

Abstract
Diabetic nephropathy (DN), a severe microvascular complication frequently associated with both type 1 and type 2 diabetes mellitus, is a leading cause of renal failure. It can also lead to accelerated cardiovascular disease and macrovascular complications. Currently available therapies have not been fully efficacious in the treatment of DN, suggesting that further understanding of the molecular mechanisms underlying the pathogenesis of DN is necessary for the improved management of this disease. Although key signal transduction and gene regulation mechanisms have been identified, especially those related to the effects of hyperglycaemia, transforming growth factor-β1 and angiotensin II, progress in functional genomics, high-throughput sequencing technology, epigenetics and systems biology approaches have greatly expanded our knowledge base and uncovered new molecular mechanisms and factors involved in DN. These mechanisms include DNA methylation, chromatin histone modifications, novel transcripts and functional noncoding RNAs such as microRNAs and long noncoding RNAs. In this Review, we will discuss the significance of these emerging mechanisms, how they mediate the actions of growth factors to augment the expression of extracellular matrix and inflammatory genes associated with DN, and their potential usefulness as diagnostic biomarkers or novel therapeutic targets for DN.
Introduction
Both type 1 and type 2 diabetes mellitus are associated with debilitating microvascular complications such as diabetic nephropathy (DN).1 Approximately 50% of patients who have end-stage renal disease (ESRD) needing painful and costly dialysis are diabetic. Furthermore, these patients are also highly susceptible to macrovascular complications.2 However, the underlying molecular mechanisms leading to DN are not fully elucidated. Although several classical mechanisms and pathways leading to DN have been described over the years, new molecular and epigenetic mechanisms are emerging, which are highlighted in this Review.
Accumulation of extracellular matrix (ECM) proteins such as collagens (leading to fibrosis) and mesangial expansion (leading to hypertrophy) in the kidney glomerular and tubular compartments are major hallmarks of DN, and contribute to renal failure in diabetes.3–5 Podocyte effacement and apoptosis are also evident, and reduction in the number of podocytes per glomerulus is suggested to be a key predictor of DN progression.6,7
Hyperglycaemia as well as complex interactions between metabolic and haemodynamic factors are linked to the development of diabetic complications including DN.4,5,8 High glucose levels adversely impact all renal cell types (mesangial cells, tubular cells, podocytes and endothelial cells), and augment monocyte and macrophage infiltration. High glucose conditions also increase the formation of advanced glycation end-products and production of growth factors such as transforming growth factor-β1 (TGF-β1) and angiotensin II in renal cells (Figure 1). TGF-β1 is a major player in DN pathogenesis, mainly because of its profibrotic actions, and its levels are increased in most renal cells in diabetes.3,5,9,10 Crosstalk between these diabetogenic factors can amplify and perpetuate the expression of pathological genes associated with the progression of DN (Figure 1). Multiple biochemical and signal transduction mechanisms, including oxidant stress, protein kinase C, Akt kinase and other kinases, have been implicated in the activation of key effector transcription factors such as Smads and nuclear factor κB downstream of high glucose conditions and growth factors. This activation leads to increased production of proinflammatory cytokines, cell-cycle genes and profibrotic and ECM genes involved in DN.4,5,8 Despite such advances, currently available therapies are still not fully effective in preventing progression to ESRD, suggesting that additional mediators and mechanisms need to be explored. Furthermore, data from clinical trials have demonstrated a ‘metabolic memory’ of prior exposure to hyperglycaemia by which complications such as DN persist despite subsequent glycaemic control. In this Review, we discuss emerging evidence from research that suggests a key role for epigenetic mechanisms and noncoding RNAs in the pathogenesis of DN and metabolic memory. Epigenetics refers to heritable patterns of gene expression that are not dependent on the DNA sequence information. The epigenetic mechanisms discussed here include DNA methylation, post-translational modifications of histones in chromatin, as well as noncoding RNAs, all of which can regulate gene expression individually and cooperatively, and thus affect physiological and pathological processes.
Figure 1. Emerging molecular mechanisms of diabetic nephropathy.
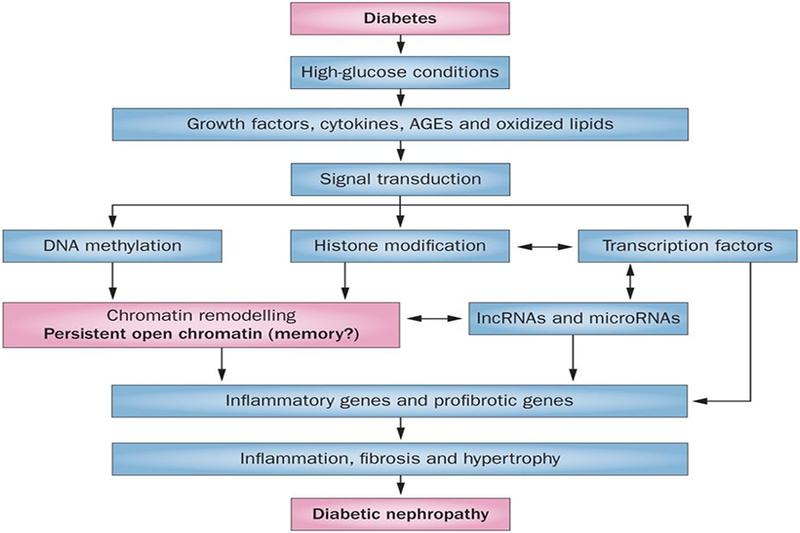
Diabetic conditions induce the expression of growth factors such as TGF-β1 and angiotensin II, cytokines and AGEs to promote inflammation, fibrosis and hypertrophy, which contribute to the progression of diabetic nephropathy. These factors stimulate various signal transduction mechanisms that activate downstream transcription factors. They can also affect DNA methylation and histone modifications, which result in increased chromatin accessibility to transcription factors near pathological genes in renal cells. Coordinated interactions between transcription factors and epigenetic mechanisms can increase the expression of not only coding RNAs, but also noncoding RNAs such as microRNAs and lncRNAs. Furthermore, microRNAs and lncRNAs can also increase the expression of pathological genes via post-transcriptional mechanisms. Notably, the induction of key coding genes and proteins, lncRNAs and microRNAs can also ‘lock’ open chromatin states to create persistent expression of genes, which could be one mechanism of metabolic memory. Abbreviations: AGE, advanced glycation end-product; lncRNA, long noncoding RNA.
Cellular mechanisms leading to DN
Growth factors and oxidant stress
Several growth factors have been shown to influence the progression of DN; in particular TGF-β1, angiotensin II and platelet-derived growth factor have been extensively studied.9–12 These factors can be induced in renal cells by high glucose levels, advanced glycation end-products and growth factors by various mechanisms to promote fibrosis and inflammation (Figure 1). TGF-β1 is induced by high glucose in mesangial cells via upstream stimulatory factors (USF1/USF2) through promoter E-boxes.13,14 Engagement of TGF-β1 to its receptors induces the phosphorylation and nuclear translocation of Smad2/Smad3 transcription factors,15,16 which regulate TGF-β1-induced gene expression.17,18 TGF-β1 can activate mitogen-activated protein kinases such as p38 and ERKs,19–21 and also Akt through inhibition of Pten.22,23 TGF-β1 plays a key role in mesangial fibrosis and hypertrophy by increasing the expression of ECM proteins such as collagen and fibronectin.3,4,9,10 Angiotensin II signalling via the type 1 angiotensin II receptor is critical in the pathogenesis of DN11 and leads to increased production of TGF-β1 and amplification of inflammatory and profibrotic gene expression. High glucose conditions and growth factors can also cause renal dysfunction by promoting oxidant stress, and activation of key NADPH oxidase enzymes has been implicated.5,24 Besides oxidant stress, other factors such as mitochondrial and endoplasmic reticulum stress and autophagy have also been implicated in DN.5,25
Cytokines, chemokines and inflammation
Increasing evidence suggests that inflammation due to proinflammatory cytokines and chemokines secreted by renal cells and macrophages infiltrating into the kidney can substantially contribute to DN.26 Thus the cytokines IL-1, IL-6, IL-18 and tumour necrosis factor-α (TNF-α) and their effectors have all been implicated in DN.27,28 Much attention has been paid to TNF-α and its receptors in DN.27 Interestingly, circulating levels of TNF receptors TNFR1 and TNFR2 predict renal function decline in type 1 diabetes.29 Elevated urinary levels of IL-18 in patients were correlated with the progression of DN.30 The chemokine C-C motif chemokine 2 (CCL2, also known as monocyte chemoattractant protein 1) can attract monocytes to injured tissues to provoke inflammation. The engagement of CCL2 to CC chemokine receptor 2 has been implicated in the progression of DN.31 Stromal cell-derived factor 1 contributes to podocyte loss, and specific inhibitors ameliorated proteinuria and glomerulosclerosis in db/db mice.32 Fractalkine and its receptor were increased in the glomeruli of diabetic rats33 and fractalkine-knockout mice were protected from DN.34 Soluble fractalkine can attract T-cells and monocytes to activated endothelial cells expressing CX3C chemokine receptor 1. Endothelial dysfunction and inflammation caused by macrophage infiltration also play important roles in DN pathogenesis.5,26
Overall, numerous excellent studies have demonstrated the involvement of key cellular factors and biochemical pathways in DN. Furthermore, advances in functional genomics, transcriptomic profiling, follow-up data mining and systems biology approaches with renal glomeruli and tubuli of patients with chronic kidney disease including DN have not only confirmed the role of many previously implicated factors, but also led to the discovery of several new players.35–37
Metabolic memory
Long-term progression of diabetic complications such as DN, despite good glycaemic control, could be due to a memory of prior exposure of target cells to high glucose levels, leading to persistence of its deleterious effects long after glucose normalization. This effect has been observed in some major clinical trials. In the landmark Diabetes Control and Complications Trial (DCCT), patients with type 1 diabetes were treated with either conventional or intensive insulin therapy and it was observed that intensive glycaemic control could clearly delay the progression of key diabetic complications. Furthermore, in the DCCT follow-up observational Epidemiology of Diabetes Interventions and Complications (EDIC) study, patients in both DCCT treatment groups were encouraged to practice intensive therapy; since year 4 of the EDIC study both groups have maintained HbA1c levels of around 8%. The results demonstrate that patients who were on intensive glycaemic control during the DCCT and encouraged to continue on intensive control during the EDIC phase had significantly slower rates of progression of microvascular complications, such as retinopathy, neuropathy, impaired glomerular filtration rates and nephropathy, than patients on prior conventional control, even though there was no longer a significant difference in HbA1c levels between the two groups several years into the EDIC phase.38–43 This phenomenon has been termed ‘metabolic memory’.39 A clinical trial of glycaemic control in patients with type 2 diabetes, the UK Prospective Diabetes Study, found a similar phenomenon referred to as a ‘legacy effect’.44 However, some clinical trials indicate that although intensive glycaemic control was effective in reducing diabetic retinopathy, in patients with diabetes and established cardiovascular disease it was associated with increased mortality, possibly owing to increased hypoglycaemic episodes.45,46 Because Akt activation is increased in diabetic kidneys and is associated with DN pathogenesis,22,23,47 intensive treatment with insulin might worsen kidney damage through Akt activation.
