

Abstract
Salmeterol is a partial agonist for the β2 adrenergic receptor (β2AR), and the first long-acting β2AR agonist (LABA) to be widely used clinically for the treatment of asthma and chronic obstructive pulmonary disease. Salmeterol has been controversial both for its safety and mechanism of action. To understand its unusual pharmacological action and partial agonism, we obtained the crystal structure of salmeterol-bound β2AR in complex with an active-state stabilizing nanobody. The structure reveals the location of the salmeterol exosite, where sequence differences between β1AR and β2AR explain the high receptor subtype selectivity. A structural comparison with the β2AR bound to the full agonist epinephrine reveals differences in the hydrogen bond network involving residues Ser 2045.43 and Asn 2936.55. Mutagenesis and biophysical studies suggest that these interactions lead to a distinct active-state conformation that is responsible for the partial efficacy of G protein activation and the limited β-arrestin recruitment for salmeterol.
Introduction
G-protein coupled receptors (GPCRs) are able to respond to a large variety of ligands with different efficacy profiles for specific signaling pathways1: full agonists that maximally stimulate; partial agonists that produce submaximal stimulation even at fully saturating concentrations; inverse agonists that suppress the basal signaling; and neutral antagonists that bind to the receptor without stimulating or inhibiting basal signaling. Among these categories, partial agonists are often better tolerated as therapeutics compared with full agonists2.
Salmeterol is a partial agonist for the human β2-adrenergic receptor (β2AR). As a potent bronchodilator, it is among the most prescribed drugs for the treatment of asthma and chronic obstructive pulmonary disease (COPD)3. Compared to some other β2AR agonists such as isoproterenol and salbutamol, salmeterol exhibits two desirable pharmacological properties. First, salmeterol is able to distinguish β2AR from β1AR for selective stimulation (1400- to 3000-fold selectivity)4, thereby minimizing cardiac toxicity5. Second, salmeterol belongs to the class of long-acting β2AR agonists (LABAs) with a duration of action up to 12 hours, in contrast to the short-acting β2AR agonists such as salbutamol with only a 4–6 hour duration of action6–8. Those pharmacological properties of salmeterol have contributed to its successful use in treating asthma and COPD for more than two decades. However, LABAs used alone, especially salmeterol, have been implicated, in several clinic trials, to be associated with increased mortality in asthmatics. This liability is mitigated when LABAs are combined with an inhaled corticosteroid9–11.
The high selectivity and long-acting properties of salmeterol have been attributed to its unusual bitopic structure. In addition to the saligenin ethanolamine pharmacophore that replaces the catecholamine structure of the endogenous β2AR ligand epinephrine (also known as adrenaline), salmeterol contains an extra moiety of an aryloxyalkyl tail consisting of a phenol ring with an eleven-atom ether chain (Fig. 1a). While the pharmacophore binds to the orthosteric site responsible for receptor activation, the aryloxyalkyl tail is proposed to bind to an additional site (exosite), providing additional interactions responsible for the high receptor-subtype selectivity and the slow dissociation rate contributing to its long duration of action12. Previous mutagenesis and biochemical studies to locate the exosite have led to conflicting results4,12–16. Salmeterol is also of interest in being a functionally selective β2AR partial agonist with a 5- to 20-fold bias towards Gs over arrestin17,18. Previous studies revealed that, compared to isoproterenol, activation of the β2AR by salmeterol leads to slower initial rates of G protein-coupled receptor kinase (GRK) phosphorylation with similar maximal degrees of phosphorylation19,20, but strongly reduced arrestin-mediated receptor internalization and desensitization17,21,22, contributing to the prolonged therapeutic effect of salmeterol in bronchial dilation as a result of β2AR stimulation. This signaling bias may contribute to the advantageous therapeutic profile of salmeterol by maintaining bronchodilation through Gs-mediated signaling while minimizing arrestin-mediated β2AR desensitization, and avoiding arrestin-dependent pro-inflammatory effects23,24.
Figure 1: Crystal structure of salmeterol-bound β2AR.
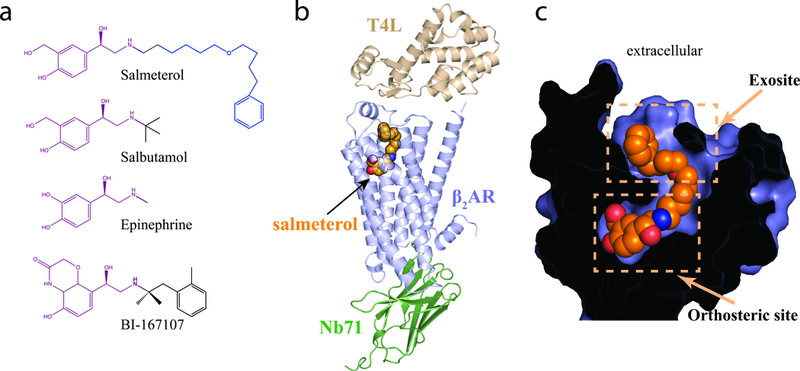
a.Chemical structures of β2AR ligands: partial agonists salmeterol and salbutamol; full agonists epinephrine and BI-16707. The respective pharmacophores that bind the orthosteric ligand binding pocket are highlighted in purple, and the aryloxyalkyl tail of salmeterol, which binds the exosite, is highlighted in blue. b. Overall ribbon representation of the salmeterol-bound β2AR – Nb71 complex structure. The T4L lysozyme fusion facilitates crystallization, while Nb71 stabilizes the active, salmeterol-bound (orange spheres) β2AR. c. Cross-section through the receptor, with the interior in black, highlighting salmeterol (orange spheres) occupying the orthosteric site and the exosite.
Recent progresses in the structural characterization of β adrenergic receptors, especially the crystal structures of active β2AR bound to full agonists BI-167107 or epinephrine (also known as adrenaline), with either a Gs protein or active-state stabilizing nanobody, as well as the crystal structures of inactive avian β1AR bound to a variety of full and partial agonists, have greatly advanced our understanding of the pharmacology and activation of β adrenergic receptors25–29. In an effort to further understand the molecular basis for the unusual pharmacological properties of salmeterol and its partial agonism, we obtained the crystal structure of salmeterol-bound human β2AR in an active state stabilized by a conformation-specific nanobody, Nb71. The structure reveals the location of the exosite and provides a structural explanation for the high receptor-subtype selectivity and distinct signaling behavior of salmeterol.
Results
Nanobody Nb71 stabilizes the salmeterol-bound β2AR
It has been shown for the β2AR and the μ-opioid receptor (μOR) that agonists alone are not sufficient to stabilize the receptors in active conformational states27,30–32. As a result, the active-state structures of the β2AR and μOR have required either a G protein or conformation-specific camelid antibody fragments (nanobodies) to stabilize the active states of the receptors25,26,29. The first active-state structure of a hormone-activated GPCR was obtained using a nanobody named Nb80, which was obtained from a llama immunized with β2AR bound to the ultra high-affinity full agonist BI-167107 reconstituted into phospholipid vesicles25. The structures of β2AR in the β2AR-Nb80 complex and the β2AR-Gs complex are very similar25,26. However, evidence from biophysical studies suggest that partial agonists may stabilize distinct states33, so Nb80 may not be the best candidate to stabilize salmeterol-bound β2AR.
While Nb80 preferentially binds to agonist-occupied β2AR over antagonist-occupied β2AR25, Octet Red studies show that Nb80 also preferentially binds to β2AR bound to BI-167107 and epinephrine over β2AR bound to salmeterol (Supplementary Table 1). Moreover, Nb80 has a greater effect on enhancing the binding affinity of the agonist isoproterenol than on the partial agonist salmeterol (Supplementary Fig. 1). We therefore selected another nanobody, Nb71, generated from the same immunization that produced Nb80. Nb71 preferentially binds to agonist-occupied β2AR34, but in contrast to Nb80, has no preference for these catecholamines over salmeterol (Supplementary Table 1, Supplementary Fig. 1). Therefore, we chose Nb71 to stabilize the salmeterol-bound β2AR for structural characterization.
