

Abstract
PURPOSE:
Genomic methods can identify homologous recombination deficiency (HRD). Rigorous evaluation of their outcome association to DNA-damage-response-targeted therapies like platinum in pancreatic cancer (PDAC) is essential in maximizing therapeutic outcome.
METHODS:
We evaluated progression-free survival (PFS) and overall survival (OS) of advanced-stage PDAC patients, who had both germline and somatic targeted-gene sequencing. Homologous recombination-gene mutations (HRm) were evaluated. (BRCA1, BRCA2, PALB2, ATM, BAP1, BARD1, BLM, BRIP1, CHEK2, FAM175A, FANCA, FANCC, NBN, RAD50, RAD51, RAD51C, RTEL1) HRm status was grouped as: (1) germline vs. somatic; (2) core (BRCAs, PALB2) vs. non-core (other HRm); and (3) monoallelic vs. biallelic. Genomic instability was compared using large-scale state transition, signature 3, and tumor mutation burden.
RESULTS:
Among 262 patients, 50 (19%) had HRD (15% germline and 4% somatic). Both groups were analyzed together due to lack of difference in their genomic instability and outcome. Median [95%CI] follow-up was 21.9 [1.4–57.0] months. Median OS and PFS were 15.5 [14.6–19] and 7 [6.1–8.1] months, respectively. Patients with HRD had improved PFS compared to no HRD when treated with first-line (1L) platinum (HR: 0.44 [95%CI: 0.29–0.67], p<0.01), but not with 1L-non-platinum. Multivariable analysis showed HRD patients had improved OS regardless of their 1L-treatment, but most had platinum exposure during their course. Biallelic HRm (11%) and core HRm (12%) had higher genomic instability, which translated to improved PFS on 1L-platinum vs. 1L-non-platinum.
CONCLUSION:
Pathogenic HRm identifies HRD in PDAC patients with the best outcome when treated with 1L-platinum. Biallelic HRm and core HRm further enriched benefit from HRD.
INTRODUCTION:
Pancreatic ductal adenocarcinoma (PDAC) is a lethal malignancy projected to become the second leading cause of cancer-related deaths in the U.S. by 2030.(1) Most PDAC patients present with advanced stage and the median overall survival (OS) for all-comers is less than a year.(2) Choosing the most effective treatment for each patient can enhance survival. Nevertheless, to date, treatment decisions have typically relied on performance status, comorbidities and patient preference. Two randomized phase 3 studies have defined FOLFIRINOX or gemcitabine plus nab-paclitaxel as superior to single-agent gemcitabine and both represent front-line standard therapies; however, neither has validated biomarkers for treatment decision-making.(3,4)
A recent randomized phase 3 trial (POLO) has highlighted a new era for precision oncology in PDAC by evaluating olaparib, the poly-ADP ribose polymerase inhibitor (PARPi), in germline BRCA-mutated metastatic PDAC.(5) Among 154 enrolled patients, the progression-free survival (PFS) was significantly longer in the olaparib group compared to the placebo group (7.4 vs. 3.8 months; hazard ratio [HR], 0.53; 95% confidence interval [CI], 0.35–0.82; p=0.004).(5) Further, a recent randomized phase II trial in germline BRCA1/2,or PALB2-mutated PDAC patients yielded very high response rates (64–75%) and OS (15–16 months) on first-line platinum chemotherapy regimen.(6) These results support that homologous recombination deficiency (HRD) can be effectively targeted in PDAC and underpins the need for a comprehensive evaluation of homologous recombination-gene mutations (HRm) beyond germline BRCA mutations for their sensitivity to DNA-damage-response (DDR)-targeted therapies including platinum.
Recent investigation of genomic profiling in large cohorts of PDAC have reported the significance of HRD/DDR in predicting sensitivity to platinum and PARPi.(7–9) In addition to mutations in canonical HR-genes, multiple groups have identified HR-deficient tumors by mutational and/or copy-number methods, which may capture a broader group of patients sensitive to DDR-targeted therapies.(7,8,10) To date, neither a consensus on the definition of HRD has been reached nor a systematic evaluation for different methods to determine HRD and their association in predicting treatment sensitivity in PDAC has been performed.(7,8,10–14)
Herein, we sought to evaluate the mutational status of HR-genes and HRD genetic signatures to determine their correlation with each other and their benefit to platinum therapy. We report on a systematic evaluation of these methodologies to identify HRD using an FDA-approved targeted-sequencing assay. The HRD subgroups defined by each method were compared for clinical outcomes (OS and PFS) when treated with or without first-line platinum-based therapies (1L-platinum).
METHODS:
Study Cohort
Written informed consent for genomic profiling was obtained (IRB# 12–245, NCT01775072) from all the patients with pathologically confirmed locally advanced or metastatic pancreatic exocrine cancers who received care at Memorial Sloan Kettering prior to this genomic analysis. The protocol was approved by the Institutional Review Board at Memorial Sloan Kettering Cancer Center, and the study was conducted in accordance with the Good Clinical Practice guidelines and the Declaration of Helsinki. Written consent was obtained from every patient. We excluded patients without germline testing.(15,16) Clinical, demographic, pathologic, genomic, treatment details, and outcome data were abstracted from the prospectively-maintained database and the electronic medical record.
Determination of Germline and Somatic Mutations
DNA was isolated from formalin-fixed, paraffin-embedded tumors and matched with normal blood, then sequenced for gene sets by MSK-IMPACT, a hybridization capture-based next-generation sequencing (NGS) assay. Sequenced data was analyzed to identify nucleotide variants, small insertions and deletions, copy number alterations (CNA), and structural rearrangements.(15,17) Both germline and somatic variants were called using previous algorithms.(13) The FACETS algorithm was used to evaluate CNA, the fraction in genome, and to identify regions with loss of heterozygosity (LOH).
