

Abstract
The worldwide outbreak of severe acute respiratory syndrome (SARS) in 2003 had caused a high rate of mortality. Main protease (Mpro) of SARS-associated coronavirus (SARS-CoV) is an important target to discover pharmaceutical compounds for the therapy of this life-threatening disease. During the course of screening new anti-SARS agents, we have identified that a series of unsymmetrical aromatic disulfides inhibited SARS-CoV Mpro significantly for the first time. Herein, 40 novel unsymmetrical aromatic disulfides were synthesized chemically and their biological activities were evaluated in vitro against SARS-CoV Mpro. These novel compounds displayed excellent IC50 data in the range of 0.516–5.954 μM. Preliminary studies indicated that these disulfides are reversible and mpetitive inhibitors. A possible binding mode was generated via molecular docking simulation and a comparative field analysis (CoMFA) model was constructed to understand the structure-activity relationships. The present research therefore has provided some meaningful guidance to design and identify anti-SARS drugs with totally new chemical structures.
Keywords: SARS-CoV Mpro, Aromatic disulfide, Molecular docking, In vitro activity
Graphical abstract

Highlights
-
•
40 novel unsymmetrical aromatic disulfides were synthesized.
-
•
The synthesized disulfide compounds are potent inhibitors of SARS main protease.
-
•
Possible binding mode and structure-activity relationships of the compounds were established.
1. Introduction
Severe acute respiratory syndrome (SARS) is a highly infective respiratory disease caused by SARS coronavirus (SARS-CoV). Its sudden emergence and rapid outbreak during 2002–2003 had resulted in ∼800 deaths among >8000 reported individual cases worldwide [1]. Although the SARS epidemic had been under control for years, reemergence of this threatening illness is still a possible risk and potentially new strains of SARS can be more dangerous than the previous ones. A number of important targets have been recognized to take part in the biological events critical to SARS-CoV replication, among which a papain-like protease (PLpro) and a chymotrypsin-like protease (3CLpro) are of significant importance to design anti-SARS inhibitors [2]. The 3CLpro, also known as the main protease (Mpro), has attracted much attention, which could be revealed from numerous publications about novel inhibitor discovery. Crystal structures of SARS-CoV Mpro, either free enzyme alone or in complex with an inhibitor, had been determined to facilitate the structural and functional investigation of this protease [3], [4]. The active site of SARS-CoV Mpro contains Cys145 and His41 to constitute a catalytic dyad, in which cysteine functions as the common nucleophile in the proteolytic process.
Biological active inhibitors against SARS-CoV Mpro have been reported mainly from two different approaches: one is screening large library to identify new active compounds using high-throughput technique, the other is novel inhibitor design based on the substrate structure or active site properties rationally [5]. The inhibitory activities of these compounds were then validated by in vitro protease assays. In most cases the kinetic study indicated that the inhibitor is involved in an irreversible process by forming a covalent bond with Cys145, while in some other cases the inhibition is actually a reversible behavior. The reported SARS-CoV Mpro inhibitors covered a variety of different chemical scaffolds, which contain peptidomimetic compounds, 3-quinoline carboxylic acid derivatives, thiophene-2-carboxylate derivatives, zinc-conjugated compounds, cinanserin, calmodulin, keto-glutamine analogues, anilide, bifunctional boronic acid compounds, isatin derivatives, etacrynic acid derivatives, serine derivatives, trifluoromethyl ketones, acetamides, pyrazolone and quercertins [5], [6], [7], [8], [9], [10], [11], [12]. It is a pity that research on drugs and vaccines towards SARS or SARS-like coronavirus has not brought any candidate for clinical use. Hence there still exists an urgent need to discover and identify new SARS-CoV Mpro agents, especially those compounds from totally new chemical families, to develop effective therapy against this fatal viral infection.
Disulfide bonds play essential roles for bioactive proteins to keep correct folding [13]. There are a few cases that simple disulfides such as diallyl disulfide and dimethyl disulfide exhibit hypochlorous acid scavenging activity and tyrosinase inhibitory activity (Fig. 1 A) [14], [15]. The unsymmetrical disulfide compounds are useful tools in the research of dynamic combinatorial chemistry [16]. These compounds have also been reported to display a variety of biological activities. For examples, Turos et al. reported that some unsymmetrical aryl-alkyl disulfides were inhibitors of methicillin-resistant Staphylococcus aureus and Bacillus anthracis (Fig. 1B) [17], while Khosla and co-workers published some unsymmetrical disulfides that could selectively inhibit extracellular thioredoxin (Fig. 1C) [18]. Yoon et al. showed that some unsymmetrical disulfide compounds were inhibitors of Mycobacterium tuberculosis and Haemophilus influenzae, by interfering with acetohydroxyacid synthase (AHAS), a key enzyme in the biosynthesis pathway of branched chain amino-acids (Fig. 1D) [19], [20]. In the past of our research, we found that some unsymmetrical aromatic disulfides could inhibit plant AHAS and was useful for herbicide research (Fig. 1E) [21], [22].
Fig. 1.

Different disulfide compounds with various biological activities from literature.
There are indeed a few reports that some aromatic disulfides exhibit antiviral activity. The virucidal activity of NSC4492 was due to targeting of arenavirus RNA synthesis (Fig. 1F) [23], while 5,5’-dithiobis-2-nitrobenzoic acid (DTNB) had antiviral propierties against T-tropic human immunodeficiency virus type 1 (HIV-1) (Fig. 1G) [24]. An antiviral disulfide NSC20625 compound could block interaction between arenavirus Z protein and cellular promyelocytic leukemia protein (Fig. 1H) [25]. However, the antiviral activities against arenavirus might be different from that against coronavirus. There is no evidence that the reported antiviral activities of aromatic disulfides have any direct relationships with SARS-CoV Mpro inhibition.
In an effort to discover novel inhibitors of SARS-CoV Mpro, we have synthesized a series of novel unsymmetrical aromatic disulfides and evaluated their biological activities in this study. The target compounds could inhibit main protease of SARS-CoV remarkably, with the best IC 50 value of 0.516 μM. Subsequent enzymatic kinetics study indicated that the aromatic disulfides acted as reversible and non-competitive inhibitors. Therefore we have demonstrated that unsymmetrical disulfide compounds with aromatic rings are novel inhibitors of SARS-CoV Mpro from a totally new chemical family, which will provide helpful information for further drug discovery.
2. Results and discussion
2.1. Chemistry of the target compounds
The target unsymmetrical disulfides were synthesized by the reaction of various substituted 2-mercapto- [1,3,4]oxadiazole, substituted 2-mercapto-thiazole, substituted 2-mercapto- 1H-imidazole or substituted 2-mercapto-pyrimidine with substituted arenesulfenyl chloride in ethyl ether under very mild condition as reported previously [21], [22]. It is a quite straightforward nucleophilic substitution, in which the thiol group in the mercapto compound serves as a nucleophilic reagent and attacks the sulfur atom in the arenesulfenyl chloride. Most yields for the reactions were satisfactory, showing that this is a simple and easy procedure to produce unsymmetrical aromatic disulfides, superior to the synthetic route developed by Hahn et al. [26]. In Hahn's paper, moist tetrahydrofuran gave better yields than dried benzene as the solvent for the reaction, so the authors added 5–10 M equivalent of water to the reaction mixture and observed satisfactory yields. Thus, the presence of water was thought to be preferable for the reaction by Hahn et al. [26]. In our synthesis experiment, only ethyl ether was used as the solvent and no additional water was added to the reaction, nevertheless, we observed very high yields for most of the target compounds.
The —S-S- bond had been confirmed in our previous paper [21]. Compound 3-4, 3-6, 3-7, 3-8, 3-9, 3-10 and 3-11 were further acetylated from corresponding parent compounds that had been published by us [21]. The molecular structures of the compounds are listed in Table 1 . The title compounds were fully characterized by 1H NMR, 13C NMR and HRMS (Part 1 of the supplementary data for the original figures).
Table 1.
