

Abstract
Chronic kidney disease is highly prevalent, affecting 10% to 15% of the adult population worldwide and is associated with increased cardiovascular morbidity and mortality. As chronic kidney disease worsens, a unique cardiovascular phenotype develops characterized by heart muscle disease, increased arterial stiffness, atherosclerosis, and hypertension. Cardiovascular risk is multifaceted, but most cardiovascular deaths in patients with advanced chronic kidney disease are caused by heart failure and sudden cardiac death. While the exact drivers of these deaths are unknown, they are believed to be caused by uremic cardiomyopathy: a specific pattern of myocardial hypertrophy, fibrosis, with both diastolic and systolic dysfunction. Although the pathogenesis of uremic cardiomyopathy is likely to be multifactorial, accumulating evidence suggests increased production of fibroblast growth factor‐23 and αKlotho deficiency as potential major drivers of cardiac remodeling in patients with uremic cardiomyopathy. In this article we review the increasing understanding of the physiology and clinical aspects of uremic cardiomyopathy and the rapidly increasing knowledge of the biology of both fibroblast growth factor‐23 and αKlotho. Finally, we discuss how dissection of these pathological processes is aiding the development of therapeutic options, including small molecules and antibodies, directly aimed at improving the cardiovascular outcomes of patients with chronic kidney disease and end‐stage renal disease.
Keywords: αKlotho, cardiorenal syndrome, FGF23, fibroblast growth factor, growth factor, kidney, treatment
Subject Categories: Cardiorenal Syndrome, Growth Factors/Cytokines, Treatment
Nonstandard Abbreviations and Acronyms
- AF
atrial fibrillation
- ESRD
end‐stage renal disease
- FGF23
fibroblast growth factor 23
- LV
left ventricular
- LVH
left ventricular hypertrophy
- SCD
sudden cardiac death
Introduction
Chronic kidney disease (CKD) and end‐stage renal disease (ESRD) requiring dialysis are complex, chronic conditions with a combined prevalence of 10% to 15% of the adult population worldwide.1, 2, 3 Cardiovascular events and mortality increase exponentially with reduced estimated glomerular filtration rate (eGFR) independent of age, sex, and other risk factors.4, 5, 6 In the early stages of CKD, the risks of occlusive atheromatous disease are increased and account for the majority of cardiovascular events observed.7 Arterial atheroma remains an important modifiable pathophysiological process in CKD, as evidenced by trials in early CKD showing benefit from lipid‐lowering therapies in modifying the risk of atherosclerotic events.8, 9, 10 However, the same treatments appear much less effective in patients with advanced stages of CKD, including ESRD.10, 11, 12 As CKD worsens, there is a shift from atherosclerotic complications to morbidity due to heart failure and sudden cardiac death (SCD).7, 13, 14 Atrial fibrillation (AF) is also common, detected in up to 41% of patients requiring hemodialysis.14 The pathophysiological basis of these events is a unique cardiovascular phenotype consisting primarily of the development of uremic cardiomyopathy with associated increased arterial stiffness and widespread atheroma.
The purpose of this article is to review the current state of the art on 2 newly postulated drivers of uremic cardiomyopathy, elevated circulating fibroblast growth factor‐23 (FGF23) and reduced αKlotho, and discuss how recent insights into the pathophysiological processes has led to development of potential therapeutic options aimed at reducing the cardiovascular risk of patients with CKD/ESRD.
Uremic Cardiomyopathy
The term uremic cardiomyopathy arose in the 1980s with reports of common abnormalities in cardiac function and structure in patients with CKD/ESRD, including increased left ventricular (LV) mass and left ventricular hypertrophy (LVH); diastolic and systolic dysfunction; as well as profound myocardial fibrosis on histology.15, 16, 17, 18, 19, 20 Uremic cardiomyopathy has been linked to conditions causing both heart failure and CKD, especially hypertension. Chronic stimulation of cardiac cells by renin, angiotensin, parathyroid hormone (PTH), cardiotonic steroids, and other uremic toxins, has also been proposed.20 The severity of uremic cardiomyopathy as measured by LV mass is a powerful predictor of cardiovascular mortality probably as a result of the factors discussed below.
Increased LV Mass and Hypertrophy
Increased LV mass and LVH are common manifestations of uremic cardiomyopathy. Forty percent of patients with an eGFR <30 mL/min per 1.73 m2 have LVH on echocardiography,21 increasing to ≈80% in ESRD.22, 23 In patients with CKD/ESRD, LVH is strongly associated with death; diastolic and systolic heart failure; and cardiac arrhythmias.22, 23 However, LV mass is a continuous variable, with a graded relationship with adverse cardiovascular outcomes.24, 25, 26, 27, 28 It is also important to emphasize that cardiac structural changes occur early in the course of CKD, with a linear association between worsening renal function and a higher prevalence of LVH.29, 30 While elevated blood pressure is an important determinant of LV mass,31 evidence from both animal and human studies supports the presence of mechanisms that are independent of pressure overload and hypertension in driving cardiac hypertrophy in CKD/ESRD.32, 33, 34, 35, 36
Diastolic and Systolic Dysfunction
Diastolic dysfunction is highly prevalent in patients with CKD, with over two thirds affected in CKD stages 2 to 430 and up to 85% in ESRD.37 Diastolic dysfunction is strongly associated with increased LV mass and LVH,7 as well as myocardial fibrosis,20, 38 and correlates with increased mortality.37, 38 Furthermore, the presence of diastolic dysfunction is considered to be a major cause for the frequent presentation of hemodialysis patients with pulmonary edema or intradialytic hypotension, despite only minor changes in fluid status.7, 38
Overt LV systolic dysfunction, as manifested by reduced ejection fraction, is uncommon in predialysis CKD with a reported prevalence of 8% and no association with eGFR.17, 30 However, several studies using echocardiography have shown changes in LV deformation in early stages of CKD, indicating the presence of subnormal LV systolic function.39, 40, 41 In ESRD, LV systolic dysfunction is very common, with a reported prevalence 10 to 30 times greater than in the general population.42, 43
Myocardial Fibrosis
It has been suggested that increased interstitial myocardial fibrosis may be the unifying pathophysiological process underlying uremic cardiomyopathy.44 In the 1990s, a postmortem study found that myocardial fibrosis was present in 91% of CKD/ESRD patients without significant flow‐limiting coronary lesions. The severity of fibrosis was related to the length of time on dialysis, but independent of hypertension, blood pressure, diabetes mellitus, or anemia.15 Over a decade later, Aoki et al.16 performed endocardial biopsies in 40 ESRD patients with reduced LV ejection fraction without coronary artery disease. The predominant pathologic findings were extensive interstitial fibrosis and cardiomyocyte hypertrophy and disarray.
Studying myocardial fibrosis in CKD/ESRD has been challenging given that myocardial biopsies are not without risk, especially in multimorbid patients and therefore are not always clinically and ethically justified.45, 46 Late gadolinium enhancement cardiac magnetic resonance imaging, a validated noninvasive method, allows in vivo quantification of myocardial fibrosis in conditions such as myocardial infarction,47 dilated48 and hypertrophic cardiomyopathies.49 This technique has also been used to characterize myocardial tissue in patients with ESRD demonstrating midwall patterns of late gadolinium enhancement consistent with replacement myocardial fibrosis not associated with large vessel coronary artery disease.17 Noncontrast myocardial native T1 relaxation time, or T1 mapping, has emerged as a novel viable technique to quantify diffuse interstitial myocardial fibrosis in CKD/ESRD50 correlating with histological interstitial fibrosis in a number of disease states, including cardiomyopathy and valvular disease.51 Indeed, native T1 times are increased in early CKD,19 increasing with worsening CKD stages52 and correlates with increased LV mass.18, 53 Native T1 mapping offers an exciting opportunity to investigate novel mechanisms of cardiac fibrosis (eg, FGF23‐mediated changes), in patients with CKD and in animal models.
Fibroblast Growth Factor‐23 and αKlotho
The hormone FGF23, first discovered in 2000, is a circulating growth factor secreted by osteocytes whose main physiological role is to increase urinary phosphate excretion.54, 55 The 4 mammalian FGF receptors (FGFR1‐4) are membrane‐bound receptor tyrosine kinases.56, 57 FGFR1 is suggested to be the primary FGF23 receptor in target organs—the kidneys and parathyroid glands.58, 59, 60, 61 Crystallography studies clearly demonstrate that the presence of αKlotho is required for the efficient binding of FGF23 to FGFR1.62 αKlotho is a cell‐surface protein, mainly expressed in the kidneys and parathyroid glands.63, 64, 65 In addition to the membrane‐associated full‐length protein, the ectodomain of αKlotho can exist in a soluble form.66, 67, 68, 69 In the presence of membrane‐bound αKlotho or soluble αKlotho, FGF23 can activate Fibroblast Growth Factor Receptor Substrate‐2 (FRS2α)/Ras/Mitogen‐Activated Protein Kinase signaling (Figure 1).62, 70, 71 Soluble αKlotho, therefore, may act as a circulating FGF23 coreceptor in cells that do not express αKlotho. Such a mechanism has been reported in osteoblasts.72 However, its role in the heart is yet to be fully characterized as neither cardiomyocytes nor cardiac fibroblasts express αKlotho.55 It is possible that FGF23 might act on these cells with circulating αKlotho as a cofactor. Treatment of cardiac myofibroblasts with full‐length αKlotho resulted in upregulated proliferation and ERK phosphorylation, which was suppressed by FGFR1 antagonism.73 This suggests the presence of FGFR1 in cardiac myofibroblasts for which soluble Klotho acts as a circulating co‐receptor, although the authors did not comment on endogenous FGFR1/FGF23 expression.
Figure 1. Potential mechanisms of fibroblast growth factor‐23 (FGF23) signal transduction and signaling pathways.
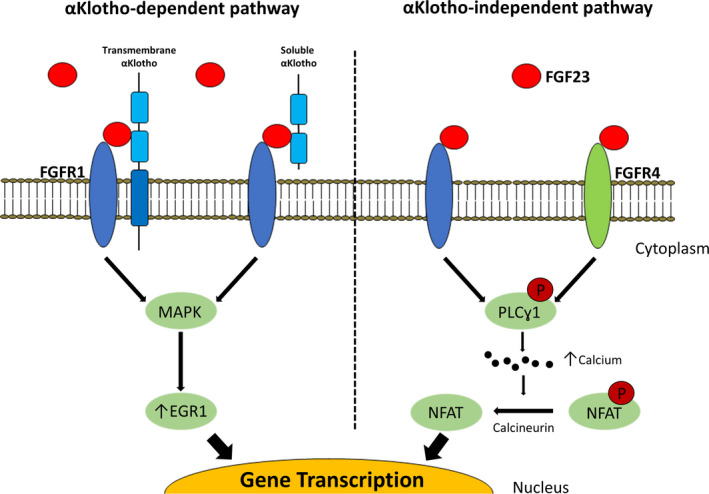
Fibroblast growth factor‐23 (FGF23) acts on cells that constitutively express fibroblast growth factor receptor‐1 (FGFR1) and its coreceptor αKlotho. In cells that do not constitutively express αKlotho, such as cardiac myocytes and fibroblasts, circulating αKlotho is thought to perform a role similar to membrane‐bound αKlotho. Circulating FGF23 can also bind to other FGFR1 and other FGFR independently of αKlotho. In cardiac myocytes, this is thought to be FGFR4 predominantly. The binding of FGF23 to an FGFR1‐αKlotho complex activates MAPK (mitogen‐activated protein kinase), upregulating early growth response protein‐1 (EGR1), thereby modulating physiological gene expression. In the absence of αKlotho, FGF23 binding to either FGFR1 or FGFR4 activates phospholipase Cγ1 (PLCγ1), increasing intracellular calcium. This in turn activates calcineurin to dephosphorylate (P, in red) nuclear factor of activated T‐cells (NFAT), which induces pathophysiological gene transcription.
