mechanisms are different. When 109 is incubated with CmlI peroxo-Fe2III/III chloramphenicol is produced. Incubation of 109 (0.3 or 1.0 equivalents) with 18O2-peroxo-Fe2III/III under an atmosphere of 16O2 resulted in production of 50% doubly-labeled and ~40% singly-labeled chloramphenicol. This result has important implications for both the reaction mechanism and stoichiometry. Doubly-labeled chloramphenicol results from successive oxygenations of 109 by 18O2-peroxo-Fe2III/III. The presence of singly labeled chloramphenicol suggests that during the course of nitro formation, a 16O2-peroxo-Fe2III/III intermediate is formed from a reduced Fe2II/II CmlI species and atmospheric 16O2 which subsequently oxygenates a pathway intermediate. Importantly, because no external electron source was provided in the in vitro reaction the oxygen-reactive, reduced Fe2II/II species is presumably generated via reduction of the oxidized μ-oxo-Fe2III/III intermediate with electrons from a substrate-derived intermediate. Other explanations for 16O incorporation, including label exchange of 18O2-peroxo-Fe2III/III with H216O or 16O2 were ruled out experimentally. Thus, this 18O2 labeling experiment suggests two important features of the CmlI-catalyzed reaction: (1) the peroxo-Fe2III/III intermediate is involved in two separate oxygenation reactions, and (2) substrate-derived electrons reduce an oxidized di-iron intermediate to provide reduced Fe2II/II for subsequent oxygenation reactions.
Details of these two aspects of CmlI catalysis were revealed upon incubation of various substrates with oxidized di-iron intermediates and analysis of reaction products. The oxidation of 109 to chloramphenicol is proposed to begin with an initial monooxygenation of 109 to the N-hydroxy intermediate, NH(OH)-Cam (111), although its short lifetime in the catalytic cycle has prevented its detection.98,290 Incubation of a 10-fold excess of the presumed intermediate 111 with μ-oxo-Fe2III/III CmlI under aerobic conditions resulted in the formation of primarily chloramphenicol (5.7 equivalents), in addition to the nitroso product, NO-Cam (112). Thus, the μ-oxo-Fe2III/III species is capable of catalytic oxidation of 111 to chloramphenicol without the requirement for an external source of electrons. This result can be explained if the oxidation of 111 to 112 provides the two electrons required to convert μ-oxo-Fe2III/III to the reduced Fe2II/II species. The reduced Fe2II/II CmlI coud then react with molecular oxygen to produce the peroxo-Fe2III/III intermediate which oxygenates 112 to chloramphenicol and regenerates the μ-oxo-Fe2III/III species. This proposal was supported by reaction monitoring via UV-Vis spectroscopy, which identified the various oxidized and reduced di-iron intermediates, and the ability of peroxo-Fe2III/III to oxidize 112 to chloramphenicol in vitro.
Taken together these experiments support the proposed catalytic cycle for CmlI N-oxygenation shown in Scheme 19B. The resting, oxidized μ-oxo-Fe2III/III species is reduced by two externally-supplied electrons to provide the oxygen-reactive Fe2II/II species. This reduced intermediate reacts with O2 to provide the non-canonical μ-η1:η2-peroxo-Fe2III/III intermediate which catalyzes oxygenation of 109 to the N-hydroxy intermediate 111 and generates the μ-oxo-Fe2III/III intermediate. From here, CmlI mediates the catalytic four-electron oxidation of 111 to chloramphenicol without the input of external electrons as discussed above, thus returning it to its resting μ-oxo-Fe2III/III state. An important feature of this four-electron oxidation is the reduction of the intermediate μ-oxo-Fe2III/III by the 111 to provide reduced Fe2II/II CmlI. Based on the unusual μ-η1:η2-peroxo-Fe2III/III intermediate, a radical-based mechanism for the initial two-electron oxidation of 109 to 111 has been put forth (Scheme 21).98 Notably, this proposal is different than the nucleophilic mechanism proposed to convert PABA to PHABA by AurF.268
Scheme 21.

Proposed radical-based mechanism for the CmlI-mediated N-hydroxylation of 109 to 111
Two crystal structures of CmlI have been obtained, one in the reduced Fe2II/II state and one in the oxidized peroxo-Fe2III/III state.289 Like AurF, the di-iron active site is located at the core of a four-helix bundle and is ligated by three histidine and four glutamate residues (Figure 11B). Surprisingly, the crystal structure of peroxo-Fe2III/III CmlI appeared to show the canonical cis-μ-1,2(μ-η1:η1)-peroxo ligand geometry for the di-iron cofactor. This is in contrast to the μ-η1:η2-peroxo geometry suggested by spectroscopic studies. Furthermore, the crystal structure of peroxo-Fe2III/III CmlI revealed an active site that resembles di-manganase reconstituted AurF in terms of its size and accessibility.264,289 It has been proposed that the smaller, closed active site of di-manganese reconstituted AurF cannot accommodate substrate, leading to a reduction in activity.266 Alternatively, it has been proposed that CmlI activity may be regulated at the structural level by this “closed” state. Unknown events may trigger structural changes that open the active site of peroxo-Fe2III/III CmlI, allowing for substrate binding and simultaneously drive conversion of the putatively inactive cis-μ-1,2(μ-η1:η1)-peroxo form to the active oxidizing μ-η1:η2-peroxo state.289
Despite the similarities in the sequences of CmlI and AurF, characterization of these enzymes has revealed striking differences not only with other non-heme di-iron oxidoreductases, but also between these two nitro-forming enzymes. The overall stoichiometry proposed for ClmI and AurF is identical, with both enzymes requiring the input of only two external electrons and two equivalents of molecular oxygen to catalyze a net six-electron oxidation. This stoichiometry results from the remarkable ability of AurF and CmlI to catalyze the reduction of one molecule of O2 using four electrons derived from a pathway intermediate. This unusual feature has not been seen in other non-heme di-iron enzymes, which require external electrons to mediate two-electron oxidations of their substrates.250 This unusual feature may have evolved to prevent the release of reactive N-hydroxyl and nitroso intermediates that could damage the cell.290
Despite many similarities, the mechanisms proposed for AurF and CmlI differ in their proposed substrate-based intermediates, the pathway intermediate that reduces the μ-oxo-Fe2III/III species, and the oxidation state of the di-iron center after a single turnover. Currently, the reason for these discrepancies remains obscure, although it may have to do with the ability of N-hydroxy intermediates 103 and 111 to react with both peroxo-Fe2III/III (AurF) and μ-oxo-Fe2III/III (CmlI) species which has led both of these reactions to be considered on-pathway.290 Assessing the contributions of these two reactions to overall product formation will be needed to establish a consensus mechanism for this class of N-oxygenases.
The unusual μ-η1:η2-peroxo or μ-1,1-hydroperoxo geometries proposed for CmlI and AurF, respectively, set these enzymes apart from other non-heme di-iron enzymes. These peroxo-Fe2III/III species may be uniquely suited to catalyzing the six-electron oxidation of an amine to a nitro group, a transformation involving several distinct intermediates.98,267,268,290 The non-canonical peroxo-Fe2III/III of CmlI has been proposed to be amphiphilic, acting as a more electrophilic species in oxygenation of 109 and a more nucleophilic species when oxidizing 112.290 Additionally, it has been suggested that the active site of CmlI may prevent protonation of oxidized di-iron intermediates, a step required to generate the high-valent oxo and bis-μ-oxo species involved in hydrocarbon oxidation.290 This may provide a strategy to prevent unwanted side-reactivity. The inability of CmlI to oxidize hydrocarbons is consistent with this proposal.98 Further characterization of these two enzymes, as well as additional non-heme di-iron N-oxygenases from this family, should provide additional insight into mechanism of this reaction and the utilization of the unusual peroxo-Fe2III/III.
2.6.1.2. Rieske non-heme mononuclear iron
Rieske non-heme mononuclear iron oxygenases perform challenging oxidative reactions in the context of xenobiotic degradation and natural product biosynthesis.291 They are members of the larger non-heme mononuclear iron enzyme family which catalyze a diverse array of oxidative reactions.292 Members of this large family use a single iron atom bound by the canonical 2-His-1-carboxylate facial triad to activate molecular oxygen and affect oxygenation.293 The Rieske-containing subfamily contain two metal centers: a Rieske [2Fe-2S] cluster, which is bound by the conserved sequence motif CXH17CX2H, and a mononuclear iron site anchored by the 2-His-1-carboxylate facial triad (Figure 12). The Rieske [2Fe-2S] center serves to transfer electrons, typically from NADH via ferredoxin partners, to the mononuclear iron site where oxygen activation and catalysis occur. The mechanism of oxygen activation occurs via binding of O2 to the FeII site followed by a one-electron transfer from the Rieske site to the mononuclear iron site, and subsequent two-electron reduction of O2 to afford a side-on (hydro)peroxo-FeIII intermediate. This intermediate has been proposed to act directly as the oxidizing agent, or alternatively, undergo O–O bond cleavage to generate the high-valent HO–FeV=O species which affects oxygenation.
Figure 12.

Depiction of a typical Rieske non-heme mononuclear iron site with the putative active oxygenating peroxo-FeIII species
Pyrrolnitrin (113) is an antifungal secondary metabolite produced by many Pseudomonads.294–299 Its nitro group was shown to be essential for activity, as the amino congener is completely inactive.300 Feeding studies and isolation of putative intermediates firmly established L-tryptophan as a biosynthetic precursor and supported a biosynthetic hypothesis involving the N-oxygenation of aminopyrrolnitrin (114) to pyrrolnitrin as the final step (Scheme 22).300–306 Identification of the pyrrolnitrin (prn) biosynthetic gene cluster, gene deletion experiments, and analysis of accumulated intermediates in blocked mutants allowed for assignment of each of the gene products to a specific step in the previously proposed pyrrolnitrin biosynthetic hypothesis.307,308 These experiments revealed PrnD as the enzyme responsible for generating the nitro group in pyrrolnitrin biosynthesis. PrnD is a member of the Rieske non-heme mononuclear iron oxygenase family and was the first enzyme discovered to catalyze nitro group formation via N-oxidation of an amine.
Scheme 22.
Proposed pathway for pyrrolnitrin biosynthesis involving nitro formation as the final step
PrnD-mediated N-oxygenation of 114 to pyrrolnitrin was demonstrated in vitro using SsuE, FMN, and NADPH as an electron transfer system, representing the first successful in vitro reconstitution of enzymatic nitro group-formation.309 NADPH, FMN, and SsuE transfer electrons one at a time to the catalytic non-heme mononuclear iron site of PrnD via the Rieske cluster to facilitate oxygen activation. Aminopyrrolnitrin oxidation is proposed to occur via a mechanism analogous to Path B described above for AurF (Scheme 17B).309,310 In this Scheme 114 is first N-hydroxylated to afford the corresponding hydroxylamine intermediate, which is then dehydrogenated to give the nitroso intermediate. Subsequent N-hydroxylation of this nitroso intermediate would furnish the nitro group of pyrrolnitrin. Overall, this six-electron oxidation of 114 requires three equivalents of O2 and six exogenous electrons. Labeling studies with the substrate mimic 4-N-hydroxybenzylamine (115) and 18O2 support this proposed mechanism.309,310 When 115 was incubated with PrnD under an 18O2 atmosphere the resulting 4-nitrosobenzylamine product had no 18O atoms, while the 4-nitrobenzylamine product showed incorporation of one 18O atom. These results argue against a dihdyroxylated intermediate, and instead provide evidence for a mechanism involving dehydrogenation. Alternatively, these results could be consistent with a dihydroxylated intermediate (116) if PrnD was capable of selective dehydration of 116 to afford the nitroso compound.
Guided by homology modeling, saturated alanine/valine-scanning mutagenesis of putative residues involved in substrate binding provided insights into the residues required for catalysis.311 As expected, mutation of many of these residues resulted in substantial loss of activity toward 114 and 115. Interestingly, several mutations resulted in improved the catalytic efficiency of PrnD toward the natural substrate 114, and lowered catalytic efficiency toward 115. These results suggest that engineering of the active site of PrnD could be a viable strategy for producing useful biocatalysts.
The flavin reductase responsible for providing electrons to PrnD in vivo (PrnF) has been identified and characterized in vitro.150 This system was studied further in order to understand the electron flow between the Rieske center and the non-heme mononuclear iron active site where oxidation occurs.312 Site-directed mutagenesis studies support two pathways for electron transfer involving either residue Asp183 or Asn180.312
2.6.1.3. P450-dependent
The pyschrophilins are nonribosomal peptides that have been isolated from various psychrotolerant Penicillum species.313–316 An unusual and defining feature of these natural products is their mode of macrocyclization, which occurs through the indole nitrogen of a tryptophan side chain. The biosynthetic gene cluster that produces pyschrophilins B (117) and C (118) was identified through genome mining of Penicillum rivulum IBT 24420 (Scheme 23A).317 In addition to the two NRPS enzymes predicted to incorporate anthranilate, L-valine, and L-tryptophan, the gene cluster encodes a P450 enzyme (PsyC) that was hypothesized to oxidize the α-amine group to the nitro group. Genetic experiments, isolation of intermediates, and feeding studies with blocked mutants support a biosynthetic sequence with the α-amino-containing psychrophilin I (119) as the immediate precursor t o 117 (Scheme 23B).
Scheme 23.

Selected psychrophilins and their proposed biosynthetic pathway involved PsyC-catalyzed nitro group installation
In this proposal the tripeptide scaffold is fully assembled by the NRPS assembly line and is then released through macrocylization to afford 119. PsyC then catalyzes the six-electron oxidation of the amino group to the nitro group to afford 117. Psychrophilin C is biosynthesized via an analgous route, but where L-valine is substituted with L-alanine in the tripeptide. This order of events imposes certain challenges on the cyclization domain of NRPS PsyB. Namely, it must promote the attack of the less nucleophilic indole nitrogen atom onto the nascent assembly line-tethered product over the α-amino group of L-tryptophan. No mechanism for the nitro-forming, six-electron oxidation of 119 to 117 has been proposed, although cytochrome P450s are known to catalyze two- and four-electron oxidations of amines to hydroxylamine and oxime products, respectively (Sections 2.1.2 and 2.2.2.). The PsyC-catalyzed reaction may utilize similar chemistry and iron-oxo intermediates in the oxidation of 119.
2.6.1.4. Flavin-dependent
Flavin-dependent enzymes are involved in the production of N-oxidized sugars, including nitro sugars. Nitro sugar biosynthesis has been studied mainly in the context of kijanimicin (120), rubradirin (121), and everninomicin (122), which contain the nitro sugars D-kijanose, D-rubranitrose, and L-evernitrose, respectively (Figure 13).318–329 As the pathways and enzymes involved in accessing these N-oxidized sugars have been reviewed previously they will not be discussed in great detail here.110 Since this review, structural studies of two flavin-dependent N-oxygenases have provided new insights into the phylogeny of these enzymes.330–332
Figure 13.

Structures of selected nitro sugar-containing natural products
Sequencing and annotation of the kijanimicin, rubradirin, and everninomicin biosynthetic gene clusters allowed a comparative genomic analysis that revealed the likely N-oxygenases involved in nitro group synthesis. 333–336 ORF36 from everninomicin, RubN8 from rubradirin, and KijD3 from kijanimicin encode homologs of flavin-dependent oxidoreductases and share very high (>60%) amino acid sequence identity to each other.337 ORF36 and RubN8 were capable of oxidizing the amino sugar substrate analogue L-TDP-epi-vancosamine (123) to the corresponding nitroso derivate 124 in vitro when supplied with NADPH, FAD, and a flavin reductase from Vibrio fisheri (Scheme 24A).337 This four-electron oxidation proceeds through a hydroxylamine intermediate (125), but mechanistic details regarding the conversion of 125 to 124 are lacking. Lastly, a related nitrososynthase, DnmZ (59% amino acid identity to ORF36), has been demonstrated in vitro to also catalyze the four electron oxidation of 123 to 124 in the biosynthesis of the baumycin family of anthracycline natural products.338
Scheme 24.

The reactions catalyzed in vitro by A) RubN8, ORF36, and DnmZ and B) KijD3
Subsequently, the predicted N-oxygenase from the kijanimicin pathway, KijD3, was structurally and biochemically characterized.330,332 The crystal structure showed an overall architecture that is exceptionally similar to flavin-dependent fatty acyl-CoA dehydrogenases. This close structural resemblance suggests a shared evolutionary history, which is interesting given the disparate reactions catalyzed by these enzymes. Given the prevalence of fatty acyl-CoA dehydrogenases, the evolution of flavin-dependent N-oxygenases may be a relatively recent addition to this enzyme family. Interestingly, KijD3 has only been shown to catalyze a single two-electron oxidation of its predicted dTDP-sugar substrate (126) to the corresponding N-hydroxylamine (127) in vitro (Scheme 24B).332 The structure of ORF36 was also determined, revealing KijD3 as its closest structural homolog.331 The biggest differences in both sequence and three-dimensional structure map to the active sites of these two enzymes.
It is currently unclear whether these three putative nitro sugar-forming N-oxygenases can catalyze the full six-electron oxidation of amines to nitro groups. The inability to fully reconstitute nitro sugar production in vitro with these enzymes raises several interesting questions about their role in these biosynthetic pathways. It is possible that the sugar substrates used in these assays are not the physiological substrates for ORF36, RubN8, or KijD3. The precise order of events in the biosynthesis of these three nitro sugars is not known, and it is conceivable that these N-oxygenases only fully oxidize their physiological substrates. Indeed, when ORF36 was assayed with additional sugar substrates the extent of amine oxidation varied, suggesting that structural differences in the substate influence the N-oxidation process.331 One particular structural difference has been attributed to the methylation state of the 4-OH in 123. In the biosynthesis of baumycin, 124 undergoes a retro oxime-aldol reaction that results in C–C bond cleavage to generate an aldehyde–oxime intermediate instead of further oxidation to the nitrosugar.338 This unstable intermediate has been characterized extensively by MS/MS. Methylation of 123 at the 4-OH to form TDP-L-evernosamine by RubN7 has been shown to prevent this oxidative cleavage, allowing further N-oxidation of the nitroso group by ORF36. Alternatively, the N-hydroxyl or nitroso sugars could be the true products of these N-oxygenases and the nitro group could arise via non-enzymatic oxidation either in vivo or during isolation and purification. It is therefore unclear whether other bioactive N-oxidized congener(s) may be produced by these pathways.
2.6.2. Direct nitration
In addition to the oxidation of amines, nature appears to install nitro groups via direct nitration of aromatic scaffolds. This C–N bond forming strategy is analogous to traditional electrophilic aromatic substitution reactions used heavily in organic synthesis, especially on the industrial scale (Figure 9). Although this strategy for nitro group installation is not as well understood as nitro group formation via the oxidation of amines, the enzymes and logic underlying this process have been characterized in the biosynthesis of thaxtomin.
2.6.2.1. P450-dependent
Thaxtomin A (128) is a phytotoxin produced by several species of Streptomyces that causes common scab disease, a globally- and economically-important root and tuber crop disease.339 This natural product was shown to be directly responsible for causing the disease by inhibiting cellulose biosynthesis.340–343 Interestingly, the nitro group of thaxtomin A was shown to be essential for bioactivity.340,341
The diketopiperazine core of 128 results from the condensation and N-methylation of L-phenylalanine and 4-nitro-L-tryptophan (129) by the NRPSs TxtA and TxtB (Scheme 25).344,345 A cytochrome P450, TxtC, has been proposed to catalyze at least one, and potentially both of the two hydroxylations to afford thaxtomin A. Shortly after the identification of the thaxtomin (txt) biosynthetic gene cluster, an encoded nitric oxide synthase (NOS) homolog (TxtD) was found to play an essential role in nitro group formation.346 Genetic disruption of txtD abolished the production of 128, and purified TxtD obtained through heterologous expression was capable of producing nitric oxide in vitro, suggesting that its activity could be involved in the nitration reaction in thaxtomin A biosynthesis. Adding NOS inhibitors to cultures of S. turbidiscabies suppressed the synthesis of 128, further supporting the participation of nitric oxide in the nitration reaction.
Scheme 25.

Proposed biosynthetic pathway for thaxtomin A
The precise role of nitric oxide this transformation remained unclear for nearly a decade. A secondary, “non-housekeeping” tryptophan synthetase (TrpRS II) from Deinocococcus radiodurans was shown to catalyze regioselective nitration at the 4-position of L-tryptophan in combination with TxtD in vitro with low efficiencies.347 However, the mechanistic details of this reaction were not elucidated and this result was later ascribed to adventitious, non-enzymatic reactivity.347,348 Eventually, the cytochrome P450 TxtE, the true nitrating enzyme, was discovered and characterized.348 TxtE was shown to be essential for thaxtomin biosynthesis in vivo and catalyzed efficient and regioselective nitration of L-tryptophan to 129 in vitro when supplied with molecular oxygen, NADPH, the spinach Fd/Fr reductase system, and the small molecule NO donor 1,1-diethyl-2-hydroxy-2-nitroso-hydrazine (DEANO).
The two proposed mechanisms for generation of the active nitrating species and subsequent nitration by TxtE are shown in Scheme 26.348 In the radical mechanism (Scheme 26A & B), NO reacts with a ferric superoxide via radical recombination to give a ferric peroxynitrite species which undergoes homolytic cleavage to generate the oxo-FeIV intermediate commonly referred to as “compound II” and nitrogen dioxide. NO2 addition at the 4-position of L-tryptophan followed by hydrogen atom abstraction by “compound II” affords 129.
Scheme 26.

Proposed mechanisms for L-tryptophan nitration by cytochrome P450 TxtE. A) Generation of potential reactive intermediates from nitric oxide. B) Radical-based mechanism C) Electrophilic aromatic substitution mechanism
In the electrophilic aromatic substitution mechanism (Scheme 26A & C), protonation of ferric peroxynitrite species and heterolytic cleavage gives an electrophilic nitronium ion. The nitronium ion is then attacked by L-tryptophan at the 4-position, which would yield 129 upon deprotonation and rearomization. In vitro assays with 18O2 resulted in incorporation of one 18O atom into 129, which is consistent with either proposed mechanism. Although neither mechanism can be ruled out based on current experimental evidence, these potential mechanisms both employ N–O bond formation between molecular oxygen and nitric oxide to generate a reactive nitrating species.
Due to its interesting chemistry and potential for biocatalysis applications, TxtE has been the focus of structural and computational investigations in order to understand and manipulate its regioselectivity and substrate scope.349–351 A crystal structure of TxtE bound with L-tryptophan revealed several key interactions between TxtE and the amino, carboxylate, and indole groups of L-tryptophan, many of which had been confirmed to be essential for catalysis by earlier mutagenesis studies based on molecular docking.350,351 Screening of a library of substrate analogs in which the amino, carboxylate, and indole moieties were all modified revealed molecular determinants for substrate binding and catalysis.350 The TxtE substrate must possess an indole ring and accepts only slight modifications to this substructure. Furthermore, the amino and carboxylate groups are essential, and act as a hydrogen bond donor and acceptor, respectively, to properly position the indole moiety in the active site. Additional insights into TxtE catalysis and engineering have been provided by examining the highly dynamic, substrate-gating F/G loop common to P450s.349 Identification of a potential substrate-binding residue (His176) within the F/G loop, and subsequent saturated mutagenesis, led to the identification of three mutants (His176Phe/Try/Trp) which all afforded a complete switch in regioselectivity to the 5-nitro-L-tryptophan isomer. Molecular dynamic simulations and X-ray crystallography revealed that in these mutants the closed-open equilibrium of the F/G loop had shifted in favor of the catalytically-competent closed state. Moreover, the residue at position 176 interacted directly with L-tryptophan providing a rationale for the switch in regioselectivity.
Currently, thaxtomin A biosynthesis represents the only pathway in which nitro group installation proceeds via a direct nitration strategy. It is currently unclear how widespread this biosynthetic logic is in natural product assembly, although several TxtE homologs have been identified in nucleotide sequence databases.349 Interestingly, a subset of these naturally contain the His176Trp polymorphism which may suggest they’re involved in the production of 5-nitro-L-tryptophan-containing natural products. Given the ability of multiple metallo and organic cofactors to activate molecular oxygen, it is conceivable that additional enzyme families may be capable of catalyzing similar nitration reactions.
2.6.3. 3-nitropropanoic acid
3-nitropropanoic acid (130) is a highly toxic metabolite that acts as a suicide inhibitor of succinate dehydrogenase, adversely affecting both the tricarboxylic acid cycle and the electron transport chain.352–354 The biosynthesis of this molecule occurs in both plants and fungi and appears to proceed via different pathways. Limited investigations into the plant-based pathways suggest malonic acid as a precursor.355 Alternatively, investigations into fungal 3-nitropropanoic acid biosynthesis have convincingly demonstrated that this metabolite is derived from L-aspartic acid.356–362 A series of detailed feeding studies with labeled precursors have mapped every carbon, oxygen, and nitrogen atom of 3-nitropropanoic acid to L-aspartic acid and molecular oxygen and strongly suggest that nitrosuccinate (131) is a late-stage biosynthetic intermediate (Scheme 27). Interestingly, 131 has been proposed as an intermediate in the conversion of L-aspartic acid to nitrite by the enzymes CreD and CreE from cremeomycin biosynthesis (Section 3.2.1).363 The fungal enzyme(s) responsible for oxidizing L-aspartic acid to 131 have not yet been identified. It will be interesting to determine whether a homolog of the flavin-dependent N-monooxygenase CreE participates in 3-nitropropanoic acid production.
Scheme 27.

Proposed biosynthetic pathway of 3-nitropropanoic acid in fungi
2.6.4. Hormaomycin
Hormaomycin (132) was isolated from Streptomyces griseoflavus and displays not only antibiotic activities against Gram-positive bacteria, but also intercellular signaling properties among Streptomyces.364 The biosynthesis of hormaomycin is of interest due to the impressive number of nonproteinogenic amino acids incorporated into its structure (Scheme 28A). One of these amino acids, nitrocyclopropyl-alanine (133), contains a nitro group. Stable isotope feeding experiments have revealed that 133 is derived from L-lysine (Scheme 28B).365 Loss of deuterium from the C4 position of this amino acid suggests that oxidation and intramolecular cyclization events take place during the biosynthesis of 133. In one proposed mechanism, an initial four-electron oxidation of the N6-amine of L-lysine affords the oxime intermediate 134 which is followed by concerted cyclopropanation and lactone-ring opening to generate the nitrosocyclopropane 135.119 A final two-electron oxidation of 135 provides 133. In an alternative mechanism, a direct six-electron oxidation of L-lysine installs the nitro group to afford the intermediate 136. Deprotonation of 136 at the α-carbon would then facilitate an intramolecular cyclization to form 133.
Scheme 28.

A) Structure of hormaomycin (132) and B) Proposed mechanisms for cyclopropanation and nitration to afford 133
The hormaomycin biosynthetic gene cluster was identified by searching the S. griseoflavus genome for NRPS genes and a homolog of the pyrrole halogenase cloN3 from chlorobiocin biosynthesis.366 Annotation of the gene cluster did not reveal any obvious candidates for either C4 oxidation or N-oxidation of L-lysine. The authors proposed that HrmI and HrmJ, both annotated as hypothetical proteins of unknown function, may be important for 133 production.
3. N–N bond forming enzymes
Natural products containing an N–N bond represent a small, yet structurally diverse class of metabolites. There are approximately 200 members of this natural product family, ranging from the simple di-amino acid azoxy compound valanimycin to the complex, diazobenzofluorene polyketide lomaiviticin.367,368 This group of natural products encompasses a wide range of functional groups, including azoxy, diazo, hydrazido, hydrazino, heterocyclic, and N-nitroso motifs. By virtue of the N–N linkage, many of these functional groups are believed to impart biological activity to the natural product, however, conclusive evidence linking these structural features to natural product function has been demonstrated only in a limited number of cases.
N–N bond formation is a chemically challenging transformation as both atoms being linked are inherently nucleophilic.15 Thus the primary difficulty that must be surmounted in enzymatic N–N bond construction is the ‘reversal’ of this reactivity for one of the amine reaction partners. Our current understanding of N–N bond formation in natural product biosynthesis points toward a predominant logic in which one of the amine partners undergoes an initial N–O bond forming reaction to facilitate the eventual generation of an electrophilic species. The reactivity of these electrophilic, oxidized nitrogen species has led to the proposal that certain N–N bonds in natural products are formed non-enzymatically, although conclusive experimental evidence is lacking in these cases.
The following section will discuss our current understanding of the enzymes and chemical logic involved in the construction of various N–N bond containing natural products. Presently, only one natural product biosynthetic enzyme that catalyzes N–N bond formation has been studied in vitro. However, investigations of additional pathways have suggested that a range of different enzymes may promote this general transformation in the context of building different functional groups. Characterization of these N–N bond forming enzymes stands as an important goal for future research efforts. This section will be organized by the type of functional group and further subdivided by the specific natural product, with certain metabolites discussed together when a shared biosynthetic logic appears operative.
3.1. Azoxy
The first natural product discovered to possess an N–N bond was the azoxy-containing compound macrozamin in 1951.369 Azoxy compounds have attracted attention given their varied bioactivities, which include antibacterial, antifungal, anticancer and carcinogenic effects.367,368 However, it is currently unknown whether the azoxy group is principally responsible for these activities due to a lack of detailed structure-activity relationship studies. While azoxy-containing metabolites are exceedingly rare among known natural products, these compounds have well-established roles as intermediates and reagents in synthetic organic chemistry.370,371 Interestingly, investigations into the biosynthesis of azoxy-containing compounds have revealed two main routes for the construction of this functional group that appear to parallel the general strategies used by synthetic chemists (Scheme 29). This functional group can be furnished by the oxidation of azo compounds, a route that involves constructing the N–N linkage first.371 Alternatively, both aliphatic and aromatic azoxides can be accessed through coupling of C-nitroso compounds and hydroxylamines, with N–O bond of the azoxy linkage installed prior to N–N bond formation.371
Scheme 29.

Logic of two azoxy group installation strategies used in biosynthesis
The natural products discussed in this section will be organized according by the logic used in azoxy group construction. Metabolites assembled by azo oxidation include valanimycin, the elaiomycins, and KA57-A, while the azoxymycins and malleobactin D are generated via oxidative dimerization. While the enzyme(s) responsible for N–N bond formation in these pathways remain to be discovered, we will review the significant advances that helped to decipher the general logic used in azoxy group biosynthesis.
3.1.1. Valanimycin, Elaiomycin, KA57A
Valanimycin (137) was isolated and structurally characterized in 1986 from the soil-dwelling microbe Streptomyces viridifaciens MG456-hF10 during a screen of a Streptomycete collection for antitumor antibiotics.372 It showed activity against both Gram-positive and Gram-negative bacteria as well as several mouse cancer cells lines.372 The increased susceptibility of a DNA-repair deficient Escherichia coli strain to valanimycin suggested DNA as its primary target, which was later supported by demonstrating the inhibition of bacterial DNA synthesis.373 Investigations into valanimycin biosynthesis have greatly advanced our understanding of the construction of this rare functional group. While the specific enzyme(s) that participate in N–N bond construction have not been characterized, a series of investigations has deciphered nearly every other biosynthetic transformation (Scheme 30).
Scheme 30.

Proposed biosynthetic pathway for valanimycin
Early feeding studies established L-valine, L-serine, isobutylamine (138), and isobutylhydroxylamine (139) as intermediates in valanimycin biosynthesis.374,375 These latter two intermediates were proposed to derive from L-valine via decarboxylation and N–hydroxylation. Subsequently a PLP-dependent L-valine decarboxylase (VlmD) and a specific flavin-dependent isobutylamine N–hydroxylase (VlmH) were identified in the valanimycin biosynthetic gene cluster and characterized in vitro.22,79,376–378 Interestingly, the flavin-dependent N–monooxygenase VlmH was found to be part of a novel two-component system in which the reduced flavin cofactor required for monooxygenation was provided by a separate NADPH:flavin reductase, VlmR. 79,375. This system showed a strong preference for FAD and NADPH over FMN and NADH, respectively. Feeding of [15N,18O]-isobutylhydroxylamine to S. viridifaciens MG456-hF10 resulted in 50% incorporation of the 15N-label into valanimcyin, but no incorporation of the 18O-label, indicating that the oxygen atom of the azoxy group does not derive from isobutylhydroxylamine.379 The conversion of 138 to 139 suggested that an oxidative logic for N–N bond construction was operative, as further oxidation or modification of this functional group could enhance the electrophilicity of this nitrogen atom, enabling subsequent nucleophilic attack.
Discovery of the valanimycin (vlm) biosynthetic gene cluster increased our understanding of valanimycin and azoxy group biosynthesis by facilitating the biochemical characterization of additional enzymes.378,380 A peculiar feature of the vlm gene cluster was the presence of a gene (vlmL) predicted to encode a seryl-tRNA synthetase. Aminoacyl-tRNA synthetases play an essential role in protein synthesis, but their roles outside of this context had only begun to be investigated.381 In vitro characterization of VlmL demonstrated that it catalyzed the aminoacylation of a pool of E. coli tRNAs with L-serine, however, the role of the seryl-tRNA intermediate in valanimycin biosynthesis was unclear.382 The details of its participation became clear with the identification of a proposed late-stage biosynthetic intermediate, O-(L-seryl)-isobutylhydroxylamine (140) and characterization of the enzyme VlmA.383 The intermediate 140 was identified by incubating 139 and enzymatically generated, radiolabeled L-[U-14C]seryl-tRNA with cell-free extracts of S. viridifaciens MG456-hF10 and analyzing assays by thin-layer chromatography (TLC) and autoradiography. The generation of 140 was dependent on VlmA, as cell-free extracts of a ΔvlmA mutant could not produce this compound. Thus, VlmA appears to catalyze the O-acylation of 139 with L-serine to afford 140, with seryl-tRNA serving as the acyl group donor. This activity could be reconstituted in vitro when VlmA was supplied with 139 and L-[U-14C]-seryl-tRNA. In accordance with this activity, VlmA shows weak sequence identity to lysylphosphatidylglycerol synthase, an enzyme that acylates a hydroxyl group of phosphotidylglycerol with L-lysine using L-lysyl-tRNA.384
The characterization of VlmA and VlmL was a crucial step forward in understanding valanimycin and azoxy biosynthesis as these enzymes connect the two halves of valanimycin. Unfortunately, the instability of 140 precluded its use in feeding studies, which could have further confirmed its role as a biosynthetic intermediate. Presuming the intermediacy of 140, the only transformations needed to afford the final natural product are azoxy formation and dehydration of the seryl moiety. Gene disruption experiments and feeding studies with radiolabelled valanimycin hydrate (141) revealed that VlmJ acts as a kinase to phosphorylate 141, generating an activated phosphoryl intermediate which is then dehydrated by VlmK to provide valanimycin.385 These studies also suggest that azoxy group formation precedes dehydration.
The enzymes involved in azoxy formation, arguably the most interesting transformation in the construction of this valanimycin, remain to be identified and characterized. Encouragingly, the work of Parry and colleagues has put constraints on both the timing of azoxy formation and the enzyme(s) involved. Only three proteins VlmO, VlmB, and VlmG remain to be investigated for their ability to catalyze azoxy formation.378 No functions for these proteins have been proposed and their closest homologs are all annotated as hypothetical proteins. Intermediate 140 may be a likely substrate for one or more of these enzymes.383,385
Two intriguing routes have been proposed that would convert 140 to the azo precursor 142, which could be subsequently oxidized and dehydrated to form valanimycin (Scheme 31).383 In Path A, the amine of the seryl moiety in 140 is oxidized to provide the α-ketoimine 143, which then cyclizes via an intramolecular attack by the nitrogen atom of the isobutylamine moeity to provide the unusual oxadiazolidine intermediate 144. Oxidation of the amine to the imine and its conjugation with the carbonyl group would increase the electrophilicity at the nitrogen atom and could potentially promote N–N bond formation. The oxadiazolidine intermediate 144 is then proposed to undergo N–O bond cleavage to form the azo intermediate 142. However, the key N–N bond forming reaction in this sequence that converts 143 to 144 is a disfavored, 5-endo-trig cyclization, potentially arguing against this route.
Scheme 31.

Proposed pathways for azoxy group installation from putative intermediate 140
Alternatively, in Path B, the nitrogen atom of the isobutylamine moiety of 140 could be hydroxylated a second time to generate the O-acylated N,N-dihydroxy intermediate 145. Paralleling the reactivity observed in oxime, nitroso, and nitro group biosynthesis, N–O cleavage would generate the nitroso compound 146 and L-serine. Subsequent condensation of these intermediates could afford the same azo compound 142. Installation of an N-hydroxyl group on 140 could therefore not only facilitate C–O bond cleavage but could also be required to sufficiently activate this nitrogen atom for subsequent nucleophilic attack.
While these two proposed mechanisms use a very different series of transformations, they both involve an oxidative logic to increase the electrophilicity at one of the two nitrogen atoms, allowing for nucleophilic attack by the second nitrogen atom. Notably, both of these scenarios are potentially inconsistent with the previous [15N, 18O]-isobutylhydroxylamine feeding studies, which observed no retention of 18O in the final natural product. Based on the mechanisms proposed in Scheme 31, the oxygen atom of isobutylhydroxylamine should end up in the carboxylic acid of valanimycin. This inconsistency may be attributed to isotope-exchange with water during the valanimycin isolation procedure, which includes several manipulations at pH 3.0 as well as the use of 2 N NH4OH. From the common azo intermediate 142, oxygenation to form the azoxy group and dehydration would afford valanimycin.
Elaiomycin (147) is a carcinogenic and tuberculostatic antibiotic isolated in 1954 from the fermentation broth of Streptomyces hepaticus (Scheme 32A).386–389 Its structure and absolute configuration were later confirmed by total synthesis.390–395 As with investigations of valanimycin assembly, our insights into elaiomycin biosynthesis originated with feeding studies using various hypothesized precursors. These experiments, in combination with the isolation of structurally related congeners, suggest a biosynthetic logic for azoxy group formation that is shared with the valanimycin pathway.
Scheme 32.

