

Abstract
Most microorganisms from all taxonomic levels are uncultured. Single-cell genomes and metagenomes continue to increase the known diversity of Bacteria and Archaea, but while ‘omics can be used to infer physiological or ecological roles for species in a community, most of those hypothetical roles remain unvalidated. Here we report an approach to capture specific microorganisms from complex communities into pure cultures using genome-informed antibody engineering. We apply our reverse genomics approach to isolate and sequence single cells and to cultivate three different species-level lineages of human oral Saccharibacteria/TM7. Using our pure cultures we show that all three saccharibacteria species are epibionts of diverse Actinobacteria. We also isolate and cultivate human oral SR1 bacteria, which are members of a lineage of previously uncultured bacteria. Reverse-genomics-enabled cultivation of microorganisms can be applied to any species from any environment and has the potential to unlock the isolation, cultivation and characterization of species from as-yet-uncultured branches of the microbial tree of life.
INTRODUCTION
Our understanding of the microbial tree of life has undergone continuous reshaping and refinement since the advent of molecular, culture-independent methods to identify and classify microorganisms. For example, 16S rRNA gene sequencing resulted in the discovery of nearly two dozen candidate Bacteria and Archaea phyla1, and more recently single-cell genomic and metagenomic assemblies (SAGs and MAGs) have led to a deluge of proposed novel lineages 2–5. Notably, however, SAGs and MAGs of specific taxa are often retrieved by chance, depending on their abundance in the community. Furthermore, although microbial cultivation has been successful for representatives of a handful of phyla that were initially discovered by 16S rRNA sequencing, the vast majority of candidate microbial taxa remain uncultured. Genome-based metabolic reconstructions might provide insights that underpin cultivation of uncultured bacteria and archaea, but so far this promise has only been realized for a select few microbes (reviewed in6).
The causes of microbial “uncultivability” are numerous7, 8 and have been associated with a requirement for factors produced by other microbes9–12, strict interspecies interactions13–15, slow growth16, competition/inhibition and dormancy17. Various high-throughput approaches have been developed to alleviate cultivation recalcitrance. Encapsulation and incubation in microdroplets under media flow, for example, enable interspecies signaling and potential cross-feeding18, 19, and integrating multiple media and cultivation conditions with rapid strain identification (“culturomics”)20–22 have both been successful in expanding the repertoire of cultured species and strains from open environments and human-associated microbiota. Restricting potential competition by stochastic dilutions of complex microbial samples to a few, or single cells, isolated in micro-compartments, while exposing them to native environmental conditions through porous membranes (ichip and similar devices), have also resulted in pure cultures of microbes that were refractory to isolation using standard techniques23, 24.
One important limitation of these cultivation approaches is stochasticity, which means that success in isolating novel organisms is driven partly by chance. Existing methods cannot target specific groups of as-yet-uncultured microbes, especially if those organisms are present at low abundance or have no readily selectable phenotypes. The availability of a plethora of sequence data from all the lineages of life presents an opportunity to design an approach to directly link a genotype to actual cells. A physical marker selected based on genomic data, and compatible with maintaining viability, would enable isolation of microbes, regardless of abundance, for potential cultivation or for selective genomic sequencing. Here we introduce an approach named ‘reverse-genomics isolation’ that is directed, and uses antibodies against predicted cell surface proteins to target efforts to cultivate selected bacteria or archaea (Fig. 1a).
Figure 1.
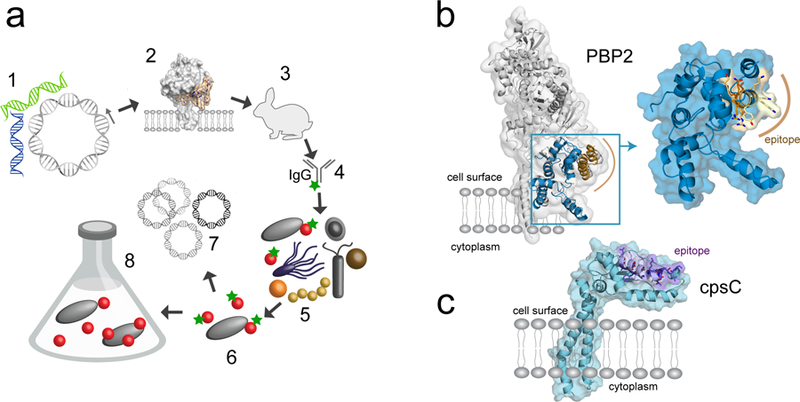
Overview of targeted microbial isolation through reverse genomics. a. Diagram showing the steps in reverse genomics-enabled microbiology: (1) Identification of membrane protein-encoding genes in SAGs and MAGs, (2) selection of predicted exposed epitopes, (3) antibody production, (4) purification and fluorescent labeling, (5) staining of target cells from microbiome samples and (6) isolation for (7) genomic sequencing or (8) cultivation. b. X-ray structure of penicillin-binding protein from E. coli, with the modeled TM7 glycosyltransferase (GT) domain colored blue and the selected epitope region highlighted in yellow-brown. c. Structural model of TM7a capsular polysaccharide biosynthesis protein (cpsC), with the peptide epitope region in purple.
TM7 was one of the first candidate bacterial phyla proposed based on 16S rRNA gene sequences25, 26 and has been found in many environments. After near-complete MAGs were obtained for several TM7 lineages from activated sludge, the group was renamed “Saccharibacteria”27. In humans approximately a dozen distinct species-level Saccharibacteria are recognized28, mostly in the oral cavity, and increased abundance of saccharibacteria is correlated with the development of periodontitis29 and inflammatory bowel disease30. Related lineages are also present in dogs, cats and dolphins31–33. The first genomic data for a human oral TM7 was generated by single-cell sequencing following micro-isolation from supragingival plaque34. Recently, the first representative (“TM7x”) was cultured as an obligate epibiont on a human oral Actinomyces odontolyticus, based on the serendipitous streptomycin resistance of both organisms35. Considering the large taxonomic diversity and ubiquitous environmental presence of these organisms, generally at low abundance (<1%), we decided to develop and test a reverse-genomics isolation method to isolate and cultivate Saccharibacteria.
RESULTS
Selection of cell-surface antibody targets
We used the single-cell genomic data from a human oral saccharibacterium named TM7a34 to identify genes coding for predicted membrane-associated proteins for which a general function could be inferred based on bacterial homologues. We avoided hypothetical proteins, because in the absence of gene expression information we considered those less likely to be present on the cell surface. However, if functional genomic data is available, any membrane protein would be a potential target. We screened for proteins with extracellular domains that could serve as antigens for antibody development, and which had homologues with solved three-dimensional structures, to facilitate searching for surface-exposed epitopes. One candidate protein was a predicted penicillin-binding protein (PBP2). In pathogenic bacteria, PBP2 is an effective vaccine target, indicating that it is expressed and recognizable by an antibody36. Crystal structures for Staphylococcus aureus (PDB entry 3DWK) and Escherichia coli (3VMA) PBP2 homologs were used to model TM7a PBP2 and identify potentially exposed epitopes. PBP2 is anchored in the membrane by a single α-helix and consists of two extracellular domains, an N-terminal glycosyltransferase (GT) domain and a C-terminal transpeptidase (TP) domain connected by a β-rich linker region37 (Fig. 1b). A 19-amino acid sequence located in the GT domain was selected as an antigen based on surface exposure, predicted immunogenicity and solubility, and was used for raising polyclonal antibodies (Supplementary Fig.1). As experimental structure data for membrane proteins is relatively scarce, computational structure prediction could was also selected be used for antigen selection. Capsular polysaccharide biosynthesis protein (CpsC) was selected as a target although it lacked close homologues with solved structures. We generated a Rosetta 3D structure model using coevolution-derived residue-residue contact restraints38 and a predicted antigenic peptide from an extracellular α-helical domain from CpsC was used to raise antibodies (Online methods).
