

Abstract
Infantile spasms (IS or epileptic spasms during infancy) were first described by Dr. William James West (aka West syndrome) in his own son in 1841. While rare by definition (occurring in 1 per 3200–3400 live births), IS represent a major social and treatment burden. The etiology of IS varies - there are many (>200) different known pathologies resulting in IS and still in about one third of cases there is no obvious reason. With the advancement of genetic analysis, role of certain genes (such as ARX or CDKL5 and others) in IS appears to be important. Current treatment strategies with incomplete efficacy and serious potential adverse effects include adrenocorticotropin (ACTH), corticosteroids (prednisone, prednisolone) and vigabatrin, more recently also a combination of hormones and vigabatrin. Second line treatments include pyridoxine (vitamin B6) and ketogenic diet. Additional treatment approaches use rapamycin, cannabidiol, valproic acid and other anti-seizure medications. Efficacy of these second line medications is variable but usually inferior to hormonal treatments and vigabatrin. Thus, new and effective models of this devastating condition are required for the search of additional treatment options as well as for better understanding the mechanisms of IS. Currently, eight models of IS are reviewed along with the ideas and mechanisms behind these models, drugs tested using the models and their efficacy and usefulness. Etiological variety of IS is somewhat reflected in the variety of the models. However, it seems that for finding precise personalized approaches, this variety is necessary as there is no “one-size-fits-all” approach possible for both IS in particular and epilepsy in general.
Keywords: Epileptic spasms, ACTH, vigabatrin, animal models, genetics
1. INTRODUCTION
Until recently, the term “infantile spasms” (IS) was interchangeably used with West syndrome (Janicot, et al., 2020). While the spasms (spasm-type seizures) are part of the West syndrome triad (see below), the adjective “infantile” fills in the additional distinguishing feature pointing to the developmental occurrence of these spasms. The latest classification of the International League Against Epilepsy (ILAE) changes the terminology to reflect the fact that the spasms, as a seizure type, may occur beyond infancy. Hence for this seizure type, the term “epileptic spasms” is now recommended (Berg, et al., 2010). ILAE also recommends the term “IS syndrome” for clustered spasms occurring during infancy (~1 year, rarely after 2 years of age) accompanied by EEG hypsarrhythmia (see below). West syndrome in this terminology represents a subset of the IS syndrome because it adds the progressive developmental arrest. A few rare forms of IS have been included: IS without hypsarrhythmia; hypsarrhythmia without IS, and single-spasms variant of IS, which lacks the clustering nature of the spasms (Kelley and Knupp, 2018; Pavone, et al., 2014).
West syndrome (the original construct) consists of the characteristic triad of IS, hypsarrhythmic EEG (hypsarrhythmia), and psychomotor regression. It is a rare syndrome, occurring in about 1 of 3200–3400 live births. There is slight predominance in boys vs. girls, approximately 1.6–1.4 to 1. The peak onset is around 6 months of age with a range between 3 months to 2 years. The spasm-type seizures (in order of frequency - flexion, extension or mixed) of proximal and truncal muscles lasting 1–2 s come in clusters ranging from a few to few hundreds of spasms per day (Pellock, et al., 2010), often appearing during awakening or feeding (Table 1). Diurnal expression of spasms seems to be a function of age: Younger patients (< 3 yrs of age) have epileptic spasms mostly between 9 am and noon and 3 pm through 6 pm, while older (>3 years of age) patients have epileptic spasms mostly between 6 am and 9 am (Ramgopal, et al., 2012). Hypsarrhythmia (from the Greek words “ὕψος” = hypsos = height and “αρρυθμία” = arrytmía) is a typical EEG pattern for IS consisting of irregular, asynchronized, large-amplitude waveforms in all EEG channels observed in between the spasms (i.e., interictally). The term was coined by the Gibbs’ in 1952 (Gibbs and Gibbs, 1952) and separately published in 1954 (Gibbs, et al., 1954). A single “r” version of “hypsarhythmia” still infrequently re-appears in the literature (Millichap and Millichap, 2015). The EEG during the ictal phase (i.e., when the spasms occur) is attenuated and described as “electrodecrement”. Finally, the severe psychomotor decline (Table 1) results in poor neurodevelopmental outcome. A recent meta-analysis (Widjaja, et al., 2015) has determined that the poor outcome of IS occurs irrespective of treatment or origin of spasms. This meta-analysis also pointed out the lack of standardized approaches to assess neurodevelopment in patients with IS. A more recent study consistent with this meta-analysis investigated predictors of poor outcome in childhood epilepsies and found that early-life epilepsies (such as IS) frequently striking prior to one year of age, carry a high risk of poor outcome, which can be further amplified by concurrent developmental delay (Berg, et al., 2019).
TABLE 1.
Features of the West Syndrome
| Typical Triad of the West Syndrome | |
| Motor expression | Spasms: flexion, extension, or mixed |
| General EEG | Hypsarrhythmia (interictal generalized irregular, large-amplitude, asynchronous waveforms. |
| Development | Psychomotor regress |
| Additional Distinguishing Features | |
| Age | 3–24 months (peaking at 6 months of age) |
| EEG during motor spasms | Electrodecrement (EEG attenuation) |
| Occurrence of spasms | Frequently in clusters, during awakening or feeding |
2. IMPORTANCE OF IS
One of the distinct features of West syndrome is the devastating effect of the IS leading to sudden developmental arrest or even regression in a previously healthy infant. The syndrome is named after a British physician, Dr. William James West, who first described the unusual features of the condition in his 1841 letter to Lancet, based on observations in his own son, James Edwin West (West, 1841). This 19th century paper was Dr. West’s cry for help not only as a physician but also as a parent. He attempted to turn to other physicians who would possibly read his report asking whether they encountered anything similar in their practices, how they managed this condition and most importantly, whether they were successful. This brief Letter to the Editor distills the pure essence of all future devastations experienced by the families who have had a child with IS. The helplessness of Dr. West highlights the extremely limited treatment options and poor prognosis of this syndrome (through current times). He consulted all available prominent physicians of his era, one of whom, Sir Charles Clarke had seen four cases (and coined the term “salaam convulsions”). A second physician, Dr. Locock, had seen two cases and Dr. Maunsell of Dublin had seen one.
The situation has not much improved over more than 150 years of the IS history. Even nowadays with current advances in diagnostic technology and therapy management, West syndrome has mostly poor prognosis (Widjaja, et al., 2015). Despite successful diagnosis and subsequent treatment, most patients with IS will develop other types of epilepsy later in life, many of them intractable (Jeavons, et al., 1973; Koo, et al., 1993). Developmental delays occur in > 75% of patients with IS and about 50% of patients with IS have cerebral palsy (Jeavons, et al., 1973; Koo, et al., 1993). Comorbidities include autism, learning (cognitive) impairments, anxiety, depression and hyperactivity (Iype, et al., 2016; Paciorkowski, et al., 2011a). Thus, IS represent an enormous burden on the patients and their families and further investigation into their etiology, mechanisms, and putative treatments is greatly warranted.
3. ETIOPATHOGENESIS OF IS
The latest classification differentiates IS into several etiopathogenetic groups: (a) structural-metabolic (Osborne, et al., 2010) including tuberous sclerosis and many inborn errors of metabolism, (b) infectious-immune (Suleiman, et al., 2011), (c) genetic (see below), and (d) unknown causes of IS (Berg, et al., 2010; Scheffer, et al., 2017).
Historically and thus, referred to in many prior studies, two major groups of IS were distinguished: symptomatic (two thirds of cases) and cryptogenic (one third of cases) IS. The symptomatic IS are secondary to an identifiable cause, while cryptogenic are those without detectable (i.e., yet unidentified) cause but with neurological deficits (symptom, sign or developmental delay) present (Pavone, et al., 2014).
According to the new classification, structural-metabolic and infectious-immune causes would encompass the original symptomatic group. Identifiable causes are represented by brain injury or malformation secondary to perinatal or neonatal ischemia, hypoxia, impact injury, brain inflammation, focal cortical dysplasia, intraventricular hemorrhage, metabolic diseases, dysmorphic syndromes, tumors or tubers (tuberous sclerosis) and identified genetic mutations (Karvelas, et al., 2009; Meencke and Gerhard, 1985; Osborne, et al., 2010; Riikonen and Donner, 1979). Hypothalamic hamartomas can also present with IS either together with gelastic seizures or as the only sign (Fox, et al., 2018; Kerrigan, et al., 2007). Table 2 shows the most common etiologies of structural-metabolic and infectious-immune IS. Approximately 200 different detrimental conditions have been associated with symptomatic IS (for review see Frost Jr. and Hrachovy, 2003).
TABLE 2. Examples of most common etiologies of structural-metabolic and infectious-immune groups of IS.
[modified from (Jellinger, 1987; Osborne, et al., 2010; Osborne, et al., 2019; Paciorkowski, et al., 2011a)]
| TIMING | Etiology | Example or explanation |
|---|---|---|
| PRENATAL | Chromosomal anomalies | 17p 13.3 microdeletion |
| 1p36 del | ||
| 22q | ||
| Del 1q36 Iptel | ||
| Down syndrome (21 trisomy) | ||
| Muscle eye brain disease | ||
| XXY | ||
| Non-chromosomal malformations | Agyria/polygyria | |
| Corpus callosum agenesis | ||
| Dandy Walker malformation | ||
| Focal cortical dysplasia | ||
| Gray matter heterotopia | ||
| Holoprosencephaly | ||
| Hydrocephalus | ||
| Lissencephaly | ||
| Microcephaly | ||
| Schizencephaly | ||
| Agyria/polygyria | ||
| Disease-specific malformations | Arachnoid cyst | |
| Cerebral artery stroke or disease | ||
| Hypoxic-ischemic encephalopathy | ||
| Hypomelanosis of Ito | ||
| Incontinentia pigmenti | ||
| Neurofibromatosis | ||
| Porencephaly | ||
| Tuberous sclerosis complex | ||
| PERINATAL | Birth trauma | Hypoxic-ischemic encephalopathy |
| Endocrine and metabolic diseases of the newborn | Hypoglycemia | |
| Infections | Cerebral tuberculomas | |
| Congenital Zika virus infection | ||
| Cytomegalovirus | ||
| Herpes | ||
| Meningitis | ||
| Toxoplasmosis | ||
| Intracranial non-traumatic hemorrage | ||
| Maternal factors | Drug abuse | |
| Periventricular leukomalacia or | ||
| periventricular hemorrhage due to pre-term injury Stroke | ||
| POSTNATAL | Brain neoplasm | Benign |
| Malignant | ||
| Cerebrovascular disease | Cerebral hemorrhage | |
| Cerebral infarct or stroke | ||
| Endocrine or metabolic | Amino acidurias | |
| Enzyme deficiencies (e.g., pyridoxine | ||
| dependency) | ||
| Hypoglycemia | ||
| Organic acidurias | ||
| Phenylketonuria | ||
| Mitochondrial disorders | ||
| External injury | Non-accidental | |
| Trauma | ||
| Nervous system impairment | Cerebral abscess | |
| Encephalitis | ||
| Meningitis | ||
| Porencephaly | ||
| Other | ||
| OTHER without timing | Arachnoid cyst | |
| Basal ganglia abnormalities | ||
| Calcifications | Postinfectious, post-bleeding | |
| Cerebral palsy | ||
| Cortical atrophy | ||
| Cortical and basal ganglia scars | Secondary to perinatal anoxia/hypoxia or vascular disorders, also to perinatal leukomalacia | |
| Hemimegaencephaly | ||
| Malformative | ||
| Micrencephaly |
Unknown causes of IS would include cases of previous “cryptogenic” IS group. It should be noted that historically, existence of the third group, idiopathic IS, was proposed. Those spasms developed without any obvious cause (including yet unidentified genetic etiologies) and without any sign, symptom or developmental delay before their onset. Nevertheless, there are causes for the spasms in this group (for example genetic) though many of them cannot be recognized with the techniques available today.
4. GENETICS
This etiologic group is constantly growing and deserves a special note since it opens the door for new treatments and contributes significantly to development of new animal models of IS. The genetic complexity of epileptic encephalopathies, which involve IS, is extremely high and in some cases there may not be enough samples for precise identification of underlying genetic issues (Takata, et al., 2019). Yet, it is essential to follow genetic pathways for the sake of development of precise treatments in the future.
Pavone in 1980 (Pavone, et al., 1980) reported simultaneous occurrence of IS in monozygotic twins within hours on the same day. Another case study described development of spasms in monozygotic twins within 5 days (Reiter, et al., 2000). Finally, a study involving three pairs of monozygotic twins supported anecdotal findings of almost simultaneous onset of spasms in these siblings (Coppola, et al., 2010). These outcomes indicate determining genetic factor as a cause in some cases of IS. Additional authors have also demonstrated familial occurrence of IS (Dulac, et al., 1993; Kato, et al., 2003; Kato, et al., 2007; Ronce, et al., 1999).
Among the first genes associated with West syndrome were ARX (Scheffer, et al., 2002) and CDKL5 (Weaving, et al., 2004). After the discovery of the first genetic mutations, an abundance of genes has been linked to IS phenotypes. For functional classification of many of those genes and respective references, see Table 3. However, there are likely many more genes associated with epileptic spasms during infancy (D’Alonzo, et al., 2018; Pavone, et al., 2014; Shbarou and Mikati, 2016; Wang, et al., 2019) and the pool of these genes increases constantly (Hengel, et al., 2020).
Table 3. Functional involvement of genes linked to IS phenotype.
(some genes may have multiple assignments)
Multiple chromosomal abnormalities may also lead to the phenotype of spasms. These include trisomy of 21 (Down’s syndrome), deletion (monosomy) 1p36 syndrome (Verrotti, et al., 2018), deletion 7q11.23 [Williams syndrome; (Morimoto, et al., 2003)], maternal duplication 15q11q13 [duplication 15q syndrome (Riikonen, et al., 2016)], tetrasomy 12p [Pallister–Killian syndrome (Candee, et al., 2012)] and deletion 17p13 [Miller–Dieker syndrome (Falsaperla, et al., 2018)] as well as the duplications or deletions of the distal segment of 16p11.2 region (Hino-Fukuyo, et al., 2015; Michaud, et al., 2014).
An interesting microdeletion associated with IS is 17q21.31 as it involves region of the receptor for corticotropin-releasing hormone 1 (CRHR1). This deletion may contribute to increased corticotropin-releasing hormone (CRH; ligand) expression and responds to treatment (possibly due to negative feedback) by ACTH (Wray, 2013). Yet not all 17q21.31 microdeletions are associated with IS (Bernardo, et al., 2016). This is further emphasized by a study, which did not find association between rare CRH1R variants and IS in a case-controlled design in the Chinese Han population. However, a relatively small sample size might have led to a false negative outcome (Yang, et al., 2015).
5. TREATMENTS IN HUMANS
5.1. Traditional treatments
5.1.1. ACTH
5.1.1.1. Discovery and overview of ACTH variants
In 1958 (~110 years after Dr. West’s article in Lancet) and by serendipity, Sorel and Dusaucy-Bauloye discovered and published in a French-speaking journal Acta Neurologica et Psychiatrica Belgica their observation of a strong effect of ACTH in 21 cases of Gibbs’ hypsarrhythmia used here in lieu of IS (Sorel and Dusaucy - Bauloye, 1958). This was the start of ACTH use as an IS therapy. However in the US, ACTH (ACTHAR® Gel) was not approved by the Food and Drug Administration (FDA) for treatment of IS till late 2010 so this use was compassionate for many years. Several forms of ACTH have been examined. In the US, the “natural” and full ACTH molecule (39 amino acids) of either bovine or porcine origin in gelatin is mostly used. In Europe and Japan, this formulation was not available. Therefore, a synthetic tetracosactide consisting of the first 24 ACTH amino acids was used (Sorel and Dusaucy - Bauloye, 1958). In Japan, if necessary, zinc salt of ACTH is utilized (Lux, et al., 2004). Tetracosactide (Cosyntropin) as well as its depot form (Synacthen Depot) became also available in the US, though the latter was made accessible for compassionate use only through a single specialty pharmacy in New York City.
Because of this geographic diversity, there never was a clinical study that would test full ACTH molecule (ACTHAR® Gel) head-to-head with tetracosactide or other short-molecule synthetic form of ACTH. So the efficacy of individual ACTH formulations can be only surmised from available clinical data (reviewed e.g. in (D’Alonzo, et al., 2018; Gettig, et al., 2009; Riikonen, 2014)).
5.1.1.2. Brief history of ACTH commercialization
For many years (since 1952), Acthar® Gel (a stabilized version of porcine ACTH in 19% gelatin) was available from its manufacturer (Sanofi Aventis) at a modest price. Eventually, the license was purchased by Questcor Pharma for mere $100,000 and the drug, although not FDA-approved for IS treatment was still widely accessible. In 2008, the board of directors of Questcor Pharma changed the business model overnight, raising the price of a single ACTHAR® gel vial from about $700 to $23,000. Soon after this pricing change, Questcor Pharma received FDA approval for this drug for treatment of IS (October 2010) as well as orphan drug protection [2010, the same formulation cannot enter the US market for additional 7 years (Doring, et al., 2016)]. Using current (i.e., $36,382 price per vial) prices, one treatment regimen for a patient with IS costs ~ $110,000-$145,000 (about 3~4 vials) and thus affecting treatment practices (Wray and Benke, 2010). The pricing changes stimulated other manufacturers in their efforts of either developing new drugs for IS or bringing formulations used to treat the IS elsewhere to the US market at much faster pace.
In 2013, Questcor Pharma procured both Synacthen and Synacthen Depot, the major functional Acthar® Gel competitors, from Novartis. And finally, in 2014 Questcor Pharma was acquired by an Irish Pharma company, Mallinckrodt Pharmaceuticals. In January 2017, this company eventually settled an antitrust lawsuit originally brought by Federal Trade Commission (FTC) against Questcor Pharma, by paying a settlement of $100 million. As part of the deal, the company had to sublicense Synacthen Depot (used in the US for IS, nephrotic syndrome and multiple sclerosis) to FTC-approved licensee (in July 2017 to West Therapeutic Development, LLC, a division of Slán Medicinal Holdings; Marathon Pharmaceuticals was originally approved for this sublicense in January 2017).
5.1.1.3. ACTH usage and adverse effects
Usage of ACTH for treatment of IS in the US was first standardized by the practice parameter published in 2004 (Mackay, et al., 2004). This comprehensive article summarized significant number of clinical studies using both high and low doses of ACTH delivered for 1–8 weeks. The parameter study recommended use of ACTH as effective short-term treatment of IS, but data were insufficient for recommending optimum dosage and duration of therapy. Yet, it appeared that short-term treatment (2 weeks) is sufficient and the effect is all or none.
The US treatment guidelines have been updated in 2010 (Go, et al., 2012) and tipped in favor of recommending a lower dose of ACTH for short treatment of IS. The final conclusions of these updated guidelines were using hormonal treatments (ACTH or prednisolone) as a first line treatment in infants with cryptogenic IS. For the first time, the recommendation of the short lag (lead) time to treatment is emphasized for a better long-term outcome. This recommendation got significantly more traction with additional and more recent studies addressing this issue (Hussain, et al., 2017a; Knupp, et al., 2016a; Knupp, et al., 2016b). It should be also mentioned that children with tuberous sclerosis who develop IS respond well to vigabatrin and thus, vigabatrin should be used as the first line treatment in those patients (Curatolo, et al., 2012).
The efficacy of ACTH in IS is not complete. Clinical studies repeatedly indicated that in long term follow-up (3 or 14 months after treatment), ACTH reached electroclinical resolution of IS (i.e., suppression of both spasms and hypsarrhythmia) in approximately 55–75% of cases (Knupp, et al., 2016a; Lux, et al., 2005; Mohamed, et al., 2011). If the first medication (here ACTH) fails, the recommendation is to use another standard IS medication with a different mechanism of action: Hormonal treatments are followed by vigabatrin and vigabatrin is followed by a hormonal treatment. This approach has approximately 55% success rate (Knupp, et al., 2016b). If hypsarrhythmia is diagnosed without IS (Lux and Osborne, 2004), it is still unclear whether these children would continue in developing IS in the future and if there would be poor developmental outcome if untreated (Kelley and Knupp, 2018). An observational study indicated that presence or absence of hypsarrhythmia does not appear to contribute to the short-term outcomes of IS (Demarest, et al., 2017). Thus, it appears that it is the resolution of spasms and especially positive psychomotor development that should govern treatment approach to IS (Iyer and Appleton, 2016).
ACTH has adverse effects that include irritability, brain atrophy with a risk of associated rupture of bridging veins and subdural hemorrhage, and hypertension (Hara, et al., 1981). Other adverse effects include obesity and electrolyte imbalance, which may even lead to renal failure (Riikonen and Donner, 1980). Chronic ACTH treatment is also associated with immune system impairments (Ohya, et al., 2009), which in some cases can result in sepsis and death (Mackay, et al., 2004; Partikian and Mitchell, 2007; Riikonen and Donner, 1980).
5.1.1.4. Landmark UKISS and ICISS reports
In 2004, a Lancet study focused on efficacy of ACTH (tetracosactide depot; 24 amino acid ACTH fragment) in a multicenter randomized controlled trial in the United Kingdom [United Kingdom Infantile Spasms Study; UKISS; (Lux, et al., 2004)]. This study determined that in infants (median age 5 months) with IS, hormonal treatments had better outcome at 2 weeks posttreatment than vigabatrin (ACTH 76%; prednisolone 70% and vigabatrin 54%; tuberous sclerosis cases responding well to vigabatrin were excluded here). Primary outcome in this study was the resolution of spasms with secondary outcomes of time to cessation of spasms and resolution of hypsarrhythmia. The cohort of infants was followed and outcome was assessed again at 12–14 months of age including Vineland adaptive behavior scales (VABS) (Lux, et al., 2005). At this age, absence of spasms was similar between hormonal (79%) and vigabatrin (77%) groups, however the VABS were higher in the group treated with hormones vs. vigabatrin indicating better neurodevelopmental outcome after hormones (in patients with no identified IS etiology = cryptogenic IS). Even longer follow up at age of 4 years confirmed higher VABS after hormonal treatments but only in those patients with cryptogenic IS (Darke, et al., 2010).
The UKISS reports provided background for the ICISS (International Collaborative Infantile Spasms Study) investigating safety and efficacy of hormonal treatment of IS versus hormonal treatment concurrent with vigabatrin. The primary outcome was suppression of spasms between days 14–42 after onset of treatment (O’Callaghan, et al., 2017). The second study reported primary outcome determined by VABS at 18 months of age and secondary outcome was presence or absence of spasms or seizures in the preceding 28 days and use of antiseizure treatment including ketogenic diet during that period (O’Callaghan, et al., 2018a). These ICISS primarily reported combination treatment of IS with hormones and vigabatrin and are more discussed in the section 5.1.5.1 below.
5.1.2. Corticosteroids
The idea of direct use of corticosteroids was developed based on the fact that the action of ACTH is mostly enhancement of adrenal corticosteroid release and production. Usually prednisone or prednisolone is given orally though intravenous pulse treatment is also an option (Yeh, et al., 2017). The opinions on the use of corticosteroids in IS differ. Older clinical studies comparing efficacy of ACTH and corticosteroids preferred ACTH (Baram, et al., 1996b; Snead, et al., 1983; Snead, et al., 1989) but recent studies did not observe a significant difference between both hormones (Gonzalez-Giraldo, et al., 2018; Gowda, et al., 2019; Kossoff, et al., 2009; Song, et al., 2017) or showed better efficacy of oral prednisolone compared to intramuscular synthetic ACTH (Wanigasinghe, et al., 2017).
Usual corticosteroid treatment consists of high dose for 2 weeks followed by slow tapering before complete withdrawal (Hani and Mikati, 2016; Knupp, et al., 2016a). A study investigating a very high dose of corticosteroids showed 63% response (cessation of spasms and EEG resolution of hypsarrhythmia). After switching to high dose of ACTH in non-responders, additional 40% responded positively (Hussain, et al., 2014). A small sample study investigated a high dose of intravenous methylprednisolone and found almost 65% efficacy (resolution of both spasms and EEG hypsarrhytmia) but the effect diminished in 45% of responders once the oral steroids were discontinued (Yeh, et al., 2017).