A) Structures of isolated elaiomycins and B) proposed biosynthesis
Early feeding studies support the biosynthetic hypothesis shown in Scheme 32B. Key biosynthetic precursors include n-octylamine (148), which is derived from fatty acid biosynthesis, L-serine, malonyl-CoA, and a SAM-derived methyl group.396,397 The results of these experiments led to the proposal that installation of the secondary hydroxyl group arises from a Claisen condensation between an activated intermediate 149 and malonyl-CoA. This would yield a β-ketoester which would be hydrolyzed, decarboxylated, and finally reduced.397 Further insights into elaiomycin biosynthesis came nearly 30 years later when several structurally related elaiomycin derivatives were isolated from Streptomyces sp. strain HKI0708 and Streptomyces sp. Tu 6399 (elaiomycin D – H, K, L; 150 – 156) (Scheme 32A).398,399 These metabolites likely represent a consortium of intermediates and analogues resulting from differential tailoring reactions (Scheme 32B). Their structures are consistent with L-serine and 148 as direct precursors to elaiomycin and support the proposal that azoxy group formation occurs early in biosynthesis potentially via a similar O-serylated intermediate (157) to that which occurs in valanimycin biosynthesis. Unfortunately, a biosynthetic gene cluster has not been reported for elaiomycin, preventing the assignment and characterization of the enzymes responsible for the proposed biosynthetic steps.
The azoxyalkene natural product KA57-A (158) was isolated from Streptomyces rochei 743AN4 during efforts to elicit the production of silent biosynthetic gene clusters.400 This natural product had previously been isolated from an Actinomadura-like fungus.401 The structure of KA57-A strongly resembles valanimycin and elaiomycin, suggesting a shared strategy for azoxy group construction and a biosynthetic route deriving from an aliphatic amine (n-hexylamine, 159) and an amino acid (L-serine) via an O-serylated intermediate 160. Insight into the biosynthesis of KA57-A derives entirely from feeding studies and supports the biosynthetic proposal outlined in Scheme 33. The generation of the intermediate 161 is proposed to follow a similar sequence to elaiomycin biosynthesis involving a Claisen condenation with malonyl-CoA followed by hydrolysis, decarboxylation, and oxidation/reduction tailoring reactions. Interestingly, analysis of the draft genome of S. rochei 743AN4 revealed homologs of vlmJ and vlmK, the kinase and dehydratase from valanimycin biosynthesis that dehydrate the serine residue. These enzymes could be involved in dehydrating the 3-aminobutane-1,2-diol moiety of 161. The authors did not comment on the genes within close physical proximity to the vlmJ and vlmK homologues and whether they might represent the KA57-A biosynthetic gene cluster.
Scheme 33.

Structure and proposed biosynthesis of KA57-A
Overall, valanimcyin, elaiomycin, and KA57-A represent a structural class of azoxyalkene natural products that appear to share a biosynthetic logic for azoxy group construction which involves O-seryl hydroxylamine and azo intermediates. The absence of biosynthetic gene clusters for both elaiomycin and KA57-A has severely hindered the characterization of these two pathways, as well as the elucidation of the enzymatic chemistry involved in azoxy group biosynthesis. If these three pathways do employ the same general logic for azoxy group assembly, the discovery of the elaiomycin and KA57-A gene clusters could aid in the identification of the azoxy group forming enzyme(s) via comparative genomics.
3.1.2. Azoxymycins, Malleobactin D
Malleobactin is a siderophore discovered in the 1990s and is produced by the pathogenic bacteria Burkholderia pseudomallei and Burkholderia mallei.402,403 The malleobactin (mba) biosynthetic gene cluster was later discovered using a combination of global transcriptional analysis and gene disruption experiments.404 The structure of malleobactin was not determined until 2013, when four structurally related nonribosomal peptides, malleobactin A – D (162 – 165), were structurally characterized (Figure 14). These structures are consistent with the tetramodular NRPS and accessory enzymes encoded within the mba gene cluster.37,405,406 Malleobactin A – C differ in the oxidation state of the N-terminal ornithine residue, ranging from the hydroxylamine to the nitro group, and malleobactin D is an azoxy-linked dimer.
Figure 14.

Structures of malleobactins A – D
The presence of the 5-nitropentanoic acid residue in malleobactin A and the azoxy group in malleobactin D is highly unusual in siderophores. Analysis of the mba gene cluster revealed only one enzyme (MbaC) likely to oxidize the N-terminal ornithine residue. MbaC is annotated as a putative flavin-dependent monooxygenase based on sequence identity and is similar to the well-studied ornithine and lysine N-monooxygenases discussed earlier in Section 2.1 in the context of hydroxylamine biosynthesis.37 In vitro characterization of MbaC revealed that this enzyme converts L-ornithine to N5-hydroxy-L-ornithine as the major product, consistent with its annotation and the reactivity of related enzymes.37 Small amounts of the nitro, nitroso and azoxy-linked dimer were also detected in this assay and were proposed to arise from autoxidation based on the known reactivity of aliphatic hydroxylamines.37
These results led to the suggestion that the hydroxylamine congener malleobactin B might be the actual product of the mba gene cluster. Indeed, incubation of malleobactin B under physiological conditions led to the non-enzymatic formation of malleobactins A, C, and D. Based on these results, it has been claimed that the azoxy-containing compound malleobactin D represents a shunt product formed non-enzymatically during the autoxidation of the hydroxylamine group of malleobactin B. This reactivity is consistent with previous reports of azoxy formation from the condensation of hydroxylamines and nitroso compounds.251,407 While its formation may be non-enzymatic, this does not rule out the possibility that malleobactin D may be generated in vivo and has a specific physiological role.
The azoxymycins are a class of dimeric aromatic azoxy compounds that were isolated from Streptomyces chattanoogensis L10 in 2015 (Figure 15).408 Azoxymycin A, B, and C (166 – 168) are bright yellow solids and analysis of these compounds using HPLC-diode array detection demonstrated that they exist as interconverting cis-trans isomers at room temperature.408 The azoxymycin (azo) biosynthetic gene cluster was discovered using a genome mining approach based upon the hypothesis that these compounds were polyketide-derived.409 Involvement of this gene cluster in azoxymycin production was confirmed by gene knockout experiments. Importantly, when azoC was disrupted two amino aromatic polyenes (169 and 170) accumulated in the mutant (Scheme 34).410 The protein encoded by azoC is a putative PABA N-oxygenase resembling the extensively studied nitro group-forming non-heme di-iron N-oxygenase AurF (Section 2.6.1.1).266 This annotation suggests that AzoC may be involved in oxidizing the amino group of 169 and 170 en route to azoxy group formation. Specifically, AzoC may oxidize these putative biosynthetic precursors to hydroxylamine and/or nitroso derivatives that could dimerize either enzymatically or non-enzymatically to generate the azoxymycins. The possibility of nonenzymatic azoxy formation during the course of AzoC-mediated oxidation of 169 and 170 was not explicitly tested, but appears possible given the known reactivity of this class of molecules.251
Figure 15.

Structures of azoxymycin A – C
Scheme 34.

Proposed biosynthesis of azoxymycin A – C involving coupling of amino precursors 169 and 170 potentially via AzoC
The examples of malleobactin D and the azoxymycins showcase a pathway for azoxy group formation that differs dramatically from that involved in the production of valanimycin, elaiomycin and KA57-A, suggesting that at least two strategies for azoxy group biosynthesis have evolved separately. Perhaps unsurprisingly, these routes rely heavily on oxidative chemistry for N–N bond formation, however, the enzymatic chemistry used in each pathway differs. A flavin-dependent N-monooxygenase is employed in malleobactin D biosynthesis, while a putative non-heme di-iron N-oxygenase is used to assemble the azoxymycins. Both types of enzymes can generate intermediates that are known to dimerize non-enzymatically to generate azoxide products (Scheme 35).251,407 Malleobactin D and azoxymycin biosynthesis therefore highlight the potential interplay between enzymatic and non-enzymatic chemistry in constructing structurally intriguing functional groups. Whether azoxy formation in these natural products is truly a non-enzymatic process in vivo and whether these azoxy congeners have physiological relevance remains unknown.
Scheme 35.

Proposed mechanisms for the non-enzymatic generation of azoxy compounds from nitroso and N-hydroxyl intermediates
3.2. Diazo
Diazo-containing natural products are a small subset of N–N bond containing natural products that exhibit a range of structural diversity and have been reviewed previously.411 This group of secondary metabolites has generated considerable interest among chemists given their potent bioactivities, interesting mechanisms of actions, intriguing structures, and unknown biosyntheses. The first diazo-containing metabolites discovered were modified α-amino acids, and many were evaluated clincally.412–418 The bioactivities of diazo-containing compounds are thought to arise from the reactivity of the diazo functional group.411 For example, the mechanism of action of the diazo-containing natural product lomaiviticin A (4) has been elucidated, and accumulation of DNA double strand breaks requires the presence of its two diazo groups.4
Although diazo-containing metabolites are rare, synthetic diazo compounds are important reagents and intermediates within organic chemistry due to their unique reactivity, which facilitates many transformations, including 1,3-dipolar cycloadditions, carbene insertions, and alkylations.419–421 Over the last two centuries synthetic chemists have developed versatile and efficient methods for installing diazo groups into organic molecules.422 Most of these approaches use a reagent that contains a pre-formed N–N bond, including trifyl azide and tosyl hydrazine. Synthetic methods for diazo construction that involve N–N bond formation are known, but the use of this disconnection in complex molecule synthesis is limited given the inherent difficulty and harsh conditions often required (Figure 16).
Figure 16.
Logic of diazo group installation in organic synthesis versus biosynthesis
Understanding the genetic and biochemical basis for diazo installation would fill a void in our understanding of enzymatic bond constructions in nature and could facilitate biocatalytic and synthetic biology efforts to access clinically relevant molecules and yield reagents for synthetic chemistry. Understanding synthetic strategies for diazo installation may be helpful in investigating hypotheses for diazo biosynthesis. Interestingly, early investigations into diazo biosynthesis clearly demonstrated that nature likely employs a strategy involving N–N bond formation to install this reactive functional group into secondary metabolites.411 However, enzymatic diazo formation has not yet been demonstrated in vitro, and the role of non-enzymatic processes in generating this functional group is unclear.
Recently, the availability of sequenced genomes has provided new insights into the enzymatic chemistry that may be involved in constructing diazo groups. Comparative analyses of gene clusters that produce diazo-containing metabolites has revealed two putative sets of biosynthetic enzymes: a cre-type diazo gene cassette currently found only in the cremeomycin gene cluster and a lom/kin-type diazo gene cassette shared by the lomaiviticin and kinamycin biosynthetic gene clusters. The following section will discuss these two types of pathways, with similarities in biosynthetic logic highlighted where appropriate.
3.2.1. Cremeomycin
Cremeomycin (171) was isolated in 1967 from the soil-dwelling bacteria Streptomyces cremeus by the Upjohn Company and showed activity against both Gram-positive and Gram-negative bacteria.423 The unusual o-diazoquinone structure of cremeomycin was determined 28 years later.424 A total synthesis of this natural product has been reported that employs a classical nitrous acid-based diazotization of 3-amino-2-hydroxy-4-methoxybenzoic acid (3,2,4-AHMBA, 172).425 As discussed below, recent characterization of the cremeomycin biosynthetic pathway suggests that nature may employ an analogous strategy for diazo group construction.363
In the report describing the structure of cremeomycin, 3-amino-4-hydroxybenzoic acid (3,4-AHBA; 91) was proposed as a biosynthetic intermediate.424,426 Based on this hypothesis, a genome mining strategy revealed a putative cremeomycin (cre) biosynthetic gene cluster in S. cremeus.427 Heterologous expression of the gene cluster, feeding studies with stable isotope-labeled intermediates, and in vitro and in vivo characterization of biosynthetic enzymes confirmed the connection to cremeomycin production and elucidated the order of the events in this pathway (Scheme 36).363,427 Biosynthesis of cremeomycin begins with the production of 3,4-AHBA by the cyclase, CreH, and aldolase, CreI, from the primary metabolites L-aspartate semialdehyde (L-ASA, 173) and dihydroxyacetone phosphate (DHAP, 174). Synthesis of 3,4-AHBA is followed by C2 hydroxylation by the flavin-dependent monooxygenase CreL to provided 3-amino-2,4-dihydroxybenzoic acid (3,2,4-ADBA; 175), which is then O-methylated at the C4 phenol to afford 3,2,4-AHMBA. At this point, the only biosynthetic transformation needed to afford cremeomycin is the enigmatic diazo group installation.
Scheme 36.

Proposed pathway for cremeomycin biosynthesis
Using a heterologous expression system in Streptomyces albus, systematic disruption of the remaining genes within the cre gene cluster revealed several enzymes that are essential for diazo group installation.363 While creA, creB, creO, and creM appear to not be involved in diazotization, ΔcreD and ΔcreE mutants both failed to produce cremeomycin and accumulated 3,2,4-AHMBA. The functions of these gene products were interrogated in vitro to elucidate their roles in diazo biosynthesis. CreD is a putative member of the fumarase/aspartase enzyme superfamily, which catalyzes the elimination of β-heteroatoms from carbonyl-containing substrates. CreE is a predicted flavin-dependent monooxygenase similar to N-hydroxylating flavin-dependent enzymes discussed earlier in hydroxylamine biosynthesis in Section 2.1.1. Based on the roles of related enzymes in N–N bond formation, CreE was postulated to oxidize the amino group of either 3,2,4-AHMBA or an amino acid. In vitro biochemical characterization of CreE demonstrated that it oxidizes L-aspartate to nitrosuccinic acid (131) (Scheme 37). CreE appeared to consume three equivalents of NADPH for every molecule of 131 produced, indicating that it catalyzes the full six-electron oxidation. This represents the first flavin-dependent monooxygenase capable of catalyzing the full six-electron oxidation required to convert an amine to a nitro group. Interestingly, there was no evidence for a non-enzymatic contribution to this reaction, in contrast to what has been observed for other flavin-dependent enzymes involved in nitro group biosynthesis (Section 2.6.1.4. and 3.1.2.).
Scheme 37.

Proposed mechanism for CreD/CreE-catalyzed nitrite formation from L-aspartate
The connection between CreE’s reactivity and diazo group formation became clear when it was hypothesized that nitrite might be a key biosynthetic intermediate. Previous investigations of azamerone biosynthesis using stable isotope feeding experiments had implicated the involvement of nitrite in diazo biosynthesis.428 Thus, it seemed plausible that nitrite is the source of the distal nitrogen atom of the cremeomycin diazo group. With this in mind, CreD was proposed to catalyze the elimination of nitrite from 131. Indeed, CreD and CreE generated nitrite and fumarate (176) from L-aspartate in a coupled enzyme assay in vitro (Scheme 37). Nitrite production required the presence of both enzymes, suggesting that CreD can accept 131 only when it is generated and provided by CreE. The ability of CreD and CreE to generate nitrite in vitro strongly suggests this oxidized nitrogen metabolite is an intermediate in diazo group biosynthesis. However, CreD and CreE are not capable of generating cremeomycin from 3,2,4-AHMBA and nitrite in vitro suggesting that they do not catalyze diazotization in vivo. The structural basis for the tight coupling between CreD and CreE functions remain to be elucidated, but suggests a highly coordinated pathway for diazo formation. This control may not be surprising given the reactivity of diazo-containing molecules.
The last challenge remaining in deciphering cremeomycin biosynthesis is identifying the enzyme(s) that catalyze N–N bond formation between nitrite and 3,2,4-AHMBA and subsequent diazo group formation. While nitrite is capable of diazotizing 3,2,4-AHMBA to form cremeomycin under mildly acidic conditions (pH 4.0), this reactivity is abolished at neutral pH.363 The buffered, pH neutral microbial fermentation media used in these biosynthetic studies suggest that cremeomycin production may not derive from this non-enzymatic reaction. Instead, the enzyme(s) responsible for diazotization may activate nitrite in analogous manner, generating an electrophilic nitrosonium ion-like intermediate. Both the mechanism by which this activation occurs and the enzyme(s) responsible are currently unknown. All remaining enzymes encoded by the cre gene cluster are non-essential for cremeomycin production perhaps indicating the diazo-forming enzyme(s) are encoded elsewhere in the genome. It is also possible that as yet undefined conditions promote non-enzymatic diazotization between nitrite and 3,2,4-AHMBA in vivo.
As noted above, nitrite had been suggested previously as a diazotizing agent in azamerone biosynthesis. Azamerone is a pyridazine-containing natural product that originates from a diazo-containing intermediate that is in turn postulated to arise from an amine precursor (Section 3.5.2.).428 Feeding 15N-labelled ammonium, nitrite, nitrate, and hydrazine to the azamerone producer resulted in robust and specific incorporation of nitrite and nitrate into the distal nitrogen atom of the diazo group, suggesting a highly oxidized nitrogen species as the potential diazotizing agent.428 The high incorporation of 15N labeled nitrate may arise from its reduction to nitrite by endogenous nitrate reductases. The azamerone biosynthetic gene cluster has been identified in Streptomyces sp. CNQ-525 and it does not contain homologs of creD and creE. However, these enzymes are encoded elsewhere in the genome.363 Similarly, the lomaiviticin and kinamycin (Section 3.2.2) biosynthetic gene clusters do not have creD and creE homologs, but these genes are located in other parts of the genome. Perhaps surprisingly, creD and creE homologs are also found in the gene cluster that produces the hydrazide-containing natural product fosfazinomycin and have been shown in vitro to produce nitrite from L-aspartate (Section 3.3.1.).429–431
Queries of publicly available sequence databases have revealed that genes resembling creD and creE are widespread in actinobacterial genomes and are often clustered in close proximity to natural product biosynthetic gene clusters.363 These findings suggest that this previously unknown pathway for nitrite generation may be important for the biosynthesis of a diverse set of N–N bond containing natural products, many of which have yet to be discovered.
3.2.2. Kinamycin, Lomaiviticin
The kinamycins (A, C, D, F; 177 – 180) are a family of anti-proliferative antibiotics first isolated in 1970 and produced by several bacterial species including Streptomyces murayamaensis ATCC 21414 (Figure 17).432–439 Initial structural characterization incorrectly assigned the kinamycins as N-cyanocarbazoles.439–441 It was only after two decades of investigations that the molecules were reassigned as the correct 5-diazobenzo[b]fluorenes.442,443 The bioactivity of these compounds is directly related to the reactive diazo group, with studies demonstrating a range of potential mechanisms for its activation and cytotoxicity (Figure 18).444–449 The kinamycins can cleave DNA in vitro under biomimetic conditions with mild reductants.446,447 Both reductive and nucleophilic activation of the diazo group have been proposed to give rise to various reactive intermediates, including a vinyl radical, o-quinone methide, acylfulvene, and a covalent adduct. In addition, the quinone moiety may undergo redox cycling to produce reactive oxygen species.
Figure 17.

Structures of several kinamycins and the previously proposed, incorrect structure for kinamycin C
Figure 18.

Reactive intermediates proposed to be relevant for the bioactivity of the kinamycins
Due to their bioactivity and complex molecular architecture, which includes a highly oxygenated and congested A-ring, redox-sensitive quinone, and a reactive diazo group, the kinamycins have been subject to a number of total synthesis efforts.450–453 In these syntheses the predominant methods for diazo group installation has been Regitz diazo transfer to activated methines or oxidation of hydrazone intermediates accessed via condensation of hydrazine reagents with ketone precursors. Notably, the diazo group in the kinamycins has not been installed using a late-stage N–N bond formation such as the nitrous acid-based diazotization used to synthesize cremeomycin.
Kinamycin biosynthesis has been studied primarily through feeding studies and the isolation of intermediates from mutant strains. The recent identification of the full kinamycin biosynthetic gene cluster has enabled a more complete understanding of genetic and biochemical basis for kinamycin biosynthesis, and laid the groundwork for understanding diazo group installation. Despite the original misassignment of their structures, much of the information gleaned from early kinamycin studies remains relevant, and this work has been reviewed previously (Scheme 38).454
Scheme 38.

Proposed pathway for kinamycin biosynthesis
Initial feeding experiments demonstrated that the kinamycins were of polyketide origin based upon incorporation of labeled acetate.455–457 The previously known benz[a]anthraquinone polyketide dehydrorabelomycin (181) was subsequently isolated from S. murayamensis ATCC 21414 and was confirmed to be a biosynthetic intermediate by feeding a deuterated analog.458 Over the next decade, additional metabolites connected to kinamycin biosynthesis were isolated, including prekinamycin (182), kinobscurinone (183) and stealthin C (184).458–461 Deuterated versions of both 183 and 184 were fed and incorporated into kinamycin D, suggesting they are intermediates in kinamycin production. These feeding studies suggest that diazo group installation occurs via a stepwise process involving N–N bond formation on the polyketide scaffold rather than through the incorporation of an intermediate containing an intact N–N bond. Kinamycin D was not labeled when deuterated 182 was fed. This result was attributed to the low solubility of 182 in fermentation media and this metabolite has still been proposed as an intermediate in kinamycin biosynthesis.454
Early efforts to identify the kinamycin (kin) biosynthetic gene cluster led to the discovery of a cosmid from a genomic library of S. murayamaensis ATCC 21414 that produced 181, 183, and 184 upon expression in S. lividans ZX7.462 The lack of kinamycin production was attributed to the incomplete capture of the biosynthetic gene cluster in a single cosmid. Recently, the full kin gene cluster (initially identified as the alp gene cluster) was identified in the genome of Streptomyces ambofaciencs ATCC 23877, a strain previously not known to produce kinamycins. This gene cluster had previously been shown to produce an antibacterial compound termed alpomycin, however its structure had not been elucidated. Genetic disruption of a transcriptional repressor (AlpW) enabled structural elucidation of the encoded natural products, revealing several members of the kinamycin family, including 178 and 179.
Recently, the enzymatic chemistry involved in constructing the benzo[b]fluorene scaffold of the kinamycins has been elucidated. A combination of genetics and in vitro and in vivo enzyme characterization identified a pair of flavin-dependent oxidases (AlpJ and AlpK) that appear to be responsible for B-ring cleavage of 181, ring contraction, and hydroxylation to yield the hydroquinone form of the intermediate 183.463
Identification of the putative diazo-forming enzymes encoded in the kin gene cluster relied upon having access to biosynthetic gene clusters that produce the lomaiviticins, a related family of diazobenzofluorenes. A discussion of these enzymes will therefore be included in the following section covering lomaiviticin biosynthesis. Additionally, this section will compare the kin/lom-type putative diazo biosynthetic machinery with that from cremeomycin assembly.
The lomaiviticins are C2-symmetric diazobenzofluorene glycosides obtained in 2001 from the ascidian-associated marine bacterium Micromonospora lomaivitiensis (later classified Salinospora) using a DNA-damage assay to guide isolation (Figure 19).464 The lomaiviticins are extraordinarily potent antitumor antibiotics with lomaiviticin A (4) and B exhibiting minimum inhibitory concentrations (MIC) of 6–25 ng/spot against several Gram-positive bacteria and lomaiviticin A exhibiting IC50 values of 0.01–98 ng/mL against a panel of 24 cancer cell lines.464 This cytotoxicity profile suggested a novel mechanism of action for 4, and extensive studies have provided both biochemical and structural evidence that this metabolite intercalates DNA and induces double strand breaks.4,444,448,449,465–467 The two diazobenzofluroene moieties are thought to insert into the DNA duplex, disrupting base pairing and positioning the reactive diazo functional groups in close proximity to each DNA strand.467 These groups are then activated nucleophilically or reductively to generate vinyl radicals that abstract hydrogen atoms from the DNA backbone. The greatly enhanced cytotoxicity of 4 compared to lomaiviticin C (185) and 178 likely arises from the presence of two diazo groups in 4 compared with one diazo group in 185 and 178, which allow a single molecule of 4 to generate a double strand break.4
Figure 19.

Structures of lomaiviticin A and C
As with the kinamycins, the lomaivitcins have been the focus of a number of total synthesis efforts due to the exciting synthetic challenges associated with their construction and the desire to evaluate their bioactivity and potential therapeutic applications. To date, no total synthesis of any lomaiviticin has been achieved, although the aglycone can be accessed in just eleven steps and 4 can be generated semi-synthetically from the more abundant metabolite 185.449,453,465,468,469 In these total synthesis and semi-synthesis efforts, the diazo groups are installed via Regitz diazo transfer onto an activated methine intermediate.
Our current understanding of lomaiviticin biosynthesis largely arises from the assumed parallels between this pathway and kinamycin production. Highly similar lomaiviticin (lom) biosynthetic gene clusters containing roughly 60 genes were identified in Salinispora tropica CNB-440, Salinispora pacifica DPJ-0019 and Salinispora pacifica DPJ-0016 using a combination of bioactivity-guided fractionation, genomic library screening, and genome mining.470,471 Annotation of these gene clusters provided the first insights into a highly complex biosynthetic pathway. Comparison with the kin gene clusters from S. murayamaensis and S. ambofaciens supported the use of an analogous route for diazobenzofluorene construction and identified putative enzymes involved in dimerization and propionate starter unit generation.470,471 The biochemistry of starter unit generation and A-ring oxidation has been investigated in vitro.472,473
This comparative analysis also revealed strong candidates for the diazo-forming enzymes (Figure 20).471 Comparison of the complete lom and kin gene clusters revealed a conserved, col-ocalized set of genes encoding homologs of several C–N bond forming and bond cleaving enzymes, as well as enzymes that are predicted to perform redox chemistry. Notably, these genes are absent from the partial kin gene cluster that was identified in S. murayamaensis.462 This observation is consistent with the inability of this partial gene cluster to generate diazo-containing compounds when heterologously expressed in S. lividans.
Figure 20.

Comparison of putative diazo- and hydrazide-forming gene cassettes from the lomaiviticin, kinamycin, and fosfazinomycin biosynthetic gene clusters
The general biochemical functions encoded within the predicted kin/lom-type diazo gene cassette are consistent with the types of transformations needed to construct a diazo group from two separate nitrogen-containing metabolites. This includes predicted C–N bond forming enzymes [glutamine synthetase (lom32/SAMT0146) and an N-acetyltransferase (lom35/SAMT0144)] and homologs of C–N bond cleaving enzymes [an amidase (lom33/SAMT0147) and an adenylosuccinate lyase (lom34/SAMT0145)]. Additionally, a hypothetical protein (lom29/SAMT0148) encoded adjacent to a predicted ferredoxin (lom30/SAMT0149) may be involved in redox chemistry. If diazo group installation in lomaiviticin and kinamycin biosynthesis occurs using similar logic to the cremeomycin pathway, this combination of enzymatic chemistry is well-suited for installing a nitrogen atom on the polyketide scaffold and generating an oxidized nitrogen-containing species, potentially nitrite, for subsequent diazotization.
It is currently unclear whether nitrite plays a role in diazotization in kinamycin or lomaiviticin biosynthesis. No feeding studies with inorganic, oxidized nitrogen species analogous to the experiments with azamerone have been performed with these natural products. Neither the lom/kin-type diazo gene cassettes nor the lom/kin gene clusters contain homologs of creD or creE. If these pathways do employ a nitrous acid-based strategy for diazo group construction it will represent an intriguing example of the convergent evolution of biosynthetic logic. Comparison of the cre, lom, and kin gene clusters does not indicate any obvious candidate for an N–N bond forming enzyme(s) involved in diazotization. Intriguingly, all but one (lom30/SAMT0149) of the kin/lom-type diazo-forming enzymes are encoded within the gene cluster that produces the hydrazide-containing natural product fosfazinomycin (fzm), further supporting their role in N–N bond formation (Scheme 19).430 The relationship between the lom/kin-type diazo-forming gene cassette and the fzm-type hydrazide-forming gene cassette will be discussed below in the section covering fosfazinomycin biosynthesis (Section 3.3.1.).
Queries of publicly available genome sequences revealed similar lom/kin-type gene cassettes in 57 gene clusters encoding putative secondary metabolites.471 The wide distribution of this putative diazo biosynthetic machinery, as well as that from the cremeomycin pathway suggests that diazo-containing natural products may be far more numerous than previously expected. Targeted genome mining approaches may now be used to identify new metabolites that contain diazo groups, expanding this family of exceptionally bioactive natural products.
3.3. Hydrazide
Hydrazides are well represented among N–N bond containing natural products and can be found in both linear and cyclic scaffolds. The first hydrazide-containing secondary metabolite was isolated in the early 1960s and since then molecules bearing this functional group have been discovered in fungi, both terrestrial and marine bacteria, and plants.367,368 These natural products exhibit a wide range of bioactivities including antidepressant, antiinflammatory, antiproliferative, antibacterial, and antifungal.14,367
Hydrazide compounds are widely used synthetic organic chemistry, both as reagents and intermediates, as well as final target scaffolds.474 In this context, hydrazides are most commonly accessed by acylation of hydrazine and hydrazine derivatives (Figure 21).14 Hydrazide natural products may be biosynthesized using related logic in which an activated carboxylic acid derivative undergoes nucleophilic attack by hydrazine or a hydrazine derivative. Currently, our understanding of the genes and enzymes involved in hydrazide biosynthesis come from studying the fosfazinomycins and piperazic acid-containing nonribosomal peptides. For both classes of molecules, hydrazide installation appears to follow the strategy described above.
Figure 21.

Logic for hydrazide construction in organic synthesis and biosynthesis
3.3.1. Fosfazinomycin
Fosfazinomycin A (186) and B (187) were isolated in 1983 from the culture filtrate of Streptomyces lavendofoliae No. 630 during a screening program for antifungal antibiotics.475–477 Investigations into these natural products were reinvigorated over 30 years later with their isolation from Streptomyces sp. WM6372 and the identification of the fosfazinomycin (fzm) biosynthetic gene cluster using genome mining and a novel enzymatic isotope labeling approach termed SILPE (stable isotope labeling of phosphonates in extracts).430 Interestingly, the fzm gene cluster contains putative diazo-forming gene cassettes from both the cremeomycin and kinamycin/lomaiviticin pathway. In vitro characterization of enzymes from both of these gene cassettes has led to proposals for their roles in hydrazide biosynthesis.
Investigation of fosfazinomycin biosynthetic enzymes in vitro has clarified the order of events in this pathway, confirmed the proposed activities of several enzymes, and elucidated key biosynthetic intermediates that are proximal to hydrazide installation.429,431 Based on this work, it is currently hypothesized that fosfazinomycin is assembled via a convergent process involving the incorporation of phosphonate and amino acid building blocks (Scheme 39). In the phosphonate side of the pathway, the enzymes FzmBCDG, which consist of an O-methyltranferase, phosphoenolpyruvate (PEP) phosphomutase, phosphonopyruvate (PnPy) decarboxylase, and α-ketoglutarate diooxygenase, respectively, convert PEP (188) to the intermediate (S)-2-hydroxy-2-phosphono-acetate (189).429 This phosphonate intermediate 189 is then coupled with methylated arginine hydrazide (190) to form 187, which can be further elaborated to 186 by ligation of L-valine. While the enzymatic chemistry involved in constructing 189 is well-precedented, the biosynthetic route to the hydrazide coupling partner 190 has not been completely elucidated.
Scheme 39.

Proposed biosynthesis of fosfazinomycin A (186) and B (187)
A preliminary hypothesis for hydrazide construction has been put forth based on the contents of the fzm gene cluster and the in vitro activities of biosynthetic enzymes (Scheme 40). Arginine hydrazide (191) is believed to be a precursor to the late-stage intermediate 190 as it is selectively methylated by FzmH to generate 190.429 The synthesis of 191 is thought to involve nitrite production as the fzm gene cluster encodes homologs of CreD and CreE (FzmL and FzmM, respectively) from cremeomycin biosynthesis (Section 3.2.1).363,431 Consistent with this proposal, these enzymes FzmM and FzmL convert L-aspartate to nitrite and fumarate in vitro. Based on the hypothesis that hydrazinosuccinate (192) is a biosynthetic intermediate, it has been proposed that nitrite diazotizes a second equivalent of L-aspartate, forging the N–N bond that appears in the final hydrazide natural product. This diazo acid intermediate (193) may then be reduced via an unknown process to generate 192.
Scheme 40.

Proposed pathway for hydrazido assembly in fosfazinomycin biosynthesis
As noted earlier, in addition to homologs of CreD and CreE the fzm gene cluster contains most of the putative diazo-forming gene cassette from the kinamycin and lomaiviticin pathways, including the glutamine synthetase (fzmN), the amidase (fzmO), the hypothetical protein (fzmP), the N-acetyltransferase (fzmQ), and the adenylosuccinate lyase (fzmR). In vitro characterization of two biosynthetic enzymes, FzmQ and FzmR, has led to the proposal that these enzymes participate in converting 192 to acetylhydrazine (194). The N-acetyltransferase FzmQ was shown to selectively acylate the terminal nitrogen atom of hydrazinosuccinate with acetyl-CoA, providing 195. The adenylosuccinate lyase homolog FzmR catalyzed the elimination of 194 from 195 forming fumarate as a co-product. One or more of the remaining enzymes encoded by the putative hydrazide gene cassette (FzmN, FzmO), the asparagine synthase FzmA, or the pyruvate carboxylase FzmF have been proposed to ligate 194 with L-arginine and subsequently remove the acetate group to afford 191. Thus, the key hydrazide-forming reaction appears to involve acylation of 194 or a related intermediate. The generation of 194 rather than direct use of hydrazine may have evolved to avoid accumulation of this toxic and reactive species.
The biosynthesis of the fosfazinomycins concludes with the methylation of 191 by methyltransferase FmzH to give 190, its condensation with the phosphonate intermediate 189 via unknown enzymes to afford fosfazinomycin B, and finally, the ligation of L-valine by the aminoacyl-tRNA peptidyl transferase FzmI to provide fosfazinomycin A. The FmzH-catalyzed methylation of 191 and appendage of L-valine to fosfazinomycin B by FzmI have been confirmed in vitro. The N–P bond forming reaction that constructs the phosphonamidate linkage remains enigmatic, but will be discussed briefly in Section 4.2.
From an evolutionary perspective, fosfazinomycin biosynthesis is particularly intriguing. The fzm gene cluster contains elements from two very different diazo-containing natural product biosynthetic pathways. The presence of functional CreD and CreE homologs suggests that nitrite is involved in hydrazide biosynthesis.363 The high degree of similarity between the putative fzm hydrazide-synthesizing gene cassette and the kin/lom diazo-forming gene cassette suggests additional parallels in biosynthetic logic. The lack of the predicted ferredoxin oxidoreductase from the lom/kin-type diazo gene cassette (Lom30) in the fzm hydrazide gene cassette is notable but its contribution to diazo biosynthesis is unclear.471 The distribution of similar biosynthetic machinery in very distinct biosynthetic pathways should help to guide future experimental efforts to decipher the elusive enzymatic chemistry involved in N–N bond formation in the context of both hydrazide and diazo functional group synthesis.
3.3.2. Piperazic–acid containing nonribosomal peptides
The hydrazide functional group also appears in a wide range of nonribosomal peptides that utilize piperazic acid as a building block, including the antibiotic monamycin F (196)478–480, the antiproliferative himastatin (197)481–483, the immunosuppressive mixed PKS-NRPS sanglifehrin (198)484,485, and the antimicrobial kutzneride 1 (7)486–488 (Figure 22). Although piperazic acid elongation by an NRPS has not yet been demonstrated in vitro, its incorporation into these scaffolds is expected to be consistent with the established logic of these assembly-line enzymes. Piperazic acid is believed to be incorporated intact rather than synthesized on the assembly line, with additional modifications (chlorination, oxidation) occurring on the piperazyl-S-ACP tethered intermediate.21,489
Figure 22.

Selected piperazic acid-containing hydrazide NRPS-derived natural products
Presumably, piperazic acid is selected and activated by the adenylation (A) domain using ATP (Scheme 41). An A domain sequence motif predictive of piperazic acid utilization has not been described; however, the A domain specificity code of the predicted piperazic acid-incorporating NRPS module SfaD is most similar to the motif for L-proline activation, which is in agreement with their similar structures.484,485 The resulting acyladenylate intermediate is then transferred to the phosphopantetheinyl arm of the thiolation (T) domain. The condensation (C) domain then likely catalyzes the nucleophilic attack of one of the amines of the tethered piperazic acid onto the thioester of an upstream tethered intermediate to form the hydrazide linkage. In most natural products, including monamycins, himasatin, and kutzernides the N1 amine acts as the nucleophile in hydrazide formation, however, in sanglifehrin A the N2 amine is the nucleophile. Therefore, use of piperazic acid as an NRPS extender unit generates a hydrazide linkage rather than a typical amide bond. The biosynthesis of the piperazic acid building block is discussed below in Section 3.4.2.
Scheme 41.