Mutational Status of Homologous Recombination-genes
Labeling priority was in the order of hotspot and pathogenic alteration, and missense variants of unknown significance (VUS). Pathogenic alterations included frameshift, truncating, splice-site, or pathogenic missense mutations, annotated by OncoKB.(18–20) Homologous recombination deficiency (HRD) was defined as germline or somatic pathogenic alterations of the following 17 homologous recombination-genes (HRm): ATM, BAP1, BARD1, BLM, BRCA1, BRCA2, BRIP1, CHEK2, FAM175A, FANCA, FANCC, NBN, PALB2, RAD50, RAD51, RAD51C, and RTEL1. (Supplementary 1) These 17 candidate HR-genes were selected as they are included in both germline and somatic gene sets by MSK-IMPACT.(12,14,21) Emerging candidate DDR genes such as ARID1A, ATR, ATRX, CHEK1, RAD51L1, and RAD51L3 were excluded.(22,23) Pathogenic HRm were interpreted as somatic HRm (sHRm) if identified only in tumor whereas HRm found in blood sample were named germline HRm (gHRm). Core HRm (cHRm) was defined as either somatic or germline HRm of the canonical genes: BRCA1, BRCA2, or PALB2. And non-core HRm (ncHRm) was defined as HRm of the other 14 HR-genes. The allelic status of HR-genes was assessed using FACETS, manual review of the copy number plots, and evaluation of somatic mutation allele frequency to confirm loss of the wild-type allele.(13,24) Based on the mutation and LOH calling, a tumor was considered to have biallelic HRm if HRm was coupled with loss of the wild-type allele, or two pathogenic mutations on one HR-gene.
Homologous Recombination Deficiency Genomic Signatures
Two signature-based methods were evaluated to identify HRD: large-scale state transition (LST) and mutational signature 3 (Sig3).(12,25,26) Copy number change profile produced by FACETS was analyzed with a custom R script to count transition break points across the genome, which were summed for LST scores. For Sig3, we first called single nucleotide variants (SNV) with MuTect2 on both IMPACT-targeted and off-target regions, filtered SNV loci with 150X coverage depth in the tumor sequence and 80X in the blood sequence, then performed Signature Multivariate Analysis (SigMA) for cases with at least four somatic synonymous or non-synonymous SNV.(26) The SigMA R library with parameters “data=msk, tumor type= panc_ad” was used.
Statistical and Survival Analysis
We measured OS and PFS from the first treatment date until death (for OS) or until progression or death (for PFS). Disease progression was determined by imaging as adjudicated by the treating physician and radiologist. Patients were censored on the last follow-up date or on July-15–2019. OS and PFS were estimated using the Kaplan-Meier method and compared between subgroups using log-rank test. Kruskal Wallis test was used to compare mutational signatures among HRm subgroups. Pairwise two-sided multiple comparison was performed using Dwass, Steel, Critchlow-Fligner method, and Spearman correlation was used to assess correlation between continuous mutational signatures. Cox proportional hazards model was used for multivariable survival analyses. To evaluate whether 1L-platinum significantly affected the outcome differentially by HRD status, an interaction product term between binary covariates of HRD and 1L-platinum was included. Multivariable OS and PFS models were constructed by including known risk factors such as age and stage at diagnosis. GraphPad Prism (GraphPad Software, La Jolla, CA), SAS Version 9.4 (SAS Institute, INC., Cary, NC. USA) and R statistical environment (v3.6.1, http://www.r-project.org) were used. All p-values were two-sided with p<0.05 indicating statistical significance.
RESULTS:
Study Population
From October 2013 to July 2019, n=411 patients with advanced PDAC were screened and a total of n=262 were identified for the final analysis who had undergone both germline and somatic MSK-IMPACT analysis. (Supplementary 2) The median age was 64 years (interquartile range: 56–73). There were n=145 (55%) males. Majority were whites (n=216, 82%). Fifty-two self-reported as Ashkenazi Jewish. The majority had adenocarcinoma (n=258, 98%). Most had de novo stage IV (n=230, 88%). First-line platinum was administered in 61% (n=160). (Table 1)
Table 1.
Patient characteristics at baseline
| Characteristic | Total n= 262 (%) |
|---|---|
| Age, median (range), y | 64 (56–73) |
| Gender, male, No. (%) | 144 (55) |
| Ethnicity, No. (%) | |
| White | 216 (82) |
| Black | 16 (6.1) |
| Asian | 15 (5.7) |
| Unknown | 15 (5.7) |
| Pathology, No. (%) | |
| Adenocarcinoma | 258 (98) |
| Adenosquamous carcinoma | 2 (1) |
| Carcinoma, NOS | 2 (1) |
| Location of primary | |
| Head | 96 (37) |
| Body | 60 (23) |
| Tail | 53 (20) |
| Overlap and NOS | 53 (20) |
| Stage at presentation | |
| Metastatic | 230 (88) |
| Locally advanced | 32 (12) |
| First-line Treatment No. (%) | |
| First-line platinum | 160 (61) |
| FOLFIRINOX | 138 (53) |
| Gemcitabine plus nab-paclitaxel | 92 (35) |
| FOLFOX | 8 (3) |
| Gemcitabine only | 8 (3) |
| Gemcitabine plus cisplatin | 5 (2) |
| Others | 11 (4) |
Genetic Landscape of Cohort and Frequency of Different HR-gene Mutations
From 262 patients, we identified 1,193 high confidence non-synonymous somatic coding mutations including 949 SNVs, 81 insertions and 163 deletions. The median SNVs per case was four on the 468-gene MSK-IMPACT. The genes most frequently altered were KRAS, TP53, CDKN2A, SMAD4, ARID1A, RNF43, RB1, and GNAS.(27–29) (Figure 1) Then, we evaluated the presence of pathogenic mutations affecting the 17 HR-gene set, and identified n=50 patients (19%) with HRm. (Table 2) Notably, one patient with initially identified somatic BRCA2 loss from outside testing later developed a reversion mutation, therefore intact BRCA2 from reversion, which was not counted towards sHRm group. There were more gHRm (n=40, 15%) than sHRm (n=10, 4%). One patient had both germline BRCA2, and somatic BRCA1 mutation. There were more cHRm (BRCA1, BRCA2, and PALB2, n=31, 12%) than ncHRm (n=19, 7%). Biallelic HRm (n=29, 58%) was more common than monoallelic HRm (n=21, 42%).
