

Abstract
Engineered biological systems such as genetic circuits and microbial cell factories have promised to solve many challenges in the modern society. However, the artisanal processes of research and development are slow, expensive, and inconsistent, representing a major obstacle in biotechnology and bioengineering. In recent years, biological foundries or biofoundries have been developed to automate design-build-test engineering cycles in an effort to accelerate these processes. This review summarizes the enabling technologies for such biofoundries as well as their early successes and remaining challenges.
INTRODUCTION
Many engineering endeavors involve design-build-test cycles to achieve optimal solutions. The execution of these cycles when engineering a biological system (i.e. a protein, a metabolic pathway, a genetic circuit, and a genome), however, has encountered multiple obstacles. One main reason is that research and development in biology is largely dependent on the artisanal work of skillful researchers, which is problematic in two aspects. First, low throughput limits the turnover rate of the design-build-test (DBT) cycles and the number of designs explored in parallel in each cycle. Second, manual operations are prone to human errors and biases making it difficult to conduct the experiments in a consistent and objective manner. Due to the complexity of biosystems, numerous iterations may be required to achieve the desirable engineering objectives. Therefore, biosystem engineering is still slow and expensive. For example, it took over 10 years and more than one hundred million dollar to develop the biosynthetic process for 1,3-propanediol (Committee on Industrialization of Biology, 2015).
Examining paths from other engineering disciplines allows new insight into possible solutions. Mechanical engineering started as a craftsmanship in ancient times when design and production were also completely artisanal. It followed a slow pace with scattered innovations throughout history until theoretical tools such as Newton’s Laws of Motion and calculus laid the groundwork for more rapid innovations (Engineers, 2016). Later, precision manufacturing enabled standardization of parts while steam and electricity powered up the rapid development of machines. Nowadays, engineers can sketch and evaluate ideas with computer-aided-design (CAD) tools without performing tedious hand calculations. The designs can then be prototyped by computerized numerical controlled (CNC) machining or 3D printing. As a result, the turnover time of the DBT cycles can be as short as a few hours, which previously took days or months. Likewise, electrical engineering and computer engineering have also advanced due to the same three factors: 1) breakthroughs in theories and technology, 2) standardization, and 3) rapid prototyping. Especially in software engineering, many DBT cycles can be executed in a timespan of minutes. As designs (code) are typed out in standard syntax, a program can be built (compiled) and tested in a split second.
Looking back to biotechnology, although a number of theories and technologies, such as the central dogma, DNA sequencing, and DNA synthesis have established a foundation for engineering applications, many properties of biosystems remain elusive. Further understanding of the biosystems, or learning, becomes an essential part of the engineering cycles (DBTL). Moreover, the lack of widely adopted standards and rapid prototyping capabilities still hinders the development of this field. Limitations in precision, speed, and cost in each step of the DBTL cycles further delays the adaptation of standards and acquisition of rapid prototyping capabilities. All these limitations can be partially attributed to unavoidable human involvement. In fact, the history of most engineering disciplines represents our endeavor to substitute humans with machines.
Automation has been proposed as a solution to improve consistency and speed, as well as to reduce labor costs and help researchers to focus more on intellectual tasks. Early automated practices focused on drug screening, genomic studies, and immunoassays (Chapman, 2003; Meldrum, 2000). While successful in performing highly defined protocols involving a limited number and types of operations, these early generation automation systems may be inadequate for emerging workflows in biological engineering with increasing complexity. The challenges are largely related to high variability in experimental protocols and high failure rates that require constant handling of exceptions. Recent advances in metabolic engineering, synthetic biology, and bioinformatics such as robust DNA assembly methods, versatile genome engineering tools, and powerful retrobiosynthesis algorithms may help to tackle these limitations. Based on these enabling technologies, academic institutions and industrial companies are starting to build industrialized biofoundries for rapid prototyping of biosystems for a variety of applications. These endeavors have been largely simulated by a recent wave of investments from government agencies such as the U.S. Defense Advanced Research Projects Agency (DARPA), the U.K. Biotechnology and Biological Sciences Research Council (BBSRC)), the Engineering and Physical Sciences Research Council, and private venture funds. Both technological and financial support in biofoundry development can potentially overcome the current barriers of biotechnology research and carry this field to the exponential growth phase. We envision such biofoundries will be a platform for integrating tools to automate each step in the DBTL cycle (Figure 1). This review seeks to summarize the enabling technologies, early successes, and future challenges in developing and applying automated biofoundries for engineering biosystems, organized around each individual step of DBTL and the integration into a complete cycle.
Figure 1. The architecture of a typical biofoundry.

A biofoundry platform is an integration of biology with software and hardware systems. The protocols, parts, and biological entities used as engineering tools should be optimized first to facilitate automated high-throughput manipulations. The software system orchestrates the automation processes and assists design as well as data processing. The hardware system conducts the build and test tasks. Through iterations of design-build-test, learning algorithms can potentially be integrated to help extend the understanding of the biosystems beyond human cognition.
DESIGN
The pipeline for engineering biosystems begins with a hierarchy of designs. On the abstract level, the objects of construction and modification need to be defined. On the practical level, the plans for experiment implementations need to be laid out (Figure 2). Designing for both the means and the ends synergistically can be a demanding task, due to the complexity of biosystems and lack of predictable models. Several computational tools have been developed to help with either abstract design such as predicting genome modification targets and metabolic pathway configurations, as well as design for experimental implementation such as determining genetic part sequences and DNA assembly strategies (Kelwick et al., 2014; Long et al., 2015; Medema et al., 2012; Shin et al., 2013).
Figure 2. Biosystems design at multi-scales.
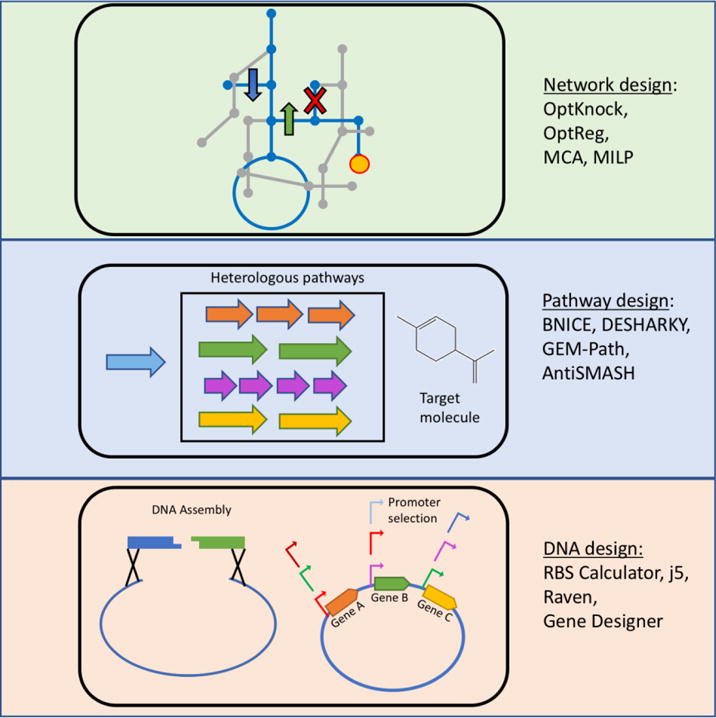
Design algorithms can be implemented at the scale of network design, pathway design and genetic construct design. At the level of network modifications, optimization algorithms such as OptKnock or OptReg (for stoichiometric models) and MCA or MILP (for kinetic models) are used to design engineering strategies for the metabolic network (Okuda et al., 2008). At the pathway level, algorithms such as BNICE are used to suggest native or heterologous pathways that can be used for synthesis of a target molecule. Having decided a specific network and/or pathway strategy, algorithms such as RBS Calculator suggest specific RBS designs for controlling expression whereas algorithms such as j5 and Raven aid in designing the method of construction of genetic elements from the user-defined parts.
Metabolic network design
To overproduce a metabolite from a living organism, the metabolic network needs to be optimized (Figure 2). The yield and productivity of the target molecule in the context of the native metabolism are first assessed using metabolic modeling on the reference (host) strain and/or mutant strains. Two prominent types of metabolic models are stoichiometric and kinetic models.
Stoichiometric models rely on the stoichiometric coefficients and steady state mass balances to calculate flux values at the steady state – this method is known as flux balance analysis (FBA). Often, multiple flux distributions are possible, and hence a cellular objective such as biomass growth or cofactor production is optimized in FBA to obtain a single flux solution (Varma and Palsson, 1993). To also include metabolic overproduction as a cellular objective, the algorithm OptKnock performs a bilevel optimization on a genome-scale stoichiometric model to highlight knockouts that will generate growth-coupled production of the target compound (Burgard et al., 2003). A number of subsequent algorithms expanded the scope of network modifications to up- and down-regulation (Pharkya and Maranas, 2006), introduction of non-native enzymatic reactions (Kim et al., 2011; Pharkya et al., 2004), elimination of competing pathways (Tepper and Shlomi, 2009), and use of faster optimization schemes (Xu et al., 2013). The MATLAB toolbox, COBRA, has provided a convenient framework to simulate and analyze the phenotypic behavior of a genome-scale stoichiometric model (Schellenberger et al., 2011). The strain design algorithms have been explored in a much more detailed manner elsewhere (Long et al., 2015).
FBA-based methods provide only steady state flux distributions. Kinetic models, on the other hand, employ kinetic parameters and rate law expressions to simulate the dynamic behavior of a metabolic network. Various metabolic design algorithms can be used in conjunction with these models to perform in silico optimization on the network and predict engineering strategies. To obtain a time-varying profile of metabolism, the dynamic Flux Balance Analysis (DFBA) was introduced which operates under a quasi-steady-state assumption (Mahadevan, Edwards and Doyle 3rd, 2002). To facilitate the shift towards integration of stoichiometric models with kinetic descriptions, the k-OptForce algorithm was developed which predicts metabolic interventions for both reactions with kinetic as well as only stoichiometric information(Chowdhury, Zomorrodi and Maranas, 2014).