NRPS-catalyzed hydrazide formation using piperazic acid as an extender unit. C = condensation, A = adenylation, T = thiolation
While the overall logic for hydrazide biosynthesis in the fosfazinomycins and piperazic-acid containing nonribosomal peptides is analogous, the enzymes used are highly distinct. The putative hydrazide-forming enzymes in fosfazinomycin biosynthesis resemble enzymes that participate in primary metabolism, while the piperazic acid containing peptides utilize the well-characterized NRPS assembly-line machinery. In both pathways the N–N bond appears to be synthesized before the acyl groups are appended.
3.4. Hydrazine
Hydrazines are relatively rare among natural products containing an N–N bond.368 Moreover, there is considerable debate as to whether small hydrazine-containing natural products are synthesized enzymatically or whether they result from abiotic processes.367 Hydrazine derivatives have been isolated from bacteria, plants, and fungi.367 These compounds exhibit varied bioactivities and can be highly toxic as is the case for N-formyl-N-methylhydrazine from the mushroom Gyromitra esculenta.490
Unlike their metabolite counterparts, hydrazine derivatives are well-represented among synthetic compounds. Most commonly, the N–N linkage in complex hydrazines is constructed through the incorporation of hydrazine and simple hydrazine derivatives into synthetic intermediates (Figure 23). Methods for synthesizing hydrazine derivatives that involve N–N bond formation are quite limited.15 While it is conceivable that hydrazine itself could be involved in the biosynthesis of more complex hydrazine-containing natural products, little experimental evidence supports this hypothesis. Although hydrazine is generated as an intermediate in the energy-yielding metabolic pathway known as anammox, it is restricted to very specialized classes of anaerobic bacteria (Scheme 42).17 Furthermore, due to the reactivity and toxicity of hydrazine, it is produced and sequestered in a highly adapted bacterial organelle called the annamoxasome, which likely prevents its interaction with other metabolic pathways.
Figure 23.
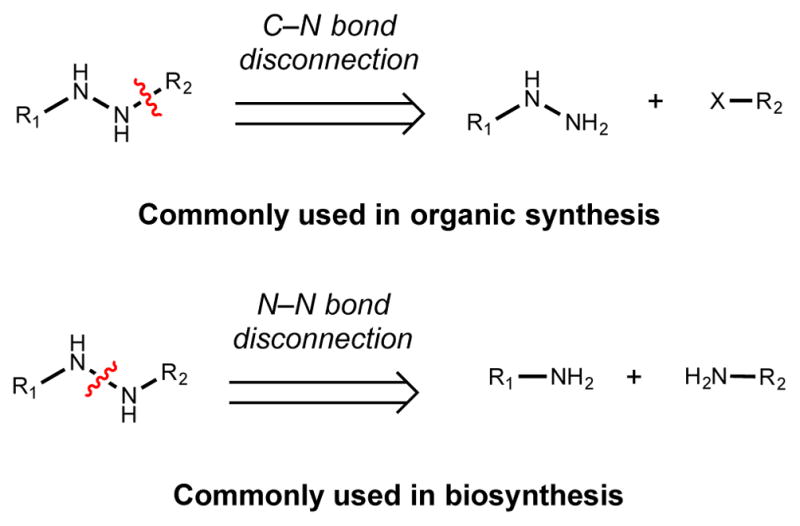
Logic of hydrazine installation in organic synthesis versus biosynthesis
Scheme 42.

Biosynthesis of hydrazine in anammox metabolism. HZS = hydrazine synthase
Our current understanding of the biosynthesis of hydrazine-containing natural products derives largely from studies of two structurally unrelated compounds, the atropisomeric dixiamycins A and B and piperazic acid. A flavin-dependent enzyme has been discovered that is capable of catalyzing late-stage N–N bond formation in the biosynthesis of the dimeric dixiamycins. In the case of piperazic acid no enzyme(s) has been uncovered that mediate N–N bond formation, although evidence suggests that its biosynthesis utilizes an N–N bond disconnection and is not derived from hydrazine or a simple derivative. A comparison of the chemical logic used to overcome the challenge in forging N–N bonds in these two different methods will be discussed.
3.4.1. Dixiamycins
Xiamycin A (199) was discovered in 2010 from Streptomyces sp. GT2002/1503 during a screen for new natural products from endophytes of mangrove plants.491 Xiamycin represented the first example of a bacterial indolosesquiterpene, a class of biologically active and medically relevant molecules previously isolated only from fungi and plants. Xiamycin exhibits both anti-HIV and antitumor activity. The xiamycin (xia) biosynthetic gene cluster was reported independently two years later in the strains Streptomyces sp. HKI0576 and Streptomyces sp. SCSIO 02999.492,493 These gene clusters were identified by genome mining and verified by genetic disruption experiments and heterologous expression. A combination of genetics, heterologous expression, and in vitro biochemical studies elucidated several of the key steps that afford the indolosesquiterpene backbone of the xiamycins. In particular, a novel type of flavin-dependent oxygenase (XiaI) was shown to be involved in the final oxidative cyclization cascade that furnishes the carbazole-decalin fused ring system of 199.492,493
Several new metabolites were also identified that were produced by the xia cluster when expressed in Streptomyces albus, including the N–N linked atropisomers dixiamycin A/B (200a/200b) (Scheme 43A).493 These dimeric compounds appeared to result from the direct N–N coupling of two xiamycin monomers through their carbazole nitrogens.494 Additionally, two regioisomeric C–N linked xiamycin dimers were isolated, including dixiamycin C (201) and the atropisomers dixiamycins D/E (202a/202b). Lastly, an oxygenated cycloether derivative of xiamycin termed oxiamycin (203) was isolated.494 Interestingly, the N–N linked analogs were the most active against panels of bacteria, filamentous fungi, and yeast. Systematic mutational analysis of the xia gene cluster revealed that XiaH, an enzyme homologous to FAD-dependent aromatic ring hydroxylases, was responsible for producing all the dimeric dixiamycins as well as oxiamycin. S. albus expressing XiaH also produced all the dimeric dixiamycins and oxiamycin when fed 199.
Scheme 43.

A) Structures of the diaxiamycins. B) Proposed biosynthesis of the dixiamycins and oxiamycin by XiaH-mediated one-electron oxidation
The ability of one enzyme to generate the dimeric dixiamycins and the cycloether oxiamycin suggested a radical-based mechanism might be operative (Scheme 43B).494 A resonance-stabilized xiamycin radical (204) produced by XiaH-mediated one-electron oxidation could combine with a second equivalent of 204 at various positions, affording both N–N- and C–N-linked dimers (200 – 202). Alternatively, 204 could react with molecular oxygen to afford 203. When 199 was incubated with the radical initiator benzoyl peroxide, all the dixiamycins and oxiamycin were produced, supporting the hypothesis that radical chemistry is used in the synthesis of these metabolites.494
XiaH is the first enzyme from natural product biosynthesis demonstrated to catalyze N–N bond formation. However, its ability to construct such a wide range of distinct products is unusual. The ability of a non-enzymatic catalyst to mediate the same chemistry suggests that XiaH may generate and then release the stabilized xiamycin radical from the active site where it could participate in further non-enzymatic reactions. As discussed in Section 2.1, flavin-dependent monooxygenases are capable of transferring single electrons during the generation of the flavin (hydro)peroxide oxygenating species.29 XiaH may catalyze the one-electron oxidation of 199 to 204 with molecular oxygen, FAD, or the flavin semiquinone serving as the oxidant, and further experiments will be needed to understand this reactivity.
It is unclear whether additional hydrazine-containing natural products are constructed using similar biosynthetic logic. This strategy can only be used when the substrate-based radical is sufficiently stabilized to allow for a one-electron oxidation within the redox potential of flavin-dependent enzymes – approximately −495 mV to +150 mV relative to standard hydrogen electrode.495 The highly conjugated nature of the carbazole moiety within xiamycin may stabilize the resulting radical enough to facilitate this chemistry. Interestingly, this biosynthetic strategy is conceptually analogous to the electrochemical-based oxidation used by Baran for their total synthesis of diaxiamycin B.15
3.4.2. Piperazic acid
Piperazic acid is a nonproteinogenic amino acid that was first identified in 1971 as a constituent of the antibiotic monamycin.478 Intense research into the activity and synthesis of piperazic acid and piperazic-acid containing natural products has resulted in several previous reviews.6–8 While the biosynthesis of piperazic acid has also been reviewed,7 recent insights have increased our understanding of this process.
Piperazic acid assembly was initially studied in the context of monamycin and polyoxypeptin A biosynthesis by feeding isotopically-labelled amino acids to Streptomyces jamaicensis and Streptomyces strain MK498-98F14, respectively.479,496 The observed incorporation rates suggested that L-glutamine and D/L-glutamate were likely precursors to piperazic acid while L-ornithine was not. Based on these experiments, two potential biosynthetic hypotheses were proposed (Scheme 44).497 These routes require reduction of the amide carbonyl of L-glutamine, oxidation of the corresponding amine of 205 or 206 to the hydroxylamine 207 or 208, respectively, and intramolecular nucleophilic displacement to construct the N–N linkage.
Scheme 44.

Original biosynthetic proposal for the formation of substituted piperazic acids
Recently, biochemical evidence has been provided that implicates N-oxygenation in piperazic acid formation in the kutzneride pathway, albeit with L-ornithine rather than L-glutamine. 21 The kutznerides are a family of piperazic acid-containing antimicrobial and antifungal cyclodepsipeptides produced by Kutzneria sp. 744 discussed earlier in Section 3.3.2. Discovery and annotation of the kutzneride (ktz) biosynthetic gene cluster revealed a putative flavin-dependent lysine/ornithine N-monooxygenase (KtzI).486 Since no N-hydroxylated lysines or ornithines were present in the kutznerides, it was postulated that KtzI generated the N-hydroxylamine intermediate previously proposed to be involved in piperazic acid biosynthesis.497 KtzI was characterized in vitro and shown to hydroxylate the N5 nitrogen of L-ornithine, with D-Orn, D/L-Lys, D/L-Glu, and D/L-Gln not accepted as substrates.
Further support for the involvement of L-ornithine and N5-hydroxy-L-ornithine in piperazic acid biosynthesis came from feeding studies with labeled amino acids and analysis of extracts by LC-MS/MS.21 Consistent with previous studies, 13C5-Glu and 13C5-Gln were incorporated into the kutznerides. However, the mass isotopic envelope was particularly wide suggesting that the 13C label was distributed across both the hydroxyglutamate and piperazic acid residues. Furthermore, mass shifts ranging from +1 Da to +5 Da suggested that extensive scrambling of the label had occurred due to entry into primary metabolism. Alternatively, feeding studies with 13C5-L-ornithine and 13C5-N5-hydroxy-L-orinithine resulted in incorporation levels as high as 35%, with the labeling largely isolated to the piperazic acid residues. These results indicate that L-ornithine and N5-hydroxy-L-orinithine are more proximal precursors to piperazic acid than glutamate or glutamine. Label suppression feedings studies with 13C5-L-ornithine and 13C5-N5-hydroxy-L-orinithine further demonstrated that 13C5-N5-hydroxy-L-orinithine is downstream of 13C5-L-ornithine in piperazic acid biosynthesis. In total, these results are consistent with L-glutamate and L-glutamine being converted to L-ornithine via L-proline in primary metabolism. It is unclear why previous studies did not detect incorporation of ornithine; this amino acid may not be taken up by these organisms or they may access piperazic acid via a different route.21 The latter explanation is unlikely given that KtzI homologs have been implicated in the biosynthesis of additional piperazic acid-containing natural products, including himastatin, sanglifehrin A, and polyoxypeptin A.481,484,498
From N5-hydroxy-L-ornithine two pathways have been proposed for piperazic acid formation (Scheme 45).21 N5-hydroxy-L-ornithine could be oxidized to the nitroso intermediate 209 which could then cyclize through condensation of the α-amine onto the nitroso group thus forging the N–N bond of piperazic acid. Reduction of the resulting hydrazone (210) would afford piperazic acid. Alternatively, the N-hydroxyl group could be activated as a leaving group to 211 via an unknown mechanism, enabling an intramolecular SN2-like displacement. Currently, no enzymes encoded in piperazic acid-containing natural product biosynthetic gene clusters have been linked to these reactions.
Scheme 45.

Characterization of KtzI in N-oxidation of ornithine and proposed biosynthesis of piperazic acid
The chemical logic used to construct the hydrazine linkages in dixiamycins A/B and piperazic acid differs markedly, with each type of pathway evolving a distinct solution to overcome the inherent difficulty of forming a bond between two nucleophilic atoms. In the case of the dixiamycins it appears that the flavoenzyme XiaH uses one-electron chemistry to generate a stabilized, substrate-based radical, while in piperazic acid biosynthesis the N-hydroxylating flavoenzyme KtzI generates N5-hydroxy-L-ornithine via a two-electron oxidation. However, the need to oxidize amine substrates is a unifying feature of these two strategies, as is the central role of flavin-dependent enzymes. The catalytic versatility of this cofactor may explain its use in diverse N–N bond forming pathways.
3.5. Aromatic Heterocycle
Aromatic heterocycles containing N–N bonds have been isolated from a wide variety of sources including terrestrial and marine bacteria, fungi, and many plants.367 Synthetic compounds containing these heterocycles, including pyrazoles and pyridazines, are often considered “privileged scaffolds” as they are well-represented in modern screening libraries and are structural elements in many important pharmaceuticals.9,10,499,500 There are many synthetic methods available for constructing these various 5- and 6-membered scaffolds.14 As noted in previous sections, synthetic methods for constructing various N–N bond containing functional groups commonly utilize reagents in which this linkage is pre-formed. Methods for constructing heterocycles that involve direct N–N bond formation are rare, yet desirable due to the drawbacks of working with hydrazine.11–13
Understanding biosynthetic routes to aromatic heterocycles containing N–N linkages could enable the discovery and development of therapeutics. Although enzymes capable of catalyzing N–N bond formation in such pathways have not yet been identified, in vivo experiments have provided insights into the logic and enzymatic chemistry used. The following sections will be divided into two structural subclasses: pyrazoles and pyridazines. Knowledge of pyrazole biosynthesis has been obtained by studying the plant metabolites pyrazole and β-pyrazole-1-ylalanine, and investigations of azamerone and pyridazomycin have informed our understanding of pyridazine assembly.
3.5.1. Pyrazole
Pyrazole (212) and β-pyrazol-1-ylalanine (213) are structurally and biosynthetically related metabolites isolated from seed extracts of the watermelon Citrullus vulgaris.501 The latter molecule has also been detected in several cucumber varieties including Cucumis sativus.502 The identification of 213 in 1959 marked the first discovery of a pyrazole-containing natural product.503 The biosynthesis of these metabolites has been studied using stable isotope feeding experiments and in vitro assays with partially purified protein extracts. These studies support a biosynthetic hypothesis for 213 in which 1,3-diaminopropane (214) undergoes a six-electron oxidation to 212 via the intermediate 2-pyrazoline (215), followed by condensation with O-acetylserine (Scheme 46).502,504,505 A partially purified protein extract from Cucumis sativus seedlings was capable of converting 214 to 212.502 Interestingly, this activity was stimulated by the addition of FAD, but not NAD+ or NADP+. Dialysis of the protein extract against EDTA did not inhibit its activity nor did the addition of various metals ions. Taken together, these results suggest that one or more flavin-dependent enzymes are involved in the six-electron oxidative cyclization and dehydrogenation that converts 214 to 212. The lack of a stimulatory effect by addition of NAD+/NADP+ and lack of an inhibitory effect of EDTA suggest that this oxidative process is not catalyzed by nicotinamide- or metal-dependent oxidases. A related assay with crude protein extracts revealed the final coupling reaction that affords 213.506,507 Although direct evidence is lacking, it is interesting to consider whether the biosynthetic logic for N–N bond formation in pyrazole biosynthesis may be similar to that of other pathways which use flavin-dependent enzymes, including the azoxy compound valanimycin and the hydrazine piperazic acid.
Scheme 46.

Proposed biosynthesis of pyrazole and β-pyrazol-1-alanine
3.5.2. Pyridazine
An understanding of the biosynthesis of pyridazine-containing natural products has come from studying the Streptomyces-derived metabolites azamerone and pyridazomycin. Insights into N–N bond formation have been gained entirely from stable isotope feedings studies. Currently, no enzyme(s) have been identified to be involved in formation of the N–N bonds in these heterocyclic natural products.
Azamerone (216) is a meroterpenoid phthalazinone which was isolated from Streptomyces sp. CNQ766 and CNQ525 during a screening program for marine sediment-derived actinomycete natural products.508 Based on this natural product’s resemblance to the diazonaphthoquinones A80915D (217) and SF2415A3 (218), it was proposed that the pyridazine ring of azamerone arises through oxidative rearrangement of a diazo intermediate (Scheme 47).428,508–510 This hypothesis was supported by feeding the isotopically-labelled diazo compound [2-15N, 9-13C]-SF2415A3, which resulted in incorporation of the 15N label into the pyridazine ring of azamerone.428 Feeding studies with various 15N-labelled inorganic nitrogen sources revealed high (>70%) and selective labeling of both N2 of azamerone and the distal nitrogen atom of the diazo intermediate 217 by nitrite and nitrate. As described earlier in Section 3.2.1 this result suggests nitrite may play a key role in diazo formation. The putative aminotransferase NapB3 has been postulated to generate the substrate 219 for diazotization. No enzymes from the azamerone gene cluster have been assigned roles in either N–N bond formation or the oxidative rearrangement. Interestingly, when this gene cluster was heterologously expressed, no nitrogenated compounds were identified. As mentioned earlier, homologs of the nitrite-generating enzymes CreD and CreE are encoded elsewhere in the genome of the producer, potentially explaining this result.363
Scheme 47.

Proposed biosynthetic pathway involving oxidative rearrangement of the diazo intermediate to afford the pyridazine ring of azamerone
The antifungal natural product pyridazomycin (220) was isolated in 1988 from Streptomyces violaceoniger sp. griseofuscus strain Tu 2557, representing the first pyridazine natural product.511 Extensive feeding studies with labeled amino acids and a detailed analysis accounting for potential scrambling suggested that glycine and L-ornithine are likely precursors to this metabolite (Scheme 48).512 Mapping these building blocks onto the pyridazomycin scaffold suggested that the remaining precursor is oxaloacetate (221). Although no enzymes from this biosynthetic pathway have been identified, the biosynthetic intermediates identified may implicate a late-stage N–N bond formation.
Scheme 48.

Biosynthetic origins of pyridazomycin based on feeding studies
3.5.3. Toxoflavin
Toxoflavin (222) is isolated from various strains of Burkholderia and is responsible for bacterial panicle blight and food poisoning in humans.513 Endogenous enzymes can reduce 222 in a manner similar to flavin, and this reduced species subsequently converts molecular oxygen to H2O2, causing oxidative damage.514 Under anaerobic conditions, toxoflavin is inactive and does not exert antibiotic activity against yeast. Transposon mutagenesis of Burkholderia glumae identified two enzymes TRP-1 (ToxA) and TRP-2 (ToxD) to be essential for toxoflavin biosynthesis.515 Subsequent sequencing around the locus revealed an 8.2 kb operon containing putative toxoflavin biosynthetic genes.516 The biosynthetic gene cluster was also recently identified in Pseudomonas protegens Pf-5 by searching for homologues of the toxoflavin genes from B. glumae.517
The gene products of toxB and toxE are annotated as a GTP cyclohydrolase II and a deaminase, two genes that are important in the biosynthesis of 223, a precursor to riboflavin. The early stages of toxoflavin biosynthesis are therefore proposed to parallel that of riboflavin assembly (Scheme 49). Cleavage of the sugar of 223 is proposed to release 5,6-diaminouracil (224), which is subsequently condensed with glycine, a known precursor based on stable isotope feeding experiments in Pseudomonas cocovenenans, to generate the N–N containing heterocycle 225.518 Finally, ToxA is a methyltransferase that can install the two methyl groups on 225.519 The N–N bond formation that forms the triazine ring remains to be studied. In this context, the functions of the remaining enzymes in the toxoflavin gene cluster ToxC and ToxD, which have no significant homology to characterized proteins, have yet to be identified. Deletion of toxC and toxD abolished toxoflavin production; however, no new metabolites or shunt products were detected.517 Future characterization of this biosynthetic pathway could provide insights into how the tox biosynthetic enzymes activate the nitrogen atom of glycine or 224 to promote N–N bond formation.
Scheme 49.

Proposed biosynthetic pathway for toxoflavin
3.6. N-Nitroso
N-nitrosation of primary metabolites has been extensively studied due to this functional group’s connection to gastric cancer and inflammation.216,520 N-nitrosation of secondary amines confers deleterious bioactivities to otherwise inactive compounds, as nitrosamines and alkylnitrosoureas are precursors to diazonium alkylating agents. Nitrite is central to nitrosamine formation in vivo, as it can be activated under acidic conditions to nitrous acid (pKa = 3.2) or reduced by metalloenzymes like nitrite reductases to afford reactive nitrogen species. Recently, nitrosation of secondary amines from nitrite-derived nitrous anhydride (N2O3) was also shown to be a physiologically relevant reaction (Figure 24).521 The N–N bond forming reaction that results in a nitrosamine has yet to be attributed to an enzymatic reaction.
Figure 24.

A) Current paradigms for N-nitrosation in living organisms. B) Mechanisms of action for nitrosamines
3.6.1. Streptozotocin
Although N-nitrosation in primary metabolism and synthetic chemistry has been extensively studied, N-nitrosation in secondary metabolism remains to be examined. Nitrous acid is proposed to play an important role by providing the nitroso nitrogen.368 Naturally occurring N-nitroso compounds isolated include streptozotocin (1) and L-alanosine. The biosynthesis of streptozotocin is hypothesized to utilize nitrous acid or a biochemical equivalent to nitrosate the corresponding urea (Scheme 50).1 Feeding studies with radioactively labeled compounds have indicated that glucosamine, L-methionine, and either L-citrulline or L-arginine are precursors to this metabolite. Future studies will be required to address the similarities and differences of N-nitrosation mechanism in primary and secondary metabolism.
Scheme 50.

Proposed biosynthesis for streptozotocin based on feeding studies
4. N–P bond forming enzymes
Exceedingly few natural products have been discovered that possess N–P bonds, however this linkage is more common in synthetic molecules. In the context of chemical biology and medicinal chemistry, the increased stability of N–P bonds toward hydrolysis in comparison with O–P bonds helps to make N–P bond containing natural and synthetic compounds promising inhibitors of important protein targets. The N–P bond is also present as a linkage in several synthetic prodrugs that help to improve the cell penetration. For example, synthetic nucleoside analogues containing N–P bonds are effective antiviral and antibacterial agents.522 These synthetic nucleosides undergo intracellular reductive activation to phosphoramidate anion followed by the cleavage of the N–P bond by endogenous phosphoramidases to release the active nucleotide.523 For the natural product microcin C7 (226), the phosphoramidate motif is present in the active metabolite after prodrug cleavage. Finally, the N–P bond plays a critical role in the modern chemical synthesis of oligonucleotides.524
Biosynthetic strategies for N–P bond construction have been studied in the biosynthetic pathways that produce the natural products microcin C7 and fosfazinomycin. In the case of 226 biosynthesis, an enzyme catalyzing N–P bond formation has been characterized. Based on investigations of fosfazinomycin biosynthesis, a biosynthetic logic and potential enzymes have been proposed to installation an N–P bond. The biosynthetic strategies used in both pathways appear to involve catalysis of the attack of a nucleophilic nitrogen atom onto an activated, electrophilic phosphorus atom mediated by an ATP-dependent enzyme.
4.1. Phosphoramidate: Microcin C7
The microcins are a group of RiPPs produced by Enterobacteriaceae that exhibit antibiotic activity against closely related bacterial competitors.525,526 An N–P bond containing phosphoramidate functional group is found in 226 produced by E. coli.527 This phosphoramidate links a C-terminal adenosine monophosphate moiety with a seven-residue peptide backbone (Scheme 51). The Asp-NH-AMP ‘warhead’ of 226 inhibits protein synthesis after it is released by proteolytic cleavage in target cells.528,529
Scheme 51.

Maturation of microcin C7 involves an N–P bond formation step catalyzed by MccB
The microcin C7 (mcc) biosynthetic gene cluster is composed of six genes (mccABCDEF). The predicted adenylating enzyme, MccB, was shown to promote formation of the C-terminal N–P bond in vitro.530 Incubation of MccB with a synthetic precursor peptide, ATP, and Mg2+ provided two products: phosphoramidate 227 and succinimide 228.531 The succinimide 228 was isolated and converted to the phosphoramidate 227 in presence of MccB and ATP, indicating its intermediacy in the N–P bond forming reaction. Replacing Asn7 in the heptapeptide substrate with doubly-labeled 15N-Asn7 resulted in retention of both 15N in 227. Based on these observations, a mechanism for N–P bond formation was proposed (Scheme 52). First the C-terminal carboxylate of the precursor peptide is activated by ATP to form an acyladenylate intermediate 229. The nitrogen atom of the β-carboxamido group then attacks the activated carbonyl group, releasing AMP and forming the succinimide intermediate 227. Subsequently, the nitrogen atom of the succinimidyl group attacks the α-phosphate group of a second equivalent of ATP to form the N–P bond and generate 230. This intermediate is then hydrolyzed to afford 228, a late-stage intermediate in 226 biosynthesis (Scheme 52).
Scheme 52.

Proposed mechanism of MccB-mediated N–P formation in microcin C7 biosynthesis
While the N-terminal region of MccB shows no detectable homology to known enzymes, its C-terminal region resembles the adenylation domain of ubiquitin-like (UBL) protein-activating enzymes (E1s).532,533 Crystal structures of MccB have been obtained in both the ligand-free and ligand-bound form.534 A domain termed the “peptide clamp domain” is essential for substrate recognition. In the adenylation domain active site, Mg2+ is coordinated by ATP and D214 in a manner similar to other UBL-activating enzymes. It is proposed that MccB uses the unique peptide clamp domain to orient the nitrogen atom of the succinimide intermediate to attack the α-phosphate of ATP, forging the unusual N–P bond.
4.2. Phosphonamidate: Fosfazinomycin
An overview of the biosynthesis of fosfazinomycin and the enzymatic chemistry involved in constructing its hydrazide linkage was covered in Section 3.3.1. The biosynthesis of the phosphonamidate-containing 187 is believed to occur by coupling the late stage intermediates 189 and 190 to form N–P bond formation. Currently, there are three enzymes from the fzm cluster that have not been assigned a specific function and could be reasonable candidates for installing the N–P bond of the phosphonamidate group: a predicted asparagine synthase FzmA, a glutamine synthetase homolog FzmN, and a putative pyruvate carboxylase FzmF (Scheme 39). The closest homologs of these enzymes all use ATP to perform C–C and C–N bond forming reactions, suggesting that N–P bond formation likely involves activation of the phosphonate with ATP, followed by nucleophilic attack by the distal nitrogen atom of the hydrazide. If operative, this logic for N–P bond construction would be analogous to that seen in microcin C7 biosynthesis. However, there is not yet experimental evidence to support this proposal.429,430
5. S–S, S–O, and S–N bond forming enzymes
Whereas sulfur is an essential and ubiquitous component of proteins in the form of cysteine and methionine, sulfur-heteroatom (S–X) bond containing natural products are relatively scarce. A range of natural products with S–X bond containing functional groups have been isolated from variety of sources including plants, fungi, bacteria, and animals.535 Although these natural products possess diverse carbon scaffolds, their biological activities are often strongly associated with the specific chemical properties of the sulfur-containing moieties. The biosynthetic logic for construction of S–X bonds varies depending upon the specific linkage, and the enzymes involved in these transformations employ several different cofactors to carry out this transformation. Here, we will discuss the biosynthesis of four sulfur-containing functional groups: disulfide-, sulfoxide-, sulfone-, and sulfate-containing natural products. Additionally, due to structural considerations thiosulfinate-, sulfonamide-, and sulfamate-containing natural products will be discussed in the sulfoxide, sulfone, and sulfate sections, respectively.
5.1. Disulfide
5.1.1. Gliotoxin, holomycin, romidepsin
Disulfide bridges are well-known as key stabilizing components of peptide and protein tertiary structures. In addition to maintaining protein structures, disulfide functional groups can also mediate and regulate protein activities. In eukaryotic organisms, protein disulfide isomerase catalyzes disulfide bond formation and removes nonnative disulfide linkages in proteins.536 Gram-negative bacteria use thioredoxin-like enzymes to ensure the correct disulfide oxidation state during protein folding.537 Similarly, disulfide moieties in secondary metabolites are essential in conferring biological activity through the generation of reactive oxygen species and by inactivating protein targets via covalent adduct formation (Scheme 53). The biosyntheses of three disulfide bond containing natural products, gliotoxin (231)538,539, holomycin (232)540, and romidepsin (233)541 have been well-studied (Figure 25), and the enzymes involved in S–S bond formation are discussed below.
Scheme 53.

Two proposed mechanisms of action for gilotoxin
Figure 25.

Chemical structures of gilotoxin, holomyin, and romidepsin
Gliotoxin was first isolated from the fungus Gliocladim fimbriatum as a potent antibiotic with high fungicidal activity.542 It belongs to a class of epipolythiodioxopiperazine (ETP) toxins that feature a disulfide functional group embedded in a diketopiperazine scaffold.543 The initially proposed structure was reassigned in 1985.544 Gliotoxin’s activity has been extensively studied in mammalian cells545 since it initially showed inhibition of viral reverse transcriptase in a disulfide bond dependent manner.546 Two pathways have been suggested for gliotoxin’s mode of action, which include protein inactivation through covalent modification of susceptible cysteine residues and the generation of reactive oxygen species via gliotoxin-induced redox cycling (Scheme 53).
The gliotoxin (gli) biosynthetic gene cluster was discovered in Aspergillus fumigatus using a genome mining strategy based on its putative biosynthetic relationship to the related ETP natural product sirodesmin.547,548 The putative gli gene cluster contains twelve genes, and its connection to biosynthesis was confirmed by quantitative real time PCR and gene disruption experiments.549 The putative FAD-dependent thioredoxin-like oxidoreductase GliT was proposed to catalyze disulfide bond formation. Phylogenetic analysis of GliT placed it within a clade of poorly characterized oxidoreductases, including several other putative S–S bond forming homologs. Gene disruption of gliT resulted in complete abolishment of gliotoxin production supporting its involvement in biosynthesis.539 Heterologous expression and purification of GliT provided a protein that co-purified with FAD. When GliT was incubated in vitro with the reduced form of 231, conversion to gliotoxin was observed. A ΔgliT knock out strain was sensitive to exogenous gliotoxin, suggesting disulfide bond formation by GliT could be related to self-resistance.538,539
Two additional, related FAD-dependent thioredoxin-like disulfide oxidases, HlmI and DepH, have been biochemically characterized and shown to catalyze disulfide bond formation in holomycin and romidepsin biosynthesis, respectively.540,541 Interestingly, while all three enzymes utilize FAD as a cofactor they use different terminal oxidants to catalyze disulfide bond formation, with DepH using NADP+ and GliT and HlmI using molecular oxygen. To better understand the reaction mechanisms of these three enzymes, the structures of GilT, HlmI, and DepH were determined by X-ray crystallography.550,551 While all three enzymes share a conserved CxxC motif believed to be involved in S–S bond formation, the substrate binding sites are quite varied in their size, perhaps reflecting differences in their respective natural product substrates. Interestingly, while GliT showed cross-reactivity with reduced holomycin,540 HlmI and DepH failed to oxidize reduced gliotoxin.550
Based on biochemical and structural studies, a general mechanism for these enzymes has been proposed that is related to the analogous disulfide bond forming reaction that occurs in proteins (Scheme 54). Deprotonation of one of the thiols of the substrate is followed by nucleophilic attack of the resulting thiolate anion on a disulfide bond in the enzyme active site. This reaction cleaves the S–S bond of the protein disulfide and forms a new mixed disulfide linkage between the substrate and enzyme. The newly-formed enzyme-based thiolate anion then attacks the C4a atom of FAD to generate a second covalent linkage. A disulfide bond rearrangement then occurs which results in C–S bond cleavage to release the reduced flavin cofactor, formation of the disulfide bond on the natural product scaffold, and regeneration of the active site disulfide linkage. Finally, the FAD cofactor is regenerated via oxidation of FADH2 by molecular oxygen (GliT and HlmI) or NADP+ (DepH). The characterization of these enzymes should now allow for genome mining efforts to uncover new S–S bond containing natural products, in addition to understanding the distribution and prevalenc of this biosynthetic logic for constructing S–S bonds.
Scheme 54.

Proposed mechanism of disulfide formation catalyzed by flavin-dependent disulfide oxidases GilT, HImI, and DepH
5.2. Sulfoxide & Thiosulfinate
5.2.1. Sulfoxide: Ergothioneine, Ovothiol, Ustiloxin
Ergothioneine (234) is produced by certain fungi, mycobacteria, and streptomycetes including Aspergillus niger, Neurospora crassa, Micrococcus pyogenes var. aureus, and Streptomyces lactamdurans.552,553 The precise function of this histidine betaine derivative is unknown, but recent research suggests that it might play an important function in the human body.554 Mutations in a human gene encoding the ergothioneine transporter OCTN1 have been linked to Crohn’s disease.554 In streptomyces, 234 plays an essential role in protecting microbes from oxidative stress555 was recently shown to be involved in the biosynthesis of the antibiotic lincomycin A.556
Early assays with cell-free extracts from Neurospora crassa suggested the involvement of two 234 biosynthetic intermediates, hercynine (235) and hercynylcysteine sulfoxide (236), which could be synthesized from the amino acids L-methionine, L-cysteine, and L-histidine. 557,558 Incorporation of L-methionone is proposed to occur through conversion to S-adenosylmethionine (SAM) which is used as a methyl group donor to convert L-histidine to 235, suggesting the involvement of a SAM-dependent methyltransferase. The dependency of FeII and O2 in generating hercynylcysteine sulfoxide from hercynine suggested an FeII-dependent oxygenase might catalyze this transformation.
Recently, comparing methyltransferases in the genomes of ergothioneine producers and non-producers led to the identification of the ergothioneine (egt) biosynthetic gene cluster in Mycobacterium avium.559 Characterization of several of these enzymes in vitro supports the proposed biosynthetic pathway showed in Scheme 55.559 The SAM-dependent methyltransferase EgtD was shown to selectively convert L-histidine to 235. Transformation of 235 to 237 catalyzed by EgtB will be discussed below. Ergothioneine biosynthesis is completed by conversion of 237 to 236 by the glutamine amidotransferase homolog EgtC, which is followed by C–S bond cleavage to afford 234. This final C–S bond cleaving transformation to afford 234 is proposed to be catalyzed by the PLP-dependent enzyme EgtE, however, this activity could not be reconstituted in vitro due to failure to access soluble recombinant protein.
Scheme 55.

EgtB and OvoA catalyze C–S bond formation coupled with formation of a sulfoxide
EgtB is proposed to catalyze an oxidative sulfurization reaction that converts 235 to 237, simultaneously forming both S–O and C–S bonds. Interestingly, EgtB contains a conserved N-terminal sequence motif (HX3HXE) that was proposed to bind iron.560 Based on this motif, EgtB was proposed to be a non-heme mononuclear iron oxidase and thus a candidate for catalyzing the oxidative sulfurization that forms the sulfoxide group of 237. In the presence of FeII, γ-glutamine cysteine (γGC), and molecular oxygen, recombinant EgtB converted 235 to 237 in vitro. The substrate γ-glutamine cysteine was chosen because EgtA showed homology to γ-glutamyl cysteine ligases. These results demonstrated that EgtB was responsible for the unique oxidative sulfurization reaction that simultaneously forms S–O and C–S linkages in 234 biosynthesis.559
Ovothiol A (238) was discovered in sea urchin eggs as a hydrogen peroxide scavenger.561 Assays with cell-extracts from Crithidia fasciculate showed that L-cysteine and L-histidine could be converted to the 5-histidylcysteine sulfoxide intermediate (239) which is analogous to intermediate 237 formed in 234 biosynthesis.562 Using the presence of a putative a C-terminal methyltransferase domain as a criterion to distinguish 5-histidylcystine sulfoxide synthases (OvoAs) from EgtB homologs, 80 potential OvoAs were identified within nucleotide sequence databases. In the presence of L-histidine, L-cysteine, FeII, and oxygen, OvoAe (from E. tasmaiensis) generated 239. These results demonstatrate that, like EgtB, the enzyme OvoAe catalyzes an oxidative sulfurization reaction. Therefore, the putative non-heme mononuclear iron enzymes EgtB and OvoAe employ similar chemical logic that involves coupled S-oxygenation and C–S bond formation to generate their respective products (Scheme 55).563
The non-heme mononuclear iron enzyme family has been shown to catalyze a diverse range of oxidative transformations.564 EgtB and OvoAe both contain the 2-His-1-carboxylate facial triad motif that is proposed to coordinate iron and is conserved in most members of this family.565 One subfamily of non-heme mononuclear iron enzymes can couple the two-electron oxidation of α-ketoglutarate to succinate and carbon dioxide to generate a high-valent iron oxo-FeIV species (Scheme 56).566 This highly reactive intermediate can promote a variety of challenging oxidative reactions.
Scheme 56.

General mechanism for α-ketoglutarate-dependent non-heme mononuclear iron enzymes
The shared logic of coupled S-oxygenation and C–S bond formation observed for EgtB and OvoAe, as well as their homology to non-heme mononuclear iron enzymes, informed initial hypotheses regarding their mechanisms.563,567 As EgtB and OvaAe do not require α-ketoglutarate, it has been suggested that an iron oxo-FeIV species might instead be generated by oxidation of the thiol of cysteine or γGC to the sulfoxide.563,567 The crystal structure of EgtB from Mycobacterium thermoresistibile has been solved which has provided insight into this coupled S-oxygenation/C–S bond forming reaction.568 Three structures were obtained, including the apo protein, the protein in complex with FeII and 235, and the protein in complex with manganese, N-α-dimethyl histidine (DMH) and γGC. Interestingly, although the enzyme contains the conserved 2-His-1-carboxylate motif predicted to bind iron, these crystal structures revealed an unusual binding mode involving three histidine residues coordinating to the iron center.
Based on the geometry of the ligands in the active site of EgtB, a mechanism for EgtB-catalyzed sulfoxidation has been put forth (Scheme 57). Notably, in this mechanism an iron oxo-FeIV species is not proposed to mediate oxidative sulfurization contrary to previous suggestions. In this proposed mechanism, the FeII center is initially bound by 235 and water. Upon γGC and oxygen binding, single electron transfer from the FeII center to oxygen generates a superoxo-FeIII species. Electron transfer from the thiol ligand to the distal oxygen of the superoxo-FeIII species results in the formation of a peroxo-FeIII intermediate and a thiyl radical. The thiyl radical then attacks the imidazole C2 of 235 to form the C–S bond and give an iminyl radical. During this step, the peroxide anion is protonated by Tyr377. The iminyl radical is then rearomatized through deprotonation by Tyr377 and electron transfer from the iminyl radical back to the FeIII center to regenerate the resting FeII state. Finally, nucleophilic attack of the newly formed thioether intermediate on the hydroperoxo-FeII species forms the S–O bond and affords 237 thus completing the catalytic cycle.
Scheme 57.

