


Abstract
Automated function prediction (AFP) of proteins is of great significance in biology. AFP can be regarded as a problem of the large-scale multi-label classification where a protein can be associated with multiple gene ontology terms as its labels. Based on our GOLabeler—a state-of-the-art method for the third critical assessment of functional annotation (CAFA3), in this paper we propose NetGO, a web server that is able to further improve the performance of the large-scale AFP by incorporating massive protein-protein network information. Specifically, the advantages of NetGO are threefold in using network information: (i) NetGO relies on a powerful learning to rank framework from machine learning to effectively integrate both sequence and network information of proteins; (ii) NetGO uses the massive network information of all species (>2000) in STRING (other than only some specific species) and (iii) NetGO still can use network information to annotate a protein by homology transfer, even if it is not contained in STRING. Separating training and testing data with the same time-delayed settings of CAFA, we comprehensively examined the performance of NetGO. Experimental results have clearly demonstrated that NetGO significantly outperforms GOLabeler and other competing methods. The NetGO web server is freely available at http://issubmission.sjtu.edu.cn/netgo/.
INTRODUCTION
As the most basic structural molecules, proteins maintain the basic cell activities and biodiversity (1). Identification of protein/gene functions is of great significance to understand the nature of biology. For this purpose, gene ontology (GO), launched in 1998, has become the most influential ontology currently (2). So far, GO contains 45 013 biological concepts (February 2019), covering three different ontologies, Molecular Function Ontology (MFO), Biological Process Ontology (BPO), and Cellular Component Ontology (CCO). Due to the advancement of sequencing technologies, the number of known protein sequences has been significantly increased. Only a very tiny part of newly obtained sequences, however, have experimental GO annotations. For example, only <0.1% of ∼146 million protein sequences in UniProKB (February 2019) have experimental GO annotations (3). This is because identifying protein functions by biological experiments is both time- and resource-consuming. As such, automated function prediction (AFP) has become increasingly important in reducing the gap between the huge number of protein sequences and very limited experimental annotations (4,5).
For advancing research on a large-scale AFP, the community-wide Critical Assessment of Functional Annotation (CAFA, http://biofunctionprediction.org/cafa/) has been held three times, i.e., CAFA1 in 2010–2011, CAFA2 in 2013–2014 and CAFA3 in 2016–2017 (4,5). By using a time-delayed evaluation procedure, CAFA assesses the accuracy of protein function prediction submitted by participants. A large set of target proteins (∼100 000 in CAFA2 and CAFA3) was first available to the participants, who were required to submit their predictions before the deadline (T0). A few months later (T1), target proteins with experimental annotations were then used as a benchmark for performance evaluation. The benchmark data in CAFA was grouped into two categories: no-knowledge and limited-knowledge. The no-knowledge benchmark proteins refer to those who do not have experimental annotations before T0, but instead have at least one experimental annotation before T1. The limited-knowledge benchmark proteins are those who have the first experimental annotations in the target domain between T0 and T1, as well as experimental annotations in at least one other domain before T0. Currently, >99.9% of all proteins have no experimental annotations. This means that AFP for no-knowledge protein is valuable to biologists.
From a machine learning viewpoint, AFP is a problem of a large-scale multilabel classification, where multiple GO terms (labels) can be assigned to a protein (instance) (6). AFP faces two main challenges from the sides of the GO (label) and protein (instance). On the GO side, one protein can be associated with multiple GO terms from all 45 000 GO terms. All of these GO terms are organized in a hierarchical structure under the three GO ontologies. If a protein is assigned by a GO term, for example, all GO terms located at its ancestor nodes (in GO) of this term should be assigned to this particular protein as well. The experimental GO annotations of human proteins in Swissprot (7) (December 2017) reveal that one human protein can be annotated by 74 GO terms on average. On the protein side, information about proteins is not limited to sequences. Sequences are just part of all information about proteins. Sequences are static and genetic, while proteins are alive and dynamic. Thus, an imperative issue is how to effectively integrate multiple types of data other than protein sequences for AFP.