5.1.3. Possible mechanisms of action of hormonal treatments
Original studies expected that in IS, ACTH worked indirectly by activating the production and release of corticosteroids from the adrenal cortex because the administration of corticosteroids also showed efficacy (see above and (Go, et al., 2012)). Then, Baram and colleagues proposed that ACTH, as a member of the peptide family derived from pro-opiomelanocortin, might work centrally, providing brain region-specific activation of melanocortin (MC) 4 receptor subtype (Brunson, et al., 2001b). Accordingly, there are several reports of decreased levels of endogenous ACTH in CSF of patients with IS (Baram, et al., 1992b; Baram, et al., 1995; Nagamitsu, et al., 2001; Nalin, et al., 1985). In contrast, a different report showed increases in endogenous ACTH levels in CSF of patients with cryptogenic IS before ACTH treatment was initiated followed by a significant drop in those levels in responders to treatment (Heiskala, 1997). This decrease might have led to the normalization of the endogenos CRF levels. The idea is also consistent with findings that ACTH is effective in patients with IS (or other responsive seizures), who have ablation of adrenal production of corticosteroids (Farwell, et al., 1984; Willig and Lagenstein, 1982). While in some patients the ablation of adrenal activity was achieved by corticosteroid administration, it should be emphasized that the exogenous corticosteroids were unable to control the spasms. Only after adding ACTH, the response to therapy occured. However, furhter research on the mechanisms of anti-spasm action of ACTH has been severely affected by lack of appropriate animal models for IS.
The ACTH molecule is large (39 amino acids peptide or 24 amino acids for tetracosactide) and may not cross the blood brain barrier easily. However, there are few interesting ideas about how ACTH can reach the brain after systemic administration. First is the idea of pharmacokinetics - the larger concentration of ACTH in the periphery, the larger (despite small) penetration to the CNS. This idea supports better outcome following treatment with a large ACTH dose proposed earlier. The second idea is that seizures and spasms may contribute to further weakening of the blood brain barrier allowing thus for enhanced central ACTH delivery after peripheral administration. The last idea suggests that ACTH while in circulation, works in the circumventricular organs with weakened or nonexistent blood brain barrier and especially areas enriched with MC receptors (main ACTH effectors) such as hypothalamic arcuate nucleus and brainstem solitary tract nucleus (Stafstrom, et al., 2011).
Blood brain barrier is not an obstacle for corticosteroids as those are cholesterol derivatives, hydrophobic and thus penetrate biological barriers readily. Thus delivery to the brain is direct. Assuming that either hormonal treatment reaches brain, the question is, what is it doing once there? One attractive hypothesis (Baram, 1993; Baram and Schultz, 1991; Baram, et al., 1996a; Brunson, et al., 2002; Korosi and Baram, 2008) suggests that both ACTH and corticosteroids provide negative CNS feedback for decreased release of the proconvulsant molecule, CRH, thus contributing to amelioration of spasms.
5.1.4. Vigabatrin (gamma vinyl-GABA, GVG)
In 2009 vigabatrin (Sabril®), manufactured by Lundbeck, was approved by the U.S. FDA to control seizures in patients with IS. Although vigabatrin was available in other countries before, the approval in the US was delayed because of the adverse effects. The most concerning adverse effect associated with vigabatrin use is the risk of retinal toxicity presenting as permanent concentric visual field constriction (Riikonen, 2004, 2015; Riikonen, et al., 2015; Vanhatalo, et al., 1999; Vanhatalo, et al., 2002). This defect has been reported in about 30% of adult patients treated with vigabatrin and in about 19% in children (Vanhatalo, et al., 2002). Further, several MRI studies showed that vigabatrin, especially in high doses and after prolonged administration, leads to occurrence of T2-weighted hyperintensities in corpus callosum, thalamus, pallidum and brainstem. However, these MRI changes seem to be reversible after treatment cessation (Dracopoulos, et al., 2010; Hussain, et al., 2017b; Pearl, et al., 2009). These MRI changes have been recently confirmed histopathologically as a diffuse white matter spongiosis in forebrain and brainstem white matter in a vigabatrin-treated patient after unexpected sudden death in epilepsy; SUDEP (Pearl, et al., 2018).
Vigabatrin as its mechanism of action increases availability of GABA by irreversible inhibition of GABA-transaminase, the enzyme responsible for degragation of GABA. This leads to increased extracellular concentration of GABA and thus, enhanced inhibition. Vigabatrin is recommended as a first line drug to control spasms in patients with symptomatic IS, especially those with tuberous sclerosis (Camposano, et al., 2008; Hussain, et al., 2018; van der Poest Clement, et al., 2018; Vigevano and Cilio, 1997). Some authors advocate using a minimal effective dose to decrease the risk of serious adverse effects (Ounissi, et al., 2019) but others found that high doses of vigabatrin decreased the risk of relapse of the spasms (Hussain, et al., 2018). Despite the high efficiency in control of spasms, vigabatrin seems to have questionable value in long-term prevention of cognitive impairment (Hancock, et al., 2013; Lux, et al., 2005; Yum, et al., 2013). In patients with cryptogenic IS or prematurely born children with perinatal hypoxic/ischemic injury, the spasms better respond to the hormonal treatment (especially ACTH) than to vigabatrin (Hancock, et al., 2013; Lux, et al., 2005; Vigevano and Cilio, 1997). In addition, patients, especially younger infants with cryptogenic IS, are more sensitive to develop vigabatrin-induced MRI abnormalities in the brainstem and in the forebrain deep nuclei (Dracopoulos, et al., 2010).
5.1.5. Combination treatments
The idea of combining treatments in patients with IS arises from original UKISS reports (Darke, et al., 2010; Lux, et al., 2004, 2005). In these and later studies (Knupp, et al., 2016b), if the initial treatment was ineffective, the patients were immediately transferred to the secondary treatment (ACTH to vigabatrin or vigabatrin to ACTH). In many cases, there was an instant response. Additionally, there was a previous ACTH and vigabatrin combination study in a small cohort of patients with IS with underlying cerebral palsy. Combination treatment in this study led to quick resolution of spasms in all 9 children with 18 months relapse in only one patient (Zafeiriou, et al., 1996). Hence the hypothesis of combining both treatments from the beginning of the treatment was tested in a prospective randomized trial.
5.1.5.1. Hormones (ACTH and/or corticosteroids) plus vigabatrin
In an attempt to improve the efficacy of control of spasms and cognitive outcome, a recent multinational randomized study (ICISS) used combination of vigabatrin and hormonal (ACTH or corticosteroids) treatment and compared to hormonal monotherapy (O’Callaghan, et al., 2017). The study found a better control of spasms with the combination therapy compared to hormonal monotherapy initially but the combination therapy did not prevent relapses or improve the long-term cognitive outcome (O’Callaghan, et al., 2017; O’Callaghan, et al., 2018b). On the other hand, the study confirmed previous observations that early diagnosis with a prompt initiation of treatment and good initial response to treatment has more favorable long-term prognosis (Hussain, et al., 2017b; O’Callaghan, et al., 2017; O’Callaghan, et al., 2018b). Another study, though not using a true combination of treatments, demonstrated that addition of prednisolone in non-responders to vigabatrin showed combined effect in 48 out of 66 patients compared to 22 vigabatrin only responders (Ko, et al., 2018a). In this study, the primary outcome was the resolution of spasms plus BASED [Burden of Amplitudes and Epileptiform Discharges (Mytinger, et al., 2015)] score equal to or less than 2, i.e. suppression of both spasms and EEG events.
5.2. Second line treatments
5.2.1. Vitamin B6 (pyridoxine)
In Japan, pyridoxine (vitamin B6) in high doses is by far the most commonly used drug for initial treatment of both cryptogenic and symptomatic IS because of the fear of adverse effects associated with ACTH or vigabatrin use (see above). Valproate is the second line of treatment, and hormones, specifically ACTH, represent the third line of treatment (Hamano, et al., 2018).
Some studies found that certain types of IS are responsive to vitamin B6 treatment, specifically those with ALDH7A1 gene mutations (Stockler, et al., 2011), yet their occurrence in Western world is very low. Mutations of the PNPO gene result in pyridoxamine 5’-phosphate oxidase malfunction leading to a deficit of pyridoxal-5-phosphate associated with IS. This deficit is responsive to pyridoxal-5-phosphate treatment or sometimes even to B6 treatment (Stockler, et al., 2011). Yet, per the 2012 evidence-based guidelines, there is insufficient evidence to determine whether B6 is effective in treatment of IS (Go, et al., 2012) despite vitamin B6 being historically proposed for IS treatment in high doses (Blennow and Starck, 1986). Interestingly, high doses of vitamin B6 are both neurotoxic in humans (Levine and Saltzman, 2004; Schaumburg, et al., 1983) and can directly induce seizures [at least in rodents, both adult (Ebadi, et al., 1983) and immature (Verešová, et al., 1998)]. Thus, B6 therapy outside Japan is typically reserved for those clearly indicated cases of IS mentioned above.
5.2.2. Ketogenic diet
The ketogenic diet (KD) has usually been used as the last resort non-pharmacological treatment option to control seizures in patients with intractable epilepsy syndromes, when pharmacologic treatment fails (Kossoff, et al., 2018). The main idea is intake of high-fat and low-carbohydrate diet to induce production of ketone bodies, which are then used by neurons as the main source of energy instead of glucose. It is not completely clear how the ketone bodies mediate the antiseizure effects but evidence from animal models mainly emphasizes changes in neurotransmitter systems, namely GABA, glutamate, and adenosine, with the final result of increased neuronal inhibition. Current advances in our understanding of the multimodal inhibitory effects of ketone bodies on neuronal excitability and neuroprotection have been reviewed recently (Fedorovich, et al., 2018; Simeone, et al., 2018).
In clinical settings, four types of KD are used. Two types of the classic KD with either the long-chain or medium-chain triglycerides are used as a major source of energy. Two additional types use the modified KD; the modified Atkins diet and low glycemic index treatment that are not so strict and thus easier to follow. In infants with IS, the initial seizure control is achieved by special ketogenic formulas, which can be tailored for the child’s needs and strictly follow the classic KD fat-to-carbohydrate ratio of 4:1 or 3:1 (Kossoff, et al., 2002a; Nordli, et al., 2001). The modified KDs are recommended for long-term use in older children and adults (Kossoff, et al., 2002b). KD is usually well tolerated by most IS patients and the adverse effects that have not been considered severe include mainly gastrointestinal problems (diarrhea, constipation, nausea), and in some patients, behavioral problems, hematuria, kidney stones, dry skin or acidosis (Hong, et al., 2010; Hussain, et al., 2016; Kayyali, et al., 2014; Kossoff, et al., 2018). Contraindications of KD include patients with certain fat metabolism and mitochondrial disorders (Kossoff, et al., 2018).
Several retrospective as well as prospective studies found promising data on the use of KD as the last resort treatment to control spasms in children with severe IS with over 50% of patients experiencing seizure reduction or even cessation of seizures within 3 – 6 months (Eun, et al., 2006; Hirano, et al., 2015; Kayyali, et al., 2014; Kossoff, et al., 2002a; Nordli, et al., 2001; Pires, et al., 2013; Than, et al., 2005; Yum, et al., 2008). EEG ictal activity including hypsarrhythmia, although not always monitored, also improved in those patients with spasms cessation [reviewed in (Prezioso, et al., 2018)]. Most importantly, a large prospective study at Johns Hopkins Hospital involving over 100 IS patients with different etiologies also included 18 patients started on the KD as first line therapy (Hong, et al., 2010). It is noteworthy that 10 of the 18 patients became spasms free within two weeks of KD initiation and their EEG normalized within two months. These patients stayed seizure free for 6 months of the KD treatment and did not relapse after KD discontinuation (Hong, et al., 2010). The study concluded that important predictors of response to KD are early diagnosis of IS and fewer anticonvulsant drugs used prior to onset of KD. This supports the findings of a recent study that in the most severe cases of intractable IS, the KD is not efficacious (Hussain, et al., 2016). An additional randomized controlled trial/parallel cohort investigated 101 children with West syndrome and compared efficacy of KD versus high dose of synthetic ACTH (150 U/m2) with further stratification by children pretreated with vigabatrin (Dressler, et al., 2019). Interestingly, there was a similar outcome for KD and ACTH in terms of electroclinical remission (primary endpoint: cessation of spasms + hypsarrhythmia) at day 28, if the children were pretreated with vigabatrin. Otherwise ACTH by itself was more effective than the KD per se but also brought significantly more frequent adverse effects (94% versus 30% with KD) requiring acute intervention (Dressler, et al., 2019). The most recent recommendations of the International KD Study Group have been published recently (Kossoff, et al., 2018).
5.3. Emerging treatments
5.3.1. Sirolimus and everolimus
Sirolimus (rapamycin) or its analogue everolimus act as mTOR inhibitors and are commonly used as an immunosuppressant in patients with kidney transplants to prevent organ rejection and as an add-on therapy in malignancies. The importance for IS rests in mTORC1 being downstream in the pathway from TSC1 and TSC2, genes responsible for tuberous sclerosis, which commonly presents with spasms during the first year of life. TSC1 or TSC2 mutations relieve suppressive effects of these proteins on mTOR leading to mTOR hyperactivation. Hence, a possible role for mTOR inhibitors in treatment of IS in the tuberous sclerosis is by downregulation of mTORC1 (Meikle, et al., 2008). In children with tuberous sclerosis complex, early screening may provide a window of opportunity for effective treatment of IS (French, et al., 2016; Gipson, et al., 2014). Most studies present successful use of mTOR inhibitors for seizure control in older patients with tuberous sclerosis with other types of seizures (focal or tonic-clonic) rather than with IS (Krueger, et al., 2013; Moavero, et al., 2016; Muncy, et al., 2009). A recent prospective observational study with everolimus as an add-on treatment in infants with IS not responding to vigabatrin showed good tolerability even during infancy (as early as at 8 months of age) and improvement in seizures and behavior in three out of four infants (Samueli, et al., 2018). Surprisingly, a recent study suggested that it is rather inhibition of mTORC2 (rather than mTORC1) that has antiseizure effects (Chen, et al., 2019).
5.3.2. Valproic acid
Valproic acid (or sodium valproate) is the second line of treatment of IS in Japan (after B6) though it is frequently followed by either hormonal therapy or vigabatrin (Hamano, et al., 2018). However, there is insufficient evidence to determine whether valproic acid is effective for treatment of IS (Go, et al., 2012).
5.3.3. Cannabidiol
There is increasing interest in the use of cannabidiol or cannabidiol enriched cannabis extracts as an add-on therapy for intractable epilepsy, including IS. There are some anecdotal reports of significant improvements after such therapy (Hussain, et al., 2015; Porter and Jacobson, 2013). A small rigorous study (9 subjects with IS; clinicaltrials.gov NCT02551731) did not show statistically significant effects of purified cannabidiol; only 1 participant responded with cessation of spasms but relapsed quickly (Hussain, 2018). A larger (estimated 190 participants), phase 3 randomized, double-blind, placebo-controlled, parallel-group study of cannabidiol oral solution as adjunctive therapy with vigabatrin in patients with IS is currently underway (clinicaltrials.gov NCT03421496).
5.3.4. Clobazam
A recent study investigated clobazam, a 1,5-benzodiazepine, administration after several failed medications (average 2.6) for treatment of IS (Hahn, et al., 2019). While effective in 22% of their 171 patient cohort, there were also serious adverse effects in almost 60% of the cohort, such as severe drowsiness, hallucinations, confusion, shallow breathing or interaction with other drugs.
5.3.5. Other and anti-seizure medications
Standard anti-seizure medications are used for treatment attempts in IS including zonisamide, topiramate, levetiracetam, lamotrigine, felbamate, ganaxolone, intravenous immunoglobulin (IVIG), thyrotropin releasing hormone and others (Tang-Wai, et al., 2017), reviewed in (Iyer and Appleton, 2016; Riikonen, 2014). For none of these compounds does sufficient evidence exist to determine efficacy in IS (Go, et al., 2012). Many studies published on the use of these drugs are retrospective, they have not used placebo controls and there is no standardized assessment of the outcome (electroclinical resolution of spasms and hypsarrhythmia) (Hussain, 2018). A very recent study (Auvin, et al., 2020) investigated a noncompetitive NMDA receptor (NR2B subunit-specific) antagonist radiprodil as some patients with IS may have gain of function mutation in the GRIN2B gene encoding this subunit (Lemke, et al., 2016; Platzer, et al., 2017). Out of three infants, radiprodil completely suppressed spasms in one and two showed clinical improvement without reaching freedom from spasms (Auvin, et al., 2020).
5.3.6. Transcranial magnetic stimulation (TMS)
Two weeks of TMS treatment (40 mins per day) has been used in seven patients with refractory epileptic spasms out of which five started in infancy (Yang, et al., 2019). All these five patients responded to transcranial magnetic stimulation with a decreased number of seizures (>50%) and were considered as responders at the end of the 28 day follow up period.
5.4. Additional treatment considerations
5.4.1. Early diagnosis and treatment onset
Initial observations suggested that early diagnosis of IS together with promptly initiated treatment may significantly improve the overall prognosis (Kivity, et al., 2004; Riikonen, 1982). Several rigorous studies confirmed that early diagnosis and good initial response to treatment are associated with more favorable long-term outcomes (Hussain, et al., 2017a; O’Callaghan, et al., 2011; O’Callaghan, et al., 2017; O’Callaghan, et al., 2018a). This was also a conclusion of a thorough meta-analysis (Widjaja, et al., 2015). One of the limitations in prompt diagnosis of IS is the difficulty to recognize accurately hypsarrhythmia on the EEG. Recently, new computerized EEG techniques are being explored to help in recognizing hypsarrhythmia patterns but none has reached clinical utility yet (Smith, et al., 2018; Smith, et al., 2017). One of these studies focused on patients with epileptic spasms developed on the tuberous sclerosis background and found that there is increased EEG connectivity over all frequency bands. This excess connectivity may reflect pathologic network synchronization resulting in generalized epileptic spasms (Davis, et al., 2019).
5.4.2. Markers of treatment efficacy
Currently, treatment efficacy of IS is evaluated on day 14 of treatment (up to 66 days) by assessing both occurrence of spasms and presence of hypsarrhythmia on the EEG [i.e., electroclinical resolution (Go, et al., 2012)]. Both spasms and hypsarrhythmia should disappear with a successful treatment (Hani and Mikati, 2016; Knupp, et al., 2016b). Some studies determine the effect of treatment based only on the presence or absence of spasms because of the inaccurate hypsarrhythmia determination and in a short-term outcome (Lagae, et al., 2010; Lee, et al., 2013; Smith, et al., 2018; Smith, et al., 2017).
For long-term outcome (commonly at 2–4 years after treatment), usually the presence of epilepsy is investigated (Guveli, et al., 2015; Lagae, et al., 2010). Behavioral evaluation of children with IS should also be included to determine accurately and in a standardized way possible developmental delays using VABS or similar (O’Callaghan, et al., 2018a). While there is a better long-term neurobehavioral outcome in cryptogenic IS [especially if treated with ACTH or corticosteroids (Go, et al., 2012)] compared to symptomatic IS, the outcome in general remains poor (Widjaja, et al., 2015).
5.4.3. Precision and personalized treatments
Without extensive repetition of the Genetic subheading above, genetic testing AND understanding the role of individual genes (or rather their products) may significantly help in tailoring treatments to the issues suffered by individual patients. As an example, the spasms may be dependent on pyridoxine or pyridoxal-phosphate treatments (defects in ALDH7A1 or and PNPO genes, respectively). The treatments counter the gene defect and lead to resolution of spasms. The expectation is that with more detailed gene screening panels we will be (or already are) able to detect many other gene mutations linked to IS. Unfortunately our exact understanding of different gene function or lack of appropriate intervention still limits the treatment possibilities. A swift approach should be exercised - with respect to known metabolic defects and available treatments. If the gene defect is unrecognized and untreated for a protracted period, it may lead to encephalopathy (with spasms or other seizure types). After this pathology is established, there is unlikely to be significant resolution even with aggressive and correct treatments.
5.5. Conclusions
Several partially effective treatments of IS are available, such as hormones (ACTH and corticosteroids) and vigabatrin. Some evidence supports that these should be rather used concurrently rather than in isolation. There are few add-on treatments such as vitamin B6 as well as some newer anti-seizure medications that may be helpful but solid evidence is still lacking. In refractory cases, KD and additional treatment means (cannabidiol) may become useful. Genetic testing will give more information but without functional understanding and means to affect the altered gene function in the correct direction (loss of function vs. gain of function gene mutations).
Thus novel treatments are desperately needed. These treatments may be brand new as well as those repurposed from other areas of medicine. However, before these treatments enter the stage of clinical trials they should be rigorously investigated in multiple models of IS. These models and their values constitute the following part.
6. ANIMAL MODELS
“The best material model of a cat is another, or preferably the same, cat.”
Norbert Wiener (1894–1964)
Or, as boldly expressed in a classic movie Some Like It Hot: “Nobody is perfect”. With this notion in mind, we should approach all existing (and future) animal models of IS. Currently, there are several models and they may be differentiated based on various points of view (acute vs. chronic; symptomatic vs. cryptogenic, responsive vs. refractory to ACTH, genetic based vs. acquired, etc.). These viewpoints however, have an actual importance for testing novel treatments and for aligning the models as well as drug efficacy with human condition. The ideal criteria of a model have been set in 2002 and reviewed again on a symposium in 2006 (Stafstrom and Holmes, 2002; Stafstrom, et al., 2006). Still, the first sentence of the paragraph is valid: Even direct genetic matches may not result in identical phenotypes between humans and rodents, e.g., Tsc1 mouse knockouts lack cortical tubers (Wang, et al., 2007) despite having age-appropriate electrographic seizures (Gataullina, et al., 2016) dependent on the cortical excess of NMDA receptor subunit GluN2C=NR2C (Lozovaya, et al., 2014). Interestingly, the mice with conditional deletion of Tsc1 gene in astrocytes and neurons (Tsc1GFAPCKO) have no spontaneous spasms despite having spontaneous focal seizures and reduced threshold and increased severity of NMDA-induced spasms during neonatal period (see 6.1 and 6.2.1 below) (Rensing, et al., 2020). One of the first reviews of emerging models of IS suggests that the primary goal of the model should be discovery and utilization of the underlying pathophysiological process (Marsh and Golden, 2009). While such effort is commendable, it may not work in the model system. The work on the models of neuropsychiatric diseases (Belzung and Lemoine, 2011; Nestler and Hyman, 2010) suggests three major benchmarks for animal models: (1) Face validity demonstrated by homology of the behavioral phenotypes. (2) Construct validity, which is mimicking the theoretical neurobiological underpinnings of the disease (i.e., pathophysiological process). (3) Predictive validity, which is arguably the most imperative and demonstrates an animal model’s expected therapeutic response to current therapies and ability to screen for novel treatments allowing the comparison with the old treatment efficacy.
In the realm of animal models of IS, the extensive clinical and experimental work of Dr. Tallie Baram is acknowledged. Her contributions have markedly increased understanding of the role of endogenous CRH as a proconvulsant molecule with a developmental twist (Baram and Schultz, 1991). Her findings even resulted in a clinical trial of a CRH antagonist (Baram and Schultz, 1991) as well as in recognition of the role of endogenous ACTH in IS (Baram, et al., 1992b; Baram, et al., 1995).
6.1. Previous models
There was a dedicated search for animal models of IS in the 80’s and 90’s as summarized in 2002 (Stafstrom and Holmes, 2002). The initial search for an animal model relevant to IS was based on a response to ACTH probing established seizure models. Holmes and Weber (Holmes and Weber, 1986) found that ACTH pretreatment delayed amygdala kindling in developing rats in a dose-dependent manner. However, once kindled, the rats were resistant to ACTH effects (Thompson and Holmes, 1987). While this may sound promising in showing a seizure model responsive to ACTH, there are several reservations in considering developmental kindling to mimic the specific features of IS, i.e., the seizure phenotype dissimilarity to spasms, identified seizure focus in the amygdala, as well as dose-response rather than all-or-none effect of ACTH.
The superior (to any other treatment) efficacy of ACTH resonated in the idea to connect IS to the stress system (Baram, 1993). The role of ACTH as a feedback molecule for the CRH initiated investigation of proconvulsant activity of CRH as a possible culprit behind IS (Brunson, et al., 2001a). Recent findings of upregulated CRH and CRH receptor type 1 in surgically removed epileptiform brain tissue samples of children with IS support this notion (Yang, et al., 2017). Indeed, CRH was known to be a proconvulsant peptide in adult brain (Ehlers, et al., 1983). In developing brain, convulsant potency of this peptide increases by thousand fold, i.e., CRH is an effective convulsant within picomolar range of concentrations (Baram and Schultz, 1991). However, CRH-induced seizures do not present as spasms, they start focally in the limbic system, namely in the amygdala and spread to the hippocampus (Baram, et al., 1992a). Further, these seizures provoked by exogenous CRH do not respond to ACTH treatment, very likely because the feedback mechanism here is impaired (Baram and Schultz, 1995).