Structural features of Nb71-stabilized β2AR
We used an engineered β2AR with T4 lysozyme (T4L) fused to its N-terminal region and a truncated intracellular loop3 (referred to as T4L-β2AR-ΔICL3 hereafter) to crystallize β2AR bound to salmeterol. Previous studies have demonstrated that T4L-β2AR-ΔICL3 exhibits similar ligand binding and G protein activating properties as the wild type β2AR37. Using this construct, we obtained crystals of the β2AR-salmeterol-Nb71 complex by the lipidic cubic phase (LCP) method35. A complete data set was obtained by merging data from 23 crystals and the structure was determined to a resolution of 3.0 Å (Fig. 1b,c; Supplementary Table 2). Like Nb80, Nb71 binds to the cytoplasmic surface of the receptor with its third complementarity-determining region (CDR3) loop inserted into a hydrophobic cavity formed by residues from transmembrane helices (TMs) 3, 5, 6 and 7 (Fig. 1b, Supplementary Fig. 2), yet Nb71 stabilizes a conformation that is distinct from that stabilized by Nb80 (Supplementary Fig. 2).
The active-state conformation of the receptor in the β2AR-salmeterol-Nb71 complex resembles that of the BI-167107-bound β2AR in complex with Nb80 (PDB ID 3P0G, rmsd = 1.0 Å) or with Gs (PDB ID 3SN6, rmsd = 1.3 Å) more than the inactive-state conformation of the receptor in the inverse agonist carazolol-bound β2AR structure (PDB ID 2RH1, rmsd = 1.9 Å)25,26. Indeed, the structural features associated with receptor activation25, including the outward movement of TM6 at the cytoplasmic side associated with the conformational changes of the core triad residues Pro 2115.50, Ile 1213.40 and Phe 2826.44, as well as the slightly contracted ligand-binding pocket at the extracellular side, are all observed in the structure of β2AR-salmeterol-Nb71 complex when compared to the inactive β2AR (Fig. 2a-c). However, the Nb71-stabilized β2AR shows some distinct structural features compared to the Nb80- and Gs-stabilized β2AR. Relative to the inactive β2AR, the Nb71-stabilized β2AR displays a smaller outward movement of the cytoplasmic end of TM6 (8Å) than the Nb80-stabilized β2AR (11Å) or Gs-stabilized β2AR (13Å) (Fig. 2a). There is also a slightly smaller counter-clockwise rotation of TM6 in the Nb71-stabilized β2AR when viewed from the cytoplasmic surface, as shown by the position of Glu268 (Fig. 2b). While this current structure clearly shows an active-state conformation of β2AR, the smaller conformational rearrangements observed for TM6 upon activation compared to the changes seen for the active-state stabilized β2AR bound to Nb80 or Gs suggest a less active or a partially active conformational state of β2AR stabilized by Nb71. It is tempting to speculate that such partially active conformational state may closely resemble a salmeterol-stabilized conformation of β2AR that is less efficient at coupling to Gs. However, the conformation of TM6 is primarily stabilized by interactions with Nb71 and may not reflect the conformation stabilized by salmeterol alone.
Figure 2: Structural features of the β2AR-salmeterol-Nb71 complex.
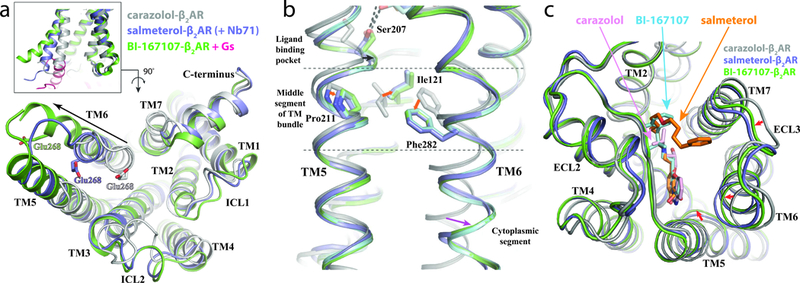
a,b,c. Overlay of inactive carazolol bound receptor (grey), Nb71 and salmeterol bound β2AR (purple) and the BI-167107 bound β2AR - Gs complex (green and pink, respectively). a. Side view (inset) and cytoplasmic view of TM6, showing the position of TM6 relative to the helical bundle. The extent of TM6 movement, indicated by black arrows, was measured using the Cα of Glu268 as reference point. b. Rearrangement of Pro2115.50, Ile1213.40 and Phe2826.44 and the associated outward movement of the cytoplasmic end of TM6. The inactive structure of β2AR is colored silver. The active structures of β2AR are colored blue (coupled with salmeterol and Nb71), cyan (coupled with epinephrine and 6B9) and green (coupled with BI-167107 and Gs). c. Extracellular (top) view of β2AR, with major conformational changes between structures indicated by red arrows and ligands in stick representation.
Exosite binding of salmeterol
The location and the molecular details of the exosite for the aryloxyalkyl tail of salmeterol are of great interest because of its association with the high receptor selectivity, high affinity and long-lasting action properties of salmeterol. The clear electron density map of salmeterol based on our structure allowed us to unambiguously define the structural basis of the exosite (Figs. 3a,b & Supplementary Figs. 3,4). The crystal structure shows that the aryloxyalkyl tail of salmeterol extends towards the extracellular surface of the receptor, occupying a cleft formed by residues from extracellular loop2 (ECL2), ECL3 and the extracellular ends of TM6 and TM7. Interactions between salmeterol and the exosite are mediated primarily through extensive van der Waals and hydrophobic interactions (Fig. 3c). The phenol ring of the tail also forms π-π interactions with the surrounding aromatic residues Phe 194ECL2, Tyr 3087.35 and His 2966.58 (Ballesteros-Weinstein numbers), in agreement with previous studies reporting a 5- to 18-fold decrease of salmeterol affinity caused by mutating those residues4. In addition, the ether oxygen atom of the tail forms a hydrogen bond with the main chain amide group of residues Phe 193 ECL2 (Fig. 3a). It acts as a ‘hinge’ point where the tail of salmeterol bends almost 90° to fit the exosite. Previous studies indicated that shifting the ether oxygen in the aryloxyalkyl tail or removing it substantially reduces the affinity of salmeterol for β2AR21, suggesting an important role of this hydrogen bond in the exosite binding (Supplementary Fig. 5). All those additional interactions in the exosite contribute to the 1000-fold higher affinity of salmeterol compared to salbutamol, a short-acting β2AR agonist (SABA) which shares the same orthosteric pharmacophore as salmeterol but lacks the long aryloxyalkyl tail36 (Fig. 1a).
Figure 3: Salmeterol exosite and receptor subtype-selectivity determinants.
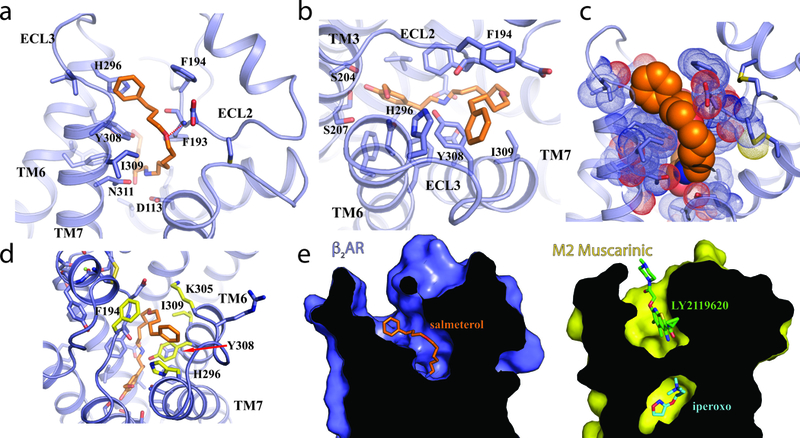
a. Side and b. top view of β2AR, with stick representation of salmeterol (orange). c. Spherical representation of the salmeterol aryloxyalkyl moiety (orange) and the β2AR (blue). d. Residues important for interaction and specificity are highlighted in yellow and labeled. With the exception of Y308 (indicated by a red arrow), the amino acids that form the orthosteric binding pocket for epinephrine are identical between β1AR and β2AR. e. Comparison of the ligand binding sites between β2AR (left panel) bound to salmeterol (orange sticks) and M2R (right panel) bound to the allosteric modulator LY2119620 (green sticks).