The results of past CAFA show that sequence-based AFP methods can be the best-performing ones. Even the simple homology-based methods by using BLAST or PSI-BLAST are very competitive (8–10). Recently, we developed a top-performing, sequence-based AFP method, called GOLabeler (11), to address the challenge on the GO term side. GOLabeler deems the AFP as a ranking problem, and utilizes a learning to rank (LTR) (12) framework to seamlessly integrate multiple types of sequence-based evidence, such as homology, domain, family, motif, amino acid k-mer, and biophysical properties. The final evaluation on CAFA3 reported in the 2018 meeting of the function special interest group at ISMB2018 (July 2018) concluded that among nearly 150 submissions by ∼50 groups from all over the world, GOLabeler scored among top-performing approaches in no-knowledge proteins under all of the three GO ontologies in terms of Fmax (see Supplementary for the definition and more detail). Despite this fact, many protein functions cannot be inferred from protein sequences only. For example, a well-accepted hypothesis of network-based methods is that interacting proteins should share similar functions under the principle of ‘guilt by association’ (13,14). A natural question then arises as to whether other types of protein information can further improve the performance of GOLabeler for AFP.
We implement a new AFP web server called NetGO. The basic idea of NetGO is to incorporate the network-based evidence into the GOLabeler framework (i.e. LTR) so as to improve the performance of a large-scale AFP. The advantages of NetGO are as follows:
NetGO addresses both sides of the challenges: (i) the label side by using LTR; and (ii) the instance (protein) side by incorporating network-based information;
NetGO is scalable to incorporate network information at a large-scale level; and
The performance of NetGO has been validated on large-scale datasets under the CAFA settings.
Experimental results have indicated that NetGO significantly outperformed GOLabeler in both BPO and CCO, with the respective 14% and 3% improvements in terms of AUPR (Area Under the Precision–Recall curve).
METHODS
Notation
Let D be a set of training data, Gi be the ith GO term, and Pj be the jth protein. Denote S(Gi, Pj) as the score (obtained by a AFP method), which quantifies the chance that Pj is associated with Gi.
For a given organism, there are m types of its protein networks PN(l) (l = 1, ⋅⋅⋅, m) from different sources, such as genomic context, gene expression, and physical interaction. Each network PN(l) consists of two sets of nodes PV(l) and edges PE(l). Each node corresponds to a protein in this organism, while an edge represents an interaction (association) between two proteins. In the l-th network, we denote PE(l)(i, j) as the edge between nodes  and
and 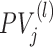 with ω(l)(i, j) ∈ [0, 1] as its weight, which measures the confidence of an association between the two nodes. Given a target protein Pj and GO term Gi, the core idea of NetGO is to estimate S(Gi, Pj) by using both sequence and all available networks PN(l)(l = 1, ⋅⋅⋅, m) of different organisms.
with ω(l)(i, j) ∈ [0, 1] as its weight, which measures the confidence of an association between the two nodes. Given a target protein Pj and GO term Gi, the core idea of NetGO is to estimate S(Gi, Pj) by using both sequence and all available networks PN(l)(l = 1, ⋅⋅⋅, m) of different organisms.
Learning to Rank
Within the framework of learning to rank (LTR) (12), NetGO integrates both protein sequence and network information effectively and efficiently to improve the performance of the large-scale AFP. As a powerful machine learning paradigm, LTR aims to rank instances in terms of their optimal ordering, rather than to produce a numerical score for each of the instances. As mentioned before, the essence of AFP lies in ranking GO terms (labels) in order of their relevance to a given query protein. The detailed method used in our LTR is a pairwise approach, which can be cast as a problem of pairwise classification. In this kind of the approach, given pairs of GO terms with respect to a specific protein, the LTR model tries to tell which GO term is more relevant by ranking more relevant GO terms at top positions in the list. During the testing, the top rank GO terms are chosen as the true labels, after they are ordered by their prediction scores.
In this study, we make use of LambdaMART, a pairwise LTR approach, for AFP (15). This is because it has demonstrated a good performance in several international machine learning competitions, such as BioASQ challenge (16,17) and Yahoo Learning to Rank competition (18).
NetGO: overview
Figure 1 illustrates the whole framework of NetGO. The basic idea of NetGO is to integrate six component methods in the framework of LTR for better AFP performance. The five components of Naive, BLAST-KNN, LR-3mer, LR-Interpro, and LR-ProFET are from GOLabeler that uses protein sequence information only (11) (note: KNN and LR stand for k-nearest neighbors and logistic regression, respectively). On the other hand, the newly developed component, called Net-KNN, makes use of network information.
Figure 1.

The framework of NetGO with seven steps. The top five component methods use sequence information, while Net-KNN relies on network information. An offline training process consists of Steps 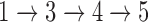 , while an online test process is Steps
, while an online test process is Steps 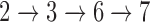 .
.