Figure 1.

Oncoprint: pathogenic alterations of homologous recombination-genes and recurrent gene alterations from 50 patients with HRD are depicted (One patient had both germline and somatic BRCA variants). For each sample, mutation profile is shown in a column including 8 recurrently mutated genes (upper panel), 9 candidate HR-genes (lower panel). Pathogenic alterations included frameshift, truncating, splice site, or known pathogenic missense mutations, annotated by OncoKB. The labeling priority was in the order of hotspot alteration, pathogenic alteration, and missense variants of unknown significance (VUS). Core HRm (BRCA1, BRCA2, and PALB2) is shown as a track with green and yellow color bar. The level of genomic instability of each sample is depicted by LST score and TMB (mutation/MB) in the bars below. Biallelic HRm is annotated with a diagonal line whereas germline mutations are annotated with circles inside the square. Abbreviations: HRD, homologous recombination defect; LST, large-scale state transition; TMB, tumor mutation burden. HRm, HR mutation(s)
Table 2.
Frequencies of different HR-gene mutational status and genetic signatures. Abbreviation: BRCA, breast cancer gene; PALB2, partner and localizer of BRCA2;
| Wild Type (no HRD) (%) | Total: 212 (81) |
| Zygosity of HR-genes (%) | Total: 50 (19) |
| Biallelic loss (biallelic HRm) | 29 (11) |
| Monoallelic loss (monoallelic HRm) | 21 (8) |
| Mutated HR-genes (%) | Total: 50 (19) |
| Core (cHRm: BRCA1, BRCA2, PALB2) | 31 (12) |
| Non-core (ncHRm: other 14 genes)* | 19 (7) |
| Homologous recombination deficiency (HRD) (%) | Total: 50 (19)** |
| Germline HR-gene mutations | 40 (15)** |
| Somatic HR-gene mutations | 10 (4)*** |
| Monoallelic HRm (%) | N=21 (8) |
| Monoallelic non-core HRm | 12 (5) |
| Monoallelic germline ncHRm | 8 (3) |
| Monoallelic somatic ncHRm | 4 (2) |
| Monoallelic core HRm | 9 (3) |
| Monoallelic germline cHRm | 9 (3) |
| Monoallelic somatic cHRm | 0 (0) |
| Biallelic HRm (%) | N=29 (11)** |
| Biallelic non-core HRm | 7 (3) |
| Biallelic germline ncHRm | 6 (2) |
| Biallelic somatic ncHRm | 1 (0.4) |
| Biallelic core HRm | 22 (8)** |
| Biallelic germline cHRm | 17 (6)** |
| Biallelic somatic cHRm | 5 (2) |
| Genetic signatures beyond HR-gene mutations (HRm), No. (%) | |
| Large-scale state transition (LST) available: 262 (100%) | Range: 0–27 |
| Signature 3 (Sig3) | Positive 49 (19%), Negative 102 (39%) |
| Available: 151 (58%), unavailable: 111 (42%) | |
Non-core HR-genes: ATM, BAP1, BARD1, BLM, BRIP1, CHEK2, FAM175A, FANCA, FANCC, NBN, RAD50, RAD51, RAD51C, RTEL1
One individual had both germline and somatic BRCA variants, thus included in both but the count for the total number of HRD, it was counted as one individual in germline.
One individual was excluded from HRD group because only negative results for BRCA2 mutation from germline and somatic MSK-IMPACT. Initially reported BRCA2 loss from outside report.
Survival Outcomes
The median follow-up among surviving patients (n=75) was 21.9 (range:1.4–57) months. The median OS of the entire cohort was 15.5 months (95% CI, 14.6–19.0). Patients with gHRm and sHRm had similar outcomes and were combined as HRD. (Supplementary 3) HRD patients treated with 1L-platinum (n=35) had a superior median OS compared to patients with no HRD treated with or without 1L-platinum (25.1 [21.6-NR] vs. 15.3 [14.2–20.3] or 13 [10.1–16.9] months, respectively). (Figure 2A). Association of HRD on OS was independent of 1L-platinum and in multivariable model, patients with HRD had significantly reduced risk of all-cause mortality compared to patients with no HRD (HR: 0.50, 95%CI: 0.33–0.75, p<0.01) after adjusting for age, 1L-platinum, and stage at diagnosis. (Table 3A)
Figure 2.
