

Abstract
BACKGROUND:
Myocardial infarction (MI) triggers myelopoiesis resulting in heightened production of neutrophils. However, the mechanisms that sustain their production and recruitment to the injured heart are unclear.
METHODS:
Using a mouse model of the permanent ligation of the left anterior descending (LAD) artery and flow cytometry, we first characterized the temporal and spatial effects of MI on different myeloid cell types. We next performed global transcriptome analysis of different cardiac cell types within the infarct to identify the drivers of acute inflammatory response and the underlying signaling pathways. Utilizing a combination of genetic and pharmacological strategies, we identified the sequalae of events that led to MI-induced myelopoiesis. Cardiac function was assessed by echocardiography. The association of early indices of neutrophilia with major adverse cardiovascular events (MACE) was studied in a cohort of acute MI patients.
RESULTS:
Induction of MI resulted in a rapid recruitment of neutrophils to the infarct, where they release specific alarmins, S100A8 and S100A9. These alarmins bind to the Toll Like Receptor (TLR) 4 and prime the Nod Like Receptor (NLR) family Pyrin Domain-Containing 3 (Nlrp3) inflammasome in naïve neutrophils and promote interleukin 1 (IL-1β) secretion. The released IL-1β interact with its receptor (Interleukin 1 Receptor Type 1, IL1R1) on hematopoietic stem and progenitor cells in the bone marrow (BM), and stimulate granulopoiesis in a cell-autonomous manner. Genetic or pharmacological strategies aimed at disruption of S100A8/A9 and its downstream signaling cascade suppress MI-induced granulopoiesis and improve cardiac function. Furthermore, in patients with acute coronary syndrome (ACS), higher neutrophil count on admission and post-revascularization correlates positively with major adverse cardiovascular disease (CVD) outcomes.
CONCLUSIONS:
Our study provides novel evidence for the primary role of neutrophil-derived alarmins (S100A8/A9) in dictating the nature of the ensuing inflammatory response following myocardial injury. Therapeutic strategies aimed at disruption of S100A8/A9 signaling or its downstream mediators (e.g. Nlrp3, IL-1β) in neutrophils suppress granulopoiesis and may improve cardiac function in ACS patients.
Keywords: Myocardial Ischemia, Neutrophils, Myelopoiesis, S100A8/A9, Inflammation
Introduction
Acute coronary syndrome (ACS) is one of the leading causes of death in the modern world. Leukocytosis is widely suspected to play a causal role in ACS1. Among leukocytes, neutrophil count is considered as an independent prognostic factor in ACS patients, both on admission and after revascularization by percutaneous coronary intervention (PCI) or coronary bypass surgery2. The number of neutrophils in the circulation is directly related to both infarct size and a decline in left ventricular ejection fraction (LVEF)3, suggesting that neutrophil count post-procedurally is an independent predictor of cardiovascular disease (CVD) outcomes. Thus, limiting the number of neutrophils in ACS patients may improve cardiac function and survival rates.
Neutrophils are the first line of defense both in septic and sterile inflammatory conditions. Attracted by the cellular debris and alarmins/ Damage Associated Molecular Patterns (DAMPs) generated by the necrotic cells, neutrophils appear in the myocardium within hours after ischemic injury4. At the ischemic microenvironment, they become hyper-activated, generate high amounts of reactive oxygen species, and release a variety of proteolytic enzymes that could exacerbate local tissue injury5. Even subtle alterations in the residency of these cells in the vicinity of injury may have a profound impact on myocardial remodeling and catastrophic consequences6. Consequently, many experimental anti-neutrophil strategies aimed at reducing neutrophil recruitment/ activation have been shown to limit tissue injury; however, the translation of this to the clinical arena has not been successful7. The reasons for this lack of success in the clinical setting range from poor efficacy of anti-neutrophil therapeutics to the improper dosing, duration of treatment, the presence of concurrent risk factors, and the failure to identify the correct therapeutic window7. However, the major limitation in identifying this so called “window of myocardial salvage” is a lack of our understanding of the signaling pathways that orchestrate neutrophil production, recruitment, and their timely removal. We propose that deciphering the signaling pathway(s) that promote neutrophil production and recruitment after MI will help identify the right therapeutic target to reduce the risk of heart failure (HF).
Methods
Detailed methods including statistics is provided in the online-only Data Supplement. All animal studies were approved by the IACUCs in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Humans studies were approved by the University of Kentucky’s Institutional Review Committee. All subjects gave informed consent. Detailed analytical methods and reagents will be made available to other investigators on a reasonable request. Key resources and reagents used in the study are listed in Table-3 (online-only Data Supplement)
Statistics
All data are expressed as mean ± SEM. Normal distribution was evaluated using Shapiro-Wilk test. For statistical comparisons of 2 groups, an unpaired t test (for normally distributed variables), a Mann-Whitney test (for non-normally distributed variables) and a Fisher’s exact test (for categorical data) was applied. For comparing 3 or more groups with normal distribution data, a 1-way or two-way analysis of variance (ANOVA) followed by either a Holm-Sidak’s or Tukey’s multiple comparison tests was applied. For non-normal data, a Kruskal-Wallis test followed by a Dunn’s multiple comparisons test was applied. For RNA-seq differential expression, analysis was performed with EdgeR and false discovery rate was controlled at 5% using the Benjamini-Hochberg method. For human data, cumulative MACE-free survival curves were derived by the Kaplan–Meier method followed by Log-rank (Mantel-Cox) and Gehan-Breslow-Wilcoxon tests to compare the survival time distributions between the groups. A P value ≤ 0.05 was used as a cut off for statistical significance throughout the analyses. All analyses were done using GraphPad Prism version 8.
Results
Neutrophils dominate the infarcted heart early during MI.
To explore the mechanisms involved in leukocytosis following myocardial infarction (MI), we employed a mouse model of permanent ligation of the left anterior descending (LAD) artery. Following MI, an increase in neutrophil recruitment occurred within 6 hours and remained elevated (peaking at 24 hours) throughout the 15-day observation period (Figure 1A). The number of neutrophils in the blood followed a similar pattern, except for a transient decline at 12 hours followed by a robust rebound at 24 hours (Figure 1B). The neutrophils in the blood were activated as assessed by cell surface expression of a key adhesion molecule CD11b which is crucial for adhesion/ transmigration to the infarct (Figure IA in the online-only Data Supplement). The initial surge in blood neutrophils (up to 12 hours) was likely a result of mass exodus of neutrophils from their reservoirs in the BM (Figure IB in the online-only Data Supplement) facilitated by raised Granulocyte- Colony Stimulating Factor (G-CSF) levels in the blood (Figure IC in the online-only Data Supplement) and suppression of neutrophil retention factors in the BM (Figure ID in the online-only Data Supplement). Despite a decrease in the overall number of neutrophils in the BM, new stocks of neutrophils were constantly emerging, and by the end of the 24 hours, the majority of the resident neutrophils were newly produced, as assessed by EdU incorporation (Figure IE in the online-only Data Supplement). Monocyte kinetics followed a similar pattern and consistent with previous reports8, showed a bi-phasic response characterized by early recruitment of pro-inflammatory Ly6C hi cells (peak, day 3) followed by anti-inflammatory Ly6C lo monocytes (peak, day 5) at the infarct site (Figure IF and IG in the online-only Data Supplement). Both monocyte subsets had higher surface expression of CD11b during their peak cell numbers in the heart (Figure IH in the online-only Data Supplement). The increase in the number of myeloid cells in the blood (24 hours) roughly coincided with a decrease in lymphocytes and their progenitors (Figure II and IJ in the online-only Data Supplement), suggesting a myeloid bias of hematopoietic stem (HSCs) and progenitor cells (HSPCs) in the BM.
Figure 1. Neutrophils dominate the heart following myocardial infarction (MI).
