

ABSTRACT
The endosomal sorting complex required for transport (ESCRT) machinery is necessary for budding of many enveloped viruses. Recently, it was demonstrated that Vps4, the key regulator for recycling of the ESCRT-III complex, is required for efficient infection by the baculovirus Autographa californica multiple nucleopolyhedrovirus (AcMNPV). However, ESCRT assembly, regulation, and function are complex, and little is known regarding the details of participation of specific ESCRT complexes in AcMNPV infection. In this study, the core components of ESCRT-I (Tsg101 and Vps28) and ESCRT-III (Vps2B, Vps20, Vps24, Snf7, Vps46, and Vps60) were cloned from Spodoptera frugiperda. Using a viral complementation system and RNA interference (RNAi) assays, we found that ESCRT-I and ESCRT-III complexes are required for efficient entry of AcMNPV into insect cells. In cells knocking down or overexpressing dominant negative (DN) forms of the components of ESCRT-I and ESCRT-III complexes, entering virions were partially trapped within the cytosol. To examine only egress, cells were transfected with the double-stranded RNA (dsRNA) targeting an individual ESCRT-I or ESCRT-III gene and viral bacmid DNA or viral bacmid DNA that expressed DN forms of ESCRT-I and ESCRT-III components. We found that ESCRT-III components (but not ESCRT-I components) are required for efficient nuclear egress of progeny nucleocapsids. In addition, we found that several baculovirus core or conserved proteins (Ac11, Ac76, Ac78, GP41, Ac93, Ac103, Ac142, and Ac146) interact with Vps4 and components of ESCRT-III. We propose that these viral proteins may form an “egress complex” that is involved in recruiting ESCRT-III components to a virus egress domain on the nuclear membrane.
IMPORTANCE The ESCRT system is hijacked by many enveloped viruses to mediate budding and release. Recently, it was found that Vps4, the key regulator of the cellular ESCRT machinery, is necessary for efficient entry and egress of Autographa californica multiple nucleopolyhedrovirus (AcMNPV). However, little is known about the roles of specific ESCRT complexes in AcMNPV infection. In this study, we demonstrated that ESCRT-I and ESCRT-III complexes are required for efficient entry of AcMNPV into insect cells. The components of ESCRT-III (but not ESCRT-I) are also necessary for efficient nuclear egress of progeny nucleocapsids. Several baculovirus core or conserved proteins were found to interact with Vps4 and components of ESCRT-III, and these interactions may suggest the formation of an “egress complex” involved in the nuclear release or transport of viral nucleocapsids.
KEYWORDS: ESCRT-I, ESCRT-III, baculovirus, AcMNPV, virus entry and egress
INTRODUCTION
The endosomal sorting complex required for transport (ESCRT) comprises five distinct protein complexes designated ESCRT-0, I, II, III, and the AAA ATPase Vps4, as well as some ESCRT-associated proteins, such as Alix (1, 2). ESCRT-0 is required for selectively sorting ubiquitinated membrane proteins and recruiting ESCRT-I. ESCRT-I, a heterotetramer complex composed of Vps23 (Tsg101), Vps28, Vps37, and MVB12/UBP1, in turn recruits the heterotetramer ESCRT-II complex. ESCRT-II is comprised of Vps22, Vps36, and two molecules of Vps25. ESCRT-I/-II interact with ubiquitinated cargo proteins and membrane phospholipids, and this larger complex is involved in generating membrane curvature and creating membrane buds. Within the membrane bud neck, ESCRT-I/-II and Alix recruit ESCRT-III and promote the formation of ESCRT-III polymers that result in filament or ring formation. It is believed that constriction of this ring results in scission of the newly budded vesicle (3, 4). ESCRT-III is a dynamic polymer complex, and its components are conserved from Archaea to mammalians. In Saccharomyces cerevisiae and humans, ESCRT-III contains a set of closely related proteins, including Vps2 (two isoforms in humans termed charged multivesicular body protein 2A [CHMP2A] and CHMP2B), Vps20 (CHMP6), Vps24 (CHMP3), Vps32/Snf7 (CHMP4A, CHMP4B, CHMP4C), Vps46 (CHMP1A, CHMP1B), Vps60 (CHMP5), and IST1. Among these components, Vps2, Vps20, Vps24, and Snf7 serve as the “core” proteins to build the ESCRT-III helical filaments (5). Following ESCRT-III-mediated membrane scission, the ESCRT-III complex is disassembled by Vps4 in an ATP-dependent manner (1, 4, 6, 7). The activity of Vps4 is regulated by its cofactor Vta1 (8). Initially, the ESCRT system was identified as an essential membrane-remodeling and scission machinery for sorting ubiquitinated membrane proteins into the intraluminal vesicles (ILVs) of multivesicular bodies (MVBs) (9). Components of the ESCRT pathway are also involved in a variety of other biological processes, including the abscission stage of cytokinesis, biogenesis of exosomes, plasma membrane wound repair, neuron pruning, extraction of defective nuclear pore complexes, nuclear envelope re-formation, and budding of virus particles (2, 10–13).
It was previously discovered that many enveloped viruses hijack components of the ESCRT pathway to mediate virus budding and release from infected cells (12). The detailed mechanism of ESCRT-mediated virus budding has been examined extensively in retroviruses, particularly HIV-1. Retroviral Gag proteins contain late assembly domains (L-domains) with consensus sequences such as PPXY, P(T/S)AP, and YPXnL. These L-domains mediate interactions of Gag with cellular proteins such as NEDD4-like ubiquitin ligases, ESCRT-I component Tsg101, and Alix. Through specific protein-protein interactions, Gag proteins bind and recruit ESCRT-I and/or Alix, which in turn recruits and directs the localization of ESCRT-III and Vps4 to regions of the plasma membrane where virion budding occurs (12, 14–16). Involvement of the ESCRT pathway in nonenveloped virus release was also observed for bluetongue virus and hepatitis A virus (17, 18). In addition to their importance in viral egress, components of the ESCRT system were also found to be required for the entry of some enveloped viruses, including Kaposi's sarcoma-associated herpesvirus (KSHV), Crimean-Congo hemorrhagic fever virus (CCHFV), vesicular stomatitis virus (VSV), Autographa californica multiple nucleopolyhedrovirus (AcMNPV), and the nonenveloped rhesus rotavirus (RRV) (19–23). Most recently, ESCRT-I/-III have been shown to function in the formation of a viral replication compartment during infection by certain positive-strand RNA viruses of plants (24).
AcMNPV is the most intensively studied baculovirus and is the type species of the virus family Baculoviridae (25). Baculoviruses are enveloped, insect-specific double-stranded DNA (dsDNA) viruses that replicate in the nuclei of infected cells. During the infection cycle, baculoviruses produce two phenotypes of enveloped virions: occlusion-derived virions (ODV) and budded virions (BV). ODV and BV appear to share identical nucleocapsids and genome content but differ in the source and composition of their envelopes and in their roles in virus infection (25). ODV initiate infection of insect midgut epithelial cells upon oral ingestion of occlusion bodies (OBs) and are responsible for spreading viral infection horizontally among insects. The nucleocapsids of ODV are enveloped in the nucleus by membranes derived from intranuclear microvesicles, which are derived from the inner nuclear membrane (26, 27). The BV transmit infection from cell to cell within and between insect tissues, and BV are highly infectious in cultured cell lines. The envelopes of BV are acquired from the plasma membrane during virion budding and release (25). Budded virions of AcMNPV enter cells via clathrin-mediated endocytosis (28). During the entry process by BV, the major viral envelope glycoprotein GP64 mediates receptor binding and low-pH-triggered membrane fusion (29, 30). After release into the cytoplasm, nucleocapsids nucleate the formation of actin filaments as a propulsion mechanism and are eventually delivered into the nucleus through nuclear pores (31, 32). In the nucleus, viral early gene transcription is followed by DNA replication and late gene transcription. At a relatively early stage of infection, progeny nucleocapsids are transported from the nucleus to the plasma membrane by a mechanism that is largely unknown. Then, the nucleocapsids bud and are pinched off from the plasma membrane to form BV (25). The cell surface-localized GP64 is also important for virion budding (33). At a late stage of infection, most of the assembled nucleocapsids appear to be retained within the nucleus to form virions of the ODV phenotype (25). In recent years, numerous gene knockout studies have reported that certain baculovirus core genes (such as those encoding Ac76, Ac78, Ac93, Ac103, Ac142, and Ac146) are required for production of infectious AcMNPV BV. However, it is not clear how these viral proteins are involved in virus infection (34–39).
Recently, we found that the ESCRT pathway is conserved in sequenced insect genomes and that the expression levels of certain components of ESCRT-I, -II, -III, Vps4, and Alix were significantly upregulated upon AcMNPV infection (40, 41). In addition, prior studies revealed that efficient entry and egress of AcMNPV BV are dependent on functional Vps4 (23). Since Vps4 is required for recycling of ESCRT III and represents a terminal step in the ESCRT pathway, this suggests that other components of the ESCRT pathway may be specifically involved in entry and egress. To investigate the potential roles of other ESCRT components in efficient production of infectious BV of AcMNPV, we cloned ESCRT-I (Vps23 [Tsg101] and Vps28) and ESCRT-III (Vps2B, Vps20, Vps24, Vps32/Snf7, Vps46, and Vps60) cDNAs and then knocked down or generated and expressed dominant negative (DN) forms of these proteins in insect cells. We found that ESCRT-I and ESCRT-III were both required for efficient entry of AcMNPV, whereas ESCRT-III was also involved in egress of AcMNPV BV. We also identified interactions of certain ESCRT pathway proteins with viral core proteins that are required for infectious BV production. We propose that these viral proteins form a complex to recruit ESCRT-III/Vps4 to virion-budding and -releasing regions at the nuclear membrane.
RESULTS
Isolation of ESCRT-I and ESCRT-III components from Sf9 cells.
Using the Spodoptera frugiperda expressed sequence tag (EST) sequence database (SpodoBase) to identify homologs of yeast ESCRT-I and ESCRT-III components, we identified full-length cDNA sequences for Vps2B, Vps20, Vps28, Vps46, Vps60, and Snf7. We also identified 5′ and 3′ ends of partial sequences for Vps23 (Tsg101 in mammalians) and only a 5′ end sequence for Vps24. To confirm and subclone cDNAs of these ESCRT components from Spodoptera frugiperda Sf9 cells, we initially designed gene-specific primers targeted to the 5′ upstream and 3′ downstream untranslated sequences. For Vps24, the sequence of the 3′ end of the gene was obtained by 3′ rapid amplification of cDNA ends (RACE). We then designed gene-specific primers containing unique restriction enzyme sites to amplify the open reading frame (ORF) for each component of ESCRT-I and ESCRT-III complexes (listed above). Nucleotide sequence analysis revealed several nucleotide sequence errors in the SpodoBase sequences, resulting in frameshift or same-sense mutations within the original EST sequences. cDNA sequences for these mRNAs were deposited into GenBank.
Amino acid sequence alignments revealed that the components of ESCRT-I and ESCRT-III from Sf9 cells have a high level of identity to their homologs from other insects, yeast, and humans (see Fig. S1 in the supplemental material). In mammalian cells, there are multiple isoforms of ESCRT-III components, including CHMP1 (CHMP1A and -B; homologs of yeast Vps46), CHMP2 (CHMP2A and -B; homologs of yeast Vps2), and CHMP4 (CHMP4A, -B, and -C; homologs of yeast Snf7) (isoforms are in parentheses). In insects, gene expansion was observed for Vps2 (Vps2A and -B) only in lepidopteran and a few other species. Most of the sequenced insect genomes contain only a single ortholog of Vps2 (40). Here, we identified a single ortholog of Vps2 from Sf9 cells, and that ortholog is most similar to human CHMP2B and to Vps2B from other insects. Thus, we designated this single identified Sf9 Vps2 gene Vps2B. Additionally, the N-terminal ubiquitin-enzyme variant (UEV) domain, middle region proline-rich region (PRD) and coiled coil (CC), and the C-terminal steadiness box (SB) of Tsg101/Vps23 (Fig. S1, Tsg101/Vps23) and the N-terminal core region and the C-terminal four-helix bundle domain (CTD) of Vps28 (Fig. S1, Vps28) are highly conserved among insects, yeast, and humans. All the isolated ESCRT-III components of Sf9 contain a predicted Snf7 domain of about 170 amino acids (Fig. S1).
Expression of wild-type and DN ESCRT-I and ESCRT-III components.
It was previously shown that overexpression of Tsg101 lacking one of its subdomains (UEV, PRD, CC, or SB) results in DN inhibition of HIV-1 budding (42–44). Also, mutations within the CTD of Vps28 (that disrupt the interaction of Vps28 and ESCRT-III component Vps20) result in lower-efficiency budding by equine infectious anemia virus (EIAV) Gag (45). To generate the predicted DN forms of Sf9 ESCRT-I components Tsg101 and Vps28, we generated constructs of Sf9 Tsg101 (Fig. 1A) consisting of residues 1 to 160 (UEV, UEV domain), 150 to 403 (dUEV, deletion of UEV domain), 250 to 403 (CC-SB, deletion of UEV and PRD domains), 330 to 403 (SB, SB domain), and a construct called Core, which lacks the C-terminal domain (CTD) of Vps28. Wild-type and modified forms of Tsg101 and Vps28 were N-terminally tagged with green fluorescent protein (GFP). DN forms of Sf9 ESCRT-III components were generated by fusing a GFP tag to the C terminus of each. As described previously, the C terminus of ESCRT-III components serves as the autoinhibitory element that interacts with the N-terminal portion and maintains ESCRT-III components as monomers in the cytoplasm. Release of the inhibitory effect of the C terminus is required for ESCRT-III assembly (46–48). In yeast and mammalian cells, fusion of a bulky tag such as GFP to the C terminus of ESCRT-III components interferes with the autoinhibition and results in the DN phenotype (49, 50). Each fusion construct was transiently expressed in Sf9 cells under the control of an AcMNPV ie1 promoter and detected by Western blotting with an anti-GFP antibody and by epifluorescence microscopy (Fig. 1B and C and 2B and C).
FIG 1.

Construction and transient expression of GFP-tagged wild-type or truncated forms of ESCRT-I components Tsg101 and Vps28. (A) Schematic representation of the domain organization of WT Tsg101 and Vps28 and truncated forms of each. Numbers on the right denote the amino acid sequence length of each construct. Abbreviations: CC, coiled coil; CTD, C-terminal four-helix bundle domain; dUEV, deletion of UEV domain; PRD, proline-rich domain; SB, steadiness box; UEV, ubiquitin-enzyme variant domain. (B, C) Expression of GFP-tagged WT or truncated forms of Tsg101 and Vps28 in transfected Sf9 cells. (B) The expression of GFP-tagged Tsg101 and Vps28 constructs was analyzed by Western blotting using a GFP-specific polyclonal antibody; gels were spliced for labeling purposes. (C) The cellular distribution of GFP-tagged Tsg101 and Vps28 constructs was visualized by epifluorescence microscopy (Epi, left panels) and confocal microscopy (Confocal, right panels). Phase-contrast images on the left side illustrate the presence of vesicles induced by Vps28 construct Core, which lacks the CTD domain. (D) Colocalization of GFP-tagged Tsg101 and Vps28 constructs with mCherry-tagged Vps4 mutant E231Q in cotransfected Sf9 cells. Cell boundaries are traced with circular dashed lines. Bar, 10 μm.
FIG 2.

Transient expression of GFP-tagged ESCRT-III components in Sf9 cells. (A) Schematic representation of the ESCRT-III components cloned from Sf9 cells. The predicted Snf7 domain of each component is shown as a black box, and the start and end amino acids of Snf7 domains in individual components are indicated. The amino acid sequence length for each protein is indicated on the right. (B, C) Expression of GFP-tagged ESCRT-III proteins in transfected Sf9 cells. (B) The expression of GFP-tagged ESCRT-III proteins was analyzed by Western blotting using a GFP-specific polyclonal antibody; gels were spliced for labeling purposes. (C) The cellular distribution of GFP-tagged ESCRT-III proteins was visualized by epifluorescence microscopy (Epi, left panels) and confocal microscopy (Confocal, right panels). The presence of vesicles induced by DN ESCRT-III constructs can be observed in phase-contrast images on the left. (D) Colocalization of GFP-tagged ESCRT-III proteins with mCherry-tagged Vps4 mutant E231Q in cotransfected Sf9 cells. Cell boundaries are traced with circular dashed lines. Bar, 10 μm.
(i) ESCRT-I components.
Transient expression of full-length Tsg101 resulted in a highly punctate, putatively endosomal pattern of expression (Fig. 1C, Tsg101). In contrast, expression of the N-terminal UEV domain or the C-terminal SB domain of Tsg101 resulted in GFP-tagged proteins that were distributed diffusely throughout the cytoplasm and nucleus, even though a small portion of the proteins from these modified constructs (especially SB) had a punctate distribution in the cytoplasm (Fig. 1A and C, UEV, SB). Transient expression of dUEV and CC-SB (both lacking the N-terminal UEV domain) resulted in the formation of large, apparently spherical structures in the cytoplasm. GFP-Vps28 exhibited a diffuse pattern of localization in both the nucleus and the cytoplasm (Fig. 1C, Vps28). Deletion of the CTD of Vps28 did not alter the distribution of Vps28. However, transient expression of the Vps28 without the CTD appears to induce the formation of vacuoles in Sf9 cells (Fig. 1C, Core, see phase image).
(ii) ESCRT-III components.
For ESCRT-III components (Fig. 2), Vps20-GFP was localized diffusely in the cytoplasm and induced a low occurrence of vacuoles (Fig. 2C, Vps20). In contrast, when the other ESCRT-III components were GFP tagged, they accumulated in punctate structures in the cytoplasm and a significantly high level of cytoplasmic vacuolation was observed (Fig. 2C, phase panels).
Previous studies indicated that overexpression of a DN form of Sf9 Vps4 (an ATP hydrolysis-defective form of Sf9 Vps4, E231Q-GFP) induced the formation of an aberrant endosomal compartment (23). To determine whether the compartment induced by Vps4 E231Q resembled that induced by overexpression of the wild-type or DN forms of ESCRT-I and ESCRT-III components, we cotransfected Sf9 cells with two plasmids, one expressing Vps4 E231Q-mCherry, and another expressing each construct of ESCRT-I and ESCRT-III, and we then examined the localization of the two proteins (DN Vps4 and ESCRT-I or -III constructs). Complete colocalization was observed between Vps4 E231Q-mCherry and GFP-tagged ESCRT-III components, Vps2B, Vps24, Snf7, Vps46, or Vps60, and the majority of the GFP-tagged Vps20 were also observed to be colocalized with E231Q-mCherry (Fig. 2D). In contrast, no colocalization was observed between Vps4 E231Q-mCherry and wild-type Tsg101 or the truncated forms of Tsg101: dUEV and CC-SB (Fig. 1D). Additionally, coexpression with Vps4 E231Q-mCherry did not change the localization pattern of modified forms of Tsg101 (UEV and SB), Vps28, and Vps28 core domain (Core) (compare Fig. 1C and D). These results suggested that overexpression of GFP-tagged ESCRT-III components resulted in the formation of the aberrant endosomal compartment similar to that induced upon Vps4 E231Q-mCherry expression or that coexpression of Vps4 E231Q-mCherry and components of ESCRT-III results in the retention of both of the proteins in the same aberrant endosomal compartment.