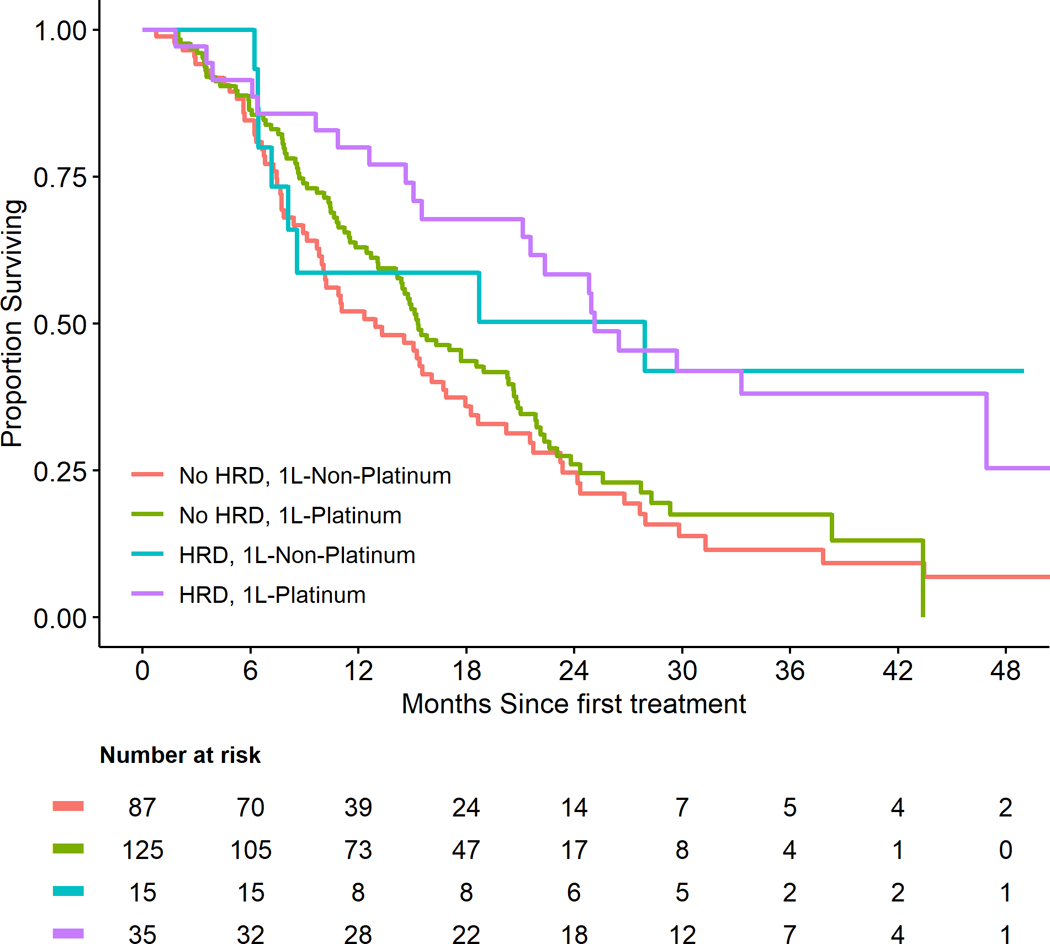

Outcome of pancreatic cancer patients with homologous recombination deficiency depending on 1L-platinum or 1L-non-platinum. Abbreviations: HDR, homologous recombination defect; HRm, homologous recombination-gene mutation(s); 1L-Platinum, first-line platinum; 1L-Non-platinum, first-line non-platinum
Table 3.
Multivariable analysis for overall survival (A) and progression-free survival (B) Abbreviations: HDR, homologous recombination defect; HRm, homologous recombination-gene mutation(s); 1L-Platinum, first-line platinum; 1L-Non-platinum, first-line non-platinum
| A. OS | |||
|---|---|---|---|
| Variables | Comparisons (n) | HR (95%CI) | p-value |
| HRD | HRD (50) vs. No HRD (212) | 0.50 (0.33,0.75) | <0.01 |
| 1L-Platinum | Yes (160) vs. No (102) | 0.93 (0.68,1.26) | 0.64 |
| Stage | IV (232) vs. III (32) | 2.26 (1.32,3.85) | <0.01 |
| Age at diagnosis | per 10-year increase | 1.05 (0.91,1.21) | 0.5 |
| B. PFS | |||
| Variables | Comparisons | HR (95%CI) | p-value |
| 1L-Platinum | HRD vs. No HRD | 0.44 (0.28,0.66) | <0.01 |
| 1L-Non-Platinum | HRD vs. No HRD | 1.42 (0.80,2.50) | 0.23 |
| Stage | IV vs. III | 1.85 (1.22,2.80) | <0.01 |
The median PFS of the entire cohort was 7.1 [95%CI: 6.1–8.1] months. Patients with HRD had a significantly improved median PFS (mPFS) when treated with 1L-platinum compared to 1L-non-platinum (12.6 [95%CI: 9.6–24.9] vs 4.4 [3.0–10.0] months). (Figure 2B) In multivariable interaction model, HRD patients treated with 1L-platinum had a significantly reduced risk of a disease progression or death (PFS) compared to non-HRD patients (HR: 0.44, 95%CI: 0.28–0.66, p<0.01). (Table 3B) In contrast, although statistically not significant, an increased risk for progression or death was observed among patients with HRD treated with 1L-non-platinum (HR: 1.42, 95%CI: 0.80–2.50).
Although 15 HRD patients did not receive 1L -platinum, 7 received platinum at second-line (2L), two had platinum exposure as a later line, and six did not have any platinum exposure. The mPFS for these 15 patients in the 1L-setting was 4.4 months. Notably, for those six patients who received 2L-platinum, the 2L mPFS (from the 2L-platinum start date until progression or death) was 8.3 months and mOS was markedly longer than the median of entire cohort at 24.2 months. More cHRm patients received platinum compared to ncHRm patients beyond first-line as part of their treatment course; six of eight (75%) cHRm patients compared to three of seven (42%) ncHRm patients.
Correlation of HR-gene Mutational Status with Genetic Signatures
We compared the magnitude of genomic instability among different mutational status of HRm (HRD: n=50 and WT, wild-type: n=212) by assessing HRD mutational signatures, namely LST, Sig3, and TMB. In view of SigMA requiring 4 mutations or higher, Sig3 score could only be derived from a subset of patients (n=151; 58%). As such, we first focused on LST and TMB, which were universally derivable.
We observed a significantly diverse level of genomic instability between different zygosity groups. Biallelic HRm (n=29) was associated with higher LST scores both when compared to WT (n=212, p=0.0006) and compared to monoallelic HRm (n=21, p=0.005). (Figure 3) Both core HRm (cHRm, n=31) and biallelic cHRm (n=22) had higher LST scores than WT (p=0.04 and p=0.0004, respectively). Biallelic germline HRm (gHRm, n=23), somatic HRm (sHRm, n=10), and biallelic sHRm (n=6) all individually had higher LST scores than WT (all p<0.05). However, no statistical difference was seen when compared to each other.