The novel unsymmetrical aromatic disulfide compounds and their SARS-CoV Mpro inhibitory activities (IC50).
| Entry no. | Chemical structure | IC50 (μM) | Entry no. | Chemical structure | IC50(μM) |
|---|---|---|---|---|---|
| 3-1 |  |
1.871 ± 0.071 | 3-21 |  |
1.250 ± 0.023 |
| 3-2 |  |
2.803 ± 0.052 | 3-22 |  |
2.211 ± 0.152 |
| 3-3 |  |
3.675 ± 0.193 | 3-23 |  |
3.321 ± 0.068 |
| 3-4 | 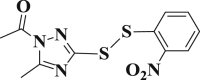 |
3.130 ± 0.052 | 3-24 |  |
2.555 ± 0.270 |
| 3-5 |  |
1.506 ± 0.184 | 3-25 | 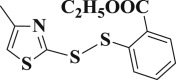 |
2.452 ± 0.126 |
| 3-6 |  |
4.344 ± 0.538 | 3-26 |  |
1.679 ± 0.042 |
| 3-7 |  |
4.100 ± 0.832 | 3-27 |  |
1.557 ± 0.116 |
| 3-8 | 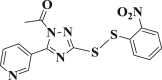 |
1.762 ± 0.044 | 3-28 |  |
1.713 ± 0.052 |
| 3-9 |  |
5.654 ± 0.259 | 3-29 | 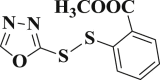 |
1.118 ± 0.132 |
| 3-10 |  |
4.511 ± 0.105 | 3-30 |  |
1.264 ± 0.033 |
| 3-11 | 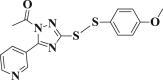 |
5.794 ± 0.050 | 3-31 |  |
0.516 ± 0.060 |
| 3-12 |  |
2.626 ± 0.082 | 3-32 | 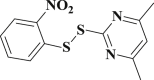 |
0.921 ± 0.060 |
| 3-13 | 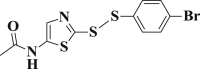 |
1.651 ± 0.048 | 3-33 | 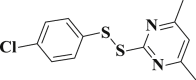 |
1.437 ± 0.053 |
| 3-14 | 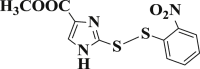 |
2.075 ± 0.016 | 3-34 | 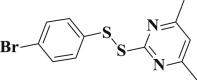 |
1.121 ± 0.060 |
| 3-15 |  |
5.954 ± 0.363 | 3-35 |  |
1.991 ± 0.086 |
| 3-16 | 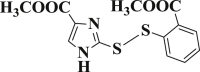 |
3.957 ± 0.190 | 3-36 | 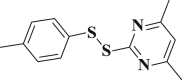 |
1.495 ± 0.055 |
| 3-17 |  |
4.126 ± 0.094 | 3-37 | 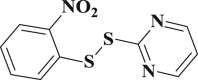 |
0.883 ± 0.028 |
| 3-18 | 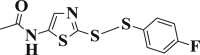 |
2.565 ± 0.075 | 3-38 | 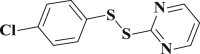 |
0.684 ± 0.012 |
| 3-19 | 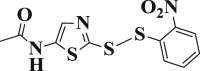 |
1.947 ± 0.508 | 3-39 |  |
0.697 ± 0.053 |
| 3-20 | 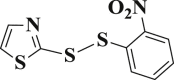 |
2.029 ± 0.488 | 3-40 |  |
1.522 ± 0.214 |
2.2. In vitro inhibitory activity of SARS-CoV Mpro
All the synthesized unsymmetrical aromatic disulfides were subjected to the in vitro assay of SARS-CoV Mpro. The results are also illustrated in Table 1, expressed by IC 50 values. It could be seen that the target compounds exhibited encouraging biological potency, with excellent IC 50 values ranging from 0.516 μM to 5.954 μM (The inhibition curves of all target compounds can be found in part 2 of the supplementary data). This was surprising due to the fact that not any research group had ever identified the disulfide compounds as inhibitors of SARS-CoV Mpro, not to mention such strong inhibition.
Inhibition type of the disulfides was determined by means of enzymatic kinetic study, for which 3-31 and 3-39 were used. From Fig. 2 it can be seen that in the plot of enzyme concentration versus reaction velocity, the lines represent different inhibitor concentrations intersect at a same point, suggesting that the inhibition is actually a reversible action. We then measured the enzymatic velocity of SARS-CoV Mpro versus substrate concentrations in the presence of either 3-31 or 3-39 (Part 3 of the supplementary data). The lines displayed in reciprocal plots intersect at a same point, indicating that both inhibitors serve as a non-competitive inhibitor with α < 1 (Part 3 of the supplementary data) [27]. On this basis, the kinetic parameters (αK i = 0.20 μM, K i = 0.24 μM) of 3-31 were determined [28] (Fig. 3 ), which clearly proved that, the non-competitive inhibitor 3-31 is characterized by smaller equilibrium-binding constant compared to some known inhibitors such as N3 (K i = 9.0 μM) and N9 (K i = 6.7 μM) [3].
Fig. 2.

Plot of enzyme concentration versus reaction velocity for enzymatic kinetic study of 3-31 (A) and 3-39 (B).
Fig. 3.

Secondary plots for the determination of the kinetic constants (Ki and αKi) of inhibitor 3-31 as a non-competitive inhibitor. The values of αKi (A)and Ki (B) are calculated from the x intercept.
Since there is a cysteine in the active site of SARS-CoV Mpro (Cys145), which plays an essential role for the biological activity of this protease, it is possible that the disulfide compound reacts with Cys145 to form a new —S-S- bond and results in a loss of enzyme activity. It is known that, if a disulfide reacts with another thiol to give a new disulfide, the thiols that are parts of the old disulfide can also react directly with this thiol to form the same new disulfide [29]. We tested the biological activities of different aryl thiols derived from our disulfides, and no inhibition of SARS-CoV Mpro could be detected for any of them even at very high concentration. Accordingly it seemed unlikely that Cys145 formed a —S-S- bond by reacting with the target disulfide compounds. Another method to rule out this possibility is to determine the change of the molecular weight of the protein before and after inhibition [30], [31]. If the disulfide compound reacts with Cys145, the molecular weight would have a shift after SARS-CoV Mpro is inhibited, and this is approximately the mass of half moiety of the unsymmetrical disulfide. Bearing this in mind, we measured the molecular weight of protease before and after inhibition by three disulfide compounds with significant structure difference (3-8, 3-31 and 3-39, data shown in part 4 of the supplementary data). However, no such assumed shift in the molecular weight was observed to support this idea.
For another possibility, Khosla et al. had reported selective inhibition of extracellular thioredoxin by unsymmetrical disulfides [18], in which two cysteine residues in a close distance form an intramolecular disulfide bond. After careful analysis of the SARS-CoV Mpro crystal structure (pdb entry 2AMD) [3], no other cysteine residue was found to be in a nearby space of Cys145. In fact not any two cysteine residues are in a reasonable distance to form possible intramolecular disulfide bond. Thus we also denied this probable inhibitory mechanism.
2.3. Molecular docking and three dimensional structure-activity relationships
Since the unsymmetrical aromatic disulfides do not react with the residues of the SARS-CoV Mpro, it means that these compounds act as intact molecules when inhibiting this protease. Therefore, in silico molecular docking technique was utilized to predict possible binding modes of the disulfide compounds with SARS-CoV Mpro. In our previous study, we had docked a small library of isatin compounds to the active site of AHAS and probable binding modes were predicted by FlexX [32], [33], [34], [35], [36], [37]. Here similar strategy was adopted to carry out database docking. After investigation of the resulting docked conformations, nineteen compounds were found to overlay one another quite well (Part 5 of the supplementary data), while all the other twenty-one compounds were in unreasonable binding space, and they failed to overlay well with one another. Thus, the docked conformations of the nineteen compounds were thought to be the possible binding conformations in this study. Compound 3-31 was chosen to depict the binding mode of the disulfide inhibitors. Fig. 4 is a two-dimensional illustration of the interactions between the inhibitor and the surrounding residues of SARS-CoV Mpro drawn by LIGPLOT [38]. The location of 3-31 has some overlap with the inhibitor N9, the inhibitor in the original pdb file. The compound binds with SARS-CoV Mpro via multiple hydrogen bonding contacts and hydrophobic contacts. Phe140, Leu141, His163, Met165, Glu166 and His172 form hydrophobic interactions with the small molecule, while Asn142, Gly143 and Cys145 form intermolecular H-bond with the inhibitor. The predicted binding mode therefore provides a useful clue to understand the possible molecular basis of these inhibitors.
Fig. 4.

LIGPLOT 2D representation of 3-31 bound with SARS CoV Mpro from FlexX docking. The hydrogen bonds between the enzyme and the inhibitor are shown as green dashed lines, and distances are in Å units. Amino acid residues that are within van der Waals contact of the inhibitor are shown as red arcs. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Comparative field analysis (CoMFA) is a tool to generate 3D contour models to quantitatively analyze the structure-activity relationships of bioactive compounds by steric and electrostatic contributions [39], [40], [41]. On the basis of the docked conformation, the molecules in the database were aligned to construct a CoMFA model. Compounds 3-8, 3-23 and 3-40 were excluded from the database because they were statistical outliers in the training set, that is, the inclusion of any of these molecules did not yield a satisfactory leave-one-out q 2. The training set without the outliers gave a leave-one-out q 2 of 0.681 when the optimum components was 6, and the non-crossvalidated r 2 was 0.916, with a standard error of estimate of 0.088 and F values of 37.968. The steric and electrostatic contributions were 43.6% and 56.4%, respectively.
