

SUMMARY
Shigella is a Gram-negative bacterium that causes bacillary dysentery worldwide. It invades the intestinal epithelium to elicit intense inflammation and tissue damage, yet the underlying mechanisms of its host selectivity and low infectious inoculum remain perplexing. Here we have reported that Shigella co-opts human α-defensin 5 (HD5), a host defense peptide important for intestinal homeostasis and innate immunity, to enhance its adhesion to and invasion of mucosal tissues. HD5 promoted Shigella infection in vitro in a structure-dependent manner. Shigella, commonly devoid of effective host-adhesion apparatus, preferentially targeted HD5 to augment its ability to colonize the intestinal epithelium through interactions with multiple bacterial membrane proteins. HD5 exacerbated infectivity and Shigella-induced pathology in a culture of human colorectal tissues and three animal models. Our findings illuminate how Shigella exploits innate immunity by turning HD5 into a virulence factor for infection, unveiling a mechanism of action for this highly proficient human pathogen.
INTRODUCTION
Intestinal colonization and epithelial adhesion is a crucial early event in the pathogenesis of many enteropathogens, which can then enable bacterial invasion of host epithelial cells and disseminated infection (Cossart and Sansonetti, 2004; Donnenberg, 2000; Pizarro-Cerdá and Cossart, 2006). Most enterobacteria use fimbriae, an adhesive filamentous organelle protruding from the outer-membrane surface of Gram-negative bacteria, for host attachment (Choudhury et al., 1999; Kline et al., 2009; Li et al., 2009). Paradoxically, Shigella, the etiological agent of bacillary dysentery, lacks such adhesion machinery in general, yet it is a remarkably infectious and contagious enteropathogen that invades and elicits intense inflammation and tissue damage of the colorectal epithelium (Carayol and Tran Van Nhieu, 2013; Perdomo et al., 1994; Phalipon and Sansonetti, 2007; Schroeder and Hilbi, 2008). Despite a continued search for mechanisms of adhesion, the question of how Shigella has acquired extraordinary infectivity without a highly efficient and more general host adhesion apparatus remains unanswered. In studying the mode of action of antimicrobial peptides against Shigella, we found that when the human enteric α-defensin 5 (HD5), an abundant and important host protective molecule produced by Paneth cells of the small intestine (Bevins and Salzman, 2011), binds Shigella, it augments infectivity via enhanced bacterial adhesion to and subsequent invasion of epithelial cells and tissues. We posited that Shigella subverts innate host defense to colonize and destroy the intestinal epithelium by turning HD5 into a molecular accomplice that imparts its infectivity and host selectivity.
RESULTS
HD5 promotes Shigella infection of epithelial cells in vitro
Antimicrobial peptides, expressed primarily in phagocytes and epithelia, play critical roles in host immune defense against pathogenic infection often through microbicidal activity (Bevins and Salzman, 2011; Ganz, 2003; Lehrer and Lu, 2012; Selsted and Ouellette, 2005; Zasloff, 2002). To study the role of antimicrobial peptides in Shigella pathogenesis, we tested a panel of six human and two murine defensin peptides against the Shigella flexneri strain Sf301 in an in vitro antibacterial activity assay (Ericksen et al., 2005; Mastroianni and Ouellette, 2009), including human neutrophil alpha-defensin 1 (HNP1), enteric alpha-defensins 5 and 6 (HD5 and HD6), beta-defensins 2 and 3 (HBD2 and HBD3), the cathelicidin peptide LL-37, and murine alpha-defensins (cryptdins) 3 and 4 (Crp3 and Crp4). While most of the human antimicrobial peptides displayed varying but weak bactericidal activity at low micromolar concentrations, HD6 and the two cryptdins showed little killing (Fig. S1A). Weak antibacterial activity was also observed for HD5 with the Shigella sonnei strain Ss86 and five clinical isolates (Fig. S1B). To test whether this antibacterial activity correlated with the ability of these peptides to inhibit Shigella infection in vitro, we quantified bacterial adhesion, invasion and intracellular replication in a conventional infection assay using HeLa cells (Fig. S2). When added to Sf301, two human alpha-defensins, HNP1 and HD5, enhanced Shigella adhesion to and invasion of HeLa cells in an inoculum-dependent manner, with the enteric alpha-defensin HD5 being markedly more active than its neutrophil counterpart HNP1 (Fig. 1A–B). In fact, HD5 at 2 μM enhanced bacterial adhesion or invasion by more than 30-fold. Our subsequent study thus focused on HD5 not only for its site of abundant expression relevant to Shigella infection, but also for its superior infection-enhancing activity.
Fig. 1. HD5 promotes Shigella infection of epithelial cells in vitro.

(A, B) The effects of eight antimicrobial peptides at sub-lethal concentrations on Shigella flexneri Sf301 adhesion (A) to and invasion (B) of HeLa cells during bacterial infection. (C, D) The effects of HD5 treatment on Sf301 invasion (C) and proliferation (D) when added during initial infection (co-incubation) or after invasion (post-infection). Adhesion, invasion and proliferation assays were performed as described in Methods. Experimental details are illustrated in Fig. S2. Data are shown as mean ± SD of at least three independent experiments. Statistical significance was calculated (for peptide-treated samples compared to vehicle controls (0 μM)) using a one-way ANOVA (Dunnett’s multiple comparison Test), and p values are as follows: *p < 0.05, **p < 0.01, and ***p < 0.001. (E) Fluorescence microscopy (left panels) and scanning electron microscopy (right panels) analysis of Sf301 adhesion to HeLa cells in the absence (control) or presence of 4 μM HD5 (MOI=50:1). GFP-expressing bacteria are green, β1 integrin is red, and nuclei are blue (DAPI). For fluorescent images, the scale bars represent 50 m; for SEM images, the bars represent 10 μm. (F) Confocal microscopy images of HeLa cells infected for 60 min with mCherry-labeled WT Sf301 harboring a GFP-expressing reporter plasmid (Campbell-Valois et al., 2014) in presence or absence of 4 μM HD5. HeLa cells are counterstained with DAPI, and GFP expression is induced upon activation of type 3 secretion system triggered by bacterial cell contact with the host. Note that red and green overlay gives rise to yellow. The scale bars represent 50 μm.
Dissection of time-dependent cellular events in Shigella infection further revealed that HD5 acted early, predominantly at the bacterial infection step (adhesion and invasion) rather than on intracellular replication (Fig. 1C–D). Similar results were obtained for HD5 with Shigella sonnei and also multiple clinical isolates (Fig. S1C–D). Consistent with these findings, immunofluorescence and scanning electron microscopy (SEM) studies showed that HD5-treated GFP-expressing or unlabeled Sf301 clustered on the surface of HeLa cells within 10 min of bacteria and HeLa coincubation (Fig. 1E). To verify the viability of adhered Shigella, we transformed mCherry-labeled Sf301 (red) with a reporter plasmid that expresses GFP (green) when the type 3 secretion system (T3SS) is activated upon host cell contact (Campbell-Valois et al., 2014), an obligate step in Shigella invasion, and found that HD5 treatment turned many clustered bacteria yellow (colocalized green and red) within 60 min of coincubation with HeLa cells (Fig. 1F). Without host cells, HD5 alone failed to change the color of bacteria as illustrated in Figs. 1F and S1G, suggesting that it did not activate the T3SS directly. Furthermore, HD5 was found to promote strong adhesion of Shigella even when the type III secretion system (T3SS), responsible for bacterial invasion and virulence, was inactivated either transcriptionally at low growth temperature (Maurelli et al., 1984) or genetically by deletion of spa33 encoding an essential component of the T3SS (Morita-Ishihara et al., 2006) (Fig. S1E). In addition, HD5-enhanced Shigella infection was not restricted to HeLa cells, albeit the standard in vitro infection model for Shigella (Philpott et al., 2000), as a wide variety of epithelial cell lines of different species and/or tissue origins were found equally susceptible (Fig. S1F and Table S1). Taken together, these results indicate that HD5 promoted Shigella infection in vitro by enhancing bacterial adhesion to epithelial cells, leading to increased bacterial invasion.
HD5 promotes Shigella infection in vivo
The impact of HD5 on the in vivo invasiveness of Shigella was extensively examined in three different animal models: (1) cornea infection in guinea pigs (the classic “Sereny test”) (Sereny, 1955), (2) colon infection in guinea pigs (Arena et al., 2015; Shim et al., 2007), and (3) ileum and colon infection in mice (Sawasvirojwong et al., 2013). For the Sereny test, Sf301 was inoculated in the eye at a density of 1×106 CFU/eye, together with HD5 at 0, 4, and 8 μM; the severity of infection graded from 1 to 3 was scored daily for one week (Fig. S3A). Eye infection worsened over time with the symptoms developing much more rapidly in the HD5-treated groups than the control group. As shown in Fig. 2A, in the absence of HD5, three out of fifteen animals developed full-blown keratoconjunctivitis and one showed signs of mild irritation, after one week. By sharp contrast, in the presence of 4 μM HD5 the number of animals with high grade keratoconjunctivitis increased to 6 in just three days (Fig. S3A) and to 11 (of 15) in one week, and three animals died in the group treated with 8 μM HD5. Of note, treatment with HD5 alone at 8 μM had no adverse effects on the animals (Fig. 2A). These data support that HD5 facilitated bacterial adhesion to and invasion of corneal epithelial cells in guinea pigs.
Fig. 2. HD5 promotes Shigella infection in vivo.

