

Abstract
Background
Clonal hematopoiesis of indeterminate potential (CHIP), defined by the presence of an expanded somatic blood cell clone in those without other hematologic abnormalities, is common in older individuals and associates with an increased risk of developing hematologic cancer. We previously found preliminary evidence for an association of CHIP with human atherosclerotic cardiovascular disease, but the nature of this association was unclear.
Methods
We used whole exome sequencing to detect the presence of CHIP in peripheral blood cells and associated this with coronary heart disease in four case-control studies together comprising 4,794 cases and 3,537 controls. To assess causality, we perturbed the function of Tet2, the second most commonly mutated gene linked to clonal hematopoiesis, in the hematopoietic cells of atherosclerosis-prone mice.
Results
In nested case-control analyses from two prospective cohorts, carriers of CHIP had a 1.9-fold (95% confidence interval 1.4–2.7) increased risk of coronary heart disease compared to non-carriers. In two retrospective case-control cohorts for early-onset myocardial infarction, those with CHIP had a 4.0-fold greater risk (95% confidence interval 2.4–6.7) of having myocardial infarction. Mutations in DNMT3A, TET2, ASXL1, and JAK2 were each individually associated with coronary heart disease. Those with clonal hematopoiesis also had increased coronary artery calcification, a marker of coronary atherosclerosis burden. Hyperlipidemic mice engrafted with Tet2−/− or Tet2+/− bone marrow developed larger atherosclerotic lesions in the aortic root and aorta than mice receiving control marrow. Analyses of Tet2−/− macrophages demonstrated elevated expression of several chemokine and cytokine genes that contribute to atherosclerosis.
Conclusions
Clonal hematopoiesis robustly associates with coronary heart disease in humans and causes accelerated atherosclerosis in mice.
Introduction
Aging associates with increased incidence of cancer and cardiovascular disease. We and others recently used whole exome sequencing data to identify a common, age-related disorder marked by an expansion of hematopoietic clones carrying recurrent somatic mutations, most frequently loss-of-function alleles in the genes DNMT3A, TET2, and ASXL11-3. These mutations, which are also common in the myelodysplastic syndrome and acute myeloid leukemia4, provide a selective advantage to the hematopoietic stem cells in which they occur5,6, and are detectable as clones in peripheral blood samples because the mutated stem cells maintain the ability to differentiate into circulating granulocytes, monocytes, and lymphocytes7,8. Individuals under the age of 40 rarely accumulate these clones, but they become common in aging, with over 10% of those over age 70 harboring such a mutation. Carriers of these mutations have a ~10-fold increased risk of developing a hematologic malignancy. Based on these findings, we provisionally defined those carrying such mutations in the absence of any other hematologic abnormalities as having clonal hematopoiesis of indeterminate potential (CHIP)9.
Our exploratory analysis revealed that those with CHIP have increased risk for all-cause mortality and, surprisingly, for developing coronary heart disease1. While traditional risk factors such as hypercholesterolemia, type 2 diabetes, hypertension, and smoking account for a large proportion of the risk for coronary heart disease, many individuals who develop atherosclerosis or coronary heart disease lack established risk factors10,11, suggesting that unknown factors may also contribute to atherosclerosis and its complications. Here, we test the hypothesis that CHIP contributes causally to atherosclerotic cardiovascular disease.
Methods
Sample Ascertainment
The primary study sample consisted of two prospective cohorts for coronary heart disease from which we selected cases that were matched to controls on the basis of age, sex, type 2 diabetes status, and smoking history without regard to follow-up time, and two retrospective case-control studies for early-onset myocardial infarction (MI)(Table S1-S2). The protocols for these studies were approved by the ethics committees at all involved institutions; written informed consent was obtained from all participants.
Whole Exome Sequencing, Variant Calling, and Statistical Analysis
DNA was obtained from individual studies, and further processing was performed at the Broad Institute of Harvard and the Massachusetts Institute of Technology. Pathogenic variants in 74 genes associated with human myeloid cancers were identified and used to define those with CHIP (Table S3). The association of the presence of CHIP with coronary heart disease was tested using Cox proportional hazards models or logistic regression. Full details are in the Extended Methods of the Supplementary Appendix.