Compound 3-31 was used to illustrate the steric and electrostatic contour maps, together with the neighboring residues in the docked binding pocket (Fig. 5 ). For the steric contour map, a bulky group is favorable for better inhibition in the green contour region and such a group is likely to decrease the activity in the yellow contour space. The green maps are mostly in three bulks: one formed by Ser1and Leu141, one formed by Asn142 and Gly143 and the last one formed by Ser144 and Cys145; whereas the yellow maps are just located in a space nearby Gly143. For the electrostatic contour map, in the blue contour region, an increase in the positive charge will lead to an increase of activity, yet in the red contour region, negative charge is favorable to enhance the activity. The blue map is in a region surrounded by Leu27, Cly143, Ser144 and Cys145, whereas the red maps are in three cavities: one formed by Leu141 and Asn142, one formed by Ser144, Cys145 and Met165 and the last one formed by Asn142 and Gly143. The 3D CoMFA maps have afforded important structural features of the unsymmetrical aromatic disulfides from steric and electrostatic views, which is valuable for further design and discovery of more potent inhibitors.
Fig. 5.

Steric contour map (A) and electrostatic contour map (B) for the CoMFA model. Sterically favored and disfavored regions are shown in green and yellow in map A. Electrostatic favored and disfavored regions are shown in blue and red in map B. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3. Conclusion
The lack of effective anti-SARS agents makes it a possible danger when SARS breaks out sometime in the future, numerous people will be killed again. Therefore it is still an urgent demand to discover novel anti-SARS inhibitors to combat this deadly disease. SARS CoV Mpro is an important target for the design of therapeutically useful drugs. In the present study, in an effort to develop non-peptidic anti-SARS inhibitors, we have identified for the first time, that some unsymmetrical aromatic disulfides are excellent inhibitors of SARS CoV Mpro, the mechanism of which seems distinct as they are reversible and mpetitive inhibitors. This suggests that the unsymmetrical disulfides are promising lead compounds identification and development of a new family of biologically active anti-SARS agents. A possible binding model of the disulfide inhibitor was built by molecular docking, and a CoMFA model was constructed subsequently to point out the structural features of these novel inhibitors of SARS CoV Mpro. Based on this information, further structural modifications are ongoing for better pharmaceutical compounds. The Lipinski rules will also be utilized to help to develop compounds with a final in vivo activity [42]. We are also trying to co-crystallize the protease and the best inhibitor, to gain insight into a real binding mode and explain the molecular basis of these compounds.
4. Experimental section
4.1. General synthesis and instruments
Various heterocyclic aromatic thiols 1 were commercial procured from 5A Pharmatech (China), Apichemical (China), Aldrich and ACES pharma, which were all >95% purity grade. All solvents and liquid reagents were dried in advance using standard methods and distilled before use. Substituted arenesulfenyl chlorides 2 were synthesized as described in our previous publications [21], [22]. Synthetic methods for compounds 3′ had been reported and these parent compounds for 3-4, 3-6. 3-7, 3-8, 3-9, 3-10 and 3-11 had also been fully characterized before [21]. Melting points were determined using an X-4 melting apparatus and were uncorrected. 1H NMR spectra and 13C NMR were obtained using a 400 MHz Varian Mercury Plus 400 spectrometer. The chemical shift values (d) for the NMR spectra were reported as parts per million (ppm), using deuterated chloroform (CDCl3) or dimethyl sulfoxide (DMSO-d 6) as the solvent and tetramethylsilane (TMS) as an internal reference standard. Mass spectra were recorded on a Thermo Finnigan LCQ Advantage LC/mass detector instrument.
4.2. Synthesis of the target compounds (Scheme 1)
Scheme 1.

Synthesis route of the target unsymmetrical aromatic disulfide compounds.
Heterocyclic aromatic thiols (1, 5 mmol) was added to a solution of freshly prepared arenesulfenyl chlorides (2, 5 mmol) in 25 mL of anhydrous ethyl ether at room temperature. The mixture was then stirred for 5 h at the same temperature, after that the solvent was removed under reduced pressure. Products 3 (3′ for 3-4, 3-6. 3-7, 3-8, 3-9, 3-10 and 3-11) were purified by column chromatography in 75–95% yields.
4.3. Synthesis of the compounds 3-4, 3-6. 3-7, 3-8, 3-9, 3-10 and 3-11 (Scheme 2)
Scheme 2.

Synthesis route of the target unsymmetrical aromatic disulfide compounds (3-4, 3-6, 3-7, 3-8, 3-9, 3-10 and 3-11).
Unsymmetrical aromatic disulfide (3′, 5 mmol) was added to 10 mL of acetic anhydride. The reaction mixture was stirred for 0.5 h at 60 °C and then 200 mL water was added to the mixture. The pH value was adjusted by sodium bicarbonate to 7–8. The product was extracted by ethyl acetate and the solvent was removed under reduced pressure. The target compounds were finally purified by column chromatography in 87–95% yields.
4.4. Characterization of the target compounds
4.4.1. 2-((4-chlorophenyl)disulfanyl)thiazole (3-1)
Yield 89%; m.p.: 114–116 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 7.62 (d, J = 8.5 Hz, 2H, ArH), 7.47 (d, J = 8.5 Hz, 2H, ArH), 7.31 (s, 1H, NCH), 6.99 (d, J = 4.3 Hz, 1H, SCH); 13C NMR (101 MHz, DMSO-d 6), δ 134.8, 132.2, 129.3, 121.9, 114.20, 99.7; HRMS(MALDI) m/z: calculated for C9H6ClNS3 259.9423, found 259.9420 [M + H] +.
4.4.2. N-(2-(p-tolyldisulfanyl)thiazol-5-yl)acetamide (3-2)
Yield 91%; m.p.: 113–115 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 11.70 (s, 1H, NH), 7.50 (d, J = 6.5 Hz, 2H, ArH), 7.47 (s, 1H, CH), 7.26 (d, J = 7.8 Hz, 2H, ArH), 2.32 (s, 3H, CH3), 2.10 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d 6), 167.6, 154.1, 138.9, 132.3, 130.6, 130.0, 128.4, 22.6, 21.1; HRMS(MALDI) m/z: calculated for C12H12N2OS3 297.0185, found 297.0185 [M + H] +.
4.4.3. Ethyl 2-((4-chlorophenyl)disulfanyl)-1H-imidazole-4-carboxylate (3-3)
Yield 87%; m.p.: 100–102 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 7.98 (s, 1H), 7.64 (d, J = 8.6 Hz, 2H, ArH), 7.49 (d, J = 8.6 Hz, 2H, ArH), 4.24 (q, J = 7.0 Hz, 2H, CH2CH3), 1.27 (t, J = 7.1 Hz, 3H, CH2CH3); 13C NMR (101 MHz, DMSO-d 6), δ 161.1, 132.9, 131.7, 128.7, 117.1, 116.9, 60.5, 14.7; HRMS(MALDI) m/z: calculated for C12H11ClN2O2S2 315.0022, found 315.0021 [M + H] +.
4.4.4. 1-(5-Methyl-3-((2-nitrophenyl)disulfanyl)-1H-1,2,4-triazol-1-yl)ethanone (3-4)
Yield 90%; m.p.: 157–159 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 8.31 (d, J = 8.2 Hz, 1H), 8.24 (d, J = 8.2 Hz, 1H), 7.91 (t, J = 7.7 Hz, 1H), 7.58 (t, J = 7.8 Hz, 1H), 2.59 (s, 3H), 2.55 (s, 3H); 13C NMR (101 MHz, DMSO-d 6), δ 169.3, 157.8, 157.3, 144.9, 135.6, 134.3, 127.9, 126.6, 23.5, 15.6; HRMS(MALDI) m/z: calculated for C11H10N4O3S2 311.0273, found 311.0261 [M + H] +.
4.4.5. N-(2-(phenyldisulfanyl)thiazol-5-yl)acetamide (3-5)
Yield 89%; m.p.: 108–110 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 11.79 (s, 1H, NH), 7.64 (t, J = 8.6 Hz, 2H, ArH), 7.53 (d, J = 8.5 Hz, 1H, ArH), 7.51–7.42 (m, 2H, ArH), 7.39 (d, J = 7.1 Hz, 1H, ArH), 2.10 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d 6), δ 167.6, 139.4, 137.2, 130.8, 130.0, 129.9, 129.0, 128.8, 128.42, 22.6; HRMS(MALDI) m/z: calculated for C11H10N2OS3 283.0029, found 283.0031 [M + H] +.
4.4.6. 1-(5-Phenyl-3-(p-tolyldisulfanyl)-1H-1,2,4-triazol-1-yl)ethanone (3-6)
Yield 92%; m.p.: 112–114 °C; white solid; 1H NMR (400 MHz, DMSO-d 6), δ 8.35 (d, J = 17.6 Hz, 1H), 8.00 (d, J = 8.1 Hz, 2H), 7.79 (d, J = 7.6 Hz, 3H), 7.56 (t, J = 7.6 Hz, 1H), 7.27 (s, 2H), 2.72 (s, 3H), 2.33 (s, 3H); 13C NMR (101 MHz, DMSO-d 6), δ 135.4, 129.4, 128.9, 128.1, 126.2, 125.8, 129.5, 22.2, 20.8; HRMS(MALDI) m/z: calculated for C17H15N3OS2 342.0729, found 342.0734 [M+ H]+.