NetGO has to be trained before accepting test queries (proteins). As shown in the figure, an offline training process consists of Steps 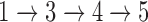 , while an online test process is Steps
, while an online test process is Steps 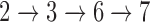 . Note that Step 6 relies on Step 5 of Ranking model that has been learned from the training data. The training data contains a number of instances that consist of protein sequences, their network information, and their associated ground-truth GO annotations. In other words, a protein is associated with a number of GO terms in the form of a pair of protein-a GO term and their score (score 1 for relevant and score 0 for irrelevant). During the training, given a training protein, NetGO first relies on each component method in Step 3 to predict the association score of each GO term to this protein. The top k (we used k = 30 in NetGO. See the Result section.) predicted GO terms by each component are combined to generate the candidate GO terms. For each candidate GO term, we use their association scores to form a six-dimensional feature vector. Second, Step 4 of LTR tries to learn a ranking model to minimize the number of incorrectly ordered pairs in the training data. This minimization of the cost function is achieved by adjusting the parameters of Steps 3, 4 and 5. In particular, LTR aims to produce an optimal ordering of GO annotations for all pairs of the proteins in the training data. As such, LTR does not care much about the exact score that each candidate obtains, but does care about the relative ordering among all pairs of the candidate in the output list.
. Note that Step 6 relies on Step 5 of Ranking model that has been learned from the training data. The training data contains a number of instances that consist of protein sequences, their network information, and their associated ground-truth GO annotations. In other words, a protein is associated with a number of GO terms in the form of a pair of protein-a GO term and their score (score 1 for relevant and score 0 for irrelevant). During the training, given a training protein, NetGO first relies on each component method in Step 3 to predict the association score of each GO term to this protein. The top k (we used k = 30 in NetGO. See the Result section.) predicted GO terms by each component are combined to generate the candidate GO terms. For each candidate GO term, we use their association scores to form a six-dimensional feature vector. Second, Step 4 of LTR tries to learn a ranking model to minimize the number of incorrectly ordered pairs in the training data. This minimization of the cost function is achieved by adjusting the parameters of Steps 3, 4 and 5. In particular, LTR aims to produce an optimal ordering of GO annotations for all pairs of the proteins in the training data. As such, LTR does not care much about the exact score that each candidate obtains, but does care about the relative ordering among all pairs of the candidate in the output list.
During a test, NetGO accepts a protein query with its network information. Again, the six components in Step 3 use their already learned parameters to extract the features of this protein, producing a score feature vector of length six. Candidate GO terms, i.e., feature vectors, are then inputted into Step 6 of the LTR model. A ranked list of GO terms is returned as the final output of NetGO for the query protein in Step 7.
NetGO: Six component models
In the following, we briefly describe the six component methods of NetGO. Note that the details of the top five component methods can be found in (11), and the formula for Net-KNN is given in the supplement.
Naive
Naive is an official baseline of CAFA. For a given Pj, the score that Pj is associated with Gi is defined as the relative frequency of Gi in D.
BLAST-KNN
For a given protein Pj and a specific GO term Gi, their S(Gi, Pj) of BLAST-KNN is computed as weighted voting by Pj’s homologous proteins in the training data. The weight of such a homologous protein is set to its bit-score (similarity score) by BLAST alignment(The E-value cutoff was set to 0.001 in our experiments). The higher the normalized sum of bit-scores of homologous proteins of Pj is associated with Gi, the bigger S(Gi, Pj) will become.
LR-3mer, LR-InterPro and LR-ProFET
We briefly compare the three LR-based components in terms of their different features used: (i) LR-3mer: The frequency of amino acid trigram (3mer) is counted for each protein to produce 8000 (=203) features in total. (ii) LR-InterPro: we run InterProScan(http://www.ebi.ac.uk/interpro/interproscan.html) to obtain 33 879 binary features that represent the absence /presence of a large number of motifs, protein families, and domains in InterPro (19). (iii) LR-ProFET: Having been used in various function predictions, ProFET (20) consists of 1170 features including elementary biophysical properties, and local potential features.
Net-KNN
Net-KNN is capable of identifying candidate GO terms for each protein by using network information. In essence, the basic idea of Net-KNN is similar to that of BLAST-KNN. The sequence similarity (bit-score by BLAST) in BLAST-KNN is replaced by the association score (edge weight) in a network for Net-KNN.
The procedure of Net-KNN is illustrated in Figure 2. Given a test protein Pj and a protein network (from STRING in our experiments), Net-KNN computes the score S(Gi, Pj) between Pj and GO term Gi by using one of the following three methods (see the supplementary materials for the detailed formulas).
