

SUMMARY
Sensory experience influences the establishment of neural connectivity through molecular mechanisms that remain unclear. Here, we employ single-nucleus RNA sequencing to investigate the contribution of sensory-driven gene expression to synaptic refinement in the dorsal lateral geniculate nucleus of the thalamus, a region of the brain that processes visual information. We find that visual experience induces the expression of the cytokine receptor Fn14 in excitatory thalamocortical neurons. By combining electrophysiological and structural techniques, we show that Fn14 is dispensable for early phases of refinement mediated by spontaneous activity, but that Fn14 is essential for refinement during a later, experience-dependent period of development. Refinement deficits in mice lacking Fn14 are associated with functionally weaker and structurally smaller retinogeniculate inputs, indicating that Fn14 mediates both functional and anatomical rearrangements in response to sensory experience. These findings identify Fn14 as a molecular link between sensory-driven gene expression and vision-sensitive refinement in the brain.
Keywords: Synapse, synaptic refinement, synapse elimination, LGN, lateral geniculate nucleus, retinogeniculate, single-cell, single-nucleus
eTOC paragraph:
Visual experience promotes the synaptic refinement of the retinogeniculate circuit by inducing the expression of the cytokine receptor Fn14 in excitatory neurons of the dorsal lateral geniculate nucleus of the thalamus.
INTRODUCTION
Neurons in the developing brain assemble into circuits through the formation and mremodeling of synaptic connections. Following their initial assembly, synapses undergo an extensive period of refinement during which they are strengthened, remodeled, or eliminated based upon their level of activity, such that stronger synapses are retained while weaker synapses are eliminated (Riccomagno and Kolodkin, 2015; Vonhoff and Keshishian, 2017). This synaptic refinement is influenced by spontaneous, intrinsically driven neural activity during early postnatal life and is remarkably sensitive to sensory experience later in postnatal development (Andreae and Burrone, 2017; Leighton and Lohmann, 2016; Wiesel and Hubel, 1963). While a significant number of proteins have been described to mediate the early stages of synapse formation, comparatively few molecular regulators of postnatal synaptic refinement have been identified. In particular, the molecular mechanisms by which sensory experience drives synaptic refinement are not yet well understood.
One challenging aspect of the identification of molecules that regulate synaptic refinement is the dependence of this process upon properly timed, physiologically relevant patterns of neuronal activity. Therefore, synaptic refinement is most effectively studied within the context of an intact circuit (Hashimoto and Kano, 2013; Sanes and Lichtman, 1999). Towards this end, the retinogeniculate pathway in which retinal ganglion cells (RGCs) synapse onto excitatory thalamocortical (TC) neurons of the dorsal lateral geniculate nucleus (dLGN) of the thalamus has emerged as a useful model for studying postnatal circuit development (Hong and Chen, 2011). The retinogeniculate synapse undergoes distinct postnatal phases of refinement that first rely upon spontaneous retinal activity between birth and P20, and subsequently require visual input (Hooks and Chen, 2006, 2008). The temporal segregation of these phases provides a unique opportunity to disentangle experience-dependent mechanisms from those driven by spontaneous activity that have been more extensively investigated. Therefore, in the present study, we investigate the mechanisms underlying the experience-dependent phase of synaptic refinement in the dLGN, specifically during the vision-sensitive period between postnatal days (P)20 and P30.
How might sensory input drive this vision-sensitive phase of retinogeniculate refinement at the molecular level? Decades of work across many brain regions have shown that neuronal activation, such as that mediated by sensory experience, has both short-term and long-term effects on synaptic connectivity. For example, in the short term, protein phosphorylation and neurotransmitter receptor trafficking can scale synaptic strength and efficacy on the order of seconds to minutes (Turrigiano, 2012; Zucker and Regehr, 2002). However, long-term changes that result in a persistent remodeling of synapses, like those that occur during refinement, rely upon the induction of gene expression programs in the nuclei of activated neurons (West and Greenberg, 2011). These transcriptional programs include immediate early genes (IEGs) such as Fos, Egr1, and Npas4 that encode broadly expressed transcriptional regulators and are induced within one hour of neurotransmitter release onto a postsynaptic neuron (Bloodgood et al., 2013; Lin et al., 2008; Malik et al., 2014). These transcription factors then bind cis-regulatory elements across the genome to drive the expression of late response genes (LRGs) whose protein products can function at synapses to regulate neuronal connectivity (Mardinly et al., 2016). While these prior studies have been conducted primarily using mouse cortical tissue, they raise the possibility that similar sensory-driven gene programs in the mouse dLGN might encode postsynaptic mediators of vision-sensitive retinogeniculate refinement. However, coordinated experience-dependent gene expression in the mouse dLGN had not yet been fully characterized, and whether induced genes are critical for vision-sensitive refinement remained to be determined.
In the present study, we applied whole-transcriptome single-nucleus RNA sequencing (snRNAseq) to characterize the genes that are acutely induced in excitatory TC neurons in response to visual stimulation during the vision-sensitive period of synaptic refinement in the dLGN. Among the hundreds of experience-dependent genes identified, the cell surface pro-inflammatory cytokine receptor Fibroblast growth factor inducible 14 (Fn14) is the most robustly induced. We focused our subsequent analysis on the function of Fn14 for several reasons, including its high level of inducibility, selective expression in excitatory neurons, subcellular localization to the cell surface, and relatedness to the tumor necrosis factor (TNF) receptor superfamily of molecules that bind ligands known to mediate synaptic composition and strength, and have the ability to remodel tissues in response to injury or disease (Bu rkly, 2014; Steinmetz and Turrigiano, 2010; Stellwagen and Malenka, 2006). Using a combination of molecular, electrophysiological, and ultrastructural techniques to compare retinogeniculate development in wild type (WT) and Fn14 knockout (KO) mice, we found that Fn14 is largely dispensable for spontaneous activity-dependent refinement at P13 and P20, but is required for vision-sensitive refinement between P20 and P27. Taken together, our experiments demonstrate that Fn14 is a sensory-dependent regulator of functional and structural refinement of the retinogeniculate synapse that serves as a molecular link between experience-dependent gene expression and synaptic refinement in the brain.
RESULTS
Sensory-driven gene expression in neurons of the dLGN
To test our hypothesis that genes induced by visual experience contribute to the vision-sensitive component of retinogeniculate refinement, we sought to characterize the experience-dependent transcriptome in neurons of the dLGN. Towards this end, we employed a previously used method to synchronize vision-dependent gene expression by rearing mice in the dark for several days then reexposing them to light. Although this manipulation is non-physiological for the mouse, it provides a robust stimulation paradigm for the detection of experience-dependent genes (Mardinly et al., 2016). One obstacle to analyzing experience-dependent transcription in vivo is the recent finding that multiple cell populations in the brain—including excitatory and inhibitory neurons, glia, and vascular cells—mount cell type-specific gene programs in response to sensory stimulation (Hrvatin et al., 2018). Therefore, to avoid obscuring the cell type-specificity of vision-dependent gene expression, we sequenced the dLGN at single-cell resolution.
Wild-type C57Bl/6J mice were dark-reared during the vision-sensitive period of retinogeniculate refinement between P20 and P27 (late-dark-reared, LDR) then reexposed to light for zero (unstimulated condition), one, or eight hours (Fig. 1A). These time points were chosen to allow for the detection of both immediate-early genes (IEGs; one hour) and late-response genes (LRGs; eight hours). As expected, this paradigm induced robust changes in gene expression, including an increase in the levels of the well-described IEGs Fos and Npas4 as measured by whole-tissue RNAseq, quantitative PCR, fluorescence in situ hybridization (FISH), and immunofluorescence (Fig. 1B,C and Fig. S1A-F). Consistent with the conclusion that these changes accurately reflect acute, activity-driven increases in gene transcription, analysis of chromatin accessibility by ATAC-seq on dLGN tissue following three hours of visual stimulation identified open chromatin at activity-dependent enhancers surrounding the Fos locus (Fig. S1G; Buenrostro et al., 2015; Su et al., 2017).
Figure 1. Single-cell transcriptomics of the dLGN following visual stimulation.

(A) Schematic of the experimental paradigm in which mice were dark-reared during the vision-sensitive period of refinement then reexposed to light for zero, one, or eight hours. RNA from single cells of the dorsal LGN (dLGN) was sequenced via inDrops.
(B) Confocal images of FISH on coronal dLGN sections from mice late-dark-reared between P20 and P27 then reexposed to light for zero or one hour. Sections were probed for the excitatory neuron marker Vglut1 (red) and the activity-dependent immediate early gene Fos (green). Scale bar, 5 μm.
(C) Quantification of the number of individual Fos mRNA molecules per Vglut1-positive neuron, as shown in (B). Unpaired t-test.
(D) Expression pattern of the excitatory neuron marker Slc17a6 across all cell clusters. Scale, 0 to 125 transcripts per cell (log2).
(E) Expression pattern of the inhibitory neuron marker Gad1 across all cell clusters. Scale, 0 to 54 transcripts per cell (log2).
(F) Bar graphs displaying the specificity of excitatory and inhibitory markers within each cell population. Left, expression of the excitatory marker Slc17a6. Right, expression of the inhibitory marker Gad1. Y-axis, normalized mean transcript count per cell.
(G-J) Scatterplots comparing gene expression, displayed as log10 values of transcripts per cell, in neuronal subpopulations of mice stimulated for one or eight hours (y-axes) versus unstimulated zero hour controls (x-axes). (G) excitatory neurons at one hour; (H) inhibitory neurons at one hour; (I) excitatory neurons at eight hours (inset shows higher magnification for comparison of Fn14 induction with other genes); and (J) inhibitory neurons at eight hours. Genes up-regulated by at least 1.5-fold, FDR < 0.05 shown in red. Genes down-regulated by at least 1.5-fold, FDR < 0.05 shown in blue. **** = p < 0.0001. All error bars represent S.E.M. See also Figure S1 and Table S1.
Having confirmed that this paradigm induces sensory-driven changes in gene expression, we subjected mice to LDR and visual stimulation, microdissected the dLGNs, and isolated the nuclei for single-nucleus RNA sequencing (snRNAseq). Previous studies have shown that RNAseq of individual nuclei as opposed to whole cells preserves a larger number of neurons across multiple subtypes, minimizes the effects of cell dissociation on gene expression, and enriches transcripts for those that are being actively transcribed in vivo (Habib et al., 2017; Lacar et al., 2016). Therefore, in the current study, whole-transcriptome RNA sequencing was performed on individual nuclei that were captured and barcoded using a recently developed technique termed inDrops (Fig. 1A; Klein et al., 2015; Zilionis et al., 2017).
After sequencing, nuclei were classified by cell type as previously described (Macosko et al., 2015; Satija et al., 2015) and gene expression in 8,398 excitatory TC neurons and 4,987 inhibitory interneurons (identified by expression of Slc17a6 and Gad1, respectively) was assessed (Fig. 1D-F). Transcript levels across all genes were compared between the one or eight hour time point and the unstimulated condition (Qiu et al., 2017), and differentially expressed genes were identified based upon at least a 1.5-fold difference with a false discovery rate (FDR) < 0.05. This strategy identified both up- and down-regulated genes at each time point (Table S1).
Overall, 43 genes were up-regulated in excitatory neurons and 16 in inhibitory neurons at one hour, while 233 genes were up-regulated in excitatory neurons and 157 in inhibitory neurons at eight hours (Fig. 1G-J). Previous studies in mouse visual cortex have shown that the early gene programs mounted in response to sensory stimulation overlap across neuronal subtypes, while the late-response programs are more neuronal subtype-specific (Hrvatin et al., 2018; Spie gel et al., 2014). However, we find that in the dLGN only four of the early-response genes induced in excitatory and inhibitory neurons overlap: the IEGs Egr1 and Nr4a1, the circadian transcription factor Per1, and an uncharacterized gene Fam13c (Fig. 1G,H). By contrast, the broadly expressed IEG Fos was highly induced in excitatory neurons but not in inhibitory neurons, while the IEG encoding the postsynaptic scaffold component Homer1 was selectively induced in inhibitory neurons but not excitatory neurons.
Since IEG transcription factors have been proposed to function as regulators of late-response activity-dependent gene programs, the observation that some IEGs are shared between excitatory and inhibitory neurons while some are activated selectively in a neuronal subtype-specific manner in the dLGN suggests that the late-response programs within these neuronal populations might also include both shared as well as neuronal subtype-specific genes. Consistent with this prediction, the late-response gene programs induced in excitatory and inhibitory neurons following eight hours of reexposure to light showed a moderate degree of overlap, with 45 genes shared between the two datasets and 300 genes displaying a neuronal subtype-specific pattern of induction (Fig. 1I,J). Shared genes of interest include the neuropeptide Vgf, which is among the most highly induced genes in both cell types, and has known roles in synaptic plasticity downstream of brain derived neurotrophic factor (BDNF) and CREB activation (Lin et al., 2014; Lin et al., 2015). Additionally, the chemokine Cx3cl1 (fractalkine), which is thought to regulate brain development and neuroinflammation, is also induced in both excitatory and inhibitory neurons (Arnoux and Audinat, 2015). The induction of these genes in both neuronal populations suggests that they may be involved in experience-dependent functions within both subtypes, including shared aspects of synapse development and remodeling.
Our observation that the late-response gene programs in excitatory and inhibitory neurons display overlap was unexpected based on recent findings in the visual cortex, where late-response programs are nuanced and subtype-specific (Hrvatin et al., 2018).This difference may reflect a unique feature of thalamic or dLGN circuit wiring. For example, excitatory and inhibitory neurons in the dLGN both receive driving input from RGCs through which they each inherit their receptive field properties, as well as modulatory input from layer VI of visual cortex that sharpens these features (Weyand, 2016). These observations suggest that the anatomical and physiological context of a neuron may significantly influence its late-response gene program. Nevertheless, we note that many genes are induced in one neuronal subtype but not the other. For example, the non-canonical Notch ligand Dlk2 is highly induced in excitatory neurons, while the RNA-binding protein Pcbp4 is selectively induced in inhibitory neurons. Overall, this atlas of single-cell transcriptomics provides a useful resource for future studies investigating visual experience-dependent gene expression in the dLGN.