4.4.7. 1-(3-((4-methoxyphenyl)disulfanyl)-5-phenyl-1H-1,2,4-triazol-1-yl)ethanone (3-7)
Yield 91%; m.p.: 133–135 °C; white solid; 1H NMR (400 MHz, DMSO-d 6), δ 8.11 (s, 2H), 7.70 (d, J = 7.3 Hz, 2H), 7.38 (s, 3), 6.73 (d, J = 7.2 Hz, 2H), 3.65 (s, 3H), 2.58 (s, 3H); 13C NMR (101 MHz, DMSO-d 6), δ 169.3, 161.9, 161.2, 158.9, 134.8, 129.9, 129.3, 128.5, 127.1, 124.8, 115.0, 55.2, 22.5; HRMS(MALDI) m/z: calculated for C17H15N3O2S2 358.0683, found 358.0685 [M+H]+.
4.4.8. 1-(3-((2-nitrophenyl)disulfanyl)-5-(pyridin-3-yl)-1H-1,2,4-triazol-1-yl)ethanone (3-8)
Yield 89%; m.p.: 186–188 °C; yellow solid; 1H NMR (400 MHz, CDCl3), δ 8.64 (d, J = 4.6 Hz, 1H), 7.95 (d, J = 4.8 Hz, 1H), 7.70–7.42 (m, 1H), 7.33 (d, J = 8.4 Hz, 2H), 7.21 (d, J = 8.7 Hz, 3H), 3.06 (s, 3H); 13C NMR (101 MHz, CDCl3), δ 174.9, 150.1, 135.3, 129.9, 129.5, 129.3, 127.5, 120.7, 46.0, 29.7; HRMS(MALDI) m/z: calculated for C15H11N5O3S2 374.0381, found 374.0380 [M+H]+.
4.4.9. Ethyl 2-((1-acetyl-5-(pyridin-3-yl)-1H-1,2,4-triazol-3-yl)disulfanyl)benzoate(3-9)
Yield 92%; m.p.: 128–130 °C; white solid; 1H NMR (400 MHz, DMSO-d 6), δ 9.10 (s,1H), 8.66 (s, 1H), 8.26 (d, J = 7.8 Hz, 2H), 8.00 (d, J = 7.5 Hz, 1H), 7.75 (s, 1H), 7.57 (d, J = 7.2 Hz, 1H), 7.41 (s, 1H), 4.22 (q, 2H), 2.50 (s, 3H), 1.33 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, DMSO-d 6), δ 150.1, 135.1, 133.6, 130.9, 129.5, 129.3, 120.7, 45.9, 29.7, 22.5, 8.7; HRMS(MALDI) m/z: calculated for C18H16N4O3S2 401.0736, found 401.0746 [M + H] +.
4.4.10. Ethyl 2-((1-acetyl-5-(pyridin-4-yl)-1H-1,2,4-triazol-3-yl)disulfanyl)benzoate (3-10)
Yield 95%; m.p.: 130–132 °C; white solid; 1H NMR (400 MHz, DMSO-d 6), δ 9.10 (s, 1H), 8.66 (s,1H), 8.26 (d, J = 7.8 Hz, 2H), 8.00 (d, J = 7.5 Hz,1H), 7.75 (t, J = 7.6 Hz,1H), 7.54 (s, 1H), 7.41 (t, J = 7.2 Hz, 1H), 4.34 (q, J = 7.1 Hz, 2H), 2.50 (s, 3H), 1.33 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, DMSO-d 6), δ 167.9, 150.1, 135.1, 133.6, 130.9, 129.5, 129.3, 120.7, 45.9, 29.7, 8.6; HRMS(MALDI) m/z: calculated for C18H16N4O3S2 401.0737, found 401.0746 [M + H] +.
4.4.11. 1-(3-((4-methoxyphenyl)disulfanyl)-5-(pyridin-3-yl)-1H-1,2,4-triazol-1-yl)ethanone (3-11)
Yield 87%; m.p.: 113–115 °C; white solid; 1H NMR (400 MHz, DMSO-d 6), δ 9.32 (d, J = 37.6 Hz, 1H), 8.53 (d, J = 5.9 Hz, 2H), 7.72 (d, J = 8.5 Hz, 1H), 7.54–7.27 (m, 2H), 6.76 (d, J = 8.8 Hz, 2H), 3.70 (s, 3H), 2.64 (s, 3H); 13C NMR (101 MHz, DMSO-d 6), δ 161.0, 160.3, 150.9, 148.3, 135.8, 134.0, 123.7, 115.0, 114.7, 55.4, 29.7; HRMS(MALDI) m/z: calculated for C16H14N4O2S2 359.0630, found 359.0639 [M + H] +.
4.4.12. N-(2-((4-chlorophenyl)disulfanyl)thiazol-5-yl)acetamide (3-12)
Yield 94%; m.p.: 127–129 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 11.73 (s, 1H, NH), 7.65 (d, J = 8.6 Hz, 2H, ArH), 7.52 (d, J = 8.5 Hz, 2H, ArH), 7.48 (s, 1H, CH), 2.10 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d 6), δ 167.6, 153.1, 152.0, 139.3, 134.4, 133.6, 130.8, 129.9, 128.5, 22.6; HRMS(MALDI) m/z: calculated for C11H9ClN2OS3 316.9639, found 316.9641 [M + H] +.
4.4.13. N-(2-((4-bromophenyl)disulfanyl)thiazol-5-yl)acetamide (3-13)
Yield 93%; m.p.: 123–125 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 11.72 (s, 1H, NH), 7.65 (d, J = 8.6 Hz, 2H, ArH), 7.58 (d, J = 8.6 Hz, 2H, ArH), 7.48 (s, 1H, CH), 2.10 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d 6), δ 167.5, 167.0, 152.0, 139.6, 132.4, 130.5, 128.0, 121.6, 22.2; HRMS(MALDI) m/z: calculated for C11H9BrN2OS3 360.9132, found 360.9128 [M + H] +.
4.4.14. Methyl 2-((2-nitrophenyl)disulfanyl)-1H-imidazole-4-carboxylate (3-14)
Yield 88%; m.p.: 160–163 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 8.51 (d, J = 8.2 Hz, 1H, ArH), 8.31 (dd, J = 8.2, 1.1 Hz, 1H, ArH), 7.98 (s, 1H), 7.96 (s, 1H, NHCH), 7.58 (dd, J = 11.4, 4.1 Hz, 1H, ArH), 3.74 (s, 3H, OCH3); 13C NMR (101 MHz, DMSO-d 6), δ 158.9, 151.1, 139.8, 137.7, 131.2, 130.5, 129.4, 125.2, 124.4, 122.2, 53.0; HRMS(MALDI) m/z: calculated for C11H9N3O4S2 312.0107, found 312.0112 [M + H] +.
4.4.15. Methyl 2-((2-(ethoxycarbonyl)phenyl)disulfanyl)-1H-imidazole-4-carboxylate (3-15)
Yield 92%; m.p.: 93–95 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 8.34 (d, J = 8.2 Hz, 1H, ArH), 8.03–7.96 (m, 1H, ArH), 7.96 (s, 1H, NHCH), 7.79–7.66 (m, 1H, ArH), 7.41 (t, J = 7.5 Hz, 1H, ArH), 4.32 (q, J = 7.1 Hz, 2H, CH2CH3), 3.74 (s, 3H, OCH3), 1.32 (t, J = 7.1 Hz, 3H, CH2CH3); 13C NMR (101 MHz, DMSO-d 6), δ 166.1, 161.8, 157.8, 141.0, 139.9, 134.1, 131.5, 126.9, 126.8, 126.5, 121.9, 61.9, 51.8, 14.5; HRMS(MALDI) m/z: calculated for C14H14N2O4S2 339.0468, found 339.0475 [M + H] +.
4.4.16. Methyl 2-((2-(methoxycarbonyl)phenyl)disulfanyl)-1H-imidazole-4-carboxylate (3-16)
Yield 89%; m.p.: 114–116 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 8.35 (d, J = 8.2 Hz, 1H, ArH), 8.00 (d, J = 7.8 Hz, 1H, ArH), 7.95 (s, 1H, NHCH), 7.76 (t, J = 7.8 Hz, 1H, ArH), 7.41 (t, J = 7.5 Hz, 1H, ArH), 3.87 (s, 3H, OCH3), 3.74 (s, 3H, OCH3); 13C NMR (101 MHz, DMSO-d 6), δ 158.9, 137.8, 134.2, 131.9, 129.6, 128.9, 127.8, 126.9, 125.2, 124.4, 53.2, 52.4; HRMS(MALDI) m/z: calculated for C13H12N2O4S2 325.0311, found 325.0314 [M + H] +.
