

Abstract
Aromatic l‐amino acid decarboxylase deficiency (AADCD) is a rare, autosomal recessive neurodevelopmental disorder characterized by impaired synthesis of dopamine, noradrenaline, adrenaline and serotonin, leading to a complex syndrome of motor, behavioral, and autonomic symptoms. This retrospective study assessed the symptoms and developmental outcome of a large international cohort of patients with AADCD via physician and/or caregiver responses to a detailed, standardized questionnaire. Sixty‐three patients (60% female; ages 6 months‐36 years, median 7 years; 58 living) from 23 individual countries participated. Common symptoms at onset (median age 3 months, range 0‐12 months) were hypotonia, developmental delay, and/or oculogyric crises. Oculogyric crises were present in 97% of patients aged 2 to 12 years, occurred in the majority of patients in all age groups, and tended to be most severe during early childhood. Prominent non‐motor symptoms were sleep disturbance, irritable mood, and feeding difficulties. The majority of subjects (70%) had profound motor impairment characterized by absent head control and minimal voluntary movement, while 17% had mild motor impairment and were able to walk independently. Dopamine agonists were the medications most likely to produce some symptomatic benefit, but were associated with dose‐limiting side effects (dyskinesia, insomnia, irritability, vomiting) that led to discontinuation 25% of the time. The age distribution of our cohort (70% of subjects under age 13 years) and the observation of a greater proportion of patients with a more severe disease phenotype in the younger compared to the older patients, both suggest a significant mortality risk during childhood for patients with severe disease.
Keywords: dystonia‐parkinsonism, gene therapy, natural history, neurotransmitter disorders, rare diseases

1. INTRODUCTION
Aromatic l‐amino acid decarboxylase deficiency (AADCD) is a rare, autosomal recessive, neurometabolic disorder characterized by impaired synthesis of the catecholamines (dopamine, epinephrine, and norepinephrine) and serotonin. AADCD typically presents in infancy with hypotonia, oculogyric crises, and developmental delay. Mood and sleep disturbance, autonomic dysfunction, and intellectual disability are additional disease features. Since the initial description of the condition 1 over 135 patients with this disorder have been described in the medical literature. 2
The majority of published case reports and case series focus on presenting clinical, biochemical and genetic disease features. Unanswered questions remain about symptom evolution, the full spectrum of developmental outcomes, and mortality. In Taiwan, where there is a higher disease prevalence due to the presence of a founder mutation in the population, 3 patients have an almost uniformly severe disease course with profound motor impairment 4 and high risk of death in early childhood. 5 Internationally, the majority of reported patients have severe symptoms and motor disability, but patients with mild disease and more favorable response to medical treatment have also been described. 6 , 7 , 8
Most patients with AADCD experience limited benefit from currently available medical therapies. 9 Gene therapy for this disorder is under investigation using two different stereotactic neurosurgical approaches to deliver gene vector either to the putamen 5 , 10 , 11 or the midbrain (ClinicalTrials.gov Identifier: NCT02852213). In the context of new methods for early diagnosis already implemented in some newborn screening programs 12 and these prospective novel therapeutic opportunities, an improved knowledge of the natural history of the disease, modified by available medical treatments, is critical.
The objective of this study is to describe the initial symptoms, symptom course, treatment response, and developmental outcomes in a large international cohort of patients with AADCD.
2. METHODS
We created a written questionnaire to collect data about disease onset, symptom course, developmental outcome, and mortality (see Supporting Information). Participants were recruited via two sources: (1) The International Working Group of Neurotransmitter‐Related Disorders (iNTD) patient registry, which includes collaborating physicians from 32 centers in North America, Europe, and Asia 13 ; and (2) The AADC Research Trust, a parent‐run foundation based in the United Kingdom. For participants recruited via the iNTD, the questionnaire was included as a module of the working group's de‐identified online registry and was completed by the physician and/or parent/caregiver. For participants recruited via the AADC Trust, an online English‐language version of the questionnaire was created using REDCap and was completed by a parent/caregiver. Families received an email invitation from the AADC Trust with a link to participate in the REDCap survey. Translations of the questionnaire were available in multiple languages to facilitate participation by non‐English speaking families. Participation was voluntary, and responses were anonymous. Limited demographic information (subject month and year of birth; country of residence) was collected to permit identification of potential duplicate responses. Questionnaires were completed by physicians and caregivers between December 2015 and October 2018.
If a respondent did not answer any questions for a given section of the questionnaire, they were excluded from that section's analysis. The total number of respondents for each section is indicated in the Results section below.
Permission to use items from Your Child's Usual Way of Moving Around to assess functional mobility was granted by Robert Palisano, CanChild Centre for Childhood Disability Research, McMaster University, Hamilton, Ontario, Canada.