STRI: If Pj appears in STRING (meaning that PVj exists), the neighbor nodes PVk of PVj in the network will be used;
HOMO: Net-KNN searches for the homologous protein in STRING of Pj with the highest bit-score by using BLAST with the cutoff E-value of 0.001. If found, this protein will be used as PVj, together with its neighboring nodes PVk; and
NOHO: Net-KNN will return zero if no homologous proteins are found.
Figure 2.

The procedure of Net-KNN.
Given m networks PN(l) (l = 1, ⋅⋅⋅, m) over the same set of nodes, the aggregated weight ω(PVj, PVk) can be computed in an ensemble way (see Equation (2) in the supplementary materials). The higher the weight of two proteins in all of the m individual networks is, the higher their aggregated weight is.
Note that the different types of networks and the various ways of their combinations affect the weights and the final performance.
RESULTS
Benchmark Datasets
The validation datasets were generated by following the procedures of CAFA1 (4), CAFA2 (5) and CAFA3. Specifically, protein sequences were downloaded from UniProt (3), while experimental annotations were extracted from SwissProt (7), GOA(http://www.ebi.ac.uk/GOA) (21), and GO (http://geneontology.org/page/download-annotations) (2). STRING is a database of protein/gene interaction, and the version 10.0a of STRING (22) was used as network information. This database covers 9 643 763 proteins from 2031 organisms with 932 553 897 interactions in total. The networks of 359 organisms appearing in the training data were used in Net-KNN. NetGO made use of the six different types of networks in STRING: 0:neighbourhood, 1:fusion, 2:co-occurrence, 3:co-expression, 4:experiment and 5:database.
Specifically, four datasets have been generated for NetGO training and testing, where the proteins are annotated at different time stamps.
-
Training: the training data for the component methods
All data annotated in October 2015 or before.
-
LTR1: training data for LTR
no-knowledge proteins, experimentally annotated from October 2015 to October 2016 and not before October 2015.
-
LTR2: training data for LTR
limited-knowledge proteins, experimentally annotated from October 2015 to October 2016 and no before October 2015.
-
Test: testing data for competing methods
All data experimentally annotated after October 2016 by October 2017 and not before October 2016.
Table 1 in the supplementary materials reports the number of proteins in the above datasets. All these datasets are available at https://drive.google.com/open?id=1HLH1aCDxlrVpu1zKvgfdQFEFnbT8gChm.
Table 1.
Performance comparisons of NetGO with its own components and competing methods against test data
| F max | AUPR | |||||
|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | |
| Naive | 0.317 | 0.255 | 0.604 | 0.169 | 0.115 | 0.610 |
| BLAST-KNN | 0.589 | 0.283 | 0.641 | 0.453 | 0.110 | 0.560 |
| Net-KNN | 0.344 | 0.306 | 0.642 | 0.158 | 0.097 | 0.568 |
| DeepGO | 0.379 | 0.243 | 0.569 | 0.239 | 0.092 | 0.535 |
| GoFDR | 0.542 | 0.271 | 0.570 | 0.338 | 0.067 | 0.329 |
| GOLabeler | 0.630 | 0.321 | 0.668 | 0.549 | 0.171 | 0.685 |
| NetGO | 0.631 | 0.341 | 0.674 | 0.557 | 0.195 | 0.706 |
Performance evaluation metrics
In our experiments, we use two measures for performance evaluation: AUPR and Fmax . As a standard evaluation metric in machine learning, AUPR punishes false positive prediction. It is suitable for highly imbalanced data. For Fmax , we give the definition of this official metric of CAFA in the supplementary materials.
Given a test set of proteins, we first obtain the predicted association scores (probabilities) of each pair of a protein and a GO term. According to these scores, we then sort all pairs of proteins and GO terms, and evaluate the performance by Fmax and AUPR. Similar to GOLabeler, we evaluate the top 100 GO terms predicted from every competing method for each ontology by considering the importance of the top GO terms.
Parameter settings
Similar to GOLabeler, both LTR1 and LTR2 were combined to train the ranking model of NetGO, since it performed better than using LTR1 only (11). The top 30 predictions from each component were merged. This was because this number provided the best performance in 5-fold cross validation over LTR training data with four values {10, 30, 50 and 70} tested (see supplementary materials for the detailed results and other settings for the components of GOLabeler and NetGO).