FGF23 can also exert cellular effects via αKlotho‐independent mechanisms.74 FGF23 has been shown to stimulate phospholipase Cγ (PLCγ)/calcineurin/nuclear factor of activated T‐cells (NFAT) via FGFR4 in cells that lack αKlotho (Figure 1).74, 75, 76, 77 Such increases in PLCγ/calcineurin/NFAT signaling appear to be important in pathological, as opposed to physiological, cardiac hypertrophy.78, 79 Clearly further mechanistic studies are warranted to delineate mechanisms that can be targeted therapeutically in patients with elevated FGF23 levels.
FGF23, αKlotho, and Kidney Disease
One of the first clinically detectable signs of CKD is an elevation in serum FGF23, probably in response to increased extracellular phosphate, although the details of the stimulus and its detection are still unclear, with levels rising steeply as kidney function worsens54, 80 (Figure 2). Indeed, elevations are observed as early as eGFR 75 mL/min per 1.73 m2, long before increased concentrations in PTH or phosphate are observed.80 Circulating FGF23 levels are 2‐ to 5‐fold above the normal range in early/intermediate CKD, but can reach levels of 1000‐fold above normal in ESRD.80, 81, 82 Increased FGF23 levels are also found in heart failure83, 84, 85, 86, 87, 88 and AF,89, 90, 91, 92, 93 and are associated with all‐cause and cardiovascular mortality in patients with and without CKD.87, 88, 94, 95, 96, 97, 98, 99 Ongoing research therefore explores FGF23 as both a potential biomarker100 and a causative factor for cardiac mechanoelectrical dysfunction. However, effective quantification of circulating FGF23 is not currently standardized. Several FGF23 assay kits utilize differing detection techniques, epitope binding regions, analytical ranges and measurement units, making direct comparisons challenging.101
Figure 2. Dynamic interplay of fibroblast growth factor‐23–αKlotho axis.
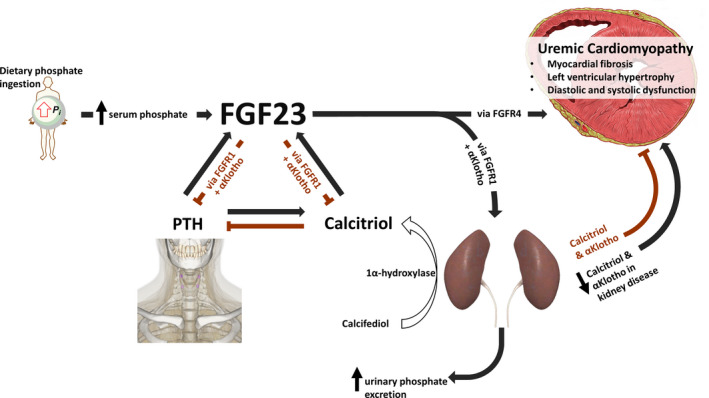
Dietary phosphate (Pi) ingestion and absorption leads to osteocytes secreting fibroblast growth factor‐23 (FGF23). It is not clear how osteocytes detect circulating phosphate, but the sensing of calciprotein particles (nanoparticles consisting of calcium, phosphate, and fetuin A) has been proposed as a potential mechanism. Increased FGF23 leads to increased phosphate excretion by downregulating sodium‐dependent phosphate cotransporter IIa/c, via mitogen‐activated protein kinase signaling, in the renal proximal tubules expressing FGF‐receptor 1 (FGFR1) in an αKlotho‐dependent manner. In addition, FGF23 lowers renal vitamin D hydroxylation and decreases parathyroid hormone (PTH) secretion, both actions also being αKlotho‐dependent, reducing calcium entry into the circulation. In the context of reducing renal function, FGF23 production increases to compensate for reduced renal phosphate excretion, decreased activated vitamin D levels, and rising PTH secretion. High levels of circulating FGF23 have been implicated in the development of uremic cardiomyopathy through αKlotho‐independent mechanisms via either the FGFR1 or, more likely, FGFR4. Circulating αKlotho may mitigate some of the pathophysiological actions of FGF23 on the myocardium. However, the kidneys are the main source of circulating αKlotho and levels decrease with reducing renal function.
The kidney is the principal source of circulating soluble αKlotho.69, 102 Its levels are downregulated in the presence of albuminuria,103 inflammation,104 and with the progression of CKD. αKlotho levels start to decline in CKD stage 2 and precede the elevation of FGF23, PTH, and serum phosphate.105 Low levels of circulating αKlotho are associated with increased cardiovascular events and mortality in patients with CKD/ESRD.106, 107, 108, 109 It is, therefore, conceivable that some of the adverse physiological effects that have been attributed to increased FGF23 may be either caused by, or compounded by, lower αKlotho (Figure 2). These mechanistic complexities require further investigation and need to be considered when developing FGF23/αKlotho‐directed therapies.
FGF23, αKlotho, and Left Ventricular Mass/Hypertrophy
The heart has been shown to respond to FGF23, increasing LV mass independently of blood pressure, promoting cardiac fibrosis and reducing LV systolic function in animal models.74, 75, 77, 110, 111 Elevations of cardiac FGFR4 and enhanced PLCγ/calcineurin/NFAT signaling have been observed in both animal models of CKD and in patients with CKD/ESRD.75, 112, 113, 114 Several studies have shown that repetitive administration of FGF23 in wild‐type mice, either intravenous or intraperitoneal, induced cardiac hypertrophy within 5 days.74, 110, 111 The signaling actions of FGF23 on the heart are still not fully characterized. However, several independent experimental approaches demonstrate the involvement of the αKlotho‐independent FGFR4‐PLCγ/calcineurin/NFAT signaling pathway in cardiomyocytes.74, 75, 77 On the other hand, independent studies demonstrate that αKlotho may be cardioprotective and that subnormal levels may be required for FGF23 to induce LVH.115, 116, 117, 118 Although many of the studies indicate that elevation of FGF23 and reduction in αKlotho are involved in the development of LVH, recent studies by Leifheit‐Nestler and Slavic have not recapitulated these findings.119, 120 Chronic FGF23 overexpression (via myocardial gene transfer), or genetic ablation of FGF23 or αKlotho on the background of transverse aortic constriction did not affect cardiac function or morphology.119, 120
Fibroblast Growth Factor‐23, αKlotho, and Myocardial Fibrosis
Every third to fourth cell in the heart is a fibroblast. Fibroblasts produce the extracellular matrix in the heart and act as regulators of the cardiac interstitium.121, 122 In the injured myocardium, inflammation and mechanical stress promote activation of fibroblasts to myofibroblasts, leading to maladaptive deposition of extensively cross‐linked extracellular matrix, which drives increased stiffness and impaired mechanoelectrical coupling of cardiomyocytes. This loss of cardiomyocyte coupling not only leads to attenuated cardiac function, but also provides a substrate for arrhythmias.123, 124, 125, 126 Although recent studies have shown that FGF23 can activate cardiac fibroblasts, neither the underlying mechanism127, 128 nor its role in the development of cardiac fibrosis are fully defined.127, 129 As cardiac fibroblasts do not express αKlotho,74, 102, 113, 114 the alternative pathway of αKlotho‐independent FGF23 signaling through FGFR4‐PLCγ/calcineurin/NFAT is likely to play a role (Figure 1). Future studies are required to examine which FGF receptors are expressed in cardiac fibroblasts, whether FGF23 contributes to cardiac fibrosis, and whether these mechanisms are dependent on αKlotho.
FGF23, αKlotho, and Cardiac Arrhythmias
Patients with CKD/ESRD are at increased risk of a wide spectrum of cardiac arrhythmias, including supraventricular tachycardias, particularly AF, and potentially lethal ventricular arrhythmias.14, 130, 131 All 3 components of uremic cardiomyopathy (increased LV mass/LVH; diastolic and systolic dysfunction; and especially myocardial fibrosis) are associated with arrhythmogenesis.131, 132 While emerging evidence from implantable loop recorder studies is beginning to implicate bradyarrhythmias as the major cause of SCD in ESRD, rather than the previously assumed tachyarrhythmias, the precise causes of SCD in ESRD are the subject of investigation.14 αKlotho has been found in sinoatrial node pacemaker cells in mice133 and αKlotho‐deficient animals exhibit sinoatrial node dysfunction, and higher rates of bradyarrhythmias and SCD.133
FGF23 distrupts intracellular calcium cycling within the cardiomyocyte, which is an important risk factor for arrhythmogenesis.134, 135, 136 Administration of FGF23 to rat ventricular cardiomyocytes caused calmodulin‐dependent protein kinase II‐dependent aberrant intracellular calcium, resulting in in vitro and in vivo arrhythmogenicity.137 Administration of recombinant αKlotho or a pan‐FGFR blocker prevented contractile dysfunction and reduced pro‐arrhythmogenic activity.137
Large observational studies in patients with CKD or AF and in the general population have found an association between elevated FGF23 and increased risk of developing AF.89, 138 High FGF23 and low αKlotho levels are associated with periods of AF in patients with paroxysmal or persistent AF.91 Increased expression of FGF23, FGFR4 mRNA, and FGFR4 protein in the right atrial appendages of patients with AF has been reported and positively correlate with atrial collagen fraction. Collectively these data/studies suggest that FGF23/FGFR4 may play a role in promoting AF through atrial fibrosis.139
Reversing or Preventing Uremic Cardiomyopathy by Targeting the FGF23 and αKlotho Axis
Several therapies exist that directly or indirectly target FGF23, αKlotho, and abnormalities in bone metabolism. These are reviewed below and summarized in Table.
Table 1.