Identification of Fn14 as a candidate regulator of vision-sensitive refinement
Although inhibitory neurons in the dLGN receive the same patterned activity as their neighboring TC neurons, and excitatory and inhibitory neurons display some overlap in LRG expression, inhibitory neurons are different from TC neurons in that they do not undergo a developmental process of retinogeniculate refinement (Seabrook et al., 2013). For this reason, we speculated that molecular regulators of sensory-driven synaptic refinement in TC neurons would include LRGs that are highly induced by experience selectively in TC neurons and not interneurons. Thus, we next focused our attention on an LRG that is specifically induced in dLGN excitatory neurons.
The most highly induced gene after eight hours of visual stimulation that is selective to excitatory neurons encodes the cell surface pro-inflammatory cytokine receptor TNF receptor superfamily member 12a, or Tnfrsf12a (Fig. 1I). The protein encoded by Tnfrsf12a, referred to as Fibroblast growth factor-inducible 14 (Fn14), promotes tissue remodeling in non-neural systems such as skeletal muscle in part by driving inflammatory gene expression through NF-ĸB activation (Brown et al., 2003; Burkly, 2014; Meighan-Mantha et al., 1999; Wiley and Winkles, 2003). Fn14 has also been shown to mediate actin remodeling by engaging the cytoskeletal regulator Rac1, suggesting that Fn14 might regulate retinogeniculate refinement by effecting vision dependent changes in synaptic architecture (Tanabe et al., 2003). While little was known about Fn14 expression or function in the brain, we hypothesized that, following its induction by sensory experience, Fn14 might remodel synaptic connections between RGCs and excitatory neurons of the dLGN. Consistent with this possibility, RNAseq of whole-tissue dLGN by previously described strategies (Fig. S2; Gray et al., 2014; McCarthy et al., 2012) shows that Fn14 is most highly expressed between P20 and P27, when visual input is required to refine the retinogeniculate circuit.
We validated the sensory-driven induction and developmental expression profile of Fn14 by qPCR (Fig. 2A,B), and further found by western blotting of dLGN extracts that Fn14 protein is up-regulated around 8 hours following reexposure of LDR animals to light, with Fn14 protein expression peaking between 24 and 48 hours after light exposure (Fig. 2C,D). Western blotting also revealed a 60% decrease in Fn14 protein expression at P27 following LDR compared to normally reared (NR), age-matched controls, indicating that visual stimulation not only acutely up-regulates Fn14 expression, but also is required for the proper expression of Fn14 in the dLGN during development (Fig. 2E,F). Similarly, probing dLGN lysates from animals at different postnatal ages revealed a protein expression pattern that is correlated with Fn14 mRNA expression, with Fn14 protein expression increasing significantly at P16 and remaining high through the vision-sensitive period between P20 and P27 (Fig. 2G,H).
Figure 2. Developmental and experience-dependent expression of Fn14 mRNA and protein.

(A) Validation by qPCR of Fn14 mRNA induction in the dLGN of mice reexposed to light following late-dark-rear (LDR), normalized to Gapdh expression.
(B) Validation by qPCR of Fn14 mRNA expression in the dLGN across postnatal development in normally reared (NR) mice, normalized to Gapdh expression.
(C) Western blot of dLGN lysates from mice following LDR and reexposure to light, probed for Fn14. GAPDH, loading control.
(D) Quantification of Fn14 protein in the dLGN following reexposure to light, as shown in (E).
(E) Western blot of dLGN lysates from NR and LDR mice at P27. Blot probed for Fn14. GAPDH, loading control.
(F) Quantification of Fn14 protein in dLGN of NR and LDR mice.
(G) Western blot of dLGN lysates from mice at multiple time points across postnatal development, probed for Fn14. GAPDH, loading control.
(H) Quantification of Fn14 protein levels across postnatal development, as shown in (G).
Statistical significance was assessed by one-way ANOVA with Dunnett’s test except for (F), which was determined by unpaired t-test. *= p < 0.05; **= p <0.01; ***= p < 0.001; ****= p < 0.0001. All error bars represent S.E.M. See also Figure S2.
We next employed multiplexed single-molecule fluorescence in situ hybridization (FISH) to characterize the regional distribution of Fn14 in coronal brain sections from P27 mice following LDR and reexposure to light for eight hours. Low magnification confocal microscopy revealed that Fn14 is expressed in the thalamus and selectively enriched in the dLGN but is undetectable in other brain regions, including visual cortex and hippocampus (Fig. 3A). This expression profile is consistent with RNAseq datasets from the visual cortex that show little Fn14 expression in cortical excitatory neurons, even after visual stimulation (Hrvatin et al., 2018).
Figure 3. Fn14 expression is enriched in excitatory TC neurons of the dLGN.

(A) Low magnification confocal images of Fn14 (green) and Vglut1 (red) mRNA expression, and DAPI (blue) in (a) dLGN (outlined), (b) visual cortex, (c) auditory cortex, and (d) hippocampus. Scale bar, 200 μm.
(B) High magnification confocal images of FISH for Fn14 (green) and molecular markers for all major cell types in the dLGN (red). White squares = insets, below (left to right: molecular marker, Fn14, merge). (a) Vglut1; (b) Gad1; (c) Olig1; (d) P2ry12; (e) Cldn5; and (f) Aldh1l1. Scale bar, 10 μm. Inset scale bar, 4 μm.
(C) Quantification of the percentage of Fn14-expressing cells labeled with listed cell type markers.
(D) High magnification confocal images of individual TC neurons in the dLGN of wild type mice following LDR and reexposure to light, or unstimulated controls (zero hours). TC neurons express Vglut1 (red) along with increasing levels of Fn14 (green). Scale bar, 5 µm.
(E) Quantification of Fn14 mRNA molecules per TC neuron at each time point.
(F) Confocal images of FISH for Fn14 (green) and Vglut1 (red) in normally reared animals at P20 and P27, the time points flanking the vision-sensitive period. Scale bar, 10 µm.
Statistical analyses, one-way ANOVA with Dunnett’s test. *** = p < 0.001; **** = p < .0001. All error bars represent S.E.M.
FISH allows for the simultaneous localization of up to three mRNA markers, enabling this strategy to identify cell type(s) that express and induce Fn14 in the dLGN. Although our snRNAseq analysis revealed robust induction of Fn14 in excitatory neurons but not inhibitory neurons, our single-cell analysis included only a small number of non-neuronal cells. Thus, we could not rule out the possibility that Fn14 is also expressed in non-neuronal cells. To further characterize the cell type-specificity of Fn14, we probed P27 dLGN sections for (1) Fn14, (2) one of several excitatory TC neuron markers including Vglut1 and Stmn2, and (3) markers of each of the other five predominant cell populations in the dLGN, including inhibitory interneurons (Gad1 and Gad2), astrocytes (Aldh1l1), oligodendrocytes (Olig1), vascular endothelial cells (Cldn5 and Pecam), and microglia (P2ry12 and Cx3cr1).
Consistent with the snRNAseq data, we found that, in response to light stimulation at the eight hour time point, the vast majority of Fn14-expressing cells (97%) also expressed high levels of the excitatory TC neuron markers Vglut1 and Stmn2 (Fig. 3Ba,C). Very few Fn14-expressing cells expressed Gad1 (interneurons; Fig. 3Bb), Olig1 (oligodendrocytes; Fig. 3Bc), P2ry12 (microglia; Fig. 3Bd), Cldn5 (endothelial cells; Fig. 3Be), or Aldh1l1 (astrocytes; Fig. 3Bf; quantification in Fig. 3C). High magnification confocal imaging of the sections confirmed that Fn14 is induced by light stimulation selectively in excitatory TC neurons (Fig. 3D,E). Similar patterns of Fn14 expression were observed at the time points flanking the vision-sensitive period of refinement in NR mice, with Fn14 enriched in TC neurons at P20 and P27 (Fig. 3F).
Fn14 regulates pre- and postsynaptic morphology
The experience-dependent and developmental up-regulation of Fn14 expression in TC neurons during postnatal refinement suggested that Fn14 might regulate neuronal or synaptic structure in the dLGN. To address this possibility, we performed Golgi and electron microscopy (E.M.) analysis of the dLGN in wild type (WT) and Fn14 knockout (KO) mice, which have been well-studied outside of the nervous system (Jakubowski et al., 2005). We confirmed that Fn14 KO mice lack Fn14 Mrna and protein in the brain (Fig. S3A-D). Further, Fn14 KO mice appear normal, have weights and brain sizes similar to those of WT mice, and the gross anatomy of the dLGN as well as general synaptic staining patterns are indistinguishable in Fn14 KO and WT littermates (Fig. S3E-I).
Analysis of dendrite and spine morphology of excitatory TC neurons by Golgi staining revealed that dendritic complexity and total spine density were the same in Fn14 KO and WT littermates at P27 (Fig. 4A, Fig. S4A,B; Sholl, 1953). However, we detected significant differences in spine morphology: spines were 37% longer and 11% more narrow in the KO compared to WT (p < 0.0001 and p <0.01, respectively; Fig. 4A C). We next classified spines by assigning them to traditional morphological categories such as mushroom-, stubby-, or thin- and filopodia-like spines, and found that Fn14 KO mice had a trend toward fewer mushroom spines (p = 0.06), significantly fewer stubby spines (p < 0.0001), and significantly more thin spines (p < 0.0001) than WT littermates (Fig. S4C; Fig.4 D,E). Since thin spines are more prevalent early in development and often do not contain postsynaptic machinery, they are thought to be less mature, while mushroom spines are the predominant spine type in the mature brain and are therefore thought to represent mature synapses (Berry and Nedivi, 2017). Thus, our data suggest that retinogeniculate connectivity is less mature in the Fn14 KO mouse.
Figure 4. Fn14 regulates pre- and postsynaptic morphology.
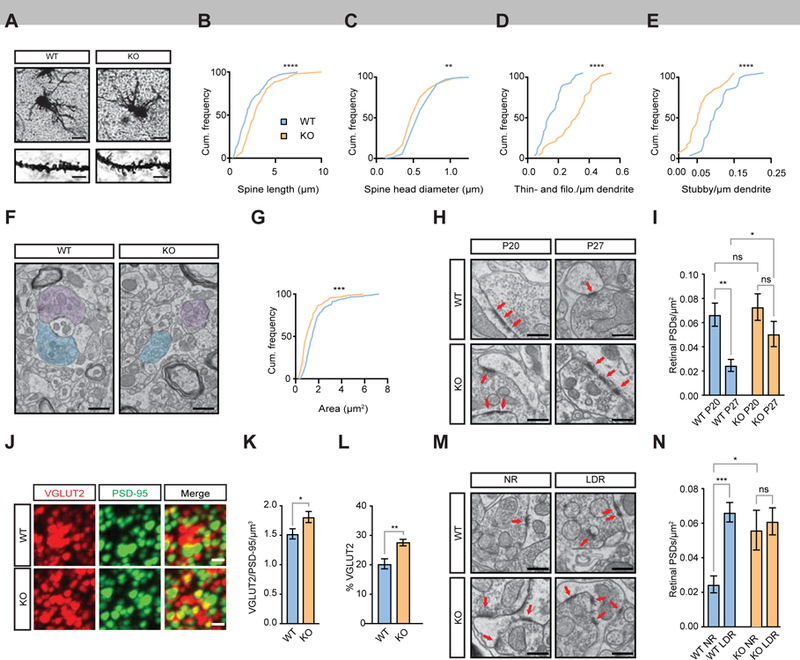
(A) Brightfield images of Golgi-stained dLGN neurons. Scale bar, 25 µm. Inset scale bar, 3.5 µm.
(B) Cumulative frequency distribution of spine length in WT and Fn14 KO neurons at P27. WT = blue; KO = orange.
(C) Cumulative frequency distribution of spine head diameter in WT and Fn14 KO neurons at P27.
(D) Cumulative frequency distribution of filopodia and thin spine density in WT and Fn14 KO neurons at P27.
(E) Cumulative frequency distribution of stubby spine density in WT and Fn14 KO neurons at P27.
(F) Electron micrographs of dLGN sections from WT and Fn14 KO mice at P27. Retinogeniculate boutons are shaded in blue and purple. Scale bar, 500 nm.
(G) Cumulative frequency distribution graph of bouton area (µm2) at P27.
(H) Electron micrographs of retinogeniculate boutons and associated PSDs at P20 and P27 in WT and Fn14 KO mice. Arrows, individual PSDs adjacent to morphologically identified retinogeniculate boutons. Scale bar, 500 nm.
(I) Quantification of retinal PSDs per µm2 in WT and Fn14 KO mice at P20 and P27. Fn14 KO dLGNs contain 39% more PSDs than WT at P27. 2-way ANOVA and Bonferroni correction.
(J) Confocal images of the dLGN following array tomography for the retinogeniculate presynaptic marker VGLUT2 and the postsynaptic marker PSD-95. Scale bar, 200 nm.
(K) Fn14 KO mice maintain significantly more colocalized synaptic puncta than WT mice. Unpaired t-test.
(L) A greater percentage of VGLUT2 puncta in the KO is associated with PSD-95. Unpaired t-test.
(M) Electron micrographs of the LGN of WT and Fn14 KO mice at P27 after normal rearing (NR) or late-dark-rearing (LDR). Arrows, individual PSDs of retinogeniculate synapses. Scale bar, 500 nm.
(N) Retinal PSD densities per µm2 across all conditions. 2-way ANOVA and Bonferroni correction.