Antibody-enabled sorting of oral Saccharibacteria (TM7)
Human oral samples (saliva, subgingival fluid) from healthy donors or individuals with periodontitis were incubated with fluorescently labeled anti-PBP2 or anti-CpsC IgGs. Stained samples were analyzed by flow cytometry, revealing a distinct population of fluorescent cells (0.1–5%, depending on the sample) (Fig. 2a, b). To determine if Saccharibacteria were labeled by either IgG, we initially sorted pools of 10–100 fluorescent cells and used multiple displacement genomic amplification (MDA) followed by PCR with specific primers (Online methods) targeting the small subunit ribosomal RNA (SSU rRNA) gene. PCR products were analysed by MiSeq amplicon sequencing of the V4 region of the SSU rRNA gene amplified with universal primers (Supplemental Table1, SRA data). Between 25–100% of the cell pools were positive for TM7, depending on individual subjects/samples and gating parameters, with both anti-PBP2 and anti-CpsC retrieving TM7 cells. To enhance the fluorescence signal and to cover as much as possible of the sequence variation space, all experiments directed at cultivation were subsequently performed by dual labelling using both anti-PBP2 and anti-CpsC antibodies. When we analyzed such labeled cell pools for taxonomic representation using MiSeq amplicon sequencing. Saccharibacteria (TM7) comprised up to 95% of the sequences and predominantly represented groups 1, 3 and 6 phylotypes (Fig. 2c, d, Supplementary Fig. 2, Supplemental Table 1, SRA data). Other bacteria were primarily members of Fusobacteria, Actinobacteria, Bacteroidetes, Haemophilus and Streptococcus. The identification of species other than the target TM7 may indicate cross-reactivity of the antibodies, free genomic DNA, or interspecies cell-cell adhesion (specific or non-specific), the latter being extensive in the oral microbiota39, 40.
Figure 2.

Isolation of TM7 cells by flow cytometry cell sorting. a-b. Flow cytometry plot (forward scatter versus green fluorescence) of native (a) and TM7 antibody-labelled oral sample (b). The sort gate area indicates selection characteristics for cell sorting. Highly fluorescent particles (RFU>103) are auto-fluorescent precipitates/minerals. c. Microbial diversity in pools of 10 sorted cells, following MDA and 16S rRNA gene amplicon sequencing. Also shown for comparison is the diversity of the original subgingival sample (far right column). d. Maximum likelihood tree of human oral TM7 SSU rRNA sequences, indicating phylotypes retrieved by antibody labeling and cell sorting (blue dots) and TM7 groups (G1-G6). Circles at nodes indicate bootstrap values (not shown if <50, black>80). Scale bar = substitutions per site. FSC, forward scatter, relative units; RFU, relative fluorescence units.
Antibody enabled cultivation of Saccharibacteria
Having demonstrated that anti-TM7 antibodies can label target cells in biological samples and enable sorting by flow cytometry, we next tested if antibody-sorted cells were viable and could be propagated in culture. Importantly, cultivation could enable identification of potential physiological interactions between various Saccharibacteria (TM7) lineages and other bacteria in the oral microbiota. Using freshly collected oral samples, we labeled bacteria with anti-PBP2 and anti-CpsC antibodies and sorted individual fluorescent cells onto different liquid and solid culture media (Online methods]. Following incubation in anaerobic or hypoxic conditions, cultured cells were assayed using 16S rRNA amplicon sequencing (Supplemental Datasets 1–3).
On solid media, we identified colonies containing five distinct TM7 phylotypes (Human Oral Taxa or HOTs, representing species to family/order taxonomic range), HOTs 346, 348, 352, 952/TM7x (group 1) and HOT351 (group 3) (Fig. 3, 4). All TM7 positive colonies also contained species of Actinobacteria and most (80%) also contained Streptococcus. We hypothesized that Actinobacteria were host organisms, as has been reported for TM7x35, and that Streptococcus, known to be highly adherent cells41, were co-isolates. Surprisingly, TM7 HOTs 346 and 348 (distinct species, based on 93% rRNA identity), were identified in the same colony with Cellulosimicrobium cellulans, an actinobacterium present in healthy saliva but occasionally associated with bacteremia and other diseases42. As it is unlikely the fluorescent particle could have contained multiple Cellulosimicrobium cells that independently also harbored different TM7 (both types of organisms are at very low abundance), multiple epibiont TM7 species may simultaneously be associated with a single host cell. A second round of labeling of the Cellulosimicrobium-TM7 culture (which also contained Streptococcus) and cell sorting resulted in a pure co-culture of C. cellulans-TM7 HOT346 (Fig. 3). In a separate experiment using an oral sample from the same donor, another TM7 HOT346 strain was co-isolated with Actinomyces sp. HOT171. This represents the first demonstration, to our knowledge, that in their natural environment (oral microbiota), TM7 species are most likely epibionts of bacteria that belong to distinct genera. Once pure, the Actinobacteria-TM7 co-cultures could be passaged repeatedly on several types of standard rich solid or liquid media under standard anaerobic conditions and revived following freezing, based on microscopy and PCR-sequencing.
Figure 3.

TM7-host colonies following single particle sorting. (a) Cells from a primary TM7 HOT346–348 positive colony were re-labeled with TM7 antibody and (b) Vertical axis shows relative fluorescence (emission filter 528–538 nm) following excitation at 488 nm. flow sorted based on the P1 gate on BHI-blood agar. The circled colonies contained pure TM7 HOT346-Cellulosimicrobium co-cultures. (c) Primary colony of the TM7 HOT351-Actinomyces sp. HOT897 co-culture on a BHI-blood agar.
Figure 4.

Diversity of cultivated and uncultivated TM7 bacteria. (a) Maximum-likelihood phylogenies of TM7 bacteria and of selected human oral Actinobacteria, based on small subunit rRNA genes. TM7 bacteria include human oral phylotypes (HOTs) as well as related host-associated lineages from open environments, identified by sequence accession numbers and associated with six operational groupings (G1–6). HOTs detected using antibodies, isolated as single cells or identified in cultures are indicated by colored stars. Circles at nodes indicate bootstrap support (>80% filled, 50–80% open, only shown for major groupings). Lines connecting TM7 and Actinobacteria species indicate epibiont-host systems identified previously (black) and in this study (green). (b) Epifluorescence microscopy images of novel TM7-host associations identified in this study using immunolabeling (TM7 HOT351-Actinomyces HOT897 and TM7 HOT346-Cellulosimicrobium cellulans) or FISH (TM7352-Actinomyces odontolyticus OR). Arrows point to green fluorescent TM7 cocci. Scale bar is 5 μm.
TM7 HOT351 is, to our knowledge, the first cultivated member of a novel family- or order- level Saccharibacteria (TM7) lineage (85% 16S RNA identity to TM7x) and was identified in pure co-culture with a previously uncultured Actinomyces species (A. sp. HOT897), most closely related to A. dentalis and A. israelii (92% identity). Actinomyces odontolyticus XH001, the known host of TM7x35 was also identified as the host of two closely related phylotypes, TM7 HOT352 and HOT952, which belong to the same species complex as TM7x. In liquid microcultures of sorted TM7 cells we also detected TM7 HOT347 (Group 1) and HOT870 (Group 6)(Supplemental Data), however those cultures could not be propagated on solid media so far, and we did not yet identify the native hosts of those lineages.
Identification of multiple oral TM7-host pairs provides additional evidence for interbacterial ectosymbiosis within this candidate phylum. Interestingly, based on four oral TM7-Actinobacteria association pairs, there is no apparent correlation in terms of phylogenetic distance between the TM7/ Saccharibacteria species and their Actinobacteria hosts that would indicate co-speciation (Fig. 4a). This suggests that these interactions evolved separately, perhaps after oral colonization, and were not necessarily linked to speciation events. Host specificity appears somewhat limited for TM7x, as closely related Actinomyces cannot substitute the strain that it was co-isolated with35. The findings from our study suggest that cells from distinct TM7 lineages that may also simultaneously attach to the same host cell and that the same TM7 species can use distinct hosts in vivo. That indicates some degree of flexibility in the establishment of the interspecies interaction. The molecular and cellular mechanisms by which different TM7 species interact with their hosts are unknown.
All of the TM7 species reported in this are epibiont cocci of <0.5 μm in diameter present on the surface of host cells. We observed these species using both fluorescence in situ hybridization (FISH) and staining with the TM7 antibody (Fig. 4b). TM7 HOT351 cells are primarily associated with the pole of the rod-shaped Actinomyces host cells, while for TM7 HOTs 346, 352 and 952, there does not appear to be a localization preference, as previously noted43. As with TM7x-A.odontolyticus XH00143 we found that growth conditions affected the relative abundance of each TM7 species in co-culture differently. Whereas TM7x abundance markedly increased in the presence of oxygen, growth of TM7 HOT351 and 346 was inhibited by micro-oxic conditions. The physiology of each TM7 lineage is linked to that of their hosts and are under current studies.