Mouse atherosclerosis
Atherosclerosis-prone Ldlr−/− mice were transplanted with bone marrow from Tet2+/+ or Tet2−/− mice, initiated on a high-fat, high-cholesterol diet, and then assessed for atherosclerosis at various time points. Macrophages cultured from Tet2+/+ or Tet2−/− bone marrow exposed to vehicle or low-density lipoprotein underwent gene expression analysis.
Results
CHIP associates with coronary heart disease
To test for an association of CHIP with coronary heart disease, we utilized a modified nested case-control study design from two prospective cohort studies (see Extended Methods in the Supplementary Appendix), BioImage and Malmö Diet and Cancer (MDC). Biolmage was selected because of the cohort’s enrichment in aged and high cardiovascular risk participants12, while MDC was selected because of its longer follow-up period and extensive phenotypic data13. Table S1 shows the baseline characteristics for each cohort. After excluding those with prevalent events, coronary heart disease cases were defined as those having Ml or coronary revascularization procedures following the time of DNA collection and were matched to event-free controls. We performed exome sequencing from blood cell DNA on 113 cases and 257 controls from Biolmage and on 320 cases and 320 controls from MDC. We identified those with CHIP based on a pre-specified list of variants in 74 genes known to be recurrently mutated in myeloid malignancies (Table S3). Similar to previous studies, we found that the most commonly mutated genes were DNMT3A, TET2, and ASXL1 and that 72/77 (93.5%) individuals with CHIP had only a single driver gene mutated1,2 (Figure S1, Table S4).
The median age of participants in BioImage at the time of DNA sample collection was 70 years and the median follow-up time was 2.6 years. We found that 19/113 (16.8%) coronary heart disease cases had CHIP as compared to 25/257 (9.7%) controls (hazard ratio (HR) 1.8, 95% confidence interval 1.1–2.9, p=0.03 from a Cox proportional hazards model adjusted for age, sex, type 2 diabetes status, total cholesterol, high density lipoprotein cholesterol, hypertension, and smoking status) (Figure 1A, Figure S2,and Table S5).
Figure 1. CHIP associates with coronary heart disease.
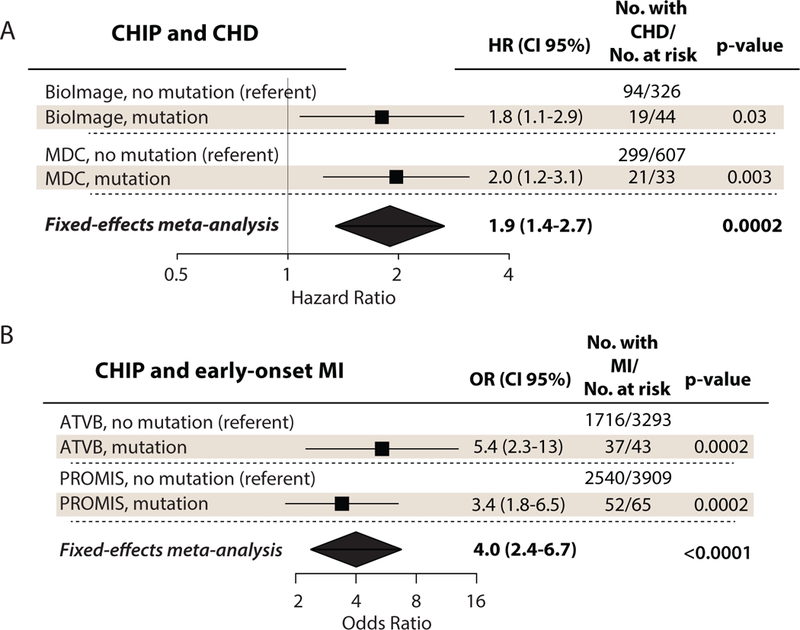
A) Forest plot for association between CHD and CHIP in BioImage and MDC. Hazard ratio for having CHD in those with mutations was obtained by a Cox proportional hazards model adjusted for age, sex, type 2 diabetes, total cholesterol, high-density lipoprotein cholesterol, smoking status, and hypertension.
B) Forest plot for association between MI and CHIP in ATVB and PROMIS. Odds ratio was obtained by a logistic regression model adjusted for age, sex, type 2 diabetes, smoking status.