Several experimental models with cells and animals have been used to document the metabolic memory phenomenon.48 Vascular smooth muscle cells derived from aortas of type 2 diabetic db/db mice showed increased proinflammatory responses compared with those from nondiabetic control db/+ mice even after in vitro culture for several passages.49 Endothelial cells cultured for short periods in high glucose continued to exhibit increased oxidant stress and inflammatory gene expression even after restoration of normoglycaemia.50 Metabolic memory has also been studied in models of diabetic retinopathy.51,52 Although not yet tested in animal models of DN, these clinical and experimental studies suggest that metabolic memory poses a major challenge in DN treatment, and has triggered intense interest in determining the underlying molecular mechanisms. Notably, epigenetic factors have been implicated in metabolic memory in which early effects of hyperglycaemia may be imprinted in target tissues susceptible to complications.
Epigenetic mechanisms underlying DN
Epigenetics and chromatin
Epigenetics is the study of heritable changes in gene expression that occur without alterations in the underlying DNA sequences. Epigenetic control of gene regulation plays a critical role in developmental processes, cell identity, genomic imprinting, X-chromosome inactivation, immune cell function, stem cell plasticity, differential disease susceptibility between monozygotic twins, and cellular responses to environmental cues.53–55
Chromosomal DNA in mammalian cells is tightly packaged into chromatin, a higher-order structure composed of subunits called nucleosomes. Each nucleosome consists of an octamer of histones, with two copies of each of the core histone proteins H2A, H2B, H3 and H4, wrapped by 147 base pairs of chromosomal DNA. Post-translational modifications of nucleosomal histones and DNA methylation represent major epigenetic modifications.53,54 These modifications, together with short noncoding RNAs (ncRNAs, for example microRNAs [miRNAs]) and long ncRNAs (lncRNAs), regulate chromatin function and gene expression. They collectively form the epigenome, which stores the epigenetic information needed for cell identity and cell-type-specific gene expression patterns. Advances in high-throughput genome-wide profiling and sequencing approaches have led to a broader understanding of various aspects of the epigenome and its correlations to phenotype. Alterations in epigenome states have profound effects on gene regulation and biological outcomes and are implicated in the pathogenesis of various human disorders.55,56 Furthermore, persistence of aberrant epigenetic marks, even after withdrawal of the original stimuli, can mediate disease progression and resistance to conventional therapies. Notably, foetal environment and maternal nutrition can promote epigenetic changes to control the onset of metabolic abnormalities in adult life.56,57 Excessive or poor nutrition can also lead to heritable changes in epigenetic marks that may predispose future generations to metabolic abnormalities56,57 which in turn could affect the development of DN. Therefore, evaluation of alterations in the epigenome under disease conditions can yield new insights into the pathogenesis of diabetic complications such as DN and metabolic memory that, in turn, could be translated to novel biomarkers and therapeutic modalities. Epigenotype information from epigenome-wide association studies may complement genotype information gleaned from genome-wide association studies.
Why study epigenetics in DN?
DN clearly has a genetic component and there have been intense efforts including genome-wide association studies to identify causal genes or single-nucleotide polymorphisms that are associated with the disease.58,59 Ongoing efforts are also aimed at determining the potential functional and pathological role(s) of these genes and single-nucleotide polymorphisms.58 On the other hand, given that environmental influences could be key contributing factors, and that epigenetics is a molecular interface between genetics and the environment, it is increasingly believed that epigenetics may also play an important role in common human diseases such as diabetes and DN.48,60,61 A second epigenetic hit derived from environmental influences may cooperate with genetics to confer functionality to disease-associated variants. An example of this type of crosstalk was provided in a study of epigenetic histone modifications at type 1 diabetes susceptibility loci.62 In another study, some of the DNA methylated genes identified in saliva samples from patients with diabetes and ESRD compared with those without DN were previously reported to have sequence variants associated with kidney disease.63 Furthermore, most disease-associated single-nucleotide polymorphisms are present in noncoding regions of the genome, including promoters, enhancers and ncRNA regions,64 which can affect gene expression by altering transcription factor binding. Thus, a cooperation between epigenetics and genetics may play a role in DN. A study of epigenetic layers, which include DNA methylation, histone modifications and ncRNAs, can therefore provide valuable information.
Epigenetic mechanisms in DN
DNA methylation, a well recognized epigenetic mark that regulates gene transcription, usually occurs at 5’-cytosines of CpG dinucleotides and is catalyzed by specific DNA methyltransferases.54 In general, DNA methylation at gene promoter regions leads to gene repression, whereas at gene bodies it can modulate transcription elongation and alternative splicing. DNA methylation is recognized by various methyl binding proteins, which can recruit transcriptional corepressors to inhibit gene expression. DNA methylation can also interfere with the binding of transcription factors at promoters to inhibit gene expression.54
Some studies have examined the role of DNA methylation in type 1 and type 2 diabetes and in inherited metabolic diseases,48 further underscoring the key role played by environment in regulating epigenetic modifications to influence phenotypic outcomes.56 In addition, there is much interest in examining how single-nucleotide polymorphisms affect DNA methylation and vice versa, and DNA methylation variations have been associated with diabetes susceptibility genes.65,66
The role of DNA methylation in DN and ESRD has elicited much interest,48,63,67 more so because most genome-wide association studies of DN have yielded few susceptibility loci. Studies of DNA methylation profiles in genomic DNA of diabetic patients with and without DN revealed differential methylation in several genes, including UNC13B, which has been suggested to mediate apoptosis in glomerular cells as a result of hyperglycaemia, and hence the association could be relevant to the initiation and pathogenesis of DN.67 Several differentially DNA methylated regions were noted in saliva of patients with ESRD compared with those with chronic kidney disease alone.63 DNA methylation has also been implicated in fibrosis and in the actions of TGF-β1. Hypermethylation of RASAL1 increased Ras activation in fibroblasts, leading to proliferation and fibrosis.68 In endothelial cells, treatment with high glucose led to changes in DNA methylation at several genes involved in endothelial cell dysfunction.69 Notably, DNA methylation profiling performed with microdissected kidney tubuli obtained from patients with chronic kidney disease (including DN) compared with controls showed significant differences in their epigenomes.70 Differentially methylated genes were related to fibrosis, and methylation was found mostly in enhancer regions, underscoring the importance of such regulatory regions. Together, these emerging reports highlight the importance of epigenetic DNA methylation in regulating fibrotic and other genes associated with DN. They also suggest that the combination of DNA methylation with genetic variants provide improved functional predictions of disease outcomes.
In addition to DNA methylation, post-translational modifications of nucleosomal histone proteins in chromatin are also part of the epigenetic layer that regulate gene expression (Figure 2).53 In fact, genome-wide profiling of modified histones using chromatin immunoprecipitation (ChIP) sequencing has demonstrated that cell-type-specific regulatory genes (promoters and enhancers) and transcribed regions can be identified by the presence of specific histone marks.71,72 Histone post-translational modifications include lysine acetylation (Kac), lysine methylation (Kme) and ubiquitination, serine and threonine phosphorylation and arginine methylation.53,72 In general, histone Kac (such as H3K9ac and H3K14ac) is enriched at gene promoters or actively transcribed regions, whereas loss of acetylation is associated with gene repression. H3K4me1/2/3 and H3K36me2/3 are generally associated with transcriptionally active genome regions, whereas H3K9me3, H3K27me3 and H4K20me3 are associated with repressed regions.53 Enhancers are enriched with H3K4me1 and H3K27ac (both active histone marks) .73 Bivalent promoters in developmental genes are enriched with both active (H3K4me3) and repressive (H3K27me3) marks and are therefore in a poised state.71,72 Interestingly, actively transcribed regions in the genome are characterized by histone H3K4me3 (promoter) and H3K36me3 (gene body) chromatin marks and are therefore valuable to identify both known and novel transcripts including genome-wide ncRNAs.74
Figure 2. Regulation of gene transcription in DN mediated by histone lysine modifications.

Diabetic conditions activate transcription factors through signal transduction mechanisms to increase gene expression. These signalling mechanisms can also alter histone lysine modifications to increase chromatin accessibility, that is from compact (suppressed) to open (active) chromatin. Two types of histone modifications are proposed: histone lysine acetylation (Kac) and histone lysine methylation (Kme). A dynamic balance between active and repressive marks can create open chromatin states near DN-related genes, increase transcription factor accessibility, and promote expression of genes associated with inflammation and fibrosis. These signalling events and epigenetic mechanisms also promote the expression of noncoding RNAs and, together, can lead to persistently open chromatin states and unchecked gene expression, which can be a mechanism underlying metabolic memory. Several approaches can interrupt these processes, which could form the basis of new therapeutic targets for DN. a | Activation of histone acetyltransferases mediates increases in H3Kac (especially H3K9ac), which increases chromatin accessibility to transcription factors. b | Kme can be induced by HMTs and erased by KDMs. Diabetic conditions can downregulate repressive H3K9 HMTs to decrease H3K9me2/3 levels, or activate permissive H3K4 HMTs to increase H3K4me1/2/3 levels near pathological genes. Activation of some H3K9 KDMs may also demethylate H3K9me3. Abbreviations: Ac, acetylation; DN, diabetic nephropathy; HDAC, histone deacetylase; HMT, histone methyltransferase; KDM, histone lysine demethylase; lncRNA, long noncoding RNA; Me, methylation; P, phosphorylation.
Histone Kac is catalyzed by histone acetyl transferases such as p300 and CREB-binding protein, which also act as transcription coactivators. Conversely, histone deacetylases (HDACs) and sirtuins remove acetylation marks and can act as corepressors with some exceptions. Kme is catalyzed by histone lysine methyltransferases (HMTs) and removed by histone lysine demethylases (KDMs).53 HMTs and KDMs are specific to the lysine residue being modified and the extent of methylation (Kme1/2/3). For example, the HMT SUV39H1 mediates H3K9me3 (a repressive mark), whereas the KDM lysine-specific demethylase 1 removes H3K4me1/2.75
The interactions and crosstalk between histone post-translational modifications, DNA methylation and ncRNAs can serve as a footprint of the environment and an epigenetic layer regulating gene transcription.76,77 Dysregulation of this fine balance and crosstalk can result in diabetes and its complications including DN.
Several studies show that key histone post-translational modifications are involved in the regulation of genes associated with the pathogenesis of diabetes, such as insulin and islet-specific transcription factors.48,60 In addition, several groups are examining the role of histone post-translational modifications in adipocytes related to type 2 diabetes, obesity and the metabolic syndrome.48,60 These endeavors highlight the increasing evidence that histone post-translational modifications can play key roles in the pathogenesis of diabetes. Logically, they can be expected to also affect chromatin structure of target genes in organs associated with complications such as the kidney.