Reverse genomics for other uncultured microbial lineages
We also applied our reverse genomics strategy to cultivate members of a microbial lineage that has no cultured representatives. We selected candidate bacterial phylum SR1/”Absconditabacteria”. SR1 are present at low abundance in a variety of environments including the human oral cavity. SR1 bacteria have highly reduced genomes, indicating likely dependence on interacting organism(s), and have recoded the opal stop codon (UGA) for glycine44. Using single cell genomic data of human oral SR1 HOT345 we selected a predicted surface exposed transglycosylase, also annotated as a surface antigen involved in cell wall biogenesis (Genbank accession RAL55607). This protein is primarily extracellular, with one predicted transmembrane helix. We chose the entire exposed ~300 amino acids domain for antibody production (Online Methods). The antibody we generated was effective for labeling and flow sorting of SR1 cells from human oral samples (Fig. 5). We independently obtained two SR1 cultures. Sequencing of SSU rRNA amplicons using with SR1-specific primers (Online Methods and Supplemental Table 1) revealed that both contained SR1 HOT875, one of three known human oral phylotypes (Fig. 5). The co-cultured bacteria were, however, different and were Fusobacterium periodonticum and Parvimonas micra, respectively. Species related to those bacteria have previously been shown to produce growth factors required by other fastidious bacteria11, 12. However, it is unknown if oral SR1 bacteria are epibiotic on other bacteria as we have not yet found conditions that could increase their abundance to a level that would enable detailed microscopic characterization. Cultivation optimization conditions as well as characterization of binding specificity of the SR1 antibody on bacteria in samples from various environments are under active study.
Figure 5.

Targeted isolation of oral SR1 bacteria. (a) Domains representation of the SR1 HOT 345 LysMpeptidoglycan-binding domain-containing protein. A heterologously expressed extracellular fragment(LysM-CHAP domains) was used as antigen. (b) Flow cytometry plot (forward scatter versus green fluorescence) of anti-SR1 antibody-labelled oral sample. The sort gate area indicates selection characteristics for cell sorting. (c) Phylogenetic diversity of human oral SR1 bacteria (GN02 bacteria is the outgroup) based on SSU rRNA gene. SR1 HOT875 (green star) was identified in enrichments of sorted cells, HOT345 genome (red) was the source for the antigen sequence.
Single cell genomics enabled by reverse genomics
Because not every organism may be readily cultivable, we also tested our targeted isolation approach to obtain single cell genomes for various oral Saccharibacteria (TM7) lineages. Conceptually, targeted collection of cells representing specific species could provide enriched genome sequence information that might underpin physiological hypotheses or support population genomic studies. Flow cytometry enables analysis of more than 5,000 cells per second, therefore taxa present at extremely low abundance (<0.01%) could be isolated. Retrieving such organisms by standard, stochastic single cell sorting and sequencing is impractical. Cell sorting combined with phylogenetic-specific labelling by fluorescence in situ hybridization has occasionally been applied including for TM7 bacteria45, but is inefficient and kills the cells. Our reverse genomics approach may be better suited for population genomic studies involving large number of samples and low abundance target organisms.
In order to study the genomic diversity of TM7 phylotypes from human oral microbiota we carried out single cell sorting and multiple displacement amplification (MDA) on antibody-labeled samples from healthy individuals and from individuals with periodontitis. From 384 sorted particles/cells we identified more than two dozen TM7 single-cell amplified genomes (SAGs), that, based on their 16S rRNA gene sequences, were assigned to six human oral taxa, HOT’s 346, 348, 349, 351, 352 and 353/952. SAGs from two other TM7 HOTs (350 and 356), obtained in a previous large scale un-targeted single-cell isolation44, 46, were also included in the comparative genomics analysis. Unlike genomes derived from metagenome assembled genomes (MAGs), which often lack 16S rRNA sequences and are difficult to match with previously proposed lineages, SAGs can enable bioinformatics analyses. In total, we sequenced, assembled and analyzed 23 SAGs representing four of the six groups of TM7/Saccharibacteria in the human oral microbiome (Groups 1, 2, 3 and 5) (Supplementary Note and Supplemental Table 1).
Comparative genomic analysis of our reconstructed SAGs with existing TM7 genomes supports the hypothesis that their small genome size (~0.6–1 Mbp) is a result of gene (and metabolic capability) loss associated with their epibiotic lifestyle, which likely extends across all oral phylotypes. However, it is not known if TM7 from open (as opposed to oral) environments are dependent on physical association with other bacteria. As with other related lineages from the “Candidate Phyla Radiation” (CPR)2,47, genome reduction has led to the loss of numerous functions, including biosynthesis of amino acids and vitamins. All TM7 genomes to date lack electron transport chains and seem instead likely to have an obligately fermentative metabolism with no respiratory functions. We identified notable differences between the various types of TM7 based on environmental origin or phylogenetic grouping (Fig. 6) and tested experimentally several predictions from our sequenced and cultured species (Supplementary Note). TM7 species (n=20) from open environments encode a nearly complete pentose phosphate pathway coupled with heterolactic fermentation, which could generate reduced NADPH and ATP via substrate-level phosphorylation. On the other hand, human oral TM7 species (n=8) lack important enzymes for the initial NADPH-generating reactions of the pentose phosphate pathway and a key heterolactic fermentation enzyme, xylulose phosphate phosphoketolase. Therefore, oral TM7 may use hexose-pentose interconversions and primarily operate glycolysis reactions for ATP synthesis, suggesting that TM7 central metabolic pathways are evolving to adapt to open environments or oral niches.
Figure 6.

Reconstruction of central metabolic pathways for TM7 bacteria, based on completed genomes and draft SAG/MAG assemblies. Blue or green circles indicate broad presence in environmental and animal-associated TM7. Semicircles indicate occasional presence, associated with specific environments. Absent reactions are in red. The absence of genes encoding three essential pentose phosphate pathway enzymes in human oral TM7 is highlighted in gray.
DISCUSSION
Genome sequences have fostered an improved understanding of microbial diversity and evolution. However, the absence of cultured representatives of many lineages hinders our ability to study the roles numerous lineages of Bacteria and Archaea have in different environments and how they interact with each other. We devised an approach that uses information present in single cell genomic or metagenomic sequences to engineer antibodies that can be used to selectively isolate bacterial or archaeal cells. We validated our approach by bringing three diverse species of TM7/Saccharibacteria into culture along with their interacting Actinobacteria hosts, including one previously uncultured Actinomyces species. Antibody enabled approaches may therefore be especially useful for microbiota whose members are refractory to traditional cultivation methods.
Although previous studies have attempted to bring TM7 into stable laboratory cultures48, 49, the only successful report achieved cultivation of TM7x based on serendipitous antibiotic resistance35. Our method, dubbed ‘reverse genomics’ exploits culture-independent sequence data for physical retrieval of specific microorganisms and does not depend on any physiological selection criteria. Therefore, it should be broadly applicable to any target organisms, as long as robust cultivation conditions can be identified for isolated cells.
We also applied our approach to isolate oral SR1 bacteria, members of a candidate phylum with no previously cultured representatives. Based on their highly reduced genomes44, oral SR1 bacteria also likely depend on other microbes. Following antibody-targeted single particle sorting, we detected growth of SR1 HOT875, one of three distinct human oral phylotypes28. The genome sequences of many putative bacterial and archaeal lineages indicate partnerships such as exo or endosymbiosis. Cultivation is essential for the characterization of such interspecies interactions. Physical retrieval of numerous live cells of even low abundance target microbial taxa from complex communities enables a high throughput search for optimal cultivation parameters and could be applicable to any organism for which a suitable surface antigen can be identified. While for the Saccharibacteria targets we opted to select short peptides, entire protein domains could also be used as antigens for generating polyclonal antibodies, which would target multiple regions of the membrane protein, That may increase the chance of binding, especially if some residues are sterically hindered (e.g. due to glycosylation). We used that approach for generating an effective antibody against SR1 bacteria. However, depending on the degree of sequence conservation, antibodies raised against large protein domains might be less specific.
Antibody-isolated cells could also funneled into other platforms that enable high throughput media selection and optimization protocols, e.g. microdroplets or ichips23, 50. Clearly, not every organism will be immediately culturable, but single cell genomics data from targeted cells could be used to underpin population studies, make additional physiological inferences and, may ultimately results in more complete genome information and ensuing successful cultivation. Applying and further developing our strategy to other yet-uncultured taxa could begin to fill the many cultivation gaps in the tree of life.
Online Methods
Ethics Statement.
Human subjects’ recruitment and sampling protocols were approved by the Ohio State University Institutional Review Board and by the Oak Ridge Site-Wide Institutional Review Board. Written informed consent was obtained from all participants. A table listing participants and samples is provided in Supplemental Table 1.
Sample Collection and Processing.