Figure 3.

Correlation of homologous recombination-gene mutational status with large-scale state transition. (A) LST and zygosity: biallelic HRm (n=29) had higher LST than monoallelic HRm (n=21) and wild type (n=212), (B) LST with core HRm: cHRm (n=31) and biallelic cHRm (n=22) had higher LST than WT (n=212), (C) LST with germline and somatic HRm: sHRm, biallelic gHRm, and biallelic sHRm all had higher LST than WT (n=212), however, no statistical difference was seen among gHRm, sHRm, biallelic gHRm, and sHRm. P-values are annotated with star symbols *, with one star indicating p<0.05, two stars indicating p<0.005, and three stars indicating p<0.0005. Abbreviation: LST, large-scale state transition; WT, wild type; HRm, homologous recombination gene mutation(s); cHRm, core HRm; ncHRm, non-core HRm; gHRm, germline HRm; sHRm, somatic HRm
Sig3 was derivable from 35 of 50 HRm tumors because of low mutation numbers. Biallelic HRm had higher Sig3 score compared to WT (p=0.01). And cHRm (n=25) had higher Sig3 score compared to WT (p=0.0047), but there was no difference when compared to ncHRm. (Supplementary 4) Biallelic cHRm (n=18, shaded) continued to have higher Sig3 score when compared to WT (p=0.001). There were limited Sig3-positive tumors among sHRm tumors (n=8). sHRm had higher Sig3 compared to WT (p=0.026), but there was no difference in Sig3between gHRm and WT. Higher Sig3 trend continued when the mutation was biallelic (n=5), but it was not statistically significant in our analysis.
Biallelic HRm also had higher TMB compared to WT (p=0.028). And cHRm had a trend of higher TMB but not significant (p=0.076). (Supplementary 5) However, among biallelic HRm, this difference was observed (biallelic-cHRm vs WT, p=0.0095). Similar to LST and Sig3, there was no difference of TMB among gHRm, sHRm, and WT.
Overall, biallelic HRm had the strongest association with genomic instability, and this trend was maintained in biallelic cHRm and both in biallelic gHRm and sHRm. These genetic signatures capturing different characteristics of HRD did not have significant association with each other except for LST and TMB. (Spearman’s coefficient: 0.19, p=0.0012) (Supplementary 6)
Platinum Affects PFS of Patients with Different HR-gene Mutational Status and Genetic Signatures
Biallelic HRm patients (n=29) had significantly improved mPFS on 1L-platnium compared to 1L-non-platinum (13.3 [95%CI: 9.57-NR] vs 3.8 [95%CI: 2.79-NR] months, p<0.0001). However, this was not observed in monoallelic HRm patients (n=21, p=0.22). (Supplementary 7), Furthermore, a sensitivity multivariable PFS analysis showed that compared to WT (=no HRD) patients, both cHRm patients (HR: 0.43, 95%CI: 0.26–0.70) as well as ncHRm (HR: 0.45, 95%CI: 0.23–0.87) had better PFS when treated with 1L-platinum (Supplementary 8). Also, in univariable analysis, among patients with gHRm (n=40), patients treated with 1L-platinum also had significantly improved PFS compared to those who received 1L-non-platinum (HR: 0.21, 95%CI: 0.10–0.50). Sig3-positive patients also had better mPFS compared to Sig3-negative patients (11.0 [95%CI: 8.5–15.7] vs 7.2 [95%CI: 6.0–9.1] months, p=0.03) when treated with 1L-platinum compared to 1L-non-platinum. Although there was a reduced risk of event between patients with 1L-platinum compared to 1L-non-platinum among Sig3-positive patients (n=49), it was not statistically significant (HR: 0.57, 95%CI: 0.29–1.10). Unexpectedly, no association between LST groups in tertiles and PFS was detected (data not shown).
Concordance and Discordance of Genetic Methods for HRD Identification
Patients with concordant HRD results had significant outcome differences depending on their first-line therapy. Sig3 was computable in 151 (58%) and 49 (19%) were Sig3-positive. There were 15 patients who had concordance for HRm and Sig3. Eleven received 1L-platinum and their mPFS was 12.6 (95%CI: 11.87-NR) months and mOS was 53.7 (95%CI: 24.8-NR) months, whereas the other four received 1L-non-platinum and had mPFS of 4.9 (95%CI: 2.8-NR) months and mOS of 18.2 (8.1-NR) months. Additionally, 82 patients (43%) had tumors that were concordantly negative for HRm and Sig3. OS and PFS distributions were not significantly different between patients treated with 1L-platinum or 1L-non-platinum (data not shown). Patients with discordant results had fewer apparent outcome differences depending on platinum treatment exposure. Interestingly, there were 33 patients who had exceptional OS over 24 months with HRm-negative tumors who had received 1L-platinum and eight (24%) of them were Sig3-positive.
Case Study of Platinum Resistance and Development of a Reversion Mutation
We emphasize the importance of understanding resistance mechanisms by presenting an exceptional case notable for extraordinary response to platinum however, eventually progressed after reversion mutation. Sixty-one-year-old male with Ashkenazi Jewish heritage and metastatic PDAC harboring a somatic BRCA2 loss responded durably to several platinum-based therapies for almost 3 years. Furthermore, the patient had a prior diagnosis of early stage colon cancer, but germline testing was negative for Lynch syndrome or germline BRCA mutation. A liver biopsy from a progressing liver metastasis following platinum revealed subclonal branching mutations of ERBB3 G1271C and an ATR-AGTR1 fusion and loss of CDKN2A and absent BRCA2 loss (i.e. reversion mutation) in addition to truncal mutations in KRAS G12D and TP53 E180K which were shared with the original biopsy.