Potential Therapies for Reversing or Preventing Uremic Cardiomyopathy by Targeting the Fibroblast Growth Factor‐23 and αKlotho Axis
| Treatment | Study | Species | CKD Status | Outcome |
|---|---|---|---|---|
| Targeting phosphate | ||||
| Dietary phosphate restriction | Burnett et al141 | Human | No renal impairment | Reduction in serum FGF23 |
| Antoniucci et al143 | Human | No renal impairment | ||
| Moe et al144 | Human | CKD Stage 3B to 4 | ||
| Di Iorio et al145 | Human | CKD Stage 3A to 4 | ||
| Sigrist et al146 | Human | No CKD & Stage 3A to 4 | ||
| Rodriguez‐Ortiz et al147 | Rat | 5/6 Nx | ||
| Calcium‐sparing phosphate binders (eg, sevelamer) | Oliveira et al148 | Human | CKD Stage 3A to 4 | |
| Block et al149 | Human | CKD Stage 3B to 4 | ||
| Chue et al150 | Human | CKD Stage 3 | ||
| Rodelo‐Haad et al151 | Human | ESRD on HD | ||
| Sprague et al152 | Human | ESRD on HD/PD | ||
| Nicotinamide | Shahbazian et al155 | Human | ESRD on HD | |
| Tenapanor | Block et al157 | Human | ESRD on HD | |
| Labonte et al156 | Rat | 5/6 Nx | ||
| Combination therapy with lanthanum and nicotinamide | Ix et al159 | Human | CKD Stage 3B to 4 | No sustained reduction in serum FGF23 |
| Targeting Vitamin D | ||||
| Calcitriol | Leifheit‐Nestler et al114 | Rat | 5/6 Nx | Reduction in LVH, cardiac FGF23 & FGFR4 expression, and NFAT/calcineurin activation |
| Leifheit‐Nestler et al114 | Rat (NRVM) | n/a | In vitro reduction in FGF23‐induced cardiomyocyte hypertrophy | |
| Calcitriol & paricalcitol | Lau et al163 | Mice | Partial renal ablation, phosphate loaded | Increase in serum αKlotho. No effect on renal/parathyroid αKlotho expression |
| Paricalcitol | Ritter et al164 | Rat | 5/6 Nx | Preservation of renal αKlotho, and increase in parathyroid αKlotho expression in uremia |
| Targeting parathyroid hormone | ||||
| Cinacalcet | Moe et al87 | Human | ESRD on HD | Reduction in serum FGF23, cardiovascular death, SCD, and heart failure |
| Charytan et al173 | Human | CKD Stage 3A to 4 | Reduction in FGF23 and PTH | |
| Chonchol et al174 | Human | CKD Stage 3A to 4 | Reduction in FGF23 and PTH; increase in hypocalcemia | |
| Other indirect targets | ||||
| Intensified (daily) hemodialysis | Zaritsky et al175 | Human | ESRD on HD | Reduction in FGF23 vs conventional hemodialysis |
| Renal transplantation | Barros et al176 | Human | ESRD (⅘ on HD) | Reduction in FGF23 and phosphate |
| Treatment of iron deficiency (eg, ferric citrate) | Block et al179, 180 | Human | CKD Stage 3A to 5 | Reduction in serum FGF32 |
| Inhibition of inflammation (eg, NFᴋB inhibitor) | Rodriguez‐Ortiz et al147 | Rat | No renal impairment | Attenuation of LPS‐induced FGF23 elevation |
| ATII receptor blockade | Yoon et al181 | Mice | CsA‐induced renal injury | Increase in renal αKlotho expression |
| Statins (eg, atorvastatin, pitavastatin) | Narumiya et al182 | Mouse (IMCD3) | n/a | In vitro upregulation of αKlotho mRNA expression |
| PPARγ agonist (eg, pioglitazone) | Yang et al183 | Rat | No renal impairment | Increase in renal αKlotho expression |
| Exercise | Matsubara et al184 | Human | No renal impairment | Increase in serum/plasma αKlotho |
| Tan et al185 | ||||
| Directly targeting FGF23 | ||||
| FGF23 neutralizing antibodies | Hasegawa et al189 | Rat | Anti‐GBM nephritis | Decrease in PTH; increase in vitamin D, calcium and phosphate |
| Shalhoub et al188 | Rat | 5/6 Nx | In addition to above, increase in mortality & aortic calcification | |
| FGFR antagonists | ||||
| FGFR4 antibody | Grabner et al75 | Rat | 5/6 Nx | Attenuation of LVH |
| Rat (NRVM) | n/a | In vitro inhibition of FGF23‐induced cardiac myocyte hypertrophy | ||
| Pan‐FGFR antibody | Faul et al74 | Rat | 5/6 Nx | Attenuation of LVH |
| Rat (NRVM) | n/a | In vitro inhibition of FGF23‐induced cardiac myocyte hypertrophy | ||
| Di Marco et al191 | Rat | 5/6 Nx | Reduction in LV mass and fibrosis; improvement in ejection fraction | |
| Yanochko et al192 | Rat | No renal impairment | Cardiac toxicity, hyperphosphatemia and ectopic calcification | |
| Sodium‐phosphate co‐transporter PiT2 knockout | Bon et al187 | Mice | No renal impairment | PiT2 regulates FGF23 synthesis; potential target for therapeutics |
| Directly targeting αKlotho | ||||
| Intravenous αKlotho transgene | Xie et al198 | Mice | 5/6 Nx±heterozygous Klotho | Attenuation of cardiac hypertrophy and fibrosis |
| Recombinant αKlotho | Hu et al196 | Mice | Uni‐nephrectomy + contralateral IR injury | Preservation of cardiac function, reduced hypertrophy and fibrosis; attenuation of renal fibrosis |
| Yang et al118 | Mice | 5/6 Nx | Inhibition of LVH and reduction in myocardial reactive oxygen species production | |
| Yu et al200 | Mice | No renal impairment | Attenuation of angiotensin II‐induced cardiac hypertrophy, fibrosis, and dysfunction | |
| Suassuna et al199 | Rat | 5/6 Nx | Reduction of uremic cardiac remodeling (hypertrophy and fibrosis) | |
| Yang et al118 | Rat (NRVM) | n/a | In vitro inhibition of uremic toxin‐induced (indoxyl sulphate) myocyte hypertrophy | |
| Small molecule αKlotho modulators | King et al201 | Human (HEK293) | n/a | In vitro elevation of αKlotho protein expression |
5/6 Nx indicates 5/6 nephrectomized; anti‐GBM, anti‐glomerular basement membrane; ATII, angiotensin II; CKD, chronic kidney disease; CM, cardiomyocyte; CsA, cyclosporine A; eGFR, estimated glomerular filtration rate; ESRD, end‐stage renal disease; FGF23, fibroblast growth factor‐23; FGFR, fibroblast growth factor receptor; HD, hemodialysis; HEK293, human embryonic kidney 293 cells; IR, ischemia‐reperfusion; LPS, lipopolysaccharide; LV, left ventricle; LVH, left ventricular hypertrophy; n∕a, not applicable; NFAT, nuclear factor of activated T‐cells; NRVM, neonatal rat ventricular myocytes; PD, peritoneal dialysis; PPARγ, peroxisome proliferator‐activated receptor γ; PTH, parathyroid hormone; and SCD, sudden cardiac death.
Targeting Phosphate Levels in the Body
Studies in healthy subjects have shown that circulating FGF23 levels are associated with dietary phosphate intake levels,140, 141 and can be further increased by acute phosphate loading.142 This can be reduced, in the short‐term, by aggressive reduction of dietary phosphate absorption141, 143, 144, 145, 146 and restriction.147 Overall, in relatively short‐term studies, noncalcium‐based phosphate binders lower FGF23 in patients with CKD/ESRD, whereas calcium‐based binders do not.148, 149, 150, 151, 152 Calcium is thought to be a secondary stimulus for FGF23 synthesis.153, 154 However, lowering intestinal phosphate absorption with dietary change, phosphate binders, nicotinamide,155 tenapanor,156, 157, 158 or combination therapy159 produces only modest decreases in FGF23 that do not appear to be sustained in the long term.150, 159 Whether this is because of increased total intestinal phosphate absorption by active phosphate transport,160, 161, 162 high pill burden, or intolerability of the medications is unknown.
Targeting Vitamin D
There is strong experimental data supporting vitamin D as a potential treatment for FGF23‐mediated uremic cardiomyopathy. Calcitriol, the synthetic analogue of vitamin D3, blocks FGF23‐induced activation of FGFR4 and cardiomyocyte growth.114 Increases in FGF23 expression, FGFR4‐induced calcineurin/NFAT signaling, and LVH in 5/6 nephrectomized rats are reduced by calcitriol.114 Vitamin D also increases αKlotho expression.163, 164 Observational studies demonstrate a survival advantage of vitamin D therapy in patients with CKD/ESRD despite raising calcium and phosphate levels.165, 166 However, in a randomized, placebo‐controlled study in patients with CKD stages 4 to 5, paricalcitol (activated vitamin D2 analogue) treatment did not reduce LV mass.167 Taken together, these data suggest combining vitamin D receptor activation with FGF23/FGFR4 signaling blockade could have beneficial synergistic actions on uremic cardiomyopathy.
Targeting Parathyroid Hormone
In patients on dialysis, the clinically available allosteric modulators (calcimimetics) of the calcium‐sensing receptor, cinacalcet and etelcalcetide, are used to treat hyperparathyroidism and consistently lower circulating FGF23.168, 169, 170, 171, 172 In secondary analyses of the large and well‐designed EVOLVE (Evaluation of Cinacalcet HCl Therapy to Lower Cardiovascular Events) trial, a >30% reduction in FGF23 in patients randomized to cinacalcet was associated with a reduction in cardiovascular mortality, SCD, and admissions for heart failure. The findings were amplified in those with a >50% reduction in FGF23.87
In CKD patients not requiring dialysis, randomized‐controlled trials of cinacalcet have reported significant reductions in FGF23, but also poor suppression of PTH as well as high rates of hypocalcemia and hyperphosphatemia.173, 174 These actions are thought to negate many of the clinical benefits of calcimimetics and these agents are not licensed for use in patients with non‐end‐stage CKD. Nevertheless, cinacalcet remains a promising therapeutic option for the treatment of uremic cardiomyopathy in ESRD.
Other Indirect Targets
Intensified dialysis treatment,175 renal transplantation,176 reduced inflammation,147, 177, 178 and treatment of iron deficiency152, 178, 179, 180 all reduce circulating FGF23 levels. Angiotensin‐receptor antagonists,181 statins,182 peroxisome proliferator‐activated receptor gamma agonists,183 and exercise184, 185 all increase αKlotho expression. The clear indications for these treatments remain and may well continue to give further insights into the pathophysiology of FGF23, but it is unlikely that these interventions will be used to directly target FGF23 and αKlotho.
Directly Targeting FGF23
The mechanism(s) regulating FGF23 synthesis are poorly understood and no “phosphate‐sensor” has yet been found in mammals.186, 187 Animal data have recently suggested that sodium‐phosphate cotransporter PiT2 found in bone might regulate phosphate‐dependent FGF23 synthesis and that targeting PiT2 could potentially reduce FGF23 synthesis.187 The development of novel small molecules against PiT2 or the yet‐to‐be characterized PiT2‐FGF23 pathway would give a proof of principle approach in animals for blocking FGF23 synthesis.
Indiscriminate FGF23 neutralization with monoclonal antibodies has been shown to worsen hyperphosphatemia, and increase vascular calcification and mortality in rat models of CKD.188, 189 Use of anti‐FGF23 monoclonal antibodies such as burosumab, currently approved for the treatment of x‐linked hypophosphatemia, causes severe side effects in patients with CKD by decreasing phosphaturia.190 Analogous to the use of calcimimetics, total blockade of FGF23 may theoretically be of benefit in ESRD. From a clinical therapeutic and drug development perspective, the ideal target would be the FGFR responsible for the adverse cardiac effects of FGF23 and not FGFR1, which is critical for maintaining normal phosphate levels. Indiscriminate blockade of FGFRs, although shown to be effective at preventing the development of,191 and reversing LVH in rodents,74 results in cardiac toxicity, hyperphosphatemia, and ectopic calcium deposition.192 Targeting cardiac FGFR4, especially its αKlotho‐independent activation of downstream signaling pathways, represents an exciting possibility. Indeed, FGFR4‐blocking antibodies have been shown to inhibit FGF23‐induced hypertrophy of isolated rat cardiomyocytes in vitro, and attenuated LVH in a 5/6 nephrectomy rat model of CKD.75 Currently, very little is known about the specific FGFRs mediating the actions of FGF23 in nonmyocyte cardiac cells including fibroblasts, or whether blocking FGFR4 prevents or reverses cardiac fibrosis.128 Several anti‐FGF small molecule tyrosine kinase inhibitors and related compounds, monoclonal antibodies, and FGFR‐analogues currently in development are mainly for use in oncology.193 Development of these agents specifically for the treatment of uremic cardiomyopathy is, therefore, a real possibility.
Directly Targeting αKlotho
In animal studies, administration of αKlotho protein has been shown to be effective in protecting against progression of CKD.194, 195, 196, 197 Intravenous administration of a transgene‐encoding soluble αKlotho reduces LVH in αKlotho‐deficient mice.198 Recombinant αKlotho also attenuates cardiac remodeling, fibrosis,195, 199 reactive oxygen species production, and LVH117 induced by CKD in mice. In another study, αKlotho improved cardiac function and reduced hypertrophy and fibrosis in a mouse model of hypertension, although decreasing FGF23 expression.200 However, it remains unclear whether αKlotho is cardioprotective in the absence of increased FGF23.