Our group has also contributed to the early search for IS model. In 1992 (Mareš and Velíšek, 1992), we have shown that intraperitoneal N-methyl-D-aspartic acid (NMDA) administration in 7–18 day old rats induces age-specific behavioral seizures resembling spasms [emprosthotonus, see (West, 1841)]. NMDA-induced spasms only rarely occur beyond postnatal day 21 and are accompanied by EEG suppression similar to electrodecrement (Kábová, et al., 1999; Mareš and Velíšek, 1992). Interictal EEG shows chaotic waves, a possible rat variant of hypsarrhythmia. There were also transient and long-term behavioral and learning deficits (Kábová, et al., 1999; Stafstrom and Sasaki-Adams, 2003). However, corticosteroids were ineffective in this model and the only non-specific efficacy was found after clonazepam pretreatment (Kábová, et al., 1999; Velíšek and Mareš, 1995). Rapamycin also did not suppress NMDA-induced spasm (Chachua, et al., 2012).
Finally, an additional study investigated ACTH efficacy in multiple seizure models in 15 day old (infant) rats. Specifically, clonic and tonic-clonic pentylenetetrazole-induced seizures, maximal electroshock-induced seizures as well as hippocampal kindling-induced seizures were tested. ACTH was not effective in any of those seizure models (Edwards, et al., 2002).
The models above were probing individual features of IS: ACTH efficacy in kindling, ACTH feedback effect on CRH and semiologic/EEG similarity. However, they lacked several additional features that the minimal model of IS should have (Stafstrom and Holmes, 2002). Thus despite a long exploration period, a satisfactory model was still lacking.
6.2. Current animal models
6.2.1. Prenatal betamethasone-postnatal NMDA model
Ideas:
In 2007 we published an expanded version of our previous NMDA model of IS (Velíšek, et al., 2007). Our main hypothesis was built on the ideas of Dr. Baram and others mentioned above: (1) Efficacy of ACTH as well as low endogenous ACTH in patients with IS indicates alteration of the hypothalamo-pituitary-adrenal (HPA) axis. (2) IS develop in infancy (as early as 3 months of age) so the HPA is impaired either perinatally or even prenatally. In search of such impairment, we found studies showing that in guinea pigs, prenatal exposure to corticosteroids during a distinct developmental period alters HPA regulation (Kapoor and Matthews, 2005; Kapoor, et al., 2006). A similar effect can be accomplished by prenatal stress [reviewed in (Kapoor, et al., 2006)]. This situation is similar to human condition of HPA impairment in patients with IS (Baram, et al., 1995; Nagamitsu, et al., 2001; Nalin, et al., 1985). Therefore, we used prenatal exposure to synthetic corticosteroid betamethasone at the end of the second trimester of rat pregnancy, specifically on gestational day 15 (Figure 1). Exposed offspring was then injected with NMDA in a similar way as in our original study (Mareš and Velíšek, 1992).
Figure 1 – Time line of the two-hit (prenatal betamethasone & postnatal NMDA) model of IS.

Priming: Pregnant Sprague-Dawley rats are injected IP with two doses of betamethasone (0.4 mg/kg of phosphate salt in saline) on gestational day 15 (G15) at 08:00 and 18:00 hours. Controls (if needed) receive equivalent volume (1 ml/kg) normal saline IP. Offspring are consistently delivered at G23. Note that different strains of rats may have different durations of pregnancy and thus, the timing of prenatal priming needs to be adjusted [see (Kábová, et al., 2000) for comparison]. Prolonged stress delivered to pregnant dams with similar timing as betamethasone on G15 has comparable priming effects (two episodes of 45 min restraint stress each) as injected betamethasone. Day of delivery (G23) in these experiments is considered as postnatal day 0 (P0).
Postnatal induction of spasms: Primed offspring are used for induction of spasms between P10-P15. Spasms are triggered by graded doses of N-methyl-D-aspartic acid (NMDA) dissolved in normal saline. For the randomized studies, spasms are triggered on P12 (7.5 mg/kg IP), P13 (12 mg/kg IP) and P15 (15 mg/kg IP) (Chern, et al., 2019). For drug treatment studies, the treatment drug vs. vehicle is started after the first bout of spasms at P12 diminishes to simulate the human condition of treatment initiation after the IS occur. Spasms can be reliably induced before P12, in primed model we used also P10 rats (Chachua, et al., 2011), previously in non-primed rats also P7 rats (Mareš and Velíšek, 1992). Note that the phenotype of NMDA-triggered spasms is fading between P25-P30 and the pattern of NMDA-induced seizures changes to the “adult” type with tonic-clonic seizures predominating (Mareš and Velíšek, 1992) so the spasms are age-specific, similar to human condition.
Success:
Our approach was successful since the spasms in prenatally betamethasone-exposed pups had significantly faster onset and occurred in larger numbers with significant clustering compared to prenatally saline-exposed offspring. The spasms are brief hyperflexions [emprosthotonus; (Mareš and Velíšek, 1992; West, 1841)] of the entire body and are preceded by additional signs (tail twisting) that confirm correct NMDA administration. However, the most important effect of prenatal betamethasone exposure was the presence of sensitivity to ACTH treatment (Velíšek, et al., 2007). We further characterized and validated the model and determined that exposure to prenatal stress also enhanced developmental spasms and carried ACTH sensitivity (Chachua, et al., 2011; Chachua, et al., 2016; Iacobas, et al., 2018; Velíšek, et al., 2010; Yum, et al., 2012). The model was independently reproduced and is used by several independent groups world-wide (Tsuji, et al., 2016; Shi, et al., 2015; Shi, et al., 2016; Baek, et al., 2016; Kim, et al., 2017; Kwon, et al., 2018). EEG during spasms is suppressed (electrodecrement), in between spasms asynchronous waves with large amplitude prevail (rat hypsarrhythmia variant; (Yum, et al., 2012)). With time, we further improved the model. Currently, we use multiple bouts of NMDA spasms in prenatally betamethasone-exposed rats: On P12, all rats go through the first bout of spasms and then are randomized (distributed) to treatment and control groups. The treatment is started after cessation of spasms on P12 and continues through P14. Additional bouts of spasms are induced on P13 and P15. Thus, we can determine both early effects of treatment on P13 spasms as well as response build up after three days of treatment (P12-P14) on P15 spasms. Finally, for each individual rat, we determine the number of spasms before randomization and after completed treatment to evaluate individual improvement (Chern, et al., 2019).
Drug effects:
The advantage of this model is that the spasms always occur and they do develop in sufficient numbers to assess the response to treatment. Thus, besides ACTH, we found that also vigabatrin and methylprednisolone are effective. In these experiments ACTH and methylprednisolone were administered after the initial induction of spasms, vigabatrin was given 24 hours prior to trigger of spasms (Chachua, et al., 2011; Chachua, et al., 2016). The spasms are also sensitive to a neurosteroid ganaxolone (Yum, et al., 2014) as well as to one of the ketogenic substances, β-hydroxybutyrate (Yum, et al., 2015) mirroring efficacy of ketogenic diet in some patients with IS (Hong, et al., 2010; Prezioso, et al., 2018). Treatment with PMX53, a potent complement C5ar1 antagonist, was also effective (Iacobas et al., 2018). In a study using a novel fusion peptide (consisting of ACTH1–24, a three amino-acid linker and a modified α-MSH molecule) called AQB-565 (Aequus Biopharma), we found almost equipotent effects to ACTH in our model (Chern, et al., 2019). Our most recent study compared efficacy of Acthar® Gel and porcine synthetic ACTH in methylcellulose (ACTON PROLONGATUM®) and found that both drugs are equipotently effective in the model (Chern, et al., 2020). Another recent study indicated that calpain-2 might become a treatment target for IS as a calpain inhibitor decreased frequency of spasms in the model (Kwon, et al., 2019). On the other hand, neonatal estradiol, a treatment increasing number of GABAergic neurons effective in the genetic Arx triplet expansion model of IS (see below) was ineffective in our model, likely because there is no impairment in GABAergic neuronal numbers (Chachua, et al., 2016). In addition, caution needs to be exercised with neonatal estrogen as it may cause irreversible male infertility (Minabe, et al., 2019).
Mechanisms:
Prenatal exposure to corticosteroids alters expression of various genes (and related proteins). The most obvious are the genes and proteins of the HPA axis [glucocorticoid and mineralocorticoid receptors; reviewed in (Hamada and Matthews, 2018; Harris and Seckl, 2011; McGowan and Matthews, 2018); Figure 2 in rats] including increased levels of CRF (Benson et al., 2020). The major transmitter systems (GABA and glutamate) are also impaired and biomarker of successful treatment of spasms be based the (partial) reversal of these impairments (Iacobas, et al., 2013; Iacobas, et al., 2018). Interestingly, similar but not identical gene and protein alterations are observed after prenatal stress exposure (Son, et al., 2006; Yaka, et al., 2007) also reviewed in (Weinstock, 2011, 2017). In support of our findings, a clinical study determined that intense prenatal maternal stress was associated with increased occurrence of IS in affected infants (Shang, et al., 2010). While this was a self-reported retrospective study, there are two important aspects that need to be considered. First, the study stratified by levels of perceived gestational stress in mothers and showed that only high levels of stress contributed to increased occurrence of IS. Second, as control, the study used age-matched children with other types of epilepsy than IS and did not find the correlation to prenatal stress.
Figure 2 – Time course of prenatal development of the CRF, melanocortin (ACTH), and corticosteroid system in the rat.
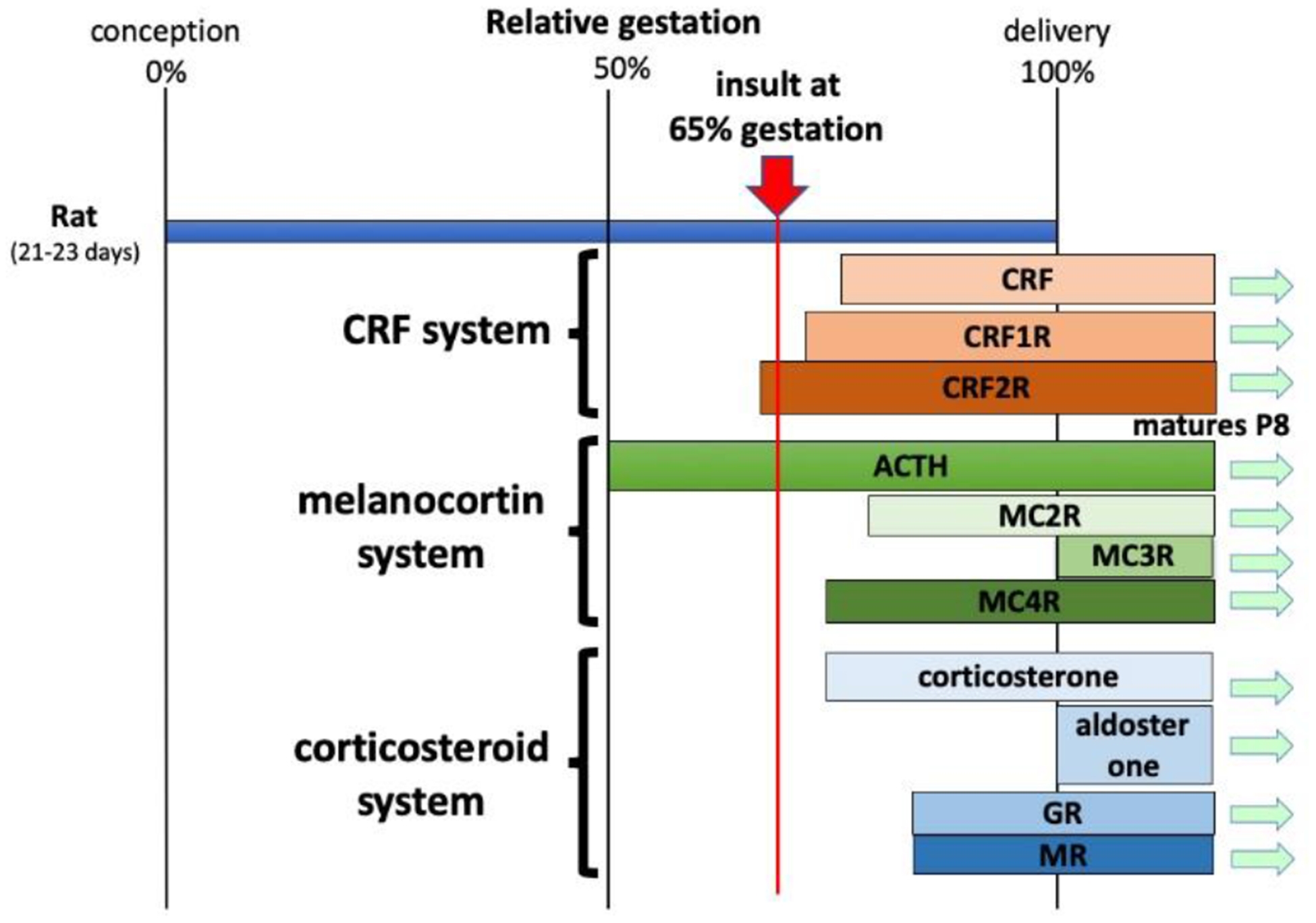
Based on the rat strain, the duration of pregnancy varies between 21–23 days. In the rat, all organ systems with available information are developing from the mid-pregnancy on. Interestingly, development of CRF receptors precedes slightly occurrence of CRF itself (Bugnon, et al., 1982; Insel, et al., 1988; Lovenberg, et al., 1995). ACTH appears early, already present at the mid pregnancy, followed by its effectors, melanocortin receptor isoforms (MCR) (Kistler-Heer, et al., 1998). Appearance of corticosterone also precedes detection of glucocorticoid and mineralocorticoid receptors (GR and MR, respectively) (Noorlander, et al., 2006). Aldosterone, by contrast, is detected around birth (Bohn, et al., 1994).
Conclusions:
Prenatal exposure to betamethasone or stress creates complex changes in gene-protein environment as well as metabolic changes (Lee, et al., 2018) that require further characterization but they significantly contribute to increased susceptibility to spasms and make the spasms sensitive to ACTH treatment. Long-term prenatal stress combined with terbutaline (not terburaline per se) may even result in spontaneous seizures in adulthood (Bercum, et al., 2015) though in different models prenatal betamethasone may even decrease seizure susceptibility (Velíšek, 2011). As the effects of prenatal stress (betamethasone) in our model are not easily detected macro- or even microscopically at the level of brain morphology, it appears that the prenatal betamethasone-postnatal NMDA model reproduces features of cryptogenic IS (Velíšek, et al., 2010).
The model is a very useful tool for testing of novel, putative treatments of the IS as its ACTH response has been demonstrated repeatedly in several independent laboratories (Baek, et al., 2016; Kim, et al., 2017; Kwon, et al., 2018; Shi, et al., 2015; Shi, et al., 2016; Tsuji, et al., 2016). While no spontaneous spasms have been described previously because the animals were not followed beyond the acute period after the NMDA exposure, our new data show that in animals experiencing severe spasms after repeated NMDA administrations (P12-P15), the spontaneous seizures (epilepsy) may develop within 7–10 days (unpublished observations). The model may also provide insights to the final common pathway responsible for genesis of spasms as well as to the variety of subtle pre- and perinatal mechanisms involved in creating an environment susceptible to development of cryptogenic IS.
6.2.2. Brain intraparenchymal tetrodotoxin model
Idea:
During the second postnatal week there is an extensive outgrow of axons in hippocampal CA3 with prominent axonal arborization further subjected to pruning (Gomez-Di Cesare, et al., 1997). Experimental silencing of the synapses during this period may lead to synaptic reorganization with possible aberrant, seizure prone circuitry (Swann, 1995). Thus local circuit activity has been blocked by a unilateral intrahippocampal or intracortical injection of tetrodotoxin (TTX) between P10–12 via osmotic minipumps.
Success:
Approximately one third of the animals (irrespective of hippocampal or cortical TTX infusion) developed brief spasms within 2–3 weeks (Lee, et al., 2008). These spasms were 1–2 s in duration, extensor or flexor, sometimes accompanied by forelimb clonus (symmetrical or asymmetrical). The spasms largely occurred in clusters and were of various intensity. At P39–41, an array of EEG electrodes has been implanted (4 ipsilaterally and 5 contralaterally to the infusion side). Ictal EEG consisted of a large-amplitude wave followed with generalized voltage attenuation (electrodecrement). Interictal EEG was abnormal in all TTX infused animals irrespective of the occurrence of spasms. The typical interictal EEG pattern consisted of multifocal spike and sharp wave discharges. Most of the animals developing spasms also showed a high voltage chaotic background pattern resembling hypsarrhythmia. Additional EEG analysis revealed prominent high-frequency oscillations at >200 Hz (Frost, et al., 2011; Frost, et al., 2012).
Drug effects:
The model is responsive to vigabatrin treatment including suppression of the high frequency EEG oscillations (Frost, et al., 2015). Further, an abstract reports about efficacy of a high dose of ACTH (32 U/kg per day) for 10 days, which eliminated spasms in two thirds of the rats and improvement in those rats displaying hypsarrhythmia (Swann, et al., 2017). In another abstract report, the authors presented long-term treatment with insulin-like growth factor peptide fragment (1–3)IGF, which significantly decreased number of spasms during the three-week treatment period and eliminated hypsarrhythmia in 2/3 of the rats (Lee, et al., 2014). Unfortunately, none of these interesting datasets have been published as original reports.
Mechanisms:
Arrest of the synaptic transmission during the most critical period of cortical and hippocampal development (Swann, 1995) introduces significant developmental disturbance leading to the occurrence of chronic spasms, i.e., an epilepsy syndrome. An older mechanistic study determined that there are no obvious presynaptic changes (besides increased size and number of presynaptic varicosities yet unrelated to postsynaptic specializations). On the other hand, that study demonstrated increased postsynaptic expression of receptor subunits for glutamate (GluR1, NR1 and NR2B) also associated with increased sensitivity of the hippocampal synaptic transmission to the NR2B subunit specific antagonist ifenprodil (Galvan, et al., 2003). Thus it seems reasonable that these postsynaptic changes are major contributors to network hyperexcitability and also plasticity.
Conclusions:
This is a promising model with strong focus on brain EEG (electrocorticogram). Yet, there are some caveats diminishing the link of this model to IS. First, the spontaneous spasms occur well beyond the developmental period of infancy in rat (Avishai-Eliner, et al., 2002; Gottlieb, et al., 1977) since they develop during the rat puberty (Ojeda and Urbanski, 1994). The prevailing electrographic pattern is multifocal spike and sharp wave discharges rather reminiscent of Lennox-Gastaut syndrome (Bourgeois, et al., 2014). The correlated rat age of this model is closer to the age range of Lennox-Gastaut syndrome (3–5 years in children) than to IS. The description of rat hypsarrhythmia may be misleading as comparison of complexity of discharges between lissencephalic (rat) and gyrencephalic (human) brain must be considered. Also the response to drug treatments is either non-specific (vigabatrin) or requires a very long treatment (ACTH or (1–3)IGF) relative to the rat lifespan.
6.2.3. IS in the Down syndrome animal model
Idea:
The 8% incidence of seizures in Down syndrome greatly exceeds incidence of seizures in general population. One third of those seizures are IS (Goldberg-Stern, et al., 2001; Stafstrom and Konkol, 1994). The authors used available Ts(1716)65Dn=Ts65Dn mouse model of distal end of chromosome 16 trisomy, which replicates phenotypes of human Down syndrome minus seizures (Salehi, et al., 2007). This mouse overexpresses the Girk2 inward rectifying potassium channel subunit (the gene is located on the triplicated chromosome arm) and GABAB receptor agonists produce significant increases in this current, clearly pointing to an association of the GABAB receptor with Girk2/KCNJ6 channel. As GABAB receptor agonists can precipitate seizures, the authors hypothesized that the combination of GABAB receptor agonists plus a mouse mutant might reveal an epilepsy (or seizure) phenotype. The authors used the Ts65Dn mice and injected them with γ-butyrolactone, a precursor of GABAB receptor agonist γ-hydroxybutyrate.
Success:
In contrast to wild type mice, in which GBL produces absence seizures, the same doses of GBL in Ts65Dn mice produced facial myoclonus and clusters of acute epileptic extensor spasms associated with bursts of epileptiform activity on the EEG with interposed EEG electrodecrements, irrespective of age (1 week to 2 months of age) and sex (Cortez, et al., 2009).
Drug effects:
As with any model of IS, one would expect to see efficacy of ACTH. Interestingly, only the 24 amino acid ACTH fragment (ACTH1–24) provided complete suppression of extensor spasms. However, the full molecule (ACTH1–39) of porcine origin was completely ineffective. Additionally, the authors found complete resolution of spasms after ethosuximide, valproic acid and CGP35348 (a specific GABAB-receptor antagonist) as well as partial suppression after vigabatrin. There was exacerbation of spasms in the model with baclofen (a GABAB receptor agonist) added on top of γ-butyrolactone or after 5-hydroxytryptophan. Indeed, administration of baclofen in patients with Down syndrome resulted in occurrence of IS (Coleman, 1971). When the effects of drugs on the EEG electrodecrement were evaluated, administration of the 24 amino acid ACTH fragment resulted in an all-or-none response. Interestingly both ethosuximide and valproic acid showed all-or-none effect on the EEG electrodecrement as well while CGP35348 showed dose-dependent protection and baclofen showed dose-dependent exacerbation of the EEG electrodecrement.
Mechanisms:
Exacerbation of absence seizures with GABAB receptor agonists such as baclofen was experimentally shown in the rats with absence seizures (Vergnes, et al., 1984) or in lh/lh mice (Hosford, et al., 1992) and this effect is also found in patients with absence epilepsy (Snodgrass, 1992). Interestingly, the Ts65Dn mice overexpress the GABAB receptor subunit 2 (Gabbr2) in the thalamus and medulla compared to the wild type mice. Mutations in this gene (GABBR2) were found as determinants of severity of the phenotype in children with Rett syndrome and epileptic encephalopathy (Yoo, et al., 2017). It is likely that overexpression of this protein and functional overexpression of GIRK channels, especially in the medulla, contributes to the spasm phenotype of seizures. In a follow-up study the authors demonstrated that Girk2−/− mice were unable to develop an IS phenotype after large doses of GABAB receptor agonists (Blichowski, et al., 2015).
Conclusions:
The authors demonstrated how the IS in the Down syndrome may develop. Indeed, in humans, the KCNJ6 potassium channel maps to chromosome 21 and in patients with Down syndrome, it is affected by triplication. There are additional interesting aspects of the model. First, the model nicely shows all-or-none effect of some treatments (ACTH fragment, valproic acid, ethosuximide). This is consistent with the effects of treatments in human IS, though some of the tested drugs lack efficacy in humans. Here it appears that in these mice, ethosuximide works through deactivation of thalamo-cortical circuitry. Interestingly, interruption of normal thalamo-cortical pathways has been proposed as one of the mechanisms for IS (Frost and Hrachovy, 2005). On the other hand, the connection between the two systems (thalamic and medullar) and its functional consequences are still unclear. Second, the model clearly demonstrates that the ACTH fragment and ACTH full molecule have different efficacies. While this may be due to interspecies difference (porcine full molecule used in mice), the differential efficacy of molecular fragments versus full molecules needs to be explained. Finally, the model does not provide age-specificity while in patients with trisomy 21, IS occur only during infancy and later different types of seizures prevail (Beatty, et al., 2017; Lott, 2012).
While the model provides some response to drugs and as such could be used for drug screening, the authors have not reconciled the induction of acute extensor epileptic spasms after injection of GABAB agonists in wild type Girk2+/+ mice (Blichowski, et al., 2015) versus unique features of their Ts65Dn model (Cortez, et al., 2009). Since the authors did not provide information on the background mouse strain (Blichowski, et al., 2015), this may be the function of the wild type background [the Ts65Dn mouse is raised on the C57BL/6J background (Cortez, et al., 2009)]. Yet the value of this IS model in the mouse model of Down syndrome is appreciated.
6.2.4. Targeted loss of Arx gene
Idea:
Aristaless-related homeobox gene (ARX) is an X-linked transcription factor important for cortical development (Kato and Dobyns, 2005). ARX guides non-radial migration of cells (in this case interneurons) into cortex, as well as radial migration of neurons and basal ganglia development. ARX is associated with occurrence of IS (Moey, et al., 2016; Stromme, et al., 2002). Male mice with Arx mutation have severe interneuron migration deficit (Kitamura, et al., 2002). Thus affecting interneuronal development with a targeted Arx mutation could lead to development of spasms relevant to IS. Arx mutant mice were created by crossing a floxed Arx allele (Arxf l) to Dlx5/6CIG mice. The purpose was to target Arx ablation in subpallial neurons (selective expression of Dlx5/6).