Furthermore, there has been a growing interest in the use of kinetic models and accompanying optimization schemes for strain design since kinetic models generate dynamic profiles of the cell’s phenotypic behavior. A mixed-integer non-linear problem framework has been applied to an E. coli kinetic model to identify enzyme modifications for achieving a pre-established objective (Nikolaev, 2010). Various studies regarding CHO cell engineering demonstrated the benefit of using a kinetic model framework to predict effective strategies involving gene expression modulation (Nolan and Lee, 2011; Nolan and Lee, 2012; Villaverde et al., 2016). The ORACLE framework enables the construction of a large-scale kinetic model that includes mechanistic descriptions of enzyme kinetics (Chakrabarti et al., 2013). With the development of predictive kinetic models, suitable optimization algorithms can now be developed to make use of the mechanistic detail for proposing strain engineering strategies. The most commonly used method for analyzing kinetic models is called the metabolic control analysis (MCA), which quantifies the effect of change in a variable (e.g. flux and concentration) on every other flux or metabolite concentration in the metabolic network (Fell, 1992; Kacser and Burns, 1995; Ke et al., 2015; Somvanshi and Venkatesh, 2013).
Heterologous pathways design
To produce a target molecule non-native to the host’s metabolism, it is necessary to introduce a heterologous pathway (Figure 2). Identification of such a pathway is not a trivial task, and software such as the retrobiosynthesis algorithm BNICE can be employed. If the target molecule is a known compound and the objective is to synthesize it using a combination of native and heterologous metabolic pathways, then several algorithms are available for designing metabolic pathways that connect the substrates to generate the final product. The algorithm BNICE (Biochemical Network Integrated Computational Explorer) predicts a large number of enzymatic steps that can possibly convert a given substrate to a desired molecule (Hatzimanikatis et al., 2005). The algorithm first formulates molecules and enzymatic transformations in the form of bond-electron matrices. The transformation of any molecule via a particular enzyme is achieved by addition of the two matrices. This process is performed iteratively until either the desired molecule is reached or the pre-established iteration limit is exceeded. The original BNICE algorithm did not account for thermodynamic parameters in its pathway search – this was added in later versions that ranked the resulting pathways according to criteria such as thermodynamic feasibility, pathway length, and achievable yields (Henry et al., 2010). In the context of natural product discovery by targeting biosynthetic gene clusters, the antiSMASH algorithm predicts regions on an organism’s genome that express biosynthetic enzymes with high probability (Blin et al., 2013; Medema et al., 2011; Weber et al., 2015). This method can be used to discover novel enzymes that synthesize natural products with valuable functions. Different criteria are followed by other predictive algorithms such as DESHARKY that is focused on employing the metabolism of the host organism (Rodrigo et al., 2008). The GEM-Path algorithm also targets pathway prediction by employing the native metabolic network of the organism and adding non-native reactions to highlight novel compounds that can be produced from a limited number of steps (Campodonico et al., 2014). To constrain the number of possibilities in heterologous pathway design, the online webserver-based RetroPath presents a ranking function based on the molecular signatures of compounds and reactions (Carbonell et al., 2014a; Carbonell et al., 2011). The ranking function also has the option of integrating different types of data such as compatibility of the heterologous genes with the host, metabolite toxicity etc. Once a heterologous pathway has been chosen, the OptStrain algorithm suggests genetic modifications in the host organism that are required to channel optimum flux towards the non-native pathway (Pharkya et al., 2004). M-Path enables the design of a synthetic metabolic pathway for producing a given chemical by utilizing an iterative random approach that searches through enzymatic reaction and chemical compound databases (Araki et al., 2015). Another novel approach known as Sensi-Path predicts multistep enzymatic reactions that can transform non-detectable compounds into detectable ones, expanding the set of biosensor-detectable compounds (Delépine et al., 2016). A molecular signatures-based ranking score is also utilized by the eXTended Metabolic Space (XTMS) webserver to predict novel pathways that can be introduced into E. coli for producing a given chemical (Carbonell et al., 2014b). The pathway design software and algorithms have been reviewed in greater detail here (Medema et al., 2012).
DNA parts design
After deciding on a network or pathway engineering strategy, the focus of the design pipeline moves towards its implementation at the DNA scale. Metabolic engineering involves introduction of heterologous enzymes/pathways and/or modifications of the native metabolism via genetic overexpression, down-regulation or knockout. These modulations can be achieved using episomal vectors, genome editing, and/or genomic integration. For genetic parts, software such as RBS (ribosome binding site) Calculator (Farasat et al., 2014; Salis et al., 2009) and Gene Designer (Villalobos et al., 2006) are useful (Figure 2). RBS Calculator utilizes a thermodynamic model to predict the effect of a ribosome binding site (RBS) on protein expression, which is used to design synthetic RBSs to precisely control protein expression in a synthetic context. When expressing heterologous genes or pathways in a host organism, the gene sequences are often codon optimized to ensure efficient transcription of the foreign DNA. Codon optimization is a feature of various gene design software packages such as Gene Designer and DNAWorks (Hoover and Lubkowski, 2002). Gene Designer also offers functionalities such as calculation of oligonucleotide annealing temperatures and design of sequencing primers. The tool Gene Design offers features for synthetic gene design such as codon optimization, restriction site insertion and removal, and oligonucleotide design (Richardson et al., 2006). An updated version of the software also includes tools for analysis and visualization of codon bias statistics, as well as design of parts and oligonucleotides for longer gene assemblies (Richardson et al., 2010). Besides the above software, databases such as Standard Biological Parts Knowledgebase (Galdzicki et al., 2011), KEGG (Kanehisa et al., 2002), and NCBI are also useful repositories for parts design. Recently, a Design-of-Experiments based method for the design of combinatorial libraries of pathway variants called Double Dutch was published, which aims to reduce human effort as well as increase the efficiency of part design (Roehner et al., 2016). The XML-based data standard for representing synthetic biology designs comprised of annotated modular parts, known as Synthetic Biology Open Language (SBOL), has been proposed to facilitate ease of sharing and collaborating on synthetic biology designs across institutions (Galdzicki et al., 2014; Roehner et al., 2015). Further, to facilitate consistent characterization of the bioparts employed in such designs, a data acquisition standard for synthetic biology parts, known as DICOM-SB, was recently published (Sainz De Murieta et al., 2016).
DNA assembly design
Once the parts are designed and synthesized, various design questions also arise regarding the method of assembly of the constructs into a vector. The design of oligonucleotides for amplification of DNA substrates, inclusion of appropriate restriction sites, and addition of complementary overhangs are all design variables that need to be considered when performing DNA assembly using established methods such as digestion-ligation, Golden Gate (Engler et al., 2008), Gibson assembly (Gibson et al., 2009), and SLIC (Li and Elledge, 2007). As the assembly process scales up to larger combinatorial libraries, the magnitude of variables can become difficult to handle on a case-to-case scenario and hence requires assembly design automation. One of such design tools, j5 enables the automated design for assembly of multiple DNA segments simultaneously using assembly methods such as Golden Gate, Gibson, and SLIC (Hillson et al., 2012). Another web based application, Raven, optimizes the cloning steps allowing solutions for shared parts to be reused to reduce the total number of steps (Appleton et al., 2014). Raven also optimizes the overhangs between DNA segments and automatically designs primers for the polymerase chain reaction (PCR) steps. Based on the successes and failures at each step, this algorithm updates its library entries, thus improving the efficiency of its assembly plans in an interactive manner. A recent publication described a publicly accessible library of modular DNA parts such as promoters, terminators, ribosomal binding sites etc., built using the MoClo assembly framework (Iverson et al., 2016). By employing assembly designs from the Raven algorithm, this library can be used to efficiently assemble combinatorial libraries. The Cello algorithm simplifies the construction of genetic circuits by borrowing from principles of design of semiconductor-electronics, and outputs a DNA sequence that codes for the circuit (Nielsen et al., 2016). DNA design algorithms have been reviewed comprehensively in these review articles (Garcia-Ruiz et al., 2016; Medema et al., 2012).
BUILD
Build usually refers to the process of generating the designated genotypes to prototype the designs through a either bottom-up or top-down approach.
Bottom-up
In the bottom-up approach, DNA parts either derived from naturally existing templates or de novo synthesized are used as building blocks to assemble designed constructs. Living organisms provide a vast source of genetic elements that can be directly amplified using PCR from the natural sequences. However, PCR amplification may be challenging, especially for long fragments or from complex templates such as high GC content genomes, highly repetitive sequences, or metagenomic samples, and hence requires continuing technical improvement (Jiang et al., 2015). On the other hand, de novo synthesis starts from oligonucleotide synthesis (a few base-pairs to a couple hundred base-pairs) and then assembled to double-stranded fragments of a few hundred to a couple thousand bps (Chao et al., 2015a; Kosuri and Church, 2014), capable of writing DNA molecules of arbitrary sequences with few constraints. This methodology enables in silico design and optimization of genetic parts in a highly flexible manner, such as codon optimization of genes (Villalobos et al., 2006), tuning of promoter (Redden and Alper, 2015) and RBS (Salis et al., 2009) strengths. Although de novo synthesis is still very costly, synthetic DNA parts have already been applied in large-scale prototyping for high-value products (Petrone, 2016). Since synthesis of DNA oligos and kilo-base-pair fragments has become a more mature process, many parts can be prefabricated in specialized commercial facilities before further processing in biofoundries.