This study was approved by the Human Research Protection Office of Washington University School of Medicine (protocol 201801179) and the local ethics committee of the University Hospital Heidelberg (iNTD coordinating center, application S‐471/2014).
3. RESULTS
3.1. Subject characteristics
Completed questionnaires were received on behalf of 64 individuals with AADCD, comprised of 24 responses through the iNTD registry, and 48 through the AADC Research Trust (response rate 66% (48/73). Data was combined for eight participants for whom we received responses through both recruitment channels. One subject was excluded due to prior participation in a gene therapy clinical trial. The final number of participants included for analysis was 63 (38 female, 60%). Fifty‐eight (58) of the 63 subjects were living at the time of questionnaire completion, and 5 were deceased.
Subjects' ages at time of questionnaire completion (or death, when applicable) ranged from 6 months to 36.8 years (median 7 years) (Figure 1A). Participants originated from 23 different countries in Europe (n = 27, 43%), North America (n = 16, 25%), Asia (n = 12, 19%), South America (n = 4, 6.5%) and the Middle East (n = 4, 6.5%).
FIGURE 1.
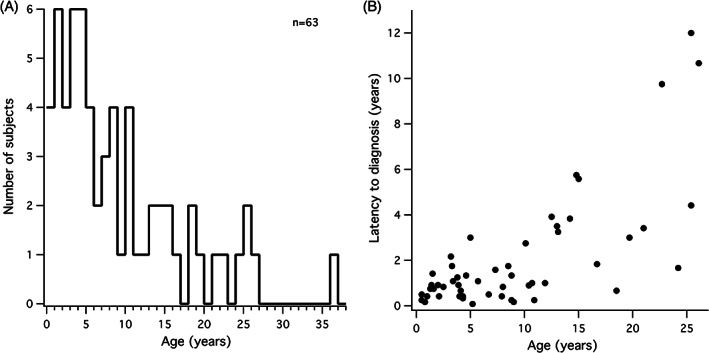
Subject age distribution and latency to diagnosis. A, Subject age ranged from 6 months to 36 years. There is a skew toward younger age (48% of subjects younger than 6 years, 70% younger than 13 years). B, Latency from symptom onset to diagnosis has decreased in the past decade; most subjects over age 10 years experienced a delay of years between symptom onset and diagnosis
3.2. Initial symptoms
The median age of onset of initial symptoms was 3 months (range: 0‐12 months), and was ≤6 months in all but three subjects. Latency from symptom onset to diagnosis varied considerably, and tended to be longer in subjects older than age 10 years (8 months to 31 years n = 17) compared to those age ≤ 10 years (mean ± SD = 0.9 ± 0.7 years) (Figure 1B).
The most commonly reported initial symptoms were hypotonia (75%), oculogyric crises (62%), developmental delay (62%), and feeding problems (54%) (Figure 2A). Non‐motor symptoms were also prominent, including sleepiness (50%), irritability (44%), excessive sweating (44%), and nasal congestion (42%). (n = 52) (Figure 2A). Initial misdiagnosis with epilepsy and/or a history treatment with anti‐epileptic medication prior to confirmation of the diagnosis of AADCD was reported for 14/52 subjects (27%), which is likely an underestimate.
FIGURE 2.
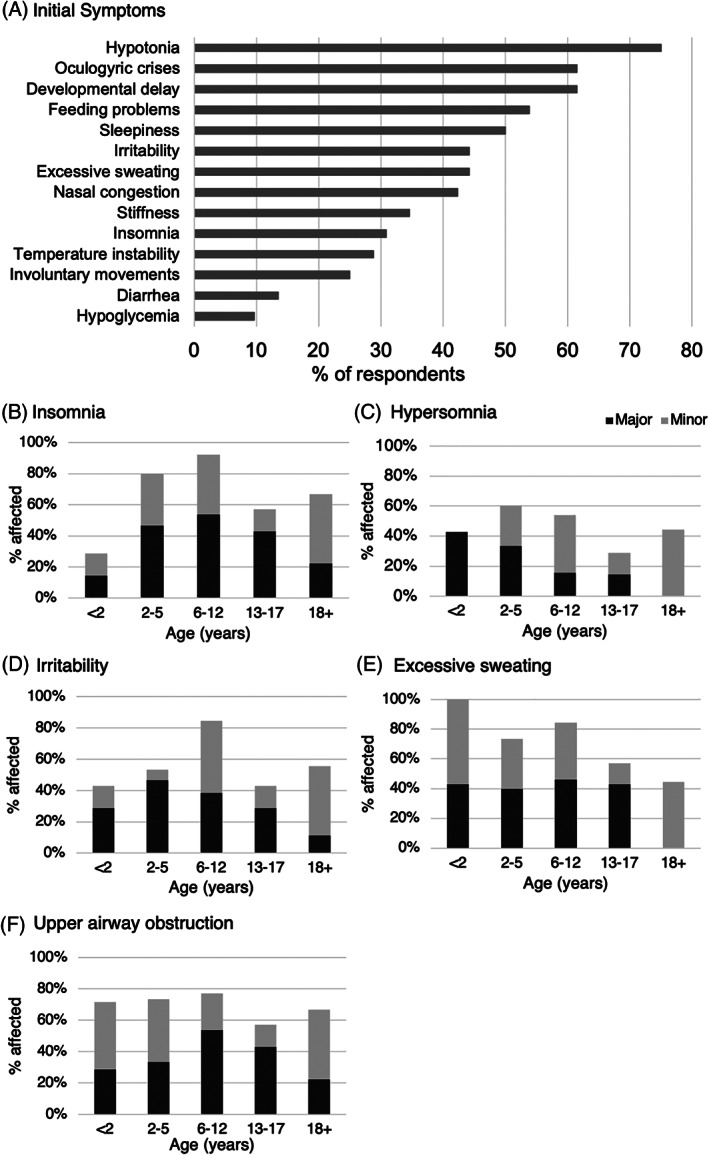
Motor and non‐motor symptoms. A, Reported prevalence of initial symptoms (n = 52). B‐F, Reported prevalence of current non‐motor symptoms across age groups (n = 51). Major (dark gray): frequent and/or severe, with significant impact on comfort or function; Minor (light gray): minor and/or infrequent
3.3. Non‐motor symptoms