Discussion
Genetic alterations associated with HRD/DDR pathways have been shown to correlate with response to PARPi and platinum.(7,8,30) Herein, we report on a comprehensive analysis of HRD detection methodologies in a cohort of patients with advanced PDAC.
The prevalence of HRD was 19%, primarily of germline origin (15%) similar to previous reports.(7,8,31) A key aim was to determine whether outcome differed by germline versus somatic HRm, as most of the data in PDAC to date is based on germline findings (POLO).(5) We observed neither differences in genomic instability nor clinical outcome between germline and somatic HRD, although the number of patients with somatic HRD was small, suggesting that both may predict for benefit to platinum-based therapies. Among mutational status of HR-genes (germline vs. somatic, core vs. non-core, and monoallelic vs. biallelic) and HRD genetic signatures (LST and Sig3) evaluated, HRD identification using a targeted-sequencing approach, a current standard for pathogenic HRm determination, appears to be the most predictive methodology evaluated herein.
We observed that patients with HRD had significantly improved PFS when treated with 1L-platinum compared to those who received 1L-non-platinum. Subgroup analyses in PFS further confirmed that biallelic HRm, cHRm, and Sig3-positive patients mostly benefited from 1L-platinum. These data suggest that patients with either pathogenic somatic or germline BRCA1, BRCA2, or PALB2 mutations as well as biallelic loss of other rarer HR-genes such as ATM, CHEK2, and perhaps Sig3-positivity, should be recommended for DDR-targeted therapies like platinum and PARPi. Collectively, each method has limitations and may complement each other in identifying platinum-sensitive HRD patients.
It is important to note that most patients with HRD who did not receive 1L-platinum, ultimately did receive platinum therapy during their treatment course and had benefit, which may explain the lack of the predictive effect of HRD to 1L-platinum on OS. Interestingly, three of six patients with no platinum exposure had very long mOS (47.2 months). This may be due to HRD being a prognostic, as well as predictive biomarker or perhaps HR-mutant tumors have a broader sensitivity to other cytotoxic agents that induce double-strand DNA breaks, such as irinotecan.(31–33)
Although the benefit of 1L-platinum in PDAC patients with HRD is apparent, these considerations need to be weighed against the turn-around time of obtaining sequencing results. Specifically, tissue-based genomic sequencing for first-line treatment decision making in PDAC remains challenging. Typically, the timeframe for these results is 3–6 weeks, outside the timeframe to determine first-line treatment.
The subgroup analyses described here are limited by relatively small numbers, are of an exploratory nature, are hypothesis-generating, and survival outcomes are retrospectively correlated. Biallelic cHRm showed higher LST, Sig3, and TMB, indicating more significant genomic instability. These data support that these patients can benefit from DDR-targeted therapies and immunotherapy. The hypothesis that synthetically lethality with PARPi can render HRm-tumors immunogenic is being investigated (34,35).
Furthermore, MSK is a high-volume referral-based cancer center and has a population enriched for certain ethnic backgrounds. There were 52 patients self-reported to be Ashkenazi Jewish and 15 of them were HRD (29%). And this study defined HR-genes as those that are included in both germline and somatic gene sets from MSK-IMPACT. Rarer candidate HR-genes (e.g., ATR, ATRX, CHEK1, RAD51L1, and RAD51L3) were excluded from the analysis. Computing mutational signatures or structural variants associated with HRD from a targeted-sequencing approach is known to have limitations and may influence our findings. Evaluating the association of Sig3 and LST with HR-gene mutations was difficult due to generally low mutation numbers in PDAC, thus limiting the clinical applicability of targeted-sequencing approach for genomic signatures. We further acknowledge there maybe additional survivorship bias in the cohort pertaining to the timepoint of consenting to MSK-IMPACT, related to the early part of this cohort (patients enrolled in 2013–2014).
Clinical outcomes vary due to the development of acquired resistance mechanisms such as drug efflux, reversion mutations, or compensatory changes in DNA Damage response.(36–40) Initially sensitive tumors develop clonal evolution and develop resistance. Identifying and delaying emergence of resistance mechanisms and developing novel strategies to overcome resistance will meaningfully improve the outcome of future PDAC patients. Broader sequencing methods (whole exome or whole genome) and serial genomic, transcriptomic, and functional analyses from patients with HRD are warranted to better understand the biology of HRD in PDAC.
Conclusion
HRD status defined as germline or somatic pathogenic HR-gene alterations, are associated with superior survival outcome to first-line platinum in advanced PDAC. Biallelic mutation of HR-genes showed the best association with this genomic characteristic independent of germline versus somatic origin. Determining the HRD status is ideally needed at the time of diagnosis to optimize survival and refine treatment selection in advanced PDAC. Short term goals are to make this clinically realizable.
Supplementary Material
TRANSLATIONAL RELEVANCE:
The primary objective of this study was to evaluate the biological and clinical significance of homologous recombination deficiency (HRD) determined by several genomic methods in canonical BRCA germline mutations and other putative HR-gene mutations in pancreas cancer (PDAC). We systematically evaluated these methods and their associated clinical outcome in PDAC from a large prospectively maintained database. We interrogated the mutational status of HR-genes (HRm) and HRD genetic signatures. We identified that biallelic HRm was associated with higher-level genomic instability and with better outcome on platinum. Somatic HRm was rarer than germline HRm and no significant difference in genomic instability and clinical outcome was observed between them. Higher degree genomic instability and superior PFS was observed in core HRm (BRCA1, BRCA2, and PALB2) compared to wild type (no HRD). These translational findings are significant for future application of DNA-damage-response-targeted treatment and immunotherapy clinical trials in broader population with HRD in PDAC.