The DNA parts can then be assembled into larger DNA molecules. DNA assembly is often intensely time consuming and has a high failure rate. In our research group researchers are estimated to spend >50% of their time constructing DNA molecules. Sometimes a few rounds of assembly are necessary to accomplish the target size and configuration. Diversity is often introduced in this process by assembling different combinations of parts in parallel to test many possible designs (Kim et al., 2013; Xu et al., 2016). Both increased rounds and combinations of assemblies make the process more tedious and error-prone. Such tasks are better suited for machines as they can programmatically perform protocols with high speed and precision. However, transferring the manual operation to a robust and high-throughput automated process is nontrivial. It is important to avoid handling of sample-specific exceptions and frequent failures to unleash the advantages of machines. A number of innovations in assembly reactions and assembly schemes were reported in recent years. These methods have greatly improved the efficiency (likelihood of generating products) and fidelity (percentage of correctly assembled products) while simplifying the procedures. Furthermore, several assembly methods have made it easier to standardize the parts and modularize the design (Figure 3). Since these advances have been extensively reviewed (Chao et al., 2015a; Chao et al., 2015b; Ellis et al., 2011), this article will only elaborate on one example. Golden Gate method utilizes Type IIs restriction enzymes to generate customizable 4-bp sticky ends and to ligate the complimentary ends between fragments (Engler et al., 2008). In the one-pot reaction, the digestion and ligation reactions happen simultaneously, which forms a highly efficient irreversible cascade reaction towards the assembled products. By optimizing the sequences of these 4-bp junctions, multiple fragments can be assembled in one step with superior fidelity. The optimized set of junctions can also be assigned to the parts according to the position or function under a predefined standard so that parts can be used in different assemblies or even different projects, like standardized fasteners used in mechanical manufacturing (Figure 3). Taking advantage of the standard parts, MoClo, a scheme was developed to modularize the assembly of multi-gene DNA constructs (Weber et al., 2011). 11 transcription units consisting of 44 parts were assembled into a 33kb plasmid in three steps. The robustness and modularity make it possible to prototype combinatorial designs of DNA constructs in a high throughput manner. Such protocols can then be converted to an automated workflow and implemented in biofoundries.
Figure 3. A typical one-pot modular DNA assembly scheme.

DNA parts are mixed and ligated to form the final constructs based on the designs. The order of parts in each assembly is designated by the linkers flanking the parts. The DNA fragments can be inserted to donor plasmids as an intermediate step. Golden Gate assembly is used as an example.
Most of the DNA assembly workflows only use limited number of operations, including pipetting, heat incubation, thermo-cycling, etc. Thus, these steps can be readily implemented with off-the-shelf equipment. State-of-the-art liquid handling stations can pipette as low as a fraction of a microliter while providing the flexibility required in combinatorial assembly. To further reduce the reagent costs, nanoliter-scale acoustic dispensing (Kanigowska et al., 2016; Woodruff et al., 2017) and microfluidic devices (Linshiz et al., 2016; Patrick et al., 2015; Shih et al., 2015) have been applied to miniaturize the reactions.
Top-down
Besides building DNA from scratch, the desired genotypes can also be derived from existing genomes by modifying certain loci. Traditionally, such editing harnessed either endogenous (Baudin et al., 1993; Stahl and Ferrari, 1984) or heterologous (Ellis et al., 2001; Fu et al., 2012; Zhang et al., 2000) recombination machineries. Although these protocols can be automated by existing or custom-built instruments (Wang et al., 2009), the yield of positive clones in each round of experiments is low unless a selection marker is used. Due to the low editing efficiency, more clones need to be selected and verified, resulting in lower throughput and higher cost. On the other hand, the use of auxotrophic or drug selection markers limits the number of loci that can be edited, reduces the cell fitness, and makes it difficult to perform scarless modifications. Although application of the Cre-LoxP system (Arakawa et al., 2001; Sauer, 1987) allows iterative modification of multiple genome loci by recycling selection markers, the process is slow and still leaves a loxP scar.
A number of new genome editing tools became available in recent years. Zinc-finger nucleases (Kim et al., 1996), transcription activator-like effector nucleases (TALENs) (Miller et al., 2011), and Clustered regularly interspaced short palindromic repeats (CRISPR) - CRISPR associated proteins (Cas) (Cong et al., 2013; Jinek et al., 2012; Mali et al., 2013) are three classes of programmable nucleases that can induce double-stranded breaks (DSBs) in specific genome loci. These nucleases have greatly improved the efficiency of genome editing as DSBs can induce either non-homologous end joining (NHEJ) or homologous recombination (HR) (Haber, 2000). NHEJ can result in loss-of-function of genetic elements while HR can be harnessed to over-write, insert, or delete genomic sequences. Moreover, these nucleases also act as effective selection pressures since the chromosomes would be constantly cleaved until the sequences were mutated. Thus genome modification is no longer dependent on the availability of auxotrophic or drug resistance markers. Among these tools, the CRISPR system is the easiest to implement as it is relatively simple to synthesize the DNA fragments coding for the guide RNAs. The CRISPR system has already been applied to create genome-wide knockout libraries (Shalem et al., 2014), simultaneously knocking out multiple genes (Bao et al., 2015; Cong et al., 2013), as well as inserting genetic elements (Shi et al., 2016; Tsai et al., 2015). Due to the high efficiency of the CRISPR system and the simplicity of the protocols, these manipulations can be potentially automated by existing instruments. With the latest advances in genome editing technology, a more or less complete toolkit has become available for biofoundries to generate desired genotypes in a high-throughput manner.
It is debatable whether the bottom-up or top-down approach represents the future of bioprototyping. Bottom-up approaches, especially de novo DNA synthesis, allow the largest freedom in design, but the cost of DNA synthesis is still high. Similarly, two strategies co-exist in mechanical engineering, i.e. subtractive and additive manufacturing. The easier or more economical route is usually chosen.
It is worthwhile to mention that phenotypes can also be manipulated by regulatory machineries. Besides applications of naturally existing regulators such as transcription activators and repressors, a few synthetic regulatory tools have also been developed. A commonly used group of regulatory engineering tools are mediated by heterologous RNA (Si et al., 2015a). Derived from endogenous RNA regulation machinery, both synthetic small RNAs (sRNA) (Kang et al., 2012; Na et al., 2013) and RNA interference (RNAi) (Si et al., 2015b) were applied in global tuning of gene expressions. Regulatory tools were also developed based on modified CRISPR systems to repress or activate gene expression (Dominguez et al., 2016). Overall, these methods have broadened the spectrum of engineering tools to facilitate the fine tuning of biosystems. Most of the RNA mediated regulation protocols are not too different from other top-down prototyping approaches in terms of operation. In fact, if an automated workflow is developed properly, different engineering approaches can be attempted in parallel on the biofoundries.
DNA transfer and clone selection
Both top-down and bottom-up approaches involve construction of DNA molecules encoding the design itself or the tool to implement the design. In most cases, these DNA molecules need to be introduced into cells to be expressed and tested. There is already a collection of well-defined protocols for transforming, transfecting, or transducing model organisms, while some of these operations has been automated (Ben Yehezkel et al., 2011; Si et al., 2017), some, however still remains challenges to be automated in high throughput. First, some protocols require strict experimental conditions, such as low temperature, precise timing, as well as quick but gentle pipetting. Second, some involve expensive consumables and reagents like electroporation cuvettes and mammalian lipofection kits. Lastly, certain strains of microorganism, especially non-model organisms still lack an efficient method to transfer DNA. Therefore, this step may become a bottleneck in certain prototyping processes. Several interdisciplinary technologies have been developed to address these issues. With the help of microfluidics, cells can be positioned onto micro channels, capillaries, or solid micro electrodes. Thus electroporation can be precisely exerted on a single cell or even a small patch of the cell membrane (Olofsson et al., 2003; Wang and Lee, 2013). There are also reports on intrusion of cells with DNA coated nano-structures merely using mechanical force (Cai et al., 2005; Han et al., 2008; Obataya et al., 2005). Although outstanding DNA transfer efficiency and cell survival rate have been reported in these works, application of such new methods and devices in synthetic biology and metabolic engineering is still rarely seen. Further development in these technologies to allow high throughput DNA transfer is nontrivial, as most of them require specially fabricated consumables and sophisticated supporting equipment.
TEST
In the DBTL cycles, test currently remains the throughput bottleneck (Delépine et al., 2016). Similar to design and build, test automation should be implemented over a range of system scales including genetic constructs and genome, transcriptome, proteome, and metabolome (Figure 4). Recent advances in high-throughput analytical technologies enable multi-omics systems biology with improved efficiency and speed.
Figure 4. Biosystems testing at different levels.
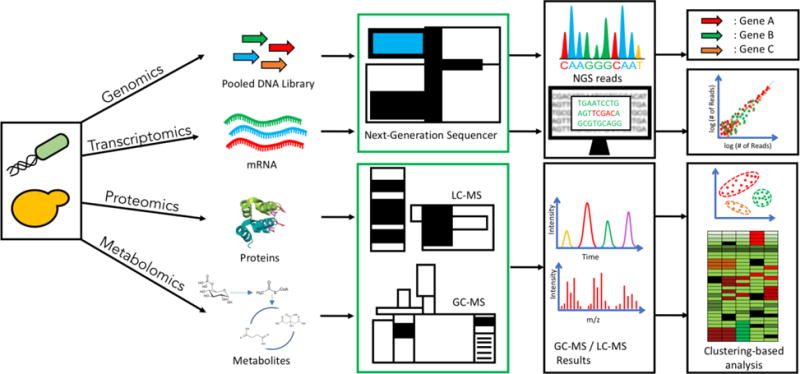
The strategies for testing a biosystem are implemented at various levels, depending on the engineering strategy being implemented. Genetic modifications to the host genome or assembly of DNA parts usually require sequencing methods such as Sanger or NGS for verification of the end product. If inquiry is focused on studying global changes on gene expression levels, influenced by either genetic or environmental modifications, then mRNA transcript levels are measured using methods such as microarray analysis and RNA-Seq. Modification strategies targeted at changing protein numbers in the biosystem are monitored using proteomics-based tools such as MALDI-TOF and LC-MS. However, if protein numbers are not solely indicative of the modified phenotype, the end-effect of the engineering strategy on the metabolic profiles is typically analyzed using LC-MS or GC-MS based metabolomics tools. Testing capabilities can also be performed for in a targeted manner for specific mRNA, protein or metabolite species.
For genotyping, gel electrophoresis and Sanger sequencing have been instrumental in verification for both synthetic DNA constructs and engineered genomes, but both involve many manual steps. Recently, liquid handlers coupled with high-throughput capillary electrophoresis has resulted in the implementation of a high-throughput genotype verification process (Dharmadi et al., 2014). For whole-genome (re-) sequencing and transcriptomic analysis, next-generation sequencing (NGS) has become routine practices. Notably, NGS is also extended to massive analysis of small genetic constructs in a parallel and cost-effective manner, using new barcoding technologies that enable alignment of pooled reads to their source samples (Metzker, 2010).