Respondents were asked to classify each of a standard list of symptoms as either “major,” (frequent and/or severe) “minor,” (infrequent and/or mild) or “absent” across different ages. A cross‐sectional analysis of symptoms for each subjects' current age was performed (n = 51, Figure 2B‐F). Insomnia was present in 86% of subjects aged 2 to 12 years (24/28), characterized as a major problem in 50% of children in this age range (Figure 2B), while excessive sleepiness was more prominent in subjects under age 2 years (Figure 2C). Irritability was reported in 85% of children aged 6‐12 years, and over 40% of subjects in all other age groups (Figure 2D). Excessive sweating was reported in all children under age 2 years, and classified as a major symptom in approximately 40% of subjects from infancy through age 17 years (Figure 2E).
3.4. Oculogyric crises
Oculogyric crises (OGC) are episodes characterized by involuntary eye deviation that may be accompanied by involuntary movements of the face, neck, trunk, or limbs. Severe OGCs may cause whole body dystonia. The occurrence of OGCs in the present, past, or both, was reported by 98% of subjects (56/57). OGCs were a present symptom in the majority of subjects across all age groups. Episodes were particularly prevalent in those age 2 to 12 years (97%, 30/31), and were less prevalent in patients over 18 years (60%, 6/10) (Figure 3A). When asked to retrospectively state the age at which OGCs had been the most problematic (frequent, severe, and/or prolonged), 94% of respondents for 6 to 12 year‐olds (15/16), and 50% of respondents for those over 12 years old (8/16) cited ages ranging from infancy to age 5 years.
FIGURE 3.
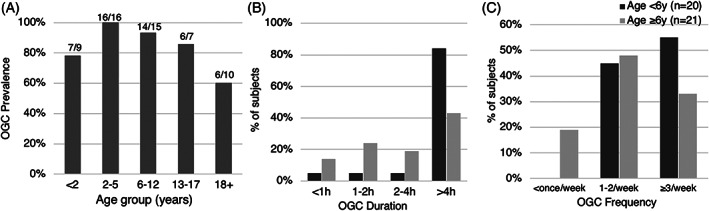
Oculogyric Crises (OGC). A, Reported current prevalence of OGC's across age groups (n = 57): 91% of subjects under 18, 60% of subjects over 18. B and C, OGC duration and frequency, respectively, in younger (age < 6 years, dark gray) vs older (age ≥ 6 years, light gray) subjects. Over 80% of younger subjects experienced prolonged episodes (> 4 hours duration), and the majority experienced episodes at least three times per week
In a more detailed cross‐sectional analysis (n = 42) of the presently experienced characteristics of oculogyric crises in younger subjects (age < 6 years) vs older subjects (age ≥ 6 years), younger patients were more likely to experience OGC episodes over 4 hours in duration (84% vs 43%), and to have episodes at least three times per week (55% vs 33%) (Figure 3B,C). A subset of patients (n = 38) provided further details about episode severity. Across all ages, the majority (29/38, 76%) experienced episodes characterized by dystonia of the trunk and/or limbs in addition to ocular deviation, while 9/28 (24%) experienced symptoms limited to the head and neck region only (eye deviation with or without involuntary movements of the lower face and head turning). A Mann‐Whitney test indicated that the group with whole‐body dystonic episodes was significantly younger (median age 5.2, range 1.3‐19.7 years) than the group with symptoms limited to the head and neck (median age 10.4, range 4.3‐26.1 years) (Z = 2.232, P = .026). Taken all together, these features suggest an association between more frequent, prolonged and severe OGCs and younger age in this cohort.
