

Abstract
Introduction: Carpal tunnel syndrome and ulnar neuropathy are such common maladies affecting the upper extremties that they often become the default diagnosis when patients complain of numbness, pain, or weakness of the hands. While often correct, there are a number of other conditions that can also cause sensory or motor loss of the hands, which should be considered when appropriate, as they can mimic upper extremity entrapment syndromes. Methods: In this review, we will discuss such mimics, including Charcot-Marie-Tooth disease, multifocal motor neuropathy, hereditary neuropathy with pressure palsies, mononeuropathy multiplex, Lewis-Sumner syndrome, brachial plexitis (Parsonage-Turner syndrome), myotonic dystrophy, inclusion body myopathy, and distal myopathy of Welander. We will discuss the clinical presentation, as well as diagnostic testing, treatment (if available), and prognosis. Conclusion: The objective is to provide a differential diagnosis for those patients who do not fit well clinically or respond to usual therapy for entrapment neuropathy of the upper extremities.
Keywords: mimics, entrapment neuropathy, neuropathy, myopathy, nerve injury, nerve, diagnosis
Introduction
Entrapment neuropathies occur when nerves are chronically compressed or mechanically injured at specific locations. Common entrapment neuropathies of the arm include carpal tunnel syndrome, ulnar neuropathy at the cubital tunnel or Guyon’s canal, ulnar neuropathy after humeral fracture (“tardy” ulnar palsy), radial nerve at the spiral groove and from humeral fractures, and superficial radial nerve at the wrist. Less common, and usually more controversial, entrapments (and this is not meant to be a comprehensive list) include the neurogenic thoracic outlet syndrome, the suprascapular nerve at the suprascapular notch, the proximal median nerve by a ligament of Struthers or at the pronator teres, the deep terminal branch of the ulnar nerve in the palm, and the posterior interosseous nerve by the Arcade of Frohse or at the extensor carpi radialis brevis (which may also entrap the superficial radial nerve). It is important to recognize the presence of entrapment neuropathies, as intervention can alleviate symptoms, prevent progression and avoid permanent damage to the nerve. Interventions may include rest from repetitive motion, change in activity, splinting, injection therapy, or surgery. Decisions on which therapy to apply are often made clinically and by electrodiagnostic testing, using factors such as the severity of symptoms, impairment of activities of daily living, degree of nerve damage, and prognosis. There are a variety of classification schemes for mononeuropathies, but the one we find most useful is the simplified scheme of Seddon (Table 1). Occasionally, other disorders of peripheral nerve and muscle (Table 2) can mimic entrapment neuropathies. Such conditions are important, as they will not respond to interventions designed to alleviate entrapment neuropathies, and failure to recognize them will delay appropriate evaluation and therapy, and inappropriate treatment may lead to worse outcomes (Figure 1).
Table 1.
Seddon’s Classification of Nerve Injury.
| Class 1 (Neurapraxia): Is used to describe compression injury to the nerve characterized by conduction block, which may be attributed to either transient ischemia or paranodal demyelination. The focal demyelination is confirmed by nerve conduction preservation below the injury. Denervation is not seen on needle electromyography. Neuropraxia is transient and recovery happens in a few weeks. |
| Class 2 (Axonotmesis): Is used to describe closed crush injury to the nerve characterized by Wallerian degeneration and loss of nerve conduction distal to the lesion. Denervation is seen on needle electromyography. There is loss of axons with retention of the integrity of Schwann cell basal lamina and endoneurial tissue, providing the basis for regeneration. Recovery is slow, occurring proximal to distal and taking months, and may be incomplete with prognosis best for distal lesions. |
| Class 3 (Neurotmesis): Describes the state of a nerve which has been completely severed at the axon, the endoneurium and the Schwann cell basal lamina. Wallerian degeneration occurs and nerve conduction is not seen below the lesion, with denervation on needle EMG. Prognosis is poor as axonal regeneration is severely limited with outcomes in the absence of surgical intervention of either neuroma formation or aberrant regeneration. |
Note. EMG = electromyography.
Table 2.
List of Possible Neuromuscular Mimics of Entrapment Neuropathies Discussed in this Review.
| Neuropathies | Myopathies |
|---|---|
| Mononeuritis multiplex | Inclusion body myositis |
| Diabetes | |
| Vasculitis | |
| Amyloidosis | Myotonic dystrophy type 1 |
| Hereditary neuropathy with liability to pressure palsies | Distal myopathy of Welander |
| Inflammatory | |
| Lewis-Sumner syndrome | |
| Multifocal motor neuropathy | |
| Sensory perineuritis | |
| Parsonage-Turner syndrome (brachial plexitis) | |
| Hereditary | |
| Charcot-Marie-Tooth types 1 and 2 | |
| Hereditary neuropathy with liability to pressure palsies | |
| CMT 2D/distal SMA 5 | |
| Infectious | |
| Lyme | |
| Leprosy | |
| Cervical radiculopathy | |
| Tumor | |
| Solitary schwannomas and neurofibromas | |
Note. Trauma and uncommon peripheral nerve tumors are other potential mimics of entrapment neuropathy, which will not be discussed. HNPP = hereditary neuropathy with liability to pressure; CMT = Charcot-Marie-Tooth.