Statistical significance of differences between cumulative frequency distributions determined by Kolmogorov-Smirnov test, other statistical analyses given above. * = p < 0.05; ** = p < 0.01; *** = p < 0.001; **** = p < .0001. Error bars represent S.E.M. See also Figures S3 and S4 and Tables S3 and S4.
We next analyzed the ultrastructure of retinogeniculate connections by performing single-section transmission E.M.. In particular, we assessed whether the ultrastructural properties of retinogeniculate inputs were different in Fn14 KO and WT mice. Measurement of the area of RGC boutons, identified by morphological parameters (Colonnier and Guillery, 1964; Guillery and Colonnier, 1970), and postsynaptic density (PSD) length by E.M. revealed that, while PSD length was unaffected by the loss of Fn14, retinogeniculate boutons were 36% smaller in the Fn14 KO dLGN than in the WT (Fig. 4F,G; Fig. S4D; p < 0.01). This change in RGC terminal structure and the maintenance of smaller spines in Fn14 KO mice may reflect a less mature retinogeniculate synapse.
Retinogeniculate terminals are large and each terminal makes multiple contacts with postsynaptic structures on both excitatory and inhibitory neurons in the dLGN (Hamos et al., 1985; Rafols and Valverde, 1973). To begin to clarify whether loss of Fn14 from postsynaptic neurons alters the number of structural PSDs that are apposed to retinogeniculate boutons, we assessed PSD number in our electron micrographs at P20 and P27. Fn14 KO and WT animals had the same number of PSDs apposed to retinogeniculate boutons at P20 (0.073 PSDs/µm2 versus 0.067 PSDs/µm2, p > 0.05). Interestingly, upon analyzing the number of PSDs in the dLGNs of Fn14 KO and WT mice at P27, we found that in WT mice, the number of PSDs apposed to retinogeniculate boutons decreased by 55% (0.067 to 0.025 PSDs/µm2, p < 0.01) across the vision-sensitive period, and while this decrease also happened in Fn14 KO mice, it was not statistically significant (0.073 to 0.05 PSDs/µm2, p > 0.05; Fig. 4H,I). As a result, there was a significantly higher number of PSDs in the KO compared to the WT at P27. This finding is consistent with the results of an analysis by array tomography which showed that the number of colocalized VGLUT2/PSD-95 puncta was greater in the Fn14 KO dLGN than in WT at this age (Fig. 4J-L).
To assess whether the difference in synapse maturation between WT and Fn14 KO neurons might reflect an experience-dependent defect in the refinement of postsynaptic structures, we also analyzed the number of PSDs in Fn14 KO and WT mice at P27 following LDR, a paradigm that is known to result in impairments in vision sensitive refinement. Compared to NR WT mice at P27, LDR of WT mice led to an approximately 2.6 fold increase in the number of PSDs directly apposed to presynaptic retinal terminals (0.025 PSDs/µm2 versus 0.066 PSDs/µm2, p < 0.001; Fig. 4M,N). However, NR Fn14 KO mice had nearly the same density of retinogeniculate PSDs as Fn14 KO mice reared under LDR conditions, suggesting that, in the absence of Fn14, light-dependent changes in the number of postsynaptic specializations do not occur (0.056 PSDs/µm2 vs. 0.061 PSDs/µm2, p > 0.05; Fig. 4M,N). Taken together, these findings indicate that Fn14 restricts the number of PSDs in a vision-dependent manner.
Fn14 does not regulate early, spontaneous activity-driven remodeling of the retinogeniculate synapse
Golgi and E.M. studies identified significant differences in pre- and postsynaptic architecture in the dLGNs of Fn14 KO compared to WT mice. To determine whether these structural changes reflect impairments in synaptic function and/or the progression of functional refinement across the vision-sensitive period, we performed acute slice electrophysiology on the dLGNs of WT and Fn14 KO mice around the time of eye opening (P12-P15) when Fn14 is first up-regulated (Fig. S2; Fig. 2B,G,H). Using a parasagittal acute slice preparation that preserves many of the retinal ganglion cell approximately 2.6 fold increase in the number of PSDs directly apposed to presynaptic retinal terminals (0.025 PSDs/µm2 versus 0.066 PSDs/µm2, p < 0.001; Fig. 4M,N). However, NR Fn14 KO mice had nearly the same density of retinogeniculate PSDs as Fn14 KO mice reared under LDR conditions, suggesting that, in the absence of Fn14, light-dependent changes in the number of postsynaptic specializations do not occur (0.056 PSDs/µm2 vs. 0.061 PSDs/µm2, p > 0.05; Fig. 4M,N). Taken together, these findings indicate that Fn14 restricts the number of PSDs in a vision-dependent manner. axons of the optic tract (Chen and Regehr, 2000), we recorded excitatory postsynaptic currents (EPSCs) in a whole cell voltage clamp configuration from TC neurons as the optic tract was stimulated at increasing intensities (Fig. S5A). Synaptic strength was assessed by measuring the peak EPSC amplitude evoked by minimal stimulation to isolate the response of a single RGC (single fiber amplitude). Additionally, maximal EPSC amplitudes were determined by stimulating the optic tract at intensities of at least 50 µA to recruit all convergent RGC inputs in the slice. Changes in retinogeniculate convergence, i.e., the number of RGCs that synapse on a TC neuron, were estimated using the fiber fraction (FF) ratio, which enumerates the contribution of a single RGC to the total retinal synaptic drive that a neuron receives in these acute slices (FF = single fiber EPSC amplitude/maximal EPSC amplitude; STAR methods; (Hooks and Chen, 2006; Litvina and Chen, 2017). AMPAR- and NMDAR-mediated responses were selectively measured by holding the cell at two different potentials: −70 mV to isolate inward AMPAR-mediated EPSCs due to Mg2+ block of the NMDAR at this potential, and +40 mV to reveal outward currents comprised of a fast, transient AMPAR-mediated current and a more slowly activating and decaying NMDAR-mediated EPSC. We validated these two components of the EPSC by testing their pharmacological sensitivity to the NMDAR antagonist CPP, and the AMPAR-selective antagonist NBQX, as previously described (Fig. S5B; Chen and Regehr, 2000).
We found that at P13, Fn14 does not play a significant role in retinogeniculate synapse refinement. All electrophysiological parameters measured at this time point were equivalent and statistically indistinguishable in WT and Fn14 KO mice, including single fiber EPSC amplitudes, maximal EPSC amplitudes, AMPAR/NMDAR ratio, FF, and decay kinetics of the EPSC (Fig. 5A-E). Furthermore, although Fn14 expression is up-regulated prior to the onset of the vision-sensitive period of refinement (Fig. 2), all the aforementioned measurements taken at P20 were also unaltered in Fn14 KO mice (Fig. S5C-G).
Figure 5. Retinogeniculate synaptic connectivity is normal in P13 Fn14 KO mice.

(A) Example recordings from P13 WT (top) and P13 Fn14 KO (bottom) mice demonstrating appropriate synaptic connectivity in Fn14 KO mice. Recordings show overlaid AMPAR-mediated inward currents recorded at −70 mV and AMPAR- and NMDAR-mediated outward currents recorded at +40 mV from the same cell. EPSCs were evoked with incremental increases in optic tract stimulation, and their peak amplitudes are plotted to the right of each recording. Arrows, single fibers. Y-axis, current (nA). X-axis, stimulus intensity (µA).
(B) AMPAR- and NMDAR-mediated single fiber strengths (top), and maximal EPSC amplitudes (bottom) are not significantly different between WT and Fn14 KO mice. Mann-Whitney U test, p > 0.05.
(C-E) AMPAR/NMDAR ratio (C), fiber fraction (D), and decay kinetics of the EPSC at - 70 mV (E, left and middle), and at +40 mV (E, right) are also not significantly different in P12-P15 WT and Fn14 KO mice. For (B), n (WT) = 31 single fibers from 5 mice; n (KO) = 33 single fibers from 8 mice. For (B-E), n (WT) = 24 cells from 5 mice; n (KO) = 30 cells from 8 mice. ns: p > 0.05, Mann-Whitney two-tailed test. Box, 25%−75% interquartile range; whiskers, 10%−90% interquartile range. See also Figure S5 and Table S2.
Developmental synaptic refinement during the vision-sensitive period requires Fn14
While retinogeniculate refinement during the first three weeks of life appears largely Fn14-independent, it remained to be determined whether Fn14 might regulate synaptic refinement later in development, when Fn14 expression is up-regulated by visual experience and structural changes in retinogeniculate connectivity in the KO emerge (Fig. 4). To assess the progression of refinement across the vision-sensitive period, we next compared FF values at P20 and P27 in Fn14 KO and WT mice. RGC inputs were eliminated in WT mice over this developmental period as indicated by a three-fold increase in the FF from P20 to P27 (0.06 to 0.18, p < 0.001). By contrast, the FF does not significantly change between P20 and P27 in Fn14 KO mice (0.09 to 0.13, p > 0.05; Fig. 6D). As a result, the FF is significantly higher in KO mice at P27 than WT littermates, indicating that although developmental refinement in Fn14 KO mice proceeds normally until P20, further synaptic refinement across the vision-sensitive period does not occur.
Figure 6. Fn14 is required for refinement of the retinogeniculate synapse during the vision-sensitive period.

(A) Cumulative probability plots of AMPAR-mediated single fiber EPSCs show a significant shift toward stronger retinal inputs from P20 to P27 in WT mice (left), but no shift in strength of retinal inputs from P20 to P27 in Fn14 KO mice (right). Mann-Whitney two-tailed test.
(B) Significant strengthening of AMPAR-mediated single fiber EPSCs from P20 to P27 in WT, but not Fn14 KO mice. Kruskal-Wallis, Dunn’s multiple comparisons test.
(C) AMPAR-mediated maximal EPSCs at −70 mV do not differ across development in WT and Fn14 KO mice, Kruskal-Wallis, Dunn’s multiple comparisons test. n (AMPAR) = P20 WT: 28 cells from 9 mice; P27 WT: 39 cells from 15 mice; P20 KO: 32 cells from 9 mice; P27 KO: 45 cells from 14 mice.
(D) The degree of retinal convergence for each TC neuron significantly decreased from P20 to P27 in WT mice, shown by the significant increase in the FF, whereas the FF did not significantly increase from P20 to P27 in Fn14 KO mice. The P27 KO FF is significantly lower than that of P27 WT mice. n = P20 WT: 21 cells from 9 mice; P27 WT: 29 cells from 15 mice; P20 KO: 21 cells from 9 mice; P27 KO: 28 cells from 14 mice; Kruskal-Wallis, Dunn’s multiple comparisons test. For (B-D), Box, 25%−75% interquartile range; whiskers, 10%−90% interquartile range.
(E) Representative recordings of evoked quantal events from P27 WT and Fn14 KO mice in an extracellular solution containing 4 mM SrCl2 and 1 mM MgCl2. The stimulus artifact is blanked and the synchronous EPSC is abridged for clarity. Arrows, time of optic tract stimulation.
(F) Representative recordings of paired-pulse depression at −70 mV from P27 WT and Fn14 KO mice measured at 50, 100, 250, and 500 ms inter-stimulus intervals (ISIs). Stimulus artifacts are blanked for clarity.
(G) Cumulative probability distributions of quantal amplitudes from P27 WT and Fn14 KO mice revealed a ~15% larger median evoked mEPSC amplitude in Fn14 KO mice (18.7 pA) relative to WT mice (16.3 pA). WT: n = 2464 events from 4 cells; KO: n = 2473 events from 4 cells, Mann-Whitney two-tailed test.
(H) Paired pulse ratio (PPR = A2/A1) did not significantly differ between WT and KO mice at P27 at all ISIs (∆t) tested, p > 0.05, Kruskal-Wallis, Dunn’s multiple comparisons test. A1 and A2 correspond to the peak amplitudes of the first and second EPSC, respectively. WT: n=15 cells from 3 mice; KO: n=15 cells from 3 mice.
ns = p > 0.05; * = p < 0.05; *** = p < 0.001; **** = p < 0.001. See also Figure S6 and Table S2.
We next asked whether Fn14 regulates other developmental changes in retinogeniculate connectivity in addition to RGC input elimination. Previous work has shown that, in addition to a reduction in the number of retinogeniculate inputs, the strength of remaining individual RGC single fibers increases over development. Indeed, our results confirm that, in WT mice, unitary EPSCs at the retinogeniculate synapse mstrengthen as mice develop from P20 to P27 (Fig. 6A,B; Fig. S6D; Chen and Regehr, 2000; Hooks and Chen, 2006). This is reflected by a three-fold increase in the AMPAR mediated EPSC from P20 to P27 in WT mice. Notably, we find that this developmental increase in AMPAR-mediated single fiber EPSC amplitude fails to occur in Fn14 KO mice (Fig. 6A,B). By contrast, the amplitudes of maximal AMPAR-mediated EPSCs were equivalent in WT and Fn14 KO mice (Fig. 6C), suggesting that total retinal drive to a particular TC neuron (contributed by the number of inputs and the total sum of their individual strengths) is unaltered upon loss of Fn14. Taken together, these experiments suggest that Fn14 regulates both retinogeniculate input number and synaptic strength during the vision-sensitive period of synaptic refinement.
We next investigated whether the changes observed in Fn14 KO mice are indicative of general synaptic dysfunction due to delayed brain development, or rather reflect a direct and specific role for Fn14 in retinogeniculate refinement. In support of a direct role, we found that the aberrant synaptic remodeling seen in the dLGNs of Fn14 KO mice is not due to total developmental stagnation, gross anatomical defects, or general synaptic dysfunction as several properties of retinogeniculate development and synaptic function are normal in Fn14 KO mice. For instance, the similar median FFs and number of retinal PSDs of Fn14 KO and WT mice at P20 suggest that refinement up to the start of the vision-sensitive period in Fn14 KO mice occurs relatively normally (p > 0.05, Fig. S5F; Fig. 4H,I). Furthermore, the number of non-retinal synapses largely arising from visual cortex in our electron micrographs is similar in WT and Fn14 KO ,dLGNs at P20 and P27 (Fig. S4E,F). In addition, NMDAR-mediated EPSCs were also similar when normally reared P27 WT and Fn14 KO mice were compared (Fig. S6A-C). Therefore, our findings indicate that Fn14 regulates the strength of AMPAR-mediated EPSCs and the number of functional inputs in mice between P20 and P27.