Saliva samples were self-collected by healthy donors by passive drool, using a SalivaBio collection aid (Salimetrics LLC, Carlsbad, CA, USA), in sterile cryovials. 1–2 ml saliva were collected at least 2 hours after eating or drinking, kept at room temperature and processed immediately or within 2 hours from collection. Recurring samples were collected from the same donors as needed for individual experiments. Gingival crevicular fluid from patients with periodontitis was collected from affected teeth using sterile endodontic paper points that were placed immediately in sterile vials with 1 ml reduced dental transport media (Anaerobe Systems, Morgan Hill, CA, USA), transported to the lab and further processed within 24 hours. Patients with periodontitis were sampled once.
Oral microbiota samples were diluted 1:5 in dental transport media (per liter: Na thioglycollate 1.0 g, Na2HPO4 1.15 g, NaCl 3.0 g, KCl 0.2 g, KH2PO4 0.2g, MgSO4 x 7H2O 0.1 g, L-cysteine HCl 0.5g, pH 7.3), mixed by vortexing for 1 minute then centrifuged at 1000 x g for 2 minutes to remove large particles. The supernatant was passed through 10-μm CellTrics filters (Sysmex, USA) and the cells were collected by centrifugation (12,000 x g for 15 minutes), followed by resuspension in 1 ml PBS. An aliquot was used for total bacterial DNA extraction using proteinase K digestion and phenol-chloroform extraction1. For fixation, an aliquot was fixed by addition of an equal volume of ice cold, 100% ethanol and stored at −20°C overnight. For live cell sorting, the processed microbiota samples were used immediately.
TM7a genome mining, epitope selection and antibody generation.
For selection of candidate proteins to serve as immunogens, we used the single cell genomic data generated by Marcy et al.2, the only oral TM7 genomes available at the time we initiated this work. We used the TM7a (6806 protein genes) and TM7c datasets (590 protein genes), based on the annotations available in IMG (https://img.jgi.doe.gov)3, with the expectation that some of the genes may represent organisms other than TM74. We first selected genes encoding proteins associated with membrane processes, based on COG categories (transport functions, cell wall/membrane/envelope biogenesis, extracellular structures), using the IMG assignments. The corresponding proteins were analyzed for the presence of transmembrane anchoring helical domains using TMHMM v.2.05. From the list of proteins with such domains, we eliminated the ones annotated as hypothetical as well as those shorter than 100 amino acids, as we aimed for final selection of relatively large proteins with known function. The proteins that passed those criteria (117 combined between TM7a and TM7c) were analyzed by blastp against the proteomes of all oral bacteria downloaded from the Human Oral Microbiome Database (http://www.homd.org/). Forty eight of the proteins had high sequence similarity (>90%) to proteins from species of Leptotrichia (Fusobacteria), indicating they are likely contaminants, as previously reported4. We then individually analyzed the remaining sequences for number of transmembrane domains, number and size of predicted extracellular domains, availability of three-dimensional structures for homologues from other bacteria, and experimental data related to abundance or prior use as immunogens. The top pick was penicillin-binding protein 2 (PBP2), which belongs to a class of proteins with important roles in bacterial peptidoglycan synthesis6. Both high resolution crystal structure data7 and multiple reports that anti-PBP2 antibodies can bind to the cell surface of pathogenic bacteria8, 9 were available. The closest homologues to the TM7a PBP2 present in genomes of human oral bacteria had 30–40% pairwise sequence identity (species of Selenomonas, Leptotrichia, Fusobacterium, Streptococcus). To test the applicability of the approach with a protein for which 3D structure or antigenic data were not available, we also selected CpsC as a target, as cellular studies indicated a cell surface-exposed domain10, 11. CpsC belongs to the polysaccharide co-polymerases (PCPs) superfamily and is involved in capsular polysaccharide biosynthesis12. We found no homologues in the oral bacterial genomes with a pairwise identity to TM7a CpsC over 30%.
For selecting peptides to serve as antigens, we analyzed the two protein sequences for antigenic regions and peptide hydrophilicity using the online servers ABCpred13, BepiPred14, IEBD Analysis Resource (http://tools.immuneepitope.org/bcell) and Bio-Synthesis peptide design tools (https://www.biosyn.com), using a 16–18 amino acid-peptide threshold. We used Bio-Synthesis (Lewisville, TX, USA) for peptide synthesis and antibody production. For PBP2, we identified 7 peptides that could serve as antigens and for CpsC we identified four peptides (Supplementary Fig. 1). We next analyzed the location of the 7 peptides from TM7a on the 3D structure of the protein based on the homologous regions of the E. coli PBPB structure (PDB 3vma) using PyMOL (https://pymol.org/2/). Further details on structural modeling and visualization are presented in the comparative structural analysis section. The selected antigen peptide (SGNIKYAKQRQKTVLTRMV) was one of the three peptides with the most exposed residues. In the case of CpsC, because no experimental 3D structure was available, we selected an antigenic peptide (ESAIQEFKEQSKSLYGNS) that is in a helical region of the predicted extracellular domain (Supplementary Fig.1).
The two selected peptides were commercially synthesized by Bio-Synthesis (Lewisville, TX, USA), with cysteine added at N (CpsC) or C terminus (PBP2), purified and injected in rabbits. For antisera production, Bio-Synthesis used a 70-day protocol, with 5 booster doses and 4 test bleeds followed by ELISA. We received serum batches confirmed by ELISA to be reactive against the antigenic peptides. For IgG purification we used HiTrap Protein A HP antibody purification columns (GE Bio-Sciences (Pittsburgh, PA, USA) according to manufacturer’s protocol. For fluorescent IgG labelling of 100 μg IgG aliquots we used the Alexa Fluor™ 488 Antibody Labeling Kit from ThermoFisher (A20181) according to manufacturer’s instructions.
Target selection for an anti-SR1 antibody.
To identify a protein target for generating an antibody against SR1 bacteria, we mined the single cell genome (SAG) of human oral SR1 HOT345 (GenBank: MPSQ00000000.1). We selected the 357 amino acids long hypothetical protein BSK20_04595, which is predicted to contain one transmembrane domain and an extracellular region composed of LysM and CHAP domains. A fragment between aminoacids 39–357 was selected as the antigen. Codon-optimized gene synthesis, overexpression, protein purification, rabbit immunization and affinity purification of IgG were performed commercially by GenScript (https://www.genscript.com/).
Immunofluorescence labeling of oral microbiota samples and flow cytometry.
Aliquots of oral microbiota samples, processed as described above and suspended in PBS were blocked for 30 minutes with 5% goat serum and a mixture of IgGs (1 μg/ml each rabbit anti-Ignicoccus IgG15, anti-Clostridium IgG16 and human IgG, I4506, Sigma, St. Louis MO, USA) for blocking potential non-specific antibody-binding sites on some oral bacteria. We also used 5% rabbit pre-bleed serum instead of the goat serum, with no observed differences. Following blocking, fluorescently labelled anti-PBP2, anti-CpsC or anti-SR1 IgG were added to the samples at 0.1–1 μg/ml. It was important to centrifuge the antibody solution (15,000 x g for 5 minutes) and carefully recover supernatant, prior to addition to the microbiota samples in order to reduce the background of fluorescent particles. Antibody labelling was conducted for 1 hour at room temperature, followed by either centrifugation (12,000 x g for 15 minutes) and resuspension in 2 ml PBS supplemented with 1% serum or directly diluting in 2 ml PBS with 1% serum (mainly for live cell sorting for cultivation experiments). After 30 minutes, the samples were analyzed by flow cytometry. Negative controls included unstained oral sample (to check for auto-fluorescence), and samples processed in parallel with the oral sample but without microbiota (to check for fluorescent antibody precipitates).
For flow cytometry and cell sorting, we used a Cytopeia/BD Influx Model 208S (Cytopeia, Seattle, now BD, San Jose, CA), in a biological safety cabinet (BSC) designed specifically for the Influx cell sorter (Baker Co). Preparation of the flow cytometer for single cell sorting for genomic amplification and sequencing was as we and other have previously described17–19. For sorting, we used a 70-μm nozzle and the 488-nm laser for forward-side scatter (FSC-SSC) analyses as well as fluorescence detection and cell sorting trigger. Gating parameters were selected based on FSC-SSC and fluoresce levels for each experiment and sample. A very low level of highly autofluorescent particles (<0.01%, RFU>103) were observed in all samples in the absence of staining with the antibody and gating parameters were set to avoid sorting such particles. For genomic and community diversity analyses single or pools of fluorescent particles were deposited in 3 μl of DNA-free, Tris–HCl (10 mM pH 8.0), 1 mM EDTA (TE) in individual wells of 96 well plates. We observed sufficient fluorescence signal with primary labelled antibody preparations. However, if the signal would be low, using a secondary labelled antibody (e.g. goat anti-rabbit IgG) would provide signal amplification.
Isolation and cultivation of oral TM7 bacteria.
