

Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder caused by loss of motor neurons from the brain and spinal cord. The ALS community has made remarkable strides over three decades by identifying novel familial mutations, generating animal models, elucidating molecular mechanisms, and ultimately developing promising new therapeutic approaches. Some of these approaches which reduce expression of mutant genes are in human clinical trials, highlighting the need to carefully consider the normal functions of these genes and potential contribution of gene loss-of-function to ALS. Here we highlight known loss-of-function mechanisms underlying ALS, potential consequences of lowering levels of gene products, and the need to consider both gain- and loss- of function to develop safe and effective therapeutic strategies.
Keywords: ALS, Loss of function, Gain of function, C9orf72, TDP-43, TARDBP, FUS, SOD1, TBK1, OPTN
In this review, Kim et al. discuss evidence for loss-of-function mutations and mechanisms in amyotrophic lateral sclerosis (ALS), and propose the need to consider both gain- and loss-of function in designing effective and safe therapeutic strategies.
INTRODUCTION
Nearly three decades following the discovery of the first amyotrophic lateral sclerosis (ALS) gene, the field has now entered a therapeutic phase. The promise of human genetics is coming to fruition, with several clinical trials underway to test therapies directly targeting mutant ALS-causing genes and with more in the process of development (Figure 1). Likewise, there is great hope that the remarkable pace of ALS gene discovery will empower the development of even more therapeutic approaches, including ones for sporadic forms of ALS.
Figure 1: ASOs Targeting Mutant ALS Genes.

A. ASOs targeting mRNA from three ALS genes—SOD1, C9ORF72, and FUS—for degradation have been administered to human ALS patients. These ASOs are at various stages of development, as indicated. B. ASOs are delivered broadly throughout the central nervous system after injection into the cerebrospinal fluid at the base of the spinal cord (intrathecal administration).
ALS is an adult onset neurodegenerative disorder that is characterized by degeneration of upper and lower motor neurons, clinically manifesting in a progressive loss of motor function and ultimately death often due to respiratory failure (Taylor et al., 2016). While the most salient clinical feature of ALS is motor involvement, ALS shares many neuropathological and genetic features with another neurodegenerative disorder—frontotemporal dementia (FTD)—the second leading cause of dementia after Alzheimer Disease. The two disorders share common genetic mutations and are often pathologically characterized by ubiquitinated inclusions of the 43-kD TAR DNA-binding protein (TDP-43) and abnormal accumulations of other RNA-binding proteins (Ferrari et al., 2011; Liscic et al., 2008; Morris et al., 2012; Neumann et al., 2006; Orr, 2011).
In the past decade, the development and clinical application of antisense oligonucleotides (ASOs) has taken the field by storm. The approach is elegantly simple: if a mutated gene causes disease because of a toxic function, then target that toxic gene product for degradation – stopping the deleterious RNAs or proteins in their tracks (Figure 1). There are variations to this theme in which other types of oligonucleotides are used to boost production of a gene product by blocking undesirable splicing events, like in treatment of the childhood neuromuscular disorder spinal muscular atrophy (Finkel et al., 2016; Hua et al., 2011; Wan and Dreyfuss, 2017; Wood et al., 2017). ASOs have shown striking efficacy in preclinical studies using animal models or human cells (Becker et al., 2017; McCampbell et al., 2018; Meng et al., 2015; Scoles et al., 2017; Smith et al., 2006; Sztainberg et al., 2015), and clinical trials are underway to test ASOs targeting at least two mutant genes in ALS patients (Miller et al., 2020; ClinicalTrials.gov identifier: NCT02623699; ClinicalTrials.gov identifier: NCT03626012) (Figure 1). There are even efforts to tailor ASOs to target patients on an individual basis (Daskalakis, Stella, 2020). Gene therapy using viral vectors to deliver RNA interference is also emerging as a powerful therapeutic approach to complement the ASO method (Bravo-Hernandez et al., 2020). There is great optimism in the field that these will provide the first disease-modifying therapies for this otherwise fatal disease.
Approximately 10% of ALS cases are considered “familial” (fALS), or transmitted within families, while the remaining are considered “sporadic” (sALS), or presenting without a clear familial history. Although less common than sALS, fALS has played an outsize role in our understanding of disease mechanisms through the discovery of ALS-causing mutations within families and subsequent experimental perturbations of these mutant genes. Since fALS occurs within the same family across multiple generations, genetic approaches can be used to pinpoint the mutated gene that tracks with people who developed ALS and away from those who did not.
This type of analysis has led to the discovery of almost all of the major ALS genes (Chia et al., 2018). After an ALS gene is discovered and validated, the next step is to define how that mutation affects the protein encoded by the gene. In other words, does the mutation cause a loss of function or some sort of gain of function (including gain of a toxic function)? If a mutation causes disease by conferring a loss of function, then a gene therapy approach to restore the gene’s function would be warranted. However, if the mutation causes disease because of a gain of function, then a therapeutic approach to inhibit the mutant gene’s functionality or lower the levels of its gene products would be more appropriate. As evident, sorting this out will be critical for guiding therapy development for ALS.
So far, most of the mutations that have been identified in fALS are thought to cause disease via gain-of-function (GOF) mechanisms, meaning the mutation endows the protein with increased wild type (WT) functionality or altered function that is deleterious. Diseases caused by toxic GOF mutations often exhibit autosomal dominant or semi-dominant inheritance patterns, such that one mutant copy of the gene is sufficient to cause disease despite the presence of another WT gene and presumably half the amount of WT protein. Many observations have implied GOF mechanisms as key drivers in ALS, such as dominant inheritance patterns, generation of toxic protein aggregates, and altered properties of molecules that impair biological processes (Taylor et al., 2016). For instance, the first ALS gene that was described in 1993—superoxide dismutase 1 (SOD1)—is widely considered to cause ALS through a GOF mechanism, not only because of compelling genetic studies in animal models but also because of its dominant inheritance patterns and ability to cause protein aggregation (Bruijn et al., 1997; Deng et al., 1993; Gurney et al., 1994; Rosen et al., 1993; Wong et al., 1995).
Other known causes of ALS, such as mutations in RNA-binding protein encoding genes like TARDBP, FUS, and HNRNPA1, might work through GOF mechanisms as alluded to by aggregation or other altered physical properties. These mutant proteins can disrupt RNA function and metabolism, proteostasis, or axonal cytoskeletal dynamics through the formation of toxic protein aggregates (Taylor et al., 2016). However, the normal functions of these RNA-binding proteins are also disturbed by mutations. They are redistributed from their normal localization in the nucleus to the cytoplasm in disease, and nuclear depletion of these proteins results in widespread alterations in the regulation of their RNA-binding targets, consistent with a loss of function (Klim et al., 2019; Lagier-Tourenne et al., 2012; Ling et al., 2015; Melamed et al., 2019; Polymenidou et al., 2011).