Elevated FGF23 and decreased circulating αKlotho are observed in both aging and in CKD,116, 197, 198 leading to speculation that both CKD and the age‐related decline in this and other physiological functions are caused in part by increased FGF23 and decreased αKlotho.55 If true, this would assume soluble αKlotho acts as an inhibitor of αKlotho‐independent actions of FGF23. Potential mechanisms include inhibiting FGF23/FGFR4 signaling by either binding first to FGFR4 or via an initial interaction with FGF23 (decoy receptor). An alternative mechanism involves FGF23 and FGFR4 forming a complex in the presence of soluble αKlotho but activating FRS2α/Ras/mitogen‐activated protein inase rather than PLCγ/calcineurin/NFAT signaling (Figure 1).55 Therefore, development of αKlotho‐mimetics, through the development of protein–protein inhibitors, provides another potential therapeutic option. Trials of such agents to prevent progression of CKD are expected to start in the next couple of years.
To date, small molecule αKlotho modulators have been identified from a high‐throughput screen of 150 000 compounds with those showing most promise being αKlotho transcription activators. Furthermore, extracellular signal‐regulated kinase phosphorylation in FGFR‐transfected cells increased, demonstrating an effect on FGF23 signaling.201 The recently discovered crystal structure of αKlotho:FGF23:FGFR1 in a 1:1:1 relationship has provided new insight into this dynamic interplay of factors and may reveal new therapeutic options.62 Although clearly at an early stage of exploration, the identification of new small molecules demonstrates the potential of drugs acting via αKlotho.
Conclusions
While patients with early stages of CKD are at increased risk of atherosclerotic complications, later stages of kidney disease are associated with heart failure and sudden death caused by uremic cardiomyopathy. Significant progress has been made over the past 2 decades in our understanding of, and ability to study the pathological basis of uremic cardiomyopathy using native T1 mapping. There are clear clinical data illustrating an association of increased FGF23 and reduced αKlotho with uremic cardiomyopathy in patients with CKD, and in heart failure and AF in subjects without known CKD. However, whether FGF23 has a truly causal relationship in uremic cardiomyopathy remains controversial. Characterization of the receptors and molecular pathways by which FGF23 might mediate LVH, cardiac fibrosis, and arrhythmias will help to identify therapeutic targets. Further work is required to identify the interplay between FGF23, cardiomyocytes and fibroblasts, and the effects of these interactions on the subsequent cardiac remodeling to reveal the molecular and cellular targets of FGF23 and αKlotho. Improved understanding is likely to enable the development of novel therapeutic interventions capable of effectively reducing the excess cardiovascular risk associated with CKD/ESRD, and perhaps even the risk of AF and heart failure in patients without CKD.
Sources of Funding
This work was supported by the British Heart Foundation Accelerator Award (AA/18/2/34218 to Kirchhof and The Institute of Cardiovascular Sciences, University of Birmingham); British Heart Foundation Clinical Research Training Fellowships (FS/19/16/34169 to Law, FS/16/73/32314 to Price, and FS/18/29/44554 to Pickup); and British Heart Foundation Grants (PG/17/55/33087 to Pavlovic, RG/17/15/33106, to Pavlovic, FS/19/12/34204 to Pavlovic).
Disclosures
None.
J Am Heart Assoc. 2020;9:e016041 DOI: 10.1161/JAHA.120.016041
References
- 1. Bruck K, Stel VS, Gambaro G, Hallan S, Volzke H, Arnlov J, Kastarinen M, Guessous I, Vinhas J, Stengel B, et al. CKD prevalence varies across the European general population. J Am Soc Nephrol. 2016;27:2135–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Levin A, Tonelli M, Bonventre J, Coresh J, Donner JA, Fogo AB, Fox CS, Gansevoort RT, Heerspink HJL, Jardine M, et al. Global kidney health 2017 and beyond: a roadmap for closing gaps in care, research, and policy. Lancet. 2017;390:1888–1917. [DOI] [PubMed] [Google Scholar]
- 3. Thomas B, Matsushita K, Abate KH, Al‐Aly Z, Arnlov J, Asayama K, Atkins R, Badawi A, Ballew SH, Banerjee A, et al. Global cardiovascular and renal outcomes of reduced GFR. J Am Soc Nephrol. 2017;28:2167–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351:1296–1305. [DOI] [PubMed] [Google Scholar]
- 5. Chronic Kidney Disease Prognosis C , Matsushita K, van der Velde M, Astor BC, Woodward M, Levey AS, de Jong PE, Coresh J, Gansevoort RT. Association of estimated glomerular filtration rate and albuminuria with all‐cause and cardiovascular mortality in general population cohorts: a collaborative meta‐analysis. Lancet. 2010;375:2073–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Methven S, Steenkamp R, Fraser S. UK Renal Registry 19th Annual Report: Chapter 5 Survival and Causes of Death in UK Adult Patients on Renal Replacement Therapy in 2015: National and Centre‐specific Analyses. Nephron. 2017;137(suppl 1):117–150. [DOI] [PubMed] [Google Scholar]
- 7. Wanner C, Amann K, Shoji T. The heart and vascular system in dialysis. Lancet. 2016;388:276–284. [DOI] [PubMed] [Google Scholar]
- 8. Tonelli M, Isles C, Curhan GC, Tonkin A, Pfeffer MA, Shepherd J, Sacks FM, Furberg C, Cobbe SM, Simes J, et al. Effect of pravastatin on cardiovascular events in people with chronic kidney disease. Circulation. 2004;110:1557–1563. [DOI] [PubMed] [Google Scholar]
- 9. Baigent C, Landray MJ, Reith C, Emberson J, Wheeler DC, Tomson C, Wanner C, Krane V, Cass A, Craig J, et al. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo‐controlled trial. Lancet. 2011;377:2181–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ferro CJ, Mark PB, Kanbay M, Sarafidis P, Heine GH, Rossignol P, Massy ZA, Mallamaci F, Valdivielso JM, Malyszko J, et al. Lipid management in patients with chronic kidney disease. Nat Rev Nephrol. 2018;14:727–749. [DOI] [PubMed] [Google Scholar]
- 11. Wanner C, Krane V, Marz W, Olschewski M, Asmus HG, Kramer W, Kuhn KW, Kutemeyer H, Mann JF, Ruf G, et al. Randomized controlled trial on the efficacy and safety of atorvastatin in patients with type 2 diabetes on hemodialysis (4D study): demographic and baseline characteristics. Kidney Blood Press Res. 2004;27:259–266. [DOI] [PubMed] [Google Scholar]
- 12. Fellstrom BC, Jardine AG, Schmieder RE, Holdaas H, Bannister K, Beutler J, Chae DW, Chevaile A, Cobbe SM, Gronhagen‐Riska C, et al. Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N Engl J Med. 2009;360:1395–1407. [DOI] [PubMed] [Google Scholar]
- 13. Thompson S, James M, Wiebe N, Hemmelgarn B, Manns B, Klarenbach S, Tonelli M; Alberta Kidney Disease N . Cause of death in patients with reduced kidney function. J Am Soc Nephrol. 2015;26:2504–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kalra PA, Green D, Poulikakos D. Arrhythmia in hemodialysis patients and its relation to sudden death. Kidney Int. 2018;93:781–783. [DOI] [PubMed] [Google Scholar]
- 15. Mall G, Huther W, Schneider J, Lundin P, Ritz E. Diffuse intermyocardiocytic fibrosis in uraemic patients. Nephrol Dial Transplant. 1990;5:39–44. [DOI] [PubMed] [Google Scholar]
- 16. Aoki J, Ikari Y, Nakajima H, Mori M, Sugimoto T, Hatori M, Tanimoto S, Amiya E, Hara K. Clinical and pathologic characteristics of dilated cardiomyopathy in hemodialysis patients. Kidney Int. 2005;67:333–340. [DOI] [PubMed] [Google Scholar]
- 17. Mark PB, Johnston N, Groenning BA, Foster JE, Blyth KG, Martin TN, Steedman T, Dargie HJ, Jardine AG. Redefinition of uremic cardiomyopathy by contrast‐enhanced cardiac magnetic resonance imaging. Kidney Int. 2006;69:1839–1845. [DOI] [PubMed] [Google Scholar]
- 18. Rutherford E, Talle MA, Mangion K, Bell E, Rauhalammi SM, Roditi G, McComb C, Radjenovic A, Welsh P, Woodward R, et al. Defining myocardial tissue abnormalities in end‐stage renal failure with cardiac magnetic resonance imaging using native T1 mapping. Kidney Int. 2016;90:845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hayer MK, Price AM, Liu B, Baig S, Ferro CJ, Townend JN, Steeds RP, Edwards NC. Diffuse myocardial interstitial fibrosis and dysfunction in early chronic kidney disease. Am J Cardiol. 2018;121:656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang X, Shapiro JI. Evolving concepts in the pathogenesis of uraemic cardiomyopathy. Nat Rev Nephrol. 2019;15:159–175. [DOI] [PubMed] [Google Scholar]
- 21. Levin A, Singer J, Thompson CR, Ross H, Lewis M. Prevalent left ventricular hypertrophy in the predialysis population: identifying opportunities for intervention. Am J Kidney Dis. 1996;27:347–354. [DOI] [PubMed] [Google Scholar]
- 22. Parfrey PS, Foley RN, Harnett JD, Kent GM, Murray DC, Barre PE. Outcome and risk factors for left ventricular disorders in chronic uraemia. Nephrol Dial Transplant. 1996;11:1277–1285. [PubMed] [Google Scholar]
- 23. Foley RN, Parfrey PS, Harnett JD, Kent GM, Martin CJ, Murray DC, Barre PE. Clinical and echocardiographic disease in patients starting end‐stage renal disease therapy. Kidney Int. 1995;47:186–192. [DOI] [PubMed] [Google Scholar]
- 24. Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–1566. [DOI] [PubMed] [Google Scholar]
- 25. Haider AW, Larson MG, Benjamin EJ, Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J Am Coll Cardiol. 1998;32:1454–1459. [DOI] [PubMed] [Google Scholar]
- 26. Schillaci G, Verdecchia P, Porcellati C, Cuccurullo O, Cosco C, Perticone F. Continuous relation between left ventricular mass and cardiovascular risk in essential hypertension. Hypertension. 2000;35:580–586. [DOI] [PubMed] [Google Scholar]
- 27. Devereux RB, Wachtell K, Gerdts E, Boman K, Nieminen MS, Papademetriou V, Rokkedal J, Harris K, Aurup P, Dahlof B. Prognostic significance of left ventricular mass change during treatment of hypertension. JAMA. 2004;292:2350–2356. [DOI] [PubMed] [Google Scholar]
- 28. Simpson HJ, Gandy SJ, Houston JG, Rajendra NS, Davies JI, Struthers AD. Left ventricular hypertrophy: reduction of blood pressure already in the normal range further regresses left ventricular mass. Heart. 2010;96:148–152. [DOI] [PubMed] [Google Scholar]
- 29. Edwards NC, Hirth A, Ferro CJ, Townend JN, Steeds RP. Subclinical abnormalities of left ventricular myocardial deformation in early‐stage chronic kidney disease: the precursor of uremic cardiomyopathy? J Am Soc Echocardiogr. 2008;21:1293–1298. [DOI] [PubMed] [Google Scholar]
- 30. Park M, Hsu CY, Li Y, Mishra RK, Keane M, Rosas SE, Dries D, Xie D, Chen J, He J, et al. Associations between kidney function and subclinical cardiac abnormalities in CKD. J Am Soc Nephrol. 2012;23:1725–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hammond IW, Devereux RB, Alderman MH, Lutas EM, Spitzer MC, Crowley JS, Laragh JH. The prevalence and correlates of echocardiographic left ventricular hypertrophy among employed patients with uncomplicated hypertension. J Am Coll Cardiol. 1986;7:639–650. [DOI] [PubMed] [Google Scholar]