For cultivation, we used saliva samples collected recurrently from three healthy donors and processed them as detailed above. Antibody staining was performed as already described. For the initial cultivation tests, in 200 μl liquid media in 96-well plates, we used as basal media commercial or home-made brain heart infusion (BHI, Difco), Oral Treponeme Enrichment Broth (OTEB, Anaerobe Systems, Morgan Hill, CA, USA), MTGE Broth (Anaerobe Systems), Tryptic Soy broth (TSB, Difco) supplemented with various additional factors (ATCC vitamins, trace minerals, clarified filtered saliva, pig gastric mucin, sugars, amino acids and nucleobases, N-acetyl muramic acid, N-acetyl glucosamine, pyruvate). Following flow sorting, performed as already described, the plates were incubated in an anaerobic chamber (COY, Grass Lake, MI, USA) at 37°C, under 85%N2, 10%CO2, 5%H2 or in anaerobic jars to which we added air via a syringe port to reach 2% O2. To test for TM7 growth, after 48–72 hours we retrieved 100 μl culture using multichannel pipettes and vacuum-filtered through 0.2 μm filter 96-well plates (Millipore, Burlington MA, USA). The cells were washed with 200 μl PBS. Then, 20 μl TE were added to the filter. The plate was placed on an orbital shaker at 100 rpm for 5 min at room temperature. 10 μl of the cell suspension was then recovered in 96 well PCR plates, to which we added 10 μl lysis solution (0.13 M KOH, 3.3 mM EDTA pH 8.0). The plate was covered with adhesive foil and heated in a PCR machine at 95°C for 5 min. The plate was immediately cooled on ice after which, 10 μl neutralization buffer (0.13 M HCl, 0.42 M Tris pH 7.0, 0.18 M Tris 8.0) was added. We then used 1.5–2 μl of the lysate in PCR reactions with universal and TM7 primers, as detailed earlier. The presence of TM7 in PCR positive cultures were further confirmed by Sanger sequencing and the cultures were transferred into fresh medium under the original growth conditions. We initially obtained TM7 positive cultures using BHI, OTEB, MTGE and TSB, primarily under microaerobic conditions. In liquid microcultures, in addition to TM7 HOT352 (Group 1), we also identified TM7 HOT347 (Group 1) and HOT870 (Group 6). However, none of positive microcultures were pure and contained various other bacteria (Streptococcus, Fusobacterium, Campylobacter, Actinomyces, Propionibacterium and Veillonella). While TM7 survived in those liquid cultures for several passages, we have not identified the hosts for HOT347 and HOT870. Because liquid cultures in 96 well plates were difficult to monitor and to prevent overgrowth, we used cell deposition and cultivation on solid media for final isolation experiments.
For cultivation on solid medium we used DMM-agar20 supplemented with 2% glucose, 0.1g/l casamino acids and 10% sheep blood as well as BHI-agar supplemented with 10% sheep blood. Single cells were deposited by flow sorting in a 10x10 array on each plate, followed by incubation at 37°C under anaerobic or microaerobic conditions. Visible colonies formed after 3–5 days of incubation were sampled with sterile tips and analyzed by direct PCR using primers 27FM-510R (TM7 specific) and 27FM-1492R (universal bacteria). Secondary screening for TM7 bacteria was also performed using primers 910F (5’CATAAAGGAATTGACGGGGAC 3’) and 1177R (5’-GACATCATCCCCTCCTTCC-3’) under the conditions 95°C for 2 minutes, 32–34 cycles of 95°C for 30 seconds, 61°C for 30 seconds, 72°C for 1 minute, followed by 72°C for 5 minutes. Single band products were directly sequenced at Eurofins Genomics (Louisville, KY, USA) using Sanger chemistry (Supplementary Datafile 3). For colonies that contained other bacteria (Streptococcus, Veilonella), the original TM7-positive colony was repeatedly streaked on fresh plates until colonies containing only TM7 and a host actinobacterium were obtained. For TM7 HOT346, a pure co-culture with Cellulosimicrobium cellulans was obtained following a second round of antibody staining and sorting. TM7 enrichments and co-cultures with hosts could be maintained as freezer stocks in media containing 10% DMSO at −80°C. For further physiological characterization of TM7 metabolism potential we supplemented solid (with 10% blood) and liquid (without blood) BHI media with 2–5mM of ribose, lactate, pyruvate, or phosphoenolpyruvate at 37°C or 39°C in duplicate and screened for TM7 relative abundance after 2 and 5 days of incubation and a subsequent passage in the same media, as described above. Compared to a BHI only control, no significant differences were observed.
For microscopic characterization of co-cultures, cells were fixed in 50% ethanol-PBS overnight at −20°C. For fluorescence in situ hybridization we used cell immobilization and dehydration on gelatin-coated slides as described21, with 30% formamide in the hybridization buffer. The probes we used were EUB338 (5’GCTGCCTCCCGTAGGAGT3’)(universal bacteria), labeled with Alexa536 or Alexa647 and TM7–567 (5’CCTACGCAACTCTTTACGCC3’) labeled with Alexa488. In some experiments we also labelled cells with CellBrite Fix 640 Membrane Stain (Biotium) prior to fixation. Following hybridization and washing, the slides were covered with EverBrite Mounting Medium (Biotium) and analyzed by epifluorescence microscopy using a Zeiss AxioImager microscope. Color photographs were taken using a Canon 7D digital SLR and were merged in Adobe Photoshop. For immunofluorescence microscopy, in addition to the anti-PBP2-CspC antibody mix (labelled with Alexa 488), we also used a broad anti-Actinomyces rat IgG, generated by immunizing rats with a mix of Actinomyces species (A. odontoliticus, A. israelii, A. naeslundii, A. gerencseriae) and labeled with Alexa 647. The slides were first blocked with the same cocktail used in antibody staining for flow cytometry and stained with a mix of anti-PBP2-CspC and anti-Actinomyces antibodies for 60 minutes. After washing with PBS five times, the slides were mounted and visualized as above.
Genomic amplification, taxonomic analysis and single cell genome sequencing.
Sorted single cells or cell pools were stored frozen at −80°C until genomic DNA amplification. For amplification by MDA we used a protocol that we and others have described in detail previously17–19. Single cells amplified genomes (SAGs) were screened for the presence of TM7 by PCR using the primers 27FM (5’AGAGTTTGATYMTGGCTCAG-3’) and 510R (5’-CTCTTTACGCCCAGTCAC-3’) by denaturation at 94°C for 3 minutes followed by 32 cycles of amplification (95°C for 30 seconds, 62°C for 30 seconds, 72°C for 30 seconds) and a final extension at 72°C for 5 minutes. PCR products were sequenced on an ABI3730 DNA Analyzer (Applied Biosystems), at the University of Tennessee Sequencing Facility or at Eurofins Genomics (Louisville, KY, USA), using the same primers. Resulting chromatograms were manually edited for low quality regions using Geneious (https://www.geneious.com/) and sequences were used for phylogenetic identifications using the HOMD SSU rRNA database (http://www.homd.org/). Samples for which the chromatograms were not homogeneous and that denoted potential heterogeneous template DNA (more than one sorted cell) were not further used. While a mixed chromatogram may indicate the presence of a symbiont of TM7, because they could not be unambiguously distinguished from either DNA contamination or non-specifically adherent cells co-sorted with TM7, so we did not pursue characterization of mixed SAGs. Also, as gram positive bacteria (such as Actinobacteria) cannot be efficiently lysed by the alkaline treatment step of the MDA amplification protocol, identifying the potential hosts of those TM7 cells would be unlikely.
To characterize the microbial diversity in cell populations sorted following antibody staining (10–100 cells), genomic amplification by MDA was conducted the same way as for single cells. The amplification product was first purified using a Zymo genomic DNA cleanup kit (Zymo Research, Irvine CA, USA). To determine the microbial composition of the original oral microbiota samples we used gDNA purified from the saliva and the subgingival samples. Microbial diversity was determined by amplification of the hypervariable V4 region of SSU rRNA gene followed by MiSeq amplicon sequencing. We followed the protocol described by Lundberg et al.22, with a modification in the universal primers. Specifically, because the canonical 515F primer sequence (5’GTGCCAGCMGCCGCGGTAA) does not recognize TM7 rRNA, we supplemented the PCR reaction mix with 5% of a 515F-derivative that can bind TM7 rRNA (5’GTGCCAGCMGCCGCGGTCA). The reverse primer, 806R (5’GGACTACHVGGGTWTCTAAT) can recognize the full diversity of oral bacterial SSU rRNA. The purified amplicons were sequenced on the MiSeq platform (Illumina, San Diego CA, USA) on v2 or v3 sequencing chips using 2x250 nt reads. Read processing and microbial diversity analysis was performed using the software programs cutadapt23, usearch24 and QIIME25 as well as the HOMD SSU rRNA taxonomy database and following the commands and parameters listed in the Supplementary Dataset 4. To determine if the 254 nt V4 amplicon provides sufficient resolution to enable distinguishing the different human oral TM7 phylotypes, we determined the pairwise sequence identity between the 13 main types of oral TM7 OTUs and performed phylogenetic analysis including the TM7 OTUs identified based on sequencing the V4 amplicon (TM7HOTs_OTUs V4.fasta supplementary file and files deposited in the SRA archive linked to NCBI Bioproject PRJNA472398). As shown in Supplementary Fig.2 (sequences provided in Supplementary Dataset 5 and in the online SRA data), with the exception of 2 OTU pairs (HOT346-HOT869 and HOT352-HOT952), all other TM7 could be readily distinguished in MiSeq amplicons, with pairwise identity values bellow 98% and sufficient phylogenetic resolution.
