

Abstract
Mutations, the fuel of evolution, are first manifested as rare DNA changes within a population of cells. Although next-generation sequencing (NGS) technologies have revolutionized the study of genomic variation between species and individual organisms, most have limited ability to accurately detect and quantify rare variants among the different genome copies in heterogeneous mixtures of cells or molecules. We describe the technical challenges in characterizing subclonal variants using conventional NGS protocols and the recent development of error correction strategies, both computational and experimental, including consensus sequencing of single DNA molecules. We also highlight major applications for low-frequency mutation detection in science and medicine, describe emerging methodologies and provide our vision for the future of DNA sequencing.
Keywords: NGS, error-correction, molecular barcoding, molecular tagging, single molecule consensus sequencing, UMI, unique molecular identifier, SMI, single molecule identifier, high accuracy, Duplex Sequencing
Table of contents blurb
Despite the remarkable throughput of next-generation sequencing technologies, standard techniques are limited by the difficulty in distinguishing sequencing errors from genuine low-frequency DNA variants within heterogeneous cellular or molecular populations. This Review discusses sequencing methodologies and bioinformatic strategies that have been devised for the reliable detection of rare mutations and describes various important applications in diverse fields including cancer, ageing and metagenomics.
Introduction
Genetic heterogeneity underlies the evolution and adaptation of all life on earth. This is equally true of stochastically generated variants in germ cells as it is of somatic cells within tumours and ageing tissues. Rare variants can selectively proliferate upon exposure to new environments through natural selection1–3. Inter-cellular genetic diversity underlies many elements of human disease, from the emergence of therapeutic resistance to antimicrobial and anticancer therapies4,5, to the development of certain inherited genetic diseases6, to ageing and age-associated pathologies7,8. Yet despite the importance, until the last decade, our tools for quantifying and studying genetic variation in heterogeneous cell populations have been limited.
Beginning in 2005 a new generation of tools, referred to by the now somewhat anachronistic moniker of next-generation sequencing (NGS), emerged and wholly reshaped genetics9. NGS technologies have reduced the cost and increased the scale of genomic investigations by many orders of magnitude. They have transformed the study of genetic variation in humans and model organisms, elucidated the genetic basis of some diseases, and advanced our understanding of the organization, regulation, and function of genomes with unprecedented granularity10. Multiple distinct NGS platforms now exist, but all share the same fundamental feature of parallel interrogation of millions of individual DNA templates. The digital nature of the approach stands in contrast to the prior gold-standard Sanger method of sequencing aggregate populations of molecules11. Although this makes NGS methods potentially more sensitive for quantifying genetic heterogeneity, the vastly higher throughput means that, in practice, the absolute number of errors to contend with is greater than ever before.
Nearly all measurements in science are limited by an assay’s signal-to-noise ratio (Figure 1), and genetic heterogeneity is no exception. The rarer the variant, the more sensitive a technique must be to find it. Historically, scientific aspirations in this field exceeded the capabilities of available tools. For germline sequencing where variants are clonal, high-confidence genotypes can still be obtained, despite the modest accuracy of standard NGS, by redundantly sequencing identical genomic copies from multiple cells of an individual and ignoring erroneous read-to-read variation. By contrast, shortcomings in accuracy fundamentally limits the sensitivity of routine NGS for detection of low-level genetic variation in subclonal populations encompassing fewer than ~1% of the DNA molecules in a sample12,13. This has been a particular challenge for the reliable identification of somatically acquired mutations in multicellular organisms and for disentangling mixed microbial populations. Several recent advances have now markedly improved NGS accuracy, and therefore our rare variant detection capabilities; the deeper we have been able to look, the more reasons we are now discovering to look even deeper.
Figure 1. The signal-to-noise problem.
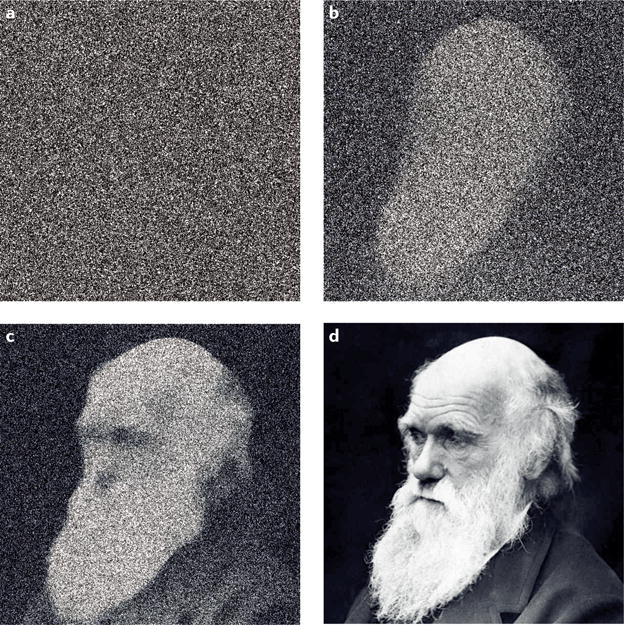
The accuracy of all analytical measurements, DNA sequencing included, depends on the ratio between true value and the precision of the detection method. This is analogous to the noisiness of a digital camera image: at a low signal-to-noise ratio, an image is indecipherable (a), but with increasing sensor quality (b–d) the image becomes progressively recognizable as a face and then a specific individual.
In this Review, we summarize the transformative impact of NGS on resolving molecular heterogeneity and survey the technical evolution of computational, biochemical and recently developed single-molecule consensus methods for error-correction that help mitigate the inherently high error rates of NGS platforms. We discuss emerging technologies that have the potential to further enhance our understanding of the role of rare genetic variants and highlight major fields of research that either have or will soon benefit from advances in sequencing accuracy for the purpose of obtaining higher sequencing sensitivity. We close with a discussion of future opportunities and the next wave of technical sequencing challenges we see on the horizon.
Subclonal mutation detection
For many areas of medicine and biology, genetic heterogeneity is the rule rather than the exception. Although several sensitive technologies for low variant allele frequency (VAF) mutation detection predated the advent of NGS, these were limited to interrogation of very small genomic regions and not easily transferred between loci14,15. A variety of methods for selective amplification of low-frequency variants facilitate detection but lack the ability to precisely quantify their relative abundance16. Digital PCR is a powerful technique that can be used for both precise molecular counting17 and, in allele-specific forms, for robust low-frequency mutation detection18. In recent years the method has become more widespread with the advent of convenient high-throughput emulsion-based platforms, referred to as digital droplet PCR19; however, variants being sought typically must be known a priori. NGS is indisputably the most generalizable method of mutation detection, but it has only been recently that technical advances have allowed it to achieve a comparable accuracy.
At the outset, we acknowledge that comparing the accuracy of different NGS protocols is challenging. The error rate of conventional NGS is about 1%, and can be as low as 0.1% in optimal scenarios20. However, the precise value varies by specific platform, chemistry version, sequence context, filtering stringency and various other factors that make for lively discussions, but few hard-and-fast agreements among researchers. The accuracy of many error-correction methodologies is similarly affected by variables that are difficult to normalize between studies, such as degree of DNA damage and whether the DNA standard being sequenced is, itself, truly free of mutations. Diluted oligonucleotide templates with defined sequences are often used to represent rare variants in published mixing experiments, but the frequency of errors during oligonucleotide synthesis may be as high as one-in-one-hundred. Similarly, although standardized cell lines are an attractive source of DNA that can be benchmarked against by different researchers, it has been shown that, in at least some situations, the mutations that accumulate during a few months in culture can exceed those accrued over an entire human lifetime21. For all these reasons, we have opted to limit our comparisons of method accuracy to order magnitude.
Improving the accuracy of NGS
Most attempts to lower the limit of detection for commercially available NGS platforms can be grouped into one of three broad categories. The first are purely computational and statistical strategies to exclude sequences of low confidence from conventionally generated NGS data. The second are library preparation protocols that either biochemically remove, or limit the formation of, mutagenically damaged nucleotides in templates before sequences are generated, both of which we review in this section. The third and most successful type of strategy is single-molecule consensus sequencing, which we describe in subsequent sections. This more contemporary latter approach involves an intertwined combination of chemical labelling prior to sequencing and informatic deconvolution thereafter, which allows for the identification and exclusion of errors that invariably occur despite all other measures.
Computational reduction of sequencing artefacts
Initial efforts to reduce the background error rate of NGS focused on data filtering schemes to discount low-confidence sequences caused by technical artefacts. Phred quality scores, originally developed for Sanger sequencing electropherograms to estimate the probability of an error at each sequenced base22, were adapted for the image-based output of NGS platforms23.
Quality score filtering does not improve accuracy if the errors are introduced prior to the sequencing, for example, during PCR amplification. A variety of bioinformatics tools such as MuTect24 and VarScan225, use additional filters, such as whether variants are biased toward the beginning or ends of reads (reflecting erroneous end-repair of fragments or mapping errors), or requiring that true variants be seen in multiple independent sequencing reads or both read orientations26. Newer alignment algorithms and read-trimming tools are better able to avoid artefacts from off-target mapping or inadvertent sequencing into artificial adapter or primer sequences used in library preparation27. Increasingly sophisticated software packages that are specifically designed for the detection of low-frequency variants in traditional NGS data are able to eliminate many false calls through rigorous statistical approaches that involve modelling the error profile of specific sequencing applications, or even individual sequencer runs, and using these to appropriately assign confidence to particular conclusions28,29.
Some sequencing errors can be identified and removed empirically by simultaneously sequencing a test sample that is thought to have subclonal mutations (for example, a tumour) alongside an apparently homogeneous control sample30. Low-level variants in the control sample are assumed to reflect sites with a higher propensity for technical errors and are discounted from all samples. As increasingly large sequencing data sets are generated, databases of specific sequence contexts that recurrently yield artefactual errors can be identified and viewed more sceptically. Particular mutation patterns, such as those resulting from oxidative DNA damage31,32 or engineered polymerases and nucleotides used during sequencing33 can be partially remedied informatically. Nevertheless, even the most careful computational data scrubbing cannot universally produce high-confidence calls of subclonal mutations much below 1% abundance13,34–36, and the more aggressive the approach, the greater the risk of excluding true rare variants.
Biochemical reduction of sequencing artefacts
Errors arising during the generation of NGS data can occur at many stages. Mistakes may occur on the sequencer itself as a result of optical imperfections, enzymatic errors during cluster formation or overlapping or polyclonal clusters, among other issues33,37. Substantial errors can also arise during pre-sequencing library preparation as a result of PCR misincorporations, chimeric PCR products, template switching or hairpin formation37–40. Although high-fidelity proofreading polymerases are typically used for amplification steps, lower-fidelity polymerases may be used for preceding repair and A-tailing of library fragments. Furthermore, all polymerases are considerably more error prone when copying across damaged nucleotide templates. Such damage may be present at the time of sampling from normal cell processes or environmental exposures, but can also occur from extrinsic manipulations such as chemical extraction, heating or clinically used stabilization methods such as formalin fixation41,42.
A common step in library preparation is ultrasonic shearing of DNA into short fragments. This produces sufficient energy to break the phosphodiester backbone, which can also oxidize bases and lead to artefactual C:G→A:T transition mutations. This can be somewhat reduced by pH buffering and cation chelation31,32. Furthermore, nicks and non-blunt-ended breaks produce regions of single-stranded DNA, which are both biochemically more susceptible to damage43 and subjected to copying by lower-accuracy polymerases during end-repair steps in adapter-ligation-based library preparation methods. Enzymatic DNA fragmentation methods avoid some of these issues, but at the same time may produce other low-level artefacts as a result of nicks, abasic sites or other incomplete cleavage products that vary by the enzymatic mechanism44.
Although some DNA damage may be prevented, other sources are unavoidable. Both formalin-fixation and heating accelerates the rate of spontaneous deamination, particularly of cytosine to uracil, to produce C:G→T:A substitutions upon PCR amplification42,45. Uracil bases in DNA can be excised by treatment with uracil-DNA glycosylase (UDG), which yields abasic sites that are resistant to amplification46,47. However, abasic sites are, themselves, somewhat mutagenic if copied: polymerases typically mis-insert an A when encountered48,49, which also generates a single nucleotide C:G→T:A substitution. Destruction of the remnant phosphodiester backbone at abasic sites with a DNA lyase, commonly endonuclease VIII, may somewhat abrogate this42.
Another common mutagenic base modification, 8-oxo-dG, is formed by oxygen free radicals and readily mis-pairs with adenine to lead to C:G→A:T mutations50. 8-oxo-dG can be biochemically excised by incubating with [fapy]-DNA glycosylase (FPG), which has both glycosylase and lyase activity42,46. As with UDG, treatment can reduce artefacts but may also render some highly damaged DNA unamplifiable. This can be a particular problem with some low-input applications described later. Combinations of glycosylases with other repair enzymes to replace the damaged base may improve amplifiability32, yet themselves may introduce errors at low levels.
Nevertheless, although biochemical approaches, in concert with computational strategies, have a positive effect on improving NGS accuracy, it is relatively modest. Not all mutagenic damage can be prevented and not all damage that occurs can be easily corrected. For example, spontaneous deamination of 5-methylcytosine generates a canonical thymine base. To achieve error rates below one-in-one-thousand, other techniques are required.