In vitro studies in cell culture have demonstrated the involvement of histone post-translational modifications in regulating key profibrotic and other pathological genes associated with DN pathogenesis. In rat mesangial cells, TGF-β1 and high glucose treatment increased H3K9/14ac enrichment near Smad and SP1 binding sites at the plasminogen activator inhibitor 1 and p21 gene promoters together with HATs p300 and CREB-binding protein. Histone H3K9/14ac was found to have a key role in transcription of these genes in response to TGF-β1.78 Furthermore, TGF-β1-induced expression of collagen α−1(I) chain, connective tissue growth factor and plasminogen activator inhibitor 1 was associated with enrichment of active Kme marks (H3K4me1/2/3) and reduced levels of repressive marks (H3K9me2/3 ) at their promoters in rat mesangial cells.79 Silencing of the H3K4me HMT SET7 using small interfering RNA attenuated TGF-β1-induced gene expression, suggesting a key role for H3K4me and SET7 in fibrotic gene expression in mesangial cells. High glucose treatment also elicited similar histone post-translational modifications at fibrotic and cell-cycle gene (p21) promoters, which were blocked by an anti-TGF-β antibody (Figure 2).78,79
In vivo animal models of DN have also demonstrated changes in histone post-translational modifications. In some studies, global histone modifications were associated with the expression of fibrotic and cell-cycle genes related to DN.48 ChIP assays with renal glomeruli of diabetic mice demonstrated that increased expression of plasminogen activator inhibitor 1 and p21 genes was associated with H3K9/14ac enrichment near their promoter Smad and SP1 binding sites compared with the nondiabetic controls.78 Because gene expression and chromatin structure can be regulated by a histone code dictated by a combination of multiple histone post-translational modifications, several candidate histone post-translational modifications were profiled by matrix ChIP assays in glomeruli from diabetic db/db mice and control db/+ mice.80 Results showed that glomeruli from db/db mice exhibited increased enrichment of RNA polymerase II and certain active histone marks, and decreased levels of key repressive marks at the promoters of plasminogen activator inhibitor 1 and AGER, suggesting cooperative mechanisms that promote chromatin accessibility to enable increased transcription factor access near these genes (Figure 2). Recently Komers et al.81 also reported that increases in RNA polymerase II recruitment and H3K4me2 levels, but decreases in H3K27me3 levels, were associated with the expression of DN-related genes in mouse and rat models, although there were some differences between the two species. Interestingly, in studies with db/db mice, it was found that treatment with losartan, an angiotensin II type 1 receptor blocker, for 10 weeks ameliorated key indices of DN, and reversed histone H3K9ac, but not other epigenetic changes observed in the db/db mice.80 Such incomplete inhibition of epigenetic changes may be one reason for the relative inefficiency of angiotensin II type 1 receptor blockade in some patients with DN.11 It also raises the question whether epigenetic therapies should be added to traditionally used drugs for DN treatment for better control of the disease.
Overall, we clearly expect to see much activity in the area of epigenetics and DN. Because of the cell-type-specific nature of epigenetics, studies need to be performed with key component renal cells, including podocytes, mesangial, endothelial and tubular cells, as well as renal tissues and biopsies obtained from animals as well as humans at various stages of DN, to detect distinct histone modifications and DNA methylation patterns specific to DN.
Epigenetics and metabolic memory
Several diabetic complications, including DN, are characterized by chronic inflammation, and increased activity of the proinflammatory transcription factor nuclear factor κB. Epigenetic mechanisms have a role in high-glucose-induced activation of nuclear factor κB and inflammatory gene expression in vascular cells and monocytes relevant to complications associated with diabetes.48,82,83 Furthermore, disease relevance was demonstrated in genome-wide profiling studies using ChIP linked to microarrays (ChIP-on-chip), which showed differential H3K4me2 and H3K9me2 enrichment at several genes related to diabetes and inflammation in high-glucose-treated THP-1 monocytes, and in blood monocytes and lymphocytes from patients with diabetes.83–85 The H3K4me HMT SET7 was identified as a nuclear factor κB coactivator at a subset of inducible inflammatory genes in monocytes.86 Induction of key inflammatory genes and histone Kac of their promoters by high glucose were attenuated by the anti-inflammatory agent curcumin,87 indicating that natural products could be evaluated for epigenetic therapies. These and other studies emphasize the role of epigenetic mechanisms in the regulation of inflammatory genes relevant to DN.
It is particularly interesting to examine a role for epigenetics in metabolic memory. A number of studies have examined how persistently altered histone Kme, a relatively stable mark, at key pathological genes might lead to their sustained upregulation and metabolic memory. Thus, increased inflammatory gene expression and migration in vascular smooth muscle cells obtained from type 2 diabetic db/db mice compared with control db/+ mice, even after being cultured for several passages in vitro, was associated with a persistent loss of the repressive mark H3K9me3 at these gene promoters. In parallel, the expression of the corresponding H3K9me3 HMT SUV39H1 was downregulated, which was attributed at least in part to increased levels of miR-125b in db/db mice, revealing a novel crosstalk between miRNA actions and epigenetic components in diabetes.88,89 The persistent expression of p65 was attributed to sustained enrichments in H3K4me1 and SET7 in endothelial cells that were previously exposed to high glucose for short time periods.50,90 The relevance of metabolic memory in human DN needs more investigation. In this regard, the first epigenome profiling of patients with type 1 diabetes experiencing metabolic memory was reported.91 Results of a case group of EDIC study participants from the DCCT former conventional treatment group experiencing progression of nephropathy and retinopathy versus a control group from the former DCCT intensive treatment group not showing progression were compared. There was significant enrichment in H3K9ac in monocytes from cases compared with controls. Furthermore, the top case hyperacetylated promoters were enriched in inflammatory genes and those related to diabetic complications. Interestingly, monocyte H3K9ac was significantly associated with the mean HbA1c during DCCT and EDIC. These results suggest an association between HbA1c and H3K9ac and a potential epigenetic explanation for metabolic memory in humans.91
The field of epigenetics has received considerable impetus owing to the development of several technologies for epigenome profiling that have led to major discoveries by consortia such as ENCODE.71,92,93 In particular, next-generation DNA sequencing platforms have been used for ChIP-seq, bisulfite-seq, FAIRE-seq and others,94–97 and have yielded high-resolution profiles of genome-wide epigenetic states. Integration of these multiple datasets with transcriptome data from RNA-seq has led to the detection of novel transcripts including lncRNAs.74 Publicly available databases together with resources from the NIH Roadmap Epigenomics Mapping consortium98 are valuable reference tools to advance research in this area. Studies with blood cells and renal tissues from patients with DN and mouse models of DN are ongoing in several laboratories. These epigenome-wide association studies are likely to greatly increase our knowledge of the epigenomic variations linked with DN and metabolic memory.
Noncoding RNAs: miRNAs
Biogenesis and actions in gene regulation
MiRNAs are endogenously produced small ncRNAs (about 20–22 nucleotides) that regulate gene expression via key post-transcriptional mechanisms. Mammalian miRNAs can silence the expression of target genes by imperfect base-pairing to the 3’-untranslated regions of target mRNAs to promote translational repression, and/or mRNA degradation.99 More than 1,000 miRNAs have now been identified which can potentially regulate nearly 60% of the human protein-coding genes.99 Thus, miRNAs can modulate the expression of numerous genes to alter key cellular functions and thereby influence the course of various diseases.
MiRNAs are transcribed in the nucleus by RNA polymerase II into primary transcripts (pri-miRNAs). Within the nucleus, the ribonuclease III Drosha cuts pri-miRNAs to precursor miRNAs (pre-miRNAs), which are ~70 nucleotide stem-loop structures.100,101 These pre-miRNAs are transferred to the cytoplasm by a protein called exportin-5. In the cytoplasm, pre-miRNAs are further processed by a second ribonuclease, Dicer, to mature miRNAs.100,101 Mature miRNA duplexes are then incorporated into the RNA-induced silencing complex, which also contains the Argonaute family of proteins. Within this complex, miRNAs interact with the 3’-untranslated region of the target mRNA species and induce translational repression or degradation.100 Whereas these are well recognized modes of action of miRNAs which can be further influenced by regulation of proteins within the miRNA microprocessor complex, other novel mechanisms have also been reported.100–102
Research of miRNAs has grown exponentially over the past 10 years owing to observations that they are dysregulated or mutated in several diseases. Because miRNAs can modulate the expression of genes associated with key pathophysiological processes, they are likely to affect the progression of diabetic complications. Studies showing that the deletion of Dicer or Drosha in mice results in severe cardiac and renal defects are supportive of this concept.103–110 These small molecules therefore present a big opportunity for evaluating new mechanisms underlying the progression of DN that in turn could lead to the identification of novel biomarkers and therapeutic targets.