Quantal size and Probability of Release in Fn14 KO Mice
The disrupted strengthening of AMPAR-mediated single fibers at the retinogeniculate synapse in Fn14 KO mice from P20 to P27 could arise from defects in several synaptic mechanisms that contribute to the EPSC amplitude: the quantal response, the probability of release, and/or the number of release sites. To determine if Fn14 regulates one or more of these features of synaptic function, we compared these synaptic properties in WT and Fn14 KO mice at P27. We first analyzed the quantal response. Because TC neurons in the dLGN receive both feed-forward RGC inputs and feedback corticothalamic inputs, analysis of spontaneous miniature EPSCs (mEPSCs) of TC neurons do not accurately measure mEPSCs that are due to synaptic vesicle release from RGCs alone. To overcome this confound, we restricted our quantal analysis to events occurring within a 250 ms window following optic tract stimulation in the presence of extracellular Sr2+ and absence of extracellular Ca2+. These conditions desynchronize evoked vesicle release, and thus allow RGC quantal events to be measured. Surprisingly, we found that evoked quantal amplitudes from Fn14 KO mice were not decreased but were instead ~15% larger than those of WT mice (16.3 pA vs. 18.7 pA, p < 0.0001; Fig. 6E,G), perhaps indicative of a homeostatic compensatory mechanism in Fn14 KO mice that is a consequence of weaker single RGC inputs in these mice.
Because we observed a small increase rather than a decrease in quantal amplitudes at the retinogeniculate synapse in Fn14 KO mice, a decrease in quantal amplitude is not the explanation for failed single fiber strengthening of Fn14 KO mice during vision-sensitive refinement. Therefore, we next asked whether a decrease in the probability of vesicle release (p) from RGCs occurs in Fn14 KO mice. As an indirect measure of p, we stimulated the optic tract twice in rapid succession with varying inter stimulus intervals to determine paired-pulse ratios (PPR) for WT and Fn14 KO mice (Chen et al., 2002; Hauser et al., 2014). PPR was not significantly different between WT and Fn14 KO mice at this age, ruling out a contribution of presynaptic release probability to the absence of single fiber strengthening in Fn14 KO mice (p > 0.15 for all ISIs; Fig. 6F,H). Thus, we conclude that the absent RGC input strengthening in Fn14 KO mice is likely the result of a reduction in the number of functional synaptic release sites from single RGC axons in Fn14 KO mice compared to their WT littermates.
Fn14-dependent refinement is driven by sensory experience
Our results thus far suggest that Fn14 regulates functional retinogeniculate refinement and synaptic morphology during the vision-sensitive period from P20 to P27. However, it is unclear if the deficits in synaptic refinement of the Fn14 KO dLGN are specifically due to loss of the experience-dependent component of Fn14 function, or if Fn14 promotes refinement during this period in an experience-independent manner. To determine if Fn14 specifically regulates the visual experience-dependent component of refinement, we examined the effects of visual deprivation on retinogeniculate connectivity in WT and Fn14 KO mice by returning to the LDR paradigm that we initially used to profile sensory-driven gene expression. Previous functional studies show that LDR disrupts retinogeniculate refinement, leading to an increase in the number of convergent RGC inputs (Hooks and Chen, 2006; Narushima et al., 2016). Because LDR also leads to a significant decrease in Fn14 protein levels (Fig. 2E,F), we considered whether this reduction in Fn14 levels in LDR WT mice might contribute to the disruption of synaptic refinement that is observed in the absence of visual input. In this case, we would predict that NR Fn14 KO mice would show a similar level of refinement as LDR WT littermates at P27, since in both cases there is an absence of Fn14 in excitatory neurons. Alternatively, since there are many other genes that are induced by experience in excitatory neurons (Table S1), it remained possible that a failure to induce other experience-regulated genes might also cause the lack of refinement during visual deprivation in LDR WT mice. In this case, LDR Fn14 KO mice should show a greater disruption of retinogeniculate connectivity compared to NR Fn14 KO mice.
To ascertain the effects of visual deprivation on retinogeniculate refinement, we first compared the FF of P27 WT mice reared under standard conditions to those undergoing LDR. Consistent with previous work, we found that LDR caused a significant decrease in the FF of WT mice (0.18 to 0.10, p < 0.05; Fig. 7A,C), suggesting that visual deprivation results in an increased number of retinal inputs to TC neurons (Hooks and Chen, 2008).
Figure 7. Fn14-dependent refinement is driven by sensory experience.

(A) Example recordings from P27 normally reared WT mice (NR WT, Left) and late dark-reared WT mice (LDR WT mice, Right) demonstrating failure of synaptic refinement after LDR. Incremental increases in optic tract stimulation evoked EPSCs of varying amplitudes, which are plotted below each recording. Stimulus artifacts are blanked for clarity. Arrows, single fibers.
(B) Example recordings from P27 NR Fn14 KO (KO NR, Left), and LDR KO mice (KOLDR, Right) and accompanying current by stimulus intensity plots showing no change in connectivity following visual deprivation. Arrows, single fibers. KO NR traces in this example display a small input that is activated at stimulus intensities higher than 50 µA. Such asynchrony in these doublets and triplets are rare, but do occur in both WT and KO mice.
(C) The degree of retinal convergence for each TC neuron is significantly lower in LDR WT mice than in normally reared P27 WT mice, shown by the significant difference in the FF, whereas the FF was not significantly different between normally reared Fn14 KO mice and LDR KO mice.
(D) Ratio of maximal AMPAR EPSC amplitude to maximal NMDAR EPSC amplitude is significantly lower in LDR WT mice than P27 normally reared WT mice, but not in LDR KO mice and P27 normally reared KO mice.
(E) Increased maximal NMDAR-mediated EPSC amplitudes in LDR WT mice relative to P27 NR WT mice, but not in LDR KO mice vs. P27 NR KO mice.
(F) Increased NMDAR decay 𝝉 values (ms) in LDR WT mice relative to P27 NR WT mice, but not in LDR KO mice and P27 NR KO mice.
For (C-F), Box, 25%−75% interquartile range; whiskers, 10%−90% interquartile range. n = P27 WT: 29 cells from 15 mice; LDR WT: 24 cells from 9 mice; P27 KO: 28 cells from 14 mice; LDR KO: 28 cells from 6 mice;* = p < 0.05, ns = p > 0.05, ** = p < 0.01, *** = p < 0.001, Kruskal-Wallis, Dunn’s multiple comparisons test. Details provided in Table S2.
To determine whether Fn14 contributes to the sensory-dependent refinement of the retinogeniculate synapse, we next measured the FF of WT and Fn14 KO mice at P27 following LDR. Because Fn14 expression in WT mice is driven by visual input (Fig. 2A,C-F), we hypothesized that light-evoked synaptic refinement would fail to occur in both WT mice and Fn14 KO mice after LDR, due to the absence of Fn14 in both genotypes: failure of Fn14 expression in WT mice due to visual deprivation, and genetic ablation of Fn14 in Fn14 KO mice. Thus, the FF would be predicted not to differ between LDR Fn14 KO mice and LDR WT mice. Consistent with these predictions, we found that the FF of Fn14 KO mice that had undergone LDR was similar to that of WT mice that had undergone LDR (0.09 vs. 0.10, p > 0.05). The FFs of NR Fn14 KO mice and LDR Fn14 KO mice were also similarly low (0.13 vs. 0.09, p > 0.05; Fig. 7A-C), suggesting that loss of Fn14 is a major contributing factor to LDR-dependent impairment of refinement in WT mice. In addition, LDR WT mice displayed decreased AMPAR/NMDAR ratios, increased maximal NMDAR-mediated EPSCs, and increased decay constants (𝝉) of NMDAR-mediated EPSCs compared to NR WT mice, consistent with previous work (Hooks and Chen, 2008). By contrast, in LDR Fn14 KO mice and NR Fn14 KO mice, the AMPAR/NMDAR ratio, maximal NMDAR-mediated EPSCs, and decay time constants were similar (Fig. 7D-F). These findings suggest a role for sensory experience-dependent induction of Fn14 in the late phase of synaptic maturation at the retinogeniculate synapse (Model, Figure 8).
Figure 8. Model of Fn14-dependent vision-sensitive refinement.
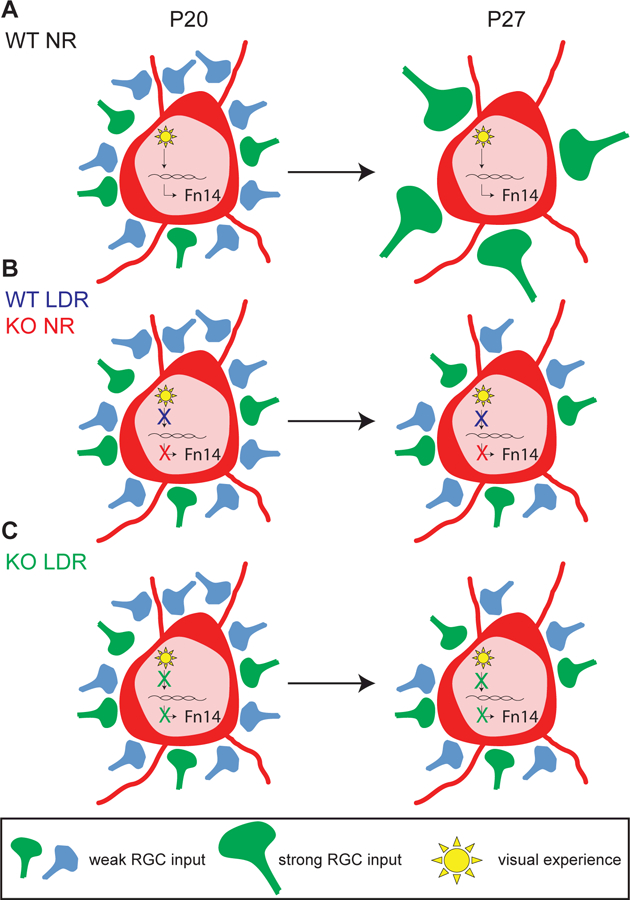
(A) In NR WT mice, visual experience induces the expression of Fn14 to drive strengthening of retinogeniculate connections and concomitant elimination of weak inputs. These aspects of experience-dependent refinement are impaired in the absence of Fn14 (B, red), visual experience (B, blue), or both (C). See also Figure S7.
DISCUSSION
While recent studies support roles for cell type-specific, sensory-driven gene expression in neural development (Hrvatin et al., 2018), the mechanistic relationship between the protein products of sensory-driven genes and the experience-dependent refinement of synapses had not been thoroughly investigated. In the present study, we directly addressed this gap in knowledge by applying snRNAseq to profile sensory driven gene expression in neurons of the dLGN as their connections with converging retinal inputs undergo vision-dependent refinement. Our data provide a dynamic transcriptomic resource of stimulus-responsive gene programs in excitatory and inhibitory thalamic neurons, which complements and extends previous molecular investigations of gene expression in the dLGN (Horng et al., 2009; Kalish et al., 2018; Monavarfeshani et al., 2018; Singh et al., 2012).
Of the hundreds of experience-regulated genes we identified, we focused on the TNF receptor superfamily member Fn14 partly because of the known roles of other immune-related molecules, including the Tumor n ecrosis factor ligand TNFα, in the regulation of synaptic plasticity and development (Goddard et al., 2007; Stellwagen and Malenka, 2006) and in retinogeniculate refinement specifically (e.g., MHC class I and Complement cascade proteins; (Datwani et al., 2009; Stevens et al., 2007). Fn14 contrasts with these other molecules in that its functions in the dLGN are largely constricted to the experience-dependent phase of development. Intriguingly, Fn14 expression is also induced in the dLGNs of adult mice following a week of dark-rearing and subsequent light exposure, suggesting that it may mediate vision-dependent remodeling even once the retinogeniculate circuit is fully mature (Fig. S7). This result additionally suggests that the developmental changes in retinogeniculate connectivity that require Fn14 may be maintained into maturity through an ongoing Fn14-dependent mechanism.
Consistent with the mechanistic distinction between vision-sensitive refinement driven by Fn14 and earlier phases promoted by molecules such as MHC and complement proteins, the expression levels of MHC and complement proteins are not sensitive to visual input. However, other immune-related molecules, including the chemokine Cx3cl1 and the Ifngr2 receptor, are induced by sensory input and may play roles in synapse refinement that are not yet appreciated (Table S1). Supporting the idea that vision-sensitive refinement is influenced by multiple molecular mechanisms, the transcription factor MeCP2, the auxiliary AMPAR subunit Stargazin, and the metabotropic glutamate receptor mGluR1 are also required for the vision-sensitive refinement of the retinogeniculate synapse (Louros e t al., 2014; Narushima et al., 2016; Noutel et al., 2011).
In contrast to their WT littermates, Fn14 KO mice exhibit various synaptic deficits in retinogeniculate connectivity during the vision-sensitive period, including diminished single-fiber strengthening and altered synaptic ultrastructure (Fig. 8). That strengthening of retinogeniculate connections is abnormal in Fn14 KO mice is consistent with our finding that retinal terminals are smaller and dendritic spines are less mature in the dLGNs of Fn14 KO mice at P27 (Hamos et al., 1985). Our functional data show that differences in quantal size or release probability cannot account for the reduced single fiber strength in Fn14 KO mice when compared to WT littermates; therefore, the number of release sites from a given axon is likely reduced in the Fn14 mutant (Chen and Regehr, 2000). However, by E.M., we find that Fn14 KO mice actually have an increased number of PSDs apposed to retinogeniculate terminals. Several possible explanations may account for the apparent discrepancy between this aspect of our E.M. and electrophysiology data. First, it is possible that non-functional PSDs persist in the absence of Fn14. Second, our electrophysiological data only measured connectivity between RGCs and excitatory neurons, while our ultrastructural analysis included PSDs on both excitatory and inhibitory neurons. Third, it is also possible that there is an excess of RGC synaptic contacts mislocated in the distal dendrites of TC neurons in Fn14 KO mice that are functionally not detectable because of dendritic filtering (Rall, 1970). In future studies, these remaining questions will begin to be addressed by performing 3D reconstructions of retinogeniculate convergence in Fn14 KO mice to analyze the number of release sites per bouton onto excitatory neurons specifically (Morgan et al., 2016).