CHIP (clonal hematopoiesis of indeterminate potential), CHD (coronary heart disease), HR (hazard ratio), MDC (Malmo Diet and Cancer Study), VAF (variant allele fraction), MI (myocardial infarction), OR (odds ratio), ATVB (Atherosclerosis, Thrombosis, and Vascular Biology Italian Study Group), PROMIS (The Pakistan Risk of Myocardial Infarction Study)
The participants in MDC had a median age of 60 years at the time of DNA sample collection and a median follow-up time of 17.7 years. CHIP occurred in 21/320 (6.5%) of coronary heart disease cases but only 12/320 (3.8%) controls (HR 2.0, 95% confidence interval 1.2–3.1, p=0.003 from a Cox proportional hazards model adjusted as above) (Figure 1A, Figure S2,and Table S5).
Combined analysis of both cohorts in a fixed-effects meta-analysis showed that those with clonal hematopoiesis had a 1.9-fold greater risk of incident coronary heart disease (95% confidence interval 1.4–2.7, p=0.0002) (Figure 1A).
CHIP associates with early-onset myocardial infarction
Having established an association between clonal hematopoiesis and coronary heart disease in older individuals, we next asked whether CHIP was a risk factor for early-onset (age <50 years) MI. We analyzed whole exome sequencing data from two case-control studies, the Atherosclerosis, Thrombosis, and Vascular Biology Italian Study Group (ATVB)14 and the Pakistan Risk of Myocardial Infarction Study (PROMIS)15,16. In these studies, cases consisted of individuals with early-onset MI events selected at the time of index presentation to hospitals, while cardiovascular disease-free controls were drawn from the same medical centers. For ATVB, cases were age 45 or younger and age-matched to controls, while for PROMIS we selected only those cases and controls between age 40 and 50. Table S2 presents the baseline characteristics for these cohorts.
The early-onset MI cases had marked enrichment of CHIP compared to controls. In ATVB, 37/1,753 (2.1%) of MI cases had CHIP as compared to 6/1,583 (0.4%) controls (odds ratio (OR) 5.4, 95% confidence interval 2.3–13, p=0.0002 from a logistic regression model adjusted for age, sex, type 2 diabetes status, and smoking status. In PROMIS, 52/2,540 (2.0%) subjects who were MI cases had CHIP as compared to 13/1,369 (0.9%) controls (OR 3.4, 95% confidence interval 1.8–6.5, p=0.0002 from a logistic regression adjusted as above).
A combined fixed-effects meta-analysis of both cohorts showed that CHIP associated with an OR of 4.0 for early-onset MI (95% confidence interval 1.8–6.5, p<0.0001) (Fig 1B).
Mutations in DNMT3A, TET2, ASXL1, and JAK2 individually associate with risk of coronary events
To understand which CHIP genes contributed to risk of coronary heart disease, we performed a gene-level analysis on BioImage, MDC, and three prospective cohorts unselected for coronary events: Jackson Heart Study (JHS), the Finland United States Study of NIDDM Genetics (FUSION), and Framingham Heart Study (FHS)17. JHS and FUSION were part of our prior association study of CHIP with coronary heart disease1, while FHS was newly analyzed for this study. We specifically tested for associations between coronary heart disease and mutations in DNMT3A, TET2, ASXL1, and JAK2. Mutations in DNMT3A, TET2, and ASXL1 associated with a 1.7-fold to 2.0-fold increased risk of incident coronary heart disease, while the JAK2 V617F mutation was associated with a 12-fold increased risk (Figure 2A).
Figure 2. Mutations in DNMT3A, TET2, ASXL1, and JAK2 associate with coronary heart disease.

A) Forest plot for risk of CHD in BioImage and MDC by mutated gene. Hazard ratio for listed mutations was obtained by a fixed-effects meta-analysis of Cox proportional hazards models adjusted for age, sex, type 2 diabetes, total cholesterol, high-density lipoprotein cholesterol, triglycerides, smoking status, and hypertension from BioImage, MDC, and JHS/FUSION/FHS.
B) Table for risk of early-onset MI in ATVB and PROMIS (combined analysis) by mutated gene. Odds ratio for having MI in those with listed mutations was obtained by Fisher’s exact test, p-values not adjusted for multiple hypothesis testing.
C) Proportion of total mutations by gene among MI cases in ATVB and PROMIS versus CHD cases in Biolmage and MDC.