MiRNAs associated with DN
Five miRNAs (miR-192, miR-194, miR-204, miR-215 and miR-216a) were shown to be enriched in the kidney compared with other organs, suggesting that miRNAs have critical renal functions.111 The earliest studies of miRNAs in diabetic complications were performed in DN models. Involvement of miRNAs was suggested by the phenotypes observed in mice with podocyte-specific deletion of Dicer,103,104,106 which included major renal abnormalities reminiscent of DN, including proteinuria, podocyte foot process effacement and apoptosis, glomerulosclerosis, and tubulointerstitial fibrosis with renal failure. In other studies, renal cortex and medulla were found to have different miRNA expression patterns, suggesting their cell-type and tissue-specific functions.112
A series of reports using renal cells and in vivo animal models of DN have demonstrated functional relationships between aberrant miRNA expression and processes related to renal fibrosis and DN. An upregulation of several miRNAs (miR-192, miR-200b/c, miR-216a and miR-217) in TGF-β1-treated mouse mesangial cells and in renal glomeruli of mouse models of diabetes (streptozotocin-injected type 1 diabetic mice and type 2 diabetic db/db mice) compared with the corresponding nondiabetic control mice were shown in early reports.14,113–116 These studies revealed the first functional role for an miRNA (miR-192) in DN, and that miR-192 actions lead to upregulation of the key fibrotic genes Col1a2 and Col4a1 in mesangial cells. In addition, miR-192 upregulates other miRNAs, including miR-216a/miR-217 and miR-200b/c, in mesangial cells by targeting the E-box repressors Zeb1 and Zeb2 to relieve repression at their promoters, and this amplifying circuit augments collagen expression. Interestingly, the miR-216a/miR-217 cluster activates Akt by targeting Pten in TGF-β1-treated mouse mesangial cells, thereby uncovering an miRNA-mediated mechanism for TGF-β1-induced Akt activation and cellular hypertrophy.115 MiR-200b/c can also regulate collagen expression and promote the autoregulation of TGF-β1 in mouse mesangial cells by inhibiting Zeb1.14 The expression levels of TGF-β1, p53 and miR-192 were all increased in the renal cortex of diabetic mice compared with nondiabetic mice, which was associated with increased glomerular expansion and fibrosis.117 In this report, miR-192-knockout mice were developed and made diabetic using streptozotocin injections. Compared with control diabetic mice, the diabetic miR-192-knockout mice were protected from key features of DN.117 Mechanistic studies in cultured mesangial cells demonstrated mutual activation of miR-192 and p53 in an amplification loop in response to TGF-β1 which also involved the miR-192 target Zeb2, leading to increased mesangial cell ECM gene expression and hypertrophy.117,118 Additional in-depth studies showed that the miR-192 promoter was regulated by Smads, protein C-ets-1 as well as chromatin remodelling via promoter histone Kac in response to TGF-β1 in renal cells.119,120 Altogether, these studies have identified miR-192 as a master miRNA regulator in TGF-β1-treated mesangial cells and in increased expression of ECM genes associated with DN.14,113–120
MiR-192 was also reported to be downregulated in cultured proximal tubular epithelial cells treated with high glucose or TGF-β1 and in diabetic ApoE−/− mice, which was associated with increased fibrosis.121,122 Another study showed, however, that upregulation of miR-192 expression in tubular cells treated with TGF-β1 increased fibrosis.119 These complexities could be due to cell-type-specific effects of miRNAs and differences in animal models studied. However, cell-specific transcription factors could also dictate the actions of miRNAs. In the case of miR-192, the cell-type-specific response to TGF-β1 might be explained by the cellular status of p53 or protein C-ets-1 as TGF-β1-induced miR-192 expression was abolished in mesangial cells from p53−/− or protein C-ets-1−/− mice.117,120 Similarly, one report showed that miR-200a levels were decreased in TGF-β1-treated proximal tubular epithelial cells, which could be associated with fibrosis because of the concomitant upregulation of the miR-200a target TGF-β2.123 The pioneering work of Gregory et al.124 demonstrated the miR-200 family as regulators of the epithelial phenotype by targeting the transcriptional repressors of E-cadherin, Zeb1 and Zeb2. These observations suggest an important link between miRNAs such as miR-200, TGF-β1 actions and epithelial–mesenchymal transition (EMT), especially in cancer cells. It is however unclear whether miRNA-regulated EMT is critical for renal fibrosis in DN owing to increasing evidence from studies that EMT may not play a major role in renal fibrosis.125,126 It has also been suggested that EMT is observed mainly in immortalized cultured cell lines and not in in vivo renal fibrosis in humans or animal models.127 Because immortalized cell lines likely have mutations in tumour suppressors such as p53 and oncogenes, the cell-type responses to TGF-β1 can differ between primary cells and immortalized cell lines depending on the status of these genes and cell-type-specific transcription factors.
Numerous other miRNAs have also been reported to regulate various genes associated with DN. In particular, much attention has been paid to miR-21 owing to its several relevant targets. MiR-21 was upregulated in the renal cortex of OVE26 type 1 diabetic mice and could target Pten and promote mTOR activation, all factors related to DN.128 An inhibitor of miR-21 has been evaluated as a therapeutic target for DN in mouse models.129 MiR-21 was upregulated in the KK-Ay mouse model of DN and contributed to renal fibrosis by targeting matrix metalloproteinase-9 and metalloproteinase inhibitor 1,130 while in db/db mice miR-21 accelerated TGF-β1 signalling by targeting Smad7.129 Chau et al.131 demonstrated that miR-21−/− mice had reduced interstitial fibrosis in response to renal injury, as did wild-type mice treated with anti-miR-21 oligonucleotides. By contrast, miR-21 was also reported in one study to be downregulated in db/db mice and its overexpression blocked mesangial cells proliferation.132 MiR-377 was found to be upregulated by high glucose or TGF-β1in mesangial cells and increased fibronectin expression and oxidant stress via repression of manganese superoxide dismutase and p21-activated kinase.133 Another study demonstrated increases in miR-192 expression in streptozotocin-injected mice fed with a high-fat diet, which was further augmented in farnesoid X-activated receptor-knockout mice.134 Long et al.135 found miR-93 to be a key miRNA that was downregulated in glomeruli of diabetic db/db mice compared with controls, and also in podocytes and renal microvascular endothelial cells treated with high glucose. Because VEGF-A was identified as a target of miR-93, these studies suggest an antiangiogenic role for miR-93. Conversely, miR-29c, miR-192, and miR-200b were upregulated under similar conditions.136 MiR-29c activated Rho kinase by targeting Spry-1 and subsequently led to ECM protein accumulation and podocyte apoptosis. By contrast, studies in diabetic mice, tubular cells, mesangial cells and podocytes treated with TGF-β1 demonstrate a downregulation of miR-29 family members, and this downregulation correlated with increased levels of collagen I, collagen III and collagen IV, which are direct targets.137,138 Studies in other nondiabetic animal models of renal fibrosis have also demonstrated a profibrotic role for miR-192 and an antifibrotic role for miR-29 family members.119,139,140 MiR-192 and miR-215 were upregulated in mesangial cells treated with TGF-β1 and glomeruli from diabetic db/db mice and were suggested to induce phenotype transition of mesangial cells by targeting β-catenin-interacting protein 1.141 In another study, miR-22 was suggested to be a master regulator of BMP-7 and BMP-6 to further increase TGF-β1 signalling.142 Decreases in expression of the miR-30 family were suggested to accelerate DN by concomitant upregulation of their target, connective tissue growth factor.143 MiR-433 also increased TGF-β1 signalling and fibrosis by targeting antizyme inhibitor 1, which regulates polyamine synthesis.144 MiR-451 was downregulated in early DN in db/db mice and induced hypertrophy by targeting Ywhaz, a protein required for activation of mitogen-activated protein kinase.145 Let-7, an interesting miRNA that is downregulated in several cancers, was downregulated by TGF-β1 in renal cells which correlated with fibrosis because of the concomitant upregulation of its targets TGF-β1 receptor type-1 and collagens.146,147
Some miRNAs have been implicated in oxidant stress, a major player in DN pathogenesis. Nox4, a key player in DN,24,148,149 was identified as an miR-25 target, suggesting that decreased miR-25 expression can upregulate Nox4 to promote oxidant stress and renal dysfunction in rats.150 Nox4 was also identified as a target of miR-146a which was downregulated in high-glucose-treated endothelial cells.151 Reduction in miR-205 expression was associated with increased production of reactive oxygen species via mechanisms that led to decreases in heme oxygenase and superoxide dismutase (SOD) in HK-2 tubular cells.152 MiRNA cascades (miR-192, miR-216a and miR-217) activate Akt and inhibit FoxO3a/SOD2 signalling in mesangial cells.22,115 A report showed that aldose reductase downregulates miR-200a-3p and miR-141–3p to modulate Keap1-Nrf2, TGF-β1/TGF-β2, and Zeb1/Zeb2 signalling in mesangial cells and kidneys of diabetic mice, with Keap1 identified as an miR-200a target.153 Further studies are needed to determine how oxidant stress regulates miRNAs and vice versa in the diabetic kidney.
Overall, it is clear that several miRNAs are involved in promoting or attenuating the progression of DN by targeting genes related to fibrosis, inflammation, oxidant stress and signal transduction (Table 1).102,118 Some of these miRNAs work in amplifying circuits, while others have autonomous effects and cell-specific roles. Thus, targeting a single miRNA may not be the best approach for DN treatment, although some miRNAs such as miR-192, miR-29 or miR-21 may be better targets than others owing to their notable actions. We anticipate seeing much progress in this field in the upcoming years aided by new technologies and conditional mouse models.
Table 1. MicroRNAs relevant to the pathogenesis of diabetic nephropathy.
| MicroRNAs | Cell types/animal models | Targets | Expression | References |
|---|---|---|---|---|
| miR-21 | OVE26 mice | Pten | Increase | 128 |
| miR-21 | db/db mice, MC | Pten | Decrease | 132 |
| miR-21 | KK-Ay mice | MMP-9, TIMP-1 | Increase | 130 |
| miR-21 | db/db mice | Smad7 | Increase | 129 |
| miR-25 | STZ rat, MC | NADPH oxidase 4 | Decrease | 150 |
| miR-29c | db/db mice, EC, podocytes | Spry-1 | Increase | 136 |
| miR-29 | ApoE−/− mice, STZ rats, PTEC, human HK-2 cells | Collagens | Decrease | 137,138,191 |
| miR-93 | STZ mice, db/db mice, EC, podocytes | Vegf-A | Decrease | 135 |
| miR-192 | STZ mice, db/db mice, MC | Zeb1/Zeb2 | Increase | 14,113–115,119,120,133,134,136,187,192 |
| miR-192 | ApoE−/− mice, PTEC | Zeb1/Zeb2 | Decrease | 121,122 |
| miR-200a | ApoE−/− mice, PTEC | TGF-β2 | Decrease | 123 |
| miR-200b/c | STZ mice, db/db mice, MC | Zeb1 | Increase | 14,116,136 |
| miR-215 | db/db mice | β-catenin-interacting protein 1 | Increase | 141 |
| miR-216a | STZ mice, db/db mice, MC | Ybx1 | Increase | 113 |
| miR-216a/217 | STZ mice, db/db mice, MC | Pten | Increase | 115 |
| miR-377 | STZ mice, MC | Pak1, Sod1/Sod2 | Increase | 133 |
| miR-451 | db/db mice | Ywhaz | Decrease | 145 |
| let-7 | ApoE−/− mice, MC, PTEC | TGF-β R-1, collagens | Decrease | 146,147 |
Abbreviations: EC, endothelial cells; MC, mesangial cells; PTEC, proximal tubular epithelial cells; STZ, streptozotocin.