Acknowledgement:
This research received MSK Cancer Center Support Grant (P30 CA008748), NIH-K12 (K12 CA184746), Parker Institute for Cancer Immunotherapy MSKCC Pilot Grant Award, and funding from David M. Rubenstein Center for Pancreatic Cancer Research. Bonnie Reiss Family Foundation. Breast Cancer Research Foundation, Sarah Jenkins Fund.
Nitya Raj, MD, Parisa Momtaz, MD, Geoffrey Ku, MD, Neil Segal, MD, PhD, Jia Li, MD, Andrew Epstein, MD, Curtis Chong, MD, PhD
Marie-Josée and Henry R. Kravis Center for Molecular Oncology, Cancer Center Core Grant, CMO, and the Molecular Diagnostics Service.
Conflicts of Interest Statement:
Wungki Park: Research funding to MSK: Merck, Astellas, Gossamerbio; Honoraria: Ipsen
Anna Varghese: Research funding to MSK: Illumina, Grail; Consultancy to Genentech.
Kenneth Yu: Research funding to MSK: BMS, Halozyme, Ipsen. Honoraria: Ipsen, QED.
Danny K. Khalil: Intellectual property interests related to CD40 and in situ vaccination
James Harding: Research and consulting fees from BMS. Consulting fees from Eisai, Exilexis, Imvax, and Elly Lilly
Ghassan K. Abou-Alfa: Research funding to MSK: Genentech, Roche, BMS, Halozyme, Celgene, MabVax Therapeutics, ActaBiologica, AstraZeneca, Silenseed, Polaris. Consulting/Advisory: Cytomx, BioLineRx, Targovax, Celgene, Bayer, Polaris, Sobi, Merck
Mark E. Robson: Research Funding to MSK: Abbvie, AstraZeneca, Invitae, Medivation, Merck, Myriad, Pfizer, Tesaro; Uncompensated consultancy to AstraZeneca, Daiichi-Sankyo, Epic Sciences, McKesson, Merck, Pfizer. Travel, Accommodation: AstraZeneca, Merck, Other transfer of value: AstraZeneca (editorial services), Pfizer (editorial services), NIH/NCI Cancer Center Support Grant P30 CA008748, Breast Cancer Research Foundation.
Liying Zhang: Honoraria from Future Technology Research LLC, Roche Diagnostics Asia Pacific, BGI, and Illumina; Family member has a leadership position and ownership interest of Shanghai Genome Center.
Michael F. Berger: Research funding to MSK: Illumina, Grail; Consultancy to Genentech.
Timothy Chan: Research Funding to MSK: AstraZeneca, Bristol-Myers Squibb, Illumina, Pfizer, An2H; Leadership: Cancer Genetics, Illumina, Bristol-Myers Squibb; Stock or other ownership: Gritstone Oncology; Patents, Royalties, Other Intellectual Property: neoantigen discovery and genomic biomarkers for immunotherapy response
Jorge S. Reis-Filho: Consultancy to Goldman Sachs and REPARE Therapeutics; Advisory board: Volition RX and Paige.AI, Ventana Medical Systems, Roche Tissue Diagnostics, Roche, Genentech, Novartis, InVicro.
Christine A. Iacobuzio-Donahue: Research funding to MSK: BMS
Nadeem Riaz: Research funding to MSK: AstraZeneca, Pfizer, BMS; Consultancy and Honoraria from Illumina, Mirati Therapeutics;
Eileen M. O’Reilly: Research funding to MSK: Genentech, Roche, BMS, Halozyme, Celgene, MabVax Therapeutics, ActaBiologica, AstraZeneca, Silenseed, Polaris. Consulting/Advisory: Cytomx, BioLineRx, Targovax, Celgene, Bayer, Polaris, Sobi, Merck,
Footnotes
No COI: Jiapeng Chen, Joanne F. Chou, Winston Wong, Vinod Balachandran, Caitlin A. McIntyre, Imane El Dika, Nima Ghalehsari, Zoe McKinell, Xin Pei, Nicolas Lecomte, Pier Selenica, David Kelsen, Vladimir Makarov, Simon Powell, Sree Chalasani, Marinela Capanu, Nikolaus Schultz.
REFERENCES
- 1.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer research 2014;74(11):2913–21 doi 10.1158/0008-5472.Can-14-0155. [DOI] [PubMed] [Google Scholar]
- 2.Cronin KA, Lake AJ, Scott S, Sherman RL, Noone AM, Howlader N, et al. Annual Report to the Nation on the Status of Cancer, part I: National cancer statistics. Cancer 2018. doi 10.1002/cncr.31551. [DOI] [PMC free article] [PubMed]
- 3.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. The New England journal of medicine 2013;369(18):1691–703 doi 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. The New England journal of medicine 2011;364(19):1817–25 doi 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 5.Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. The New England journal of medicine 2019. doi 10.1056/NEJMoa1903387. [DOI] [PMC free article] [PubMed]
- 6.O’Reilly EM, Lee JW, Zalupski M, Capanu M, Park J, Golan T, et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin With or Without Veliparib in Patients With Pancreas Adenocarcinoma and a Germline BRCA/PALB2 Mutation.0(0):JCO.19.02931 doi 10.1200/jco.19.02931. [DOI] [PMC free article] [PubMed]
- 7.Aguirre AJ, Nowak JA, Camarda ND, Moffitt RA, Ghazani AA, Hazar-Rethinam M, et al. Real-time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer discovery 2018;8(9):1096–111 doi 10.1158/2159-8290.Cd-18-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pishvaian MJ, Bender RJ, Halverson D, Rahib L, Hendifar AE, Mikhail S, et al. Molecular Profiling of Patients with Pancreatic Cancer: Initial Results from the Know Your Tumor Initiative. Clinical cancer research : an official journal of the American Association for Cancer Research 2018;24(20):5018–27 doi 10.1158/1078-0432.Ccr-18-0531. [DOI] [PubMed] [Google Scholar]
- 9.Pishvaian MJ, Blais EM, Brody JR, Rahib L, Lyons E, Arbeloa PD, et al. Outcomes in Patients With Pancreatic Adenocarcinoma With Genetic Mutations in DNA Damage Response Pathways: Results From the Know Your Tumor Program. 2019(3):1–10 doi 10.1200/po.19.00115. [DOI] [PubMed] [Google Scholar]