- 32. Wolf WC, Yoshida H, Agata J, Chao L, Chao J. Human tissue kallikrein gene delivery attenuates hypertension, renal injury, and cardiac remodeling in chronic renal failure. Kidney Int. 2000;58:730–739. [DOI] [PubMed] [Google Scholar]
- 33. McMahon AC, Greenwald SE, Dodd SM, Hurst MJ, Raine AE. Prolonged calcium transients and myocardial remodelling in early experimental uraemia. Nephrol Dial Transplant. 2002;17:759–764. [DOI] [PubMed] [Google Scholar]
- 34. Suzuki H, Nakamoto H, Okada H, Sugahara S, Kanno Y. A selective angiotensin receptor antagonist, Valsartan, produced regression of left ventricular hypertrophy associated with a reduction of arterial stiffness. Adv Perit Dial. 2003;19:59–66. [PubMed] [Google Scholar]
- 35. Tai DJ, Lim TW, James MT, Manns BJ, Tonelli M, Hemmelgarn BR; Alberta Kidney Disease N . Cardiovascular effects of angiotensin converting enzyme inhibition or angiotensin receptor blockade in hemodialysis: a meta‐analysis. Clin J Am Soc Nephrol. 2010;5:623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hammer F, Malzahn U, Donhauser J, Betz C, Schneider MP, Grupp C, Pollak N, Stork S, Wanner C, Krane V, et al. A randomized controlled trial of the effect of spironolactone on left ventricular mass in hemodialysis patients. Kidney Int. 2019;95:983–991. [DOI] [PubMed] [Google Scholar]
- 37. Farshid A, Pathak R, Shadbolt B, Arnolda L, Talaulikar G. Diastolic function is a strong predictor of mortality in patients with chronic kidney disease. BMC Nephrol. 2013;14:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pecoits‐Filho R, Bucharles S, Barberato SH. Diastolic heart failure in dialysis patients: mechanisms, diagnostic approach, and treatment. Semin Dial. 2012;25:35–41. [DOI] [PubMed] [Google Scholar]
- 39. Rakhit DJ, Zhang XH, Leano R, Armstrong KA, Isbel NM, Marwick TH. Prognostic role of subclinical left ventricular abnormalities and impact of transplantation in chronic kidney disease. Am Heart J. 2007;153:656–664. [DOI] [PubMed] [Google Scholar]
- 40. Edwards NC, Ferro CJ, Townend JN, Steeds RP. Aortic distensibility and arterial‐ventricular coupling in early chronic kidney disease: a pattern resembling heart failure with preserved ejection fraction. Heart. 2008;94:1038–1043. [DOI] [PubMed] [Google Scholar]
- 41. Hensen LCR, Goossens K, Delgado V, Abou R, Rotmans JI, Jukema JW, Bax JJ. Prevalence of left ventricular systolic dysfunction in pre‐dialysis and dialysis patients with preserved left ventricular ejection fraction. Eur J Heart Fail. 2018;20:560–568. [DOI] [PubMed] [Google Scholar]
- 42. Stack AG, Bloembergen WE. A cross‐sectional study of the prevalence and clinical correlates of congestive heart failure among incident US dialysis patients. Am J Kidney Dis. 2001;38:992–1000. [DOI] [PubMed] [Google Scholar]
- 43. Sood MM, Pauly RP, Rigatto C, Komenda P. Left ventricular dysfunction in the haemodialysis population. NDT Plus. 2008;1:199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zannad F, Rossignol P. Cardiorenal syndrome revisited. Circulation. 2018;138:929–944. [DOI] [PubMed] [Google Scholar]
- 45. Becker AE, Heijmans CD, Essed CE. Chronic non‐ischaemic congestive heart disease and endomyocardial biopsies. Worth the extra? Eur Heart J. 1991;12:218–223. [DOI] [PubMed] [Google Scholar]
- 46. Cooper LT, Baughman KL, Feldman AM, Frustaci A, Jessup M, Kuhl U, Levine GN, Narula J, Starling RC, Towbin J, et al. The role of endomyocardial biopsy in the management of cardiovascular disease: a scientific statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology. Circulation. 2007;116:2216–2233. [DOI] [PubMed] [Google Scholar]
- 47. Simonetti OP, Kim RJ, Fieno DS, Hillenbrand HB, Wu E, Bundy JM, Finn JP, Judd RM. An improved MR imaging technique for the visualization of myocardial infarction. Radiology. 2001;218:215–223. [DOI] [PubMed] [Google Scholar]
- 48. McCrohon JA, Moon JC, Prasad SK, McKenna WJ, Lorenz CH, Coats AJ, Pennell DJ. Differentiation of heart failure related to dilated cardiomyopathy and coronary artery disease using gadolinium‐enhanced cardiovascular magnetic resonance. Circulation. 2003;108:54–59. [DOI] [PubMed] [Google Scholar]
- 49. Moon JC, Reed E, Sheppard MN, Elkington AG, Ho SY, Burke M, Petrou M, Pennell DJ. The histologic basis of late gadolinium enhancement cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;43:2260–2264. [DOI] [PubMed] [Google Scholar]
- 50. Edwards NC, Moody WE, Chue CD, Ferro CJ, Townend JN, Steeds RP. Defining the natural history of uremic cardiomyopathy in chronic kidney disease: the role of cardiovascular magnetic resonance. JACC Cardiovasc Imaging. 2014;7:703–714. [DOI] [PubMed] [Google Scholar]
- 51. Bull S, White SK, Piechnik SK, Flett AS, Ferreira VM, Loudon M, Francis JM, Karamitsos TD, Prendergast BD, Robson MD, et al. Human non‐contrast T1 values and correlation with histology in diffuse fibrosis. Heart. 2013;99:932–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hayer M, Price A, Liu B, Baig S, Ferro CJ, Townend JN, Steeds RP, Edwards NC. Progression of myocardial fibrosis in chronic kidney disease. Nephrol Dial Transplant. 2019;34:i35–i36. [Google Scholar]
- 53. Graham‐Brown MP, March DS, Churchward DR, Stensel DJ, Singh A, Arnold R, Burton JO, McCann GP. Novel cardiac nuclear magnetic resonance method for noninvasive assessment of myocardial fibrosis in hemodialysis patients. Kidney Int. 2016;90:835–844. [DOI] [PubMed] [Google Scholar]
- 54. Ito M, Sakai Y, Furumoto M, Segawa H, Haito S, Yamanaka S, Nakamura R, Kuwahata M, Miyamoto K. Vitamin D and phosphate regulate fibroblast growth factor‐23 in K‐562 cells. Am J Physiol Endocrinol Metab. 2005;288:E1101–1109. [DOI] [PubMed] [Google Scholar]
- 55. Richter B, Faul C. FGF23 actions on target tissues‐with and without klotho. Front Endocrinol (Lausanne). 2018;9:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol. 2001;2:REVIEWS3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–149. [DOI] [PubMed] [Google Scholar]
- 58. Ben‐Dov IZ, Galitzer H, Lavi‐Moshayoff V, Goetz R, Kuro‐o M, Mohammadi M, Sirkis R, Naveh‐Many T, Silver J. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007;117:4003–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Krajisnik T, Bjorklund P, Marsell R, Ljunggren O, Akerstrom G, Jonsson KB, Westin G, Larsson TE. Fibroblast growth factor‐23 regulates parathyroid hormone and 1alpha‐hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol. 2007;195:125–131. [DOI] [PubMed] [Google Scholar]
- 60. Gattineni J, Bates C, Twombley K, Dwarakanath V, Robinson ML, Goetz R, Mohammadi M, Baum M. FGF23 decreases renal NaPi‐2a and NaPi‐2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Renal Physiol. 2009;297:F282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Olauson H, Lindberg K, Amin R, Sato T, Jia T, Goetz R, Mohammadi M, Andersson G, Lanske B, Larsson TE. Parathyroid‐specific deletion of Klotho unravels a novel calcineurin‐dependent FGF23 signaling pathway that regulates PTH secretion. PLoS Genet. 2013;9:e1003975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen G, Liu Y, Goetz R, Fu L, Jayaraman S, Hu MC, Moe OW, Liang G, Li X, Mohammadi M. alpha‐Klotho is a non‐enzymatic molecular scaffold for FGF23 hormone signalling. Nature. 2018;553:461–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kuro‐o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. [DOI] [PubMed] [Google Scholar]
- 64. Kato Y, Arakawa E, Kinoshita S, Shirai A, Furuya A, Yamano K, Nakamura K, Iida A, Anazawa H, Koh N, et al. Establishment of the anti‐Klotho monoclonal antibodies and detection of Klotho protein in kidneys. Biochem Biophys Res Commun. 2000;267:597–602. [DOI] [PubMed] [Google Scholar]
- 65. Lim K, Groen A, Molostvov G, Lu T, Lilley KS, Snead D, James S, Wilkinson IB, Ting S, Hsiao LL, et al. alpha‐Klotho expression in human tissues. J Clin Endocrinol Metab. 2015;100:E1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc Natl Acad Sci USA. 2007;104:19796–19801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bloch L, Sineshchekova O, Reichenbach D, Reiss K, Saftig P, Kuro‐o M, Kaether C. Klotho is a substrate for alpha‐, beta‐ and gamma‐secretase. FEBS Lett. 2009;583:3221–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen CD, Tung TY, Liang J, Zeldich E, Tucker Zhou TB, Turk BE, Abraham CR. Identification of cleavage sites leading to the shed form of the anti‐aging protein klotho. Biochemistry. 2014;53:5579–5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hu MC, Shi M, Zhang J, Addo T, Cho HJ, Barker SL, Ravikumar P, Gillings N, Bian A, Sidhu SS, et al. Renal production, uptake, and handling of circulating alphaklotho. J Am Soc Nephrol. 2016;27:79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–774. [DOI] [PubMed] [Google Scholar]
- 71. Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, et al. Regulation of fibroblast growth factor‐23 signaling by klotho. J Biol Chem. 2006;281:6120–6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hum JM, O'Bryan LM, Tatiparthi AK, Cass TA, Clinkenbeard EL, Cramer MS, Bhaskaran M, Johnson RL, Wilson JM, Smith RC, et al. Chronic hyperphosphatemia and vascular calcification are reduced by stable delivery of soluble Klotho. J Am Soc Nephrol. 2017;28:1162–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liu X, Chen Y, McCoy CW, Zhao T, Quarles DL, Pi M, Bhattacharya SK, King G, Sun Y. Differential regulatory role of soluble klothos on cardiac fibrogenesis in hypertension. Am J Hypertens. 2016;29:1140–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, Gutierrez OM, Aguillon‐Prada R, Lincoln J, Hare JM, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121:4393–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Grabner A, Amaral AP, Schramm K, Singh S, Sloan A, Yanucil C, Li J, Shehadeh LA, Hare JM, David V, et al. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab. 2015;22:1020–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Singh S, Grabner A, Yanucil C, Schramm K, Czaya B, Krick S, Czaja MJ, Bartz R, Abraham R, Di Marco GS, et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney Int. 2016;90:985–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Grabner A, Schramm K, Silswal N, Hendrix M, Yanucil C, Czaya B, Singh S, Wolf M, Hermann S, Stypmann J, et al. FGF23/FGFR4‐mediated left ventricular hypertrophy is reversible. Sci Rep. 2017;7:1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Samak M, Fatullayev J, Sabashnikov A, Zeriouh M, Schmack B, Farag M, Popov AF, Dohmen PM, Choi YH, Wahlers T, et al. Cardiac hypertrophy: an introduction to molecular and cellular basis. Med Sci Monit Basic Res. 2016;22:75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Molkentin JD. Calcineurin‐NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc Res. 2004;63:467–475. [DOI] [PubMed] [Google Scholar]