Proposed mechanism for the C–S/S–O bond forming non-heme mononuclear iron enzyme EgtB
The non-heme mononuclear iron enzymes EgtB and OvoA catalyze both C–S bond formation and S-oxygenation in an O2-dependent manner. This reactivity is new for the non-heme mononuclear iron enzyme family. Interestingly, a single point mutation Tyr377Phe in EgtB alters the enzyme from a sulfoxide synthase producing 237 into a thiol dioxygenase which installs the C–S bond into 235 and oxidizes it to the sulfinic acid.569 This ability to alter the reactivity of EgtB indicates it is a candidate for further protein engineering.
The ustiloxins were first isolated from Ustilaginoidea virens, a pathogenic fungus that causes rice false smut.570 Ustiloxin H possesses a sulfoxide functional group (Scheme 58). Gene inactivation of ustF1, a gene from the ustiloxin biosynthesis gene cluster that encodes a putative FAD-dependent oxidoreductase, led to accumulation of the thioether S-deoxyustiloxin H (240), suggesting ustiloxin H arises from direct S-oxygenation of 240. In vitro assays with UstF1, which co-purified with FAD, resulted in the conversion of 240 to 51, thus demonstrating that UstF1 is involved in sulfoxide formation in 51 biosynthesis (Scheme 58).571 Based on the known modes of reactivity of flavin monooxygenase, it seems plausible that S-oxygenation of 240 involves attack of the nucleophilic S atom on the electrophilic distal oxygen of a flavin C4a-hydroperoxide intermediate.
Scheme 58.

Proposed biosynthetic pathway of ustiloxin H
5.2.2. Thiosulfinate: Allicin, Leinamycin
The thiosulfinate-containing molecule allicin (241) is the active ingredient of garlic, and this metabolite exhibits antibacterial and antifungal activities. Allicin is derived form the sulfoxide-containing molecule alliin (242) (Scheme 59).572 Using activity-guided protein purification and N-terminal protein sequence analysis, a pyridoxal 5′-phosphate (PLP) dependent C–S lyase/S–S bond forming enzyme, termed alliinase, was identified that catalyzed the transformation of alliin to allicin and generated ammonia and pyruvate as co-products.573 Two structures of alliinase from Alliium sativum L. were determined by X-ray crystallography.573 An aminoacrylate-PLP complex (243) was observed in the crystal structure of alliinase when incubated with PLP and the substrate analogue (+S)-allyl-L-cysteine. The aminoacrylate-PLP complex is believed to be an on-pathway intermediate when alliinase utilizes its natural substrate 242. This snapshot of alliinase in an intermediate stage of catalysis led to a proposed mechanism for C–S bond cleavage during conversion of 242 to 241 (Scheme 59).
Scheme 59.

Proposed biosynthesis of allicin from precursor alliin
In the presence of the substrate alliin, a covalent aldimine complex is formed (244). The α-proton of alliin is then abstracted affording the intermediate (245), which leads to C–S bond cleavage and the production of the intermediates allylsulfenic acid (246) and aminoacrylic acid (247) after hydrolysis from PLP. Two molecules of sulfenic acid condense to afford allicin via S–S bond formation. The observed pyruvate and ammonia by-products result from the hydrolysis of aminoacrylic acid.
Interestingly, the non-active substance 242 and the aliinase enzyme are contained in different cell compartments within the garlic plant.574 Only during mechanical injury or pathogen invasion, does alliinase encounter 242, leading to the hypothesis that 241 production is a chemical defense mechanism. This logic has been exploited for site-directed anticancer therapy, with system administration of alliin and an alliinase-conjugated antibody that targets specific tumor cells, thus localizing the allicin-generating reaction to cancerous tissue.575
Leinamycin (DC107) (248) was first isolated from a Streptomyces species in 1989 and exhibited potent antitumor activity against P388 lymphocytic leukemia in mice.576,577 The chemical structure of leinamycin was determined by NMR and X-ray crystallography, which revealed a thiosulfinate moiety.577 This unusual structural motif was later verified as the DNA-damaging “warhead”.578 A thiol-triggered DNA alkylation mechanism has been proposed (Scheme 60), involving initial attack of a thiol on this thiosulfinate warhead leading to formation of a cyclic 1,2-oxathiolan-5-one (249) via a sulfenic acid intermediate (250). The cyclic oxathiolanone then undergoes an intramolecular ring rearrangement to produce an episulfonium ion (251), which serves as an alkylating agent. Additionally, the release of a hydrodisulfide could cause further oxidative DNA damage via generation of reactive oxygen species.
Scheme 60.

Proposed mechanism of DNA alkylation by leinamycins
The leinamycin (lnm) biosynthetic gene cluster was identified using degenerate primers for thiazole-forming NRPS encoding genes.579,580 Leinamycin biosynthesis has been proposed to proceed through the dithiolane intermediate, although no enzymes have been identified that catalyze the S–S bond formation needed to install this ring system (Scheme 61). However, two cytochrome P450 oxidases, LnmA and LnmZ, were proposed to be involved in the oxidation of this fused diothiolane ring to give the thiosulfinate moiety.580
Scheme 61.

Proposal for installation of a key sulfur atom in leinamycin biosynthesis
Recently, a PLP-dependent domain (SH) and a domain of unknown function (DUF), both contained within the PKS LnmJ, have been characterized in vitro and shown to catalyze incorporation of one of the sulfur atoms of the thiosulfinate group (Scheme 61).581 In this reaction the DUF is proposed to catalyze C–S bond formation between L-cysteine and an ACP-tethered intermediate (249) to generate 250. The SH domain then catalyzes C–S bond cleavage to produce the thiol-containing intermediate 251, as well as ammonia and pyruvate co-products. While the precise mechanisms of the DUF and SH domain remain to be characterized, these results establish the origin of one of the sulfur atoms in the thiosulfinate moiety of leinamycin.
5.3. Sulfone & Sulfonamide
5.3.1. Sulfadixiamycins
The biosynthesis of the N–N bond containing natural product dixiamycin B was discussed in Section 3.4.1. A putative FAD-dependent oxidoreductase XiaH was characterized in vivo and show to be capable of catalyzing C–N, N–N, and C–O bond formation to generate a total of five distinct family members. Using a S. albus-based heterologous expression system, three additional molecules, termed the sulfadixiamycins, were isolated and structurally characterized.582 Two of these analogues contain a diaryl sulfone linkage, while the third has a bridging sulfonamide functional group (Figure 26).
Figure 26.

Chemical structures of the sulfadixiamycins A – C
To elucidate the genetic basis of the unusual sulfone and sulfonamide groups, putative oxygenase encoding genes from the xia gene cluster were disrupted in the S. albus expression system. Sulfadixiamycin production was abolished only in the xiaH mutant strain suggesting this enzyme is critical for producing these metabolites. Feeding xiamycin to a XiaH expression strain also led to formation of both the dixiamycins and sulfadixiamycins. It has been reported that bacterial cells generate small amounts of sulfur dioxide via the non-enzymatic oxidation of sulfite generated during sulfur assimilation.583 A radical-based mechanism involving sulfur dioxide has been proposed for the XiaH-mediated formation of the sulfadixiamycins (Scheme 62). Similar to its role in dixiamycin formation, XiaH may react with xiamycin to form a stabilized, substrate-based radical that can react with sulfur dioxide at multiple positions. Attack of an N-centered xiamycin radical on sulfur dioxide would forge the N–S bond of sulfadixiamycin A. This intermediate can undergo radical recombination with a C-centered xiamycin radical to afford sulfadixiamycin A (252). The production of the sulfone-containing sulfadixiamycins results from the reaction of C-centered xiamycin radicals with sulfur dioxide to form the C–S bonds of sulfadixiamycins B (253) and C (254). Sulfone and sulfonamide natural products are extremely rare with only one other known example – the sulfone-containing metabolite echinosulfone A, a bromoindole derivative isolated from a marine sponge.584 Currently, it is unclear if echinosulfone A or other unknown sulfone and sulfonamide natural products are biosynthesized using a similar strategy involving sulfur dioxide and radical chemistry.
Scheme 62.

Proposed mechanism for the formation of three sulfadixiamycins from xiamycin and sulfur dioxide by XiaH
5.4. Sulfamate & Sulfate
5.4.1. Sulfamate: Ascamycin, Dealanylascamycin
The nucleoside antibiotics ascamycin (ACM, 255) and dealanylascamycin (DACM, 256) are produced by Streptomyces sp. JCM9888.585 DACM exhibits broader spectrum antibiotic activities than 255, indicating that installation of the L-alanyl group might serve as a resistance mechanism in the native producer. 256 acts through the inhibition of protein synthesis.586 Both 255 and 256 contain an unusual N–S bond containing 5′-O-sulfamate moiety.
Recently, the gene cluster that encodes the biosynthesis of both 255 and 256 was discovered.587 Annotation of the acm gene cluster led to a biosynthetic proposal and identification of specific enzymes that may be responsible for sulfamate group installation (Scheme 63). The putative adenylsulfate kinase AcmB may use ATP to activate sulfate ions, forming 3′-phosphoadenosine-5′-phosphosulfate (PAPS). The sulfotransferase AcmK or the sulfate adenyltransferase complex AcmA-AcmW is then proposed to catalyze the attack of the 5′-OH of adenosine onto the activated sulfate donor, forming adenosine 5′-sulfonate (257). From here, the putative aminotransferase AcmN is proposed to catalyze the key S–N bond forming reaction to generate 2-deschloro-dealanylascamycin (258), thus installing the sulfamate group. The acm gene cluster encodes two additional enzymes that could be involved in sulfamate construction, a sulfatase AcmG and an acyldulfatase AcmI. The biosynthesis of 255 may be completed by chlorination of 258 and ligation of L-alanine catalyzed by AcmXY (flavin-dependent halogenases) and AcmDEF (alanyl-tRNA synthetase, esterase, alanyl-tRNA synthetase), respectively.587
Scheme 63.

Proposed biosynthetic pathway for ascamycin
5.4.2. Sulfate: Caprazamycins
The caprazamycins (CPZ) are liponucleoside antibiotics produced by Streptomyces sp. MK730-62F2 that display potent anti-Mycobacterium tuberculosis activity.588 Several newly discovered 2′-O-sulfated CPZ analogs raised interesting questions regarding the installation of the sulfate group. Enzymatic sulfation is usually catalyzed by a group of PAPS-dependent sulfotransferases. Consistent with this known pathway for sulfation, a homolog of these PAPS-dependent sulfotransferases (Cpz8) is encoded within the cpz gene cluster.589 A type III PKS (Cpz6) was also encoded in the cpz gene cluster, but this enzyme could not be readily linked to any structural features in the caprazamycins.590 Using a combination of bioinformatics, genetics, and chemical analysis, it was shown that Cpz6 participates in an unusual two-step sulfation mechanism (Scheme 64). This enzyme uses an iso-acyl starter unit, one molecule of malonyl-CoA, and one molecule of methylmalonyl-CoA to form pyrone products (259). The PAPS-dependent sulfotransferase Cpz8 then transfers a sulfate group from PAPS to these pyrones. The sulfated pyrones then serve as sulfate carriers, delivering the sulfate group to a pyrone-dependent sulfotransferase (Cpz4). Cpz4 catalyzes the S–O bond forming sulfation reaction of the CPZ scaffold to afford the final 2″-O-sulfated CPZ analogs.591 Given that sulfate groups are usually installed using PAPS as the sulfate donor, this pathway represents a novel mechanism for generating this functional group.
Scheme 64.