- 10.Shroff RT, Hendifar A, McWilliams RR, Geva R, Epelbaum R, Rolfe L, et al. Rucaparib Monotherapy in Patients With Pancreatic Cancer and a Known Deleterious BRCA Mutation. 2018(2):1–15 doi 10.1200/po.17.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shahda S, Timms KM, Ibrahim AA, Reid JE, Cramer HM, Radovich M, et al. Homologous Recombination Deficiency in Patients With Pancreatic Ductal Adenocarcinoma and Response to Chemotherapy. 2018(2):1–11 doi 10.1200/po.17.00087. [DOI] [PubMed] [Google Scholar]
- 12.Riaz N, Blecua P, Lim RS, Shen R, Higginson DS, Weinhold N, et al. Pan-cancer analysis of biallelic alterations in homologous recombination DNA repair genes. Nat Commun 2017;8(1):857 doi 10.1038/s41467-017-00921-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jonsson P, Bandlamudi C, Cheng ML, Srinivasan P, Chavan SS, Friedman ND, et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature 2019;571(7766):576–9 doi 10.1038/s41586-019-1382-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lord CJ, Ashworth A. BRCAness revisited. Nature reviews Cancer 2016;16(2):110–20 doi 10.1038/nrc.2015.21. [DOI] [PubMed] [Google Scholar]
- 15.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nature medicine 2017;23(6):703–13 doi 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling 2013;6(269):pl1 doi 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research 2010;20(9):1297–303 doi 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chakravarty D, Gao J, Phillips S, Kundra R, Zhang H, Wang J, et al. OncoKB: A Precision Oncology Knowledge Base. 2017(1):1–16 doi 10.1200/po.17.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nature biotechnology 2016;34(2):155–63 doi 10.1038/nbt.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao J, Chang MT, Johnsen HC, Gao SP, Sylvester BE, Sumer SO, et al. 3D clusters of somatic mutations in cancer reveal numerous rare mutations as functional targets. Genome medicine 2017;9(1):4 doi 10.1186/s13073-016-0393-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer discovery 2015;5(11):1137–54 doi 10.1158/2159-8290.Cd-15-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. 2015;5(7):752–67 doi 10.1158/2159-8290.CD-14-0849 %J Cancer Discovery. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heeke AL, Pishvaian MJ, Lynce F, Xiu J, Brody JR, Chen WJ, et al. Prevalence of Homologous Recombination-Related Gene Mutations Across Multiple Cancer Types. JCO precision oncology 2018;2018 doi 10.1200/po.17.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic acids research 2016;44(16):e131 doi 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Popova T, Manie E, Rieunier G, Caux-Moncoutier V, Tirapo C, Dubois T, et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer research 2012;72(21):5454–62 doi 10.1158/0008-5472.Can-12-1470. [DOI] [PubMed] [Google Scholar]
- 26.Gulhan DC, Lee JJ, Melloni GEM, Cortes-Ciriano I, Park PJ. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nature genetics 2019;51(5):912–9 doi 10.1038/s41588-019-0390-2. [DOI] [PubMed] [Google Scholar]
- 27.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015;518(7540):495–501 doi 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531(7592):47–52 doi 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 29.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nature genetics 2015;47(10):1168–78 doi 10.1038/ng.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Reilly EM, Lee JW, Lowery MA, Capanu M, Stadler ZK, Moore MJ, et al. Phase 1 trial evaluating cisplatin, gemcitabine, and veliparib in 2 patient cohorts: Germline BRCA mutation carriers and wild-type BRCA pancreatic ductal adenocarcinoma. Cancer 2018;124(7):1374–82 doi 10.1002/cncr.31218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yurgelun MB, Chittenden AB, Morales-Oyarvide V, Rubinson DA, Dunne RF, Kozak MM, et al. Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genetics in medicine : official journal of the American College of Medical Genetics 2019;21(1):213–23 doi 10.1038/s41436-018-0009-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olivieri M, Cho T, Álvarez-Quilón A, Li K, Schellenberg MJ, Zimmermann M, et al. A genetic map of the response to DNA damage in human cells. 2019:845446 doi 10.1101/845446 %J bioRxiv. [DOI] [PMC free article] [PubMed]
- 33.Lowery MA, Wong W, Jordan EJ, Lee JW, Kemel Y, Vijai J, et al. Prospective Evaluation of Germline Alterations in Patients With Exocrine Pancreatic Neoplasms. Journal of the National Cancer Institute 2018;110(10):1067–74 doi 10.1093/jnci/djy024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncology 2019;5(8):1141–9 doi 10.1001/jamaoncol.2019.1048 %J JAMA Oncology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vinayak S, Tolaney SM, Schwartzberg L, Mita M, McCann G, Tan AR, et al. Open-label Clinical Trial of Niraparib Combined With Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncology 2019;5(8):1132–40 doi 10.1001/jamaoncol.2019.1029 %J JAMA Oncology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008;451(7182):1111–5 doi 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 37.Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer research 2008;68(8):2581–6 doi 10.1158/0008-5472.Can-08-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol 2011;29(22):3008–15 doi 10.1200/jco.2010.34.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer discovery 2013;3(1):68–81 doi 10.1158/2159-8290.Cd-12-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burger H, Loos WJ, Eechoute K, Verweij J, Mathijssen RH, Wiemer EA. Drug transporters of platinum-based anticancer agents and their clinical significance. Drug resistance updates : reviews and commentaries in antimicrobial and anticancer chemotherapy 2011;14(1):22–34 doi 10.1016/j.drup.2010.12.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.