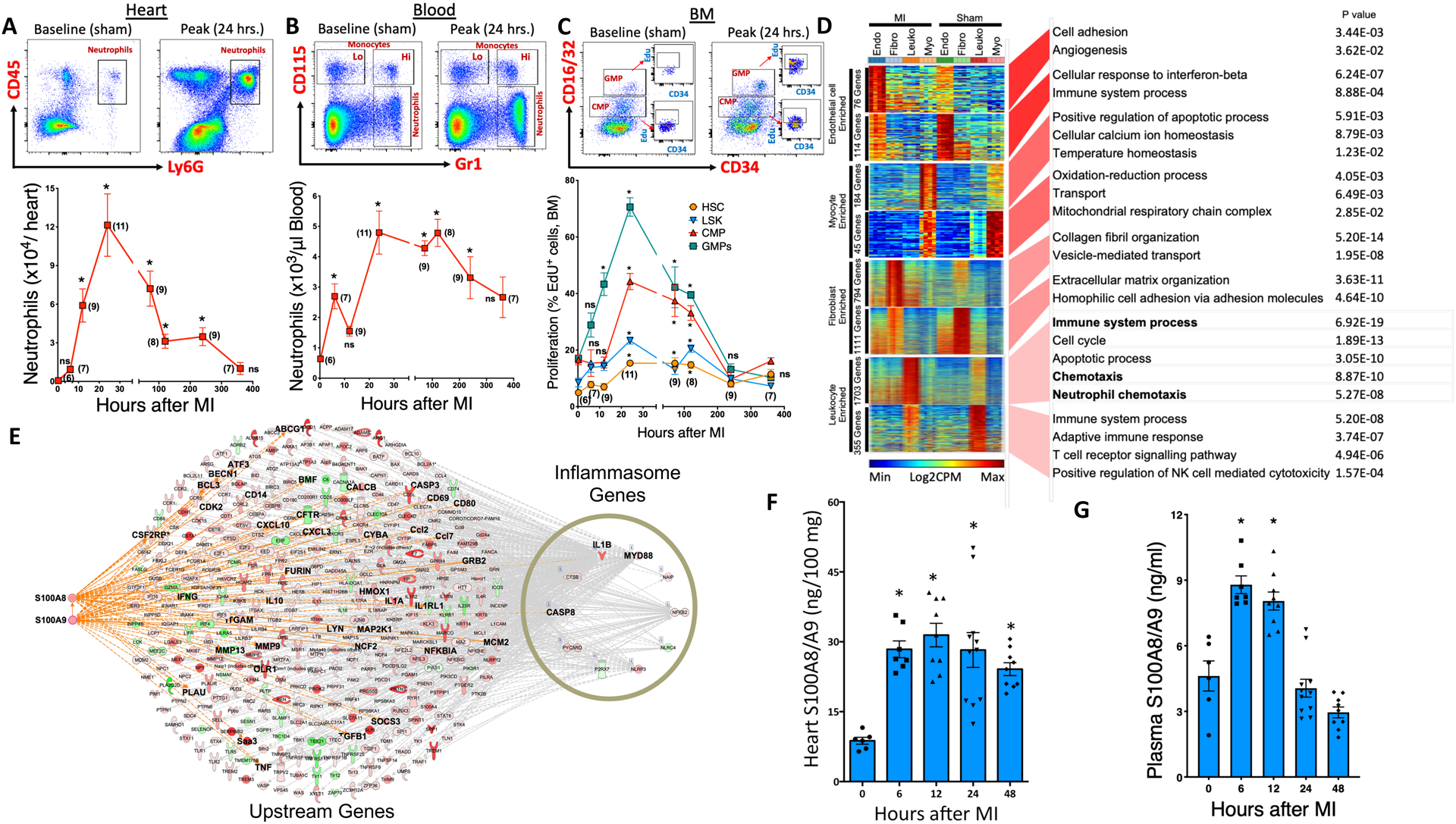
C57BL6 WT mice were subjected to MI by permanent ligation of the left anterior descending (LAD) artery and their myeloid progenitor (BM) and leukocyte cell populations (blood and heart) were quantified at 6, 12, 24, 72, 120, 240 and 360 hours following MI. Flow cytometry plots (top panel) representing neutrophil and /or monocyte populations in the heart (A) and blood (B) at baseline (sham) and peak along with their quantification (bottom panel). Cells in the hearts are normalized to 100 mg of tissue. C, Flow cytometric gating (top panel) and quantification of proliferation rates (as assessed by EdU incorporation) of myeloid progenitor cells in the BM at baseline (sham) and peak along with their quantification (bottom panel). MI, Myocardial infarction; Lo, Ly6C Lo monocytes; Hi, Ly6C Hi monocytes; CMP, Common myeloid progenitors; GMP, Granulocyte macrophage progenitors; LSK, Lineage−, ckit+, Sca1+ cells and HSC, Hematopoietic stem cells. All cells were gated after eliminating debris, dead and clustered cells. Monocytes and neutrophils in the blood were gated on CD45+ cells, myeloid progenitor cells (CMP, GMP) on Lin−, ckit+, Sca1− cells while HSCs were gated on LSK cells. The number in parenthesis next to each data symbol indicates sample size. Mean± SEM, n=6–11/ group. Statistical tests for (A) through (C): Kruskal-Wallis test and Dunn’s multiple comparison test, *P<0.05 compared to the baseline/ sham control in each figure, ns; not significant. D, Gene ontologies of cell-specific differentially expressed genes. The heatmaps depict genes ranked by the degree of cell-type specific enrichment. Endo, Endothelial cells; Fibro, Fibroblasts; Leuko, Leukocytes and Myo, Cardiomyocytes. The P value of GO enrichment in each gene set is reported. n=4 per sample. E, Immune response gene network connecting S100A8/9, Nlrp3 inflammasome and IL-1β in CD45 cells. The upstream connections of the inflammasome genes identified S100A8 and S100A9 as upstream drivers. The interconnections of S100A8/A9 and IL-1β in the inflammasome network are highlighted. Red or green nodes denote upregulation during MI or Sham, respectively. Quantification of S100A8/A9 protein levels (by ELISA) in the (F) heart lysates and (G) plasmas of WT sham and MI mice. Mean± SEM, n=6–11/ group. Statistical tests for (F) and (G): 1-way ANOVA and Holm-Sidak’s post-hoc test, *P<0.05 compared to the baseline/ sham controls in each figure.
Granulopoiesis facilitates the supply and recruitment of neutrophils to the infarcted heart.
Myeloid bias of hematopoietic stem and progenitor cells was evident both in the BM and spleen as demonstrated by a robust increase in the number of HSCs, common myeloid (CMP) and granulocyte macrophage progenitors (GMP) in the BM (Figure IIA in the online-only Data Supplement) and spleen (Figure IIB in the online-only Data Supplement) at 24 hours post-MI. Furthermore, these cells were proliferative, as assessed by incorporation of EdU in the BM (Figure 1C) and spleen (Figure IIC in the online-only Data Supplement) along with other markers (Figure IID and IIE in the online-only Data Supplement). The cells in the BM (Figure IIF in the online-only Data Supplement) and spleen (Figure IIG in the online-only Data Supplement) also showed a myeloid lineage, as demonstrated by an increase in the expression of PU.1 and its target genes, Gmcsfr (Csf2ra) and Mcsfr (Csf1r). This is consistent with previous studies that found a critical role for PU.1-dependent gene program during emergency myelopoiesis9. Taken together, these data suggest that both medullary and extramedullary myelopoiesis programs were activated to meet the excessive demand of neutrophils from the injured heart.
Global transcriptome analysis suggests hierarchical activation of acute inflammatory response during MI in cardiac leukocytes.
To identify the signaling molecules that are critical in driving myelopoiesis, we performed a global transcriptome analysis on cardiomyocytes, endothelial cells, fibroblasts, and CD45+ leukocytes isolated from sham and ischemic mouse hearts as described previously10. First, we obtained an unbiased cross-sectional snapshot of changes in the transcriptomes of all the groups. Specifically, we identified the non-overlapping and cell-type specific transcripts that were enriched in each cell type and also differentially regulated between sham and MI (Figure 1D). A number of unique transcripts were significantly altered in endothelial cells (76), myocytes (184), and fibroblasts (794) after MI. However, the highest number of differentially-expressed genes (1703) were found in leukocytes, a clear indication that leukocyte-specific processes dominate the cellular landscape, with many of their pathways perturbed after MI. Canonical pathway analysis of leukocyte-enriched genes identified upregulation of key signaling pathways representing acute inflammation. These included activation of chemokine, leukocyte extravasation, NF-κB, and Inflammasome pathways (Figure IIIA in the online-only Data Supplement). Among the transcripts that are known to mediate these actions, a robust upregulation of cytokines and DAMPs (Figure IIIB in the online-only Data Supplement), along with their downstream receptors/ mediators (e.g. Toll Like Receptors (TLR), Inflammasome and NF-κB) (Figure IIIC in the online-only Data Supplement) were found, suggesting an intense inflammatory response driven by leukocytes. Analysis of leukocyte-enriched gene networks identified two major DAMPs, S100A8 and S100A9, as upstream drivers of a gene network that included IL-1β (Figure IIID in the online-only Data Supplement). We next generated regulatory networks that link these key upstream regulators with the dominant biological processes in the dataset, representing a strong and hierarchical activation of immune responses. We found that S100A8 and S100A9 were among the top three most consistently associated gene networks linking IL-1β with its signaling regulators, such as Myd88 and inflammasomes (Figure 1E). Furthermore, multiple measures of centrality suggested IL-1β as one of the highly connected nodes and a major regulator of the immune response gene network in leukocytes (Figure IIIE in the online-only Data Supplement). These data led us to postulate that IL-1β, which is an effector molecule downstream of S100A8/A9, TLR4, and inflammasome, is likely a master regulator of leukocytosis following MI. Although the genes representing the S100A8/A9-Nlrp3-IL1β network were also significantly upregulated in other cell types in response to MI, their expression profiles were several hundred-fold lower compared to leukocytes (Figure IVA to IVD in the online-only Data Supplement).
We next sought to identify the specific immune cell subtype(s) within the CD45+ leukocyte fraction that represented the above signaling network. Genes that were specifically enriched in leukocytes from each group were compared to expression profiles of different cell types available from the ImmGen compendium (https://www.immgen.org). There was no enrichment of any particular cell type in the steady state, but in response to MI, a large increase in the proportional representation of neutrophils was observed (Figure IVE in the online-only Data Supplement). Further enrichment of CD45+ leukocytes from the infarct by cell sorting (mRNA) and flow cytometry (Mean Fluorescence Intensity, MFI) suggested neutrophils as the predominant source of S100A8/A9, Nlrp3, and IL-1β (Figure IVF and IVG in the online-only Data Supplement). Among the members of the inflammasome family, Nlrp3 was the most abundant transcript in both monocytes and neutrophils and increased further in neutrophils after MI (Figure IVH in the online-only Data Supplement). Together, these results suggest that the S100A8/A9-NLRP3-IL-1β signaling axis is housed mainly in cardiac neutrophils.
MI-induced granulopoiesis was not due to circulating DAMPs (S100A8/A9).