- 80. Isakova T, Wahl P, Vargas GS, Gutierrez OM, Scialla J, Xie H, Appleby D, Nessel L, Bellovich K, Chen J, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79:1370–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gutierrez OM, Mannstadt M, Isakova T, Rauh‐Hain JA, Tamez H, Shah A, Smith K, Lee H, Thadhani R, Juppner H, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359:584–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Shimada T, Urakawa I, Isakova T, Yamazaki Y, Epstein M, Wesseling‐Perry K, Wolf M, Salusky IB, Juppner H. Circulating fibroblast growth factor 23 in patients with end‐stage renal disease treated by peritoneal dialysis is intact and biologically active. J Clin Endocrinol Metab. 2010;95:578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kestenbaum B, Sachs MC, Hoofnagle AN, Siscovick DS, Ix JH, Robinson‐Cohen C, Lima JA, Polak JF, Blondon M, Ruzinski J, et al. Fibroblast growth factor‐23 and cardiovascular disease in the general population: the Multi‐Ethnic Study of Atherosclerosis. Circ Heart Fail. 2014;7:409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lutsey PL, Alonso A, Selvin E, Pankow JS, Michos ED, Agarwal SK, Loehr LR, Eckfeldt JH, Coresh J. Fibroblast growth factor‐23 and incident coronary heart disease, heart failure, and cardiovascular mortality: the Atherosclerosis Risk in Communities study. J Am Heart Assoc. 2014;3:e000936 DOI: 10.1161/JAHA.114.000936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Scialla JJ, Xie H, Rahman M, Anderson AH, Isakova T, Ojo A, Zhang X, Nessel L, Hamano T, Grunwald JE, et al. Fibroblast growth factor‐23 and cardiovascular events in CKD. J Am Soc Nephrol. 2014;25:349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Seiler S, Rogacev KS, Roth HJ, Shafein P, Emrich I, Neuhaus S, Floege J, Fliser D, Heine GH. Associations of FGF‐23 and sKlotho with cardiovascular outcomes among patients with CKD stages 2‐4. Clin J Am Soc Nephrol. 2014;9:1049–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Moe SM, Chertow GM, Parfrey PS, Kubo Y, Block GA, Correa‐Rotter R, Drueke TB, Herzog CA, London GM, Mahaffey KW, et al. Cinacalcet, fibroblast growth factor‐23, and cardiovascular disease in hemodialysis: the evaluation of cinacalcet hcl therapy to lower cardiovascular events (EVOLVE) trial. Circulation. 2015;132:27–39. [DOI] [PubMed] [Google Scholar]
- 88. Marthi A, Donovan K, Haynes R, Wheeler DC, Baigent C, Rooney CM, Landray MJ, Moe SM, Yang J, Holland L, et al. Fibroblast growth factor‐23 and risks of cardiovascular and noncardiovascular diseases: a meta‐analysis. J Am Soc Nephrol. 2018;29:2015–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mathew JS, Sachs MC, Katz R, Patton KK, Heckbert SR, Hoofnagle AN, Alonso A, Chonchol M, Deo R, Ix JH, et al. Fibroblast growth factor‐23 and incident atrial fibrillation: the Multi‐Ethnic Study of Atherosclerosis (MESA) and the Cardiovascular Health Study (CHS). Circulation. 2014;130:298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mehta R, Cai X, Lee J, Scialla JJ, Bansal N, Sondheimer JH, Chen J, Hamm LL, Ricardo AC, Navaneethan SD, et al. Association of fibroblast growth factor 23 with atrial fibrillation in chronic kidney disease, from the chronic renal insufficiency cohort study. JAMA Cardiol. 2016;1:548–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mizia‐Stec K, Wieczorek J, Polak M, Wybraniec MT, Wozniak‐Skowerska I, Hoffmann A, Nowak S, Wikarek M, Wnuk‐Wojnar A, Chudek J, et al. Lower soluble Klotho and higher fibroblast growth factor 23 serum levels are associated with episodes of atrial fibrillation. Cytokine. 2018;111:106–111. [DOI] [PubMed] [Google Scholar]
- 92. Dong Q, Li S, Wang W, Han L, Xia Z, Wu Y, Tang Y, Li J, Cheng X. FGF23 regulates atrial fibrosis in atrial fibrillation by mediating the STAT3 and SMAD3 pathways. J Cell Physiol. 2019;234:19502–19510. [DOI] [PubMed] [Google Scholar]
- 93. Chua W, Purmah Y, Cardoso VR, Gkoutos GV, Tull SP, Neculau G, Thomas MR, Kotecha D, Lip GYH, Kirchhof P, et al. Data‐driven discovery and validation of circulating blood‐based biomarkers associated with prevalent atrial fibrillation. Eur Heart J. 2019;40:1268–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Arnlov J, Carlsson AC, Sundstrom J, Ingelsson E, Larsson A, Lind L, Larsson TE. Higher fibroblast growth factor‐23 increases the risk of all‐cause and cardiovascular mortality in the community. Kidney Int. 2013;83:160–166. [DOI] [PubMed] [Google Scholar]
- 95. Westerberg PA, Tivesten A, Karlsson MK, Mellstrom D, Orwoll E, Ohlsson C, Larsson TE, Linde T, Ljunggren O. Fibroblast growth factor 23, mineral metabolism and mortality among elderly men (Swedish MrOs). BMC Nephrol. 2013;14:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Brandenburg VM, Kleber ME, Vervloet MG, Tomaschitz A, Pilz S, Stojakovic T, Delgado G, Grammer TB, Marx N, Marz W, et al. Fibroblast growth factor 23 (FGF23) and mortality: the Ludwigshafen Risk and Cardiovascular Health Study. Atherosclerosis. 2014;237:53–59. [DOI] [PubMed] [Google Scholar]
- 97. Masson S, Agabiti N, Vago T, Miceli M, Mayer F, Letizia T, Wienhues‐Thelen U, Mureddu GF, Davoli M, Boccanelli A, et al. The fibroblast growth factor‐23 and Vitamin D emerge as nontraditional risk factors and may affect cardiovascular risk. J Intern Med. 2015;277:318–330. [DOI] [PubMed] [Google Scholar]
- 98. Souma N, Isakova T, Lipiszko D, Sacco RL, Elkind MS, DeRosa JT, Silverberg SJ, Mendez AJ, Dong C, Wright CB, et al. Fibroblast growth factor 23 and cause‐specific mortality in the general population: the Northern Manhattan study. J Clin Endocrinol Metab. 2016;101:3779–3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Bergmark BA, Udell JA, Morrow DA, Cannon CP, Steen DL, Jarolim P, Budaj A, Hamm C, Guo J, Im K, et al. Association of fibroblast growth factor 23 with recurrent cardiovascular events in patients after an acute coronary syndrome: a secondary analysis of a randomized clinical trial. JAMA Cardiol. 2018;3:473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Chua W, Easter CL, Guasch E, Sitch A, Casadei B, Crijns H, Haase D, Hatem S, Kaab S, Mont L, et al. Development and external validation of predictive models for prevalent and recurrent atrial fibrillation: a protocol for the analysis of the CATCH ME combined dataset. BMC Cardiovasc Disord. 2019;19:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Smith ER. The use of fibroblast growth factor 23 testing in patients with kidney disease. Clin J Am Soc Nephrol. 2014;9:1283–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Olauson H, Mencke R, Hillebrands JL, Larsson TE. Tissue expression and source of circulating alphaKlotho. Bone. 2017;100:19–35. [DOI] [PubMed] [Google Scholar]
- 103. Fernandez‐Fernandez B, Izquierdo MC, Valino‐Rivas L, Nastou D, Sanz AB, Ortiz A, Sanchez‐Nino MD. Albumin downregulates Klotho in tubular cells. Nephrol Dial Transplant. 2018;33:1712–1722. [DOI] [PubMed] [Google Scholar]
- 104. Moreno JA, Izquierdo MC, Sanchez‐Nino MD, Suarez‐Alvarez B, Lopez‐Larrea C, Jakubowski A, Blanco J, Ramirez R, Selgas R, Ruiz‐Ortega M, et al. The inflammatory cytokines TWEAK and TNFalpha reduce renal klotho expression through NFkappaB. J Am Soc Nephrol. 2011;22:1315–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lu X, Hu MC. Klotho/FGF23 axis in chronic kidney disease and cardiovascular disease. Kidney Dis. 2017;3:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Otani‐Takei N, Masuda T, Akimoto T, Honma S, Watanabe Y, Shiizaki K, Miki T, Kusano E, Asano Y, Kuro OM, et al. Association between serum soluble Klotho levels and mortality in chronic hemodialysis patients. Int J Endocrinol. 2015;2015:406269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Marcais C, Maucort‐Boulch D, Drai J, Dantony E, Carlier MC, Blond E, Genet L, Kuentz F, Lataillade D, Legrand E, et al. Circulating Klotho associates with cardiovascular morbidity and mortality during hemodialysis. J Clin Endocrinol Metab. 2017;102:3154–3161. [DOI] [PubMed] [Google Scholar]
- 108. Memmos E, Sarafidis S, Pateinakis P, Tsiantoulas A, Faitatzidou D, Giamalis P, Vasilikos V, Papagianni A. Low plasma Klotho is associated with cardiovascular events in hemodialysis independently of major risk factors, FGF23, arterial stiffness and intima‐medial‐thickness. Nephrol Dial Transplant. 2019;34:i38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Gao S, Xu J, Zhang S, Jin J. Meta‐analysis of the association between fibroblast growth factor 23 and mortality and cardiovascular events in hemodialysis patients. Blood Purif. 2019;47(suppl 1):24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Andrukhova O, Slavic S, Smorodchenko A, Zeitz U, Shalhoub V, Lanske B, Pohl EE, Erben RG. FGF23 regulates renal sodium handling and blood pressure. EMBO Mol Med. 2014;6:744–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Han X, Ross J, Kolumam G, Pi M, Sonoda J, King G, Quarles LD. Cardiovascular effects of renal distal tubule deletion of the FGF receptor 1 gene. J Am Soc Nephrol. 2018;29:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Di Marco GS, Reuter S, Kentrup D, Ting L, Ting L, Grabner A, Jacobi AM, Pavenstadt H, Baba HA, Tiemann K, et al. Cardioprotective effect of calcineurin inhibition in an animal model of renal disease. Eur Heart J. 2011;32:1935–1945. [DOI] [PubMed] [Google Scholar]
- 113. Leifheit‐Nestler M, Grosse Siemer R, Flasbart K, Richter B, Kirchhoff F, Ziegler WH, Klintschar M, Becker JU, Erbersdobler A, Aufricht C, et al. Induction of cardiac FGF23/FGFR4 expression is associated with left ventricular hypertrophy in patients with chronic kidney disease. Nephrol Dial Transplant. 2016;31:1088–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Leifheit‐Nestler M, Grabner A, Hermann L, Richter B, Schmitz K, Fischer DC, Yanucil C, Faul C, Haffner D. Vitamin D treatment attenuates cardiac FGF23/FGFR4 signaling and hypertrophy in uremic rats. Nephrol Dial Transplant. 2017;32:1493–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Xie J, Cha SK, An SW, Kuro OM, Birnbaumer L, Huang CL. Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat Commun. 2012;3:1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Hu MC, Shi M, Cho HJ, Adams‐Huet B, Paek J, Hill K, Shelton J, Amaral AP, Faul C, Taniguchi M, et al. Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. J Am Soc Nephrol. 2015;26:1290–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Grabner A, Faul C. The role of fibroblast growth factor 23 and Klotho in uremic cardiomyopathy. Curr Opin Nephrol Hypertens. 2016;25:314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Yang K, Wang C, Nie L, Zhao X, Gu J, Guan X, Wang S, Xiao T, Xu X, He T, et al. Klotho protects against indoxyl sulphate‐induced myocardial hypertrophy. J Am Soc Nephrol. 2015;26:2434–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Slavic S, Ford K, Modert M, Becirovic A, Handschuh S, Baierl A, Katica N, Zeitz U, Erben RG, Andrukhova O. Genetic Ablation of Fgf23 or Klotho does not modulate experimental heart hypertrophy induced by pressure overload. Sci Rep. 2017;7:11298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Leifheit‐Nestler M, Wagner MA, Nowak J, Richter B, Böckmann I, Foinquinos A, Thum T, Meier M, Müller OJ, Haffner D. Fo083chronic Fgf23 overload fails to induce cardiac dysfunctions. Nephrol Dial Transplant. 2019;34:i37. [Google Scholar]