For phenotyping, transcriptomic analysis is often used, which provides a snapshot of the machinery regulating gene expression inside the organism (Figure 4). Common methods for transcriptome analysis are microarrays (Heller, 2002) and RNA-Seq (Sharma and Vogel, 2014). RNA-seq has been used in various studies for testing strains with refactored pathways (Smanski et al., 2014), or the characterization of strong promoters in different organisms (Creecy and Conway, 2015; Luo et al., 2015). The resulting data can also be used to ‘reverse engineer’ the most statistically significant regulatory interactions, using a range of methods such as Bayesian Networks (Heckerman, 1995) or information-theoretic approaches (Margolin et al., 2006). Reconstructing these networks can help identify major players that influence significant control over the regulatory processes, also known as ‘hubs’. In the context of metabolic engineering, reconstruction of regulatory networks across various mutant strains can be used to compare the effects of modified genotypes on the global transcriptional machinery of the host organism. This knowledge can, in turn, be used to design microorganisms with better biochemical production capabilities.
Other commonly used phenotyping tools include proteomics and metabolomics. Given the large number and chemical complexity of analytes in proteomes and metabolomes, mass spectrometry should be the method-of-choice to obtain sufficient coverage and identification. Currently, gas and liquid chromatography (GC and LC) is often required before MS analysis to reduce competition for ionization at a given time for better separation, at the cost of longer analysis time and more complex sample preparation. Given the ever-increasing resolution, scanning speed, and quantification capability in MS analyzers, direct infusion methods may be used for higher throughput. For example, Agilent RapidFire platform couples solid-phase extraction with HT-ESI-MS for rapid and automated MS analysis (Woodruff et al., 2016). Moreover, MALDI mass spectrometry imaging has also been increasingly used for high-throughput screening thanks to its rapid speed, high tolerance to salts, and simple sample preparation (Zhou et al., 2015). Targeted proteomics was successfully applied to identify bottle-necks in the mevalonate pathway, enabling an improvement in the production of a sesquiterpene (Redding-Johanson et al., 2011). Recently, studies have utilized proteomics and metabolomics in combination for analyzing engineered strains, resulting in improved production of target molecules (Brunk et al., 2016; George et al., 2014). Testing capabilities have been reviewed more comprehensively here (Petzold et al., 2015).
Difficulty in automating the above-mentioned analytical techniques has resulted in test capabilities still posing a significant bottleneck in the turnover cycles of strain engineering. A review paper on metabolomics pre-treatment techniques highlighted Liquid-Liquid-Extraction based methods as amenable to automation (Raterink et al., 2014). Focused technology development in this field is anticipated to lead towards greater levels of test automation, enabling even higher throughputs in turnover cycles.
LEARN
The essential goal of most biosystems engineering is to understand the mechanism or discover a statistical pattern. Learning through DBT cycles is crucial for both fundamental science and practical applications. Biofoundries have demonstrated their capability of substantially accelerating and parallelizing the DBT cycles. Although the learning tasks are still mainly performed by human brains, with high dimension of the systems of interests today and the complexity of experiments, the data generated from biofoundries can quickly expand beyond the processing power of human minds. Assistance from computer algorithms will become a necessity when analyzing or at least preprocessing these data. Since the number of samples to be built and tested grows geometrically with the dimensions, exhaustive approaches will quickly fail after adding more variables. Thus, more efficient research algorithms are needed to reduce the time and complexity when exploring large variant spaces, which will rely on learning as well.
Data science has demonstrated its potential in many disciplines (Dhar, 2013). A wide range of data mining tools, statistical models, and machine learning algorithms have become available to extract patterns from data and to solve prediction problems. With appropriate abstraction, these tools can be readily used to guide the design and engineering of biosystems. Some early applications have already been reported. Lee et al. and Xu et al. trained regression models for violacein production as a function of gene expression levels (Lee et al., 2013; Xu et al., 2016) and predicted the optimal combination of promoter strength. Xu and colleagues implemented a Design-of-Experiment (DOE) approach to identify enzymes in a 5-gene pathway that exercised the most significant control, and subsequently used to identify promoter combinations that resulted in optimal expression of the pathway. The final strain has a 3.2-fold increase in violacein titer compared to the baseline strain. A study to improve taxol production employed the use of a multivariate search method that changed the expression of smaller modules of the pathway simultaneously (Ajikumar et al., 2010). This enabled an efficient scheme to sample the parameter space of the pathway flux, obviating the need for a high-throughput screen. A DOE-based approach was also adopted in a study exploring the combined role of genetic and environmental factors to bring down the sampling size from a library size of 29 to just 32 sampled combinations (Zhou et al., 2015). Application of modeling in these works has resulted in significant reduction of the amount of experiments to identify the improved producers, if not optimum.
INTEGRATION
As more enabling tools emerge for design, build, and test, it has become important to integrate these steps to form industrialized pipelines. Only by automating time-consuming operations, the overall cost of the DBT cycles in time and labor has been greatly decreased. As a result, the paradigm of research starts to change. In conventional biological engineering, heuristic approaches are usually adopted since the cost of each turnover is high. When the cost drops down to a critical point, exhaustive approaches can be adopted, i.e. many design variants can be automatically generated, prototyped, and tested. Likewise, the invention of modern computers changed the way we solve many problems. For example, ordinary differential equations no longer have to be solved by tedious algebraic transformations. Instead, numerical solutions can be obtained by more generally applicable but more computation-intensive methods like Runge-Kutta. Intrigued by this new paradigm, a few academic institutions have attempted to build integrated research facilities with a goal of studying and engineering biological systems with industrial efficiency. These facilities has adopted the name “foundry”, a term from the semiconductor industry. Early foundry adopters include MIT-Broad Foundry (http://web.mit.edu/foundry/), Edinburgh Genome Foundry (Fletcher et al., 2016), Manchester Foundry (Ra et al., 2016), DNA synthesis and construction foundry at the Imperial College London (Chambers et al., 2016), GeneMill at the University of Liverpool (https://genemill.liv.ac.uk/), DNA Foundry at Earlham Institute (http://www.earlham.ac.uk/automated-dna-assembly), NUS Synthetic Biology Foundry (http://syncti.org/research/synthetic-biology-foundry/), and Illinois Biological Foundry for Advanced Biomanufacturing (iBioFAB) (Chao et al., 2017). A number of venture capital backed companies are also trying to capitalize on these advanced research tools (Benjamin et al., 2016; Check Hayden, 2015; Davison and Lievense, 2016; Zanghellini, 2016).
Semi-automated integration
Among the academic and industrial biofoundries, many researchers have adopted a partially automated system, i.e. using machines to replace humans in certain steps while humans are still in control of the workflows. In the early stages of biofoundry development, partially automated systems have clear advantages: a) The system can be quickly setup with off-the-shelf equipment and software; b) the human researcher gets instant feedback; c) Equipment can be used in different workflows as long as the operators fully understand the different tasks in these workflows; d) Systems can easily recover from interruptions due to hardware or software malfunction. With the current state of the art, there are also procedures such as next generation sequencing that are difficult to operate without manual operation. However, human interventions are still time-consuming and error-prone. There are also fully automated systems developed to run complete workflows (Dorr et al., 2016; King et al., 2009; King et al., 2004; Wang et al., 2009), but they are extensively customized towards specific applications, and hence are difficult to reconfigure for other tasks.
Fully automated integration
In today’s biological engineering practice, designs at different levels are implemented and tested by a continuously advancing collection of methods in a variety of biosystems. Therefore, an integrated biofoundry needs to be a highly reconfigurable flexible manufacturing system (FMS). On the other hand, such an FMS should be highly automated to improve efficiency and reduce human influence. Establishing a biofoundry that can handle long, complex, and highly diversified workflows with minimal human intervention is very challenging. To decompose the problem, some fundamental principles of chemical engineering can be adopted. To facilitate design and analysis, various chemical processes are usually broken up into basic types of unit operations. If we break up the routine biological engineering workflows into a series of shared process modules and unit operations, many workflows can be found to share finite number of process modules and unit operations. During the hardware design process, the unit operations can be matched with appropriate instruments. In principle, any workflow can be performed by linking available unit operations in customized sequences. To facilitate such linkage, the input and output sample format for each unit operation should be standardized. Society for Biomolecular Sciences (SBS) multi-well plate format is a widely adopted standard in this industry and most of the high-throughput instruments are SBS compliant. Integrating microfluidic devices with SBS standard will require certain sample format conversion mechanisms. To allow reconfiguration, a high degree-of-freedom central robotic system controlled by a computational framework can be integrated to form and manage a virtual network of unit operations. Notably, in a biological foundry, N unit operations are linked by high-degree-of-freedom robotics, forming NP theoretical configurations in a workflow of P steps. In biofoundries based on this architecture, a roadmap for workflow implementation may generally apply to various projects. First, all necessary biological transformations are listed for producing target constructs. Then, individual steps and the overall scheme are optimized for modularity, standardization, and robustness to facilitate automated and high-throughput operations. Finally, the optimized workflow is converted into a sequence of unit operations for implementation in the foundry (Figure 1). This way, the versatility of the biological foundry is only limited by its collection of automatable unit operations, which can be readily expanded by modifying existing instruments or introducing new ones. In case of unit operations that cannot be automated, human interventions can still be added. With the current technologies, it is already feasible to automate the majority of the commonly used unit operations and compose a variety of workflows for engineering biosystems.