Purified DNAs of TM7 SAGs selected for genomic sequencing were used to generate libraries and were sequenced on Illumina HiSeq (100 nt paired reads) at the Hudson Alpha Institute for Biotechnology (Huntsville, AL, USA) or on MiSeq (250 nt paired reads) at ORNL. Approximately 100 million reads were obtained for each SAG sequenced on HiSeq and 10 million for the MiSeq SAGs.
SAG assembly, annotation and comparative genomic analysis.
For individual assembly, the reads for each SAG were trimmed using Trim Galore 0.4 (https://github.com/FelixKrueger/TrimGalore) and assembled using SPADES (v3.9.0)26. Removal of non-TM7 contigs (human and other bacterial sequences) was performed by a combination of GC-content-based binning, and Emergent Self-Organizing Maps (ESOM) based on kmer frequency 27. Further contamination was removed using DNA and protein BLAST and single-copy gene analysis28, 29. Lastly, further assembly in Geneious (v8–10)30 consolidated some contigs and the final cleaned genomes were analyzed with Prokka (v1.11)31 for gene prediction and annotation. Completion and contamination estimates were calculated using CheckM (v1.0.11)29 using the lineage workflow and a manually adapted gene-list workflow containing 42 single copy genes conserved in CPR Bacteria32 (Table S1, Supplementary Note, Supplementary Fig. 3, 4). Pfam (v31)33, eggNOG (v4.5.1)34 and Interproscan (v5.27–66.0)35 databases were used for protein assignment to families and orthologous groups. Anvi’o (v5.1) was used for core genome analysis and identification of conserved COGs following the pangenomics pipeline with mapping of eggNOGs36, 37 (Table S1 and Supplementary Fig 3, 5). NCBI, IMG and Geneious were used for basic homology searches and for obtaining genomic data for further metabolic inference analyses (Supplementary Note). SSU rRNA and concatenated single-copy protein gene trees were generated in Geneious using RAxML, with estimation of all free parameters and 100 bootstraps (Supplementary Fig. 6). The TM7 SAGs sequences and associated metadata were deposited in Genbank under Bioproject PRJNA472398. SSU rRNA sequence alignments that include reference strains as well as detected phylotypes, SAGs and isolates obtained in this study are provided as supplemental datasets files 1–3.
Protein structure modelling.
The glycosyltransferase domain of PBP2 from TM7a (residues 95–278) and TM7x (residues 145–334) was modeled using a combination of homology modeling and coevolution-based residue-residue contact prediction. We used the GREMLIN web server38–40 to identify structural templates, predict residue contacts based on coevolution analysis, and generate contact restraints for Rosetta structural modeling41. Briefly, the server uses HHblits42 and to generate a multiple sequence alignment (MSA), which is then filtered to include only sequences that cover at least 75% of the query sequence, have fewer than 75% gaps, and have a maximum mutual sequence identity of 90%. From this MSA, GREMLIN learns a global statistical model that accounts for both sequence conservation and coevolution. Next, HHsearch43 was used to identify potential templates for homology modeling from the Protein Data Bank (PDB)44. More than 9,300 sequences of PBP2 homologs were identified from the sequence database search. For the ~190 residue proteins this number corresponds to a sequence/length ratio of ~51, indicating the robustness of the predicted residue contacts.
For both TM7a and TM7x, the following five templates identified by HHsearch were used as templates for modeling PBP2: 3VMA45, which is missing part of jaw subdomain, 3UDF46 (also missing the jaw subdomain), 2OQO47, 2OLV7, and 3VMT48 (Supplementary Fig.7). Although multiple template structures are available, we included coevolution information to provide additional benefits compared to standard homology modeling. We used map_align (https://github.com/gjoni/map_align) to align the contact maps predicted from the coevolution analysis to selected templates identified by HHsearch. The initial threaded models generated by map_align were then used as input for Rosetta hybrid modeling, and all models were given weights of 1.0. Fragment files were obtained with the Robetta server49. We generated 10,000 models with both sigmoidal and bounded restraints during coarse-grained sampling and only sigmoidal restraints during full-atom refinement40. Models were ranked on the basis of the sum of their Rosetta energy and the restraint score. The final models are shown in Supplementary Fig.7. PyMOL (https://pymol.org/2/) and pdb format files of the models are also provided as Supplementary datasets 6–8.
CpsC (residues 1–197) was modeled with Rosetta using coevolution-derived residue-residue contact restraints. More than 3,700 sequences obtained with the GREMLIN webserver were used to perform the coevolution analysis. A PyMOL file of the 3D model structure is provided as Supplementary dataset 9.
We analyzed the SAGs for the presence of the genes encoding the two proteins used as epitopes for raising antibodies. For PBP2, we found the gene in SAGs representing all of the TM7 lineages isolated by antibody capture. For Cbp2, we found the gene in only three of the lineages. This gene is also missing in the complete genome of TM7x, so the Cbp2 protein is not present across all TM7. The level of sequence similarity at the epitopes level varied between the different TM7 as compared to the original TM7a sequence but there were multiple conserved amino acids across all lineages. To better understand the structural basis of epitope recognition in the different TM7 PBP2, we modeled the three-dimensional structure of the TM7a/HOT348 and TM7x/HOT952 proteins based on crystal structures available for several homologues (Supplementary Fig. 7). In addition to structural templates, we included coevolutionary information in the form of residue-residue contacts 39 predicted from a deep multiple sequence alignment to improve the structural models. Because we were primarily interested in identifying structural determinants of epitope recognition, we generated structural models only of the extracellular, N-terminal glycosyltransferase domain (residues 95–278 in the TM7a PBP2 sequence). The final models share a similar overall fold and structure between TM7a and TM7x PBP2 even though they are only 37% identical. The α-helix containing most of the epitope is located on the surface of the protein, with some of the conserved residues buried and others exposed or partially exposed (Figure 1b). A similar epitope topology was observed in the Cbp2 protein model (Figure 1c). The exposed residues include multiple polar and charged amino acids, which may be responsible for antibody recognition and binding. It is important to note that although the sequence similarity in the epitope region is as low as 35–50 % across the TM7 lineages, IgG variants were able to bind their target on the cell surface. Although the specificity breadth cannot be predicted, especially when only a single target sequence is available (as was the case with TM7a), polyclonal antibodies can therefore recognize multiple related species and increase the diversity in targeted isolation of microbial cells. Broader specificity can also result in cross reactivity with more distant, non-target organisms, potentially explaining some of the additional taxa we observed in pooled cell populations.
Sorting and cultivation of SR1 bacteria.
Fresh healthy saliva samples were processed and labelled with the anti-SR1 antibody as described above. Fluorescent particles selected based on the gate shown in Figure 5 were deposited (1–5 per spot) on BHI-blood agar plates and were incubated at 37C in an anaerobic chamber for 5 days, when distinct colonies were observed. To screen for the presence of SR1 bacteria, we used specific SSU rRNA primers SR1–31F, 5’GATGAACGCTAGCGAAAT and SR1–32R, 5’CTTAACCCCAGTCACTGATT3’. Direct Sanger sequencing of the PCR products obtained from two cultures revealed the presence of SR1 HOT875 (99% sequence identity) (Supplementary Figure 8). Sequencing of PCR products obtained using universal primers (27F-1492R) from the same cultures identified Fusobacterium periodonticum and Parvimonas micra as dominant species. We prepared freezer stocks from those cultures for conditions and media optimization in order to increase the abundance of SR1 bacteria.
Statistics and Reproducibility.