4.4.17. Methyl 2-((4-chlorophenyl)disulfanyl)-1H-imidazole-4-carboxylate (3-17)
Yield 89%; m.p.: 138–140 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 8.00 (s, 1H, NHCH), 7.64 (d, J = 8.6 Hz, 2H, ArH), 7.49 (d, J = 8.7 Hz, 2H, ArH), 3.76 (s, 3H, OCH3); 13C NMR (101 MHz, DMSO-d 6), δ 158.7, 147.5, 137.8, 133.5, 129.9, 128.2, 127.9, 125.2, 124.4, 53.2; HRMS(MALDI) m/z: calculated for C11H9ClN2O2S2 300.9867, found 300.9894 [M + H] +.
4.4.18. N-(2-((4-fluorophenyl)disulfanyl)thiazol-5-yl)acetamide (3-18)
Yield 95%; m.p.: 119–122 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 11.78 (s, 1H, NH), 7.68 (d, J = 8.6 Hz 2H, ArH), 7.50 (s, 1H, CH), 7.32 (t, J = 8.3 Hz, 2H, ArH), 2.11 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d 6), δ 167.4, 153.8, 139.4, 132.6, 132.5, 131.5, 128.5, 117.3, 117.1, 22.6; HRMS(MALDI) m/z: calculated for C11H9FN2OS3 300.9934, found 300.9934 [M + H] +.
4.4.19. N-(2-((2-nitrophenyl)disulfanyl)thiazol-5-yl)acetamide (3-19)
Yield 93%; m.p.: 130–133 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 11.76 (s, 1H, NH), 8.41 (d, J = 7.9 Hz, 1H, ArH), 8.35 (d, J = 8.5 Hz, 1H, ArH), 7.96 (d, J = 7.4 Hz, 1H, ArH), 7.61 (t, J = 7.4 Hz, 1H, ArH), 7.49 (s, 1H CH), 2.09 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d 6), δ 167.6, 151.4, 145.3, 139.4, 135.8, 134.8, 128.4, 128.3, 127.8, 126.8, 22.5; HRMS(MALDI) m/z: calculated for C11H9N3O3S3 327.9879, found 327.9876 [M + H] +.
4.4.20. 2-((2-nitrophenyl)disulfanyl)thiazole (3-20)
Yield 90%; m.p.: 105–107 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 8.37 (d, J = 8.2 Hz, 1H, ArH), 8.23 (d, J = 8.2 Hz, 1H, ArH), 7.93 (t, J = 7.7 Hz, 1H, ArH), 7.87–7.74 (m, 2H, CH), 7.62 (t, J = 7.7 Hz, 1H, ArH); 13C NMR (101 MHz, DMSO-d 6), δ 163.8, 145.6, 144.9, 136.2, 133.9, 128.7, 127.6, 126.9, 124.5; HRMS(MALDI) m/z: calculated for C9H6N2O2S3 270.9664, found 270.9663 [M+H]+.
4.4.21. 2-(p-tolyldisulfanyl)thiazole (3-21)
Yield 75%; m.p.: 60–62 °C; yellow solid; 1H NMR (400 MHz, CDCl3), δ 7.80 (d, J = 6.1 Hz, 2H, ArH), 7.53 (d, J = 7.9 Hz, 2H, ArH), 7.25 (d, J = 7.7 Hz, 2H, ArH), 2.30 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3), δ 136.5, 136.3, 130.2, 129.3, 127.6, 124.9, 21.5; HRMS(MALDI) m/z: calculated for C10H9NS3 239.9969, found 239.9970 [M+H]+.
4.4.22. 2-((4-fluorophenyl)disulfanyl)thiazole (3-22)
Yield 82%; m.p.: 89–91 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 8.37 (d, J = 8.2 Hz, 1H, ArH), 8.23 (d, J = 8.2 Hz, 1H, ArH), δ 7.66 (d, J = 8.7 Hz, 2H, ArH), 7.52 (d, J = 8.7 Hz, 2H, ArH); 13C NMR (101 MHz, DMSO-d 6), δ 144.9, 132.7, 132.6, 130.6, 125.0, 123.9, 117.3, 117.1, 40.1, 39.9, 39.7; HRMS(MALDI) m/z: calculated for C9H6FNS3 243.9719, found 243.9721 [M+H]+.
4.4.23. 2-((4-bromophenyl)disulfanyl)thiazole (3-23)
Yield 80%; m.p.: 92–94 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 7.61 (d, J = 8.5 Hz, 2H, ArH), 7.48 (d, J = 8.5 Hz, 2H, ArH), 7.30 (s, 1H, NCH), 6.99 (d, J = 4.3 Hz, 1H, SCH); 13C NMR (101 MHz, DMSO-d 6), δ 134.9, 132.3, 129.2, 121.9, 114.2, 99.4; HRMS(MALDI) m/z: calculated for C9H6BrNS3 303.8918, found 303.8914 [M+H]+.
4.4.24. 4-Methyl-2-((2-nitrophenyl)disulfanyl)thiazole (3-24)
Yield 92%; m.p.: 61–63 °C; yellow solid; 1H NMR (400 MHz, CDCl3), δ 8.36 (d, J = 8.2 Hz, 1H, ArH), 8.20 (d, J = 8.2Hz, 1H, ArH), 8.01–7.84 (m, 1H, ArH), 7.64–7.53 (m, 1H, ArH), 7.33 (s, 1H, CH), 2.33 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3), δ 162.8, 154.6, 145.7, 136.0, 133.9, 128.7, 127.6, 126.9, 118.4, 17.3; HRMS(MALDI) m/z: calculated for C10H8N2O2S3 284.9821, found 284.9823 [M+H]+.
4.4.25. Ethyl 2-((4-methylthiazol-2-yl)disulfanyl)benzoate (3-25)
Yield 91%; m.p.: 103–105 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 8.09–8.03 (m, 1H, ArH), 8.01 (d, J = 7.9 Hz, 1H, ArH), 7.74 (t, J = 7.7 Hz, 1H, ArH), 7.45 (t, J = 7.5 Hz, 1H, ArH), 7.28 (s, 1H), 4.41 (s, 2H, CH2CH3), 2.32 (s, 3H, CH3), 1.36 (t, J = 7.1 Hz, 3H, CH2CH3); 13C NMR (101 MHz, DMSO-d 6), δ 166.2, 156.3, 154.5, 147.4, 138.4, 134.4, 131.8, 127.4, 125.9, 117.6, 62.2, 17.4, 14.5; HRMS(MALDI) m/z: calculated for C13H13NO2S3 312.0181, found 312.0188 [M+H]+.
4.4.26. Methyl 2-((5-methyl-1,3,4-oxadiazol-2-yl)disulfanyl)benzoate (3-26)
Yield 93%; m.p.: 80–82 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 8.14 (d, J = 7.4 Hz, 1H, ArH), 8.03 (d, J = 6.7 Hz, 1H, ArH), 7.78 (s, 1H, ArH), 7.47 (d, J = 6.3 Hz, 1H, ArH), 3.89 (s, 3H, OCH3), 2.49 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d 6), δ 167.3, 166.7, 161.7, 138.7, 134.4, 131.6, 127.4, 127.1, 126.5, 53.3, 11.3; HRMS(MALDI) m/z: calculated for C11H10N2O3S2 283.0206, found 283.0209 [M+H]+.
4.4.27. Ethyl 2-((5-methyl-1,3,4-oxadiazol-2-yl)disulfanyl)benzoate (3-27)
Yield 88%; m.p.: 73–74 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 8.14 (d, J = 8.0 Hz, 1H, ArH), 8.04 (d, J = 7.7 Hz, 1H, ArH), 7.78 (t, J = 7.8 Hz,1H, ArH), 7.46 (t, J = 7.5 Hz, 1H, ArH), 4.35 (q, J = 7.1 Hz, 2H, CH2CH3), 2.50 (s, 3H,CH3), 1.34 (t, J = 7.1 Hz, 3H, CH2CH3); 13C NMR (101 MHz, DMSO-d 6), δ 167.3, 166.3, 161.7, 138.6, 134.3, 131.6, 127.4, 126.5, 62.2, 14.5, 11.3; HRMS(MALDI) m/z: calculated for C12H12N2O3S2 297.0362, found 297.0365 [M+H]+.
4.4.28. 2-Methyl-5-((2-nitrophenyl)disulfanyl)-1,3,4-oxadiazole (3-28)
Yield 92%; m.p.: 91–93 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 8.34 (d, J = 8.2 Hz, 1H, ArH), 8.30 (d, J = 8.2 Hz, 1H, ArH), 7.97 (t, J = 7.7 Hz, 1H, ArH), 7.63 (t, J = 7.7 Hz, 1H, ArH), 2.51 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d 6), δ 167.5, 161.2, 145.4, 135.9, 134.0, 128.7, 128.1, 126.8, 11.3; HRMS(MALDI) m/z: calculated for C9H7N3O3S2 270.0002, found 270.0000 [M+H]+.