We next sought to examine how S100A8/A9, the NLRP3 inflammasome, and IL-1β could form a signaling network to drive granulopoiesis. S100A9 naturally forms a hetero-oligomer with S100A8 in the presence of calcium and the complex is capable of provoking myelopoiesis by interacting directly with its receptors (e.g. TLR4, Receptor for Advanced Glycation End Products (RAGE)) on cells in the BM11, or indirectly by priming the NOD-like receptors (NLRs), first, via interaction with TLR4 on myeloid cells (signal 1), followed by secretion of IL-1β in response to signal 212. For the latter mechanism, activation of caspase 1 is critical for maturation and release of IL-1β. The released IL-1β can then interact with its receptor (IL-1R1) on hematopoietic progenitor cells in the BM to stimulate myelopoiesis12, 13. Although there was a marked increase in S100A8/A9 protein in the heart (Figure 1F) and briefly in the circulation (Figure 1G), the expression of its receptors (i.e. TLR4 and RAGE) on stem/ myeloid progenitor cells in the BM did not increase at any time point after MI (Figure VA in the online-only Data Supplement). Furthermore, direct stimulation of Wild Type (WT) BM cells with S100A8/A9 did not induce myelopoiesis (Figure VB in the online-only Data Supplement) and production of new neutrophils (Figure VC in the online-only Data Supplement) ruling out the possibility of a direct interaction between soluble S100A8/A9 and its receptors in the BM.
We next pursued the second possibility where neutrophil-derived S100A8/A9 interacted locally with TLR4 on resident and/ or naïve neutrophils to prime the NLRP3 inflammasome and release IL-1β. To test this hypothesis, we first primed the naïve neutrophils with S100A8/A9 or lipopolysaccharide (LPS) followed by stimulation with a potassium ionophore (nigericin, signal 2). This led to a robust release of IL-1β in a TLR4, NLRP3, and Caspase 1-dependent manner (Figure VIA and VIB in the online-only Data Supplement), confirming that S100A8/A9, similar to LPS, is a strong inflammasome priming agent. Similar data were also obtained for another potassium ionophore, ATP, although, surprisingly, the response was less robust compared to nigericin (Figure VIC in the online-only Data Supplement). Furthermore, neutrophils from the heart produced far more IL-1β than neutrophils from blood and spleen, while immature BM neutrophils were resistant to IL-1β secretion (Figure VID in the online-only Data Supplement). Again, only cardiac neutrophils had the highest cell-surface expression of TLR4 post-MI (Figure VIE in the online-only Data Supplement), suggesting a local priming effect within the infarct.
Neutrophil-derived S100A8/A9 is indispensable for MI-induced granulopoiesis.
Placing S100A8/A9-priming of neutrophils as an upstream process in the sequalae of events leading to granulopoiesis, we tested the hypothesis that deletion of S100a9 or pharmacological inhibition of S100A8/A9 binding to TLR4 should suppress MI-induced granulopoiesis. As expected, global deletion of S100a9 significantly dampened MI-induced granulopoiesis with reduced number of neutrophils and monocytes in the blood (Figure 2A) and heart (Figure 2B). The number (Figure VIIA in the online-only Data Supplement) and proliferation of HSCs and granulocyte precursors in BM (Figure 2C) and spleen (Figure VIIB and VIIC in the online-only Data Supplement) was also decreased. This led to significant downregulation of Nlrp3 and Il1β transcripts in cardiac neutrophils (Figure VIID in the online-only Data Supplement), suggesting that induction of Nlrp3 and Il1β depends on S100A8/A9. In other words, S100A8/A9 priming of neutrophils is an upstream event in the overall sequalae of events. If this is true, then administration of IL-1β to S100a9-/ mice should produce a phenotype similar to one induced by IL-1β injection to normal WT mice. To verify this, we injected IL-1β13 to WT and S100a9−/− mice and examined myelopoiesis (Figure VIIE in the online-only Data Supplement). Administration of IL-1β to both WT and S100a9−/− mice resulted in a significant increase in the number of circulating neutrophils (Figure VIIF in the online-only Data Supplement). This was associated with elevated myelopoiesis in the BM, as shown by increased incorporation of Edu in HSCs and HSPCs (Figure VIIG in the online-only Data Supplement). The leukocytosis and myeloproliferation in WT and S100a9−/− mice was indistinguishable suggesting that S100A8/A9 is an upstream molecule.
Figure 2. Genetic deletion or pharmacological inhibition of S100A8/A9 suppress MI-induced granulopoiesis.
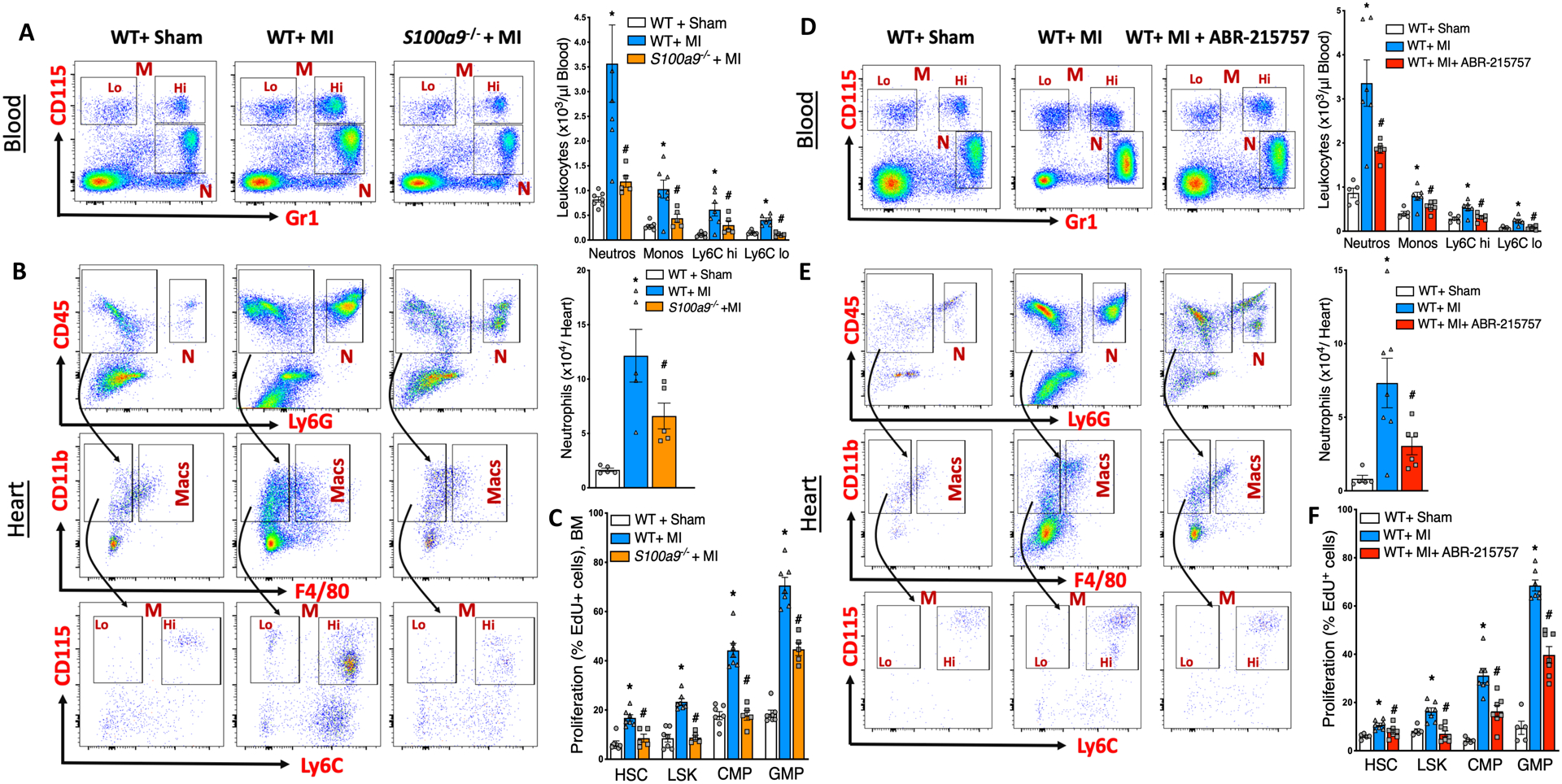
Flow cytometric gating (left panel) and quantification (right panel) of neutrophils in the blood (A) and heart (B) at ~ 18 hours post-MI in WT+ Sham, WT+ MI and S100a9−/−+ MI mice. N, Neutrophils; M, Monocytes; Lo, Ly6C Lo monocytes; Hi, Ly6C Hi monocytes and Macs, Macrophages. C, Effect of MI on the proliferation of hematopoietic stem and progenitor cells in the BM as assessed by EdU incorporation and flow cytometry. CMP, Common myeloid progenitors; GMP, Granulocyte macrophage progenitors; LSK, Lin−, ckit+, Sca1+ cells and HSC, Hematopoietic stem cells. Cells in the hearts are normalized to 100 mg of tissue. Mean± SEM, n=5–7 per group. Statistical tests for (A) and (B): 1-way ANOVA and Holm-Sidak’s post-hoc test, * P<0.05 compared to all other groups in each cell type, # vs WT+ MI for each cell type. Statistical tests for (C): Kruskal Wallis test and Dunn’s multiple comparison test, * P<0.05 compared to all other groups in each cell type, # vs WT+ MI for each cell type. Flow cytometric gating (left panel) and quantification (right panel) of neutrophils in the blood (D) and heart (E) at ~18 hours post-MI in WT+ Sham, WT+ MI and WT+ MI+ ABR-215757 treated mice. ABR-215757 was administered orally once (10 mg /kg) 4 hours before LAD ligation and the treatment was continued (10 mg /kg/ day) in drinking water until termination. F, Effect of MI on the proliferation of hematopoietic stem and progenitor cells in the BM as assessed by EdU incorporation and flow cytometry. Cells in the hearts are normalized to 100 mg of tissue. Mean± SEM, n=5–7 per group. Statistical tests for (D) and (F): 1-way ANOVA and Holm-Sidak’s post-hoc test, * P<0.05 compared to all other groups in each cell type, # vs WT+ MI for each cell type. Statistical tests for (E): Kruskal Wallis test and Dunn’s multiple comparison test, P<0.05 compared to all other groups in each cell type, # vs WT+ MI for each cell type.