(A) Sereny test of Shigella infection using guinea pigs. Hartley guinea pigs (6–8 weeks of age) were inoculated with 106 CFU/eye of mid-log phase Sf301 either in the absence (n=15) and presence of HD5 (either 4 μM (n=14) or 8 μM (n=14)). A control group (n=6) was inoculated with HD5 alone (8 μM). Animals were observed and scored for the development of conjunctivitis over 7 consecutive days. Eye pathology was independently scored by three individuals (blinded to treatment group) on a scale of 0–3, grade 0 (no disease or mild irritation), grade 1 (mild conjunctivitis or late development and/or rapid clearing of symptoms), grade 2 (keratoconjunctivitis without purulence), and grade 3 (fully developed keratoconjunctivitis with purulence) as indicated in Fig. S3A. The data are representative of three independent experiments. Each point represents a single animal. Statistical significance compared with inoculation of 106 CFU in the absence of HD5 was determined using a Mann-Whitney test, *p < 0.05, **p < 0.01. Please also see Fig. S3A for daily scoring. (B–H) Colon infection model with guinea pigs. Hartley guinea pigs (6–8 weeks of age) were inoculated intrarectally by 1×108 CFU of HD5-treated (8 μM), 1×108 CFU of mock-treated, 1×109 CFU of mock-treated GFP-expressing Sf301 or medium alone, with 20 animals in each of the three treatment groups and 8 in the negative control group. Animals were monitored for 48h, and the distal 10 cm of colon tissue from groups of euthanized animals was harvested for analysis at 4, 8, 24 and 48 h post-challenge. (B) Confocal microscopy images of representative colon sections at 4 h post-challenge, counterstained with DAPI. The scale bars represent 50 μm. Please also see Fig. S3B for images at 8h and 24h post-challenge. The ten most bacteria-enriched fields from each experimental group were analyzed at 4 h (n=3), 8 h (n=3), 24 h (n=5) and 48 h (n=5) post-challenge, followed by automated enumeration of individual bacteria (C). Results are representative of three independent experiments and are shown as mean ± SD. Statistical significance in comparison with 1×108 CFU of mock-treated group at each time point was calculated using a one-way ANOVA (Dunnett’s multiple comparison Test), and p values are as follows: *p < 0.05, **p < 0.01. (D) SEM analysis of bacterial infection of the colonic mucosa at 2h post-challenge. The scale bars represent 5 μm. (E) Histopathology analysis of representative colon sections at 24 h and 48 h post-challenge by HE staining. The scale bars represent 100 μm. Colon histopathology scores at 24 h (F) or 48 h (G) (n=5, each) were assigned as follows: 0, intact colonic architecture, no acute inflammation or epithelial injury; 1, focal minimal acute inflammation; 2, focal mild acute inflammation; 3, severe acute inflammation with multiple crypt abscesses and/or focal ulceration; 4, severe acute inflammation, multiple crypt abscesses, epithelial loss, and extensive ulceration. Results are representative of three independent experiments. Indicated are mean ± SEM. Each point represents a single animal. Statistical significance in comparison with 1×108 CFU of mock-treated group was determined using a Mann-Whitney test, *p < 0.05, **p < 0.01. (H) Core body temperature of animals 48 h post-challenge. Results are representative of three independent experiments. Indicated are mean ± SD. Each point represents a single animal (n=9). Statistical significance between indicated groups was determined using a one-way ANOVA (Tukey’s multiple comparison Test), *p < 0.05, **p < 0.01. (I–J) Ileum and colon infection model in mice. For the colon lops (I), a small abdominal incision was made in fasted, anesthetized mice and two separate loops of colon were isolated by suture (2–3 cm in length). For each animal, one isolated colonic loop was instilled with 1×107 CFU of HD5-treated Sf301, and the second loop with mock-treated Sf301 (both in 100 μl medium). For the ileal loop model (J), the same experimental approach was employed except that the two sutured loops were with the distal ileum. Two-hours post inoculation, mice were euthanized, and loops were harvested for quantitative fluorescence imaging of Shigella infection. The scale bars represent 50 μm. Results are shown as mean ± SD. Statistical significance in comparison with group challenged with 1×107 CFU of mock-treated bacteria was calculated using a t-test, and “*” indicates p < 0.05.
In a second guinea pig model, anaesthetized animals were inoculated intrarectally with 1×108 CFU of HD5-treated, 1×108 CFU of mock-treated, 1×109 CFU of mock-treated GFP-expressing Sf301 in 200 μl medium, or medium alone at equal volume (negative control). Animals were euthanized at 4, 8, 24 and 48 h post-challenge, and the distal 10 cm of colon tissue was harvested for quantitative fluorescence imaging of Shigella infection as previously described (Arena et al., 2015). Analysis of tissue-associated bacteria revealed that HD5-treated Shigella achieved a much greater early adhesion and colonization (at both 4 and 8 h post-challenge time points), when compared to the corresponding 1×108 CFU of mock-treated bacteria, but also when compared to a higher inoculum (1×109 CFU) of mock-treated bacteria (Figs. 2B–C, S3B). SEM imaging corroborated these results by showing extensive colonization of colonic crypts by HD5-treated Shigella (Fig. 2D).
Histological changes by bacterial infection in the colon at the later time points (24 h and 48 h) were examined by HE staining (Fig. 2E–G). All Shigella-inoculated animals showed some evidence of histopathology at 24 h and 48 h post-challenge, albeit to different extents. Without HD5 treatment, 1×108 CFU of Sf301 caused mild disruption of luminal surface adjacent to the initial inoculating site 24 h post-challenge, whereas 1×109 CFU caused much more severe tissue damage during the same time period. HD5 potentiated the tissue damage caused by 1×108 CFU of Sf301, characterized by thickened submucosa and disruption of mucosal and submucosal layers, with edema, erosion, and crypt distortion comparable to 1×109 CFU. By 48 h after initial inoculation, most of the histopathology caused by 1×108 CFU of Sf301 inoculum had healed, and the integrity of mucosa was almost restored. In contrast, the infection by 1×108 CFU of HD5-treated Sf301 bacteria showed worsened pathology at 48 h, comparable to infection with 10-fold higher inoculum (1×109 CFU) Sf301 Shigella. The initial infection loci expanded and there was nearly complete destruction of mucosal and submucosal layers. Moreover, rather than a single major infection locus caused by Sf301 bacteria in each animal of the mock treated groups, multiple (3–5) infection loci were identified in colonic tissue of each animal infected by HD5-treated Shigella 48 h post-challenge. These data are consistent with the conclusion that HD5-treated Shigella disseminated more extensively along the colonic tissue mucosa within 8 h after initial inoculation than did mock-treated Sf301 bacteria. Of note, core body temperature of challenged animals, a representative sign of bacillary dysentery, showed a dramatic HD5-dependent effect. While inoculation of 1×108 CFU of mock-treated Sf301 Shigella resulted in a marginal increase in body temperature within 48 h, 1×108 CFU of HD5-treated bacteria caused severe fever in inoculated animals (an average increase by 1.3 °C) from 24 h, comparable to the high-inoculum (1×109 CFU) group (Fig. 2H).
To extend the findings from the two guinea pig models, we adopted a well-established murine ileal and colonic loop model to investigate the impact of HD5 on the in vivo colonization and pathogenesis of Shigella (Sawasvirojwong et al., 2013). Analysis of tissue-associated bacteria in mouse colonic and ileal loops confirmed that HD5-treated Shigella was much more robust in adhesion and colonization than mock-treated bacteria (Fig. 2I–J). Taken together, our findings from three different animal models provide compelling evidence that HD5 can promote Shigella infection in vivo by enhancing bacterial adhesion to and invasion of epithelial tissues.
HD5 exacerbates Shigella-elicited human tissue damage ex vivo
Human colorectal explants were cultured as an ex vivo model to further investigate the role of HD5 in pathogenesis with human tissue (Fig. S3C). SEM analysis revealed that HD5 significantly enhanced the adhesion of clustered Shigella cells to the colonic mucosa (Fig. 3A–B). Histologically, human colonic tissues mock-treated with medium or treated with HD5 alone at 8 μM displayed an intact lumenal epithelial layer and normal tissue architecture (Fig. 3C–D). In the absence of HD5, samples inoculated with Shigella (1×106 CFU) showed disruption of the lumenal surface of epithelia, but the tissue architecture was largely maintained (Fig. 3C–D). Co-inoculation of Shigella and HD5 led to a complete destruction of not only the epithelium, but also its underlying tissue structure (Fig. 3D). These results indicate that Shigella was capable of invading the human colonic epithelium and did so much more efficiently in the presence of HD5, thus further corroborating the proposed model of pathogenesis.
Fig. 3. HD5 promotes Shigella infection ex vivo.

(A) SEM analysis of the bacterial adhesion (red arrows) to human colonic mucosa 30 min after inoculation of 106 CFU Sf301 in the absence (control) and presence of HD5 (8 μM). The scale bars represent 10 μm. (B) SEM analysis of bacterial invasion of human colonic tissue at 2 h post-inoculation in presence of 8 μM HD5. Clustered bacteria are indicated by red circles in the upper panel (the scale bar represents 100 μm), and individual bacteria by red arrows in the lower panel (the scale bar represents 5 μm). (C, D) Analysis of histopathology of human colorectal explants 2 h after the inoculation either with or without Sf301 in either the absence or presence of HD5 (8 μM). Untreated (control) and HD5-treated specimens are included for comparison. Experimental details are described in Methods. Colon histopathology scores (C) were assigned as described above. Results are representative of three independent experiments. Indicated are mean ± SEM. Each point represents a colon sample. Statistical significance was determined using a Mann-Whitney test, *p < 0.05. Representative images of HE staining (the scale bars represent 100 μm) and SEM analysis (the scale bars represent 500 μm) of colonic mucosa at 2h post-challenge are shown in D. Please also see Fig. S3C for experimental procedure.
To provide additional support of the ex vivo study, we established an in vitro infection model using a polarized epithelial monolayer of human Caco-2 cells as previously described (Mounier et al., 1992). As shown in Fig. 4A, addition of 8 μM HD5 to the Caco-2 monolayer had limited impact over a period of 24 h on its integrity and permeability as measured by trans-epithelial electrical resistance (TEER). While Shigella alone was slowly destructive, a dramatically accelerated disintegration of the monolayer was evident in the presence of HD5. In fact, at 6 h, HD5 treatment increased intracellular CFU by over 300-fold (compared with the mock treatment group) due presumably to highly efficient bacterial invasion and cell-cell spreading of amplifying Shigella (Fig. 4B). These results are consistent with fluorescence and SEM imaging studies showing that HD5 potentiated Shigella destruction of the tight junction of the epithelium (Fig. 4C).