4.4.29. Methyl 2-((1,3,4-oxadiazol-2-yl)disulfanyl)benzoate (3-29)
Yield 87%; m.p.: 99–101 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 9.37 (s, 1H, CH), 8.14 (d, J = 8.2, Hz, 1H, ArH), 8.05 (d, J = 7.8, Hz, 1H, ArH), 7.87 (t, J = 7.8, Hz, 1H, ArH), 7.48 (t, J = 7.7, Hz, 1H, ArH), 3.91 (s, 3H, OCH3); 13C NMR (101 MHz, DMSO-d 6), δ 166.8, 162.3, 157.5, 138.5, 134.5, 131.7, 127.5, 127.2, 126.6, 53.3; HRMS(MALDI) m/z: calculated for C10H8N2O3S2 290.9869, found 290.9868 [M+H]+.
4.4.30. Methyl 2-((4-methyloxazol-2-yl)disulfanyl)benzoate (3-30)
Yield 90%; m.p.: 68–70 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 8.16 (d, J = 8.0 Hz, 1H, ArH), 8.04 (d, J = 7.7, Hz, 1H, ArH), 7.97 (s, 1H, CH), 7.88 (t, J = 7.2 Hz, 1H, ArH), 7.45 (t, J = 7.2 Hz, 1H, ArH), 3.90 (s, 3H, OCH3), 2.07 (s 3H, CH3); 13C NMR (101 MHz, DMSO-d 6), δ 166.7, 139.6, 139.3, 138.8, 134.3, 131.6, 127.2, 126.9, 126.4, 53.2, 11.7; HRMS(MALDI) m/z: calculated for C12H11NO3S2 282.0253, found 282.0257 [M+H]+.
4.4.31. 2-((4-chlorophenyl)disulfanyl)-1,3,4-oxadiazole (3-31)
Yield 90%; m.p.: 69–72 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 9.46 (s, 1H, CH), 7.71 (d, J = 8.5 Hz, 2H, ArH), 7.54 (d, J = 8.3 Hz, 2H, ArH); 13C NMR (101 MHz, DMSO-d 6), δ 157.7, 134.7, 133.8, 132.5, 131.4, 130.1; HRMS(MALDI) m/z: calculated for C8H5ClN2OS2 261.9871, found 261.9872 [M + NH4] +.
4.4.32. 4,6-Dimethyl-2-((2-nitrophenyl)disulfanyl)pyrimidine (3-32)
Yield 94%; m.p.: 151–153 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 8.34 (d, 6.5 Hz, 1H, ArH), 7.99–7.69 (m, 2H, ArH), 7.55 (d, J = 8.4 Hz, 1H, ArH), 7.12 (d, J = 25.8 Hz, 1H, ArH), 2.35 (s, 6H, CH3); 13C NMR (101 MHz, DMSO-d 6), δ 168.7, 168.3, 135.5, 128.5, 128.0, 127.4, 127.0, 126.3, 118.7, 118.1, 23.8; HRMS(MALDI) m/z: calculated for C12H11N3O2S2 294.0365, found 294.0364 [M + H] +.
4.4.33. 2-((4-chlorophenyl)disulfanyl)-4,6-dimethylpyrimidine (3-33)
Yield 80%; m.p.: 84–86 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 7.60 (d, J = 8.6 Hz, 2H, ArH), 7.44 (d, J = 8.6 Hz, 2H, ArH), 7.14 (s, 1H, ArH), 2.40 (s, 6H, CH3); 13C NMR (101 MHz, DMSO-d 6), δ 168.6, 167.8, 135.6, 133.1, 130.9, 129.6, 118.4, 23.8; HRMS(MALDI) m/z: calculated for C12H11ClN2S2 283.0125, found 283.0130 [M + H] +.
4.4.34. 2-((4-bromophenyl)disulfanyl)-4,6-dimethylpyrimidine (3-34)
Yield 84%; m.p.: 76–79 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 7.53 (dd, J = 15.6, 7.2 Hz, 4H, ArH), 7.15 (s, 1H, ArH), 2.38 (s, 6H, CH3); 13C NMR (101 MHz, DMSO-d 6), δ 168.6, 167.7, 136.1, 132.5, 131.0, 121.5, 118.4, 23.8; HRMS(MALDI) m/z: calculated for C12H11BrN2S2 326.9620, found 326.9622 [M + H] +.
4.4.35. 4,6-Dimethyl-2-(phenyldisulfanyl)pyrimidine (3-35)
Yield 81%; m.p.: 61–63 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 7.67 (t, J = 8.0 Hz, 2H, ArH), 7.60–7.31 (m, 3H, ArH), 7.22 (s, 1H, ArH), 2.48 (s, 6H, CH3); 13C NMR (101 MHz, DMSO-d 6), δ 168.6, 130.9, 129.7, 129.6, 129.2, 128.3, 118.4, 23.8; HRMS(MALDI) m/z: calculated for C12H12N2S2 249.0515, found 249.0515 [M + H] +.
4.4.36. 4,6-Dimethyl-2-(p-tolyldisulfanyl)pyrimidine (3-36)
Yield 84%; m.p.: 72–75 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 7.50 (d, J = 8.2 Hz, 2H, ArH), 7.18 (d, J = 8.0 Hz, 2H, ArH), 7.12 (s, 1H, ArH), 2.40 (s, 6H, CH3), 2.28 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d 6), δ 168.4, 138.4, 133.1, 131.6, 130.3, 130.2, 118.2, 23.86, 21.1; HRMS(MALDI) m/z: calculated for C13H14N2S2 263.0671, found 263.0674 [M + H] +.
4.4.37. 2-((2-nitrophenyl)disulfanyl)pyrimidine (3-37)
Yield 86%; m.p.: 130–132 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 8.63 (s, 2H, ArH), 8.31 (d, J = 8.1 Hz, 2H, ArH), 7.96 (s, 1H, ArH), 7.59 (s, 1H, ArH), 7.38 (s, 1H, ArH), 7.17 (s, 1H, ArH); 13C NMR (101 MHz, DMSO-d 6), δ 169.4, 158.6, 136.3, 134.3, 128.1, 126.7, 125.9, 118.9, 99.9; HRMS(MALDI) m/z: calculated for C10H7N3O2S2 265.0052, found 266.0053 [M + H] +.
4.4.38. 2-((4-chlorophenyl)disulfanyl)pyrimidine (3-38)
Yield 80%; m.p.: 79–80 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 8.66–8.47 (m, 2H), 7.47 (t, J = 9.0 Hz, 2H), 7.29–7.16 (m, 2H), 7.07 (d, J = 4.9 Hz, 1H); 13C NMR (101 MHz, DMSO-d 6), δ 158.0, 134.9, 133.8, 130.2, 129.3, 129.1, 118.3; HRMS(MALDI) m/z: calculated for C10H7ClN2S2 254.9812, found 254.9816 [M + H] +.
4.4.39. 2-((4-bromophenyl)disulfanyl)pyrimidine (3-39)
Yield 83%; m.p.: 75–78 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 8.63 (dd, J = 22.9, 4.7 Hz, 2H), 7.43 (dd, J = 20.0, 12.8 Hz, 4H), 7.10 (d, J = 4.0 Hz, 1H); 13C NMR (101 MHz, DMSO-d 6), δ 158.1, 135.5, 132.0, 130.3, 128.9, 121.8, 118.3; HRMS(MALDI) m/z: calculated for C10H7BrN2S2 298.9307, found 298.9307 [M + H] +.
4.4.40. 2-(p-tolyldisulfanyl)pyrimidine (3-40)
Yield 86%; m.p.: 45–48 °C; yellow solid; 1H NMR (400 MHz, DMSO-d 6), δ 8.56 (d, J = 4.9 Hz, 2H), 7.44 (d, J = 8.2 Hz, 2H), 7.13–6.90 (m, 3H), 2.24 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d 6), δ 171.2, 157.9, 138.1, 132.9, 129.8, 129.7, 118.0, 21.1; HRMS(MALDI) m/z: calculated for C11H10N2S2 235.0358, found 235.0361 [M + H] +.
4.5. In vitro enzyme inhibition assay
The expression and purification of SARS CoV Mpro was described by Rao et al. [43]. Basically, the sequence of SARS-CoV Mpro cloned into the pGEX-6P-1 vector was transformed into E. coli BL21 (DE3) cells. The GST fusion protein, GST-SARS-CoV Mpro, was purified by GST-glutathione affinity chromatography, cleaved with PreScission protease, and the recombinant SARS-CoV Mpro was further purified by using anion-exchange chromatography. Eventually purified protein was of high purity (>95%) as judged by SDSPAGE analysis and the concentration is 0.5 μM, and the buffer contains 50 mM Tris-HCl, pH 7.3 and 1 mM EDTA. The fluorogenic substrate with consensus sequence of CoV Mpro, MCA-AVLQSGFR-Lys(Dnp)-Lys-NH2 (95% purity), was synthesized in Shanghai Biological Engineering Company. The substrate was dissolved in DMSO in 0.8 mM liquid storage for use.