Sometimes the ALS-causing mutations are not located in the coding portion of the gene. For example, the most common genetic cause of ALS is a mutation in the C9ORF72 gene (DeJesus-Hernandez et al., 2011; Renton et al., 2011). This mutation is a hexanucleotide repeat expansion that is located in C9ORF72’s intron or promoter. The expanded GGGGCC repeat gets transcribed into repeat-containing RNAs and then translated into aberrant proteins, both of which could cause disease via GOF toxicity (Gitler and Tsuiji, 2016) (Figure 2A). These two mechanisms seem unrelated to the normal function of the C9ORF72 gene and have motivated therapeutic strategies to target these mutant RNAs and/or proteins (Donnelly et al., 2013; Jiang et al., 2016; Lagier-Tourenne et al., 2013).
Figure 2: C9ORF72.
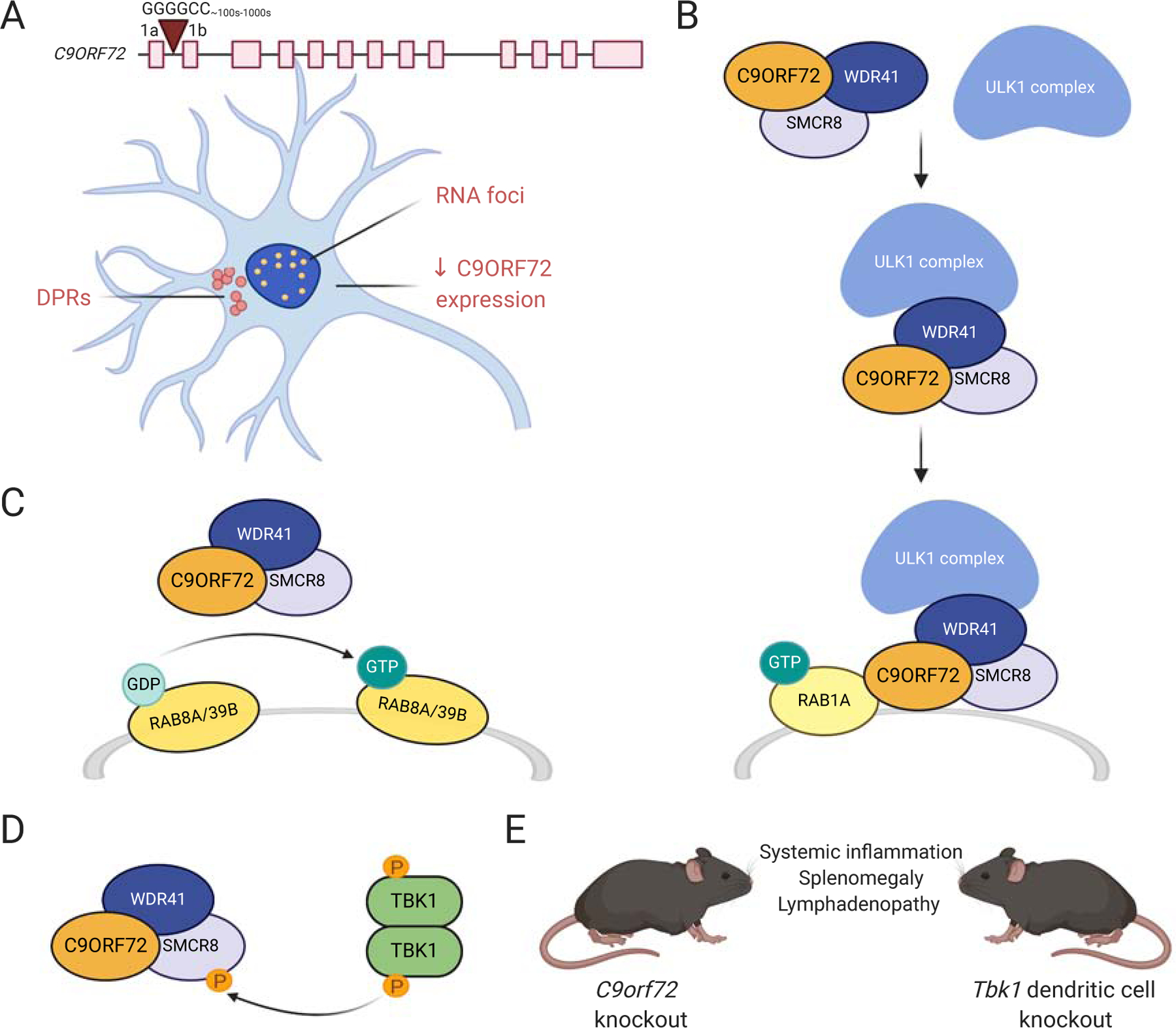
A. G4C2 repeat expansions in a non-coding region of C9ORF72 are hypothesized to cause ALS by at least one of three main mechanisms: 1) RNA foci-mediated toxicity, 2) dipeptide repeat protein (DPR)-mediated toxicity, and/or 3) reduced levels of C9ORF72 protein. B. C9ORF72 forms a complex with SMCR8 and WDR41 and plays a role in autophagy initiation via Rab1a-dependent recruitment of the ULK1 complex to phagophore assembly sites. C. The C9ORF72/SMCR8/WDR41 complex acts as a GDP/GTP exchange factor for Rab8a and Rab39b GTPases, which are involved in autophagy. D. TBK1 interacts with the C9ORF72/SMCR8/WDR41 complex and regulates autophagy by phosphorylating SMCR8. E. C9orf72 knockout mice and Tbk1 dendritic cell knockout mice have similar phenotypes affecting the immune system.
Loss-of-function (LOF) mutations, also called inactivating mutations, result in a gene product that is partially or wholly inactivated. LOF diseases are often caused by recessive mutations, meaning both alleles must carry the mutation in order for the mutant phenotype to occur. Consequently, effects from a mutant gene copy are normally masked in the presence of a normal copy in individuals heterozygous for the mutation. Dominantly inherited LOF diseases can result from haploinsufficiency, which means that having half as much of a functional gene product is not enough to prevent disease. Pathogenic mutations considered to be primarily LOF have been identified in ALS, such as mutations in OPTN and TBK1. As alluded to above, however, even genetic mutations that are considered to work predominantly through GOF mechanisms—such as SOD1 and C9ORF72—have also demonstrated LOF properties. Moreover, genetic variants can contribute to disease even if LOF is not itself sufficient to cause ALS, as exemplified by the immune phenotypes present in patients with mutations in C9ORF72 (Fredi et al., 2019; Ismail et al., 2013; Miller et al., 2016; Turner et al., 2013), or in mice harboring mutations in Tbk1 (Xu et al., 2018) or Optn (Ito et al., 2016).