ESCRT-I and ESCRT-III components are required for production of infectious AcMNPV BV.
To first ask whether the components of ESCRT-I and ESCRT-III are required for production of AcMNPV BV, we initially used a viral complementation assay to examine viral replication in transfected-infected cells (Fig. 3A). Because all cells do not become transfected and express the constructs of ESCRT-I and ESCRT-III in transient-transfection assays, the complementation assay ensures that productive viral replication can occur only in cells that are productively transfected and express both the constructs of ESCRT-I or ESCRT-III and AcMNPV GP64 (which complements infection by a gp64 knockout virus and permits the production of infectious BV) (Fig. 3A and B). In this assay, Sf9 cells were initially cotransfected with two plasmids separately expressing the essential viral envelope protein GP64 and a GFP-tagged ESCRT-I or ESCRT-III protein. After a 16-h period of expression, the cells were infected with the gp64 knockout virus mCherryGUS-gp64ko that was produced in Sf9Op1D cells (the reporter genes mCherry and GUS are controlled by the Orgyia pseudotsugata multicapsid nuclear polyhedrosis virus [OpMNPV] ie2 immediate early promoter and AcMNPV p6.9 late promoter, respectively). Supernatants were harvested at 24 h postinfection (p.i.), and infectious AcMNPV BV titers were determined by 50% tissue culture infectious dose (TCID50) assays on Sf9OP1D cells that stably express OpMNPV GP64. Sf9 cells cotransfected with plasmids expressing GP64 and GFP served as a negative control, whereas cotransfection of cells with plasmids expressing the DN Vps4 construct, Vps4 E231Q-GFP, and GP64 (which result in dramatically reduced infectious virus titers) served as a positive control for inhibition of AcMNPV production (23).
FIG 3.

Transient expression of GFP-tagged ESCRT-I and ESCRT-III proteins significantly inhibits the production of infectious AcMNPV in a viral complementation assay. (A) Schematic representation of the viral complementation assay. (a). In cells transfected with a gp64 knockout AcMNPV bacmid, virus budding is defective. When the gp64 knockout bacmid DNA is transfected into Sf9Op1D cells that stably express OpMNPV GP64, virus budding and infectivity are complemented by OpMNPV GP64. (b) Sf9 cells are cotransfected with two plasmids separately expressing AcMNPV GP64 (pBieGP64) and GFP or a GFP-tagged ESCRT protein. At 16 h p.t., the cells are infected with a gp64 knockout AcMNPV virus that was produced in Sf9Op1D cells and containing the OpMNPV GP64 protein on its surface. Because all cells do not become transfected, the gp64 knockout AcMNPV can only bud and propagate in cells that are productively transfected, expressing both GFP or the GFP-tagged ESCRT protein and AcMNPV GP64, which complements the gp64 knockout. In nontransfected cells, the gp64 knockout AcMNPV virus can enter the cells, but budding of progeny virions is defective. (B, C) Sf9 cells were cotransfected with a plasmid expressing GP64 together with a plasmid encoding GFP-tagged ESCRT-I and ESCRT-III proteins, E231Q-GFP, or GFP. At 16 h p.t., the cells were infected with a gp64 knockout AcMNPV at an MOI of 1 or 5. At 24 h p.i., the titers of progeny viruses from cell culture medium were determined by TCID50 assay on a GP64-complementing cell line (Sf9OP1D). Error bars indicate the standard deviations from the means of results for triplicate samples. (D) The expressions of GP64 and GFP-tagged ESCRT-I and ESCRT-III proteins in cotransfected and infected cells were analyzed by Western blotting using anti-GP64 MAb (AcV5) and an anti-GFP polyclonal antibody; gels were spliced for labeling purposes. *, P < 0.005 (by unpaired t test).
The overexpression of full-length GFP-tagged ESCRT-I components Tsg101 and Vps28 significantly reduced the production of infectious AcMNPV, with reductions of approximately 85 to 90% (Fig. 3B). The reduction in BV titer was typically more severe in the presence of the truncated forms of Tsg101 and Vps28, and that effect was more clearly evident when infections were performed at a multiplicity of infection (MOI) of 1 (Fig. 3B). DN ESCRT-III components also caused a substantial inhibition in BV production, with the majority of ESCRT-III constructs reducing the infectious BV titers to <10% of the control (Fig. 3C). The most dramatic reduction in BV production (approximately 98%) was detected in cells expressing Vps24-GFP (Fig. 3C). Western blot analysis showed that GP64 and each of the ESCRT-I and ESCRT-III constructs were expressed at similar levels in transfected-infected cells (Fig. 3D), indicating that variations in transiently expressed proteins were not responsible for the observed effects. In addition, when parallel experiments were performed in another lepidopteran cell line (Trichoplusia ni High5 cells; data not shown), similar reductions in BV production were also observed.
To extend our observations, we used a double-stranded RNA (dsRNA)-based RNA interference (RNAi) approach to knock down the expression of individual components of ESCRT-I, ESCRT-III, or Vps4 and evaluated the effects on infectious AcMNPV production. Sf9 cells were mock transfected or transfected with dsRNA targeting the specific component of ESCRT-I, ESCRT-III, or Vps4 or a dsRNA targeting GFP. Knockdown efficiencies ranged from 71.5 to 94.3% (Fig. 4A), and transfection with the dsRNA of the ESCRT components or GFP caused no notable change in the viability of Sf9 cells at 24, 48, and 72 h posttransfection (h p.t.) (data not shown). Similarly, we observed that knockdown of individual components of ESCRT-I (Tsg101, Vps28), ESCRT-III (Vps2B, Vps20, Vps24, Snf7, Vps46, Vps60), or Vps4 resulted in a dramatic reduction of infectious AcMNPV production (Fig. 4B). Taken together, these results suggest that when cells are infected with BV, functional ESCRT-I and ESCRT-III complexes are required for production of infectious AcMNPV BV progeny.
FIG 4.

RNAi knockdown of ESCRT-I or ESCRT-III proteins inhibits production of infectious AcMNPV. (A) Sf9 cells were transfected with a plasmid expressing HA- or c-Myc-tagged ESCRT-I and ESCRT-III proteins or Vps4 or were cotransfected with a plasmid expressing individual HA- or c-Myc-tagged ESCRT protein and a dsRNA specific for an ESCRT gene or GFP. At 48 h p.t., the transfected cells were collected and expression of the HA- or c-Myc-tagged ESCRT protein was detected by Western blotting with an anti-HA monoclonal antibody or an anti-Myc polyclonal antibody. Actin was detected (using anti-β-actin) as a loading control. (B) Sf9 cells were mock transfected or transfected with the dsRNA specific for GFP or for an individual ESCRT gene. At 48 h p.t., the transfected cells were infected with control AcMNPV. At 24 h p.i., the cell culture supernatants were collected and virus titers were determined by TCID50. **, P < 0.0005 (by unpaired t test).
Overexpression of ESCRT-I and ESCRT-III components affects early stages of AcMNPV infection.
Since AcMNPV budded virions enter host cells by clathrin-mediated endocytosis (28), the inhibitory effect of GFP-tagged ESCRT-I and ESCRT-III proteins on AcMNPV infection could occur at an early stage of virus infection, by inhibiting virus entry or transport to the cell nucleus. To address this possibility, we used the virus complementation system described above but with a virus that expresses early and late phase reporter genes. Sf9 cells were first cotransfected with two plasmids expressing (i) GP64 and (ii) one of the ESCRT-I or ESCRT-III constructs or the control GFP or control Vps4 E231Q-GFP. At 16 h p.t., the cotransfected cells were infected with a gp64 knockout virus (LacZGUS-gp64ko), which contains the reporter genes encoding LacZ (beta-galactosidase) under an AcMNPV ie1 early promoter and GUS (beta-glucuronidase) under the AcMNPV p6.9 late promoter. We used these two reporter genes to monitor early and late events in the AcMNPV infection cycle. In comparison with the GFP control, we found that the expression of full-length or truncated forms of ESCRT-I proteins (Tsg101 and Vps28) or DN forms of ESCRT-III proteins significantly decreased the beta-Gal- and GUS-positive cells (data not shown). In the transfected-infected cell lysates, the activities of beta-Gal and GUS were generally suppressed by more than 35% to 60% by expressing these ESCRT-I and -III constructs (Fig. 5A to D). Both Vps2B-GFP and Snf7-GFP reduced the detection of the late reporter (GUS) to a level similar to that of the control Vps4 DN construct (E231Q-GFP) (Fig. 5D). To confirm these results, we further examined viral DNA replication in Sf9 cells cotransfected with the above-described plasmids and infected with the gp64 knockout virus (LacZGUS-gp64ko; MOI, 5). Viral genomic DNA was extracted at 24 h p.i. and analyzed by quantitative real-time PCR (qPCR). Consistent with the reporter gene expression results, viral genomic DNA was substantially reduced in cells expressing full-length or DN proteins of ESCRT-I and ESCRT-III (Fig. 5E and F). Overall, these data suggest that overexpression of GFP-tagged full-length or truncated forms of ESCRT-I and ESCRT-III proteins in Sf9 cells interfere with AcMNPV infection at an early stage such as virus entry or transport to the cell nucleus.
FIG 5.

Effects of overexpression of GFP-tagged ESCRT-I and ESCRT-III proteins on early stages of AcMNPV replication. Sf9 cells were cotransfected with two plasmids separately expressing (i) GP64 and (ii) one of the GFP-tagged ESCRT-I or ESCRT-III proteins, E231Q-GFP, or GFP. (A, B) At 16 h p.t., the cells were infected with a gp64 knockout virus, LacZGUS-gp64ko (MOI = 5). At 6 h p.i., the infected cells were collected and the early reporter (beta-galactosidase) activity was measured using CPRG as the substrate. (C to F) At 24 h p.i., the parallel transfected and infected cells were lysed and the late reporter (GUS) activity was measured (C, D) and viral genomic DNA replication efficiency was evaluated by real-time PCR (E, F). Error bars indicate standard deviations of the means from three replicates. *, P < 0.005; **, P < 0.0005; ***, P < 0.00005 (by unpaired t test).
ESCRT-I and ESCRT-III components are required for efficient AcMNPV budded virion entry.
During endocytosis, trafficking, and multivesicular body formation, ESCRT-I is required for cargo sorting and promoting membrane curvature. In contrast, ESCRT-III functions at a late step to catalyze scission and membrane fission (1, 6) to release the newly formed vesicle. To determine whether ESCRT-I and ESCRT-III are important for AcMNPV internalization or transport during entry, we used two strategies: (i) overexpression of GFP-tagged ESCRT-I and ESCRT-III proteins, and (ii) RNAi knockdown of individual components of ESCRT-I and ESCRT-III. For these studies, one set of Sf9 cells were transfected with a plasmid expressing GFP-tagged ESCRT-I or ESCRT-III proteins or the control GFP or E231Q-GFP, and the other set of Sf9 cells were transfected with dsRNA targeting the component of ESCRT-I or ESCRT-III, or Vps4, or the control GFP. At 16 h p.t. (for the first set of cells, transfected with plasmids) or 48 h p.t. (for the second set of cells, transfected with dsRNA), the cells were chilled at 4°C and infected with control AcMNPV (AcMNPV-LacZGUS) or a previously described AcMNPV virus (AcMNPV-3mC), which contains an mCherry-tagged major capsid protein VP39 (VP39-mCherry). After low-temperature binding to cells for 1 h, the infected cells were incubated at 27°C for 90 min to allow the viruses to enter the cells. To quantitatively analyze viruses that had entered the cells, DNA was extracted from control AcMNPV-infected cells and viral genomic DNA was quantified by real-time PCR. As shown in Fig. 6 and 7, viral genomic DNA levels were substantially decreased in cells expressing the DN Vps4 protein, E231Q-GFP (Fig. 6A and B), or in cells with an RNAi knockdown of Vps4 (Fig. 7A). Cells transfected with the DN constructs of ESCRT-III (Fig. 6B) or cells transfected with dsRNA targeting specific ESCRT-III components (Fig. 7A) showed a similar reduction of viral genomic DNA. In contrast, prior expression of full-length or truncated forms of ESCRT-I proteins Tsg101 or Vps28 or targeted knockdown of Tsg101 or Vps28 did not appear to affect the amount of virus that entered the cells (Fig. 6A and 7A). To track virion entry more directly, cells infected with the AcMNPV-3mC virus were analyzed by confocal microscopy. As shown in Fig. 6C and 7B, entering mCherry-labeled nucleocapsids (NCs) are found distributed uniformly in the cytoplasm of cells transiently expressing control GFP or cells transfected with the dsRNA of GFP, and some virus particles were observed in the nucleus of cells. In contrast, similar to what was observed for Vps4 E231Q-GFP, virus NCs were observed mostly aggregated within the cytosol in cells expressing the constructs of ESCRT-I and ESCRT-III (Fig. 6C) or in cells transfected with dsRNA specific for components of ESCRT-I, ESCRT-III, or Vps4 (Fig. 7B). Similar results were also observed in parallel experiments using High5 cells expressing GFP-tagged ESCRT-I and ESCRT-III proteins (data not shown). Together, these data indicate that during AcMNPV BV entry, the ESCRT-I complex is required for virion or nucleocapsid trafficking, whereas the ESCRT-III complex is required for efficient internalization and transport of virions.
FIG 6.

Analysis of the effects of overexpression of GFP-tagged ESCRT-I and ESCRT-III proteins on entry of AcMNPV. Sf9 cells were cotransfected with two plasmids separately expressing (i) GP64 and (ii) one of the GFP-tagged ESCRT-I or ESCRT-III proteins, E231Q-GFP, or GFP. At 16 h p.t., cells were infected with prechilled control AcMNPV or an mCherry-labeled AcMNPV virus (3mC) (MOI = 10 TCID50) at 4°C for 1 h, and then the cells were incubated at 27°C for 90 min. The control AcMNPV-infected cells were lysed, and the internalized viral genomic DNAs were determined by real-time PCR (A, B). The 3mC virus-infected cells were fixed and analyzed by confocal microscopy (C). Cell boundaries are traced with circular dashed lines. Bar, 10 μm. *, P < 0.005; **, P < 0.0005 (by unpaired t test).
FIG 7.
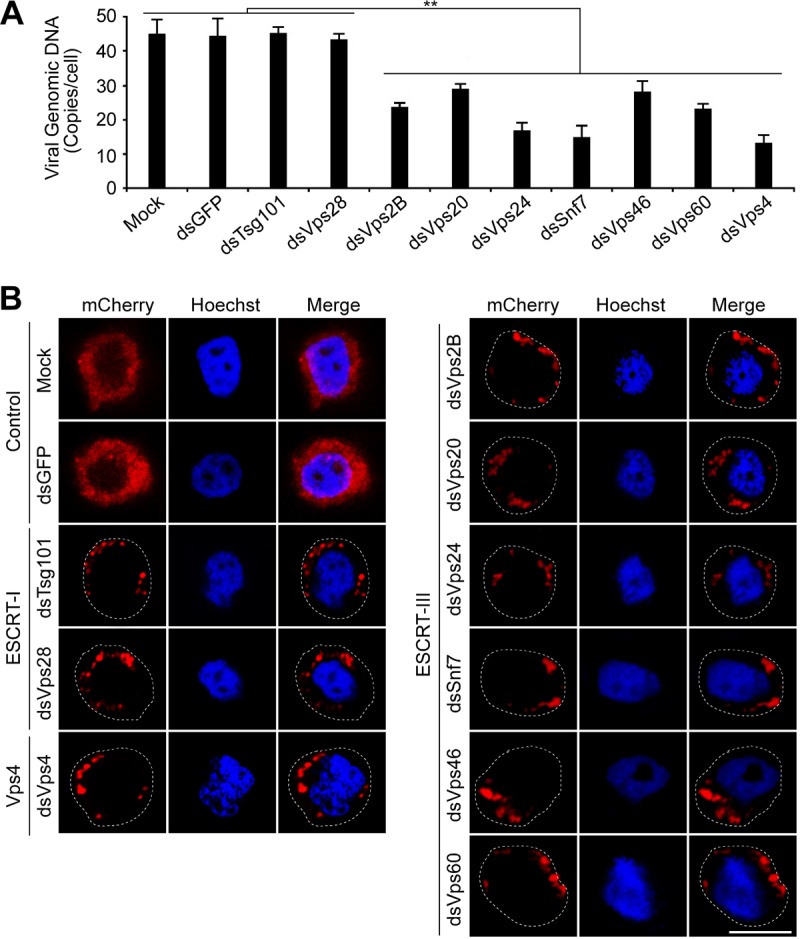
Analysis of the effects of RNAi knockdowns targeting specific ESCRT-I and ESCRT-III genes on entry of AcMNPV. Sf9 cells were mock transfected or transfected with the dsRNA targeting an individual ESCRT-I or ESCRT-III gene, Vps4, or GFP. At 48 h p.t., cells were infected with prechilled control AcMNPV or an mCherry-labeled AcMNPV virus (3mC) (MOI = 10) at 4°C for 1 h, and then the cells were incubated at 27°C for 90 min. The control AcMNPV-infected cells were lysed, and the internalized viral genomic DNAs were determined by quantitative real-time PCR (A). The 3mC virus infected cells were fixed and analyzed by confocal microscopy (B). Cell boundaries are traced with dashed lines. Bar, 10 μm. Error bars represent standard deviations from the means of results from three replicates. **, P < 0.0005 (by unpaired t test).
ESCRT-III but not ESCRT-I components are required for efficient egress of infectious AcMNPV.