Human volunteer subjects were recruited to provide oral various sample types (saliva, subgingival crevicular fluid) in order to broaden the diversity of target TM7 bacteria that could be found across a small sampled population. Age, sex, race, ethnic origin, oral health or other factors were not part of the study and were not linked to any of the results. Sampling was conducted based on subjects’ availability and none of the results (microbial diversity, genomic data or cultivated strains) were used for correlations to those samples or individuals.
Flow cytometry profiles of oral samples stained with TM7 antibodies (unstained cells and the fluorescent population identified to contain TM7 bacteria) (Figure 2) were highly reproducible across over two dozen independent experiments performed on oral samples from the different individuals.
We performed 9 independent cultivation experiments in 96-well plates using a variety of liquid culture media (detailed above), aimed at narrowing down conditions permissive for TM7 propagation from single sorted cells/particles. In six of those experiments, we obtained TM7-positive cultures (1–5 positives per experiment) (Figure 3). We subsequently performed seven independent cultivation experiments on solid media (100 cells/particles per plate, 2–4 plates per experiment). Between 8 and >50 colonies were obtained per plate, following incubation under anaerobic or microaerophilic conditions. Following screening by PCR, we identified 1–4 positive colonies in every experiment. Each of the TM7 phylotypes was independently identified in at least two separate experiments, using independent oral samples. Because the abundance of TM7 in colonies could be below detection, depending on growth rate and competing co-isolated bacteria, TM7-negative colonies cannot be automatically ruled out as representing bacteria that cross reacted with the TM7 antibody.
Fluorescence imaging of TM7 bacteria and their Actinobacteria hosts was performed in two separate experiments, Figure 4 showing a combination of panels from both experiments. The cell size, morphology and observable interaction between the TM7 and their host cells are similar to those reported for TM7x50.
Supplementary Material
Acknowledgements
We thank S. Allman, S. Kauffman. S. Lebreux, L. Sukharnikov and M. Robeson for technical assistance. Support for this work was provided by grants R56DE021567 and R01DE024463 from the National Institute of Dental and Craniofacial Research of the U.S. National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. J.M.P. was supported by the Laboratory Directed Research and Development program at Oak Ridge National Laboratory (ORNL), which is managed by UT-Battelle, LLC for the U.S. Department of Energy under Contract No. DE-AC05-00OR22725. C.J.C. was supported by a National Science Foundation Graduate Research Fellowship under Grant No. 2017219379. This work used resources of the Compute and Data Environment for Science (CADES) at ORNL.
Footnotes
Online Methods, supplementary data and associated references are available in the online version of the paper.
Ethical Compliance. Human subjects’ recruitment and sampling protocols were approved by the Ohio State University Institutional Review Board and by the Oak Ridge Site-Wide Institutional Review Board. Written informed consent was obtained from all participants. The use of commercial custom antibodies was approved by the Oak Ridge National Laboratory Animal Care and Use Committee.
Data Availability. Annotated TM7 SAGs are deposited in GenBank under the Bioproject PRJNA472398. Raw and aligned SSU rRNA sequences are provided as supplementary files (Datasets 1–3). MiSeq amplicon data are deposited in SRA linked to Bioproject PRJNA472398.
Competing Interests. The authors declare no competing interests.
References:
- 1.DeLong EF & Pace NR Environmental diversity of bacteria and archaea. Syst Biol 50, 470–478 (2001). [PubMed] [Google Scholar]
- 2.Hug LA et al. A new view of the tree of life. Nature microbiology 1, 16048 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Zaremba-Niedzwiedzka K et al. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature 541, 353–358 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Parks DH et al. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nature microbiology 2, 1533–1542 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Rinke C et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Gutleben J et al. The multi-omics promise in context: from sequence to microbial isolate. Critical reviews in microbiology, 1–18 (2017). [DOI] [PubMed]
- 7.Epstein SS The phenomenon of microbial uncultivability. Curr Opin Microbiol 16, 636–642 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Overmann J, Abt B & Sikorski J Present and Future of Culturing Bacteria. Annu Rev Microbiol 71, 711–730 (2017). [DOI] [PubMed] [Google Scholar]
- 9.D’Onofrio A et al. Siderophores from neighboring organisms promote the growth of uncultured bacteria. Chemistry & biology 17, 254–264 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strandwitz P et al. GABA-modulating bacteria of the human gut microbiota. Nature microbiology (2018). [DOI] [PMC free article] [PubMed]
- 11.Vartoukian SR et al. In Vitro Cultivation of ‘Unculturable’ Oral Bacteria, Facilitated by Community Culture and Media Supplementation with Siderophores. PLoS One 11, e0146926 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cross KL et al. Insights into the Evolution of Host Association through the Isolation and Characterization of a Novel Human Periodontal Pathobiont, Desulfobulbus oralis. mBio 9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huber H et al. A new phylum of Archaea represented by a nanosized hyperthermophilic symbiont. Nature 417, 63–67 (2002). [DOI] [PubMed] [Google Scholar]
- 14.St. John E et al. A new symbiotic nanoarchaeote (Candidatus Nanoclepta minutus) and its host (Zestosphaera tikiterensis gen. nov., sp. nov.) from a New Zealand hot spring. Systematic and applied microbiology (2018). [DOI] [PubMed]
- 15.Wurch L et al. Genomics-informed isolation and characterization of a symbiotic Nanoarchaeota system from a terrestrial geothermal environment. Nature communications 7, 1–10 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pulschen AA et al. Isolation of Uncultured Bacteria from Antarctica Using Long Incubation Periods and Low Nutritional Media. Front Microbiol 8, 1346 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oliver JD Recent findings on the viable but nonculturable state in pathogenic bacteria. FEMS Microbiol Rev 34, 415–425 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Terekhov SS et al. Microfluidic droplet platform for ultrahigh-throughput single-cell screening of biodiversity. Proc Natl Acad Sci U S A 114, 2550–2555 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zengler K et al. High-throughput cultivation of microorganisms using microcapsules. Methods Enzymol 397, 124–130 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Browne HP et al. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature 533, 543–546 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lagier JC et al. Culture of previously uncultured members of the human gut microbiota by culturomics. Nature microbiology 1, 16203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Oberhardt MA et al. Harnessing the landscape of microbial culture media to predict new organism-media pairings. Nature communications 6, 8493 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berdy B, Spoering AL, Ling LL & Epstein SS In situ cultivation of previously uncultivable microorganisms using the ichip. Nat Protoc 12, 2232–2242 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Sizova MV et al. New approaches for isolation of previously uncultivated oral bacteria. Appl Environ Microbiol 78, 194–203 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rheims H, Rainey FA & Stackebrandt E A molecular approach to search for diversity among bacteria in the environment. Journal of Industrial Microbiology 17, 159–169 (1996). [Google Scholar]
- 26.Hugenholtz P, Goebel BM & Pace NR Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180, 4765–4774 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Albertsen M et al. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31, 533–538 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Dewhirst FE et al. The Human Oral Microbiome. Journal of Bacteriology 192, 5002–5017 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brinig MM, Lepp PW, Ouverney CC, Armitage GC & Relman DA Prevalence of bacteria of division TM7 in human subgingival plaque and their association with disease. Appl Environ Microbiol 69, 1687–1694 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuehbacher T et al. Intestinal TM7 bacterial phylogenies in active inflammatory bowel disease. J Med Microbiol 57, 1569–1576 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Dewhirst FE et al. The feline oral microbiome: a provisional 16S rRNA gene based taxonomy with full-length reference sequences. Veterinary microbiology 175, 294–303 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dewhirst FE et al. The canine oral microbiome. PLoS One 7, e36067 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dudek NK et al. Novel Microbial Diversity and Functional Potential in the Marine Mammal Oral Microbiome. Curr Biol (2017). [DOI] [PubMed]
- 34.Marcy Y et al. Dissecting biological “dark matter” with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. Proc Natl Acad Sci U S A 104, 11889–11894 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He X et al. Cultivation of a human-associated TM7 phylotype reveals a reduced genome and epibiotic parasitic lifestyle. Proc Natl Acad Sci U S A 112, 244–249 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haghighat S, Siadat SD, Sorkhabadi SMR, Sepahi AA & Mahdavi M A novel recombinant vaccine candidate comprising PBP2a and autolysin against Methicillin Resistant Staphylococcus aureus confers protection in the experimental mice. Mol Immunol 91, 1–7 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Lovering AL, de Castro LH, Lim D & Strynadka NC Structural insight into the transglycosylation step of bacterial cell-wall biosynthesis. Science 315, 1402–1405 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Simons KT et al. Improved recognition of native-like protein structures using a combination of sequence-dependent and sequence-independent features of proteins. Proteins 34, 82–95 (1999). [DOI] [PubMed] [Google Scholar]