Success:
Repeated EEG recordings were performed in both immature and adult animals. Recording in adult mice (90–120 days old), showed that both male mutants Arx−/Y;Dlx5/6CIG and heterozygous females Arx−/+; Dlx5/6CIG had abnormal EEG activity. The males had missing theta frequency during exploration, while the females had periods of faster rhythms with higher amplitude. In contrast to control mice, the male Arx-/Y;Dlx5/6CIG developed seizures in 100% of subjects (3/3) and heterozygous females Arx−/+; Dlx5/6CIG in 53% of subjects (9/17). Clinically, males had flexion or extension spasms, while females had rather generalized clonic seizures, freezing and some spasms (Marsh, et al., 2009). The spasms occurred in adult mice only. When the mice were recorded between P15–17, a more developmentally appropriate period for IS occurrence, only clonic seizures were detected in both affected males (3/3) and heterozygous females (4/7). The authors also examined the brains for immunohistochemical expression of interneuron markers, namely calbindin, calretinin, and parvalbumin as well as for the presence of GFP (the Dlx5/6 tag) and Arx. The Arx-/Y;Dlx5/6CIG had prominent decreases in neocortical calbindin positive interneurons in both KO males and heterozygous females. No changes in parvalbumin positive neurons were found.
Drug effects:
No established or putative IS treatments have been reported.
Mechanisms:
While the Arx deficit is obvious, the authors also followed on gene pathway analysis in the hippocampal parvalbumin-expressing neurons of the Arx KO mice compared to wild type mice. They analyzed 110 genes and demonstrated that on top of the nervous system development, Arx KO affected several biological processes including synaptic transmission and regulation of ion transmembrane transport. Additionally, molecular pathways of the differentially expressed transcripts included calcium ion binding, ion channel activity, and voltage-gated channel activity as well as transcripts related to the synaptic membrane, ionotropic glutamate receptor complex, and extracellular matrix. These findings indicate that Arx affects basic neuronal functions such as synaptic transmission and ion channel activity, which relates to the network and cellular phenotypes (von Deimling, et al., 2017). Additional work (pertinent also for the following model) determined that polyalanine expansion mutation of the Arx gene has no effect on any diencephalic neuronal nuclei. On the other hand, deletion of Arx resulted in loss of specific markers (tyrosine hydroxylase) in zona incerta and reticular thalamic nucleus (Sunnen, et al., 2014). Focused Arx removal from pallial cells that give rise to cortical projection neurons led to (without any seizure phenotype) cortical thinning and interesting behaviors (more active, less anxious, less social) (Sunnen, et al., 2014). Finally, the authors determined that Arx loss in subpallial neurons (identical mutation to their IS model paper) is associated with increased perinatal lethality in Arx−/Ymice. Interestingly, the Arx expression in mature wild type, Arx+/− and Arx−/Y mice is variable. Conditional KO mice with a more complete loss of Arx in the interneuron precursors have a loss of all interneurons (co-stained with calretinin, calbindin, somatostatin, parvalbumin, and neuropeptide Y) in the cortex and hippocampus on embryonic day 18.5 and postnatal day 14. There was also no survival of males with complete loss of Arx (Marsh, et al., 2016). Interestingly, GABA, synthesized from glutamate by glutamic acid decarboxylase (GAD) in pancreatic β cells, is an extracellular signaling molecule acting on the pancreatic islets (Adeghate and Ponery, 2002) and a complete loss of Arx leads to death due to pancreatic failure (Collombat, et al., 2007).
Conclusions:
This is an interesting model indicating that some but not all ARX mutations may be associated with IS. Additional ARX mutations are obviously associated with other epileptic encephalopathies (Sherr, 2003) such as Ohtahara syndrome (Bettella, et al., 2013; Fullston, et al., 2010; Kato, et al., 2010), yet other ARX mutations are also associated with muscular spasticity and abundance of different signs and symptoms (Scheffer, et al., 2002). This is probably happening because of incomplete penetration of the mutations, mosaicism, and stochastic inactivation of the X chromosome. As such, the work on this transcription factor provides an illustrative example of multiple ways in which a gene mutation may manifest. Yet, currently, the contribution of this model to the treatment of IS or epileptic encephalopathies or seizures in general is limited.
6.2.5. Triplet repeat Arx expansion mouse model
Idea:
A similar rationale as in 6.2.4 above was utilized. The triplet repeat expansion of the first polyalanine tract in the X-linked ARX gene is commonly associated with human IS and mental retardation (Poirier, et al., 2008). The triplet expansion is not lethal in mice in contrast to the complete loss of Arx (Marsh, et al., 2016).
Success:
In humans, the ARX gene triplet expansion (GCG) for alanine adds 7 codons in the first polyalanine tract for final 10 consecutive alanine codons (Stromme, et al., 2002). In the mouse Arx gene, the expansion using knock-in mutation inserted GCT repeats results in a total of 23 alanine codons in tract 1. This mouse Arx knock-in reproduces the 23 alanine codons in human ISSX-ARX (GCG)10+7 and thus the mouse line was designated as Arx(GCG)10+7. Mating heterozygous Arx(GCG)10+7 females to wild-type males resulted in 25% of males carrying Arx(GCG)10+7 according to Mendelian inheritance. The mutant mice developed normally, had normal fertility, and had normal pancreatic function [which is the cause of prenatal death in mutants with complete Arx deletion (Collombat, et al., 2007)]. Starting on postnatal day 7, mutant pups had severe spasms (1–11 per 30 min period) while the nonmutant pups (background strain) had maximum of 2 of those spasms. At postnatal day 9, the contrast was better as 31% of mutants had 1–2 spasms while only a single non-mutant had one episode during the same 30 min period. At postnatal day 11, 38% of mutant pups had 1–3 severe spasm-like movements while there were no spasms in nonmutants (Price, et al., 2009). The EEG of mutants younger than 21 days was associated with a sharp spike slow wave discharge with attenuation of background activity during the myoclonic twitches with a sudden head drop (= a spasm). Behavioral analysis at age 2 months determined no difference between mutants and non-mutants in the pre-pulse inhibition test. Mutant mice were more sensitive to heat-induced pain and they also performed better on an accelerated rotarod compared to wild type mice. Mutants were also less anxious in the light-dark test and spent significantly more time in the center of the open field compared to the wild types while there was no effect of mutation on other open field parameters. Finally, in fear learning (a foot shock applied with a conditioning sound cue), freezing responses to sound only were significantly reduced in mutants comparedto wild type mice, yet in the tube test of aggression, the mutants demonstrated non-aggressive behavior by retreating in 79% of trials in comparison with wild types. In this test two mice enter a narrow plexiglass tube against each other. They cannot turn and they cannot fight. Thus, the more aggressive mouse pushes the less aggressive one out of the tube (Chachua, et al., 2014).
Drug effects:
The authors did not report responses to drugs used to treat IS. Instead, they referred back to the Arx mutation mechanism of action. The model they developed, the Arx(GCG)10+7, has severe deficits in specific interneuronal populations in the cortex and striatum since Arx as a transcription factor regulates interneuronal migration and maturation (Colasante, et al., 2008; Friocourt and Parnavelas, 2011). Interestingly, estradiol is capable to enhance neuronal migration in songbirds (Williams, et al., 1999). Thus, estradiol was injected in mutants and the outcome on seizures and EEG discharges was determined. Long-term estradiol (P5-P40) treatment in mutants significantly decreased the number of discharges and eliminated seizures including the severe spasms (Olivetti, et al., 2014). Further refinement of estradiol delivery has indicated that the critical window of estradiol efficacy is between P3 and P10, corresponding to the end of migration of GABAergic neurons in mice (Wonders and Anderson, 2006). Indeed, if the expression and number of specific GABAergic neuronal subtypes were evaluated, there was significant increase in neuropeptide Y-positive neurons and also calbindin-positive neurons in mutants with estradiol compared to wild type. The effect was mediated by combined activation of ERα and ERβ receptors.
Mechanisms:
There are some differences compared to the Arx deletion. Arx(GCG)10+7 mutations decreased the amount of Arx protein as well as number of interneurons. The decrease in GABAergic interneuron numbers in mutants was particular for somatomotor cortex (at 68% of the wild type numbers) and more profound in the deep layers V and VI (only at 53% of the wild type). No difference in Arx expression was found in parietal cortex (Price, et al., 2009) suggesting further region-specific modifications. The authors additionally studied individual types of interneurons, as there might have been subtype-specific changes (Marsh, et al., 2009). In the mutants, there was a profound loss of calbindin-expressing GABA interneurons in all brain areas where decrease in Arx protein was found. There was a deficit (~30% loss) of NPY positive interneurons as well as 40% loss of cholinergic interneurons, but only in the mutant striatum compared to wild type mice. There were no differences in numbers of parvalbumin and calretinin subtypes of GABAergic interneurons between the two genotypes.
Conclusions:
This model is a variant of the previous one with spontaneous spasms and additional developmental seizures. This situation is similar to human condition when children with IS later during childhood develop other types of epilepsy. The effects of gene mutation are described in details and the authors were able to address the mechanism by restoring the number of GABAergic inhibitory neurons with early neonatal estradiol treatment. While the effect of estradiol restoration of GABAergic inhibition confirms the role of these neurons in spasms and other seizure phenotypes in this mutation, this treatment is not universal (Chachua, et al., 2016; Galanopoulou, et al., 2017).
Thus, neonatal estradiol treatment is potentially appropriate only in children with IS who have ARX mutations, where deficits in the GABAergic system are expected. This is especially important since neonatal administration of estradiol severely interferes with gonadal development leading to cryptorchism in males (Chachua, et al., 2016) and may cause irreversible infertility (Minabe, et al., 2019). Finally, it would be interesting to determine whether a simple GABAergic system enhancing treatment (i.e., vigabatrin, barbiturates, ganaxolone) would be effective in this type of mutation, with less severe side effects compared to neonatal estradiol.
6.2.6. Triple hit model of IS syndrome
Idea:
There are structural and functional abnormalities in cortical and brain stem structures in patients with symptomatic IS (Frost Jr. and Hrachovy, 2003; Juhasz, et al., 2002). Positron emission tomography revealed specific hyperactivation in basal ganglia as well as in serotonergic nuclei of dorsal raphe (Chugani, et al., 1992). Further, in patients with epilepsy, there is a significant contribution of inflammatory processes and molecules (Vezzani and Granata, 2005; Vezzani, et al., 2013). Thus, the authors decided to address all three components: brain morphology, inflammation and the serotonergic system together.
Success:
The model is subject to the US patent number US20080216183A1 issued on Jan 4, 2011, to M.H. Scantlebury and S.L. Moshé after provisional application No 60/900,487 filed on February 8, 2007 (Scantlebury and Moshé, 2011). The authors injected P3 rats intracerebroventricularly (icv) with doxorubicin, an anti-cancer agent producing diffuse brain abnormalities and intraparenchymally with lipopolysaccharide (LPS, activating inflammatory cascades). On P5, rats received i.p. injection of p-chorphenylalanine (PCPA) to deplete serotonin by blocking its synthetic enzyme, tryptophan hydroxylase. Spasms (flexion, extension of mixed) were observed in all injected pups; some of those pups developed spasms already on P4, before injection of PCPA (Scantlebury, et al., 2010). Indeed, higher PCPA doses contributed to increased occurrence of spasms, which were observed between P4 and P13 with a peak between P4-P6. Two thirds of the pups injected with all three drugs developed additional seizure types between P9 and P20 including freezing, tonic seizures, sudden drops as well as forelimb clonus with rearing and falling. About 25% of pups displayed EEG electrodecrement along with spasms; an additional 50% had spikes or sharp waves. Yet, about half of spasms did not have any clear EEG correlate. There was also a significant mortality in these pups. Significant behavioral impairments were also recorded: affected pups spent less time exploring the subject-occupied space in the three-chamber sociability apparatus. There was also increased grooming, and abnormal latencies to localize escape box in the Barnes maze test (Scantlebury, et al., 2010).
Drug effects:
The model is responsive to vigabatrin, which significantly decreases occurrence of spasms though leading eventually to 100% mortality due to over-sedation. There was no effect of chronically administered Synacthen Retard (ACTH1–24) (Scantlebury, et al., 2010). Carisbamate, a broad spectrum anti-seizure drug with potential antiepileptogenic effects preclinically (Francois, et al., 2011) and with some efficacy in focal-onset seizures in patients with epilepsy (Sperling, et al., 2010), suppressed spasms but not interictal EEG discharges, while phenytoin had no effect (Ono, et al., 2011). Since IS are frequently associated with tuberous sclerosis (mutations in TSC1 or TSC2), treatment with rapamycin may be an effective approach (mTORC1 is downstream from TSC1 and TSC2). A brief (two-day) administration of rapamycin to the rats decreased frequency of spasms not only during the treatment but also on the following days. There was no effect on other seizure types (occurring later) or on associated behavioral problems (Raffo, et al., 2011). Further, NAX-5055, an analogue of an inhibitory peptide galanin has been tested, yet in single administration had no effect on spasms (Jequier Gygax, et al., 2014). A more effective analogue of vigabatrin, CPP-115, was also investigated. The drug significantly reduced occurrence of spasms but did not improve learning after the underlying brain damage was factored in. Similar to vigabatrin, the largest dose of CPP-115 (5 mg/kg) contributed to 100% lethality at P8 (Briggs, et al., 2014). The model was further used to determine the effects of new putative treatments for IS: The caspase-1 inhibitor VX-765 (blocks pro-inflammatory interleukin IL-1β), GABAB receptor blocker CGP-35348, and neonatal estradiol (see above for rationale) were administered between P3-P10. Unfortunately, none of these treatments was effective (Galanopoulou, et al., 2017).
Thus, this model showed efficacy of several drugs, which rather suggests that the model responds more like a model of symptomatic IS. Indeed, severe brain structural damage is apparent in the rats injected with the three drugs (Scantlebury, et al., 2010).
Mechanisms:
In this model, multiple drugs were used to harm the developing brain. The result is diffuse brain damage consisting of hemispheric and periventricular injury with thinningof the cortex with diffuse damage to corpus callosum, striatum, septum, hippocampus and thalamus on the side of the injections (Scantlebury, et al., 2010). Indeed, the changes will be significantly more complex and will include multiple gene transcripts [as illustrated by changes in galanin receptors (Jequier Gygax, et al., 2014)]. There is a significant reduction of parvalbumin-expressing interneurons with preserved populations of those expressing calretinin and somatostatin. This may mechanistically explain positive effects of vigabatrin and its analogue CPP-115 (Katsarou, et al., 2018).
Conclusions:
This is an interesting yet complex model of spasms that occur within very close time span of drug administration during very early developmental stage and diminish later in development. Other seizure types emerge later, similar to humans with IS. However, the very early developmental occurrence of spasms is rather akin to human early infantile epileptic encephalopathies [EIEE, such as Ohtahara syndrome (Tharp, 2002; Yamatogi and Ohtahara, 2002)]. Indeed, similar to EIEE, this model is tainted by significant mortality. Regarding drug efficacy, the model is most similar to symptomatic IS in humans. Intraparenchymal or intracerebroventricular vehicles for doxorubicin and LPS were not administered to test for possible occurrence of spontaneous seizures as reported previously by others (Knuth and Etgen, 2004). US patent rights generally prevent other laboratories from independent replication and widespread use (together with significant complexity of approach and necessity to use pup-in-the-cup approach for some treatments) so the model is used only in Dr. Moshé’s lab at Einstein and in Dr. Scantlebury’s lab in Calgary, Alberta, Canada.
6.2.7. APC conditional knock-out mouse model of IS
Idea:
The model is based on the findings that β-catenin pathways are tightly regulated developmentally. Increased β-catenin levels lead to enhanced dendritic branching, altered maturation of excitatory synapses, and changes in synaptic function (Yu and Malenka, 2004). Additionally, the β-catenin pathway is closely linked to many genes associated with IS such as MAGI2, GRIN1, GRIN2A, TSC1/2, FOXG1, ARX, DCX, NRF1, LIS1 and STXBP1 (Boutry-Kryza, et al., 2015) and Table 3. Thus, β-catenin can be a convergent target of various IS-linked genes.
Success:
The authors intended to upregulate β-catenin expression by knocking out the Apc gene, which is upstream of β-catenin and a major negative regulator of β-catenin expression. In their knockout, Apc was floxed and the Apc-floxed mice were crossed with CamKII-Cre mice. This approach created an Apc knockout and ergo β-catenin overexpression only in the regions with enriched CamKII, which are excitatory neurons in the forebrain (but not the cerebellum) (Mohn, et al., 2014). P9 mice developed robust clustered spasms involving flexions or extensions of limbs, trunk flexion and curling (Pirone, et al., 2017). Interestingly, the spasms peaked at P9 and disappeared by P14, mimicking a window of susceptibility to spasms in humans. EEG telemetry demonstrated high-amplitude EEG discharges (a single spike followed by a wave or multiple waves) and their peak occurrence temporally corresponded with the behavioral phenotype. No correlate of hypsarrhythmia was observed in the model. Interestingly, 85% of 2-month-old APC cKO mice developed further EEG seizures accompanied by freezing, head bobbing, and forelimb clonus (Pirone, et al., 2017). This feature parallels quite well progression from spasms to other seizure types seen in humans.
Drug effects:
No effects of drugs, established or novel, have been reported.
Mechanisms:
Since CamKII-Cre recombinase activation increases during development, at P9, β-catenin expression was enhanced in layer V of neocortex, while at P14 the overexpression was widespread in the forebrain. At P9, recordings of the spontaneous excitatory postsynaptic currents (ePSCs) in layer V pyramidal neurons confirmed increased activity of these neurons. Thus, it seems that a correlate to spasms was increased excitability in specific cortical layer, namely layer V. Once the β-catenin expression spread to other parts of the forebrain, such as infralimbic prefrontal cortex (Pirone, et al., 2018), the spasms first disappeared and then other seizure types developed. The mice have also certain autistic traits such as reduced social interest, increased repetitive behaviors, and cognitive impairments (Mohn, et al., 2014; Pirone, et al., 2017).
Conclusions:
This is a unique model of genetically based IS at a developmentally appropriate period with progression to other seizure types in adulthood. This pattern mimics IS in humans quite well especially since autistic traits are also present. However, the EEG pattern lacks the correlation to human IS. Further, the authors have not reported any treatments, which limits the model’s validity at present.
6.2.8. Prenatal methylazoxymethanol-postnatal NMDA
Idea:
Many IS are associated with or result from preexisting brain malformations, with a prominent standing of malformations of cortical development (Blumcke, et al., 2009; Frost Jr. and Hrachovy, 2003; Osborne, et al., 2010). Experimental cortical dysplasias and microcephaly, especially those induced during embryonic development with a Cycas toxin, methylazoxymethanol (MAM) (Balduini, et al., 1984; Haddad, et al., 1972) are associated with proconvulsant effects (Baraban and Schwartzkroin, 1996; Chevassus-au-Louis, et al., 1998a). These effects appear to be model-related and age-specific (de Feo, et al., 1995; Germano, et al., 1993). Thus, the idea was to use prenatal MAM for induction of cortical malformations and then, in the immature offspring, trigger epileptic spasms with NMDA (Mareš and Velíšek, 1992). In essence such a model would be the symptomatic variant of the model of cryptogenic IS based on prenatal exposure to betamethasone and postnatal trigger of spasms with NMDA (Velíšek, et al., 2007).
Success:
After prenatal exposure to MAM on gestational day 15, magnetic resonance images showed significant cortical thinning in two developmental groups (P16 and P30) of the offspring, enlargement of lateral and third ventricles as well as dysplasias of both hippocampi and amygdalae (Kim, et al., 2017). Behaviorally at P30, MAM-exposed rats had decreased movement velocity in the central area of the open field compared to controls prenatally exposed to saline. Also, response to a sound stimulus in a conditioned freezing response was shorter in MAM-exposed P35 rats compared to age-matched controls. The number of spasms triggered by NMDA (either a single trigger or repetitive triggers over P12, P13 and P15) was increased in MAM-exposed rats vs. controls (prenatal saline plus postnatal NMDA). In the EEG recordings, the rats prenatally exposed to MAM showed more prominent power in the range of beta waves (13–24 Hz) as well as in the range of fast oscillations (defined here as frequencies between 25–100 Hz) following postnatal NMDA challenge yet with attenuated background compared to controls (Kim, et al., 2017).
Drug effects:
The authors tested both ACTH (a synthetic human molecule) and vigabatrin (Kim, et al., 2017). Interestingly, only vigabatrin suppressed the number of spasms. This finding further supports the notion of prenatal MAM-postnatal NMDA representing a model of symptomatic IS. In a follow-up study, the authors also determined that methylprednisolone is effective in a dose-dependent manner (Lee, et al., 2019).
Mechanisms:
MAM interferes with the cell cycle by DNA methylation (Dibble, et al., 2016; Matsumoto and Higa, 1966; Van den Berg and Ball, 1972) during the G1 and M phases (Chanda, et al., 1973) resulting in antiproliferative effects, which may be very precisely timed in terms of development of particular brain areas (Dibble, et al., 2016). Thus, specific arrests in cell division and eventually migration lead to brain dysplasias and ectopias, which are integrated, yet with abnormal connections, to local neuronal circuits (Chevassus-au-Louis, et al., 1999; Chevassus-au-Louis, et al., 1998b). Besides dysplasias there are additional effects on the expression of striatal inhibitory neurons (Balduini, et al., 1984), changes in cortical somatostatin expression (Cattabeni, et al., 1989; Chevassus-au-Louis, et al., 1998b) as well as noradrenergic and serotonergic markers (Johnston and Coyle, 1980). Thus, all these changes may be contributing to the enhanced sensitivity to NMDA producing spasms.
Conclusions:
The authors created a model of symptomatic IS based on prenatal cortical malformations with a treatment response similar to the human condition. Future experiments will show whether this model can contribute with a novel treatment approach for this type of relatively refractory IS.
7. CONCLUSIONS
7.1. Current state of IS treatment and possible anti-epileptogenesis
What treatment are available for IS in humans? Partially effective treatments include ACTH in its various forms, glucocorticoids, vigabatrin, in some patients, the KD or possibly pyridoxine. ACTH and related peptides derived from pro-opiomelanocortin will probably remain a staple of the IS treatment. Yet their side effects occurring especially with high dose treatment should not be neglected. Further, experimental evidence and clinical studies indicate that not all varieties of ACTH are equally effective. It seems that for some patients, the full ACTH molecule may work better, while for others the synthetic fragment is sufficient. Glucocorticoids are also effective, though the magnitude of their efficacy in comparison to ACTH is still under debate. Combination of ACTH (or corticosteroids) and vigabatrin may become increasingly used in the years to come. Yet, irreversible peripheral retinopathy as well as myelinopathy as common side effects of vigabatrin represent a significant concern. Sirolimus (rapamycin) or everolimus are finding their way to the treatment of tuberous sclerosis frequently associated with IS (Franz, et al., 2018; French, et al., 2016) as they are being repurposed from cancer therapy.
It may be wise here to reconsider the common final pathway of IS as proposed by Dr. Baram and others, based on the observations that no matter what the etiology is, the final phenotype is spasm-type seizures (in West syndrome it is also presence of EEG hypsarrhythmia and developmental delays) (Brunson, et al., 2001a). This indicates that eventually many if not most of the etiologies funnel through limited brain circuitry capable of generation of spasms (and hypsarrhythmia and developmental delays). While we may not know the full pathogenetic pathway for many genetic based IS yet, it seems that the process of epileptogenesis starts with the first occurrence of spasms or EEG hypsarrhythmia because: (1) Spasms occur first (at least in the cryptogenic form of IS) and developmental delay follows (if present). (2) If ACTH treatment is applied very early (within two weeks of the occurrence of the first spasm), it is significantly more effective than if treatments starts later (Hussain, et al., 2017a). (3) Alternatively if hypsarrhythmia occurs first and is treated with ACTH/vigabatrin, the spasms may never develop (Lux and Osborne, 2004). If the above information is combined, we may speculate that ACTH (or vigabatrin; clinicaltrials.gov: NCT02849457) may be able to affect epileptogenesis in IS (if applied early) and thus become a disease modifying treatment in an epilepsy syndrome. This is also a topic of the European EPISTOP study (clinicaltrials.gov: NCT02098759; www.epistop.eu) aiming on discovery of clinical and molecular biomarkers useful for early diagnosis of tuberous sclerosis and early preventative treatment (Auvin, et al., 2017). The disease modifying effect may be due to the interference of ACTH or vigabatrin within the common final pathway of IS that may not remove the cause of IS but may significantly affect progression of the EEG signature and development of motor spasms, both of which may be needed for co-morbidities to eventually occur.