As described above, overexpression of GFP-tagged forms or knockdown of the ESCRT-I or ESCRT-III components substantially impaired virion entry and transport of nucleocapsids to the nuclei of cells. Therefore, to avoid this negative effect and ask whether ESCRT-I and ESCRT-III are also required for efficient budding of AcMNPV, we used two strategies: (i) viral bacmid DNA expressing GFP-tagged ESCRT proteins was used to transfect cells to eliminate virion entry effects, and (ii) RNAi knockdowns were used in addition to overexpression of wild-type (WT) and DN constructs. For the first strategy, Sf9 cells were infected by transfecting cells with AcMNPV bacmid DNAs that individually express a GFP-tagged ESCRT-I or ESCRT-III protein plus a reporter GUS protein. In each bacmid, the ESCRT component was expressed under the ie1 early promoter and GUS was expressed under a p6.9 late promoter. For the second strategy, Sf9 cells were transfected with the dsRNA specific for the component of ESCRT-I or ESCRT-III, and at 48 h p.t., the cells were transfected again with control AcMNPV bacmid DNA, which contains the reporter genes LacZ and GUS under the OpMNPV ie2 early promoter and the AcMNPV p6.9 late promoter, respectively. For both strategies, after transfection with the bacmid DNAs and incubation for 24 h, we determined the GFP fluorescence or beta-galactosidase (beta-Gal) and GUS activities to evaluate the transfection efficiencies and monitor progression of the virus infection, respectively. In the first strategy, the percentage of GFP-positive cells at 24 h p.t. ranged from 30.4 to 37.5% (Fig. 8A), and GUS activities at 24 h p.t. were similar among cells transfected with the different bacmids (Fig. 8B). For cells transfected with dsRNA and the AcMNPV-LacZGUS bacmid, the beta-Gal and GUS activities were similar (Fig. 9A and B). These results indicated that the transfection efficiencies for different bacmids or for the control AcMNPV bacmid and various dsRNAs were equivalent and the virus infection cycle progressed into the late phase of infection. As shown in Fig. 8C and 9C, the expression of full-length and truncated forms of ESCRT-I components Tsg101 or Vps28 or knockdown of the expression of these two components did not reduce infectious virus titers compared to those for the GFP control. Similarly, the expression of the ESCRT-III construct Vps20-GFP (Fig. 8C) or the Vps20 knockdown (Fig. 9C) had no substantial effect on the levels of infectious virus produced. In contrast, however, the expression of the other two DN ESCRT-III proteins (Vps24 and Snf7) (Fig. 8C, ESCRT-III) and the corresponding Vps24 and Snf7 knockdowns (Fig. 9C) resulted in a strong inhibition of infectious AcMNPV BV production (>650-fold) similar to the reduction observed when DN Vps4 construct E231Q-GFP was expressed or when Vps4 was knocked down by RNAi (Fig. 8C and 9C). Virus titers were also reduced substantially in supernatants from cells expressing Vps2B-GFP, Vps46-GFP, or Vps60-GFP and from cells with RNAi knockdowns of Vps2B, Vps46, or Vps60 (Fig. 8C and 9C). Similar results were observed in T. ni High5 cells expressing DN forms of ESCRT-III proteins (data not shown).
FIG 8.
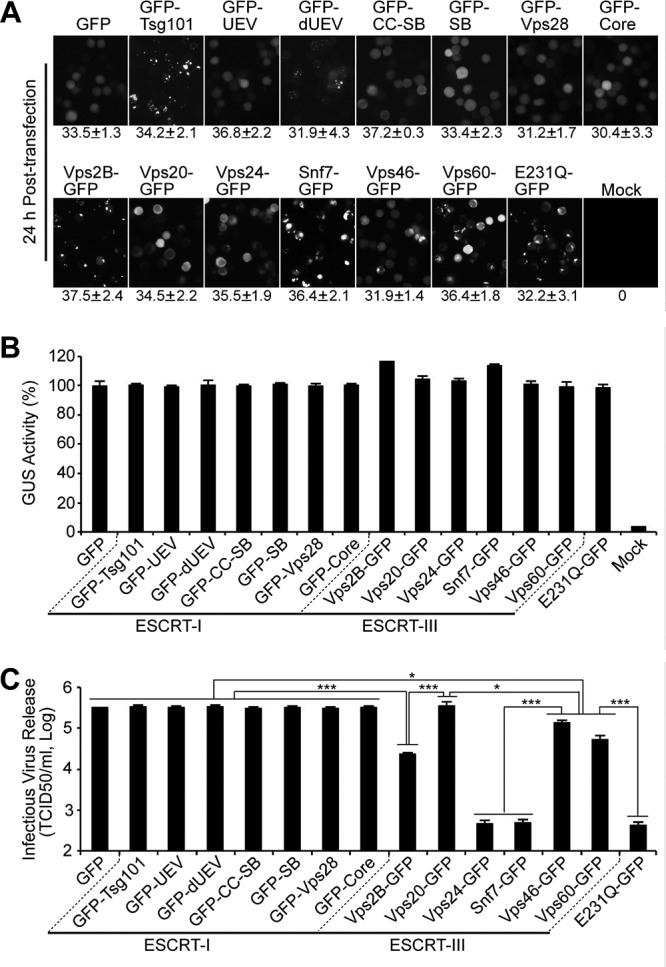
Infectious BV production in the presence of GFP-tagged ESCRT-I and ESCRT-III proteins expressed from AcMNPV bacmids. Sf9 cells were transfected with AcMNPV bacmids expressing either (i) one of the GFP-tagged ESCRT-I or ESCRT-III proteins, (ii) E231Q-GFP, or (iii) GFP. At 24 h p.t., the percentage of GFP-expressing cells was determined for each treatment, and percentages are shown below each panel as an estimate of transfection efficiency (A). A parallel group of transfected cells were also lysed at 24 h p.t., and GUS activity (expressed from late GUS reporter gene, driven by a p6.9 late promoter in each bacmid) was determined (B). The production of infectious BV from each treatment was determined by TCID50 assay of the cell supernatant (C). Error bars represent standard deviations from the means for three replicates. *, P < 0.005; ***, P < 0.00005 (by unpaired t test).
FIG 9.
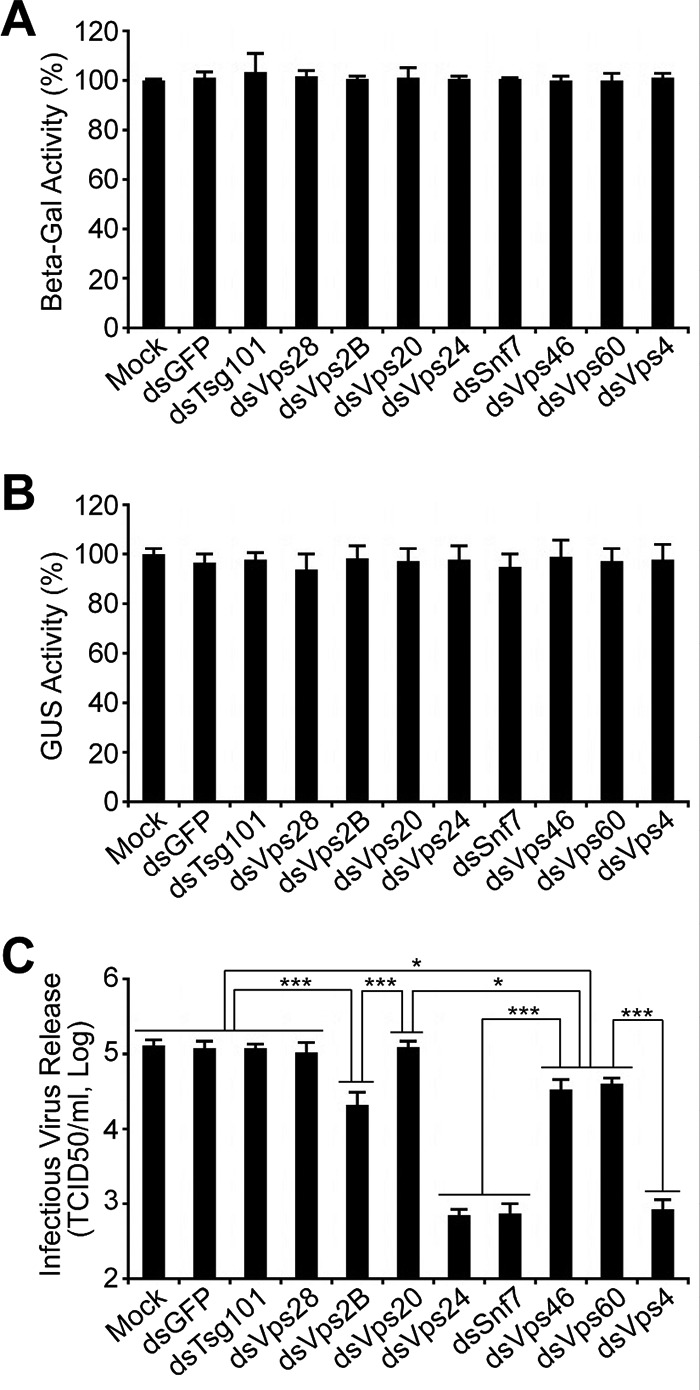
Analysis of the effects of RNAi knockdowns targeting specific ESCRT-I and ESCRT-III genes on infectious AcMNPV BV release. Sf9 cells were mock transfected or transfected with the dsRNA targeting an individual ESCRT-I or ESCRT-III gene or the control GFP gene. At 48 h p.t., the cells were transfected again with control AcMNPV bacmid DNA (AcMNPV-LacZGUS). After transfection with the viral bacmid DNA for 24 h, the transfected cells were lysed, and beta-Gal and GUS activities (separately expressed from early LacZ and late GUS reporter genes, driven by an ie2 early promoter and a p6.9 late promoter, respectively, in each bacmid) were determined (A, B). The production of infectious BV from each treatment was determined by TCID50 assay of the cell supernatant (C). Error bars represent standard deviations from the means for three replicates. *, P < 0.005; ***, P < 0.00005 (by unpaired t test).
Since the viral envelope glycoprotein GP64 is important for budding and release of BV (33), the effect of DN ESCRT-III proteins on infectious BV production might result from the disruption of transport or cell surface localization of GP64. To examine this possibility, Sf9 cells transfected with AcMNPV bacmids expressing GFP, GFP-tagged ESCRT-III proteins, or Vps4 E231Q-GFP (as described above) were fixed at 24 h p.t., and relative cell surface levels of GP64 were analyzed by cell surface enzyme-linked immunosorbent assay (cELISA). In the presence of DN ESCRT-III proteins or Vps4 E231Q, cell surface levels of GP64 were similar to that observed in the presence of control GFP protein (data not shown). Analysis of syncytium formation also revealed that in the presence of DN ESCRT-III proteins or Vps4 E231Q, GP64 efficiently induced membrane fusion (data not shown). These results suggested that DN ESCRT-III proteins and Vps4 E231Q had no apparent effect on transport or cell surface localization and membrane fusion activity of GP64. Together, these data provide evidence that ESCRT-III is required for efficient egress of infectious AcMNPV, and that effect does not appear to result from a defect in GP64 transport.
ESCRT-III/Vps4 is involved in nuclear egress of nucleocapsids of AcMNPV.
During viral egress of BV, progeny nucleocapsids of AcMNPV (which assemble in the nucleus) cross the nuclear membrane and are transported to the plasma membrane. There, nucleocapsids bud and virions pinch off to form infectious budded virus (BV) (25). The inhibitory effect of RNAi or DN ESCRT-III and Vps4 on infectious AcMNPV BV release could result from defect(s) in nucleocapsid egress across the nuclear membrane, transport through the cytoplasm, and/or budding and fission at the plasma membrane. To track nucleocapsid release, Sf9 cells were transfected with AcMNPV bacmids expressing mCherry-tagged major capsid protein VP39 (VP39-mCherry) plus one of the following: Vps24-GFP, Snf7-GFP, Vps60-GFP, Vps4 E231Q-GFP, or the control GFP. At 24 h p.t., the transfected cells were subjected to confocal microscopy. In cells expressing Vps60-GFP, Vps60 was found predominantly in the cytoplasm. While mCherry-labeled nucleocapsids were found predominantly in the nuclei as expected, we also observed a substantial amount of mCherry fluorescence in the cytosol, suggesting detection of substantial amounts of NCs in the cytoplasm (Fig. 10, Vps60-GFP). In contrast, mCherry-labeled VP39 appeared notably absent in the cytoplasm of cells expressing Vps24-GFP, Snf7-GFP, or E231Q-GFP (Fig. 10, mCherry column), suggesting that the inhibitory effect of DN ESCRT-III proteins and Vps4 on infectious AcMNPV production during release may result from blocking progeny nucleocapsid egress through or from host cell nuclear membranes. It was of note, additionally, that mCherry-labeled VP39 also appears to colocalize with Vps60-GFP at or near the apparent nuclear ring zone region (Fig. 10, Vps60-GFP). To examine these results in more detail, a parallel set of transfected cells were analyzed by transmission electronic microscopy (TEM) at 72 h p.t. As shown in Fig. 11, we observed a typical electron-dense virogenic stroma (VS), and progeny nucleocapsids had a normal morphology in cells expressing GFP, DN ESCRT-III constructs, or DN Vps4 (Fig. 11A to E). Typical bundles of nucleocapsids were observed in the ring zone region (white triangles), and progeny nucleocapsids were observed budding through the nuclear membrane, in vesicles and free in the cytoplasm, and budding at the cytoplasmic membrane. Nucleocapsids in these locations were observed to various degrees in the cells expressing the control GFP and DN ESCRT-III constructs (Fig. 11F). Numbers of nucleocapsids found in the postnuclear locations (within the cytoplasm and budding at the cytoplasmic membrane) were substantially reduced in cells expressing Vps24-GFP and Snf7-GFP and slightly reduced in Vps60-GFP-expressing cells compared with cells expressing the control GFP (Fig. 11F). In Snf7-GFP-expressing cells, we observed progeny nucleocapsids aggregated and localized in large spaces between inner and outer nuclear membranes (Fig. 11C). A similar defect in nucleocapsid budding was also observed in E231Q-GFP-expressing cells (Fig. 11F). In addition, it was noted that in Snf7-GFP and E231Q-GFP-expressing cells, nucleocapsid bundles in the nuclear ring zone region were rarely observed (Fig. 11C and E). Together, our analyses of infectious virus release, mCherry-tagged nucleocapsid protein, and TEM of nucleocapsids suggest that the inhibitory effect of DN ESCRT-III constructs on infectious AcMNPV production may result from a lower efficiency of progeny nucleocapsid egress from host cell nuclear membranes.
FIG 10.
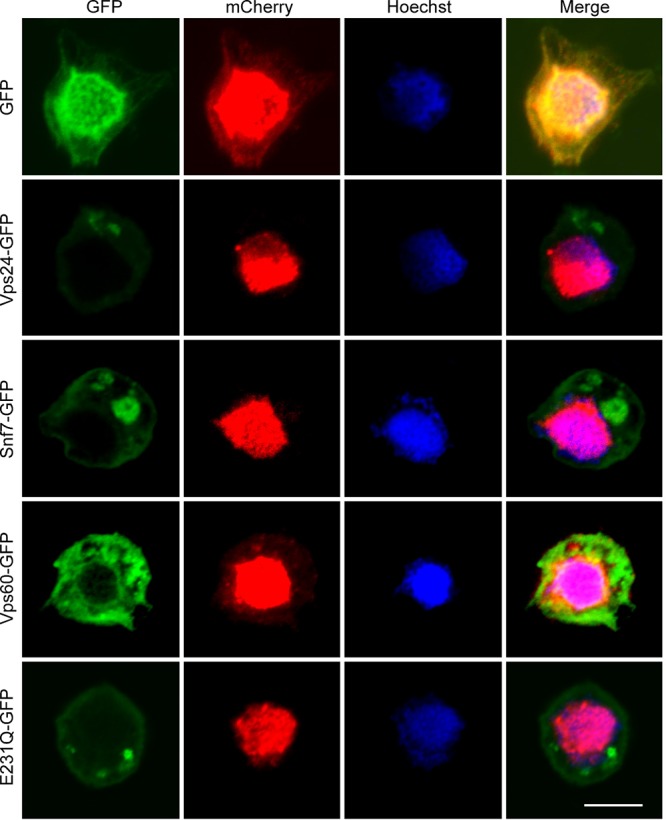
DN ESCRT-III and Vps4 proteins appear to inhibit the nuclear release of nucleocapsids. Sf9 cells were transfected with AcMNPV bacmids expressing VP39-mCherry and either Vps24-GFP, Snf7-GFP, Vps60-GFP, E231Q-GFP, or the control GFP. At 24 h p.t., the transfected cells were fixed and analyzed by confocal microscopy. Bar, 10 μm.
FIG 11.

TEM analysis of Sf9 cells transfected with AcMNPV bacmid DNAs expressing DN ESCRT-III and Vps4 proteins. (A to E) Sf9 cells were transfected with AcMNPV bacmids expressing VP39-mCherry and either GFP (A), Vps24-GFP (B), Snf7-GFP (C), Vps60-GFP (D), or E231Q-GFP (E). At 72 h p.t., the transfected cells were fixed and analyzed by transmission electron microscopy. The nuclear membrane (nm), cytoplasmic membrane (cm), and nucleocapsids (white arrows) are indicated. Multiple aggregated nucleocapsids localized in the space between the inner and outer nuclear membrane are indicated by closed triangles. (F) The numbers of postnuclear nucleocapsids were determined, and these include those residing in the cytoplasm and budding through the cytoplasmic membrane. Numbers were calculated from 13 cells for each construct. Bar, 1 μm. **, P < 0.0005 (by unpaired t test).
Interaction of ESCRT-III with viral proteins necessary for efficient budded virus production.
A number of AcMNPV proteins (Ac11, Ac76, Ac78, Ac80 or GP41, Ac93, Ac103, Ac142, and Ac146) have been identified as important or essential for infectious budded virion production (34–39, 51, 52). Individual knockouts of the genes encoding these proteins have no effect on viral DNA replication, but progeny nucleocapsids appear to be restricted to the nucleus in some or many cases, and egress from the nuclear membrane may be inhibited in many cases. Because the defects caused by these AcMNPV gene knockouts are similar to the defects caused by DN ESCRT-III proteins, we hypothesized that some of these viral proteins might interact with cellular ESCRT-III proteins. To address this hypothesis, an immunoprecipitation assay was used to examine the potential interaction between the components of ESCRT-III and each of the above viral proteins. We selected AcMNPV GP41 and Lef3 as control proteins, as it was previously demonstrated that GP41 interacts with itself but does not interact with Lef3 (53). For these interaction studies, Sf9 cells were cotransfected with two plasmids: one plasmid expressing a hemagglutinin (HA)-tagged viral protein and the other plasmid expressing a c-Myc epitope-tagged ESCRT-III protein, GP41, or Lef3. Transfected cell lysates were used for immunoprecipitation with anti-HA monoclonal antibody (MAb) and protein G agarose. Western blot analysis of the transfected cell lysates using anti-HA MAb and anti-Myc polyclonal antibody confirmed the expression of each tagged viral and ESCRT-III protein. HA-tagged viral proteins were immunoprecipitated with an anti-HA MAb, and then precipitates were challenged with an anti-Myc polyclonal antibody in Western blot analysis. As described previously (53), GP41 coimmunoprecipitated (Co-IP) with itself but not with Lef3 (data not shown). Of the 8 viral proteins examined, we found that 6 (Ac11, Ac76, Ac78, Ac80 or GP41, Ac93, and Ac146) coimmunoprecipitated with all or most of the components of ESCRT-III. Two (Ac103 and Ac142) coimmunoprecipitated with only one (Ac103 with Vps24) or two (Ac142 with Vps24 and Vps46) ESCRT-III proteins (Table 1 and data not shown). The results suggest that these viral proteins either may interact directly with the ESCRT-III proteins or may be found in a complex that includes the identified ESCRT-III proteins.