For all forest plots, black square represents the point estimate of hazard ratio, horizontal lines are the 95% confidence interval, vertical line signifies hazard ratio of 1, and results that were from a meta-analysis are shown as diamonds.
CHIP (clonal hematopoiesis of indeterminate potential), Ml (myocardial infarction), Inf. (infinity), CHD (coronary heart disease), HR (hazard ratio), OR (odds ratio), ATVB (Atherosclerosis, Thrombosis, and Vascular Biology Italian Study Group), PROMIS (The Pakistan Risk of Myocardial Infarction Study), MDC (Malmo Diet and Cancer Study), FHS (Framingham Heart Study), JHS (Jackson Heart Study), FUSION (Finland United States Study of NIDDM Genetics)
Mutations in TET2, JAK2, and ASXL1 also showed significant enrichment in early-onset MI cases in ATVB and PROMIS (12/13, 16/16, and 8/8 of individuals with these mutations were myocardial infarction cases, respectively, Figure 2B). JAK2 V617F accounted for 19.3% of the total mutations in MI cases in ATVB/PROMIS, but only 3.7% in BioImage/MDC (Figure 2C).
CHIP associates with increased coronary artery calcification
We hypothesized that increased atherosclerosis burden drives the association between CHIP and coronary heart disease, rather than other factors that might cause MI such as increased thrombosis or vasospasm. In participants from the BioImage study, we assessed data on coronary artery calcification, a non-invasive measure of atherosclerosis detected by cardiac computed tomography. The presence of CHIP associated with increased median coronary artery calcification scores by 3.3-fold amongst those without incident coronary heart disease, and by 1.8-fold amongst those who subsequently developed coronary heart disease (overall p for CHIP=0.03 in a linear regression model adjusted for incident coronary heart disease) (Figure 3A).
Figure 3. CHIP associates with greater coronary artery calcification.

A) Coronary artery calcification scores in those with or without CHIP, stratified by incident CHD. P-value obtained by a linear regression model adjusted for incident CHD status. Median score for each group is shown.
B) Forest plot for association between coronary artery calcification score ≥615 and mutation status stratified by VAF in those without incident CHD in BioImage. Odds ratio was obtained by a logistic regression model adjusted for age, sex, type 2 diabetes, total cholesterol, high-density lipoprotein cholesterol, smoking status, and hypertension.
CHIP (clonal hematopoiesis of indeterminate potential), CAC (coronary artery calcification), VAF (variant allele fraction), odds ratio (OR)
A coronary artery calcification score of greater than 615 Agatston units has served as an empiric cutoff for identifying older individuals at high-risk for coronary events18. Amongst those without incident coronary heart disease, those with CHIP were 3.0-fold more likely to have a coronary artery calcification score ≥615 (p=0.05 in a logistic regression model adjusted for age, sex, type 2 diabetes status, total cholesterol, high density lipoprotein cholesterol, hypertension, and smoking status) (Figure 3B).
We previously found that having a CHIP clone with a variant allele fraction ≥10% (corresponding to ≥20% of nucleated blood cells harboring the mutation) associated with greater risk for developing hematological malignancy than a CHIP clone below this size1. Therefore, we tested whether CHIP with larger clone size also associated with a greater burden of atherosclerosis. Those without incident coronary heart disease but with CHIP with a variant allele fraction ≥10% had 12-fold increased risk of having a coronary artery calcification score ≥615 compared to those without mutations (p=0.002 in a logistic regression model adjusted as above), while those with variant allele fraction <10% had no increased risk (p for test of heterogeneity=0.02). (Figure 3B and Figure S3).
We hypothesized that individuals with a higher proportion of mutated cells might also have greater risk of incident coronary heart disease. In a meta-analysis of BioImage and JHS/FUSION/FHS, we found that the risk for incident coronary heart disease amongst those with a variant allele fraction ≥10% was 2.2-fold increased compared to those without mutations (p=0.0004 from a Cox proportional hazards model adjusted for age, sex, type 2 diabetes status, total cholesterol, high density lipoprotein cholesterol, hypertension, and smoking status), while those with a variant allele fraction <10% had 1.4-fold increased risk compared to those without mutations (p=0.16, p for test of heterogeneity=0.24) (Figure S4). MDC was not included because DNA in this cohort was obtained from granulocytes as opposed to whole blood, which likely inflates the variant allele fraction.