Fig. 4. (A–C) HD5 potentiates destruction of the epithelium by Shigella.

(A) Trans-epithelial electrical resistance (TEER) analysis of epithelial integrity of the monolayer of polarized Caco-2 cells infected with Sf301 in either the absence or presence of HD5 (8 μM). Percent TEER values were normalized against values of each treatment group at time 0. Data are mean ± SD from at least three independent experiments. Statistical significance was determined using a two-way ANOVA, **, p < 0.01; ***, p < 0.001. (B) Intracellular CFU of the polarized Caco-2 monolayer 6 h post-inoculation of Sf301 in either the absence or presence of HD5 (8 μM). Data are mean ± SD from at least three independent experiments. Statistical significance was determined using a t test ****, p < 0.0001. (C) Disruption of the tight junction (as shown by immunofluorescence) and impairment of the integrity (as shown by SEM) of the epithelium. Polarized Caco-2 cells grown in transwell inserts were infected with Sf301 in either the absence or presence of HD5 (8 μM) for 6 h, followed by immunostaining with antibody against the tight junction marker ZO-1 (green). Nuclei are stained with DAPI (blue). The scale bars represent 50 μm. Invading bacteria in the polarized Caco-2 monolayer are indicated by red arrows in the SEM image. The scale bars represent 2 μm. (D, E) Effects of concentrated ileal fluid aspirates on Shigella Sf301 adhesion (D) and invasion (E) in comparison with DMEM either without or with HD5 (4 μM). Anti-HD5 antiserum was added to ileal fluids and HD5-containing DMEM at a dilution titer of 1:100 to examine its neutralizing activity against Shigella infection promoted by endogenous HD5. Data are shown as mean ± SD of at least three independent experiments. Statistical significance between indicated groups was determined using a one-way ANOVA (Tukey’s multiple comparison test), and p values are as follows: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Human luminal fluids from the small intestine promote Shigella infection in vitro
When maximally secreted, the concentration of HD5 in the lumen of the small intestine is estimated to be in the millimolar range (Ayabe et al., 2000; Ghosh et al., 2002; Ouellette, 2011). However, unstimulated the quantity of HD5 is far lower in aspirated small intestinal fluids from healthy donors undergoing routine screening colonoscopy, in part due to the large volumes of electrolyte solution to enable sampling. When such clinical aspirates collected from 17 individuals were analyzed directly, none were found active in enhancing Shigella adhesion to and invasion of HeLa cells in vitro. When concentrated and tested for activity in the infection assay, the ileal fluids became active in promoting Shigella infection in vitro (Fig. 4D–E). The infection-enhancing activity was largely neutralized by anti-HD5 serum, indicating that HD5 in human luminal fluids from the small intestine contributed to Shigella infection in vitro. In addition, if prior to concentration exogenous HD5 (4 μM) was added to the intestinal fluid samples, they gained the ability to promote Shigella infection (Fig. S3E), albeit to various degrees. These data support that the activity shown for 1–8 μM concentrations of HD5 in the in vitro and in vivo models of Shigella infection reported in the current investigation reflects similar activity reasonably anticipated during clinical infection in the human intestine.
Structural basis of HD5-enhanced Shigella infection
The primary structures of epithelial defensins vary remarkably from species to species (Bevins and Salzman, 2011; Ganz, 2003; Lehrer and Lu, 2012; Selsted and Ouellette, 2005). To elucidate the structural basis underlying HD5-promoted Shigella infection of host cells, we functionally analyzed a panel of 23 analogs of HD5 in an infection assay using Sf301 and HeLa cells (Fig. 5A–B). Our data showed that: (1) the native tertiary structure of HD5 was absolutely required, as replacement of the six Cys residues by isosteric aminobutyric acid (Abu) to remove the three intra-molecular disulfide bonds of HD5 abolished adhesion and invasion enhancement; (2) while native HD5 readily dimerizes, the dimerization-debilitating analog E21Me-HD5, where the amide peptide bond is methylated at Glu21 to impair HD5 dimerization (Rajabi et al., 2012), was largely inactive in promoting Shigella infection; (3) Ala replacement of bulky hydrophobic residues, particularly those in the C-terminal region of HD5, such as Leu26, Tyr27 and Leu29, was functionally detrimental; (4) Arg28 was a key residue for activity, although the other cationic Arg residues were functionally dispensable. Because Tyr27 and Arg28 appeared to be critically important residues, we crystallized both Y27A-HD5 and R28A-HD5 and determined their structures to 1.75 and 2.4 Å, respectively (Fig. 5C–D). The functionally inactive Y27A-HD5 existed as a monomer (Fig. 5E), consistent with the fact that this residue, but not Arg28, is part of a contiguous hydrophobic core (along with Leu29) mediating HD5 dimerization. The R28A-HD5 formed a canonical dimer similar to wild-type HD5 (Fig. 5F), but was inactive, indicating dimerization alone was not sufficient for activity. Rather, for the importance of Arg28, mutational and structural analysis identified two amphipathic surfaces in the HD5 dimer (Fig. 5G), comprising residues Leu16, Val19, Leu26 and Arg28, which likely operate in tandem in interactions with molecular and cellular targets of the defensin. Taken together, these results highlight hydrophobicity and selective cationicity that are structurally segregated in a stable dimer as the most important molecular determinants of HD5 function. Of note, these structure-function determinants are in agreement with previous studies of antibacterial and antiviral activities of HD5 (Lehrer and Lu, 2012; Rajabi et al., 2012; Tenge et al., 2014).
Fig. 5. Structural determinants of HD5 function.
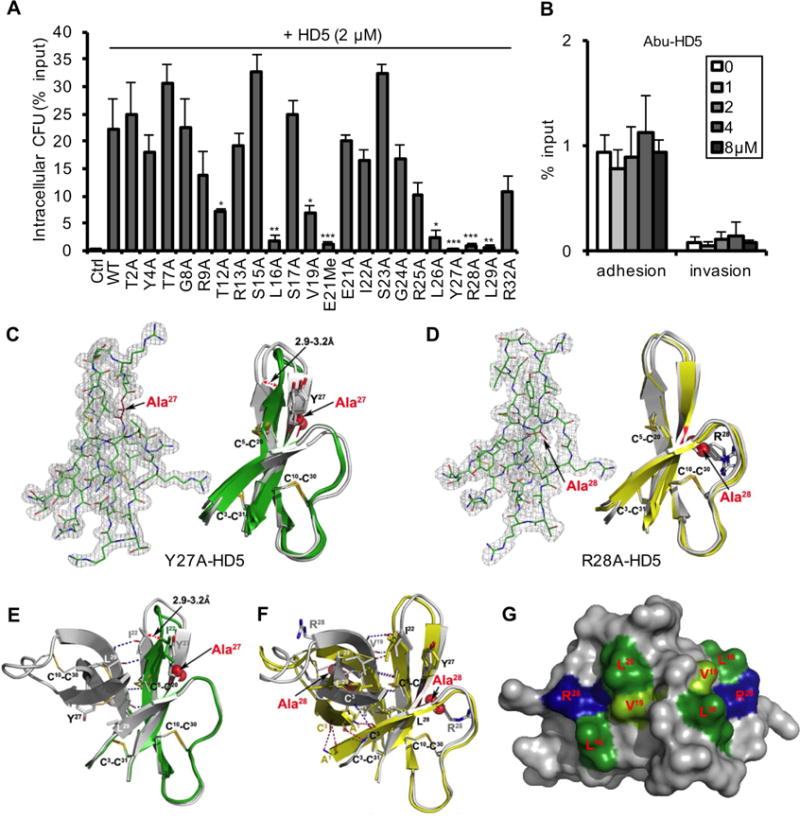
(A, B) Activities of native and alanine mutants of HD5 (A) and Abu-HD5 (B) on Shigella adhesion and invasion (for one hour). The adhesion and invasion assays were as in Fig. 1A, and data are expressed as the number of intracellular (A, B) and adhering (B) bacteria in HeLa cells relative to the input. Data are shown as mean ± SD of at least three independent experiments. Statistical significance in comparison with wildtype HD5 in A and in comparison with solvent control group in B was determined using a one-way ANOVA (Dunnett’s multiple comparison Test), and p values are as follows: *p < 0.05, **p < 0.01, ***p < 0.001. (C, D) The 2Fo-Fc electron density map contoured at 1.0σ of molecule A of Y27A-HD5 (C) and R28A-HD5 crystal (D) and the superimposition of defensin molecules present in the asymmetric units of analogs’ crystals with the wildtype HD5 monomers (shown in grey, from PDB code: 1ZMP (Szyk et al., 2006)). Side chains of cysteines forming disulfide bridges and mutated residues are shown as sticks. Structural analysis of Y27A-HD5 and R28A-HD5 analogs confirms that both mutant monomers assume the same fold as the wildtype HD5 monomer with no major changes to the overall structure and the network of three disulfide bridges. When superimposed, the root-mean-square deviations (RMSD) between 128 equivalent main chain atoms of wildtype HD5 and Y27A-HD5 and R28A-HD5 are in the range of 0.91–1.33 Å and 0.35–0.95 Å, respectively. (E, F) Crystal structures of the Y27A-HD5 monomer (E, green) and the R28A-HD5 dimer (F, yellow) superimposed on the wildtype HD5 dimer in grey (PDB code: 1ZMP). Mutated residues and alanine substitutions are shown as spheres. (G) Key functional residues of HD5 forming putative binding surfaces for interactions with bacterial and host proteins. Positively charged Arg28 residues are colored in blue, and hydrophobic residues Leu16, Val19, and Leu26 in green. Shades of green depict differences in activity with residues in light green being less important than those in dark green. Important residues not depicted in this view are Tyr27 and Leu29.