Long noncoding RNAs
LncRNAs and their actions
Data from genomic consortia such as ENCODE, and whole transcriptome sequencing (RNA-seq), have demonstrated that the majority of the genome is transcribed into RNA, much of which is noncoding.71,74 This ncRNA includes not only small ncRNAs, such as miRNAs, but also lncRNAs (>200 nucleotides up to ~100kb), both of which have regulatory functions. LncRNAs are long transcripts similar to mRNAs but lack protein coding (translation) potential.74,154 Increasing evidence suggests that lncRNAs function in various biological processes, including gene expression and transcription, epigenetic regulation, cell-cycle control, differentiation and immune responses.155,156 LncRNAs can regulate the expression of local and distal genes by various mechanisms that include recruiting histone modifying complexes to affect chromatin states, and modulating the activities of transcription factors.155–157 Interestingly, lncRNAs also serve as hosts for miRNAs. LncRNAs act as modular molecules with individual domains that enable them to specifically associate with DNA, RNA and/or protein.157,158 LncRNAs can also influence the activity and localization of the proteins they bind; for example, by serving as key coactivators of proteins involved in transcriptional regulation.156–158 LncRNA research is in its infancy, but has elicited a lot of interest because dysregulated expression of lncRNAs, as well as the presence of primary sequence mutations, are highly correlated with human diseases including cardiovascular disease.155–159
LncRNAs in DN
Only a few reports have emerged with respect to the involvement of lncRNAs in DN. An early report showed that key miRNAs (miR-216a and miR-217) were induced by TGF-β1 together with their host ncRNA, RP23, in mesangial cells.115 Angiotensin II induced several lncRNAs in vascular smooth muscle cells, of which one novel lncRNA was a host gene of miR-221 and miR-222, and could mediate vascular smooth muscle cell proliferation.159 MiR-192 is co-regulated in mesangial cells with its host ncRNA CJ241444, which is induced by TGF-β1 through promoter Smad binding elements and epigenetic regulation via protein C-ets-1 and histone acetylation.120 Evidence shows that an lncRNA, plasmacytoma variant translocation 1 (PVT1), may be involved in DN pathogenesis. This lncRNA increased expression of plasminogen activator inhibitor 1 and TGF-β1 in mesangial cells.160 PVT1 was identified in a potential locus for diabetic ESRD in a genome-wide single-nucleotide polymorphism genotyping study.161 PVT1 levels were increased in mesangial cells treated with high glucose and PVT1 small interfering RNA attenuated the expression of key ECM proteins.160 Several miRNAs, including miR-1204, miR-1205, miR-1206, miR-1207 and miR-1208, mapped to the PVT1 locus162 and were upregulated by high glucose in human mesangial cells and found to modulate expression of ECM.163 Another study found 21 lncRNAs upregulated in two models of renal fibrosis, but downregulated in Smad3-knockout mice.164 Overall, this emerging field is expected to uncover additional lncRNAs related to DN in the near future.
NcRNAs as biomarkers for DN
Studies of miRNA signatures in biofluids and tissues as biomarkers and diagnostics constitute a very active area of clinical translational research. Early detection of DN can be extremely useful to clinicians to prevent progression to renal failure, and hence prediction biomarkers can have a major clinical impact. Several biomarkers of DN progression have been reported165 with most being proteins, peptides, growth factors and cytokines. MiRNAs are gaining interest as sensitive, noninvasive and quantitative diagnostic biomarkers for DN, especially owing to their inherent stability in biofluids such as urine and plasma, and in exosomes, and improved methods of detection (including by sequencing) and quantification.166–169 A flurry of reports have demonstrated profiles of key miRNAs in urine, urinary sediment and in the circulation of patients with various kidney diseases and showed varying levels of correlation with nephropathy or fibrosis.170–177 In general, these reports were performed on small cohorts, focused on a few miRNAs and did not determine their cellular source. Urinary exosomes, which can originate from most renal cells, can be extremely valuable for miRNA profiling in renal disorders.176 MiR-145 was enriched in urinary exosomal miRNAs from type 1 diabetic patients with microalbuminuria and in mesangial cells treated with high glucose.178 A much more comprehensive study was performed in which over 700 miRNAs were profiled in urine obtained from patients with type 1 diabetes at different stages of DN, and provided strong support for the use of miRNA profiles as molecular predictors of DN.179 Notably, miRNAs are quite stable in paraffin-embedded sections, which presents another exciting opportunity for biomarker development. However, more validations in independent cohorts are needed. Determining whether miRNAs in biofluids originate from dead cells or diseased cells is also important. Notwithstanding, miRNAs are likely to become valuable biomarkers, particularly for kidney diseases. With respect to lncRNAs, it is not clear whether they are stable in urine and plasma, and hence their value and potential as noninvasive biomarkers needs further investigation.
Epigenetic and ncRNA-based therapy
Numerous drugs are already in clinical use or under development for the treatment of DN and are not discussed here. Anti-TGF-β antibody was partly effective in animal models of DN180 and some trials targeting TGF-β itself and with antifibrotic drugs are ongoing.181 Clinical studies show that type 1 angiotensin II receptor blockers can slow down the progression of DN.11,182 In diabetic db/db mice, the type 1 angiotensin II receptor blocker losartan reversed most of the physiological and histological parameters of DN, and changes in expression of key genes. However, it only blocked some, and not all the epigenetic changes observed in db/db mice,80 indicating that the relative inefficacy of losartan in some patients with DN may be due to its inefficient blockade of epigenetic marks. Thus, in the future, combination therapies with standard plus epigenetic therapies might be more effective in controlling DN and possibly metabolic memory.
As discussed earlier in this Review, epigenetic modification of DNA and histones play important roles in renal fibrosis and DN. Therefore, epigenetic interventions are the focus of intensive research. Studies screening for small molecules to accelerate recovery after acute kidney injury identified the HDAC inhibitor, methyl-4-(phenylthio) butanoate, as a promising therapeutic candidate.183 HDAC inhibitors have also been tested in animal models of DN.184 However, because most HDAC inhibitors are quite nonselective, the exact mechanisms of their action are unclear and further work with selective HDAC inhibitors is warranted. Sirt1, also an HDAC, suppressed advanced glycation end-product-induced profibrotic gene expression via activation of the Nrf2 pathway, followed by upregulation of antioxidant gene expression in glomerular mesangial cells.185 Several HAT, HDAC and HMT inhibitors are currently being developed for cancer and other diseases where the potential for epigenetic therapy has been recognized. Thus they could also be evaluated for DN treatment. Epigenetic marks are in general reversible, at least more so than the genetic code, so they are amenable to therapeutic targeting.
Because several miRNAs have now been shown to be dysregulated in DN, targeting them could be a valuable approach for therapeutic intervention. In order to determine their potential, the effects of downregulating or upregulating them in vivo are being assessed in preclinical models of translational research using numerous delivery approaches.102,176 Traditionally, miRNA levels are manipulated with miRNA mimics or antisense miRNAs, and there have been several technical advances in the development of stable nuclease-resistant oligonucleotides for miRNA inhibitors and mimics. MiRNAs that are upregulated can be inactivated by using miRNA sponges, antisense oligonucleotides, or miRNA knockout. Locked nucleic acid (LNA) and modified anti-miRNAs or antagomirs can specifically inhibit miRNAs of interest115,186,187 and are also being evaluated in some clinical trials.188 LNA-modified anti-miR-192 was effective in inhibiting miR-192, downstream miRNAs (miR-216a, miR-217 and miR-200 family) and p53 in the renal cortex of normal as well as streptozotocin-injected diabetic mice, and reduced key parameters of DN.14,115,117,187 This finding suggests that such anti-miRNA therapies can be developed in the future for human DN. In other studies, db/db mice injected with 2’-O-methyl antisense oligonucleotides targeting miR-29c showed reduced rates of DN.136 Other in vivo delivery modalities such as adeno-associated virus vectors, and miRNA sponges are also being used,176 while bacteriophage MS2 virus-like particles have been tested for overexpressing miRNAs that are downregulated.189 In general, approaches to overexpress miRNAs to achieve functional replacement of mature miRNA in vivo with mimics have been more challenging than with inhibition.
MiRNA-based therapies are being actively researched188 and some clinical trials are already ongoing. Several synthetic stable, biodegradable miRNA carriers have been assessed, as well as optimal delivery methods to ensure efficiency without toxicity. Considerable challenges also need to be overcome to avoid undesired adverse effects, off-target and off-tissue effects, especially with a heterogeneous organ such as the kidney. Notwithstanding, given the considerable ongoing activity in this area, we can anticipate ample progress in the field of DN.
With respect to lncRNAs, it is worth harnessing their vast unexplored potential and more recently, they have also been evaluated as therapeutic targets.190 LNA modification of anti-lncRNAs could also be evaluated for efficient inhibition of lncRNAs.
Conclusions
The pathogenesis of DN involves complex interactions between metabolic and haemodynamic factors. In this Review, we have highlighted some emerging mechanisms by which they might operate, with emphasis on epigenetics, miRNAs and lncRNAs. Long-term persistence of epigenetic modifications and ncRNAs triggered by diabetic stimuli could be key mechanisms underlying metabolic memory. Similarities as well as differences with regards to the epigenetic factors and ncRNAs regulated in DN associated with type 1 versus type 2 diabetes could exist; apart from hyperglycaemia, type 2 diabetes is also associated with insulin resistance and dyslipidaemia, and these additional diabetogenic factors can augment or synergize with the effects of hyperglycaemia. Several HMTs and the corresponding histone post-translational modifications, as well as numerous miRNAs have been implicated in the expression of fibrotic and inflammatory genes associated with DN (Figure 2). Controlling these molecules might provide new therapeutics for DN and potentially even for reversing metabolic memory. Furthermore, profiling miRNAs in biofluids and exosomes could be used to meet the critical need for improved and noninvasive biomarkers for the early detection and diagnosis of DN. This need has triggered numerous ongoing efforts to evaluate epigenetic changes and miRNA profiles in archived or freshly obtained biosamples, blood cells and renal tissues from clinical cohorts of patients with DN. Integration of these datasets with available genetic data from these patients using experimental and in silico methods might accelerate the identification of new mediators of DN progression.
Clearly this rapidly expanding field is aided by technological and scientific advances that can also lead to new related discoveries in the field of DN. Epigenomics or epigenome-wide association studies in conjunction with other genomic profiling could also help determine the functional significance of genetic variants, especially the large numbers occurring in noncoding regulatory regions. Unlike genetics, epigenetics is reversible and thus provides important opportunities for developing much needed improved biomarkers and treatments for DN.
Key points.
-
➢
Diabetic conditions induce inflammation, fibrosis and hypertrophy in renal cells through various cytokines and growth factors such as transforming growth factor-β1, angiotensin II and platelet-derived growth factor
-
➢
The engagement of cytokines and growth factors to their receptors triggers signal transduction cascades that result in the activation of transcription factors to increase expression of inflammatory and fibrotic genes
-
➢
These signalling mechanisms affect epigenetic states such as DNA methylation and chromatin histone modifications to augment the expression of profibrotic and inflammatory genes, as well as noncoding RNAs
-
➢
Noncoding RNAs induced by diabetic conditions can also promote the expression of pathological genes via various post-transcriptional and post-translational mechanisms
-
➢
These epigenetic mechanisms and noncoding RNAs can lead to persistently open chromatin structures at pathological genes and sustained gene expression, which can also be a mechanism for ‘metabolic memory’
-
➢
Key epigenetic regulators, microRNAs and long noncoding RNAs could serve as new therapeutic targets for diabetic nephropathy
Acknowledgements
The authors gratefully acknowledge funding from the National Institutes of Health (NIDDK and NHLBI), the Juvenile Diabetes Research Foundation and the American Diabetes Association.