We next tested whether pharmacological inhibition of S100A8/A9 would recapitulate the phenotype similar to genetic deletion of S100a8/a9. ABR-215757 (paquinimod) is an orally-active drug that binds to S100A8/A9 in a Ca2+/Zn2+ -dependent manner and blocks its interaction with TLR4 and RAGE14. Pharmacological blockade of S100A8/A9 led to a phenotype similar to that of S100a9−/− mice, with decreased number of neutrophils in the blood (Figure 2D) and heart (Figure 2E), as well as reduced number (Figure VIIH in the online-only Data Supplement) and proliferation (Figure 2F) of granulocyte precursors in the BM. These data suggest that neutrophil-derived S100A8/A9 prime the NLRP3 inflammasome early during the injury.
Neutrophil-specific NLRP3 and IL-1β are critical for MI-induced granulopoiesis.
Given the ability of S100A8/A9 in stimulating NLRP3-dependent IL-1β release, we hypothesized that MI-induced granulopoiesis requires the NLRP3 inflammasome in cardiac neutrophils. If this is true, then myeloid cell-specific deletion of S100a8/a9 or its downstream signaling mediators, Nlrp3 or Il1β, should also suppress MI-induced granulopoiesis. To confirm this, we transplanted BM from WT, S100a9−/−, Nlrp3−/− or Il1β−/− mice into WT mice and gave them MI (Experimental design in Figure 3A). As expected, deletion of the above genes in the myeloid compartment resulted in a dramatic suppression of myelopoiesis in the BM (Figure VIIIB and VIIIC in the online-only Data Supplement), with fewer of the newly-formed neutrophils in the BM (Figure 3B) and thus, in the blood and heart (Figure 3C and 3D). Consistent with our proposed sequelae of events, neutrophils from all knockout mice carried less pro-IL-1β without any change in S100A8/A9 protein (Figure VIIID in the online-only Data Supplement). Together, these data suggested that S100A8/A9-priming of the Nlrp3 inflammasome is an early event in MI-induced myelopoiesis.
Figure 3. MI-induced granulopoiesis requires a functional S100A8/A9-Nlrp3-IL-1β signaling axis in neutrophils.
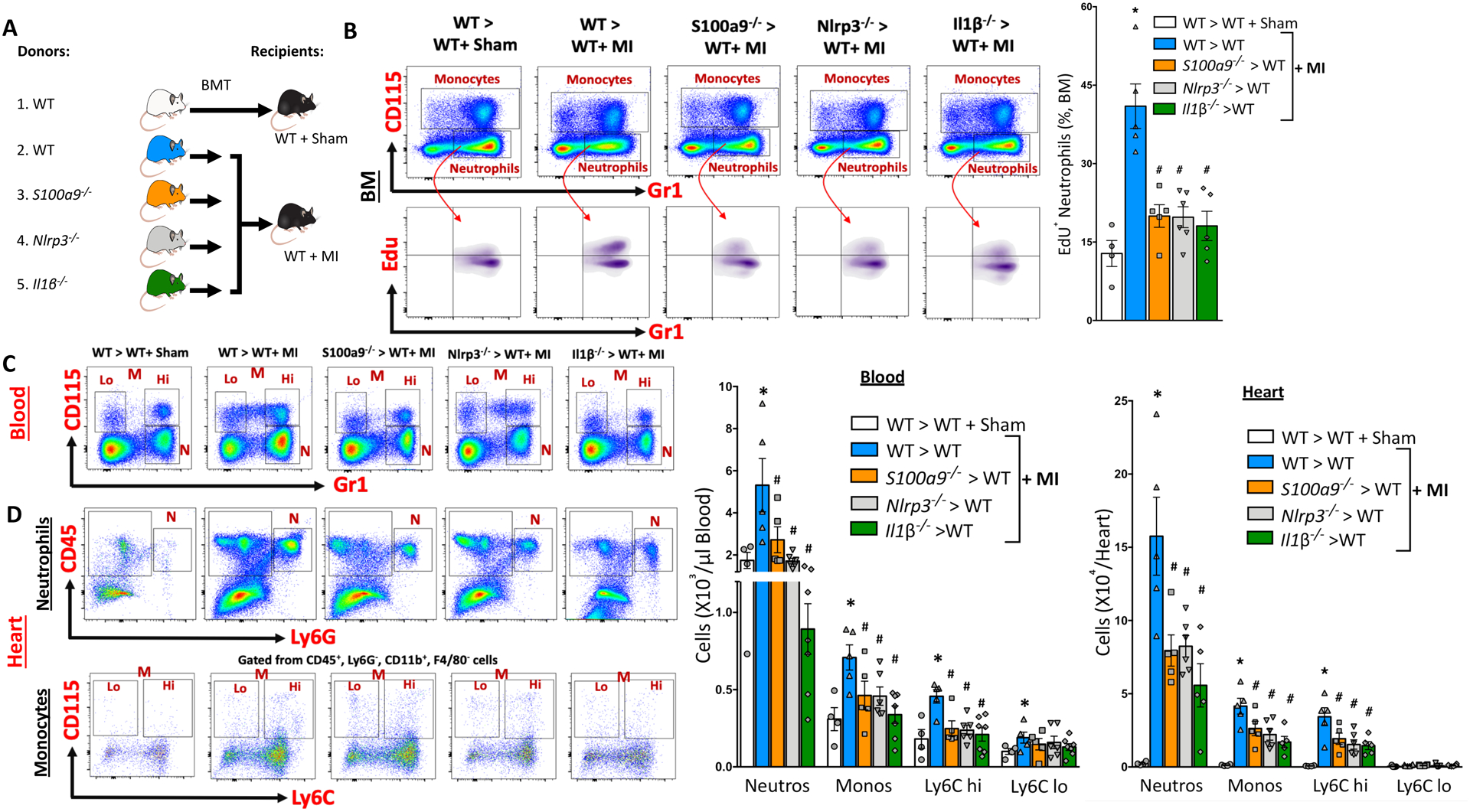
(A) Experimental overview: Bone marrow (BM) from C57BL6 WT, S100a9−/−, Nlrp3−/− and Il1β−/− mice in BL6 background was transplanted to WT recipients and allowed to reconstitute for 6 weeks following which MI was induced by LAD ligation. Approximately 18 hours later, the effect of S100a9, Nlrp3 and Il1β gene deletion on MI-induced granulopoiesis was determined. B, Representative flow cytometry plots (left panel) and quantification (right panel) of newly formed neutrophils (EdU) in the BM of recipient mice (18 hr. post-MI) transplanted with BM from WT, S100a9−/−, Nlrp3−/− or Il1β−/− mice. Mean ± SEM, n= 4–6 per group. Statistical tests for (B):1-way ANOVA and Holm-Sidak’s post-hoc test, *P<0.05 compared to all other groups, # P<0.05 compared to WT> WT+ MI group. Representative flow cytometry plots (left panels) and quantification (right panels) of leukocytes in the blood (C) and heart (D) at ~18 hours post-MI. All cells were gated after eliminating debris, dead and clustered cells. Monocytes and neutrophils were gated on CD45+ cells (blood) or CD45+, CD11b+, F4/80− cells (heart). N, Neutrophils; M, Monocytes; Lo, Ly6C Lo monocytes and Hi, Ly6C Hi monocytes. Cells in the hearts are normalized to 100 mg of tissue. Mean ± SEM, n= 4–7 per group. Statistical tests for (C) and (D): 1-way ANOVA and Holm-Sidak’s post-hoc test, *P<0.05 compared to all other groups in each cell type, # P<0.05 compared to WT> WT+ MI group for each cell type.
TLR4 is the proximal and Interleukin 1 Receptor Type 1 (IL1R1) is the distal receptor in MI-induced granulopoiesis.
To explore if IL-1β could be mediating myelopoiesis through its interaction with BM progenitor cells, we first mapped IL-1R1 on cell surface (MFI) and mRNA expression in BM cells. We found a marked elevation in both the protein (Figure 4A) and mRNA expression (Figure IXA in the online-only Data Supplement) in myeloid progenitor cells in MI mice. Since S100A8/A9 is not a ligand for IL1R1 but to TLR4, we hypothesized that neutrophil-derived S100A8/A9 first binds to TLR4 on cardiac/ circulating neutrophils and facilitate the release of IL-1β. This prompted us to further explore the role of TLR4 (i.e. priming receptor on neutrophils) and IL-1R1 (i.e. end receptor on HSPCs) in MI-driven myelopoiesis. This was achieved by creating mice with hematopoietic deficiencies in either TLR4 (i.e. including neutrophils) or IL-1R1 (i.e. including progenitors) by performing BM transplant (BMT) studies (Experimental design in Figure 4B). Deletion of either gene resulted in fewer myeloid cells in the blood (Figure 4C) and heart (Figure 4D). Most importantly, MI did not provoke myelopoiesis in Tlr4−/− and Il1r−/− BMT mice, as shown by decreased number of HPSCs (Figure IXB in the online-only Data Supplement) and their proliferation (Figure 4E) in the BM.
Figure 4. TLR4 on cardiac neutrophils and IL1R1 on HSPCs in the BM is critical for MI-induced granulopoiesis.