We find that these functional and structural changes to connectivity in Fn14 KO mice are accompanied by changes in the molecular composition of the dLGN, such that the dLGNs of Fn14 KO mice express significantly lower levels of genes critical for aspects of later stages of neural development such as myelination and synaptic transmission (Table S4). The functions of these misregulated genes may provide hints regarding mechanisms underlying Fn14-dependent refinement. For example, the complement protein C4, which is required for synaptic pruning in the dLGN, is down regulated by about 4-fold in the Fn14 KO dLGN. This down-regulation of C4 may contribute to the refinement deficits in Fn14 KO mice described here.
Based upon studies of Fn14 function in other systems, additional potential downstream mechanisms of Fn14-dependent refinement include: (1) cytoskeletal regulation, which is supported by evidence that Fn14 can bind to the small GTPase Rac1 in PC12 cells (Tanabe et al., 2003), and (2) regulation of pro-inflammatory gene expression through NF-ĸB and Mapk signaling pathways (Locksley et al., 2001; Winkles, 2008). Moreover, determining whether Fn14 mediates refinement following binding to its only known ligand, TNF-related weak inducer of apoptosis (Tweak; Wiley and Winkles, 2003), or by binding to an as-yet unidentified ligand will be priorities for investigation in future studies. Through further i nvestigation of the upstream and downstream mechanisms of Fn14-dependent retinogeniculate refinement, it should be possible to obtain increased molecular insight into sensory experience-dependent aspects of neural development.
STAR Methods:
Visual experience-dependent expression of Fn14 is required for retinogeniculate refinement.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael E. Greenberg (meg@hms.harvard.edu). B6.Tnfrsf12atm1(KO)Biogen (Fn14 KO; Jakubowski et al, 2005) mice are subject to restrictions imposed in an MTA by Biogen (Cambridge, MA).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal experiments were performed in compliance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Harvard Medical School. Experiments used male and female C57Bl6/J (Cat #000664; RRID:IMSR_JAX:000664) mice supplied by the Jackson Laboratory and male and female B6.Tnfrsf12atm1(KO)Biogen (Fn14 KO; Jakubowski et al, 2005) mice supplied by Biogen (Cambridge, MA). Animals younger than P28 were housed with their mothers and sometimes their fathers in individually ventilated cages, and mice were provided food and water ad libitum. Developmental ages between E18 and P90 were used, and ages of animals are stated in the figures and figure legends, and under “Method Details.” Fn14 KO and WT mice were bred as heterozygotes, and littermate KO and WT mice were used for experiments.
Unless otherwise specified, mice were housed under standard conditions according to a 12 hour light/dark cycle, a condition referred to in this manuscript as “normally reared” (NR). For late-dark-rear (LDR) experiments, mice were reared under standard conditions until P20, then housed in a custom-built, ventilated light-proof cabinet and handled when necessary by an investigator using night vision goggles (Pulsar). For reexposure to light at P27, mice were moved to an upper chamber in the cabinet and exposed to uninterrupted white light for one or eight hours. Conversely, unstimulated control mice were housed in the dark chamber then euthanized by isofluorane and the brain removed in the dark by an investigator using night vision goggles, to avoid the aberrant induction of experience-dependent gene expression upon exposure to light. The eyes of most of the mice used in electrophysiology experiments between the ages of P12 and P15 had not yet opened, but in some cases they had.
METHOD DETAILS
Isolation of dLGN tissue
Coronal slices (300 µm) were prepared from C57Bl6/J mice or Fn14 KO and WT littermates at a range of ages (as stated in the text) in ice cold PBS using a Leica VT1000S vibratome. Dorsal LG Ns were microdissected following visual identification using a Nikon SMZ-10A brightfield dissection microscope. Following microdissection, dLGNs were flash-frozen in liquid nitrogen and stored at −80°C until further processing.
Single-nucleus RNA sequencing (inDrops)
Dorsal LGNs from three to four mice per condition (LDR and no light; LDR and one hour of light; LDR and eight hours of light) were rapidly thawed, transferred to a dounce homogenizer, and dounced 15 times with a tight pestle in 1 mL homogenization buffer containing 0.25 M sucrose, 25 mM KCl, 5 mM MgCl2, 20 mM Tricine-KOH pH 7.8, 1 mM DTT, 0.15 mM spermine, 0.5 mM spermidine, protease inhibitor cocktails 2 and 3, and 2.5% IGEPAL CA-630 (Sigma). Each sample was filtered through a 40 μm cell strainer and 5 mL of 50% iodixanol (Sigma) was added. Each sample was then layered onto a 30%−40% iodixanol gradient and centrifuged at 10,000xg for 18 minutes at 4°C. Roughly 1 mL of filtered sample was recovered from the 30%−40% iodixanol interface and transferred to an Eppendorf tube. Each sample was diluted to a desired concentration of 80–100,000 nuclei/mL in 30% iodixanol and individual nuclei were captured and barcoded via inDrops as previously described (Klein et al., 2015). The experiment was performed a total of four times such that four independent samples of each of the three conditions were collected, resulting in a total of 12 samples. For each of the 12 samples, approximately 3000 nuclei were encapsulated into microfluidic droplets containing polyacrylamide gels with embedded barcoded reverse transcription primers. Reverse transcription was carried out in intact droplets to generate barcoded cDNA from single nuclei. Following droplet lysis, inDrops libraries were prepared as previously described (Klein et al., 2015; Zilionis et al, 2017). All 12 libraries were indexed, pooled and sequenced (Read 1: 54 cycles, Read 2: 21 cycles, Index 1: 8 cycles, Index 2: 8 cycles) across 2 runs on a NextSeq 500 (Illumina).
inDrops sequencing - data processing
Sequenced reads were processed according to a previously published pipeline (Macosko et al., 2015). Briefly, this pipeline was used to build a custom transcriptome from Ensembl GRCm38 genome and GRCm38.84 annotation using Bowtie 1.1.1, after filtering the annotation gtf file (gencode.v17.annotation.gtf filtered for feature_type=“gene”, gene_type=“protein_coding” and gene_status=“KNOWN”). Read quality control and mapping against this transcriptome were performed. Unique molecular identifiers (UMIs) were used to link sequence reads back to captured molecules. All steps of the pipeline were run using default parameters unless explicitly stated.
Quality control and clustering of nuclei
All cells were combined into a single dataset. Nuclei with >10% mitochondrial content were excluded from the dataset. Cells with fewer than 500 or more than 15,000 UMI counts were excluded. Cells were then clustered using the Seurat R package (Satija et al., 2015). The data were log normalized and scaled to 10,000 transcripts per cell. Variable genes were identified using the following parameters: x.low.cutoff = 0.0125, x.high.cutoff = 3, y.cutoff = 0.5. We limited the analysis to the top 30 principal components (PCs). Clustering resolution was set to 0.6. Clusters containing fewer than 100 cells were discarded. The expression of known marker genes was used to assign each cluster to one of the main cell types. Snap25, Olig1, Aqp4, Cx3cr1, Cldn5, and Vtn were used to identify neurons, oligodendrocytes, astrocytes, microglia, endothelial cells, and pericytes, respectively. Slc17a6 and Gad1 were used to distinguish excitatory and inhibitory neurons, respectively, which comprised the majority of cells analyzed. Clusters with significant expression of two or more markers were removed, as they likely represented doublet clusters resulting from simultaneous capture of two or more nuclei in a single droplet. In total, the final dataset included 8,398 excitatory neurons and 4,987 inhibitory neurons.
Identification of stimulus-dependent genes
To identify induced genes in the dLGN, we performed a differential gene expression analysis using Monocle2 (Qiu et al., 2017). Monocle2 is an R package developed for analysis of differential gene expression across single-cell data. The analysis was conducted on excitatory and inhibitory neurons, comparing each time point of light reexposure (one or eight hours) to gene expression in unstimulated controls (zero hour). The data were modeled using a negative binomial distribution consistent with data generated by high-throughput single-cell RNA-seq platforms such as inDrops. Unlike deep single-cell sequencing, inDrops probabilistically captures/samples the transcriptome of each cell and retrieves only a small fraction of all the present transcripts. Genes whose differential gene expression false discovery rate (FDR) was less than 0.05 (FDR < 0.05) were considered statistically significant.
To narrow down the list of genes to those with the largest fold change in gene expression, we next depth-normalized the transcript counts (each cell normalized to contain 10,000 transcripts) and averaged these depth-normalized counts across all the cells comprising a cell type. Log2 fold changes were calculated from the averaged depth-normalized data after adding 0.1 to the expression of each gene: Fold Change=Log2(Mean1+0.1)-Log2(Mean2+0.1). Genes whose fold change in expression in either direction was greater than 1.5-fold were considered to be either up- or downregulated by experience. Genes that were up- or downregulated are listed in Supplemental Table S1.
Whole-tissue RNA sequencing
Total RNA was extracted from flash-frozen dLGNs using Trizol reagent and purified using the Qiagen RNeasy Micro kit with on-column DNAse digestion. For the LDR and light reexposure paradigm shown in Figure S1, three mice were included for each condition (LDR and no light; LDR and one hour of light; LDR and eight hours of light) and each mouse was considered an individual bioreplicate (n = 3). For the developmental time course shown in Figure S2 and the Fn14 KO analysis in Supplemental Table S4, four mice were included at each of seven ages (E18, P4, P10, P16, P20, P27, and P32) or each genotype, and each mouse represented an individual bioreplicate (n = 4). Total RNA was depleted of ribosomal RNA, heat-fragmented to ~ 350 bp using the NEBNext kit, and processed with adapters to allow either strand specific (for the LDR samples) or -nonspecific (for the developmental samples) sequencing on an Illumina NextSeq 500 System. Reads of fragment ends were nominally 75 bp in all cases, although for about one third of the reads not all sequencing cycles were completed. Generally, over 99.9% of reported sequences were at least 70 bp long, so longer reads were uniformly 3’-trimmed to that length and shorter ones were discarded, as were reads having any base with a sequencing Phred score below 13. Most samples yielded a population of 35–80 million 70-bp reads.
Alignment and mapping of whole-tissue data
Reads were aligned to the mouse genome (GRCm38/mm10 assembly, Dec. 2011) using the Burrows-Wheeler Aligner (bwa) tool. Two sets of target sequences were incorporated into the bwa index in addition to the usual 21 chromosomal targets: (1) the 16,299-bp mouse mitochondrial genome (GenBank accession NC_005089.1) and (2) a set of ~ 8 million short (≤ 138 bp) exon-exon splice-junction sequences (see below). Typically ~70–75% of all LDR reads and ~80–90% of all developmental reads were mappable in each sample, allowing up to 2 mismatches, and of these ~75–90% aligned uniquely.
The splice-junction target sequences were based on the NCBI RefSeq database for GRCm38. For each annotated transcript, we noted all subsets of two or more exons, ordered but not necessarily consecutive, that could be spliced together to produce a sequence at least as long as the read length (70 bp). Each of these sequences was then trimmed to the maximum number of bases such that a read mapping to the sequence would necessarily cross these exons’ splice junction(s). This procedure produced a library of all unique sets of exons whose intragenic splice junctions could possibly be covered by a read of the given length, based on the RefSeq annotation of exonic loci. Aligned reads thus had the opportunity to align either to genomic (chromosomal) sequences or to exon-junction-crossing sequences found only in mature mRNA. Multiple reads whose 5' ends were assigned to the same locus on the same strand were not flattened to a single count.
An in-house software tool, MAPtoFeatures, was used to quantify expression levels for individual genes as follows (Gray et al., 2014): a database of genic features (CDSs and UTRs) was constructed from all 95,023 genomic and 37 mitochondrial transcripts annotated in RefSeq for GRCm38. Merged genes were constructed by joining all exons in all transcripts assigned to each distinct gene; the resulting segments defined the gene’s exonic coordinates used here (with the gaps between them defining introns). Genes with zero CDS exons were labeled “noncoding”. These 33,102 genes were supplemented with 1,563 additional nonc oding genes specified by the loci of all ribosomal RNA genes obtained from RepeatMasker (where the options Variations and Repeats, rmsk.repFamily=“rRNA” yielded 480 LSU-rRNA_Hsa elements; 45 SSU rRNA_Hsa; and 1,038 5S). The purpose of this step was to allow the filtering out of reads stemming from transcription of repeats and rRNA genes, which tend to get populated to inconsistent degrees from sample to sample depending on variability in the quality of rRNA depletion.
Differential gene expression analysis
For the developmental time course, our differential expression analysis calculated mean fold change ratios and their significance between (adjacent) pairs of time points for every expressed gene. For the late-dark-rear study, we compared samples from mice reexposed to light for one or eight hours to the unstimulated control using the three replicates for each pair. Statistical significance of each fold change was evaluated by Benjamini-Hochberg (BH) corrected p-values of all genes based on the biological variability implied by the replicates for each pair of conditions with a false discovery rate of 5% (FDR=0.05). Differential-expression between sets of replicates was analyzed using the R Bioconductor package edgeR (version 3.14.0) (McCarthy et al., 2012).