It is the ultimate tautology that all genes have functions. But given that several therapies under development aim to reduce toxicity by knocking down expression of mutant ALS genes (sometimes by indiscriminately targeting both WT and mutant alleles), it is important to also consider the normal functions of these genes and the potential consequences of losing or reducing their levels. In this review, we critically discuss evidence for LOF mechanisms in ALS via known genetic mutations or loss of normal protein function. We highlight that considering both GOF and LOF mechanisms will be crucial in understanding the molecular underpinnings of ALS, as the field works to develop novel therapeutic strategies for this currently untreatable disorder.
1. SOD1
The first ALS gene was identified in 1993, when mutations causing fALS were linked to the gene encoding the Cu/Zn superoxide dismutase, SOD1 (Deng et al., 1993; Rosen et al., 1993). To date, 187 SOD1 variants have been described in patients with ALS (http://alsod.iop.kcl.ac.uk/), mutations which present in approximately 18.9% and 1.2% of fALS and sALS cases (SOD1-ALS), respectively (Zou et al., 2017), and typically display an autosomal dominant inheritance pattern (Andersen et al., 1998, 1996, 1995). SOD1 is ubiquitously expressed (Fridovich, 1995) and localizes to several cellular compartments including the cytoplasm, nucleus, mitochondrial intermembrane space, lysosomes, and peroxisomes (Chang et al., 1988; Crapo et al., 1992; Keller et al., 1991; Sturtz et al., 2001). The enzyme normally protects cells from toxic reactive oxygen species by catalyzing the dismutation of superoxide anion to oxygen and hydrogen peroxide (Fridovich, 1986).
While some ALS-causing SOD1 variants result in decreased SOD1 enzymatic activity in transformed human lymphocytes, others retain activity comparable to that of WT SOD1 (Borchelt et al., 1994; Hayward et al., 2002). A systematic analysis of many different ALS-causing SOD1 mutants found no correlation between SOD1 enzymatic activity and ALS onset or disease progression in humans (Cleveland et al., 1995), arguing against a causal role of SOD1 LOF in SOD1-ALS. There is now also ample evidence supporting GOF in SOD1-ALS. For example, mice expressing the human ALS-associated variants SOD1G93A, SOD1G37R, and SOD1G85R exhibit overt ALS-like phenotypes including mitochondrial dysfunction, SOD1 aggregation, motor neuron death, and paralysis (Bruijn et al., 1997; Gurney et al., 1994; Wong et al., 1995). Furthermore, the presence or absence of endogenous mouse Sod1 has no effect on the decreased survival times of mice expressing the SOD1G85R transgene (Bruijn et al., 1998), demonstrating that this phenotype is due to a toxic GOF, as opposed to a LOF, mechanism.
With the overwhelmingly compelling role of GOF mechanisms in SOD1-ALS, ASOs targeting SOD1 are being developed and tested as potential therapeutics for humans (Figure 1A) (McCampbell et al., 2018; Miller et al., 2013). But because this therapy will reduce SOD1 levels (the ASOs do not selectively target the mutant gene), we should consider the resulting LOF effects as they relate to ALS and other disorders. For instance, even though Sod1 knockout mice develop normally and do not exhibit overt ALS-like phenotypes such as motor neuron death and paralysis (Reaume et al., 1996), they do show noteworthy deficits of the motor system including decreased mitochondrial density in motor neurons (Fischer et al., 2011; Hayes et al., 2019), denervation and dysfunction at neuromuscular junctions (NMJs) (Fischer et al., 2012, 2011; Flood et al., 1999; Ivannikov and Van Remmen, 2015; Larkin et al., 2011; Shi et al., 2014), decreased peripheral nerve conduction velocity and reduced myelin thickness (Flood et al., 1999; Hamilton et al., 2013), increased motor neuron death after axotomy (Reaume et al., 1996), and secondary muscle pathology (Flood et al., 1999; Sakellariou et al., 2014). These animals also exhibit accelerated loss of skeletal muscle mass with age accompanied by decreased stride length, rotarod performance, and voluntary wheel running, with no change in spontaneous activity (Flood et al., 1999; Muller et al., 2006). Some of these animals also develop abnormal gait and tremors (Muller et al., 2006) that are indicative of neuromuscular impairments. Sod1 knockout mice also exhibit wide-ranging neuronal and non-neuronal abnormalities outside of the motor system (Saccon et al., 2013). Sod1 heterozygous mice—which leave one WT allele intact—do not exhibit death of motor neurons. But they display a wide range of phenotypes that may directly relate to ALS, such as increased susceptibility to glutamate toxicity (Schwartz et al., 1998) and axonal damage (Reaume et al., 1996), denervation (Fischer et al., 2012, 2011), and loss of select neuronal subtypes with aging (Keithley et al., 2005). Together these observations suggest that SOD1 LOF may not be causal but could modify aspects of ALS pathogenesis or instigate other unwanted effects (Saccon et al., 2013).
Additionally, two recent human genetics studies suggest that SOD1 homozygous LOF can cause debilitating phenotypes, even though it alone does not cause ALS (Andersen et al., 2019; Park et al., 2019). Human patients carrying homozygous truncating mutations in SOD1 (c.335dupG) did not produce any detectable SOD1 activity in erythrocytes (Andersen et al., 2019) or leukocytes (Park et al., 2019). Heterozygous carriers had approximately half the SOD1 activity of WT subjects. Both patients with homozygous SOD1 c.335dupG variants had similar symptoms including progressive loss of motor abilities, tetraspacticity, and hyperreflexia with symptom onset beginning in early (Miller et al., 2020; Mueller et al., 2020)childhood, while heterozygous carriers were unaffected (Andersen et al., 2019; Park et al., 2019). Additionally, a sibling of the patient described by Andersen et al. had similar symptoms to the patient and died at the age of six (Andersen et al., 2019).
Importantly, SOD1 LOF in humans is distinct from ALS; for ALS the key observation is that heterozygous carriers of the LOF mutation are unaffected, indicating that chronic partial LOF does not cause ALS or other diseases. On the other hand, complete SOD1 LOF does result in significant, debilitating abnormalities of the motor system, such that we must consider potential LOF effects when silencing SOD1 as a potential treatment for ALS, and perhaps target the mutant allele specifically. Current clinical efforts to target SOD1 result in incomplete suppression of SOD1 and preliminary clinical evidence indicates that this level of gene knockdown is safe for SOD1-ALS patients (Miller et al., 2020; Mueller et al., 2020, ClinicalTrials.gov identifier: NCT02623699).