- 39.Palmer RJ Jr. et al. Interbacterial Adhesion Networks within Early Oral Biofilms of Single Human Hosts. Appl Environ Microbiol 83 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mark Welch JL, Rossetti BJ, Rieken CW, Dewhirst FE & Borisy GG Biogeography of a human oral microbiome at the micron scale. Proc Natl Acad Sci U S A 113, E791–800 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jakubovics NS, Yassin SA & Rickard AH Community interactions of oral streptococci. Adv Appl Microbiol 87, 43–110 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Heym B et al. Molecular detection of Cellulosimicrobium cellulans as the etiological agent of a chronic tongue ulcer in a human immunodeficiency virus-positive patient. J Clin Microbiol 43, 4269–4271 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bor B et al. Phenotypic and Physiological Characterization of the Epibiotic Interaction Between TM7x and Its Basibiont Actinomyces. Microb Ecol 71, 243–255 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Campbell JH et al. UGA is an additional glycine codon in uncultured SR1 bacteria from the human microbiota. PNAS 110, 5540–5545 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Podar M et al. Targeted access to the genomes of low-abundance organisms in complex microbial communities. Appl Environ Microbiol 73, 3205–3214 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Campbell AG et al. Diversity and genomic insights into the uncultured Chloroflexi from the human microbiota. Environ Microbiol 16, 2635–2643 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Castelle CJ et al. Biosynthetic capacity, metabolic variety and unusual biology in the CPR and DPANN radiations. Nature Reviews Microbiology 16, 629–645 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Ferrari BC, Binnerup SJ & Gillings M Microcolony cultivation on a soil substrate membrane system selects for previously uncultured soil bacteria. Appl Environ Microbiol 71, 8714–8720 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Soro V et al. Axenic culture of a candidate division TM7 bacterium from the human oral cavity and biofilm interactions with other oral bacteria. Appl Environ Microbiol 80, 6480–6489 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zengler K et al. Cultivating the uncultured. Proc Natl Acad Sci U S A 99, 15681–15686 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1.Wurch L et al. Genomics-informed isolation and characterization of a symbiotic Nanoarchaeota system from a terrestrial geothermal environment. Nature communications 7, 1–10 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marcy Y et al. Dissecting biological “dark matter” with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. Proc Natl Acad Sci U S A 104, 11889–11894 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Markowitz VM et al. The integrated microbial genomes (IMG) system. Nucleic Acids Res 34, D344–348 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Albertsen M et al. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31, 533–538 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Krogh A, Larsson B, von Heijne G & Sonnhammer EL Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305, 567–580 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Sauvage E, Kerff F, Terrak M, Ayala JA & Charlier P The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiology Reviews 32, 234–258 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Lovering AL, de Castro LH, Lim D & Strynadka NC Structural insight into the transglycosylation step of bacterial cell-wall biosynthesis. Science 315, 1402–1405 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Haghighat S, Siadat SD, Sorkhabadi SM, Sepahi AA & Mahdavi M Cloning, Expression and Purification of Penicillin Binding Protein2a (PBP2a) from Methicillin Resistant Staphylococcus aureus: A Study on Immunoreactivity in Balb/C Mouse. Avicenna J Med Biotechnol 5, 204–211 (2013). [PMC free article] [PubMed] [Google Scholar]
- 9.Zarantonelli ML et al. Immunogenicity of meningococcal PBP2 during natural infection and protective activity of anti-PBP2 antibodies against meningococcal bacteraemia in mice. J Antimicrob Chemother 57, 924–930 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Byrne JP, Morona JK, Paton JC & Morona R Identification of Streptococcus pneumoniae Cps2C residues that affect capsular polysaccharide polymerization, cell wall ligation, and Cps2D phosphorylation. J Bacteriol 193, 2341–2346 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toniolo C et al. Streptococcus agalactiae capsule polymer length and attachment is determined by the proteins CpsABCD. J Biol Chem 290, 9521–9532 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morona R, Purins L, Tocilj A, Matte A & Cygler M Sequence-structure relationships in polysaccharide co-polymerase (PCP) proteins. Trends Biochem Sci 34, 78–84 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Saha S & Raghava GP Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 65, 40–48 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Larsen JE, Lund O & Nielsen M Improved method for predicting linear B-cell epitopes. Immunome Res 2, 2 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heimerl T et al. A Complex Endomembrane System in the Archaeon Ignicoccus hospitalis Tapped by Nanoarchaeum equitans. Front Microbiol 8, 1072 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller LD et al. Establishment and metabolic analysis of a model microbial community for understanding trophic and electron accepting interactions of subsurface anaerobic environments. BMC Microbiol 10, 149 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campbell AG et al. Multiple single-cell genomes provide insight into functions of uncultured Deltaproteobacteria in the human oral cavity. PLoS One 8, e59361 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Podar M et al. Insights into archaeal evolution and symbiosis from the genomes of a nanoarchaeon and its inferred crenarchaeal host from Obsidian Pool, Yellowstone National Park. Biol Direct 8, 9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rinke C et al. Obtaining genomes from uncultivated environmental microorganisms using FACS-based single-cell genomics. Nat Protoc 9, 1038–1048 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Wong L & Sissons C A comparison of human dental plaque microcosm biofilms grown in an undefined medium and a chemically defined artificial saliva. Arch Oral Biol 46, 477–486 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Pernthaler A & Pernthaler J Fluorescence in situ hybridization for the identification of environmental microbes. Methods Mol Biol 353, 153–164 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Lundberg DS, Yourstone S, Mieczkowski P, Jones CD & Dangl JL Practical innovations for high-throughput amplicon sequencing. Nat Methods 10, 999–1002 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Martin M Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMBnet.journal. [Google Scholar]
- 24.Edgar RC Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Caporaso J, Kuczynski J, Stombaugh J & Bittinger K QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–336 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bankevich A et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of computational biology : a journal of computational molecular cell biology 19, 455–477 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown CT et al. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 523, 208–211 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Hug LA et al. Critical biogeochemical functions in the subsurface are associated with bacteria from new phyla and little studied lineages. Environ Microbiol (2015). [DOI] [PubMed] [Google Scholar]
- 29.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P & Tyson GW CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25, 1043–1055 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kearse M et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seemann T Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Anantharaman K et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nature communications 7, 13219 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finn RD et al. Pfam: the protein families database. Nucleic Acids Res 42, D222–230 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huerta-Cepas J et al. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res 44, D286–293 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones P et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delmont TO & Eren AM Linking pangenomes and metagenomes: the Prochlorococcus metapangenome. PeerJ 6, e4320 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eren AM et al. Anvi'o: an advanced analysis and visualization platform for 'omics data. PeerJ 3, e1319 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balakrishnan S, Kamisetty H, Carbonell JG, Lee SI & Langmead CJ Learning generative models for protein fold families. Proteins 79, 1061–1078 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Kamisetty H, Ovchinnikov S & Baker D Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era. Proc Natl Acad Sci U S A 110, 15674–15679 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ovchinnikov S et al. Protein structure determination using metagenome sequence data. Science 355, 294–298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simons KT et al. Improved recognition of native-like protein structures using a combination of sequence-dependent and sequence-independent features of proteins. Proteins 34, 82–95 (1999). [DOI] [PubMed] [Google Scholar]
- 42.Remmert M, Biegert A, Hauser A & Soding J HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods 9, 173–175 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Johnson LS, Eddy SR & Portugaly E Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics 11, 431 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burley SK et al. Protein Data Bank (PDB): The Single Global Macromolecular Structure Archive. Methods Mol Biol 1607, 627–641 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sung MT et al. Crystal structure of the membrane-bound bifunctional transglycosylase PBP1b from Escherichia coli. Proc Natl Acad Sci U S A 106, 8824–8829 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han S et al. Distinctive attributes of beta-lactam target proteins in Acinetobacter baumannii relevant to development of new antibiotics. Journal of the American Chemical Society 133, 20536–20545 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Yuan Y et al. Crystal structure of a peptidoglycan glycosyltransferase suggests a model for processive glycan chain synthesis. Proc Natl Acad Sci U S A 104, 5348–5353 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang CY et al. Crystal structure of Staphylococcus aureus transglycosylase in complex with a lipid II analog and elucidation of peptidoglycan synthesis mechanism. Proc Natl Acad Sci U S A 109, 6496–6501 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gront D, Kulp DW, Vernon RM, Strauss CE & Baker D Generalized fragment picking in Rosetta: design, protocols and applications. PLoS One 6, e23294 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He X et al. Cultivation of a human-associated TM7 phylotype reveals a reduced genome and epibiotic parasitic lifestyle. Proc Natl Acad Sci U S A 112, 244–249 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.