TABLE 1.
Co-IP analysis of interactions of ESCRT-III/Vps4 and AcMNPV proteinsa
| AcMNPV protein | ESCRT-III |
Vps4 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Vps2B | Vps20 | Vps24 | Snf7 | Vps46 | Vps60 | Vps4 | K176Q | E231Q | |
| Ac11 | + | − | + | − | + | + | − | − | − |
| Ac76 | + | + | + | + | + | + | − | − | − |
| Ac78 | − | + | + | + | + | + | − | − | − |
| GP41 | − | + | + | + | + | + | + | + | + |
| Ac93 | + | + | + | + | + | + | + | + | + |
| Ac103 | − | − | + | − | − | − | − | − | − |
| Ac142 | − | − | + | − | + | − | − | − | − |
| Ac146 | − | − | + | + | + | + | − | − | − |
AcMNPV proteins were tagged with an HA epitope. ESCRT-III and Vps4 proteins were tagged with a c-Myc epitope. + and −, positive and negative Co-IP signal, respectively. The original Co-IP data for the interaction of Vps4 and viral proteins are shown in Fig. 13, and the Co-IP data for the interaction of ESCRT-III and viral proteins are not shown.
To extend the results from coimmunoprecipitation studies, we further examined the possible interactions of ESCRT-III components and viral proteins using a bimolecular fluorescent complementation (BiFC) analysis in living cells. For these studies, Sf9 cells were cotransfected with two plasmids expressing separately the bait and prey proteins, each fused to the N- or C-terminal domain of mCherry (Nm and Cm). Initially, to verify the specificity of the mCherry-based BiFC system in our experimental system, we also selected AcMNPV GP41 and Lef3 as candidate bait and prey proteins. By coexpressing GP41-Nm with either GP41-Cm or Lef3-Cm, we observed mCherry fluorescence complementation in approximately 50% of the cells cotransfected with GP41-Nm and GP41-Cm plasmids. In contrast, fluorescence was not detected in cells cotransfected with GP41-Nm and Lef3-Cm plasmids (data not shown), as expected. Next, we examined interactions among ESCRT-III proteins or between ESCRT-III proteins and Vps4 by coexpressing Nm- and Cm-fused proteins in transfected Sf9 cells. Because ESCRT-III proteins are closely associated in a complex and associate with Vps4 during disassembly, BiFC fluorescence was observed in many combinations of ESCRT-III proteins or ESCRT-III proteins and Vps4 (data not shown). The percentages of cells with mCherry fluorescence detected ranged from 18.7 to 69.8% (data not shown). To examine the interaction of ESCRT-III components and viral proteins, we first added Nm to the C terminus of ESCRT-III proteins and Cm to the N terminus of Ac146 or the C termini of Ac11, Ac76, Ac78, GP41, Ac93, Ac103, and Ac142. Western blot analysis with anti-HA MAb or an anti-Myc polyclonal antibody showed that all the constructs were expressed in transfected Sf9 cells (Fig. 12A). In the cases of Ac76-Cm and Cm-Ac146, two bands were detected for each construct, and this has been observed previously (39, 54).
FIG 12.

BiFC analysis of the interaction of ESCRT-III and AcMNPV proteins. (A) Sf9 cells were transfected with plasmids expressing each construct consisting of the N- or C-terminal domain of mCherry (Nm and Cm) fused with each ESCRT-III or viral protein. At 36 h p.t., the expression of each fusion protein construct was analyzed by Western blotting with an anti-HA MAb (ESCRT-III proteins) or an anti-Myc polyclonal antibody (viral proteins); gels were spliced for labeling purposes. (B) Fluorescence complementation in cells expressing Nm and Cm fused with ESCRT-III and viral proteins. Sf9 cells were cotransfected with two plasmids separately expressing Nm or Cm fused with ESCRT-III or viral proteins. At 36 h p.t., the cells were photographed using epifluorescence microscopy. Labels on the left and top identify the cotransfected construct pairs in each panel. Cell boundaries are traced with circular dashed lines. (C) The bar graphs show the percentages of mCherry-positive cells in cotransfected Sf9 cells expressing Nm- and Cm-fused ESCRT-III and viral proteins. The pairs of cotransfected constructs are indicated below the x axis of each graph. Error bars represent standard deviations from the means for three replicates.
To identify interactions between viral proteins and ESCRT-III proteins, we examined each viral protein (Ac11, Ac76, Ac78, GP41, Ac93, Ac103, Ac142, and Ac146) against each of the ESCRT-III complex proteins (Vps2B, Vps20, Vps24, Snf7, Vps46, or Vps60) in the BiFC analysis (Fig. 12B and C). Three of the viral proteins (Ac76, Ac78, and Ac93) showed strong BiFC with all the ESCRT-III proteins examined (Fig. 12B and C). Three additional viral proteins (Ac11, GP41, and Ac146) were positive for BiFC with 5 of the 6 ESCRT-III proteins examined, although the specific groups of ESCRT-III proteins that interacted were different. The percentage of fluorescent cells detected in most of these combinations ranged from 5% to 35% but reached 50% in one combination (Ac76-Cm and Snf7-Nm). One of the viral proteins (Ac103) showed BiFC only with Vps24 (Fig. 12B and C).
While analysis of each ESCRT-III complex protein resulted in BiFC with several viral proteins, Vps2B showed a weaker fluorescence complementation signal than that of other ESCRT-III proteins (Fig. 12B, column Vps2B-Nm). Vps24, on the other hand, had some degree of BiFC with all the viral proteins examined (Fig. 12B, column Vps24-Nm). Several control experiments were performed to support and confirm these results. Western blot analysis revealed that all the constructs were expressed in cotransfected cells (data not shown). Also, similar BiFC fluorescence was observed by performing reciprocal fusions. i.e., fusing Nm with viral proteins and Cm with ESCRT-III proteins (data not shown). Taken together, the interactions identified in coimmunoprecipitation assays were consistent with those detected in the complementation (BiFC) studies. However, a few viral protein-ESCRT-III protein interactions that were negative in coimmunoprecipitation analysis were detected by BiFC assays. These included the following combinations: Ac11 and Vps20, Ac78 and Vps2B, Ac142 and Vps20, and Ac146 and Vps20 (Fig. 12; Table 1; data not shown).
Interactions of Vps4 with GP41, Ac93, and Ac103.
Vps4 functions in disassembly and recycling of the ESCRT-III complex (7), and prior studies showed that Vps4 is required for efficient egress of AcMNPV budded virions (23). Because we found evidence of interactions between certain viral proteins and ESCRT-III proteins, we also asked whether cellular Vps4 might interact with those viral proteins. To examine this question, we first used an immunoprecipitation assay. HA-tagged viral proteins as well as Myc-tagged Vps4 and Vps4 mutants were coexpressed in Sf9 cells and then analyzed by coimmunoprecipitation. Myc-tagged Vps4 and the two DN forms of Vps4 (K176Q and E231Q) were efficiently coimmunoprecipitated when HA-tagged proteins GP41-HA and Ac93-HA were immunoprecipitated with the anti-HA MAb (Fig. 13B and C). In contrast, other HA-tagged viral proteins (Ac11, Ac76, Ac78, Ac103, Ac142, and Ac146) did not coimmunoprecipitate with Vps4-Myc, K176Q-Myc, or E231Q-Myc (Fig. 13A and D; Table 1). Expression of all proteins was confirmed by Western blotting, as described earlier (data not shown). To confirm the immunoprecipitation results, we also used the BiFC assay as described earlier, in which we fused the N terminus of mCherry (Nm) to the C terminus of Vps4. In Sf9 cells coexpressing Vps4-Nm in combination with each of the Cm-fused viral proteins described above, we detected fluorescence complementation (BiFC) with viral proteins GP41-Cm, Ac93-Cm, and Ac103-Cm (BiFC was detected in 25%, 35%, and 5% of the cells, respectively) (Fig. 13F and G). Similar levels of fluorescent cells were also observed in cells coexpressing Nm-fused DN Vps4 proteins (K176Q-Nm and E231Q-Nm). Swapping the Nm and Cm domains between bait and prey proteins had no significant effect on the BiFC fluorescence observed (data not shown). Combined, these results suggest that GP41, Ac93, and possibly Ac103 interact or may be found in complexes with Vps4 and that the interaction does not depend on the ATPase activity of Vps4 since the ATPase-deficient DN Vps4 proteins also interacted with these viral proteins.
FIG 13.

Coimmunoprecipitation and BiFC analysis of interactions of Vps4 and AcMNPV proteins. (A to D) Sf9 cells were transfected with the indicated (+ or −) plasmids or combinations of plasmids expressing either HA-tagged viral proteins or Myc-tagged Vps4 or modified Vps4 constructs (E231Q and K176Q). At 36 h p.t., the transfected and cotransfected cells were separately lysed and subjected to immunoprecipitation with anti-HA monoclonal antibodies and protein G agarose. The precipitates (Co-IP) were detected on Western blots with an anti-Myc polyclonal antibody (right panel in each group). The cell lysates (Lysate) were also examined on Western blots with an anti-HA monoclonal antibody (top panels) or an anti-Myc polyclonal antibody (bottom panels). Abs, antibodies. (E) Sf9 cells were transfected with a plasmid expressing the N- or C-terminal domain of mCherry (Nm and Cm) fused with Vps4, Vps4 with DN mutations (E231Q and K176Q), or viral proteins (Ac11, Ac93, Ac103, or GP41). At 36 h p.t., expression of the fusion proteins was analyzed by Western blotting using anti-HA MAb (Vps4 and its DN mutations K176Q and E231Q) or an anti-Myc polyclonal antibody (viral proteins); gels were spliced for labeling purposes. (F) BiFC analysis of cells coexpressing Vps4 and viral protein pairs. Sf9 cells were cotransfected with two plasmids: one that expressed Nm-fused Vps4, E231Q, or K176Q and a second plasmid that expressed Cm-fused viral proteins Ac11, Ac93, Ac103, or GP41. At 36 h p.t., the cells were photographed using epifluorescence microscopy. Labels on the left and top identify the cotransfected construct pairs in each panel. Cell boundaries are traced with circular dashed lines. (G) The bar graphs show the percentages of mCherry-positive cells in cotransfected Sf9 cells expressing Nm- and Cm-fused Vps4 and viral proteins. The pairs of cotransfected constructs are indicated below the x axis of each graph. Error bars represent standard deviations from the means for three replicates.
Interactions among viral proteins.
To also examine interactions among these viral proteins, we expressed each of these HA- or Myc-tagged viral proteins (Ac11, Ac76, Ac78, GP41, Ac93, Ac103, Ac142, and Ac146) and then coexpressed (homologous or heterologous) combinations of these proteins in Sf9 cells and analyzed the combinations by coimmunoprecipitation. As shown in Table 2, each of the viral proteins Ac11, Ac76, Ac78, GP41, Ac93, and Ac146 appears to interact with itself and was immunoprecipitated in homologous combinations. For heterologous combinations, coimmunoprecipitation was observed between the following pairs: Ac11-GP41, Ac11-Ac93, Ac76-Ac78, Ac76-Ac93, Ac76-Ac103, Ac78-Ac103, Ac93-Ac103, and Ac103-Ac146 (Table 2, data not shown). The immunoprecipitation results were also confirmed by BiFC analysis. Nm and Cm fragments of mCherry were fused to the N terminus of Ac146 and to the C termini of the other viral proteins (Ac11, Ac76, Ac78, GP41, Ac93, Ac103, and Ac142). Expression of all fusion protein constructs was confirmed by Western blotting (Fig. 14A [note that Ac142-Nm and Ac142-Cm are not shown]). In cells cotransfected with plasmids expressing the same protein but fused with the Nm and Cm fragments of mCherry, from 20% to 45% of the cells showed fluorescence complementation, and this was true in all cases (Ac11, Ac76, Ac78, GP41, Ac93, and Ac146) except for Ac103 (Fig. 14B and C), suggesting the self-association of these viral proteins. For heterologous combinations of viral proteins, fluorescence complementation was observed only in cells coexpressing certain combinations (Fig. 14B and C), as summarized in Fig. 15 (right panel, center circle). Reciprocal fusions of Nm and Cm with each viral protein did not significantly affect the BiFC detected from the combinations of viral proteins (data not shown). Additionally, no BiFC fluorescence was observed in cells coexpressing Ac142-Nm or Ac142-Cm, with Nm or Cm fused to other viral proteins (data not shown). Thus, we found that the interactions or associations suggested by the coimmunoprecipitating pairs of viral proteins were confirmed by BiFC in transfected cells coexpressing Nm- and Cm-fused proteins (Fig. 14).
TABLE 2.
Co-IP analysis of interactions of AcMNPV proteinsa
| AcMNPV protein | Ac11 | Ac76 | Ac78 | GP41 | Ac93 | Ac103 | Ac142 | Ac146 |
|---|---|---|---|---|---|---|---|---|
| Ac11 | + | − | − | + | + | − | − | − |
| Ac76 | − | + | + | − | + | + | − | − |
| Ac78 | − | + | + | − | − | + | − | − |
| GP41 | + | − | − | + | − | − | − | − |
| Ac93 | + | + | − | − | + | + | − | − |
| Ac103 | − | + | + | − | + | − | − | + |
| Ac142 | − | − | − | − | − | − | − | − |
| Ac146 | − | − | − | − | − | + | − | + |
AcMNPV proteins were tagged with HA and c-Myc epitopes. + and −, positive and negative Co-IP signal, respectively. The original Co-IP data are not shown.
FIG 14.

BiFC analysis of interactions of AcMNPV proteins. (A) Sf9 cells were transfected with plasmids expressing the N- or C-terminal domain of mCherry (Nm or Cm) fused to viral proteins (Ac11, Ac76, Ac78, Ac93, Ac103, Ac146, and GP41). At 36 h p.t., the expression of each fusion protein was analyzed by Western blotting using an anti-HA MAb (Nm-fused viral proteins) or an anti-Myc polyclonal antibody (Cm-fused viral proteins) for detection; gels were spliced for labeling purposes. (B) BiFC analysis of cells coexpressing Nm- and Cm-fused viral proteins. Sf9 cells were cotransfected with two plasmids, separately expressing Nm- or Cm-fused viral proteins. The pairs of cotransfected constructs are indicated at the top and left of each panel. At 36 h p.t., cells were photographed using epifluorescence microscopy and analyzed. Cell boundaries are traced with circular dashed lines. (C) Percentages of mCherry-positive cells in transfected Sf9 cells expressing Nm- and Cm-fused viral proteins. The pairs of cotransfected constructs are indicated below the x axis of each graph. Error bars represent standard deviations from the means for three replicates.
FIG 15.

Schematic representation of the protein-protein interaction network of ESCRT-III proteins and Vps4 and viral proteins and ESCRT-III/Vps4. ESCRT-III components and viral proteins that interact with themselves are shown as shaded circles. The top left panel shows interactions among ESCRT-III proteins (Vps2B, Vps20, Vps24, Vps46, Vps60, and Snf7) and Vps4. The panel on the right shows interactions among the viral proteins (inner circle, Ac11, Ac76, Ac78, Ac93, Ac103, Ac142, Ac146, and GP41) and interactions between each viral protein (inner group) and ESCRT-III proteins (outer group, Vps2B, Vps20, Vps24, Vps46, Vps60, and Snf7). The lower left panel shows interactions between cellular Vps4 and viral proteins (Ac93, Ac103, and GP41).
DISCUSSION
The ESCRT machinery is a highly conserved system of protein complexes that mediate membrane budding and scission (2). In addition to its important role in budding and scission of retroviruses and many other RNA and DNA viruses, several studies have demonstrated that the ESCRT system is also sometimes involved in efficient viral entry, assembly, and replication compartment formation (12, 24). Comparatively little is known about the roles of the components of the cellular ESCRT pathway in baculovirus infection. In the current study, we found that functional ESCRT-I and ESCRT-III complexes were required for efficient entry and transport of AcMNPV budded virions early in infection. In addition, we found that ESCRT-III but not ESCRT-I played important roles in efficient egress of infectious AcMNPV. These results extend our previous studies using a DN Vps4 protein to show that the ESCRT pathway was involved in efficient infection by AcMNPV (23).
Isolation and expression of ESCRT-I and ESCRT-III components of Sf9.
Insect genomes encode many of the same ESCRT system components as those found in yeast and humans. However, the human genome contains a number of gene expansions in the ESCRT-III complex that are not present in insect genomes (40, 41). In insect genomes, gene expansions of the ESCRT-III complex appear to be limited to Vps2 (Vps2A and 2B), as identified in insect species from Phthiraptera, Lepidoptera, and Coleoptera (40). To determine the importance and roles of the cellular ESCRT pathway in AcMNPV infections, we first cloned the ESCRT-I components Tsg101 and Vps28 and ESCRT-III components Vps20, Vps24, Snf7, Vps46, and Vps60. We also isolated an ortholog of Vps2B from Sf9 cells. Amino acid sequence alignment and domain architecture analysis indicated that these ESCRT components of Sf9 are highly conserved with those of other insects and humans. In almost all cases, overexpression of ESCRT-I proteins or DN forms of ESCRT-I or ESCRT-III proteins resulted in cellular localization or aberrant vesicles that were similar to those previously reported in human cells (42–44, 55–57). ESCRT-III proteins contain a basic N terminus and an acidic C-terminal region (48, 58). Without stimulation, the interaction of the C termini of ESCRT-III subunits with their amino-terminal cores closes the conformation in an autoinhibited monomer state. Removal of the intramolecular interaction activates ESCRT-III proteins to assemble as polymers (47, 58). Several prior studies demonstrated that fusion of a bulky tag, such as GFP, to the C terminus of ESCRT-III proteins blocked their autoinhibition and activated ESCRT-III proteins to polymerize. The unregulated assembly of ESCRT-III complexes resulted in the formation of aberrant endosomes (46, 50). We used the same strategy for DN ESCRT-III proteins in the current study, and the colocalization of these GFP-tagged ESCRT-III proteins and Vps4 E231Q-mCherry in Sf9 cells suggests that they form a similar aberrant endosome.