2. C9ORF72
The most common known genetic cause underlying both ALS and FTD is a GGGGCC (G4C2) hexanucleotide repeat expansion in the chromosome 9 open reading frame 72 (C9ORF72) gene. The mutation accounts for approximately 34% of familial ALS and nearly a quarter of familial FTD (C9ALS/FTD) in populations of European ancestry, where it is most prevalent (van Blitterswijk et al., 2013; Zou et al., 2017). While healthy individuals can have from ~2 to 25 G4C2 repeats, C9ALS/FTD patients harbor hundreds to even thousands of these repeats (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Rohrer et al., 2015). Depending on the transcript variant, the expansion is located in intron 1 or the promoter region of C9ORF72, bidirectionally transcribed (Kumar et al., 2017; Mizielinska et al., 2013; Niblock et al., 2016), and then translated via a non-canonical form of translation (repeat-associated non-AUG (RAN) translation) (Ash et al., 2013; Mori et al., 2013c; Zu et al., 2013) (Figure 2A). The C9ORF72 gene is expressed not only in the central nervous system, but also in the periphery, such as in lymphocytes, bone marrow, and spleen (McCauley and Baloh, 2019; Suzuki et al., 2013), and its function is still not fully understood.
On one hand, pathogenic mechanisms underlying the C9ORF72 repeat expansion have been attributed to GOF mechanisms such as (1) repeat-RNA-mediated toxicity in the form of RNA foci and (2) production of toxic homopolymeric dipeptide repeat proteins (DPRs) through RAN translation (Freibaum and Taylor, 2017; Kwon et al., 2014; Mizielinska et al., 2013; Mori et al., 2013c; Swinnen et al., 2018; Taylor et al., 2016; Wen et al., 2014) (Figure 2A). Briefly, sense and antisense RNA foci have been shown to sequester RNA-binding proteins (RBPs) in induced pluripotent stem cell (iPSC)-differentiated neurons (Conlon et al., 2016; Donnelly et al., 2013; Mori et al., 2013b; Sareen et al., 2013; Xu et al., 2013), which may lead to LOF in one or more of those RBPs. However, there has not been conclusive evidence directly proving that phenotypes caused by C9orf72 mutations are due to altered function in these RBPs. Additionally, DPRs can cause toxicity in neurons (Kwon et al., 2014; Lee et al., 2016; May et al., 2014), with arginine-rich GR and PR repeats being considered particularly toxic. Expression of these causes significant cell death in vitro (Tao et al., 2015; Wen et al., 2014), severe degeneration in Drosophila eyes (Freibaum et al., 2015; Lee et al., 2016; Mizielinska et al., 2014), and neurodegeneration and behavioral abnormalities in mouse models (Choi et al., 2019; Hao et al., 2019; Zhang et al., 2018; Y. J. Zhang et al., 2019). The suggestion of a GOF mechanism contributing to disease has led to the current development of ASOs against C9ORF72 for potential human therapy (Figure 1B) (Donnelly et al., 2013; Jiang et al., 2016; Lagier-Tourenne et al., 2013). Given that this would still result in reduced levels of C9ORF72 (even in the case of a mutant allele-specific targeting approach), we must also consider the potentially deleterious effects of C9ORF72 LOF. This brings us to another prominent—but not mutually exclusive—hypothesis for repeat expansion toxicity: (3) haploinsufficiency owing to the partial loss of normal C9ORF72 protein expression in repeat expansion carriers (Renton et al., 2011; DeJesus-Hernandez et al., 2011) (Figure 2A).
The possibility of a LOF mechanism is exemplified by immune phenotypes in heterozygous or homozygous C9orf72 knockout mice. For instance, mice from multiple studies in which C9orf72 was ablated from all tissues have shown signs of immune system dysregulation in the form of enlarged lymph nodes, splenomegaly, autoimmunity, increased cytokine release, and massive infiltration of histiocytes/macrophages and lymphocytes in multiple organs post-mortem (Figure 2E) (Atanasio et al., 2016; Burberry et al., 2020, 2016; Jiang et al., 2016; O’Rourke et al., 2016; Shao et al., 2019; Sudria-Lopez et al., 2016; Sullivan et al., 2016). While some studies have shown no effects of C9orf72 loss on survival (O’Rourke et al., 2016), several have demonstrated higher premature mortality upon C9orf72 loss (Burberry et al., 2020, 2016; Jiang et al., 2016), with further exacerbation of premature death when DPRs are expressed (Zhu et al., 2020). These phenotypes are perhaps not surprising given the high levels of expression of C9orf72 in myeloid cells (McCauley and Baloh, 2019). In fact, most recently, mice with myeloid cell-specific knockout of C9orf72 were shown to exhibit immune defects similar to global C9orf72-deficient mice via a STING (stimulator of interferon genes)-dependent mechanism, implying that the immune defects are in large part due to a cell-type specific loss of C9orf72’s normal ability to suppress STING-induced inflammation (McCauley et al., 2020). Interestingly, mice with a dendritic cell knockout of Tbk1—a gene in which LOF mutations can cause ALS (Freischmidt et al., 2015)—have similar phenotypes to C9orf72 knockout mice (Xiao et al., 2017) (Figure 2E), suggesting a potential connection between C9ORF72 and TBK1 in immune system regulation (McCauley and Baloh, 2019).
In addition, autoimmunity and premature mortality phenotypes of C9orf72 deficient mice can be mitigated upon transplantation of WT mouse bone marrow into mutant mice, and vice versa: transplanting mutant bone marrow into WT was sufficient to cause autoimmunity (Burberry et al., 2016). A similar effect was seen with fecal transplants, such that transplanting gut microflora from C9orf72 mutant mice grown in a protective environment (i.e. one that causes less inflammation), or reducing gut microbial burden with broad spectrum antibiotics, attenuated inflammatory phenotypes in C9orf72 knockout mice grown in a proinflammatory environment (Burberry et al., 2020). Despite accruing evidence for LOF, it is worth noting that these C9orf72-deficient mouse lines have failed to recapitulate ALS pathology or significant changes in motor neuron degeneration/function (e.g., using grip strength and accelerating rotarod tests) (Jiang et al., 2016; Sudria-Lopez et al., 2016), unless crossed with gain-of-toxic-function mouse models (Shao et al., 2019; Zhu et al., 2020). When C9orf72 ablation is neuron- and glia-specific, the LOF is insufficient to cause behavioral or pathologic phenotypes, although these mice show decreased body weight compared to WT mice (Koppers et al., 2015).
Immune deficiency associated with reduced levels of C9ORF72 has also been observed in humans, including precedence and/or concurrence of C9ALS/FTD with other autoimmune or immune-mediated conditions such as systemic lupus erythematosus (commonly referred to as lupus), myasthenia gravis, and multiple sclerosis (MS) (Fredi et al., 2019; Ismail et al., 2013; Miller et al., 2016; Turner et al., 2013). For instance, a study of 650 ALS patients from North England reported that the C9ORF72 expansion was present in 4 of 5 individuals with MS—an immune-mediated demyelinating disorder that results in progressive motor and other disabilities—that preceded ALS (MS-ALS), but not in those with MS alone (Ismail et al., 2013). Moreover, if MS preceded ALS, the C9-ALS patients were more rapidly progressing (Ismail et al., 2013). However, given the small number of MS-ALS patients described and other population-based studies that report MS-ALS cases presenting at a rate expected by chance, the possibility of misdiagnoses cannot be ruled out (van Doormaal et al., 2013).