The exosite binding also explains the very high selectivity of salmeterol for β2AR over β1AR (>3000-fold)36. β1AR and β2AR share a very high overall structural similarity (rmsd of carazolol-bound avian β1AR and human β2AR = 0.58 Å, PDB IDs 2YCW & 2RH1, respectively) and sequence similarity (92% in TM segments for human β1AR and β2AR, Supplementary Fig. 6). In particular, all residues that form the orthosteric binding pocket, with the exception of Y308 are identical in β1AR and β2AR. In contrast, the exosite is relatively divergent in those two receptors. Salmeterol achieves high receptor selectivity by sampling this divergent region with its long aryloxyalkyl tail (Fig. 3d, Supplementary Fig. 6). Such selectivity determinants do not apply to salbutamol, which only has about 20-fold selectivity for β2AR over β1AR36. Our results are in agreement with extensive chimeric receptor and site-directed mutagenesis studies recently reported by Baker et al.7, who observed the K305D and H296K mutations in the β2AR to have a substantial effect on salmeterol affinity while having little effect on salbutamol affinity.
The salmeterol exosite in β2AR is reminiscent of the well-defined allosteric site in the M2 muscarinic receptor (M2R)37. The M2R represents one of the most extensively characterized model systems for allosteric regulation. Fig. 3e shows a comparison of the exosite in the β2AR and the binding site for the positive allosteric modulator LY2119620 revealed by the active-state crystal structure of M2R bound to the orthosteric agonist iperoxo and LY211962037. The similarity between those two sites suggests the potential for allosterically regulating β2AR activity by small molecules that target the exosite. On the other hand, the bitopic nature of salmeterol with two-site binding on β2AR supports the feasibility of developing highly selective bitopic compounds for other GPCRs including muscarinic receptors38.
Polar interactions within the orthosteric binding site
While the interactions between the aryloxyalkyl tail of salmeterol and the exosite in the extracellular vestibule of β2AR confer the high affinity and selectivity of salmeterol, they are not responsible for its efficacy and signaling bias. Salmeterol and salbutamol exhibit similar signaling properties including partial agonism and the selective activation of Gs over arrestin and Gi (Supplementary Fig. 7)17, despite their differences in affinity and receptor selectivity. Considering that salmeterol and salbutamol only share the saligenin ethanolamine pharmacophore (Fig. 1a), the interactions between this shared pharmacophore and the receptor within the orthosteric binding pocket are likely to be responsible for their shared signaling properties. In the orthosteric binding site, the alkylamine and β-hydroxyl groups of salmeterol form hydrogen-bonding interactions with Asp 1133.32 and Asn 3127.39 and the saligenin group forms hydrogen-bonding interactions with Ser 2035.42 and Ser2075.46, similar to those observed for β2AR bound to the full agonist epinephrine (Figs. 4a,b). In the structure of β2AR bound to epinephrine, the hydrogen bonding interactions with Ser 2035.42 and Ser2075.46 are associated with the inward movement of TM5 around those two serine residues, which is further linked to the rearrangement of the core triad residues and the outward movement of the cytoplasmic end of TM629. For salmeterol, the two saligenin hydroxyl groups also form direct hydrogen bonding interactions with Ser 2035.42 and Ser 2075.46. However, compared to epinephrine, these interactions with salmeterol may have a weaker effect on stabilizing the inward movement of TM5 because of the additional methylene between the meta position hydroxyl group and the phenyl ring.
Figure 4: Hydrogen bond interactions in the orthosteric site and re-arrangement of the ligand binding pocket.
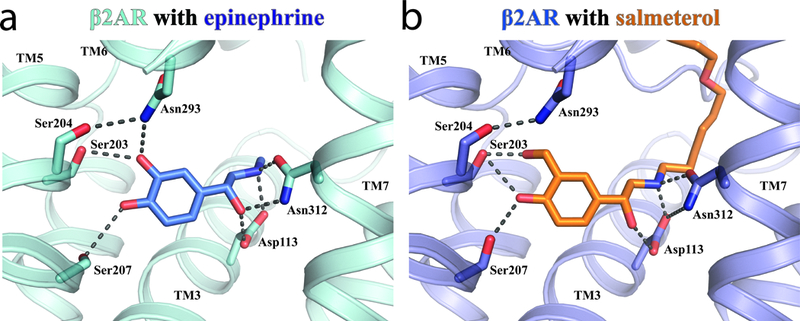
Comparison of ligand-mediated hydrogen bonds (black dashed lines) in a. epinephrine-bound and b. salmeterol-bound β2AR.
In epinephrine-bound β2AR, Asn 2936.55 forms a hydrogen bond with Ser 2045.43 and the meta-hydroxyl of epinephrine. This polar network is not observed in salmeterol-bound β2AR. Asn 2936.55 and Ser 2045.43 have previously been shown to be important for the binding of epinephrine but not for the binding of antagonists39,40. To further investigate the possible role of these polar networks in ligand efficacy, we used molecular dynamics simulations to characterize the stability of specific ligand-receptor hydrogen bonds that would be expected to stabilize the inward movement of TM5 and the rotation of TM6 observed in active state structures of the β2AR (Fig. 5a). These simulations show that salmeterol forms less stable hydrogen bonds with Asn2936.55 and Asn3127.39 than does epinephrine. Salmeterol forms a more stable hydrogen bond with Ser2035.42, but apparently only because its longer hydroxymethyl group can maintain this hydrogen bond better than epinephrine’s hydroxyl group even when TM5 moves away from its crystallographic position. Thus, while the salmeterol-bound crystal structure captured a binding pocket conformation similar to that observed for full agonist, molecular dynamics simulations suggest that this active conformation is stabilized less by salmeterol than by epinephrine, with the most dramatic difference observed for Asn2936.55. To further characterize the role of Asn 2936.55 and Ser 2045.43 interactions, we examined the effects of mutating these residues on β-arrestin2 recruitment and Gs activation by bioluminescence resonance energy transfer (BRET) between membrane anchored GFP and luciferase-tagged arrestin or luciferase-tagged Gs and GFP-tagged Gβ1/Gγ1, respectively41,42(Fig. 5b,c & Supplementary Table 3). For all the mutations we tested in the presence of isoproterenol, a catecholamine similar to epinephrine, we observe a dramatic reduction in β-arrestin2 recruitment (Fig. 5b, left panel), and a more moderate reduction in Gs activation (Fig. 5c, left panel). This suggests an important role of these two residues in regulating bias, either by directly modulating arrestin coupling, or by altering GRK phosphorylation and hence arrestin binding. Interestingly, the same mutations, with the exception of S204T, had a less pronounced effect on the ability of salmeterol to activate Gs (Fig. 5c, right panel), and all mutants showed very little or no salmeterol-induced arrestin recruitment, similar to wild-type β2AR (Fig. 5b, right panel). Thus, the absence of the hydrogen bonds between Asn 2936.55 and salmeterol may account for the weak β-arrestin recruitment, as well as the lower efficacy in Gs activation of salmeterol-bound β2AR compared to β2AR bound to full agonist.
Figure 5: Ligands and specific residues in the orthosteric site modulate hydrogen bonding and signaling outcome.