Mice with loss of Tet2 function in hematopoietic cells display accelerated atherosclerosis
Having established a correlation between CHIP and coronary heart disease, we next sought to assess causality experimentally. We selected Tet2 for further study because it is the second most commonly mutated gene in CHIP and associated with risk of coronary heart disease in both older and younger individuals. Previous studies have demonstrated that hematopoietic stem cells from mice with loss-of-function of Tet2 in all hematopoietic cells (Tet2−/−; Vav1-Cre mice) recapitulate the clonal advantage of TET2 mutant hematopoietic cells seen in humans6. We transplanted bone marrow from these mice, or control mice, into irradiated, atherosclerosis-prone Ldlr−/− recipient mice19 and initiated high cholesterol diet after allowing time for hematopoietic reconstitution.
Compared to Ldlr−/−mice receiving control marrow, recipients of Tet2−/− marrow had a median lesion size in the aortic root that was 2.0-fold (p=0.02 by Wilcoxon rank sum test), 1.7-fold (p=0.01 by Wilcoxon rank sum test), and 1.4-fold (p=0.03 by Dunn’s test) larger after 5 weeks, 9 weeks, and 13 weeks on diet, respectively (Figure 4A-B and Figure S5A-B). By 17 weeks on diet, mice receiving Tet2−/− marrow also had a median lesion size in the descending aorta that was 2.7-fold larger than mice receiving control marrow (p=0.02 by Dunn’s test) (Figure 4B and 4D).
Figure 4. Loss of Tet2 in hematopoietic cells accelerates atherosclerosis in a mouse model.

A) Aortic root sections in female Ldlr−/− mice transplanted with either Tet2+/+; Vav1-Cre (WT) or Tet2−/−; Vav1-Cre (KO) marrow after 5 and 9 weeks of feeding on high cholesterol diet. Oil red O (left) and Masson’s trichrome (right) images are shown (40X magnification). Dashed lines indicate lesion area.
B) Quantification of aortic root lesions in female Ldlr−/− mice transplanted with either Tet2+/+; Vav1-Cre (WT), Tet2+/−; Vav1-Cre (HET), or Tet2−/−; Vav1-Cre (KO) marrow at 5 and 9 weeks on diet. P-values obtained by Wilcoxon rank-sum test.
C) Descending aorta lesions stained with oil red O at 17 weeks in female Ldlr−/− mice transplanted with either Tet2+/+; Vav1-Cre (WT), Tet2+/−; Vav1-Cre (HET), or Tet2−/−; Vav1-Cre (KO) marrow.
D) Quantification of descending aorta lesions at 17 weeks in female Ldlr−/− mice transplanted with either Tet2+/+; Vav1-Cre (WT), Tet2+/−; Vav1-Cre (HET), or Tet2−/−; Vav1-Cre (KO) marrow. P-values obtained by Dunn’s Kruskal-Wallis test for multiple comparisons using Benjamini-Hochberg correction.black horizontal line represents the median value.
Most humans with TET2 associated CHIP have only a single allele of the gene mutated1,2. Therefore, we also tested the phenotype of Tet2 haploinsufficiency. Ldlr−/− mice receiving Tet2+/− bone marrow had a median aortic root lesion size that was 1.4-fold larger after 13 weeks on diet (p=0.05 by Dunn’s test) (Figure S5A-B), and a median lesion size in the descending aorta that was 2.7-fold larger after 17 weeks on diet than mice receiving control marrow (p=0.03 by Dunn’s test) (Figure 4C and 4D).
Mice receiving Tet2−/− marrow had normal peripheral white blood cell counts and differential during the study period (Table S6). Fasting serum lipoprotein levels in each group also showed no statistically significant differences after 17 weeks on diet (Table S6).
Loss of Tet2 function in myeloid cells enhances atherosclerosis in mice and alters macrophage inflammatory gene expression in vitro and in vivo
The earliest stages of atherosclerosis involve monocyte infiltration into vessel walls and differentiation into macrophages20. We hypothesized that Tet2 loss alters monocyte/macrophage function in plaques to enhance atherosclerosis. We tested this hypothesis by generating mice that lacked Tet2 in the majority of myeloid cells, but not other lineages (Tet2−/−; Lyz2-Cre)21. In Ldlr−/− mice transplanted with marrow from these mice, the mean aortic root lesion size was 1.7-fold larger than mice receiving control marrow after 10 weeks on diet (p=0.003 by Wilcoxon rank sum test) (Figure S5C).