Biography
Mitsuo Kato is an Assistant Research Professor in the Department of Diabetes, and was trained (in RNA biology) in the Department of Molecular Cellular Biology, Beckman Research Institute of City of Hope, CA, USA. He received his PhD (Medical Science, Cancer Genetics and Epigenetics) from Kyoto University, Kyoto, Japan. His main fields of recent interest are epigenetic mechanisms of diabetic complications, especially nephropathy and its prevention by controlling noncoding RNAs, such as microRNAs.
Rama Natarajan is an Endowed Professor in the Department of Diabetes, Beckman Research Institute of City of Hope, Los Angeles, CA, USA. She is well known for her research pertaining to the molecular mechanisms responsible for the accelerated cardiovascular and renal complications of diabetes with over 160 publications. Her major interests are to examine the role of epigenetics and noncoding RNAs in the augmented expression of pathological genes under diabetic conditions. Dr Natarajan is funded by grants from the NIH, and the Juvenile Diabetes Research Foundation. She is on the Editorial Boards of Diabetes, Kidney International, ATVB, and the American Journal of Physiology—Renal Physiology [Au: change okay?].
Footnotes
Review criteria
A search for original published articles focusing on diabetic nephropathy, signal transduction, noncoding RNAs, microRNAs, long noncoding RNAs, genetics and epigenetics was performed in MEDLINE and PubMed. All articles identified were English-language, full-text papers. We also searched the reference lists of identified articles for further relevant papers.
Competing interests
The authors declare no competing interests.
References
- 1.Jones CA et al. Epidemic of end-stage renal disease in people with diabetes in the United States population: do we know the cause? Kidney Int. 67, 1684–1691 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Sarnak MJ et al. Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation 108, 2154–2169 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Chen S, Jim B & Ziyadeh FN Diabetic nephropathy and transforming growth factor-β: transforming our view of glomerulosclerosis and fibrosis build-up. Semin. Nephrol 23, 532–543 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Qian Y, Feldman E, Pennathur S, Kretzler M & Brosius FC 3rd From fibrosis to sclerosis: mechanisms of glomerulosclerosis in diabetic nephropathy. Diabetes 57, 1439–1445 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanwar YS, Sun L, Xie P, Liu FY & Chen S A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu. Rev. Pathol 6, 395–423 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pagtalunan ME et al. Podocyte loss and progressive glomerular injury in type II diabetes. J. Clin. Invest 99, 342–348 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meyer TW, Bennett PH & Nelson RG Podocyte number predicts long-term urinary albumin excretion in Pima Indians with Type II diabetes and microalbuminuria. Diabetologia 42, 1341–1344 (1999). [DOI] [PubMed] [Google Scholar]
- 8.Brownlee M Biochemistry and molecular cell biology of diabetic complications. Nature 414, 813–820 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Sharma K & Ziyadeh FN Hyperglycemia and diabetic kidney disease. The case for transforming growth factor-β as a key mediator. Diabetes 44, 1139–1146 (1995). [DOI] [PubMed] [Google Scholar]
- 10.Yamamoto T, Nakamura T, Noble NA, Ruoslahti E & Border WA Expression of transforming growth factor β is elevated in human and experimental diabetic nephropathy. Proc. Natl Acad. Sci. USA 90, 1814–1818 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruggenenti P, Cravedi P & Remuzzi G The RAAS in the pathogenesis and treatment of diabetic nephropathy. Nat. Rev. Nephrol 6, 319–330 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Abboud HE Role of platelet-derived growth factor in renal injury. Annu. Rev. Physiol 57, 297–309 (1995). [DOI] [PubMed] [Google Scholar]
- 13.Zhu Y, Casado M, Vaulont S & Sharma K Role of upstream stimulatory factors in regulation of renal transforming growth factor-β1. Diabetes 54, 1976–1984 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Kato M et al. A microRNA circuit mediates transforming growth factor-β1 autoregulation in renal glomerular mesangial cells. Kidney Int. 80, 358–368 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Feng X, We R & Derynck R Receptor-associated Mad homologues synergize as effectors of the TGF-β response. Nature 383, 168–172 (1996). [DOI] [PubMed] [Google Scholar]
- 16.Roberts AB, McCune BK & Sporn MB TGF-β: regulation of extracellular matrix. Kidney Int. 41, 557–559 (1992). [DOI] [PubMed] [Google Scholar]
- 17.Poncelet AC & Schnaper HW Sp1 and Smad proteins cooperate to mediate transforming growth factor-β1-induced α2(I) collagen expression in human glomerular mesangial cells. J. Biol. Chem 276, 6983–6992 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Tsuchida K, Zhu Y, Siva S, Dunn SR & Sharma K Role of Smad4 on TGF-β-induced extracellular matrix stimulation in mesangial cells. Kidney Int. 63, 2000–2009 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Kim YS et al. Novel interactions between TGF-β1 actions and the 12/15-lipoxygenase pathway in mesangial cells. J. Am. Soc. Nephrol 16, 352–362 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Chin BY, Mohsenin A, Li SX, Choi AM & Choi ME Stimulation of pro-α1(I) collagen by TGF-β1 in mesangial cells: role of the p38 MAPK pathway. Am. J. Physiol. Renal Physiol 280, F495–F504 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Hayashida T, Poncelet AC, Hubchak SC & Schnaper HW TGF-β1 activates MAP kinase in human mesangial cells: a possible role in collagen expression. Kidney Int. 56, 1710–1720 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Kato M et al. Role of the Akt/FoxO3a pathway in TGF-β1-mediated mesangial cell dysfunction: a novel mechanism related to diabetic kidney disease. J. Am. Soc. Nephrol 17, 3325–3335 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Mahimainathan L, Das F, Venkatesan B & Choudhury GG Mesangial cell hypertrophy by high glucose is mediated by downregulation of the tumor suppressor PTEN. Diabetes 55, 2115–2125 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Sedeek M et al. Oxidative stress, Nox isoforms and complications of diabetes—potential targets for novel therapies. J. Cardiovasc. Transl. Res 5, 509–518 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Inagi R, Shoji K & Nangaku M Oxidative and endoplasmic reticulum (ER) stress in tissue fibrosis. Curr. Pathobiol. Rep 1, 283–289 (2013). [Google Scholar]
- 26.Wang Y & Harris DC Macrophages in renal disease. J. Am. Soc. Nephrol 22, 21–27 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Navarro-González JF, Mora-Fernández C, Muros de Fuentes M & Garcia-Pérez J Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat. Rev. Nephrol 7, 327–340 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Berthier CC et al. Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes 58, 469–477 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gohda T et al. Circulating TNF receptors 1 and 2 predict stage 3 CKD in type 1 diabetes. J. Am. Soc. Nephrol 23, 516–524 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujita T et al. Interleukin-18 contributes more closely to the progression of diabetic nephropathy than other diabetic complications. Acta Diabetol. 49, 111–117 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Kanamori H et al. Inhibition of MCP-1/CCR2 pathway ameliorates the development of diabetic nephropathy. Biochem. Biophys. Res. Commun 360, 772–777 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Sayyed SG et al. Podocytes produce homeostatic chemokine stromal cell-derived factor-1/CXCL12, which contributes to glomerulosclerosis, podocyte loss and albuminuria in a mouse model of type 2 diabetes. Diabetologia 52, 2445–2454 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Kikuchi Y et al. Fractalkine and its receptor, CX3CR1, upregulation in streptozotocin-induced diabetic kidneys. Nephron Exp. Nephrol 97, e17–e25 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Song KH, Park J, Park JH, Natarajan R & Ha H Fractalkine and its receptor mediate extracellular matrix accumulation in diabetic nephropathy in mice. Diabetologia 56, 1661–1669 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Komorowsky CV, Brosius FC 3rd, Pennathur S & Kretzler M Perspectives on systems biology applications in diabetic kidney disease. J. Cardiovasc. Transl. Res 5, 491–508 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brosius FC 3rd & Alpers CE New targets for treatment of diabetic nephropathy: what we have learned from animal models. Curr. Opin. Nephrol. Hypertens 22, 17–25 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woroniecka KI et al. Transcriptome analysis of human diabetic kidney disease. Diabetes 60, 2354–2369 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nathan DM et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N. Engl. J. Med 353, 2643–2653 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.et al. Sustained effect of intensive treatment of type 1 diabetes mellitus on development and progression of diabetic nephropathy: the Epidemiology of Diabetes Interventions and Complications (EDIC) study. JAMA 290, 2159–2167 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.et al. Effect of intensive therapy on the microvascular complications of type 1 diabetes mellitus. JAMA 287, 2563–2569 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pop-Busui R et al. Effects of prior intensive insulin therapy on cardiac autonomic nervous system function in type 1 diabetes mellitus: the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications study (DCCT/EDIC). Circulation 119, 2886–2893 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Boer IH et al. Intensive diabetes therapy and glomerular filtration rate in type 1 diabetes. N. Engl. J. Med 365, 2366–2376 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Boer IH for the DCCT/EDIC Research Group. Kidney disease and related findings in the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications study. Diabetes Care 37, 24–30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Colagiuri S, Cull CA & Holman RR for the UKPDS Group. Are lower fasting plasma glucose levels at diagnosis of type 2 diabetes associated with improved outcomes?: U.K. Prospective Diabetes Study 61. Diabetes Care 25, 1410–1417 (2002). [DOI] [PubMed] [Google Scholar]
- 45.ADVANCE Collaborative Group. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med 358, 2560–2572 (2008). [DOI] [PubMed] [Google Scholar]
- 46.The Action to Control Cardiovascular Risk in Diabetes Study Group. Effects of intensive glucose lowering in type 2 diabetes. N. Engl. J. Med 358, 2545–2559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu Q et al. The Akt-FoxO3a-manganese superoxide dismutase pathway is involved in the regulation of oxidative stress in diabetic nephropathy. Exp. Physiol 98, 934–945 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Reddy MA, Tak Park J & Natarajan R Epigenetic modifications in the pathogenesis of diabetic nephropathy. Semin. Nephrol 33, 341–353 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li SL et al. Enhanced proatherogenic responses in macrophages and vascular smooth muscle cells derived from diabetic db/db mice. Diabetes 55, 2611–2619 (2006). [DOI] [PubMed] [Google Scholar]
- 50.El-Osta A et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med 205, 2409–2417 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Engerman RL & Kern TS Progression of incipient diabetic retinopathy during good glycemic control. Diabetes 36, 808–812 (1987). [DOI] [PubMed] [Google Scholar]
- 52.Kowluru RA, Abbas SN & Odenbach S Reversal of hyperglycemia and diabetic nephropathy: effect of reinstitution of good metabolic control on oxidative stress in the kidney of diabetic rats. J. Diabetes Complications 18, 282–288 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Kouzarides T Chromatin modifications and their function. Cell 128, 693–705 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Jones PA Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet 13, 484–492 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Portela A & Esteller M Epigenetic modifications and human disease. Nat. Biotechnol 28, 1057–1068 (2010). [DOI] [PubMed] [Google Scholar]