Figure 1.

Algorithm for evaluating neuromuscular cause of arm weakness and numbness.
Note. HNPP = hereditary neuropathy with pressure palsies; NCV/EMG = nerve conduction velocity/electromyography.
Diagnostic Tests
In addition to careful history taking and detailed physical examination (Figures 2 and 3), there are several tests available which may help delineate entrapment neuropathies of the upper extremity from potential mimics. We briefly mention them here as we will be referring to them later.
Figure 2.

(a) Photograph of hand of patient with myotonic dystrophy type 1 showing typical swan neck posture of all fingers. (b) Photograph of hand of same patient attempting to relax fingers after grip, illustrating grip myotonia.
Figure 3.
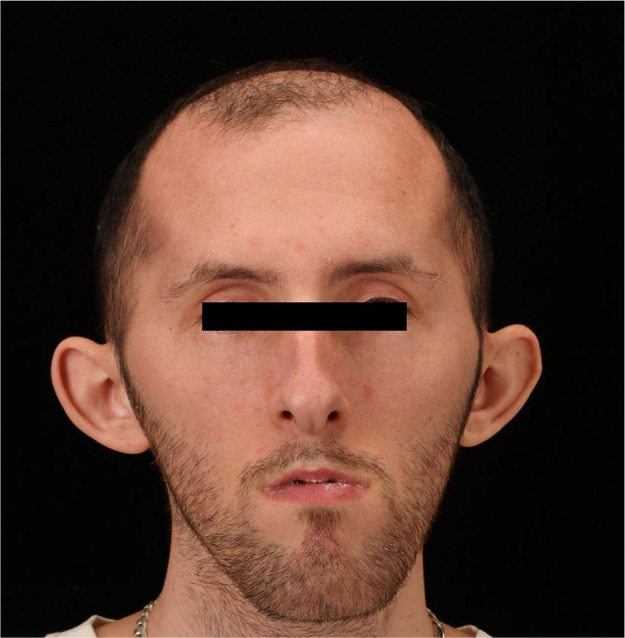
The face of a patient with myotonic dystrophy type 1 illustrating typical features of frontal balding, hatchet shape, mild bilateral ptosis, temporal wasting, and facial diparesis.
Laboratory tests for mononeuritis multiplex should include erythrocyte sedimentation rate (ESR), anti-nuclear antibody (ANA), C-reactive protein (CRP) serum protein electrophoresis (SPEP), and immunoflorescent electrophoresis (IFE), serum glucose and HgbA1c, and Lyme enzyme linked immunosorbent assay (ELISA). If myopathy is in question, creatine phosphokinase (CPK), aldolase, ESR, and thyroid profile should be done. Anti-ganglioside antibodies, such as anti-GM1, should be evaluated if multifocal motor neuropathy (MMN) or Lewis-Sumner syndromes are under consideration. Lumbar puncture for cerebrospinal fluid evaluation is important in evaluating inflammatory neuropathies such as chronic inflammatory demyelinating polyneuropathy (CIDP) and Lewis-Sumner syndrome, and should include at least cell count, glucose, and protein. Genetic testing is available and may be appropriate for several of the hereditary conditions, such as Charcot-Marie-Tooth (CMT) 1 and 2, hereditary neuropathy with liability to pressure palsies (HNPP), neurofibromatosis, and myotonic dystrophy.
Electrodiagnostic testing evaluates the electrical integrity of the peripheral nervous system by utilizing nerve conduction testing to evaluate peripheral nerves, and needle electromyography (EMG) to evaluate muscles. It is routinely available and performed by either neurologists or physiatrists. It can be very useful not only for diagnosis but also can provide information on extent of injury, type, and degree of injury, severity of the underlying condition, prognosis, and potential for recovery. There are very few mimics of entrapment neuropathy which would not be appropriate for electrodiagnostic testing early in the evaluation.
Imaging may be helpful, not only to make a diagnosis but to also exclude alternative diagnoses. Ultrasound of peripheral nerve and muscle is a developing field which can provide useful information on entrapment neuropathies as well as inform about other possibilities. Magnetic resonance imaging (MRI) of the spine is very important in the evaluation for cervical radiculopathy, neurofibromatosis, and peripheral nerve tumors. MRI of muscle can indicate muscle pathology as well as more precisely indicate which muscles are involved. MRI neurography can help with localization of injury, degree of injury, and diagnosis of tumors or nerve hypertrophy.