On a subcellular level, decreased autophagy and enlarged and/or defective lysosomes have been described in C9orf72-null cell lines and mice (Ji et al., 2017; O’Rourke et al., 2016; Zhu et al., 2020). In human and mouse knockdown/knockout cell models and primary neurons from mice, C9ORF72 has also been shown to play a mechanistic role in early stages of autophagy signaling pathways by participating in heteromeric complexes (e.g. with SMCR8, WDR41, ATG13, ULK1) involved in autophagy initiation and/or regulation (Amick et al., 2016; Ho et al., 2019; Ji et al., 2017; Lan et al., 2019; Sellier et al., 2016; Webster et al., 2016; Yang et al., 2016) (Figure 2B, 2C). Moreover, TBK1 has been shown to interact with SMCR8, suggesting that they may belong to a common autophagy regulatory pathway (Sellier et al., 2016) (Figure 2D). In both human cells and C. elegans, C9ORF72 also regulates TFEB, a transcription factor and master regulator of autophagic and lysosomal genes (Ji et al., 2020). Defects in autophagosome formation have also been demonstrated in iPSC-derived motor neuron models from C9ALS/FTD patients, along with abnormal accumulation of glutamate receptors and altered axonal trafficking (Abo-Rady et al., 2020; Selvaraj et al., 2018; Shi et al., 2019, 2018). Given that formation of cytoplasmic protein aggregates due to defective protein quality control is a key pathologic hallmark in ALS and numerous other neurodegenerative disorders, these impairments in protein degradation and clearance seem particularly relevant in driving disease (Ramesh and Pandey, 2017).
Despite supportive evidence for LOF mechanisms contributing to C9ALS, human ALS patients with LOF mutations in C9ORF72 other than the G4C2 hexanucleotide repeats have not yet been identified. Moreover, homozygous carriers of C9ORF72 mutations seem to exhibit clinical phenotypes that are no worse than heterozygous individuals (Fratta et al., 2013). Together, the above observations suggest that multiple mechanisms are at play: one could imagine that the reduced levels of normal C9ORF72 and subsequent immune deficiency/subcellular defects increase a neuron’s susceptibility to GOF effects of DPRs or RNA foci. We suspect that a multi-targeted therapeutic approach that not only targets and mitigates G4C2 repeat-containing RNAs but also increases C9ORF72 levels will likely be necessary to treat C9ALS/FTD patients (Figure 5A). Alternatively (or concomitantly), a STING inhibitor to suppress the increased interferon response seen in whole blood and brain tissue from C9ALS/FTD patients could perhaps be used to mitigate immune effects caused by lowered C9ORF72 levels (McCauley et al., 2020). We must also consider environmental contributions, as C9orf72 knockout mice with identical genetic backgrounds have exhibited strikingly different survival depending on their environment (Burberry et al., 2020), potentially raising the importance of boosting reduced C9ORF72 levels. Given that much of our current understanding of C9ALS/FTD pathogenesis relies on cultured cells and animal models, it will be critical for future studies to further investigate disease-relevant models and tissues derived from patients to more fully understand the underlying pathogenic mechanisms. This will allow the field to not only gain a more relevant perspective on the mechanistic contributions of C9ORF72 deficiency to disease, but also to better address the gap in knowledge pertaining to selective regional and cell-type-specific vulnerabilities in C9ALS/FTD.
Figure 5: Next-Generation Therapies: Targeting LOF and GOF Effects.

A. G4C2 repeat expansions in C9ORF72 cause production of sense and antisense RNA foci and DPRs, as well as reduced levels of C9ORF72, all of which may contribute to disease. A combination therapy that targets G4C2 repeat-containing RNAs and increases levels of C9ORF72 could mitigate both the LOF and toxic GOF effects resulting from this mutation. B. TDP-43 is depleted from the nucleus and forms cytoplasmic inclusions in most ALS cases. Nuclear depletion of TDP-43 alters mRNA metabolism and results in decreased levels of STMN2, which may contribute to ALS pathogenesis. A combination therapy that targets toxic cytoplasmic TDP-43 and restores STMN2 levels through gene therapy or an ASO that blocks cryptic exon inclusion could be more beneficial to patients than either treatment alone.
3. TDP-43
Transactive response (TAR) DNA-binding protein 43 (TDP-43), encoded by the TARDBP gene, is a DNA/RNA-binding protein normally localized in the nucleus (Polymenidou et al., 2011; Tollervey et al., 2011) (Figure 3A). TDP-43 regulates various steps of RNA metabolism, including mRNA splicing, RNA transport, translation, and microRNA biogenesis (Alami et al., 2014; Buratti et al., 2001; Deshaies et al., 2018; Kawahara and Mieda-Sato, 2012; McDonald et al., 2011; Neelagandan et al., 2019). It is also the most widespread and characterized pathologic protein in the ALS/FTD spectrum; in disease states, TDP-43 is depleted from the nucleus and found as hyperphosphorylated, aggregated cytoplasmic inclusions in ~97% of ALS and ~50% of FTD patients (Dormann and Haass, 2011; Giordana et al., 2010) (Figure 3B). Although causal mutations in TARDBP are rare and account for only ~4% of fALS and <1% of sALS (Zou et al., 2017), the nearly ubiquitous presence of pathologic TDP-43 inclusions and nuclear TDP-43 depletion in ALS indicates that ALS-causing mechanisms eventually converge on this central pathological axis.
Figure 3: TDP-43.

A. Under physiological conditions, TDP-43 predominantly localizes to the nucleus and plays a role in RNA metabolism. B. In most cases of ALS, TDP-43 is depleted from the nucleus and forms cytoplasmic aggregates. TDP-43 pathology may contribute to ALS through 1) a loss of normal function in the nucleus and/or 2) a toxic GOF via formation of cytoplasmic aggregates. C. Nuclear TDP-43 binds to STMN2 pre-mRNA transcripts and suppresses inclusion of the cryptic exon 2a. This mRNA isoform encodes functional STMN2, a microtubule-associated protein involved in neurite outgrowth and repair. D. Reduced levels of nuclear TDP-43 cause inclusion of cryptic exon 2a and premature polyadenylation of STMN2 mRNA, resulting in decreased levels of STMN2 and impaired axonal regeneration in vitro.
On one hand, the presence of pathologic inclusions and their correlation with affected brain regions suggests a toxic GOF mechanism (Arai et al., 2006; Neumann et al., 2006). On the other hand, the clearance of TDP-43 from its usual nuclear localization suggests a loss of TDP-43’s canonical nuclear function. Given the ubiquity of both phenotypes, naturally many have attempted to directly alter TDP-43 mRNA or protein levels to investigate the resulting effects—yet, evidence suggests that directly targeting its protein or mRNA levels for therapy is probably not favorable.