Molecular consensus sequencing strategies
Following the release of the first commercial NGS platform in 2005, efforts to improve accuracy initially focused on refining the core elements of sequencers themselves. During the next several years, iterative improvements to optics, polony formation methods, sequencing-by-synthesis chemistry and on-machine data filtering boosted the accuracy of raw outputted data by about an order of magnitude. Paired end sequencing allowed additional confirmation of the identity of bases sequenced from both ends of a molecule. During the last several years, although NGS throughput has increased dramatically, improvements to raw accuracy have largely plateaued; on some high-output platforms it has even decreased51. Recognizing that some biochemical mistakes are unavoidable, around 2009 an innovative solution for improving accuracy was developed that focused on identifying and ignoring errors, rather than preventing them entirely52. The approach, eventually becoming known as “single-molecule consensus sequencing”, “tag-based error correction” or “molecular barcoding”, rapidly emerged, through the work of multiple investigators, to become a new standard for high-accuracy NGS applications53–55. We begin with reviewing different embodiments of this concept that apply to the short-read platforms, which currently comprise most of the NGS market. In the section that follows, we discuss consensus-sequencing approaches that apply to the commercially available long-read platforms that rely on direct sequencing of single DNA molecules.
[H3] Barcoding of individual DNA molecules
During conventional short-read platform NGS, a DNA library is typically PCR amplified before sequencing. It is impossible to definitively know whether two identical sequence reads arose from copies of the same starting molecule or from two independent molecules. However, if a unique tag (i.e. a molecular barcode) is applied to each molecule before amplification, this label will be propagated to all derivative copies and independent sequence reads can thus be recognized as having arisen from a common founder. It is worth noting that the concept of a molecular barcode (also known as a unique molecular identifier (UMI), a single molecule identifier (SMI) or simply a tag) is different from that of an index sequence. Molecular barcodes serve to uniquely label individual molecules within a sample whereas index sequences are identical DNA labels that are affixed to all molecules in a given sample for the purpose of sample multiplexing.
Molecular barcodes can be used to improve the accuracy of counting DNA or RNA molecules in mixtures by eliminating biases from variable amplification54,56–59. More importantly, because when designed carefully, all identically tagged reads will have derived from a common founder, any variation between their actual sequences must necessarily reflect technical errors53–55. Tag-based error-correction relies on this principle: independent reads sharing a common tag are recognized and grouped as amplicon copies of the same starting molecule; any sites of sequence differences among the reads are discounted as errors when forming a consensus sequence (Figure 2). A fundamental element of the approach is the need to intentionally produce and sequence redundant molecular copies, which requires relatively higher raw sequencing depth than conventional NGS, and thus, additional costs.
Figure 2. Methods of consensus-based error correction on short-read platforms.
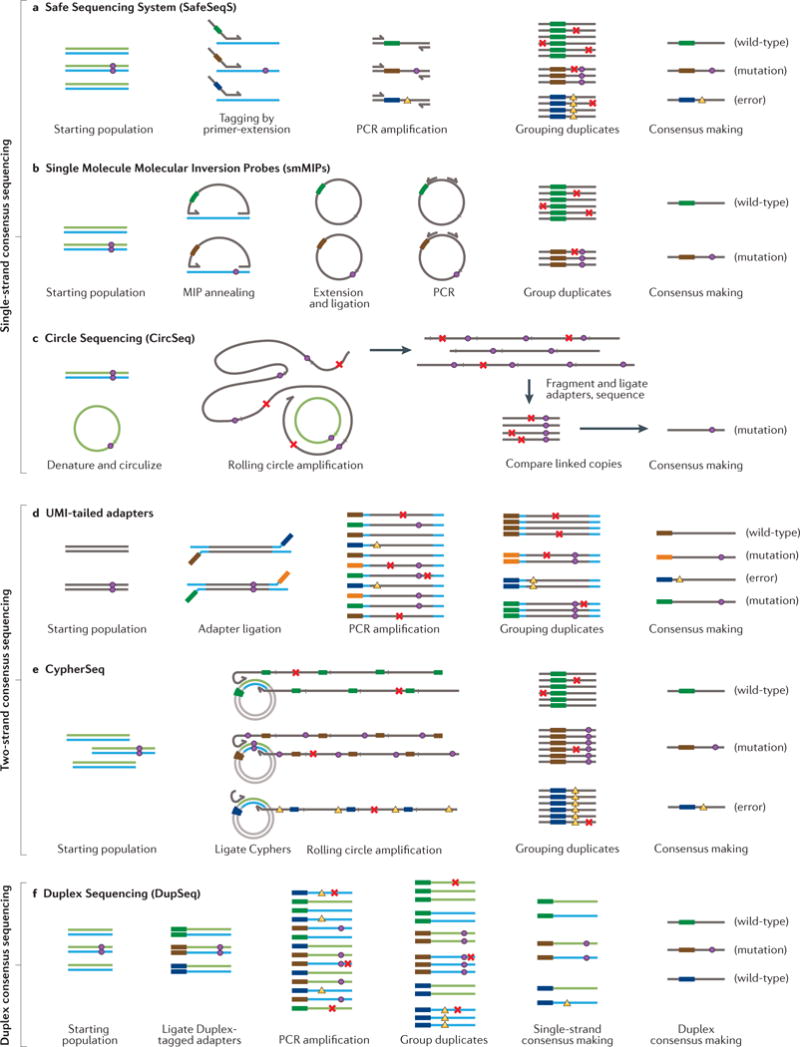
a| Safe Sequencing System (SafeSeqS) uses randomly generated molecular barcodes carried by PCR primers (coloured thick bars) to reduce errors by independently labelling each single-stranded DNA molecule, thus allowing identification of derivative copies. True mutations (circles) can be discerned from sequencing errors or late PCR errors (crosses) because the latter occur only in a subset of identically labelled duplicate reads. PCR errors that occur during the first cycle of amplification (triangles), can be propagated to all duplicates and escape error correction. b | Single-Molecule Molecular Inversion Probes (smMIPs) entail two targeting arms joined by a linker that contains a molecular barcode. The molecules are hybridized with single-stranded DNA and then extended and ligated to form closed loops which are amplified and sequenced. Consensus-based error correction is similar to SafeSeqS, and similarly susceptible to first-cycle amplification artefacts. c | Circular Sequencing (CircSeq) entails circularization of single-stranded DNA fragments without any molecular barcodes, followed by rolling-circle amplification, fragmentation and sequencing of short stretches of concatemerized fragments. The molecular fragmentation points of the starting molecules serve as unique molecular identifiers (UMIs) for consensus-based error correction. As with other single-stranded consensus methods, recurrent amplification errors may fail to be identified and corrected. d | UMI-tailed adapters can be ligated to a library to uniquely mark each single strand. Despite both strands in a complex being tagged, no means is provided to relate the consensus of one strand to that of its mate for comparison and early PCR errors (triangles) may go unrecognized. e | CypherSeq circularizes double-stranded DNA molecules using a single adapter molecule containing double-stranded molecular barcodes. Targeted enrichment is achieved with rolling-circle amplification using primers directed to each DNA strand. Although information from both strands may be contribute to consensus making, lack of asymmetry between the two strands makes it impossible to discern whether one or both strands successfully amplified. Recurrent early amplification errors (triangles) can escape error correction when only one strand worth of data is successfully recovered because this cannot be recognized. f | Duplex Sequencing (DupSeq) allows true duplex error correction on high-throughput short-read sequencing platforms by applying molecular barcodes to each double-stranded DNA molecule in such a way that amplification products of the two strands can be informatically related to each other (thick colored bars), but also distinguished (blue versus green strands). After tagging, derivative PCR products are grouped by molecular barcode and by strand. Consensuses are made for each strand group and then compared to that of the complementary strand. True mutations (circles) can be confidently distinguished from both sequencing errors and late PCR errors (crosses) as well as first-round PCR errors (triangles), because complementary errors are extremely unlikely to occur by chance at the same position on both DNA strands. See the main text for a detailed description of each method.
Molecular barcodes come in two forms: exogenous and endogenous53,55. Exogenous barcodes entail random or semi-random artificial sequences that are incorporated into either sequencing adapters or PCR primers. Endogenous barcodes describe the randomly or semi-randomly generated fragmentation points at the ends of DNA molecules in ligation-based library preparation methods. The two approaches can be used either alone or in combination60.
With either approach it is important that a sufficient variety of possible tag sequences exist such that the probability of two independent molecules being tagged the same way is low. With low molecular depth sequencing, the chance of two independent DNA fragments having the same shear points by chance is small and these endogenous sequences alone suffice as tags60,61. At the other extreme is deep sequencing following an amplicon-based library preparation. In this case, molecular ends are defined by invariant primer sites, not random fragmentation, so all tag information must come from degenerate tags55. A similar problem arises with targeted enzymatic fragmentation62. If barcode diversity is inadequate, tag clashes can occur, whereby independent molecules are identically labelled. In this scenario, true low-frequency variants can be erroneously discarded as errors. If barcodes are too complex, they may develop errors themselves and artificially create false families that incorrectly appear as arising from distinct molecules. Both problems can be mitigated with careful design and strategies for tolerating errors in barcodes63–65.
Over the past five years molecular consensus sequencing has proven itself as the most impactful means for reducing NGS errors. Different implementations variably reduce sequencing error rates from ~10−2 to 10−4–10−7 or lower. The variety of approaches developed to date can be grouped into three basic categories: single-strand consensus sequencing; two-strand consensus sequencing; and duplex consensus sequencing (Figure 2).
Single-strand consensus sequencing
One of the most widely cited early implementations of tag-based NGS error correction is the SafeSeqS technique, which applies tags via PCR primers carrying a degenerate sequence tail55. In this method, after a small number of PCR cycles with the barcoded primers, additional amplification with a second set of universal primers is carried out to generate multiple copies of each tag-defined founding molecule, which are then sequenced and grouped into families for consensus-based error correction (Figure 2a). A substantial challenge to widespread implementation was the difficulty of sequencing large numbers of targets at the same time because of PCR multiplexing problems caused by the random tags. Newer variations on the technique that protect the tags from nonspecific binding through the use of a hairpin design have somewhat reduced this problem66,67.
Primer-based tagging is convenient insofar as it facilitates sequence targeting at the same time as tagging for error correction, but because several PCR tagging cycles are needed, occasionally the same DNA molecule will be labelled by more than one tag and create false families. Another tagging method that circumvents this issue is that of single-molecule molecular inversion probes (smMIPs)68,69. Instead of a pair of primers, a single oligonucleotide with two targeting arms connected by a linker region with a molecular barcode is hybridized to a DNA sample and then extended and ligated to form tagged, closed-loop products that can then be enriched, amplified and sequenced (Figure 2b). With smMIPs, many targets can be easily multiplexed together and there is little risk of double-tagging the same molecule. Design constraints around the narrow proximity window for the targeting arms add a challenge, but improved software algorithms have recently made the method more tractable70.
Another consensus sequencing method that takes a very different approach is CircSeq46,71. In this technique, DNA is fragmented and melted into very short single-stranded pieces that are then circularized and copied into concatemers via rolling-circle amplification. The concatemers are further amplified and then sequenced (Figure 2c). Instead of exogenously applied barcodes, the unique genomic coordinates of fragmentation points serve as molecular identifiers to define which sequence reads derived from a given starting molecule. The fact that tandem copies of the sequence are physically joined means that each sequencing read contains the necessary information for an initial level of consensus calling. In contrast to tag-based barcoding of unlinked copies, which may have either too few or an excess of copies present, this linkage improves cost-efficiency by keeping the duplicate rate more uniform. The two major challenges to the approach are the very limited length of sequences that can be genotyped as tandem copies on short-read platforms (some solutions have since been developed72) and the risk of tag clashes that stems from the use of the inherently limited number of possible shear points as identifiers73. Nevertheless, the concept of sequencing tandem linked copies is powerful and will undoubtedly become more relevant as the performance of long-read platforms improves.
Most of applications for NGS do not require an entire genome to be sequenced and targeting of specific regions is important to reduce costs. NGS library preparation workflows involving DNA fragmentation, adapter ligation and then hybrid capture enrichment of loci of interest are slightly more time consuming than amplicon-based methods, but are generally easier to design, and thus, are more widely used. One of the most easily implemented and popular consensus sequencing approaches that has made its way into many commercial products in the past two years incorporates a degenerate UMI sequence into one adapter strand74 Depending in the implementation, either one or both (the version shown in Figure 2d) of the library fragment strands are thus labelled. The combination of variable shear points and high-diversity exogenous UMIs substantially reduces the risk of tag-clashes.
All single-strand consensus techniques reduce errors by 2–3 orders of magnitude, which is far greater than any prior computational or biochemical approach, and make it possible to accurately identify rare variants below 0.1%. However, certain errors persist. All four methods rely on consensus sequencing of tagged copies derived from just one strand of what are natively double-stranded DNA molecules. Mistakes that occur during the first round of amplification can be propagated to all other copies as ‘jackpot errors’ that escape correction (i.e. the yellow triangles in Figure 2a). This is particularly true of misincorporation errors at sites of mutagenic DNA damage, especially 8-oxo-guanine adducts and deaminated cytosine bases. This is clearly apparent in the spectrum of background errors from any single-stranded tagging method: G>T and C>T mutations, respectively stemming from oxidation and deamination, are far more frequent than the reciprocal mutations C>A and G>A. True mutations should be present in equal proportions of complements60. The developers of CircSeq noted that this mutational bias could be partially abrogated by treatment with damage-removing glycosylases46.