ESCRT-I and ESCRT-III are required for efficient entry of AcMNPV.
Budded virions of AcMNPV enter host cells via clathrin-dependent endocytosis (28). During entry, membrane fusion mediated by the viral envelope glycoprotein GP64 occurs within the endosome, and nucleocapsids are released into the cytosol (25). Nucleocapsids are then transported to the nucleus and through nuclear pore complexes in a process that is mediated by actin polymerization (31, 32). The initial endosomal trafficking of AcMNPV is not well understood, although prior studies (23) found that expression of DN Vps4 resulted in an inhibition of both AcMNPV entry and egress. To understand in more detail the role of the ESCRT pathway in viral infection, we examined viral replication and budded virus production using full-length and truncated forms of ESCRT-I, DN ESCRT-III proteins, and a dsRNA-based RNAi approach. We detected a significantly reduced production of infectious AcMNPV budded virions when these forms of ESCRT-I or ESCRT-III proteins were expressed or when ESCRT-I and ESCRT-III proteins were knocked down by RNAi. We used qPCR and mCherry-labeled virions to examine particle entry, and we analyzed early and late reporter gene expression and viral genomic DNA replication to monitor subsequent events in infection. From these studies, we concluded that the reduced production of infectious AcMNPV resulted from the disruption of AcMNPV infection at an early step, prior to early gene expression. Because overexpression or knockdown of ESCRT-I and ESCRT-III proteins resulted in aberrant intracellular compartments, it is likely that entering virions are trafficked through compartments that require functional ESCRT-I or ESCRT-III. In yeast and mammalian cells, both endosomal cargo trafficking and formation of MVBs are dependent on ESCRT-I and ESCRT-III (1, 6, 59).
Tsg101 (UEV domain) is known to interact with so-called viral late domains such as “P(T/S)AP,” and genome-wide analysis of AcMNPV revealed that several conserved baculovirus proteins contain a typical late-domain motif. These viral proteins include Ac71 (IAP2), Ac83, and Ac104. Deletion of these genes from the AcMNPV genome has various effects. Deletion of Ac71 has no effect on infectious virion production (60), but in contrast, deletion of Ac83 or Ac104 each significantly reduces infectious AcMNPV production (61, 62). However, the functional role of the “P(T/S)AP” domain in these viral proteins has not been examined. How the various ESCRT-I constructs interfere with entry/viral infection is unknown, but based on prior work we can propose the following possible hypotheses. Because of the presence of the CC domain, Tsg101 constructs dUEV and CC-SB could potentially multimerize with endogenous Tsg101, and all 3 C-terminal forms of Tsg101 (Fig. 1A, dUEV, CC-SB, and SB) might also compete with endogenous Tsg101 for binding with Vps28. Additionally, overexpression of Vps28 might also interfere with the proper assembly of ESCRT-II, which is required for cargo trafficking and intraluminal vesicle (ILV) formation (6). Previously, it has been demonstrated that the CTD of Vps28 is not directly involved in ESCRT-I complex assembly but was able to function as an adaptor module for the interaction of Vps28 with the ESCRT-III component Vps20 (45). The effect of Vps28 construct Core (lacking the CTD) on AcMNPV entry might therefore result from its effect on the assembly of ESCRT-III or recruitment of Vps20. Further studies will be necessary to understand the precise roles of the full-length and truncated forms of Tsg101 and Vps28 on endosomal trafficking and virus entry into insect cells.
In yeast, the ESCRT-III complex contains four core components, Vps2, Vps20, Vps24, and Snf7 (corresponding mammalians homologs are CHMP2, CHMP6, CHMP3, and CHMP4, respectively) (6, 50). These components assemble in a sequential manner. Vps20 recruits and initiates oligomerization of Snf7, and Vps24 caps the oligomer of Snf7 and terminates its oligomerization by recruiting Vps2, which in turn recruits Vps4 for disassembly and recycling of ESCRT-III (3, 4, 50). The other two ESCRT-III components, Vps46 and Vps60 (CHMP1 and CHMP5 in mammals), are involved in promoting Vps4 localization and activation (63–65). Our BiFC results indicated that the ESCRT-III components of Sf9 cells interact with each other and these components all interact with Vps4, as might be expected. Overexpression of DN ESCRT-III proteins or Vps4 or RNAi knockdowns of these proteins likely affect the assembly or disassembly of ESCRT-III, which in turn disrupts endosomal cargo trafficking and ILV formation. Several studies have demonstrated that host ESCRT factors are involved in efficient entry for enveloped viruses such as rhabdoviruses, arenaviruses, flaviviruses, herpesviruses, and bunyaviruses (12, 20–22, 66), as well as the nonenveloped rotavirus (19). Similar to AcMNPV, these viruses enter host cells via receptor-mediated endocytosis and the nucleocapsids are released into the cytoplasm through the limiting membrane of the endosome. The roles of cellular ESCRT complexes or components in the entry of these viruses are not clear, and the roles of ESCRT components may differ for different viruses. In the case of VSV, in which transport during entry has been examined in some detail, interactions may be complex, with virions fusing in some cases with the intraluminal vesicles of MVBs, followed by back fusion of ILVs with the limiting membrane of the endosome to release nucleocapsids into the cytoplasm (67). It is unclear whether this process occurs in the same manner during entry of BV of AcMNPV, but it is possible that disruption of ESCRT-I or ESCRT-III functions could disrupt the successful release of nucleocapsids by interfering with this or other processes in vesicular transport.
Roles of ESCRT-III in efficient egress of AcMNPV.
In AcMNPV-infected Sf9 cells, substantial quantities of infectious progeny budded viruses are produced at 24 h postinfection (25). Observations from transmission electron microscopy suggest that the progeny nucleocapsids destined to form BV exit the nucleus and transiently obtain an envelope derived from the nuclear membrane (68). In the cytoplasm, the membranes of these vesicles (containing nucleocapsids) are lost by an unknown mechanism (25, 68), and the nucleocapsids are subsequently transported to the plasma membrane, where they interact with the plasma membrane and bud to acquire an envelope, forming the budded virions (25). Egress of BV requires kinesin, suggesting that vesicles involved in nucleocapsid egress move along microtubules (69). Because of the importance of ESCRT-I and ESCRT-III components in AcMNPV entry, we could not use the same viral complementation system to study the role of these ESCRT factors in virus egress. Therefore, we transfected insect cells with AcMNPV bacmid DNA encoding and expressing individual ESCRT-I or ESCRT-III protein constructs. The effect of each ESCRT component on virus replication was analyzed at 24 h p.t. Using this method for initiating infection, expression of full-length and truncated forms of ESCRT-I components Tsg101 and Vps28 and DN ESCRT-III proteins did not appear to affect or inhibit the early stage of AcMNPV infection, as the virus infection progressed into the late phase. In the presence of overexpressed ESCRT-I proteins (full-length and truncated forms), we identified no substantial effects on infectious AcMNPV production. However, when DN ESCRT-III proteins were expressed, we observed a reduced production of infectious BV. A substantial reduction in BV production was observed in the presence of either Vps24-GFP, Snf7-GFP, or the control DN Vps4 construct E231Q-GFP (Fig. 8C). A less dramatic reduction was observed when either Vps2B-GFP, Vps46-GFP, or Vps60-GFP was expressed. In one case (overexpression of Vps20-GFP), no apparent reduction in BV production was observed. Similar results were observed from RNAi knockdowns of these ESCRT-III proteins or Vps4 in AcMNPV bacmid DNA-transfected cells (Fig. 9C). In total, these data suggest that most of the ESCRT-III proteins are necessary for infectious BV release. Further analysis by confocal microscopy and transmission electronic microscopy suggested that the DN ESCRT-III proteins Vps24-GFP and Snf7-GFP as well as Vps4 E231Q-GFP may block nucleocapsid egress from nuclear membrane (Fig. 10 and 11). In contrast, an apparently lower-level inhibition of nucleocapsids released into the cytoplasm was observed in Vps60-GFP-expressing cells. These results suggest that ESCRT-III components Vps24, Snf7, and Vps4 (and possibly Vps2B) may be important for nuclear egress of progeny nucleocapsids.
The role of host ESCRT complex proteins in the context of virus budding has been studied most intensively for retroviruses, particularly HIV-1 (12), which serves as an important paradigm for understanding the roles of the cellular ESCRT pathway in the budding and release of other enveloped viruses (12, 70). Similar to the requirement in HIV-1 budding, we demonstrated that Snf7 (the homolog of human CHMP4) is critical for AcMNPV BV egress (71). In contrast, however, we found that Vps20 (the homolog of human CHMP6) was not necessary for egress of AcMNPV BV. ESCRT protein requirements for egress of AcMNPV differ from those for HIV-1 budding in two other aspects: (i) ESCRT-I components Tsg101 and Vps28 were dispensable for AcMNPV egress, while both are required for HIV-1 budding (72, 73); (ii) ESCRT-III proteins Vps24 and Vps60 were both required for efficient AcMNPV egress, but HIV-1 virions are released efficiently in the absence of the human orthologs of Vps24 and Vps60 (CHMP3 and CHMP5) (71). Similar to our observations in AcMNPV egress, Tsg101 is not required for herpes simplex virus (HSV-1) budding, although CHMP3 and CHMP5 are critical for HSV-1 production (74). ESCRT-III is also required for efficient budding of a variety of other viruses, including Epstein-Barr virus and hepatitis A virus (12, 18, 75). While the conserved mechanism of membrane fission by the ESCRT-III complex (2) may be utilized by many viruses in the budding process, the different requirements for subunits of ESCRT-III suggest that the mechanisms of their recruitment to and assembly at the virus budding sites likely differ between AcMNPV, HIV-1, and other viruses (12).
AcMNPV proteins involved in ESCRT-III recruitment.
Several studies have demonstrated that a variety of conserved AcMNPV genes (including Ac11, Ac76, Ac78, Ac80 [GP41], Ac93, Ac103 [p48], Ac142, and Ac146 genes) are essential for the production of infectious budded virions. Deletion of these genes individually from the AcMNPV genome does not affect viral DNA replication, but when infections are initiated by transfection with bacmids containing knockouts in most of these genes, progeny nucleocapsids are not efficiently released from the nucleus (34–39, 51, 52). Western blot analysis indicated that Ac76, Ac78, and Ac93 are all present on the envelope of BV and ODV (35, 37, 54). Ac142 and Ac146 are associated with the nucleocapsid of BV (38, 39), and Ac80 (GP41) is an ODV tegument protein (25). To determine whether these viral proteins may interact with ESCRT-III/Vps4, we examined combinations of viral and host proteins in coimmunoprecipitation (Co-IP) and BiFC assays. We found that these viral proteins interacted or were associated with each other and with ESCRT-III subunits and Vps4 in a complex manner (Fig. 15). Intriguingly, Ac76, Ac78, and Ac93 (viral proteins found on both BV and ODV envelopes) appear to interact with all ESCRT-III proteins, highlighting the potentially central role of these viral proteins in either recruiting or functionally interacting with ESCRT-III components. Ac11, Ac146, and GP41 also interacted broadly, with 5 of the 6 ESCRT-III proteins, suggesting that they may also be involved in recruiting or in functional interactions in the ESCRT-III fission machine. For viral proteins Ac103, and Ac142, we identified interactions with only one (Ac103) or a few (Ac142) ESCRT-III proteins, although this does not imply that they may not play an important role. Our data suggested that three viral proteins (Ac93, Ac103, and GP41) interact directly or indirectly with Vps4, and these viral proteins could play a role in recruiting or activating Vps4. These three viral proteins also interacted with the modified (ATPase-defective) form of Vps4, suggesting that their interactions did not depend on Vps4 ATPase activity.
In yeast and mammals, Snf7 (CHMP4) is the most abundant ESCRT-III component, and it plays central roles in ESCRT-III polymer formation and membrane fission (76). The detection of interactions between multiple AcMNPV proteins and Vps20 or Snf7 may indicate redundancies in recruiting Snf7, i.e., via viral proteins interacting with Snf7 or viral proteins interacting with Vps20, which recruits Snf7. These possibly redundant interactions might explain why DN Vps20 or RNAi knockdown of this protein did not block production of infectious BV.
In addition to their roles in egress of progeny nucleocapsids from the nuclear membrane, viral proteins Ac11, Ac76, Ac93, Ac103, and Ac142 are also required for envelopment of nucleocapsids in the nucleus to form ODV (34, 36–38, 52). Deletion of Ac76 or Ac93 resulted in a reduced formation of the virus-induced intranuclear microvesicles (34, 37), which are derived from the inner nuclear membrane and are the source for ODV envelopes (26, 27, 68). We found that in addition to their roles in ODV formation, they also have a complex web of interactions with host ESCRT-III proteins and Vps4. Viral genes encoding Ac76, Ac78, GP41, Ac93, Ac103, and Ac142 are core baculovirus genes that are present in most or all sequenced baculovirus genomes (note that Ac76 was not identified in the dipteran virus genome but is present in all other sequenced baculovirus genomes.) (25, 77). Because these genes are conserved across baculovirus genomes and serve critical roles in BV egress and ODV formation, this suggests a long and important evolutionary association with cellular pathways critical for production of BV and ODV.
Based on our observations and prior studies of these viral proteins, we developed a hypothetical model of the coordinated action of viral proteins and ESCRT-III/Vps4 in efficient budding of progeny nucleocapsids from the nuclear membrane (Fig. 13). In this model, we hypothesize that the viral core protein Ac76 (which is encoded by one of the most highly expressed late genes) accumulates in the inner nuclear membrane (54, 78) to form a shell and initiates the nuclear membrane protrusion. To this process, the interaction of viral core proteins Ac78 and Ac93 with Ac76 might also contribute. Another viral core protein, Ac103, bridges the Ac76-Ac78-Ac93 complex and Ac146, which is present in the nucleocapsid. Through these interactions, the progeny nucleocapsid might be directed to the budding region on the nuclear membrane (Fig. 16A). The core viral capsid protein Ac142 might also be involved in targeting the nucleocapsid. In addition to initiating nuclear membrane remodeling, the complex Ac76-Ac78-Ac93 may interact with Ac11 and GP41 to recruit the core components of ESCRT-III (which include Vps2, Vps20, Vps24, and Snf7). These ESCRT-III proteins may then form a complex to build the filament of Snf7 that constricts the nuclear membrane (Fig. 16B). After releasing the vesicle containing the nucleocapsid from the nuclear membrane, the viral protein complex would further recruit Vps4 and the ESCRT-III proteins Vps46 and Vps60, which are required for activation of Vps4. The disassembly and recycling of the ESCRT-III complex would then be catalyzed by the Vps4 complex (Fig. 16C). While highly speculative, this hypothetical model for nucleocapsid trafficking is based on prior and current results and provides a framework for future experimental analysis. Validation of this or other models may require high-resolution microscopy to localize viral and host protein complexes and cellular compartments associated with virus entry and egress.
FIG 16.

A hypothetical model of the interaction of the viral proteins and ESCRT-III/Vps4 in nuclear egress of progeny nucleocapsids. (A) In AcMNPV-infected cells, the nuclear membrane-associated Ac76 may initiate the nuclear membrane protrusion. Ac76 interacts with Ac93 and Ac78, which may form a complex that interacts with Ac103, which in turn interacts with nucleocapsid-associated protein Ac146, to target the progeny nucleocapsids to the budding region on the nuclear membrane. (B) A viral protein complex (Ac76, Ac93, Ac78, and possibly Ac142) may recruit the core components of ESCRT-III to the budding region and result in the formation of the Snf7 filament that constricts the nuclear membrane, releasing a double-membraned vesicle containing nucleocapsids. (C) After pinching off the double-membraned vesicle, the viral protein complex within the nucleus recruits Vps4 and its regulatory ESCRT-III proteins (Vps46 and Vps60) to form the activated Vps4 complex, which disassembles and recycles the ESCRT-III complex. BV, budded virions; CCV, clathrin-coated vesicle; DN, dominant negative; EE, early endosome; ER, endoplasmic reticulum; INM, inner nuclear membrane; LE/MVBs, late endosome/multivesicular bodies; NPC, nuclear pore complex; ONM, outer nuclear membrane; VS, virogenic stroma.
MATERIALS AND METHODS
Cells, transfections, and infections.
Spodoptera frugiperda (Sf9), Trichoplusia ni (High 5), and Sf9Op1D (a cell line expressing the Orgyia pseudotsugata MNPV [OpMNPV] GP64 protein) cells (79) were cultured at 27°C in TNMFH medium (Sigma-Aldrich) containing 10% fetal bovine serum (FBS; Gibco). Transfection of plasmid DNAs or double-stranded RNA in 12-well plate (2 × 105 cells per well) was performed using a standard CaPO4 precipitation procedure (29), and viral bacmid transfections in 6-well plates (1 × 106 cells/well) using Cellfectin II reagent (Invitrogen). For viral infections, the virus was incubated on cells for 1 h, and then cells were washed once in TNMFH. Times postinfection (p.i.) were calculated from the time the viral inoculum was added.
Cloning and mutagenesis of ESCRT-I and ESCRT-III components.
Total RNA was isolated from Sf9 cells by using TRIzol reagent RNAiso plus (TaKaRa), and the first-strand cDNA synthesis was performed with AMV reverse transcriptase using the RNA LA PCR kit (TaKaRa). Gene-specific primers (Table 3) targeted to the outside regions of the open reading frame (ORF) of ESCRT-I components Tsg101 and Vps28 and ESCRT-III components Vps2B, Vps20, Snf7, Vps46, and Vps60 were designed based on the EST sequences at the SpodoBase database (http://bioweb.ensam.inra.fr/spodobase/) (80) and used to amplify the complete ORF and partial 5′ and 3′ ends of each gene, from the cDNA. Another set of gene-specific primers were further designed and used to PCR amplify the complete ORF of the above-listed ESCRT components. To obtain the ESCRT-III component Vps24, 3′ RACE (rapid amplification of cDNA ends) was conducted with the 3′-Full RACE Core Set with PrimeScript RTase (TaKaRa) and gene-specific primers Vps24SP1 and Vps24SP2 (Table 3). Two primers specific for 5′ and 3′ ends of Vps24 and two primers specific for the ORF of Vps24 were further designed according to the SpodoBase database and 3′RACE sequences and used to PCR amplify the ORF of Vps24.
TABLE 3.