Five (5) of 17 subjects aged ≥13 years do not presently experience OGCs. Four (4) of these 5 subjects have a mild phenotype characterized by the ability to walk independently. Therefore, the reduced prevalence of OGCs in adolescents and adults may be attributable either to older age, milder disease phenotype (see “Developmental and Functional Outcome” below), or a combination of these factors.
3.5. Developmental and functional outcome
3.5.1. Motor development
Forty‐nine (49) respondents for subjects ≥12 months in age (range 1‐36 years) reported information about attainment of motor developmental milestones. Head control was reported in 33% (16/49), and independent sitting and walking was reported in 22% (11/49). All 11 subjects who could sit independently could also walk; there were none who reported the ability to sit only. Motor function differed notably across age groups in this cohort: younger subjects had more severe motor impairment than older subjects. None of the 19 subjects under 6 years of age could sit or walk independently. In contrast, 4/14 subjects (29%) aged 6‐12 years, and 7/16 subjects (44%) over age 12 years could sit and walk. This does not reflect simply a delay in the attainment of walking, since all but one of the 11 subjects who could walk had gained the ability before age 6 years (range 22 months‐7 years). Rather, the data suggest that a greater proportion of the older patients in this cohort had a milder disease phenotype.
An early regression in motor or developmental skills was reported in 15/63 patients (24%), typically occurring during infancy at the onset of oculogyric crises and other disease symptoms. The most commonly described lost skill was the ability to feed by mouth (n = 8). Others were loss of head control or other early motor skills such as rolling (n = 6), and speech (n = 2).
Feeding difficulties were common, reported in 75% of respondents (40/53) as either a past or current problem. Swallowing difficulties and vomiting were the most frequent problems reported (43% and 26%, respectively). In this cohort, 45% of subjects (24/53) had a gastrostomy tube placed for feeding.
3.5.2. Activities of daily living and adaptive behavior
Functional independence for routine activities of daily living including mobility, feeding, bathing, and dressing was analyzed for 38 subjects age 5 years and older. Respondents classified 11% (4/38) as completely independent, 18% (7/38) as partially independent, and 71% (27/38) as completely dependent. Of 29 subjects age 5 to 18 years, 62% (18/29) attended school. Of 8 young adults over age 18 years, 2 participated in work outside the home.
3.6. Molecular genetic results and genotype‐phenotype correlation
Molecular genetic data was known for 43 subjects, 11 of whom carried 14 variants that were not previously reported (Table 1). Overall, 71% of variants were classified as missense, 19% as splice site, 8% as frameshift, and 1% as nonsense. The most frequent variant was the known common splice site variant, 714+4A>T (15% of alleles in this cohort), which was detected in either homozygous (n = 3) or compound heterozygous (n = 7) form in 10 patients of southern Chinese ethnicity.
TABLE 1.
Patient demographics and genotype
| ID | Age (y) | Sex | Geographic region | Allele 1 genotype | Allele 2 genotype | Motor phenotypic severity c |
|---|---|---|---|---|---|---|
| 1 | 8 | F | North America | c.179T>C (p.V60A) | 714+4A>T | Moderate |
| 2 | 26.1 | M | Europe | c.476C>T (p. A159V) | ? | Moderate |
| 3 | 6.6 | F | North America | Severe | ||
| 4 | 10.9 | M | Europe | c.73G>A (p.E25K) | c.1073G>A (p. R358H) | Severe |
| 5 | 15 | M | North America | Moderate | ||
| 6 | 4 | M | North America | Severe | ||
| 7 a | 18.5 | F | North America | c.140C>A (p. P47H) | Homozygous | Severe |
| 8 | 8.8 | M | North America | Mild | ||