- 121. Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res. 2009;105:1164–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair. 2012;5:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. de Bakker JM, van Capelle FJ, Janse MJ, Tasseron S, Vermeulen JT, de Jonge N, Lahpor JR. Fractionated electrograms in dilated cardiomyopathy: origin and relation to abnormal conduction. J Am Coll Cardiol. 1996;27:1071–1078. [DOI] [PubMed] [Google Scholar]
- 124. Weber KT. Fibrosis and hypertensive heart disease. Curr Opin Cardiol. 2000;15:264–272. [DOI] [PubMed] [Google Scholar]
- 125. Espira L, Czubryt MP. Emerging concepts in cardiac matrix biology. Can J Physiol Pharmacol. 2009;87:996–1008. [DOI] [PubMed] [Google Scholar]
- 126. Chaturvedi RR, Herron T, Simmons R, Shore D, Kumar P, Sethia B, Chua F, Vassiliadis E, Kentish JC. Passive stiffness of myocardium from congenital heart disease and implications for diastole. Circulation. 2010;121:979–988. [DOI] [PubMed] [Google Scholar]
- 127. Hao H, Li X, Li Q, Lin H, Chen Z, Xie J, Xuan W, Liao W, Bin J, Huang X, et al. FGF23 promotes myocardial fibrosis in mice through activation of beta‐catenin. Oncotarget. 2016;7:64649–64664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Leifheit‐Nestler M, Haffner D. Paracrine effects of FGF23 on the heart. Front Endocrinol. 2018;9:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Faul C. Cardiac actions of fibroblast growth factor 23. Bone. 2017;100:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Roberts PR, Green D. Arrhythmias in chronic kidney disease. Heart. 2011;97:766–773. [DOI] [PubMed] [Google Scholar]
- 131. Boriani G, Savelieva I, Dan GA, Deharo JC, Ferro C, Israel CW, Lane DA, La Manna G, Morton J, Mitjans AM, et al. Chronic kidney disease in patients with cardiac rhythm disturbances or implantable electrical devices: clinical significance and implications for decision making‐a position paper of the European Heart Rhythm Association endorsed by the Heart Rhythm Society and the Asia Pacific Heart Rhythm Society. Europace. 2015;17:1169–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Turakhia MP, Schiller NB, Whooley MA. Prognostic significance of increased left ventricular mass index to mortality and sudden death in patients with stable coronary heart disease (from the Heart and Soul Study). Am J Cardiol. 2008;102:1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Takeshita K, Fujimori T, Kurotaki Y, Honjo H, Tsujikawa H, Yasui K, Lee JK, Kamiya K, Kitaichi K, Yamamoto K, et al. Sinoatrial node dysfunction and early unexpected death of mice with a defect of klotho gene expression. Circulation. 2004;109:1776–1782. [DOI] [PubMed] [Google Scholar]
- 134. Touchberry CD, Green TM, Tchikrizov V, Mannix JE, Mao TF, Carney BW, Girgis M, Vincent RJ, Wetmore LA, Dawn B, et al. FGF23 is a novel regulator of intracellular calcium and cardiac contractility in addition to cardiac hypertrophy. Am J Physiol Endocrinol Metab. 2013;304:E863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Hsueh CH, Chen NX, Lin SF, Chen PS, Gattone VH 2nd, Allen MR, Fishbein MC, Moe SM. Pathogenesis of arrhythmias in a model of CKD. J Am Soc Nephrol. 2014;25:2812–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Glukhov AV, Kalyanasundaram A, Lou Q, Hage LT, Hansen BJ, Belevych AE, Mohler PJ, Knollmann BC, Periasamy M, Gyorke S, et al. Calsequestrin 2 deletion causes sinoatrial node dysfunction and atrial arrhythmias associated with altered sarcoplasmic reticulum calcium cycling and degenerative fibrosis within the mouse atrial pacemaker complex1. Eur Heart J. 2015;36:686–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Navarro‐Garcia JA, Delgado C, Fernandez‐Velasco M, Val‐Blasco A, Rodriguez‐Sanchez E, Aceves‐Ripoll J, Gomez‐Hurtado N, Bada‐Bosch T, Merida‐Herrero E, Hernandez E, et al. Fibroblast growth factor‐23 promotes rhythm alterations and contractile dysfunction in adult ventricular cardiomyocytes. Nephrol Dial Transplant. 2019;34:1864–1875. [DOI] [PubMed] [Google Scholar]
- 138. Mehta R, Cai X, Hodakowski A, Lee J, Leonard M, Ricardo A, Chen J, Hamm L, Sondheimer J, Dobre M, et al. Fibroblast growth factor 23 and anemia in the chronic renal insufficiency cohort study. Clin J Am Soc Nephrol. 2017;12:1795–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Dong QB, Tang YH, Wang WX, Wu YB, Han L, Li JX, Hong K, Wu YQ, Wu QH, Cheng XS. [Relationship between FGF23/FGFR4 expression in atrial tissue and atrial fibrosis in patients with atrial fibrillation]. Zhonghua Yi Xue Za Zhi. 2018;98:1003–1007. [DOI] [PubMed] [Google Scholar]
- 140. Ferrari SL, Bonjour JP, Rizzoli R. Fibroblast growth factor‐23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab. 2005;90:1519–1524. [DOI] [PubMed] [Google Scholar]
- 141. Burnett SM, Gunawardene SC, Bringhurst FR, Juppner H, Lee H, Finkelstein JS. Regulation of C‐terminal and intact FGF‐23 by dietary phosphate in men and women. J Bone Miner Res. 2006;21:1187–1196. [DOI] [PubMed] [Google Scholar]
- 142. Scanni R, vonRotz M, Jehle S, Hulter HN, Krapf R. The human response to acute enteral and parenteral phosphate loads. J Am Soc Nephrol. 2014;25:2730–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Antoniucci DM, Yamashita T, Portale AA. Dietary phosphorus regulates serum fibroblast growth factor‐23 concentrations in healthy men. J Clin Endocrinol Metab. 2006;91:3144–3149. [DOI] [PubMed] [Google Scholar]
- 144. Moe SM, Zidehsarai MP, Chambers MA, Jackman LA, Radcliffe JS, Trevino LL, Donahue SE, Asplin JR. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol. 2011;6:257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Di Iorio B, Di Micco L, Torraca S, Sirico ML, Russo L, Pota A, Mirenghi F, Russo D. Acute effects of very‐low‐protein diet on FGF23 levels: a randomized study. Clin J Am Soc Nephrol. 2012;7:581–587. [DOI] [PubMed] [Google Scholar]
- 146. Sigrist M, Tang M, Beaulieu M, Espino‐Hernandez G, Er L, Djurdjev O, Levin A. Responsiveness of FGF‐23 and mineral metabolism to altered dietary phosphate intake in chronic kidney disease (CKD): results of a randomized trial. Nephrol Dial Transplant. 2013;28:161–169. [DOI] [PubMed] [Google Scholar]
- 147. Rodriguez‐Ortiz ME, Diaz‐Tocados JM, Munoz‐Castaneda JR, Herencia C, Pineda C, Martinez‐Moreno JM, Montes de Oca A, Lopez‐Baltanas R, Alcala‐Diaz J, Ortiz A, et al. Inflammation both increases and causes resistance to FGF23 in normal and uremic rats. Clin Sci. 2020;134:15–32. [DOI] [PubMed] [Google Scholar]
- 148. Oliveira RB, Cancela AL, Graciolli FG, Dos Reis LM, Draibe SA, Cuppari L, Carvalho AB, Jorgetti V, Canziani ME, Moyses RM. Early control of PTH and FGF23 in normophosphatemic CKD patients: a new target in CKD‐MBD therapy? Clin J Am Soc Nephrol. 2010;5:286–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Block GA, Wheeler DC, Persky MS, Kestenbaum B, Ketteler M, Spiegel DM, Allison MA, Asplin J, Smits G, Hoofnagle AN, et al. Effects of phosphate binders in moderate CKD. J Am Soc Nephrol. 2012;23:1407–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Chue CD, Townend JN, Moody WE, Zehnder D, Wall NA, Harper L, Edwards NC, Steeds RP, Ferro CJ. Cardiovascular effects of sevelamer in stage 3 CKD. J Am Soc Nephrol. 2013;24:842–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Rodelo‐Haad C, Rodriguez‐Ortiz ME, Martin‐Malo A, Pendon‐Ruiz de Mier MV, Aguera ML, Munoz‐Castaneda JR, Soriano S, Caravaca F, Alvarez‐Lara MA, Felsenfeld A, et al. Phosphate control in reducing FGF23 levels in hemodialysis patients. PLoS One. 2018;13:e0201537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Sprague SM, Ketteler M, Covic AC, Floege J, Rakov V, Walpen S, Rastogi A. Long‐term efficacy and safety of sucroferric oxyhydroxide in African American dialysis patients. Hemodial Int. 2018;22:480–491. [DOI] [PubMed] [Google Scholar]
- 153. Shimada T, Yamazaki Y, Takahashi M, Hasegawa H, Urakawa I, Oshima T, Ono K, Kakitani M, Tomizuka K, Fujita T, et al. Vitamin D receptor‐independent FGF23 actions in regulating phosphate and vitamin D metabolism. Am J Physiol Renal Physiol. 2005;289:F1088–1095. [DOI] [PubMed] [Google Scholar]
- 154. Rodriguez‐Ortiz ME, Lopez I, Munoz‐Castaneda JR, Martinez‐Moreno JM, Ramirez AP, Pineda C, Canalejo A, Jaeger P, Aguilera‐Tejero E, Rodriguez M, et al. Calcium deficiency reduces circulating levels of FGF23. J Am Soc Nephrol. 2012;23:1190–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Shahbazian H, Zafar Mohtashami A, Ghorbani A, Abbaspour MR, Belladi Musavi SS, Hayati F, Lashkarara GR. Oral nicotinamide reduces serum phosphorus, increases HDL, and induces thrombocytopenia in hemodialysis patients: a double‐blind randomized clinical trial. Nefrologia. 2011;31:58–65. [DOI] [PubMed] [Google Scholar]
- 156. Labonte ED, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo‐McCoy S, He L, Dy E, Black D, Zhong Z, et al. Gastrointestinal inhibition of sodium‐hydrogen exchanger 3 reduces phosphorus absorption and protects against vascular calcification in CKD. J Am Soc Nephrol. 2015;26:1138–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Block GA, Rosenbaum DP, Yan A, Greasley PJ, Chertow GM, Wolf M. The effects of tenapanor on serum fibroblast growth factor 23 in patients receiving hemodialysis with hyperphosphatemia. Nephrol Dial Transplant. 2019;34:339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Block GA, Rosenbaum DP, Yan A, Chertow GM. Efficacy and safety of tenapanor in patients with hyperphosphatemia receiving maintenance hemodialysis: a randomized phase 3 trial. J Am Soc Nephrol. 2019;30:641–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Ix JH, Isakova T, Larive B, Raphael KL, Raj DS, Cheung AK, Sprague SM, Fried LF, Gassman JJ, Middleton JP, et al. Effects of nicotinamide and lanthanum carbonate on serum phosphate and fibroblast growth factor‐23 in CKD: the COMBINE trial. J Am Soc Nephrol. 2019;30:1096–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Marks J, Churchill LJ, Srai SK, Biber J, Murer H, Jaeger P, Debnam ES, Unwin RJ; Epithelial T, Cell Biology G . Intestinal phosphate absorption in a model of chronic renal failure. Kidney Int. 2007;72:166–173. [DOI] [PubMed] [Google Scholar]