PCR primers
| Primer purpose and name | Sequence (5′ to 3′) |
|---|---|
| Amplification of the ORF of AcMNPV genes ac11, ac76, ac78, ac80 (gp41), ac93, ac103, ac142, ac146, and lef-3 | |
| Ac11XF | ATATCTAGAATGTCTCTCGCTGCAAAGTTAAT |
| Ac11ER | ATAGAATTCTTGTAAATGTTTATTATTTAAAAACG |
| Ac76XF | AATTCTAGAATGAATTTATATTTGTTGTTG |
| Ac76ER | AATGAATTCATCTATTGAGCTGGTATTTTTGT |
| Ac78XF | ATATCTAGAATGAATTTGGACGTGCCCT |
| Ac78ER | ATAGAATTCATCAAATTTATTAAAAACAAAAGGA |
| Ac80XF | ATAATCTAGAATGACAGATGAACGTGGCA |
| Ac80ER | ATAAGAATTCTGCACTGCGCCCTTTCGTGTT |
| Ac93XF | AATTCTAGAATGGCGACTAGCAAAACGAT |
| Ac93ER | AATGAATTCATTTACAATTTCAATTCCAATG |
| Ac103XF | AATTCTAGAATGTGCGCTTACAGATTACAATAC |
| Ac103ER | AATGAATTCTTTATTGAAGCAATCATGGTTGAG |
| Ac142XF | AATTCTAGAATGAGTGGTGGCGGCAACTTGT |
| Ac142ER | AATGAATTCTTGTACCGAGTCGGGGATTAATAA |
| Ac146BF | AATGGATCCATGAACGTCAATTTATACTGC |
| Ac146ER | AATGAATTCCTATGAAGAGCGGGTTTC |
| Lef3XF | ATATCTAGAATGGCGACCAAAAGATCTTTG |
| Lef3ER | ATTGAATTCCAAAAATTTATATTCATTTTC |
| Amplification of the ORF of ESCRT-I components Tsg101 and Vps28 and their truncated forms | |
| Tsg101F | GGGATGTGATTCTGTGATTT |
| Tsg101R | ACATCATCCGAGTATGACTCA |
| Tsg101BF | ATAGGATCCATGGCTAACGACGATGTAGTG |
| Tsg101ER | ATAGAATTCTTAGCACGCCAACTGAGCCTTCT |
| UEVER | ATAAGAATTCTGGCATGAAAGAGTTTACTGGGT |
| dUEVXF | AAGTAATCTAGAATGAGAGCGCCTTACCCAGTAAACT |
| CC-SBXF | ATAATCTAGAATGGTAGAAGATAAACTACGAAGGAG |
| SBXF | ATAATCTAGAATGGACGAAGCTGTTGTGACCACTG |
| Vps28F | AACCTTAGCCTTGCCTTAACAAT |
| Vps28R | TTGAGCTGGTCACATCGATGAC |
| Vps28BF | AATGGATCCATGCAGGACACAAGACCAGAA |
| Vps28ER | ATAGAATTCTCAGTCCTTGTGCAGGAACTTG |
| CoreER | ATTGAATTCTCAGCCCTTGTCGTCCTTGATGAGGT |
| Amplification of the ORF of the ESCRT-III components Vps2B, Vps20, Vps24, Snf7, Vps46, and Vps60 | |
| Vps2BF | ATCGGGAAGTGGTAGTTATA |
| Vps2BR | AGAGATTATATTTCATGTGCGCG |
| Vps2BXF | AATTCTAGAATGGATTTCTTCTTTGGCAAGCA |
| Vps2BER | AATGAATTCGGACTTTAGCTTAGCTAATTG |
| Vps20F | TATGTAGATAAGGCTACAACAT |
| Vps20R | TATACTTTAAAGCCTATATACA |
| Vps20XF | AATTCTAGAATGGGTTCCTTATTCGGTAAAC |
| Vps20ER | AATGAATTCAGCTTCGGCTGCTAATTTGA |
| Vps24SP1 | AGAGTTGCTGGGTCATTGCAGA |
| Vps24SP2 | TGGCATCATTGAGGAGATGCT |
| Vps24F | AATAGGTAATTGTTATATTATAAC |
| Vps24R | GCAATAGTCAATCCGTGGCGGCT |
| Vps24XF | AATTCTAGAATGGGCCTGTTTGGTAAATCACC |
| Vps24ER | AATGAATTCCGAAGACCTGAGTGCCTCTAACC |
| Snf7F | TCTCTTGCAATACGTTGTTT |
| Snf7R | ACCAGTATACATCGACGTGCTGTG |
| Snf7mF | ACAAGAGTTTCTGGAGAAGAAAATCGAT |
| Snf7mR | ATCGATTTTCTTCTCCAGAAACTCTTGT |
| Snf7XF | ATATCTAGAATGAGTTTTCTGGGGAAGTTATT |
| Snf7ER | ATAGAATTCTGTGGCCCAAGACTGCAACTGTG |
| Vps46F | ACCCTGTGCTTAGTGCTAAGCTT |
| Vps46R | ACATGCATCATTTAGGTCTTACA |
| Vps46XF | AATTCTAGAATGTCTTCATCCGCTATGGAA |
| Vps46ER | ATAGAATTCTTCGGCTTGTCGTAATCGAGC |
| Vps60F | TCACGATCCGGGGCAATGAGGAT |
| Vps60R | GTTTCCCAGTCACGATCT |
| Vps60XF | AATTCTAGAATGAACAGAATATTCGGAAG |
| Vps60ER | AATCTGCAGCGACGATCTACCGCGGGAAGT |
| Amplification of the promoter and ORF of AcMNPV vp39 | |
| VP39pF | AATGAGCTCGGTACCTTGTTCGCCATCGTGGAATCA |
| VP39pR | AATTCTAGAATTGTTGCCGTTATAAATATG |
| VP39XF | AATTCTAGAATGGCGCTAGTGCCCGTGGGT |
| VP39ER | AATGAATTCGACGGCTATTCCTCCACCTG |
| Amplification of the dsDNA of GFP, the components of ESCRT-I (Tsg101, Vps28) and ESCRT-III (Vps2B, Vps20, Vps24, Snf7, Vps46, Vps60), and Vps4 | |
| GFPiF | TAATACGACTCACTATAGGGACGTAAACGGCCACAAGTTC |
| GFPiR | TAATACGACTCACTATAGGGTGTTCTGCTGGTAGTGGTCG |
| Tsg101iF | TAATACGACTCACTATAGGGAGTGGACACAGAATGGCTCC |
| Tsg101iR | TAATACGACTCACTATAGGGTTCCTTCAGCCTCCTTCGTA |
| Vps28iF | TAATACGACTCACTATAGGGAGCATGACAACATGGCAGAG |
| Vps28iR | TAATACGACTCACTATAGGGGCTTGTCCACCCACTCTTGT |
| Vps2BiF | TAATACGACTCACTATAGGGACGATGGGAGCAAACATAGC |
| Vps2BiR | TAATACGACTCACTATAGGGCCGCATCTTTGGTTGATTCT |
| Vps20iF | TAATACGACTCACTATAGGGCCTGCAAGCAGAGTGACTGA |
| Vps20iR | TAATACGACTCACTATAGGGTTGGTCACCCACATCAAGAA |
| Vps24iF | TAATACGACTCACTATAGGGAAGCTGCAGCCAAGAATGAT |
| Vps24iR | TAATACGACTCACTATAGGGCATGCCTGACATGGTCTCAT |
| Snf7iF | TAATACGACTCACTATAGGGTCTGGGGAAGTTATTCGGTG |
| Snf7iR | TAATACGACTCACTATAGGGTATGAGCCAATTTCATGGCA |
| Vps46iF | TAATACGACTCACTATAGGGGCACGGATACATGCAGAGAA |
| Vps46iR | TAATACGACTCACTATAGGGAACCAGCCTCATCAGCAACT |
| Vps60iF | TAATACGACTCACTATAGGGTATTCGGAAGGGGAAAACCT |
| Vps60iR | TAATACGACTCACTATAGGGCTGTGTGACTCCGTCCTTCA |
| Vps4iF | TAATACGACTCACTATAGGGGGAAACACGAGGCATCAACT |
| Vps4iR | TAATACGACTCACTATAGGGAACTTCCTAAGCCGGACCAT |
| Construction of pIEnGFP and pIEcGFP | |
| nGFPF | AATTCTAGAATGGTGAGCAAGGGCGAGGAG |
| GFPR | AATGGATCCCTTGTACAGCTCGTCCATGCC |
| cGFPF | AATGAATTCATGGTGAGCAAGGGCGAGGAG |
| GFPpAF | CATGGACGAGCTGTACAAGTAAATGTAATAATAAAAATTGTATCA |
| GFPpAR | TGATACAATTTTTATTATTACATTTACTTGTACAGCTCGTCCATG |
| 64pAR | ATTAAGCTTCACACTCGCTATTTGGAACAT |
To remove the XbaI site within the ORF of Snf7 for subsequent cloning, a silent mutation was introduced by overlap PCR using the pair primers Snf7XF-Snf7mR and Snf7mF-Snf7ER (Table 3). The truncated forms of Tsg101 and Vps28 were generated by PCR. All the PCR products were cloned into pMD18-T vector (TaKaRa) and sequenced with M13-47 and M13-48 primers. The pMD18-T vector containing the ORF or the truncated forms of ESCRT-I and ESCRT-III components was designated X-pMD18-T or Y-pMD18-T (X and Y represent ESCRT-I and ESCRT-III components, respectively).
Construction of plasmids, bacmids, and viruses.
All expression plasmids are listed in Table 4. Initially, to generate the transient-expression vectors pIEnGFP and pIEcGFP, the ORF of enhanced green fluorescent protein (EGFP) and a fragment containing the ORF of EGFP and the poly(A) signal of the AcMNPV gp64 gene were amplified by PCR using Vps4-gfppBlue (23) as the template and separately inserted between the XbaI and BamHI or the EcoRI and HindIII sites of the plasmid pIE (81). For generating ESCRT-I components Tsg101- and Vps28-derived expression plasmids, the ORF and truncated forms of Tsg101 and Vps28 were isolated from X-pMD18-T with enzymes BamHI and EcoRI and then inserted into pIEnGFP vector. The ORFs of ESCRT-III components were isolated from Y-pMD18-T using restriction enzymes XbaI and EcoRI (Vps2B, Vps20, Vps24, Snf7, Vps46) or XbaI and PstI (Vps60) and then cloned into the same enzyme sites of pIEcGFP or pIE-MCS-Myc (81) to produce the target genes fused with GFP or a c-Myc tag at the C terminus. The plasmids expressing HA- or c-Myc-tagged AcMNPV genes (Ac11, Ac76, Ac78, GP41, Ac93, p48, Ac142, Ac146, Lef3 genes) and the mCherry-based bimolecular fluorescent complementation (BiFC) system were constructed as described previously (81, 82). The gene-specific BiFC plasmids Y-HA-NmpBlue, Z-HA-NmpBlue, U-HA-NmpBlue, Y-Myc-CmpBlue, Z-Myc-CmpBlue, U-Myc-CmpBlue (where Y, Z, U, Nm, and Cm represent ESCRT-III components, AcMNPV genes except Ac146, Vps4, and its mutants E231Q and K176Q, and the N and C termini of mCherry, respectively) were generated by insertion of the XbaI-EcoRI fragment isolated from Y-pMD18-T, pIE-Z-Myc, Vps4-gfppBlue, E231Q-gfppBlue, and K176Q-gfppBlue (23) into HA-NmpBlue or Myc-CmpBlue (81), respectively. Nm-Ac146pBlue and Cm-Ac146pBlue were generated by insertion of the PCR products of the ORF of Ac146 digested with BamHI and EcoRI into Nm-HApBlue or Cm-MycpBlue (81), respectively.
TABLE 4.
Plasmids constructed in this studya
| Purpose | Construct name(s) |
|---|---|
| Expression of ESCRT-I and ESCRT-III proteins | GFP-Tsg101pBlue, GFP-UEVpBlue, GFP-dUEVpBlue, GFP-CC-SBpBlue, GFP-SBpBlue, GFP-Vps28pBlue, GFP-CorepBlue, HA-Tsg101pBlue, HA-Vps28pBlue, Vps2B-GFPpBlue, Vps20-GFPpBlue, Vps24-GFPpBlue, Snf7-GFPpBlue, Vps46-GFPpBlue, Vps60-GFPpBlue |
| Coimmunoprecipitation | Ac11-HApBlue, Ac76-HApBlue, Ac78-HApBlue, GP41-HApBlue, Ac93-HApBlue, Ac103-HApBlue, Ac142-HApBlue, HA-Ac146pBlue, Ac11-MycpBlue, Ac76-MycpBlue, Ac78-MycpBlue, GP41-MycpBlue, Ac93-MycpBlue, Ac103-MycpBlue, Ac142-MycpBlue, Myc-Ac146pBlue, Lef3-MycpBlue, Vps2B-MycpBlue, Vps20-MycpBlue, Vps24-MycpBlue, Snf7-MycpBlue, Vps46-MycpBlue, Vps60-MycpBlue, Vps4-MycpBlue, K176Q-MycpBlue, E231Q-MycpBlue |
| BiFC assay | Ac11-CmpBlue, Ac76-CmpBlue, Ac78-CmpBlue, GP41-CmpBlue, Ac93-CmpBlue, Ac103-CmpBlue, Ac142-CmpBlue, Cm-Ac146pBlue, Lef3-CmpBlue, Ac11-NmpBlue, Ac76-NmpBlue, Ac78-NmpBlue, GP41-NmpBlue, Ac93-NmpBlue, Ac103-NmpBlue, Ac142-NmpBlue, Nm-Ac146pBlue, Vps2B-CmpBlue, Vps20-CmpBlue, Vps24-CmpBlue, Snf7-CmpBlue, Vps46-CmpBlue, Vps60-CmpBlue, Vps4-CmpBlue, K176Q-CmpBlue, E231Q-CmpBlue, Vps2B-NmpBlue, Vps20-NmpBlue, Vps24-NmpBlue, Snf7-NmpBlue, Vps46-NmpBlue, Vps60-NmpBlue, Vps4-NmpBlue, K176Q-NmpBlue, E231Q-NmpBlue |
Nm and Cm, N and C termini of mCherry, respectively.
Recombinant AcMNPV bacmids expressing GFP, GFP-tagged ESCRT-I or ESCRT-III components, or Vps4 mutant E231Q-GFP were constructed by inserting a cassette containing GFP, GFP-tagged ESCRT-I or ESCRT-III components, or E231Q-GFP under the control of the AcMNPV ie1 immediate early promoter, into either (i) a pFastbac plasmid (GUSpFB) that contains a β-glucuronidase (GUS) gene under the control of the AcMNPV p6.9 late promoter or (ii) a pFastbac plasmid (VP39-mCherrypFB) that contains AcMNPV vp39 gene fused with mCherry at its C terminus under the control of the vp39 native promoter. The resulting pFastbac constructs (GFPpFB, GFP-XpFB, Y-GFPpFB, E231Q-GFPpFB, GFP-VP39-mCherrypFB, Y-GFP-VP39-mCherrypFB, and E231Q-GFP-VP39-mCherrypFB, where X or Y represents ESCRT-I or ESCRT-III components, respectively) were each inserted into the polyhedrin locus of an AcMNPV bacmid (bMON14272) by Tn7-mediated transposition (83). The resulting recombinant bacmids were separately named GFPBac, GFP-XBac, Y-GFPBac, E231Q-GFPBac, GFP-VP39-mCherryBac, Y-GFP-VP39-mCherryBac, and E231Q-GFP-VP39-mCherryBac. All constructs were confirmed by restriction enzyme analysis and DNA sequencing. The control AcMNPV bacmid AcMNPV-LacZGUS and gp64 knockouts AcMNPV bacmid LacZGUS-gp64ko and mCherryGUS-gp64ko were constructed as described earlier (23). In these bacmids, the expression of the LacZ or mCherry reporter gene is directed by an OpMNPV ie2 immediate early promoter, and GUS is directed by the AcMNPV p6.9 late promoter. The plasmids and bacmids were purified by using the Midiprep kit (Invitrogen). The gp64 knockout virus LacZGUS-gp64ko and mCherryGUS-gp64ko were grown in Sf9OP1D cells, and their titers were determined. Wild-type AcMNPV encoding VP39-triple mCherry (3mC) was kindly provided by Taro Ohkawa and Matthew Welch (University of California, Berkeley) (31).
Infectivity complementation assay.
Sf9 cells and High5 cells in 12-well plates were transfected with a total of 4 μg of plasmid DNA per well comprising 2 μg of pBieGP64 (84) expressing AcMNPV GP64 and 2 μg of plasmid encoding either GFP, GFP-tagged ESCRT-I or ESCRT-III components, or E231Q-GFP. At 16 h p.t., the cells were infected with the gp64 knockout AcMNPV virus mCherryGUS-gp64ko (MOI, 1 or 5) that was produced in Sf9Op1D cells (79). At 24 h p.i., the infected cells and medium were collected separately. Titers of infectious viruses in the medium were determined by 50% tissue culture infective dose (TCID50) on Sf9OP1D cells. Cell samples were subjected to Western blot analysis.
RNAi assay.
The dsRNA-based RNAi assay was performed as described previously with modifications (81, 85). A fragment (305 to 495 bp) of the coding sequence of the components of ESCRT-I (Tsg101, Vps28) or ESCRT-III (Vps2B, Vps20, Vps24, Snf7, Vps46, and Vps60), Vps4, or GFP was amplified by PCR. PCR primers were designed with the SnapDragon tool (http://www.flyrnai.org/cgi-bin/RNAi_find_primers.pl), and each primer contained the T7 RNA polymerase promoter sequence (5′-TAATACGACTCACTATAGGG-3′) at the 5′-end (Table 3). The PCR products were purified using the QIAEXII gel extraction kit (Qiagen). The purified PCR products were used as the templates to produce dsRNA by using the T7 RiboMAX Express RNAi system (Promega). The dsRNA products were purified with RNeasy minikit (Qiagen) and analyzed by 1.2% agarose gel electrophoresis.