The iBioFAB platform followed the architecture referred to previously (Chao et al., 2017) (Figure 1). A 6-degree-of-freedom robotic arm and a 4-degree-of-freedom robotic arm interlink unit operations into various process modules and workflows by transporting samples and consumables between 20 instruments of 12 types. To manage virtual networks of unit operations, we developed iScheduler, a high-level modular programming environment. Although off-the-shelf automation graphical user interfaces were available to control the automation hardware, programs were scripted by joining individual device commands. This flat programming practice was capable of handling simple repetitive jobs, but became chaotic and incompetent when implementing complex workflows that involved hundreds of steps. Using iScheduler, instead, commonly used process modules such as DNA assembly and cell density measurements were first scripted and validated, and then logically orchestrated at a high level. This modular and hierarchical approach has several merits: a) Programming and debugging were performed for each module separately without engaging the entire workflow. Since modules were tested individually, little debugging is needed at the workflow level. b) The system could recover from errors in situ and resume the program. c) Programming was expedited by reusing existing modules or building on prior workflows. In theory, such a biofoundry is capable of fully automating various workflows yet match up the advantages of the semi-automated biofoundries.
Preliminary applications
There are few published applications of biofoundries so far. Although industrial engineering progresses are often not well publicized, this new biological engineering methodology has already been shown to generate commercial values. Amyris is one of the pioneers in streamlining the DBTL cycles for engineering industrial microbes. A few technologies have been developed to improve the robustness and lower the cost of DNA assembly (de Kok et al., 2014) and construct verification (Dharmadi et al., 2014; Shapland et al., 2015). In the same space, startup companies such as Ginkgo Bioworks and Zymergen are also growing rapidly. Due to differences in funding structure, some academic institutions have started taking more risks to develop fully automated and flexible systems, including the iBioFAB and Edinburgh Genome Foundry. Two proof-of-concept studies on the iBioFAB have been published. A generalized DNA assembly workflow based on the Golden Gate method (Engler et al., 2008) has been developed (Chao et al., 2017). Following the roadmap mentioned earlier, the robustness of the Golden Gate assembly protocol was improved by optimizing reaction formulations and introducing a clean-up step to degrade residual linear fragments. A pretested set of 4-bp junctions was used to standardize the parts. Then the protocol was converted to an automatable process and a sequence of executable unit operations. As a demonstration, this workflow can assemble up to 400 pairs of TALENs per day with a success rate of 96.2% at a material cost of $2.1/pair. Each assembly involves 16 DNA fragments encoding 28 highly repetitive protein domains, which is known to be tedious to synthesize. This DNA assembly workflow was also applied to pathway engineering in different organisms for various products. In addition to bottom-up assembly of synthetic DNA constructs, a genome engineering workflow for Saccharomyces cerevisiae was also implemented on the iBioFAB (Si et al., 2017). According to the above-mentioned roadmap, two bottlenecks were identified as lack of standardized and modular methods to introduce genetic modulations, and low genome modification efficiency during each engineering cycle. To solve the first obstacle, a collection of modulation parts cloned from full-length cDNA libraries were created to up- and down-regulate each gene. These parts were constructed as standardized integration donors into repetitive sequences in the yeast genome, so that genetic modifications can be accumulated in a scalable manner. On the other hand, low integration efficiency was improved using CRISPR-Cas9, allowing genome-wide coverage and multiplex modifications to be introduced at the population level and the protocol became much more robust. These genetic and process designs enable us to devise an automated workflow on the iBioFAB to iteratively create and screen yeast strain libraries. In the course of 3 rounds of evolution, 10100 theoretical genetic variants were explored, resulting in a strain that has the highest acetate tolerance reported.
Algorithm-driven DBTL cycles
With the unprecedented prototyping efficiency in biofoundries, orders of magnitude more data can be generated at a reasonable cost and better consistency, which will further improve the fidelity of modeling. Furthermore, it becomes possible to apply some advanced learning strategies. Up to date, we have preliminary success on automating complex workflows in each round of DBT as well as modeling certain traits of biosystems. These proceedings make it possible for the design of subsequent rounds of experiments to be driven by algorithms based on analysis of the preceding rounds of data. A complete integration of the design-build-test-learn cycles will open the door to active learning (Settles, 2012) (Figure 1). By allowing the modeling algorithm to directly drive the biofoundries, the total amount of experiments required to make a prediction can be greatly reduced when compared to random sampling. Such algorithms were applied in functional genomics research (King et al., 2009; King et al., 2004) while no biosystems engineering work driven by active learning has been reported yet.
CONCLUSIONS
With the recent technology advances, it has become feasible to establish automated and versatile engineering platforms to accelerate both fundamental and applied research and development of biosystems. A number of biofoundries have been established around the world and have accomplished some proof-of-concept studies.
Several challenges need to be addressed for future biofoundry development. The success rate of DNA assembly is still largely sequence dependent with no reliable modeling. Many of them also impose certain constraints onto the design step. Most automation instruments and corresponding software are not well developed due to the small served market, exhibiting suboptimal performance and reliability compared with consumer products or more common industrial processes. No ready-to-integrate instruments are currently available off-the-shelf for a few unit operations, which limits the development of fully automated workflows. In addition, new technology may be integrated. For example, a wide application of microfluidics and nanoliter scale liquid handling is anticipated to greatly reduce reagent cost and increase throughput. For system integration, more powerful dynamic and multiplex scheduling algorithms need to be developed to maximize throughput and resolve resource conflicts without human intervention. Last but not least, more rapid and parallelized analytical technologies are highly desirable because testing can easily become a throughput bottleneck when involving chromatography and DNA sequencing.
We envision an ideal biological foundry should not only be able to parallelize and automate the steps in the design-build-test cycles, but can also autonomously learn about the biosystems. This will allow scientists to focus on conceptual innovations and implement their ideas rapidly. Furthermore, it can extend human intelligence or even intuition when handling complex data. On the other hand, security and ethics of such a machine capable of “creating” lives should not be neglected, as it will most likely become a concern in the near future.
Highlights.
This review summarizes the enabling technologies, early successes, and existing challenges in developing and applying automated biofoundries for engineering biosystems.
Iterative rounds of the Design, Build and Test cycle are implemented in the biofoundries for biosystems engineering.
Various Design tools at parts-, pathway-, and network-levels and for DNA assembly have been developed.
Both bottom-up and top-down automated strategies are used for the Build step.
Test automation should be implemented over a range of system scales including genetic constructs and genome, transcriptome, proteome, and metabolome.
As more enabling tools emerge for Design, Build, and Test, it has become important to integrate these steps to form industrialized pipelines.
Learning through DBT cycles is crucial for both fundamental science and practical applications.
Acknowledgments
This work was supported by the Roy J. Carver Charitable Trust, Carl R. Woese Institute for Genomic Biology at the University of Illinois at Urbana-Champaign (UIUC), Defense Advanced Research Program Agency, and National Institutes of Health (1U54DK107965). R.C. acknowledges fellowship support from 3M Corporation. T. S. acknowledges postdoctoral fellowship support from Carl R. Woese Institute for Genomic Biology (UIUC). We specially thank Dr. Christopher V. Rao and Dr. Weicong Ding for insightful discussions and suggestions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ajikumar PK, Xiao WH, Tyo KEJ, Wang Y, Simeon F, Leonard E, Mucha O, Phon TH, Pfeifer B, Stephanopoulos G. Isoprenoid pathway optimization for Taxol precursor overproduction in Escherichia coli. Science (New York, NY) 2010;330:70–4. doi: 10.1126/science.1191652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleton E, Tao JH, Haddock T, Densmore D. Interactive assembly algorithms for molecular cloning. Nat Methods. 2014;11:657–+. doi: 10.1038/nmeth.2939. [DOI] [PubMed] [Google Scholar]