Fimbria deficiency in Shigella confers its sensitivity to HD5-mediated enhancement in bacterial infection
Type I fimbriae are the major components that impart host adhesiveness for many enterobacteria such as E. coli and Salmonella (Edwards and Puente, 1998; Jones et al., 1995; Pizarro-Cerdá and Cossart, 2006), yet they are conspicuously absent from most Shigella strains, including Sf301 and many clinical isolates, due to independent mutations in the fim gene clusters (Bravo et al., 2015; Snellings et al., 1997). To investigate the role of fimbriae in HD5-enhanced Shigella infection, we restored fimbria production in Sf301 by expressing the fim cassette from E. coli JM103 (Fig. 6A). While fimbria-expressing Sf301 showed much stronger basal adhesion to HeLa cells than wild-type Sf301, its improved adhesion capacity became largely insensitive to HD5 treatment (Fig. 6B). Results paralleling these were found for E. coli BL21, the fimbria-deficient counterpart of JM103 of the same genetic background, as well as the fimA-deleted strain JM103 ΔfimA (Fig. 6C). Similar to observations with Shigella, the fimbria-expressing E. coli showed stronger basal adhesion to HeLa cells, but the enhanced adhesion became largely insensitive to HD5 treatment. Consistent with these results, HD5 promoted a substantial increase in adhesion to HeLa cells of the mutant strain SNP494 of Salmonella enterica serovar Typhimurium, whose fimbria and flagella were deleted (Chu et al., 2012), despite a lower basal adhesion level as compared with its wild type counterpart IR715 (Fig. S4H–I). Taken together, our findings demonstrate that although fimbria deficiency in Shigella conferred its poor ability to adhere intrinsically, this deficiency greatly augmented the ability for HD5 to mediate adhesion to host epithelial cells.
Fig. 6. Bacterial determinants of HD5-promoted Shigella infection.

(A) TEM analysis of fimbriae expression in Sf301. The Fim cassette from E. coli JM103 was expressed from arabinose-inducible pBAD vector in Sf301. The scale bars represent 1 μm. (B) Influence of fimbriae-expression on the adhesion and invasion of Sf301 in the absence or presence of HD5. Data are shown as mean ± SD of at least three independent experiments. Statistical significance was determined using a two-way ANOVA, and p values are as follows: *p < 0.05, **p < 0.01 and ***p < 0.001. (C) Host cell-adhesion activity of fimbriated and non-fimbriated E. coli strains in the absence or presence of HD5. Adhesion assays to HeLa cells were carried out with fimbriated E. coli (JM103), its fimbriaedeficient mutant (JM103 Δfim) and non-fimbriated E. coli (BL21). TEM images of these strains are shown on the right panel. The scale bars represent 500 nm. Statistical significance between indicated groups was determined using a t test, and p values are as follows: *p < 0.05, **p < 0.01 and ***p < 0.001. (D) Either Sf301 bacteria, or the HeLa cells, were pre-treated with HD5 at the indicated concentrations for 30 min, washed once with DMEM, and the adhesion and invasion assays were performed. Data are shown as mean ± SD of at least three independent experiments. Statistical significance compared with solvent control (0 μM) group at each time point was determined using a one-way ANOVA (Dunnett’s multiple comparison test), and p values are as follows: *p < 0.05, **p < 0.01, and ***p < 0.001. (E) SEM and TEM analysis of Sf301 treated with HD5 or its linear analogue (Abu-HD5). The scale bars represent 2 μM for SEM and 500 nm for TEM. (F) Immunogold-TEM analysis of HD5 localization in Sf301-HeLa interaction in presence of HD5. HD5 was labeled by ~12 nm colloidal gold particles and TEM were performed as described in Methods. B, bacterium; C, Cell. The scale bars represent 500 nm. Please also see Fig. S5B for more Immunogold-TEM images showing HD5 bridging single bacterium to host and HD5 clustering multiple bacteria. (G) Relative adhesion ability of different Sf301 mutants in the presence of 4 μM HD5. spa33, icsA, ompA, ompC, ompF genes and some combinations of two were ablated as described in Methods. Shigella strains were transformed with pBAD plasmids carrying the OmpC coding sequence and induced with 10 mM L-arabinose for OmpC expression. Please also see Fig. S6F for SDS-PAGE analysis of the genetic ablations and recompletions. Adhesion assays were performed as in Fig. 1A. Data are normalized to the input and shown as the percentage of the adherent bacteria of wild-type Sf301 in the presence of 4 μM HD5. Data are shown as mean ± SD of at least three independent experiments. Statistical significance compared with wild type was determined using a one-way ANOVA (Dunnett’s multiple comparison test), and p values are as follows: *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
HD5 targets multiple bacterial membrane proteins to promote Shigella adhesion to host cells
To better understand the mechanism of HD5-mediated adherence, we found that in the presence of HD5, Shigella preferably attached to the periphery of adhered, but not suspended, host cells, where the dynamic cell-substratum contacts occur (Fig. S4A–C). siRNA silencing studies coupled with immunofluorescence staining suggest that integrins are involved as host factors in HD5-promoted Shigella adhesion (Fig. S4D–G), consistent with their known ability to interact with α-defensins (Chavakis et al., 2004; Economopoulou et al., 2005). These findings notwithstanding, HD5 primarily targeted the pathogen rather than host cells to enhance infection. Although HD5 can bind to both host and bacterial cells efficiently (Fig. S5A), HD5 was substantially more effective in promoting bacterial infection when pre-incubated with Sf301, compared to pre-incubation with HeLa cells (Fig. 5D). SEM, TEM and immunogold-TEM studies revealed that HD5, but not its unstructured analog Abu-HD5, bound to the Shigella surface and formed patches of an “adhesive” structure, leading to the clustering of Shigella cells and their adhesion to host cells (Fig. 6E–F, S5B).
To identify the bacterial targets with which HD5 directly interacts, we first investigated whether HD5, a known lectin capable of binding to glycosylated proteins (Lehrer et al., 2009), could interact with the bacterial LPS to promote host adhesion by characterizing several LPS truncation mutants of Sf301. Our data (Fig. S6A–C), however, showed that LPS was not targeted by HD5 for bacterial adhesion promotion. We next focused on potential proteinaceous targets of HD5 on the Shigella surface. Trypsin is the proteolytic processing enzyme of pro-HD5 and mature HD5 is stable to its hydrolytic activity (Ghosh et al., 2002). Pre-treatment of Sf301 with trypsin, while maintaining bacterial viability, lost HD5-augmented host adhesion capacity (Fig. S6D–E), consistent with a proteinaceous target. HD5 enhanced bacterial adhesion to host cells within minutes of co-incubation, thus likely targeting preexisting surface components without involving the de novo protein synthesis and/or membrane shuttling machinery – a notion also supported by an adhesion assay using heat and paraformaldehyde-inactivated Sf301 (Fig. S6G–H).
Using the λ red mutagenesis system (Datsenko and Wanner, 2000), we genetically ablated Spa33, an essential component of the T3SS (Morita-Ishihara et al., 2006), IcsA, an autotransporter protein reported to function as an adhesin in hyperadhesive Shigella flexneri mutants lacking the T3SS component IpaD (Brotcke Zumsteg et al., 2014), and three most abundant outermembrane proteins, OmpA, OmpC and OmpF (Ambrosi et al., 2012; Bernardini et al., 1993). Deletion of Spa33, IcsA, OmpA or OmpC in Sf301 led uniformly to a moderate drop in HD5-mediated adhesion (Fig. 6G), except for OmpF whose expression in Sf301 was undetectable (Fig. S6F). As expected, a double genetic ablation of a combination of Spa33, OmpA and OmpC, and ΔOmpAΔOmpC in particular, further reduced bacterial adhesion. OmpC expression via plasmids not only restored adhesion of the OmpC-null mutant above that of the wild-type, but also increased HD5-mediated adhesion of the wild-type, OmpA-null and OmpF-null strains (Fig. 6G). Thus, our data indicate that multiple bacterial surface proteins collectively contributed to HD5-mediated adhesion.
DISCUSSION
The human α-defensin HD5 contributes to innate host defense against enteropathogens in the gut (Bevins and Salzman, 2011; Chu et al., 2012; Salzman et al., 2003), and helps maintain intestinal homeostasis by forming a chemical barrier that segregates the gut microbiota from host epithelium to limit tissue inflammation and microbial translocation (Belkaid and Hand, 2014; Bevins and Salzman, 2011). Our biochemical and structural data, mechanistic and functional studies at the molecular and cellular levels, and in vivo and ex vivo findings all support that Shigella exploits HD5 for virulence and host infection. These results highlight that host defense factors such as α-defensins, which are vitally host protective (Bevins and Salzman, 2011; Selsted and Ouellette, 2005; Zasloff, 2002), can be important contributors to pathogenesis when exploited by pathogens. Our work thus provides a noteworthy example of how immunity can serve as a “double-edged sword” in health and disease (Hansson and Libby, 2006).
Our findings not only shed light on how Shigella adheres to and invade host cells despite its lack of fimbriae, but also offer a clue on host-range selectivity of Shigella infection. Enteropathogens such as Salmonella and E. coli have a sophisticated adhesion apparatus, including fimbriae, flagella and other adhesins to ensure efficient bacterial colonization of the intestinal epithelium and subsequent invasion and infection (Donnenberg, 2000; Pizarro-Cerdá and Cossart, 2006). Shigella, on the contrary, lacks such adhesion machinery in general (Bravo et al., 2015; Schroeder and Hilbi, 2008) and has a poor inherent ability to colonize host epithelium in vitro, confounding its extraordinary in vivo infectivity – 10–100 bacterial cells are sufficient to induce clinical symptoms in humans (DuPont et al., 1989). Despite recent reports of two adhesion molecules of Shigella, IcsA and MAM, which operate only in specific biological settings (Brotcke Zumsteg et al., 2014; Mahmoud et al., 2016), no efficient and general Shigella adhesin has been reported thus far. In fact, Shigella had long been thought to initially breach intestinal epithelial barriers through M cell-mediated transcytosis, followed by dissemination to epithelial cells from the basolateral side (Carayol and Tran Van Nhieu, 2013; Cossart and Sansonetti, 2004; Phalipon and Sansonetti, 2007). Our findings provide a compelling alternative hypothesis that secreted HD5 enables a mode of direct and active invasion by Shigella from the apical surface of the intestinal epithelium. We propose that Shigella, while in transit, encounters HD5 molecules in the small intestine and becomes highly adhesive and invasive as it reaches more distal sites in the colon. Since HD5 is expressed by Paneth cells in the small intestine (Bevins and Salzman, 2011), its high local concentration at the luminal surface of the small intestine is likely lethal to Shigella, which might then largely restrict bacterial adhesion and invasion to the downstream colonic epithelium where HD5 is greatly reduced in abundance. Moreover, the finding that Concanavalin A, a lectin that interacts with diverse host proteins containing mannose carbohydrates, failed to inhibit HD5-enhanced Shigella infection in vitro suggests a limited role played by mucin and/or surface glycoproteins in attenuating this HD5 activity in vivo. This direct invasion model reinforces a recent finding that Shigella primarily targets colonic crypts during the initial stages of mucosal invasion (Arena et al., 2015).