In certain cases, tissue diagnosis by way of nerve or muscle biopsies may be necessary. Muscle biopsy can assist with both myopathic and neuropathic disease, and is required to make a diagnosis of inclusion body myositis. Non-specific pathologic abnormalities are common but can help differentiate between muscle or nerve disease. Sampling error is possible, as the amount of tissue is small and the number of muscles which can be biopsied is limited. Best choice is a clinically involved muscle though not severely weak or atrophic, and which may also have abnormalities on EMG or imaging.
Nerve biopsy can be very helpful in diagnosing certain causes of neuropathy, such as vasculitis, inflammation, demyelination, and amyloidosis. For most other causes of neuropathy, nerve biopsy is less helpful due to non-specific abnormalities which may confirm neuropathy but little else. Due to the consequences of nerve biopsy, motor nerves are rarely biopsied and never in the upper extremity. Sensory nerves are preferred but due to the consequent sensory loss, a distal foot source such as the sural nerve is preferable to any sensory nerve in the hand or arm, though rarely the superficial radial sensory nerve may be the best choice. The lack of biopsy of a nerve in an affected limb, such as an arm, also adversely impacts the usefulness of nerve biopsy.
Neuropathies
Neuropathies can be divided into mononeuropathy or polyneuropathy. Mononeuropathy is involvement of individual peripheral nerves causing weakness, sensory loss, and deep tendon reflex loss within the distribution of that nerve. Involvement of multiple individual nerves, often in an asymmetric anatomic and chronologic pattern, is called mononeuropathy multiplex. Polyneuropathy, on the other hand, is a diffuse involvement of peripheral nerve, often from a systemic process. The cell body is the manufacturing engine of the peripheral nerve, with distribution of virtually everything needed for growth, maintenance and function of a nerve fiber needing to be distributed distally via axoplasmic transport. It is the vulnerability of this axoplasmic transport by which many diseases affect peripheral nerves. Thus, the distal portion of nerve axons are affected earliest and most severely. Polyneuropathy begins distally, in the feet and ascends over time, often not affecting the hands until symptoms and signs have reached the level of the knee. For the purposes of our discussion, that is, neuromuscular mimics of upper extremity entrapment neuropathies, entities involving polyneuropathy will be less germane than those causing mononeuropathy.
Diabetes
The most common cause of peripheral neuropathy in the United States and worldwide is diabetes mellitus. Diabetes can cause a variety of neuropathic syndromes, including distal symmetric neuropathy, proximal motor neuropathy (also called diabetic amyotrophy), cervical, thoraco-abdominal and lumbar radiculopathies, mononeuropathy, and mononeuropathy multiplex.1 The most typical form of diabetic neuropathy is distal symmetric polyneuropathy which affects up to 60% of diabetics. It is chronic, distal, symmetric, sensory predominant, and often painful. In a minority of patients, the motor involvement can be severe. The forms of diabetic neuropathy most likely to mimic entrapment neuropathy of the arm are mononeuropathy and mononeuropathy multiplex.
Diabetic mononeuropathy may present variously as cranial, axial, or appendicular neuropathy. The most commonly involved cranial nerve is the third nerve, which results in partial oculomotor palsy, typically with pupillary sparing. In the arm, any of the major nerves may be involved, both singly and multiply, in an asymmetric fashion both anatomically and chronologically. The etiology is believed to be due to microvascular disease causing occlusion of the vasa nervosum and small focal infarcts of the nerve. Sites of infarction are often not at common sites of compression, and can often be more precisely localized by electrodiagnostic testing. The prognosis is usually good, with recovery noted over the course of weeks to months. Diabetes also predisposes to compressive mononeuropathies at sites of entrapment, and this needs to be recognized as it affects wound healing, decreases surgical success at symptom relief, and increases the likelihood of recurrence.
Vasculitis
Vasculitis causing neuropathy may be primary or secondary in etiology, depending on whether coexisting systemic autoimmune disease is present.2 Polyarteritis nodosa is the most common cause of peripheral nerve vasculitis but small vessel vasculitis such as Churg-Strauss, microscopic polyangiitis, Wegener’s granulomatosis, essential mixed cryoglobinuria, and hepatitis C are other frequent causes. Connective tissue diseases such as rheumatoid arthritis, systemic sclerosis, lupus, and scleroderma can also cause vasculitis of peripheral nerve. Peripheral nerve vasculitis may manifest as acute single or multiple mononeuropathies, less acute overlapping mononeuropathies or subacute distal symmetric sensorimotor polyneuropathy. In those presenting with mononeuropathies, the onset of neuropathic symptoms may be quite sudden, with deficits appearing within a few hours. The clinical course may be stepwise or progressive. Though any nerve may be involved, legs are often affected first. Pain is common. Laboratory tests should include complete blood count, ESR, comprehensive metabolic panel, SPEP, CRP, and markers for other systemic autoimmune diseases. Nerve conduction velocity (NCV)/EMG show abnormalities, which are asymmetric within and between limbs. Motor and sensory nerve amplitudes are reduced while conduction velocities are preserved, and denervation is often seen on needle EMG, consistent with axonal loss. The definitive diagnosis is through tissue diagnosis with combined nerve/muscle biopsy advisable. Steroids remain the mainstay of treatment with other agents indicated depending on the underlying disease process.