Validation results
Table 1 reports the test results of NetGO, GOLabeler, and other compared methods. In the upper part of this table, we report the results of the three component methods: Naive, BLAST-KNN, and Net-KNN. Among these component methods, BLAST-KNN performed best for MFO, while Net-KNN did the best for BPO in terms of Fmax. For example, BLAST-KNN achieved the Fmax of 0.589 in MFO, followed by Net-KNN (0.344), and Naive (0.317). In the middle part, we show the results of two competing methods, GoFDR (23) and DeepGO (24). GoFDR achieved the good performance in the recent CAFA (5), while DeepGO was a recently developed deep learning based methods. Using the same training data as NetGO, we trained these models with their recommended parameters. Note that DeepGO made predictions on only MFO, BPO and CCO terms that appeared more than 50, 250 and 50 times in the training data, respectively. The experimental results demonstrate that NetGO outperformed both GoFDR and DeepGO in all of three GO ontologies. The under-performing DeepGO exposes the weakness of deep learning based methods that work only on a small number of GO terms. This fact results from the insufficient training data and high computational complexity.
In the lower part of the table, we compare NetGO with the state-of-the-art method of GOLabeler. Experimental results show that NetGO outperformed GOLabeler in all three GO Ontologies. The improvement is especially significant in BPO and CCO. In particular, NetGO achieved 14% improvements over GOLabeler in terms of AUPR in BPO, and around 3% improvements in CCO. This demonstrates the advantages of incorporating network information into the functional annotation of BPO and CCO. In addition, similar to the CAFA overview paper (5), we plotted precision-recall curves for comparing all methods for BPO in Figure 3. Finally, we used 100 bootstrapped datasets with replacement to further validate the superiority of NetGO by a paired t-test (P-values < 0.01 for all cases. See supplement materials for the details).
Figure 3.

Precision-recall curves of NetGO compared with its own components and competing methods over BPO.
THE NETGO WEB SERVER
Implementation
NetGO was implemented by using Python. Careful tests had been performed to ensure the compatibility of common browsers on different operating systems. The FASTA-format data was processed by biopython(http://biopython.org/), and sklearn (http://scikit-learn.org/stable/index.html) was used to run logistic regression and xgboost (25) for LTR. Training LR classifiers is time-consuming, so we store all the trained LR classifiers on the server in order to make fast predictions. NetGO is updated annually by (i) downloading new datasets on annotations and networks; (ii) updating the InterProScan version and (iii) generating new LR classifiers. New components will also be added to the existing framework.
Input
The NetGO web server is available at http://issubmission.sjtu.edu.cn/netgo/. It has a simple user-friendly interface, together with a detailed help page. Accepting protein sequences in the FASTA format, the NetGO web server is able to process up to 1000 proteins for each job. The length of each sequence and its species are not limited, but all sequences have to be amino acids specified in a single letter code (ACDEFGHIKLMNPQRSTVWYVBZX*). Any other non-white space characters in a query will be rejected by the input processor with notification.
Output
For all query proteins, NetGO outputs prediction results in MFO, BPO and CCO. By forming scores (of each of candidate GO terms) predicted by all of the six component methods as features, NetGO relies on LTR to rank all candidate GO terms of each query protein. In particular, the top number of m (m = 20 by default, and can be set to 30, 50 or 100) predicted GO terms in three ontologies and their obtained scores are displayed in a result table for each query protein, as shown in Figure 4. Besides the total score of NetGO, the respective prediction scores and ranks of the three main component models, BLAST-KNN, Net-KNN and LR-InterPro, are also listed in the table. In fact, these scores and ranks correspond to those by using sequence alignment, protein-protein network, and protein domain information, respectively. Comparing these results, users can easily understand the contributions of different types of information to the final score of NetGO for a particular query protein. Except for displaying query results in a table, the top predictions of GO terms are visualized by using the AmiGO API (http://amigo.geneontology.org/) (2), according to the hierarchical structure of GO. All GO terms with prediction scores higher than 0.6 are highlighted with colors. It is impossible to display all prediction results of the submitted proteins within one web page for a large-scale AFP. Therefore, only the top 10 (20 or 30 depending on the user’s choice) proteins are shown on a result page. The full list of all returned results, however, can be retrieved by an URL from the same page.
Figure 4.

An example of query results (UniProt: O74486) from the NetGO web server. The top 30 predicted GO terms in three ontologies and their assigned scores are displayed in a table. The result visualization is also provided to show the overall GO structure. All GO terms with prediction scores higher than 0.6 are highlighted with colors.
Predicted GO terms in the three different ontologies can be retrieved through the buttons of ‘Result:MF’, ‘Result:BP’, and ‘Result:CC’. As an example, Figure 4 shows the prediction results on the sequence of an uncharacterized protein (UniProt: O74486) in BPO. More specific terms with higher predicted scores are considered to be more informative, which are easily found from the visualized results. The name of each GO term is provided, and a detailed description in AmiGO can be displayed by simply clicking it in the table.