| 9 | 7.3 | M | South America | c.1040G>A (p. R347Q) | Homozygous | Severe |
| 10 | 3.7 | M | Middle East | Severe | ||
| 11 | 6.7 | M | Europe | Mild | ||
| 12 | 2.1 | M | North America | c.19C>T (p. R7*) | c.214C>T (p. H72Y) | Severe |
| 13 | 21 b | F | North America | Severe | ||
| 14 | 1.6 | F | South America | c.330_334dupCGATC (p.Q112fs*13) | Homozygous | Severe |
| 15 | 4.3 | M | Europe | c.231C>A (p. F77L) | Homozygous | Severe |
| 16 | 3.9 | F | North America | c.286G>A (p. G96R) | c.665T>C (p. L222P) | Severe |
| 17 | 2.5 b | F | Asia | Severe | ||
| 18 | 4.1 | F | Europe | c.323G>A (p. S108N) | c.1041+1G>C | Severe |
| 19 | 5.2 | F | North America | 714+4A>T | Homozygous | Severe |
| 20 | 2.4 b | F | Europe | Severe | ||
| 21 | 5.3 | F | Europe | c.73G>A (p. E25K) | c.315G>C (p. W105C) | Severe |
| 22 | 12.5 | M | Europe | c.782G>T (p. C261F) | c.1060G>A (p. G354S) | Severe |
| 23 | 1.4 | F | Middle East | Severe | ||
| 24 | 16.7 | F | Middle East | c.242C>T (p. P81L) | Homozygous | Severe |
| 25 | 10.7 | F | North America | Severe | ||
| 26 | 8.5 | M | South America | c.568_569insCGAT (p. Q190Pfs) | c.1040G>A (p. R347Q) | Severe |
| 27 | 0.8 | M | South America | Severe | ||
| 28 | 11.9 | F | Middle East | c.1040G>A (p. R347Q) | Homozygous | Severe |
| 29 | 7.9 | F | Europe | c.139C>G (p. P47A) | ? | Severe |
| 30 | 24.2 | F | Europe | c.367G>A (p. G123R) | c.876G>A (p. E292E) | Mild |
| 31 | 4 | M | North America | Severe | ||
| 32 | 7 b | M | North America | Severe | ||
| 33 | 5 | M | Europe | Moderate | ||
| 34 | 1.3 | M | Europe | c.73G>A (p. E25K) | c.624delC (p. I209Sfs*26) | Severe |
| 35 | 13.1 | M | Europe | c.367G>A (p. G123R) | c.876G>A (p. E292E) | Mild |
| 36 | 9 | M | Europe | c.1A>G (p. M1V) | c.181G>A (p. E61K) | Severe |
| 37 | 13 b | F | Europe | Severe | ||
| 38 | 14.8 | M | Europe | c.1073G>A (p. R358H) | Homozygous | Severe |
| 39 | 3.4 | M | Asia | 714+4A>T | Homozygous | Severe |
| 40 | 3.8 | F | Asia | 714+4A>T | c.1234C>T (p. R412W) | Severe |
| 41 | 1.4 | F | Asia | c.179T>C (p. V60A) | c.1234C>T (p. R412W) | Severe |
| 42 | 3.3 | F | Asia | 714+4A>T | c.1297dupA (p. I433Nfs*60) | Severe |
| 43 | 3.2 | F | Asia | c.106G>A (p. G36R) | 714+4A>T | Severe |
| 44 | 4.6 | F | Asia | c.170A>G (p. I57T) | c.1234C>T (p. R412W) | Severe |
| 45 | 2 | M | Asia | c.106G>A (p. G36R) | 714+4A>T | Severe |
| 46 | 1 | F | Asia | 714+4A>T | Homozygous | Severe |
| 47 | 10.1 | F | Europe | c.206C>T (p. T69M) | 1337T>C (p. L446P) | Mild |
| 48 | 25.4 | F | Europe | c.206C>T (p. T69M) | c.439A>C (p. S147R) | Severe |
| 49 | 5.7 | F | Europe | c.260C>T (p. P87L) | c.799T>C (p. W267R) | Severe |
| 50 a | 19.7 | F | Europe | c.214C>T (p. H72Y) | Homozygous | Severe |
| 51 | 1.5 | F | Europe | c.201+5G>C | Homozygous | Moderate |
| 52 | 8.8 | F | Europe | c.367G>A (p. G123R) | c.734C>T (p. T245I) | Mild |
| 53 | 0.5 | F | Europe | Severe | ||
| 54 a | 36.8 | F | Europe | c.105delC (p. Y37T fs*5) | c.710T>C (p. F237S) | Mild |
| 55 | 15 | M | Europe | c.843C>G (p. C281W) | c.1085T>C (p. M362T) | Mild |
| 56 | 0.5 | F | Europe | c.322A>C (p. S108R) | c.812A>T (p. D271V) | Severe |
| 57 | 0.8 | M | Europe | Moderate | ||
| 58 a | 18.2 | M | Asia | Moderate | ||
| 59 a | 25.4 | F | Asia | 714+4A>T | c.853C>T (p. R285W) | Mild |
| 60 a | 22.7 | F | Asia | 714+4A>T | c.853C>T (p. R285W) | Mild |
| 61 | 14.2 | F | North America | c.260C>T (p. P87L) | c.446G>C (p. S149T) | Mild |
| 62 | 4.3 | F | North America | c.260C>T (p. P87L) | c.446G>C (p. S149T) | Moderate |
| 63 | 10.4 | M | North America | Severe |
Note: Novel variants are italicized.
Deceased.
Phenotypic severity: mild = able to walk independently; severe = minimal or no attainment of developmental milestones; moderate = intermediate.
*Denoting a nonsense variant in the gene.