- Arakawa H, Lodygin D, Buerstedde JM. Mutant loxP vectors for selectable marker recycle and conditional knock-outs. BMC Biotechnol. 2001;1:7. doi: 10.1186/1472-6750-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki M, Cox RS, Makiguchi H, Ogawa T, Taniguchi T, Miyaoku K, Nakatsui M, Hara KY, Kondo A. M-path: A compass for navigating potential metabolic pathways. Bioinformatics. 2015;31:905–911. doi: 10.1093/bioinformatics/btu750. [DOI] [PubMed] [Google Scholar]
- Bao ZH, Xiao H, Lang J, Zhang L, Xiong X, Sun N, Si T, Zhao HM. Homology-Integrated CRISPR-Cas (HI-CRISPR) System for One-Step Multigene Disruption in Saccharomyces cerevisiae. ACS Synth Biol. 2015;4:585–594. doi: 10.1021/sb500255k. [DOI] [PubMed] [Google Scholar]
- Baudin A, Ozier-Kalogeropoulos O, Denouel A, Lacroute F, Cullin C. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res. 1993;21:3329–30. doi: 10.1093/nar/21.14.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Yehezkel T, Nagar S, Mackranrs D, Marx Z, Linshiz G, Shabi U, Shapiro E. Computer-aided high-throughput cloning of bacteria in liquid medium. Biotechniques. 2011;50:124–127. doi: 10.2144/000113514. [DOI] [PubMed] [Google Scholar]
- Benjamin KR, Hill PW, Meadows AL, Singh AH, Cherry JR. SBE Supplement: Commercializing Industrial Biotechnology - Use Cost Models to Guide R&D. Chem Eng Prog. 2016;112:44–50. [Google Scholar]
- Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, Weber T. antiSMASH 2.0–a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013;41:204–212. doi: 10.1093/nar/gkt449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunk E, George KW, Alonso-Gutierrez J, Thompson M, Baidoo E, Wang G, Petzold CJ, McCloskey D, Monk J, Yang L, O’Brien EJ, Batth TS, Martin HG, Feist A, Adams PD, Keasling JD, Palsson BO, Lee TS. Characterizing Strain Variation in Engineered E. coli Using a Multi-Omics-Based Workflow. Cell Systems. 2016;2:335–346. doi: 10.1016/j.cels.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgard AP, Pharkya P, Maranas CD. OptKnock: A Bilevel Programming Framework for Identifying Gene Knockout Strategies for Microbial Strain Optimization. Biotechnol Bioeng. 2003;84:647–657. doi: 10.1002/bit.10803. [DOI] [PubMed] [Google Scholar]
- Cai D, Mataraza JM, Qin ZH, Huang ZP, Huang JY, Chiles TC, Carnahan D, Kempa K, Ren ZF. Highly efficient molecular delivery into mammalian cells using carbon nanotube spearing. Nat Methods. 2005;2:449–454. doi: 10.1038/nmeth761. [DOI] [PubMed] [Google Scholar]
- Campodonico MA, Andrews BA, Asenjo JA, Palsson BO, Feist AM. Generation of an atlas for commodity chemical production in Escherichia coli and a novel pathway prediction algorithm, GEM-Path. Metabolic Engineering. 2014;25:140–158. doi: 10.1016/j.ymben.2014.07.009. [DOI] [PubMed] [Google Scholar]
- Carbonell P, Parutto P, Baudier C, Junot C, Faulon JL. Retropath: Automated pipeline for embedded metabolic circuits. ACS Synth Biol. 2014a;3:565–577. doi: 10.1021/sb4001273. [DOI] [PubMed] [Google Scholar]
- Carbonell P, Parutto P, Herisson J, Pandit SB, Faulon JL. XTMS: Pathway design in an eXTended metabolic space. Nucleic Acids Res. 2014b;42:389–394. doi: 10.1093/nar/gku362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonell P, Planson AG, Fichera D, Faulon JL. A retrosynthetic biology approach to metabolic pathway design for therapeutic production. BMC Systems Biology. 2011;5:122–122. doi: 10.1186/1752-0509-5-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti A, Miskovic L, Soh KC, Hatzimanikatis V. Towards kinetic modeling of genome-scale metabolic networks without sacrificing stoichiometric, thermodynamic and physiological constraints. Biotechnology Journal. 2013;8:1043–1057. doi: 10.1002/biot.201300091. [DOI] [PubMed] [Google Scholar]
- Chambers S, Kitney R, Freemont P. The Foundry: the DNA synthesis and construction Foundry at Imperial College. Biochemical Society Transactions. 2016;44:687–688. doi: 10.1042/BST20160007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao R, Liang J, Tasan I, Si T, Ju L, Zhao H. Fully Automated One-Step Synthesis of Single-Transcript TALEN Pairs Using a Biological Foundry. ACS Synth Biol. 2017;6:678–685. doi: 10.1021/acssynbio.6b00293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao R, Yuan YB, Zhao HM. Building biological foundries for next-generation synthetic biology. Sci China Life Sci. 2015a;58:658–665. doi: 10.1007/s11427-015-4866-8. [DOI] [PubMed] [Google Scholar]
- Chao R, Yuan YB, Zhao HM. Recent advances in DNA assembly technologies. Fems Yeast Res. 2015b;15 doi: 10.1111/1567-1364.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman T. Lab automation and robotics: Automation on the move. Nature. 2003;421:661–663. 665–6. doi: 10.1038/421661a. [DOI] [PubMed] [Google Scholar]
- Check Hayden E. Synthetic biology lures Silicon Valley investors. Nature. 2015;527:19. doi: 10.1038/527019a. [DOI] [PubMed] [Google Scholar]
- Committee on Industrialization of Biology, N. R. C. Industrialization of Biology: A Roadmap to Accelerate the Advanced Manufacturing of Chemicals. The National Academies Press; Washington, DC: 2015. [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin SL, Barretto R, Habib N, Hsu PD, Wu XB, Jiang WY, Marraffini LA, Zhang F. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creecy JP, Conway T. Quantitative bacterial transcriptomics with RNA-seq. Current Opinion in Microbiology. 2015;23:133–140. doi: 10.1016/j.mib.2014.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison BH, Lievense JC. SBE Supplement: Commercializing Industrial Biotechnology - Technology Challenges and Opportunities. Chem Eng Prog. 2016;112:35–42. [Google Scholar]
- de Kok S, Stanton LH, Slaby T, Durot M, Holmes VF, Patel KG, Platt D, Shapland EB, Serber Z, Dean J, Newman JD, Chandran SS. Rapid and reliable DNA assembly via ligase cycling reaction. ACS Synth Biol. 2014;3:97–106. doi: 10.1021/sb4001992. [DOI] [PubMed] [Google Scholar]
- Delépine B, Libis V, Carbonell P, Faulon J-L. SensiPath: computer-aided design of sensing-enabling metabolic pathways. Nucleic Acids Res. 2016;44:W226–W231. doi: 10.1093/nar/gkw305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar V. Data Science and Prediction. Commun Acm. 2013;56:64–73. [Google Scholar]
- Dharmadi Y, Patel K, Shapland E, Hollis D, Slaby T, Klinkner N, Dean J, Chandran SS. High-throughput, cost-effective verification of structural DNA assembly. Nucleic Acids Res. 2014;42:e22. doi: 10.1093/nar/gkt1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez AA, Lim WA, Qi LS. Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat Rev Mol Cell Biol. 2016;17:5–15. doi: 10.1038/nrm.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorr M, Fibinger MPC, Last D, Schmidt S, Santos-Aberturas J, Bottcher D, Hummel A, Vickers C, Voss M, Bornscheuer UT. Fully automatized high-throughput enzyme library screening using a robotic platform. Biotechnol Bioeng. 2016;113:1421–1432. doi: 10.1002/bit.25925. [DOI] [PubMed] [Google Scholar]
- Ellis HM, Yu DG, DiTizio T, Court DL. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. P Natl Acad Sci USA. 2001;98:6742–6746. doi: 10.1073/pnas.121164898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis T, Adie T, Baldwin GS. DNA assembly for synthetic biology: from parts to pathways and beyond. Integr Biol. 2011;3:109–118. doi: 10.1039/c0ib00070a. [DOI] [PubMed] [Google Scholar]
- Engineers, T. A. S. o. M. History of Mechanical Engineering. The American Society of Mechanical Engineers. 2016 [Google Scholar]
- Engler C, Kandzia R, Marillonnet S. A one pot, one step, precision cloning method with high throughput capability. PLoS One. 2008;3:e3647. doi: 10.1371/journal.pone.0003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farasat I, Kushwaha M, Collens J, Easterbrook M, Guido M, Salis HM. Efficient search, mapping, and optimization of multi-protein genetic systems in diverse bacteria. Molecular Systems Biology. 2014;10 doi: 10.15252/msb.20134955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell DA. Metabolic control analysis: a survey of its theoretical and experimental development. Biochem J. 1992;286(Pt 2):313–30. doi: 10.1042/bj2860313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher L, Rosser S, Elfick A. Exploring Synthetic and Systems Biology at the University of Edinburgh. Biochemical Society Transactions. 2016;44:692–695. doi: 10.1042/BST20160006. [DOI] [PubMed] [Google Scholar]
- Fu J, Bian X, Hu S, Wang H, Huang F, Seibert PM, Plaza A, Xia L, Muller R, Stewart AF, Zhang Y. Full-length RecE enhances linear-linear homologous recombination and facilitates direct cloning for bioprospecting. Nat Biotechnol. 2012;30:440–6. doi: 10.1038/nbt.2183. [DOI] [PubMed] [Google Scholar]
- Galdzicki M, Clancy KP, Oberortner E, Pocock M, Quinn JY, Rodriguez CA, Roehner N, Wilson ML, Wilson Y, Adam L, Anderson JC, Bartley BA, Beal J, Chandran D, Ch D, Chen J, ran Densmore D, Endy D, Grünberg R, Hallinan J, Hillson NJ, Johnson JD, Kuchinsky A, Lux M, Misirli G, Peccoud J, Plahar HA, Sirin E, Stan G-B, Villalobos A, Wipat A, Gennari JH, Myers CJ, Sauro HM. The Synthetic Biology Open Language (SBOL) provides a community standard for communicating designs in synthetic biology. Nat Biotechnol. 2014;32:545–550. doi: 10.1038/nbt.2891. [DOI] [PubMed] [Google Scholar]
- Galdzicki M, Rodriguez C, Chandran D, Sauro HM, Gennari JH. Standard Biological Parts Knowledgebase. PLoS One. 2011;6 doi: 10.1371/journal.pone.0017005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Ruiz E, HamediRad M, Zhao H. Pathway Design, Engineering, and Optimization. Springer Berlin Heidelberg; Berlin, Heidelberg: 2016. pp. 1–40. [Google Scholar]