The inhibition assay was similar to Yang's procedure [11]. The SARS CoV Mpro inhibition assays were conducted by fluorescence resonance energy transfer (FRET). The natural substrate amino acid sequence (AVLQSGFRKK) of SARS-CoV Mpro started with the MCA fluorescent group and connected the Dnp fluorescence quenching group with penultimate K. The tested compounds were dissolved by sterilized DMSO and diluted to various concentrations. The settled concentrations of proteins, compounds and substrate were preheated at 37 °C and oscillated. The excitation/emission light was 320/405 nm, and the test was carried out every 5 s for 200 times. Drawing curves, the maximum value of the negative control curve slope is V 0, and the largest compound curve slope is V1. The inhibition ratio can be defined (1−V 1/V 0). And the IC 50 value was calculated by equation (1) using CraphPad Prism5:
| V0/V=1+[I]/IC50 | (1) |
V 0 shows the initial rate of the reaction without inhibitor, V means the initial rate of the reaction with the inhibitor at various concentrations and [I] indicates the concentration of the inhibitor.
The determination of the inhibitor as a valent inhibitor employs the above methods as well, albeit with two modifications. Firstly, the inhibitor concentration was set to 2 or 4 μM. And for each inhibitor concentration, we measured the enzymatic activity of Mpro whose concentration spans 0–2 μM. Secondly, Mpro and inhibitor were first incubated for 20 min to ensure a thorough ‘Mpro-inhibitor’ reaction and then the inhibition assay was initiated by adding substrate and characterized by fluorescence monitoring.
The further characterization of the inhibitor as a non-competitive inhibitor employs the methods described in earlier work [28]. Basically, the enzymatic velocity of SARS-CoV Mpro versus substrate concentrations with presence of inhibitors is depicted by equation (2) [27], where K i is the dissociation constant for the SARS-CoV Mpro complexed with inhibitor 3-31; factor α reflects the effect of inhibitor 3-31 on the affinity of the enzyme for its substrate; V max and K m represent the maximum velocity and Michaelis-Menten constant, respectively.
| (2) |
The values of V max and K m at different inhibitor concentrations were apparent V max and K m, called and, respectively. According to equation (2), and can be calculated by equation (3).
| (3) |
The kinetic parameters of and, were determined by adding 1 μM SARS-CoV Mpro to 20 μM substrate containing varying concentrations of inhibitor 3-31 (0–3 μM). The value of αK i was then calculated from plots of 1/ versus 1/[I]. Similarly, the value of K i was calculated from plots of and versus 1/[I].
Mass spectra were recorded on Waters Xevo G2-XS Q-TOF mass spectrometry. Mass spectra were acquired in positive ion mode using a capillary voltage of 3 kV, a sampling cone voltage of 40 V and a source offset voltage of 80 V. The cone gas flow was set up to 50 L/h and desolvation gas flow was 800 L/h. Desolvation temperature and source temperature were set to 400 °C and 100 °C, respectively. The mass of intact protein was obtained by deconvolution of the raw data using MaxEnt1 tool. The samples were prepared at the similar condition with the IC 50 determination, except that the concentration of the inhibitors was 20 times of the concentration of SARS Mpro.
4.6. Molecular docking and comparative field analysis
Chemical structures of the compounds were built within Sybyl 7.3 (Tripos Inc., St Louis, MO). All the molecules were assigned Gasteiger-Hückel charges and minimized by the Tripos force field when convergence reached 0.001 kcal/mol/Å.
Molecular docking of the unsymmetrical aromatic disulfides to the active site of SARS-CoV Mpro was performed by FlexX. The crystal structure of SARS-CoV Mpro in complex with inhibitor (pdb code 2AMD) was retrieved from the pdb databank. All water molecules were removed, and hydrogen atoms were added in the standard geometry. Any amino acid residue within 6.5 Å of the location of the original inhibitor N9 was considered to be in the binding pocket. Cscore calculation was enabled and set to serial mode. Database docking and subsequent scoring procedures were performed using the default parameters in the program.
For CoMFA, The molecules were superimposed using 3-31 from the molecular docking result as the template. All the parameters were used the default value within CoMFA module and the column filtering was set to 2.0 kcal/mol. The “leave-one-out” (LOO) cross validation method was applied to determine the optimum number of partial least squares (PLS) components. The non-cross validated method was used to derive the final model to explain the quantitative structure-activity relationship in a three dimensional manner.
Acknowledgment
This work was financially supported by the Natural Science Foundation of China (No. 21272128 and 21672114), the “111” Project of Ministry of Education of China (No. B06005) and the National Basic Research Program of China (No. 2013CB734004). We appreciate Professor Chuanzheng Zhou in Nankai University for his kind assistance for the mass spectrometry of SARS CoV main protease.
Footnotes
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmech.2017.05.045.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.World Health Organization . December 2003. Summary of Probable SARS Cases with Onset of Illness from 1 November 2002 to 31 July 2003.http://www.who.int/csr/sars/country/table2004_04_21/en/ Based on data as of the 31. [Google Scholar]
- 2.Goetz D.H., Choe Y., Hansell E., Chen Y.T., McDowell M., Jonsson C.B., Roush W.R., McKerrow J., Craik C.S. Substrate specificity profiling and identification of a new class of inhibitor for the major protease of the SARS coronavirus. Biochemistry. 2007;46:8744–8752. doi: 10.1021/bi0621415. [DOI] [PubMed] [Google Scholar]
- 3.Yang H., Xie W., Xue X., Yang K., Ma J., Liang W., Zhao Q., Zhou Z., Pei D., Ziebuhr J., Hilgenfeld R., Yuen K.Y., Wong L., Gao G., Chen S., Chen Z., Ma D., Bartlam M., Rao Z. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005;3:1741–1752. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 5.Zhou L., Liu Y., Zhang W., Wei P., Huang C., Pei J., Yuan Y., Lai L. Isatin compounds as non-covalent SARS coronavirus 3C-like protease inhibitors. J. Med. Chem. 2006;49:3440–3443. doi: 10.1021/jm0602357. [DOI] [PubMed] [Google Scholar]
- 6.Chen L., Li J., Luo C., Liu H., Xu W., Chen G., Liew O.W., Zhu W., Puah C.M., Shen X., Jiang H. Binding interaction of quercetin-3-β-galactoside and its synthetic derivatives with SARS-CoV 3CLpro: structure-activity relationship studies reveal salient pharmacophore features. Bioorg. Med. Chem. 2006;14:8295–8306. doi: 10.1016/j.bmc.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramajayam R., Tan K.P., Liu H.G., Liang P.H. Synthesis and evaluation of pyrazolone compounds as SARS-coronavirus 3C-like protease inhibitors. Bioorg. Med. Chem. 2010;18:7849–7854. doi: 10.1016/j.bmc.2010.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shao Y.M., Yang W.B., Kuo T.H., Tsai K.C., Lin C.H., Yang A.S., Liang P.H., Wong C.H. Design, synthesis, and evaluation of trifluoromethyl ketones as inhibitors of SARS-CoV 3CL protease. Bioorg. Med. Chem. 2008;16:4652–4660. doi: 10.1016/j.bmc.2008.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobs J., Grum-Tokars V., Zhou Y., Turlington M., Saldanha S.A., Chase P., Eggler A., Dawson E.S., Baez-Santos Y.M., Tomar S., Mielech A.M., Baker S.C., Lindsley C.W., Hodder P., Mesecar A., Stauffer S.R. Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent non-covalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem. 2013;56:534–546. doi: 10.1021/jm301580n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thanigaimalai P., Konno S., Yamamoto T., Koiwai Y., Taguchi A., Takayama K., Yakushiji F., Akaji K., Chen S.E., Naser-Tavakolian A., Schön A., Freire E., Hayashi Y. Development of potent dipeptide-type SARS-CoV 3CL protease inhibitors with novel P3 scaffolds: design, synthesis, biological evaluation, and docking studies. Eur. J. Med. Chem. 2013;68:372–384. doi: 10.1016/j.ejmech.