Tet2 catalyzes DNA hydroxymethylation22, an epigenetic modification that can influence gene transcription. Therefore, we hypothesized that Tet2 modulated gene expression in macrophages exposed to excess cholesterol. We cultured bone marrow-derived macrophages from Tet2−/− or control mice, incubated them with either vehicle or a pathophysiologically relevant dose of native low-density lipoprotein (LDL, 200 mg/dL)23,24, and analyzed the transcriptome by RNA-sequencing. Gene set enrichment analysis revealed that the most significantly up-regulated functional class of genes in Tet2−/− macrophages contained cytokines/chemokines and receptors, while the most significantly suppressed class contained genes involved in lysosomal function (gene class annotations obtained from the KEGG database, Table S7–8 and Figure S6).
We focused on the set of 217 genes that showed differential regulation by both loss of Tet2 and by LDL treatment. Cxcl1, Cxcl2, Cxcl3, Pf4, Il1b, and Il6 transcript levels were among the most highly induced in Tet2−/− macrophages in this set (Figure S7A–B). Cxcl1, Cxcl2, Cxcl3, and Pf4 belong to a single C-X-C motif (CXC) chemokine gene cluster, while Il1b and Il6 are classic pro-inflammatory cytokine genes. Tet2−/− macrophages also secreted more of these proteins in vitro in response to LDL loading or endotoxin exposure than control macrophages. While either LDL or endotoxin strongly induced the CXC chemokines, endotoxin but not LDL caused robust secretion of Il-1b and Il-6 (Figure S7C).
To assess the in vivo significance of these observations, we measured CXC chemokine levels in the transplanted mice after 13–17 weeks on an atherogenic diet. We found that Cxcl1, Cxcl2, Cxcl3, Pf4, and Ppbp increased ~2–4 fold in the serum of Ldlr−/− mice receiving Tet2−/− marrow compared to mice receiving control marrow, while mice receiving Tet2+/− marrow showed intermediate levels (Figure S7D).
We also sought evidence of increased inflammation in other tissues. Mice that received Tet2−/− marrow developed prominent xanthomas in the spleen and middle ear, marked foam cell accumulation and glomerulosclerosis in the kidney, and large inflammatory infiltrates in the liver and lung (Figure S8A-B).
Because we found increased levels of CXC chemokines in the serum of mice receiving Tet2−/− marrow, we sought an analogous increase in humans with TET2 clonal hematopoiesis. The prototypical CXC chemokine in humans is IL-8, which mice lack. Plasma IL-8 levels were available on 2,689 PROMIS controls (age range 40–82). The 12 individuals with TET2 mutations in this cohort had significantly higher circulating IL-8 levels than those without mutations (median 50 versus 21 ng/mL, p=0.02 by Wilcoxon rank sum test on log transformed values) (Figure S8C).
Discussion
In four distinct human studies, we show that somatic mutations leading to CHIP are associated with risk for coronary heart disease or early-onset MI. In a mouse model of atherosclerosis, loss of Tet2 function in hematopoietic cells accelerated atherogenesis.
These results support several conclusions. First, the relationship of CHIP with coronary heart disease appears to be a causal one. Experimental manipulation of one of the genes most frequently mutated in CHIP – Tet2 – worsened atherosclerosis in mice. In humans, coronary events increased in relation to clone size and there was also a dose-response relationship between clone size and atherosclerosis by imaging.
Second, mutations in multiple CHIP-associated genes are linked to coronary heart disease. We suggest that these mutations may increase risk of coronary events due to altered transcriptional output of macrophages. These cells mediate many inflammatory responses, and prominently populate atherosclerotic plaques25,26. In support of this model, we find that loss of Tet2 augments expression of inflammatory chemokines in macrophages exposed to native LDL, an effect that is similar to Tet2-deficient macrophages exposed to bacterial endotoxin27.