(A) Cell surface expression of IL1R1 (MFI) on hematopoietic stem and progenitor cells in the Bone marrow (BM) of WT+ sham and WT+ MI mice (~18 Hrs. post-MI) as assessed by flow cytometry. Histograms representing IL1R1 expression on LSK, CMP and GMPs in the BM of sham and MI mice. Mean ± SEM, n= 5 per group. Statistical tests for (A): Mann-Whitney test, *P<0.05 compared to the corresponding sham group in each cell type. B, Experimental overview: BM from C57BL6 WT, Tlr4−/−, and Il1r1−/− mice was transplanted to WT recipients and allowed to reconstitute for 6 weeks. MI was induced by LAD ligation following which (~18 Hrs. later) the effect of Tlr4 and Il1r1 gene deletion on MI-induced granulopoiesis was examined. Representative flow cytometry plots (left panels) and quantification (right panels) of leukocytes in the blood (C) and heart (D) of sham and MI mice. Monocytes and neutrophils were gated on CD45+ cells (blood) or CD45+, CD11b+, F4/80− cells (heart). N, Neutrophils; M, Monocytes; Lo, Ly6C Lo monocytes and Hi, Ly6C Hi monocytes. E, Quantification of hematopoietic stem and progenitor cell proliferation (EdU incorporation) in the BM. Mean ± SEM, n= 4–6 per group. Statistical tests for (C) through (E): 1-way ANOVA and Holm-Sidak’s post-hoc test, *P<0.05 compared to the corresponding sham group in each cell type, # P<0.05 compared to WT> WT+ MI group in each cell type. F, Overview of competitive BMT study: Equal portions of BM from WT CD45.1 and WT CD45.2 (Group A) or WT CD45.1 and Il1r1−/− CD45.2 mice (Group B) were transplanted to WT CD45.2 mice (recipients). After the 6-week reconstitution period, MI was induced in the recipient mice and 12–14 hours later the ratio of CD45.1 to CD45.2 leukocytes in the blood (G), heart (H) and BM (I) along with the (J) proliferation (EdU incorporation) of LSK and myeloid progenitor cells (MPC) was determined. Cells in the hearts are normalized to 100 mg of tissue. Numbers in parentheses indicate the ratio of CD45.1 to CD45.2 cells. Ratios higher than 1 indicates competitive outgrowth of WT CD45.1 over Il1r1−/− CD45.2 cells. Mean ± SEM, n= 4–6 per group. Statistical tests for (G) through (J): 2-tailed unpaired t test, *P<0.05 vs. the corresponding CD45.1 (white bars) group in each cell type. n.s., not significant.
IL1R1-dependent granulopoiesis is cell autonomous.
Binding of IL-1β to IL-1R1 could directly stimulate myelopoiesis in a cell-intrinsic fashion, or alternatively induce a signaling cascade leading to the production of proliferative cytokines (e.g. G-CSF, M-CSF), which, in turn, act on other hematopoietic or non-hematopoietic cells (e.g., endothelial cells) in a cell-extrinsic fashion. To distinguish between these two possibilities, we performed a competitive BMT (cBMT) where equal proportions of BM from WT CD45.1 and WT CD45.2 mice (Group A), or WT CD45.1 and Il1r1−/− CD45.2 (Group B) were transplanted into WT recipients, and after reconstitution, gave MI (Experimental design in Figure 4F). In group A recipients (i.e. WT CD45.2 BM), the contribution of both genotypes to the blood (Figure 4G), heart (Figure 4H), and BM (Figure 4I and 4J) leukocytes was equal. However, in Group B recipients (i.e. Il1r1−/−CD45.2 BM), the WT CD45.1 BM significantly outcompeted the Il1r1−/−CD45.2 BM, producing more leukocytes as a result of enhanced myelopoiesis (Figure 4G through 4J) in the BM. These data confirmed that IL-1R on HSPCs was responsible for cell-intrinsic proliferation that is reminiscent of IL-1β-driven myelopoiesis in chronic conditions such as obesity12 and during emergencies9. We next performed similar competitive BMT studies using Tlr4−/− competing BM (Figure IXC in the online-only Data Supplement), which revealed cell-extrinsic blunting of myelopoiesis (Figure IXD through IXG in the online-only Data Supplement), supporting a role for TLR4 in the initiation of the myelopoietic response. These data confirmed that IL-1β promotes myelopoiesis by inducing cell-intrinsic proliferation of the stem and progenitor cells in the BM via its interaction with IL-1R1.
Disruption of S100A8/A9 or its downstream signaling mediators improve cardiac function.
Since elevated neutrophil counts are directly related to infarct size and decline in LVEF3, we asked if suppression of granulopoiesis and, in turn, reduced neutrophil recruitment to the infarct would improve cardiac function. To answer this question, we performed a comprehensive evaluation of cardiac function and histology in WT mice that received BM from WT, S100a9, Nlrp3, or Il1β knockout mice 30 days post-MI (Experimental design in Figure 5A). At 30 days post-MI, a significantly smaller scar was observed in mice transplanted with BM from Nlrp3−/− or Il1β−/− mice but not S100a9−/− mice compared to WT (Figure 5B). Furthermore, WT mice displayed profound hypokinesia (Figure 5C) and a marked reduction in LVEF (Figure 5D), stroke volume (Figure 5E), and cardiac output (Figure 5F). Despite similar initial (day 2) left ventricular parameters, hematopoietic deletion of S100a9, Nlrp3, or Il1β led to a marked improvement in heart function and cardiac characteristics by 30 days post-MI (Figure 5C through 5F). These data suggest that suppression of granulopoiesis by targeting myeloid cell-specific S100a9, Nlrp3, or Il1β is likely to have favorable outcomes on the heart post-MI.
Figure 5. Genetic disruption of S100A8/A9-Nlrp3-IL1β signaling in neutrophils improves cardiac function.

(A) Experimental overview: Bone marrow (BM) from C57BL6 WT, S100a9−/−, Nlrp3−/− and Il1β−/− mice in BL6 background was transplanted to WT recipients and allowed to reconstitute for 6 weeks following which MI was induced by LAD ligation. B, Scar/infarct size was determined at termination (30 days post-MI). Mean ± SEM, n= 5–6 per group. Statistical tests for (B): 1-way ANOVA and Holm-Sidak’s post-hoc test, *P<0.05 compared to the sham group, # P<0.05 compared to WT> WT+ MI group. Effect of MI on cardiac function was assessed by echocardiography at day 0 (pre-MI), 2- and 30-days post-MI. C, Vector diagrams showing the direction and magnitude of myocardial contraction. Three-dimensional regional wall displacement illustrations, demonstrating contraction (yellow–red) or relaxation (blue) of consecutive cardiac cycle results at 30 days post-MI. Measurement of left ventricular functional parameters including (D) Ejection fraction, (E) Stroke volume and (F) Cardiac output. Mean ± SEM, n= 5–6 per group. Statistical tests for (D) through (F): 1-way ANOVA and Holm-Sidak’s post-hoc test, *P<0.05 compared to all other groups at each time point, # P<0.05 compared to WT+ MI group at each time point and for each parameter, n.s, not significant.
Neutrophils correlate with major adverse cardiovascular events (MACE) in ACS patients.
Finally, we sought clinical evidence to support our mechanistic findings in mice. In a cohort of 51 subjects with acute ST-elevation myocardial infarction (STEMI), we initially sought to determine if acute myocardial injury promotes neutrophilia and if the circulating neutrophil number correlates with MACE. We assessed leukocytes in fresh blood obtained from a subgroup of these patients at baseline (i.e. on admission, 0 Hrs.), 6, 12 and 24 hours post-cardiac revascularization by percutaneous coronary intervention (PCI) and compared with leukocytes from non-ACS control subjects (n=20). The baseline patient-specific and clinical characteristics are presented in Table 1 (online-only Data Supplement). As observed in mouse models, STEMI markedly increased the number of circulating leukocytes (Figure 6A and 6B), particularly the neutrophils (Figure 6A and 6C) and monocytes (Figure 6A and 6D), on admission (compared with controls) that remain elevated for 24 hours post-revascularization. Among the monocyte subsets, all three cell types tended to increase on admission but only the classical monocytes continued to rise after PCI (Figure 6A and 6E), with increased abundance of C-C Chemokine Receptor 2 (CCR2) (Figure 6F), a chemotactic receptor that is critical for recruitment of inflammatory monocytes to the infarct8. Furthermore, circulating neutrophils of ACS patients carried more pro-IL-1β, the effector molecule in MI-induced myelopoiesis (Figure 6G). In order to recapitulate the above findings in vitro, we isolated neutrophils from healthy donors and stimulated them with S100A8/A9. Similar to mouse neutrophils, human neutrophils also secreted IL-1β (Figure 6H). Most importantly, the patients with higher peak neutrophil counts from the time of admission to 24 hr. post revascularization had significantly higher incidence of MACE during the one year follow up period (Figure 6I). No other difference except a marginal increase in troponin I levels (in high neutrophil group) was observed between the low and high neutrophil groups (Table 2 in the online-only Data Supplement).
Figure 6. Leukocytosis and correlation between neutrophil numbers and major adverse cardiovascular events in ACS patients.

(A) Representative flow cytometry plots (top panels) and quantification (bottom panels) of leukocytes in fresh blood obtained from controls and ACS patients on admission and 6-, 12- and 24-hours post-revascularization. Numbers next to gates indicate population frequencies. The number of (B) white blood cells (WBCs), (C) neutrophils, (D) total monocytes and (E) monocyte subsets in the peripheral blood as assessed by either complete blood count (CBC) or flow cytometry. F, Mean fluorescence intensity (MFI) of CCR2 on monocyte subsets. All cells were gated after eliminating debris, dead and clustered cells. Neutrophils were identified as CD45+, HLA-DR−, CD193−, CD14− and CD15+ cells. Monocytes were identified as CD45+, HLA-DR+ and further differentiated as classical (C, CD14++, CD16−), alternative (A, CD14−, CD16++) and intermediate (I, CD14+, CD16+) subsets. Sample size for each group is indicated in parenthesis. Mean ± SEM. Statistical test for (B) through (F): Mann-Whitney test, *P<0.05 compared to the healthy control group in each cell type. G, Color-coded histograms representing intracellular expression (MFI) of pro- IL-1β and their quantification in circulating neutrophils from control and ACS patients. Statistical tests for (G): Mann-Whitney test, *P<0.05 compared to the healthy control group. H, IL-1β release from human neutrophils (healthy donors) in response to stimulation by S100A8/A9 and nigericin in the presence of Nlrp3 inhibitor. Mean ± SEM, n=6. Statistical tests: 1- way ANOVA and Holm-Sidak’s post-hoc test, P<0.05 compared to vehicle treated group, # P<0.05 compared to S100A8/A9+ nigericin group. I, Cumulative MACE-free survival curves of patients with peak-low and peak-high neutrophil counts over a period of 1 year. Statistical test for (I): Log-rank (Mantel-Cox) and Gehan-Breslow- Wilcoxon test, *P<0.05 compared to peak-low neutrophil group.