Two-strand consensus sequencing
DNA is a double-stranded molecule for a reason. The ability to unwind and independently copy each half facilitates cell division. The biochemically enforced rule-based pairing of nucleotides is relied upon by cellular machinery to ensure high fidelity of strand replication: any mismatches are quickly identified and repaired. Conceptually, a tag-based sequencing approach that takes into account the genotype of both DNA strands should achieve a higher degree of error correction for analogous reasons. One effort to improve upon the SafeSeqS method used barcoded PCR primers targeted against both the reference and anti-reference strands of regions of interest75. In concept, if a mutation is seen in both PCR products, it can be viewed with greater confidence. Although theoretically higher accuracy than SafeSeqS, because the two PCR products that derive from the individual strands of any particular molecule will carry different random barcodes, the resulting sequences cannot be directly related to each other. Although both the reference and anti-reference strands of molecules in a population can be genotyped, there is no way to compare the sequence of one strand of a particular double-stranded molecule with that of the other, so true double-stranded error correction is impossible. The same is true of the adapter-based UMI approach shown in Figure 2d.
The first reported tag-based NGS error-correction method relied on a ligation-based approach where both strands of double-stranded molecules were labelled with identical molecular tags followed by PCR amplification then concatemerization of amplicons and sequencing53. After amplification, PCR products derived from both the reference and anti-reference strands carry the same tags and can be grouped to produce an error-corrected consensus. A more recent variant upon this, known as CypherSeq76, incorporates rolling-circle amplification from primers targeting both strands after ligation into a circularized adapter sequence to achieve a degree of target enrichment prior to PCR (Figure 2e). With both methods, PCR products derived from each strand of individual molecules can be used to form a consensus sequence; however, as the amplicons of the two strands are indistinguishable, it is impossible to tell whether the resulting consensus is based on single-strand or double-strand data. Because one strand often fails to amplify, either due to DNA damage or stochastic factors, or is simply not sampled60, jackpot amplification errors can still escape detection (i.e. yellow triangles in Figure 2e). A recent preliminary technique known as Pro-Seq involves concurrent amplification of both the reference and anti-reference strands of individual DNA duplexes with physically linked primers in emulsion droplets77. This enables genotyping of both strands within the same flow-cell cluster for cost savings, but has an identical limitation in that amplification of both strands cannot be ensured.
Duplex consensus sequencing
In 2012 our group described Duplex Sequencing, a technique that uses a special form of molecular tagging to independently barcode each strand of individual DNA duplexes in such a way that sequence reads derived from one strand can be related to, but also distinguished from that of the other60. Exogenous tags within each strand of the sequencing adapters and/or DNA fragment shear points serve as UMIs that informatically relate reads from the two strands. A non-complementary portion of the adapters introduces strand asymmetry that allows the products to be distinguished from each other and, importantly, allows confirmation that both strands have been sequenced (Figure 2f). To achieve true duplex error correction when the strands are separately amplified, the adapted molecule must contain both a UMI and an asymmetric strand-defining element (SDE). Together these pieces allow a separate consensus sequence to be produced for each strand for comparison to that of its mate78. In this way, early PCR jackpot errors can be confidently recognized and discounted (i.e. yellow triangles in Figure 2f).
The theoretical Duplex Sequencing error rate of <10−9 reflects the low probability of a complementary jackpot error occurring at the same position on both strands60. In fact, one challenge to experimentally verifying this error rate is identifying a gold-standard source of DNA that is truly devoid of mutations: the lowest frequency of single-base substitutions we have measured in the DNA of healthy newborns is between 10−7 and 10−8, which is consistent with mutation frequencies extrapolated from differences between human generations79. We and others have applied Duplex Sequencing to measuring variations in the spontaneous mutation frequency of microbial populations60 and in mammalian tissues in the setting of ageing61,80,81 neurodegeneration82,83 inherited DNA repair defects84 and genotoxic exposures61,85. In all cases, the mutagenic effect of near or below one mutation per million basepairs could only be recognized because of the extremely low error rate.
In theory a variety of other molecular tools could serve as Duplex Sequencing UMIs and SDEs. Other than shear points and DNA-based tags, single-molecule compartmentalization methods that keep paired strands in physical proximity77 or other non-nucleic acid tagging methods could serve the strand-relating function. Similarly, assymetric chemical labelling of the adapter strands in a way that they can be physically separated can serve an SDE role. A recently described variation of Duplex Sequencing uses bisulfite conversion to transform naturally occurring strand asymmetries in the form of cytosine methylation into sequence differences that distinguish the two strands86,87. Although this implementation limits the types of mutations that can be detected, the concept of capitalizing on native asymmetry is noteworthy in the context of emerging sequencing technologies that can directly detect modified nucleotides88.
Having distinct elements that both relate and distinguish strands in Duplex Sequencing reflects the need to add molecular identity information about an original molecule that is lost when the paired strands are physically uncoupled and copies are made. Whereas this is currently the most practical approach with short-read platforms, for newer single-molecule long-read sequencing technologies, strand uncoupling and/or DNA amplification are not required.
Direct single-molecule consensus sequencing
At the present time only two types of single-molecule sequencers are commercially available: those manufactured by Pacific Biosciences (PacBio), which rely on detection of fluorescent nucleotide incorporation by single immobilized polymerases within zero-mode wave guides89 and those of Oxford Nanopore Technologies, which capitalize on the differential voltage changes by nucleotides as a single-stranded DNA molecule traverses a molecular nanopore between two chambers to record the sequence90. These platforms currently make up a relatively small portion of all sequencing, largely because of lower raw accuracy and throughput than prevailing short-read technologies, but both have gained an increasing following as the technologies have matured.
Because of their long reads, the most common use for single-molecule sequencers is de novo genome assembly and sequencing of complex repetitive regions or structural rearrangements90,91. However, long-read capability can be repurposed to improve genotyping accuracy of shorter regions through sequencing of tandem copies. One example is the INC-Seq method whereby the terminal 5 and 3 ends of duplex molecules are intra-molecularly ligated to form closed loops that can be subjected to rolling-circle amplification followed by nanopore sequencing. The resulting reads comprise a long string of linked sense or antisense strands, akin to the CircSeq technique, but with many more copies of much longer fragments92 (Figure 3a). Although better than simple nanopore sequencing, the very low raw nanopore accuracy coupled with amplification errors leads to a final consensus error rate that is still inferior to the best short-read platforms.
Figure 3. Methods of single-molecule sequencing consensus-based error correction.
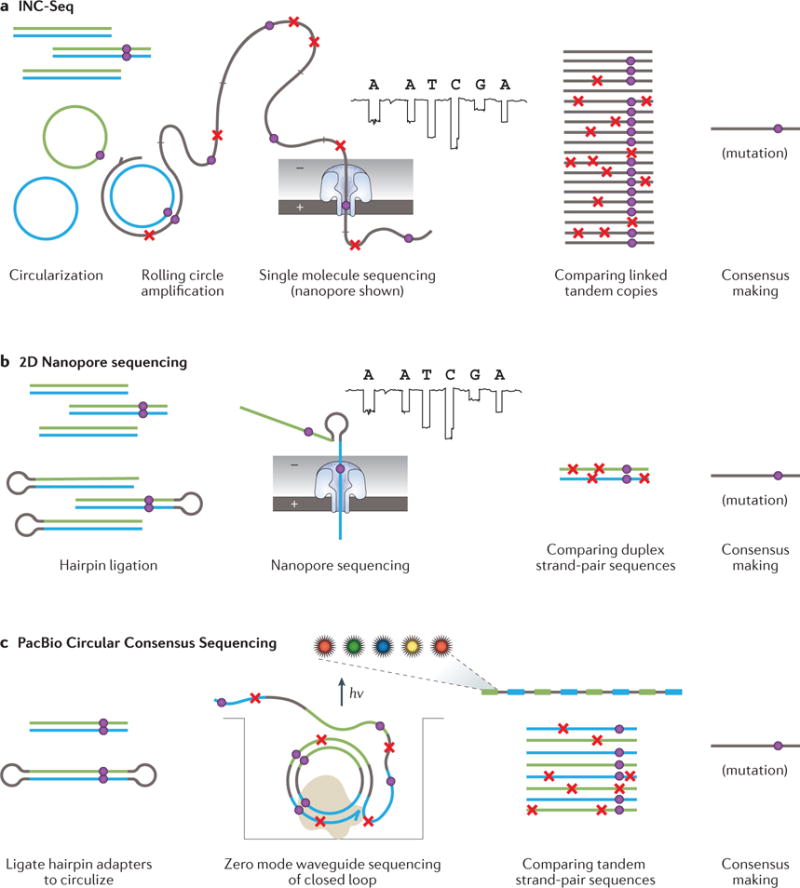
a| The INC-Seq method begins by circularizing double-stranded DNA fragments followed by rolling-circle amplification of the loop; each product is a long DNA strand comprising concatenated copies of one of the strands of the starting molecule. This is sequenced on a long-read platform. b | 2D nanopore sequencing involves ligation of a hairpin adapter to one end of a duplex DNA molecule followed by tandem nanopore sequencing of the linked original strands. c | SMRTbell sequencing entails ligation of hairpin adapters to each end of a molecule, followed by direct sequencing of the closed loop on the long-read Pacific Biosciences (PacBio) platform. Both strands are sequenced together in multiple passes. In all cases, consensus sequences incorporate data from both DNA strands.
A unique feature of single-molecule sequencers is that consensus sequencing can be achieved without any amplification. For the Oxford Nanopore platform, use of a hairpin adapter to link the two strands of an individual DNA duplex is the most simple form of amplification-free consensus sequencing, which requires neither an SDE, nor a UMI93. (Figure 3b). The incremental error correction achieved by sequencing both strands of a duplex (2D) as opposed to only one strand (1D) is still dwarfed by the substantial baseline nanopore error rate. In the future it should be possible for the same linked pair of molecules to be repeatedly passed back and forth through a nanopore for more rigorous consensus building88.
The first consensus sequencing method to verifiably error-correct using both strands of an individual DNA molecule was the Circular Consensus Sequencing (CCS) SMRTbell technique on the PacBio platform.94 This approach relies on ligation of hairpin-shaped adapters to either end of a double-stranded template to form a closed loop, which is directly sequenced and produces multiple passes of data from each strand without prior amplification (Figure 3c). CCS has been applied to high-accuracy sequencing of error-prone repetitive sequences95, noninvasive detection of low-frequency cancer-derived mutations,96, metagenomic deconvolution97 and direct measurement of DNA polymerase error rates38,98. Under optimal conditions, the method can achieve accuracies on the order of 10−7 because jackpot artefacts from one strand are unlikely to occur as complementary changes on the other. However, because of lower raw accuracy, a dozen or more sequencer passes across the tandem copies, as well as treatment with DNA repair enzymes, are needed to achieve the maximum resolution38. The lower nucleotide output of the PacBio system as compared to more standard short-read platforms means that ultra-rare variant detection is more challenging from a cost and time perspective.
Other emerging single-molecule technologies involving direct electrical base detection on microchips99 may eventually allow similar opportunities. A recently announced single-molecule sequencing-by-hybridization platform repeatedly interrogates individual bases by iterative hybridization with different overlapping probes, which is effectively another embodiment of consensus sequencing100; the system should allow complete duplex error-correction using UMIs or physical linkage of complementary DNA strands. With further developed consensus-based error-correction approaches, the unique benefits of all these newest-generation platforms, namely, very long reads, rapid library preparations and, in some cases, easy portability101, can be meaningfully realized in low-frequency variant detection applications.
Considerations in choice of consensus method
Tradeoffs between accuracy, cost, recovery, speed and read length mean that no single consensus-based error-correction method is optimal for every application. Because most sequencers in use are short read, techniques designed for these platforms have generally been favoured. Duplex approaches are by far the most accurate but also generally the most expensive per error-corrected base because of the need to sequence more duplicates to have a reasonable probability of recovering copies of both strands. Put another way, the consensus-making efficiency is lower, because a greater raw sequencing depth is required to obtain a similar molecular (consensus) depth than with single-stranded methods. Because the sensitivity for detecting a rare variant is influenced by both error rate and the number of consensus families generated at a given locus, what duplex methods gain in error rates comes at a greater cost for attaining sufficient molecular depth. For small genomic targets this tradeoff is acceptable but may become an important consideration with large multi-gene panels.
One substantial source of consensus-making inefficiency is non-uniformity of the amplification that is used to generate molecular copies. With duplex techniques, if one strand replicates better than the other, this may prevent a consensus from forming, or require an inordinately large number of raw reads to achieve a consensus. With single-stranded methods, if one locus amplifies better than another, a similar problem arises. Careful attention to factors that bias PCR efficiency, such as fragment length, help to abrogate the issue. For example, in a preliminary report we recently described the use of targeted CRISPR–Cas9 digestion to generate Duplex Sequencing libraries of uniform length, and further capitalized on this size specificity to enrich for genomic loci of interest, thus further reducing competition during PCR62. Together these features can improve consensus-making efficiency by an order of magnitude. A clever approach to eliminate amplicon competition and normalize yield per founding molecule using digital emulsion amplification has been demonstrated with both PCR and rolling-circle amplification71,77,102. Ultimately, the most efficient consensus sequencing approaches are those that do not require any amplification, but single-molecule technologies will need to improve considerably before this becomes a major consideration.