Prior studies have found that CXC chemokine interaction with the receptor CXCR2 can mediate firm monocyte adhesion to inflamed endothelium28,29, and this interaction promotes atherogenesis30,31. We propose that a major consequence of TET2 deficiency in tissue macrophages is enhanced recruitment of monocytes and other blood cells to peripheral sites, including the arterial intima, due to elevated expression of CXC chemokines. Indeed, several organs of mice lacking hematopoietic Tet2 harbored large leukocyte aggregates, and TET2 mutations in humans associated with increased levels of plasma IL-8. A recent study also identified augmented Il1b and inflammasome activation as a major mediator of atherosclerosis in mice with hematopoietic Tet2-deficiency32. It is unclear which particular inflammatory mediator(s) predominate in driving atherosclerosis associated with TET2-deficiency, and whether mutations in DNMT3A, ASXL1, and JAK2 also influence risk of coronary events through increased inflammation, issues which invite further experimentation.
An alternative explanation for the observed association is that CHIP-associated mutations provide a proliferative advantage to hematopoietic progenitors and a consequent increase in circulating myeloid cells. Elevations in peripheral blood granulocytes and monocytes have been linked to coronary outcomes in the general population33, while those with JAK2-mutated myeloproliferative neoplasms have increased risk of venous and coronary thrombosis, which is associated with the degree of leukocytosis34,35. However, nearly all individuals with clonal hematopoiesis due to DNMT3A, TET2, and ASXL1 mutations have a normal white blood cell count and differential1,36, and JAK2 mutations account for only a small percentage of CHIP. Furthermore, mice that lacked hematopoietic Tet2−/− had normal blood counts in this study, and others32,37. Nonetheless, we cannot fully rule out a role for leukocytosis in human coronary heart disease, as CHIP could lead to a myeloproliferative state over several years.
In summary, our data support the hypothesis that somatic mutations in hematopoietic cells contribute to the development of human atherosclerosis. We propose that clonal hematopoiesis might be a modifiable risk factor, perhaps through the use of cholesterol lowering medications or targeting of specific inflammatory pathways.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (R01HL082945), the Edward P. Evans Foundation, the Leukemia and Lymphoma Society, and the Howard Hughes Faculty Scholars Program to B.L.E. S.J. is supported by NIH 5T32HL116324 Training Grant and Burroughs Wellcome Career Award for Medical Sciences. P.N. is supported by the John S. LaDue Memorial Fellowship in Cardiology at Harvard Medical School. P.L. is supported by NIH-R01 HL080472 and the RRM Charitable Fund. J.D. reports funding from the UK Medical Research Council (G0800270), British Heart Foundation (SP/09/002), UK National Institute for Health Research Cambridge Biomedical Research Centre, European Research Council (268834), and European Commission Framework Programme 7 (HEALTH-F2–2012-279233). D.S. reports grants from National Heart, Lung and Blood Institute, Pfizer, Regeneron, Eli Lilly and Genentech during the conduct of the study. Fieldwork and biochemical assays in PROMIS have been funded through funding provided by the University of Cambridge from the British Heart Foundation, UK Medical Research Council, Wellcome Trust, EU Framework 6–funded Bloodomics Integrated Project, Pfizer, Novartis, Merck, the Center for Non-Communicable Diseases, Pakistan, and through project grants from the US National Institutes of Health (RC2HL101834 and RC1TW008485) and Fogarty International (RC1TW008485).