- 161. Sabbagh Y, O'Brien SP, Song W, Boulanger JH, Stockmann A, Arbeeny C, Schiavi SC. Intestinal npt2b plays a major role in phosphate absorption and homeostasis. J Am Soc Nephrol. 2009;20:2348–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Giral H, Caldas Y, Sutherland E, Wilson P, Breusegem S, Barry N, Blaine J, Jiang T, Wang XX, Levi M. Regulation of rat intestinal Na‐dependent phosphate transporters by dietary phosphate. Am J Physiol Renal Physiol. 2009;297:F1466–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Lau WL, Leaf EM, Hu MC, Takeno MM, Kuro‐o M, Moe OW, Giachelli CM. Vitamin D receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney Int. 2012;82:1261–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Ritter CS, Zhang S, Delmez J, Finch JL, Slatopolsky E. Differential expression and regulation of Klotho by paricalcitol in the kidney, parathyroid, and aorta of uremic rats. Kidney Int. 2015;87:1141–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Teng M, Wolf M, Ofsthun MN, Lazarus JM, Hernan MA, Camargo CA Jr, Thadhani R. Activated injectable vitamin D and hemodialysis survival: a historical cohort study. J Am Soc Nephrol. 2005;16:1115–1125. [DOI] [PubMed] [Google Scholar]
- 166. Shoben AB, Rudser KD, de Boer IH, Young B, Kestenbaum B. Association of oral calcitriol with improved survival in nondialyzed CKD. J Am Soc Nephrol. 2008;19:1613–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Thadhani R, Appelbaum E, Pritchett Y, Chang Y, Wenger J, Tamez H, Bhan I, Agarwal R, Zoccali C, Wanner C, et al. Vitamin D therapy and cardiac structure and function in patients with chronic kidney disease: the PRIMO randomized controlled trial. JAMA. 2012;307:674–684. [DOI] [PubMed] [Google Scholar]
- 168. Wetmore JB, Liu S, Krebill R, Menard R, Quarles LD. Effects of cinacalcet and concurrent low‐dose vitamin D on FGF23 levels in ESRD. Clin J Am Soc Nephrol. 2010;5:110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Koizumi M, Komaba H, Nakanishi S, Fujimori A, Fukagawa M. Cinacalcet treatment and serum FGF23 levels in haemodialysis patients with secondary hyperparathyroidism. Nephrol Dial Transplant. 2012;27:784–790. [DOI] [PubMed] [Google Scholar]
- 170. Kim HJ, Kim H, Shin N, Na KY, Kim YL, Kim D, Chang JH, Song YR, Hwang YH, Kim YS, et al. Cinacalcet lowering of serum fibroblast growth factor‐23 concentration may be independent from serum Ca, P, PTH and dose of active vitamin D in peritoneal dialysis patients: a randomized controlled study. BMC Nephrol. 2013;14:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Sprague SM, Wetmore JB, Gurevich K, Da Roza G, Buerkert J, Reiner M, Goodman W, Cooper K. Effect of cinacalcet and Vitamin D analogs on fibroblast growth factor‐23 during the treatment of secondary hyperparathyroidism. Clin J Am Soc Nephrol. 2015;10:1021–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Block GA, Bushinsky DA, Cunningham J, Drueke TB, Ketteler M, Kewalramani R, Martin KJ, Mix TC, Moe SM, Patel UD, et al. Effect of etelcalcetide vs placebo on serum parathyroid hormone in patients receiving hemodialysis with secondary hyperparathyroidism: two randomized clinical trials. JAMA. 2017;317:146–155. [DOI] [PubMed] [Google Scholar]
- 173. Charytan C, Coburn JW, Chonchol M, Herman J, Lien YH, Liu W, Klassen PS, McCary LC, Pichette V. Cinacalcet hydrochloride is an effective treatment for secondary hyperparathyroidism in patients with CKD not receiving dialysis. Am J Kidney Dis. 2005;46:58–67. [DOI] [PubMed] [Google Scholar]
- 174. Chonchol M, Locatelli F, Abboud HE, Charytan C, de Francisco AL, Jolly S, Kaplan M, Roger SD, Sarkar S, Albizem MB, et al. A randomized, double‐blind, placebo‐controlled study to assess the efficacy and safety of cinacalcet HCl in participants with CKD not receiving dialysis. Am J Kidney Dis. 2009;53:197–207. [DOI] [PubMed] [Google Scholar]
- 175. Zaritsky J, Rastogi A, Fischmann G, Yan J, Kleinman K, Chow G, Gales B, Salusky IB, Wesseling‐Perry K. Short daily hemodialysis is associated with lower plasma FGF23 levels when compared with conventional hemodialysis. Nephrol Dial Transplant. 2014;29:437–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Barros X, Torregrosa JV, Martinez de Osaba MJ, Casals G, Paschoalin R, Duran CE, Campistol JM. Earlier decrease of FGF‐23 and less hypophosphatemia in preemptive kidney transplant recipients. Transplantation. 2012;94:830–836. [DOI] [PubMed] [Google Scholar]
- 177. David V, Francis C, Babitt JL. Ironing out the cross talk between FGF23 and inflammation. Am J Physiol Renal Physiol. 2017;312:F1–F8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Babitt JL, Sitara D. Crosstalk between fibroblast growth factor 23, iron, erythropoietin, and inflammation in kidney disease. Curr Opin Nephrol Hypertens. 2019;28:304–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Block GA, Fishbane S, Rodriguez M, Smits G, Shemesh S, Pergola PE, Wolf M, Chertow GM. A 12‐week, double‐blind, placebo‐controlled trial of ferric citrate for the treatment of iron deficiency anemia and reduction of serum phosphate in patients with CKD Stages 3‐5. Am J Kidney Dis. 2015;65:728–736. [DOI] [PubMed] [Google Scholar]
- 180. Block GA, Pergola PE, Fishbane S, Martins JG, LeWinter RD, Uhlig K, Neylan JF, Chertow GM. Effect of ferric citrate on serum phosphate and fibroblast growth factor 23 among patients with nondialysis‐dependent chronic kidney disease: path analyses. Nephrol Dial Transplant. 2019;34:1115–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Yoon HE, Ghee JY, Piao S, Song JH, Han DH, Kim S, Ohashi N, Kobori H, Kuro‐o M, Yang CW. Angiotensin II blockade upregulates the expression of Klotho, the anti‐ageing gene, in an experimental model of chronic cyclosporine nephropathy. Nephrol Dial Transplant. 2011;26:800–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Narumiya H, Sasaki S, Kuwahara N, Irie H, Kusaba T, Kameyama H, Tamagaki K, Hatta T, Takeda K, Matsubara H. HMG‐CoA reductase inhibitors up‐regulate anti‐aging klotho mRNA via RhoA inactivation in IMCD3 cells. Cardiovasc Res. 2004;64:331–336. [DOI] [PubMed] [Google Scholar]
- 183. Yang HC, Deleuze S, Zuo Y, Potthoff SA, Ma LJ, Fogo AB. The PPARgamma agonist pioglitazone ameliorates aging‐related progressive renal injury. J Am Soc Nephrol. 2009;20:2380–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Matsubara T, Miyaki A, Akazawa N, Choi Y, Ra SG, Tanahashi K, Kumagai H, Oikawa S, Maeda S. Aerobic exercise training increases plasma Klotho levels and reduces arterial stiffness in postmenopausal women. Am J Physiol Heart Circ Physiol. 2014;306:H348–355. [DOI] [PubMed] [Google Scholar]
- 185. Tan SJ, Chu MM, Toussaint ND, Cai MM, Hewitson TD, Holt SG. High‐intensity physical exercise increases serum alpha‐klotho levels in healthy volunteers. J Circ Biomark. 2018;7:1849454418794582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Bergwitz C, Juppner H. Phosphate sensing. Adv Chronic Kidney Dis. 2011;18:132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Bon N, Frangi G, Sourice S, Guicheux J, Beck‐Cormier S, Beck L. Phosphate‐dependent FGF23 secretion is modulated by PiT2/Slc20a2. Mol Metab. 2018;11:197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Shalhoub V, Shatzen EM, Ward SC, Davis J, Stevens J, Bi V, Renshaw L, Hawkins N, Wang W, Chen C, et al. FGF23 neutralization improves chronic kidney disease‐associated hyperparathyroidism yet increases mortality. J Clin Invest. 2012;122:2543–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Hasegawa H, Nagano N, Urakawa I, Yamazaki Y, Iijima K, Fujita T, Yamashita T, Fukumoto S, Shimada T. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early‐stage chronic kidney disease. Kidney Int. 2010;78:975–980. [DOI] [PubMed] [Google Scholar]
- 190. Perwad F, Portale AA. Burosumab therapy for X‐linked hypophosphatemia and therapeutic implications for CKD. Clin J Am Soc Nephrol. 2019;14:1097–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Di Marco GS, Reuter S, Kentrup D, Grabner A, Amaral AP, Fobker M, Stypmann J, Pavenstadt H, Wolf M, Faul C, et al. Treatment of established left ventricular hypertrophy with fibroblast growth factor receptor blockade in an animal model of CKD. Nephrol Dial Transplant. 2014;29:2028–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Yanochko GM, Vitsky A, Heyen JR, Hirakawa B, Lam JL, May J, Nichols T, Sace F, Trajkovic D, Blasi E. Pan‐FGFR inhibition leads to blockade of FGF23 signaling, soft tissue mineralization, and cardiovascular dysfunction. Toxicol Sci. 2013;135:451–464. [DOI] [PubMed] [Google Scholar]
- 193. Katoh M. Therapeutics targeting FGF signaling network in human diseases. Trends Pharmacol Sci. 2016;37:1081–1096. [DOI] [PubMed] [Google Scholar]
- 194. Doi S, Zou Y, Togao O, Pastor JV, John GB, Wang L, Shiizaki K, Gotschall R, Schiavi S, Yorioka N, et al. Klotho inhibits transforming growth factor‐beta1 (TGF‐beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J Biol Chem. 2011;286:8655‐8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Shi M, Flores B, Gillings N, Bian A, Cho HJ, Yan S, Liu Y, Levine B, Moe OW, Hu MC. alphaKlotho mitigates progression of AKI to CKD through activation of autophagy. J Am Soc Nephrol. 2016;27:2331–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Hu MC, Shi M, Gillings N, Flores B, Takahashi M, Kuro OM, Moe OW. Recombinant alpha‐Klotho may be prophylactic and therapeutic for acute to chronic kidney disease progression and uremic cardiomyopathy. Kidney Int. 2017;91:1104–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Kuro OM. The Klotho proteins in health and disease. Nat Rev Nephrol. 2019;15:27–44. [DOI] [PubMed] [Google Scholar]
- 198. Xie J, Yoon J, An SW, Kuro‐o M, Huang CL. Soluble Klotho protects against uremic cardiomyopathy independently of fibroblast growth factor 23 and phosphate. J Am Soc Nephrol. 2015;26:1150–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Suassuna PGA, Cherem PM, de Castro BB, Maquigussa E, Cenedeze MA, Lovisi JCM, Custodio MR, Sanders‐Pinheiro H, de Paula RB. Highlight article: alphaKlotho attenuates cardiac hypertrophy and increases myocardial fibroblast growth factor 21 expression in uremic rats. Exp Biol Med (Maywood). 2020;245:66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Ding J, Tang Q, Luo B, Zhang L, Lin L, Han L, Hao M, Li M, Yu L, Li M. Klotho inhibits angiotensin II‐induced cardiac hypertrophy, fibrosis, and dysfunction in mice through suppression of transforming growth factor‐beta1 signaling pathway. Eur J Pharmacol. 2019;859:172549. [DOI] [PubMed] [Google Scholar]
- 201. King GD, Chen C, Huang MM, Zeldich E, Brazee PL, Schuman ER, Robin M, Cuny GD, Glicksman MA, Abraham CR. Identification of novel small molecules that elevate Klotho expression. Biochem J. 2012;441:453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]