Each subject's motor phenotypic severity was broadly classified based on responses to questions about mobility and development (mild: able to walk independently without an assistive device; severe: minimal or no attainment of developmental milestones; moderate: intermediate; Table 1). Nine (9) of the 43 subjects with known genotype had a mild motor phenotype. These nine individuals all carried compound heterozygous variants that included at least one missense variant. Specific variants that were detected only in patients with mild or moderate phenotypic severity were: c.367G>A (p. G123R), c.446G>C (p. S149T), c.734 C>T (p. T245I), c.853C>T (p. R285W), c.876G>A (p. E292E), and c.1337T>C (p. L446P).
3.7. Treatment
Fifty‐nine (59) respondents answered questions about observed benefits and side effects of medications. Pyridoxine was the most commonly tried medication (78%, 46/59), but only 3/46 respondents (7%) reported noticeable improvement (“increased energy” in all cases). No significant side effects were reported.
At least one dopamine agonist (DA) was tried by 83% of subjects (49/59), including bromocriptine (46%, 27/59), pramipexole (41%, 24/59), rotigotine (12/59, 20%), and ropinirole (14%, 8/59). The DA most commonly reported to be beneficial was the rotigotine patch, judged to be beneficial in 82% of those who had tried it (10/12). Reported rates of benefit among the other DA's were 29% for pramipexole (7/24), 26% for bromocriptine (7/27), and 13% for ropinirole (1/8). The most commonly described benefits (by caregivers who volunteered additional details) were improvements in tone or spontaneous movements, improved alertness, and decreased oculogyric crises. However, caregivers reported adverse effects in association with all DA's: 30% with bromocriptine, 38% with pramipexole, and 50% with rotigotine and ropinirole. Dyskinesia and insomnia were the two most commonly reported side effects (27% and 14%, respectively), followed by vomiting and irritability. These adverse effects were reported at similar frequencies across all the DA's, and prompted discontinuation of the medication in 25% of subjects.
A trial of therapy with at least one monoamine oxidase inhibitor (MAO‐I) was reported in 62% of subjects (37/59): either selegiline (n = 20), tranylcypromine (n = 11), or both (n = 6). Beneficial effects were reported in 35% for selegiline and 18% for tranylcypromine (better alertness, improved tone or spontaneous movements). The reported frequency of side effects was lower for both medications than was reported for any DA (27% for selegiline, 6% for tranylcypromine), and the most commonly described side effect was insomnia. Side effects led to discontinuation of the medication in a small proportion of subjects (12% for selegiline, 6% for tranylcypromine).
3.8. Mortality
The parents of five individuals who had died responded to the survey. Ages of death were 2 years (n = 2), 7 years, 13 years, and 21 years, respectively. Causes of death reported by parents were pneumonia (n = 2), acute complications during an OGC (n = 2), and “myocardial infarction” (n = 1).
4. DISCUSSION
This retrospective analysis of disease‐related symptoms and developmental outcome in an international cohort of patients, aged 6 months to 36 years, revealed that AADCD is associated with a variety of complex motor and non‐motor symptoms throughout the lifespan, including oculogyric crises, sleep disorder, mood disturbance, gastrointestinal symptoms, and autonomic dysfunction. It also confirmed that, while the majority of currently diagnosed patients have profound motor developmental impairment, some (20% of patients over age 12 months in this cohort) have milder motor impairment and the ability to walk independently.
Symptom onset occurred in early infancy, almost always within the first 6 months. The most commonly reported initial symptoms were hypotonia, oculogyric crises, delayed motor development, and feeding difficulties, consistent with previous descriptions in the literature. 2 The most prominent non‐motor symptoms were sleep disturbance and irritability, both of which typically emerge during infancy, but are experienced by patients of all ages. The sleep disturbance seems to evolve with age: infants have excessive sleepiness, while many children and adolescents exhibit prominent insomnia.
Our findings confirm that OGCs are a near‐universal disease feature, and that they occur in the majority of patients of all ages. In our cohort, they were most prevalent (97%) in children between ages 2 and 12 years. Episodes were reported to peak in duration, frequency and severity before age 6 years in many patients. Indeed, episodes lasting longer than 4 hours were typical for 80% of subjects under age 6 years. Of note, for 2 of the deceased subjects, parents reported an OGC as the apparent proximate cause of death, highlighting that OGCs represent not only a weekly burden of hours of symptom management, but may also have the potential to be life‐threatening.
Our results provided additional insight into patients' experiences with medications, previously described to be of limited benefit for most patients. 2 , 9 Pyridoxine was well‐tolerated, but not clearly observed to be beneficial. Dopamine agonists, particularly rotigotine, were associated with observed benefits in tone, movement, alertness, and OGCs, but side effects including dyskinesia, insomnia, irritability, and vomiting were dose‐limiting, leading to discontinuation of the medication about 25% of the time. MAO‐inhibitors were associated with a lower rate of side effects than DAs, but were also reported to be beneficial in only a minority of subjects.