References
- 1.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371:2477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014;20:1472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer 2017;17:5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Challen GA, Sun D, Jeong M, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nature genetics 2012;44:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moran-Crusio K, Reavie L, Shih A, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer cell 2011;20:11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jan M, Snyder TM, Corces-Zimmerman MR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Science translational medicine 2012;4:149ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shlush LI, Zandi S, Mitchell A, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014;506:328–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015;126:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baber U, Mehran R, Sartori S, et al. Prevalence, impact, and predictive value of detecting subclinical coronary and carotid atherosclerosis in asymptomatic adults: the Biolmage study. J Am Coll Cardiol 2015;65:1065–74. [DOI] [PubMed] [Google Scholar]
- 11.Wilkins JT, Ning H, Berry J, Zhao L, Dyer AR, Lloyd-Jones DM. Lifetime risk and years lived free of total cardiovascular disease. JAMA 2012;308:1795–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muntendam P, McCall C, Sanz J, Falk E, Fuster V, High-Risk Plaque I. The Biolmage Study: novel approaches to risk assessment in the primary prevention of atherosclerotic cardiovascular disease--study design and objectives. Am Heart J 2010;160:49–57 e1. [DOI] [PubMed] [Google Scholar]
- 13.Berglund G, Elmstahl S, Janzon L, Larsson SA. The Malmo Diet and Cancer Study. Design and feasibility. J Intern Med 1993;233:45–51. [DOI] [PubMed] [Google Scholar]
- 14.Atherosclerosis T, Vascular Biology Italian Study G. No evidence of association between prothrombotic gene polymorphisms and the development of acute myocardial infarction at a young age. Circulation 2003;107:1117–22. [DOI] [PubMed] [Google Scholar]
- 15.Saleheen D, Zaidi M, Rasheed A, et al. The Pakistan Risk of Myocardial Infarction Study: a resource for the study of genetic, lifestyle and other determinants of myocardial infarction in South Asia. Eur J Epidemiol 2009;24:329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Do R, Stitziel NO, Won HH, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature 2015;518:102–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feinleib M, Kannel WB, Garrison RJ, McNamara PM, Castelli WP. The Framingham Offspring Study. Design and preliminary data. Prev Med 1975;4:518–25. [DOI] [PubMed] [Google Scholar]
- 18.Elias-Smale SE, Proenca RV, Koller MT, et al. Coronary calcium score improves classification of coronary heart disease risk in the elderly: the Rotterdam study. J Am Coll Cardiol 2010;56:1407–14. [DOI] [PubMed] [Google Scholar]
- 19.Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest 1993;92:883–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Libby P, Nahrendorf M, Swirski FK. Leukocytes Link Local and Systemic Inflammation in Ischemic Cardiovascular Disease: An Expanded “Cardiovascular Continuum”. J Am Coll Cardiol 2016;67:1091–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abram CL, Roberge GL, Hu Y, Lowell CA. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J Immunol Methods 2014;408:89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009;324:930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith EB. Transport, interactions and retention of plasma proteins in the intima: the barrier function of the internal elastic lamina. Eur Heart J 1990;11 Suppl E:72–81. [DOI] [PubMed] [Google Scholar]
- 24.Kruth HS. Receptor-independent fluid-phase pinocytosis mechanisms for induction of foam cell formation with native low-density lipoprotein particles. Curr Opin Lipidol 2011;22:386–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swirski FK, Libby P, Aikawa E, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest 2007;117:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol 2013;13:709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Q, Zhao K, Shen Q, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015;525:389–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gerszten RE, Garcia-Zepeda EA, Lim YC, et al. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature 1999;398:718–23. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz D, Andalibi A, Chaverri-Almada L, et al. Role of the GRO family of chemokines in monocyte adhesion to MM-LDL-stimulated endothelium. J Clin Invest 1994;94:1968–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boisvert WA, Santiago R, Curtiss LK, Terkeltaub RA. A leukocyte homologue of the IL-8 receptor CXCR-2 mediates the accumulation of macrophages in atherosclerotic lesions of LDL receptor-deficient mice. J Clin Invest 1998;101:353–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huo Y, Weber C, Forlow SB, et al. The chemokine KC, but not monocyte chemoattractant protein-1, triggers monocyte arrest on early atherosclerotic endothelium. J Clin Invest 2001;108:1307–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fuster JJ, MacLauchlan S, Zuriaga MA, et al. Clonal hematopoiesis associated with Tet2 deficiency accelerates atherosclerosis development in mice. Science 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Madjid M, Awan I, Willerson JT, Casscells SW. Leukocyte count and coronary heart disease: implications for risk assessment. J Am Coll Cardiol 2004;44:1945–56. [DOI] [PubMed] [Google Scholar]
- 34.Carobbio A, Finazzi G, Guerini V, et al. Leukocytosis is a risk factor for thrombosis in essential thrombocythemia: interaction with treatment, standard risk factors, and Jak2 mutation status. Blood 2007;109:2310–3. [DOI] [PubMed] [Google Scholar]
- 35.Landolfi R, Di Gennaro L, Barbui T, et al. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood 2007;109:2446–52. [DOI] [PubMed] [Google Scholar]
- 36.Busque L, Patel JP, Figueroa ME, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nature genetics 2012;44:1179–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen E, Schneider RK, Breyfogle LJ, et al. Distinct effects of concomitant Jak2V617F expression and Tet2 loss in mice promote disease progression in myeloproliferative neoplasms. Blood 2015;125:327–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.