It is relatively fast (<2 h for 1000 proteins) for NetGO to make a prediction. An URL for tracking a job status is returned after each submission, together with a notification email when results are available.
Case study
The wtf (for with Tf) gene families in the fission yeast are functionally uncharacterized (26). Here, we used our server to predict potential GO terms associated with the wtf19 protein (UniProtKB: O74486) (27). As given in our results (at http://issubmission.sjtu.edu.cn/netgo/result/1555046134), considering more specific predicted GO terms, the core component Net-KNN of NetGO predicted that the protein may be involved in the biological process of ‘regulation of transcription from RNA polymerase II promoter (GO:0006357)’. Specifically, this prediction is supported by the predicted MFO term of ‘transcription factor activity, RNA polymerase II core promoter proximal region sequence-specific binding (GO:0000982)’, as well as the predicted CCO term of ‘nuclear chromosome part (GO:0044454)’. Therefore, our NetGO speculates that the wtf19 protein may act as an upstream regulatory element, required for the regulation of transcription from RNA polymerase II promoter.
Comparisons with other web servers
A number of AFP methods used in CAFA can be publicly accessed as web servers now, such as DcGO (28), CombFunc (29), INGA (30), SIFTER (31), FunFam (32), Argot2.5 (33), BAR 3.0 (34), DeepGO (24), PANNZER2 (35), MetaGO (36), BUSCA (37) and Phylo-PFP (38). We compare the main characteristics of NetGO with some existing AFP web servers on three aspects, as reported in Table 2. In particular, NetGO has at least three main advantages: (i) it exploits a wide range of features or components such as sequence-based and protein-protein interaction network-based information; (ii) it employs a powerful learning to rank framework to integrate diverse components for AFP and (iii) it provides users with the large-scale prediction of protein function at the cost of reasonable running time. It can accept up to 1000 sequences in one online job, or even unlimited for one offline job.
Table 2.
Comparisons of our web server NetGO with some other similar web servers for AFP
| Web server | Feature/component | Integrated method | Maximum number of sequences in one job |
|---|---|---|---|
| CombFunc (29) | Protein sequence; protein–protein interactions; gene co-expression; protein domain | Support vector machine | Up to 1 |
| INGA (30) | Sequence similarity; somain; protein interaction network (PIN) | The consensus score calculated as a joint probability | Up to 10 |
| SIFTER (31) | A protein family’s phylogenetic tree of each specific domain | / | Up to 10 or all proteins in a given species |
| FunFam (32) | Functional classification of the domain superfamilies | / | Up to 1 |
| Argot2.5 (33) | The e-values from BLAST and HMMER searches | Weighted scheme | Interactive (up to 100) or batch (up to 10 001) |
| BAR 3.0 (34) | A graph-based clustering of UniProtKB sequences | / | Up to 1 |
| DeepGO (24) | Sequence-based information; PIN | Convolutional neural network | Up to 10 |
| PANNZER2 (35) | Sequence similarity; enrichment statistics from the sequence neighborhood | Weighted k-nearest neighbor classifier | Interactive (up to 10) or batch (100 000) |
| MetaGO (36) | Structure; sequence and sequence-profile; PIN | Logistic regression | Up to 1 |
| BUSCA (37) | Signal and transit peptides; GPI-anchors; transmembrane domains | A rooted computation graph | Up to 500 |
| Phylo-PFP (38) | Sequence similarity based on homology by considering their phylogenetic distance | / | Up to 10 |
| GOLabeler (11) | GO term frequency; sequence alignment; amino acid trigram; protein families, domains and motifs; sequence-derived features | Learning to rank | Up to 1000 |
| NetGO | The same five components in GOLabeler; PIN | Learning to rank | Up to 1000 |
DISCUSSION AND CONCLUSION
In this paper, we have presented NetGO—a new AFP web server that incorporates massive network information. A combination of network information with other types of data for better AFP has previously been reported including sequence information, gene expression, and domain information (such as Jones-UCL CAFA submissions (39) and CombFunc (29)). So the use of network information presented in this study is not a totally new idea. However, we integrate several components into an effective framework that has achieved the best performance on comprehensive experiments with massive networks. Experimental results have demonstrated that under the CAFA settings, NetGO significantly outperformed GOLabeler in two GO ontologies, BPO and CCO, and other competing methods of DeepGO and GoFDR. The reasons for such a good performance of NetGO are threefold: (i) a powerful LTR integration framework; (ii) the massive and comprehensive network information from STRING and (iii) the various sequence information.