- George KW, Chen A, Jain A, Batth TS, Baidoo EEK, Wang G, Adams PD, Petzold CJ, Keasling JD, Lee TS. Correlation analysis of targeted proteins and metabolites to assess and engineer microbial isopentenol production. Biotechnol Bioeng. 2014;111:1648–1658. doi: 10.1002/bit.25226. [DOI] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Haber JE. Partners and pathways - repairing a double-strand break. Trends Genet. 2000;16:259–264. doi: 10.1016/s0168-9525(00)02022-9. [DOI] [PubMed] [Google Scholar]
- Han SW, Nakamura C, Kotobuki N, Obataya I, Ohgushi H, Nagamune T, Miyake J. High-efficiency DNA injection into a single human mesenchymal stem cell using a nanoneedle and atomic force microscopy. Nanomed-Nanotechnol. 2008;4:215–225. doi: 10.1016/j.nano.2008.03.005. [DOI] [PubMed] [Google Scholar]
- Hatzimanikatis V, Li C, Ionita JA, Henry CS, Jankowski MD, Broadbelt LJ. Exploring the diversity of complex metabolic networks. Bioinformatics. 2005;21:1603–1609. doi: 10.1093/bioinformatics/bti213. [DOI] [PubMed] [Google Scholar]
- Heckerman D. A tutorial on learning Bayesian networks. Technical Report MSR-TR-95-6. 1995:41. [Google Scholar]
- Heller MJ. DNA Microarray Technology: Devices, Systems, and Applications. Annual Review of Biomedical Engineering. 2002;4:129–153. doi: 10.1146/annurev.bioeng.4.020702.153438. [DOI] [PubMed] [Google Scholar]
- Henry CS, Broadbelt LJ, Hatzimanikatis V. Discovery and analysis of novel metabolic pathways for the biosynthesis of industrial chemicals: 3-hydroxypropanoate. Biotechnol Bioeng. 2010;106:462–473. doi: 10.1002/bit.22673. [DOI] [PubMed] [Google Scholar]
- Hillson NJ, Rosengarten RD, Keasling JD. j5 DNA assembly design automation software. ACS Synth Biol. 2012;1:14–21. doi: 10.1021/sb2000116. [DOI] [PubMed] [Google Scholar]
- Hoover DM, Lubkowski J. DNAWorks: an automated method for designing oligonucleotides for PCR-based gene synthesis. Nucleic Acids Res. 2002;30:e43. doi: 10.1093/nar/30.10.e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iverson SV, Haddock TL, Beal J, Densmore DM. CIDAR MoClo: Improved MoClo Assembly Standard and New E. coli Part Library Enable Rapid Combinatorial Design for Synthetic and Traditional Biology. ACS Synth Biol. 2016;5:99–103. doi: 10.1021/acssynbio.5b00124. [DOI] [PubMed] [Google Scholar]
- Jiang W, Zhao X, Gabrieli T, Lou C, Ebenstein Y, Zhu TF. Cas9-Assisted Targeting of CHromosome segments CATCH enables one-step targeted cloning of large gene clusters. Nat Commun. 2015;6:8101. doi: 10.1038/ncomms9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kacser H, Burns JA. The control of flux. Biochemical Society Transactions. 1995;23:341–66. doi: 10.1042/bst0230341. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Kawashima S, Nakaya A. The KEGG databases at GenomeNet. Nucleic Acids Res. 2002;30:42–6. doi: 10.1093/nar/30.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Z, Wang XR, Li YK, Wang Q, Qi QS. Small RNA RyhB as a potential tool used for metabolic engineering in Escherichia coli. Biotechnol Lett. 2012;34:527–531. doi: 10.1007/s10529-011-0794-2. [DOI] [PubMed] [Google Scholar]
- Kanigowska P, Shen Y, Zheng YJ, Rosser S, Cai YZ. Smart DNA Fabrication Using Sound Waves: Applying Acoustic Dispensing Technologies to Synthetic Biology. Jala-J Lab Autom. 2016;21:49–56. doi: 10.1177/2211068215593754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke W, Chang S, Chen X, Luo S, Jiang S, Yang P, Wu X, Zheng Z. Metabolic control analysis of L-lactate synthesis pathway in Rhizopus oryzae As 3.2686. Bioprocess and biosystems engineering. 2015;38:2189–2199. doi: 10.1007/s00449-015-1458-8. [DOI] [PubMed] [Google Scholar]
- Kelwick R, MacDonald JT, Webb AJ, Freemont P. Developments in the tools and methodologies of synthetic biology. Frontiers in bioengineering and biotechnology. 2014;2:60–60. doi: 10.3389/fbioe.2014.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B, Du J, Eriksen DT, Zhao H. Combinatorial design of a highly efficient xylose-utilizing pathway in Saccharomyces cerevisiae for the production of cellulosic biofuels. Appl Environ Microbiol. 2013;79:931–41. doi: 10.1128/AEM.02736-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Reed JL, Maravelias CT. Large-Scale Bi-Level strain design approaches and Mixed-Integer programming solution techniques. PLoS One. 2011;6 doi: 10.1371/journal.pone.0024162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. P Natl Acad Sci USA. 1996;93:1156–60. doi: 10.1073/pnas.93.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RD, Rowland J, Oliver SG, Young M, Aubrey W, Byrne E, Liakata M, Markham M, Pir P, Soldatova LN, Sparkes A, Whelan KE, Clare A. The automation of science. Science. 2009;324:85–9. doi: 10.1126/science.1165620. [DOI] [PubMed] [Google Scholar]
- King RD, Whelan KE, Jones FM, Reiser PGK, Bryant CH, Muggleton SH, Kell DB, Oliver SG. Functional genomic hypothesis generation and experimentation by a robot scientist. Nature. 2004;427:247–252. doi: 10.1038/nature02236. [DOI] [PubMed] [Google Scholar]
- Kosuri S, Church GM. Large-scale de novo DNA synthesis: technologies and applications. Nat Methods. 2014;11:499–507. doi: 10.1038/nmeth.2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ME, Aswani A, Han AS, Tomlin CJ, Dueber JE. Expression-level optimization of a multi-enzyme pathway in the absence of a high-throughput assay. Nucleic Acids Res. 2013;41:10668–78. doi: 10.1093/nar/gkt809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MZ, Elledge SJ. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat Methods. 2007;4:251–256. doi: 10.1038/nmeth1010. [DOI] [PubMed] [Google Scholar]
- Linshiz G, Jensen E, Stawski N, Bi C, Elsbree N, Jiao H, Kim J, Mathies R, Keasling JD, Hillson NJ. End-to-end automated microfluidic platform for synthetic biology: from design to functional analysis. Journal of Biological Engineering. 2016;10:15. doi: 10.1186/s13036-016-0024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long MR, Ong WK, Reed JL. Computational methods in metabolic engineering for strain design. Curr Opin Biotechnol. 2015;34:135–141. doi: 10.1016/j.copbio.2014.12.019. [DOI] [PubMed] [Google Scholar]
- Luo Y, Zhang L, Barton KW, Zhao H. Systematic Identification of a Panel of Strong Constitutive Promoters from Streptomyces albus. ACS Synth Biol. 2015;4:1001–1010. doi: 10.1021/acssynbio.5b00016. [DOI] [PubMed] [Google Scholar]
- Mali P, Yang LH, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-Guided Human Genome Engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Favera R, Califano A. ARACNE: An Algorithm for the Reconstruction of Gene Regulatory Networks in a Mammalian Cellular Context. BMC Bioinformatics. 2006;7:S7. doi: 10.1186/1471-2105-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema MH, Blin K, Cimermancic P, De Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R. AntiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011;39:339–346. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema MH, van Raaphorst R, Takano E, Breitling R. Computational tools for the synthetic design of biochemical pathways. Nature reviews Microbiology. 2012;10:191–202. doi: 10.1038/nrmicro2717. [DOI] [PubMed] [Google Scholar]
- Meldrum D. Automation for genomics, part one: preparation for sequencing. Genome Res. 2000;10:1081–92. doi: 10.1101/gr.101400. [DOI] [PubMed] [Google Scholar]
- Metzker ML. Sequencing technologies - the next generation. Nature reviews Genetics. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, Dulay GP, Hua KL, Ankoudinova I, Cost GJ, Urnov FD, Zhang HS, Holmes MC, Zhang L, Gregory PD, Rebar EJ. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol. 2011;29:143–8. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- Na D, Yoo SM, Chung H, Park H, Park JH, Lee SY. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat Biotechnol. 2013;31:170–174. doi: 10.1038/nbt.2461. [DOI] [PubMed] [Google Scholar]
- Nielsen AAK, Der BS, Shin J, Vaidyanathan P, Paralanov V, Strychalski EA, Ross D, Densmore D, Voigt CA. Genetic circuit design automation. Science (New York, NY) 2016;352:aac7341–aac7341. doi: 10.1126/science.aac7341. [DOI] [PubMed] [Google Scholar]
- Nikolaev EV. The elucidation of metabolic pathways and their improvements using stable optimization of large-scale kinetic models of cellular systems. Metabolic Engineering. 2010;12:26–38. doi: 10.1016/j.ymben.2009.08.010. [DOI] [PubMed] [Google Scholar]
- Nolan RP, Lee K. Dynamic model of CHO cell metabolism. Metabolic Engineering. 2011;13:108–124. doi: 10.1016/j.ymben.2010.09.003. [DOI] [PubMed] [Google Scholar]
- Nolan RP, Lee K. Dynamic model for CHO cell engineering. Journal of Biotechnology. 2012;158:24–33. doi: 10.1016/j.jbiotec.2012.01.009. [DOI] [PubMed] [Google Scholar]
- Obataya I, Nakamura C, Han S, Nakamura N, Miyake J. Nanoscale operation of a living cell using an atomic force microscope with a nanoneedle. Nano Lett. 2005;5:27–30. doi: 10.1021/nl0485399. [DOI] [PubMed] [Google Scholar]
- Okuda S, Yamada T, Hamajima M, Itoh M, Katayama T, Bork P, Goto S, Kanehisa M. KEGG Atlas mapping for global analysis of metabolic pathways. Nucleic Acids Res. 2008;36:W423–6. doi: 10.1093/nar/gkn282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olofsson J, Nolkrantz K, Ryttsen F, Lambie BA, Weber SG, Orwar O. Single-cell electroporation. Curr Opin Biotechnol. 2003;14:29–34. doi: 10.1016/s0958-1669(02)00003-4. [DOI] [PubMed] [Google Scholar]
- Patrick WG, Nielsen AAK, Keating SJ, Levy TJ, Wang W, Rivera JJ, Mondragn-Palomino O, Carr PA, Voigt CA, Oxman N, Kong DS. DNA Assembly in 3D Printed Fluidics. PLoS One. 2015;10 doi: 10.1371/journal.pone.0143636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrone J. DNA writers attract investors. Nat Biotechnol. 2016;34:363–4. doi: 10.1038/nbt0416-363. [DOI] [PubMed] [Google Scholar]