First, TDP-43 is not only essential for early embryonic development in mice, but is also required for normal neuronal function—such as the ability to control muscle movement—in mice, Drosophila, and zebrafish (Feiguin et al., 2009; Kraemer et al., 2010; Yang et al., 2014). Conditional TDP-43 knockout mice crossed to various tissue-specific Cre lines similarly display diverse cellular and behavioral phenotypes ranging from electrophysiological abnormalities to motor movement deficits (Wu et al., 2019), and knocking down TDP-43 in cultured mammalian cells leads to reduced dendritic complexity (Herzog et al., 2017). Moreover, TDP-43 autoregulates its own synthesis by binding to an alternatively spliced intron in the 3’ UTR of its pre-mRNA, rendering modification of its levels difficult (Ayala et al., 2010; Fukushima et al., 2019; Polymenidou et al., 2011). Such perplexities have led many to investigate its interactors, regulators, and binding targets.
As a DNA/RNA-binding protein, TDP-43 has been shown to interact with RNA transcripts of more than 6,000 genes in mice, where it binds preferentially to UG-rich sequences that are located in introns, non-coding RNAs, and intergenic regions (Polymenidou et al., 2011; Tollervey et al., 2011). Thus, it is not surprising that knocking down TDP-43 levels in adult mouse brains using ASOs leads to down- and up-regulation of some ~240 and ~360 genes, respectively—many of which encode proteins involved in synaptic activity and neuronal development—and alteration of nearly 1,000 splicing events (Polymenidou et al., 2011). One method by which TDP-43 affects RNA splicing events is by suppressing the inclusion of cryptic exons (Ling et al., 2015), which are non-conserved exons included in some but not all transcripts (Zavolan et al., 2003). Cryptic exons often cause frameshift mutations or introduce premature stop codons, alternative transcriptional start sites, or alternative polyadenylation sites, which can lead to nonsense-mediated decay (NMD) or disrupted translation and, consequently, the downregulation of critical proteins (Tan et al., 2016). In Drosophila and mouse motor neuron models of TDP-43 LOF, expressing an engineered chimeric protein that mimics TDP-43’s role as a splicing regulator not only decreases aberrant splicing, but also rescues other ALS-related phenotypes such as motor deficits, axon degeneration, and motor neuron loss caused by TDP-43 reduction (Donde et al., 2019). Therefore, exploring proteins downregulated by TDP-43 due to splicing defects in neurons may reveal critical pathways downstream of TDP-43.
One particular splicing target of TDP-43, Stathmin-2 (STMN2), seems likely to play a key role in ALS. Studies in human SH-SY5Y neuroblastoma cells revealed that knocking down TDP-43 levels leads to changes in expression of hundreds of genes, among which STMN2, a regulator of microtubule stability, is one of the most downregulated genes (Klim et al., 2019; Melamed et al., 2019). ALS patient fibroblast-derived neurons expressing an ALS-causing TDP-43 mutation display reduced STMN2 expression (Klim et al., 2019; Melamed et al., 2019). The STMN2 gene harbors a cryptic exon (exon 2a) that is normally not included in the mature STMN2 mRNA (Figure 3C) (Klim et al., 2019; Melamed et al., 2019). TDP-43 functions to repress inclusion of this cryptic exon but when TDP-43 is lost or its function is impaired, this exon gets incorporated into the mature mRNA (Figure 3D). Unfortunately, this exon harbors a stop codon and a poly-adenylation signal, which results in truncated STMN2 mRNA and protein (Melamed et al., 2019). Aberrant splicing and reduced STMN2 protein levels seem to be a major feature of ALS (except those with SOD1 mutations) (Klim et al., 2019; Melamed et al., 2019). Importantly, upregulation of STMN2 is able to rescue axonal regeneration defects caused by TDP-43 depletion in human iPSC-derived neurons, supporting the idea that TDP-43-dependent neurodegeneration acts, at least in part, via a LOF mechanism (Klim et al., 2019; Melamed et al., 2019). While STMN2 is a potentially attractive therapeutic target (e.g., by preventing its mis-splicing or by upregulating STMN2 levels) (Figure 5B), further studies investigating alterations in STMN2 levels in larger patient populations—whether that be through transcriptomic data or post-mortem brain specimens—will be crucial. Notably, the cryptic exon in STMN2 is not conserved to the mouse gene (Ling et al., 2015), requiring studies in human cell lines and/or the generation of humanized mouse models.
In addition to acting as a regulator of alternative splicing, TDP-43 can also regulate transposable elements, which are mobile DNA sequences that can hop around to different positions within the genome (Krug et al., 2017; Li et al., 2012). These include retrotransposons, transposable elements that copy themselves via an RNA intermediate. Several compelling studies have provided evidence that TDP-43 binds to and represses retrotransposon transcripts (Krug et al., 2017; Li et al., 2012); suppression of the Drosophila TDP-43 homolog leads to activation of certain transposable elements, whereas genetic rescue with TDP-43 expression suppresses this activation, indicating that the endogenous function of TDP-43 is required for transposable element repression (Romano et al., 2020). Moreover, nuclear loss of TDP-43 has been associated with increased chromatin accessibility to transposable elements in neurons derived from FTD-ALS patients (Liu et al., 2019). Induction of retrotransposon expression in normal human cells promotes a senescence-like phenotype (Belancio et al., 2010), and re-insertion of active retrotransposons can alter the genome of individual cells and affect normal cellular functions/lead to cell death (Savage et al., 2019). Thus, the expression of retrotransposons is tightly repressed in most somatic tissues to prevent DNA damage and ensure genomic integrity (Goodier, 2016). Taken together, these results suggest that dysregulation of transposable elements due to TDP-43 LOF may contribute to ALS pathogenesis.
In sum, despite evidence for TDP-43 GOF mechanisms in ALS, accruing evidence suggests an important, perhaps primary role of TDP-43 LOF in ALS. Further investigation of genes dysregulated in the absence of TDP-43 are required to shed light on the role of TDP-43 in regulating biological processes, which could lead to therapies that compensate for the loss of nuclear TDP-43 (Figure 5B). Given that TDP-43 is not itself a favorable target, it will be important that future studies aim to more deeply characterize pivotal RNAs and proteins that are regulated by TDP-43 as well as the potential role of TDP-43 as a transposable element regulator. These and other underexplored nuclear functions of TDP-43 may open up critical new avenues for elucidating disease pathogenesis and generating new therapeutic opportunities.