DISCUSSION
Despite significant advances in revascularization techniques, patients who experience ACS eventually develop some form of HF15. This is mainly due to abnormal cardiac remodeling induced by MI but largely dictated by the nature of the inflammatory response4. Clinically, neutrophil-to-lymphocyte ratio is one of the strongest predictors of death in ACS patients16, implying that reducing the number of neutrophils may have favorable outcomes. However, the mechanisms that regulate neutrophil production and recruitment to the ischemic heart are unclear. Our study provides novel evidence that neutrophils, being the first responders to a sterile injury, are the key determinants that decide the nature of the ensuing inflammatory response. They achieve this, first, by infiltrating the injured sites and releasing large amounts of S100A8/A9 that may act as chemo-attractants, fueling recruitment of other cell types including monocytes17. Second, S100A8/A9 primes the NLRP3 inflammasome of incoming naïve neutrophils and stimulates them to produce IL-1β, the effector molecule in MI-induced myelopoiesis13 (proposed model, Figure 7). This discovery revealed a previously unknown crosstalk between cardiac neutrophils and the granulocyte precursors in the BM and provides a rationale to target neutrophils post-MI.
Figure 7. Schematic representation of the proposed signaling pathway.
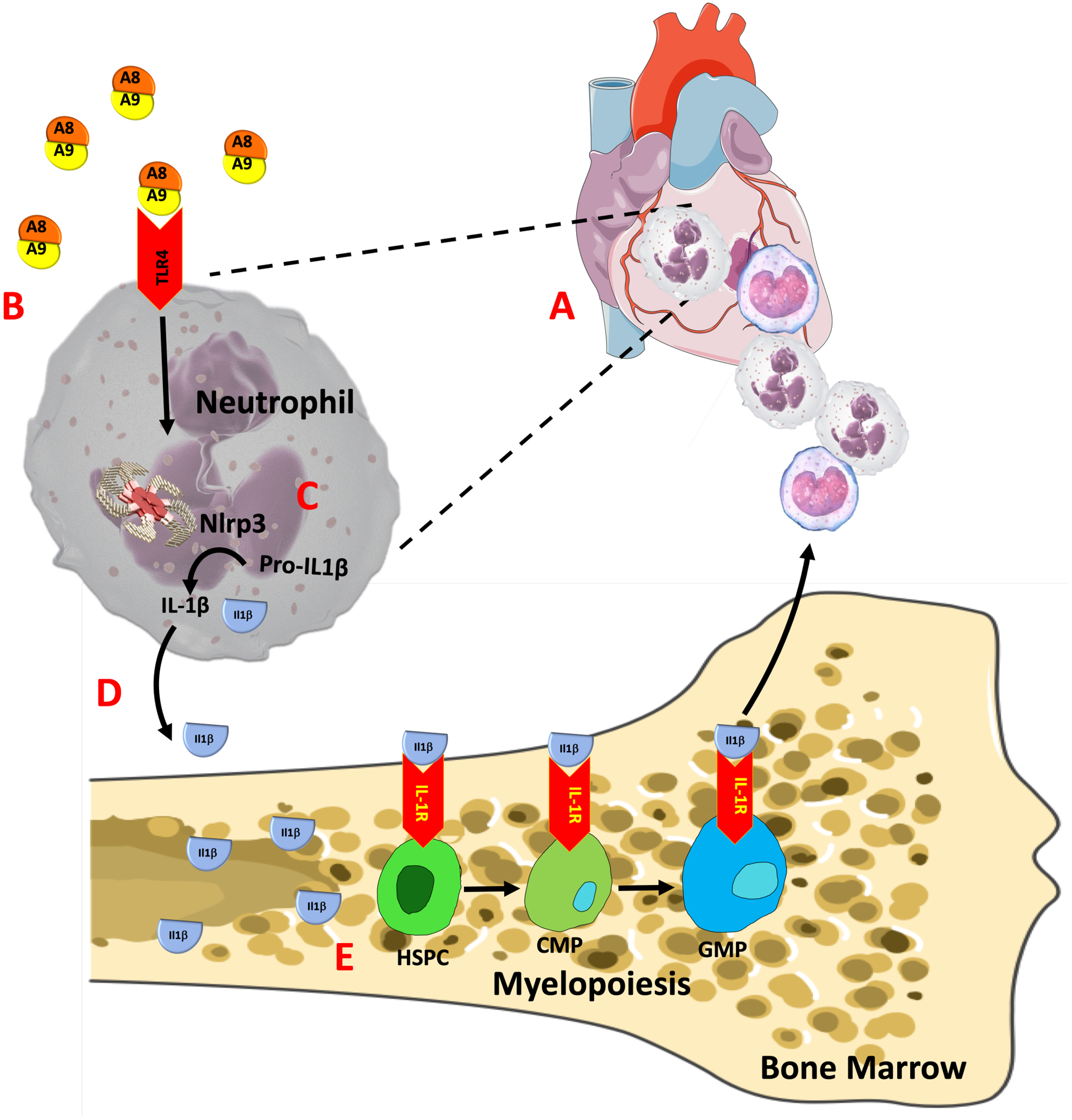
In response to myocardial infarction, circulating neutrophils are attracted to the ischemic heart (A), where, they undergo rapid priming and release S100A8/A9 locally within the ischemic microenvironment. B, S100A8/A9 binds to TLR4 on resident/ incoming neutrophils to (C) induce/ prime the Nlrp3 inflammasome and secretion of IL-1β which then (D) interact with its receptor (IL-1R) on hematopoietic stem and progenitor cells in the bone marrow to stimulate (E) myelopoiesis in a cell-intrinsic manner.
The ability of the hematopoietic system to produce such a colossal number of neutrophils in short notice points to a specialized event stemming from the infarct. Multiple levels of interrogation led to identification of two important molecules, S100A8 and S100A9 that are crucial in stimulating myelopoiesis. We previously have shown that S100A8/A9 has the ability to stimulate myelopoiesis in chronic conditions such as diabetes and obesity by interacting directly with RAGE on cells in the BM11. However, during acute MI, we found no increase in the expression of any receptors for S100A8/A9 on cells in the BM but IL1R, the receptor for IL-1β was markedly elevated. These data suggest that mechanisms by which S100A8/A9 induce myelopoiesis in MI is different from that other conditions. Further investigations led to identification of a distinctive set of mechanisms within the neutrophils by which S100A8/A9 induce myelopoiesis via the involvement of Nlrp3 and IL-1β. We found that neutrophil-derived IL-1β interacts with its receptor on HSCs and HSPCs in the BM to stimulate myelopoiesis in a cell-intrinsic manner, a mechanism that likely involve PU.1-dependent gene program9.
The cellular source of S100A8/A9 at the site of MI and moreover, the general plasma pool is unclear. The data from our studies have provided irrefutable evidence that neutrophils are the major source of S100A8/A9 because they not only accumulate in greater numbers in the infarct but also carry large amounts in their cytoplasms (~45% vs. 5% in monocytes)18. Previous studies have shown that platelets may also contribute to the S100A8/A9 plasma pool in experimental models of vascular injury19 and MI patients20, 21. Since, the infiltration of platelets precedes the recruitment of neutrophils to the sites of sterile injury22, it is possible that platelet-derived S1008/A9 may act as the initial chemoattractant for neutrophils, and, thus may contribute to the serum pool of S100A8/A9. To clarify this, we performed a cell-to-cell comparison for abundance of S100A8/A9 proteins in different leukocytes in the blood. Although S100A8/A9 was detectable in platelets, the expression was almost negligible compared to neutrophils both at steady state and after MI (data not shown), suggesting that platelets are not a major source of plasma S100A8/A9.
The mechanisms that led to release of S100A8/A9 at the site of infarct are also not clear. Despite all known extracellular functions, the secretion of S100A8/A9 lacks signal peptides essential for the classical style Golgi-mediated secretion pathway23 but seems to require reactive oxygen species (ROS) and K+ fluxes24. Previous studies have shown that activation of neutrophils induces translocation of S100A8/A9 from cytosol to the membrane before secretion, but neither neutrophil extracellular trap formation (NETosis) nor degranulation promoted their secretion24. However, these studies were performed in non-reductive conditions using isolated human neutrophils and, therefore the conditions were unfavorable for cell death. Interestingly, the abundance of S100A8/A9 protein at the site of inflammation25 correlated with neutrophil necrosis, and S100A8/A9 proteins formed the bulk of NET-bound cytosolic proteins after NETosis26. Furthermore, NETosis requires generation of ROS by NADPH oxidase27 that is also essential for S100A8/A9 secretion24. These data suggest that the death of some neutrophils, either by necrosis or NETosis, may have contributed to S100A8/A9 release at the infarct. Further studies are warranted to understand the mechanisms that regulate S100A8/A9 release at the sites of myocardial injury.