Among the genotypes of our subjects were 14 novel variants (base upon the recent review by. 14 Those novel variants were associated with a severe phenotype in all but one subject with a mild phenotype who had the heterozygous novel c.734C>T (p. T245I) variant and a known heterozygous c.367G>A (p. G123R) variant. In subjects of southern Chinese ethnicity, the known common c. 714+4A>T variant was the most frequently detected. A second recurrent variant (c.1234C>T; p.R412W), recently reported in multiple patients of Chinese ethnicity 15 was detected in three subjects in our cohort.
The age distribution of subjects in this study raises questions about survival. Younger subjects outnumbered older subjects: nearly half were younger than 6 years, and 71% were younger than 13 years. Furthermore, a significant proportion of the adult patients in our cohort exhibited a milder disease phenotype than the younger patients, characterized by milder motor impairment and the absence of oculogyric crises. These observations suggest a significant childhood mortality risk for patients with severe disease, as has been previously described for patients in Taiwan. 5 Five families shared information about the end of their child's life, providing some insight into potential causes of death. Two of the subjects died of complications of pneumonia, while the other three suffered an acute life‐terminating event that occurred in the context of an OGC in two cases.
The retrospective nature of this study prohibited precise analysis of the childhood mortality risk associated with AADCD. An additional limitation of the study design was the possibility of under‐reporting some disease features due to partially incomplete responses for some subjects.
Our findings illustrate that AADCD is associated with severe functional impairment in the majority of patients, and that prominent non‐motor and autonomic symptoms occur throughout the lifespan. Disease features are refractory to existing medical therapies, particularly in severe cases, which motivates the search for novel effective therapeutic strategies.
CONFLICT OF INTEREST
Dr. Pearson reports that she is an investigator on a NIH‐funded gene therapy clinical trial for AADC deficiency. Dr. Opladen reports grants from Dietmar Hopp Foundation, during the conduct of the study. Dr. Garcia‐Cazorla reports personal fees from PTC Therapeutics, outside the submitted work. Dr. Kato reports grants from Nobelpharma Co., Ltd., outside the submitted work. Dr. Gilbert, Dr. Mastrangelo, Dr. Leuzzi, Dr. Tay, Dr. Sykut‐Cegielska, Dr. Pons, Dr. Mercimek‐Andrews, Dr. Lücke, Dr. Oppebøen, Dr. Kurian, Dr. Steel, Dr. Manti, Dr. Jeltsch, Ms. Black, and Ms. Flint declare that they have no conflict of interest.
AUTHOR CONTRIBUTIONS
Toni S. Pearson: Study design, data analysis, manuscript initial draft, manuscript revision. Thomas Opladen, Lisa Flint: Study design, data interpretation, manuscript revision. Laura Gilbert, Kathleen Meeks: Data analysis and interpretation, manuscript revision. Angeles Garcia‐Cazorla, Mario Mastrangelo, Vincenzo Leuzzi, Stacy K. H. Tay, Jolanta Sykut‐Cegielska, Roser Pons, Saadet Mercimek‐Andrews, Mitsuhiro Kato, Thomas Lücke, Mari Oppebøen, Manju Kurian, Dora Steel, Filippo Manti, Kathrin Jeltsch: Data collection (completed questionnaire) for at least subject, data interpretation, manuscript revision. All authors approved the final version of the manuscript.
ETHICS STATEMENTS
This study was approved by the Human Research Protection Office of Washington University School of Medicine (Protocol 201 801 179, approved 26 March 2018), and by the local ethics committees of the University Hospital Heidelberg (iNTD coordinating center, application S‐471/2014, approved December 22, 2014), and Great Ormond Street Hospital (protocol 19NM23). SM‐A received an institutional case report consent form to share her patient's information. This consent is available upon request. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study. This consent is available upon request.
Supporting information
APPENDIX S1: Supporting Information.
ACKNOWLEDGMENT
This study was supported by the Pediatric Neurotransmitter Disease Association and NIH/NINDS (5 R01‐NS094292). AGC is funded by FIS: PI18/0111 (Instituto de Salud Carlos III: ISCIII and Fondo Europeo de desarrollo regional, FEDER).