Sf9 cells in 12-well plates were transfected with 7.5 μg of dsRNA targeting the individual gene of ESCRT-I, ESCRT-III, or Vps4. Also, 7.5 μg of the GFP dsRNA was transfected as a negative control. The cell viability was determined using the CellTiter96 AQueous One solution cell proliferation assay (MTS; Promega) according to the manufacturer's recommendations. Briefly, at 24, 48, and 72 h p.t., the cells were incubated with CellTiter 96 AQueous One solution reagent for 2 h at 27°C, and absorbance at 490 nm was monitored using a 96-well plate reader (Tecan iControl Reader; Tecan Group Ltd., Mannedorf, Switzerland). The specific gene expression knockdown efficiency was determined by transfecting Sf9 cells with 1 μg of the plasmid expressing HA-tagged ESCRT-I, c-Myc-tagged ESCRT-III components, or Vps4, in combination either with 7.5 μg of dsRNA individually targeting a component of ESCRT-I, ESCRT-III, or Vps4 or with 7.5 μg dsRNA of GFP as a control dsRNA. At 48 h p.t., the transfected and cotransfected cells were collected and the expression of each of the HA- or c-Myc-tagged ESCRT-I, ESCRT-III, or Vps4 proteins was determined by Western blotting. Western blot results were quantified by using Quantity One software. For analysis of virus infection, Sf9 cells were transfected with 7.5 μg of dsRNA targeting the components of ESCRT-I, ESCRT-III, or Vps4 or 7.5 μg of the GFP dsRNA. At 48 h p.t., the transfected cells were infected with control AcMNPV at an MOI of 5. At 24 h p.i., the supernatants were collected and virus titers were measured by TCID50 assays on Sf9 cells.
Analysis of viral gene expression and DNA replication.
To determine the effects of DN ESCRT-I and ESCRT-III proteins on viral gene expression, Sf9 cells in a 12-well plate were cotransfected with 2 μg of pBieGP64 and 2 μg of the plasmid expressing GFP or GFP-tagged ESCRT-I or ESCRT-III proteins. At 16 h p.t., the cells were infected with the gp64 knockout virus AcMNPV LacZGUS-gp64ko at an MOI of 5. At 6 and 24 h p.i., the infected cells were fixed and stained or lysed, and reporter proteins were quantified as described previously (23). Briefly, the infected cells were washed once with phosphate-buffered saline (PBS; pH 7.4), fixed with 2% paraformaldehyde and 0.2% glutaraldehyde in PBS (pH 7.4) for 10 min, and then washed twice with PBS and permeabilized with a solution of 2 mM MgCl2, 0.01% deoxycholate, and 0.1% Nonidet P-40 (NP-40) for 15 min. The fixed and permeabilized cells were then stained with the beta-Gal substrate X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside; Gold Biotechnology) or stained with the GUS substrate X-Gluc (5-bromo-4-chloro-3-indolyl-β-d-glucuronic acid; Gold Biotechnology). Alternatively, the infected cells were solubilized in PBS containing 0.5% NP-40, and beta-Gal or GUS activities were quantified using the substrate chlorophenol red-β-d-galactopyranoside (CPRG; Roche Diagnostics GmbH) or 4-nitrophenyl β-d-glucuronide (PNPG; Sigma-Aldrich) and spectrometry (optical density [OD] at 570 nm or 405 nm, respectively).
To examine the effects of DN ESCRT-I and ESCRT-III proteins on viral DNA replication, real-time PCR was performed as described previously (23). In brief, Sf9 cells were cotransfected with 2 μg of pBieGP64 and 2 μg the plasmid expressing GFP or GFP-tagged ESCRT-I or ESCRT-III proteins. At 16 h p.t., cells were infected with the gp64 knockout virus LacZGUS-gp64ko (MOI = 5). At 24 h p.i., the infected cells were harvested and the total DNA was extracted with a DNeasy Blood & Tissue kit (Qiagen). The viral genomic DNA was determined by real-time PCR (23). Each PCR mixture contained 5 μl SYBR green PCR master mix (TaKaRa), 1.25 μM each primer (forward primer, 5′-GATCTTCCTGCGGGCCAAACACT-3′; reverse primer, 5′-AACAAGACCGCGCCTATCAACAAA-3′), and 300 pg of the total DNA. A 183-bp fragment of the AcMNPV ODV-e56 gene was amplified by PCR. A control plasmid ODV-e56pGEM-T containing the ORF of ODV-e56 was used to generate the standard curve. The amount of AcMNPV genomic DNA was calculated and expressed as the number of viral DNA copies in each cell.
Analysis of virus entry.
For analysis of the effects on virus entry of overexpression of ESCRT-I and ESCRT-III components or RNAi knockdown of ESCRT-I and ESCRT-III proteins, one set of Sf9 and High5 cells in 12-well plates were transfected with 2 μg of the plasmid expressing GFP, GFP-tagged ESCRT-I and ESCRT-III components, or E231Q-GFP. The other set of Sf9 cells in 12-well plates were transfected with 7.5 μg dsRNA targeting the component of ESCRT-I, ESCRT-III, Vps4, or the control GFP gene. At 16 h p.t. (for cells transfected with the plasmid) or 48 h p.t. (for cells transfected with dsRNA), the transfected cells were chilled at 4°C for 45 min and then inoculated with prechilled control AcMNPV (MOI = 10 TCID50) or 3mC virus (MOI = 20 TCID50). After 1 h of attachment at 4°C, the viral inoculum was removed and the cells were washed twice with chilled TNMFH medium and shifted to 27°C in TNMFH medium. After incubation at 27°C for 90 min, the wild-type AcMNPV-infected cells were collected and the amount of viral genomic DNA was measured by real-time PCR using the same primers and conditions as described above and expressed as viral DNA genome copies per cell. The 3mC virus-infected cells were fixed with 3.7% paraformaldehyde in PBS (pH 7.4) and examined by confocal microscopy as described below.
Analysis of infectious AcMNPV production and release.
For analysis of overexpression or knockdown of ESCRT-I and ESCRT-III components on infectious AcMNPV release, one set of Sf9 and High5 cells in 6-well plates were transfected with recombinant AcMNPV bacmid DNA (6 μg per well) expressing either GFP, GFP-tagged ESCRT-I or ESCRT-III, or E231Q-GFP. The other set of Sf9 cells in 12-well plates were transfected with 7.5 μg dsRNA targeting the component of ESCRT-I, ESCRT-III, Vps4, or GFP and were transfected again at 48 h p.t. with 3 μg AcMNPV bacmid DNA (AcMNPV-LacZGUS). After transfection with AcMNPV bacmid DNA and incubation for 24 h, the GFP-positive cells in the first set of transfected Sf9 and High5 cells were scored by epifluorescence microscope (Nikon Eclipse Ti) to estimate the transfection efficiency, and wells containing more than 1 × 104 cells were selected for analysis. Also, two sets of the transfected cells were lysed, and GUS (in the first set of transfected cells) or beta-Gal and GUS (in the second set of transfected cells) activities were quantified as described above. For each transfection, the cell culture medium from 3 wells of transfected cells was collected and centrifuged (3,000 × g, 10 min, 4°C) to remove the cellular debris. Infectious virus titers were determined by TCID50 assays on Sf9 or High5 cells. To track the effects of overexpression of ESCRT-III components Vps24, Snf7, and Vps60 and Vps4 mutant E231Q on budded virion release, Sf9 cells in 6-well plates were transfected with AcMNPV bacmid DNA (6 μg per well) expressing VP39-mCherry and either GFP, Vps24-GFP, Snf7-GFP, Vps60-GFP, or E231Q-GFP (GFP-VP39-mCherryBac, Vps24-GFP-VP39-mCherryBac, Snf7-GFP-VP39-mCherryBac, Vps60-GFP-VP39-mCherryBac, and E231Q-GFP-VP39-mCherryBac, respectively). At 24 h p.t., the transfected cells were fixed with 3.7% paraformaldehyde in PBS (pH 7.4) and examined by confocal microscopy as described below.
Confocal microscopy.
Sf9 or High5 cells that were plated on glass coverslips and transfected and/or infected were fixed with 3.7% paraformaldehyde in PBS (pH 7.4) for 10 min. Cells were then washed three times with PBS (pH 7.4) and permeabilized with 0.05% Triton X-100 in PBS (pH 7.4). The nuclei were stained with 1 μg/ml Hoechst 33258 (Invitrogen) for 8 min. After washing three times with PBS (pH 7.4), the cells were mounted on slides in Fluoromount-G reagent (Southern Biotech). Images were collected on a Nikon A1R+ confocal microscope (Nikon Instruments Inc., Melville, NY, USA) using a 63× oil immersion objective; numerical aperture (NA), 1.4. GFP was excited with a blue argon ion laser (488 nm), and emitted light was collected between 480 nm and 520 nm. mCherry was excited with an orange helium-neon laser (594 nm), and emitted light was collected from 580 nm to 650 nm. Hoechst 33258 was excited with UV light at approximately 350 nm, and emitted light was collected from 400 nm to 450 nm. GFP and mCherry signals were collected separately from the Hoechst 33258 signal and later superimposed. Images were processed using NIS-Elements Viewer software (version 4.0) and Adobe Photoshop CS5 (Adobe systems).
Transmission electron microscopy.
Sf9 cells in 6-well plates were transfected with AcMNPV bacmid DNA (6 μg per well) expressing VP39-mCherry and either GFP, Vps24-GFP, Snf7-GFP, Vps60-GFP, or E231Q-GFP (GFP-VP39-mCherryBac, Vps24-GFP-VP39-mCherryBac, Snf7-GFP-VP39-mCherryBac, Vps60-GFP-VP39-mCherryBac, or E231Q-GFP-VP39-mCherryBac, respectively). At 72 h p.t., the transfected cells were harvested by centrifugation (500 × g, 10 min) and fixed with 2.5% glutaraldehyde in PBS (pH 7.4) at 4°C overnight. Then, the cells were washed five times with PBS buffer (0.1 M, pH 7.2) and stained with 1% osmium tetroxide in PBS buffer (0.2 M, pH 7.2) for 2 h at 4°C. After washing five times in PBS buffer (0.1 M, pH 7.2), the fixed cells were dehydrated with a gradient of ethanol from 30% to 100%. The cells were then embedded in Epon-812 and dried for 48 h at 55°C. Ultrathin sections were prepared and stained with lead citrate and uranyl acetate. Images were collected using an HT7700 transmission electron microscope (Hitachi, Ltd. Japan).
Syncytium formation and cELISA analysis.
Sf9 cells in each well of a 6-well plate were transfected with 6 μg of a bacmid expressing GFP, GFP-tagged ESCRT-III components, or E231Q-GFP. At 24 h p.t., one set of the transfected cells were incubated with PBS at pH 5.0 for 3 min to induce syncytium formation, and another set of the transfected cells were used to quantify the cell surface level of the major viral envelope protein GP64 by cell surface enzyme-linked immunosorbent assay (cELISA). The syncytium formation and cELISA analyses were carried out as described previously (84, 86). Briefly, in the syncytium formation assay, Sf9 cells were fixed with methanol and stained with 0.1% eosin Y and 0.1% methylene blue. For cELISA analysis, Sf9 cells were fixed in 0.5% glutaraldehyde and the relative levels of GP64 localized at the cell surface were measured using primary MAb AcV5, a secondary goat anti-mouse antibody conjugated to beta-galactosidase, and the substrate chlorophenol red-beta-d-galactopyranoside (CPRG).
Coimmunoprecipitation.
Sf9 cells in 12-well plates were transfected or cotransfected with the plasmids expressing c-Myc-tagged ESCRT-III components, Vps4 or viral proteins, or HA-tagged viral proteins (2 μg DNA for each plasmid). At 36 h posttransfection, the cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (0.1% SDS, 50 mM Tris [pH 8.0], 150 mM NaCl, 5 mM EDTA, 0.5% sodium deoxycholate, 1% NP-40) containing protein inhibitor cocktail (Roche), and the supernatant was collected after centrifugation (15,000 × g, 15 min, 4°C). For immunoprecipitation, the lysate supernatants were mixed with protein G agarose beads (Pierce) and anti-HA MAb overnight at 4°C. After pelleting and washing twice with RIPA buffer, the agarose beads were resuspended with 1× SDS gel loading buffer (2% SDS, 10% glycerol, 2% 2-mercaptoethanol, 0.02% bromophenol blue, 0.05 M Tris [pH 6.8]) and analyzed by 10% or 15% SDS-PAGE and Western blotting.
Bimolecular fluorescent complementation assay.
Sf9 cells in 12-well plates were cotransfected with the BiFC paired plasmids (2 μg DNA for each construct). At 36 h p.t., bimolecular fluorescent complementation was assessed by imaging mCherry fluorescence in transfected Sf9 cells expressing pairs of N-terminal or C-terminal gene fusions to either the N or C terminus of mCherry (Nm or Cm, respectively). Fluorescence was observed with a Nikon Eclipse Ti epifluorescence microscope. The fluorescent cells in five randomly selected representative fields were scored for each pair of constructs. The paired proteins' interaction was evaluated as described previously (5) by the ratio of the number of fluorescent cells in one field with the total number of cells in that field. The transfected cells were also collected for Western blot analysis of the target proteins expression.
Western blot analysis.
Proteins were separated by 10% or 15% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membrane (Millipore). GFP and GFP-tagged proteins were detected on Western blots with an anti-GFP polyclonal antibody (GenScript), HA- or c-Myc-tagged proteins were detected with anti-HA MAb or an anti-Myc polyclonal antibody (GenScript), and GP64 or actin was detected using MAb AcV5 (Santa Cruz Biotechnology) or anti-β-actin monoclonal antibodies (Abbkine). Immunoreactive proteins were visualized using alkaline phosphatase-conjugated anti-mouse or anti-rabbit IgG antibody and nitroblue tetrazolium/5-bromo-4-chloro-3-indolylphosphate (NBT/BCIP; Promega).
Accession number(s).
The Spodoptera frugiperda ESCRT-I component Vps23/Tsg101 and Vps28 and ESCRT-III component Vps2B, Vps20, Vps24, Vps32/Snf7, and Vps60 genes were deposited under GenBank accession numbers KY694523, KY694524, KY694525, KY694526, KY694527, KY694528, and KY694529.
Supplementary Material
ACKNOWLEDGMENTS
We thank Taro Ohkawa and Matthew Welch for the kind gift of virus 3mC.
This work was supported by grants from the National Natural Science Foundation of China (NSFC; 31672082 and 31272088) and the National Key R&D Program of China (2017YFC1200605) to Z.L. and grants from the National Science Foundation (NSF; IOS-1354421) and the U.S. Department of Agriculture (USDA; 2015-67013-23281) to G.W.B.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01636-17.