Amyloidosis
Amyloidosis can be familial or secondary, usually due to a systemic illness such as multiple myeloma, Waldenstrom’s macroglobulinemia or another form of gammopathy, including isolated amyloidomas. The hereditary forms often affect the gene for transthyretin protein (TTR)3 which is a thyroid transporter and results in a severe progressive axonal sensorimotor polyneuropathy, often associated with entrapment neuropathies such as carpal tunnel syndrome, and cataracts. Secondary amyloidosis often presents as mononeuropathy multiplex, localizing to both common entrapment sites and other, non-entrapment sites. Again, carpal tunnel syndrome is a frequent site of neuropathy in these patients. This disease often takes a malicious course, in part due to the systemic nature and the underlying disease. Amyloidosis can also present with small fiber neuropathy, with progressive distal symmetric sensory neuropathy with associated autonomic neuropathy and pain. The particular presentation in an individual depends on the type of amyloid deposition. The AA type of amyloid protein is not usually associated with neuropathy. Red flags in the history to look for are chronic progressive disease process, absence of diabetes, positive family history in the case of familial amyloid polyneuropathy, pain, autonomic dysfunction, and involvement of other organ systems like heart, kidneys, and cornea. Laboratory testing must include SPEP and usually, IFE, and if familial form suspected, TTR, and genetic testing. Urine and SPEP and Bence-Jones proteins should also be assessed. Definite diagnosis may be obtained by abdominal fat pad biopsy and/or nerve biopsy. Genetic testing should be undertaken if familial disease is suspected. Treatment may range from non-specific, such as pain medications to more specific therapies like liver transplantation to prevent disease progression. Steroids and other immunomodulating therapies are usually of little benefit. If there is an underlying malignant process, treatment for that may assist.
HNPP
HNPP is inherited as an autosomal dominant trait due to deletions of the PMP-22 gene on chromosome 17.4 It can present with distal sensory/motor polyneuropathy and is also characterized by repeated focal pressure neuropathies such as carpal tunnel syndrome and peroneal palsy with foot drop. The first attack usually occurs in the second or third decade. Recovery from acute mononeuropathy is often complete; when recovery is not complete, the resulting disability is usually mild. Frequently, affected individuals also have signs of a mild to moderate peripheral neuropathy, which does not remit. The peripheral neuropathy is primarily demyelinating in nature, with slowing of nerve conduction velocity, particularly distally and at common sites of entrapment. It can be confused with acquired forms of demyelinating neuropathy such as CIDP. A family history of neuropathy and/or of entrapment neuropathies is an important clue. The diagnosis of HNPP should be suspected in any adult with recurrent focal compression neuropathies who has a family history consistent with autosomal dominant inheritance. PMP-22 is the only gene known to be associated with HNPP and genetic testing is commercially available though expensive. Nerve conduction studies will show prolonged distal motor latencies in nerves not associated with distal entrapment, such as tibial and peroneal, as well as typical features of entrapment neuropathies such as carpal tunnel syndrome, ulnar neuropathy at the elbow, and peroneal neuropathy at the fibular head. A wrist splint may alleviate carpal tunnel syndrome; ankle-foot orthoses (AFOs) may alleviate foot drop. Surgical decompression of nerves can be beneficial, but the entrapment neuropathies often improve by themselves, particularly the peroneal neuropathies. The entrapment neuropathies often recur, even if operated upon, and persistent pain syndromes are not uncommon. A conservative approach to surgical decompression is advised.
Lewis-Sumner Syndrome
This syndrome is also called multifocal acquired demyelinating sensory and motor neuropathy, and is a variant of CIDP.5 It is characterized by an asymmetric pattern of sensory and motor deficits corresponding to particular nerves, unlike CIDP which is a diffuse, symmetric proximal and distal progressive polyneuropathy. It affects arms much more frequently than legs. It can be asymmetric both in distribution and timing with some patients having symptoms in 1 limb for weeks to months before other limbs are affected. Proximal nerves, such as phrenic and suprascapular can be affected. NCV/EMG display features of demyelination and conduction block affecting various nerves, again in an asymmetric pattern. Cerebrospinal fluid (CSF) protein is elevated in some individuals without pleocytosis. Anti-ganglioside (GM1) antibodies are normal. Nerve biopsy, which is rarely done of upper extremity nerves, can show demyelination. Corticosteroids and intravenous immunoglobulin (IVIG) are the mainstay of treatment.