Another important factor when selecting a method is the molecular conversion efficiency—that is, the fraction of input DNA molecules that are recovered as consensus sequences. This is typically lower with duplex than single-strand approaches because both halves of a molecule must successfully amplify, which may be impossible if one is damaged or missing. Amplicon-based library preparation methods offer higher recovery and more rapid workflows than ligation followed by hybridization capture methods, but do not retain complete duplex information. Simplicity and speed are important in clinical and some commercial settings. In situations where available DNA is limited, for example, in forensics or liquid biopsy applications, maximum recovery is important to detect low-frequency variants. However, greater recovery does not necessarily portend higher sensitivity if substantially more errors are introduced. Figure 4 illustrates the relationship between sequencing accuracy and positive predictive value for standard NGS, single-strand consensus sequencing and Duplex Sequencing. Although single-strand consensus methods have an absolute accuracy on the order of 10−5, when attempting to detect variants that are present at a frequency of 1/100,000, about 80% of called mutations will be errors. Higher accuracy duplex methods are necessary to reliably call mutations at this level. Increasingly sophisticated computational techniques are being developed to statistically integrate information from different types of consensus sequences and proportionally weight the certainty of called variants as a way to maximize data recovery while also retaining accuracy103,104. In addition, emerging hybrid biochemical methods that combine the benefits of PCR-based targeting on library preparation speed and conversion efficiency with the advantages of adapter-based molecular tagging are likely to further narrow current performance tradeoffs 105,106.
Figure 4. Impact of error correction technology on detection sensitivity.
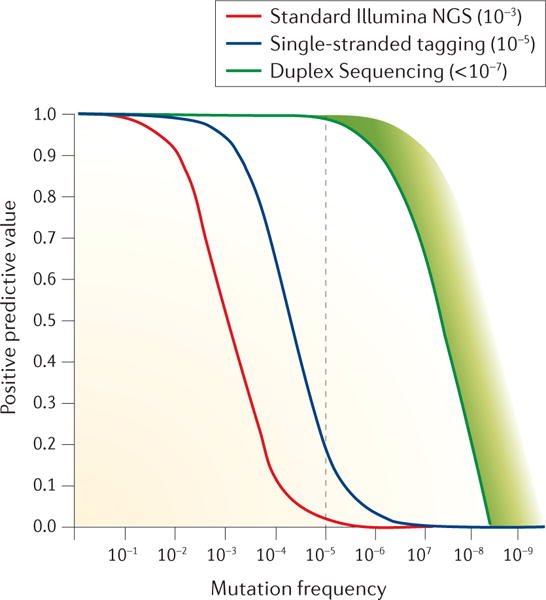
The positive predicted value (the expected number of correct positive calls divided by the total number of positive calls) is plotted as a function of the variant allele frequency in a molecular population for each sequencing method of a specified error rate. As seen by curve overlap, nearly all mutant calls will be correct using any method if the frequency of detected variants is greater than 1/10. However, the error rates of standard Illumina Sequencing and single-stranded tag-based error correction result in critical losses in positive-predictive value at variant frequencies of ~ 1/100 and 1/1000 respectively. The extremely low error rate conferred by Duplex Sequencing enables confident identification of variants below 1/100,000 (dotted line).
An important caveat is that with extreme accuracy comes new challenges from non-sequencing sources of errors. Certain artefacts that may be negligibly uncommon with standard NGS become significant when the sequencing background is reduced. For example, rare mapping errors of closely related pseudogenes or low-level cross contamination among samples during tissue processing can artificially appear as low-frequency variants. Slight carry-over between sequencer runs or amplification chimeras that lead to index shuffling37,51 can falsely make a clonal variant from one sample appear as a low-frequency mutation in another. Furthermore, when technical background is reduced, newly apparent rare variants resulting from one process can easily be mis-assumed to be the result of another. For example, mutations commonly found in cancers can be detected at very low levels as part of normal ageing in healthy individuals and be a source of false positives in sensitive diagnostic tests107. High-accuracy sequencing is a powerful tool but necessitates particularly thorough controls and well thought out experimental designs and interpretations.
As a final note, we wish to emphasize that the above descriptions and illustrations in Figures 2 and 3 are highly simplified representations of complex techniques and do not fully convey many of the informatic subtleties of analysis. For a better appreciation, we refer interested readers to a select list of relevant software packages at the end of this article and those included as supplementary information in cited papers.
Applications of rare-variant detection
The history of innovations in NGS, and technologies for improving its accuracy are best understood in the context of scientific questions they have been developed to answer. In this section we highlight major fields in which the detection of minority genetic variants is important for medicine, biology and industry and provide examples of where the different approaches described above have been (or could be) applied (Figure 5).
Figure 5. Applications of rare variant detection.
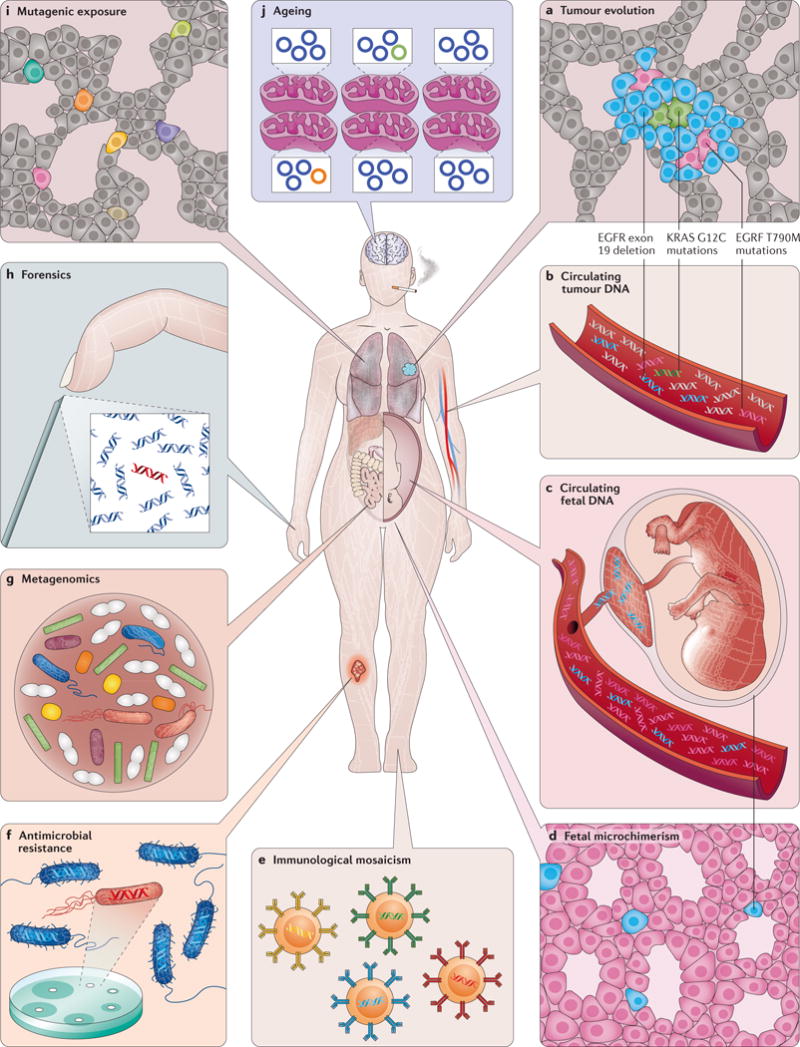
a| Cancer. Genetic heterogeneity within tumours is thought to be responsible for the emergence of therapeutic resistance. In lung adenocarcinomas with certain epidermal growth factor receptor (EGFR) mutations, under treatment with targeted inhibitors, drug resistance mutations arise at low levels then clonally expand. b | Cell-free tumour DNA. Tumour cells release fragments of DNA into plasma and other body fluids that can be sampled via ‘liquid biopsy’. This serves as a non-invasive means of determining the genetic makeup of a tumour without a physical biopsy and is a sensitive way to detect minimal residual disease and early relapse. c | Circulating fetal DNA. Placental-derived DNA in the maternal circulation can be used to non-invasively detect fetal genetic traits or abnormalities. d | Fetal microchimerism. Fetal cells that engraft into a mother may persist many years after birth. These have important immunological consequences. e | Immunological mosaicism. Somatic V(D)J recombination and hypermutation in B and T cells create heterogeneity that helps the body adapt defences to new infectious and neoplastic threats. f | Antimicrobial resistance. Low-frequency variants in single-cell populations can be responsible for drug-resistance outbreaks g | Metagenomics. Complex mixtures of microorganisms exist throughout the living world. The human body is colonized with symbiotic microbes and in some diseases, health problems can arise from disrupted microbial diversity. h | Forensics. Mixtures of human tissues are routinely recovered at crime scenes or natural disasters. In some scenarios the abundance of one individual’s DNA may be much greater than the other. i | Mutational exposure. DNA damage can be caused by normal ageing as well as carcinogens. Very-low-frequency mutation load may be proportional to future cancer risk. j | Ageing. DNA damage occurs throughout life from exogenous and endogenous processes. Low-frequency mutations in both the nuclear and mitochondrial genome (the latter is shown here) may play a role in certain age-related pathologies besides cancer, such as neurodegeneration and autoimmunity. Subclonal mutations might serve as a biomarker of disease risk or even longevity.
Cancer
Cancer is the ultimate disease of genetic heterogeneity3,108. In the past decade, an understanding of how it arises, progresses and spreads in the context of somatic evolution has been increasingly recognized109–113. The cells in a tumour are not uniform. Mutations arising during cell division under the influence of selective pressures as well as random drift, can lead to the outgrowth of genetically divergent clones in spatially distinct areas of a primary tumour114–117 and derivative metastases118,119. Minority clones, which may be present at frequencies below the detection limits of conventional NGS techniques, can both drive tumour growth120, and be an important source of resistance to therapy and basis for relapse5,121–123; in fact a higher degree of subclonal heterogeneity portends a worse prognosis in multiple tumour types124–126.
A particular challenge has been developing technologies that are robust enough for resolving heterogeneity in the clinical setting. Pre-existing subclonal drug-resistance mutations in blood cancers have implications for the choice of initial therapy127,128. After treatment, detection of rare cells with leukaemia-associated mutations using consensus sequencing indicates minimal residual disease (MRD) and the need for further treatment129. In solid tumours, subclonal drug-resistance mutations are similarly relevant, but may be missed by physical biopsy130. An intense area of research is use of ‘liquid biopsies’ to non-invasively genotype cell-free DNA (cfDNA) shed by tumours into plasma, which allows genetic sampling of more than one region of a cancer in a way that can be readily repeated as a tumour evolves104,131–133. Liquid biopsies are being used to detect both drug-sensitizing and drug-resistance mutations134, low-level MRD after surgery135,136, and to follow response to treatment137. Tumour DNA can be found at low abundance in many body fluids138–142. In each case, consensus sequencing-based error-correction approaches (either single-strand, duplex or both) have been demonstrated to improve the detection of rare subclonal mutants. Mutations in a variety of body fluids have been used for specific cancer type screening in asymptomatic individuals for some time140,141,143, but an even more ambitious prospect is the possibility of a universal pan-cancer screening blood test144. To achieve the exceptionally low false-positive rate needed for use in healthy populations, especially given the breadth of the genome that must be examined at high depth to identify very early tumours, near-perfect technical accuracy is required using the most robust error-corrected sequencing methods possible.
Ageing
Mutations occur with each cell division, thus it is no surprise that they increase with age8. A long-standing and unanswered question is whether these reflect cause or effect of ageing; regardless, the association that has been found using duplex sequencing approaches is strong61,80. Subclonal mutations have been associated with the onset of age-associated pathologies such as neurodegeneration7,82. The greatest risk factor for cancer, a disease caused by mutations, is ageing145. Studies using conventional NGS have found clones bearing leukaemia-associated mutations in the blood of a subset of healthy individuals, at a size and frequency that increases with age146,147. Higher-accuracy single-strand consensus sequencing methods have identified these mutations at lower frequency in nearly all adults148. Using ultrahigh-accuracy duplex consensus sequencing approaches, low-frequency age-associated mutations have been directly measured in multiple human tissues61, many of which are common to cancers149,150. This highlights both the novel discoveries that comes with greater accuracy and the new challenges the knowledge brings, for example, in mutation-based cancer screening107. Whether age-associated subclonal mutation patterns will be able to predict future cancer risk or longevity remains to be explored.