7.2. Short term treatment needs
Considering the above summary of IS and its treatment, what is needed in the short term for IS treatment in humans? It would be ideal to have an agent that prevents peripheral retinopathy caused by vigabatrin. Some attempts have been already made to administer concurrently NKCC1 inhibitors such as bumetanide to attenuate retinal neurotoxicity and intramyelinic edema (Jensen, 2014). Additional studies are needed to reveal the mechanisms of action of the KD, which are confounded by potential differences in mechanisms at different ages and in different species (Choudhary, et al., 2018); modification of the gut microbiome is an attractive possibility (Mu, et al., 2018). Once compound(s) responsible for positive effects are identified, less intrusive and restrictive treatment, with possibly higher compliance, may be possible. Finally, in terms of personalized precision medicine, more complete dissection of underlying genetic abnormalities are needed in patients with IS. Only then will tailored, specific treatments be possible. An example here is the Arx gene triplet expansion resulting in decline in GABAergic neuronal populations that can be replenished by neonatal estradiol. Indeed, the path to human therapy is still long and every treatment may not suit all patients.
How is translational research helping in the search of new IS treatments? Currently, there are approximately eight models of IS available. The variability of the models reflects the human condition of the IS, which comprises a large variety of genetic and epigenetic CNS alterations leading to a common syndrome (Janicot, et al., 2020). Some of them may be closer to the early infantile epileptic encephalopathies, others to Lennox-Gastaut syndrome. Some are models of symptomatic IS as the damage or identified mutation are clear and present; others may be models of cryptogenic IS as there is no obvious damage but an array of mis-expressed molecules is present. Some models are genetics based, other rely on epigenetic effects. Despite these differences, the models may be used for testing novel therapies. A concerted effort has been applied to investigate neonatal estradiol treatment of IS, which clearly showed that only certain cases (with true GABAergic deficits) of IS may benefit from this therapy (Chachua, et al., 2016; Galanopoulou, et al., 2017; Olivetti, et al., 2014). In other cases, adverse estradiol effects may prevail (Chachua, et al., 2016). Thus, use of multiple models for testing a single treatment may sort out the applicability and make preclinical results more robust.
7.3. The need for further research in personalized therapy
What is needed for future basic/translational research of IS? Perhaps more new models, but these novel models should specifically target genes linked to or associated with IS. These new genetic models can improve understanding of individual IS syndromes and their mechanisms and thus, contribute to mechanistic, personalized treatments of these IS variants (Perucca and Perucca, 2019). Yet, caution should be exercised, as the rodent gene mutations may be significantly phenotypically different from human gene mutations. Transgenic animals may help here but at a cost. Current models should be stringently applied for the search of novel and repurposed treatments. Even with those simple models, it is a difficult, tedious and time-consuming job, if it is aimed to be done right – and it should be done since faulty preclinical studies besides giving false hope of drug discovery also bear significant costs in Phase 2 and especially Phase 3 clinical trials (Parasrampuria, et al., 2018). Thus, the testing should be carried rigorously to add strength to the preclinical studies and avoid clinical trial disappointments as seen recently in some nervous system diseases such as stroke or Alzheimer’s disease (Bosetti, et al., 2017; Mehta, et al., 2017). Despite possibly having a drug with disease modifying features (ACTH as mentioned above), this treatment has so many adverse effects that it is worth of looking for as effective (or better) drug with fewer adverse effects.
7.4. The complexity of epilepsy
The example of IS charted here should also serve as an illustration of diversity in epilepsy. In this rare epilepsy syndrome, there are numerous sub-types and considerations, each requiring specific research and specific clinical attention in a manner of true personalized and precision medicine. This is not only the story of IS but the story of epilepsy as a disease.
Figure 3 – Time course of prenatal development of the CRF, melanocortin (ACTH), and corticosteroid system in humans.
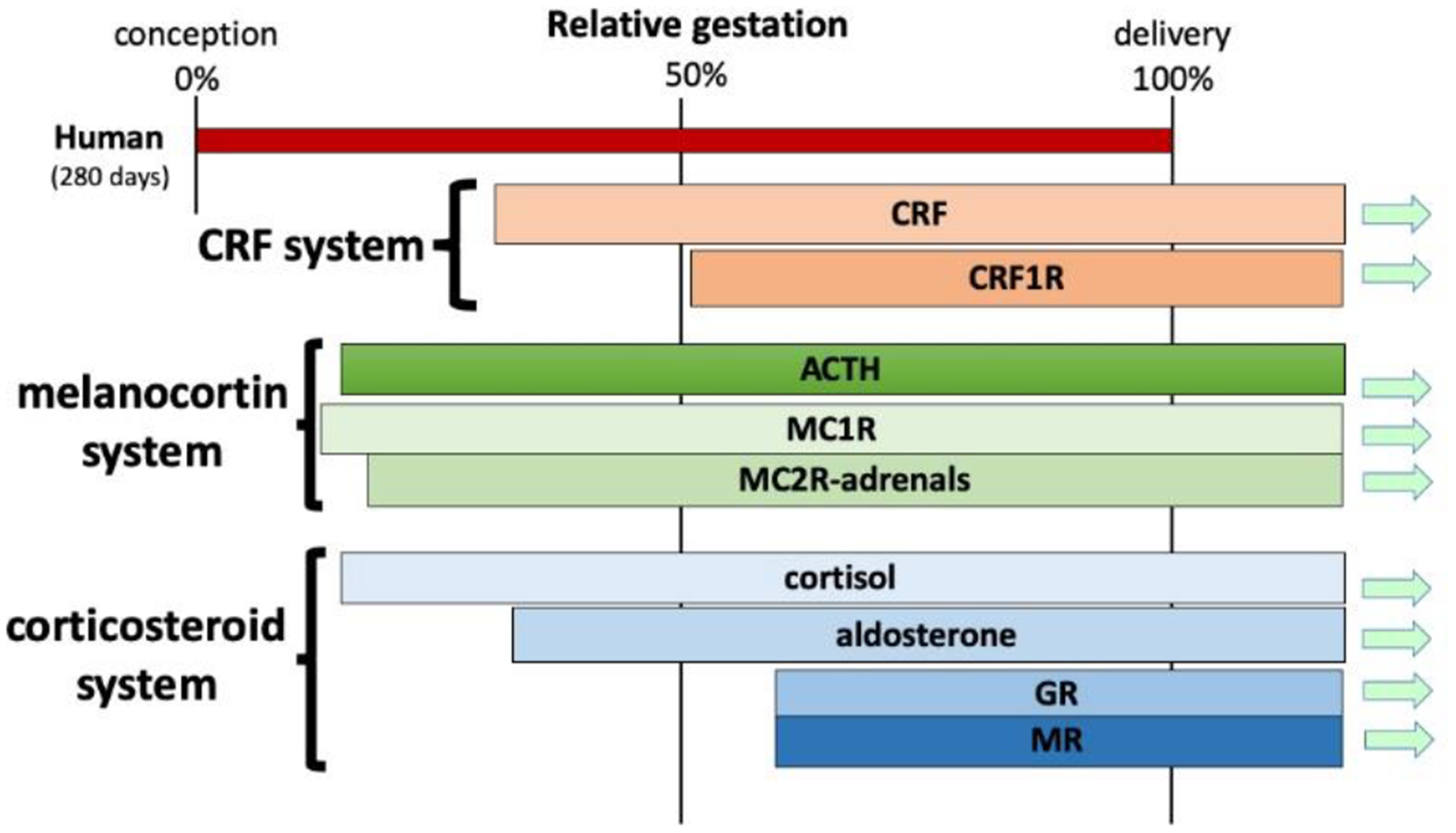
In contrast to rodents, in humans, elements of both melanocortin and corticosteroid systems appear early during embryonic life (Taylor, et al., 1953; Thomas, et al., 2018). The melanocortin system appears to be functional by the first trimester of intrauterine development. Later, the CRF system appears as do glucocorticoid and mineralocorticoid receptors (Ackland, et al., 1986; Bresson, et al., 1987; Goto, et al., 2006; Kempna and Fluck, 2008).
Abbreviations:
- ACTH
adrenocorticotropin, corticotropin
- CRF=CRH
corticotropin releasing factor=hormone
- EEG
electroencephalogram
- GABA
gamma amino-butyric acid
- HPA
hypothalamo-pituitary-adrenal axis
- ILAE
International League Against Epilepsy
- IS
infantile spasms
- KD
ketogenic diet
- MAM
methylazoxy-methanol
- MRI
magnetic resonance imaging
- mTOR
mechanistic target of rapamycin
- NMDA
N-methyl-D-aspartic acid
- TTX
tetrodotoxin
- VABS
Vineland adaptive behavioral scales
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST STATEMENT
Dr. Jana Velíšková is partially supported by the NS-092786 from the NIH. Dr. Libor Velíšek is supported by the Institute for Organic Chemistry and Biochemistry, Czech Academy of Sciences, and AMZELL Biopharma. None of the sponsors had any influence on data collection, analysis, and interpretation of data presented here or the writing of this report or intent to publish it.
9. REFERENCES
- Ackland JF, Ratter SJ, Bourne GL, & Rees LH (1986). Corticotrophin-releasing factor-like immunoreactivity and bioactivity of human fetal and adult hypothalami. J Endocrinol, 108, 171–180. [DOI] [PubMed] [Google Scholar]
- Adeghate E, & Ponery AS (2002). GABA in the endocrine pancreas: Cellular localization and function in normal and diabetic rats. Tissue Cell, 34, 1–6. [DOI] [PubMed] [Google Scholar]
- Auvin S, Walker L, Gallentine W, Jozwiak S, Tombini M, & Sills GJ (2017). Prospective clinical trials to investigate clinical and molecular biomarkers. Epilepsia, 58 Suppl 3, 20–26. [DOI] [PubMed] [Google Scholar]
- Auvin S, Dozieres-Puyravel B, Avbersek A, Sciberras D, Collier J, Leclercq K, et al. (2020). Radiprodil, a NR2B negative allosteric modulator, from bench to bedside in infantile spasm syndrome. Ann Clin Transl Neurol, 7, 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avishai-Eliner S, Brunson KL, Sandman CA, & Baram TZ (2002). Stressed-out, or in (utero)? Trends Neurosci, 25, 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek H, Yi MH, Pandit S, Park JB, Kwon HH, Zhang E, et al. (2016). Altered expression of KCC2 in GABAergic interneuron contributes prenatal stress-induced epileptic spasms in infant rat. Neurochem Int, 97, 57–64. [DOI] [PubMed] [Google Scholar]
- Balduini W, Abbracchio MP, Lombardelli G, & Cattabeni F (1984). Loss of intrinsic striatal neurons after methylazoxymethanol acetate treatment in pregnant rats. Brain Res, 317, 133–136. [DOI] [PubMed] [Google Scholar]
- Baraban SC, & Schwartzkroin PA (1996). Flurothyl seizure susceptibility in rats following prenatal methylazoxymethanol treatment. Epilepsy Res, 23, 189–194. [DOI] [PubMed] [Google Scholar]
- Baram TZ (1993). Pathophysiology of massive infantile spasms: perspective on the putative role of the brain adrenal axis. Ann Neurol, 33, 231–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram TZ, & Schultz L (1991). Corticotropin-releasing hormone is a rapid and potent convulsant in the infant rat. Dev Brain Res, 61, 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram TZ, & Schultz L (1995). ACTH does not control neonatal seizures induced by administration of exogenous corticotropin-releasing hormone. Epilepsia, 36, 174–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram TZ, Mitchell WG, & Haden E (1996a). Inhibition of pituitary-adrenal secretion by a corticotropin releasing hormone antagonist in humans. Mol Psychiatry, 1, 320–324. [PMC free article] [PubMed] [Google Scholar]
- Baram TZ, Hirsch E, Snead OC, & Schultz L (1992a). Corticotropin-releasing hormone-induced seizures in infant rats originate in the amygdala. Ann Neurol, 31, 488–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram TZ, Mitchell WG, Snead OC, Horton EJ, & Saito M (1992b). Brain-adrenal axis hormones are altered in the CSF of infants with massive infantile spasms. Neurology, 42, 1171–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram TZ, Mitchell WG, Hanson RA, Snead OC, & Horton EJ (1995). Cerebrospinal fluid corticotropin and cortisol are reduced in infantile spasms. Pediatr Neurol, 13, 108–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram TZ, Mitchell WG, Tournay A, Snead OC, Hanson RA, & Horton EJ (1996b). High-dose corticotropin (ACTH) versus prednisone for infantile spasms: a prospective, randomized, blinded study. Pediatrics, 97, 375–379. [PMC free article] [PubMed] [Google Scholar]
- Beatty CW, Wrede JE, & Blume HK (2017). Diagnosis, treatment, and outcomes of infantile spasms in the Trisomy 21 population. Seizure, 45, 184–188. [DOI] [PubMed] [Google Scholar]
- Belzung C, & Lemoine M (2011). Criteria of validity for animal models of psychiatric disorders: focus on anxiety disorders and depression. Biol Mood Anxiety Disord, 1, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bercum FM, Rodgers KM, Benison AM, Smith ZZ, Taylor J, Kornreich E, et al. (2015). Maternal stress combined with terbutaline leads to comorbid autistic-like behavior and epilepsy in a rat model. J Neurosci, 35, 15894–15902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, et al. (2010). Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia, 51, 676–685. [DOI] [PubMed] [Google Scholar]
- Berg AT, Wusthoff C, Shellhaas RA, Loddenkemper T, Grinspan ZM, Saneto RP, et al. (2019). Immediate outcomes in early life epilepsy: A contemporary account. Epilepsy Behav, 97, 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg AT, Chakravorty S, Koh S, Grinspan ZM, Shellhaas RA, Saneto RP, et al. (2018). Why West? Comparisons of clinical, genetic and molecular features of infants with and without spasms. PLoS One, 13, e0193599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo P, Madia F, Santulli L, Del Gaudio L, Caccavale C, Zara F, et al. (2016). 17q21.31 microdeletion syndrome: Description of a case further contributing to the delineation of Koolen-de Vries syndrome. Brain Dev, 38, 663–668. [DOI] [PubMed] [Google Scholar]
- Bettella E, Di Rosa G, Polli R, Leonardi E, Tortorella G, Sartori S, et al. (2013). Early-onset epileptic encephalopathy in a girl carrying a truncating mutation of the ARX gene: rethinking the ARX phenotype in females. Clin Genet, 84, 82–85. [DOI] [PubMed] [Google Scholar]
- Blennow G, & Starck L (1986). High dose B6 treatment in infantile spasms. Neuropediatrics, 17, 7–10. [DOI] [PubMed] [Google Scholar]
- Blichowski M, Shephard A, Armstrong J, Shen L, Cortez MA, Eubanks JH, et al. (2015). The GIRK2 subunit is involved in IS-like seizures induced by GABA(B) receptor agonists. Epilepsia, 56, 1081–1087. [DOI] [PubMed] [Google Scholar]
- Blumcke I, Vinters HV, Armstrong D, Aronica E, Thom M, & Spreafico R (2009). Malformations of cortical development and epilepsies: neuropathological findings with emphasis on focal cortical dysplasia. Epileptic Disord, 11, 181–193. [DOI] [PubMed] [Google Scholar]
- Bohn MC, Dean D, Hussain S, & Giuliano R (1994). Development of mRNAs for glucocorticoid and mineralocorticoid receptors in rat hippocampus. Brain Res Dev Brain Res, 77, 157–162. [DOI] [PubMed] [Google Scholar]
- Bosetti F, Koenig JI, Ayata C, Back SA, Becker K, Broderick JP, et al. (2017). Translational Stroke Research: Vision and Opportunities. Stroke, 48, 2632–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeois BF, Douglass LM, & Sankar R (2014). Lennox-Gastaut syndrome: A consensus approach to differential diagnosis. Epilepsia, 55 Suppl 4, 4–9. [DOI] [PubMed] [Google Scholar]
- Boutry-Kryza N, Labalme A, Ville D, de Bellescize J, Touraine R, Prieur F, et al. (2015). Molecular characterization of a cohort of 73 patients with infantile spasms syndrome. Eur J Med Genet, 58, 51–58. [DOI] [PubMed] [Google Scholar]
- Bresson JL, Clavequin MC, Fellmann D, & Bugnon C (1987). Human corticoliberin hypothalamic neuroglandular system: Comparative immunocytochemical study with anti-rat and anti-ovine corticotropin-releasing factor sera in the early stages of development. Brain Res, 429, 241–246. [DOI] [PubMed] [Google Scholar]
- Briggs SW, Mowrey W, Hall CB, & Galanopoulou AS (2014). CPP-115, a vigabatrin analogue, decreases spasms in the multiple-hit rat model of infantile spasms. Epilepsia, 55, 94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunson KL, Eghbal-Ahmadi M, & Baram TZ (2001a). How do the many etiologies of West syndrome lead to excitability and seizures? The corticotropin releasing hormone excess hypothesis. Brain Dev, 23, 533–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunson KL, Avishai-Eliner S, & Baram TZ (2002). ACTH treatment of infantile spasms: mechanisms of its effects in modulation of neuronal excitability. Int Rev Neurobiol, 49, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunson KL, Khan N, Eghbal-Ahmadi M, & Baram TZ (2001b). Corticotropin (ACTH) acts directly on amygdala neurons to down-regulate corticotropin-releasing hormone gene expression. Ann Neurol, 49, 304–312. [PMC free article] [PubMed] [Google Scholar]
- Bugnon C, Fellmann D, Gouget A, & Cardot J (1982). Ontogeny of the corticoliberin neuroglandular system in rat brain. Nature, 298, 159–161. [DOI] [PubMed] [Google Scholar]
- Campeau PM, Kasperaviciute D, Lu JT, Burrage LC, Kim C, Hori M, et al. (2014). The genetic basis of DOORS syndrome: an exome-sequencing study. Lancet Neurol, 13, 44–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camposano SE, Major P, Halpern E, & Thiele EA (2008). Vigabatrin in the treatment of childhood epilepsy: a retrospective chart review of efficacy and safety profile. Epilepsia, 49, 1186–1191. [DOI] [PubMed] [Google Scholar]
- Candee MS, Carey JC, Krantz ID, & Filloux FM (2012). Seizure characteristics in Pallister-Killian syndrome. Am J Med Genet A, 158A, 3026–3032. [DOI] [PubMed] [Google Scholar]
- Castronovo P, Baccarin M, Ricciardello A, Picinelli C, Tomaiuolo P, Cucinotta F, et al. (2020). Phenotypic spectrum of NRXN1 mono- and bi-allelic deficiency: A systematic review. Clin Genet, 97, 125–137. [DOI] [PubMed] [Google Scholar]
- Cattabeni F, Abbracchio MP, Cimino M, Cocchi D, Di Luca M, Mennuni L, et al. (1989). Methylazoxymethanol-induced microencephaly: Persistant increase of cortical somatostatin-like immunoreactivity. Dev Brain Res, 47, 156–159. [DOI] [PubMed] [Google Scholar]
- Chachua T, Yum M-S, Velíšková J, & Velíšek L (2011). Validation of the rat model of cryptogenic infantile spasms. Epilepsia, 52, 1666–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chachua T, Poon KL, Yum MS, Nesheiwat L, DeSantis K, Velíšková J, et al. (2012). Rapamycin has age-, treatment paradigm-, and model-specific anticonvulsant effects and modulates neuropeptide Y expression in rats. Epilepsia, 53, 2015–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chachua T, Di Grazia P, Chern CR, Johnkutty M, Hellman B, Lau HA, et al. (2016). Estradiol does not affect spasms in the betamethasone-NMDA rat model of infantile spasms. Epilepsia, 57, 1326–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chachua T, Goletiani C, Maglakelidze G, Sidyelyeva G, Daniel M, Morris E, et al. (2014). Sex-specific behavioral traits in the Brd2 mouse model of juvenile myoclonic epilepsy. Genes Brain Behav, 13, 702–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda R, Woodward DJ, & Griffin S (1973). Cerebellar development in the rat after postnatal damage by methylazoxymethanol: DNA, RNA and protein during recovery. J Neurochem, 21, 547–555. [DOI] [PubMed] [Google Scholar]
- Chen CJ, Sgritta M, Mays J, Zhou H, Lucero R, Park J, et al. (2019). Therapeutic inhibition of mTORC2 rescues the behavioral and neurophysiological abnormalities associated with Pten-deficiency. Nat Med, 25, 1684–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Giri M, Xia Z, Subedi YN, & Li Y (2017). Genetic and epigenetic mechanisms of epilepsy: a review. Neuropsychiatric disease and treatment, 13, 1841–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chern CR, Chern CJ, Velíšková J, & Velíšek L (2019). AQB-565 shows promise in preclinical testing in the model of epileptic spasms during infancy: Head-to-head comparison with ACTH. Epilepsy Res, 152, 31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chern CR, Chern CJ, Velíšková J, & Velíšek L (2020). ACTON PROLONGATUM(R) suppresses spasms head to head with Acthar(R) Gel in the model of infantile spasms. Epilepsy Behav, 105, 106950. [DOI] [PubMed] [Google Scholar]
- Chevassus-au-Louis N, Ben-Ari Y, & Vergnes M (1998a). Decreased seizure threshold and more rapid rate of kindling in rats with cortical malformation induced by prenatal treatment with methylazoxymethanol. Brain Res, 812, 252–255. [DOI] [PubMed] [Google Scholar]
- Chevassus-au-Louis N, Jorquera I, Ben-Ari Y, & Represa A (1999). Abnormal connections in the malformed cortex of rats with prenatal treatment with methylazoxymethanol may support hyperexcitability. Dev Neurosci, 21, 385–392. [DOI] [PubMed] [Google Scholar]
- Chevassus-au-Louis N, Rafiki A, Jorquera I, Ben-Ari Y, & Represa A (1998b). Neocortex in the hippocampus: An anatomical and functional study of CA1 heterotopias after prenatal treatment with methylazoxymethanol in rats. J Comp Neurol, 394, 520–536. [DOI] [PubMed] [Google Scholar]
- Choudhary A, Charkhad B, Barrett K, & Scantlebury M (2018). Effectiveness of the Ketogenic Diet for the Control of Seizures in the Triple-Hit Model of the Infantile Spasms Syndrome. In American Epilepsy Society Annual Meeting; (pp. 1.119). New Orleans, LA. [Google Scholar]
- Chugani HT, Shewmon DA, Sankar R, Chen BC, & Phelps ME (1992). Infantile spasms: II. Lenticular nuclei and brain stem activation on positron emission tomography. Ann Neurol, 31, 212–219. [DOI] [PubMed] [Google Scholar]
- Colasante G, Collombat P, Raimondi V, Bonanomi D, Ferrai C, Maira M, et al. (2008). Arx is a direct target of Dlx2 and thereby contributes to the tangential migration of GABAergic interneurons. J Neurosci, 28, 10674–10686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman M (1971). Infantile spasms associated with 5-hydroxytryptophan administration in patients with Down’s syndrome. Neurology, 21, 911–919. [DOI] [PubMed] [Google Scholar]
- Collombat P, Hecksher-Sorensen J, Krull J, Berger J, Riedel D, Herrera PL, et al. (2007). Embryonic endocrine pancreas and mature beta cells acquire alpha and PP cell phenotypes upon Arx misexpression. J Clin Invest, 117, 961–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola G, Grosso S, Verrotti A, D’Aniello A, & Pascotto A (2010). Simultaneous onset of infantile spasms in monozygotic twins. Pediatr Neurol, 43, 127–130. [DOI] [PubMed] [Google Scholar]
- Cortez MA, Shen L, Wu Y, Aleem IS, Trepanier CH, Sadeghnia HR, et al. (2009). Infantile spasms and Down syndrome: A new animal model. Pediatr Res, 65, 499–503. [DOI] [PubMed] [Google Scholar]
- Curatolo P, Jozwiak S, Nabbout R, TSC Consensus Meeting for SEGA and Epilepsy Management (2012). Management of epilepsy associated with tuberous sclerosis complex (TSC): clinical recommendations. Eur J Paediatr Neurol, 16, 582–586.22695035 [Google Scholar]
- D’Alonzo R, Rigante D, Mencaroni E, & Esposito S (2018). West syndrome: A review and guide for paediatricians. Clin Drug Investig, 38, 113–124. [DOI] [PubMed] [Google Scholar]
- Darke K, Edwards SW, Hancock E, Johnson AL, Kennedy CR, Lux AL, et al. (2010). Developmental and epilepsy outcomes at age 4 years in the UKISS trial comparing hormonal treatments to vigabatrin for infantile spasms: a multi-centre randomised trial. Arch Dis Child, 95, 382–386. [DOI] [PubMed] [Google Scholar]
- Davis PE, Kapur K, Filip-Dhima R, Trowbridge SK, Little E, Wilson A, et al. (2019). Increased electroencephalography connectivity precedes epileptic spasm onset in infants with tuberous sclerosis complex. Epilepsia, 60, 1721–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Feo MR, Mecarelli O, & Ricci GF (1995). Seizure susceptibility in immature rats with micrencephaly induced by prenatal exposure to methylazoxymethanol acetate. Pharmacol Res, 31, 109–114. [DOI] [PubMed] [Google Scholar]
- de Palma L, Boniver C, Cassina M, Toldo I, Nosadini M, Clementi M, et al. (2012). Eating-induced epileptic spasms in a boy with MECP2 duplication syndrome: insights into pathogenesis of genetic epilepsies. Epileptic Disord, 14, 414–417. [DOI] [PubMed] [Google Scholar]
- de Pontual L, Mathieu Y, Golzio C, Rio M, Malan V, Boddaert N, et al. (2009). Mutational, functional, and expression studies of the TCF4 gene in Pitt-Hopkins syndrome. Hum Mutat, 30, 669–676. [DOI] [PubMed] [Google Scholar]
- Demarest ST, Shellhaas RA, Gaillard WD, Keator C, Nickels KC, Hussain SA, et al. (2017). The impact of hypsarrhythmia on infantile spasms treatment response: Observational cohort study from the National Infantile Spasms Consortium. Epilepsia, 58, 2098–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibble SM, Du Y, & Grace AA (2016). Dysregulation of dopamine systems in a developmental disruption model of schizophrenia: Implications for pathophysiology, treatment, and prevention In Pletnikov MV & Waddington JL (Eds.), Handbook of Behavioral Neuroscience (Vol. 23, pp. 107–124): Elsevier. [Google Scholar]