Two-step, polyketide-mediated sulfation strategy involved in CPZ biosynthesis
Concluding Remarks
A variety of enzymatic chemistry has evolved to construct X–X bond containing functional groups within all major classes of natural products, reflecting biological significance of these structural motifs. While natural products containing X–X bonds have been known for many decades, it has been only relatively recently that we have begun to decipher the biosynthetic logic involved in the installation of these linkages. Rapid progress in understanding the enzymatic chemistry of X–X bond construction has been made in the last ten years, particularly in the context of N–O bonds. These advances have been possible due to rapidly increasing access to genome sequencing data, which has allowed for the discovery and characterization of biosynthetic gene clusters associated with these rare natural products and facilitated comparative genomic analyses that have assisted in identifying key enyzmes. Given the importance of these functional groups in conferring biological activity, further investigation into the biosynthesis of this class of natural products is warranted. We anticipate that progress in the coming years will provide insights into the mechanisms of additional classes of X–X bond forming enzymes and see the application of these enzymes and biosynthetic pathways in biocatalysis and synthetic biology.
Acknowledgments
We gratefully acknowledge financial support from Harvard University, the Searle Scholars Program and the NIH (DP2 GM105434). A.J.W. acknowledges fellowship support from the NIH (GM095450) and the Bristol-Myers Squibb Graduate Fellowship.
Biographies
Abraham J. Waldman attended the University of Pennsylvania where he graduated summa cum laude and Phi Beta Kappa with a B.A. in Biochemistry. In 2012 he started his Ph.D. in Chemistry at Harvard University where he works in the lab of Prof. Emily P. Balskus. His graduate studies have focused on elucidating the biosynthetic logic and enzymatic chemistry involved in constructing diazo-containing natural products. Abraham’s interests include traveling, cooking, being outdoors, and staying active.
Tai L. Ng received his B.S. in bioengineering from University of California, Berkeley in 2014. He then started his Ph.D. in chemistry at Harvard University under the supervision of Prof. Emily P. Balskus. He is currently studying the biosynthesis of N–N bond containing compounds and the mechanisms of novel enzymes implicated in N–N bond formation.
Peng Wang obtained his B.S. degree from the School of Life Sciences at East China Normal University and his Ph.D. degree from the Department of Chemical and Biomolecular Engineering at University of California, Los Angeles under the mentorship of Prof. Yi Tang. In 2013, he started at Harvard University, where he is currently a Postdoctoral Research Associate in Prof. Emily P. Balskus’s group. He is currently studying the biosynthesis of unusual natural products.
Emily P. Balskus received her B.A. in chemistry from Williams College in 2002. After obtaining an M.Phil from the University of Cambridge working with Prof. Steven Ley, she pursued graduate studies at Harvard University under the direction of Prof. Eric Jacobsen and received her Ph.D. in 2008. Emily was then an NIH postdoctoral fellow at Harvard Medical School in the lab of Prof. Christopher T. Walsh. Emily joined the faculty at Harvard University in 2011 where she is currently the Morris Kahn Associate Professor of Chemistry and Chemical Biology. Her research interests lie at the interface of chemistry and microbiology and include the discovery of new microbial enzymes, metabolites, and metabolic pathways, as well as the development of chemical approaches to study microbes and microbial communities.
Footnotes
Notes
The authors declare no competing financial interests.
References
- 1.Singaram S, Lawrence RS, Hornemann U. Studies on the Biosynthesis of the Antibiotic Streptozotocin (Streptozocin) by Streptomyces achromogenes var. streptozoticus. Feeding Experiments with Carbon-14 and Tritium Labelled Precursors. J Antibiot. 1979;32:379–385. doi: 10.7164/antibiotics.32.379. [DOI] [PubMed] [Google Scholar]
- 2.Lambert MP, Neuhaus FC. Mechanism of D-cycloserine Action: Alanine Racemase from Escherichia coli W. J Bacteriol. 1972;110:978–987. doi: 10.1128/jb.110.3.978-987.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Westlake DWS. Biosynthesis of Chloramphenicol. Biotech Bioeng. 1969;11:1125–1134. doi: 10.1002/bit.260110609. [DOI] [PubMed] [Google Scholar]
- 4.Colis LC, Woo CM, Hegan DC, Li Z, Glazer PM, Herzon SB. The Cytotoxicity of (−)-Lomaiviticin A Arises from Induction of Double-Strand Breaks in DNA. Nat Chem. 2014;6:504–510. doi: 10.1038/nchem.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walker S, Gange D, Gupta V, Kahne D. Analysis of Hydroxylamine Glycosidic Linkages: Structural Consequences of the NO Bond in Calicheamicin. J Am Chem Soc. 1994;116:3197–3206. [Google Scholar]
- 6.Ciufolini MA, Xi N. Synthesis, Chemistry and Conformational Properties of Piperazic Acids. Chem Soc Rev. 1998;27:437–445. [Google Scholar]
- 7.Oelke AJ, France DJ, Hofmann T, Wuitschik G, Ley SV. Piperazic Acid-containing Natural Products: Isolation, Biological Relevance and Total Synthesis. Nat Prod Rep. 2011;28:1445–1471. doi: 10.1039/c1np00041a. [DOI] [PubMed] [Google Scholar]
- 8.Xi N, Alemany LB, Ciufolini MA. Elevated Conformational Rigidity in Dipeptides Incorporating Piperazic Acid Derivatives. J Am Chem Soc. 1998;120:80–86. [Google Scholar]
- 9.Welsch ME, Snyder SA, Stockwell BR. Privileged Scaffolds for Library Design and Drug Discovery. Curr Opin Chem Biol. 2010;14:347–361. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGrath NA, Brichacek M, Njardarson JT. A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J Chem Educ. 2010;87:1348–1349. [Google Scholar]
- 11.Evans LE, Cheeseman MD, Jones K. N–N Bond-Forming Cyclization for the One-Pot Synthesis of N-Aryl[3:4-d]-pyrazolopyrimidines. Org Lett. 2012;14:3546–3549. doi: 10.1021/ol301561a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stokes BJ, Vogel CV, Urnezis LK, Pan M, Driver TG. Intramolecular Fe(II)-Catalyzed N–O or N–N Bond Formation from Aryl Azides. Org Lett. 2010;12:2884–2887. doi: 10.1021/ol101040p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Correa A, Tellitu I, Dominguez E, SanMartin R. Novel Alternative for the N–N Bond Formation Through a PIFA-Mediated Oxidative Cyclization and Its Application to the Synthesis of Indazol-3-ones. J Org Chem. 2006;71:3501–3505. doi: 10.1021/jo060070+. [DOI] [PubMed] [Google Scholar]
- 14.Majumdar P, Pati A, Patra M, Behera RK, Behera AK. Acid Hydrazides, Potent Reagents for Synthesis of Oxygen-, Nitrogen-, and/or Sulfur-containing Heterocyclic Rings. Chem Rev. 2014;114:2942–2977. doi: 10.1021/cr300122t. [DOI] [PubMed] [Google Scholar]
- 15.Rosen BR, Werner EW, O’Brien AG, Baran PS. Total Synthesis of Dixiamycin B by Electrochemical Oxidation. J Am Chem Soc. 2014;136:5571–5574. doi: 10.1021/ja5013323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heil J, Vereecken H, Brüggemann N. A Review of Chemical Reactions of Nitrification Intermediates and Their Role in Nitrogen Cycling and Nitrogen Trace Gas Formation in Soil. Eur J Soil Sci. 2016;67:23–39. [Google Scholar]
- 17.Kuenen JG. Anammox Bacteria: From Discovery to Application. Nat Rev Microbiol. 2008;6:320–326. doi: 10.1038/nrmicro1857. [DOI] [PubMed] [Google Scholar]
- 18.Alderton WK, Cooper CE, Knowles RG. Nitric Oxide Synthases: Structure, Function, and Inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adams JA. Kinetic and Catalytic Mechanisms of Protein Kinases. Chem Rev. 2001;101:2771–2290. doi: 10.1021/cr000230w. [DOI] [PubMed] [Google Scholar]
- 20.Daigle DM, McKay GA, Thompson PR, Wright GD. Aminoglycoside Antibiotic Phosphotransferases are also Serine Protein Kinases. Chem Biol. 1998;6:11–18. doi: 10.1016/S1074-5521(99)80016-7. [DOI] [PubMed] [Google Scholar]
- 21.Neumann CS, Jiang W, Heemstra JR, Jr, Gontang EA, Kolter R, Walsh CT. Biosynthesis of Piperazic Acid via N5-hydroxy-ornithine in Kutzneria spp. 744. ChemBioChem. 2012;13:972–976. doi: 10.1002/cbic.201200054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parry RJ, Li W. An NADPH:FAD Oxidoreductase from the Valanimycin Producer, Streptomyces viridifaciens. J Biol Chem. 1997;272:23303–23311. doi: 10.1074/jbc.272.37.23303. [DOI] [PubMed] [Google Scholar]
- 23.Hider RC, Kong X. Chemistry and Biology of Siderophores. Nat Prod Rep. 2010;27:637–657. doi: 10.1039/b906679a. [DOI] [PubMed] [Google Scholar]
- 24.Rani R, Granchi C. Bioactive Heterocycles Containing Endocyclic N-hydroxy Groups. Eur J Med Chem. 2015;97:505–524. doi: 10.1016/j.ejmech.2014.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Structures of a Histone Deacetylase Homologue Bound to the TSA and SAHA Inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 26.Melman A. The Chemistry of Hydroxylamines, Oximes and Hydroxamic Acids. John Wiley & Sons, Ltd; 2008. [DOI] [Google Scholar]
- 27.Chaiyen P, Fraaije MW, Mattevi A. The Enigmatic Reaction of Flavins with Oxygen. Trends Biochem Sci. 2012;37:373–380. doi: 10.1016/j.tibs.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 28.Van Berkel WJ, Kamerbeek NM, Fraaije MW. Flavoprotein Monooxygenases, A Diverse Class of Oxidative Biocatalysts. J Biotechnol. 2006;124:670–689. doi: 10.1016/j.jbiotec.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 29.Walsh CT, Wencewicz TA. Flavoenzymes: Versatile Catalysts in Biosynthetic Pathways. Nat Prod Rep. 2013;30:175–200. doi: 10.1039/c2np20069d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bak S, Beisson F, Bishop G, Hamberger B, Hofer R, Paquette S, Werck-Reichhart D. Cytochromes p450. Arabidopsis Book. 2011;9:e0144. doi: 10.1199/tab.0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meunier B, de Visser SP, Shaik S. Mechanism of Oxidation Reactions Catalyzed by Cytochrome p450 Enzymes. Chem Rev. 2004;104:3947–3979. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 32.Ortiz de Montellano PR, De Voss JJ. Oxidizing Species in the Mechanism of Cytochrome p450. Nat Prod Rep. 2002;19:477–493. doi: 10.1039/b101297p. [DOI] [PubMed] [Google Scholar]
- 33.Thibodeaux CJ, Chang WC, Liu HW. Enzymatic Chemistry of Cyclopropane, Epoxide, and Aziridine Biosynthesis. Chem Rev. 2012;112:1681–1709. doi: 10.1021/cr200073d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewis JC, Coelho PS, Arnold FH. Enzymatic Functionalization of Carbon–Hydrogen Bonds. Chem Soc Rev. 2011;40:2003–2021. doi: 10.1039/c0cs00067a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandy MBA. Microbial Iron Acquisition Marine and Terrestrial Siderophores. Chem Rev. 2009;109:4580–4595. doi: 10.1021/cr9002787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Challis GL. A Widely Distributed Bacterial Pathway for Siderophore Biosynthesis Independent of Nonribosomal Peptide Synthetases. ChemBioChem. 2005;6:601–611. doi: 10.1002/cbic.200400283. [DOI] [PubMed] [Google Scholar]
- 37.Franke J, Ishida K, Ishida-Ito M, Hertweck C. Nitro versus Hydroxamate in Siderophores of Pathogenic Bacteria: Effect of Missing Hydroxylamine Protection in Malleobactin Biosynthesis. Angew Chem Int Ed. 2013;52:8271–8275. doi: 10.1002/anie.201303196. [DOI] [PubMed] [Google Scholar]
- 38.Sokol PADP, Woods DE, Mahenthiralingam E, Kooi C. Role of Ornibactin Biosynthesis in the Virulence of Burkholderia cepacia: Characterization of pvdA the Gene Encoding L-ornithine N(5)-oxygenase. Infect Immun. 1999;67:4443–4455. doi: 10.1128/iai.67.9.4443-4455.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cendrowski S, MacArthur W, Hanna P. Bacillus anthracis Requires Siderophore Biosynthesis for Growth in Macrophages and Mouse Virulence. Mol Microbiol. 2004;51:407–417. doi: 10.1046/j.1365-2958.2003.03861.x. [DOI] [PubMed] [Google Scholar]
- 40.Dale SE, Doherty-Kirby A, Lajoie G, Heinrichs DE. Role of Siderophore Biosynthesis in Virulence of Staphylococcus aureus: Identification and Characterization of Genes Involved in Production of a Siderophore. Infect Immun. 2003;72:29–37. doi: 10.1128/IAI.72.1.29-37.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schrettl M, Bignell E, Kragl C, Joechl C, Rogers T, Arst HN, Jr, Haynes K, Haas H. Siderophore Biosynthesis But Not Reductive Iron Assimilation is Essential for Aspergillus fumigatus Virulence. J Exp Med. 2004;200:1213–1219. doi: 10.1084/jem.20041242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frederick RE, Mayfield JA, DuBois JL. Iron Trafficking As An Antimicrobial Target. Biometals. 2009;22:583–593. doi: 10.1007/s10534-009-9236-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hissen AH, Wan AN, Warwas ML, Pinto LJ, Moore MM. The Aspergillus fumigatus Siderophore Biosynthetic Gene sidA, Encoding L-ornithine N5-oxygenase, is Required for Virulence. Infect Immun. 2005;73:5493–5503. doi: 10.1128/IAI.73.9.5493-5503.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kang HY, Brickman TJ, Beaumont FC, Armstrong SK. Identification and Characterization of Iron-regulated Bordetella pertussis Alcaligin Siderophore Biosynthesis Genes. J Bacteriol. 1996;178:4877–4884. doi: 10.1128/jb.178.16.4877-4884.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pohlmann V, Marahiel MA. Delta-Amino Group Hydroxylation of L-ornithine During Coelichelin Biosynthesis. Org Biomol Chem. 2008;6:1843–1848. doi: 10.1039/b801016a. [DOI] [PubMed] [Google Scholar]
- 46.Kadi N, Song L, Challis GL. Bisucaberin Biosynthesis: An Adenylating Domain of the BibC Multi-enzyme Catalyzes Cyclodimerization of N-hydroxy-N-succinylcadaverine. Chem Commun (Camb) 2008:5119–5121. doi: 10.1039/b813029a. [DOI] [PubMed] [Google Scholar]
- 47.Barona-Gómez F, Wong U, Giannakopulos AE, Derrick PJ, Challis GL. Identification of a Cluster of Genes that Directs Desferrioxamine Biosynthesis in Streptomyces coelicolor M145. J Am Chem Soc. 2004;126:16282–16283. doi: 10.1021/ja045774k. [DOI] [PubMed] [Google Scholar]
- 48.Robbel L, Helmetag V, Knappe TA, Marahiel MA. Consecutive Enzymatic Modification of Ornithine Generates the Hydroxamate Moieties of the Siderophore Erythrochelin. Biochemistry. 2011;50:6073–6080. doi: 10.1021/bi200699x. [DOI] [PubMed] [Google Scholar]
- 49.Dimise EJ, Widboom PF, Bruner SD. Structure Elucidation and Biosynthesis of Fuscachelins, Peptide Siderophores from the Moderate Thermophile Thermobifida fusca. Proc Natl Acad Sci U S A. 2008;105:15311–15316. doi: 10.1073/pnas.0805451105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thariath A, Socha D, Valvano MA, Viswanatha T. Construction and Biochemical Characterization of Recombinant Cytoplasmic Forms of the IucD Protein (Lysine:N6-Hydroxylase) Encoded by the pColV-K30 Aerobactin Gene Cluster. J Bacteriol. 1993;175:589–596. doi: 10.1128/jb.175.3.589-596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robinson RM, Rodriguez PJ, Sobrado P. Mechanistic Studies on the Flavin-dependent N(6)-lysine Monooxygenase MbsG Reveal An Unusual Control for Catalysis. Arch Biochem Biophys. 2014;550–551:58–66. doi: 10.1016/j.abb.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 52.Binda C, Robinson RM, Martin Del Campo JS, Keul ND, Rodriguez PJ, Robinson HH, Mattevi A, Sobrado P. An Unprecedented NADPH Domain Conformation in Lysine Monooxygenase NbtG Provides Insights into Uncoupling of Oxygen Consumption from Substrate Hydroxylation. J Biol Chem. 2015;290:12676–12688. doi: 10.1074/jbc.M114.629485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ambrosi C, Leoni L, Putignani L, Orsi N, Visca P. Pseudobactin Biogenesis in the Plant Growth-Promoting Rhizobacterium Pseudomonas strain B10: Identification and Functional Analysis of the L-ornithine N(5)-Oxygenase (psbA) Gene. J Bacteriol. 2000;182:6233–6238. doi: 10.1128/jb.182.21.6233-6238.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ge L, Seah SY. Heterologous Expression, Purification, and Characterization of an L-ornithine N(5)-Hydroxylase Involved in Pyoverdine Siderophore Biosynthesis in Pseudomonas aeruginosa. J Bacteriol. 2006;188:7205–7210. doi: 10.1128/JB.00949-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lynch D, O’Brien J, Welch T, Clarke P, Cuiv PO, Crosa JH, O’Connell M. Genetic Organization of the Region Encoding Regulation, Biosynthesis, and Transport of Rhizobactin 1021: a Siderophore Produced by Sinorhizobium meliloti. J Bacteriol. 2001;183:2576–2585. doi: 10.1128/JB.183.8.2576-2585.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bosello M, Mielcarek A, Giessen TW, Marahiel MA. An Enzymatic Pathway for the Biosynthesis of the Formylhydroxyornithine Required for Rhodochelin Iron Coordination. Biochemistry. 2012;51:3059–3066. doi: 10.1021/bi201837f. [DOI] [PubMed] [Google Scholar]
- 57.Chocklett SW, Sobrado P. Aspergillus fumigatus SidA is a Highly Specific Ornithine Hydroxylase with Bound Flavin Cofactor. Biochemistry. 2010;49:6777–6783. doi: 10.1021/bi100291n. [DOI] [PubMed] [Google Scholar]
- 58.RHJ, Walsh CT, Sattely ES. Enzymatic Tailoring of Ornithine in the Biosynthesis of the Rhizobium Cyclic Trihydroxamate Siderophore Vicibactin. J Am Chem Soc. 2009;131:15317–15329. doi: 10.1021/ja9056008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Crosa JH, Walsh CT. Genetics and Assembly Line Enzymology of Siderophore Biosynthesis in Bacteria. Microbiol Mol Biol Rev. 2002;66:223–249. doi: 10.1128/MMBR.66.2.223-249.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barry SM, Challis GL. Recent Advances in Siderophore Biosynthesis. Curr Opin Chem Biol. 2009;13:205–215. doi: 10.1016/j.cbpa.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 61.Franceschini S, Fedkenheuer M, Vogelaar NJ, Robinson HH, Sobrado P, Mattevi A. Structural Insight into the Mechanism of Oxygen Activation and Substrate Selectivity of Flavin-dependent N-hydroxylating Monooxygenases. Biochemistry. 2012;51:7043–7045. doi: 10.1021/bi301072w. [DOI] [PubMed] [Google Scholar]
- 62.Olucha J, Meneely KM, Chilton AS, Lamb AL. Two Structures of an N-hydroxylating Flavoprotein Monooxygenase: Ornithine Hydroxylase from Pseudomonas aeruginosa. J Biol Chem. 2011;286:31789–31798. doi: 10.1074/jbc.M111.265876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meneely KM, Barr EW, Bollinger JM, Jr, Lamb AL. Kinetic Mechanism of Ornithine Hydroxylase (PvdA) from Pseudomonas aeruginosa: Substrate Triggering of O2 Addition but not Flavin Reduction. Biochemistry. 2009;48:4371–4376. doi: 10.1021/bi900442z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mayfield JA, Frederick RE, Streit BR, Wencewicz TA, Ballou DP, DuBois JL. Comprehensive Spectroscopic, Steady State, and Transient Kinetic Studies of a Representative Siderophore-associated Flavin Monooxygenase. J Biol Chem. 2010;285:30375–30388. doi: 10.1074/jbc.M110.157578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Olucha J, Lamb AL. Mechanistic and Structural Studies of the N-hydroxylating Flavoprotein Monooxygenases. Bioorg Chem. 2011;39:171–177. doi: 10.1016/j.bioorg.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meneely KM, Lamb AL. Biochemical characterization of a Flavin Adenine Dinucleotide-dependent Monooxygenase, Ornithine Hydroxylase from Pseudomonas aeruginosa, Suggests a Novel Reaction Mechanism. Biochemistry. 2007;46:11930–11937. doi: 10.1021/bi700932q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Robinson R, Franceschini S, Fedkenheuer M, Rodriguez PJ, Ellerbrock J, Romero E, Echandi MP, Martin Del Campo JS, Sobrado P. Arg279 is the Key Regulator of Coenzyme Selectivity in the Flavin-dependent Ornithine Monooxygenase SidA. Biochim Biophys Acta. 2014;1844:778–784. doi: 10.1016/j.bbapap.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 68.Baron R, Riley C, Chenprakhon P, Thotsaporn K, Winter RT, Alfieri A, Forneris F, van Berkel WJ, Chaiyen P, Fraaije MW, et al. Multiple Pathways Guide Oxygen Diffusion into Flavoenzyme Active Sites. Proc Natl Acad Sci U S A. 2009;106:10603–10608. doi: 10.1073/pnas.0903809106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Badieyan S, Bach RD, Sobrado P. Mechanism of N-hydroxylation Catalyzed by Flavindependent Monooxygenases. J Org Chem. 2015;80:2139–2147. doi: 10.1021/jo502651v. [DOI] [PubMed] [Google Scholar]
- 70.Romero E, Fedkenheuer M, Chocklett SW, Qi J, Oppenheimer M, Sobrado P. Dual Role of NADP(H) in the Reaction of a Flavin Dependent N-hydroxylating Monooxygenase. Biochim Biophys Acta. 2012;1824:850–857. doi: 10.1016/j.bbapap.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 71.Alfieri A, Malito E, Orru R, Fraaije MW, Mattevi A. Revealing the Moonlighting Role of NADP in the Structure of a Flavin-containing Monooxygenase. Proc Natl Acad Sci U S A. 2008;105:6572–6577. doi: 10.1073/pnas.0800859105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shirey C, Badieyan S, Sobrado P. Role of Ser-257 in the Sliding Mechanism of NADP(H) in the Reaction Catalyzed by the Aspergillus fumigatus Flavin-dependent Ornithine N5-Monooxygenase SidA. J Biol Chem. 2013;288:32440–32448. doi: 10.1074/jbc.M113.487181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Orru R, Pazmiño DE, Fraaije MW, Mattevi A. Joint Functions of Protein Residues and NADP(H) in Oxygen Activation by Flavin-containing Monooxygenase. J Biol Chem. 2010;285:35021–35028. doi: 10.1074/jbc.M110.161372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sucharitakul J, Wongnate T, Chaiyen P. Hydrogen Peroxide Elimination from C4ahydroperoxyflavin in a Flavoprotein Oxidase Occurs through a Single Proton Transfer from Flavin N5 to a Peroxide Leaving Group. J Biol Chem. 2011;286:16900–16909. doi: 10.1074/jbc.M111.222976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Macheroux P, Plattner HJ, Romaguera A, Diekmann H. FAD and Substrate Analogs as Probes for Lysine N6-hydroxylase from Escherichia coli EN 222. Eur J Biochem. 1993;213:995–1002. doi: 10.1111/j.1432-1033.1993.tb17846.x. [DOI] [PubMed] [Google Scholar]
- 76.Frederick RE, Ojha S, Lamb A, Dubois JL. How pH Modulates the Reactivity and Selectivity of a Siderophore-associated Flavin Monooxygenase. Biochemistry. 2014;53:2007–2016. doi: 10.1021/bi401256b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jalal MAF, Hossain MB, Van der Helm D. Structure of Anticancer Antibiotic LAlanosine. Acta Cryst. 1986;42:733–738. [Google Scholar]
- 78.Iinuma H, Kondo S, Takeuchi T, Umezawa H. Production and Isolation of Dopastin, an Inhibitor of Dopamine and beta-Hydroxylase. Agric Biol Chem. 1974;38:2093–2097. [Google Scholar]
- 79.Parry RJ, Li W. Purification and Characterization of Isobutylamine N-hydroxylase from the Valanimycin Producer Streptomyces viridifaciens MG456-hF10. Arch Biochem Biophys. 1997;339:47–54. doi: 10.1006/abbi.1996.9857. [DOI] [PubMed] [Google Scholar]
- 80.Sono M, Roach MP, Coulter ED, Dawson JH. Heme-Containing Oxygenases. Chem Rev. 1996;96:2841–2888. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 81.Bentzien J, Hickey ER, Kemper RA, Brewer ML, Dyekjaer JD, East SP, Whittaker M. An in silico Method for Predicting Ames Activities of Primary Aromatic Amines by Calculating the Stabilities of Nitrenium Ions. J Chem Inf Model. 2010;50:274–297. doi: 10.1021/ci900378x. [DOI] [PubMed] [Google Scholar]
- 82.Ripa L, Mee C, Sjo P, Shamovsky I. Theoretical Studies of the Mechanism of Nhydroxylation of Primary Aromatic Amines by Cytochrome p450 1A2: Radicaloid or Anionic? Chem Res Toxicol. 2014;27:265–278. doi: 10.1021/tx400376u. [DOI] [PubMed] [Google Scholar]
- 83.Ji L, Schuurmann G. Model and Mechanism: N-hydroxylation of Primary Aromatic Amines by Cytochrome P450. Angew Chem Int Ed. 2013;52:744–748. doi: 10.1002/anie.201204116. [DOI] [PubMed] [Google Scholar]
- 84.Crane BR, Sudhamsu J, Patel BA. Bacterial Nitric Oxide Synthases. Annu Rev Biochem. 2010;79:445–470. doi: 10.1146/annurev-biochem-062608-103436. [DOI] [PubMed] [Google Scholar]
- 85.Davydov R, Hoffman BM. Active Intermediates in Heme Mono Reactions as Revealed by Cryoreduction/Annealing, EPR/ENDOR Studies. Arch Biochem Biophys. 2011;507:36–43. doi: 10.1016/j.abb.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Davydov R, Ledbetter-Rogers A, Martasek P, Larukhin M, Sono M, Dawson JH, Masters BS, Hoffman BM. EPR and ENDOR Characterization of Intermediates in the Cryoreduced Oxy-Nitric Oxide Synthase Heme Domain with Bound L-arginine or N(G)-hydroxyarginine. Biochemistry. 2002;41:10375–10381. doi: 10.1021/bi0260637. [DOI] [PubMed] [Google Scholar]
- 87.Davydov R, Sudhamsu J, Lees NS, Crane BR, Hoffman BM. EPR and ENDOR Characterization of the Reactive Intermediates in the Generation of NO by Cryoreduced Oxy-Nitric Oxide Synthase from Geobacillus stearothermophilus. J Am Chem Soc. 2009;131:14493–14507. doi: 10.1021/ja906133h. [DOI] [PubMed] [Google Scholar]
- 88.Jansen Labby K, Li H, Roman LJ, Martasek P, Poulos TL, Silverman RB. Methylated N(omega)-hydroxy-L-arginine Analogues as Mechanistic Probes for the Second Step of the Nitric Oxide Synthase-catalyzed Reaction. Biochemistry. 2013;52:3062–3073. doi: 10.1021/bi301571v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Johnson HD, Thorson JS. Characterization of CalE10, the N-oxidase Involved in Calicheamicin Hydroxyaminosugar Formation. J Am Chem Soc. 2008;130:17762–17763. doi: 10.1021/ja807557a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim D, Guengerich FP. Cytochrome P450 Activation of Arylamines and Heterocyclic Amines. Annu Rev Pharmacol Toxicol. 2005;45:27–49. doi: 10.1146/annurev.pharmtox.45.120403.100010. [DOI] [PubMed] [Google Scholar]
- 91.Skipper PL, Kim MY, Sun HL, Wogan GN, Tannenbaum SR. Monocyclic Aromatic Amines as Potential Human Carcinogens: Old is New Again. Carcinogenesis. 2010;31:50–58. doi: 10.1093/carcin/bgp267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Knize MG, Felton JS. Formation and Human Risk of Carcinogenic Heterocyclic Amines Formed from Natural Precursors in Meat. Nutr Rev. 2005;63:158–165. doi: 10.1111/j.1753-4887.2005.tb00133.x. [DOI] [PubMed] [Google Scholar]
- 93.Stolzenberg-Solomon RZ, Cross AJ, Silverman DT, Schairer C, Thompson FE, Kipnis V, Subar AF, Hollenbeck A, Schatzkin A, Sinha R. Meat and Meat-Mutagen Intake and Pancreatic Cancer Risk in the NIH-AARP Cohort. Cancer Epidemiol Biomarkers Prev. 2007;16:2664–2675. doi: 10.1158/1055-9965.EPI-07-0378. [DOI] [PubMed] [Google Scholar]
- 94.Cross AJ, Peters U, Kirsh VA, Andriole GL, Reding D, Hayes RB, Sinha R. A Prospective Study of Meat and Meat Mutagens and Prostate Cancer Risk. Cancer Res. 2005;65:11779–11784. doi: 10.1158/0008-5472.CAN-05-2191. [DOI] [PubMed] [Google Scholar]
- 95.Bolleddula J, DeMent K, Driscoll JP, Worboys P, Brassil PJ, Bourdet DL. Biotransformation and Bioactivation Reactions of Alicyclic Amines in Drug Molecules. Drug Metab Rev. 2014;46:379–419. doi: 10.3109/03602532.2014.924962. [DOI] [PubMed] [Google Scholar]
- 96.Iyanagi T. Structure and Function of NADPH-Cytochrome P450 Reductase and Nitric Oxide Synthase Reductase Domain. Biochem Biophys Res Commun. 2005;338:520–528. doi: 10.1016/j.bbrc.2005.08.043. [DOI] [PubMed] [Google Scholar]
- 97.Kersten RD, Dorrestein PC. Metalloenzymes: Natural Product Nitrosation. Nat Chem Biol. 2010;6:636–637. doi: 10.1038/nchembio.425. [DOI] [PubMed] [Google Scholar]
- 98.Makris TM, Vu VV, Meier KK, Komor AJ, Rivard BS, Munck E, Que L, Jr, Lipscomb JD. An Unusual Peroxo Intermediate of the Arylamine Oxygenase of the Chloramphenicol Biosynthetic Pathway. J Am Chem Soc. 2015;137:1608–1617. doi: 10.1021/ja511649n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhu Y, Silverman RB. Revisiting Heme Mechanisms. A Perspective on the Mechanisms of Nitric Oxide. Biochemistry. 2008;47:2231–2243. doi: 10.1021/bi7023817. [DOI] [PubMed] [Google Scholar]
- 100.Maiese WM, Lechevalier MP, Lechevalier HA, Korshalla J, Kuck N, Fantini A, Wildey MJ, Thomas J, Greenstein M. Calicheamicins, a Novel Family of Antitumor Antibiotics: Taxonomy, Fermentation and Biological Properties. J Antibiot. 1989;42:558–563. doi: 10.7164/antibiotics.42.558. [DOI] [PubMed] [Google Scholar]
- 101.Konishi M, Ohkuma H, Saitoh K, Kawaguchi H, Golik J, Dubay G, Groenewold G, Krishnan B, Doyle TW. Esperamicins, a Novel Class of Potent Antitumor Antibiotics. I. Physico-chemical Data and Partial Structure. J Antibiot. 1985;38:1605–1609. doi: 10.7164/antibiotics.38.1605. [DOI] [PubMed] [Google Scholar]
- 102.Golik J, Clardy J, Dubay G, Groenewold G, Kawaguchi H, Konishi M, Krishnan B, Ohkuma H, Saitoh K, Doyle TW. Esperamicins, a Novel Class of Potent Antitumor Antibiotics. 2. Structure of Esperamicin X. J Am Chem Soc. 1987;109:3461–3462. [Google Scholar]
- 103.Maeda H, Glaser CB, Czombos J, Meienhoffer J. Structure of the Antitumor Protein Neocarzinostatin. Purification, Amino Acid Composition, Disulfide Reduction, and Isolation and Composition of Tryptic Peptides. Arch Biochem Biophys. 1974;164:369–378. doi: 10.1016/0003-9861(74)90045-9. [DOI] [PubMed] [Google Scholar]
- 104.Nicolaou KC, Smith AL, Yue EW. Chemistry and Biology of Natural and Designed Enediynes. Proc Natl Acad Sci U S A. 1993;90:5881–5888. doi: 10.1073/pnas.90.13.5881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.De Voss JJ, Hangeland JJ, Townsend CA. Characterization of the in vitro Cyclization Chemistry of Calicheamicin and Its Relation to DNA Cleavage. J Am Chem Soc. 1990;112:4554–4556. [Google Scholar]
- 106.Zein N, Sinha AM, McGahren WJ, Ellestad GA. Calicheamicin Gamma 1I: An Antitumor Antibiotic that Cleaves Double-stranded DNA Site Specifically. Science. 1988;240:1198–1201. doi: 10.1126/science.3240341. [DOI] [PubMed] [Google Scholar]
- 107.Long BH, Golik J, Forenza S, Ward B, Rehfuss R, Dabrowiak JC, Catino JJ, Musial ST, Brookshire KW, Doyle TW. Esperamicins, a Class of Potent Antitumor Antibiotics: Mechanism of Action. Proc Natl Acad Sci U S A. 1989;86:2–6. doi: 10.1073/pnas.86.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Galm U, Hager MH, Van Lanen SG, Ju J, Thorson JS, Shen B. Antitumor Antibiotics: Bleomycin, Enediynes, and Mitomycin. Chem Rev. 2005;105:739–758. doi: 10.1021/cr030117g. [DOI] [PubMed] [Google Scholar]
- 109.Drak J, Iwasawa N, Danishefsky S, Crothers DM. The Carbohydrate Domain of Calicheamicin Gamma I1 Determines Its Sequence Specificity for DNA Cleavage. Proc Natl Acad Sci U S A. 1991;88:7464–7468. doi: 10.1073/pnas.88.17.7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Timmons SC, Thorson JS. Increasing Carbohydrate Diversity via Amine Oxidation: Aminosugar, Hydroxyaminosugar, Nitrososugar, and Nitrosugar Biosynthesis in Bacteria. Curr Opin Chem Biol. 2008;12:297–305. doi: 10.1016/j.cbpa.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kumar RA, Ikemoto N, Patel DJ. Solution Structure of the Esperamicin A1-DNA Complex. J Mol Bio. 1997;265:173–186. doi: 10.1006/jmbi.1996.0719. [DOI] [PubMed] [Google Scholar]
- 112.Golik J, Wong H, Krishnan B, Vyas DM, Doyle TW. Stereochemical Studies on Esperamicins: Determination of the Absolute Configuration of Hydroxyamino Sugar Fragment. Tetrahedron. 1991;32:1851–1854. [Google Scholar]
- 113.Ahlert JSE, Lomovskaya N, Zazopoulos E, Staffa A, Bachmann BO, Huang K, Fonstein L, Czisny A, Whitwam RE, Farnet CM, et al. The Calicheamicin Gene Cluster and Its Iterative Type I Enediyne PKS. Science. 2002;297:1173–1176. doi: 10.1126/science.1072105. [DOI] [PubMed] [Google Scholar]
- 114.Liu W, Christenson SD, Standage S, Shen B. Biosynthesis of the Enediyne Antitumor Antibiotic C-1027. Science. 2002;297:1170–1173. doi: 10.1126/science.1072110. [DOI] [PubMed] [Google Scholar]
- 115.Gao Q, Thorson JS. The Biosynthetic Genes Encoding for the Production of the Dynemicin Enediyne Core in Micromonospora chersina ATCC53710. FEMS Microbiol Lett. 2008;282:105–114. doi: 10.1111/j.1574-6968.2008.01112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.McCoy JG, Johnson HD, Singh S, Bingman CA, Lei IK, Thorson JS, Phillips GN., Jr Structural Characterization of CalO2: A Putative Orsellinic Acid P450 Oxidase in the Calicheamicin Biosynthetic Pathway. Proteins. 2009;74:50–60. doi: 10.1002/prot.22131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang C, Bitto E, Goff RD, Singh S, Bingman CA, Griffith BR, Albermann C, Phillips GN, Jr, Thorson JS. Biochemical and Structural Insights of the Early Glycosylation Steps in Calicheamicin Biosynthesis. Chem Biol. 2008;15:842–853. doi: 10.1016/j.chembiol.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chang A, Singh S, Helmich KE, Goff RD, Bingman CA, Thorson JS, Phillips GN., Jr Complete Set of Glycosyltransferase Structures in the Calicheamicin Biosynthetic Pathway Reveals the Origin of Regiospecificity. Proc Natl Acad Sci U S A. 2011;108:17649–174654. doi: 10.1073/pnas.1108484108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Walsh CT, O’Brien RV, Khosla C. Nonproteinogenic Amino Acid Building Blocks for Nonribosomal Peptide and Hybrid Polyketide Scaffolds. Angew Chem Int Ed. 2013;52:7098–7124. doi: 10.1002/anie.201208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bell E, Lackey JA, Polhill RM. Systematic Significance of Canavanine in the Papilionoideae (faboideae) Biochem Sys Ecol. 1978;6:201–212. [Google Scholar]
- 121.Vickery HB. The Biochemistry of the Nitrogenous Constituents of the Green Plants. Annu Rev Biochem. 1934;3:475–484. [Google Scholar]
- 122.Damodaran M, Narayanan KGA. The Preparation of Canavanine from Canavalia obtusifolia. Biochem J. 1939;33:1740–1742. doi: 10.1042/bj0331740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rosenthal GA. The Biochemical Basis for the Deleterious Effects of L-canavanine. Phytochemistry. 1991;30:1055–1058. [Google Scholar]
- 124.Boyar A, Marsh RE. L-canavanine, a Paradigm for the Structures of Substituted Guanidines. J Am Chem Soc. 1982;104:1995–1998. [Google Scholar]
- 125.Rosenthal GA, Lambert J, Hoffmann D. Canavanine Incorporation Into the Antibacterial Proteins of the Fly, Phormia terranovae (Diptera), and Its Effect on Biological Activity. J Biol Chem. 1989;264:9768–9771. [PubMed] [Google Scholar]
- 126.Kamo T, Sakurai S, Yamanashi T, Todoroki Y. Cyanamide is Biosynthesized from Lcanavanine in Plants. Sci Rep. 2015;5:10527. doi: 10.1038/srep10527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rosenthal GA. Investigations of Canavanine Biochemistry in the Jack Bean Plant, Canavalia ensiformis (L.) DC: II. Canavanine Biosynthesis in the Developing Plant. Plant Physiol. 1972;50:328–331. doi: 10.1104/pp.50.3.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rosenthal GA. L-Canavanine Metabolism in Jack Bean, Canavalia ensiformis (L.) DC. (Leguminosae) Plant Physiol. 1982;69:1066–1069. doi: 10.1104/pp.69.5.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Inami K, Shiba T. Syntheses of Althiomycin Analogs in Relation to Antibacterial Activities. J Antibiot. 1986;59:2185–2189. [Google Scholar]
- 130.Sonderby IE, Geu-Flores F, Halkier BA. Biosynthesis of Glucosinolates--Gene Discovery and Beyond. Trends Plant Sci. 2010;15:283–290. doi: 10.1016/j.tplants.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 131.Dinkova-Kostova AT, Kostov RV. Glucosinolates and Isothiocyanates in Health and Disease. Trends Mol Med. 2012;18:337–347. doi: 10.1016/j.molmed.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 132.Halkier B, Gershenzon J. Biology and Biochemistry of Glucosinolates. Annu Rev Plant Biol. 2006;57:303–333. doi: 10.1146/annurev.arplant.57.032905.105228. [DOI] [PubMed] [Google Scholar]
- 133.Kelly WL, Townsend CA. Role of the Cytochrome P450 NocL in Nocardicin A Biosynthesis. J Am Chem Soc. 2002;124:8186–8187. doi: 10.1021/ja025926g. [DOI] [PubMed] [Google Scholar]
- 134.Zhu Y, Zhang Q, Li S, Lin Q, Fu P, Zhang G, Zhang H, Shi R, Zhu W, Zhang C. Insights into Caerulomycin A Biosynthesis: A Two-Component Monooxygenase CrmH-catalyzed Oxime Formation. J Am Chem Soc. 2013;135:18750–18753. doi: 10.1021/ja410513g. [DOI] [PubMed] [Google Scholar]
- 135.Cortina NS, Revermann O, Krug D, Muller R. Identification and Characterization of the Althiomycin Biosynthetic Gene Cluster in Myxococcus xanthus DK897. ChemBioChem. 2011;12:1411–1416. doi: 10.1002/cbic.201100154. [DOI] [PubMed] [Google Scholar]