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu W., Zhu H.M., Niu G.J., Shi E.Z., Chen J., Sun B., Chen W.Q., Zhou H.G., Yang C. Synthesis, modification and docking studies of 5-sulfonyl isatin derivatives as SARS-CoV 3C-like protease inhibitors. Bioorg. Med. Chem. 2014;22:292–302. doi: 10.1016/j.bmc.2013.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thanigaimalai P., Konno S., Yamamoto T., Koiwai Y., Taguchi A., Takayama K., Yakushiji F., Akaji K., Kiso Y., Kawasaki Y., Chen S.E., Naser-Tavakolian A., Schön A., Freire E., Hayashi Y. Design, synthesis, and biological evaluation of novel dipeptide-type SARS-CoV 3CL protease inhibitors: structure-activity relationship study. Eur. J. Med. Chem. 2013;65:436–447. doi: 10.1016/j.ejmech.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L., Chou C.P., Moo-Young M. Disulfide bond formation and its impact on the biological activity and stability of recombinant therapeutic proteins produced by Escherichia coli expression system. Biotechnol. Adv. 2011;29:923–929. doi: 10.1016/j.biotechadv.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 14.Argüello-García R., Medina-Campos O.N., Pérez-Hernández N., Pedraza-Chaverrí J., Ortega-Pierres G. Hypochlorous acid scavenging activities of thioallyl compounds from garlic. J. Agric. Food Chem. 2010;58:11226–11233. doi: 10.1021/jf102423w. [DOI] [PubMed] [Google Scholar]
- 15.Chu H.L., Wang B.S., Duh P.D. Effects of selected organosulfur compounds on melanin formation. J. Agric. Food Chem. 2009;57:7072–7077. doi: 10.1021/jf9005824. [DOI] [PubMed] [Google Scholar]
- 16.Otto S. Dynamic molecular networks: from synthetic receptors to self-replicators. Acc. Chem. Res. 2012;45:2200–2210. doi: 10.1021/ar200246j. [DOI] [PubMed] [Google Scholar]
- 17.Turos E., Revell K.D., Ramaraju P., Gergeres D.A., Greenhalgh K., Young A., Sathyanarayan N., Dickey S., Lim D., Alhamadsheh M.M., Reynolds K. Unsymmetric aryl-alkyl disulfide growth inhibitors of methicillin-resistant Staphylococcus aureus and Bacillus anthracis. Bioorg. Med. Chem. 2008;16:6501–6508. doi: 10.1016/j.bmc.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DiRaimondo T.R., Plugis N.M., Jin X., Khosla C. Selective inhibition of extracellular thioredoxin by asymmetric disulfides. J. Med. Chem. 2013;56:1301–1310. doi: 10.1021/jm301775s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi K.J., Yu Y.G., Hahn H.G., Choi J.D., Yoon M.Y. Characterization of acetohydroxyacid synthase from Mycobacterium tuberculosis and the identification of its new inhibitor from the screening of a chemical library. FEBS Lett. 2005;579:4903–4910. doi: 10.1016/j.febslet.2005.07.055. [DOI] [PubMed] [Google Scholar]
- 20.Choi K.J., Noh K.M., Kim D.E., Ha B.H., Kim E.E., Yoon M.Y. Identification of the catalytic subunit of acetohydroxyacid synthase in Haemophilus influenzae and its potent inhibitors. Arch. Biochem. Biophys. 2007;466:24–30. doi: 10.1016/j.abb.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 21.Shang J., Wang W.M., Li Y.H., Song H.B., Li Z.M., Wang J.G. Synthesis, crystal structure, in vitro acetohydroxyacid synthase inhibition, in vivo herbicidal activity, and 3D-QSAR of new asymmetric aryl disulfides. J. Agric. Food Chem. 2012;60:8286–8293. doi: 10.1021/jf302206x. [DOI] [PubMed] [Google Scholar]
- 22.Li Z.S., Wang W.M., Lu W., Niu C.W., Li Y.H., Li Z.M., Wang J.G. Synthesis and biological evaluation of nonsymmetrical aromatic disulfides as novel inhibitors of acetohydroxyacid synthase. Bioorg. Med. Chem. Lett. 2013;23:3723–3727. doi: 10.1016/j.bmcl.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 23.Sepúlveda C.S., García C.C., Levingston Macleod J.M., López N., Damonte E.B. Targeting of arenavirus RNA synthesis by a carboxamide-derivatized aromatic disulfide with virucidal activity. PLoS One. 2013;8:e81251. doi: 10.1371/journal.pone.0081251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lara H.H., Ixtepan-Turrent L., Garza-Treviño E.N., Flores-Teviño S.M., Borkow G., Rodriguez-Padilla C. Antiviral propierties of 5, 5'-dithiobis-2- nitrobenzoic acid and bacitracin against T-tropic human immunodeficiency virus type 1. Virol. J. 2011;8:137. doi: 10.1186/1743-422X-8-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.García C.C., Topisirovic I., Djavani M., Borden K.L., Damonte E.B., Salvato M.S. An antiviral disulfide compound blocks interaction between arenavirus Z protein and cellular promyelocytic leukemia protein. Biochem. Biophys. Res. Commun. 2010;393:625–630. doi: 10.1016/j.bbrc.2010.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han M., Lee J.T., Hahn H.G. A traceless, one-pot preparation of unsymmetric disulfides from symmetric disulfides through a repeated process involving sulfenic acid and thiosulfinate intermediates. Tetrahedron. Lett. 2011;52:236–239. [Google Scholar]
- 27.Copeland R. Reversible inhibitors. In: Copeland R., Enzymes, editors. A Practical Introduction to Structure, Mechanism, and Data Analysis. second ed. Wiley-VCH, Inc.; New York: 2000. pp. 266–304. [Google Scholar]
- 28.Wang F., Chen C., Liu X., Yang K., Xu X., Yang H. Crystal structure of feline infectious peritonitis virus main protease in complex with synergetic dual inhibitors. J. Virol. 2015;90:1910–1917. doi: 10.1128/JVI.02685-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corbett P.T., Leclaire J., Vial L., West K.R., Wietor J.L., Sanders J.K., Otto S. Dynamic combinatorial chemistry. Chem. Rev. 2006;106:3652–3711. doi: 10.1021/cr020452p. [DOI] [PubMed] [Google Scholar]
- 30.Christopeit T., Albert A., Leiros H.K. Discovery of a novel covalent non-β-lactam inhibitor of the metallo-β-lactamase NDM-1. Bioorg. Med. Chem. 2016;24:2947–2953. doi: 10.1016/j.bmc.2016.04.064. [DOI] [PubMed] [Google Scholar]
- 31.Zhang J., Pettersson H.I., Huitema C., Niu C., Yin J., James M.N., Eltis L.D., Vederas J.C. Design, synthesis, and evaluation of inhibitors for severe acute respiratory syndrome 3C-like protease based on phthalhydrazide ketones or heteroaromatic esters. J. Med. Chem. 2007;50:1850–1864. doi: 10.1021/jm061425k. [DOI] [PubMed] [Google Scholar]
- 32.Rarey M., Kramer B., Lengauer T., Klebe G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 33.Wang J., Tan H., Li Y., Ma Y., Li Z., Guddat L.W. Chemical synthesis, in vitro acetohydroxyacid synthase (AHAS) inhibition,herbicidal activity, and computational studies of isatin derivatives. J. Agric. Food Chem. 2011;59:9892–9900. doi: 10.1021/jf2021607. [DOI] [PubMed] [Google Scholar]
- 34.Jug G., Anderluh M., Tomašič T. Comparative evaluation of several docking tools for docking small molecule ligands to DC-SIGN. J. Mol. Model. 2015;21:164. doi: 10.1007/s00894-015-2713-2. [DOI] [PubMed] [Google Scholar]
- 35.Mishra R.K., Singh J. A structure guided QSAR: a rapid and accurate technique to predict IC50: a case study. Curr. Comput. Aided Drug Des. 2015;11:152–163. doi: 10.2174/1573409911666150702100839. [DOI] [PubMed] [Google Scholar]
- 36.Liu H., Shen Q., Zhang J., Fu W. Evaluation of various inverse docking schemes in multiple targets identification. J. Mol. Graph. Model. 2010;29:326–330. doi: 10.1016/j.jmgm.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 37.Hou X., Du J., Zhang J., Du L., Fang H., Li M. How to improve docking accuracy of AutoDock4.2: a case study using different electrostatic potentials. J. Chem. Inf. Model. 2013 Jan 28;53(1):188–200. doi: 10.1021/ci300417y. [DOI] [PubMed] [Google Scholar]
- 38.Wallace A.C., Laskowski R.A., Thornton J.M. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 39.Cramer R.D., Patterson D.E., Bunce J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988;110:5959–5967. doi: 10.1021/ja00226a005. [DOI] [PubMed] [Google Scholar]
- 40.Jiang D.P., Zhu C.C., Shao X.S., Cheng J.G., Li Z. Bioactive conformation analysis of anthranilic diamide insecticides: DFT-based potential energy surface scanning and 3D-QSAR investigations. Chin. Chem. Lett. 2015;26:662–666. [Google Scholar]
- 41.Yue X.L., Li H., Liu S.S., Zhang Q.Y., Yao J.J., Wang F.Y. N-Fluorinated phenyl-N′-pyrimidyl urea derivatives: synthesis, biological evaluation and 3D-QSAR study. Chin. Chem. Lett. 2014;25:1069–1072. [Google Scholar]
- 42.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 43.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.