Mutagenesis
Our bodies are exposed to endogenous and exogenous mutagens throughout life. It has been long known, using artificial selection-based assays, that mutations can be induced by genotoxic chemicals, but only recently has it been possible to detect these directly60,61. Instead of being limited to reporter genes, the highest accuracy duplex sequencing approaches can evaluate mutagenesis at any genomic site of any organism, and has the potential to become a new standard of genotoxicity evaluation85,102,151–153. New methods of therapeutic genome modification can lead to rare off-target mutagenesis through mechanisms that are not easily recapitulated in model organisms154. The same holds true for treatments that are not intended to modify the genome but with the theoretical potential for mutagenesis155. Emerging cellular therapies where stem cells are harvested and propagated ex vivo can introduce substantial numbers of mutations21. Given the rapid emergence of new medical technologies, it is important that we have equally powerful tools to carefully monitor their genetic consequences.
Maternal–fetal biology
Pregnancy is a state of chimerism: two genomes inhabit the same body and intermix. Even after birth, a mother and child remain genetically intertwined. Extremely rare populations of fetal cells can persist in a woman for decades156. This interesting state of prolonged immunotolerance may play a contributory role in autoimmune disease, miscarriages and pre-eclampsia, but also appears to confer certain benefits, particularly cancer protection. Until recently, genetic techniques for studying these rare populations of cells have been inadequate157. During pregnancy itself, apoptosis of placental cells releases fetal DNA into the maternal circulation where it can be collected for non-invasive prenatal testing (NIPT). Fetal aneuploidies can be clinically detected by NGS with relative technical ease by simply counting the relative proportions of different chromosomes, even though fetal DNA is in the minority158,159. In fact, one early indication of the feasibility of DNA-based cancer screening was the incidental detection of maternal cancers from non-fetal chromosomal imbalances found during NIPT160. Complete fetal genomes have been assembled from sequencing of cfDNA in maternal blood based on paternally inherited single nucleotide polymorphisms (SNPs), albeit using low-accuracy conventional NGS. To become viable as a clinical means of prenatally detecting de novo point mutations, ultra-accurate methods will need to be employed. This is particularly true of disease-causing mutations that arise mosaically during embryogenesis161,162. A distinct but related application of identifying foreign SNPs in cell-free DNA is the early detection of organ transplant rejection163.
The immune system
An individual’s immune system is finely tuned for generating genetic heterogeneity through sanctioned V(D)J recombination and somatic hypermutation; a single blood sample may contain hundreds of thousands of different T-cell receptor and immunoglobulin sequences164. This genetic profile changes in response to infections, immunization and age, among many other states165,166. The adaptive immune system plays a role in defence against neoplasia, and the genetic pattern of tumour-infiltrating lymphocytes (TILs) can reflect both prognosis and the likelihood of response to therapy166,167. On the other end of the immune spectrum is overactivity. It has been theorized that some autoimmune disease may develop in response to subclonal somatic mutations in non-immune tissues168. Chronic stimulation of the immune system also causes low-frequency off-target somatic hypermutation in lymphocytes themselves, leading to lymphoma169. In each category, high-accuracy consensus sequencing methods of multiple varieties are proving critical for disentangling the immunologic heterogeneity of both physiological and pathological states.
Microbial populations
Our bodies contain at least as many microbes as human cells. Although some can cause disease, a greater number serve symbiotic functions such as building or metabolizing molecules, outcompeting pathogenic organisms and training our immune systems170,171. Colonization begins even before birth, and the stability of the ecosystem that develops influences health processes including allergy and autoimmunity172, body weight173 and response to medications174, among others. Early successes with therapeutic microbiota transplantation further indicates the importance of these populations175,176.
The study of the composition of metagenomics ensembles with traditional techniques is not trivial. Some organisms grow poorly in culture or not at all. Viruses, fungi, protozoa and highly divergent phyla of bacteria that co-exist in vivo often require unique growth conditions in vitro. In the case of suspected infections, being able to quickly identify the causative agent(s) and predict which therapies will be most efficacious is clinically important, but not always possible. Whereas clinical decisions about antimicrobials often must be made in minutes to hours, traditional culture takes hours to days, and in some cases, even weeks177. NGS has emerged as a powerful and universal means of characterizing pan-kingdom microbial populations178. High-speed NGS workflows can now identify organisms causing sepsis and predict their drug sensitivity in a matter of hours179,180.
However, speed is not the only challenge in clinical microbial sequencing. When the genomes of organisms in a polymicrobial sample are very distinct, standard NGS can readily identify minority populations. When a population’s constituents are genetically similar, however, sequencing errors prevent variant detection below about 1%. Because the evolutionary success of many pathogens is predicated on rapid evolvability to circumvent host immune responses, many mutate readily to form heterogeneous populations of closely related, but genetically distinct members, collectively known as a quasispecies181,182. Deep sequencing has shown that the emergence of low-level drug-resistance mutations in multiple types of infections including HIV4,13,183, viral hepatitis184,185, and tuberculosis186 can predict therapeutic failure. As in oncology, consensus-based NGS error-correction approaches enables detection of rarer drug-resistance mutations56, potentially affording the opportunity for earlier interventions if found. Perhaps just as importantly, the ability to confidently recognize the absence of such mutations might allow one to avoid unnecessary use of broad-spectrum agents.
Other applications
The utility of deconvolving heterogeneous, closely related mixtures of nucleic acids extends beyond medicine. Of the innumerable free-living microorganisms in the natural world, the majority have never been isolated or cultured187. Direct high-accuracy NGS of environmental samples is an important tool for identifying novel protein variants with potential industrial applications188, for monitoring ecological health189 and facilitating food safety testing, including the detection of low-frequency drug-resistant microbial strains190. In forensics, the ability to confidently distinguish mixtures of DNA contributed from different individuals is critical, given the consequence of mistakes106,191. High-accuracy sequencing methods are especially important when heterogeneous mixed samples are degraded, and therefore more error-prone, as may occur at both modern crime scenes and with ancient DNA from archeological sites192.
Conclusions
Within the span of a single human lifetime we have gone from recognizing DNA as genetic material193 to sequencing the human genome194. Despite this remarkable progress, many opportunities remain. For example, the structure of certain highly repetitive regions in many organisms is still unknown. In fact, for this reason we have yet to assemble even one genuinely complete human genome. Both emerging long-read technologies and techniques for phasing and accurately assembling short-read data using fragment barcoding methods72,195 or DNA cross-linking techniques196 are bringing us closer, but are not yet accurate enough to reliably detect very-low-frequency subclonal heterogeneity of structural variation, despite the clinical importance in both the somatic and neoplastic settings197,198. Another substantial challenge on the horizon is combining ultra-accurate single-cell genomic information with other types of ‘omic’ technologies and contextualizing with higher-order topological relationships between individual cells within a tissue (Box 1).
We began this Review by enumerating NGS technologies that have had a major impact on the ability to detect subclonal variants, and discussed the many fields in which NGS has or will soon benefit from this high accuracy. Biological and clinical questions that were previously intractable can now be approached. A meaningful vision of the future should not only focus on improvements to established areas, but also address the role of high-accuracy DNA sequencing in delineating new and important hypotheses. Sequencing technology is advancing more quickly than ever before. Just as evolution is the driving force behind the progression of all life on earth, so too is it with technology: the popularity of new sequencing methods will continue to rise and fall but the discoveries that come from each will remain immortal. It is important to remember that methods are merely tools, not themselves answers; it is on the creative and responsible applications of these new technologies where we should focus our greatest attention.
Box 1. The future of DNA sequencing: complete genomics and multi-omics.
Being able to resolve heterogeneity amongst an aggregate collection of molecules is, itself, not sufficient to deeply understand evolving populations. Mutations do not act in isolation: the phenotype they confer often depends on the sequence of other genes and regulatory elements sharing the same cell. Methods for high-throughput identical barcoding of all the molecules from single cells using flow sorting or droplet compartmentalization are becoming widespread for both DNA and RNA199,200. Use of endogenous cell membranes as individual barcoding reaction chambers is another high-throughput means of single-cell molecular labelling201. We anticipate that the combination of high-throughput compartmentalization methods with ultra-long-read single-molecule sequencing technologies that incorporate duplex consensus-based error correction will enable truly whole-genome sequencing of large populations of single cells in the foreseeable future.
Single-cell transcriptomics200,201 and proteomics202 can be achieved using high-throughput sequencing technologies. DNA carries important epigenetic information beyond its primary sequence in the form of methylation, other non-canonical nucleotides, chromatin structure and nuclear co-localization patterns203. In RNA, More than 100 types of naturally occurring chemical modifications have been described204. All of these plus other emerging ‘omic’ technologies have been applied to single cells but have yet to be meaningfully combined. Amassing this parallel data from individual cells while retaining information about their relative spatial relationships in their native 3D tissue context will be a herculean undertaking. Perhaps the greatest challenge to consider is simultaneously capturing the fourth dimension: nearly all current genomic methods represent finite snapshots in time, rather than a temporally dynamic measurement. Whether we will ever be able to non-disruptively determine the sequence the complete in vivo genomes and epigenomes of billions of intact cells in a living organism is probably question for our children, or perhaps our children’s children.
Key points.
The ability to identify low frequency genetic variants amongst heterogeneous populations of cells or DNA molecules is important in many fields of basic science, clinical medicine and other applications, yet current high-throughput DNA sequencing technologies have an error rate between one-per-hundred and one-per-thousand, which obscures their presence below this level.
As next-generation-sequencing technologies evolved over the decade, throughput has improved markedly, but raw accuracy has remained generally flat. Those with high accuracy needs developed data filtering methods and incremental biochemical improvements that improve low frequency variant detection modestly, but background errors remain limiting in many fields.
The most profoundly impactful means for reducing errors, first developed about 7 years ago, has been the concept of single molecule consensus sequencing. This entails redundant sequencing of multiple copies of a given specific DNA molecule and discounting of variants that are not present in all or most of the copies as likely errors.
Consensus sequencing can be achieved by labeling of each molecule with a unique molecular barcode before generating copies, to allow subsequent and comparison of these copies, or schemes whereby copies are physically joined and sequenced together. Because of tradeoffs in cost, time and accuracy, no single method is optimal for every application and each should be considered on a case-by-case basis.
Major applications for high accuracy DNA sequencing include non-invasive cancer diagnostics, cancer screening, early detection of cancer relapse or impending drug resistance, infectious disease applications, prenatal diagnostics, forensics and mutagenesis assessment.
Future advances in ultra-high accuracy sequencing are likely to be driven by an emerging generation of single molecule sequencers, particularly those which allow independent sequence comparison of both strands if native DNA duplexes
Acknowledgments
We thank Rosana Risques, Joe Hiatt and Aaron Boswell for critical review, Edward Fox, Michael Schmitt and Eun Hyun Ahn for contributions to early drafts, Katy Loubet-Senear, Rosana Risques and Mary Emond for graphics ideas, Nils Homer and Clint Valentine for software information and members of the Loeb, Kennedy and Risques labs at the University of Washington for many lively discussions. This work was supported by NIH grants T32CA009515 (J.J.S) and R01CA193649, P01CA77852, and R33CA181771 (L.A.L.).
GLOSSARY DEFINITIONS
- Clonal
When referring to a genetic variant or mutation, it is one that is present in all or most molecules in a population being sequenced. The term typically implies that it arose from a common ancestor, such as fertilized egg in the case of germline variation, or the earliest founder cell of tumour
- Subclonal
When referring to a genetic variant or mutation, it is one that is present in only a subset of molecules being sequenced. This may refer to either a variant carried by a subpopulation that arose and expanded within a larger population, or through mixing of two or more distinct populations
- Sequencing accuracy
The number of errors made per basepair sequenced. It may be stratified by subtype of error, such as a specific type of base substitution
- Sequencing sensitivity
The ability to detect a variant at a particular variant allele frequency. This depends on both the sequencing accuracy and the number of independent DNA molecules successfully sequenced that include the genomic position (or positions) of interest
- Variant allele frequency
(VAF). The fraction of all molecules being sequenced that carry a specific genetic change or mutation at a particular genomic position
- Digital PCR
DNA amplification carried out in single-molecule reaction chambers. Recently this has most often entailed microscopic aqueous droplets immersed in oil. When DNA input is sufficiently low, only one molecule will seed each reaction. When allele-specific amplification conditions are used, the number of droplets that successfully amplify can be digitally tabulated to determine the variant allele frequency
- Polony
A population of identical amplification copies that originated from a single founder molecule and are spatially co-localized, such as on the surface of a microbead or as a spot on a surface. It is the biochemical analogue of a bacterial colony on a petri dish
- Short-read platforms
Next-generation sequencing systems that generate reads that are dozens to several hundred nucleotides in length. For example the current Illumina and Thermo Ion Torrent platforms and previously manufactured Roche 454 and ABI SOLiD. Current versions sequence amplified polonies, not single molecules
- Long-read platforms
Next-generation sequencing systems that generate reads that are thousands to tens of thousands of nucleotides in length. These currently include Pacific Biosciences (PacBio) and Oxford Nanopore which sequence single molecules, not polonies, and therefore have a higher error-rate than short-read platforms
- Molecular barcode
A set of DNA nucleotide codes where each is affixed to only one or a subset of individual DNA molecules within a sample. The purpose is to uniquely label single molecules for consensus-based error correction or molecular counting. These may be informatically combined with molecule fragmentation points for greater label diversity
- Index sequence
A particular DNA nucleotide code affixed to all molecules within a given DNA sample. The purpose is for multiplexing samples on a single sequencer run
- Tag-based error correction
Also known as consensus sequencing, an approach for error correction whereby individual DNA molecules are uniquely labelled prior to amplification and sequencing and then the sequences of the related derivative copies are compared with each other to exclude errors
- Sequencing depth
The number of sequencing reads that include a particular genomic position in their sequence. Some may be simply PCR copies of the same molecule
- Molecular depth.