ATAC sequencing
ATAC sequencing was performed according to previously described methods (Buenrostro et al., 2015) on the dLGNs of mice subjected to LDR followed by three hours of reexposure to light. The dLGNs of two mice were included in the analysis (n = 2). Briefly, tissue was thawed and lysed in ice cold Lysis Buffer containing 10 mM Tris- HCl, 10 mM NaCl, 3 mM MgCl2, and 0.1% NP40, and then homogenized by douncing. Tissue was then pelleted by centrifugation at 500xg for 10 minutes at 4°C. The pellet was then resuspended in ice cold dH2O to a final volume of 20 µL. The transposase reaction was carried out at 37oC for 30 minutes, followed by DNA purification. Index primers were added to each sample. For PCR amplification, the appropriate number of PCR cycles was determined by qPCR. After amplification, DNA was purified and quantified by Qubit. DNA product size was evaluated by Bioanalyzer. Samples were sequenced on a Nextseq 500 System. Sequencing yielded about 71 million raw reads with lengths in the range of 35–75 bp; reads shorter than 37 bp were discarded. The remaining reads were 3’-trimmed to 37 bp and aligned to the mouse genome (GRCm38/mm10 assembly) using the bwa tool. The resulting ~53 million uniquely mapped reads were piled up in tiles of width 20 bp to produce WIG-formatted ATAC-seq tracks, as shown in Fig. S1G.
Quantitative PCR
Total RNA from microdissected dLGNs was extracted with Trizol reagent and purified using the RNeasy Micro kit with on-column DNAse digestion. Reverse transcription was performed using the High Capacity cDNA Reverse Transcription kit. Real-time quantitative PCR analysis was performed using the StepOnePlus qPCR system and Pwer SYBR Green mix. Reactions were run in triplicate and Gapdh levels were used as an endogenous control for normalization. The sequences of real-time PCR primers used were selected from an existing database (Origene) or were described previously (Spiegel et al., 2014; Bloodgood et al, 2013). The sequences of the primers are given in the Key Resources Table. For qPCR analysis of mRNA samples shown in Figure 2, n = 4 bioreplicates per time point or age. For qPCR analysis of mRNA shown in Figure S1, 3 bioreplicates were included.
Western blotting
For analyzing levels of Fn14 protein, western blotting was performed on dLGN lysates. Flash-frozen dLGNs of three mice per condition were rapidly thawed, pooled, and homogenized in ice cold RIPA buffer containing 50 mM Tris-HCl pH 8.0, 1% triton- X100, 0.5% Na-deoxycholate, 0.1% sodium dodecyl sulfate (SDS), and 150 mM NaCl, including complete protease inhibitor cocktail tablet and phosphatase cocktails two and three. Tissue was resuspended in roughly 400 µL of buffer and dounced 20X in a 2 mL dounce homogenizer on ice. After complete homogenization, samples were rotated at 4°C for 10 minutes then spun at 14,000 RPM for 20 minutes to spin out the insoluble fraction. The supernatant was transferred to a new tube and Nupage LDS 4X Sample Buffer was added to a total concentration of 1X including 10% 2-mercaptoethanol, freshly added. Each sample was then boiled at 95°C for one minute prior to being run on a 12% Bis-tris gel then transferred to nitrocellulose membrane. Immunoblotting was performed using the Odyssey platform and IR dye secondaries, which allow for quantitation of protein expression by western blot. Blocking and antibody incubations were performed in 5% dry milk in TBS-T. The amount of protein run per well was 15 µg. Antibodies used for western blotting include our custom polyclonal rabbit anti-Fn14 #1074 (1:100), rabbit anti-Fn14 4403s (1:100), and rabbit anti-GAPDH (1:5000).
Quantification of protein levels was performed using the Li-Cor Odyssey system by comparing the fluorescence intensity of bands representing Fn14, as validated in lysates from Fn14 KO mice, in different samples run on the same blot. After background subtraction, the fluorescence intensity of each Fn14 band was normalized to that of the loading control GAPDH to control for technical variability between conditions not representing biological variation. For each experiment, three bioreplicates comprised of the pooled dLGNs of three mice were performed. Separate western blots were run for each bioreplicate with samples in duplicate, and protein levels across conditions were normalized to the first sample on a given blot.
Immunoprecipitation
For validating loss of Fn14 protein in Fn14 KO mice, whole forebrains from adult Fn14 KO, Heterozygous, or WT mice were homogenized in buffer containing final concentrations of 10 mM HEPES-KOH pH 7.5, 2 5 mM KAc, 320 mM sucrose, 1% triton- X100, and 250 mM NaCl, including a complete protease inhibitor cocktail tablet and phosphatase cocktails two and three. Each brain was homogenized in 1 mL of buffer in a 2 mL dounce homogenizer then transferred to an eppendorf tube and spun at 14,000 RPM for 20 minutes at 4° C to spin out the insolubl e fraction. After setting aside 100 µl of input, the supernatant was pre-cleared by rotating for 1 hour at 4° C with 25 µl of washed protein A dynabeads. Beads were then collected on a magnet and the sample split into two samples of equal volume, around 300 µl. Polyclonal anti-Fn14 (4403s, 1:50) or anti-Fn14 #1074 (1:50) was added to one sample, and a negative control rabbit IgG was added to the other. Samples were rotated at 4° C for 1.5 hours. 25 µl pre washed Protein A dynabeads were then added to each tube, and rotated at 4° C for 1 hour. Beads were then collected on a magnet and the supernatant discarded. Beads were washed 4X in 1 mL homogenization buffer without sucrose by rotating for 10 minutes per wash at 4° C. Finally, proteins were el uted from the beads in 100 µl 1X Nupage LDS 4X Sample Buffer with 10% 2-mercaptoethanol by boiling at 95°C for one minute. Western blotting of samples was performed as described above.
Single-molecule FISH
C57Bl6/J mice or Fn14 KO and WT littermate mice were euthanized with isofluorane and their brains were rapidly dissected and embedded in OCT (Optimum Cutting Temperature) on dry ice. 20 µm thick sections were made on a Leica CM 1950 cryostat, mounted on Superfrost Plus slides, and stored at −80° C until use. Single molecule multiplexed FISH was performed using the RNAscope platform according to the manufacturer’s protocol for fresh-frozen sections. Commercial probes obtained from ACD detected the following genes: Fos, Npas4, Fn14, Vglut1, Vglut2, Gad1, Gad2, Olig1, Mbp, P2ry12, Cx3cr1, Cldn5, Pecam, Aldh1l1, and Aldoc. Please note that we validated multiple excitatory neuron markers that worked well for identifying TC neurons of the dLGN by FISH, including Vglut1 and Vglut2, and we used them interchangeably.
For the quantification of the number of mRNA molecules per cell (Figure 1B,C, Figure 3D,E, and Figure S7), sections analyzed were derived from three mice per condition (n = 3). Sample processing and imaging were performed in parallel and the image acquisition parameters were constant between conditions. Commercially available negative control probes were used to confirm that background was similar for each condition. Excitatory neurons were identified based upon expression of Vglut1 overlapping a DAPI-stained nucleus. The number of Fos or Fn14 mRNA puncta overlapping with the neuron were counted, and average numbers of mRNA molecules +/− SEM were plotted. Sample processing, imaging, and quantification were performed by two investigators blinded to experimental conditions.
To determine the cell type-specificity of Fn14 expression (Figure 3B,C), sections analyzed were derived from four mice per condition (n = 4). Per bioreplicate, four dLGN sections were quantified for a given cellular marker. As above, samples within a given bioreplicate were processed in parallel with co nstant image acquisition settings, and the investigator was blinded to conditions at all times. After processing and imaging all bioreplicates, the single frame merged images containing all fluorescence channels were loaded into the Multiplex RNA FISH module in Halo software (Indica labs) and analyzed. Briefly, the software identified each cell by nuclear detection in the DAPI channel, and the number of puncta for each probe was measured within the cell boundary. The software then reported the number of molecules within each cell based upon detection of pixels meeting a standardized minimum fluorescence intensity that was held constant across all conditions and for all markers. Cells were then categorized by type based upon the expression of marker genes, and cells containing fewer than five marker gene puncta were removed from the analysis. Cells expressing at least five Fn14 mRNA puncta were counted as Fn14-positive. The percentages of Fn14-positive cells expressing at least five puncta of a given marker gene were quantified.
Immunofluorescence
To obtain sections for immunofluorescence, animals were perfused with 4% paraformaldehyde (PFA) in PBS, and the brains were incubated in 4% PFA at 4° C overnight. The next day, the brains were washed 3X in cold PBS then sectioned at 50 µm on a Leica VT1000S vibratome. Free-floating sections were stained at 4° C overnight with primary antibody. The next day, sections were washed 3X in PBS then incubated with AlexaFluor secondary dyes (1:500; Invitrogen), washed 3X in PBS, then mounted on slides in Fluoromount G plus DAPI (SouthernBiotech). Commercial antibodies used for staining in this study are rabbit anti-Fos (1:1000), guinea pig anti- VGLUT2 (1:1000, EMD Millipore) rabbit anti-VGLUT1 (1:1000, Synaptic Systems), and mouse anti-SMI-32 (1:1000, EMD Millipore).
For the quantification of immunofluorescence, sections representing each condition were processed in parallel, and z-stacks were obtained on an Olympus FluoView 1000 laser scanning confocal microscope with image acquisition settings held constant across all conditions. For Fos staining shown in Figure S1E,F and VGLUT1 and VGLUT2 staining shown in Figure S3G-I, three mice of each condition were analyzed (n = 3). Images were opened in ImageJ (NIH) and maximum projections were assembled from the z-stacks. For counting Fos-positive neurons, the number of SMI-32- positive excitatory neurons (not shown) was counted within a given section. Next, Fos expression in the red channel was analyzed, and SMI-32-positive cells whose fluorescence intensity reached two-fold higher than image background were assigned Fos-positive. Data were plotted as the percentage of Fos-positive neurons per condition. For the quantification of VGLUT1 and VGLUT2 in Fn14 KO and WT mice, ImageJ was used to calculate the proportion of the 2-dimensional image area occupied by either the green channel (VGLUT2) or the red channel (VGLUT1), and these values were normalized to the area occupied by either marker in the WT mouse. Normalized average areas occupied were plotted +/− SEM.
Array tomography
Animals were perfused with 4% PFA and the brains subsequently sectioned at 100 µm on a Leica VT1000S vibratome. The dLGNs were microdissected and further processed by the HMS array tomography core: sections were dehydrated in a series of increasing ethanol solutions then embedded in LRWhite embedding medium (Electron Microscopy Sciences) overnight at 4°C. Serial secti ons were cut at 70 nm per section and immunostained. Briefly, sections were blocked for five minutes in blocking buffer and then incubated with primary antibody overnight at 4°C. Sections were washed several times in Tris (Sigma) then stained with secondary antibody in blocking buffer for 30 minutes at room temperature. Sections were washed and mounted on slides in SlowFade Gold antifade reagent with DAPI (Invitrogen). Primary antibodies used were anti-VGLUT2 (guinea pig, 1:100, Millipore) and anti-PSD-95 (rabbit, 1:100, Cell Signaling Technologies 3450). Secondary antibodies used were AlexaFluors (Invitrogen). Images were taken on a Zeiss Axio Imager Z2 with all sections of a bioreplicate processed and imaged in parallel with constant image acquisition settings and the experimenter blinded to the conditions at all stages. For the quantitative analysis, three separate stacks were obtained from four separate Fn14 KO or Fn14 WT animals per condition (n = 4).
Analyses of colocalized VGLUT2/PSD-95 puncta were performed using the ImarisColoc analysis tool in Imaris software (Bitplane) with consultation from the Image Analysis Core at Harvard Medical School. Briefly, automated intensity thresholds for each channel were calculated and these thresholds were applied to optimize signal-to noise and to filter out spatial regions of high background fluorescence. These thresholds were constant across conditions. After thresholding, the number of voxels positive for signal in both green (PSD-95) and red (VGLUT2) channels was calculated and plotted as co-localized puncta density within the three-dimensional volume of the stack, +/− SEM.
Electron microscopy
Animals were perfused with 2.5% glutaraldehyde and 2% PFA in 0.1 M sodium cacodylate buffer, pH 7.4 (Electron Microscopy Sciences). Vibratome sections were cut coronally at 100 µm and the dLGNs microdissected. Floating sections were washed in 0.1 M cacodylate buffer and post-fixed with 1% Osmiumtetroxide (OsO4)/1.5% Potassiumferrocyanide (KFeCN6) for one hour, washed in water 3X and incubated in 1% aqueous uranyl acetate for one hour followed by two washes in water and subsequent dehydration in grades of alcohol (10 min each; 50%, 70%, 90%, 2× 10 min 100%). The samples were then infiltrated for 15 min in a 1:1 mixture of propyleneoxide and TAAB Epon (Marivac Canada Inc. St. Laurent, Canada). The samples were embedded in drops of TAAB Epon between two sheets of aclar plastic (Electron Microscopy Sciences) and polymerized at 60° C for 48 hours.
Ultrathin sections of 80 nm were cut on a Reichert Ultracut- S microtome, placed onto copper grids, stained with uranyl acetate and lead citrate, and examined in a JEOL 1200EX transmission electron microscope. For quantification, ~20 images per brain at a magnification of 10,000X were taken by a blinded EM core facility staff member. The entire coronal plane of the dLGN was sampled. Coded images at 10,000X were then analyzed for bouton and PSD densities, bouton area, and PSD length in ImageJ by an investigator blinded to conditions. For quantitative analyses shown in Figures 4 and S4, n = P20 WT: 80 micrographs from four mice; P27 WT: 100 micrographs from five mice; P20 KO: 120 micrographs from six mice; P27 KO: 100 micrographs from five mice; LDR WT: 80 micrographs from four mice; LDR KO: 120 micrographs from six mice. For quantifying bouton area, n = 229 boutons per condition.