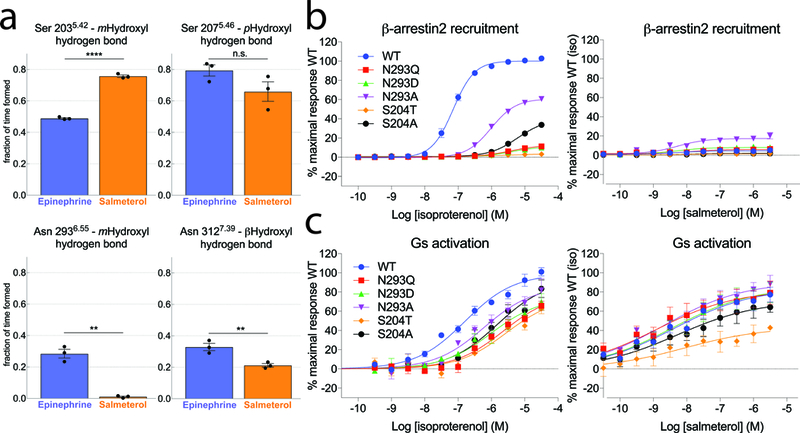
a. Frequency of hydrogen bonding between Ser2035.42 / Asn2936.55 and the ligand m-OH group, Ser2075.46 and the ligand p-OH group, and Asn3127.39 and the ligand β-OH group in MD simulations of receptor bound to epinephrine or salmeterol. Individual datapoints (black dots) and SEM (error bars) of 3 independent experiments are shown. Statistical significance was assessed using the one-side Welch’s unequal variances t-test (p values: Ser2035.42=0.00003; Ser2075.46=0.08; Asn2936.55=0.004; Asn3127.39=0.01). The actual statistical significance of the differences (e.g. for Ser207) may be better than computed, as each of the data points is based on a trajectory with thousands of samples. b,c. BRET-based assays monitoring β-arrestin2 recruitment and G-protein activation on wild-type and mutant β2AR constructs. Data represent the mean (symbols) ± SEM (error bars) of 3 independent experiments. Error bars shorter than the height of the symbol are not shown.
Spectroscopic insights into salmeterol-bound β2AR
The differences in efficacy and signaling bias between salmeterol and the full agonist epinephrine suggest that salmeterol may stabilize a distinct conformation. Previous single molecule Förster resonance energy transfer (FRET) studies suggested a smaller outward displacement of TM6 in β2AR bound to salmeterol compared to β2AR bound to epinephrine or BI16710743. In addition, cellular assays using a BRET-based β2AR conformational sensor44 confirmed the reduced propensity of salmeterol to promote the outward movement of TM6 (Supplementary Fig. 8). Nevertheless, these RET studies cannot distinguish the following two mechanistic possibilities: 1- salmeterol stabilizes the same conformation of TM6 as epinephrine, but for a smaller receptor population; 2- salmeterol stabilizes a distinct TM6 conformation. To further investigate the differences at the cytoplasmic end of TM6 upon activation between salmeterol-bound and epinephrine-bound β2AR in the absence of conformation-stabilizing nanobodies, we performed steady-state spectroscopic studies on purified, labeled receptor in detergent with two different reporter systems (Supplementary Fig. 9a). We used β2AR labeled at Cys 265 with the fluorophore monobromobimane (mBBr or bimane), which we previously used as a conformational reporter of β2AR activation31,45, and developed a new fluorescent reporter system using an engineered β2AR labeled at residue 266 with the fluorophore Atto655.
The bimane fluorophore has been extensively used to report on activation of β2AR25,45,46 as well as other GPCRs47,48, as it is very sensitive to its chemical environment. In the inactive state, bimane attached at the cytoplasmic end of TM6 is in a hydrophobic environment. Upon receptor activation, the intracellular end of TM6 undergoes an outward and “unwinding” motion (Fig. 6a) that shifts bimane to a more polar and solvent-exposed environment, resulting in a decrease in fluorescence intensity and a red-shift in λmax. Agonists alone produce a 10–20% decrease in intensity and ∼10 nm red-shift in λmax. Further changes are observed upon G protein coupling. Using this reporter system, we find that even though salmeterol has a higher binding affinity to β2AR than epinephrine6, epinephrine causes a larger reduction in intensity and red-shift in λmax of bimane compared to salmeterol when both ligands are used at saturating concentrations (Fig. 6b). This suggests that salmeterol stabilizes β2AR in conformations with on average a smaller outward movement or rotation of TM6. In addition, we observe that Gs induces a similar bimane response with salmeterol as with epinephrine (Fig. 6b, “+Gs” curves), indicating that once coupled to Gs, the β2AR adopts a similar TM6 conformation no matter what agonist is bound at the extracellular region, consistent with previous single molecule FRET studies33. Nevertheless, the extent to which salmeterol-bound receptor couples to Gs appears to be slightly lower, as the change in λmax and fluorescence intensity is slightly less pronounced compared to epinephrine-bound receptor.
Figure 6: Spectroscopic interrogation of ligand-induced changes in TM6 conformation on detergent-solubilized, purified labeled receptor.
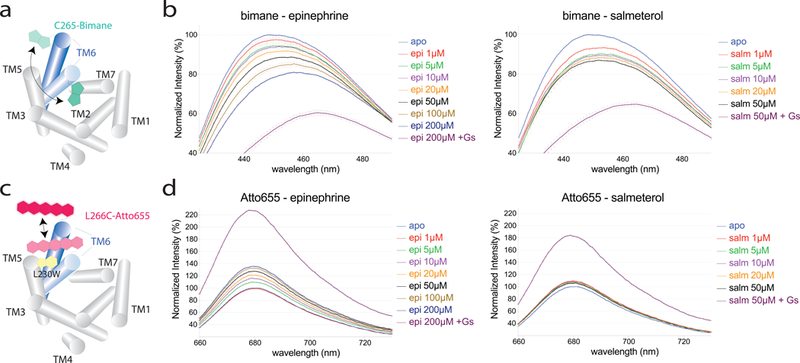
a,c. Schematic representation of labeled receptor constructs used to probe the outward motion of TM6. The β2AR transmembrane helices 1–5 and 7 are shown as grey cylinders. The blue cylinders represent TM6 in the inactive (light blue) and active (dark blue) conformation, with the labels attached at the intracellular end of TM6 depicted according to their overall structure. The bimane fluorophore (teal) is an environment-sensitive reporter, while the Atto655 dye (pink) is quenched in a distance-dependent way by an engineered tryptophan (L230W, in yellow). b,d. Steady-state fluorescence emission spectra of bimane-labeled and Atto655-labeled receptor in the presence of epinephrine or salmeterol. The spectra are normalized relative to unliganded receptor (apo, light blue curve) and show the fluorescence dose-response of receptor up to saturating ligand concentrations, in the absence and presence of G protein. Data is plotted as mean (curves) and standard deviation (error bars) of triplicate measurements.
To complement the bimane-based measurements, we developed a new, distance-sensitive fluorescent reporter. Rather than relying on FRET, where stoichiometric and specific labeling of donor and acceptor is required to faithfully report on distance changes in bulk measurements, we employed a single-dye reporter system. In photo induced electron transfer (PET), fluorophores such as Atto655 can be quenched by a tryptophan in close proximity, through the formation of weakly fluorescent or non-fluorescent dye-tryptophan ground-state complexes49 (Fig. 6c). Owing to the strong distance-dependence of PET, small-scale conformational changes result in “on–off” switching of fluorescence, yielding a change in fluorescence intensity but no shift in λmax over a small distance range, typically about 5–10Å49.
To report only on the outward motion of TM6, we chose to label Leu 266 mutated to cysteine instead of the native Cys 265, as the former does not undergo inward to outward rotations upon activation, but points outwards in both active and inactive structures (Supplementary Fig. 9a). Based on the inactive and active β2AR crystal structures and dye simulations (Supplementary Fig. 9b), we speculated that a tryptophan residue introduced at the intracellular end of TM5 (L230W) would quench the fluorescence of Atto655 in the inactive, but considerably less in the active state, as the distance range and change between TM5 and TM6 that occurs upon activation should be compatible with PET-induced quenching. By simulating the conformational ensembles of Atto655 bound to Cys 266 in the inactive and active β2AR structures, we found an average dye-Trp 230 distance change of about ∼5 Å upon receptor activation (Supplementary Fig. 9b), compatible with the reported quenching distance. We thus introduced mutations L230W and L266C into a minimal cysteine background β2AR33 and verified that the detergent-purified, Atto655-labeled β2ARΔ6 L230W:L266C construct retained wild-type ligand binding properties (Supplementary Fig. 10).