4. FUS/TLS
Mutations in FUS/TLS (fused in sarcoma/translocated in liposarcoma) can cause ALS and rare forms of FTD (Broustal et al., 2010; Kwiatkowski et al., 2009; Van Langenhove et al., 2010; Vance et al., 2009). To date, more than 50 different FUS mutations have been described in patients with ALS (Deng et al., 2014), many of which disrupt the nuclear localization signal and result in mislocalization of FUS to the cytoplasm (Deng et al., 2014; Dormann et al., 2010; Ito et al., 2011; Lagier-Tourenne et al., 2010). FUS mutations are present in ~4% of fALS patients and <1% of sALS patients (Deng et al., 2014; Zou et al., 2017) and typically display an autosomal dominant inheritance pattern. In post-mortem brains of FUS-ALS patients, FUS-positive inclusions are present in the cytoplasm and, less frequently, in the nucleus of neurons and glia (Hewitt et al., 2010; Kwiatkowski et al., 2009; Neumann et al., 2009; Vance et al., 2009).
The ubiquitously expressed DNA/RNA-binding protein FUS localizes predominantly to the nucleus under physiological conditions (Zinszner et al., 1997), and is normally involved in DNA repair (Wang et al., 2008) and several aspects of RNA metabolism including transcription (Masuda et al., 2015; Tan and Manley, 2010), alternative splicing (Hoell et al., 2011; Ishigaki et al., 2012; Lagier-Tourenne et al., 2012; Rogelj et al., 2012), mRNA transport (Fujii et al., 2005), mRNA stability (Kapeli et al., 2016; Udagawa et al., 2015; Yokoi et al., 2017), and microRNA biogenesis (Gregory et al., 2004; Morlando et al., 2012). The protein also localizes to the neuromuscular junction (So et al., 2018) as well as presynaptic terminals (Schoen et al., 2015) and postsynaptic dendrites in neurons (Aoki et al., 2012; Belly et al., 2005; Fujii et al., 2005; Yasuda et al., 2013), where it plays a role in local translation (López-Erauskin et al., 2018; Yasuda et al., 2013) and synaptic structure/function (Fujii et al., 2005; Fujii and Takumi, 2005).
FUS pathogenesis in ALS, similar to TDP-43 pathogenesis, is thought to arise from 1) a toxic GOF due to FUS aggregation and/or 2) a LOF resulting from cytoplasmic mislocalization of FUS and subsequent loss of nuclear function. The GOF hypothesis has been supported by studies demonstrating that overexpression of WT human FUS causes progressive motor neuron degeneration in mice (Ling et al., 2019; López-Erauskin et al., 2018; Mitchell et al., 2013), and that mice expressing exogenous FUS with ALS-associated mutations (Sharma et al., 2016) or without a nuclear localization signal (ΔNLS-FUS) exhibit ALS-like phenotypes such as motor neuron degeneration (Shiihashi et al., 2016). However, LOF of the FUS homolog caz in Drosophila causes reduced lifespan (Wang et al., 2011), neuromuscular junction disruption (Sasayama et al., 2012; Wang et al., 2011; Xia et al., 2012), and impaired locomotive ability (Sasayama et al., 2012; Xia et al., 2012). In zebrafish, fus knockdown similarly results in impaired motor activity (Armstrong and Drapeau, 2013; Kabashi et al., 2011), shortened axonal projections from motor neurons (Kabashi et al., 2011), and neuromuscular junction dysfunction (Armstrong and Drapeau, 2013).
Yet it seems that LOF alone is insufficient to cause FUS-ALS. In mice, loss of one Fus allele (Fus+/−) does not cause an overt phenotype (Hicks et al., 2000; Kuroda et al., 2000), whereas global Fus knockout causes perinatal death in inbred C57BL/6 mice (Hicks et al., 2000). In outbred mice, Fus knockout causes male sterility and reduced female fertility (Kuroda et al., 2000) as well as hyperactivity, reduced anxiety-like behavior, and hippocampal vacuolation, but no overt ALS-like phenotype (Kino et al., 2015). Additionally, conditional knockout of Fus in mice postnatally does not lead to motor neuron degeneration (Sharma et al., 2016). Together, these results suggest that FUS LOF alone does not cause FUS-ALS but may cause other deleterious phenotypes.
In brains of mice and humans, FUS interacts with the RNA transcripts of over 5,500 genes (Lagier-Tourenne et al., 2012), where it binds primarily through a GUGGU-binding sequence or AU-rich stem loops in a saw-tooth pattern (Hoell et al., 2011; Ishigaki et al., 2012; Lagier-Tourenne et al., 2012; Rogelj et al., 2012). Knockdown of Fus with ASOs in adult mouse brains leads to altered expression and splicing of 610 and 374 mRNAs, respectively (Lagier-Tourenne et al., 2012), including Fus itself, resulting in autoregulation through NMD (Zhou et al., 2013). The subsets of RNAs with altered expression after FUS or TDP-43 knockdown are largely non-overlapping; however, both FUS and TDP-43 knockdown result in decreased expression of mRNAs that contain long introns, many of which encode proteins important for the function and activity of synapses (Lagier-Tourenne et al., 2012).
As suggested by the mRNA targets found in the studies described above, Fus knockdown results in synaptic dysfunction in vitro and in vivo. For example, Fus knockdown causes enlarged growth cones (Orozco et al., 2012), decreased neurite length (Ishigaki et al., 2017; Orozco et al., 2012), abnormal dendritic spines (Fujii et al., 2005; Yokoi et al., 2017), and decreased synaptic transmission (Udagawa et al., 2015) in rodent primary neurons. These phenotypes are mediated in part by downregulation of the AMPA receptor subunit GluA1 (Udagawa et al., 2015) and the dendritic spine maturation protein SynGAP (Yokoi et al., 2017) as well as upregulation of tauopathy-associated 4-repeat tau (4R-T) due to inclusion of Mapt exon 10 (Ishigaki et al., 2017; Orozco et al., 2012).
In vivo, knockdown of Fus in mouse hippocampus results in accumulation of phosphorylated tau, hippocampal neuron loss, and decreased adult neurogenesis (Ishigaki et al., 2017). Moreover, a novel FUS variant (p.Q140H) was identified in two sisters who exhibited a familial form of FTD characterized by astrocyte-predominant tauopathy and altered tau splicing (Ferrer et al., 2015), suggesting a potential link between FUS mutations and tau pathology in human FTD (Ishigaki et al., 2017). Knockdown of Fus in mouse hippocampus also causes FTD-like behavioral phenotypes such as social interaction defects, hyperactivity, and disinhibition (Ishigaki et al., 2017; Udagawa et al., 2015), which can be partially rescued by 4R-T suppression (Ishigaki et al., 2017) or increased GluA1 expression (Udagawa et al., 2015). Similarly, forebrain-specific knockout of Fus causes FTD-like behaviors in mice, which are partially rescued by increased SynGAP α2 expression (Yokoi et al., 2017).