Previous studies have found increased S100A8/A9 levels both systematically and at sites of coronary occlusion28, 29. Plasma levels of S100A8/A9 is also associated with LV dysfunction and HF30 and predicts the risk of future CVD events21. However, these studies do not provide evidence whether this is driven directly by S100A8/A9 or through the involvement of downstream signaling pathways. Our mouse data supports the notion that S100A8/A9 stimulates the immune-cell stress sensor (the Nlrp3 inflammasome) to alert the innate immune system via the release of IL-1β. In order to validate these findings in humans, we assessed leukocytosis in ACS patients at the time of admission and at different time points following revascularization. Similar to mouse models, our patient population was relatively of higher risk, with larger MI and LAD being the culprit artery (in 42% of patients). As observed in mouse models, MI markedly increased the number of circulating neutrophils that remained elevated despite revascularization. Among the monocytes, the classical monocytes displayed increased expression of CCR2, a chemotactic receptor that is vital for leukocyte recruitment to the infarct8. Although we could not reliably measure plasma IL-1β levels, the circulating neutrophils carried greater amounts of IL-1β following MI, a clear indication of inflammasome priming. Most importantly and similar to mouse neutrophils, human neutrophils also responded to S100A8/A9-induced IL-1β secretion albeit at a smaller magnitude. The specific reason for this suboptimal response in human neutrophils is unclear but could be attributed to the differences in the receptor density (e.g. TLR4) and /or desensitization of the receptors and, thus, their affinity to bind to S100A8/A931. Furthermore, the physiological concentration of S100A8/A9 in human serum is several thousand-fold higher than mouse serum11, and this may reduce the overall sensitivity of TLR4s to S100A8/A9 in human neutrophils.
There are a few limitations to this study, including the choice of infarct model. Although permanent ligation of LAD mouse model is less reflective of an ideal clinical scenario where most patients are timely revascularized, it does reflect acute STEMI patients who do not receive successful reperfusion at the right time32, 33. These patients could be as high as 50% particularly in disadvantaged populations and /or those residing in regional and remote locations34, 35. Furthermore, the magnitude and the type of injury resulting from revascularization is inherently different from that caused by permanent occlusion. While the mechanisms of injury may be different, both types of injuries attract neutrophils to the infarct30, 36, 37 and, most importantly, inhibition of S100A8/A9 or NLRP3 reduces leukocyte recruitment to the myocardium and improves cardiac function30, 38. Notably, and similar to our mouse data, we found a significant increase in the number of circulating neutrophils in humans after revascularization. Furthermore, in stroke patients administration of Anakinra, a IL1R1 antagonist, resulted in decreased number of blood leukocytes (particularly neutrophils) and inflammatory markers (i.e. C-Reactive Protein, CRP)39. These data suggest that similar mechanisms (i.e. S100A9/A9-Nlrp3-IL1β) may play a role in myelopoiesis in other ischemic vascular diseases. A second limitation is failure of our S100a9−/− model to completely prevent MI-induced leukocytosis by targeting S100A8/A9. Despite near normalization of neutrophil numbers in S100a9−/− BMT mice, its effect on cardiac functional indices were not as positive as those observed for Nlrp3−/− and Il1β−/− BMT mice. Although the reasons for these discrepancies are unclear, it’s likely that S100A8/A9 may have additional cardioprotective effects independent of its role in myelopoiesis. For example, data from recent studies suggest that certain neutrophil-derived proteins may foster infarct healing by polarizing macrophages towards a reparative phenotype36. These findings caution that the duration and severity of neutrophil depletion at the infarct site should be carefully considered in formulating anti-neutrophil strategies. As such, indiscriminate blockade of S100A8/A9 signaling over prolonged periods of time may have negative repercussions. Perhaps a moderate and short-term suppression of granulopoiesis particularly during the inflammatory response phase (1–3 days post-MI), may be most beneficial in the setting of acute MI. In support of this idea, a recent study found that short-term inhibition of S100A8/A9 (3-day treatment) significantly improved cardiac function30. Whether this is due to a reduction in the number of leukocytes alone or through mechanisms independent of leukocytosis needs further investigations.
In summary, we have elucidated a novel mechanism by which neutrophils regulate their own production in response to MI. As therapies aimed at neutralizing IL-1β signaling are gaining momentum in managing CVDs40, it would be reasonable to consider targeting upstream signaling events, such as priming of the NLRP3 by DAMPs41 or disruption of NLRP3 signaling. In line with this, a recent study demonstrated marked improvement in cardiac function after pharmacological inhibition of NLRP3 in a pig model of MI. This was also associated with a significant decrease in myocardial IL-1β and neutrophil content in the heart38. Similarly, targeting IL-1β also diminishes heart failure post-MI13. Canakinumab Antiinflamatory Thrombosis outcomes study (CANTOS) trials targeting IL-1β have also shown major improvements in recurrent CVD events, and, most importantly, this was associated with a significant decrease in neutrophil number40. Thus, neutrophils and/ or their derived-proteins may represent as viable targets in the management of ischemic vascular disease.
Supplementary Material
Clinical Perspective.
What is new?
Cardiac neutrophils are critical in sustaining and amplifying the acute inflammatory response elicited by myocardial infarction (MI).
In response to MI, the initial wave of infiltrating neutrophils sets the tone for the ensuing inflammatory response by releasing key alarmins (e.g. S100A8/A9) which subsequently interact with TLR4, prime the Nlrp3 inflammasome and, promote the secretion of IL-1β.
The released IL-1β interact with IL1R1 on myeloid progenitor cells in the bone marrow and stimulate granulopoiesis in a cell-autonomous manner.
Genetic and pharmacological strategies aimed at disruption of the S100A8/A9-Nlrp3- IL-1β signaling axis dampen granulopoiesis and, improve cardiac function in mouse models.
What are the clinical implications?
To date, there are no clinical therapies aimed at reducing acute inflammation to manage chronic heart failure (HF) and associated mortality.
Because neutrophils are at the forefront in amplifying the acute inflammatory response after MI, pharmacological strategies aimed at reducing their production and/ or blocking their signaling pathways may be a viable approach to reduce chronic HF.
Acute coronary syndrome (ACS) patients have increased amounts of IL-1β in circulating neutrophils both on admission and revascularization, suggesting that targeting IL-1β or its upstream regulators (e.g. S100A8/A9, Nlrp3) could dampen leukocytosis and improve HF.
Acknowledgements
P.N., A.A.L. and A.M. conceived and designed the study, analyzed the data, created figures and wrote the main manuscript that was revised and approved by all authors. P.N., G.S., B.A., R.H., and A.D performed the animal experiments, surgeries, BMTs, Fluorescence activated cell sorting (FACS), mouse echocardiography and histological analysis. A.A.L and R.H performed the human studies. G.QR., G.P., J.H and E.P. performed the RNA sequencing and bioinformatics analysis. A.A., D.D., H.L., B.H., C.R., K.S. S.N. and S.S contributed to manuscript writing, analyzed the data, discussed results, strategy and provided critical inputs.
We thank Drs. Yoichiro Iwakura (Tokyo University of Science, Japan) and Helena Eriksson (Active Biotech., Sweden) for providing mice/ reagents, Dr. Hanamanthu (UAB) for assistance with cell sorting, Drs. Gao, Ye and Zhang for LAD surgeries and, Alexander Smith (Baker) for statistics. We thank www.creativecommons.org for unrestricted use of figure templates.
Funding
This work was supported by NIH funding to PRN (HL122505, HL137799) and AAL (HL124266 and HL138488). A.J.M is supported by the National Health and Medical Research Council (NHMRC) project grants (APP1106154 and APP1142938). BYH is supported by a K23 grant from the NIH (AR068450).