- Dimassi S, Labalme A, Ville D, Calender A, Mignot C, Boutry-Kryza N, et al. (2016). Whole-exome sequencing improves the diagnosis yield in sporadic infantile spasm syndrome. Clin Genet, 89, 198–204. [DOI] [PubMed] [Google Scholar]
- Doring JH, Lampert A, Hoffmann GF, & Ries M (2016). Thirty years of orphan drug legislation and the development of drugs to treat rare seizure conditions: A cross sectional analysis. PLoS One, 11, e0161660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dracopoulos A, Widjaja E, Raybaud C, Westall CA, & Snead OC (2010). Vigabatrin-associated reversible MRI signal changes in patients with infantile spasms. Epilepsia, 51, 1297–1304. [DOI] [PubMed] [Google Scholar]
- Dressler A, Benninger F, Trimmel-Schwahofer P, Groppel G, Porsche B, Abraham K, et al. (2019). Efficacy and tolerability of the ketogenic diet versus high-dose adrenocorticotropic hormone for infantile spasms: A single-center parallel-cohort randomized controlled trial. Epilepsia, 60, 441–451. [DOI] [PubMed] [Google Scholar]
- Dulac O, Feingold J, Plouin P, Chiron C, Pajot N, & Ponsot G (1993). Genetic predisposition to West syndrome. Epilepsia, 34, 732–737. [DOI] [PubMed] [Google Scholar]
- Ebadi M, Metzler DE, & Christenson WR (1983). Convulsant activity of pyridoxal sulphate and phosphonoethyl pyridoxal: antagonism by GABA and its synthetic analogues. Neuropharmacology, 22, 865–873. [DOI] [PubMed] [Google Scholar]
- Edvardson S, Baumann AM, Muhlenhoff M, Stephan O, Kuss AW, Shaag A, et al. (2013). West syndrome caused by ST3Gal-III deficiency. Epilepsia, 54, e24–27. [DOI] [PubMed] [Google Scholar]
- Edwards HE, Vimal S, & Burnham WM (2002). The effects of ACTH and adrenocorticosteroids on seizure susceptibility in 15-day-old male rats. Exp Neurol, 175, 182–190. [DOI] [PubMed] [Google Scholar]
- Ehlers CL, Henriksen SJ, Wang M, Rivier J, Vale W, & Bloom FE (1983). Corticotropin releasing factor produces increases in brain excitability and convulsive seizures in rats. Brain Res, 278, 332–336. [DOI] [PubMed] [Google Scholar]
- Epi4K Consortium; Epilepsy Phenome/Genome Project, Allen AS, Berkovic SF, Cossette P, Delanty N, et al. (2013). De novo mutations in epileptic encephalopathies. Nature, 501, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eun SH, Kang HC, Kim DW, & Kim HD (2006). Ketogenic diet for treatment of infantile spasms. Brain Dev, 28, 566–571. [DOI] [PubMed] [Google Scholar]
- EuroEPINOMICS-RES Consortium; Epilepsy Phenome/Genome Project; Epi4K Consortium. (2014). De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet, 95, 360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falsaperla R, Marino SD, Marino S, & Pavone P (2018). Electroclinical pattern and epilepsy evolution in an infant with Miller-Dieker syndrome. J Pediatr Neurosci, 13, 302–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farwell J, Milstein J, Opheim K, Smith E, & Glass S (1984). Adrenocorticotropic hormone controls infantile spasms independently of cortisol stimulation. Epilepsia, 25, 605–608. [DOI] [PubMed] [Google Scholar]
- Fedorovich SV, Voronina PP, & Waseem TV (2018). Ketogenic diet versus ketoacidosis: what determines the influence of ketone bodies on neurons? Neural Regen Res, 13, 2060–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox J, Hussain S, Sankar R, & Kerrigan JF (2018). Hypothalamic hamartoma with infantile spasms: Case report with surgical treatment. Semin Pediatr Neurol, 26, 115–118. [DOI] [PubMed] [Google Scholar]
- Francois J, Germe K, Ferrandon A, Koning E, & Nehlig A (2011). Carisbamate has powerful disease-modifying effects in the lithium-pilocarpine model of temporal lobe epilepsy. Neuropharmacology, 61, 313–328. [DOI] [PubMed] [Google Scholar]
- Franz DN, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, et al. (2018). Everolimus for treatment-refractory seizures in TSC: Extension of a randomized controlled trial. Neurol Clin Pract, 8, 412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French JA, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, et al. (2016). Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet, 388, 2153–2163. [DOI] [PubMed] [Google Scholar]
- Friocourt G, & Parnavelas JG (2011). Identification of Arx targets unveils new candidates for controlling cortical interneuron migration and differentiation. Front Cell Neurosci, 5, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost JD Jr., & Hrachovy RA (2005). Pathogenesis of infantile spasms: a model based on developmental desynchronization. J Clin Neurophysiol, 22, 25–36. [DOI] [PubMed] [Google Scholar]
- Frost JD Jr., Lee CL, Hrachovy RA, & Swann JW (2011). High frequency EEG activity associated with ictal events in an animal model of infantile spasms. Epilepsia, 52, 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost JD Jr., Lee CL, Le JT, Hrachovy RA, & Swann JW (2012). Interictal high frequency oscillations in an animal model of infantile spasms. Neurobiol Dis, 46, 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost JD Jr., Le JT, Lee CL, Ballester-Rosado C, Hrachovy RA, & Swann JW (2015). Vigabatrin therapy implicates neocortical high frequency oscillations in an animal model of infantile spasms. Neurobiol Dis, 82, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost JD Jr., & Hrachovy RA (2003). Infantile spasms: diagnosis, management and prognosis. Boston/Dordrecht/London: Kluwer Academic Publishers. [Google Scholar]
- Fullston T, Brueton L, Willis T, Philip S, MacPherson L, Finnis M, et al. (2010). Ohtahara syndrome in a family with an ARX protein truncation mutation (c.81C>G/p.Y27X). Eur J Hum Genet, 18, 157–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanopoulou AS, Mowrey WB, Liu W, Li Q, Shandra O, & Moshe SL (2017). Preclinical screening for treatments for infantile spasms in the multiple hit rat model of infantile spasms: An update. Neurochem Res, 42, 1949–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan CD, Wenzel JH, Dineley KT, Lam TT, Schwartzkroin PA, Sweatt JD, et al. (2003). Postsynaptic contributions to hippocampal network hyperexcitability induced by chronic activity blockade in vivo. Eur J Neurosci, 18, 1861–1872. [DOI] [PubMed] [Google Scholar]
- Gataullina S, Lemaire E, Wendling F, Kaminska A, Watrin F, Riquet A, et al. (2016). Epilepsy in young Tsc1(+/−) mice exhibits age-dependent expression that mimics that of human tuberous sclerosis complex. Epilepsia, 57, 648–659. [DOI] [PubMed] [Google Scholar]
- Germano IM, Sperber EF, & Moshé SL (1993). Do migrational disorders lower the seizure susceptibility in the immature rat brain? Epilepsia, 34, 21. [Google Scholar]
- Gettig J, Cummings JP, & Matuszewski K (2009). H.P. Acthar Gel and Cosyntropin review: Clinical and financial implications. P&T, 34, 250–257. [PMC free article] [PubMed] [Google Scholar]
- Gibbs EL, Fleming MM, & Gibbs FA (1954). Diagnosis and prognosis of hypsarhythmia and infantile spasms. Pediatrics, 13, 66–73. [PubMed] [Google Scholar]
- Gibbs FA, & Gibbs EL (1952). Atlas of Electroencephalography. Cambridge, Mass: Addison-Wesley. [Google Scholar]
- Gipson TT, Gerner G, Srivastava S, Poretti A, Vaurio R, Hartman A, et al. (2014). Early neurodevelopmental screening in tuberous sclerosis complex: a potential window of opportunity. Pediatr Neurol, 51, 398–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go CY, Mackay MT, Weiss SK, Stephens D, Adams-Webber T, Ashwal S, et al. (2012). Evidence-based guideline update: medical treatment of infantile spasms. Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology, 78, 1974–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg-Stern H, Strawsburg RH, Patterson B, Hickey F, Bare M, Gadoth N, et al. (2001). Seizure frequency and characteristics in children with Down syndrome. Brain Dev, 23, 375–378. [DOI] [PubMed] [Google Scholar]
- Gomez-Di Cesare CM, Smith KL, Rice FL, & Swann JW (1997). Axonal remodeling during postnatal maturation of CA3 hippocampal pyramidal neurons. J Comp Neurol, 384, 165–180. [PubMed] [Google Scholar]
- Gonzalez-Giraldo E, Stafstrom CE, Stanfield AC, & Kossoff EH (2018). Treating infantile spasms with high-dose oral corticosteroids: A retrospective review of 87 children. Pediatr Neurol, 87, 30–35. [DOI] [PubMed] [Google Scholar]
- Goto M, Piper Hanley K, Marcos J, Wood PJ, Wright S, Postle AD, et al. (2006). In humans, early cortisol biosynthesis provides a mechanism to safeguard female sexual development. J Clin Invest, 116, 953–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb A, Keydar I, & Epstein HT (1977). Rodent brain growth stages: an analytical review. Biol Neonate, 32, 166–176. [DOI] [PubMed] [Google Scholar]
- Gowda VK, Narayanaswamy V, Shivappa SK, Benakappa N, & Benakappa A (2019). Corticotrophin-ACTH in comparison to prednisolone in West syndrome - A randomized study. Indian J Pediatr, 86, 165–170. [DOI] [PubMed] [Google Scholar]
- Guveli BT, Cokar O, Dortcan N, Benbir G, Demirbilek V, & Dervent A (2015). Long-term outcomes in patients with West syndrome: an outpatient clinical study. Seizure, 25, 68–71. [DOI] [PubMed] [Google Scholar]
- Haddad RK, Rabe A, & Dumas R (1972). Comparison of effects of methylazoxymethanol acetate on brain development in different species. Fed Proc, 31, 1520–1523. [PubMed] [Google Scholar]
- Hahn J, Lee H, Kang HC, Lee JS, Kim HD, Kim SH, et al. (2019). Clobazam as an adjunctive treatment for infantile spasms. Epilepsy Behav, 95, 161–165. [DOI] [PubMed] [Google Scholar]
- Hamada H, & Matthews SG (2018). Prenatal programming of stress responsiveness and behaviours: Progress and perspectives. J Neuroendocrinol, 10.1111/jne.12674, e12674. [DOI] [PubMed] [Google Scholar]
- Hamano SI, Nagai T, Matsuura R, Hirata Y, Ikemoto S, Oba A, et al. (2018). Treatment of infantile spasms by pediatric neurologists in Japan. Brain Dev, 40, 685–692. [DOI] [PubMed] [Google Scholar]
- Hamici S, Bastaki F, & Khalifa M (2017). Exome sequence identified a c.320A > G ALG13 variant in a female with infantile epileptic encephalopathy with normal glycosylation and random X inactivation: Review of the literature. Eur J Med Genet, 60, 541–547. [DOI] [PubMed] [Google Scholar]
- Hancock EC, Osborne JP, & Edwards SW (2013). Treatment of infantile spasms. Cochrane Database Syst Rev, 10.1002/14651858.CD001770.pub3, CD001770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hani AJ, & Mikati MA (2016). Current and emerging therapies of severe epileptic encephalopathies. Semin Pediatr Neurol, 23, 180–186. [DOI] [PubMed] [Google Scholar]
- Hansen J, Snow C, Tuttle E, Ghoneim DH, Yang CS, Spencer A, et al. (2015). De novo mutations in SIK1 cause a spectrum of developmental epilepsies. Am J Hum Genet, 96, 682–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Watanabe K, Miyazaki S, Hakamada S, Yamada H, & Nakamura S (1981). Apparent brain atrophy and subdural hematoma following ACTH therapy. Brain Dev, 3, 45–49. [DOI] [PubMed] [Google Scholar]
- Harris A, & Seckl J (2011). Glucocorticoids, prenatal stress and the programming of disease. Horm Behav, 59, 279–289. [DOI] [PubMed] [Google Scholar]
- Heiskala H (1997). CSF ACTH and beta-endorphin in infants with West syndrome and ACTH therapy. Brain Dev, 19, 339–342. [DOI] [PubMed] [Google Scholar]
- Hengel H, Bosso-Lefevre C, Grady G, Szenker-Ravi E, Li H, Pierce S, et al. (2020). Loss-of-function mutations in UDP-Glucose 6-Dehydrogenase cause recessive developmental epileptic encephalopathy. Nat Commun, 11, 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hino-Fukuyo N, Kikuchi A, Arai-Ichinoi N, Niihori T, Sato R, Suzuki T, et al. (2015). Genomic analysis identifies candidate pathogenic variants in 9 of 18 patients with unexplained West syndrome. Human Genet, 134, 649–658. [DOI] [PubMed] [Google Scholar]
- Hirano Y, Oguni H, Shiota M, Nishikawa A, & Osawa M (2015). Ketogenic diet therapy can improve ACTH-resistant West syndrome in Japan. Brain Dev, 37, 18–22. [DOI] [PubMed] [Google Scholar]
- Holmes GL, & Weber DA (1986). Effects of ACTH on seizure susceptibility in the developing brain. Ann Neurol, 20, 82–88. [DOI] [PubMed] [Google Scholar]
- Hong AM, Turner Z, Hamdy RF, & Kossoff EH (2010). Infantile spasms treated with the ketogenic diet: prospective single-center experience in 104 consecutive infants. Epilepsia, 51, 1403–1407. [DOI] [PubMed] [Google Scholar]
- Hosford DA, Clark S, Cao Z, Wilson WA, Lin F-H, Morrisett RA, et al. (1992). The role of GABAB receptor activation in absence seizures of lethargic (lh/lh) mice. Science, 257, 398–401. [DOI] [PubMed] [Google Scholar]
- Howell KB, McMahon JM, Carvill GL, Tambunan D, Mackay MT, Rodriguez-Casero V, et al. (2015). SCN2A encephalopathy: A major cause of epilepsy of infancy with migrating focal seizures. Neurology, 85, 958–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain SA (2018). Treatment of infantile spasms. Epilepsia Open, 3, 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain SA, Lay J, Cheng E, Weng J, Sankar R, & Baca CB (2017a). Recognition of infantile spasms is often delayed: The ASSIST study. J Pediatr, 190, 215–221 e211. [DOI] [PubMed] [Google Scholar]
- Hussain SA, Shinnar S, Kwong G, Lerner JT, Matsumoto JH, Wu JY, et al. (2014). Treatment of infantile spasms with very high dose prednisolone before high dose adrenocorticotropic hormone. Epilepsia, 55, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain SA, Shin JH, Shih EJ, Murata KK, Sewak S, Kezele ME, et al. (2016). Limited efficacy of the ketogenic diet in the treatment of highly refractory epileptic spasms. Seizure, 35, 59–64. [DOI] [PubMed] [Google Scholar]
- Hussain SA, Tsao J, Li M, Schwarz MD, Zhou R, Wu JY, et al. (2017b). Risk of vigabatrin-associated brain abnormalities on MRI in the treatment of infantile spasms is dose-dependent. Epilepsia, 58, 674–682. [DOI] [PubMed] [Google Scholar]
- Hussain SA, Zhou R, Jacobson C, Weng J, Cheng E, Lay J, et al. (2015). Perceived efficacy of cannabidiol-enriched cannabis extracts for treatment of pediatric epilepsy: A potential role for infantile spasms and Lennox-Gastaut syndrome. Epilepsy Behav, 47, 138–141. [DOI] [PubMed] [Google Scholar]
- Hussain SA, Schmid E, Peters JM, Goyal M, Bebin EM, Northrup H, et al. (2018). High vigabatrin dosage is associated with lower risk of infantile spasms relapse among children with tuberous sclerosis complex. Epilepsy Res, 148, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobas DA, Iacobas S, Chachua T, Goletiani C, Sidyelyeva G, Velíšková J, et al. (2013). Prenatal corticosteroids modify glutamatergic and GABAergic synapse genomic fabric: insights from a novel animal model of infantile spasms. J Neuroendocrinol, 25, 964–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobas DA, Chachua T, Iacobas S, Benson MJ, Borges K, Velíšková J, et al. (2018). ACTH and PMX53 recover synaptic transcriptome alterations in a rat model of infantile spasms. Sci Rep, 8, 5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel TR, Battaglia G, Fairbanks DW, & De Souza EB (1988). The ontogeny of brain receptors for corticotropin-releasing factor and the development of their functional association with adenylate cyclase. J Neurosci, 8, 4151–4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A, Kang JQ, Schornak CC, Hernandez CC, Shen W, Watkins JC, et al. (2017). A de novo missense mutation of GABRB2 causes early myoclonic encephalopathy. J Med Genet, 54, 202–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer A, & Appleton R (2016). Improving outcomes in infantile spasms: Role of pharmacotherapy. Paediatr Drugs, 18, 357–366. [DOI] [PubMed] [Google Scholar]
- Iype M, Saradakutty G, Kunju PA, Mohan D, Nair MK, George B, et al. (2016). Infantile spasms: A prognostic evaluation. Ann Indian Acad Neurol, 19, 228–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicot R, Shao LR, & Stafstrom CE (2020). Infantile Spasms: An Update on Pre-Clinical Models and EEG Mechanisms. Children (Basel), 7, pii: E5, doi: 10.3390/children7010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janve VS, Hernandez CC, Verdier KM, Hu N, & Macdonald RL (2016). Epileptic encephalopathy de novo GABRB mutations impair gamma-aminobutyric acid type A receptor function. Ann Neurol, 79, 806–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeavons PM, Bower BD, & Dimitrakoudi M (1973). Long-term prognosis of 150 cases of “West syndrome”. Epilepsia, 14, 153–164. [DOI] [PubMed] [Google Scholar]
- Jellinger K (1987). Neuropathological aspects of infantile spasms. Brain Dev, 9, 349–357. [DOI] [PubMed] [Google Scholar]
- Jensen FE (2014). Combination therapies: inhibitors of GABA transaminase and NKCC1 In USPTO; (Ed.), Google Patents (Vol. US8822539B2). United States: Children s Medical Center Corp. [Google Scholar]
- Jequier Gygax M, Klein BD, White HS, Kim M, & Galanopoulou AS (2014). Efficacy and tolerability of the galanin analog NAX 5055 in the multiple-hit rat model of symptomatic infantile spasms. Epilepsy Res, 108, 98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston MV, & Coyle JT (1980). Ontogeny of neurochemical markers for noradrenergic, GABAergic, and cholinergic neurons in neocortex lesioned with methylazoxymethanol acetate. J Neurochem, 34, 1429–1441. [DOI] [PubMed] [Google Scholar]
- Juhasz C, Chugani HT, Muzik O, & Chugani DC (2002). Hypotheses from functional neuroimaging studies. Int Rev Neurobiol, 49, 37–55. [DOI] [PubMed] [Google Scholar]
- Kábová R, Velíšková J, & Velíšek L (2000). Prenatal methotrexate exposure decreases seizure susceptibility in young rats of two strains. Exp Neurol, 161, 167–173. [DOI] [PubMed] [Google Scholar]
- Kábová R, Liptáková S, Šlamberová R, Pometlová M, & Velíšek L (1999). Age-specific N-methyl-D-aspartate-induced seizures: perspectives for the West syndrome model. Epilepsia, 40, 1357–1369. [DOI] [PubMed] [Google Scholar]
- Kapoor A, & Matthews SG (2005). Short periods of prenatal stress affect growth, behaviour and hypothalamo-pituitary-adrenal axis activity in male guinea pig offspring. J Physiol, 566, 967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A, Dunn E, Kostaki A, Andrews MH, & Matthews SG (2006). Fetal programming of hypothalamo-pituitary-adrenal function: prenatal stress and glucocorticoids. J Physiol, 572, 31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karvelas G, Lortie A, Scantlebury MH, Duy PT, Cossette P, & Carmant L (2009). A retrospective study on aetiology based outcome of infantile spasms. Seizure, 18, 197–201. [DOI] [PubMed] [Google Scholar]
- Kato K, Miya F, Hori I, Ieda D, Ohashi K, Negishi Y, et al. (2017). A novel missense mutation in the HECT domain of NEDD4L identified in a girl with periventricular nodular heterotopia, polymicrogyria and cleft palate. J Hum Genet, 62, 861–863. [DOI] [PubMed] [Google Scholar]
- Kato M (2015). Genotype-phenotype correlation in neuronal migration disorders and cortical dysplasias. Front Neurosci, 9, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, & Dobyns WB (2005). X-linked lissencephaly with abnormal genitalia as a tangential migration disorder causing intractable epilepsy: proposal for a new term, “interneuronopathy”. J Child Neurol, 20, 392–397. [DOI] [PubMed] [Google Scholar]
- Kato M, Das S, Petras K, Sawaishi Y, & Dobyns WB (2003). Polyalanine expansion of ARX associated with cryptogenic West syndrome. Neurology, 61, 267–276. [DOI] [PubMed] [Google Scholar]
- Kato M, Koyama N, Ohta M, Miura K, & Hayasaka K (2010). Frameshift mutations of the ARX gene in familial Ohtahara syndrome. Epilepsia, 51, 1679–1684. [DOI] [PubMed] [Google Scholar]
- Kato M, Saitoh S, Kamei A, Shiraishi H, Ueda Y, Akasaka M, et al. (2007). A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome). Am J Hum Genet, 81, 361–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Saitsu H, Murakami Y, Kikuchi K, Watanabe S, Iai M, et al. (2014). PIGA mutations cause early-onset epileptic encephalopathies and distinctive features. Neurology, 82, 1587–1596. [DOI] [PubMed] [Google Scholar]
- Katsarou AM, Li Q, Liu W, Moshe SL, & Galanopoulou AS (2018). Acquired parvalbumin-selective interneuronopathy in the multiple-hit model of infantile spasms: A putative basis for the partial responsiveness to vigabatrin analogs? Epilepsia Open, 3, 155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayyali HR, Gustafson M, Myers T, Thompson L, Williams M, & Abdelmoity A (2014). Ketogenic diet efficacy in the treatment of intractable epileptic spasms. Pediatr Neurol, 50, 224–227. [DOI] [PubMed] [Google Scholar]
- Kelley SA, & Knupp KG (2018). Infantile spasms-have we made progress? Curr Neurol Neurosci Rep, 18, 27. [DOI] [PubMed] [Google Scholar]
- Kempna P, & Fluck CE (2008). Adrenal gland development and defects. Best Pract Res Clin Endocrinol Metab, 22, 77–93. [DOI] [PubMed] [Google Scholar]
- Kerrigan JF, Ng YT, Prenger E, Krishnamoorthy KS, Wang NC, & Rekate HL (2007). Hypothalamic hamartoma and infantile spasms. Epilepsia, 48, 89–95. [DOI] [PubMed] [Google Scholar]
- Kim EH, Yum MS, Lee M, Kim EJ, Shim WH, & Ko TS (2017). A New Rat Model of Epileptic Spasms Based on Methylazoxymethanol-Induced Malformations of Cortical Development. Front Neurol, 8, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimizu T, Takahashi Y, Oboshi T, Horino A, Koike T, Yoshitomi S, et al. (2017). A case of early onset epileptic encephalopathy with de novo mutation in SLC35A2: Clinical features and treatment for epilepsy. Brain Dev, 39, 256–260. [DOI] [PubMed] [Google Scholar]
- Kistler-Heer V, Lauber ME, & Lichtensteiger W (1998). Different developmental patterns of melanocortin MC3 and MC4 receptor mRNA: predominance of Mc4 in fetal rat nervous system. J Neuroendocrinol, 10, 133–146. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Yanazawa M, Sugiyama N, Miura H, Iizuka-Kogo A, Kusaka M, et al. (2002). Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet, 32, 359–369. [DOI] [PubMed] [Google Scholar]
- Kivity S, Lerman P, Ariel R, Danziger Y, Mimouni M, & Shinnar S (2004). Long-term cognitive outcomes of a cohort of children with cryptogenic infantile spasms treated with high-dose adrenocorticotropic hormone. Epilepsia, 45, 255–262. [DOI] [PubMed] [Google Scholar]