Multifocal Motor Neuropathy
This is another variant of CIDP with subacute onset of asymmetric weakness without sensory symptoms or signs. It usually starts with focal weakness involving distal upper extremities before spreading to involve other nerves in the same limb, then the contralateral limb.6 Weakness predominates and is elicitable on examination even in asymptomatic limbs. Fasciculations and cramps may be present with muscle wasting manifesting late into the disease process. Deep tendon reflexes are often preserved. CSF protein and serum CPK are normal. Serum antibodies against gangliosides, especially GM1, are elevated. NCV/EMG shows focal demyelination with conduction block and normal sensory responses. Periodic IVIG is the mainstay of treatment with corticosteroids and plasmapheresis actually causing worsening in certain cases.
Sensory Perineuritis
This is a pure sensory form of mononeuritis multiplex,7 which is diagnosed by sensory nerve biopsy showing focal thickening of the perineurium around the nerve fascicles with associated inflammation. It most commonly presents as a mononeuritis multiplex after the age of 50 years, though can also present as a distal demyelinating sensorimotor polyneuropathy. Perineuritis is associated with a variety of systemic diseases, most commonly, diabetes. Treatment with immunomodulation provides a variable response.
Parsonage-Turner Syndrome/Brachial Plexitis
Also known as idiopathic brachial plexopathy or neuralgic amyotrophy, it presents with sudden onset of constant unilateral severe shoulder pain. The pain may involve the rest of the limb. The pain may persist for several weeks before abating as weakness and atrophy ensue, often involving a pattern of nerves more so than the brachial plexus.8 The long thoracic nerve is often involved, causing scapular winging, and the phrenic nerve can be involved. It is self-limiting though recovery can be incomplete and take several months. The other side can subsequently be affected. Sensory symptoms are frequent. Etiopathogenesis remains unclear but is thought to be inflammatory due to its self-limited course. A familial form of brachial plexitis is inherited as a dominant trait on chromosome 17q25. NCV/EMG shows normal velocities and distal latencies but small motor and sensory amplitudes with widespread denervation, indicative of axonal loss. The pain, subsequent improvement, and axonal pattern on NCV/EMG allow differentiation from Lewis-Sumner syndrome and MMN. Steroids and IVIG may help shorten the period of pain but do not affect the weakness or atrophy. Despite this, prognosis is good. Treatment comprising non-narcotic and narcotic pain management is appropriate, as are passive and active range of motion exercises, occupational and physical therapy.
CMT Disease
There are 2 major types of CMT, with type 1 having a demyelination pattern on nerve conduction studies and nerve biopsy, and type 2 having an axonal pattern on nerve conduction study and nerve biopsy. Both are inherited as an autosomal dominant trait.9 An intermediate type is X linked CMT. There are rarer autosomal recessive forms. The usual phenotype has individuals start walking at an appropriate age but then develop distal leg then arm weakness and atrophy, with impaired ability to ambulate over the next couple of decades. All start in the feet, with progressive ascension of weakness and atrophy. Hands are not affected for at least 1 to 2 decades after onset, with weakness primarily affecting intrinsic hand muscles and finger extensors. Finger contracture is common, as are associated entrapment neuropathies of median and ulnar nerves. Tendon reflexes may or may not be lost. Sensation is affected to a lesser degree than strength. The disease is very slowly progressive and patients may need ambulatory aids by their 4th or 5th decade. Life expectancy is not affected. NCV/EMG is an important part of the diagnostic workup demonstrating demyelination versus axonal loss depending on the type of CMT. Duplication of the PMP-22 gene on chromosome 17p is the cause of 70% of CMT type 1. Genetic testing is available for several other genes causing types 1 and 2. Genetic defects in Connexin-32 are the cause for the x-linked form. Nerve biopsy is rarely indicated but when done shows evidence of chronic demyelination and remyelination in type 1, with onion bulbs, thinly myelinated axons and short intermodal lengths. Axonal dropout is the primary pathologic finding in type 2. No treatment exists for the underlying disease, but bracing with AFOs, appropriate ambulatory aids, physical therapy (PT), and occupational therapy (OT) are very important to maintaining functional independence. Surgical management of contractures may be necessary, and occasionally tendon transfer may provide important benefits for a few years. Surgical release of entrapment neuropathies is rarely of long term benefit as recurrence is common.