Running fast with a visualization interface, the NetGO web server is suitable for large-scale protein function predictions. We believe that biologists would benefit from our high performance web server.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
S. Z. is supported by National Natural Science Foundation of China (No. 61872094 and No. 61572139) and Shanghai Municipal Science and Technology Major Project (No. 2017SHZDZX01). R. Y. and S. Y. are supported by the 111 Project (NO. B18015), the key project of Shanghai Science & Technology (No. 16JC1420402), Shanghai Municipal Science and Technology Major Project (No. 2018SHZDZX01) and ZJLab. Y. X. is supported by National Natural Science Foundation of China (No. 31601074 and No. 61832019) and National Key Research and Development Program of China (No. 2016YFA0501703). H. M. has been supported in part by JST ACCEL (grant number JPMJAC1503), MEXT Kakenhi (grant numbers 16H02868 and 19H04169), FiDiPro by Tekes (currently Business Finland) and AIPSE program by Academy of Finland.
Conflict of interest statement. None declared.
REFERENCES
- 1. Weaver R.F. Molecular Biology (WCB Cell & Molecular Biology). 2011; 5 edn. McGraw-Hill Education. [Google Scholar]
- 2. Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T. et al.. Gene Ontology: tool for the unification of biology. Nat. Genet. 2000; 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Consortium U. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2018; 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Radivojac P., Clark W.T., Oron T.R., Schnoes A.M., Wittkop T., Sokolov A., Graim K., Funk C., Verspoor K., Ben-Hur A. et al.. A large-scale evaluation of computational protein function prediction. Nat. Methods. 2013; 10:221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jiang Y., Oron T.R., Clark W.T., Bankapur A.R., D'Andrea D., Lepore R., Funk C.S., Kahanda I., Verspoor K.M., Ben-Hur A. et al.. An expanded evaluation of protein function prediction methods shows an improvement in accuracy. Genome Biol. 2016; 17:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang M., Zhou Z.. A review on multi-label learning algorithms. IEEE Trans. Knowledge Data Eng. 2014; 26:1819–1837. [Google Scholar]
- 7. Boutet E., Lieberherr D., Tognolli M., Schneider M., Bansal P., Bridge A.J., Poux S., Bougueleret L., Xenarios I.. Edwards D. UniProtKB/Swiss-Prot, the manually annotated section of the UniProt KnowledgeBase: how to use the entry view. Plant Bioinformatics: Methods and Protocols. 2016; NY: Springer; 23–54. [DOI] [PubMed] [Google Scholar]
- 8. Altschul S., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J.. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gillis J., Pavlidis P.. Characterizing the state of the art in the computational assignment of gene function: lessons from the first critical assessment of functional annotation (CAFA). BMC Bioinf. 2013; 14:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hamp T., Kassner R., Seemayer S., Vicedo E., Schaefer C., Achten D., Auer F., Boehm A., Braun T., Hecht M. et al.. Homology-based inference sets the bar high for protein function prediction. BMC Bioinf. 2013; 14:S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. You R., Zhang Z., Xiong Y., Sun F., Mamitsuka H., Zhu S.. GOLabeler: improving sequence-based large-scale protein function prediction by learning to rank. Bioinformatics. 2018; 34:2465–2473. [DOI] [PubMed] [Google Scholar]
- 12. Li H. A Short Introduction to Learning to Rank. IEICE Trans. 2011; 94:1854–1862. [Google Scholar]
- 13. Oliver S. Proteomics: guilt-by-association goes global. Nature. 2000; 403:601–603. [DOI] [PubMed] [Google Scholar]
- 14. Schwikowski B., Uetz P., Fields S.. A network of protein protein interactions in yeast. Nat. Biotechnol. 2000; 18:1257–1261. [DOI] [PubMed] [Google Scholar]
- 15. Burges C.J. From RankNet to LambdaRank to LambdaMart: an overview. Technical report, Microsoft Research. 2010; MSR-TR-2010-82. [Google Scholar]
- 16. Liu K., Peng S., Wu J., Zhai C., Mamitsuka H., Zhu S.. MeSHLabeler: improving the accuracy of large-scale MeSH indexing by integrating diverse evidence. Bioinformatics. 2015; 31:i339–i347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Peng S., You R., Wang H., Zhai C., Mamitsuka H., Zhu S.. DeepMeSH: deep semantic representation for improving large-scale MeSH indexing. Bioinformatics. 2016; 32:i70–i79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chapelle O., Chang Y.. Yahoo! Learning to rank challenge overview. Proceedings of the Yahoo! Learning to Rank Challenge, held at ICML 2010, Haifa, Israel, June 25, 2010. 2011; 1–24. [Google Scholar]