The number of collapsed consensus reads derived from an independent DNA molecule that include a particular genomic position.
- Tag clashes
The occurrence of two independent molecules being identically labelled by random chance. This may happen if the diversity of applied molecular barcodes is too low for the number of DNA molecules sequenced. True mutations may erroneously be excluded
- False families
Sets of related molecules where an error has occurred during amplification that mutates the common tag sequence to erroneously make it appear that two independent molecules gave rise to a these molecules
- Consensus-making efficiency
The number of raw sequencing reads that are required to form a consensus read. This typically refers to an average: total raw reads divided by total consensus reads
- Molecular conversion efficiency
The fraction of inputted DNA molecules of interest that are recovered as consensus sequences. This is often described in terms of genome-equivalents
- Aneuploidy
An abnormal number of chromosomes in a cell. This may be inherited, such as trisomy 21, the basis of Down syndrome, or somatically acquired, such as in cancer
- Metagenomics
The study of complex microbial populations encompassing many co-mingling species that form an ecosystem; for examplean individual’s gut microbiota
- Phasing
The proper assignment of two or more variants at spatially distant genomic locations to the derivative nucleic acid molecule; for examplethe maternal or paternal allele
Footnotes
FURTHER INFORMATION:
Aryee-Lab: https://github.com/aryeelab/umi
IDES: https://cappseq.stanford.edu/ides/download.php
Connor tools: https://github.com/umich-brcf-bioinf/Connor
CRISPR-DS: https://github.com/risqueslab/CRISPR-DS
DuNovo: https://github.com/galaxyproject/dunovo
DuplexSeq: https://github.com/loeblab/Duplex-Sequencing
Ferm-lite: https://github.com/lh3/fermi-lite
FgBio: https://github.com/fulcrumgenomics/fgbio
MAGERI: https://github.com/mikessh/mageri
Presto: https://bitbucket.org/kleinstein/presto/src
Strand: http://www.strand-ngs.com
UMI-tools: https://github.com/CGATOxford/UMI-tools
Author contributions
Both authors contributed to discussion of content, and reviewing and editing the manuscript before submission. J.J.S. was primarily responsible for researching data and writing the manuscript.
References
- 1.Darwin C. On the origin of species. John Murray Press; 1859. [Google Scholar]
- 2.Luria SE, Delbrück M. Mutations of Bacteria from Virus Sensitivity to Virus Resistance. Genetics. 1943;28:491–511. doi: 10.1093/genetics/28.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cairns J. Mutation selection and the natural history of cancer. Nature. 1975;255:197–200. doi: 10.1038/255197a0. [DOI] [PubMed] [Google Scholar]
- 4.Fisher R, et al. Deep sequencing reveals minor protease resistance mutations in patients failing a protease inhibitor regimen. J Virol. 2012;86:6231–6237. doi: 10.1128/JVI.06541-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmitt MW, Loeb LA, Salk JJ. The influence of subclonal resistance mutations on targeted cancer therapy. Nat Rev Clin Oncol. 2016;13:335–347. doi: 10.1038/nrclinonc.2015.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maher GJ, et al. Visualizing the origins of selfish de novo mutations in individual seminiferous tubules of human testes. Proc Natl Acad Sci USA. 2016;113:2454–2459. doi: 10.1073/pnas.1521325113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kennedy SR, Loeb LA, Herr AJ. Somatic mutations in aging, cancer and neurodegeneration. Mech Ageing Dev. 2012;133:118–126. doi: 10.1016/j.mad.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vijg J. Somatic mutations, genome mosaicism, cancer and aging. Curr Opin Genet Dev. 2014;26:141–149. doi: 10.1016/j.gde.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shendure J, et al. DNA sequencing at 40: past, present and future. Nature. 2017;550:345–353. doi: 10.1038/nature24286. [DOI] [PubMed] [Google Scholar]
- 10.Goodwin S, Mcpherson JD, Mccombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17:333–351. doi: 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. Sanger 1977: One of two Nobel prize-winning DNA sequencing methodologies published in 1977 (the other being that of Maxam and Gilbert). The Sanger approach formed the basis of The Human Genome Project. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ley TJ, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zagordi O, Klein R, Däumer M, Beerenwinkel N. Error correction of next-generation sequencing data and reliable estimation of HIV quasispecies. Nucleic Acids Research. 2010;38:7400–7409. doi: 10.1093/nar/gkq655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parsons BL, Heflich RH. Genotypic selection methods for the direct analysis of point mutations. Mutat Res. 1997;387:97–121. doi: 10.1016/s1383-5742(97)00026-4. [DOI] [PubMed] [Google Scholar]
- 15.Bielas JH, Loeb LA. Quantification of random genomic mutations. Nat Methods. 2005;2:285–290. doi: 10.1038/nmeth751. [DOI] [PubMed] [Google Scholar]
- 16.Li J, et al. Replacing PCR with COLD-PCR enriches variant DNA sequences and redefines the sensitivity of genetic testing. Nat Med. 2008;14:579–584. doi: 10.1038/nm1708. [DOI] [PubMed] [Google Scholar]
- 17.Sykes PJ, et al. Quantitation of targets for PCR by use of limiting dilution. BioTechniques. 1992;13:444–449. [PubMed] [Google Scholar]
- 18.Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci USA. 1999;96:9236–9241. doi: 10.1073/pnas.96.16.9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hindson BJ, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fox EJ, Reid-Bayliss KS, Emond MJ, Loeb LA. Accuracy of Next Generation Sequencing Platforms. Next Gener Seq Appl. 2014;1 doi: 10.4172/jngsa.1000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blokzijl F, et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature. 2016;538:260–264. doi: 10.1038/nature19768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. Ewing 1998: Among the first and most important uses of rigorous statistical methods to assign degree of certainty to DNA sequencing data. [PubMed] [Google Scholar]
- 23.Cock PJA, Fields CJ, Goto N, Heuer ML, Rice PM. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Research. 2010;38:1767–1771. doi: 10.1093/nar/gkp1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cibulskis K, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koboldt DC, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Q, et al. Detecting somatic point mutations in cancer genome sequencing data: a comparison of mutation callers. Genome Med. 2013;5:91. doi: 10.1186/gm495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013 doi:arXiv:1303.3997v2 [q-bio.GN] [Google Scholar]
- 28.Wei Z, Wang W, Hu P, Lyon GJ, Hakonarson H. SNVer: a statistical tool for variant calling in analysis of pooled or individual next-generation sequencing data. Nucleic Acids Research. 2011;39:e132–e132. doi: 10.1093/nar/gkr599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilm A, et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Research. 2012;40:11189–11201. doi: 10.1093/nar/gks918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gerstung M, et al. Reliable detection of subclonal single-nucleotide variants in tumour cell populations. Nat Commun. 2012;3:811. doi: 10.1038/ncomms1814. [DOI] [PubMed] [Google Scholar]
- 31.Costello M, et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Research. 2013;41:e67–e67. doi: 10.1093/nar/gks1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen L, Liu P, Evans TC, Ettwiller LM. DNA damage is a pervasive cause of sequencing errors, directly confounding variant identification. Science. 2017;355:752–756. doi: 10.1126/science.aai8690. [DOI] [PubMed] [Google Scholar]
- 33.Schirmer M, D’Amore R, Ijaz UZ, Hall N, Quince C. Illumina error profiles: resolving fine-scale variation in metagenomic sequencing data. BMC Bioinformatics. 2016;17:125. doi: 10.1186/s12859-016-0976-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martincorena I, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. 2015;348:880–886. doi: 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Welch JS, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nik-Zainal S, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534:47–54. doi: 10.1038/nature17676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kircher M, Sawyer S, Meyer M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Research. 2012;40:e3–e3. doi: 10.1093/nar/gkr771. Kircher 2012: An important description of the commonness of PCR chimeras that form during NGS library preparation. This contributed to the now common practice of dual indexing for error-sensitive applications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Potapov V, Ong JL. Examining Sources of Error in PCR by Single-Molecule Sequencing. PLoS ONE. 2017;12:e0169774. doi: 10.1371/journal.pone.0169774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brodin J, et al. PCR-induced transitions are the major source of error in cleaned ultra-deep pyrosequencing data. PLoS ONE. 2013;8:e70388. doi: 10.1371/journal.pone.0070388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Star B, et al. Palindromic sequence artifacts generated during next generation sequencing library preparation from historic and ancient DNA. PLoS ONE. 2014;9:e89676. doi: 10.1371/journal.pone.0089676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Allen EM, et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat Med. 2014;20:682–688. doi: 10.1038/nm.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arbeithuber B, Makova KD, Tiemann-Boege I. Artifactual mutations resulting from DNA lesions limit detection levels in ultrasensitive sequencing applications. DNA Res. 2016;23:547–559. doi: 10.1093/dnares/dsw038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lindahl T, Nyberg B. Rate of depurination of native deoxyribonucleic acid. Biochemistry. 1972;11:3610–3618. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- 44.Knierim E, Lucke B, Schwarz JM, Schuelke M, Seelow D. Systematic comparison of three methods for fragmentation of long-range PCR products for next generation sequencing. PLoS ONE. 2011;6:e28240. doi: 10.1371/journal.pone.0028240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Do H, Dobrovic A. Sequence artifacts in DNA from formalin-fixed tissues: causes and strategies for minimization. Clinical Chemistry. 2015;61:64–71. doi: 10.1373/clinchem.2014.223040. [DOI] [PubMed] [Google Scholar]
- 46.Lou DI, et al. High-throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing. Proc Natl Acad Sci USA. 2013;110:19872–19877. doi: 10.1073/pnas.1319590110. Lou 2013: The first significant description of consensus sequencing by tandem duplication of library molecules. Although challenging on short-read sequencers, this concept is likely to become very important as single-molecule sequencers improve in the coming years. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen G, Mosier S, Gocke CD, Lin MT, Eshleman JR. Cytosine deamination is a major cause of baseline noise in next-generation sequencing. Mol Diagn Ther. 2014;18:587–593. doi: 10.1007/s40291-014-0115-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schaaper RM, Kunkel TA, Loeb LA. Infidelity of DNA synthesis associated with bypass of apurinic sites. Proc Natl Acad Sci USA. 1983;80:487–491. doi: 10.1073/pnas.80.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sagher D, Strauss B. Insertion of nucleotides opposite apurinic/apyrimidinic sites in deoxyribonucleic acid during in vitro synthesis: uniqueness of adenine nucleotides. Biochemistry. 1983;22:4518–4526. doi: 10.1021/bi00288a026. [DOI] [PubMed] [Google Scholar]
- 50.Nishimura S. 8-Hydroxyguanine: a base for discovery. DNA repair. 2011;10:1078–1083. [PubMed] [Google Scholar]
- 51.Sinha R, et al. Index Switching Causes ‘Spreading-Of-Signal’ Among Multiplexed Samples In Illumina HiSeq 4000 DNA Sequencing. 2017 doi: 10.1101/125724. [DOI] [Google Scholar]
- 52.Hiatt JB, Turner EH, Patwardhan RP, Caperton L, Shendure J. Next-generation DNA Sequencing for De Novo Genome Assembly. Western Student Medical Research Forum. 2009 [Google Scholar]
- 53.Hiatt JB, Patwardhan RP, Turner EH, Lee C, Shendure J. Parallel, tag-directed assembly of locally derived short sequence reads. Nat Methods. 2010;7:119–122. doi: 10.1038/nmeth.1416. Hiatt 2010: The first description of consensus sequencing PCR duplicates for error correction, both with UMIs and without. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Casbon JA, Osborne RJ, Brenner S, Lichtenstein CP. A method for counting PCR template molecules with application to next-generation sequencing. Nucleic Acids Research. 2011;39:e81–e81. doi: 10.1093/nar/gkr217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108:9530–9535. doi: 10.1073/pnas.1105422108. Kinde 2011: A key early description of single-strand tag-based error correction for rare variant detection. This publication put the significance in clinical context and was probably the most important launch for the field. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jabara CB, Jones CD, Roach J, Anderson JA, Swanstrom R. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc Natl Acad Sci USA. 2011;108:20166–20171. doi: 10.1073/pnas.1110064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fu GK, Hu J, Wang PH, Fodor SPA. Counting individual DNA molecules by the stochastic attachment of diverse labels. Proc Natl Acad Sci USA. 2011;108:9026–9031. doi: 10.1073/pnas.1017621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kivioja T, et al. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods. 2011;9:72–74. doi: 10.1038/nmeth.1778. [DOI] [PubMed] [Google Scholar]
- 59.Shiroguchi K, Jia TZ, Sims PA, Xie XS. Digital RNA sequencing minimizes sequence-dependent bias and amplification noise with optimized single-molecule barcodes. Proc Natl Acad Sci USA. 2012;109:1347–1352. doi: 10.1073/pnas.1118018109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schmitt MW, et al. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci USA. 2012;109:14508–14513. doi: 10.1073/pnas.1208715109. Schmitt 2012: The initial description of Duplex Sequencing and the concept of labelling copies of both strands of individual double-stranded molecules to allow them to be sequenced and compared for even greater accuracy. This technique opened the door to investigations of ultra-rare variants, such as those that occur in ageing and with mutagenic chemical exposure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hoang ML, et al. Genome-wide quantification of rare somatic mutations in normal human tissues using massively parallel sequencing. Proc Natl Acad Sci USA. 2016;113:9846–9851. doi: 10.1073/pnas.1607794113. Hoang 2016: A duplex sequencing approach at very low depth and not requiring exogenous UMIs. An excellent example of genotoxicity and ageing applications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nachmanson D, et al. CRISPR-DS: an efficient, low DNA input method for ultra-accurate sequencing. bioRxiv. 2017 doi: 10.1101/207027. [DOI]