Another, rarer, form of CMT can cause bilateral thenar weakness and wasting. CMT 2D, also sometimes classified as hereditary distal motor neuropathy type 5 (dSMA5), has been described in only a handful of families and is an autosomal dominant disorder caused by a mutation in the glycyl tRNA synthetase gene on 7p14.3. The disease results in slow, progressive weakness and atrophy of the thenar muscles and first dorsal interosseous, as well as the intrinsic toe muscles, particularly involving the big toe. Sensation can be preserved, as it was in the one case we have seen. Thumb opposition and abduction is lost though flexion of the distal phalangeal joint of the thumb is preserved.
Lyme
Lyme disease is caused by Borrelia burgdoferi and is acquired in the United States from the bite of the deer tick, Ixodes ixodes. A different species of Borrelia causes a related disease in Europe, usually manifesting as Bannworths disease, a myeloradiculitis. Acute Lyme is usually seen from spring to fall. It is characterized by focal rash, low grade fever, and malaise. Neuropathy is generally acute in onset and occurs within a couple of months of initial infection. It commonly causes cranial neuropathies and radiculopathies, but may also present with mononeuropathy multiplex.10 Pain is common along with sensory and motor symptoms. The most commonly affected nerve is the facial nerve. Lyme ELISA is a sensitive screening test though may be negative very early in the course. Lyme Western Blot should be used to confirm any abnormal ELISA result. CSF may show evidence of meningitis. NCV/EMG shows evidence of axonal loss in the distribution of the affected nerve(s). Early and local disease should be treated with oral antibiotics (doxycycline or amoxicillin) but other neurologic disease may require IV treatment (ceftriaxone). The prognosis for Lyme mononeuropathy is quite good, even without antibiotics, but failure to treat may result in subsequent manifestations of late Lyme infection.
Leprosy
Leprosy is caused by Mycobacterium leprae and is also called Hansen disease. It is not common in United States but remains a differential of entrapment neuropathy. It causes multifocal sensory neuropathies along with characteristic skin lesions. Cooler areas of the body are more prone to infection, such as fingers, nose and toes. Diagnosis is confirmed by demonstrating acid-fast bacilli in skin smear or biopsy.11 Multi-drug anti-bacterials are used for treatment, which may cause initial worsening of neurologic symptoms. This may be counteracted by use of steroids in the initial treatment phase.
Cervical Radiculopathy
Radiculopathy is caused by impingement, infarction, or inflammation of exiting nerve roots by a variety of causes such as intervertebral disk herniation, spondylosis, osteophyte formation, tumors, diabetes, Lyme disease, or meningitis. It may mimic entrapment neuropathy, but symptoms and signs should be limited to particular myotomes and dermatomes. Detailed history and examination may help differentiate from mononeuropathy and definitive diagnosis can be made by NCV/EMG. Imaging, such as with MRI, can determine the anatomical nature of the injury but the prevalence of non-specific degenerative disk and spine disease often requires bedside and electrodiagnostic exam to determine extent and severity of root involvement. Surgical treatment is an option in many cases with impingement. The so-called “double-crush” phenomenon12 is an oft-discussed and controversial notion that symptoms may be due to dual impingements upon nerve fibers, such as at the neuro-foramen and at a more distal entrapment site. This would seemingly require both lesions be alleviated to provide relief and recovery though many patients have substantial improvement after a single procedure.
Isolated Neurofibromas
Neurofibromas are peripheral nerve tumors most notoriously associated with the hereditary disorder, neurofibromatosis type 1 (“The Elephant Man” disease). More germane here is that neurofibromas can occur in the absence of neurofibromatosis, as isolated tumors. They are commonly benign, presenting with weakness, sensory loss, and pain within the distribution of the affected nerve. The tumors arise from neural elements and so, the nerve fibers stream into and throughout the tumor, composed of a mixture of Schwann cells, perineural-like cells, and fibroblasts, such that surgical resection usually requires sacrificing the nerve. There are different varieties of neurofibromas—dermal, intraneural, plexiform, and massive soft tissue neurofibromas. Most worrisome are malignant peripheral nerve tumors.13 Features which suggest malignancy include rapid growth, increasing pain, and certain imaging features. NCV/EMG can be helpful in defining the nerve involved but do not help in determining neoplastic state or anatomy. Imaging, particularly MRI, is invaluable in diagnosis and provides important clues to extent of tumor, involvement of extraneural tissues and potential malignancy. Biopsy of any suspicious tumor is appropriate and removal of malignant tumors is required. Surgery becomes more relevant if there are features indicative of malignancy.