It is plausible that Shigella may have undertaken a different evolutionary trajectory from Salmonella or E. coli to manifest its pathogenicity. Hijacking a host innate immune molecule to facilitate bacterial adhesion and invasion might provide evolutionary advantage for Shigella, as lack of adhesive appendages such as fimbriae or flagella (Bravo et al., 2015) should help the pathogen evade host immune surveillance and thrive in the gut with significantly less anabolic burden. However, such a strategy that depends on a specific host factor would restrict host range, especially when targeting an epithelial α-defensin, where primary structures vary markedly from species to species. While it is not unusual for a microbial pathogen to exploit host components to promote its pathogenicity, our findings contribute a striking example of this phenomenon.
In addition, human neutrophil granular proteins containing α-defensins HNPs 1–4 can enhance Shigella adhesion in vitro at sub-lethal concentrations (Eilers et al., 2010), in accordance with our results on HNP1. Furthermore, both HD5 and HD6 have been reported to enhance HIV-1 infectivity in vitro by promoting virion attachment to target cells (Rapista et al., 2011). However, both the molecular mechanisms and physiological implications of those findings remain to be determined. A very recent report found that a mouse adenovirus to promotes its host entry in a receptor-independent manner by binding to mouse alpha-defensins (cryptdins), resulting in enhanced enteric viral infection (Wilson et al., 2017) and suggesting that pathogen exploitation of defensins for infection is not restricted to humans.
The precise molecular details underlying HD5-enhanced Shigella adhesion to epithelial cells and tissues need to be further clarified. HD5 is capable of binding to diverse molecular, bacterial and viral targets in a multivalent, somewhat promiscuous fashion (Lehrer et al., 2009; Lehrer and Lu, 2012; Rajabi et al., 2012; Tenge et al., 2014). It may serve as a bridging molecule to directly crosslink bacterial and host cells as reported for HD5 and other mammalian defensins (Lehrer et al., 2009; Leikina et al., 2005). Alternatively, by clustering Shigella cells, HD5 could endow the pathogen with a much-enhanced ability to adhere through multivalent high-avidity interactions between bacterial and host surface proteins. Cell-cell contact activates the T3SS, leading to Shigella invasion orchestrated by bacterial virulent effector proteins delivered by the T3SS into host cells (Cossart and Sansonetti, 2004; Donnenberg, 2000; Hauser, 2009; Pizarro-Cerdá and Cossart, 2006). In the absence of an efficient host-adhesion apparatus of its own, HD5-promoted Shigella adhesion and colonization could potentiate T3SS activation, thus indirectly facilitating bacterial invasion and infection. Whether or not HD5 is capable of directly enabling Shigella invasion remains to be examined, though. Of note, shortening the LPS of Shigella increases accessibility of its T3SS tip to the host cell surface, thus augmenting T3SS activation and bacterial invasion (West et al., 2005). Although HD5 did not target LPS directly to promote adhesion, the possibility that HD5 perturbs the LPS structure for enhanced T3SS activation and Shigella invasion cannot be formally excluded.
Finally, although Shigella is highly infectious in humans at an extremely low inoculum, it does not readily infect other animals. In fact, no suitable animal model is available to accurately recapitulate the pathogenesis of Shigella (Phalipon and Sansonetti, 2007). Mice express abundant quantities of α-defensins (cryptdins) in the intestine (Ouellette, 2011), yet they are relatively very resistant to oral Shigella challenge. While this resistance may be due at least in part to the lack of IL-8 (Singer and Sansonetti, 2004), the finding that mouse cryptdins, in contrast to HD5, are incapable of promoting Shigella adhesion and colonization suggests an alternative explanation for host specificity of this important human enteropathogen. Whether the lack of expression in other animals of an ortholog of HD5 capable of enhancing Shigella pathogenesis is sufficient to confer resistance to infection warrants additional investigation.
STAR Methods section
Wuyuan Lu (wlu@ihv.umaryland.edu) is the lead contact author responsible for providing reagents upon request.
Ethics Approval
All the animals used in this study were acquired from the Experimental Animal Center of Xi’an Jiaotong University. The animal studies were approved by the Committee on Animal Research and Ethics, Xi’an Jiaotong University. All the animals were maintained in animal care facilities in the School of Life Science and Technology, and provided with food and water ad libitum. The use of healthy colonic tissues from patients undergoing surgery for colon cancer and the use of human small intestinal fluid aspirates from healthy donors undergoing routine screening colonoscopy were approved by the Ethics Committee of the Second Affiliated Hospital of Xi’an Jiaotong University School of Medicine. Informed consent was obtained from all patients.
Reagents
All peptides used in this study were chemically synthesized, correctly folded and highly purified as previously described (Pazgier et al., 2013; Rajabi et al., 2012; Szyk et al., 2006; Wei et al., 2010; Wu et al., 2004; Wu et al., 2003). Anti-human integrin β1 monoclonal antibody (MAB1959Z, P5D2) and anti-human integrin α5β1 monoclonal antibody (MAB1969, JBS5) were purchased from Millipore. Anti-human integrin β5 monoclonal antibody (#3629, D24A5) was purchased from Cell Signaling Technology. All common chemicals and reagents were purchased from Sigma-Aldrich unless indicated otherwise.
Bacterial strains
Bacterial strains used in this study are listed in Table S2. Shigella strains were cultured aerobically at 37 °C in Tryptic Soy (TS) broth (Aob oxing, Beijing, China) or on TS agar plates with addition of 0.1% Congo Red. E. coli strains in this study were cultured in Luria-Bertani (LB) broth (Aoboxing, Beijing, China) or on LB agar plates. Antibiotics (Sigma) were used as follows: ampicillin 200 μg/ml; kanamycin 100 μg/ml.
Strain construction
Genetic ablation of Bacterial genes was performed using the λ Red recombination system (Datsenko and Wanner, 2000). Briefly, bacterial cells transformed with pKD46 were grown in the presence of L-arabinose to induce the expression of the lambda Red recombinase. A linear PCR product, amplified using the primers listed in Table S2, containing a kanamycin-resistance cassette (KRC) flanked by FLP and 50 bp of the 5’-and 3’-end homologous sequences of the target gene was electroporated into the bacterial cells and kanamycin was used to select the transformants. The plasmid pKD46 was eliminated by incubation at 37 °C. To cure the kanamycin maker, pCP20 was introduced into kanamycin-resistant cells to elicit the recombination of flanking FLP sequences at both ends of the kanamycin cassettes. PCR screening for cured colonies were performed using specific primers listed in Table S2. For OmpC and fimbriae re- expression experiments, the OmpC coding sequence from Sf301 and the fimbriae-expression cassette from E. coli JM103 (encoding FimA, I, C, D, F, G, H) were separately cloned into Nco I and Xho I sites of pBAD vector using the primers listed in Table S2. Bacterial cells transformed with pBAD-OmpC or pBAD-Fim were cultured in LB with 10 mM L-arabinose to induce the expression of OmpC or production of fimbriae in Sf301.
Bactericidal assays
Different bacterial strains (~106 CFU) were treated with the indicated concentrations of peptide in 500 μl DMEM (without serum) for 40 min at 37 °C with mil d agitation. After washing, the bacteria were diluted and plated. Bacterial viability was determined by colony counting and normalized against the viability observed with mock (PBS) treatment. Results are reported as the mean percentage of input bacteria of three independent experiments ± SD.
In vitro adhesion and invasion assay
The cell lines and their culture medium used in this study are listed in Table S1. One day before the assays, cells were seeded into 24-well plates at a density of ~105 cells per well. One hour before the infection, cell culture medium was changed into serum-free medium and ~106 CFU Sf301 from mid-exponential phase was added to the cells together with a titration of HD5 (0–8 μM). Bacteria were centrifuged (2000 rpm, 10 min, RT) onto HeLa cells (MOI 10:1, or indicated MOI) to synchronize the infection. For the adhesion assay, after washing, the cells were lysed with 0.1 % Triton/H2O and the CFU were enumerated after plating. For invasion and proliferation assays, bacteria/HeLa mixtures were incubated for 40 min after centrifugation and then washed, treated with gentamicin-containing (25 μg/ml) medium for another 1 hour (invasion) or 4 hours (proliferation) before being lysed for plating. Adhesion was defined as the total number of HeLa cell-associated bacteria and is shown as the percentage of input. Invasion and proliferation were defined as the total number of intracellular bacteria in cells (extracellular bacteria were killed by gentamicin, a cell-impermeable antibiotic). Average results of three independent experiments are reported as mean ± SD. For pre-treatment experiments, cells or bacteria were pre-incubated with HD5 at the indicated concentrations for 30 min, washed once with DMEM and then mixed to allow infection. Adhesion assays, invasion assays and proliferation assays were performed as stated above. Schematic illustration of the assays is shown in Fig. S2.