Classification of inputs
Boutons and associated PSDs were categorized as retinal or non-retinal based upon well-described morphological features of retinal inputs. These inputs are particularly large and densely packed with round vesicles. Further, they contain large, pale mitochondria (Colonnier and Guillery, 1964; Guillery and Colonnier, 1970) In our analysis, we only considered inputs to be derived from the retina if they contained at least one recognizable pale mitochondrion, as this is the most distinctive feature of these inputs. This conservative approach decreased the likelihood of errantly categorizing non-retinal inputs as retinal, although it likely contributed to an underestimation of the absolute numbers of retinal boutons and PSDs in our quantification.
Golgi staining and Sholl analysis
Four Fn14 KO and four WT littermate mice at P27 were euthanized with isofluorane and their brains rapidly dissected and subjected to Golgi staining using the FD Rapid GolgiStain kit (FD Neurotechnologies, Inc) according to the manufacturer’s protocol. Rapidly dissected brains were immersed in impregnation solution for 10 days in the dark at room temperature then transferred to Solution C at 4° C for two days. Brains were quickly frozen in a butanol dry ice bath and coronally sectioned at 200 µm on a Leica CM 1950 cryostat, then mounted onto Superfrost Plus slides. Sections containing the dLGN were dried at room temperature for 24 hours, and then stained, dehydrated in ethanol, cleared in Xylene, and mounted in Permount (Fisher). This procedure resulted in sparsely stained neurons throughout the dLGN. Golgi-stained neurons in the dLGN were simultaneously imaged and traced in x, y, and z planes using a Zeiss Axioskop microscope and Neurolucida (Microbrightfield Bioscience).
For quantification of dendritic branching and morphology by Sholl analysis (Sholl, 1953), 22 Golgi-stained neurons from Fn14 KO and WT dLGNs from four animals per genotype were traced in x,y, and z planes in Neurolucida at 40X, and the traces were subsequently opened in Neurolucida Explorer. Sholl analysis was performed by applying a series of radiating concentric circles beginning at the soma with 10 µm spacing between circles, and the number of dendrites intersecting each circle was calculated and plotted. This analysis focused on the more proximal dendrites, as dendrites extending beyond the 200 µm border of the section were not traced.
Analysis of dendritic spines
The dendritic spine morphologies of Golgi-stained neurons were assessed in two ways following the tracing of dendritic segments, within the same sections described above, at 63X resolution. First, spines were categorized into three well-described structural classifications based upon the following parameters: spines with a bulbous head whose diameter was at least 2X that of the shaft were classified as “mushroom”; spines with a length-to-width ratio of less than one and a length of less than 0.3 µm were classified as “stubby”; and spines with a length-to-width ratio greater than one, without a bulbous head, and with a length greater than 0.3 µm were co-classified as thin spines or filopodia. The densities of each spine subtype per length of dendrite in Fn14 KO and WT neurons were quantified and the cumulative frequency distributions plotted. For this analysis, n = WT: 35 dendrites from 22 neurons from four mice; KO: 31 dendrites from 22 neurons from four mice.
The second method of spine analysis took into account the biological reality that spine structure exists along a continuum, such that categorization of spines into separate morphological subclasses is, in some ways, arbitrary. Therefore, the analysis was expanded to include measurements of spine head width and total spine length of all dendritic protrusions regardless of spine “subtype.” These measurements were performed on the Golgi-stained sections described above, and 225 spines from each condition, Fn14 KO or WT, derived from sections from three mice per genotype, were included in the analysis. These data are plotted as cumulative frequency distributions in Figures 4 and S4.
Fluorescence imaging
Imaging of immunofluorescence and FISH was performed on an Olympus FluoView 1000 laser scanning confocal microscope equipped with 405 nm, 440 nm, 488 nm, 515 nm, 559 nm, and 635 nm excitation lasers, and 10x air 0.4NA, 20x air 0.75NA, 40x oil 1.3NA, 60x oil 1.42NA, and 100x oil 1.4NA objectives.
dLGN slice preparation for electrophysiology
Acute brain slices containing the optic tract and dLGN were prepared as previously described (Chen and Regehr, 2000; Litvina and Chen, 2017). Mice were anesthetized by inhaling isofluorane then immediately decapitated, after which the head was immersed into an oxygenated ice-cold solution containing (in mM): 130 mM K gluconate, 15 mM KCl, 0.05 mM E GTA, 20 mM HEPES, and 25 mM glucose (pH 7.4 with NaOH; Sigma). For each mouse, a single 250 µm-thick parasagittal slice was cut in this ice-cold solution using a sapphire blade (Delaware Diamond Knives, Wilmington, DE) on a vibratome (VT1000S; Leica, Deerfield, IL). Slices then recovered at 32º C for 20–30 minutes in oxygenated artificial cerebrospinal fluid (ACSF) in mM: 125 NaCl, 26 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 1.0 MgCl2, 2.0 CaCl2, and 25 glucose (Sigma), adjusted to 310–312 mOsm with water.
Electrophysiology
Whole-cell voltage clamp recordings of thalamocortical TC neurons in dLGN were performed as previously described (Chen and Regehr, 2000) using borosilicate glass pipettes (1–2.5 MOhms, Sutter Instrument, Novato, CA) filled with an internal solution (in mM): 35 CsF, 100 CsCl, 10 EGTA, 10 HEPES, and 0.1 methoxyverapamil (290–300 mOsm, pH 7.3; Sigma). All experiments were performed at room temperature in oxygenated ACSF containing 50 µM of the GABAA receptor antagonist picrotoxin (Tocris, Ellisville, MO). Recordings from synapses that were silent (no response at −70 mV, but EPSC evident at +40 mV) were averaged for at least 3–5 trials. Single RGC fiber EPSCs were obtained using a threshold method as previously described (Hooks and Chen, 2006; Noutel et al., 2011). See following section for detailed description of fiber fraction determination.
Desynchronized, evoked AMPAR-mediated miniature EPSCs (mEPSCs) were resolved in the presence of an oxygenated extracellular solution containing (in mM): 4 SrCl2, 1 MgCl2, 0 CaCl2, 125 NaCl, 26 NaHCO3, 1.25 NaH2PO4•H2O, 2.5 KCl, 25 glucose, and 50 µM picrotoxin, 20 µM (R)-CPP.
Paired Pulse Ratio (PPR) was determined by stimulating the optic tract twice with randomized inter-stimulus intervals (ISIs) of 50 ms, 100 ms, 250 ms, and 500ms while holding the cell at −70 mV. PPR for each cell was calculated by dividing the peak amplitude of the second EPSC (A2) by the peak amplitude of the first EPSC (A1). For ISIs of 50, 100, and 250 ms, the average waveform of the first EPSC at an ISI of 500 was subtracted from the second EPSC to obtain a more accurate measure of A2. Averages of three to six trials of each ISI were used to calculate PPR. To inhibit modulatory and postsynaptic contributions to short-term synaptic plasticity at the retinogeniculate synapse (Chen et al., 2002; Hauser et al., 2014), recordings were done in the presence of (in µM): 50 cyclothiazide (AMPAR desensitization inhibitor), 5 CGP- 55845 (GABAB-receptor antagonist), 10 DPCPX (A1 adenosine receptor antagonist), 50 LY-341495 (mGluR antagonist), and 10 cyanopindolol (5-HT1 receptor antagonist).
Fiber fraction calculation
The fiber fraction (“FF”) ratio [single fiber current amplitude/ maximal current amplitude] was used to estimate afferent RGC convergence onto each TC neuron. Maximal current amplitudes (“Max”) were first determined by stimulating at increasing intensity until the synaptic current amplitude reaches a plateau (at least 50–100 µA for durations of 0.2 ms using an A365 stimulus isolator from WPI). For each cell, two electrodes filled with ACSF were moved along the optic tract in a few locations, in order to determine a site that would activate as many RGC axons as possible and yield the largest maximal EPSC current. One electrode was inserted into the optic tract while the other electrode was positioned just above the surface of the slice to serve as a local ground. Once the stimulating electrode was optimally placed, it was not moved until after the recording on a given cell was completed. The stimulus intensity was decreased systematically until a failure of transmission was observed, then incrementally increased by 0.25 µA steps until an EPSC occurred, whose amplitude is the single fiber amplitude (“SF”). To determine the fiber fraction, the single fiber amplitude was divided by the maximal amplitude (FF = SF/ Max). For each stimulus intensity, we recorded the synaptic response, or lack thereof, at both −70 mV and +40 mV with an inter-trial interval of 20 seconds, yielding two fiber fraction values for each cell. AMPAR antagonists were not used to determine NMDAR amplitudes; rather, the more slowly rising peak NMDAR mediated EPSCs were measured following the initial decay of the AMPAR transient (see Figure S5B). Cells that were lost before the determination of FF values at both holding potentials were discarded from our analysis. Only the minimal threshold response is quantified for single fiber amplitude because it is difficult to distinguish between the recruitment of an additional fiber from trial-to-trial variation of the same fiber. An exception is made if an incremental step in stimulation intensity of 0.25 μA recruits an EPSC with five times the amplitude of the initial single fiber EPSC at either - 70 mV OR +40 mV (as it would be difficult to attribute this increased EPSC as arising from stochastic variations in vesicular release of the initial single fiber). In this case, the increased current was counted as a second single fiber and included in our fiber fraction ratio. The 5x cutoff was determined after subtracting the first single fiber amplitude from the second single fiber amplitude. For example, if SF1 = 100pA and SF2 = 500 pA, “SF2” was not counted because 500pA – 100pA = 400pA, which is only 4x the amplitude of SF1. In cases where the second single fiber amplitude was five times the amplitude of the first single fiber, the fiber fraction was determined as follows: FF = [(SF1/Max) + (SF2/Max)]/2. For “silent synapses” described in the previous section, SF2 was counted regardless of the amplitude because SF1 at −70mV in these cases is 0. For further details and justification of the FF assay, please see the Supplemental Methods of Hooks and Chen, 2008 and Noutel et al., 2011.
QUANTIFICATION AND STATISTICAL ANALYSIS
Blinding
Specific blinding schemes for quantitative experiments are described in Method Details. For sequencing experiments, samples were blinded subsequent to initial collection but prior to library preparation and sequencing. For FISH, S.R. performed hybridizations and blinded L.C. to the identity of the cell type markers. L.C. performed imaging and analysis and was unblinded following quantification. For comparison of Fn14 KO and WT littermates via E.M., array tomography, immunofluorescence, and Golgi staining, genotyping of heterozygous breeding litters was performed by S.R. and tissue was processed by L.C. who was blinded to the genotypes of the mice. Genotypes of the mice were revealed only after the analysis and quantification were completed. In the case of Golgi staining and analysis, L.C. performed genotyping and blinding, and S.R. performed analysis and quantification.
For blinding of electrophysiology experiments, after tails were cut from litters of Fn14 hetereozygous breeding, C.P.T. prepared DNA, ran PCRs, and loaded 1.5% agarose gels for genotyping, after which E.L. imaged the gels and informed C.P.T. of heterozygous mice and non-heterozygous mice by eartag number without revealing whether such mice were WT or KO. E.L. revealed genotypes to C.P.T. after recordings and analyses for a particular experiment were completed, and were double-checked by re-genotyping a newly cut tail collected at the time of the acute slice dissection.
Image analysis, quantification, and presentation
In all cases, when images were used to compare two or more conditions, the tissues were processed and imaged in parallel using the same detection parameters at the time of image acquisition. Post-acquisition, images were processed in parallel and any minor alterations in contrast or brightness were applied uniformly to the entire image and repeated on the relevant corresponding image for comparison.
Analysis and statistics
For sequencing experiments, statistical methods are described under Method Details. For molecular and structural studies, statistical significance was calculated by unpaired t-tests when two conditions were being compared. When more than two conditions were being compared, one-way ANOVA with Dunnett’s or Tukey’s post test was used. When multiple variables were involved, 2-way ANOVA with Bonferonni correction was applied. The statistical analyses for each experiment are given in the figure legends. Differences between cumulative frequency distributions were calculated by Kolmogorov-Smirnov test. All statistical analyses were performed using Prism software. Statistics and “n” numbers for all experiments are given in figure legends. Error bars represent SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Statistical analysis of structural data is detailed in Table S3.
For electrophysiological experiments, data acquisition of voltage-clamp experiments was performed using Clampex10.2, an Axopatch 200B amplifier, and digitized with a DigiData 1440 data acquisition board (Molecular Devices, Sunnyvale, CA). Analysis of EPSCs was done using custom software written in IgorPro (Wave- Metrics, Portland, OR), Excel (Microsoft, Redmond, WA), Clampfit (Molecular Devices, Sunnyvale, CA) and Prism 7 (GraphPad Software, La Jolla, CA). For recordings at +40 mV in which the rapid AMPAR transient was visible, the second, slower peak of NMDAR EPSCs was measured.
Data in all figures with electrophysiology experiments (Figures 5–7, S5, and S6) are plotted as medians, with boxes displaying the interquartile range (IQR = 25th to 75th percentile), and whiskers displaying 10th to 90th percentile. One of two two-tailed nonparametric tests were used to analyze all electrophysiology data: Mann-Whitney test or Kruskal-Wallis ANOVA followed by Dunn’s post-test comparing ranks of selected groups. Statistical significance in plots were indicated (* p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001), and exact p-values for all comparisons are listed in Table S2.
DATA AND SOFTWARE AVAILABILITY
Lists of visual experience-dependent genes (S1) and Fn14-regulated genes (S4) can be found in the Supplemental Information along with tables S2 and S3 which detail statistical analyses and other parameters for the electrophysiological and structural studies, respectively. In addition, both raw and processed sequencing data of four data sets will be uploaded to the GEO database: snRNAseq following visual stimulus; whole tissue RNAseq following visual stimulus; whole-tissue RNAseq across development; and whole-tissue RNAseq in the Fn14 KO mouse.