Non-standard Abbreviations and Acronyms
- BM
Bone Marrow
- CANTOS
Canakinumab Antiinflammatory Thrombosis Outcome Study
- cBMT
Competitive BMT
- CCR2
C-C Chemokine Receptor Type 2
- CMP
Common Myeloid Progenitors
- CVD
Cardiovascular Disease
- DAMP
Damage Associated Molecular Patterns
- FACS
Fluorescence-Activated Cell Sorting
- GMP
Granulocyte Macrophage Progenitors
- HF
Heart Failure
- HSPC
Hematopoietic Stem and Progenitor Cells
- IL1R1
Interleukin 1 Receptor Type I
- LPS
Lipopolysaccharide
- LSK
Lineage- Sca1+ Kit+
- MACE
Major Adverse Cardiovascular Events
- MFI
Mean Fluorescence Intensity
- NETosis
Neutrophil Extracellular Trap Formation
- NLRP3
Nod Like Receptor Family Pyrin Domain-Containing 3
- RAGE
Receptor for Advanced Glycation End Products
- ROS
Reactive Oxygen Species
- TLR
Toll Like Receptor
- WT
Wild Type
Footnotes
Disclosures
None
References
- 1.Coller BS. Leukocytosis and ischemic vascular disease morbidity and mortality: is it time to intervene? Arterioscler Thromb Vasc Biol. 2005;25:658–670. doi: 10.1161/01.ATV.0000156877.94472.a5. [DOI] [PubMed] [Google Scholar]
- 2.Guasti L, Dentali F, Castiglioni L, Maroni L, Marino F, Squizzato A, Ageno W, Gianni M, Gaudio G, Grandi AM, et al. Neutrophils and clinical outcomes in patients with acute coronary syndromes and/or cardiac revascularisation. A systematic review on more than 34,000 subjects. Thromb Haemost. 2011;106:591–599. doi: 10.1160/TH11-02-0096. [DOI] [PubMed] [Google Scholar]
- 3.Chia S, Nagurney JT, Brown DF, Raffel OC, Bamberg F, Senatore F, Wackers FJ, Jang IK. Association of leukocyte and neutrophil counts with infarct size, left ventricular function and outcomes after percutaneous coronary intervention for ST-elevation myocardial infarction. Am J Cardiol. 2009;103:333–337. doi: 10.1016/j.amjcard.2008.09.085. [DOI] [PubMed] [Google Scholar]
- 4.Soehnlein O, Steffens S, Hidalgo A, Weber C. Neutrophils as protagonists and targets in chronic inflammation. Nat Rev Immunol. 2017;17:248–261. doi: 10.1038/nri.2017.10. [DOI] [PubMed] [Google Scholar]
- 5.Ma Y, Yabluchanskiy A, Lindsey ML. Neutrophil roles in left ventricular remodeling following myocardial infarction. Fibrogenesis Tissue Repair. 2013;6:11. doi: 10.1186/1755-1536-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159–173. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vinten-Johansen J Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc Res. 2004;61:481–497. doi: 10.1016/j.cardiores.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 8.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, Lakshminarasimhan R, Chin CP, Techner JM, Will B, et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol. 2016;18:607–618. doi: 10.1038/ncb3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quaife-Ryan GA, Sim CB, Ziemann M, Kaspi A, Rafehi H, Ramialison M, El-Osta A, Hudson JE, Porrello ER. Multicellular Transcriptional Analysis of Mammalian Heart Regeneration. Circulation. 2017;136:1123–1139. doi: 10.1161/CIRCULATIONAHA.117.028252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, Ramkhelawon B, Distel E, Westerterp M, Huang LS, et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013;17:695–708. doi: 10.1016/j.cmet.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagareddy PR, Kraakman M, Masters SL, Stirzaker RA, Gorman DJ, Grant RW, Dragoljevic D, Hong ES, Abdel-Latif A, Smyth SS, et al. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab. 2014;19:821–835. doi: 10.1016/j.cmet.2014.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sager HB, Heidt T, Hulsmans M, Dutta P, Courties G, Sebas M, Wojtkiewicz GR, Tricot B, Iwamoto Y, Sun Y, et al. Targeting Interleukin-1beta Reduces Leukocyte Production After Acute Myocardial Infarction. Circulation. 2015;132 (20):1880–1890. doi: 10.1161/CIRCULATIONAHA.115.016160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bjork P, Bjork A, Vogl T, Stenstrom M, Liberg D, Olsson A, Roth J, Ivars F, Leanderson T. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol. 2009;7:e97. doi: 10.1371/journal.pbio.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prabhu SD, Frangogiannis NG. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ Res. 2016;119:91–112. doi: 10.1161/CIRCRESAHA.116.303577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhat T, Teli S, Rijal J, Bhat H, Raza M, Khoueiry G, Meghani M, Akhtar M, Costantino T. Neutrophil to lymphocyte ratio and cardiovascular diseases: a review. Expert Rev Cardiovasc Ther. 2013;11:55–59. doi: 10.1586/erc.12.159. [DOI] [PubMed] [Google Scholar]
- 17.Ryckman C, Vandal K, Rouleau P, Talbot M, Tessier PA. Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J Immunol. 2003;170:3233–3242. doi: [DOI] [PubMed] [Google Scholar]
- 18.Edgeworth J, Gorman M, Bennett R, Freemont P, Hogg N. Identification of p8,14 as a highly abundant heterodimeric calcium binding protein complex of myeloid cells. J Biol Chem. 1991;266:7706–7713. doi: [PubMed] [Google Scholar]
- 19.Wang Y, Fang C, Gao H, Bilodeau ML, Zhang Z, Croce K, Liu S, Morooka T, Sakuma M, Nakajima K, et al. Platelet-derived S100 family member myeloid-related protein-14 regulates thrombosis. J Clin Invest. 2014;124:2160–2171. doi: 10.1172/JCI70966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morrow DA, Wang Y, Croce K, Sakuma M, Sabatine MS, Gao H, Pradhan AD, Healy AM, Buros J, McCabe CH, et al. Myeloid-related protein 8/14 and the risk of cardiovascular death or myocardial infarction after an acute coronary syndrome in the Pravastatin or Atorvastatin Evaluation and Infection Therapy: Thrombolysis in Myocardial Infarction (PROVE IT-TIMI 22) trial. Am Heart J. 2008;155:49–55. doi: 10.1016/j.ahj.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Healy AM, Pickard MD, Pradhan AD, Wang Y, Chen Z, Croce K, Sakuma M, Shi C, Zago AC, Garasic J, et al. Platelet expression profiling and clinical validation of myeloid-related protein-14 as a novel determinant of cardiovascular events. Circulation. 2006;113:2278–2284. doi: 10.1161/CIRCULATIONAHA.105.607333. [DOI] [PubMed] [Google Scholar]
- 22.Slaba I, Wang J, Kolaczkowska E, McDonald B, Lee WY, Kubes P. Imaging the dynamic platelet-neutrophil response in sterile liver injury and repair in mice. Hepatology. 2015;62:1593–1605. doi: 10.1002/hep.28003. [DOI] [PubMed] [Google Scholar]
- 23.Rammes A, Roth J, Goebeler M, Klempt M, Hartmann M, Sorg C. Myeloid-related protein (MRP) 8 and MRP14, calcium-binding proteins of the S100 family, are secreted by activated monocytes via a novel, tubulin-dependent pathway. J Biol Chem. 1997;272:9496–9502. doi: [DOI] [PubMed] [Google Scholar]
- 24.Tardif MR, Chapeton-Montes JA, Posvandzic A, Page N, Gilbert C, Tessier PA. Secretion of S100A8, S100A9, and S100A12 by Neutrophils Involves Reactive Oxygen Species and Potassium Efflux. J Immunol Res. 2015;2015:296149. doi: 10.1155/2015/296149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Voganatsi A, Panyutich A, Miyasaki KT, Murthy RK. Mechanism of extracellular release of human neutrophil calprotectin complex. J Leukoc Biol. 2001;70:130–134. doi: [PubMed] [Google Scholar]
- 26.Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, Brinkmann V, Jungblut PR, Zychlinsky A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009;5:e1000639. doi: 10.1371/journal.ppat.1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altwegg LA, Neidhart M, Hersberger M, Muller S, Eberli FR, Corti R, Roffi M, Sutsch G, Gay S, von Eckardstein A, et al. Myeloid-related protein 8/14 complex is released by monocytes and granulocytes at the site of coronary occlusion: a novel, early, and sensitive marker of acute coronary syndromes. Eur Heart J. 2007;28:941–948. doi: 10.1093/eurheartj/ehm078. [DOI] [PubMed] [Google Scholar]
- 29.Katashima T, Naruko T, Terasaki F, Fujita M, Otsuka K, Murakami S, Sato A, Hiroe M, Ikura Y, Ueda M, et al. Enhanced expression of the S100A8/A9 complex in acute myocardial infarction patients. Circ J. 2010;74:741–748. doi: [DOI] [PubMed] [Google Scholar]
- 30.Marinkovic G, Grauen Larsen H, Yndigegn T, Szabo IA, Mares RG, de Camp L, Weiland M, Tomas L, Goncalves I, Nilsson J, et al. Inhibition of pro-inflammatory myeloid cell responses by short-term S100A9 blockade improves cardiac function after myocardial infarction. Eur Heart J. 2019;40:2713–2723. doi: 10.1093/eurheartj/ehz461. [DOI] [PubMed] [Google Scholar]
- 31.Soehnlein O Neutrophil Research, Quo Vadis? Trends Immunol. 2019;40:561–564. doi: 10.1016/j.it.2019.04.011. [DOI] [PubMed] [Google Scholar]
- 32.Cohen M, Boiangiu C, Abidi M. Therapy for ST-segment elevation myocardial infarction patients who present late or are ineligible for reperfusion therapy. J Am Coll Cardiol. 2010;55:1895–1906. doi: 10.1016/j.jacc.2009.11.087. [DOI] [PubMed] [Google Scholar]
- 33.Gharacholou SM, Alexander KP, Chen AY, Wang TY, Melloni C, Gibler WB, Pollack CV Jr., Ohman EM, Peterson ED, Roe MT. Implications and reasons for the lack of use of reperfusion therapy in patients with ST-segment elevation myocardial infarction: findings from the CRUSADE initiative. Am Heart J. 2010;159:757–763. doi: 10.1016/j.ahj.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 34.Randall DA, Jorm LR, Lujic S, O’Loughlin AJ, Eades SJ, Leyland AH. Disparities in revascularization rates after acute myocardial infarction between aboriginal and non-aboriginal people in Australia. Circulation. 2013;127:811–819. doi: 10.1161/CIRCULATIONAHA.112.000566. [DOI] [PubMed] [Google Scholar]
- 35.Kotwal S, Ranasinghe I, Brieger D, Clayton P, Cass A, Gallagher M. Long-term Outcomes of Patients with Acute Myocardial Infarction Presenting to Regional and Remote Hospitals. Heart Lung Circ. 2016;25:124–131. doi: 10.1016/j.hlc.2015.07.019. [DOI] [PubMed] [Google Scholar]
- 36.Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, Weber C, Soehnlein O, Steffens S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J. 2017;38:187–197. doi: 10.1093/eurheartj/ehw002. [DOI] [PubMed] [Google Scholar]
- 37.Chatelain P, Latour JG, Tran D, de Lorgeril M, Dupras G, Bourassa M. Neutrophil accumulation in experimental myocardial infarcts: relation with extent of injury and effect of reperfusion. Circulation. 1987;75:1083–1090. doi: [DOI] [PubMed] [Google Scholar]
- 38.van Hout GP, Bosch L, Ellenbroek GH, de Haan JJ, van Solinge WW, Cooper MA, Arslan F, de Jager SC, Robertson AA, Pasterkamp G, et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur Heart J. 2017;38:828–836. doi: 10.1093/eurheartj/ehw247. [DOI] [PubMed] [Google Scholar]
- 39.Emsley HC, Smith CJ, Georgiou RF, Vail A, Hopkins SJ, Rothwell NJ, Tyrrell PJ, Acute Stroke I. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J Neurol Neurosurg Psychiatry. 2005;76:1366–1372. doi: 10.1136/jnnp.2004.054882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 41.O’Neill LA, Cooper MA. Canakinumab for Atherosclerotic Disease. N Engl J Med. 2018;378:198–199. doi: 10.1056/NEJMc1714635. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.