- 56.Jirtle RL & Skinner MK Environmental epigenomics and disease susceptibility. Nat. Rev. Genet 8, 253–262 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Simmons R Epigenetics and maternal nutrition: nature v. nurture. Proc. Nutr. Soc 70, 73–81 (2011). [DOI] [PubMed] [Google Scholar]
- 58.Thomas MC, Groop PH & Tryggvason K Towards understanding the inherited susceptibility for nephropathy in diabetes. Curr. Opin. Nephrol. Hypertens 21, 195–202 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Sandholm N et al. New susceptibility loci associated with kidney disease in type 1 diabetes. PLoS Genet. 8, e1002921 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Villeneuve LM, Reddy MA & Natarajan R Epigenetics: deciphering its role in diabetes and its chronic complications. Clin. Exp. Pharmacol. Physiol 38, 401–409 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cooper ME & El-Osta A Epigenetics: mechanisms and implications for diabetic complications. Circ. Res 107, 1403–1413 (2010). [DOI] [PubMed] [Google Scholar]
- 62.Miao F et al. Profiles of epigenetic histone post-translational modifications at type 1 diabetes susceptible genes. J. Biol. Chem 287, 16335–16345 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sapienza C et al. DNA methylation profiling identifies epigenetic differences between diabetes patients with ESRD and diabetes patients without nephropathy. Epigenetics 6, 20–28 (2011). [DOI] [PubMed] [Google Scholar]
- 64.Maurano MT et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Toperoff G et al. Genome-wide survey reveals predisposing diabetes type 2-related DNA methylation variations in human peripheral blood. Hum. Mol. Genet 21, 371–383 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bell CG et al. Integrated genetic and epigenetic analysis identifies haplotype-specific methylation in the FTO type 2 diabetes and obesity susceptibility locus. PLoS One 5, e14040 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bell CG et al. Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med. Genomics 3, 33 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bechtel W et al. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat. Med 16, 544–550 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pirola L et al. Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res. 21, 1601–1615 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ko YA et al. Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development. Genome Biol. 14, R108 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou VW, Goren A & Bernstein BE Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet 12, 7–18 (2011). [DOI] [PubMed] [Google Scholar]
- 73.Jin F, Li Y, Ren B & Natarajan R Enhancers: multi-dimensional signal integrators. Transcription 2, 226–230 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guttman M et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223–227 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Klose RJ & Zhang Y Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol 8, 307–318 (2007). [DOI] [PubMed] [Google Scholar]
- 76.Cedar H & Bergman Y Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet 10, 295–304 (2009). [DOI] [PubMed] [Google Scholar]
- 77.Guttman M & Rinn JL Modular regulatory principles of large non-coding RNAs. Nature 482, 339–346 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yuan H et al. Involvement of p300/CBP and epigenetic histone acetylation in TGF-β1-mediated gene transcription in mesangial cells. Am. J. Physiol Renal Physiol 304, F601–613 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sun G et al. Epigenetic histone methylation modulates fibrotic gene expression. J. Am. Soc. Nephrol 21, 2069–2080 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reddy MA et al. Losartan reverses permissive epigenetic changes in renal glomeruli of diabetic db/db mice. Kidney Int. 85, 362–373 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Komers R et al. Epigenetic changes in renal genes dysregulated in mouse and rat models of type 1 diabetes. Lab. Invest 93, 543–552 (2013). [DOI] [PubMed] [Google Scholar]
- 82.Giacco F & Brownlee M Oxidative stress and diabetic complications. Circ. Res 107, 1058–1070 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Miao F, Gonzalo IG, Lanting L & Natarajan R In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions. J. Biol. Chem 279, 18091–18097 (2004). [DOI] [PubMed] [Google Scholar]
- 84.Miao F et al. Genome-wide analysis of histone lysine methylation variations caused by diabetic conditions in human monocytes. J. Biol. Chem 282, 13854–13863 (2007). [DOI] [PubMed] [Google Scholar]
- 85.Miao F et al. Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: an epigenetic study in diabetes. Diabetes 57, 3189–3198 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li Y et al. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-κB-dependent inflammatory genes. Relevance to diabetes and inflammation. J. Biol. Chem 283, 26771–26781 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yun JM, Jialal I & Devaraj S Epigenetic regulation of high glucose-induced proinflammatory cytokine production in monocytes by curcumin. J. Nutr. Biochem 22, 450–458 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Villeneuve LM et al. Enhanced levels of microRNA-125b in vascular smooth muscle cells of diabetic db/db mice lead to increased inflammatory gene expression by targeting the histone methyltransferase Suv39h1. Diabetes 59, 2904–2915 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Villeneuve LM et al. Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. Proc. Natl Acad. Sci. USA 105, 9047–9052 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tonna S, El-Osta A, Cooper ME & Tikellis C Metabolic memory and diabetic nephropathy: potential role for epigenetic mechanisms. Nat. Rev. Nephrol 6, 332–341 (2010). [DOI] [PubMed] [Google Scholar]
- 91.Miao F et al. Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes 63, 1748–1762 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mouse ENCODE Consortium. An encyclopedia of mouse DNA elements (Mouse ENCODE). Genome Biol 13, 418 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rosenbloom KR et al. ENCODE whole-genome data in the UCSC Genome Browser: update 2012. Nucleic Acids Res. 40, D912–D917 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Park PJ ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet 10, 669–680 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mortazavi A, Williams BA, McCue K, Schaeffer L & Wold B Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008). [DOI] [PubMed] [Google Scholar]
- 96.Laird PW Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet 11, 191–203 (2010). [DOI] [PubMed] [Google Scholar]
- 97.Simon JM, Giresi PG, Davis IJ & Lieb JD Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat. Protoc 7, 256–267 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bernstein BE et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol 28, 1045–1048 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bartel DP MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Filipowicz W, Bhattacharyya SN & Sonenberg N Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat. Rev. Genet 9, 102–114 (2008). [DOI] [PubMed] [Google Scholar]
- 101.Kim VN MicroRNA biogenesis: coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol 6, 376–385 (2005). [DOI] [PubMed] [Google Scholar]
- 102.Kato M, Castro NE & Natarajan R MicroRNAs: potential mediators and biomarkers of diabetic complications. Free Radic. Biol. Med 64, 85–94 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Harvey SJ et al. Podocyte-specific deletion of dicer alters cytoskeletal dynamics and causes glomerular disease. J. Am. Soc. Nephrol 19, 2150–2158 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ho J et al. Podocyte-specific loss of functional microRNAs leads to rapid glomerular and tubular injury. J. Am. Soc. Nephrol 19, 2069–2075 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ho J et al. The pro-apoptotic protein Bim is a microRNA target in kidney progenitors. J. Am. Soc. Nephrol 22, 1053–1063 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shi S et al. Podocyte-selective deletion of dicer induces proteinuria and glomerulosclerosis. J. Am. Soc. Nephrol 19, 2159–2169 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nagalakshmi VK et al. Dicer regulates the development of nephrogenic and ureteric compartments in the mammalian kidney. Kidney Int. 79, 317–330 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhdanova O et al. The inducible deletion of Drosha and microRNAs in mature podocytes results in a collapsing glomerulopathy. Kidney Int. 80, 719–730 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhao Y et al. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1–2. Cell 129, 303–317 (2007). [DOI] [PubMed] [Google Scholar]
- 110.Chen JF et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc. Natl Acad. Sci. USA 105, 2111–2116 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sun Y et al. Development of a micro-array to detect human and mouse microRNAs and characterization of expression in human organs. Nucleic Acids Res. 32, e188 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tian Z, Greene AS, Pietrusz JL, Matus IR & Liang M MicroRNA-target pairs in the rat kidney identified by microRNA microarray, proteomic, and bioinformatic analysis. Genome Res. 18, 404–411 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kato M et al. Post-transcriptional up-regulation of Tsc-22 by Ybx1, a target of miR-216a, mediates TGF-β-induced collagen expression in kidney cells. J. Biol. Chem 285, 34004–34015 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kato M et al. MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-β-induced collagen expression via inhibition of E-box repressors. Proc. Natl Acad. Sci. USA 104, 3432–3437 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kato M et al. TGF-β activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat. Cell Biol 11, 881–889 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Park JT et al. FOG2 protein down-regulation by transforming growth factor-β1-induced microRNA-200b/c leads to Akt kinase activation and glomerular mesangial hypertrophy related to diabetic nephropathy. J. Biol. Chem 288, 22469–22480 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Deshpande SD et al. Transforming growth factor-β-induced cross talk between p53 and a microRNA in the pathogenesis of diabetic nephropathy. Diabetes 62, 3151–3162 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kato M & Natarajan R MicroRNA circuits in transforming growth factor-β actions and diabetic nephropathy. Semin. Nephrol 32, 253–260 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chung AC, Huang XR, Meng X & Lan HY miR-192 mediates TGF-β/Smad3-driven renal fibrosis. J. Am. Soc. Nephrol 21, 1317–1325 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kato M et al. TGF-β induces acetylation of chromatin and of Ets-1 to alleviate repression of miR-192 in diabetic nephropathy. Sci. Signal 6, ra43 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Krupa A et al. Loss of microRNA-192 promotes fibrogenesis in diabetic nephropathy. J. Am. Soc. Nephrol 21, 438–447 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang B et al. E-cadherin expression is regulated by miR-192/215 by a mechanism that is independent of the profibrotic effects of transforming growth factor-β. Diabetes 59, 1794–1802 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wang B et al. miR-200a prevents renal fibrogenesis through repression of TGF-β2 expression. Diabetes 60, 280–287 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gregory PA et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol 10, 593–601 (2008). [DOI] [PubMed] [Google Scholar]
- 125.Ren S & Duffield JS Pericytes in kidney fibrosis. Curr. Opin. Nephrol. Hypertens 22, 471–480 (2013). [DOI] [PubMed] [Google Scholar]