- Pharkya P, Burgard AP, Maranas CD. OptStrain : A computational framework for redesign of microbial production systems OptStrain : A computational framework for redesign of microbial production systems. Genome Res. 2004:2367–2376. doi: 10.1101/gr.2872004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pharkya P, Maranas CD. An optimization framework for identifying reaction activation/inhibition or elimination candidates for overproduction in microbial systems. Metabolic Engineering. 2006;8:1–13. doi: 10.1016/j.ymben.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Ra LF, P C, A C, M D, D F, Aj J, Njw R, Cj R, N S, M V, A W, C Y, P B, R B, Gg C, Jl F, C G, R G, Db K, J M, Ns S, P S, E T, Nj T. SYNBIOCHEM Synthetic Biology Research Centre, Manchester - A UK foundry for fine and speciality chemicals production. Synthetic and Systems Biotechnology. 2016 doi: 10.1016/j.synbio.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raterink RJ, Lindenburg PW, Vreeken RJ, Ramautar R, Hankemeier T. Recent developments in sample-pretreatment techniques for mass spectrometry-based metabolomics. TrAC - Trends in Analytical Chemistry. 2014;61:157–167. [Google Scholar]
- Redden H, Alper HS. The development and characterization of synthetic minimal yeast promoters. Nat Commun. 2015;6:7810. doi: 10.1038/ncomms8810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redding-Johanson AM, Batth TS, Chan R, Krupa R, Szmidt HL, Adams PD, Keasling JD, Soon Lee T, Mukhopadhyay A, Petzold CJ. Targeted proteomics for metabolic pathway optimization: Application to terpene production. Metabolic Engineering. 2011;13:194–203. doi: 10.1016/j.ymben.2010.12.005. [DOI] [PubMed] [Google Scholar]
- Richardson SM, Nunley PW, Yarrington RM, Boeke JD, Bader JS. GeneDesign 3.0 is an updated synthetic biology toolkit. Nucleic Acids Res. 2010;38:2603–2606. doi: 10.1093/nar/gkq143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson SM, Wheelan SJ, Yarrington RM, Boeke JD. GeneDesign : Rapid, automated design of multikilobase synthetic genes GeneDesign : Rapid, automated design of multikilobase synthetic genes. Genome Res. 2006:550–556. doi: 10.1101/gr.4431306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigo G, Carrera J, Prather KJ, Jaramillo A. DESHARKY: Automatic design of metabolic pathways for optimal cell growth. Bioinformatics. 2008;24:2554–2556. doi: 10.1093/bioinformatics/btn471. [DOI] [PubMed] [Google Scholar]
- Roehner N, Oberortner E, Pocock M, Beal J, Clancy K, Madsen C, Misirli G, Wipat A, Sauro H, Myers CJ. Proposed data model for the next version of the synthetic biology open language. ACS Synth Biol. 2015;4:57–71. doi: 10.1021/sb500176h. [DOI] [PubMed] [Google Scholar]
- Roehner N, Young EM, Voigt CA, Gordon DB, Densmore D. Double Dutch: A Tool for Designing Combinatorial Libraries of Biological Systems. ACS Synth Biol. 2016;5:507–517. doi: 10.1021/acssynbio.5b00232. [DOI] [PubMed] [Google Scholar]
- Sainz De Murieta IA, Bultelle M, Kitney RI. Toward the First Data Acquisition Standard in Synthetic Biology. ACS Synth Biol. 2016;5:817–826. doi: 10.1021/acssynbio.5b00222. [DOI] [PubMed] [Google Scholar]
- Salis HM, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol. 2009;27:946–50. doi: 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer B. Functional expression of the cre-lox site-specific recombination system in the yeast Saccharomyces cerevisiae. Mol Cell Biol. 1987;7:2087–96. doi: 10.1128/mcb.7.6.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenberger J, Que R, Fleming RMT, Thiele I, Orth JD, Feist AM, Zielinski DC, Bordbar A, Lewis NE, Rahmanian S, Kang J, Hyduke DR, Palsson BØ. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat Protoc. 2011;6:1290–1307. doi: 10.1038/nprot.2011.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settles B. Active Learning. Synthesis Lectures on Artificial Intelligence and Machine Learning. 2012;6:1–114. [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapland EB, Holmes V, Reeves CD, Sorokin E, Durot M, Platt D, Allen C, Dean J, Serber Z, Newman J, Chandran S. Low-Cost, High-Throughput Sequencing of DNA Assemblies Using a Highly Multiplexed Nextera Process. ACS Synth Biol. 2015;4:860–866. doi: 10.1021/sb500362n. [DOI] [PubMed] [Google Scholar]
- Sharma CM, Vogel J. Differential RNA-seq: The approach behind and the biological insight gained. Current Opinion in Microbiology. 2014;19:97–105. doi: 10.1016/j.mib.2014.06.010. [DOI] [PubMed] [Google Scholar]
- Shi SB, Liang YY, Zhang MZM, Ang EL, Zhao HM. A highly efficient single-step, markerless strategy for multi-copy chromosomal integration of large biochemical pathways in Saccharomyces cerevisiae. Metabolic Engineering. 2016;33:19–27. doi: 10.1016/j.ymben.2015.10.011. [DOI] [PubMed] [Google Scholar]
- Shih SC, Goyal G, Kim PW, Koutsoubelis N, Keasling JD, Adams PD, Hillson NJ, Singh AK. A Versatile Microfluidic Device for Automating Synthetic Biology. ACS Synth Biol. 2015;4:1151–64. doi: 10.1021/acssynbio.5b00062. [DOI] [PubMed] [Google Scholar]
- Shin JH, Kim HU, Kim DI, Lee SY. Production of bulk chemicals via novel metabolic pathways in microorganisms. Biotechnology Advances. 2013;31:925–935. doi: 10.1016/j.biotechadv.2012.12.008. [DOI] [PubMed] [Google Scholar]
- Si T, Chao R, Min Y, Wu Y, Ren W, Zhao H. Automated multiplex genome-scale engineering in yeast. Nat Commun. 2017;8:15187. doi: 10.1038/ncomms15187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si T, HamediRad M, Zhao H. Regulatory RNA-assisted genome engineering in microorganisms. Curr Opin Biotechnol. 2015a;36:85–90. doi: 10.1016/j.copbio.2015.08.003. [DOI] [PubMed] [Google Scholar]
- Si T, Luo Y, Bao Z, Zhao H. RNAi-assisted genome evolution in Saccharomyces cerevisiae for complex phenotype engineering. ACS Synth Biol. 2015b;4:283–91. doi: 10.1021/sb500074a. [DOI] [PubMed] [Google Scholar]
- Smanski MJ, Bhatia S, Zhao D, Park Y, B A, Woodruff L, Giannoukos G, Ciulla D, Busby M, Calderon J, Nicol R, Gordon DB, Densmore D, Voigt CA. Functional optimization of gene clusters by combinatorial design and assembly. Nat Biotechnol. 2014;32:1241–1249. doi: 10.1038/nbt.3063. [DOI] [PubMed] [Google Scholar]
- Somvanshi PR, Venkatesh KV. Metabolic Control Analysis. Encyclopedia of Systems Biology. 2013:1229–1233. [Google Scholar]
- Stahl ML, Ferrari E. Replacement of the Bacillus subtilis subtilisin structural gene with an In vitro-derived deletion mutation. J Bacteriol. 1984;158:411–8. doi: 10.1128/jb.158.2.411-418.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper N, Shlomi T. Predicting metabolic engineering knockout strategies for chemical production: Accounting for competing pathways. Bioinformatics. 2009;26:536–543. doi: 10.1093/bioinformatics/btp704. [DOI] [PubMed] [Google Scholar]
- Tsai CS, Kong II, Lesmana A, Million G, Zhang GC, Kim SR, Jin YS. Rapid and marker-free refactoring of xylose-fermenting yeast strains with Cas9/CRISPR. Biotechnol Bioeng. 2015;112:2406–2411. doi: 10.1002/bit.25632. [DOI] [PubMed] [Google Scholar]
- Villalobos A, Ness JE, Gustafsson C, Minshull J, Govindarajan S. Gene Designer: a synthetic biology tool for constructing artificial DNA segments. BMC bioinformatics. 2006;7:285. doi: 10.1186/1471-2105-7-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villaverde AF, Bongard S, Mauch K, Balsa-Canto E, Banga JR. Metabolic engineering with multi-objective optimization of kinetic models. Journal of Biotechnology. 2016;222:1–8. doi: 10.1016/j.jbiotec.2016.01.005. [DOI] [PubMed] [Google Scholar]
- Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009;460:894–8. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SN, Lee LJ. Micro-/nanofluidics based cell electroporation. Biomicrofluidics. 2013;7 doi: 10.1063/1.4774071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber E, Engler C, Gruetzner R, Werner S, Marillonnet S. A modular cloning system for standardized assembly of multigene constructs. PLoS One. 2011;6:e16765. doi: 10.1371/journal.pone.0016765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, Lee SY, Fischbach MA, M??ller R, Wohlleben W, Breitling R, Takano E, Medema MH. AntiSMASH 3.0-A comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43:W237–W243. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff LBA, Gorochowski TE, Roehner N, Mikkelsen TS, Densmore D, Gordon DB, Nicol R, Voigt CA. Registry in a tube: multiplexed pools of retrievable parts for genetic design space exploration. Nucleic Acids Res. 2016;45:gkw1226–gkw1226. doi: 10.1093/nar/gkw1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff LBA, Gorochowski TE, Roehner N, Mikkelsen TS, Densmore D, Gordon DB, Nicol R, Voigt CA. Registry in a tube: multiplexed pools of retrievable parts for genetic design space exploration (vol 45, pg 1567, 2017) Nucleic Acids Res. 2017;45:1567–1568. doi: 10.1093/nar/gkx032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Rizzoni EA, Sul SY, Stephanopoulos G. Improving Metabolic Pathway Efficiency by Statistical Model-Based Multivariate Regulatory Metabolic Engineering. ACS Synth Biol. 2016 doi: 10.1021/acssynbio.6b00187. [DOI] [PubMed] [Google Scholar]
- Xu Z, Zheng P, Sun J, Ma Y. ReacKnock: Identifying reaction deletion strategies for microbial strain optimization based on genome-scale metabolic network. PLoS One. 2013;8 doi: 10.1371/journal.pone.0072150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanghellini A. Synthetic Biology for Chemical Production. Chem Eng-New York. 2016;123:44–49. [Google Scholar]
- Zhang Y, Muyrers JP, Testa G, Stewart AF. DNA cloning by homologous recombination in Escherichia coli. Nat Biotechnol. 2000;18:1314–7. doi: 10.1038/82449. [DOI] [PubMed] [Google Scholar]
- Zhou H, Vonk B, Roubos JA, Bovenberg RAL, Voigt CA. Algorithmic co-optimization of genetic constructs and growth conditions: Application to 6-ACA, a potential nylon-6 precursor. Nucleic Acids Res. 2015;43:10560–10570. doi: 10.1093/nar/gkv1071. [DOI] [PMC free article] [PubMed] [Google Scholar]