When measuring the steady-state fluorescence emission of Atto655-labeled β2ARΔ6 L230W:L266C, referred to as Atto-β2AR from here on, in the presence of saturating concentrations of epinephrine, we observe a ∼30% increase in fluorescence intensity compared to unliganded receptor, compatible with an outward motion of TM6 and an increased distance between Atto655 and the Trp 230 quencher on TM5 (Fig. 6d, Supplementary Fig. 11a). Given the short distance over which PET-quenching occurs, this observation likely reflects a TM6 displacement of ∼5Å or more in approximately 30% of epinephrine-bound receptor. This is consistent with the fraction of receptor in an active conformation observed in double electron-electron resonance spectroscopy studies31. Interestingly, we observed a much smaller difference (<10%) between unliganded and salmeterol-bound receptor (Fig. 6d, Supplementary Fig. 11a), suggesting that TM6 did not move sufficiently far away from TM5 in salmeterol-bound β2AR to reduce Atto655 quenching by Trp230. Based on the efficacy of salmeterol in G protein activation (60% of isoproterenol, Supplementary Fig. 7c) we would expect a much larger change in Atto-β2AR fluorescence in response to salmeterol if salmeterol and isoproterenol stabilized the same active state. Addition of Gs to salmeterol- and isoproterenol-bound Atto-β2AR led to a 1.8- and 2.2-fold increase in fluorescence intensity, respectively, compared to unliganded receptor (Fig. 6d, “+Gs” curves). This is consistent with the G protein stabilizing an active conformation in a larger fraction of receptor. Control measurements on a construct without the engineered L230W mutation showed little intensity change upon addition of agonists alone or together with Gs (Supplementary Fig 11b). In line with the difference in TM6 outward motion between the Nb80- and Nb71-bound structures, the Atto response in the presence of Nb80 is much larger than in the presence of Nb71, both for epinephrine and salmeterol (Supplementary Fig. 11c). This is similar to what we observed in the presence of Gs (Fig. 6b,d): when both ligand and Gs or nanobody are present, the TM6 receptor conformation is mainly stabilized by the intracellular binding protein and not the ligand. However, both of our fluorescence-based approaches in the absence of nanobody clearly suggest a difference in TM6 conformation between epinephrine- and salmeterol-bound receptors.
Discussion
β2ARs expressed in airway smooth muscle and epithelial cells mediate bronchodilation and fluid clearance, respectively, and are thus well-established targets for the treatment of asthma and COPD50. The unusual pharmacological characteristics of salmeterol including the extremely high selectivity for β2AR and the long duration of action, which can be attributed to its long aryloxyalkyl tail, make it among the most commonly prescribed LABAs for treating asthma and COPD. Our structure revealed an additional site in the extracellular vestibule of β2AR, the exosite, for the binding of the aryloxyalkyl tail, thus providing a structural basis for the prominent pharmacological action of salmeterol. It also resolves a long-standing debate as to the location of the exosite.
Our results also provide structural insights into the partial agonism and the biased signaling property of salmeterol, which are attributed to its saligenin ethanolamine group bound in the orthosteric binding pocket. Although the interactions with the receptor in the orthosteric binding pocket are very similar for the full agonist epinephrine and the partial agonist salmeterol, we observe subtle differences in hydrogen bonding interactions. Most notable is the absence of interactions between salmeterol and Asn 2936.55, which is further supported by molecular dynamics simulations. The meta-hydroxyl of epinephrine forms a hydrogen bond with Asn 2936.55, which is also hydrogen bonded with Ser 2045.43. Our mutagenesis studies suggest that this hydrogen bond network may be important for stabilizing TM6 in a conformation necessary for efficient G protein coupling as well as GRK phosphorylation and/or arrestin coupling. Therefore, the less extensive polar interactions between salmeterol and Asn 2936.55 may contribute to the weaker efficacy of salmeterol in activating Gs and the near absence of β-arrestin recruitment, possibly due to inefficient coupling as a result of reduced GRK phosphorylation19.
The structure of salmeterol-bound β2AR revealed a smaller outward movement of TM6 compared to the epinephrine-bound β2AR. While this conformation implies a ‘partially active’ conformation of the receptor, it is likely imposed by the nanobody Nb71 rather than by the partial agonist salmeterol. Previous studies have suggested relatively weak allosteric coupling between the orthosteric site and the cytoplasmic site31. Thus it is difficult to capture the ligand-specific conformation of β2AR by protein crystallography. We took spectroscopic approaches using two different reporter systems to interrogate the conformational changes of β2AR associated with salmeterol, and showed that indeed TM6 did not achieve the same extent of outward motion compared to the full agonist epinephrine. This correlates with previous single-molecule investigations of TM6 motion in the β2AR33, and provides a structural basis for salmeterol’s weaker Gs efficacy and possibly ligand bias. Whether this mechanism applies to other GPCRs needs further investigation.
Online Methods
Receptor Constructs
All constructs have an N-terminal Flag tag. The T4L-β2AR-ΔICL3, consists of an N-terminal T4 lysozyme fusion to β2AR(29–365) ΔICL3, i.e. Δ(235–263) with mutations M96T, M98T and N187E. The β2AR-PN1 consists of β2(1–24) – TEV – β2(25–365) – 3C – β2(366–413), with “TEV” and “3C” indicating TEV protease and 3C protease cut sites, respectively. Additionally, mutations M96T, M98T, C378A, N187E and C406A were introduced, as previously described51. The β2ARΔ6 L230W:L266C consists of full-length WT receptor with a minimal cysteine background33 (C77V, C265A, C327S, C341L, C378A, C406A), as well as mutations L230W and L266C and a C-terminal hexahistidine tag.
Expression, purification and labeling of β2AR constructs
Receptor constructs were expressed in Sf9 insect cell cultures infected with recombinant baculovirus (BestBac, Expression Systems), and solubilized in n-dodecyl-β-d-maltoside (DDM) according to methods described previously51. The solubilized receptor was purified through M1 FLAG chromatography followed by alprenolol-sepharose chromatography to remove non-functional receptor51. A second M1 FLAG chromatography was applied such that the receptor-bound alprenolol could be removed for unliganded protein or exchanged for salmeterol.
For the T4L-β2AR-ΔICL3, after the protein was eluted from the M1 resin, the FLAG epitope tag of T4L-β2AR-Δ-ICL3 was removed by the treatment of tobacco etch virus (TEV) protease (Invitrogen) for 3h at room temperature or overnight at 4°C. When necessary, a 50kDa MWCO Vivaspin concentrator (GE Healthcare) was used to concentrate the receptor. The β2AR constructs used for fluorescence studies were concentrated and flash-frozen in the presence of 20% glycerol at a final concentration of 200μM. Aliquots were then stored at –80 °C until use.
Full-length PN1–β2AR was labeled with monobromobimane as previously described52. Briefly, FLAG pure receptor (∼2μM) was incubated overnight on ice with 100μM TCEP and 20μM monobromobimane (Thermo Fisher Scientific). Subsequently, 5mM cysteine was added to quench the labeling reaction followed by alprenolol-sepharose chromatography as described above. A similar procedure was followed for labeling β2ARΔ6 L230W:L266C with Atto655-iodoacetamide (Atto-TEC).
G protein expression and purification
Wild-type Gs heterotrimer was expressed and purified as previously described33. Following purification, the protein was dialyzed into 20mM HEPES pH 7.5, 100mM NaCl, 0.02%DDM, 100μM TCEP and 20μM GDP, concentrated, and flash-frozen in the presence of 20% glycerol at a final concentration of 200μM. Aliquots were then stored at –80 °C until use.
Preparation of Nb71 and Nb80
The recombinant Nb71 was generated in the same way as Nb8025. These two nanobodies were screened from a same library of single-chain nanobody clones after immunizing a llama with purified agonist-bound β2AR reconstituted at high density into phospholipid vesicles. Nb71 was expressed in E. coli and purified by nickel affinity chromatography in a same manner as Nb8025. The protein was then further purified by cation exchange using a Mono-S column (GE Healthcare), loading the protein at 20mM NaCl in 20mM MES pH 6.0 and eluting with a linear gradient from 50 to 500mM NaCl. In order to minimize severe precipitation of Nb71 over time, the purified protein was stored at a concentration below 5 mg/mL in 20mM Hepes, 1M NaCl.
Crystallization
Salmeterol bound receptor (~40mg/ml) was incubated with a 5.5-fold molar excess of Nb71 (~50mg/mL) for 1 hour on ice. Size exclusion chromatography was then performed to remove free Nb71. The purified complex was concentrated to ~60mg/mL for crystallization using the lipid cubic phase (LCP) method as previously described35. The protein complex was reconstituted in monoolein containing 10% cholesterol at a 1:1.5 protein to lipid ratio (w/w). Reconstituted protein-lipid mixture drops of 30 nL were deposited in each well of a 96-well glass sandwich plate (Molecular Dimensions). The drop was then overlaid with 650nL of precipitant and the wells sealed with a glass coverslip. Diffraction-quality crystals were grown at 20°C in 31–34% PEG 400, 100mM HEPES pH 7.5, 1% 1,2,3-heptanetriol following 3 days of incubation at 20°C.