- 126.LeBleu VS et al. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med 19, 1047–1053 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kriz W, Kaissling B & Le Hir M Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy? J. Clin. Invest 121, 468–474 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dey N et al. MicroRNA-21 orchestrates high glucose-induced signals to TOR complex 1, resulting in renal cell pathology in diabetes. J. Biol. Chem 286, 25586–25603 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhong X et al. miR-21 is a key therapeutic target for renal injury in a mouse model of type 2 diabetes. Diabetologia 56, 663–674 (2013). [DOI] [PubMed] [Google Scholar]
- 130.Wang J et al. Effect of miR-21 on renal fibrosis by regulating MMP-9 and TIMP1 in kk-ay diabetic nephropathy mice. Cell Biochem. Biophys 67, 537–546 (2013). [DOI] [PubMed] [Google Scholar]
- 131.Chau BN et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci. Transl. Med 4, 121ra18 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang Z et al. MicroRNA-21 protects from mesangial cell proliferation induced by diabetic nephropathy in db/db mice. FEBS Lett. 583, 2009–2014 (2009). [DOI] [PubMed] [Google Scholar]
- 133.Wang Q et al. MicroRNA-377 is up-regulated and can lead to increased fibronectin production in diabetic nephropathy. FASEB J. 22, 4126–4135 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang XX et al. Diabetic nephropathy is accelerated by farnesoid X receptor deficiency and inhibited by farnesoid X receptor activation in a type 1 diabetes model. Diabetes 59, 2916–2927 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Long J, Wang Y, Wang W, Chang BH & Danesh FR Identification of microRNA-93 as a novel regulator of vascular endothelial growth factor in hyperglycemic conditions. J. Biol. Chem 285, 23457–23465 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Long J, Wang Y, Wang W, Chang BH & Danesh FR MicroRNA-29c is a signature microRNA under high glucose conditions that targets Sprouty homolog 1, and its in vivo knockdown prevents progression of diabetic nephropathy. J. Biol. Chem 286, 11837–11848 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang B et al. Suppression of microRNA-29 expression by TGF-β1 promotes collagen expression and renal fibrosis. J. Am. Soc. Nephrol 23, 252–265 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chen HY et al. MicroRNA-29b inhibits diabetic nephropathy in db/db mice. Mol. Ther 22, 842–853 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Qin W et al. TGF-β/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J. Am. Soc. Nephrol 22, 1462–1474 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sun L et al. Low-dose paclitaxel ameliorates fibrosis in the remnant kidney model by down-regulating miR-192. J. Pathol 225, 364–377 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mu J et al. Functional implications of microRNA-215 in TGF-β1-induced phenotypic transition of mesangial cells by targeting CTNNBIP1. PLoS One 8, e58622 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Long J et al. MicroRNA-22 is a master regulator of bone morphogenetic protein-7/6 homeostasis in the kidney. J. Biol. Chem 288, 36202–36214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wu J et al. Downregulation of microRNA-30 facilitates podocyte injury and is prevented by glucocorticoids. J. Am. Soc. Nephrol 25, 92–104 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Li R et al. The microRNA miR-433 promotes renal fibrosis by amplifying the TGF-β/Smad3-Azin1 pathway. Kidney Int. 84, 1129–1144 (2013). [DOI] [PubMed] [Google Scholar]
- 145.Zhang Z et al. MicroRNA-451 regulates p38 MAPK signaling by targeting of Ywhaz and suppresses the mesangial hypertrophy in early diabetic nephropathy. FEBS Lett. 586, 20–26 (2012). [DOI] [PubMed] [Google Scholar]
- 146.Brennan EP et al. Lipoxins attenuate renal fibrosis by inducing let-7c and suppressing TGFβR1. J. Am. Soc. Nephrol 24, 627–637 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang B et al. Transforming growth factor-β1-mediated renal fibrosis is dependent on the regulation of transforming growth factor receptor 1 expression by let-7b. Kidney Int. 85, 352–361 (2014). [DOI] [PubMed] [Google Scholar]
- 148.Babelova A et al. Role of Nox4 in murine models of kidney disease. Free Radic. Biol. Med 53, 842–853 (2012). [DOI] [PubMed] [Google Scholar]
- 149.Zhu Y, Usui HK & Sharma K Regulation of transforming growth factor-beta in diabetic nephropathy: implications for treatment. Semin. Nephrol 27, 153–160 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Fu Y et al. Regulation of NADPH oxidase activity is associated with miRNA-25-mediated NOX4 expression in experimental diabetic nephropathy. Am. J. Nephrol 32, 581–589 (2010). [DOI] [PubMed] [Google Scholar]
- 151.Feng B et al. miR-146a-mediated extracellular matrix protein production in chronic diabetes complications. Diabetes 60, 2975–2984 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Muratsu-Ikeda S et al. Downregulation of miR-205 modulates cell susceptibility to oxidative and endoplasmic reticulum stresses in renal tubular cells. PLoS One 7, e41462 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wei J et al. Aldose reductase regulates miR-200a-3p/141–3p to coordinate Keap1–Nrf2, Tgfβ1/2, and Zeb1/2 signaling in renal mesangial cells and the renal cortex of diabetic mice. Free Radic. Biol. Med 67, 91–102 (2014). [DOI] [PubMed] [Google Scholar]
- 154.Cabili MN et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 25, 1915–1927 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wapinski O & Chang HY Long noncoding RNAs and human disease. Trends Cell Biol. 21, 354–361 (2011). [DOI] [PubMed] [Google Scholar]
- 156.Taft RJ, Pang KC, Mercer TR, Dinger M & Mattick JS Non-coding RNAs: regulators of disease. J. Pathol 220, 126–139 (2010). [DOI] [PubMed] [Google Scholar]
- 157.Moran VA, Perera RJ & Khalil AM Emerging functional and mechanistic paradigms of mammalian long non-coding RNAs. Nucleic Acids Res. 40, 6391–6400 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wilusz JE, Sunwoo H & Spector DL Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 23, 1494–1504 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Leung A et al. Novel long noncoding RNAs are regulated by angiotensin II in vascular smooth muscle cells. Circ. Res 113, 266–278 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Alvarez ML & DiStefano JK Functional characterization of the plasmacytoma variant translocation 1 gene (PVT1) in diabetic nephropathy. PLoS One 6, e18671 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hanson RL et al. Identification of PVT1 as a candidate gene for end-stage renal disease in type 2 diabetes using a pooling-based genome-wide single nucleotide polymorphism association study. Diabetes 56, 975–983 (2007). [DOI] [PubMed] [Google Scholar]
- 162.Huppi K et al. The identification of microRNAs in a genomically unstable region of human chromosome 8q24. Mol. Cancer Res 6, 212–221 (2008). [DOI] [PubMed] [Google Scholar]
- 163.Alvarez ML, Khosroheidari M, Eddy E & Kiefer J Role of microRNA 1207–5P and its host gene, the long non-coding RNA Pvt1, as mediators of extracellular matrix accumulation in the kidney: implications for diabetic nephropathy. PLoS One 8, e77468 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zhou Q et al. Identification of novel long noncoding RNAs associated with TGF-β/Smad3-mediated renal inflammation and fibrosis by RNA sequencing. Am. J. Pathol 184, 409–417 (2014). [DOI] [PubMed] [Google Scholar]
- 165.Fassett RG et al. Biomarkers in chronic kidney disease: a review. Kidney Int. 80, 806–821 (2011). [DOI] [PubMed] [Google Scholar]
- 166.Farazi TA, Spitzer JI, Morozov P & Tuschl T miRNAs in human cancer. J. Pathol 223, 102–115 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Fabbri M miRNAs as molecular biomarkers of cancer. Expert Rev. Mol. Diagn 10, 435–444 (2010). [DOI] [PubMed] [Google Scholar]
- 168.Wang K et al. Circulating microRNAs, potential biomarkers for drug-induced liver injury. Proc. Natl Acad. Sci. USA 106, 4402–4407 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Tijsen AJ et al. MiR423–5p as a circulating biomarker for heart failure. Circ. Res 106, 1035–1039 (2010). [DOI] [PubMed] [Google Scholar]
- 170.Szeto CC et al. Micro-RNA expression in the urinary sediment of patients with chronic kidney diseases. Dis. Markers 33, 137–144 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Neal CS et al. Circulating microRNA expression is reduced in chronic kidney disease. Nephrol. Dial. Transplant 26, 3794–3802 (2011). [DOI] [PubMed] [Google Scholar]
- 172.Luk CC et al. Urinary biomarkers for the prediction of reversibility in acute-on-chronic renal failure. Dis. Markers 34, 179–185 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Wang G et al. Urinary sediment miRNA levels in adult nephrotic syndrome. Clin. Chim. Acta 418, 5–11 (2013). [DOI] [PubMed] [Google Scholar]
- 174.Yang Y et al. Urine miRNAs: potential biomarkers for monitoring progression of early stages of diabetic nephropathy. Med. Hypotheses 81, 274–278 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ichii O et al. Altered expression of microRNA miR-146a correlates with the development of chronic renal inflammation. Kidney Int. 81, 280–292 (2012). [DOI] [PubMed] [Google Scholar]
- 176.DiStefano JK, Taila M & Alvarez ML Emerging roles for miRNAs in the development, diagnosis, and treatment of diabetic nephropathy. Curr. Diab. Rep 13, 582–591 (2013). [DOI] [PubMed] [Google Scholar]
- 177.Cai X et al. Serum microRNAs levels in primary focal segmental glomerulosclerosis. Pediatr. Nephrol 28, 1797–1801 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Barutta F et al. Urinary exosomal microRNAs in incipient diabetic nephropathy. PLoS One 8, e73798 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Argyropoulos C et al. Urinary microRNA profiling in the nephropathy of type 1 diabetes. PLoS One 8, e54662 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ziyadeh FN et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-β antibody in db/db diabetic mice. Proc. Natl Acad. Sci. USA 97, 8015–8020 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Declèves AE & Sharma K New pharmacological treatments for improving renal outcomes in diabetes. Nat. Rev. Nephrol 6, 371–380 (2010). [DOI] [PubMed] [Google Scholar]
- 182.Brenner BM et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med 345, 861–869 (2001). [DOI] [PubMed] [Google Scholar]
- 183.Cianciolo Cosentino C et al. Histone deacetylase inhibitor enhances recovery after AKI. J. Am. Soc. Nephrol 24, 943–953 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Advani A et al. Long-term administration of the histone deacetylase inhibitor vorinostat attenuates renal injury in experimental diabetes through an endothelial nitric oxide synthase-dependent mechanism. Am. J. Pathol 178, 2205–2214 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Huang K et al. Sirt1 resists advanced glycation end products-induced expressions of fibronectin and TGF-β1 by activating the Nrf2/ARE pathway in glomerular mesangial cells. Free Radic. Biol. Med 65, 528–540 (2013). [DOI] [PubMed] [Google Scholar]
- 186.Krützfeldt J et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 438, 685–689 (2005). [DOI] [PubMed] [Google Scholar]
- 187.Putta S et al. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J. Am. Soc. Nephrol 23, 458–469 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lindow M & Kauppinen S Discovering the first microRNA-targeted drug. J. Cell Biol 199, 407–412 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Pan Y et al. MS2 VLP-based delivery of microRNA-146a inhibits autoantibody production in lupus-prone mice. Int. J. Nanomedicine 7, 5957–5967 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wahlestedt C Targeting long non-coding RNA to therapeutically upregulate gene expression. Nat. Rev. Drug Discov 12, 433–446 (2013). [DOI] [PubMed] [Google Scholar]
- 191.Du B et al. High glucose down-regulates miR-29a to increase collagen IV production in HK-2 cells. FEBS Lett. 584, 811–816 (2010). [DOI] [PubMed] [Google Scholar]
- 192.Jenkins RH et al. miR-192 induces G/M growth arrest in aristolochic acid nephropathy. Am. J. Pathol 184, 996–1009 (2014). [DOI] [PubMed] [Google Scholar]