Sereny test
Female specific pathogen-free Hartley guinea pigs, aged 6–8 weeks, weighing 120–250 g, were inoculated via conjunctival route with 106 CFU/eye of mid-log phase Sf301 in the absence or presence of 4 μM or 8 μM HD5 as described (Labrec et al., 1964), with 15 animals in each group. The protocol was approved by the Committee on Animal Research and Ethics of Xi’an Jiaotong University. Inoculated animals were observed and scored for 7 consecutive days for the development of conjunctivitis. Eye tissues were scored by three individuals (DX, YS, YC) who were kept unaware of the treatment group on a scale of 0–3 defined as follows: grade 0 (no disease or mild irritation), grade 1 (mild conjunctivitis or late development and/or rapid clearing of symptoms), grade 2 (keratoconjunctivitis without purulence), grade 3 (fully developed keratoconjunctivitis with purulence). Statistical significance was calculated using a Mann-Whitney test.
Colon infection model in guinea pigs
Female pathogen-free Hartley guinea pigs, aged 6–8 weeks, weighing 120–250 g, were fasted for 24 h and anesthetized by intraperitoneal injection of nembutal (30 mg/kg). Animals were inoculated intrarectally with either 1×108 CFU of HD5-treated, 1×108 CFU of mock-treated (low-inoculum positive control) or 1×109 CFU (high-inoculum positive control) of mock-treated GFP-expressing Sf301 in 200 μl medium. Animals inoculated with medium containing 8 μM HD5 at equal volume served as negative controls. HD5-treated Shigella bacteria were prepared as following: 1×108 CFU of Sf301 were incubated with 8 μM HD5 in 50ml DMEM for 20 minutes followed by centrifugation for 10-min. Most supernatant was decanted, leaving 200 μl to resuspend the bacteria. Temporally representative samples from colonic tissues were obtained at 4, 8, 24 and 48h post-intrarectal challenge by euthanizing animals using nembutal. The distal 10 cm of colon was harvested and flushed with 4% (vol/vol) paraformaldehyde (PFA) in 1×PBS, inverted on wooden skewers, and kept in 4% PFA 1×PBS for 1–2 h to complete fixation of the tissue and then incubated in 1×PBS glycine (100 mM) for 30 min to quench the PFA. Tissues were then immersed successively in 15% and 30% (wt/vol) sucrose at 4 °C overnight. Tissues were removed from the skewers by a longitudinal incision and prepared as Swiss rolls (Arena et al., 2015). Swiss rolls were then embedded in Tissue-Tek OCT compound (Sakura) using a flash-freeze protocol and frozen at −80 °C. These OCT-frozen tissue preparati ons were cut as 20-μm-thick transversal sections. Tissues on slides were fixed with 4% PFA for 10 min at room temperature and permeabilized in PBS/0.2% Triton X-100 for 10 min. The slides were washed, mounted with Anti-Fade solution (Invitrogen) containing DAPI onto glass slides and visualized under a Zeiss confocal microscope. Shigella infection foci were identified by GFP fluorescence microscopy. The ten most bacteria-enriched fields from each group were analyzed, followed by automatic enumeration of individual Shigella bacteria using ImageJ 1.51k software (from http://imagej.nih.gov/ij).
Murine ileal and colonic loop
Eight-week-old pathogen-free Balb/c mice (weight 20–30 g) were fasted for 24 h and anesthetized by intraperitoneal injection of nembutal (60 mg/kg). While maintaining the body temperature at 37° using a heating pad, a small abdominal incision was made and two adjacent loops of either distal ileum or colon were isolated by suture (2–3 cm in length) in each animal. For each animal, one loop was instilled with 1×107 CFU of HD5-treated Sf301 (GFP-expressing), and the other was instilled with mock-treated Sf301 (both in 100 μl medium). HD5-treated Shigella bacteria were prepared as following: 1×107 CFU of Sf301 were incubated with 8 μM HD5 in 5ml DMEM for 20 minutes followed by centrifugation for 10-min. Most supernatant was decanted leaving 100 μl to resuspend the bacteria. The ileum (or colon) tissues were obtained at 2h post-inoculation, processed and analysized as described above for the colon infection model of guinea pigs.
Human small intestinal fluid aspirates
Ileal aspirates were obtained from healthy individuals who were undergoing routine screening colonoscopy for colon polyps. Prior to colonoscopy (~24h beforehand), patients were administered a routine polyethylene glycol-electrolyte solution to purge the bowel of contents, and the patients remained on clear fluids until the procedure was completed. During the colonoscopy procedure, the terminal ileum was intubated. Approximately 5–15 ml of ileal lumenal fluid was aspirated and immediately placed on ice. Specimens were clarified by centrifugation and filtered through a 0.22 μm filter. For some specimens, the fluid was further concentration by centrifugal filtration. The aliquots were stored in a freezer at −80° prior to analysis. The clarified fluid was used as the medium for Shigella infection assays.
Human colorectal explants
The human colorectal explants were established in accordance to a protocol recently developed by Tisilingiri et al. (Tsilingiri et al., 2012). Briefly, healthy colonic tissues were obtained from patients undergoing surgery for colon cancer. Tissue samples, maintained in Hank’s balanced salt solution on ice, were transported to the laboratory and processed within two hours. The mucosa layer was separated from the underlying tissues, and then divided into pieces (~2–3 cm by ~2–3 cm) and placed on soft agar (1% in PRMI 1640 medium) with the mucosal surface facing upward. A sterile cylinder (8 mm in inner diameter) was used to remove small pieces of soft agar under the center of the tissue segments and fresh medium was added to the small holes so that a small dip was formed at the center of the segment (Fig. S3C). An aliquot (5×105 CFU) of Shigella from logarithmic growth culture in 10 μl of PRMI 1640 medium was added to the dip of the colon tissue and the explants were cultured in medium with or without 8 μM HD5 at 37 °C for various times (from 30 min to 2 hours) in a cell culture incubator. Bacteria-free segments served as negative controls for each experiment and time point. Following incubation, infected explants were fixed in 10% buffered formalin, paraffin embedded and HE-stained for histopathological examination.
Measurement of trans-epithelial electrical resistance (TEER)
Caco-2 cells (3×104 cells per well) were grown on 24-well transwell inserts with a 0.4 μm pore size (Corning) in 10% FBS DMEM. Five days after seeding, the polarized cells were placed in serum-free DMEM, and an aliquot of Sf301 bacteria (MOI=50:1) was inoculated to the apical chamber either with or without HD5. The infected CaCo-2 cells were incubated for 60 min and then washed, treated with gentamicin-containing (50 μg/ml) medium. TEER values of the polarized epithelial monolayer were measured by Millicell ERS-2 Voltohmmeter (Merck Millipore) for 24 hours. Monolayers with TEER values within, 800–1200 Ω.cm2 were considered to have an appropriate barrier function and were used in the study.
Crystallization, Data collection and Structure Determination
Lyophilized HD5 mutant proteins were dissolved in water (20 mg/ml), mixed in a 1:1 ratio with precipitant solutions composed as listed in Table S4 and equilibrated at 22° in a hanging drop crystallization format. Crystals were flash frozen in liquid nitrogen after a brief soak in cryoprotectant solution (Table S4). Data were collected at the Stanford Synchrotron Radiation Light Source (SSRL) beamlines BL7-1 (Y27A mutant crystal form 1) and BL12-2 (Y27A mutant crystal form 2 and the R28A mutant). All data were processed and reduced with HKL2000 (Otwinowski and Minor, 1997). Structures were solved by molecular replacement with the program Phaser (McCoy et al., 2007) from the CCP4 suite based on the coordinates of the 1ZMP wild type HD5 monomer (Szyk et al., 2006). Refinement was carried out with Refmac (Murshudov et al., 1997) and/or Phenix (Adams et al., 2010) and model building was done with COOT (Emsley et al., 2010). Data collection and refinement statistics are shown in Table S5. Ramachandran statistics were calculated with Molprobity (Chen et al., 2010) and illustrations were prepared with Pymol Molecular graphics (http://pymol.org). Crystallographic data were collected at the Stanford Synchrotron Radiation Lightsource (SSRL), a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the US Department of Energy Office of Biological and Environmental Research, by the National Institutes of Health (NIH) National Center for Research Resources, Biomedical Technology Program (P41RR001209), and by the National Institute of General Medical Sciences.
Immunofluorescence microscopy
Cells were plated onto glass coverslips and adhesion assays were performed as described using Sf301 harboring the GFP-expressing plasmid. Cells were fixed in 3% paraformaldehyde at room temperature for 15 min followed permeablization with 0.1%Trtion (in PBS) for 3–5min, washed in PBS and blocked with 5% BSA (in PBS) for 30 min at room temperature. The coverslips were incubated with Actin, α5β1 or β1 antibodies at 4 °C overnight followed by PBS washin g and incubation with Alexa Fluor 594 secondary antibody (Invitrogen Molecular Probes, Carlsbad, CA) for 1 h. The coverslips were washed, mounted with Anti-Fade solution (Invitrogen) containing DAPI onto glass slides and visualized under Zeiss confocal microscope.
Electron microscopy
For scanning electron microscopy (SEM), adhesion assay was performed in the presence of 4 μM HD5 (MOI 50:1) as described above. Cells were fixed with paraformaldehyde (15min) and glutaraldehyde (overnight). The fixed specimens were dehydrated in graded ethanol, critical point dried with CO2 and coated with gold-palladium beads with a diameter of 15 nm. Samples were photographed using a Philips XL-30 scanning electron microscope at 20 kV.
For transmission electron microscopy (TEM), bacteria (treated with or without HD5) were stained with 1.5% phosphotungstic acid for 90s and examined by TEM (H-7650, HITACHI).
For immunogold-TEM, the adhesion assay was performed in the presence of 4 μM HD5 (MOI 50:1) as previously described. Cells were fixed in 3% paraformaldehyde at room temperature for 15 min followed permeablization with 0.1%Trtion (in PBS) for 3–5min, washed in PBS and blocked with 5% BSA (in PBS) for 30 min at room temperature. Cells were then incubated with rabbit anti-HD5 antibody at 1:100 dilution at 4 °C overnight, followed by PBS washing and incubation with 12nm colloidal gold-conjugated donkey anti-rabbit IgG (H+L) for 2h at room temperature. Cells were washed and further fixed in 3% paraformaldehyde at room temperature for 15 min. Cells were collected by scratching and centrifugation. Further fixation was performed in 3% paraformaldehyde and 0.25% glutaraldehyde at 4 °C overnight. Cell ma ss were embedded and cut into ultrathin sections as described, colloidal gold particles were recognized as dark spots under TEM (H-7650, HITACHI).
siRNA silencing
The siRNA oligonucleotides were synthesized by Shanghai GenePharma (Shanghai, China) and their sequences are shown in Table S6. Cells were transfected with siRNAs according to the recommended procedures of Lipofectamine™2000 Transfection Reagent (Invitrogen, Carlsbad, CA).