- 136.Almeida C, Part N, Bouhired S, Kehraus S, Konig GM. Stachylines A-D from the Sponge-derived Fungus Stachylidium sp. J Nat Prod. 2011;74:21–25. doi: 10.1021/np1005345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ingavat N, Dobereiner J, Wiyakrutta S, Mahidol C, Ruchirawat S, Kittakoop P. Aspergillusol A, an Alpha-Glucosidase Inhibitor from the Marine-derived Fungus Aspergillus aculeatus. J Nat Prod. 2009;72:2049–2052. doi: 10.1021/np9003883. [DOI] [PubMed] [Google Scholar]
- 138.Moya P, Castillo M, Primo-Yúfera E, Couillaud F, Martínez-Máñez R, Garcerá MD, Miranda MA, Primo J, Martínez-Pardo R. Brevioxime: A New Juvenile Hormone Biosynthesis Inhibitor Isolated from Penicillium brevicompactum. J Org Chem. 1997;62:8544–8545. doi: 10.1021/jo970397y. [DOI] [PubMed] [Google Scholar]
- 139.Chen HP, Zhao ZZ, Li ZH, Dong ZJ, Wei K, Bai X, Zhang L, Wen CN, Feng T, Liu JK. Novel Natural Oximes and Oxime Esters with a Vibralactone Backbone from the Basidiomycete Boreostereum vibrans. ChemistryOpen. 2016;5:142–149. doi: 10.1002/open.201500198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Funk A, Divekar PV. Caerulomycin, a New Antibiotic from Streptomyces caeruleus Baldacci. I. Production, Isolation, Assay, and Biological Properties. Can J Microbiol. 1959;5:317–321. doi: 10.1139/m59-039. [DOI] [PubMed] [Google Scholar]
- 141.Mcinnes AG, Smith DG, Wright JCL, Vining LC. Caerulomycins B and C, New 2:2′-Dipyridyl Derivatives from Streptomyces caeruleus. Can J Chem. 1977;55:4159–4165. [Google Scholar]
- 142.Mcinnes AG, Smith DG, Walter JA, Wright JLC, Vining LC, Arsenault GP. Caerulomycin D, a Novel Glycosidic Derivative of 3:4-Dihydroxy-2:2′-Dipyridyl 6-Aldoxime from Streptomyces caeruleus. Can J Chem. 1978;56:1836–1842. [Google Scholar]
- 143.Divekar PV, Read G, Vining LC. Caerulomycin, a New Antibiotic from Streptomyces caeruleus Baldacci. II. Structure. Can J Chem. 1967;45:1215–1223. [Google Scholar]
- 144.Fu P, Wang S, Hong K, Li X, Liu P, Wang Y, Zhu W. Cytotoxic Bipyridines from the Marine-derived Actinomycete Actinoalloteichus cyanogriseus WH1-2216-6. J Nat Prod. 2011;74:1751–1756. doi: 10.1021/np200258h. [DOI] [PubMed] [Google Scholar]
- 145.Fu P, Liu P, Li X, Wang Y, Wang S, Hong K, Zhu W. Cyclic Bipyridine Glycosides from the Marine-derived Actinomycete Actinoalloteichus cyanogriseus WH1-2216-6. Org Lett. 2011;13:5948–5951. doi: 10.1021/ol202245s. [DOI] [PubMed] [Google Scholar]
- 146.Qu X, Pang B, Zhang Z, Chen M, Wu Z, Zhao Q, Zhang Q, Wang Y, Liu Y, Liu W. Caerulomycins and Collismycins Share a Common Paradigm for 2,2′-Bipyridine Biosynthesis Via an Unusual Hybrid Polyketide-Peptide Assembly Logic. J Am Chem Soc. 2012;134:9038–9041. doi: 10.1021/ja3016457. [DOI] [PubMed] [Google Scholar]
- 147.Tsuge N, Furihata K, Shin-Ya K, Hayakawa Y, Seto H. Novel Antibiotics Pyrisulfoxin A and B Produced by Streptomyces californicus. J Antibiot. 1999;52:505–507. doi: 10.7164/antibiotics.52.505. [DOI] [PubMed] [Google Scholar]
- 148.Zhu Y, Fu P, Lin Q, Zhang G, Zhang H, Li S, Ju J, Zhu W, Zhang C. Identification of Caerulomycin A Gene Cluster Implicates a Tailoring Amidohydrolase. Org Lett. 2012;14:2666–2669. doi: 10.1021/ol300589r. [DOI] [PubMed] [Google Scholar]
- 149.Zhu Y, Xu J, Mei X, Feng Z, Zhang L, Zhang Q, Zhang G, Zhu W, Liu J, Zhang C. Biochemical and Structural Insights into the Aminotransferase CrmG in Caerulomycin Biosynthesis. ACS Chem Biol. 2016;11:943–952. doi: 10.1021/acschembio.5b00984. [DOI] [PubMed] [Google Scholar]
- 150.Lee JK, Zhao H. Identification and Characterization of the Flavin:NADH Reductase (PrnF) Involved in a Novel Two-Component Arylamine Oxygenase. J Bacteriol. 2007;189:8556–8563. doi: 10.1128/JB.01050-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Shindo K, Yamagishi Y, Okada Y, Kawai H. Collismycins A and B, Novel Non-Steroidal Inhibitors of Dexamethasone-Glucocorticoid Receptor Binding. J Antibiot. 1994;47:1072–1074. doi: 10.7164/antibiotics.47.1072. [DOI] [PubMed] [Google Scholar]
- 152.Garcia I, Vior NM, Brana AF, Gonzalez-Sabin J, Rohr J, Moris F, Mendez C, Salas JA. Elucidating the Biosynthetic Pathway for the Polyketide-Nonribosomal Peptide Collismycin A: Mechanism for Formation of the 2:2′-Bipyridyl Ring. Chem Biol. 2012;19:399–413. doi: 10.1016/j.chembiol.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 153.Vior NM, Olano C, Garcia I, Mendez C, Salas JA. Collismycin A Biosynthesis in Streptomyces sp. CS40 is Regulated by Iron Levels Through Two Pathway-specific Regulators. Microbiology. 2014;160:467–478. doi: 10.1099/mic.0.075218-0. [DOI] [PubMed] [Google Scholar]
- 154.Garcia I, Vior NM, Gonzalez-Sabin J, Brana AF, Rohr J, Moris F, Mendez C, Salas JA. Engineering the Biosynthesis of the Polyketide-Nonribosomal Peptide Collismycin A for Generation of Analogs with Neuroprotective Activity. Chem Biol. 2013;20:1022–1032. doi: 10.1016/j.chembiol.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 155.Liu N, Song L, Liu M, Shang F, Anderson Z, Fox DJ, Challis GL, Huang Y. Unique Post-Translational Oxime Formation in the Biosynthesis of the Azolemycin Complex of Novel Ribosomal Peptides from Streptomyces sp. FXJ1.264. Chem Sci. 2016;7:482–488. doi: 10.1039/c5sc03021h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ye Y, Minami A, Igarashi Y, Izumikawa M, Umemura M, Nagano N, Machida M, Kawahara T, Shin-Ya K, Gomi K, et al. Unveiling the Biosynthetic Pathway of the Ribosomally Synthesized and Post-translationally Modified Peptide Ustiloxin B in Filamentous Fungi. Angew Chem Int Ed. 2016;55:8072–8075. doi: 10.1002/anie.201602611. [DOI] [PubMed] [Google Scholar]
- 157.Umemura M, Nagano N, Koike H, Kawano J, Ishii T, Miyamura Y, Kikuchi M, Tamano K, Yu J, Shin-ya K, et al. Characterization of the Biosynthetic Gene Cluster for the Ribosomally Synthesized Cyclic Peptide Ustiloxin B in Aspergillus flavus. Fungal Genet Biol. 2014;68:23–30. doi: 10.1016/j.fgb.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 158.Irmisch S, McCormick AC, Boeckler GA, Schmidt A, Reichelt M, Schneider B, Block K, Schnitzler JP, Gershenzon J, Unsicker SB, et al. Two Herbivore-induced Cytochrome P450 Enzymes CYP79D6 and CYP79D7 Catalyze the Formation of Volatile Aldoximes Involved in Poplar Defense. Plant Cell. 2013;25:4737–4754. doi: 10.1105/tpc.113.118265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bak S, Nielsen HL, Halkier BA. The Presence of CYP79 Homologues in Glucosinolate-producing Plants Shows Evolutionary Conservation of the Enzymes in the Conversion of Amino Acid to Aldoxime in the Biosynthesis of Cyanogenic Glucosides and Glucosinolates. Plant Mol Biol. 1998;38:725–734. doi: 10.1023/a:1006064202774. [DOI] [PubMed] [Google Scholar]
- 160.Kahn RA, Bak S, Svendsen I, Halkier BA, Møller BL. lsolation and Reconstitution of Cytochrome P450ox and in vitro Reconstitution of the Entire Biosynthetic Pathway of the Cyanogenic Glucoside Dhurrin from Sorghum. Plant Physiol. 1997;115:1661–1670. doi: 10.1104/pp.115.4.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Halkier BA, Olsen CE, Møller BL. The Biosynthesis of Cyanogenic Glucosides in Higher Plants. The (E)- and (Z)-Isomers of p-hydroxyphenylacetaldehyde Oxime As Intermediates in the Biosynthesis of Dhurrin in Sorghum bicolor (L.) Moench. J Biol Chem. 1989;264:19487–19494. [PubMed] [Google Scholar]
- 162.Hansen CH, Wittstock U, Olsen CE, Hick AJ, Pickett JA, Halkier BA. Cytochrome P450 CYP79F1 from Arabidopsis Catalyzes the Conversion of Dihomomethionine and Trihomomethionine to the Corresponding Aldoximes in the Biosynthesis of Aliphatic Glucosinolates. J Biol Chem. 2001;276:11078–11085. doi: 10.1074/jbc.M010123200. [DOI] [PubMed] [Google Scholar]
- 163.Reintanz B, Lehnen M, Reichelt M, Gershenzon J, Kowalczyk M, Sandberg G, Godde M, Uhl R, Palme K. Bus, A Bushy Arabidopsis CYP79F1 Knockout Mutant with Abolished Synthesis of Short-Chain Aliphatic Glucosinolates. Plant Cell. 2001;13:351–367. doi: 10.1105/tpc.13.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chen SG, Jørgensen E, Naur K, Jørgensen P, Olsen B, Hansen C, Rasmussen C, Pickett H, Halkier JA, BA CYP79F1 and CYP79F2 Have Distinct Functions in the Biosynthesis of Aliphatic Glucosinolates in Arabidopsis. Plant J. 2003;33:923–937. doi: 10.1046/j.1365-313x.2003.01679.x. [DOI] [PubMed] [Google Scholar]
- 165.Sibbesen OKB, Halkier BA, Møller BL. Isolation of the Heme-Thiolate Enzyme Cytochrome P-450TYR which Catalyzes the Committed Step in the Biosynthesis of the Cyanogenic Glucoside Dhurrin in Sorghum bicolor (L.) Moench. Proc Natl Acad Sci U S A. 1994;91:9740–9744. doi: 10.1073/pnas.91.21.9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nielsen JS, Møller BL. Cloning and Expression of Cytochrome P450 Enzymes Catalyzing the Conversion of Tyrosine to p-Hydroxyphenylacetaldoxime in the Biosynthesis of Cyanogenic Glucosides in Triglochin maritima. Plant Physiol. 2000;122:1311–1321. doi: 10.1104/pp.122.4.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wittstock U, Halkier BA. Cytochrome P450 CYP79A2 from Arabidopsis thaliana L. Catalyzes the Conversion of L-Phenylalanine to Phenylacetaldoxime in the Biosynthesis of Benzylglucosinolate. J Biol Chem. 2000;275:14659–14666. doi: 10.1074/jbc.275.19.14659. [DOI] [PubMed] [Google Scholar]
- 168.Hull AK, Vij R, Celenza JL. Arabidopsis Cytochrome P450s that Catalyze the First Step of Tryptophan-dependent Indole-3-Acetic Acid Biosynthesis. Proc Natl Acad Sci U S A. 2000;97:2379–2384. doi: 10.1073/pnas.040569997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mikkelsen MD, Hansen CH, Wittstock U, Halkier BA. Cytochrome P450 CYP79B2 from Arabidopsis Catalyzes the Conversion of Tryptophan to Indole-3-Acetaldoxime, a Precursor of Indole Glucosinolates and Indole-3-Acetic Acid. J Biol Chem. 2000;275:33712–33717. doi: 10.1074/jbc.M001667200. [DOI] [PubMed] [Google Scholar]
- 170.Andersen MD, Busk PK, Svendsen I, Møller BL. Cytochromes P-450 from Cassava (Manihot esculenta Crantz) Catalyzing the First Steps in the Biosynthesis of the Cyanogenic Glucosides Linamarin and Lotaustralin. Cloning, Functional Expression in Pichia Pastoris, and Substrate Specificity of the Isolated Recombinant Enzymes. J Biol Chem. 2000;275:1966–1975. doi: 10.1074/jbc.275.3.1966. [DOI] [PubMed] [Google Scholar]
- 171.Aoki H, Sakai H, Kohsaka M, Konomi T, Hosoda J. Nocardicin A, a new monocyclic betalactam antibiotic. I. Discovery, Isolation and Characterization. J Antibiot. 1976;29:492–500. doi: 10.7164/antibiotics.29.492. [DOI] [PubMed] [Google Scholar]
- 172.Kurita M, Jomon K, Komori T, Miyairi N, Aoki H. Isolation and Characterization of Nocardicin B. J Antibiot. 1976;29:1243–1245. doi: 10.7164/antibiotics.29.1243. [DOI] [PubMed] [Google Scholar]
- 173.Hashimoto M, Komori TA, Kamiya T. Letter: Nocardicin A and B, Novel Monocyclic Beta-Lactam Antibiotics from a Nocardia Species. J Am Chem Soc. 1976;98:3023–3025. doi: 10.1021/ja00426a063. [DOI] [PubMed] [Google Scholar]
- 174.Hashimoto M, Komori T, Kamiya T. Nocardicin A, a New Monocyclic Beta-Lactam Antibiotic II. Structure Determination of Nocardicins A and B. J Antibiot. 1976;29:890–901. doi: 10.7164/antibiotics.29.890. [DOI] [PubMed] [Google Scholar]
- 175.Nishida M, Mine Y, Nonoyama S, Kojo H. Nocardicin A, a New Monocyclic Beta-Lactam Antibiotic III. In vitro Evaluation. J Antibiot. 1977;30:917–925. doi: 10.7164/antibiotics.30.917. [DOI] [PubMed] [Google Scholar]
- 176.Kojo H, Mine Y, Nishida M. Nocardicin A, a New Monocyclic Beta-Lactam Antibiotic IV. Factors Influencing the in vitro Activity of Nocardicin A. J Antibiot. 1977;30:926–931. doi: 10.7164/antibiotics.30.926. [DOI] [PubMed] [Google Scholar]
- 177.Mine Y, Nonoyama S, Kojo H, Fukada S, Nishida M. Nocardicin A, a New Monocyclic Beta-Lactam Antibiotic V. In vivo Evaluation. J Antibiot. 1977;30:932–937. doi: 10.7164/antibiotics.30.932. [DOI] [PubMed] [Google Scholar]
- 178.Mine Y, Nonoyama S, Kojo H, Fukada S, Nishida M. Nocardicin A, a New Monocyclic Beta-Lactam Antibiotic VI. Absorption, Excretion and Tissue Distribution in Animals. J Antibiot. 1977;30:938–944. doi: 10.7164/antibiotics.30.938. [DOI] [PubMed] [Google Scholar]
- 179.Gunsior M, Breazeale SD, Lind AJ, Ravel J, Janc JW, Townsend CA. The Biosynthetic Gene Cluster for a Monocyclic Beta-Lactam Antibiotic, Nocardicin A. Chem Biol. 2004;11:927–938. doi: 10.1016/j.chembiol.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 180.Davidsen JM, Townsend CA. In vivo Characterization of Nonribosomal Peptide Synthetases NocA and NocB in the Biosynthesis of Nocardicin A. Chem Biol. 2012;19:297–306. doi: 10.1016/j.chembiol.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gaudelli NM, Long DH, Townsend CA. Beta-Lactam Formation by a Non-Ribosomal Peptide Synthetase during Antibiotic Biosynthesis. Nature. 2015;520:383–387. doi: 10.1038/nature14100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Gaudelli NM, Townsend CA. Epimerization and Substrate Gating by a TE Domain in Beta-Lactam Antibiotic Biosynthesis. Nat Chem Biol. 2014;10:251–258. doi: 10.1038/nchembio.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Reeve AM, Breazeale SD, Townsend CA. Purification, Characterization, and Cloning of an S-adenosylmethionine-dependent 3-amino-3-carboxypropyltransferase in Nocardicin Biosynthesis. J Biol Chem. 1998;273:30695–30703. doi: 10.1074/jbc.273.46.30695. [DOI] [PubMed] [Google Scholar]
- 184.Davidsen JM, Bartley DM, Townsend CA. Non-Ribosomal Propeptide Precursor in Nocardicin A Biosynthesis Predicted from Adenylation Domain Specificity Dependent on the MbtH Family Protein NocI. J Am Chem Soc. 2013;135:1749–1759. doi: 10.1021/ja307710d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Townsend CA, Salituro GM. Fate of [15N]-p-Hydroxyphenyl)glycine in Nocardicin A Biosynthesis. J Chem Soc, Chem Commun. 1984;24:1631–1632. [Google Scholar]
- 186.Kelly WL, Townsend CA. Mutational Analysis of nocK and nocL in the Nocardicin A Producer Nocardia uniformis. J Bacteriol. 2005;187:739–746. doi: 10.1128/JB.187.2.739-746.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Fujimoto H, Kinoshita T, Suzuki H, Umezawa H. Studies on the Mode of Action of Althiomycin. J Antibiot. 1970;23:271–275. doi: 10.7164/antibiotics.23.271. [DOI] [PubMed] [Google Scholar]
- 188.Yamaguchi H, Nakayama Y, Takeda K, Tawara K, Maeda K, Takeuchi T, Umezawa H. A New Antibiotic, Althiomycin. J Antibiot. 1957;10:195–200. [PubMed] [Google Scholar]
- 189.Zarantonello P, Leslie CP, Ferritto R, Kazmierski WM. Total Synthesis and Semi-Synthetic Approaches to Analogues of Antibacterial Natural Product Althiomycin. Bioorg Med Chem Lett. 2002;12:561–565. doi: 10.1016/s0960-894x(01)00802-2. [DOI] [PubMed] [Google Scholar]
- 190.Gerc AJ, Song L, Challis GL, Stanley-Wall NR, Coulthurst SJ. The Insect Pathogen Serratia marcescens Db10 Uses a Hybrid Non-Ribosomal Peptide Synthetase-Polyketide Synthase to Produce the Antibiotic Althiomycin. PLoS One. 2012;7:e44673. doi: 10.1371/journal.pone.0044673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Bézière N, Hardy M, Poulhès F, Karoui H, Tordo P, Ouari O, Frapart YM, Rockenbauer A, Boucher JL, Mansuy D, et al. Metabolic Stability of Superoxide Adducts Derived from Newly Developed Cyclic Nitrone Spin Traps. Free Radic Biol Med. 2014;67:150–158. doi: 10.1016/j.freeradbiomed.2013.10.812. [DOI] [PubMed] [Google Scholar]
- 192.Floyd RA, Chandru HK, He T, Towner R. Anti-Cancer Activity of Nitrones and Observations on Mechanism of Action. Anticancer Agents Med Chem. 2011;11:373–379. doi: 10.2174/187152011795677517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Floyd RA, Kopke RD, Choi CH, Foster SB, Doblas S, Towner RA. Nitrones as Therapeutics. Free Radic Biol Med. 2008;45:1361–1374. doi: 10.1016/j.freeradbiomed.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Myers AG, Herzon SB. Identification of a Novel Michael Acceptor Group for the Reversible Addition of Oxygen- and Sulfur-Based Nucleophiles. Synthesis and Reactivity of the 3-Alkylidene-3H-indole 1-Oxide Function of Avrainvillamide. J Am Chem Soc. 2003;125:12080–12081. doi: 10.1021/ja0372006. [DOI] [PubMed] [Google Scholar]
- 195.Mukherjee H, Chan KP, Andresen V, Hanley ML, Gjertsen BT, Myers AG. Interactions of the Natural Product (+)-Avrainvillamide with Nucleophosmin and Exportin-1 Mediate the Cellular Localization of Nucleophosmin and Its AML-Associated Mutants. ACS Chem Biol. 2015;10:855–863. doi: 10.1021/cb500872g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Scott PM, Merrien MA, Polonsky J. Roquefortine and Isofumigaclavine A, Metabolites from Penicillium roqueforti. Experientia. 1976;32:140–142. [PubMed] [Google Scholar]
- 197.de Jesus AE, Steyn PS, van Heerden FR, Vleggaar R, Wessels PL, Hull WE. Tremorgenic Mycotoxins from Penicillium crustosum: Isolation of Penitrems A–F and the Structure Elucidation and Absolute Configuration of Penitrem A. J Chem Soc, Perkin Trans 1. 1983:1847–1856. [Google Scholar]
- 198.Wagener RE, Davis ND, Diener UL. Penitrem A and Roquefortine Production by Penicillium commune. Appl Environ Microbiol. 1980;39:882–887. doi: 10.1128/aem.39.4.882-887.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Rand TG, Giles S, Flemming J, Miller JD, Puniani E. Inflammatory and Cytotoxic Responses in Mouse Lungs Exposed to Purified Toxins from Building Isolated Penicillium brevicompactum Dierckx and P. chrysogenum Thom. Toxicol Sci. 2005;87:213–222. doi: 10.1093/toxsci/kfi223. [DOI] [PubMed] [Google Scholar]
- 200.Koizumi Y, Arai M, Tomoda H, Ômura S. Oxaline, A Fungal Alkaloid, Arrests the Cell Cycle in M Phase by Inhibition of Tubulin Polymerization. Biochim Biophys Acta. 2004;1693:47–55. doi: 10.1016/j.bbamcr.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 201.Steyn PS. The Isolation, Structure and Absolute Configuration of Secalonic Acid D, the Toxic Metabolite of Penicillium oxalicum. Tetrahedron. 1970;26:51–57. doi: 10.1016/0040-4020(70)85006-2. [DOI] [PubMed] [Google Scholar]
- 202.Steyn PS, Vleggaar R. Roquefortine, An Intermediate in the Biosynthesis of Oxaline in Cultures of Penicillium oxalicum. J Chem Soc, Chem Commun. 1983;10:560–561. [Google Scholar]
- 203.García-Estrada C, Ullán Ricardo V, Albillos Silvia M, Fernández-Bodega María Á, Durek P, von Döhren H, Martín Juan F. A Single Cluster of Coregulated Genes Encodes the Biosynthesis of the Mycotoxins Roquefortine C and Meleagrin in Penicillium chrysogenum. Chem Biol. 2011;18:1499–1512. doi: 10.1016/j.chembiol.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 204.Ries MI, Ali H, Lankhorst PP, Hankemeier T, Bovenberg RAL, Driessen AJM, Vreeken RJ. Novel Key Metabolites Reveal Further Branching of the Roquefortine/Meleagrin Biosynthetic Pathway. J Biol Chem. 2013;288:37289–37295. doi: 10.1074/jbc.M113.512665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Newmister SA, Gober CM, Romminger S, Yu F, Tripathi A, Parra LLL, Williams RM, Berlinck RGS, Joullié MM, Sherman DH. OxaD: A Versatile Indolic Nitrone Synthase from the Marine-Derived Fungus Penicillium oxalicum F30. J Am Chem Soc. 2016;138:11176–11184. doi: 10.1021/jacs.6b04915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kaur K, Kumar V, Sharma AK, Gupta GK. Isoxazoline Containing Natural Products as Anticancer Agents: A Review. Eur J Med Chem. 2014;77:121–133. doi: 10.1016/j.ejmech.2014.02.063. [DOI] [PubMed] [Google Scholar]
- 207.Jin Z. Muscarine, Imidazole, Oxazole, and Thiazole Alkaloids. Nat Prod Rep. 2011;28:1143–1191. doi: 10.1039/c0np00074d. [DOI] [PubMed] [Google Scholar]
- 208.Harris DA, Ruger M, Reagan MA, Wolf FJ, Peck RL, Wallick H, Wodruff HB. Discovery, Development, and Antimicrobial Properties of D-4-amino-3-Isoxazolidone (Oxamycin), a New Antibiotic Produced by Streptomyces garyphalus n. sp. Antibiot Chemother. 1955;5:183–190. [PubMed] [Google Scholar]
- 209.Svensson M, Valeria K, Gatenbeck S. Hydroxyurea, a Natural Metabolite and an Intermediate in D-cycloserine Biosynthesis in Streptomyces garyphalus. Arch Microbiol. 1981;129:210–212. [Google Scholar]
- 210.Lowther J, Yard BA, Johnson KA, Carter LG, Bhat VT, Raman MC, Clarke DJ, Ramakers B, McMahon SA, Naismith JH, et al. Inhibition of the PLP-Dependent Enzyme Serine Palmitoyltransferase by Cycloserine: Evidence for a Novel Decarboxylative Mechanism of Inactivation. Mol Biosyst. 2010;6:1682–1693. doi: 10.1039/c003743e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Prosser GA, de Carvalho LPS. Kinetic Mechanism and Inhibition of Mycobacterium tuberculosis D-alanine:D-alanine Ligase by the Antibiotic D-cycloserine. FEBS J. 2013;280:1150–1166. doi: 10.1111/febs.12108. [DOI] [PubMed] [Google Scholar]
- 212.Matsuo H, Kumagai T, Mori K, Sugiyama M. Molecular Cloning of a D-cycloserine Resistance Gene from D-cycloserine-Producing Streptomyces garyphalus. J Antibiot. 2003;56:762–767. doi: 10.7164/antibiotics.56.762. [DOI] [PubMed] [Google Scholar]
- 213.Kumagai T, Koyama Y, Oda K, Noda M, Matoba Y, Sugiyama M. Molecular Cloning and Heterologous Expression of a Biosynthetic Gene Cluster for the Antitubercular Agent Dcycloserine produced by Streptomyces lavendulae. Antimicrob Agents Chemother. 2010;54:1132–1139. doi: 10.1128/AAC.01226-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kumagai T, Ozawa T, Tanimoto M, Noda M, Matoba Y, Sugiyama M. High-Level Heterologous Production of D-cycloserine by Escherichia coli. Appl Environ Microbiol. 2015;81:7881–7887. doi: 10.1128/AEM.02187-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Kumagai T, Takagi K, Koyama Y, Matoba Y, Oda K, Noda M, Sugiyama M. Heme Protein and Hydroxyarginase Necessary for Biosynthesis of D-cycloserine. Antimicrob Agents Chemother. 2012;56:3682–3689. doi: 10.1128/AAC.00614-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Lundberg JO, Weitzberg E, Gladwin MT. The Nitrate–Nitrite–Nitric Oxide Pathway in Physiology and Therapeutics. Nat Rev Drug Discov. 2008;7:156–167. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- 217.Sudhamsu J, Crane BR. Bacterial Nitric Oxide Synthases: What are They Good for? Trends in Microbiology. 2009;17:212–218. doi: 10.1016/j.tim.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 218.Uda N, Matoba Y, Oda K, Kumagai T, Sugiyama M. The Structural and Mutational Analyses of O-ureido-L-serine Synthase Necessary for D-cycloserine Biosynthesis. FEBS J. 2015;282:3929–3944. doi: 10.1111/febs.13386. [DOI] [PubMed] [Google Scholar]
- 219.Dietrich D, van Belkum MJ, Vederas JC. Characterization of DcsC, a PLP-Independent Racemase Involved in the Biosynthesis of D-cycloserine. Org Biomol Chem. 2012;10:2248–2254. doi: 10.1039/c2ob06864h. [DOI] [PubMed] [Google Scholar]
- 220.Uda N, Matoba Y, Kumagai T, Oda K, Noda M, Sugiyama M. Establishment of an in vitro D-cycloserine Synthesizing System by using O-ureido-L-serine Synthase and D-cycloserine Synthetase Found in the Biosynthetic Pathway. Antimicrob Agents Chemother. 2013;57:2603–2612. doi: 10.1128/AAC.02291-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Kreuzer J, Bach NC, Forler D, Sieber SA. Target Discovery of Acivicin in Cancer Cells Elucidates Its Mechanism of Growth Inhibition: Synthesis, Cloning, Protein Expression, Purification and Biochemical Assays. Chem Sci. 2014;6:237–245. doi: 10.1039/c4sc02339k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Gould SJ, Ju S. The Biosynthesis of Acivicin and 4-Hydroxyacivicin from N-Hydroxyornithine. J Am Chem Soc. 1989;111:2329–2331. [Google Scholar]
- 223.Gould SJ, Ju S. Biosynthesis of Acivicin. 3. Incorporation of Ornithine and N-delta-Hydroxyornithine. J Am Chem Soc. 1992;114:10166–10172. [Google Scholar]
- 224.Ranger CM, Winter RE, Singh AP, Reding ME, Frantz JM, Locke JC, Krause CR. Rare Excitatory Amino Acid from Flowers of Zonal Geranium Responsible for Paralyzing the Japanese Beetle. Proc Natl Acad Sci U S A. 2011;108:1217–1221. doi: 10.1073/pnas.1013497108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Murakoshi I, Kaneko M, Koide C, Ikegami F. Enzymatic Synthesis of the Neuroexcitatory Amino Acid Quisqualic Acid by Cysteine Synthase. Phytochemistry. 1986;25:2759–2763. [Google Scholar]
- 226.Gowenlock BG, Richter-Addo GB. Preparations of C-Nitroso Compounds. Chem Rev. 2004;104:3315–3340. doi: 10.1021/cr030450k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Douglass ML, Kabacoff BL, Anderson GA, Cheng MC. The Chemistry of Nitrosamine Formation, Inhibition and Destruction. J Soc Cosmet Chem. 1978;29:581–606. [Google Scholar]
- 228.Blinova IN, Egorova SA, Marchenko IV, Saulina LI, Blinov NO, Khokhlov AS. New Iron-Containing Antibiotics Isolation and Properties of Viridomycins A, B, and C. Chem Nat Compd. 1975;11:506–512. [Google Scholar]
- 229.Marchenko IV, Egorova SA, Blinov NO, Krassilnikov NA. The Role of Iron in the Formation of Green Pigments by Actinomycetes. Z Allg Mikrobiol. 1968;8:437–439. [PubMed] [Google Scholar]
- 230.Yang CC, Leong J. Production of Deferriferrioxamines B and E from a Ferroverdin-producing Streptomyces Species. J Bacteriol. 1982;149:381–383. doi: 10.1128/jb.149.1.381-383.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Noguchi A, Kitamura T, Onaka H, Horinouchi S, Ohnishi Y. A Copper-containing Oxidase Catalyzes C-Nitrosation in Nitrosobenzamide Biosynthesis. Nat Chem Biol. 2010;6:641–643. doi: 10.1038/nchembio.418. [DOI] [PubMed] [Google Scholar]
- 232.Suzuki H, Furusho Y, Higashi T, Ohnishi Y, Horinouchi S. A Novel o-Aminophenol Oxidase Responsible for Formation of the Phenoxazinone Chromophore of Grixazone. J Biol Chem. 2006;281:824–833. doi: 10.1074/jbc.M505806200. [DOI] [PubMed] [Google Scholar]
- 233.Itoh S, Fukuzumi S. Monooxygenase Activity of Type 3 Copper Proteins. Acc Chem Res. 2007;40:592–600. doi: 10.1021/ar6000395. [DOI] [PubMed] [Google Scholar]
- 234.Sánchez-Ferrer Á, Neptuno Rodríguez-López J, García-Cánovas F, García-Carmona F. Tyrosinase: A Comprehensive Review of Its Mechanism. Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology. 1995;1247:1–11. doi: 10.1016/0167-4838(94)00204-t. [DOI] [PubMed] [Google Scholar]
- 235.Solomon EI, Sundaram UM, Machonkin TE. Multicopper Oxidases and Oxygenases. Chem Rev. 1996;96:2563–2606. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]
- 236.Kovacic P, Somanathan R. Nitroaromatic Compounds: Environmental Toxicity, Carcinogenicity, Mutagenicity, Therapy and Mechanism. J Appl Toxicol. 2014;34:810–824. doi: 10.1002/jat.2980. [DOI] [PubMed] [Google Scholar]
- 237.Yan G, Yang M. Recent Advances in the Synthesis of Aromatic Nitro Compounds. Org Biomol Chem. 2013;11:2554–2566. doi: 10.1039/c3ob27354g. [DOI] [PubMed] [Google Scholar]
- 238.Parry RJ, Nishino S, Spain J. Naturally-occurring Nitro Compounds. Nat Prod Rep. 2011;28:152–167. doi: 10.1039/c0np00024h. [DOI] [PubMed] [Google Scholar]
- 239.Winkler R, Hertweck C. Biosynthesis of Nitro Compounds. Chembiochem. 2007;8:973–977. doi: 10.1002/cbic.200700042. [DOI] [PubMed] [Google Scholar]
- 240.Kulkarni M, Chaudhari A. Microbial Remediation of Nitro-Aromatic Compounds: An Overview. J Environ Manage. 2007;85:496–512. doi: 10.1016/j.jenvman.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 241.Boelsterli UA, Ho HK, Zhou S, Leow KY. Bioactivation and Hepatotoxicity of Nitroaromatic Drugs. Curr Drug Metab. 2006;7:715–727. doi: 10.2174/138920006778520606. [DOI] [PubMed] [Google Scholar]
- 242.Padda RS, Wang C, Hughes JB, Kutty R, Bennett GN. Mutagenicity of Nitroaromatic Degradation Compounds. Environ Microbiol. 2003;22:2293–2297. doi: 10.1897/02-220. [DOI] [PubMed] [Google Scholar]
- 243.Spain JC. Biodegradation of Nitroaromatic Compounds. Annu Rev Microbiol. 1995;49:523–555. doi: 10.1146/annurev.mi.49.100195.002515. [DOI] [PubMed] [Google Scholar]
- 244.Marvin-Sikkema FD, de Bont JAM. Degradation of Nitroaromatic Compounds by Microorganisms. Appl Microbiol Biotechnol. 1994;42:499–507. doi: 10.1007/BF00173912. [DOI] [PubMed] [Google Scholar]
- 245.White PA, Claxton LD. Mutagens in Contaminated Soil: A Review. Mutat Res. 2004;567:227–345. doi: 10.1016/j.mrrev.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 246.Badgujar DM, Talawar MB, Mahulikar PP. Review on Greener and Safer Synthesis of Nitro Compounds. Propellants Explos Pyrotech. 2016;41:24–34. [Google Scholar]
- 247.Roldan MD, Perez-Reinado E, Castillo F, Moreno-Vivian C. Reduction of Polynitroaromatic Compounds: The Bacterial Nitroreductases. FEMS Microbiol Rev. 2008;32:474–500. doi: 10.1111/j.1574-6976.2008.00107.x. [DOI] [PubMed] [Google Scholar]
- 248.Ryan A, Kaplan E, Laurieri N, Lowe E, Sim E. Activation of Nitrofurazone by Azoreductases: Multiple Activities in One Enzyme. Sci Rep. 2011;1:63–67. doi: 10.1038/srep00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Leahy JG, Batchelor PJ, Morcomb SM. Evolution of the Soluble Diiron Monooxygenases. FEMS Microbiol Rev. 2003;27:449–479. doi: 10.1016/S0168-6445(03)00023-8. [DOI] [PubMed] [Google Scholar]
- 250.Krebs C, Bollinger JM, Jr, Booker SJ. Cyanobacterial Alkane Biosynthesis Further Expands the Catalytic Repertoire of the Ferritin-like ‘Di-iron-Carboxylate’ Proteins. Curr Opin Chem Biol. 2011;15:291–303. doi: 10.1016/j.cbpa.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Winkler R, Hertweck C. Sequential Enzymatic Oxidation of Aminoarenes to Nitroarenes via Hydroxylamines. Angew Chem, Int Ed. 2005;44:4083–4087. doi: 10.1002/anie.200500365. [DOI] [PubMed] [Google Scholar]
- 252.He J, Hertweck C. Biosynthetic Origin of the Rare Nitroaryl Moiety of the Polyketide Antibiotic Aureothin: Involvement of an Unprecedented N-oxygenase. J Am Chem Soc. 2004;126:3694–3695. doi: 10.1021/ja039328t. [DOI] [PubMed] [Google Scholar]
- 253.Maeda K. A Crystalline Toxic Substance of Streptomyces thioluteus Producing Aureothricin. J Antibiot (Tokyo) 1953;4:137–138. [PubMed] [Google Scholar]
- 254.Hirata Y, Nakata H, Yamada K, Okurhara K, Naito T. The Structure of Aureothin, a Nitro Compound Obtained from Streptomyces thioluteus. Tetrahedron. 1961;14:252–274. [Google Scholar]
- 255.Schwartz JL, Tishler M, Arison BH, Shafer HM, Omura S. Identification of Mycolutein and Pulvomycin as Aureothin and Labilomycin Respectively. J Antibiot (Tokyo) 1976;29:236–241. doi: 10.7164/antibiotics.29.236. [DOI] [PubMed] [Google Scholar]
- 256.Yamazaki M, Maebayashi Y, Katoh H, Ohishi JI, Koyama Y. Application of 13C-NMR to the Biosynthetic Investigations. II. Biosynthesis of Aureothin and Related Nitro-containing metabolites of Streptomyces luteoreticuli. Chem Pharm Bull. 1975;23:569–574. [Google Scholar]
- 257.Cardillo R, Fuganti C, Ghiringhelli D, Giangrasso D, Grasselli P, Santopietro-Amisano A. The Biosynthesis of Aureothin. Tetrahedron. 1974;30:459–461. [Google Scholar]
- 258.Yamazaki M, Katoh H, Ohishi JI, Koyama Y. Study on Biosynthesis of Aureothin, A Nitro-containing Metabolite from Streptomyces luteoreticuli Using 13C-NMR Spectroscopy. Tetrahedron Lett. 1972;26:2701–2704. [Google Scholar]
- 259.Kawai S, Kobayashi K, Oshima T, Egami F. Studies on the Oxidation of p-Aminobenzoate to p-Nitrobenzoate by Streptomyces thioluteus. Arch Biochem Biophys. 1965;112:537–543. doi: 10.1016/0003-9861(65)90092-5. [DOI] [PubMed] [Google Scholar]
- 260.Cardillo R, Fuganti C, Ghiringhelli D, Giangrasso D, Grasselli P. On the Biological Origin of the Nitroaromatic Unit of the Antibiotic Aureotine. Tetrahedron Lett. 1972;48:4875–4878. [Google Scholar]
- 261.He J, Hertweck C. Iteration as Programmed Event during Polyketide Assembly; Molecular Analysis of the Aureothin Biosynthesis Gene Cluster. Chem Biol. 2003;10:1225–1232. doi: 10.1016/j.chembiol.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 262.Simurdiak M, Lee J, Zhao H. A New Class of Arylamine Oxygenases: Evidence that p-Aminobenzoate N-oxygenase (AurF) is a Di-iron Enzyme and Further Mechanistic Studies. Chembiochem. 2006;7:1169–1172. doi: 10.1002/cbic.200600136. [DOI] [PubMed] [Google Scholar]
- 263.Winkler R, Zocher G, Richter I, Friedrich T, Schulz GE, Hertweck C. A Binuclear Manganese Cluster that Catalyzes Radical-mediated N-oxygenation. Angew Chem, Int Ed. 2007;46:8605–8608. doi: 10.1002/anie.200703089. [DOI] [PubMed] [Google Scholar]
- 264.Zocher G, Winkler R, Hertweck C, Schulz GE. Structure and Action of the N-oxygenase AurF from Streptomyces thioluteus. J Mol Biol. 2007;373:65–74. doi: 10.1016/j.jmb.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 265.Winkler R, Richter ME, Knupfer U, Merten D, Hertweck C. Regio- and Chemoselective Enzymatic N-oxygenation in vivo, in vitro, and in flow. Angew Chem, Int Ed. 2006;45:8016–8018. doi: 10.1002/anie.200603060. [DOI] [PubMed] [Google Scholar]
- 266.Choi YS, Zhang H, Brunzelle JS, Nair SK, Zhao H. In vitro Reconstitution and Crystal Structure of p-aminobenzoate N-oxygenase (AurF) Involved in Aureothin Biosynthesis. Proc Natl Acad Sci US A. 2008;105:6858–6863. doi: 10.1073/pnas.0712073105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Li N, Korboukh VK, Krebs C, Bollinger JM., Jr Four-electron Oxidation of p-Hydroxylaminobenzoate to p-Nitrobenzoate by a Peroxodiferric Complex in AurF from Streptomyces thioluteus. Proc Natl Acad Sci USA. 2010;107:15722–15727. doi: 10.1073/pnas.1002785107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Korboukh VK, Li N, Barr EW, Bollinger JM, Jr, Krebs C. A Long-Lived Substrate-Hydroxylating Peroxodiiron(III/III) Intermediate in the Amine Oxygenase, AurF from Streptomyces thioluteus. J Am Chem Soc. 2009;131:13608–13609. doi: 10.1021/ja9064969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Krebs C, Matthews ML, Jiang W, Bollinger JM., Jr AurF from Streptomyces thioluteus and a Possible New Family of Manganese/Iron Oxygenases. Biochemistry. 2007;46:10413–10418. doi: 10.1021/bi701060g. [DOI] [PubMed] [Google Scholar]
- 270.Chanco E, Choi YS, Sun N, Vu M, Zhao H. Characterization of the N-oxygenase AurF from Streptomyces thioletus. Bioorg Med Chem. 2014;22:5569–5577. doi: 10.1016/j.bmc.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 271.Fries A, Bretschneider T, Winkler R, Hertweck C. A Ribonucleotide Reductase-like Electron Transfer System in the Nitroaryl-forming N-oxygenase AurF. Chembiochem. 2011;12:1832–1835. doi: 10.1002/cbic.201100138. [DOI] [PubMed] [Google Scholar]
- 272.Vazquez D. Binding of Chloramphenicol to Ribosomes: The Effect of a Number of Antibiotics. Biochim Biophys Acta. 1966;114:277–288. doi: 10.1016/0005-2787(66)90309-1. [DOI] [PubMed] [Google Scholar]
- 273.Rebstock MC, Crooks HMJ, Controulis J, Bartz QR. Chloramphenicol (Chloromycetin). IV. Chemical Studies. J Am Chem Soc. 1949;71:2458–2462. [Google Scholar]
- 274.Schlunzen F, Zarlvach R, Harms J, Bashan A, Tocilj A, Albrecht R, Yonath A, Franceshi F. Structural Basis for the Interaction of Antibiotics with the Peptidyl Transferase Centre in Eubacteria. Nature. 2001;413:813–821. doi: 10.1038/35101544. [DOI] [PubMed] [Google Scholar]
- 275.Ehrlich J, Bartz QR, Smith RM, Joslyn DA, Burkholder PR. Chloromycetin, a New Antibiotic from a Soil Actinomycete. Science. 1947;106:417. doi: 10.1126/science.106.2757.417. [DOI] [PubMed] [Google Scholar]
- 276.Gottlieb D, Bhattacharyya PK, Anderson HW, Carter HE. Some Properties of an Antibiotic Obtained from a Species of Streptomyces. J Bacteriol. 1948;55:410–417. [PMC free article] [PubMed] [Google Scholar]
- 277.Siddiqueullah M, McGrath R, Vining LC. Biosynthesis of Chloramphenicol. II. p-Aminophenylalanine as a Precursor of the p-Nitrophenylserinol Moiety. Can J Biochem. 1967;45:1881–1889. doi: 10.1139/o67-221. [DOI] [PubMed] [Google Scholar]
- 278.Vining LC, Westlake DWS. Biosyntheis of the Phenylpropanoid Moiety of Chloramphenicol. Can J Microbiol. 1964;10:705–716. doi: 10.1139/m64-090. [DOI] [PubMed] [Google Scholar]
- 279.Gottlieb D, Robbins PW, Carter HE. The Biosynthesis of Chloramphenicol. II. Acetylation of p-Nitrophenylserinol. J Bacteriol. 1956;72:153–156. doi: 10.1128/jb.72.2.153-156.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Gottlieb D, Carter HE, Legator M, Gallicchio V. The Biosynthesis of Chloramphenicol. I. Precursors Stimulating the Synthesis. J Bacteriol. 1954;68:243–251. doi: 10.1128/jb.68.2.243-251.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Vining LC, Stuttard C. Genetics and Biochemistry of Antibiotic Production. Butterworth-Heinemann; Boston: 1995. [Google Scholar]