Data collection, structure determination and analysis
Crystals were harvested and frozen in liquid nitrogen directly without using additional cryo-protectant. Diffraction data from 24 different crystals were measured using the GM/CA-CAT microfocus beam at 23-ID-D (Advanced Photon Source, Argonne National Labs). The data were processed with HKL2000 and the structure solved by molecular replacement using Phaser. Further model rebuilding was performed by using Coot and the structure was refined with Phenix. The validation of the final structural model was performed using Molprobity. The overall MolProbity score is 1.35. For the Ramachandran analysis, 96.8% atoms are in Ramachandran favored regions and 3.2% atoms are in Ramachandran allowed regions. Data processing and refinement statistics are shown in Supplementary Table 2.
The structure of the inactive β2AR (PDB ID 2RH1), the β2AR complexed with Nb80 (PDB ID 3P0G), the β2AR complexed with Gs (PDB ID 3SN6), the chain A of sabultamol bound β1AR (PDB ID 2Y04) were used for structural alignments in PYMOL based on Cα only. The secondary structure of β2AR ICL2 was assigned by PYMOL and DSSP.
Reconstitution of β2AR in HDL particles for Octet Red measurements
The β2AR was reconstituted in high-density lipoprotein (HDL) particles using established methods51. The scaffold protein ApoA-I used for reconstitution was purified as previously described51. Purified ApoA-I was biotinylated for 30 min at room temperature using NHS-PEG4-biotin (Thermo) at a 1:1 molar ratio in a buffer composed of 20 mM HEPES, pH 8.0, 100mM NaCl, 1mM EDTA, 5 mM sodium cholate. Following the labeling reaction, unreacted biotin was quenched by the addition of Tris-HCl, pH 8.0 to a final concentration of 20 mM. Biotinylated protein was separated from free biotin by size exclusion chromatography on Superdex 75 HR 10/30. FLAG-tagged β2AR was incorporated into HDL particles using biotinylated ApoA-I and receptor-containing HDL particles were isolated using M1 anti-FLAG immunoaffinity chromatography.
Nanobody binding to β2AR in biotinylated HDL particles was monitored using the OctetRED biolayer interferometry system (Pall FortéBio) using streptavidin-coated biosensors (Pall FortéBio). Sensors were hydrated for at least 10 min at room temperature in assay buffer (20 mM Tris-HCl pH 7.4, 136 mM NaCl, 2.7 mM KCl, 1 mM EDTA, 0.02% w/v ascorbic acid, 0.05% w/v BSA), then incubated with biotinylated β2AR-HDL (~100 nM) for 10 min at room temperature prior to loading the sensors onto the OctetRED instrument. All steps on the OctetRED were performed at 25 °C with the assay plate shaking at 1,000 rpm. After an initial baseline reading, sensors were dipped into wells containing assay buffer with a saturating concentration of agonist (100μM Epinephrine, 0.1μM BI-167107, or 1μM Salmeterol) and incubated for 20 min to equilibrate β2AR with agonist. The sensors were transferred to wells containing assay buffer plus agonist and nanobody (1, 3.2, 10, 32, 100, or 320 nM) for 5 min to monitor nanobody association. Nanobody dissociation was monitored by then transferring the sensors to wells containing assay buffer plus agonist for 30 min. Nonspecific nanobody binding at each concentration was measured in a parallel experiment in which sensors were loaded with empty HDL. Buffer-only controls were also included in each experiment to correct for baseline drift. Data were first analyzed using Octet Data Analysis 7.0 software (Pall FortéBio) to remove baseline and nonspecific binding, and the processed data were exported to Prism 6 (GraphPad) for curve fitting. All association and dissociation curves were fit using single-phase exponential association or decay.
BRET measurements for Gs activation, β-arrestin recruitment and β2AR conformational change in live cells.
HEK293T cells used for the BRET assays were grown in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% newborn calf serum (NCS), at 37 ⁰C with 5% CO2. Cells were detached with trypsin-EDTA and transfected with 2.5 μg of total DNA per 106 cells, using linear polyethyleneimine (PEI, Polysciences) as transfecting agent, at a ratio of 3:1 (PEI:DNA). Gs activation was evaluated with Gs-117-RLucII/Gβ1/Gγ1-GFP1041 and β-arrestin recruitment with CAAX-rGFP/β-arr2-RLucII42 sensors, in presence of WT 3HA-β2AR or mutant receptors. The conformational changes were detected with the NY-β2AR sensor53. Directly after transfection, cells were plated in 96-well white plates (Greiner) at a density of 50,000 cells/well, and incubated for 48 h. After that period, the plates were washed with PBS and assay buffer (Hank’s balanced salt solution (HBSS)) was added. Cells were stimulated with the ligands during 5 or 15 min for evaluating, respectively, Gs activation or β-arrestin recruitment and conformational changes. Coelenterazine 400a (2.5 μM) was added 5 min before the reads. BRET was monitored using a TriStar LB942 microplate reader (Berthold) equipped with a 410/80 nm donor filter and a 515/40 nm acceptor filter (for Gs activation and β-arrestin recruitment) or a 485/20 nm donor filter and a 530/25 nm acceptor filter (for receptor conformational changes). BRET ratio was calculated by dividing the acceptor emission by the donor emission. The data were analyzed with Prism (Graphpad) using “dose-response- stimulation log(agonist) vs normalized response- variable slope” with the constraint of sharing the hill slope across all data sets.
Molecular Dynamics Simulations
We simulated the β2-adrenergic receptor bound to the partial agonist salmeterol as well as the full agonist epinephrine. We initiated these simulations from crystal structures of the receptor bound to each of these ligands. Each of the crystal structures we used includes a nanobody that binds to the intracellular side of the receptor and stabilizes an active or intermediate receptor state: Nb71 for salmeterol and Nb6B9 for epinephrine29.
We performed three simulations of β2AR–salmeterol–Nb71 and three simulations of β2AR–epinephrine–Nb6B9. Each of the crystallized constructs was a β2AR–T4 lysozyme (T4L) fusion protein, with T4L replacing the receptor’s N-terminus. T4L was omitted from all simulations, while all other resolved residues were included. The majority of ICL3 was absent in all simulations, because it was deleted from the crystallized construct in β2AR–salmeterol–Nb71 and β2AR–epinephrine–Nb6B9. Because the simulations were performed before the published coordinates had been finalized, they were initiated using coordinates that differed very slightly from the published ones (root mean squared deviation below 0.3 Å, computed over all resolved receptor Cα atoms); these differences were much smaller than the typical motions of the atoms in simulation. In all simulations, a palmitoyl group not resolved but presumed to be present in the crystallized constructs was added to Cys341 using Maestro (Schrödinger LLC, New York, NY).
The β2AR was embedded in a hydrated lipid bilayer in all simulations; all atoms (including those in lipids and water) were represented explicitly. Hydrogen atoms were added to the crystal structures using Maestro, as described previously, and receptor chain termini were capped with neutral groups (acetyl and methylamide). Titratable residues other than Glu1223.41 were left in their dominant protonation state at pH 7.0. Glu1223.41, which faces the lipid bilayer, was neutral (protonated) in all simulations.
Prepared protein structures were inserted into an equilibrated bilayer solvated with 0.15 M NaCl, using a previously described protocol54. The bilayer consisted of palmitoyl-oleoyl-phosphatidylcholine, with 24% palmitoyl-oleoyl-phosphatidylserine in the inner membrane leaflet. Simulated systems initially measured roughly 71 × 71 × 115 Å3 and contained approximately 140 lipid molecules, 12,000 water molecules, 32 chloride ions, and 40 sodium ions.
We also performed three simulations of β2AR–salmeterol with the nanobodies removed (but otherwise starting from the same structures as above). Simulation setup was as described above, except that the removal of the nanobody allowed the simulated volume to be somewhat smaller. These simulations initially measured roughly 69 × 69 × 85 Å3 and contained approximately 120 lipid molecules, 7200 water molecules, 19 chloride ions, and 30 sodium ions.