Statistical analysis
The data were collected from at least three independent experiments in triplicate or quadruplicate, unless otherwise indicated. Data were combined and represented as mean ± SEM or mean ± SD as indicated. Results were analyzed by various statistical tests using GraphPad Prism version 7. p < 0.05 was considered statistically significant. Microscopy images are representative of at least two independent experiments.
Data availability
The data that support the findings of this study are available from the corresponding author upon request.
Supplementary Material
Highlights.
HD5 exacerbates Shigella infectivity by enhancing bacterial adhesion.
HD5 activity in promoting Shigella infection is sequence and structure-dependent.
Fimbria deficiency in Shigella confers its sensitivity to HD5-enhanced infection
HD5 targets bacterial membrane proteins to promote Shigella adhesion and infection
Acknowledgments
This work was supported in part by Program 985 of XJTU (to WL) and grants from the Natural Science Foundation of China (31401211 & 31770146 to DX), and the National Institutes of Health of USA (R01GM106710 and R01CA219150 to WL and R37AI032738 to CLB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
WL, YS and DX conceived and designed the study. DX, CL, BZ, WDT, WH, WZ, WY and MP performed the experiments. ZD provided human colorectal tissue samples and performed histological analysis. JY, PJS and CLB provided bacterial strains, helped with study design, and edited the manuscript. DX, YS and WL wrote the paper. All authors read and approved the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosi C, Pompili M, Scribano D, Zagaglia C, Ripa S, Nicoletti M. Outer Membrane Protein A (OmpA): A New Player in Shigella flexneri Protrusion Formation and Inter-Cellular Spreading. PLoS ONE. 2012;7:e49625. doi: 10.1371/journal.pone.0049625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arena ET, Campbell-Valois FX, Tinevez JY, Nigro G, Sachse M, Moya-Nilges M, Nothelfer K, Marteyn B, Shorte SL, Sansonetti PJ. Bioimage analysis of Shigella infection reveals targeting of colonic crypts. Proceedings of the National Academy of Sciences. 2015a;112:E3282–E3290. doi: 10.1073/pnas.1509091112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arena ET, Campbell-Valois FX, Tinevez JY, Nigro G, Sachse M, Moya-Nilges M, Nothelfer K, Marteyn B, Shorte SL, Sansonetti PJ. Bioimage analysis of Shigella infection reveals targeting of colonic crypts. Proc Natl Acad Sci U S A. 2015b;112:E3282–3290. doi: 10.1073/pnas.1509091112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ. Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol. 2000;1:113–118. doi: 10.1038/77783. [DOI] [PubMed] [Google Scholar]
- Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157:121–141. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardini ML, Sanna MG, Fontaine A, Sansonetti PJ. OmpC is involved in invasion of epithelial cells by Shigella flexneri. Infection and Immunity. 1993;61:3625–3635. doi: 10.1128/iai.61.9.3625-3635.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol. 2011;9:356–368. doi: 10.1038/nrmicro2546. [DOI] [PubMed] [Google Scholar]
- Bravo V, Puhar A, Sansonetti P, Parsot C, Toro CS. Distinct Mutations Led to Inactivation of Type 1 Fimbriae Expression in Shigella spp. PLoS ONE. 2015;10:e0121785. doi: 10.1371/journal.pone.0121785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brotcke Zumsteg A, Goosmann C, Brinkmann V, Morona R, Zychlinsky A. IcsA Is a Shigella flexneri Adhesin Regulated by the Type III Secretion System and Required for Pathogenesis. Cell Host & Microbe. 2014;15:435–445. doi: 10.1016/j.chom.2014.03.001. [DOI] [PubMed] [Google Scholar]
- Brunger AT. Free R value: Cross-validation in crystallography. Methods in Enzymology (Academic Press) 1997:366–396. doi: 10.1016/s0076-6879(97)77021-6. [DOI] [PubMed] [Google Scholar]
- Campbell-Valois FX, Schnupf P, Nigro G, Sachse M, Sansonetti PJ, Parsot C. A fluorescent reporter reveals on/off regulation of the Shigella type III secretion apparatus during entry and cell-to-cell spread. Cell Host Microbe. 2014;15:177–189. doi: 10.1016/j.chom.2014.01.005. [DOI] [PubMed] [Google Scholar]
- Carayol N, Tran Van Nhieu G. Tips and tricks about Shigella invasion of epithelial cells. Curr Opin Microbiol. 2013;16:32–37. doi: 10.1016/j.mib.2012.11.010. [DOI] [PubMed] [Google Scholar]
- Chart H, Smith HR, La Ragione RM, Woodward MJ. An investigation into the pathogenic properties of Escherichia coli strains BLR, BL21, DH5alpha and EQ1. J Appl Microbiol. 2000;89:1048–1058. doi: 10.1046/j.1365-2672.2000.01211.x. [DOI] [PubMed] [Google Scholar]
- Chavakis T, Cines DB, Rhee JS, Liang OD, Schubert U, Hammes HP, Higazi AA, Nawroth PP, Preissner KT, Bdeir K. Regulation of neovascularization by human neutrophil peptides (alpha-defensins): a link between inflammation and angiogenesis. FASEB J. 2004;18:1306–1308. doi: 10.1096/fj.03-1009fje. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallographica Section D. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury D, Thompson A, Stojanoff V, Langermann S, Pinkner J, Hultgren SJ, Knight SD. X-ray Structure of the FimC-FimH Chaperone-Adhesin Complex from Uropathogenic Escherichia coli. Science. 1999;285:1061–1066. doi: 10.1126/science.285.5430.1061. [DOI] [PubMed] [Google Scholar]
- Chu H, Pazgier M, Jung G, Nuccio SP, Castillo PA, de Jong MF, Winter MG, Winter SE, Wehkamp J, Shen B, et al. Human alpha-defensin 6 promotes mucosal innate immunity through self-assembled peptide nanonets. Science. 2012;337:477–481. doi: 10.1126/science.1218831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart P, Sansonetti PJ. Bacterial Invasion: The Paradigms of Enteroinvasive Pathogens. Science. 2004;304:242–248. doi: 10.1126/science.1090124. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proceedings of the National Academy of Sciences. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnenberg MS. Pathogenic strategies of enteric bacteria. Nature. 2000;406:768–774. doi: 10.1038/35021212. [DOI] [PubMed] [Google Scholar]
- DuPont HL, Levine MM, Hornick RB, Formal SB. Inoculum Size in Shigellosis and Implications for Expected Mode of Transmission. Journal of Infectious Diseases. 1989;159:1126–1128. doi: 10.1093/infdis/159.6.1126. [DOI] [PubMed] [Google Scholar]
- Economopoulou M, Bdeir K, Cines DB, Fogt F, Bdeir Y, Lubkowski J, Lu W, Preissner KT, Hammes HP, Chavakis T. Inhibition of pathologic retinal neovascularization by alpha-defensins. Blood. 2005;106:3831–3838. doi: 10.1182/blood-2005-03-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards RA, Puente JL. Fimbrial expression in enteric bacteria: a critical step in intestinal pathogenesis. Trends in Microbiology. 1998;6:282–287. doi: 10.1016/s0966-842x(98)01288-8. [DOI] [PubMed] [Google Scholar]
- Eilers B, Mayer-Scholl A, Walker T, Tang C, Weinrauch Y, Zychlinsky A. Neutrophil antimicrobial proteins enhance Shigella flexneri adhesion and invasion. Cellular Microbiology. 2010;12:1134–1143. doi: 10.1111/j.1462-5822.2010.01459.x. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallographica Section D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericksen B, Wu Z, Lu W, Lehrer RI. Antibacterial activity and specificity of the six human {alpha}-defensins. Antimicrob Agents Chemother. 2005;49:269–275. doi: 10.1128/AAC.49.1.269-275.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol. 2003;3:710–720. doi: 10.1038/nri1180. [DOI] [PubMed] [Google Scholar]
- Ghosh D, Porter E, Shen B, Lee SK, Wilk D, Drazba J, Yadav SP, Crabb JW, Ganz T, Bevins CL. Paneth cell trypsin is the processing enzyme for human defensin-5. Nat Immunol. 2002;3:583–590. doi: 10.1038/ni797. [DOI] [PubMed] [Google Scholar]
- Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- Hauser AR. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol. 2009;7:654–665. doi: 10.1038/nrmicro2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Q, Yuan Z, Xu J, Wang Y, Shen Y, Lu W, Wang J, Liu H, Yang J, Yang F, et al. Genome sequence of Shigella flexneri 2a: insights into pathogenicity through comparison with genomes of Escherichia coli K12 and O157. Nucleic Acids Res. 2002;30:4432–4441. doi: 10.1093/nar/gkf566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CH, Pinkner JS, Roth R, Heuser J, Nicholes AV, Abraham SN, Hultgren SJ. FimH adhesin of type 1 pili is assembled into a fibrillar tip structure in the Enterobacteriaceae. Proceedings of the National Academy of Sciences. 1995;92:2081–2085. doi: 10.1073/pnas.92.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline KA, Fälker S, Dahlberg S, Normark S, Henriques-Normark B. Bacterial Adhesins in Host-Microbe Interactions. Cell Host & Microbe. 2009;5:580–592. doi: 10.1016/j.chom.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Labrec EH, Schneider H, Magnani TJ, Formal SB. Epithelial cell penetration as an essential step in the pathogenesis of bacillary dysentery. Journal of Bacteriology. 1964;88:1503–1518. doi: 10.1128/jb.88.5.1503-1518.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrer RI, Jung G, Ruchala P, Andre S, Gabius HJ, Lu W. Multivalent binding of carbohydrates by the human alpha-defensin, HD5. J Immunol. 2009;183:480–490. doi: 10.4049/jimmunol.0900244. [DOI] [PubMed] [Google Scholar]