Pearson TS, Gilbert L, Opladen T, et al. AADC deficiency from infancy to adulthood: Symptoms and developmental outcome in an international cohort of 63 patients. J Inherit Metab Dis. 2020;43:1121–1130. 10.1002/jimd.12247
Communicating Editor: Nenad Blau
Funding information Fondo Europeo de desarrollo regional; Instituto de Salud Carlos III; Pediatric Neurotransmitter Disease Association and NIH/NINDS
REFERENCES
- 1. Hyland K, Clayton PT. Aromatic amino acid decarboxylase deficiency in twins. J Inherit Metab Dis. 1990;13:301‐304. [DOI] [PubMed] [Google Scholar]
- 2. Wassenberg T, Molero‐Luis M, Jeltsch K, et al. Consensus guideline for the diagnosis and treatment of aromatic l‐amino acid decarboxylase (AADC) deficiency. Orphanet J Rare Dis. 2017;12:12 10.1186/s13023-016-0522-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee HF, Tsai CR, Chi CS, et al. Aromatic L‐amino acid decarboxylase deficiency in Taiwan. Eur J Paediatr Neurol. 2009;13:135‐140. 10.1016/j.ejpn.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 4. Hwu WL, Chien YH, Lee NC, Li MH. Natural history of aromatic L‐amino acid decarboxylase deficiency in Taiwan. JIMD Rep. 2018;40:1–6. 10.1007/8904_2017_54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hwu WL, Muramatsu S, Tseng SH, et al. Gene therapy for aromatic L‐amino acid decarboxylase deficiency. Sci Transl Med. 2012;4:134ra61 10.1126/scitranslmed.3003640. [DOI] [PubMed] [Google Scholar]
- 6. Leuzzi V, Mastrangelo M, Polizzi A, et al. Report of two never treated adult sisters with aromatic L‐amino acid decarboxylase deficiency: a portrait of the natural history of the disease or an expanding phenotype? JIMD Rep. 2015;15:39‐45. 10.1007/8904_2014_295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mastrangelo M, Manti F, Patanè L, et al. Successful pregnancy in a patient with L‐amino acid decarboxylase deficiency: therapeutic management and clinical outcome. Mov Disord Clin Pract. 2018;5:446‐447. 10.1002/mdc3.12622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tay SK, Poh KS, Hyland K, et al. Unusually mild phenotype of AADC deficiency in 2 siblings. Mol Genet Metab. 2007;91:374‐378. 10.1016/j.ymgme.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 9. Brun L, Ngu LH, Keng WT, et al. Clinical and biochemical features of aromatic L‐amino acid decarboxylase deficiency. Neurology. 2010;75:64‐71. 10.1212/WNL.0b013e3181e620ae. [DOI] [PubMed] [Google Scholar]
- 10. Chien Y‐H, Lee N‐C, Tseng S‐H, et al. Efficacy and safety of AAV2 gene therapy in children with aromatic L‐amino acid decarboxylase deficiency: an open‐label, phase 1/2 trial. Lancet Child Adolesc Health. 2017;1:265‐273. 10.1016/S2352-4642(17)30125-6. [DOI] [PubMed] [Google Scholar]
- 11. Kojima K, Nakajima T, Taga N, et al. Gene therapy improves motor and mental function of aromatic l‐amino acid decarboxylase deficiency. Brain. 2019;142:322‐333. 10.1093/brain/awy331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brennenstuhl H, Kohlmüller D, Gramer G, et al. High throughput newborn screening for aromatic l‐amino‐acid decarboxylase deficiency by analysis of concentrations of 3‐O‐methyldopa from dried blood spots. J Inherit Metab Dis.2020;43(3):602–610. 10.1002/jimd.12208. [DOI] [PubMed] [Google Scholar]
- 13. Opladen T, Cortès‐Saladelafont E, Mastrangelo M, et al. The international working group on neurotransmitter related disorders (iNTD): a worldwide research project focused on primary and secondary neurotransmitter disorders. Mol Genet Metab Rep. 2016;9:61‐66. 10.1016/j.ymgmr.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Himmelreich N, Montioli R, Bertoldi M, et al. Aromatic amino acid decarboxylase deficiency: molecular and metabolic basis and therapeutic outlook. Mol Genet Metab. 2019;127:12‐22. 10.1016/j.ymgme.2019.03.009. [DOI] [PubMed] [Google Scholar]
- 15. Dai W, Lu D, Gu X, et al. Aromatic L‐amino acid decarboxylase deficiency in 17 mainland China patients: clinical phenotype, molecular spectrum, and therapy overview. Mol Genet Genomic Med. 2020;8(3):e1143 10.1002/mgg3.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pons R, Ford B, Chiriboga CA, et al. Aromatic L‐amino acid decarboxylase deficiency: clinical features, treatment, and prognosis. Neurology. 2004;62:1058‐1065. [DOI] [PubMed] [Google Scholar]
- 17. Manegold C, Hoffmann GF, Degen I, et al. Aromatic L‐amino acid decarboxylase deficiency: clinical features, drug therapy and follow‐up. J Inherit Metab Dis. 2009;32:371‐380. 10.1007/s10545-009-1076-1. [DOI] [PubMed] [Google Scholar]
- 18. Ide S, Sasaki M, Kato M, et al. Abnormal glucose metabolism in aromatic L‐amino acid decarboxylase deficiency. Brain Dev. 2010;32:506‐510. 10.1016/j.braindev.2009.05.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
APPENDIX S1: Supporting Information.