We used the CHARMM-h force field for proteins. We used the CHARMM36 lipid force field, along with standard CHARMM salt ion parameters and the CHARMM TIP3P model for water. Parameters for palmitoyl-cysteine were as described previously27,54, and parameters for epinephrine were obtained by adapting previously published parameters for isoproterenol55. Parameters for salmeterol were obtained from the ParamChem server. Full parameter sets are available upon request.
Each simulation consisted of a 50-ns equilibration run followed by a longer production run. Systems were equilibrated in the NPT ensemble (310 K, 1 bar, Martyna-Tuckerman-Klein Nosé-Hoover chain coupling scheme using a multigrator, with initial velocities sampled from the Boltzmann distribution and with 5 kcal mol−1 Å−2 harmonic position restraints applied to the protein and ligand atoms, which were tapered off linearly over 50 ns. Production simulations used the same integrator, pressure and temperature, and were initiated from the final snapshot of the corresponding equilibration simulation. All simulations were performed on an Anton 1 computer56.
All bond lengths to hydrogen atoms were constrained using M-SHAKE. An r-RESPA integrator was used with a time step of 2.5 fs, and long-range electrostatics were computed every 7.5 fs. Long-range electrostatics were computed in reciprocal space with the u-series method57.
Simulation snapshots were saved every 180 ps. For the purposes of evaluating the fraction of time a hydrogen bond was formed in simulation, such a bond was considered to be formed in snapshots in which the relevant non-hydrogen atoms were within 3.0 Å of one another. Because the bound ligands shifted pose during the later parts of certain simulations (possibly as a result of motion of the nanobodies, which are not held in place by crystallographic contacts), we used the first microsecond of each simulation for these analyses. Each error bar was calculated as standard error of the mean across three simulations.
Dye modeling
The structure of the iodoacetamide derivative of the Atto655 dye (Atto-TEC GmbH) was obtained from PubChem (CID 16218785) and optimized using Avogadro and the GAFF force field. The structures of β2AR in the active (PDB ID 3SN6) and inactive conformations (PDB ID 2RH1) were retrieved from the RCSB PDB and mutated (L230W and L266C) using PyMOL. Parameters for the dye molecule attached to a cysteine residue were obtained using PRODRG and manually edited to enforce planarity of the ring system of the dye. The Crystallography and NMR System (CNS) software, version 1.358, was used to attach the dye to L266C and simulate its positions using rigid-body docking and multi-trial simulated annealing, respectively, as done previously by Brunger and co-workers59. The entire receptor structure was kept rigid, except for the side-chain atoms of L230W and L266C. Distances between the centers of mass of the tryptophan side-chain atoms and center of mass of the ring system of the dye were calculated for each generated model (300 for each conformation of β2AR).
Fluorescence measurements on purified receptor in detergent solution
Bimane-labeled and Atto-labeled receptors were used at respective concentrations of 0.2μM and 0.1μM in buffer containing 20mM Hepes pH 7.4, 100mM NaCl, 0.02% (w/v) DDM and 0.002% (w/v) cholesterol hemisuccinate. Salmeterol and epinephrine stocks were prepared as 50mM and 100mM solutions in DMSO and added at indicated final concentrations. Ligand concentrations were chosen to achieve saturation of detergent-solubilized receptor. To avoid any non-specific vehicle effects, care was taken to obtain the same final concentration of DMSO in all samples. Where indicated, Gs was added to a final concentration of 2μM. To allow efficient ligand binding and full equilibration, samples were incubated at the final concentrations used for 1 hour in the dark prior to measurements. Fluorescence data was collected in a quartz cuvette with 500μL of sample using FluorEssence v3.8 software on a Fluorolog instrument (Horiba) in photon counting mode. Bimane fluorescence was measured by excitation at 370nm with excitation and emission bandwidth passes of 4nm, and emission spectra were recorded from 410 to 510 nm in 1nm increments and 1s integration time. Atto fluorescence was measured by excitation at 650nm with excitation and emission bandwidth passes of 5nm, and emission spectra were recorded from 660 to 730 nm in 1nm increments and 1s integration time. Measurements were performed in triplicate.
Radioligand binding assays
Binding curves were obtained by incubating the DDM purified wild-type, bimane- and Atto655-labeled receptors in the presence of M1 FLAG–sepharose and 2 mM Ca2+, under shaking at room temperature for 1.5h. The antagonist [3H]-dihydroalprenolol (DHA) (PerkinElmer) was used to obtain saturation binding curves. Competition binding measurements for salmeterol and epinephrine were performed in a similar way, using DHA at a concentration slightly above Kd, as determined by saturation binding. Non-specific binding was accounted for by measuring in the presence of 10μM cold alprenolol. Beads were harvested using a 48-well Brandell harvester and counted in a LS6000TA scintillator (Beckman) using Cytoscint (MP Biomedical).
Statistical Analysis.
Statistical significance of hydrogen bond duration from MD trajectories (Fig. 5a) was assessed using the one-sided Welch’s unequal variances t-test. Statistical significance of the binding affinities (Supplementary Table 1) was assessed using Ordinary One-way ANOVA (Tukey) test at a multiplicity adjusted P value = 0.05. Statistical significance of the fitted mean logKi values (Supplementary Fig. 1) was assessed using a one-way ANOVA Tukey test at a P values of 0.05 (adjusted for multiple comparisons).
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants R01NS028471 (B.K.K.), the Canadian Institute for Health Research foundation grant FDN-148431 (M.B.), the American Heart Association Postdoctoral fellowship (17POST33410958) (M.M.) and Predoctoral Fellowship (13PRE17110027) (J.P.M.), a studentship from the FRQ-S (L.P.P.) and the NIH Pharmacological Sciences Training Program (T32GM007767) (J.P.M.). B.K.K. is supported by the Chan Zuckerberg Biohub. M.B. Holds a Canada Research Chair in Signal Transduction and Molecular Pharmacology. The authors thank J. Gullingsrud for assistance with MD software.
Footnotes
Competing Interests Statement
The BRET-based biosensors used in the present study are licensed to Domain Therapeutics but are freely available from M.B. for non-commercial academic use. M.B. is the chair of the Scientific Advisory Board of Domain Therapeutics. B.K.K. is a co-founder of and consultant for ConfometRx.
Data Availability.
Atomic coordinates and the structure factors for the crystal structure have been deposited with the Protein Data Bank under accession code 6CSY. Other data and results are available upon request.