Supplementary Material
HIGHLIGHTS:
Visual stimulation induces a unique gene program in excitatory neurons of mthe dLGN
Fn14 is the most inducible molecule specific to the excitatory population
Vision-sensitive refinement is impaired in mice lacking Fn14
Changes in synaptic morphology accompany physiological deficits in mice lacking Fn14
ACKNOWLEDGEMENTS
We thank Drs. S. Ashrafi, B. Bean, G. Boulting, E. Griffith, P. Kaeser, S. Paradis, G. Pouchelon, T. Schwarz, S. Su, T. Vierbuchen, and E. Fisher as well as additional members of the Greenberg and Chen labs for helpful discussions and input on the project and manuscript. We thank Dr. A. Thompson, Dr. L. Litvina, and Ee-Lynn Yap for input on electrophysiology. We thank Vance Soares for design work on schematics, and Dr. E. Pollina, Dr. L. Boxer, D. Gilliam, C. Cowley, K. Mei, and M. Yang for blinding of electrophysiology experiments. We also thank the following cores: Neurobiology Imaging Facility and Array Tomography cores supported by NINDS P30 Core Center Grant #NS072030, the HMS Electron Microscopy Facility, the Imaging and Data Analysis Core of HMS, the Vision Core supported by grant P30 EY12196, and the HMS Single Cell Core. This work was funded by R37 NS028829 to M.E.G., R01EY013613 to C.C., and 5T32AG000222–23 and Goldenson Postdoctoral Fellowship to L.C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers
DESIGNATION OF INTERESTS
L.B. is an employee and shareholder of Biogen.
REFERENCES
- Andreae LC, and Burrone J (2017). The role of spontaneous neurotransmission in synapse and circuit development. J Neurosci Res [DOI] [PMC free article] [PubMed]
- Arnoux I, and Audinat E (2015). Fractalkine Signaling and Microglia Functions in the Developing Brain. Neural Plast 2015, 689404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry KP, and Nedivi E (2017). Spine Dynamics: Are They All the Same? Neuron 96, 43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloodgood BL, Sharma N, Browne HA, Trepman AZ, and Greenberg ME (2013). The activity-dependent transcription factor NPAS4 regulates domain-specific inhibition. Nature 503, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SA, Richards CM, Hanscom HN, Feng SL, and Winkles JA (2003). The Fn14 cytoplasmic tail binds tumour-necrosis-factor-receptor-associated factors 1, 2, 3 and 5 and mediates nuclear factor-kappaB activation. Biochem J 371, 395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, and Greenleaf WJ (2015). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkly LC (2014). TWEAK/Fn14 axis: the current paradigm of tissue injury-inducible function in the midst of complexities. Semin Immunol 26, 229–236. [DOI] [PubMed] [Google Scholar]
- Chen C, Blitz DM, and Regehr WG (2002). Contributions of receptor desensitization and saturation to plasticity at the retinogeniculate synapse. Neuron 33, 779–788. [DOI] [PubMed] [Google Scholar]
- Chen C, and Regehr WG (2000). Developmental remodeling of the retinogeniculate synapse. Neuron 28, 955–966. [DOI] [PubMed] [Google Scholar]
- Colonnier M, and Guillery RW (1964). Synaptic Organization in the Lateral Geniculate Nucleus of the Monkey. Z Zellforsch Mikrosk Anat 62, 333–355. [DOI] [PubMed] [Google Scholar]
- Datwani A, McConnell MJ, Kanold PO, Micheva KD, Busse B, Shamloo M, Smith SJ, and Shatz CJ (2009). Classical MHCI molecules regulate retinogeniculate refinement and limit ocular dominance plasticity. Neuron 64, 463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard CA, Butts DA, and Shatz CJ (2007). Regulation of CNS synapses by neuronal MHC class I. Proc Natl Acad Sci U S A 104, 6828–6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JM, Harmin DA, Boswell SA, Cloonan N, Mullen TE, Ling JJ, Miller N, Kuersten S, Ma YC, McCarroll SA, et al. (2014). SnapShot-Seq: a method for extracting genome-wide, in vivo mRNA dynamics from a single total RNA sample. PLoS One 9, e89673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillery RW, and Colonnier M (1970). Synaptic patterns in the dorsal lateral geniculate nucleus of the monkey. Z Zellforsch Mikrosk Anat 103, 90–108. [DOI] [PubMed] [Google Scholar]
- Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, Choudhury SR, Aguet F, Gelfand E, Ardlie K, et al. (2017). Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat Methods 14, 955–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamos JE, Van Horn SC, Raczkowski D, Uhlrich DJ, and Sherman SM (1985). Synaptic connectivity of a local circuit neurone in lateral geniculate nucleus of the cat. Nature 317, 618–621. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, and Kano M (2013). Synapse elimination in the developing cerebellum. Cell Mol Life Sci 70, 4667–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser JL, Liu X, Litvina EY, and Chen C (2014). Prolonged synaptic currents increase relay neuron firing at the developing retinogeniculate synapse. J Neurophysiol 112, 1714–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong YK, and Chen C (2011). Wiring and rewiring of the retinogeniculate synapse. Curr Opin Neurobiol 21, 228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooks BM, and Chen C (2006). Distinct roles for spontaneous and visual activity in remodeling of the retinogeniculate synapse. Neuron 52, 281–291. [DOI] [PubMed] [Google Scholar]
- Hooks BM, and Chen C (2008). Vision triggers an experience-dependent sensitive period at the retinogeniculate synapse. J Neurosci 28, 4807–4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horng S, Kreiman G, Ellsworth C, Page D, Blank M, Millen K, and Sur M (2009). Differential gene expression in the developing lateral geniculate nucleus and medial geniculate nucleus reveals novel roles for Zic4 and Foxp2 in visual and auditory pathway development. J Neurosci 29, 13672–13683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrvatin S, Hochbaum DR, Nagy MA, Cicconet M, Robertson K, Cheadle L, Zilionis R, Ratner A, Borges-Monroy R, Klein AM, et al. (2018). Single-cell analysis of experience-dependent transcriptomic states in the mouse visual cortex. Nat Neurosci 21, 120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubowski A, Ambrose C, Parr M, Lincecum JM, Wang MZ, Zheng TS, Browning B, Michaelson JS, Baetscher M, Wang B, et al. (2005). TWEAK induces liver progenitor cell proliferation. J Clin Invest 115, 2330–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalish BT, Cheadle L, Hrvatin S, Nagy MA, Rivera S, Crow M, Gillis J, Kirchner R, and Greenberg ME (2018). Single-cell transcriptomics of the developing lateral geniculate nucleus reveals insights into circuit assembly and refinement. Proc Natl Acad Sci U S A 115, E1051–E1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, and Kirschner MW (2015). Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacar B, Linker SB, Jaeger BN, Krishnaswami S, Barron J, Kelder M, Parylak S, Paquola A, Venepally P, Novotny M, et al. (2016). Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat Commun 7, 11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leighton AH, and Lohmann C (2016). The Wiring of Developing Sensory Circuits-From Patterned Spontaneous Activity to Synaptic Plasticity Mechanisms. Front Neural Circuits 10, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P, Wang C, Xu B, Gao S, Guo J, Zhao X, Huang H, Zhang J, Chen X, Wang Q, et al. (2014). The VGF-derived peptide TLQP62 produces antidepressant-like effects in mice via the BDNF/TrkB/CREB signaling pathway. Pharmacol Biochem Behav 120, 140–148. [DOI] [PubMed] [Google Scholar]
- Lin WJ, Jiang C, Sadahiro M, Bozdagi O, Vulchanova L, Alberini CM, and Salton SR (2015). VGF and Its C-Terminal Peptide TLQP-62 Regulate Memory Formation in Hippocampus via a BDNF-TrkB-Dependent Mechanism. J Neurosci 35, 10343–10356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Bloodgood BL, Hauser JL, Lap an AD, Koon AC, Kim TK, Hu LS, Malik AN, and Greenberg ME (2008). Activity-dependent regulation of inhibitory synapse development by Npas4. Nature 455, 1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvina EY, and Chen C (2017). Functional Convergence at the Retinogeniculate Synapse. Neuron 96, 330–338 e335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locksley RM, Killeen N, and Lenardo MJ (2001). The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104, 487–501. [DOI] [PubMed] [Google Scholar]
- Louros SR, Hooks BM, Litvina L, Carvalho AL, and Chen C (2014). A role for stargazin in experience-dependent plasticity. Cell Rep 7, 1614–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik AN, Vierbuchen T, Hemberg M, Rubin AA, Ling E, Couch CH, Stroud H, Spiegel I, Farh KK, Harmin DA, et al. (2014). Genome-wide identification and characterization of functional neuronal activity-dependent enhancers. Nat Neurosci 17, 1330–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardinly AR, Spiegel I, Patrizi A, Centofante E, Bazinet JE, Tzeng CP, Mandel-Brehm C, Harmin DA, Adesnik H, Fagiolini M, et al. (2016). Sensory experience regulates cortical inhibition by inducing IGF1 in VIP neurons. Nature 531, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, and Smyth GK (2012). Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40, 4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meighan-Mantha RL, Hsu DK, Guo Y, Brown SA, Feng SL, Peifley KA, Alberts GF, Copeland NG, Gilbert DJ, Jenkins NA, et al. (1999). The mitogen inducible Fn14 gene encodes a type I transmembrane protein that modulates fibroblast adhesion and migration. J Biol Chem 274, 33166–33176. [DOI] [PubMed] [Google Scholar]
- Monavarfeshani A, Stanton G, Van Name J, Su K, Mills WA 3rd, Swilling K, Kerr A, Huebschman NA, Su J, and Fox MA (2018). LRRTM1 underlies synaptic convergence in visual thalamus. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JL, Berger DR, Wetzel AW, and Lichtman JW (2016). The Fuzzy Logic of Network Connectivity in Mouse Visual Thalamus. Cell 165, 192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narushima M, Uchigashima M, Yagasaki Y, Harada T, Nagumo Y, Uesaka N, Hashimoto K, Aiba A, Watanabe M, Miyata M, et al. (2016). The Metabotropic Glutamate Receptor Subtype 1 Mediates Experience-Dependent Maintenance of Mature Synaptic Connectivity in the Visual Thalamus. Neuron 91, 1097–1109. [DOI] [PubMed] [Google Scholar]
- Noutel J, Hong YK, Leu B, Kang E, and Chen C (2011). Experience-dependent retinogeniculate synapse remodeling is abnormal in MeCP2-deficient mice. Neuron 70, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat Methods [DOI] [PMC free article] [PubMed]
- Rafols JA, and Valverde F (1973). The structure of the dorsal lateral geniculate nucleus in the mouse. A Golgi and electron microscopic study. J Comp Neurol 150, 303–332. [DOI] [PubMed] [Google Scholar]
- Rall W (1970). Cable properties of dendrites and effects of synaptic location. In Excitatory Synaptic Mechanisms, pp. 175–187.
- Riccomagno MM, and Kolodkin AL (2015). Sculpting neural circuits by axon and dendrite pruning. Annu Rev Cell Dev Biol 31, 779–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, and Lichtman JW (1999). Development of the vertebrate neuromuscular junction. Annu Rev Neurosci 22, 389–442. [DOI] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seabrook TA, Krahe TE, Govindaiah G, and Guido W (2013). Interneurons in the mouse visual thalamus maintain a high degree of retinal convergence throughout postnatal development. Neural Dev 8, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sholl DA (1953). Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat 87, 387–406. [PMC free article] [PubMed] [Google Scholar]
- Singh R, Su J, Brooks J, Terauchi A, Umemori H, and Fox MA (2012). Fibroblast growth factor 22 contributes to the development of retinal nerve terminals in the dorsal lateral geniculate nucleus. Front Mol Neurosci 4, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel I, Mardinly AR, Gabel HW, Bazinet JE, Couch CH, Tzeng CP, Harmin DA, and Greenberg ME (2014). Npas4 regulates excitatory-inhibitory balance within neural circuits through cell-type-specific gene programs. Cell 157, 1216–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz CC, and Turrigiano GG (2010). Tumor necrosis factor-alpha signaling maintains the ability of cortical synapses to express synaptic scaling. J Neurosci 30, 14685–14690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, and Malenka RC (2006). Synaptic scaling mediated by glial TNF alpha. Nature 440, 1054–1059. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. [DOI] [PubMed] [Google Scholar]
- Su Y, Shin J, Zhong C, Wang S, Roychowdhury P, Lim J, Kim D, Ming GL, and Song H (2017). Neuronal activity modifies the chromatin accessibility landscape in the adult brain. Nat Neurosci 20, 476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe K, Bonilla I, Winkles JA, and Strittmatter SM (2003). Fibroblast growth factor-inducible-14 is induced in axotomized neurons and promotes neurite outgrowth. J Neurosci 23, 9675–9686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G (2012). Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol 4, a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonhoff F, and Keshishian H (2017). Activity-Dependent Synaptic Refinement: New Insights from Drosophila. Front Syst Neurosci 11, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, and Greenberg ME (2011). Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb Perspect Biol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyand TG (2016). The multifunctional lateral geniculate nucleus. Rev Neurosci 27, 135–157. [DOI] [PubMed] [Google Scholar]
- Wiesel TN, and Hubel DH (1963). Single-Cell Responses in Striate Cortex of Kittens Deprived of Vision in One Eye. J Neurophysiol 26, 1003–1017. [DOI] [PubMed] [Google Scholar]
- Wiley SR, and Winkles JA (2003). TWEAK, a member of the TNF superfamily, is a multifunctional cytokine that binds the TweakR/Fn14 receptor. Cytokine Growth Factor Rev 14, 241–249. [DOI] [PubMed] [Google Scholar]
- Winkles JA (2008). The TWEAK-Fn14 cytokine-receptor axis: discovery, biology and therapeutic targeting. Nat Rev Drug Discov 7, 411–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, and Regehr WG (2002). Short-term synaptic plasticity. Annu Rev Physiol 64, 355–405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.