REFERENCES
- 1.Henne WM, Buchkovich NJ, Emr SD. 2011. The ESCRT pathway. Dev Cell 21:77–91. doi: 10.1016/j.devcel.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 2.Hurley JH. 2015. ESCRTs are everywhere. EMBO J 34:2398–2407. doi: 10.15252/embj.201592484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guizetti J, Gerlich DW. 2012. ESCRT-III polymers in membrane neck constriction. Trends Cell Biol 22:133–140. doi: 10.1016/j.tcb.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 4.Henne WM, Stenmark H, Emr SD. 2013. Molecular mechanisms of the membrane sculpting ESCRT pathway. Cold Spring Harb Perspect Biol 5(9):a016766. doi: 10.1101/cshperspect.a016766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Webster BM, Colombi P, Jager J, Lusk CP. 2014. Surveillance of nuclear pore complex assembly by ESCRT-III/Vps4. Cell 159:388–401. doi: 10.1016/j.cell.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCullough J, Colf LA, Sundquist WI. 2013. Membrane fission reactions of the mammalian ESCRT pathway. Annu Rev Biochem 82:663–692. doi: 10.1146/annurev-biochem-072909-101058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang B, Stjepanovic G, Shen Q, Martin A, Hurley JH. 2015. Vps4 disassembles an ESCRT-III filament by global unfolding and processive translocation. Nat Struct Mol Biol 22:492–498. doi: 10.1038/nsmb.3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Norgan AP, Davies BA, Azmi IF, Schroeder AS, Payne JA, Lynch GM, Xu Z, Katzmann DJ. 2013. Relief of autoinhibition enhances Vta1 activation of Vps4 via the Vps4 stimulatory element. J Biol Chem 288:26147–26156. doi: 10.1074/jbc.M113.494112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wollert T, Hurley JH. 2010. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature 464:864–869. doi: 10.1038/nature08849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jimenez AJ, Maiuri P, Lafaurie-Janvore J, Divoux S, Piel M, Perez F. 2014. ESCRT machinery is required for plasma membrane repair. Science 343:1247136. doi: 10.1126/science.1247136. [DOI] [PubMed] [Google Scholar]
- 11.Vietri M, Schink KO, Campsteijn C, Wegner CS, Schultz SW, Christ L, Thoresen SB, Brech A, Raiborg C, Stenmark H. 2015. Spastin and ESCRT-III coordinate mitotic spindle disassembly and nuclear envelope sealing. Nature 522:231–235. doi: 10.1038/nature14408. [DOI] [PubMed] [Google Scholar]
- 12.Votteler J, Sundquist WI. 2013. Virus budding and the ESCRT pathway. Cell Host Microbe 14:232–241. doi: 10.1016/j.chom.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakayama K. 2016. Regulation of cytokinesis by membrane trafficking involving small GTPases and the ESCRT machinery. Crit Rev Biochem Mol Biol 51:1–6. doi: 10.3109/10409238.2015.1085827. [DOI] [PubMed] [Google Scholar]
- 14.Prescher J, Baumgartel V, Ivanchenko S, Torrano AA, Brauchle C, Muller B, Lamb DC. 2015. Super-resolution imaging of ESCRT-proteins at HIV-1 assembly sites. PLoS Pathog 11:e1004677. doi: 10.1371/journal.ppat.1004677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Engelenburg SB, Shtengel G, Sengupta P, Waki K, Jarnik M, Ablan SD, Freed EO, Hess HF, Lippincott-Schwartz J. 2014. Distribution of ESCRT machinery at HIV assembly sites reveals virus scaffolding of ESCRT subunits. Science 343:653–656. doi: 10.1126/science.1247786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bleck M, Itano MS, Johnson DS, Thomas VK, North AJ, Bieniasz PD, Simon SM. 2014. Temporal and spatial organization of ESCRT protein recruitment during HIV-1 budding. Proc Natl Acad Sci U S A 111:12211–12216. doi: 10.1073/pnas.1321655111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wirblich C, Bhattacharya B, Roy P. 2006. Nonstructural protein 3 of bluetongue virus assists virus release by recruiting ESCRT-I protein Tsg101. J Virol 80:460–473. doi: 10.1128/JVI.80.1.460-473.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng Z, Hensley L, McKnight KL, Hu F, Madden V, Ping L, Jeong SH, Walker C, Lanford RE, Lemon SM. 2013. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 496:367–371. doi: 10.1038/nature12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silva-Ayala D, Lopez T, Gutierrez M, Perrimon N, Lopez S, Arias CF. 2013. Genome-wide RNAi screen reveals a role for the ESCRT complex in rotavirus cell entry. Proc Natl Acad Sci U S A 110:10270–10275. doi: 10.1073/pnas.1304932110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shtanko O, Nikitina RA, Altuntas CZ, Chepurnov AA, Davey RA. 2014. Crimean-Congo hemorrhagic fever virus entry into host cells occurs through the multivesicular body and requires ESCRT regulators. PLoS Pathog 10:e1004390. doi: 10.1371/journal.ppat.1004390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Veettil MV, Kumar B, Ansari MA, Dutta D, Iqbal J, Gjyshi O, Bottero V, Chandran B. 2016. ESCRT-0 component Hrs promotes macropinocytosis of Kaposi's sarcoma-associated herpesvirus in human dermal microvascular endothelial cells. J Virol 90:3860–3872. doi: 10.1128/JVI.02704-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pasqual G, Rojek JM, Masin M, Chatton JY, Kunz S. 2011. Old world arenaviruses enter the host cell via the multivesicular body and depend on the endosomal sorting complex required for transport. PLoS Pathog 7:e1002232. doi: 10.1371/journal.ppat.1002232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Z, Blissard GW. 2012. Cellular VPS4 is required for efficient entry and egress of budded virions of Autographa californica multiple nucleopolyhedrovirus. J Virol 86:459–472. doi: 10.1128/JVI.06049-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diaz A, Zhang J, Ollwerther A, Wang X, Ahlquist P. 2015. Host ESCRT proteins are required for bromovirus RNA replication compartment assembly and function. PLoS Pathog 11:e1004742. doi: 10.1371/journal.ppat.1004742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rohrmann G. 2013. Baculovirus molecular biology, 3rd ed National Center for Biotechnology Information, Bethesda, MD. [PubMed] [Google Scholar]
- 26.Shi Y, Li K, Tang P, Li Y, Zhou Q, Yang K, Zhang Q. 2015. Three-dimensional visualization of the Autographa californica multiple nucleopolyhedrovirus occlusion-derived virion envelopment process gives new clues as to its mechanism. Virology 476:298–303. doi: 10.1016/j.virol.2014.11.030. [DOI] [PubMed] [Google Scholar]
- 27.Braunagel SC, Elton DM, Ma H, Summers MD. 1996. Identification and analysis of an Autographa californica nuclear polyhedrosis virus structural protein of the occlusion-derived virus envelope: ODV-E56. Virology 217:97–110. doi: 10.1006/viro.1996.0097. [DOI] [PubMed] [Google Scholar]
- 28.Long G, Pan X, Kormelink R, Vlak JM. 2006. Functional entry of baculovirus into insect and mammalian cells is dependent on clathrin-mediated endocytosis. J Virol 80:8830–8833. doi: 10.1128/JVI.00880-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blissard GW, Wenz JR. 1992. Baculovirus gp64 envelope glycoprotein is sufficient to mediate pH-dependent membrane fusion. J Virol 66:6829–6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou J, Blissard GW. 2008. Identification of a GP64 subdomain involved in receptor binding by budded virions of the baculovirus Autographica californica multicapsid nucleopolyhedrovirus. J Virol 82:4449–4460. doi: 10.1128/JVI.02490-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohkawa T, Volkman LE, Welch MD. 2010. Actin-based motility drives baculovirus transit to the nucleus and cell surface. J Cell Biol 190:187–195. doi: 10.1083/jcb.201001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Au S, Pante N. 2012. Nuclear transport of baculovirus: revealing the nuclear pore complex passage. J Struct Biol 177:90–98. doi: 10.1016/j.jsb.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 33.Oomens AG, Blissard GW. 1999. Requirement for GP64 to drive efficient budding of Autographa californica multicapsid nucleopolyhedrovirus. Virology 254:297–314. doi: 10.1006/viro.1998.9523. [DOI] [PubMed] [Google Scholar]
- 34.Hu Z, Yuan M, Wu W, Liu C, Yang K, Pang Y. 2010. Autographa californica multiple nucleopolyhedrovirus ac76 is involved in intranuclear microvesicle formation. J Virol 84:7437–7447. doi: 10.1128/JVI.02103-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tao XY, Choi JY, Kim WJ, Lee JH, Liu Q, Kim SE, An SB, Lee SH, Woo SD, Jin BR, Je YH. 2013. The Autographa californica multiple nucleopolyhedrovirus ORF78 is essential for budded virus production and general occlusion body formation. J Virol 87:8441–8450. doi: 10.1128/JVI.01290-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuan M, Wu W, Liu C, Wang Y, Hu Z, Yang K, Pang Y. 2008. A highly conserved baculovirus gene p48 (ac103) is essential for BV production and ODV envelopment. Virology 379:87–96. doi: 10.1016/j.virol.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 37.Yuan M, Huang Z, Wei D, Hu Z, Yang K, Pang Y. 2011. Identification of Autographa californica nucleopolyhedrovirus ac93 as a core gene and its requirement for intranuclear microvesicle formation and nuclear egress of nucleocapsids. J Virol 85:11664–11674. doi: 10.1128/JVI.05275-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCarthy CB, Dai X, Donly C, Theilmann DA. 2008. Autographa californica multiple nucleopolyhedrovirus ac142, a core gene that is essential for BV production and ODV envelopment. Virology 372:325–339. doi: 10.1016/j.virol.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 39.Dickison VL, Willis LG, Sokal NR, Theilmann DA. 2012. Deletion of AcMNPV ac146 eliminates the production of budded virus. Virology 431:29–39. doi: 10.1016/j.virol.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 40.Li Z, Blissard G. 2015. The vacuolar protein sorting genes in insects: a comparative genome view. Insect Biochem Mol Biol 62:211–225. doi: 10.1016/j.ibmb.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 41.Chen YR, Zhong S, Fei Z, Gao S, Zhang S, Li Z, Wang P, Blissard GW. 2014. Transcriptome responses of the host Trichoplusia ni to infection by the baculovirus Autographa californica multiple nucleopolyhedrovirus. J Virol 88:13781–13797. doi: 10.1128/JVI.02243-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goila-Gaur R, Demirov DG, Orenstein JM, Ono A, Freed EO. 2003. Defects in human immunodeficiency virus budding and endosomal sorting induced by TSG101 overexpression. J Virol 77:6507–6519. doi: 10.1128/JVI.77.11.6507-6519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martin-Serrano J, Zang T, Bieniasz PD. 2003. Role of ESCRT-I in retroviral budding. J Virol 77:4794–4804. doi: 10.1128/JVI.77.8.4794-4804.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson MC, Spidel JL, Ako-Adjei D, Wills JW, Vogt VM. 2005. The C-terminal half of TSG101 blocks Rous sarcoma virus budding and sequesters Gag into unique nonendosomal structures. J Virol 79:3775–3786. doi: 10.1128/JVI.79.6.3775-3786.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pineda-Molina E, Belrhali H, Piefer AJ, Akula I, Bates P, Weissenhorn W. 2006. The crystal structure of the C-terminal domain of Vps28 reveals a conserved surface required for Vps20 recruitment. Traffic 7:1007–1016. doi: 10.1111/j.1600-0854.2006.00440.x. [DOI] [PubMed] [Google Scholar]
- 46.Zamborlini A, Usami Y, Radoshitzky SR, Popova E, Palu G, Gottlinger H. 2006. Release of autoinhibition converts ESCRT-III components into potent inhibitors of HIV-1 budding. Proc Natl Acad Sci U S A 103:19140–19145. doi: 10.1073/pnas.0603788103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bajorek M, Schubert HL, McCullough J, Langelier C, Eckert DM, Stubblefield WM, Uter NT, Myszka DG, Hill CP, Sundquist WI. 2009. Structural basis for ESCRT-III protein autoinhibition. Nat Struct Mol Biol 16:754–762. doi: 10.1038/nsmb.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muziol T, Pineda-Molina E, Ravelli RB, Zamborlini A, Usami Y, Gottlinger H, Weissenhorn W. 2006. Structural basis for budding by the ESCRT-III factor CHMP3. Dev Cell 10:821–830. doi: 10.1016/j.devcel.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 49.Strack B, Calistri A, Craig S, Popova E, Gottlinger HG. 2003. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114:689–699. doi: 10.1016/S0092-8674(03)00653-6. [DOI] [PubMed] [Google Scholar]
- 50.Teis D, Saksena S, Emr SD. 2008. Ordered assembly of the ESCRT-III complex on endosomes is required to sequester cargo during MVB formation. Dev Cell 15:578–589. doi: 10.1016/j.devcel.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 51.Olszewski J, Miller LK. 1997. A role for baculovirus GP41 in budded virus production. Virology 233:292–301. doi: 10.1006/viro.1997.8612. [DOI] [PubMed] [Google Scholar]
- 52.Tao XY, Choi JY, Kim WJ, An SB, Liu Q, Kim SE, Lee SH, Kim JH, Woo SD, Jin BR, Je YH. 2015. Autographa californica multiple nucleopolyhedrovirus ORF11 is essential for budded-virus production and occlusion-derived-virus envelopment. J Virol 89:373–383. doi: 10.1128/JVI.01742-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peng K, Wu M, Deng F, Song J, Dong C, Wang H, Hu Z. 2010. Identification of protein-protein interactions of the occlusion-derived virus-associated proteins of Helicoverpa armigera nucleopolyhedrovirus. J Gen Virol 91:659–670. doi: 10.1099/vir.0.017103-0. [DOI] [PubMed] [Google Scholar]
- 54.Wei D, Wang Y, Zhang X, Hu Z, Yuan M, Yang K. 2014. Autographa californica nucleopolyhedrovirus Ac76: a dimeric type II integral membrane protein that contains an inner nuclear membrane-sorting motif. J Virol 88:1090–1103. doi: 10.1128/JVI.02392-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kostelansky MS, Schluter C, Tam YY, Lee S, Ghirlando R, Beach B, Conibear E, Hurley JH. 2007. Molecular architecture and functional model of the complete yeast ESCRT-I heterotetramer. Cell 129:485–498. doi: 10.1016/j.cell.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee HH, Elia N, Ghirlando R, Lippincott-Schwartz J, Hurley JH. 2008. Midbody targeting of the ESCRT machinery by a noncanonical coiled coil in CEP55. Science 322:576–580. doi: 10.1126/science.1162042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gill DJ, Teo H, Sun J, Perisic O, Veprintsev DB, Emr SD, Williams RL. 2007. Structural insight into the ESCRT-I/-II link and its role in MVB trafficking. EMBO J 26:600–612. doi: 10.1038/sj.emboj.7601501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lata S, Roessle M, Solomons J, Jamin M, Gottlinger HG, Svergun DI, Weissenhorn W. 2008. Structural basis for autoinhibition of ESCRT-III CHMP3. J Mol Biol 378:818–827. doi: 10.1016/j.jmb.2008.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schuh AL, Audhya A. 2014. The ESCRT machinery: from the plasma membrane to endosomes and back again. Crit Rev Biochem Mol Biol 49:242–261. doi: 10.3109/10409238.2014.881777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Griffiths CM, Barnett AL, Ayres MD, Windass J, King LA, Possee RD. 1999. In vitro host range of Autographa californica nucleopolyhedrovirus recombinants lacking functional p35, iap1 or iap2. J Gen Virol 80(Part 4):1055–1066. [DOI] [PubMed] [Google Scholar]
- 61.Zhu S, Wang W, Wang Y, Yuan M, Yang K. 2013. The baculovirus core gene ac83 is required for nucleocapsid assembly and per os infectivity of Autographa californica nucleopolyhedrovirus. J Virol 87:10573–10586. doi: 10.1128/JVI.01207-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marek M, van Oers MM, Devaraj FF, Vlak JM, Merten OW. 2011. Engineering of baculovirus vectors for the manufacture of virion-free biopharmaceuticals. Biotechnol Bioeng 108:1056–1067. doi: 10.1002/bit.23028. [DOI] [PubMed] [Google Scholar]
- 63.Lottridge JM, Flannery AR, Vincelli JL, Stevens TH. 2006. Vta1p and Vps46p regulate the membrane association and ATPase activity of Vps4p at the yeast multivesicular body. Proc Natl Acad Sci U S A 103:6202–6207. doi: 10.1073/pnas.0601712103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shim JH, Xiao C, Hayden MS, Lee KY, Trombetta ES, Pypaert M, Nara A, Yoshimori T, Wilm B, Erdjument-Bromage H, Tempst P, Hogan BL, Mellman I, Ghosh S. 2006. CHMP5 is essential for late endosome function and down-regulation of receptor signaling during mouse embryogenesis. J Cell Biol 172:1045–1056. doi: 10.1083/jcb.200509041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang Z, Vild C, Ju J, Zhang X, Liu J, Shen J, Zhao B, Lan W, Gong F, Liu M, Cao C, Xu Z. 2012. Structural basis of molecular recognition between ESCRT-III-like protein Vps60 and AAA-ATPase regulator Vta1 in the multivesicular body pathway. J Biol Chem 287:43899–43908. doi: 10.1074/jbc.M112.390724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Luyet PP, Falguieres T, Pons V, Pattnaik AK, Gruenberg J. 2008. The ESCRT-I subunit TSG101 controls endosome-to-cytosol release of viral RNA. Traffic 9:2279–2290. doi: 10.1111/j.1600-0854.2008.00820.x. [DOI] [PubMed] [Google Scholar]
- 67.Le Blanc I, Luyet PP, Pons V, Ferguson C, Emans N, Petiot A, Mayran N, Demaurex N, Faure J, Sadoul R, Parton RG, Gruenberg J. 2005. Endosome-to-cytosol transport of viral nucleocapsids. Nat Cell Biol 7:653–664. doi: 10.1038/ncb1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fraser M. 1986. Ultrastructural observations of virion maturation in Autographa californica nuclear polyhedrosis virus infected Spodoptera frugiperda cell cultures. J Ultrastruct Mol Struct Res 95:189–195. doi: 10.1016/0889-1605(86)90040-6. [DOI] [Google Scholar]
- 69.Biswas S, Blissard GW, Theilmann DA. 2016. Trichoplusia ni kinesin-1 associates with AcMNPV nucleocapsid proteins and is required for the production of budded virus. J Virol 90:3480–3495. doi: 10.1128/JVI.02912-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Martin-Serrano J, Neil SJ. 2011. Host factors involved in retroviral budding and release. Nat Rev Microbiol 9:519–531. doi: 10.1038/nrmicro2596. [DOI] [PubMed] [Google Scholar]
- 71.Morita E, Sandrin V, McCullough J, Katsuyama A, Baci Hamilton I, Sundquist WI. 2011. ESCRT-III protein requirements for HIV-1 budding. Cell Host Microbe 9:235–242. doi: 10.1016/j.chom.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Demirov DG, Ono A, Orenstein JM, Freed EO. 2002. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc Natl Acad Sci U S A 99:955–960. doi: 10.1073/pnas.032511899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morita E, Sundquist WI. 2004. Retrovirus budding. Annu Rev Cell Dev Biol 20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- 74.Pawliczek T, Crump CM. 2009. Herpes simplex virus type 1 production requires a functional ESCRT-III complex but is independent of TSG101 and ALIX expression. J Virol 83:11254–11264. doi: 10.1128/JVI.00574-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee CP, Liu PT, Kung HN, Su MT, Chua HH, Chang YH, Chang CW, Tsai CH, Liu FT, Chen MR. 2012. The ESCRT machinery is recruited by the viral BFRF1 protein to the nucleus-associated membrane for the maturation of Epstein-Barr Virus. PLoS Pathog 8:e1002904. doi: 10.1371/journal.ppat.1002904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chiaruttini N, Redondo-Morata L, Colom A, Humbert F, Lenz M, Scheuring S, Roux A. 2015. Relaxation of loaded ESCRT-III spiral springs drives membrane deformation. Cell 163:866–879. doi: 10.1016/j.cell.2015.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Afonso CL, Tulman ER, Lu Z, Balinsky CA, Moser BA, Becnel JJ, Rock DL, Kutish GF. 2001. Genome sequence of a baculovirus pathogenic for Culex nigripalpus. J Virol 75:11157–11165. doi: 10.1128/JVI.75.22.11157-11165.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen YR, Zhong S, Fei Z, Hashimoto Y, Xiang JZ, Zhang S, Blissard GW. 2013. The transcriptome of the baculovirus Autographa californica multiple nucleopolyhedrovirus in Trichoplusia ni cells. J Virol 87:6391–6405. doi: 10.1128/JVI.00194-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Plonsky I, Cho MS, Oomens AG, Blissard G, Zimmerberg J. 1999. An analysis of the role of the target membrane on the Gp64-induced fusion pore. Virology 253:65–76. doi: 10.1006/viro.1998.9493. [DOI] [PubMed] [Google Scholar]
- 80.Negre V, Hotelier T, Volkoff AN, Gimenez S, Cousserans F, Mita K, Sabau X, Rocher J, Lopez-Ferber M, d'Alencon E, Audant P, Sabourault C, Bidegainberry V, Hilliou F, Fournier P. 2006. SPODOBASE: an EST database for the lepidopteran crop pest Spodoptera. BMC Bioinformatics 7:322. doi: 10.1186/1471-2105-7-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Guo Y, Yue Q, Gao J, Wang Z, Chen YR, Blissard GW, Liu TX, Li Z. 2017. Roles of cellular NSF protein in entry and nuclear egress of budded virions of Autographa californica multiple nucleopolyhedrovirus. J Virol 91: doi: 10.1128/JVI.01111-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fan JY, Cui ZQ, Wei HP, Zhang ZP, Zhou YF, Wang YP, Zhang XE. 2008. Split mCherry as a new red bimolecular fluorescence complementation system for visualizing protein-protein interactions in living cells. Biochem Biophys Res Commun 367:47–53. doi: 10.1016/j.bbrc.2007.12.101. [DOI] [PubMed] [Google Scholar]
- 83.Luckow VA, Lee SC, Barry GF, Olins PO. 1993. Efficient generation of infectious recombinant baculoviruses by site-specific transposon-mediated insertion of foreign genes into a baculovirus genome propagated in Escherichia coli. J Virol 67:4566–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li Z, Blissard GW. 2008. Functional analysis of the transmembrane (TM) domain of the Autographa californica multicapsid nucleopolyhedrovirus GP64 protein: substitution of heterologous TM domains. J Virol 82:3329–3341. doi: 10.1128/JVI.02104-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Deng Z, Huang Z, Yuan M, Yang K, Pang Y. 2014. Baculovirus induces host cell aggregation via a Rho/Rok-dependent mechanism. J Gen Virol 95:2310–2320. doi: 10.1099/vir.0.066811-0. [DOI] [PubMed] [Google Scholar]
- 86.Yu Q, Blissard GW, Liu TX, Li Z. 2016. Autographa californica multiple nucleopolyhedrovirus GP64 protein: analysis of domain I and V amino acid interactions and membrane fusion activity. Virology 488:259–270. doi: 10.1016/j.virol.2015.11.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.