Schwannomas
Schwannomas are the most common peripheral nerve tumor in adults. They are also called neurilemmomas. They arise from Schwann cells and display a benign natural history. They are incidentally diagnosed, often characterized by painful swelling and shooting pain on nerve palpation.14 Motor or sensory symptoms are rare unless the tumor is in a confined space such as the carpal tunnel. Imaging may help define the extent of the tumor but often fails to differentiate from neurofibromas. Surgical resection is indicated in such cases. Outcome after surgery is better when compared to neurofibromas as the tumor grows around the nerve.
Myopathies
Muscle disease, in contrast to peripheral neuropathy, tends to affect proximal muscles more than distal, and often lower extremity muscles before upper. Most myopathic disorders do not mimic entrapment neuropathies of the hand and arm. But a few myopathic diseases characteristically affect distal muscles in the upper extremity exclusively or relatively early in the course of disease and it will be those disorders we discuss here.
Inclusion Body Myositis
Inclusion body myositis (IBM) is an inflammatory myopathy and is the most common myopathic disorder of the elderly. IBM presents with insidious asymmetric weakness involving the quadriceps muscles and the wrist and finger flexors. This distribution of atrophy and weakness is so characteristic as to almost be pathognomic. The disease is uncommon before the age of 50 and predominantly affects men. It is very slowly progressive and ambulation is usually not substantially impaired for at least a decade. Serum CPK may be elevated but usually not above 10 fold normal and can be normal. ESR is normal and other inflammatory markers are also negative. NCV/EMG demonstrates a non-specific pattern of abnormality usually most consistent with an active destructive myopathy. The distribution of affected muscles can be helpful in diagnosis but may be most helpful in assisting selection of which muscle(s) to biopsy. This is a pathologic diagnosis with the characteristic abnormalities including rimmed vacuoles and intermysial lymphocytic inflammation (which can be absent or not impressive) on light microscopy, and intranuclear filamentous inclusions on EM.15 Various immunohistochemical markers have been proposed but remain less than definitive. The pathologic abnormalities can be difficult to find and so, dual muscle biopsies may increase yield. Antibodies against the cytosolic 5’ nucleotidase 1A are positive in many but not all cases.16 In a patient with typical distribution of weakness and atrophy, failure to find pathologic confirmation should encourage consideration of further muscle biopsy. Unfortunately, no treatment has been shown to be effective in halting or slowing down the disease, despite the presumed inflammatory nature. This, and the presence of numerous proteins also seen in neurons and neuronal plaques of Alzheimer’s disease, has led some investigators to consider this the muscle equivalent of a neurodegenerative disease.
Myotonic Dystrophy Type I
Myotonic dystrophy type 1 is one of the most prevalent inherited neuromuscular diseases in adults and is inherited as an autosomal dominant trait.17 It is caused by a triplet repeat within the gene for a protein kinase, Myotonin, located on chromosome 19. There is marked variation in severity within families and anticipation is seen. A very accurate genetic test is available to confirm the diagnosis. The triple repeat causes a toxic gain of function, with transcription and splicing factors bound into inclusions and thus unavailable. This leads to multi-system disease as multiple enzymes, receptors, channels, and other proteins are dysfunctional. One of the more common manifestations is myotonia due to defective chloride channels in muscle. Myotonia is the persistent contraction of muscle fibers as the fiber cannot maintain a stable re-polarization potential. This results in grip myotonia, in which patients can only very slowly relax their grip, and percussion myotonia, in which a muscle will continue to contract after a brief tap on the muscle. This is most easily detected by tapping on the opponens pollicis and observing for progressive thenar adduction. Another characteristic feature is distal weakness, involving finger extensors and intrinsic hand muscles as well as ankle dorsiflexion (Figure 2). Weakness slowly progresses over decades, involving proximal muscles only late in the disease. Muscle wasting is common and seen early in dorsal aspect of forearm. There are multiple other characteristic features, including temporal wasting, bilateral facial weakness, ptosis, dysarthria and dysphagia, and cataracts which allow a high index of suspicion at the bedside (Figure 3). A family history often is very helpful in making the diagnosis. Serum CPK may be normal to mildly elevated. NCV/EMG demonstrates myotonia, including otherwise asymptomatic patients. Muscle biopsy provides non-specific changes indicative of myopathy. The facial features are so characteristic as to often enable diagnosis, especially in the presence of grip or percussion myotonia. Definite diagnosis arises from genetic testing. There is no effective medical treatment.
Distal Myopathy of Welander
The distal myopathies are a group of inherited diseases of muscle, which affect distal muscles initially and predominantly.18 All are rare. They are usually classified by age of onset, inheritance pattern and location of muscle onset. Only one, distal myopathy of Welander, has usual onset in the hands. It is inherited as an autosomal dominant trait, usually presenting in late adulthood (mean age of 47 years at onset). It has only been described in patients of Scandinavian descent. The disease begins with clumsiness of fine finger movements, often involving abductor pollicis longus and brevis moreso than adductor pollicis, and index finger extension before spreading to wrist and other finger extension. Distal leg muscles become involved eventually. It does not shorten lifespan. Serum creatine kinase may be normal or mildly elevated. NCV/EMG may reveal myopathic features. There is no effective medical treatment.