- Lehrer RI, Lu W. alpha-Defensins in human innate immunity. Immunol Rev. 2012;245:84–112. doi: 10.1111/j.1600-065X.2011.01082.x. [DOI] [PubMed] [Google Scholar]
- Leikina E, Delanoe-Ayari H, Melikov K, Cho MS, Chen A, Waring AJ, Wang W, Xie Y, Loo JA, Lehrer RI, Chernomordik LV. Carbohydrate-binding molecules inhibit viral fusion and entry by crosslinking membrane glycoproteins. Nat Immunol. 2005;6:995–1001. doi: 10.1038/ni1248. [DOI] [PubMed] [Google Scholar]
- Li YF, Poole S, Nishio K, Jang K, Rasulova F, McVeigh A, Savarino SJ, Xia D, Bullitt E. Structure of CFA/I fimbriae from enterotoxigenic Escherichia coli. Proceedings of the National Academy of Sciences. 2009;106:10793–10798. doi: 10.1073/pnas.0812843106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud RY, Stones DH, Li W, Emara M, El-domany RA, Wang D, Wang Y, Krachler AM, Yu J. The Multivalent Adhesion Molecule SSO1327 plays a key role in Shigella sonnei pathogenesis. Molecular Microbiology. 2016;99:658–673. doi: 10.1111/mmi.13255. [DOI] [PubMed] [Google Scholar]
- Mastroianni JR, Ouellette AJ. Alpha-defensins in enteric innate immunity: functional Paneth cell alpha-defensins in mouse colonic lumen. J Biol Chem. 2009;284:27848–27856. doi: 10.1074/jbc.M109.050773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurelli AT, Blackmon B, Curtiss R. Temperature-dependent expression of virulence genes in Shigella species. Infection and Immunity. 1984;43:195–201. doi: 10.1128/iai.43.1.195-201.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of Applied Crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy MP, Herbert BR, Slade MB, Rabilloud T, Nouwens AS, Williams KL, Gooley AA. Proteomic analysis of the Escherichia coli outer membrane. European Journal of Biochemistry. 2000;267:2871–2881. doi: 10.1046/j.1432-1327.2000.01296.x. [DOI] [PubMed] [Google Scholar]
- Morita-Ishihara T, Ogawa M, Sagara H, Yoshida M, Katayama E, Sasakawa C. Shigella Spa33 Is an Essential C-ring Component of Type III Secretion Machinery. Journal of Biological Chemistry. 2006;281:599–607. doi: 10.1074/jbc.M509644200. [DOI] [PubMed] [Google Scholar]
- Mounier J, Vasselon T, Hellio R, Lesourd M, Sansonetti PJ. Shigella flexneri enters human colonic Caco-2 epithelial cells through the basolateral pole. Infection and Immunity. 1992;60:237–248. doi: 10.1128/iai.60.1.237-248.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallographica Section D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. [20] Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology (Academic Press) 1997:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Ouellette A. Paneth cell α-defensins in enteric innate immunity. Cell Mol Life Sci. 2011;68:2215–2229. doi: 10.1007/s00018-011-0714-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazgier M, Ericksen B, Ling M, Toth E, Shi J, Li X, Galliher-Beckley A, Lan L, Zou G, Zhan C, et al. Structural and functional analysis of the pro-domain of human cathelicidin, LL-37. Biochemistry. 2013;52:1547–1558. doi: 10.1021/bi301008r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdomo OJ, Cavaillon JM, Huerre M, Ohayon H, Gounon P, Sansonetti PJ. Acute inflammation causes epithelial invasion and mucosal destruction in experimental shigellosis. J Exp Med. 1994;180:1307–1319. doi: 10.1084/jem.180.4.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phalipon A, Sansonetti PJ. Shigella’s ways of manipulating the host intestinal innate and adaptive immune system: a tool box for survival? Immunol Cell Biol. 2007;85:119–129. doi: 10.1038/sj.icb7100025. [DOI] [PubMed] [Google Scholar]
- Philpott DJ, Edgeworth JD, Sansonetti PJ. The pathogenesis of Shigella flexneri infection: lessons from in vitro and in vivo studies. Philos Trans R Soc Lond B Biol Sci. 2000;355:575–586. doi: 10.1098/rstb.2000.0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro-Cerdá J, Cossart P. Bacterial Adhesion and Entry into Host Cells. Cell. 2006;124:715–727. doi: 10.1016/j.cell.2006.02.012. [DOI] [PubMed] [Google Scholar]
- Rajabi M, Ericksen B, Wu X, de Leeuw E, Zhao L, Pazgier M, Lu W. Functional Determinants of Human Enteric α-Defensin HD5: CRUCIAL ROLE FOR HYDROPHOBICITY AT DIMER INTERFACE. Journal of Biological Chemistry. 2012;287:21615–21627. doi: 10.1074/jbc.M112.367995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapista A, Ding J, Benito B, Lo YT, Neiditch M, Lu W, Chang T. Human defensins 5 and 6 enhance HIV-1 infectivity through promoting HIV attachment. Retrovirology. 2011;8:45. doi: 10.1186/1742-4690-8-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman NH, Ghosh D, Huttner KM, Paterson Y, Bevins CL. Protection against enteric salmonellosis in transgenic mice expressing a human intestinal defensin. Nature. 2003;422:522–526. doi: 10.1038/nature01520. [DOI] [PubMed] [Google Scholar]
- Sawasvirojwong S, Srimanote P, Chatsudthipong V, Muanprasat C. An Adult Mouse Model of Vibrio cholerae-induced Diarrhea for Studying Pathogenesis and Potential Therapy of Cholera. PLoS Negl Trop Dis. 2013;7:e2293. doi: 10.1371/journal.pntd.0002293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder GN, Hilbi H. Molecular Pathogenesis of Shigella spp.: Controlling Host Cell Signaling, Invasion, and Death by Type III Secretion. Clinical Microbiology Reviews. 2008;21:134–156. doi: 10.1128/CMR.00032-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selsted ME, Ouellette AJ. Mammalian defensins in the antimicrobial immune response. Nat Immunol. 2005;6:551–557. doi: 10.1038/ni1206. [DOI] [PubMed] [Google Scholar]
- Sereny B. Experimental shigella keratoconjunctivitis; a preliminary report. Acta Microbiol Acad Sci Hung. 1955;2:293–296. [PubMed] [Google Scholar]
- Shim DH, Suzuki T, Chang SY, Park SM, Sansonetti PJ, Sasakawa C, Kweon MN. New animal model of shigellosis in the Guinea pig: its usefulness for protective efficacy studies. J Immunol. 2007;178:2476–2482. doi: 10.4049/jimmunol.178.4.2476. [DOI] [PubMed] [Google Scholar]
- Singer M, Sansonetti PJ. IL-8 is a key chemokine regulating neutrophil recruitment in a new mouse model of Shigella-induced colitis. J Immunol. 2004;173:4197–4206. doi: 10.4049/jimmunol.173.6.4197. [DOI] [PubMed] [Google Scholar]
- Snellings NJ, Tall BD, Venkatesan MM. Characterization of Shigella type 1 fimbriae: expression, FimA sequence, and phase variation. Infection and Immunity. 1997;65:2462–2467. doi: 10.1128/iai.65.6.2462-2467.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szyk A, Wu Z, Tucker K, Yang D, Lu W, Lubkowski J. Crystal structures of human α-defensins HNP4, HD5, and HD6. Protein Science. 2006;15:2749–2760. doi: 10.1110/ps.062336606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenge VR, Gounder AP, Wiens ME, Lu W, Smith JG. Delineation of Interfaces on Human Alpha-Defensins Critical for Human Adenovirus and Human Papillomavirus Inhibition. PLoS Pathog. 2014;10:e1004360. doi: 10.1371/journal.ppat.1004360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsilingiri K, Barbosa T, Penna G, Caprioli F, Sonzogni A, Viale G, Rescigno M. Probiotic and postbiotic activity in health and disease: comparison on a novel polarised ex-vivo organ culture model. Gut. 2012;61:1007–1015. doi: 10.1136/gutjnl-2011-300971. [DOI] [PubMed] [Google Scholar]
- Wei G, Pazgier M, de Leeuw E, Rajabi M, Li J, Zou G, Jung G, Yuan W, Lu WY, Lehrer RI, Lu W. Trp-26 imparts functional versatility to human alpha-defensin HNP1. J Biol Chem. 2010;285:16275–16285. doi: 10.1074/jbc.M110.102749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West NP, Sansonetti P, Mounier J, Exley RM, Parsot C, Guadagnini S, Prevost MC, Prochnicka-Chalufour A, Delepierre M, Tanguy M, Tang CM. Optimization of virulence functions through glucosylation of Shigella LPS. Science. 2005;307:1313–1317. doi: 10.1126/science.1108472. [DOI] [PubMed] [Google Scholar]
- Wilson SS, Bromme BA, Holly MK, Wiens ME, Gounder AP, Sul Y, Smith JG. Alpha-defensin-dependent enhancement of enteric viral infection. PLoS Pathog. 2017;13:e1006446. doi: 10.1371/journal.ppat.1006446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Ericksen B, Tucker K, Lubkowski J, Lu W. Synthesis and characterization of human alpha-defensins 4-6. J Pept Res. 2004;64:118–125. doi: 10.1111/j.1399-3011.2004.00179.x. [DOI] [PubMed] [Google Scholar]
- Wu Z, Hoover DM, Yang D, Boulegue C, Santamaria F, Oppenheim JJ, Lubkowski J, Lu W. Engineering disulfide bridges to dissect antimicrobial and chemotactic activities of human beta-defensin 3. Proc Natl Acad Sci U S A. 2003;100:8880–8885. doi: 10.1073/pnas.1533186100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.