- 63.Liang RH, et al. Theoretical and experimental assessment of degenerate primer tagging in ultra-deep applications of next-generation sequencing. Nucleic Acids Research. 2014;42:e98–e98. doi: 10.1093/nar/gku355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang TH, Wu NC, Sun R. A benchmark study on error-correction by read-pairing and tag-clustering in amplicon-based deep sequencing. BMC Genomics. 2016;17:108. doi: 10.1186/s12864-016-2388-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith T, Heger A, Sudbery I. UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017;27:491–499. doi: 10.1101/gr.209601.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ståhlberg A, et al. Simple, multiplexed, PCR-based barcoding of DNA enables sensitive mutation detection in liquid biopsies using sequencing. Nucleic Acids Research. 2016;44:e105–e105. doi: 10.1093/nar/gkw224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ståhlberg A, et al. Simple multiplexed PCR-based barcoding of DNA for ultrasensitive mutation detection by next-generation sequencing. Nature Protocols. 2017;12:664–682. doi: 10.1038/nprot.2017.006. [DOI] [PubMed] [Google Scholar]
- 68.Hiatt JB, Pritchard CC, Salipante SJ, O’Roak BJ, Shendure J. Single molecule molecular inversion probes for targeted, high accuracy detection of low frequency variation. Genome Res. 2013 doi: 10.1101/gr.147686.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carlson KD, et al. MIPSTR: a method for multiplex genotyping of germline and somatic STR variation across many individuals. Genome Res. 2015;25:750–761. doi: 10.1101/gr.182212.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Boyle EA, O’Roak BJ, Martin BK, Kumar A, Shendure J. MIPgen: optimized modeling and design of molecular inversion probes for targeted resequencing. Bioinformatics. 2014;30:2670–2672. doi: 10.1093/bioinformatics/btu353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang K, et al. Ultra-precise detection of mutations by droplet-based amplification of circularized DNA. BMC Genomics. 2016;17:214. doi: 10.1186/s12864-016-2480-1. Wang 2016: An important description of several biochemical techniques to improve consensus making efficiency and reduce cost. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hong LZ, et al. BAsE-Seq: a method for obtaining long viral haplotypes from short sequence reads. Genome Biol. 2014;15:517. doi: 10.1186/s13059-014-0517-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schmitt MW, Fox EJ, Salk JJ. Risks of double-counting in deep sequencing. Proc Natl Acad Sci USA. 2014;111:E1560–E1560. doi: 10.1073/pnas.1400941111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hong J, Gresham D. Incorporation of unique molecular identifiers in TruSeq adapters improves the accuracy of quantitative sequencing. BioTechniques. 2017;63:221–226. doi: 10.2144/000114608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Narayan A, et al. Ultrasensitive measurement of hotspot mutations in tumor DNA in blood using error-suppressed multiplexed deep sequencing. Cancer Res. 2012;72:3492–3498. doi: 10.1158/0008-5472.CAN-11-4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gregory MT, et al. Targeted single molecule mutation detection with massively parallel sequencing. Nucleic Acids Research. 2016;44:e22–e22. doi: 10.1093/nar/gkv915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pel J, et al. Duplex Proximity Sequencing (Pro-Seq): A method to improve DNA sequencing accuracy without the cost of molecular barcoding redundancy. bioRxiv. 2017;163444 doi: 10.1101/163444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kennedy SR, et al. Detecting ultralow-frequency mutations by Duplex Sequencing. Nature Protocols. 2014;9:2586–2606. doi: 10.1038/nprot.2014.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Roach JC, et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kennedy SR, Salk JJ, Schmitt MW, Loeb LA. Ultra-Sensitive Sequencing Reveals an Age-Related Increase in Somatic Mitochondrial Mutations That Are Inconsistent with Oxidative Damage. PLoS Genet. 2013;9:e1003794. doi: 10.1371/journal.pgen.1003794. Kennedy 2013: The first description of high-accuracy consensus sequencing to measure the effect of human ageing on somatic mutation load. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Taylor PH, Cinquin A, Cinquin O. Quantification of in vivo progenitor mutation accrual with ultra-low error rate and minimal input DNA using SIP-HAVA-seq. Genome Res. 2016;26:1600–1611. doi: 10.1101/gr.200501.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hoekstra JG, Hipp MJ, Montine TJ, Kennedy SR. Mitochondrial DNA mutations increase in early stage Alzheimer disease and are inconsistent with oxidative damage. Ann Neurol. 2016;80:301–306. doi: 10.1002/ana.24709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pickrell AM, et al. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron. 2015;87:371–381. doi: 10.1016/j.neuron.2015.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Reid-Bayliss KS, Arron ST, Loeb LA, Bezrookove V, Cleaver JE. Why Cockayne syndrome patients do not get cancer despite their DNA repair deficiency. Proc Natl Acad Sci USA. 2016;113:10151–10156. doi: 10.1073/pnas.1610020113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chawanthayatham S, et al. Mutational spectra of aflatoxin B1 in vivo establish biomarkers of exposure for human hepatocellular carcinoma. Proc Natl Acad Sci USA. 2017;114:E3101–E3109. doi: 10.1073/pnas.1700759114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mattox AK, et al. Bisulfite-converted duplexes for the strand-specific detection and quantification of rare mutations. Proc Natl Acad Sci USA. 2017;114:4733–4738. doi: 10.1073/pnas.1701382114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kumar V, et al. Partial bisulfite conversion for unique template sequencing. Nucleic Acids Research. 2017 doi: 10.1093/nar/gkx1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Deamer D, Akeson M, Branton D. Three decades of nanopore sequencing. Nat Biotechnol. 2016;34:518–524. doi: 10.1038/nbt.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eid J, et al. Real-time DNA sequencing from single polymerase molecules. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 90.Madoui MA, et al. Genome assembly using Nanopore-guided long and error-free DNA reads. BMC Genomics. 2015;16:327. doi: 10.1186/s12864-015-1519-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schüle B, et al. Parkinson’s disease associated with pure ATXN10 repeat expansion. NPJ Parkinsons Dis. 2017;3:27. doi: 10.1038/s41531-017-0029-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li C, et al. INC-Seq: accurate single molecule reads using nanopore sequencing. Gigascience. 2016;5:34. doi: 10.1186/s13742-016-0140-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jain M, Olsen HE, Paten B, Akeson M. The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol. 2016;17:239. doi: 10.1186/s13059-016-1103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Travers KJ, Chin CS, Rank DR, Eid JS, Turner SW. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic Acids Research. 2010;38:e159. doi: 10.1093/nar/gkq543. Travers 2010: The first description of consensus sequencing based on iterative resequencing of both strands of individual molecules. This concept, although challenging currently, will probably become very important as single-molecule DNA sequencers improve. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Loomis EW, et al. Sequencing the unsequenceable: expanded CGG-repeat alleles of the fragile X gene. Genome Res. 2013;23:121–128. doi: 10.1101/gr.141705.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Russo G, et al. Highly sensitive, non-invasive detection of colorectal cancer mutations using single molecule, third generation sequencing. Appl Transl Genom. 2015;7:32–39. doi: 10.1016/j.atg.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Frank JA, et al. Improved metagenome assemblies and taxonomic binning using long-read circular consensus sequence data. Sci Rep. 2016;6:25373. doi: 10.1038/srep25373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hestand MS, Van Houdt J, Cristofoli F, Vermeesch JR. Polymerase specific error rates and profiles identified by single molecule sequencing. Mutat Res. 2016;784–785:39–45. doi: 10.1016/j.mrfmmm.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 99.Heerema SJ, Dekker C. Graphene nanodevices for DNA sequencing. Nature nanotechnology. 2016;11:127–136. doi: 10.1038/nnano.2015.307. [DOI] [PubMed] [Google Scholar]
- 100.Beechem J. Library free targeted sequencing of native genomic DNA FFPE samples using Hyb & Seq technology-the hybridization based single molecule sequencing system. Advances in Genome Biology and Technology Annual Meeting. 2017 [Google Scholar]
- 101.Johnson SS, Zaikova E, Goerlitz DS, Bai Y, Tighe SW. Real-Time DNA Sequencing in the Antarctic Dry Valleys Using the Oxford Nanopore Sequencer. J Biomol Tech. 2017;28:2–7. doi: 10.7171/jbt.17-2801-009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang K, et al. Using ultra-sensitive next generation sequencing to dissect DNA damage-induced mutagenesis. Sci Rep. 2016;6:25310. doi: 10.1038/srep25310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stoler N, Arbeithuber B, Guiblet W, Makova KD, Nekrutenko A. Streamlined analysis of duplex sequencing data with Du Novo. Genome Biol. 2016;17:180. doi: 10.1186/s13059-016-1039-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Newman AM, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34:547–555. doi: 10.1038/nbt.3520. Newman 2016: An important early comprehensive description of a cell-free DNA ‘liquid biopsy’ approach using tag-based error correction techniques. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zheng Z, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20:1479–1484. doi: 10.1038/nm.3729. [DOI] [PubMed] [Google Scholar]
- 106.Kennedy S. Removing Sequencer and PCR Artifacts for Forensic DNA Analysis on Massively Parallel Sequencing Platforms: International Symposium on Human Indentification. 2017 [Google Scholar]
- 107.Krimmel JD, Salk JJ, Risques R-A. Cancer-like mutations in non-cancer tissue: towards a better understanding of multistep carcinogenesis. Translational Cancer Research. 2016 [Google Scholar]
- 108.Loeb LA, Springgate CF, Battula N. Errors in DNA replication as a basis of malignant changes. Cancer Res. 1974;34:2311–2321. [PubMed] [Google Scholar]
- 109.Merlo LMF, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6:924–935. doi: 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- 110.Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nat Rev Cancer. 2008;8:56–61. doi: 10.1038/nrc2255. [DOI] [PubMed] [Google Scholar]
- 111.Salk JJ, Fox EJ, Loeb LA. Mutational heterogeneity in human cancers: origin and consequences. Annu Rev Pathol. 2010;5:51–75. doi: 10.1146/annurev-pathol-121808-102113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338–345. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 114.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sottoriva A, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA. 2013;110:4009–4014. doi: 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhang J, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science. 2014;346:256–259. doi: 10.1126/science.1256930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.de Bruin EC, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346:251–256. doi: 10.1126/science.1253462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Naxerova K, et al. Hypermutable DNA chronicles the evolution of human colon cancer. Proc Natl Acad Sci USA. 2014;111:E1889–98. doi: 10.1073/pnas.1400179111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Reiter JG, et al. Reconstructing metastatic seeding patterns of human cancers. Nat Commun. 2017;8:14114. doi: 10.1038/ncomms14114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Marusyk A, et al. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature. 2014;514:54–58. doi: 10.1038/nature13556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yates LR, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21:751–759. doi: 10.1038/nm.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ding L, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sequist LV, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Science Translational Medicine. 2011;3:75ra26–75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jamal-Hanjani M, et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N Engl J Med NEJMoa1616288. 2017 doi: 10.1056/NEJMoa1616288. [DOI] [PubMed] [Google Scholar]
- 125.Andor N, et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med. 2016;22:105–113. doi: 10.1038/nm.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mroz EA, et al. High intratumor genetic heterogeneity is related to worse outcome in patients with head and neck squamous cell carcinoma. Cancer. 2013;119:3034–3042. doi: 10.1002/cncr.28150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Parker WT, Ho M, Scott HS, Hughes TP, Branford S. Poor response to second-line kinase inhibitors in chronic myeloid leukemia patients with multiple low-level mutations, irrespective of their resistance profile. Blood. 2012;119:2234–2238. doi: 10.1182/blood-2011-08-375535. [DOI] [PubMed] [Google Scholar]
- 128.Landau DA, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152:714–726. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Klco JM, et al. Association Between Mutation Clearance After Induction Therapy and Outcomes in Acute Myeloid Leukemia. JAMA. 2015;314:811–822. doi: 10.1001/jama.2015.9643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Misale S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Stroun M, Anker P, Lyautey J, Lederrey C, Maurice PA. Isolation and characterization of DNA from the plasma of cancer patients. Eur J Cancer Clin Oncol. 1987;23:707–712. doi: 10.1016/0277-5379(87)90266-5. [DOI] [PubMed] [Google Scholar]