- 282.Fernandez-Martinez LT, Borsetto C, Gomez-Escribano JP, Bibb MJ, Al-Bassam MM, Chandra G, Bibb MJ. New Insights into Chloramphenicol Biosynthesis in Streptomyces venezuelae ATCC 10712. Antimicrob Agents Chemother. 2014;58:7441–7450. doi: 10.1128/AAC.04272-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Makris TM, Chakrabarti M, Munck E, Lipscomb JD. A Family of Diiron Monooxygenases Catalyzing Amino Acid Beta-hydroxylation in Antibiotic Biosynthesis. Proc Natl Acad Sci US A. 2010;107:15391–15396. doi: 10.1073/pnas.1007953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Pacholec M, Sello JK, Walsh CT, Thomas MG. Formation of an Aminoacyl-S-enzyme Intermediate is a Key Step in the Biosynthesis of Chloramphenicol. Org Biomol Chem. 2007;5:1692–1694. doi: 10.1039/b703356g. [DOI] [PubMed] [Google Scholar]
- 285.He J, Magarvey N, Piraee M, Vining LC. The Gene Cluster for Chloramphenicol Biosynthesis in Streptomyces venezuelae ISP5230 Includes Novel Shikimate Pathway Homologues and a Monomodular Non-Ribosomal Peptide Synthetase Gene. Microbiology. 2001;147:2817–2829. doi: 10.1099/00221287-147-10-2817. [DOI] [PubMed] [Google Scholar]
- 286.Brown MP, Aidoo KA, Vining LC. A Role for pabAB, a p-Aminobenzoate Synthase Gene of Streptomyces venezuelae ISP5230; in Chloramphenicol Biosynthesis. Microbiology. 1996;142:1345–1355. doi: 10.1099/13500872-142-6-1345. [DOI] [PubMed] [Google Scholar]
- 287.Teng CY, Ganem B, Doktor S, Nichols BP, Bhatnagar RK, Vining LC. Total Biosynthesis of 4-Amino-4-deoxychorismic Acid: A Key Intermediate in the Biosynthesis of p-Aminobenzoic Acid and L-p-aminophenylalanine. J Am Chem Soc. 1985;107:5008–5009. [Google Scholar]
- 288.Lu H, Chanco E, Zhao H. CmlI is an N-oxygenase in the Biosynthesis of Chloramphenicol. Tetrahedron. 2012;68:7651–7654. doi: 10.1016/j.tet.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Knoot CJ, Kovaleva EG, Lipscomb JD. Crystal Structure of CmlI, the Arylamine Oxygenase from the Chloramphenicol Biosynthetic Pathway. J Biol Inorg Chem. 2016;21:589–603. doi: 10.1007/s00775-016-1363-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Komor AJ, Rivard BS, Fan R, Guo Y, Que L, Jr, Lipscomb JD. Mechanism for Six-Electron Aryl-N-Oxygenation by the Non-Heme Diiron Enzyme CmlI. J Am Chem Soc. 2016;138:7411–7421. doi: 10.1021/jacs.6b03341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Barry SM, Challis GL. Mechanism and Catalytic Diversity of Rieske Non-Heme Iron-Dependent Oxygenases. ACS Catal. 2013;3:2362–2370. doi: 10.1021/cs400087p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Bruijnincx PC, van Koten G, Klein Gebbink RJ. Mononuclear Non-Heme Iron Enzymes with the 2-His-1-Carboxylate Facial Triad: Recent Developments in Enzymology and Modeling Studies. Chem Soc Rev. 2008;37:2716–2744. doi: 10.1039/b707179p. [DOI] [PubMed] [Google Scholar]
- 293.Koehntop KD, Emerson JP, Que L., Jr The 2-His-1-Carboxylate Facial Triad: A Versatile Platform for Dioxygen Activation by Mononuclear Non-Heme Iron(II) Enzymes. J Biol Inorg Chem. 2005;10:87–93. doi: 10.1007/s00775-005-0624-x. [DOI] [PubMed] [Google Scholar]
- 294.Lambowitz AM, Slayman CW. Effect of Pyrrolnitrin on Electron Transport and Oxidative Phosphorylation in Mitochondria Isolated from Neurospora crassa. J Bacteriol. 1972;112:1020–1022. doi: 10.1128/jb.112.2.1020-1022.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Elander RP, Mabe JA, Hamill RH, Gorman M. Metabolism of Tryptophans by Pseudomonas aureofaciens. VI. Production of Pyrrolnitrin by Selected Pseudomonas species. Appl Environ Microbiol. 1968:753–758. doi: 10.1128/am.16.5.753-758.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Arima K, Imanaka H, Kousaka M, Fukuda A, Tamura G. Studies on Pyrrolnitrin, a New Antibiotic. I. Isolation and Properties of Pyrrolnitrin. J Antibiot (Tokyo) 1965;18:201–204. [PubMed] [Google Scholar]
- 297.Nishida M, Matsubara T, Watanabe N. Pyrrolnitrin, a New Antifungal Antibiotic. Microbiological and Toxicological Observations. J Antibiot (Tokyo) 1965;18:211–219. [PubMed] [Google Scholar]
- 298.Imanaka H, Kousaka M, Tamura G, Arima K. Studies on Pyrrolnitrin, a New Antibiotic. 3. Structure of Pyrrolnitrin. J Antibiot (Tokyo) 1965;18:207–210. [PubMed] [Google Scholar]
- 299.Arima K, Imanaka H, Kousaka M, Fukuda A, Tamura G. Pyrrolnitrin, a New Antibiotic Substance, Produced by Pseudomonas. Argric Biol Chem. 1964;28:575–576. [Google Scholar]
- 300.Hamill RH, Elander RP, Mabe JA, Gorman M. Metabolism of Tryptophan by Pseudomonas aureofaciens. III. Production of Substituted Pyrrolnitrins from Tryptophan Analogues. Appl Microbiol. 1970;19:721–725. doi: 10.1128/am.19.5.721-725.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.van Pée KH, Ligon JM. Biosynthesis of Pyrrolnitrin and Other Phenylpyrrole Derivatives by Bacteria. Nat Prod Rep. 2000;17:157–164. doi: 10.1039/a902138h. [DOI] [PubMed] [Google Scholar]
- 302.Zhou P, Mocek U, Seisel B, Floss HG. Biosynthesis of Pyrrolnitrin. Incorporation of 13C,15N double-labelled D- and L-Tryptophan. J Basic Microbiol. 1992;32:209–214. doi: 10.1002/jobm.3620320312. [DOI] [PubMed] [Google Scholar]
- 303.Chang CJ, Floss HG, Hook DJ, Mabe JA, Manni PE, Martin LL, Schroder K, Shieh TL. The Biosynthesis of the Antibiotic Pyrrolnitrin by Psuedomonas aureofaciens. J Antibiot (Tokyo) 1981;24:555–566. doi: 10.7164/antibiotics.34.555. [DOI] [PubMed] [Google Scholar]
- 304.van Pee KH, Salcher O, Lingens F. Formation of Pyrrolnitrin and 3-(2-Amino-3-chlorophenyl)pyrrole from 7-Chlorotryptophan. Angew Chem, Int Ed. 1980;19:828–829. [Google Scholar]
- 305.Hamill RH, Elander RP, Mabe JA, Gorman M. Metabolism of Tryptophans by Pseudomonas aureofaciens. V. Conversion of Tryptophan to Pyrrolnitrin. Antimicrob Agents Chemother. 1967:388–396. [PubMed] [Google Scholar]
- 306.Ively LDH, Gorman M, Haney ME, Mabe JA. Metabolism of Tryptophans by Pseudomonas aureofaciens. I. Biosynthesis fo Pyrrolnitrin. Antimicrob Agents Chemother. 1966:462–469. [PubMed] [Google Scholar]
- 307.Hammer PE, HIll DS, Lam ST, van Pee KH, Ligon JM. Four Genes from Pseudomonas fluorescens that Encode the Biosynthesis of Pyrrolnitrin. Appl Environ Microbiol. 1997;63:2147–2154. doi: 10.1128/aem.63.6.2147-2154.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Kirner S, Hammer PE, HIll DS, Altmann A, Fischer I, Weislo LJ, Lanahan M, van Pee KH, Ligon JM. Functions Encoded by Pyrrolnitrin Biosynthetic Genes from Pseudomonas fluorescens. J Bacteriol. 1998;180:1939–1943. doi: 10.1128/jb.180.7.1939-1943.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Lee J, Simurdiak M, Zhao H. Reconstitution and Characterization of Aminopyrrolnitrin Oxygenase, a Rieske N-oxygenase that Catalyzes Unusual Arylamine Oxidation. J Biol Chem. 2005;280:36719–36727. doi: 10.1074/jbc.M505334200. [DOI] [PubMed] [Google Scholar]
- 310.Lee J, Zhao H. Mechanistic Studies on the Conversion of Arylamines into Arylnitro Compounds by Aminopyrrolnitrin Oxygenase: Identification of Intermediates and Kinetic Studies. Angew Chem, Int Ed. 2006;45:622–625. doi: 10.1002/anie.200502903. [DOI] [PubMed] [Google Scholar]
- 311.Lee JK, Ang EL, Zhao H. Probing the Substrate Specificity of Aminopyrrolnitrin Oxygenase (PrnD) by Mutational Analysis. J Bacteriol. 2006;188:6179–6183. doi: 10.1128/JB.00259-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Tiwari MK, Lee JK, Moon HJ, Zhao H. Further Biochemical Studies on Aminopyrrolnitrin Oxygenase (PrnD) Bioorg Med Chem Lett. 2011;21:2873–2876. doi: 10.1016/j.bmcl.2011.03.087. [DOI] [PubMed] [Google Scholar]
- 313.Dalsgaard PW, Larsen TO, Frydenvang K, Christophersen C. Psychrophilin A and Cycloaspeptide D, Novel Cyclic Peptides from the Psychrotolerant Fungus Penicillum ribeum. J Nat Prod. 2004;67:878–881. doi: 10.1021/np0303714. [DOI] [PubMed] [Google Scholar]
- 314.Dalsgaard PW, Blunt JW, Munro MHG, Larsen TO, Christophersen C. Psychrophilin B and C: Cyclic Nitropeptides from the Psychrotolerant Fungus Penicillum rivulum. J Nat Prod. 2004;67:1950–1952. doi: 10.1021/np0497954. [DOI] [PubMed] [Google Scholar]
- 315.Gao X, Haynes SW, Ames BD, Wang P, Vien LP, Walsh CT, Tang Y. Cyclization of Fungal Nonribosomal Peptides by a Terminal Condensation-like Domain. Nat Chem Biol. 2012;8:823–830. doi: 10.1038/nchembio.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Gao X, Chooi YH, Ames BD, Wang P, Walsh CT, Tang Y. Fungal Indole Alkaloid Biosynthesis: Genetic and Biochemical Investigation of the Tryptoquialanine Pathway in Penicillium aethiopicum. J Am Chem Soc. 2011;133:2729–2741. doi: 10.1021/ja1101085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Zhao M, Lin HC, Tang Y. Biosynthesis of the Alpha-Nitro-Containing Cyclic Tripeptide Psychrophilin. J Antibiot (Tokyo) 2016;69:571–573. doi: 10.1038/ja.2016.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Bradner WT, Claridge CA, Huftalen JB. Antitumor Activity of Kijanimicin. J Antibiot (Tokyo) 1983;36:1078–1079. doi: 10.7164/antibiotics.36.1078. [DOI] [PubMed] [Google Scholar]
- 319.Mallams AK, Puar MS, Rossman RR, McPhail AT, Macfarlane RD, Stephens RL. Kijanimicin Part 3. Structure and Absolute Stereochemistry of Kijanimicin. J Chem Soc Perkin Trans 1. 1983:1497–1534. [Google Scholar]
- 320.Waitz JA, Horan AC, Kalyanpur M, Lee BK, Loebenberg D, Marquez JA, Miller G, Patel MG. Kijanimicin (Sch 25663), A Novel Antibiotic Produced by Actinomadura kijaniata SCC 1256. Fermentation, Isolation, Characterization and Biological Properties. J Antibiot (Tokyo) 1981;34:1101–1106. doi: 10.7164/antibiotics.34.1101. [DOI] [PubMed] [Google Scholar]
- 321.Hoeksema H, Mizsak SA, Baczynskyj L, Pshigoda LM. Structure of Rubradirin. J Am Chem Soc. 1982;104:5173–5181. [Google Scholar]
- 322.Reusser F. Rubradirin, an Inhibitor of Ribosomal Polypeptide Biosynthesis. Biochemistry. 1973;12:1136–1142. doi: 10.1021/bi00730a019. [DOI] [PubMed] [Google Scholar]
- 323.Bhuyan BK, Owen SP, Dietz A. Rubradirin, a New Antibiotic. I. Fermentation and Biological Properties. Antimicrob Agents Chemother. 1964;10:91–96. [PubMed] [Google Scholar]
- 324.Jones RN, Hare RS, Sabatelli FJ. In Vitro Gram-Positive Antimicrobial Activity of Evernimicin (SCH 27899), a Novel Oligosaccharide, Compared with Other Antimicrobials: A Multicenter International Trial. J Antimicrob Chemother. 2001;47:15–25. doi: 10.1093/jac/47.1.15. [DOI] [PubMed] [Google Scholar]
- 325.McNicholas PM, Najarian DJ, Mann PA, Hesk D, Hare RA, Shaw KJ, Black TA. Evernimicin Binds Exclusively to the 50S Ribosomal Subunit and Inhibits Translation in Cell-Free Systems Derived from both Gram-Positive and Gram-Negative Bacteria. Antimicrob Agents Chemother. 2000;44:1121–1126. doi: 10.1128/aac.44.5.1121-1126.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Ganguly AK, Sarre OZ, Greeves D, Morton J. Structure of Everninomicin D. J Am Chem Soc. 1975;97:1982–1985. doi: 10.1021/ja00840a078. [DOI] [PubMed] [Google Scholar]
- 327.Ganguly AK, Sarre OZ, Reimann H. Evernitrose, a Naturally Occuring Nitro Sugar from Everninomicins. J Am Chem Soc. 1968;90:7129–7130. doi: 10.1021/ja01027a047. [DOI] [PubMed] [Google Scholar]
- 328.Wagman GH, Luedemann GM, Weinstein MJ. Fermentation and Isolation of Everninomicin. Antimicrob Agents Chemother. 1964;10:33–37. [PubMed] [Google Scholar]
- 329.Weinstein MJ, Luedemann GM, Oden EM, Wagman GH. Everninomicin, A New Antibiotic Complex from Micromonospora carbonacea. Antimicrob Agents Chemother. 1964;10:24–32. [PubMed] [Google Scholar]
- 330.Thoden JB, Branch MC, Zimmer AL, Bruender NA, Holden HM. Active Site Architecture of a Sugar N-oxygenase. Biochemistry. 2013;52:3191–3193. doi: 10.1021/bi400407x. [DOI] [PubMed] [Google Scholar]
- 331.Vey JL, Al-Mestarihi A, Hu Y, Funk MA, Bachmann BO, Iverson TM. Structure and Mechanism of ORF36: an Amino Sugar Oxidizing Enzyme in Everninomicin Biosynthesis. Biochemistry. 2010;49:9306–9317. doi: 10.1021/bi101336u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Bruender NA, Thoden JB, Holden HM. X-ray Structure of Kijd3: a Key Enzyme Involved in the Biosynthesis of D-kijanose. Biochemistry. 2010;49:3517–3524. doi: 10.1021/bi100318v. [DOI] [PubMed] [Google Scholar]
- 333.Hosted TJ, Wang TX, Alexander DC, Horan AC. Characterization of the Biosynthetic Gene Cluster for the Oligosaccharide Antibiotic, Evernimicin, in Micromonospora carbonacae var. africana ATCC3919. J Ind Microbiol Biotechnol. 2001;27:386–392. doi: 10.1038/sj.jim.7000189. [DOI] [PubMed] [Google Scholar]
- 334.Zhang H, White-Phillip JS, Melancon CE, Kwon HJ, Yu WL, Liu HW. Elucidation of the Kijanimicin Gene Cluster: Insights into the Biosynthesis of Spirotetronate Antibiotics and Nitrosugars. J Am Chem Soc. 2007;129:14670–14683. doi: 10.1021/ja0744854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Kim CG, Lamichhane J, Song KI, Nguyen VD, Kim DH, Jeong TS, Kang SH, Kim KW, Maharjan J, Hong YS, et al. Biosynthesis of Rubradirin as an Ansamycin Antibiotic from Streptomyces achromogenes var. rubradiris NRRL3061. Arch Microbiol. 2008;189:463–473. doi: 10.1007/s00203-007-0337-3. [DOI] [PubMed] [Google Scholar]
- 336.Sohng JK, Oh TJ, Lee JJ, Kim CG. Identification of a Gene Cluster of Biosynthetic Genes of Rubradirin Substructures in S. achromogenes var. rubradiris NRRL3061. Mol Cells. 1997;7:674–681. [PubMed] [Google Scholar]
- 337.Hu Y, Al-Mestarihi A, Grimes CL, Kahne D, Bachmann BO. A Unifying Nitrososynthase Involved in Nitrosugar Biosynthesis. J Am Chem Soc. 2008;130:15756–15757. doi: 10.1021/ja8051415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Al-Mestarihi A, Romo A, Liu HW, Bachmann BO. Nitrososynthase-triggered Oxidative Carbon–Carbon Bond Cleavage in Baumycin Biosynthesis. J Am Chem Soc. 2013;135:11457–11460. doi: 10.1021/ja404987r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Loria R, Bukhalid RA, Fry BA, King RR. Plant Pathogenicity in the Genus Streptomyces. Plant Dis. 1997;81:836–846. doi: 10.1094/PDIS.1997.81.8.836. [DOI] [PubMed] [Google Scholar]
- 340.King RR, Lawrence CH, Calhoun LA. Chemistry of Phytotoxins Associated with Streptomyces scabies, the Causal Organism of Potato Common Scab. J Agric Food Chem. 1992;40:834–837. [Google Scholar]
- 341.King RR, Lawrence CH, Clark MC, Calhoun LA. Isolation and Characterization of Phytotoxins Associated with Streptomyces scabies. J Chem Soc Commun. 1989;13:849–850. [Google Scholar]
- 342.Scheible WR, Fry BA, Kochevenko A, Schindelasch D, Zimerli L, Somerville S, Loria R, Somerville CR. An Arabidopsis Mutant Resistant to Thaxtomin A, a Cellulose Synthesis Inhibitor from Streptomyces scabies. Plant Cell. 2003;15:1781–1794. doi: 10.1105/tpc.013342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Lawrence CH, Clark MC, King RR. Induction of Common Scab Symptoms in Aseptically Cultured Potato Tubers by the Vivotoxin, Thaxtomin. Phytopathology. 1990;80:606–608. [Google Scholar]
- 344.Healy FG, Krasnoff SB, Wach M, Gibson DM, Loria R. Involvement of a Cytochrome P450 Monooxygenase in Thaxtomin A Biosynthesis by Streptomyces acidiscabies. J Bacteriol. 2002;184:2019–2029. doi: 10.1128/JB.184.7.2019-2029.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Healy FG, Wach M, Krasnoff SB, Gibson DM, Loria R. The txtAB Genes of the Plant Pathogen Streptomyces acidscabies Encode a Peptide Synthetase Required for Phytotoxin Thaxtomin A Production and Pathogenicity. Mol Microbiol. 2000;38:794–804. doi: 10.1046/j.1365-2958.2000.02170.x. [DOI] [PubMed] [Google Scholar]
- 346.Kers JA, Wach MJ, Krasnoff SB, Widom J, Cameron KD, Bukhalid RA, Gibson DM, Crane BR, Loria R. Nitration of a Peptide Phytotoxin by Bacterial Nitric Oxide Synthase. Nature. 2004;429:79–82. doi: 10.1038/nature02504. [DOI] [PubMed] [Google Scholar]
- 347.Buddha MR, Tao T, Parry RJ, Crane BR. Regioselective Nitration of Tryptophan by a Complex Between Bacterial Nitric Oxide Synthase and Tryptophanyl-tRNA Synthetase. J Biol Chem. 2004;279:49567–49570. doi: 10.1074/jbc.C400418200. [DOI] [PubMed] [Google Scholar]
- 348.Barry SM, Kers JA, Johnson EG, Song L, Aston PR, Patel B, Krasnoff SB, Crane BR, Gibson DM, Loria R, et al. Cytochrome P450-Catalyzed L-Tryptophan Nitration in Thaxtomin Phytotoxin Biosynthesis. Nat Chem Biol. 2012;8:814–816. doi: 10.1038/nchembio.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Dodani SC, Kiss G, Cahn JK, Su Y, Pande VS, Arnold FH. Discovery of a Regioselectivity Switch in Nitrating P450s Guided by Molecular Dynamics Simulations and Markov Models. Nat Chem. 2016;8:419–425. doi: 10.1038/nchem.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Dodani SC, Cahn JK, Heinisch T, Brinkmann-Chen S, McIntosh JA, Arnold FH. Structural, Functional, and Spectroscopic Characterization of the Substrate Scope of the Novel Nitrating Cytochrome P450 TxtE. Chembiochem. 2014;15:2259–2267. doi: 10.1002/cbic.201402241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Yu F, Li M, Xu C, Wang Z, Zhou H, Yang M, Chen Y, Tang L, He J. Structural Insights into the Mechanism for Recognizing Substrate of the Cytochrome P450 Enzyme TxtE. PLoS One. 2013;8:e81526. doi: 10.1371/journal.pone.0081526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Coles CJ, Edmondson DE, Singer TP. Inactivation of Succinate Dehydrogenase by 3-Nitropropionate. J Biol Chem. 1979;254:5161–5167. [PubMed] [Google Scholar]
- 353.Alston TA, Mela L, Bright HJ. 3-Nitropropionate, the Toxic Substance of Indigofera, is a Suicide Inactivator of Succinate Dehydrogenase. Proc Natl Acad Sci USA. 1977;74:3767–3771. doi: 10.1073/pnas.74.9.3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Hamilton BF, Gould DH, Gustine DL. Mitochondrial Inhibitors and Neurodegenerative Disorders. Humana Press; Totowa, NJ: 1999. [Google Scholar]
- 355.Candlish E, La Croix LJ, Unrau AM. The Biosynthesis of 3-Nitropropionic Acid in Creeping Indigo (Indigofera spicata) Biochemistry. 1969;8:182–186. doi: 10.1021/bi00829a026. [DOI] [PubMed] [Google Scholar]
- 356.Baxter RL, Smith SL, Martin JR, Hanley AB. The Fungal Biosynthesis of 3-Nitropropanoic Acid: Is the Decarboxylation of L-Nitrosuccinate an Enzymatic Reaction? J Chem Soc Perkin Trans 1. 1994:2297–2299. [Google Scholar]
- 357.Baxter RL, Hanley AB, Chan HWS, Greenwood SL, Abbot EM, McFarlane IJ, Milne K. Fungal Biosynthesis of 3-Nitropropanoic Acid. J Chem Soc Perkin Trans 1. 1992:2495–2502. [Google Scholar]
- 358.Baxter RL, Hanley AB, Chan HWS. Identification of L-Nitrosuccinate as an Intermediate in the Fungal Biosynthesis of 3-Nitropropanoic Acid. J Chem Soc, Chem Commun. 1988;12:757–758. [Google Scholar]
- 359.Baxter RL, Greenwood SL. Application of the 18O Isotope Shift in 15N NMR Spectra to a Biosynthetic Problem: Experimental Evidence for the Origin of the Nitro Group Oxygen Atoms of 3-Nitropropanoic Acid. J Chem Soc, Chem Commun. 1986:175–176. [Google Scholar]
- 360.Baxter RL, Abbot EM, Greenwood SL, McFarlane IJ. Conservation of the Carbon–Nitrogen Bond of Aspartic Acid in the Biosynthesis of 3-Nitropropanoic Acid. J Chem Soc, Chem Commun. 1985:564–566. [Google Scholar]
- 361.Shaw PD, McCloskey JA. Biosynthesis of Nitro Compounds. II. Studies on Potential Precursors for the Nitro Group of B-Nitropropanoic Acid. Biochemistry. 1967;6:2247–2253. doi: 10.1021/bi00859a047. [DOI] [PubMed] [Google Scholar]
- 362.Hylin JW, Matsumoto H. The Biosynthesis of 3-Nitropropanoic Acid by Penicillium atrovenetum. Arch Biochem Biophys. 1960;93:542–545. doi: 10.1016/s0003-9861(61)80049-0. [DOI] [PubMed] [Google Scholar]
- 363.Sugai Y, Katsuyama Y, Ohnishi Y. A Nitrous Acid Biosynthetic Pathway for Diazo Group Formation in Bacteria. Nat Chem Biol. 2016;12:73–75. doi: 10.1038/nchembio.1991. [DOI] [PubMed] [Google Scholar]
- 364.Andres N, Wolf H, Zahner H, Rossner E, Zeeck A, Konig WA, Sinnwell V. Hormaomycin, a Novel Peptide Lactone with Morphogenetic Activity on Streptomyces. Helv Chim Acta. 1989;72:426–437. [Google Scholar]
- 365.Brandl M, Kozhushkov SI, Zlatopolskiy BD, Alvermann P, Geers B, Zeeck A, de Meijere A. The Biosynthesis of 3-(trans-2-Nitrocyclopropyl)alanine, a Constituent of the Signal Metabolite Hormaomycin. Eur J Org Chem. 2005:123–135. [Google Scholar]
- 366.Hofer I, Crusemann M, Radzom M, Geers B, Flachshaar D, Cai X, Zeeck A, Piel J. Insights into the Biosynthesis of Hormaomycin, an Exceptionally Complex Bacterial Signaling Metabolite. Chem Biol. 2011;18:381–391. doi: 10.1016/j.chembiol.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 367.Blair LM, Sperry J. Natural Products Containing a Nitrogen–Nitrogen Bond. J Nat Prod. 2013;76:794–812. doi: 10.1021/np400124n. [DOI] [PubMed] [Google Scholar]
- 368.Le Goff G, Ouazzani J. Natural Hydrazine-containing Compounds: Biosynthesis, Isolation, Biological Activities and Synthesis. Bioorg Med Chem. 2014;22:6529–6544. doi: 10.1016/j.bmc.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 369.Langley BW, Lythgoe B, Riggs NV. J Chem Soc. 1951:2309–2316. [Google Scholar]
- 370.Hydrazo, Azo and Azoxy Groups. 1 and 2. John Wiley & Sons; 1975. [Google Scholar]
- 371.Sandler SR, Karo W. Organic Functional Group Preparations, Volume 2. Elsevier; 1986. [Google Scholar]
- 372.Yamato M, Iinuma H, Naganawa H, Yamagishi Y, Hamada M, Masuda T, Umezawa H, Abe Y, Hori M. Isolation and Properties of Valanimycin, a New Azoxy Antibiotic. J Antibiot (Tokyo) 1986;39:184–194. doi: 10.7164/antibiotics.39.184. [DOI] [PubMed] [Google Scholar]
- 373.Yamato M, Umezawa H. Valanimycin Acts on DNA in Bacterial Cells. J Antibiot (Tokyo) 1987;40:558–560. doi: 10.7164/antibiotics.40.558. [DOI] [PubMed] [Google Scholar]
- 374.Parry RJ, Li Y, Lii FL. Biosynthesis of Azoxy Compounds. Investigations of Valanimycin Biosynthesis. J Am Chem Soc. 1992;114:10062–10064. [Google Scholar]
- 375.Parry RJ, Li W. The Biosynthesis of Valanimycin. Further Evidence for the Intermediacy of a Hydroxylamine in N–N Bond Formation. J Chem Soc, Chem Commun. 1994;8:995–996. [Google Scholar]
- 376.Skae P, Parry RJ. Determination of the Stereochemistry of Hydride Transfer from NADPH to FAD catalyzed by VlmR, a Flavin Reductase from the Valanimycin Biosynthetic Pathway. Org Lett. 2001;3:1117–1119. doi: 10.1021/ol000389v. [DOI] [PubMed] [Google Scholar]
- 377.Parry RJ, Li W, Cooper HN. Cloning, Analysis, and Overexpression of the Gene Encoding Isobutylamine N-hydroxylase from the Valanimycin Producer, Streptomyces viridifaciens. J Bacteriol. 1997;179:409–416. doi: 10.1128/jb.179.2.409-416.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Garg RP, Ma Y, Hoyt JC, Parry RJ. Molecular Characterization and Analysis of the Biosynthetic Gene Cluster for the Azoxy Antibiotic Valanimycin. Mol Microbiol. 2002;46:505–517. doi: 10.1046/j.1365-2958.2002.03169.x. [DOI] [PubMed] [Google Scholar]
- 379.Tao T, Alemany LB, Parry RJ. Valanimycin Biosynthesis: Investigations of the Mechanism of Isobutylhydroxylamine Incorporation. Org Lett. 2003;5:1213–1215. doi: 10.1021/ol0340989. [DOI] [PubMed] [Google Scholar]
- 380.Ma Y, Patel J, Parry RJ. A Novel Valanimycin-resistance Determinant (vlmF) from Streptomyces viridifaciens MG456-hF10. Microbiology. 2000;146:345–352. doi: 10.1099/00221287-146-2-345. [DOI] [PubMed] [Google Scholar]
- 381.Jahn D, Verkamp E, Soll D. Glutamyl-transferase RNA: A Precursor of Heme and Chlorophyll Biosynthesis. Trends Biochem Sci. 1992;17:215–218. doi: 10.1016/0968-0004(92)90380-r. [DOI] [PubMed] [Google Scholar]
- 382.Garg RP, Gonzalez JM, Parry RJ. Biochemical Characterization of VlmL, a Seryl-tRNA Synthetase Encoded by the Valanimycin Biosynthetic Gene Cluster. J Biol Chem. 2006;281:26785–26791. doi: 10.1074/jbc.M603675200. [DOI] [PubMed] [Google Scholar]
- 383.Garg RP, Qian XL, Alemany LB, Moran S, Parry RJ. Investigations of Valanimycin Biosynthesis: Elucidation of the Role of Seryl-tRNA. Proc Natl Acad Sci US A. 2008;105:6543–6547. doi: 10.1073/pnas.0708957105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Oku Y, Kurokawa K, Ichihashi N, Sekimizu K. Characterization of the Staphylococcus aureus mprF Gene Involved in Lysinylation of Phosphatidylglycerol. Microbiology. 2004;150:45–51. doi: 10.1099/mic.0.26706-0. [DOI] [PubMed] [Google Scholar]
- 385.Garg RP, Alemany LB, Moran S, Parry RJ. Identification, Characterization, and Bioconversion of a New Intermediate in Valanimycin Biosynthesis. J Am Chem Soc. 2009;131:9608–9609. doi: 10.1021/ja901243p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Haskell TH, Ryder A, Bartz QR. Elaiomycin, a New Tuberculostatic Antibiotic. Isolation and Chemical Characterization. Antibiot Chemother. 1954;4:141–144. [PubMed] [Google Scholar]
- 387.Ehrlich J, Anderson LE, Coffey GL, Feldman WH, Fisher MW, Hillegas AB, Karlson AG, Knudsen MP, Weston JK, Youmans AS, et al. Elaiomycin, a New Tuberculostatic Antibiotic. Biological Studies. Antibiot Chemother. 1954;4:338–342. [PubMed] [Google Scholar]
- 388.Karlson AG. Specific Inhibitory Effect of Elaiomycin In Vitro upon Mycobacterium tuberculosis. Antibiot Chemother. 1962;12:446–449. [PubMed] [Google Scholar]
- 389.Schoental R. Carcinogenic Action of Elaiomycin in Rats. Nature. 1969;221:765–766. doi: 10.1038/221765a0. [DOI] [PubMed] [Google Scholar]
- 390.Stevens CL, Gillis BT, French JC, Haskell TH. The Structure of Elaiomycin, a Tuberculostatic Antibiotic. J Am Chem Soc. 1956;78:3229–3230. [Google Scholar]
- 391.Stevens CL, Gillis BT, French JC, Haskell TH. Elaiomycin. An Aliphatic a, B-Unsaturated Azoxy Compound. J Am Chem Soc. 1958;80:6088–6092. [Google Scholar]
- 392.Stevens CL, Gillis BT, Haskell TH. Elaiomycin II. Determination of the D-threo Configuration. J Am Chem Soc. 1959;81:1435–1437. [Google Scholar]
- 393.McGahren WJ, Kunstmann MP. Circular Dichroism Studies on the Azoxy Chromophore of the Antibiotics LL-BH872a and Elaiomycin. J Am Chem Soc. 1970;92:1587–1590. doi: 10.1021/ja00709a027. [DOI] [PubMed] [Google Scholar]
- 394.McGahren WJ, Kunstmann MP. Revision of Azoxy Assignments in LL-BH872a and Elaiomycin Based on Circular Dichroism Studies on Synthetic Azoxy Compounds. J Org Chem. 1972;37:902–906. doi: 10.1021/jo00971a019. [DOI] [PubMed] [Google Scholar]
- 395.Moss RA, Matsuo M. The Synthesis of Elaiomycin, a Naturally Occuring Azoxyalkene. J Am Chem Soc. 1977;99:1643–1645. doi: 10.1021/ja00447a060. [DOI] [PubMed] [Google Scholar]
- 396.Parry RJ, Rao HSP, Mueller JV. Biosynthesis of Elaiomycin. 1. Incorporation of Labeled Forms of n-Octylamine. J Am Chem Soc. 1982;104:339–340. [Google Scholar]
- 397.Parry RJ, Mueller JV. Biosynthesis of Elaiomycin. 2. An Unusual Origin for the (Methoxyamino)butanol Moiety. J Am Chem Soc. 1984;106:5764–5765. [Google Scholar]
- 398.Ding L, Ndejouong BLST, Maier A, Fiebig HH, Hertweck C. Elaiomycins D-F, Antimicrobial and Cytotoxic Azoxides from Streptomyces sp. Strain HKI0708. J Nat Prod. 2012;75:1729–1734. doi: 10.1021/np300329m. [DOI] [PubMed] [Google Scholar]
- 399.Manderscheid N, Helaly SE, Kulik A, Wiese J, Imhoff JF, Fiedler HP, Sussmuth RD. Elaiomycins K and L, New Azoxy Antibiotics from Streptomyces sp. Tu 6399. J Antibiot (Tokyo) 2013;66:85–88. doi: 10.1038/ja.2012.99. [DOI] [PubMed] [Google Scholar]
- 400.Kunitake H, Hiramatsu T, Kinashi H, Arakawa K. Isolation and Biosynthesis of an Azoxyalkene Compound Produced by a Multiple Gene Disruptant of Streptomyces rochei. Chembiochem. 2015;16:2237–2243. doi: 10.1002/cbic.201500393. [DOI] [PubMed] [Google Scholar]
- 401.Bianchi G, Dallavalle S, Merlini L, Nasini G, Quaroni S. A New Azoxyalkene from a Strain of an Actinomadura-Like Fungus. Planta Med. 2003;69:574–576. doi: 10.1055/s-2003-40633. [DOI] [PubMed] [Google Scholar]
- 402.Yang H, Chaowagul W, Sokol PA. Siderophore Production by Pseudomonas pseudomallei. Infect Immun. 1991;59:776–780. doi: 10.1128/iai.59.3.776-780.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Yang H, Kooi C, Sokol PA. Ability of Pseudomonas pseudomallei Malleobactin to Acquire Transferrin-bound, Lactoferrin-bound, and Cell-derived Iron. Infect Immun. 1993;61:656–662. doi: 10.1128/iai.61.2.656-662.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Alice AF, Lopez CS, Lowe CA, Ledesma MA, Crosa JH. Genetic and Transcriptional Analysis of the Siderophore Malleobactin Biosynthesis and Transport Genes in the Human Pathogen Burkholderia pseudomallei K96243. J Bacteriol. 2006;188:1551–1566. doi: 10.1128/JB.188.4.1551-1566.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Agnoli K, Lowe CA, Farmer KL, Husnain SI, Thomas MS. The Ornibactin Biosynthesis and Transport Genes of Burkholderia cenocepacia are Regulated by an Extracytoplasmic Function Sigma Factor Which is Part of the Fur Regulon. J Bacteriol. 2006;188:3631–3644. doi: 10.1128/JB.188.10.3631-3644.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Stephan H, Freund S, Beck W, Jung G, Meyer JM, Winkelmann G. Ornibactins - A New Family of Siderophores from Pseudomonas cepacia. BioMetals. 1993;6:93–100. doi: 10.1007/BF00140109. [DOI] [PubMed] [Google Scholar]
- 407.Lindeke B. The Non- and Post-Enzymatic Chemistry of N-Oxygenated Molecules. Drug Metab Rev. 1982;13:71–121. doi: 10.3109/03602538209002232. [DOI] [PubMed] [Google Scholar]
- 408.Guo YY, Li H, Zhou ZX, Mao XM, Tang Y, Chen X, Jiang XH, Liu Y, Jiang H, Li YQ. Identification and Biosynthetic Characterization of Natural Aromatic Azoxy Products from Streptomyces chattanoogensis L10. Org Lett. 2015;17:6114–6117. doi: 10.1021/acs.orglett.5b03137. [DOI] [PubMed] [Google Scholar]
- 409.Rui Z, Petrickova K, Skanta F, Pospisil S, Yang Y, Chen CY, Tsai SF, Floss HG, Petricek M, Yu TW. Biochemical and Genetic Insights in Asukamycin Biosynthesis. J Biol Chem. 2010;285:24915–24924. doi: 10.1074/jbc.M110.128850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Potterat O, Zahner H, Metzger JW, Freund S. Metabolic Products of Microorganisms. 269. 5-Phenylpentadienoic Acid Derivatives from Streptomyces sp. Helv Chim Acta. 1994;77:569–574. [Google Scholar]
- 411.Nawrat CC, Moody CJ. Natural Products Containing a Diazo Group. Nat Prod Rep. 2011;28:1426–1444. doi: 10.1039/c1np00031d. [DOI] [PubMed] [Google Scholar]
- 412.Fusari SA, Frohardt RP, Ryder A, Haskell TH, Johannes DW, Elder CC, Bartz QR. Azaserine, a New Tumor-inhibitory Substance. Isolation and Characterization. J Am Chem Soc. 1954;76 [Google Scholar]
- 413.Dion HW, Fusari SA, Jakubowski ZL, Zora JG, Bartz QR. 6-Diazo-5-oxo-Lnorleucine, A New Tumor-Inhibitory Substance. II. Isolation and Characterization. J Am Chem Soc. 1956;78 [Google Scholar]
- 414.Rao KV, Brooks SC, Kugelman M, Romano AA. Antibiotics Ann. 1959;93 [PubMed] [Google Scholar]
- 415.Stock CC, Clarke DA, Reilly HC, Rhoads CP, Buckley SM. Azaserine, A New Tumor–Inhibitory Substance: Studies with Crocker Mouse Sarcoma 180. Nature. 1954;173 doi: 10.1038/173071a0. [DOI] [PubMed] [Google Scholar]
- 416.Ellison RR, Karnofsky DA, Sternberg SS, Murphy ML, Burchenal JH. Clinical Trials of O-diazoacetyl-L-serine (Azaserine) in Neoplastic Disease. Cancer. 1954;7 doi: 10.1002/1097-0142(195407)7:4<801::aid-cncr2820070419>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 417.Catane R, Von Hoff DD, Glaubiger DL, Muggia FM. Azaserine, DON, and Azotomycin: Three Diazo Analogs of L-Glutamine with Clinical Antitumor Activity. Cancer Treat Rep. 1979;63:1033–1038. [PubMed] [Google Scholar]
- 418.Ahluwalia GS, Grem JL, Hao Z, Cooney DA. Metabolism and Action of Amino Acid Analog Anti-Cancer Agents. Pharmacol Ther. 1990;46:243–271. doi: 10.1016/0163-7258(90)90094-i. [DOI] [PubMed] [Google Scholar]
- 419.Ye T, Mckervey MA. Organic Synthesis with a-Diazo Carbonyl Compounds. Chem Rev. 1994;94:1091–1160. [Google Scholar]
- 420.Johnston JN, Muchalski H, Troyer TL. To Protonate or Alkylate? Stereoselective Bronsted Acid Catalysis of C–C Bond Formation Using Diazoalkanes. Angew Chem, Int Ed. 2010;49:2290–2298. doi: 10.1002/anie.200904828. [DOI] [PubMed] [Google Scholar]
- 421.Davies HML, Manning JR. Catalytic C–H Functionalization by Metal Carbenoid and Nitrenoid Insertion. Nature. 2008;451:417–424. doi: 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Maas G. New Syntheses fo Diazo Compounds. Angew Chem, Int Ed. 2009;48:8186–8195. doi: 10.1002/anie.200902785. [DOI] [PubMed] [Google Scholar]
- 423.Bergy ME, Pyke TR. Cremeomycin and Process for Making. 3350269. US. 1967 Oct 31;
- 424.McGuire JN, Wilson SR, Rinehart KL. Cremeomycin, a Novel Cytotoxic Antibiotic from Streptomyces cremeus. Structure Elucidation and Biological Activity. J Antibiot (Tokyo) 1995;48:516–519. doi: 10.7164/antibiotics.48.516. [DOI] [PubMed] [Google Scholar]
- 425.Varley LM, Moody CJ. First Synthesis of the Naturally Occurring Diazocarbonyl Compound Cremeomycin. Synthesis. 2008;22:3601–3604. [Google Scholar]
- 426.Suzuki H, Ohnishi Y, Furusho Y, Sakuda S, Horinouchi S. Novel Benzene Ring Biosynthesis from C3 and C4 Primary Metabolites by Two Enzymes. J Biol Chem. 2006;281:36944–36951. doi: 10.1074/jbc.M608103200. [DOI] [PubMed] [Google Scholar]
- 427.Waldman AJ, Pechersky Y, Wang P, Wang JX, Balskus EP. The Cremeomycin Biosynthetic Gene Cluster Encodes a Pathway for Diazo Formation. Chembiochem. 2015;16:2172–2175. doi: 10.1002/cbic.201500407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Winter JM, Jansma AL, Handel TM, Moore BS. Formation of the Pyridazine Natural Product Azamerone by Biosynthetic Rearrangement of an Aryl Diazoketone. Angew Chem, Int Ed. 2009;48:767–770. doi: 10.1002/anie.200805140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Huang Z, Wang KK, Lee J, van der Donk WA. Biosynthesis of Fosfazinomycin is a Convergent Process. Chem Sci. 2015;6:1282–1287. doi: 10.1039/c4sc03095h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Gao J, Ju KS, Yu X, Velasquez JE, Mukherjee S, Lee J, Zhao C, Evans BS, Doroghazi JR, Metcalf WW, et al. Use of a Phosphonate Methyltransferase in the Identification of the Fosfazinomycin Biosynthetic Gene Cluster. Angew Chem, Int Ed. 2014;53:1334–1337. doi: 10.1002/anie.201308363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Huang Z, Wang KKA, van der Donk WA. New Insights into the Biosynthesis of Fosfazinomycin. Chem Sci. 2016;7:5219–5223. doi: 10.1039/c6sc01389a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Ito S, Matsuya T, Omura S, Otani M, Nakagawa A, Takeshima H, Iwai Y, Ohtani M, Hata T. A New Antibiotic, Kinamycin. J Antibiot (Tokyo) 1970;23:315–317. doi: 10.7164/antibiotics.23.315. [DOI] [PubMed] [Google Scholar]
- 433.Isshiki K, Sawa T, Naganawa H, Matsuda N, Hattori S, Hamada M, Takeuchi T, Oosono M, Ishizuka M, Yang Z, et al. 3-O-isobutyrylkinamycin C and 4-deacetyl-4-Oisobutyrylkinamycin C, New Antibiotics Produced by a Saccharothrix species. J Antibiot (Tokyo) 1989;42:467–469. doi: 10.7164/antibiotics.42.467. [DOI] [PubMed] [Google Scholar]
- 434.Smitka TA, Bonjouklian R, Perun TJ, Hunt AH, Foster RS, Mynderse JS, Yao RC. A80316A, A New Kinamycin Type Antibiotic. J Antibiot (Tokyo) 1992;45:581–583. doi: 10.7164/antibiotics.45.581. [DOI] [PubMed] [Google Scholar]
- 435.Lin HC, Chang SC, Wang NL, Chang LR. FL-120A D’, New Products Related to Kinamycin from Streptomyces chattanoogensis subsp. taitungensis subsp. nov. I. Taxonomy, Fermentation, and Biological Properties. J Antibiot (Tokyo) 1994;47:675–680. doi: 10.7164/antibiotics.47.675. [DOI] [PubMed] [Google Scholar]
- 436.Young JJ, Ho SN, Ju WM, Chang LR. FL-120A D’, New Products Related to Kinamycin from Streptomyces chattanoogensis subsp. taitungensis subsp. nov. II. Isolation and Structure Determination. J Antibiot (Tokyo) 1994;47:681–687. doi: 10.7164/antibiotics.47.681. [DOI] [PubMed] [Google Scholar]
- 437.Bunet R, Song L, Mendes MV, Corre C, Hotel L, Rouhier N, Framboisier X, Leblond P, Challis GL, Aigle B. Characterization and Manipulation of the Pathway-Specific Late Regulator AlpW Reveals Streptomyces ambofaciens as a New Producer of Kinamycins. J Bacteriol. 2011;193:1142–1153. doi: 10.1128/JB.01269-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Hata T, Omura S, Iwai Y, Nakagawa A, Otani M, Ito S, Matsuya T. A New Antibiotic, Kinamycin: Fermentation, Isolation, Purification and Properties. J Antibiot (Tokyo) 1971;24:353–359. doi: 10.7164/antibiotics.24.353. [DOI] [PubMed] [Google Scholar]