- Knupp KG, Coryell J, Nickels KC, Ryan N, Leister E, Loddenkemper T, et al. (2016a). Response to treatment in a prospective national infantile spasms cohort. Ann Neurol, 79, 475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knupp KG, Leister E, Coryell J, Nickels KC, Ryan N, Juarez-Colunga E, et al. (2016b). Response to second treatment after initial failed treatment in a multicenter prospective infantile spasms cohort. Epilepsia, 57, 1834–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuth ED, & Etgen AM (2004). Neural and hormonal consequences of neonatal 5,7-dihydroxytryptamine may not be associated with serotonin depletion. Brain Res Dev Brain Res, 151, 203–208. [DOI] [PubMed] [Google Scholar]
- Ko A, Youn SE, Chung HJ, Kim SH, Lee JS, Kim HD, et al. (2018a). Vigabatrin and high-dose prednisolone therapy for patients with West syndrome. Epilepsy Res, 145, 127–133. [DOI] [PubMed] [Google Scholar]
- Ko A, Youn SE, Kim SH, Lee JS, Kim S, Choi JR, et al. (2018b). Targeted gene panel and genotype-phenotype correlation in children with developmental and epileptic encephalopathy. Epilepsy Res, 141, 48–55. [DOI] [PubMed] [Google Scholar]
- Kodera H, Ohba C, Kato M, Maeda T, Araki K, Tajima D, et al. (2016). De novo GABRA1 mutations in Ohtahara and West syndromes. Epilepsia, 57, 566–573. [DOI] [PubMed] [Google Scholar]
- Komoike Y, Fujii K, Nishimura A, Hiraki Y, Hayashidani M, Shimojima K, et al. (2010). Zebrafish gene knockdowns imply roles for human YWHAG in infantile spasms and cardiomegaly. Genesis, 48, 233–243. [DOI] [PubMed] [Google Scholar]
- Koo B, Hwang PA, & Logan WJ (1993). Infantile spasms: outcome and prognostic factors of cryptogenic and symptomatic groups. Neurology, 43, 2322–2327. [DOI] [PubMed] [Google Scholar]
- Korosi A, & Baram TZ (2008). The central corticotropin releasing factor system during development and adulthood. Eur J Pharmacol, 583, 204–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossoff EH, Hartman AL, Rubenstein JE, & Vining EP (2009). High-dose oral prednisolone for infantile spasms: an effective and less expensive alternative to ACTH. Epilepsy Behav, 14, 674–676. [DOI] [PubMed] [Google Scholar]
- Kossoff EH, Pyzik PL, McGrogan JR, Vining EP, & Freeman JM (2002a). Efficacy of the ketogenic diet for infantile spasms. Pediatrics, 109, 780–783. [DOI] [PubMed] [Google Scholar]
- Kossoff EH, Pyzik PL, Furth SL, Hladky HD, Freeman JM, & Vining EP (2002b). Kidney stones, carbonic anhydrase inhibitors, and the ketogenic diet. Epilepsia, 43, 1168–1171. [DOI] [PubMed] [Google Scholar]
- Kossoff EH, Zupec-Kania BA, Auvin S, Ballaban-Gil KR, Christina Bergqvist AG, Blackford R, et al. (2018). Optimal clinical management of children receiving dietary therapies for epilepsy: Updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open, 3, 175–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger DA, Wilfong AA, Holland-Bouley K, Anderson AE, Agricola K, Tudor C, et al. (2013). Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol, 74, 679–687. [DOI] [PubMed] [Google Scholar]
- Kurian MA, Meyer E, Vassallo G, Morgan NV, Prakash N, Pasha S, et al. (2010). Phospholipase C beta 1 deficiency is associated with early-onset epileptic encephalopathy. Brain, 133, 2964–2970. [DOI] [PubMed] [Google Scholar]
- Kwon HH, Lee T, Hong J, Kim DW, & Kang JW (2018). Long-term prenatal stress increases susceptibility of N-methyl-D-aspartic acid-induced spasms in infant rats. Korean J Pediatr, 61, 150–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HH, Neupane C, Shin J, Gwon DH, Yin Y, Shin N, et al. (2019). Calpain-2 as a treatment target in prenatal stress-induced epileptic spasms in infant rats. Exp Neurobiol, 28, 529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagae L, Verhelst H, Ceulemans B, De Meirleir L, Nassogne MC, De Borchgrave V, et al. (2010). Treatment and long term outcome in West syndrome: the clinical reality. A multicentre follow up study. Seizure, 19, 159–164. [DOI] [PubMed] [Google Scholar]
- Lee C, Frost JJ, Le JT, Hrachovy R, & Swann JW (2014). (1–3)insulin-like growth factor 1 suppresses seizures and hypsarrhythmia in an animal model of infantile spasms In American Epilepsy Society Annual Meeting (pp. C04). Seattle, WA. [Google Scholar]
- Lee CL, Frost JD Jr., Swann JW, & Hrachovy RA (2008). A new animal model of infantile spasms with unprovoked persistent seizures. Epilepsia, 49, 298–307. [DOI] [PubMed] [Google Scholar]
- Lee J, Lee JH, Yu HJ, & Lee M (2013). Prognostic factors of infantile spasms: Role of treatment options including a ketogenic diet. Brain Dev, 35, 821–826. [DOI] [PubMed] [Google Scholar]
- Lee M, Yum MS, Woo DC, Shim WH, Ko TS, & Velíšek L (2018). In vivo multimodal magnetic resonance imaging changes after N-Methyl-d-Aspartate-triggered spasms in infant rats. Front Neurol, 9, 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Kim MJ, Kim EJ, Woo DC, Yum MS, & Ko TS (2019). How can methylprednisolone work on epileptic spasms with malformation of cortical development? Eur J Neurosci, 10.1111/ejn.14539. [DOI] [PubMed] [Google Scholar]
- Lemke JR, Hendrickx R, Geider K, Laube B, Schwake M, Harvey RJ, et al. (2014). GRIN2B mutations in West syndrome and intellectual disability with focal epilepsy. Ann Neurol, 75, 147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke JR, Geider K, Helbig KL, Heyne HO, Schutz H, Hentschel J, et al. (2016). Delineating the GRIN1 phenotypic spectrum: A distinct genetic NMDA receptor encephalopathy. Neurology, 86, 2171–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev D, Weigl Y, Hasan M, Gak E, Davidovich M, Vinkler C, et al. (2007). A novel missense mutation in the NDP gene in a child with Norrie disease and severe neurological involvement including infantile spasms. Am J Med Genet A, 143A, 921–924. [DOI] [PubMed] [Google Scholar]
- Levine S, & Saltzman A (2004). Pyridoxine (vitamin B6) neurotoxicity: enhancement by protein-deficient diet. J Appl Toxicol, 24, 497–500. [DOI] [PubMed] [Google Scholar]
- Li D, Yuan H, Ortiz-Gonzalez XR, Marsh ED, Tian L, McCormick EM, et al. (2016). GRIN2D recurrent de novo dominant mutation causes a severe epileptic encephalopathy treatable with NMDA receptor channel blockers. Am J Hum Genet, 99, 802–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Li Z, Zhi X, Du Y, Lin Z, & Wu J (2018). Identification of de novo DNMT3A mutations that cause West syndrome by using whole-exome sequencing. Mol Neurobiol, 55, 2483–2493. [DOI] [PubMed] [Google Scholar]
- Lott IT (2012). Neurological phenotypes for Down syndrome across the life span. Prog Brain Res, 197, 101–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovenberg TW, Liaw CW, Grigoriadis DE, Clevenger W, Chalmers DT, De Souza EB, et al. (1995). Cloning and characterization of a functionally distinct corticotropin-releasing factor receptor subtype from rat brain. Proc Natl Acad Sci U S A, 92, 836–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozovaya N, Gataullina S, Tsintsadze T, Tsintsadze V, Pallesi-Pocachard E, Minlebaev M, et al. (2014). Selective suppression of excessive GluN2C expression rescues early epilepsy in a tuberous sclerosis murine model. Nat Commun, 5, 4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lux AL, & Osborne JP (2004). A proposal for case definitions and outcome measures in studies of infantile spasms and West syndrome: consensus statement of the West Delphi group. Epilepsia, 45, 1416–1428. [DOI] [PubMed] [Google Scholar]
- Lux AL, Edwards SW, Hancock E, Johnson AL, Kennedy CR, Newton RW, et al. (2004). The United Kingdom Infantile Spasms Study comparing vigabatrin with prednisolone or tetracosactide at 14 days: a multicentre, randomised controlled trial. Lancet, 364, 1773–1778. [DOI] [PubMed] [Google Scholar]
- Lux AL, Edwards SW, Hancock E, Johnson AL, Kennedy CR, Newton RW, et al. (2005). The United Kingdom Infantile Spasms Study (UKISS) comparing hormone treatment with vigabatrin on developmental and epilepsy outcomes to age 14 months: a multicentre randomised trial. Lancet Neurol, 4, 712–717. [DOI] [PubMed] [Google Scholar]
- Mackay MT, Weiss SK, Adams-Webber T, Ashwal S, Stephens D, Ballaban-Gill K, et al. (2004). Practice parameter: medical treatment of infantile spasms: report of the American Academy of Neurology and the Child Neurology Society. Neurology, 62, 1668–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mareš P, & Velíšek L (1992). N-methyl-D-aspartate (NMDA)-induced seizures in developing rats. Brain Res Dev Brain Res, 65, 185–189. [DOI] [PubMed] [Google Scholar]
- Marsh ED, & Golden JA (2009). Developing an animal model for infantile spasms: pathogenesis, problems and progress. Dis Model Mech, 2, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh ED, Nasrallah MP, Walsh C, Murray KA, Nicole Sunnen C, McCoy A, et al. (2016). Developmental interneuron subtype deficits after targeted loss of Arx. BMC Neurosci, 17, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh ED, Fulp C, Gomez E, Nasrallah I, Minarcik J, Sudi J, et al. (2009). Targeted loss of Arx results in a developmental epilepsy mouse model and recapitulates the human phenotype in heterozygous females. Brain, 132, 1563–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CR, Young EJ, Pani AM, Freckmann ML, Lacassie Y, Howald C, et al. (2008). Infantile spasms is associated with deletion of the MAGI2 gene on chromosome 7q11.23-q21.11. Am J Hum Genet, 83, 106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto H, & Higa HH (1966). Studies on methylazoxymethanol, the aglycone of cycasin: Methylation of nucleic acids in vitro. Biochem J, 98, 20c–22c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan PO, & Matthews SG (2018). Prenatal stress, glucocorticoids, and developmental programming of the stress response. Endocrinology, 159, 69–82. [DOI] [PubMed] [Google Scholar]
- Meencke HJ, & Gerhard C (1985). Morphological aspects of aetiology and the course of infantile spasms (West-syndrome). Neuropediatrics, 16, 59–66. [DOI] [PubMed] [Google Scholar]
- Mehta D, Jackson R, Paul G, Shi J, & Sabbagh M (2017). Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin Investig Drugs, 26, 735–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, et al. (2008). Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci, 28, 5422–5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud JL, Lachance M, Hamdan FF, Carmant L, Lortie A, Diadori P, et al. (2014). The genetic landscape of infantile spasms. Hum Mol Genet, 23, 4846–4858. [DOI] [PubMed] [Google Scholar]
- Millichap JJ, & Millichap JG (2015). Hypsarhythmia or Hypsarrhythmia? Pediatr Neurol Briefs, 29, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millichap JJ, Miceli F, De Maria M, Keator C, Joshi N, Tran B, et al. (2017). Infantile spasms and encephalopathy without preceding neonatal seizures caused by KCNQ2 R198Q, a gain-of-function variant. Epilepsia, 58, e10–e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minabe S, Sato M, Inoue N, Watanabe Y, Magata F, Matsuda F, et al. (2019). Neonatal estrogen causes irreversible male infertility via specific suppressive action on hypothalamic Kiss1 neurons. Endocrinology, 160, 1223–1233. [DOI] [PubMed] [Google Scholar]
- Miyatake S, Koshimizu E, Fujita A, Fukai R, Imagawa E, Ohba C, et al. (2015). Detecting copy-number variations in whole-exome sequencing data using the eXome Hidden Markov Model: an ‘exome-first’ approach. J Hum Genet, 60, 175–182. [DOI] [PubMed] [Google Scholar]
- Moavero R, Napolitano A, Cusmai R, Vigevano F, Figa-Talamanca L, Calbi G, et al. (2016). White matter disruption is associated with persistent seizures in tuberous sclerosis complex. Epilepsy Behav, 60, 63–67. [DOI] [PubMed] [Google Scholar]
- Moey C, Topper S, Karn M, Johnson AK, Das S, Vidaurre J, et al. (2016). Reinitiation of mRNA translation in a patient with X-linked infantile spasms with a protein-truncating variant in ARX. Eur J Hum Genet, 24, 681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed BP, Scott RC, Desai N, Gutta P, & Patil S (2011). Seizure outcome in infantile spasms--a retrospective study. Epilepsia, 52, 746–752. [DOI] [PubMed] [Google Scholar]
- Mohn JL, Alexander J, Pirone A, Palka CD, Lee SY, Mebane L, et al. (2014). Adenomatous polyposis coli protein deletion leads to cognitive and autism-like disabilities. Mol Psychiatry, 19, 1133–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto M, An B, Ogami A, Shin N, Sugino Y, Sawai Y, et al. (2003). Infantile spasms in a patient with williams syndrome and craniosynostosis. Epilepsia, 44, 1459–1462. [DOI] [PubMed] [Google Scholar]
- Mu C, Choudhary A, Charkhad B, Barrett K, Shearer J, Rho JM, et al. (2018). The ketogenic diet alters the gut microbiome in the triple-hit rodent model of infantile spasms In American Epilepsy Society Annual Meeting (pp. 3.361). New Orleans, LA. [Google Scholar]
- Muir AM, Myers CT, Nguyen NT, Saykally J, Craiu D, De Jonghe P, et al. (2019). Genetic heterogeneity in infantile spasms. Epilepsy Res, 156, 106181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muncy J, Butler IJ, & Koenig MK (2009). Rapamycin reduces seizure frequency in tuberous sclerosis complex. J Child Neurol, 24, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers KA (2019). PLCB1 biallelic point mutations cause West syndrome. Pediatr Neurol, 91, 62–64. [DOI] [PubMed] [Google Scholar]
- Mytinger JR, Hussain SA, Islam MP, Millichap JJ, Patel AD, Ryan NR, et al. (2015). Improving the inter-rater agreement of hypsarrhythmia using a simplified EEG grading scale for children with infantile spasms. Epilepsy Res, 116, 93–98. [DOI] [PubMed] [Google Scholar]
- Nagamitsu S, Matsuishi T, Yamashita Y, Shimizu T, Iwanaga R, Murakami Y, et al. (2001). Decreased cerebrospinal fluid levels of beta-endorphin and ACTH in children with infantile spasms. J Neural Transm, 108, 363–371. [DOI] [PubMed] [Google Scholar]
- Nakashima M, Kouga T, Lourenco CM, Shiina M, Goto T, Tsurusaki Y, et al. (2016). De novo DNM1 mutations in two cases of epileptic encephalopathy. Epilepsia, 57, e18–23. [DOI] [PubMed] [Google Scholar]
- Nalin A, Facchinetti F, Galli V, Petraglia F, Storchi R, & Genazzani AR (1985). Reduced ACTH content in cerebrospinal fluid of children affected by cryptogenic infantile spasms with hypsarrhythmia. Epilepsia, 26, 446–449. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, & Hyman SE (2010). Animal models of neuropsychiatric disorders. Nat Neurosci, 13, 1161–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonoda Y, Saito Y, Nagai S, Sasaki M, Iwasaki T, Matsumoto N, et al. (2013). Progressive diffuse brain atrophy in West syndrome with marked hypomyelination due to SPTAN1 gene mutation. Brain Dev, 35, 280–283. [DOI] [PubMed] [Google Scholar]
- Noorlander CW, De Graan PN, Middeldorp J, Van Beers JJ, & Visser GH (2006). Ontogeny of hippocampal corticosteroid receptors: effects of antenatal glucocorticoids in human and mouse. J Comp Neurol, 499, 924–932. [DOI] [PubMed] [Google Scholar]
- Nordli DR Jr., Kuroda MM, Carroll J, Koenigsberger DY, Hirsch LJ, Bruner HJ, et al. (2001). Experience with the ketogenic diet in infants. Pediatrics, 108, 129–133. [DOI] [PubMed] [Google Scholar]
- O’Callaghan FJ, Lux AL, Darke K, Edwards SW, Hancock E, Johnson AL, et al. (2011). The effect of lead time to treatment and of age of onset on developmental outcome at 4 years in infantile spasms: evidence from the United Kingdom Infantile Spasms Study. Epilepsia, 52, 1359–1364. [DOI] [PubMed] [Google Scholar]
- O’Callaghan FJ, Edwards SW, Alber FD, Hancock E, Johnson AL, Kennedy CR, et al. (2017). Safety and effectiveness of hormonal treatment versus hormonal treatment with vigabatrin for infantile spasms (ICISS): a randomised, multicentre, open-label trial. Lancet Neurol, 16, 33–42. [DOI] [PubMed] [Google Scholar]
- O’Callaghan FJ, Edwards SW, Alber FD, Cortina Borja M, Hancock E, Johnson AL, et al. (2018a). Vigabatrin with hormonal treatment versus hormonal treatment alone (ICISS) for infantile spasms: 18-month outcomes of an open-label, randomised controlled trial. Lancet Child Adolesc Health, 2, 715–725. [DOI] [PubMed] [Google Scholar]
- O’Callaghan FJK, Edwards SW, Alber FD, Cortina Borja M, Hancock E, Johnson AL, et al. (2018b). Vigabatrin with hormonal treatment versus hormonal treatment alone (ICISS) for infantile spasms: 18-month outcomes of an open-label, randomised controlled trial. Lancet Child Adolesc Health, 2, 715–725. [DOI] [PubMed] [Google Scholar]
- Ohya T, Nagai T, Araki Y, Yanagawa T, Tanabe T, Iyoda K, et al. (2009). A pilot study on the changes in immunity after ACTH therapy in patients with West syndrome. Brain Dev, 31, 739–743. [DOI] [PubMed] [Google Scholar]
- Ojeda SR, & Urbanski HF (1994). Puberty in the rat In Knobil E (Ed.), The Physiology of Reproduction (Vol. I, pp. 363–411). New York: Raven Press. [Google Scholar]
- Olivetti PR, Maheshwari A, & Noebels JL (2014). Neonatal estradiol stimulation prevents epilepsy in Arx model of X-linked infantile spasms syndrome. Sci Transl Med, 6, 220ra212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono T, Moshe SL, & Galanopoulou AS (2011). Carisbamate acutely suppresses spasms in a rat model of symptomatic infantile spasms. Epilepsia, 52, 1678–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne JP, Lux AL, Edwards SW, Hancock E, Johnson AL, Kennedy CR, et al. (2010). The underlying etiology of infantile spasms (West syndrome): information from the United Kingdom Infantile Spasms Study (UKISS) on contemporary causes and their classification. Epilepsia, 51, 2168–2174. [DOI] [PubMed] [Google Scholar]
- Osborne JP, Edwards SW, Dietrich Alber F, Hancock E, Johnson AL, Kennedy CR, et al. (2019). The underlying etiology of infantile spasms (West syndrome): Information from the International Collaborative Infantile Spasms Study (ICISS). Epilepsia, 60, 1861–1869. [DOI] [PubMed] [Google Scholar]
- Ounissi M, Rodrigues C, Bienayme H, Duhamel P, Pons G, Dulac O, et al. (2019). Proposition of a minimal effective dose of vigabatrin for the treatment of infantile spasms using pediatric and adult pharmacokinetic data. J Clin Pharmacol, 59, 177–188. [DOI] [PubMed] [Google Scholar]
- Paciorkowski AR, Thio LL, & Dobyns WB (2011a). Genetic and biologic classification of infantile spasms. Pediatr Neurol, 45, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paciorkowski AR, Shafrir Y, Hrivnak J, Patterson MC, Tennison MB, Clark HB, et al. (2011b). Massive expansion of SCA2 with autonomic dysfunction, retinitis pigmentosa, and infantile spasms. Neurology, 77, 1055–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paciorkowski AR, Traylor RN, Rosenfeld JA, Hoover JM, Harris CJ, Winter S, et al. (2013). MEF2C Haploinsufficiency features consistent hyperkinesis, variable epilepsy, and has a role in dorsal and ventral neuronal developmental pathways. Neurogenetics, 14, 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papandreou A, McTague A, Trump N, Ambegaonkar G, Ngoh A, Meyer E, et al. (2016). GABRB3 mutations: a new and emerging cause of early infantile epileptic encephalopathy. Dev Med Child Neurol, 58, 416–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parasrampuria DA, Benet LZ, & Sharma A (2018). Why drugs fail in late stages of development: Case study analyses from the last decade and recommendations. AAPS J, 20, 46. [DOI] [PubMed] [Google Scholar]
- Partikian A, & Mitchell WG (2007). Major adverse events associated with treatment of infantile spasms. J Child Neurol, 22, 1360–1366. [DOI] [PubMed] [Google Scholar]
- Pavone L, Mollica F, Incorpora G, & Pampiglione G (1980). Infantile spasms syndrome in monozygotic twins. Arch Dis Child, 55, 870–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavone P, Striano P, Falsaperla R, Pavone L, & Ruggieri M (2014). Infantile spasms syndrome, West syndrome and related phenotypes: what we know in 2013. Brain Dev, 36, 739–751. [DOI] [PubMed] [Google Scholar]
- Pearl PL, Poduri A, Prabhu SP, Harini C, Goldstein R, Atkinson RM, et al. (2018). White matter spongiosis with vigabatrin therapy for infantile spasms. Epilepsia, 59, e40–e44. [DOI] [PubMed] [Google Scholar]
- Pearl PL, Vezina LG, Saneto RP, McCarter R, Molloy-Wells E, Heffron A, et al. (2009). Cerebral MRI abnormalities associated with vigabatrin therapy. Epilepsia, 50, 184–194. [DOI] [PubMed] [Google Scholar]
- Pellock JM, Hrachovy R, Shinnar S, Baram TZ, Bettis D, Dlugos DJ, et al. (2010). Infantile spasms: a U.S. consensus report. Epilepsia, 51, 2175–2189. [DOI] [PubMed] [Google Scholar]
- Perrault I, Hamdan FF, Rio M, Capo-Chichi JM, Boddaert N, Decarie JC, et al. (2014). Mutations in DOCK7 in individuals with epileptic encephalopathy and cortical blindness. Am J Hum Genet, 94, 891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perucca P, & Perucca E (2019). Identifying mutations in epilepsy genes: Impact on treatment selection. Epilepsy Res, 152, 18–30. [DOI] [PubMed] [Google Scholar]
- Pires ME, Ilea A, Bourel E, Bellavoine V, Merdariu D, Berquin P, et al. (2013). Ketogenic diet for infantile spasms refractory to first-line treatments: an open prospective study. Epilepsy Res, 105, 189–194. [DOI] [PubMed] [Google Scholar]
- Pirone A, Alexander J, Lau LA, Hampton D, Zayachkivsky A, Yee A, et al. (2017). APC conditional knock-out mouse is a model of infantile spasms with elevated neuronal beta-catenin levels, neonatal spasms, and chronic seizures. Neurobiol Dis, 98, 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirone A, Alexander JM, Koenig JB, Cook-Snyder DR, Palnati M, Wickham RJ, et al. (2018). Social Stimulus Causes Aberrant Activation of the Medial Prefrontal Cortex in a Mouse Model With Autism-Like Behaviors. Front Synaptic Neurosci, 10, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platzer K, Yuan H, Schutz H, Winschel A, Chen W, Hu C, et al. (2017). GRIN2B encephalopathy: novel findings on phenotype, variant clustering, functional consequences and treatment aspects. J Med Genet, 54, 460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier K, Eisermann M, Caubel I, Kaminska A, Peudonnier S, Boddaert N, et al. (2008). Combination of infantile spasms, non-epileptic seizures and complex movement disorder: a new case of ARX-related epilepsy. Epilepsy Res, 80, 224–228. [DOI] [PubMed] [Google Scholar]