- 132.Bettegowda C, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Science Translational Medicine. 2014;6:224ra24–224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wan JCM, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17:223–238. doi: 10.1038/nrc.2017.7. [DOI] [PubMed] [Google Scholar]
- 134.Murtaza M, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497:108–112. doi: 10.1038/nature12065. [DOI] [PubMed] [Google Scholar]
- 135.Garcia-Murillas I, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Science Translational Medicine. 2015;7:302ra133. doi: 10.1126/scitranslmed.aab0021. [DOI] [PubMed] [Google Scholar]
- 136.Tie J, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Science Translational Medicine. 2016;8:346ra92. doi: 10.1126/scitranslmed.aaf6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Newman AM, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20:548–554. doi: 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fujii T, et al. Mutation-Enrichment Next-Generation Sequencing for Quantitative Detection of KRAS Mutations in Urine Cell-Free DNA from Patients with Advanced Cancers. Clin Cancer Res. 2017 doi: 10.1158/1078-0432.CCR-16-2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang Y, et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc Natl Acad Sci USA. 2015;112:9704–9709. doi: 10.1073/pnas.1511694112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kinde I, et al. Evaluation of DNA from the Papanicolaou test to detect ovarian and endometrial cancers. Science Translational Medicine. 2013;5:167ra4–167ra4. doi: 10.1126/scitranslmed.3004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Maritschnegg E, et al. Lavage of the Uterine Cavity for Molecular Detection of Müllerian Duct Carcinomas: A Proof-of-Concept Study. J Clin Oncol. 2015;33:4293–4300. doi: 10.1200/JCO.2015.61.3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang Y, et al. Detection of somatic mutations and HPV in the saliva and plasma of patients with head and neck squamous cell carcinomas. Science Translational Medicine. 2015;7:293ra104–293ra104. doi: 10.1126/scitranslmed.aaa8507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sidransky D, et al. Identification of ras oncogene mutations in the stool of patients with curable colorectal tumors. Science. 1992;256:102–105. doi: 10.1126/science.1566048. [DOI] [PubMed] [Google Scholar]
- 144.Aravanis AM, Lee M, Klausner RD. Next-Generation Sequencing of Circulating Tumor DNA for Early Cancer Detection. Cell. 2017;168:571–574. doi: 10.1016/j.cell.2017.01.030. [DOI] [PubMed] [Google Scholar]
- 145.Armitage P, Doll R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer. 1954;8:1–12. doi: 10.1038/bjc.1954.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Genovese G, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jaiswal S, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016;7:12484. doi: 10.1038/ncomms12484. Young 2016: This paper used a single-strand tag-based error correction technique to identify preneoplastic clones in nearly all adults, which had only two years earlier been believed to occur in only a subset of the very elderly. It is an important example of how a fundamental biological understanding can change quickly with improved discovery technologies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Krimmel JD, et al. Ultra-deep sequencing detects ovarian cancer cells in peritoneal fluid and reveals somatic TP53 mutations in noncancerous tissues. PNAS. 2016;113:6005–6010. doi: 10.1073/pnas.1601311113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Salk JJ, et al. Duplex Sequencing detects cancer-associated mutations arising during normal aging: Clonal evolution over a century of human lifetime: American Association for Cancer Research. 2017 [Google Scholar]
- 151.Jee J, et al. Rates and mechanisms of bacterial mutagenesis from maximum-depth sequencing. Nature. 2016;534:693–696. doi: 10.1038/nature18313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Maslov AY, Quispe-Tintaya W, Gorbacheva T, White RR, Vijg J. High-throughput sequencing in mutation detection: A new generation of genotoxicity tests? Mutat Res. 2015;776:136–143. doi: 10.1016/j.mrfmmm.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Fielden MR, et al. Modernizing Human Cancer Risk Assessment of Therapeutics. Trends Pharmacol Sci. 2017 doi: 10.1016/j.tips.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 154.Kim D, Kim S, Kim S, Park J, Kim JS. Genome-wide target specificities of CRISPR-Cas9 nucleases revealed by multiplex Digenome-seq. Genome Res. 2016;26:406–415. doi: 10.1101/gr.199588.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Caperton L, et al. Assisted reproductive technologies do not alter mutation frequency or spectrum. Proc Natl Acad Sci USA. 2007;104:5085–5090. doi: 10.1073/pnas.0611642104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nelson JL. The otherness of self: microchimerism in health and disease. Trends Immunol. 2012;33:421–427. doi: 10.1016/j.it.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Eun JK, Guthrie KA, Zirpoli G, Gadi VK. In situ breast cancer and microchimerism. Sci Rep. 2013;3:2192. doi: 10.1038/srep02192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fan HC, Blumenfeld YJ, Chitkara U, Hudgins L, Quake SR. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc Natl Acad Sci USA. 2008;105:16266–16271. doi: 10.1073/pnas.0808319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chiu RWK, et al. Non-invasive prenatal assessment of trisomy 21 by multiplexed maternal plasma DNA sequencing: large scale validity study. BMJ. 2011;342:c7401. doi: 10.1136/bmj.c7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bianchi DW, et al. Noninvasive Prenatal Testing and Incidental Detection of Occult Maternal Malignancies. JAMA. 2015;314:162–169. doi: 10.1001/jama.2015.7120. [DOI] [PubMed] [Google Scholar]
- 161.Jamuar SS, Walsh CA. Somatic mutations in cerebral cortical malformations. N Engl J Med. 2014;371:2038–2038. doi: 10.1056/NEJMc1411784. [DOI] [PubMed] [Google Scholar]
- 162.Poduri A, Evrony GD, Cai X, Walsh CA. Somatic mutation, genomic variation, and neurological disease. Science. 2013;341:1237758–1237758. doi: 10.1126/science.1237758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.De Vlaminck I, et al. Circulating cell-free DNA enables noninvasive diagnosis of heart transplant rejection. Science Translational Medicine. 2014;6:241ra77–241ra77. doi: 10.1126/scitranslmed.3007803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Shugay M, et al. Towards error-free profiling of immune repertoires. Nat Methods. 2014;11:653–655. doi: 10.1038/nmeth.2960. [DOI] [PubMed] [Google Scholar]
- 165.DeWitt WS, et al. Dynamics of the cytotoxic T cell response to a model of acute viral infection. J Virol. 2015;89:4517–4526. doi: 10.1128/JVI.03474-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hsu MS, et al. TCR Sequencing Can Identify and Track Glioma-Infiltrating T Cells after DC Vaccination. Cancer Immunol Res. 2016;4:412–418. doi: 10.1158/2326-6066.CIR-15-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tumeh PC, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Goodnow CC. Multistep pathogenesis of autoimmune disease. Cell. 2007;130:25–35. doi: 10.1016/j.cell.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 169.Qian J, et al. B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell. 2014;159:1524–1537. doi: 10.1016/j.cell.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lynch SV, Pedersen O. The Human Intestinal Microbiome in Health and Disease. N Engl J Med. 2016;375:2369–2379. doi: 10.1056/NEJMra1600266. [DOI] [PubMed] [Google Scholar]
- 172.Van de Wiele T, Van Praet JT, Marzorati M, Drennan MB, Elewaut D. How the microbiota shapes rheumatic diseases. Nat Rev Rheumatol. 2016;12:398–411. doi: 10.1038/nrrheum.2016.85. [DOI] [PubMed] [Google Scholar]
- 173.Rosenbaum M, Knight R, Leibel RL. The gut microbiota in human energy homeostasis and obesity. Trends Endocrinol Metab. 2015;26:493–501. doi: 10.1016/j.tem.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Alexander JL, et al. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat Rev Gastroenterol Hepatol. 2017;1805:105. doi: 10.1038/nrgastro.2017.20. [DOI] [PubMed] [Google Scholar]
- 175.Vindigni SM, Surawicz CM. Fecal Microbiota Transplantation. Gastroenterol Clin North Am. 2017;46:171–185. doi: 10.1016/j.gtc.2016.09.012. [DOI] [PubMed] [Google Scholar]
- 176.Dominguez-Bello MG, et al. Partial restoration of the microbiota of cesarean-born infants via vaginal microbial transfer. Nat Med. 2016;22:250–253. doi: 10.1038/nm.4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Roach DJ, et al. A Year of Infection in the Intensive Care Unit: Prospective Whole Genome Sequencing of Bacterial Clinical Isolates Reveals Cryptic Transmissions and Novel Microbiota. PLoS Genet. 2015;11:e1005413. doi: 10.1371/journal.pgen.1005413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Cummings LA, et al. Clinical Next Generation Sequencing Outperforms Standard Microbiological Culture for Characterizing Polymicrobial Samples. Clinical Chemistry. 2016;62:1465–1473. doi: 10.1373/clinchem.2016.258806. [DOI] [PubMed] [Google Scholar]
- 179.Grumaz S, et al. Next-generation sequencing diagnostics of bacteremia in septic patients. Genome Med. 2016;8:73. doi: 10.1186/s13073-016-0326-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kim S, et al. High-throughput automated microfluidic sample preparation for accurate microbial genomics. Nat Commun. 2017;8:13919. doi: 10.1038/ncomms13919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Acevedo A, Brodsky L, Andino R. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature. 2014;505:686–690. doi: 10.1038/nature12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Eigen M. The concept of the quasispecies will soon be 50 years old. Introduction. Curr Top Microbiol Immunol. 2016;392:vii. [PubMed] [Google Scholar]
- 183.Henn MR, et al. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog. 2012;8:e1002529. doi: 10.1371/journal.ppat.1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Solmone M, et al. Use of massively parallel ultradeep pyrosequencing to characterize the genetic diversity of hepatitis B virus in drug-resistant and drug-naive patients and to detect minor variants in reverse transcriptase and hepatitis B S antigen. J Virol. 2009;83:1718–1726. doi: 10.1128/JVI.02011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Svarovskaia ES, Martin R, McHutchison JG, Miller MD, Mo H. Abundant drug-resistant NS3 mutants detected by deep sequencing in hepatitis C virus-infected patients undergoing NS3 protease inhibitor monotherapy. J Clin Microbiol. 2012;50:3267–3274. doi: 10.1128/JCM.00838-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Daum LT, et al. Next-generation ion torrent sequencing of drug resistance mutations in Mycobacterium tuberculosis strains. J Clin Microbiol. 2012;50:3831–3837. doi: 10.1128/JCM.01893-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Katz M, Hover B, Brady S. Culture-independent discovery of natural products from soil metagenomes. J Ind Microbiol Biotechnol. 2016;43:129–141. doi: 10.1007/s10295-015-1706-6. [DOI] [PubMed] [Google Scholar]
- 188.Bassil NM, Bryan N, Lloyd JR. Microbial degradation of isosaccharinic acid at high pH. ISME J. 2015;9:310–320. doi: 10.1038/ismej.2014.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Yamamoto S, et al. Environmental DNA metabarcoding reveals local fish communities in a species-rich coastal sea. Sci Rep. 2017;7:40368. doi: 10.1038/srep40368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Mayo B, et al. Impact of next generation sequencing techniques in food microbiology. Curr Genomics. 2014;15:293–309. doi: 10.2174/1389202915666140616233211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Jäger AC, et al. Developmental validation of the MiSeq FGx Forensic Genomics System for Targeted Next Generation Sequencing in Forensic DNA Casework and Database Laboratories. Forensic Sci Int Genet. 2017;28:52–70. doi: 10.1016/j.fsigen.2017.01.011. [DOI] [PubMed] [Google Scholar]
- 192.Stiller M, et al. Patterns of nucleotide misincorporations during enzymatic amplification and direct large-scale sequencing of ancient DNA. Proc Natl Acad Sci USA. 2006;103:13578–13584. doi: 10.1073/pnas.0605327103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Avery OT, Macleod CM, McCarty M. STUDIES ON THE CHEMICAL NATURE OF THE SUBSTANCE INDUCING TRANSFORMATION OF PNEUMOCOCCAL TYPES : INDUCTION OF TRANSFORMATION BY A DESOXYRIBONUCLEIC ACID FRACTION ISOLATED FROM PNEUMOCOCCUS TYPE III. J Exp Med. 1944;79:137–158. doi: 10.1084/jem.79.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 195.Mostovoy Y, et al. A hybrid approach for de novo human genome sequence assembly and phasing. Nat Methods. 2016;13:587–590. doi: 10.1038/nmeth.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Bickhart DM, et al. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat Genet. 2017;49:643–650. doi: 10.1038/ng.3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.King DA, et al. Mosaic structural variation in children with developmental disorders. Hum Mol Genet. 2015;24:2733–2745. doi: 10.1093/hmg/ddv033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011:1–6. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Vitak SA, et al. Sequencing thousands of single-cell genomes with combinatorial indexing. Nat Methods. 2017;14:302–308. doi: 10.1038/nmeth.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Zheng GXY, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Rosenberg AB, et al. Scaling single cell transcriptomics through split pool barcoding. 2017 doi: 10.1101/105163. [DOI] [Google Scholar]
- 202.Ullal AV, et al. Cancer cell profiling by barcoding allows multiplexed protein analysis in fine-needle aspirates. Science Translational Medicine. 2014;6:219ra9–219ra9. doi: 10.1126/scitranslmed.3007361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Sun WJ, et al. RMBase: a resource for decoding the landscape of RNA modifications from high-throughput sequencing data. Nucleic Acids Research. 2016;44:D259–65. doi: 10.1093/nar/gkv1036. [DOI] [PMC free article] [PubMed] [Google Scholar]