Summary
Entrapment neuropathies are due to compression or stretching of peripheral nerves. Surgical management is often indicated for relief of symptoms. Prior to committing an individual to surgery, careful history, detailed examination, and adequate testing should be carried out to ascertain that there are no non-compressive etiologies for the presentation, some of which may be adversely affected by surgery. As figure 1 indicates, a fairly simple diagnostic algorithm can be useful in determining the differential diagnosis for unilateral or bilateral upper extremity weakness which may mimic entrapment neuropathy.
Acknowledgments
No funding was provided by any source to support this manuscript
Footnotes
Ethical Approval: This study is exempt from institutional review board approval.
Statement of Human and Animal Rights: This is a review article which contains no original research.
Statement of Informed Consent: This is a review article which contains no original research. Thus, informed consent was not required.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iD: James M. Gilchrist  https://orcid.org/0000-0003-4460-2807
https://orcid.org/0000-0003-4460-2807
References
- 1. Callaghan BC, Cheng HT, Stables CL, et al. Diabetic neuropathy: clinical manifestations and current treatments. Lancet Neurol;11(6):521-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gwathmey KG, Burns TM, Collins MP, et al. Vasculitic neuropathies. Lancet Neurol. 2014;13:67-82. [DOI] [PubMed] [Google Scholar]
- 3. Shin SC, Robinson-Papp J. Amyloid neuropathies. Mt Sinai J Med. 2012;79:733-748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lewis RA, Sumner AJ, Shy ME. Electrophysiologic features of inherited demyelinating neuropathies: a reappraisal in the era of molecular diagnosis. Muscle Nerve. 2000;23:1472-1487. [DOI] [PubMed] [Google Scholar]
- 5. Lewis RA, Sumner AJ, Brown MJ, et al. Multifocal demyelinating neuropathy with persistent conduction block. Neurology. 1982;32:958-964. [DOI] [PubMed] [Google Scholar]
- 6. Muley SA, Parry GJ. Multifocal motor neuropathy. J Clin Neurosci. 2012;19:1201-1209. [DOI] [PubMed] [Google Scholar]
- 7. Sorenson EJ, Sima AA, Blaivas M, et al. Clinical features of perineuritis. Muscle Nerve. 1997;20:1153-1157. [DOI] [PubMed] [Google Scholar]
- 8. Feinberg JH, Radecki J. Parsonage-turner syndrome. HSS J. 2010;6:199-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pareyson D, Marchesi C. Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol. 2009;8(7):654-667. [DOI] [PubMed] [Google Scholar]
- 10. Halperin JJ, Little BW, Coyle PK, et al. Lyme disease: cause of a treatable peripheral neuropathy. Neurology. 1987;37(11):1700-1706. [DOI] [PubMed] [Google Scholar]
- 11. Ooi WW, Srinivasan J. Leprosy and the peripheral nervous system: basic and clinical aspects. Muscle Nerve. 2004;30(4):393-409. [DOI] [PubMed] [Google Scholar]
- 12. Morgan G, Wilbourn AJ. Cervical radiculopathy and coexisting distal entrapment neuropathies: double-crush syndromes. Neurology. 1998;50(1):78-83. [DOI] [PubMed] [Google Scholar]
- 13. Bhattacharyya AK, Perrin R, Guha A. Peripheral nerve tumors: management strategies and molecular insights. J Neurooncol. 2004;69(1-3):335-349. [DOI] [PubMed] [Google Scholar]
- 14. MacCollin M, Woodfin W, Kronn D, et al. Schwannomatosis: a clinical and pathologic study. Neurology. 1996;46(4):1072-1079. [DOI] [PubMed] [Google Scholar]
- 15. Engel WK, Askanas V. Inclusion-body myositis: clinical, diagnostic, and pathologic aspects. Neurology. 2006;66(2 suppl 1):S20-S29. [DOI] [PubMed] [Google Scholar]
- 16. Larman HB, Salajegheh M, Nazareno R, et al. Cytosolic 5’-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann Neurol. 2013;73(3):408-418. [DOI] [PubMed] [Google Scholar]
- 17. Udd B, Krahe R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012;11(10):891-905. [DOI] [PubMed] [Google Scholar]
- 18. Kraya T, Zierz S. Distal myopathies: from clinical classification to molecular understanding. J Neural Transm (Vienna). 2013;120(suppl 1):S3-S7. [DOI] [PubMed] [Google Scholar]