References
- 1.Kenakin T Drug efficacy at G protein-coupled receptors. Annu Rev Pharmacol Toxicol 42, 349–79 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Zhu BT Rational design of receptor partial agonists and possible mechanisms of receptor partial activation: a theory. J Theor Biol 181, 273–91 (1996). [DOI] [PubMed] [Google Scholar]
- 3.Cazzola M & Donner CF Long-acting beta2 agonists in the management of stable chronic obstructive pulmonary disease. Drugs 60, 307–20 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Baker JG, Proudman RG & Hill SJ Salmeterol’s extreme beta2 selectivity is due to residues in both extracellular loops and transmembrane domains. Mol Pharmacol 87, 103–20 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Ferguson GT, Funck-Brentano C, Fischer T, Darken P & Reisner C Cardiovascular safety of salmeterol in COPD. Chest 123, 1817–24 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Twentyman OP, Finnerty JP, Harris A, Palmer J & Holgate ST Protection against allergen-induced asthma by salmeterol. Lancet 336, 1338–42 (1990). [DOI] [PubMed] [Google Scholar]
- 7.Ball DI et al. Salmeterol, a novel, long-acting beta 2-adrenoceptor agonist: characterization of pharmacological activity in vitro and in vivo. Br J Pharmacol 104, 665–71 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson M et al. The pharmacology of salmeterol. Life Sci 52, 2131–43 (1993). [DOI] [PubMed] [Google Scholar]
- 9.Calverley PM et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 356, 775–89 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Wijesinghe M, Perrin K, Harwood M, Weatherall M & Beasley R The risk of asthma mortality with inhaled long acting beta-agonists. Postgrad Med J 84, 467–72 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Weatherall M, Wijesinghe M, Perrin K, Harwood M & Beasley R Meta-analysis of the risk of mortality with salmeterol and the effect of concomitant inhaled corticosteroid therapy. Thorax 65, 39–43 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Coleman RA, Johnson M, Nials AT & Vardey CJ Exosites: their current status, and their relevance to the duration of action of long-acting beta 2-adrenoceptor agonists. Trends Pharmacol Sci 17, 324–30 (1996). [PubMed] [Google Scholar]
- 13.Clark RB, Allal C, Friedman J, Johnson M & Barber R Stable activation and desensitization of beta 2-adrenergic receptor stimulation of adenylyl cyclase by salmeterol: evidence for quasi-irreversible binding to an exosite. Mol Pharmacol 49, 182–9 (1996). [PubMed] [Google Scholar]
- 14.Green SA, Spasoff AP, Coleman RA, Johnson M & Liggett SB Sustained activation of a G protein-coupled receptor via “anchored” agonist binding. Molecular localization of the salmeterol exosite within the 2-adrenergic receptor. J Biol Chem 271, 24029–35 (1996). [DOI] [PubMed] [Google Scholar]
- 15.Isogaya M et al. Identification of a key amino acid of the beta2-adrenergic receptor for high affinity binding of salmeterol. Mol Pharmacol 54, 616–22 (1998). [PubMed] [Google Scholar]
- 16.Rong Y et al. Probing the salmeterol binding site on the beta 2-adrenergic receptor using a novel photoaffinity ligand, [(125)I]iodoazidosalmeterol. Biochemistry 38, 11278–86 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Gimenez LE, Baameur F, Vayttaden SJ & Clark RB Salmeterol Efficacy and Bias in the Activation and Kinase-Mediated Desensitization of beta2-Adrenergic Receptors. Mol Pharmacol 87, 954–64 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Westhuizen ET, Breton B, Christopoulos A & Bouvier M Quantification of ligand bias for clinically relevant beta2-adrenergic receptor ligands: implications for drug taxonomy. Mol Pharmacol 85, 492–509 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Tran TM et al. Characterization of agonist stimulation of cAMP-dependent protein kinase and G protein-coupled receptor kinase phosphorylation of the beta2-adrenergic receptor using phosphoserine-specific antibodies. Mol Pharmacol 65, 196–206 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Drake MT et al. beta-arrestin-biased agonism at the beta2-adrenergic receptor. J Biol Chem 283, 5669–76 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Carter AA & Hill SJ Characterization of isoprenaline- and salmeterol-stimulated interactions between beta2-adrenoceptors and beta-arrestin 2 using beta-galactosidase complementation in C2C12 cells. J Pharmacol Exp Ther 315, 839–48 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Moore RH et al. Salmeterol stimulation dissociates beta2-adrenergic receptor phosphorylation and internalization. Am J Respir Cell Mol Biol 36, 254–61 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walker JK & DeFea KA Role for beta-arrestin in mediating paradoxical beta2AR and PAR2 signaling in asthma. Curr Opin Pharmacol 16, 142–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Billington CK, Penn RB & Hall IP beta2 Agonists. Handb Exp Pharmacol 237, 23–40 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rasmussen SG et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 469, 175–80 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rasmussen SG et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–55 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenbaum DM et al. Structure and function of an irreversible agonist-beta(2) adrenoceptor complex. Nature 469, 236–40 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warne T et al. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature 469, 241–4 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ring AM et al. Adrenaline-activated structure of beta2-adrenoceptor stabilized by an engineered nanobody. Nature 502, 575–579 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nygaard R et al. The dynamic process of beta(2)-adrenergic receptor activation. Cell 152, 532–42 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manglik A et al. Structural Insights into the Dynamic Process of β2-Adrenergic Receptor Signaling. Cell 161, 1101–1111 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sounier R et al. Propagation of conformational changes during mu-opioid receptor activation. Nature 524, 375–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gregorio GG et al. Single-molecule analysis of ligand efficacy in β2AR-G-protein activation. Nature 547, 68–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Staus DP et al. Regulation of beta2-adrenergic receptor function by conformationally selective single-domain intrabodies. Mol Pharmacol 85, 472–81 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caffrey M Crystallizing membrane proteins for structure determination: use of lipidic mesophases. Annual review of biophysics 38, 29–51 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Baker JG The selectivity of beta-adrenoceptor agonists at human beta1-, beta2- and beta3-adrenoceptors. Br J Pharmacol 160, 1048–61 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kruse AC et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fronik P, Gaiser BI & Sejer Pedersen D Bitopic Ligands and Metastable Binding Sites: Opportunities for G Protein-Coupled Receptor (GPCR) Medicinal Chemistry. J Med Chem 60, 4126–4134 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Wieland K, Zuurmond HM, Krasel C, Ijzerman AP & Lohse MJ Involvement of Asn-293 in stereospecific agonist recognition and in activation of the beta 2-adrenergic receptor. Proc Natl Acad Sci U S A 93, 9276–81 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liapakis G, Chan WC, Papadokostaki M & Javitch JA Synergistic contributions of the functional groups of epinephrine to its affinity and efficacy at the beta2 adrenergic receptor. Mol Pharmacol 65, 1181–90 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Thomsen ARB et al. GPCR-G Protein-β-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell 166, 907–919 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Namkung Y et al. Monitoring G protein-coupled receptor and beta-arrestin trafficking in live cells using enhanced bystander BRET. Nat Commun 7, 12178 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gregorio GG et al. Single-molecule analysis of ligand efficacy in beta2AR-G-protein activation. Nature 547, 68–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Picard L-P, Schönegge AM, Lohse MJ & Bouvier M Bioluminescence resonance energy transfer-based biosensors allow monitoring of ligand- and transducer-mediated GPCR conformational changes. Communications Biology 1(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yao XJ et al. The effect of ligand efficacy on the formation and stability of a GPCR-G protein complex. Proc Natl Acad Sci U S A 106, 9501–6 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dawaliby R et al. Allosteric regulation of G protein-coupled receptor activity by phospholipids. Nat Chem Biol 12, 35–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schafer CT, Fay JF, Janz JM & Farrens DL Decay of an active GPCR: Conformational dynamics govern agonist rebinding and persistence of an active, yet empty, receptor state. Proc Natl Acad Sci U S A 113, 11961–11966 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fay JF & Farrens DL Purification of Functional CB1 and Analysis by Site-Directed Fluorescence Labeling Methods. Methods Enzymol 593, 343–370 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Doose S, Neuweiler H & Sauer M A close look at fluorescence quenching of organic dyes by tryptophan. Chemphyschem 6, 2277–85 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Cazzola M, Calzetta L & Matera MG beta(2) -adrenoceptor agonists: current and future direction. Br J Pharmacol 163, 4–17 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dawaliby R et al. Allosteric regulation of G protein–coupled receptor activity by phospholipids. Nature Chemical Biology 12, 35–41 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosenbaum DM et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science (New York, N.Y.) 318, 1266–73 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Picard LP, Schonegge AM, Lohse MJ & Bouvier M BRET-based biosensors to monitor ligand- and transducer-mediated GPCR conformational changes. (Communications Biology, 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dror RO et al. Identification of two distinct inactive conformations of the beta2-adrenergic receptor reconciles structural and biochemical observations. Proc Natl Acad Sci U S A 106, 4689–94 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dror RO et al. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proceedings of the National Academy of Sciences of the United States of America 108, 13118–23 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shaw DE et al. Millisecond-scale molecular dynamics simulations on Anton. in Proceedings of the Conference on High Performance Computing Networking, Storage and Analysis 1–11 (ACM, Portland, Oregon, 2009). [Google Scholar]
- 57.Shaw DE et al. Anton 2: Raising the bar for performance and programmability in a special-purpose molecular dynamics supercomputer. Sc14: International Conference for High Performance Computing, Networking, Storage and Analysis, 41–53 (2014). [Google Scholar]
- 58.Brunger AT Version 1.2 of the Crystallography and NMR system. Nature Protocols 2, 2728 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Choi UB et al. Single-molecule FRET-derived model of the synaptotagmin 1-SNARE fusion complex. Nat Struct Mol Biol 17, 318–24 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.