- 439.Omura S, Nakagawa A, Yamada H, Hata T, Furusaki A, Watanabe T. Structures and Biological Properties of Kinamycin A, B, C, and D. Chem Pharm Bull. 1973;21:931–940. doi: 10.1248/cpb.21.931. [DOI] [PubMed] [Google Scholar]
- 440.Omura S, Nakagawa A, Yamada H, Hata T, Furusaki A, Watanabe T. Structures of Kinamycin C, and the Structural Relationship among Kinamycin A, B, C, and D. Chem Pharm Bull. 1971;19:2428–2430. [Google Scholar]
- 441.Furusaki A, Matsuo M, Watanabe T, Omura S, Nakagawa A, Hata T. The Crystal and Molecular Structure of Kinamycin C p-Bromobenzoate. Isr J Chem. 1972;10:173–187. [Google Scholar]
- 442.Gould SJ, Tamayo N, Melville CR, Cone MC. Revised Structures for the Kinamycin Antibiotics: 5-diazobenzo[b]fluorenes Rather than Benzo[b]carbazole Cyanamides. J Am Chem Soc. 1994;116:2207–2208. [Google Scholar]
- 443.Mithani S, Weeratunga G, Taylor NJ, Dmitrienko GI. The Kinamycins are Diazofluorenes and Not Cyanocarbazoles. J Am Chem Soc. 1994;116:2209–2210. [Google Scholar]
- 444.Laufer RS, Dmitrienko GI. Diazo Group Electrophilicity in Kinamycins and Lomaiviticin A: Potential Insights into the Molecular Mechanisim of Antibacterial and Antitumor Activity. J Am Chem Soc. 2002;124:1854–1855. doi: 10.1021/ja0167809. [DOI] [PubMed] [Google Scholar]
- 445.Feldman KS, Eastman KJ. Studies on the Mechanism of Action of Prekinamycin, a Member of the Diazoparaquinone Family of Natural Products: Evidence for Both sp2 Radical Orthoquinonemethide Intermediates. J Am Chem Soc. 2006;128:12562–12573. doi: 10.1021/ja0642616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.O’Hara KA, Wu X, Patel D, Liang H, Yalowich JC, Chen N, Goodfellow V, Adedayo O, Dmitrienko GI, Hasinoff BB. Mechanism of the Cytotoxicity of the Diazoparaquinone Antitumor Antibiotic Kinamycin F. Free Radic Biol Med. 2007;43:1132–1144. doi: 10.1016/j.freeradbiomed.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Ballard TE, Melander C. Kinamycin-mediated DNA Cleavage Under Biomimetic Conditions. Tetrahedron Lett. 2008;49:3157–3161. [Google Scholar]
- 448.Mulcahy SP, Woo CM, Ding W, Ellestad GA, Herzon SB. Characterization of a Reductively-Activated Elimination Pathway Relevant to the Biological Chemistry of the Kinamycins and Lomaiviticins. Chem Sci. 2012;3:1070–1074. [Google Scholar]
- 449.Herzon SB, Woo CM. The Diazofluorene Antitumor Antibiotics: Structural Elucidation, Biosynthetic, Synthetic, and Chemical Biological Studies. Nat Prod Rep. 2012;29:87–118. doi: 10.1039/c1np00052g. [DOI] [PubMed] [Google Scholar]
- 450.Lei X, Porco JA. Total Synthesis of the Diazobenzofluorene Antibiotic (−)-Kinamycin C. J Am Chem Soc. 2006;128:14790–14791. doi: 10.1021/ja066621v. [DOI] [PubMed] [Google Scholar]
- 451.Nicolaou KC, Li H, Nold AL, Pappo D, Lenzen A. Total Synthesis of Kinamycins C, F, J. J Am Chem Soc. 2007;129:10356–10357. doi: 10.1021/ja074297d. [DOI] [PubMed] [Google Scholar]
- 452.Woo CM, Lu L, Gholap SL, Smith DR, Herzon SB. Development of a Convergent Entry to the Diazofluorene Antitumor Antibiotics: Enantioselective Synthesis of Kinamycin F. J Am Chem Soc. 2010;132:2540–2541. doi: 10.1021/ja910769j. [DOI] [PubMed] [Google Scholar]
- 453.Woo CM, Gholap SL, Lu L, Kaneko M, Li Z, Ravikumar PC, Herzon SB. Development of Enantioselective Synthetic Routes to (−)-Kinamycin F and (−)-Lomaiviticin Aglycon. J Am Chem Soc. 2012;134:17262–17273. doi: 10.1021/ja307497h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Gould SJ. Biosynthesis of the Kinamycins. Chem Rev. 1997;97:2499–2509. doi: 10.1021/cr9600215. [DOI] [PubMed] [Google Scholar]
- 455.Sato Y, Gould SJ. Biosynthesis of Kinamycin D. Incorporation of [1,2-13C]Acetate and of [2-2H3,-13C]Acetate. Tetrahedron Lett. 1985;26:4023–4026. [Google Scholar]
- 456.Sato Y, Gould SJ. Biosynthesis of the Kinamycin Antibiotics by Streptomyces murayamaensis. Determination of the Origin of Carbon, Hydrogen, and Oxygen Atoms by 13C NMR Spectroscopy. J Am Chem Soc. 1986;108:4625–4631. [Google Scholar]
- 457.Ajisaka K, Takeshima H, Omura S. Application in Biosynthetic Studies of 13C Isotope Shifts in Infared Spectroscopy. J Chem Soc Chem Commun. 1976;14:571–572. [Google Scholar]
- 458.Seaton PJ, Gould SJ. Kinamycin Biosynthesis. Derivation by Excision of an Acetate Unit from a Single-Chain Decaketide Intermediate. J Am Chem Soc. 1987;109:5282–5284. [Google Scholar]
- 459.Seaton PJ, Gould SJ. New Products Related to Kinamycin from Streptomyces murayamaensis. II. Structures of Pre-kinamycin, Keto-anhydrokinamycin, and Kinamycins E and F. J Antibiot (Tokyo) 1989;42:189–197. doi: 10.7164/antibiotics.42.189. [DOI] [PubMed] [Google Scholar]
- 460.Gould SJ, Melville CR. Kinamycin Biosynthesis. Synthesis, Detection, and Incorporation of Kinobscurinone, a Benzo[b]fluorenone. Bioorg Med Chem. 1995;5:51–54. [Google Scholar]
- 461.Gould SJ, Melville CR, Cone MC, Chen J, Carney JR. Kinamycin Biosynthesis. Synthesis, Isolation and Incorporation of Stealthin C, an Aminobenzo[b]fluorene. J Org Chem. 1997;62:320–324. doi: 10.1021/jo961486y. [DOI] [PubMed] [Google Scholar]
- 462.Gould SJ, Hong ST, Carney JR. Cloning and Heterologous Expression of Genes from the Kinamycin Biosynthetic Pathway of Streptomyces murayamaensis. J Antibiot (Tokyo) 1998;51:50–57. doi: 10.7164/antibiotics.51.50. [DOI] [PubMed] [Google Scholar]
- 463.Wang B, Ren J, Li L, Guo F, Pan G, Ai G, Aigle B, Fan K, Yang K. Kinamycin Biosynthesis Employs a Conserved Pair of Oxidases for B-ring Contraction. Chem Commun. 2015;51:8845–8848. doi: 10.1039/c5cc01986a. [DOI] [PubMed] [Google Scholar]
- 464.He H, Ding WD, Bernan VS, Richardson AD, Ireland CM, Greenstein M, Ellestad GA, Carter GT. Lomaiviticins A and B, Potent Antitumor Antibiotics from Micromonospora lomaivitiensis. J Am Chem Soc. 2001;123:5362–5363. doi: 10.1021/ja010129o. [DOI] [PubMed] [Google Scholar]
- 465.Woo CM, Beizer NE, Janso JE, Herzon SB. Isolation of Lomaiviticins C-E, Transformation of Lomaiviticin C to Lomaiviticin A, Complete Structure Elucidation of Lomaiviticin A, and Structure-Activity Analyses. J Am Chem Soc. 2012;134:15285–15288. doi: 10.1021/ja3074984. [DOI] [PubMed] [Google Scholar]
- 466.Woo CM, Ranjan N, Arya DP, Herzon SB. Analysis of Diazofluorene DNA Binding and Damaging Activity: DNA Cleavage by a Synthetic Monomeric Diazofluorene. Angew Chem, Int Ed. 2014;53:9325–9328. doi: 10.1002/anie.201404137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Woo CM, Li Z, Paulson EK, Herzon SB. Structural Basis for DNA Cleavage by the Potent Antiproliferative Agent (−)-Lomaiviticin A. Proc Natl Acad Sci USA. 2016;113:2851–2856. doi: 10.1073/pnas.1519846113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Krygowski ES, Murphy-Benenato K, Shair MD. Enantioselective Synthesis of the Central Ring System of Lomaiviticin A in the Form of an Unusually Stable Cyclic Hydrate. Angew Chem, Int Ed. 2008;47:1680–1684. doi: 10.1002/anie.200704830. [DOI] [PubMed] [Google Scholar]
- 469.Herzon SB, Lu L, Woo CM, Gholap SL. 11-Step Enantioselective Synthesis of (−)- Lomaiviticin Aglycon. J Am Chem Soc. 2011;133:7260–7263. doi: 10.1021/ja200034b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Kersten RD, Lane AL, Nett M, Richter TKS, Duggan BM, Dorrestein PC, Moore BS. Bioactivity-Guided Genome Mining Reveals the Lomaiviticin Biosynthetic Gene Cluster in Salinispora tropica. Chembiochem. 2013;14:955–962. doi: 10.1002/cbic.201300147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Janso JE, Haltli BA, Eustaquio AS, Kulowski K, Waldman AJ, Zha L, Nakamura H, Bernan VS, He H, Carter GT, et al. Discovery of the Lomaiviticin Biosynthetic Gene Cluster in Salinispora pacifica. Tetrahedron. 2014;70:4156–4164. doi: 10.1016/j.tet.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Waldman AJ, Balskus EP. Lomaiviticin Biosynthesis Employs a New Strategy for Starter Unit Generation. Org Lett. 2014;16:640–643. doi: 10.1021/ol403714g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Wang B, Guo F, Ren J, Ai G, Aigle B, Fan K, Yang K. Identification of Alp1U and Lom6 as Epoxy Hydrolases and Implications for Kinamycin and Lomaiviticin Biosynthesis. Nat Commun. 2015;6:7674–7678. doi: 10.1038/ncomms8674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Narang R, Narasimhan B, Sharma S. A Review on Biological Activities and Chemical Synthesis of Hydrazide Derivatives. Curr Med Chem. 2012;19:569–612. doi: 10.2174/092986712798918789. [DOI] [PubMed] [Google Scholar]
- 475.Ogita T, Gunji S, Fukazawa Y, Terahara A, Kinoshita T, Nagaki H, Beppu T. The Structures of Fosfazinomycins A and B. Tetrahedron Lett. 1983;24:2283–2286. [Google Scholar]
- 476.Schiessl K, Roller A, Hammerschmidt F. Determination of Absolute Configuration of the Phosphonic Acid Moiety of Fosfazinomycins. Org Biomol Chem. 2013;11:7420–7426. doi: 10.1039/c3ob41574k. [DOI] [PubMed] [Google Scholar]
- 477.Kuroda Y, Tanaka H, Okamoto M, Goto T, Kohsaka M, Aoki H, Imanaka H. FR-900137. A New Antibiotic II. Structure Determination of FR-900137. J Antibiot (Tokyo) 1980;33:280–283. doi: 10.7164/antibiotics.33.280. [DOI] [PubMed] [Google Scholar]
- 478.Bevan K, Davies JS, Hassall CH, Morton RB, Phillips DAS. Amino-acids and Peptides. Part X. Characterisation of the Monamycins, Members of a New Family of Cyclodepsipeptide Antibiotics. J Chem Soc C. 1971:514–522. [Google Scholar]
- 479.Arroyo V, Hall MJ, Hassall CH, Yamasaki K. Incorporation of Amino Acids into the Cyclohexadepsipeptide, Monamycin. J Chem Soc, Chem Commun. 1976:845–846. [Google Scholar]
- 480.Hassall CH, Magnus KE. Monamycin: a New Antibiotic. Nature. 1959;184:1223–1224. [Google Scholar]
- 481.Ma J, Wang Z, Huang H, Luo M, Zuo D, Wang B, Sun A, Cheng YQ, Zhang C, Ju J. Biosynthesis of Himastatin: Assembly Line and Characterization of Three Cytochrome P450 Enzymes Involved in the Post-Tailoring Oxidative Steps. Angew Chem, Int Ed. 2011;50:7797–7802. doi: 10.1002/anie.201102305. [DOI] [PubMed] [Google Scholar]
- 482.Lam KS, Hesler GA, Mattei JM, Mamber SW, Forenza S. Himastatin, A New Antitumor Antibiotic from Streptomyces Hygroscopicus. I. Taxonomy of Producing Organism, Fermentation and Biological Activity. J Antibiot (Tokyo) 1990;43:956–960. doi: 10.7164/antibiotics.43.956. [DOI] [PubMed] [Google Scholar]
- 483.Leet JE, Schroeder DR, Golik J, Matson JA, Doyle TW, Lam KS, Hill SE, Lee MS, Whitney JL, Krishnan BS. Himastatin, a New Antitumor Antibiotic from Streptomyces hygroscopicus. III. Structural Elucidation. J Antibiot (Tokyo) 1996;49:299–311. doi: 10.7164/antibiotics.49.299. [DOI] [PubMed] [Google Scholar]
- 484.Qu X, Jiang N, Xu F, Shao L, Tang G, Wilkinson B, Liu W. Cloning, Sequencing and Characterization of the Biosynthetic Gene Cluster of Sanglifehrin A, a Potent Cyclophilin Inhibitor. Mol BioSyst. 2011;7:852–861. doi: 10.1039/c0mb00234h. [DOI] [PubMed] [Google Scholar]
- 485.Fehr T, Kallen J, Oberer L, Sanglier JJ, Schilling W. Sanglifehrins A, B, C, and D, Novel Cyclophilin-binding Compounds Isolated from Streptomyces sp. A92-308110. J Antibiot (Tokyo) 1999;52:474–479. doi: 10.7164/antibiotics.52.474. [DOI] [PubMed] [Google Scholar]
- 486.Fujimori DG, Hrvatin S, Neumann CS, Strieker M, Marahiel MA, Walsh CT. Cloning and Characterization of the Biosynthetic Gene Cluster for Kutznerides. Proc Natl Acad Sci US A. 2007;104:16498–16503. doi: 10.1073/pnas.0708242104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Broberg A, Menkis A, Vasiliauskas R. Kutznerides 1–4, Depsipeptides from the Actinomycete Kutzneriasp. 744 Inhabiting Mycorrhizal Roots of Picea abies Seedlings. J Nat Prod. 2006;69:97–102. doi: 10.1021/np050378g. [DOI] [PubMed] [Google Scholar]
- 488.Pohanka A, Menkis A, Levenfors J, Broberg A. Low-Abundance Kutznerides from Kutzneria sp. 744. J Nat Prod. 2006;69:1776–1781. doi: 10.1021/np0604331. [DOI] [PubMed] [Google Scholar]
- 489.Jiang W, Heemstra JR, Jr, Forseth RR, Neumann CS, Manaviazar S, Schroeder FC, Hale KJ, Walsh CT. Biosynthetic Chlorination of the Piperazate Residue in Kutzneride Biosynthesis by KthP. Biochemistry. 2011;50:6063–6072. doi: 10.1021/bi200656k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Michelot D, Toth B. Poisoning by Cyromitra esculenta - A Review. J Appl Toxicol. 1991;11 doi: 10.1002/jat.2550110403. [DOI] [PubMed] [Google Scholar]
- 491.Ding L, Munch J, Goerls H, Maier A, Fiebig HH, Lin WH, Hertweck C. Xiamycin, a Pentacyclic Indolosesquiterpene with Selective Anti-HIV Activity from a Bacterial Mangrove Endophyte. Bioorg Med Chem Lett. 2010;20:6685–6687. doi: 10.1016/j.bmcl.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 492.Li H, Zhang Q, Li S, Zhu Y, Zhang G, Zhang H, Tian X, Zhang S, Ju J, Zhang C. Identification and Characterization of Xiamycin A and Xiamycin Gene Cluster Reveals an Oxidative Cyclization Strategy Tailoring Indolosesquiterpene Biosynthesis. J Am Chem Soc. 2012;134:8996–9005. doi: 10.1021/ja303004g. [DOI] [PubMed] [Google Scholar]
- 493.Xu Z, Baunach M, Ding L, Hertweck C. Bacterial Synthesis of Diverse Indole Terpene Alkaloids by an Unparalleled Cyclization Sequence. Angew Chem, Int Ed. 2012;51:10293–10297. doi: 10.1002/anie.201204087. [DOI] [PubMed] [Google Scholar]
- 494.Baunach M, Ding L, Bruhn T, Bringmann G, Hertweck C. Regiodivergent N–C and N–N Aryl Coupling Reactions of Indoloterpenes and Cycloether Formation Mediated by a Single Bacterial Flavoenzyme. Angew Chem, Int Ed. 2013;52:9040–9043. doi: 10.1002/anie.201303733. [DOI] [PubMed] [Google Scholar]
- 495.Mander L, Liu HW. Comprehensive Natural Products II: Chemistry and Biology. Elsevier Ltd; Kidlington, United Kingdom: 2010. [Google Scholar]
- 496.Umezawa K, Ikeda Y, Kawase O, Naganawa H, Kondo S. Biosynthesis of Polyoxypeptin A: Novel Amino Acid 3-Hydroxy-3-Methylproline Derived from Isoleucine. J Chem Soc, Perkin Trans 1. 2001:1550–1553. [Google Scholar]
- 497.Miller ED, Kauffman CA, Jensen PR, Fenical W. Piperazimycins: Cytotoxic Hexadepsipeptides from a Marine-Derived Bacterium of the Genus Streptomyces. J Org Chem. 2007;72:323–330. doi: 10.1021/jo061064g. [DOI] [PubMed] [Google Scholar]
- 498.Du Y, Wang Y, Huang T, Tao M, Deng Z, Lin S. Identification and Characterization of the Biosynthetic Gene Cluster of Polyoxypeptin A, a Potent Apoptosis Inducer. BMC Microbiol. 2014;14:1–12. doi: 10.1186/1471-2180-14-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Kramer CS. Privileged Scaffolds in Medicinal Chemistry: Design, Synthesis, Evaluation. The Royal Society of Chemistry; 2015. [DOI] [Google Scholar]
- 500.Wermuth CG. Are Pyridazines Privileged Structures? MedChemComm. 2011;2:935–941. [Google Scholar]
- 501.LaRue TA, Child JJ. Pyrazole in Citrullis vulgaris (Cucurbitacae) Phytochemistry. 1975;14:2513–2514. [Google Scholar]
- 502.Brown EG, Diffin FM. Biosynthesis and Metabolism of Pyrazole by Cucumis sativus: Enzymic Cyclization and Dehydrogenation of 1,3-diaminopropane. Phytochemistry. 1990;29:469–478. [Google Scholar]
- 503.Noe FF, Fowden L. a-Amino-B-(pyrazolyl-N) Propionic Acid: a New Amino-Acid from Citrullus vulgaris (Watermelon) Nature. 1959;184:69–70. [PubMed] [Google Scholar]
- 504.Brown EG, Flayeh KAMR, GJ The Biosynthetic Origin of the Pyrazole Moiety of B-pyrazol-1-yl-L-alanine. Phytochemistry. 1982;21:863–867. [Google Scholar]
- 505.Flayeh KAM, Najafi SI, Al-Delymi AM, Hajar MA. 1,3-Diaminopropane and Spermidine in Cucumis sativus (Cucumber) Phytochemistry. 1984;23:989–990. [Google Scholar]
- 506.Dunnill PM, Fowden L. The Biosynthesis of B-pyrazol-1-ylalanine. J Exp Bot. 1963;14:237–248. [Google Scholar]
- 507.Murakoshi I, Kuramoto H, Haginiwa J, Fowden L. The Enzymic Synthesis of B-Substituted Alanines. Phytochemistry. 1972;11:177–182. [Google Scholar]
- 508.Cho JY, Kwon HC, Williams PG, Jensen PR, Fenical W. Azamerone, a Terpenoid Phthalazinone from a Marine-Derived Bacterium Related to the Genus Streptomyces (Actinomycetales) Org Lett. 2006;8:2471–2474. doi: 10.1021/ol060630r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Fukuda DS, Mynderse JS, Baker PJ, Berry DM, Boeck LD, Yao RC, Mertz FP, Nakatsukasa WM, Mabe J, Ott J, et al. A80915, A New Antibiotic Complex Produced by Streptomyces aculeolatus. Discovery, Taxonomy, Fermentation, Isolation, Characterization, and Antibacterial Evaluation. J Antibiot (Tokyo) 1989;43:623–633. doi: 10.7164/antibiotics.43.623. [DOI] [PubMed] [Google Scholar]
- 510.Gomi S, Ohuchi S, Sasaki T, Itoh J, Sezaki M. Studies on New Antibiotics SF2415. II. The Structural Elucidation. J Antibiot (Tokyo) 1987;40 doi: 10.7164/antibiotics.40.740. [DOI] [PubMed] [Google Scholar]
- 511.Grote R, Chen Y, Zeeck A. Metabolic Products of Microorganisms. 243. Pyridazomycin, A New Antifungal Antibiotic Produced by Streptomyces violaceoniger. J Antibiot (Tokyo) 1988;41:595–601. doi: 10.7164/antibiotics.41.595. [DOI] [PubMed] [Google Scholar]
- 512.Bockhold H, Beale JM, Rohr J. Biosynthetic Investigations on Pyridazomycin. Angew Chem, Int Ed. 1994;33:1648–1651. [Google Scholar]
- 513.Jeong Y, Kim J, Kim S, Kang Y, Nagamatsu T, Hwang I. Toxoflavin Produced by Burkholderia glumae Causing Rice Grain Rot is Responsible for Inducing Bacterial Wilt in Many Field Crops. Plant Dis. 2003;87:890–895. doi: 10.1094/PDIS.2003.87.8.890. [DOI] [PubMed] [Google Scholar]
- 514.Latuasan HE, Berends W. On the Origin of the Toxicity of Toxoflavin. Biochim Biophys Acta. 1961;52:502–508. doi: 10.1016/0006-3002(61)90408-5. [DOI] [PubMed] [Google Scholar]
- 515.Suzuki F, Zhu Y, Sawada H, Matsuda I. Identification of Proteins Involved in Toxin Production by Pseudomonas glumae. Ann Phytopathol Soc Jpn. 1998;64:75–79. [Google Scholar]
- 516.Suzuki F, Sawada H, Azegami K, Tsuchiya K. Molecular Characterization of the tox Operon Involved in Toxoflavin Biosynthesis of Burkholderia glumae. Journal of General Plant Pathology. 2004;70:97–107. [Google Scholar]
- 517.Philmus B, Shaffer BT, Kidarsa TA, Yan Q, Raaijmakers JM, Begley TP, Loper JE. Investigations into the Biosynthesis, Regulation, and Self-Resistance of Toxoflavin in Pseudomonas protegens Pf-5. ChemBioChem. 2015;16:1782–1790. doi: 10.1002/cbic.201500247. [DOI] [PubMed] [Google Scholar]
- 518.Levenberg B, Linton SN. On the Biosynthesis of Toxoflavin, an Azapteridine Antibiotic Produced by Pseudomonas cocovenenans. J Biol Chem. 1966;241:846–852. [PubMed] [Google Scholar]
- 519.Fenwick MK, Philmus B, Begley TP, Ealick SE. Burkholderia glumae ToxA is a Dual-Specificity Methyltransferase that Catalyzes the Last Two Steps of Toxoflavin Biosynthesis. Biochemistry. 2016;55:2748–2759. doi: 10.1021/acs.biochem.6b00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Lundberg JO, Weitzberg E, Cole JA, Benjamin N. Nitrate, Bacteria and Human Health. Nat Rev Micro. 2004;2:593–602. doi: 10.1038/nrmicro929. [DOI] [PubMed] [Google Scholar]
- 521.Vitturi DA, Minarrieta L, Salvatore SR, Postlethwait EM, Fazzari M, Ferrer-Sueta G, Lancaster JR, Jr, Freeman BA, Schopfer FJ. Convergence of Biological Nitration and Nitrosation via Symmetrical Nitrous Anhydride. Nat Chem Biol. 2015;11:504–510. doi: 10.1038/nchembio.1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Freel Meyers CL, Borch RF. Activation Mechanisms of Nucleoside Phosphoramidate Prodrugs. J Med Chem. 2000;43:4319–4327. doi: 10.1021/jm000302b. [DOI] [PubMed] [Google Scholar]
- 523.Tobias SC, Borch RF. Synthesis and Biological Studies of Novel Nucleoside Phosphoramidate Prodrugs. J Med Chem. 2001;44:4475–4480. doi: 10.1021/jm010337r. [DOI] [PubMed] [Google Scholar]
- 524.Russell MA, Laws AP, Atherton JH, Page MI. The Mechanism of the Phosphoramidite Synthesis of Polynucleotides. Org Biomol Chem. 2008;6:3270–3275. doi: 10.1039/b808999j. [DOI] [PubMed] [Google Scholar]
- 525.Duquesne S, Destoumieux-Garzon D, Peduzzi J, Rebuffat S. Microcins, Gene-encoded Antibacterial Peptides from Enterobacteria. Nat Prod Rep. 2007;24:708–734. doi: 10.1039/b516237h. [DOI] [PubMed] [Google Scholar]
- 526.Severinov K, Semenova E, Kazakov A, Kazakov T, Gelfand MS. Low-Molecular-Weight Post-Translationally Modified Microcins. Mol Microbiol. 2007;65:1380–1394. doi: 10.1111/j.1365-2958.2007.05874.x. [DOI] [PubMed] [Google Scholar]
- 527.Guijarro JI, Gonzalez-Pastor JE, Baleux F, San Millan JL, Castilla MA, Rico M, Moreno F, Delepierre M. Chemical Structure and Translation Inhibition Studies of the Antibiotic Microcin C7. J Biol Chem. 1995;270:23520–23532. doi: 10.1074/jbc.270.40.23520. [DOI] [PubMed] [Google Scholar]
- 528.Metlitskaya A, Kazakov T, Kommer A, Pavlova O, Praetorius-Ibba M, Ibba M, Krasheninnikov I, Kolb V, Khmel I, Severinov K. Aspartyl-tRNA Synthetase is the Target of Peptide Nucleotide Antibiotic Microcin C. J Biol Chem. 2006;281:18033–18042. doi: 10.1074/jbc.M513174200. [DOI] [PubMed] [Google Scholar]
- 529.Novikova M, Metlitskaya A, Datsenko K, Kazakov T, Kazakov A, Wanner B, Severinov K. The Escherichia coli Yej Transporter is Required for the Uptake of Translation Inhibitor Microcin C. J Bacteriol. 2007;189:8361–8365. doi: 10.1128/JB.01028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530.Gonzalez-Pastor JE, San Millan JL, Castilla MA, Moreno F. Structure and Organization of Plasmid Genes Required to Produce the Translation Inhibitor Microcin C7. J Bacteriol. 1995;177:7131–7140. doi: 10.1128/jb.177.24.7131-7140.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Roush RF, Nolan EM, Lohr F, Walsh CT. Maturation of an Escherichia coli Ribosomal Peptide Antibiotic by ATP-consuming N–P bond Formation in Microcin C7. J Am Chem Soc. 2008;130:3603–3609. doi: 10.1021/ja7101949. [DOI] [PubMed] [Google Scholar]
- 532.Hochstrasser M. Evolution and Function of Ubiquitin-like Protein-conjugation Systems. Nat Cell Biol. 2000;2:E153–157. doi: 10.1038/35019643. [DOI] [PubMed] [Google Scholar]
- 533.Hochstrasser M. Origin and Function of Ubiquitin-like Proteins. Nature. 2009;458:422–429. doi: 10.1038/nature07958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Regni CA, Roush RF, Miller DJ, Nourse A, Walsh CT, Schulman BA. How the MccB Bacterial Ancestor of Ubiquitin E1 Initiates Biosynthesis of the Microcin C7 Antibiotic. EMBO J. 2009;28:1953–1964. doi: 10.1038/emboj.2009.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Jacob C. A Scent of Therapy: Pharmacological Implications of Natural Products Containing Redox-active Sulfur Atoms. Nat Prod Rep. 2006;23:851–863. doi: 10.1039/b609523m. [DOI] [PubMed] [Google Scholar]
- 536.Givol D, Goldberger RF, Anfinsen CB. Oxidation and Disulfide Interchange in the Reactivation of Reduced Ribonuclease. J Biol Chem. 1964;239:3114–3116. [PubMed] [Google Scholar]
- 537.Denoncin K, Collet JF. Disulfide Bond Formation in the Bacterial Periplasm: Major Achievements and Challenges Ahead. Antioxid Redox Sign. 2013;19:63–71. doi: 10.1089/ars.2012.4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Scharf DH, Remme N, Heinekamp T, Hortschansky P, Brakhage AA, Hertweck C. Transannular Disulfide Formation in Gliotoxin Biosynthesis and its Role in Self-resistance of the Human Pathogen Aspergillus fumigatus. J Am Chem Soc. 2010;132:10136–10141. doi: 10.1021/ja103262m. [DOI] [PubMed] [Google Scholar]
- 539.Schrettl M, Carberry S, Kavanagh K, Haas H, Jones GW, O’Brien J, Nolan A, Stephens J, Fenelon O, Doyle S. Self-Protection against Gliotoxin-A Component of the Gliotoxin Biosynthetic Cluster, GliT, Completely Protects Aspergillus Fumigatus Against Exogenous Gliotoxin. Plos Pathog. 2010;6 doi: 10.1371/journal.ppat.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Li B, Walsh CT. Streptomyces clavuligerus HlmI is an Intramolecular Disulfide-forming Dithiol Oxidase in Holomycin Biosynthesis. Biochemistry. 2011;50:4615–4622. doi: 10.1021/bi200321c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Wang C, Wesener SR, Zhang H, Cheng YQ. An FAD-dependent Pyridine Nucleotide-disulfide Oxidoreductase is Involved in Disulfide Bond Formation in FK228 Anticancer Depsipeptide. Chem Biol. 2009;16:585–593. doi: 10.1016/j.chembiol.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 542.Johnson JR, Bruce WF, Dutcher JD. Gliotoxin, the Antibiotic Principle of Gliocladium Fimbriatum. I. Production, Physical and Biological Properties. J Am Chem Soc. 1943;65:2005–2009. [Google Scholar]
- 543.Gardiner DM, Waring P, Howlett BJ. The Epipolythiodioxopiperazine (ETP) Class of Fungal Toxins: Distribution, Mode of action, Functions and Biosynthesis. Microbiology. 2005;151:1021–1032. doi: 10.1099/mic.0.27847-0. [DOI] [PubMed] [Google Scholar]
- 544.Bell MR, Johnson JR, Wildi BS, Woodward RB. The Structure of Gliotoxin. J Am Chem Soc. 1958;80:1001–1001. [Google Scholar]
- 545.Waring P, Sjaarda A, Lin QH. Gliotoxin Inactivates Alcohol-Dehydrogenase by Either Covalent Modification or Free-Radical Damage Mediated by Redox Cycling. Biochem Pharmacol. 1995;49:1195–1201. doi: 10.1016/0006-2952(95)00039-3. [DOI] [PubMed] [Google Scholar]
- 546.De Clercq E, Billiau A, Ottenheijm HC, Herscheid JD. Antireverse Transcriptase Activity of Gliotoxin Analogs. Biochem Pharmacol. 1978;27:635–639. doi: 10.1016/0006-2952(78)90497-5. [DOI] [PubMed] [Google Scholar]
- 547.Nierman WC, Pain A, Anderson MJ, Wortman JR, Kim HS, Arroyo J, Berriman M, Abe K, Archer DB, Bermejo C, et al. Genomic Sequence of the Pathogenic and Allergenic Filamentous Fungus Aspergillus fumigatus. Nature. 2005;438:1151–1156. doi: 10.1038/nature04332. [DOI] [PubMed] [Google Scholar]
- 548.Gardiner DM, Howlett BJ. Bioinformatic and Expression Analysis of the Putative Gliotoxin Biosynthetic Gene Cluster of Aspergillus fumigatus. FEMS Microbiol Lett. 2005;248:241–248. doi: 10.1016/j.femsle.2005.05.046. [DOI] [PubMed] [Google Scholar]
- 549.Cramer RA, Gamcsik MP, Brooking RM, Najvar LK, Kirkpatrick WR, Patterson TF, Balibar CJ, Graybill JR, Perfect JR, Abraham SN, et al. Disruption of a Nonribosomal Peptide Synthetase in Aspergillus Fumigatus Eliminates Gliotoxin Production. Eukaryot Cell. 2006;5:972–980. doi: 10.1128/EC.00049-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550.Scharf DH, Groll M, Habel A, Heinekamp T, Hertweck C, Brakhage AA, Huber EM. Flavoenzyme-catalyzed Formation of Disulfide Bonds in Natural Products. Angew Chem, Int Ed. 2014;53:2221–2224. doi: 10.1002/anie.201309302. [DOI] [PubMed] [Google Scholar]
- 551.Li J, Wang C, Zhang ZM, Cheng YQ, Zhou J. The Structural Basis of an NADP(+)-independent Dithiol Oxidase in FK228 Biosynthesis. Sci Rep. 2014;4 doi: 10.1038/srep04145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Genghof DS, Inamine E, Kovalenko V, Melville DB. Ergothioneine in Microorganisms. J Biol Chem. 1956;223:9–17. [PubMed] [Google Scholar]
- 553.Newton GL, Fahey RC, Cohen G, Aharonowitz Y. Low-molecular-weight Thiols in Streptomycetes and Their Potential Role as Antioxidants. J Bacteriol. 1993;175:2734–2742. doi: 10.1128/jb.175.9.2734-2742.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554.Taubert D, Jung N, Goeser T, Schomig E. Increased Ergothioneine Tissue Concentrations in Carriers of the Crohn’s Disease Risk-associated 503F Variant of the Organic Cation Transporter OCTN1. Gut. 2009;58:312–314. doi: 10.1136/gut.2008.164418. [DOI] [PubMed] [Google Scholar]
- 555.Nakajima S, Satoh Y, Yanashima K, Matsui T, Dairi T. Ergothioneine Protects Streptomyces coelicolor A3(2) from Oxidative Stresses. J Biosci Bioeng. 2015;120:294–298. doi: 10.1016/j.jbiosc.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 556.Zhao Q, Wang M, Xu D, Zhang Q, Liu W. Metabolic Coupling of Two Small-molecule Thiols Programs the Biosynthesis of Lincomycin A. Nature. 2015;518:115–119. doi: 10.1038/nature14137. [DOI] [PubMed] [Google Scholar]
- 557.Ishikawa Y, Israel SE, Melville DB. Participation of an Intermediate Sulfoxide in the Enzymatic Thiolation of the Imidazole Ring of Hercynine to Form Ergothioneine. J Biol Chem. 1974;249:4420–4427. [PubMed] [Google Scholar]
- 558.Ishikawa Y, Melville DB. The Enzymatic Alpha-N-methylation of Histidine. J Biol Chem. 1970;245:5967–5973. [PubMed] [Google Scholar]
- 559.Seebeck FP. In Vitro Reconstitution of Mycobacterial Ergothioneine Biosynthesis. J Am Chem Soc. 2010;132:6632–6633. doi: 10.1021/ja101721e. [DOI] [PubMed] [Google Scholar]
- 560.Dierks T, Dickmanns A, Preusser-Kunze A, Schmidt B, Mariappan M, von Figura K, Ficner R, Rudolph MG. Molecular Basis for Multiple Sulfatase Deficiency and Mechanism for Formylglycine Generation of the Human Formylglycine-generating Enzyme. Cell. 2005;121:541–552. doi: 10.1016/j.cell.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 561.Turner E, Hager LJ, Shapiro BM. Ovothiol Replaces Glutathione Peroxidase as a Hydrogen Peroxide Scavenger in Sea Urchin Eggs. Science. 1988;242:939–941. doi: 10.1126/science.3187533. [DOI] [PubMed] [Google Scholar]
- 562.Steenkamp DJ, Weldrick D, Spies HS. Studies on the Biosynthesis of Ovothiol A. Identification of 4-mercaptohistidine as an Intermediate. Eur J Biochem. 1996;242:557–566. doi: 10.1111/j.1432-1033.1996.0557r.x. [DOI] [PubMed] [Google Scholar]
- 563.Braunshausen A, Seebeck FP. Identification and Characterization of the First Ovothiol Biosynthetic Enzyme. J Am Chem Soc. 2011;133:1757–1759. doi: 10.1021/ja109378e. [DOI] [PubMed] [Google Scholar]
- 564.Bruijnincx PC, van Koten G, Klein Gebbink RJ. Mononuclear Non-Heme Iron Enzymes with the 2-His-1-carboxylate Facial Triad: Recent Developments in Enzymology and Modeling Studies. Chem Soc Rev. 2008;37:2716–2744. doi: 10.1039/b707179p. [DOI] [PubMed] [Google Scholar]
- 565.Hegg EL, Que L., Jr The 2-His-1-carboxylate Facial Triad -- An Emerging Structural Motif in Mononuclear Non-heme Iron(II) Enzymes. Eur J Biochem. 1997;250:625–629. doi: 10.1111/j.1432-1033.1997.t01-1-00625.x. [DOI] [PubMed] [Google Scholar]
- 566.Costas M, Mehn MP, Jensen MP, Que L. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 567.Price JC, Barr EW, Glass TE, Krebs C, Bollinger JM., Jr Evidence for Hydrogen Abstraction from C1 of Taurine by the High-spin Fe(IV) Intermediate Detected during Oxygen Activation by Taurine:alpha-ketoglutarate Dioxygenase (TauD) J Am Chem Soc. 2003;125:13008–13009. doi: 10.1021/ja037400h. [DOI] [PubMed] [Google Scholar]
- 568.Goncharenko KV, Vit A, Blankenfeldt W, Seebeck FP. Structure of the Sulfoxide Synthase EgtB from the Ergothioneine Biosynthetic Pathway. Angew Chem, Int Ed. 2015;54:2821–2824. doi: 10.1002/anie.201410045. [DOI] [PubMed] [Google Scholar]
- 569.Goncharenko KV, Seebeck FP. Conversion of a Non-heme Iron-dependent Sulfoxide Synthase into a Thiol Dioxygenase by a Single Point Mutation. Chem Commun. 2016;52:1945–1948. doi: 10.1039/c5cc07772a. [DOI] [PubMed] [Google Scholar]
- 570.Koiso Y, Natori M, Iwasaki S, Sato S, Sonoda R, Fujita Y, Yaegashi H, Sato Z. Ustiloxin - a Phytotoxin and a Mycotoxin from False Smut Balls on Rice Panicles. Tetrahedron Lett. 1992;33:4157–4160. [Google Scholar]
- 571.Ye Y, Minami A, Igarashi Y, Izumikawa M, Umemura M, Nagano N, Machida M, Kawahara T, Shin-Ya K, Gomi K, et al. Unveiling the Biosynthetic Pathway of the Ribosomally Synthesized and Post-translationally Modified Peptide Ustiloxin B in Filamentous Fungi. Angew Chem, Int Ed. 2016;55:8072–8075. doi: 10.1002/anie.201602611. [DOI] [PubMed] [Google Scholar]
- 572.Munchberg U, Anwar A, Mecklenburg S, Jacob C. Polysulfides as Biologically Active Ingredients of Garlic. Org Biomol Chem. 2007;5:1505–1518. doi: 10.1039/b703832a. [DOI] [PubMed] [Google Scholar]
- 573.Rabinkov A, Zhu XZ, Grafi G, Galili G, Mirelman D. Alliin Lyase (Alliinase) from Garlic (Allium Sativum). Biochemical Characterization and cDNA Cloning. Appl Biochem Biotechnol. 1994;48:149–171. doi: 10.1007/BF02788739. [DOI] [PubMed] [Google Scholar]
- 574.Lancaster JE, Collin HA. Presence of Alliinase in Isolated Vacuoles and of Alkyl Cysteine Sulfoxides in the Cytoplasm of Bulbs of Onion (Allium cepa) Plant Sci Lett. 1981;22:169–176. [Google Scholar]
- 575.Miron T, Mironchik M, Mirelman D, Wilchek M, Rabinkov A. Inhibition of Tumor Growth by a Novel Approach: In situ Allicin Generation Using Targeted Alliinase Delivery. Mol Cancer Ther. 2003;2:1295–1301. [PubMed] [Google Scholar]
- 576.Hara M, Asano K, Kawamoto I, Takiguchi T, Katsumata S, Takahashi K, Nakano H. Leinamycin, a New Antitumor Antibiotic from Streptomyces: Producing Organism, Fermentation and Isolation. J Antibiot. 1989;42:1768–1774. doi: 10.7164/antibiotics.42.1768. [DOI] [PubMed] [Google Scholar]
- 577.Hara M, Takahashi I, Yoshida M, Asano K, Kawamoto I, Morimoto M, Nakano H. DC 107, a Novel Antitumor Antibiotic Produced by a Streptomyces sp. J Antibiot. 1989;42:333–335. doi: 10.7164/antibiotics.42.333. [DOI] [PubMed] [Google Scholar]
- 578.Gates KS. Mechanisms of DNA Damage by Leinamycin. Chem Res Toxicol. 2000;13:953–956. doi: 10.1021/tx000089m. [DOI] [PubMed] [Google Scholar]
- 579.Cheng YQ, Tang GL, Shen B. Identification and Localization of the Gene Cluster Encoding Biosynthesis of the Antitumor Macrolactam Leinamycin in Streptomyces atroolivaceus S-140. J Bacteriol. 2002;184:7013–7024. doi: 10.1128/JB.184.24.7013-7024.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Tang GL, Cheng YQ, Shen B. Leinamycin Biosynthesis Revealing Unprecedented Architectural Complexity for a Hybrid Polyketide Synthase and Nonribosomal Peptide Synthetase. Chem Biol. 2004;11:33–45. doi: 10.1016/j.chembiol.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 581.Ma M, Lohman JR, Liu T, Shen B. C–S Bond Cleavage by a Polyketide Synthase Domain. Proc Natl Acad Sci U S A. 2015;112:10359–10364. doi: 10.1073/pnas.1508437112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Baunach M, Ding L, Willing K, Hertweck C. Bacterial Synthesis of Unusual Sulfonamide and Sulfone Antibiotics by Flavoenzyme-Mediated Sulfur Dioxide Capture. Angew Chem, Int Ed. 2015;54:13279–13283. doi: 10.1002/anie.201506541. [DOI] [PubMed] [Google Scholar]
- 583.Fischer M, Schmidt C, Falke D, Sawers RG. Terminal Reduction Reactions of Nitrate and Sulfate Assimilation in Streptomyces coelicolor A3(2): Identification of Genes Encoding Nitrite and Sulfite Reductases. Res Microbiol. 2012;163:340–348. doi: 10.1016/j.resmic.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 584.Ovenden SPB, Capon RJ. Echinosulfonic Acids A–C and Echinosulfone A: Novel Bromoindole Sulfonic Acids and a Sulfone from a Southern Australian Marine Sponge, Echinodictyum. J Nat Prod. 1999;62:1246–1249. doi: 10.1021/np9901027. [DOI] [PubMed] [Google Scholar]
- 585.Isono K, Uramoto M, Kusakabe H, Miyata N, Koyama T, Ubukata M, Sethi SK, McCloskey JA. Ascamycin and Dealanylascamycin, Nucleoside Antibiotics from Streptomyces sp. J Antibiot (Tokyo) 1984;37:670–672. doi: 10.7164/antibiotics.37.670. [DOI] [PubMed] [Google Scholar]
- 586.Florini JR, Bird HH, Bell PH. Inhibition of Protein Synthesis In Vitro and In Vivo by Nucleocidin, an Antitrypanosomal Antibiotic. J Biol Chem. 1966;241:1091–1098. [PubMed] [Google Scholar]
- 587.Zhao C, Qi J, Tao W, He L, Xu W, Chan J, Deng Z. Characterization of Biosynthetic Genes of Ascamycin/Dealanylascamycin Featuring a 5′-O-sulfonamide Moiety in Streptomyces sp. JCM9888. PLoS One. 2014;9:e114722. doi: 10.1371/journal.pone.0114722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588.Igarashi M, Nakagawa N, Doi N, Hattori S, Naganawa H, Hamada M. Caprazamycin B, a Novel Anti-tuberculosis Antibiotic, from Streptomyces sp. J Antibiot. 2003;56:580–583. doi: 10.7164/antibiotics.56.580. [DOI] [PubMed] [Google Scholar]
- 589.Malojcic G, Glockshuber R. The PAPS-independent Aryl Sulfotransferase and the Alternative Disulfide Bond Formation System in Pathogenic Bacteria. Antioxid Redox Sign. 2010;13:1247–1259. doi: 10.1089/ars.2010.3119. [DOI] [PubMed] [Google Scholar]
- 590.Kaysser L, Lutsch L, Siebenberg S, Wemakor E, Kammerer B, Gust B. Identification and Manipulation of the Caprazamycin Gene Cluster Lead to New Simplified Liponucleoside Antibiotics and Give Insights into the Biosynthetic Pathway. J Biol Chem. 2009;284:14987–14996. doi: 10.1074/jbc.M901258200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591.Tang X, Eitel K, Kaysser L, Kulik A, Grond S, Gust B. A Two-step Sulfation in Antibiotic Biosynthesis Requires a Type III Polyketide Synthase. Nat Chem Biol. 2013;9:610–615. doi: 10.1038/nchembio.1310. [DOI] [PubMed] [Google Scholar]