- 19. Mitchell A.L., Attwood T.K., Babbitt P.C., Blum M., Bork P., Bridge A., Brown S.D., Chang H.Y., El-Gebali S., Fraser M.I. et al.. InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019; 47:D351–D360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ofer D., Linial M.. ProFET: feature engineering captures high-level protein functions. Bioinformatics. 2015; 31:3429–3436. [DOI] [PubMed] [Google Scholar]
- 21. Huntley R., Sawford T., Mutowo-Meullenet P., Shypitsyna A., Bonilla C., Martin M.J., O'Donovan C.. The GOA database: gene ontology annotation updates for 2015. Nucleic Acids Res. 2015; 43:1057–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Szklarczyk D., Morris J.H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N.T., Roth A., Bork P. et al.. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017; 45:D326–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gong Q., Ning W., Tian W.. GoFDR: a sequence alignment based method for predicting protein functions. Methods. 2016; 93:3–14. [DOI] [PubMed] [Google Scholar]
- 24. Kulmanov M., Khan M.A., Hoehndorf R., Wren J.. DeepGO: predicting protein functions from sequence and interactions using a deep ontology-aware classifier. Bioinformatics. 2018; 34:660–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen T., Guestrin C.. XGBoost: a scalable tree boosting system. Proceedings of the 22Nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. 2016; NY: ACM; 785–794. [Google Scholar]
- 26. Hu W., Jiang Z.D., Suo F., Zheng J.X., He W.Z., Du L.L.. A large gene family in fission yeast encodes spore killers that subvert Mendel’s law. eLife. 2017; 6:e26057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lock A., Rutherford K., Harris M.A., Hayles J., Oliver S.G., Bähler J., Wood V.. PomBase 2018: user-driven reimplementation of the fission yeast database provides rapid and intuitive access to diverse, interconnected information. Nucleic Acids Res. 2018; 47:D821–D827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fang H., Gough J.. DcGO: database of domain-centric ontologies on functions, phenotypes, diseases and more. Nucleic Acids Res. 2012; 41:D536–D544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wass M.N., Barton G., Sternberg M.J.. CombFunc: predicting protein function using heterogeneous data sources. Nucleic Acids Res. 2012; 40:W466–W470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Piovesan D., Giollo M., Leonardi E., Ferrari C., Tosatto S.C.. INGA: protein function prediction combining interaction networks, domain assignments and sequence similarity. Nucleic Acids Res. 2015; 43:W134–W140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sahraeian S.M., Luo K.R., Brenner S.E.. SIFTER search: a web server for accurate phylogeny-based protein function prediction. Nucleic Acids Res. 2015; 43:W141–W147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Das S., Sillitoe I., Lee D., Lees J.G., Dawson N.L., Ward J., Orengo C.A.. CATH FunFHMMer web server: protein functional annotations using functional family assignments. Nucleic Acids Res. 2015; 43:W148–W153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lavezzo E., Falda M., Fontana P., Bianco L., Toppo S.. Enhancing protein function prediction with taxonomic constraints - the Argot2.5 web server. Methods. 2016; 93:15–23. [DOI] [PubMed] [Google Scholar]
- 34. Profiti G., Martelli P.L., Casadio R.. The Bologna Annotation Resource (BAR 3.0): improving protein functional annotation. Nucleic Acids Res. 2017; 45:W285–W290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Törönen P., Medlar A., Holm L.. PANNZER2: a rapid functional annotation web server. Nucleic Acids Res. 2018; 46:W84–W88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang C., Zheng W., Freddolino P.L., Zhang Y.. MetaGO: Predicting Gene Ontology of non-homologous proteins through low-resolution protein structure prediction and protein–protein network mapping. J. Mol. Biol. 2018; 430:2256–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Savojardo C., Martelli P.L., Fariselli P., Profiti G., Casadio R.. BUSCA: an integrative web server to predict subcellular localization of proteins. Nucleic Acids Res. 2018; 46:W459–W466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jain A., Kihara D.. Phylo-PFP: improved automated protein function prediction using phylogenetic distance of distantly related sequences. Bioinformatics. 2019; 35:753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cozzetto D., Buchan D.W.A., Bryson K., Jones D.T.. Protein function prediction by massive integration of evolutionary analyses and multiple data sources. BMC Bioinf. 2013; 14:S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.