- Poke G, King C, Muir A, de Valles-Ibanez G, Germano M, Moura de Souza CF, et al. (2019). The epileptology of GNB5 encephalopathy. Epilepsia, 60, e121–e127. [DOI] [PubMed] [Google Scholar]
- Porter BE, & Jacobson C (2013). Report of a parent survey of cannabidiol-enriched cannabis use in pediatric treatment-resistant epilepsy. Epilepsy Behav, 29, 574–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prezioso G, Carlone G, Zaccara G, & Verrotti A (2018). Efficacy of ketogenic diet for infantile spasms: A systematic review. Acta Neurol Scand, 137, 4–11. [DOI] [PubMed] [Google Scholar]
- Price MG, Yoo JW, Burgess DL, Deng F, Hrachovy RA, Frost JD Jr., et al. (2009). A triplet repeat expansion genetic mouse model of infantile spasms syndrome, Arx(GCG)10+7, with interneuronopathy, spasms in infancy, persistent seizures, and adult cognitive and behavioral impairment. J Neurosci, 29, 8752–8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffo E, Coppola A, Ono T, Briggs SW, & Galanopoulou AS (2011). A pulse rapamycin therapy for infantile spasms and associated cognitive decline. Neurobiol Dis, 43, 322–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramgopal S, Shah A, Zarowski M, Vendrame M, Gregas M, Alexopoulos AV, et al. (2012). Diurnal and sleep/wake patterns of epileptic spasms in different age groups. Epilepsia, 53, 1170–1177. [DOI] [PubMed] [Google Scholar]
- Reiter E, Tiefenthaler M, Freillinger M, Bernert G, Seidl R, & Hauser E (2000). Familial idiopathic West syndrome. J Child Neurol, 15, 249–252. [DOI] [PubMed] [Google Scholar]
- Rensing N, Johnson KJ, Foutz TJ, Friedman JL, Galindo R, & Wong M (2020). Early developmental electroencephalography abnormalities, neonatal seizures, and induced spasms in a mouse model of tuberous sclerosis complex. Epilepsia, 10.1111/epi.16495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riikonen R (1982). A long-term follow-up study of 214 children with the syndrome of infantile spasms. Neuropediatrics, 13, 14–23. [DOI] [PubMed] [Google Scholar]
- Riikonen R (2004). Infantile spasms: therapy and outcome. J Child Neurol, 19, 401–404. [DOI] [PubMed] [Google Scholar]
- Riikonen R (2014). Recent advances in the pharmacotherapy of infantile spasms. CNS Drugs, 28, 279–290. [DOI] [PubMed] [Google Scholar]
- Riikonen R (2015). Long-term outcome in children with infantile spasms treated with vigabatrin: A cohort of 180 patients. Epilepsia, 56, 807–809. [DOI] [PubMed] [Google Scholar]
- Riikonen R, & Donner M (1979). Incidence and aetiology of infantile spasms from 1960 to 1976: a population study in Finland. Dev Med Child Neurol, 21, 333–343. [DOI] [PubMed] [Google Scholar]
- Riikonen R, & Donner M (1980). ACTH therapy in infantile spasms: side effects. Arch Dis Child, 55, 664–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riikonen R, Rener-Primec Z, Carmant L, Dorofeeva M, Hollody K, Szabo I, et al. (2015). Does vigabatrin treatment for infantile spasms cause visual field defects? An international multicentre study. Dev Med Child Neurol, 57, 60–67. [DOI] [PubMed] [Google Scholar]
- Riikonen R, Wallden T, & Kokkonen H (2016). Infantile spasms and 15q11.2q13.1 chromosome duplication in two successive generations. Eur J Paediatr Neurol, 20, 164–167. [DOI] [PubMed] [Google Scholar]
- Ronce N, Raynaud M, Toutain A, Moizard MP, Colleaux L, Gendrot C, et al. (1999). Evidence for a new X-linked mental retardation gene in Xp21-Xp22: clinical and molecular data in one family. Am J Med Genet, 83, 132–137. [DOI] [PubMed] [Google Scholar]
- Ruggieri M, Iannetti P, Clementi M, Polizzi A, Incorpora G, Spalice A, et al. (2009). Neurofibromatosis type 1 and infantile spasms. Childs Nerv Syst, 25, 211–216. [DOI] [PubMed] [Google Scholar]
- Salehi A, Faizi M, Belichenko PV, & Mobley WC (2007). Using mouse models to explore genotype-phenotype relationship in Down syndrome. Ment Retard Dev Disabil Res Rev, 13, 207–214. [DOI] [PubMed] [Google Scholar]
- Samueli S, Dressler A, Groppel G, Scholl T, & Feucht M (2018). Everolimus in infants with tuberous sclerosis complex-related West syndrome: First results from a single-center prospective observational study. Epilepsia, 59, e142–e146. [DOI] [PubMed] [Google Scholar]
- Scantlebury MH, & Moshé SL (2011). Model of infantile spasm syndrome In USPTO; (Ed.), (Vol. US7863499B2). United States: Albert Einstein College of Medicine. [Google Scholar]
- Scantlebury MH, Galanopoulou AS, Chudomelova L, Raffo E, Betancourth D, & Moshe SL (2010). A model of symptomatic infantile spasms syndrome. Neurobiol Dis, 37, 604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaumburg H, Kaplan J, Windebank A, Vick N, Rasmus S, & Pleasure D (1983). Sensory neuropathy from pyridoxine abuse. A new megavitamin syndrome. New Engl J Med, 309, 445–448. [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Wallace RH, Phillips FL, Hewson P, Reardon K, Parasivam G, et al. (2002). X-linked myoclonic epilepsy with spasticity and intellectual disability: mutation in the homeobox gene ARX. Neurology, 59, 348–356. [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. (2017). ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia, 58, 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seltzer LE, Ma M, Ahmed S, Bertrand M, Dobyns WB, Wheless J, et al. (2014). Epilepsy and outcome in FOXG1-related disorders. Epilepsia, 55, 1292–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang NX, Zou LP, Zhao JB, Zhang F, & Li H (2010). Association between prenatal stress and infantile spasms: a case-control study in China. Pediatr Neurol, 42, 181–186. [DOI] [PubMed] [Google Scholar]
- Shbarou R, & Mikati MA (2016). The expanding clinical spectrum of genetic pediatric epileptic encephalopathies. Semin Pediatr Neurol, 23, 134–142. [DOI] [PubMed] [Google Scholar]
- Sherr EH (2003). The ARX story (epilepsy, mental retardation, autism, and cerebral malformations): one gene leads to many phenotypes. Curr Opin Pediatr, 15, 567–571. [DOI] [PubMed] [Google Scholar]
- Shi XY, Yang XF, Tomonoh Y, Hu LY, Ju J, Hirose S, et al. (2015). Development of a mouse model of infantile spasms induced by N-methyl-D-aspartate. Epilepsy Res, 118, 29–33. [DOI] [PubMed] [Google Scholar]
- Shi XY, Ju J, Zou LP, Wang J, Shang NX, Zhao JB, et al. (2016). Increased precipitation of spasms in an animal model of infantile spasms by prenatal stress exposure. Life Sci, 152, 171–177. [DOI] [PubMed] [Google Scholar]
- Simeone TA, Simeone KA, Stafstrom CE, & Rho JM (2018). Do ketone bodies mediate the anti-seizure effects of the ketogenic diet? Neuropharmacology, 133, 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RJ, Shrey DW, Hussain SA, & Lopour BA (2018). Quantitative characteristics of hypsarrhythmia in infantile spasms. Conf Proc IEEE Eng Med Biol Soc, 2018, 538–541. [DOI] [PubMed] [Google Scholar]
- Smith RJ, Sugijoto A, Rismanchi N, Hussain SA, Shrey DW, & Lopour BA (2017). Long-range temporal correlations reflect treatment response in the electroencephalogram of patients with infantile spasms. Brain Topogr, 30, 810–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snead OC 3rd., Benton JW, & Myers GJ (1983). ACTH and prednisone in childhood seizure disorders. Neurology, 33, 966–970. [DOI] [PubMed] [Google Scholar]
- Snead OC 3rd., Benton JW Jr., Hosey LC, Swann JW, Spink D, Martin D, et al. (1989). Treatment of infantile spasms with high-dose ACTH: efficacy and plasma levels of ACTH and cortisol. Neurology, 39, 1027–1031. [DOI] [PubMed] [Google Scholar]
- Snodgrass SR (1992). GABA and epilepsy: Their complex relationship and the evolution of our understanding. J Child Neurol, 7, 77–86. [DOI] [PubMed] [Google Scholar]
- Son GH, Geum D, Chung S, Kim EJ, Jo JH, Kim CM, et al. (2006). Maternal stress produces learning deficits associated with impairment of NMDA receptor-mediated synaptic plasticity. J Neurosci, 26, 3309–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JM, Hahn J, Kim SH, & Chang MJ (2017). Efficacy of treatments for infantile spasms: A systematic review. Clin Neuropharmacol, 40, 63–84. [DOI] [PubMed] [Google Scholar]
- Sorel L, & Dusaucy - Bauloye A (1958). A propos de 21 cas d’hypsarythmie de Gibbs. Son traitement spectaculaire par l’ACTH. Acta Neurol Belg, 58, 130 – 141. [PubMed] [Google Scholar]
- Sperling MR, Greenspan A, Cramer JA, Kwan P, Kalviainen R, Halford JJ, et al. (2010). Carisbamate as adjunctive treatment of partial onset seizures in adults in two randomized, placebo-controlled trials. Epilepsia, 51, 333–343. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, & Konkol RJ (1994). Infantile spasms in children with Down syndrome. Dev Med Child Neurol, 36, 576–585. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, & Holmes GL (2002). Infantile spasms: criteria for an animal model. Int Rev Neurobiol, 49, 391–411. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, & Sasaki-Adams DM (2003). NMDA-induced seizures in developing rats cause long-term learning impairment and increased seizure susceptibility. Epilepsy Res, 53, 129–137. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, Moshé SL, Swann JW, Nehlig A, Jacobs MP, & Schwartzkroin PA (2006). Models of pediatric epilepsies: strategies and opportunities. Epilepsia, 47, 1407–1414. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, Arnason BG, Baram TZ, Catania A, Cortez MA, Glauser TA, et al. (2011). Treatment of infantile spasms: emerging insights from clinical and basic science perspectives. J Child Neurol, 26, 1411–1421. [DOI] [PubMed] [Google Scholar]
- Stockler S, Plecko B, Gospe SM Jr., Coulter-Mackie M, Connolly M, van Karnebeek C, et al. (2011). Pyridoxine dependent epilepsy and antiquitin deficiency: clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up. Mol Genet Metab, 104, 48–60. [DOI] [PubMed] [Google Scholar]
- Stromme P, Mangelsdorf ME, Shaw MA, Lower KM, Lewis SM, Bruyere H, et al. (2002). Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat Genet, 30, 441–445. [DOI] [PubMed] [Google Scholar]
- Suleiman J, Brenner T, Gill D, Troedson C, Sinclair AJ, Brilot F, et al. (2011). Immune-mediated steroid-responsive epileptic spasms and epileptic encephalopathy associated with VGKC-complex antibodies. Dev Med Child Neurol, 53, 1058–1060. [DOI] [PubMed] [Google Scholar]
- Sunnen CN, Simonet JC, Marsh ED, & Golden JA (2014). Arx is required for specification of the zona incerta and reticular nucleus of the thalamus. J Neuropathol Exp Neurol, 73, 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swann JW (1995). Synaptogenesis and epileptogenesis in developing neural networks In Schwartzkroin PA, Moshé SL, Noebells JL & Swann JW (Eds.), Brain Development and Epilepsy (pp. 195–233). New York-Oxford: Oxford University Press. [Google Scholar]
- Swann JW, Le JT, & Frost JD Jr. (2017). Infantile spasms: ACTH dose response relationships In American Epilepsy Society Annual Meeting (pp. 1.023). Washington, DC. [Google Scholar]
- Takata A, Nakashima M, Saitsu H, Mizuguchi T, Mitsuhashi S, Takahashi Y, et al. (2019). Comprehensive analysis of coding variants highlights genetic complexity in developmental and epileptic encephalopathy. Nat Commun, 10, 2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi A, Okamoto N, Fujinaga S, Morita H, Shimizu J, Akiyama T, et al. (2015). Progressive brain atrophy in Schinzel-Giedion syndrome with a SETBP1 mutation. Eur J Med Genet, 58, 369–371. [DOI] [PubMed] [Google Scholar]
- Tang-Wai R, Mailo J, & Rosenblatt B (2017). Breaking the cycle: A comparison between intravenous immunoglobulins and high dosage prednisone in the treatment of medically intractable epilepsy in children. Seizure, 47, 34–41. [DOI] [PubMed] [Google Scholar]
- Taylor NR, Loraine JA, & Robertson HA (1953). The estimation of ACTH in human pituitary tissue. J Endocrinol, 9, xxiii–xxiv. [PubMed] [Google Scholar]
- Than KD, Kossoff EH, Rubenstein JE, Pyzik PL, McGrogan JR, & Vining EP (2005). Can you predict an immediate, complete, and sustained response to the ketogenic diet? Epilepsia, 46, 580–582. [DOI] [PubMed] [Google Scholar]
- Tharp BR (2002). Neonatal seizures and syndromes. Epilepsia, 43 Suppl 3, 2–10. [DOI] [PubMed] [Google Scholar]
- Thomas AC, Heux P, Santos C, Arulvasan W, Solanky N, Carey ME, et al. (2018). Widespread dynamic and pleiotropic expression of the melanocortin-1-receptor (MC1R) system is conserved across chick, mouse and human embryonic development. Birth Defects Res, 110, 443–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J, & Holmes GL (1987). Failure of ACTH to alter transfer kindling in the immature brain. Epilepsia, 28, 17–19. [DOI] [PubMed] [Google Scholar]
- Tsuji M, Takahashi Y, Watabe AM, & Kato F (2016). Enhanced long-term potentiation in mature rats in a model of epileptic spasms with betamethasone-priming and postnatal N-methyl-d-aspartate administration. Epilepsia, 57, 495–505. [DOI] [PubMed] [Google Scholar]
- Van den Berg HW, & Ball CR (1972). The effect of methylazoxymethanol acetate on DNA synthesis and cell proliferation of synchronous HeLa cells. Mutat Res, 16, 381–390. [DOI] [PubMed] [Google Scholar]
- van der Poest Clement EA, Sahin M, & Peters JM (2018). Vigabatrin for Epileptic Spasms and Tonic Seizures in Tuberous Sclerosis Complex. J Child Neurol, 33, 519–524. [DOI] [PubMed] [Google Scholar]
- Vanhatalo S, Paakkonen L, & Nousiainen I (1999). Visual field constriction in children treated with vigabatrin. Neurology, 52, 1713–1714. [DOI] [PubMed] [Google Scholar]
- Vanhatalo S, Nousiainen I, Eriksson K, Rantala H, Vainionpaa L, Mustonen K, et al. (2002). Visual field constriction in 91 Finnish children treated with vigabatrin. Epilepsia, 43, 748–756. [DOI] [PubMed] [Google Scholar]
- Velíšek L (2011). Prenatal corticosteroid exposure alters early developmental seizures and behavior. Epilepsy Res, 95, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velíšek L, & Mareš P (1995). Age-dependent anticonvulsant action of clonazepam in the N-methyl-D-aspartate model of seizures. Pharmacol Biochem Behav, 52, 291–296. [DOI] [PubMed] [Google Scholar]
- Velíšek L, Jehle K, Asche S, & Velíšková J (2007). Model of infantile spasms induced by N-methyl-D-aspartic acid in prenatally impaired brain. Ann Neurol, 61, 109–119. [DOI] [PubMed] [Google Scholar]
- Velíšek L, Chachua T, Yum MS, Poon KL, & Velíšková J (2010). Model of cryptogenic infantile spasms after prenatal corticosteroid priming. Epilepsia, 51 Suppl 3, 145–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verešová S, Kábová R, & Velíšek L (1998). Proconvulsant effects induced by pyridoxine in young rats. Epilepsy Res, 29, 259–264. [DOI] [PubMed] [Google Scholar]
- Vergnes M, Marescaux C, Micheletti G, Depaulis A, Rumbach L, & Warter JM (1984). Enhancement of spike and wave discharges by GABAmimetic drugs in rats with spontaneous petit-mal-like epilesy. Neurosci Lett, 27, 91–94. [DOI] [PubMed] [Google Scholar]
- Verrotti A, Greco M, Varriale G, Tamborino A, Savasta S, Carotenuto M, et al. (2018). Electroclinical features of epilepsy monosomy 1p36 syndrome and their implications. Acta Neurol Scand, 138, 523–530. [DOI] [PubMed] [Google Scholar]
- Vezzani A, & Granata T (2005). Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia, 46, 1724–1743. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Aronica E, Mazarati A, & Pittman QJ (2013). Epilepsy and brain inflammation. Exp Neurol, 244, 11–21. [DOI] [PubMed] [Google Scholar]
- Vigevano F, & Cilio MR (1997). Vigabatrin versus ACTH as first-line treatment for infantile spasms: a randomized, prospective study. Epilepsia, 38, 1270–1274. [DOI] [PubMed] [Google Scholar]
- von Deimling M, Risbud R, Joseph DJ, & Marsh ED (2017). Transcriptional profiling of adult parvalbumin interneurons in a conditional knock-out model of Arx In American Epilepsy Society Annual Meeting (pp. 1.012). Washington, DC. [Google Scholar]
- von Spiczak S, Helbig KL, Shinde DN, Huether R, Pendziwiat M, Lourenco C, et al. (2017). DNM1 encephalopathy: A new disease of vesicle fission. Neurology, 89, 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Liu Z, Lin Z, Zhang R, Lu Y, Su W, et al. (2019). De novo germline mutations in SEMA5A associated with infantile spasms. Front Genet, 10, 605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Greenwood JS, Calcagnotto ME, Kirsch HE, Barbaro NM, & Baraban SC (2007). Neocortical hyperexcitability in a human case of tuberous sclerosis complex and mice lacking neuronal expression of TSC1. Ann Neurol, 61, 139–152. [DOI] [PubMed] [Google Scholar]
- Wanigasinghe J, Arambepola C, Ranganathan SS, & Sumanasena S (2017). Randomized, single-blind, parallel clinical trial on efficacy of oral prednisolone versus intramuscular corticotropin: A 12-month assessment of spasm control in West syndrome. Pediatr Neurol, 76, 14–19. [DOI] [PubMed] [Google Scholar]
- Weaving LS, Christodoulou J, Williamson SL, Friend KL, McKenzie OL, Archer H, et al. (2004). Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet, 75, 1079–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstock M (2011). Sex-dependent changes induced by prenatal stress in cortical and hippocampal morphology and behaviour in rats: an update. Stress, 14, 604–613. [DOI] [PubMed] [Google Scholar]
- Weinstock M (2017). Prenatal stressors in rodents: Effects on behavior. Neurobiol Stress, 6, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West WJ (1841). On a peculiar form of infantile convulsions. Lancet, i, 724–725. [Google Scholar]
- Widjaja E, Go C, McCoy B, & Snead OC 3rd (2015). Neurodevelopmental outcome of infantile spasms: A systematic review and meta-analysis. Epilepsy Res, 109, 155–162. [DOI] [PubMed] [Google Scholar]
- Williams S, Leventhal C, Lemmon V, Nedergaard M, & Goldman SA (1999). Estrogen promotes the initial migration and inception of NgCAM-dependent calcium-signaling by new neurons of the adult songbird brain. Mol Cell Neurosci, 13, 41–55. [DOI] [PubMed] [Google Scholar]
- Willig RP, & Lagenstein I (1982). Use of ACTH fragments of children with infantile spasms. Neuropediatrics, 13, 55–58. [DOI] [PubMed] [Google Scholar]
- Wonders CP, & Anderson SA (2006). The origin and specification of cortical interneurons. Nat Rev Neurosci, 7, 687–696. [DOI] [PubMed] [Google Scholar]
- Wong M (2013). Mammalian target of rapamycin (mTOR) pathways in neurological diseases. Biomed J, 36, 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray CD (2013). 17q21.31 microdeletion associated with infantile spasms. Eur J Med Genet, 56, 59–61. [DOI] [PubMed] [Google Scholar]
- Wray CD, & Benke TA (2010). Effect of price increase of adrenocorticotropic hormone on treatment practices of infantile spasms. Pediatr Neurol, 43, 163–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaka R, Salomon S, Matzner H, & Weinstock M (2007). Effect of varied gestational stress on acquisition of spatial memory, hippocampal LTP and synaptic proteins in juvenile male rats. Behav Brain Res, 179, 126–132. [DOI] [PubMed] [Google Scholar]
- Yamatogi Y, & Ohtahara S (2002). Early-infantile epileptic encephalopathy with suppression-bursts, Ohtahara syndrome; its overview referring to our 16 cases. Brain Dev, 24, 13–23. [DOI] [PubMed] [Google Scholar]
- Yang D, Du Q, Huang Z, Li L, Zhang Z, Zhang L, et al. (2019). Transcranial Direct Current Stimulation for Patients With Pharmacoresistant Epileptic Spasms: A Pilot Study. Front Neurol, 10, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Zou LP, Wang J, Shi XY, Yang XF, Wang B, et al. (2015). Association analysis of polymorphisms of the CRHR1 gene with infantile spasms. Mol Med Rep, 12, 2539–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XL, Chen B, Zhang XQ, Chen X, Yang MH, Zhang W, et al. (2017). Upregulations of CRH and CRHR1 in the epileptogenic tissues of patients with intractable infantile spasms. CNS Neurosci Ther, 23, 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh HR, Kim MJ, Ko TS, Yum MS, & You SJ (2017). Short-term outcome of intravenous methylprednisolone pulse therapy in patients with infantile spasms. Pediatr Neurol, 71, 50–55. [DOI] [PubMed] [Google Scholar]
- Yoo Y, Jung J, Lee YN, Lee Y, Cho H, Na E, et al. (2017). GABBR2 mutations determine phenotype in rett syndrome and epileptic encephalopathy. Ann Neurol, 82, 466–478. [DOI] [PubMed] [Google Scholar]
- Yu X, & Malenka RC (2004). Multiple functions for the cadherin/catenin complex during neuronal development. Neuropharmacology, 47, 779–786. [DOI] [PubMed] [Google Scholar]
- Yum MK, Jung KY, Kang HC, Kim HD, Shon YM, Kang JK, et al. (2008). Effect of a ketogenic diet on EEG: analysis of sample entropy. Seizure, 17, 561–566. [DOI] [PubMed] [Google Scholar]
- Yum MS, Lee EH, & Ko TS (2013). Vigabatrin and mental retardation in tuberous sclerosis: infantile spasms versus focal seizures. J Child Neurol, 28, 308–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yum MS, Chachua T, Velíšková J, & Velíšek L (2012). Prenatal stress promotes development of spasms in infant rats. Epilepsia, 53, e46–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yum MS, Lee M, Ko TS, & Velíšek L (2014). A potential effect of ganaxolone in an animal model of infantile spasms. Epilepsy Res, 108, 1492–1500. [DOI] [PubMed] [Google Scholar]
- Yum MS, Lee M, Woo DC, Kim DW, Ko TS, & Velíšek L (2015). beta-Hydroxybutyrate attenuates NMDA-induced spasms in rats with evidence of neuronal stabilization on MR spectroscopy. Epilepsy Res, 117, 125–132. [DOI] [PubMed] [Google Scholar]
- Zafeiriou DI, Kontopoulos EE, & Tsikoulas IG (1996). Adrenocorticotropic hormone and vigabatrin treatment of children with infantile spasms underlying cerebral palsy. Brain Dev, 18, 450–452. [DOI] [PubMed] [Google Scholar]
- Zehavi Y, Mandel H, Eran A, Ravid S, Abu Rashid M, Jansen EEW, et al. (2019). Severe infantile epileptic encephalopathy associated with D-glyceric aciduria: report of a novel case and review. Metab Brain Dis, 34, 557–563. [DOI] [PubMed] [Google Scholar]
- Zhou P, He N, Zhang JW, Lin ZJ, Wang J, Yan LM, et al. (2018). Novel mutations and phenotypes of epilepsy-associated genes in epileptic encephalopathies. Genes Brain Behav, 17, e12456. [DOI] [PubMed] [Google Scholar]