In humans, the FUS nonsense mutation (p.Q290X) is associated with essential tremor, a neurodegenerative disorder characterized by postural or motion tremor (Merner et al., 2012). In lymphoblastoid cells derived from essential tremor-affected individuals, FUS mRNA is largely degraded by NMD, suggesting that complete FUS LOF may cause the clinical phenotype in humans (Merner et al., 2012). Importantly, a 26-year-old ALS patient was recently given a personalized ASO treatment targeting her FUS P525L mutation, which causes an aggressive form of ALS with juvenile onset (Figure 1C) (Arnold, 2019). The patient began treatment after the disease had already begun to cause respiratory problems and passed away less than a year after being given her first injection (Carrie Arnold, 2019). Thus, it is crucial to examine the effects of decreased FUS expression, as potential therapeutics targeting FUS are being further explored and used in the clinic.
5. OPTN and TBK1
Three ALS-causing mutations in the optineurin gene (OPTN), also known as FIP-2, were first discovered by homozygosity mapping in six Japanese individuals from consanguineous marriages (Maruyama et al., 2010). These mutations were 1) a homozygous exon 5 deletion causing a frameshift and premature stop codon, 2) a homozygous nonsense mutation (p.Q398X) that generates a truncated protein, and 3) a heterozygous missense mutation (p.E478G) that causes reduced expression likely due to NMD, all of which suggest LOF (Maruyama et al., 2010). Other heterozygous nonsense and missense mutations in OPTN have subsequently been identified in sALS and fALS patients (Tümer et al., 2012; van Blitterswijk et al., 2012), suggesting that OPTN haploinsufficiency contributes to ALS (Del Bo et al., 2011; Li et al., 2015; van Blitterswijk et al., 2012). Mutations in OPTN seem to be rare in sALS, corresponding to 0.4% of cases in Japanese/Chinese and 0.3% of European populations, respectively, while their incidence in fALS is about 4% and 1.5% in the same populations (Fifita et al., 2017).
Optineurin (OPTN) is ubiquitously expressed in most tissues and harbors a ubiquitin-binding domain that is required for recognition of ubiquitinated proteins and for its role as an autophagy adaptor (Markovinovic et al., 2017; Slowicka et al., 2016; Toth and Atkin, 2018). Consistent with a LOF mechanism in ALS, knockout of optn leads to alteration in axonal trafficking dynamics and increased cell death in zebrafish embryos (Paulus and Link, 2014), and knockdown of Optn in N2a and retinal ganglion cells (RGCs) from mice results in enhanced cell death (Akizuki et al., 2013; Sippl et al., 2011). Additionally, studies of the E478G mutant in human cancer cell lines and mouse embryonic fibroblasts have reported impairments in pathogen clearance (xenophagy) (Ivannikov and Van Remmen, 2015; Noad et al., 2017; van Wijk et al., 2017), protein aggregate clearance (aggrephagy) (Korac et al., 2013; Shen et al., 2015) and degradation of damaged mitochondria (mitophagy) (Heo et al., 2015; Lazarou et al., 2015; Moore and Holzbaur, 2016; Richter et al., 2016; Wong and Holzbaur, 2014; Zachari et al., 2019), further supporting a loss of normal OPTN function in autophagy. Its role in autophagy is also regulated by another known ALS gene, TANK-binding kinase 1 (TBK1), which via phosphorylation can enhance clearance of autophagic cargos (Moore and Holzbaur, 2016; Padman et al., 2019; Richter et al., 2016; Wild et al., 2011) by increasing the affinity of OPTN for ubiquitinated substrates and its translocation to damaged mitochondria (Heo et al., 2015; Li et al., 2018; Richter et al., 2016) (Figure 4A). In addition to overlaps in autophagy-related pathways, OPTN and TBK1 share roles in the innate immune system (Figure 4B).
Figure 4: TBK1 and OPTN.

A. TBK1 phosphorylates several autophagy receptors including OPTN. Phosphorylation of OPTN by TBK1 increases its interactions with ATG8 proteins/LC3 and ubiquitinated cargo. B. TBK1 plays a role in inflammatory pathways by acting downstream of proteins that sense bacterial lipopolysaccharides and viral RNA/DNA. OPTN promotes TBK1 activation in response to viral RNA. These pathways result in expression of proinflammatory cytokines and type I interferons. C. Several proteins, including TBK1 and OPTN, have been shown to inhibit RIPK1-dependent inflammation and cell death. D. Tbk1+/− mice with reduced myeloid Tak1 expression and mice with oligodendrocyte or myeloid-specific knockout of Optn exhibit neuroinflammatory and ALS/FTD-like phenotypes.
TBK1, also known as T2K or NAK, is a serine/threonine kinase that plays a crucial role in the innate immune system by phosphorylating IRF3 and inducing the production of type-I interferon (IFN) and proinflammatory cytokines after viral infection (Figure 4B) (Cai et al., 2014; Fitzgerald et al., 2003; Ishii et al., 2006; Liu et al., 2015; Tanaka and Chen, 2012; C. Zhang et al., 2019). It also plays an important role in regulating autophagy by interacting with and phosphorylating autophagy adaptors, such as NDP52, p62, and OPTN (Figure 4A) as well as the C9ORF72/SMCR8/WDR41 autophagy complex (Ahmad et al., 2016; Heo et al., 2015; Korac et al., 2013; Moore and Holzbaur, 2016; Noad et al., 2017; Richter et al., 2016; Thurston et al., 2016; van Wijk et al., 2017; Vargas et al., 2019; Wild et al., 2011; Yang et al., 2016) (Figure 2D). TBK1 was identified as an ALS gene in two whole-exome sequencing (WES) studies from North American and European populations (Cirulli et al., 2015; Freischmidt et al., 2015), revealing eight mutations. Introducing these mutant alleles in human cell lines confirmed loss of TBK1 expression for most mutants, supporting a LOF mechanism in ALS (Brenner et al., 2019; Freischmidt et al., 2015). Over 80 ALS-associated mutations in TBK1 have been discovered to date (de Majo et al., 2018; Freischmidt et al., 2015).
In mice, Tbk1 LOF alone does not seem to cause ALS-like phenotypes, as heterozygous Tbk1+/− animals do not exhibit signs of motor neuron degeneration or impaired autophagy at 200 days of age (Brenner et al., 2019). However, Tbk1+/− mice with reduced expression of Tak1 in myeloid cells exhibit ALS/FTD-like phenotypes including TDP-43 aggregation, neuroinflammation, axonal degeneration, and neuron loss (Xu et al., 2018). Additionally, Tbk1+/− mice overexpressing the ALS-causing SOD1 G93A mutation show accelerated clinical onset, muscular denervation, and impaired autophagy (Brenner et al., 2019). Point mutations that decrease Tbk1’s kinase activity also accelerate disease onset in SOD1G39A mice but surprisingly extend lifespan (Gerbino et al., 2020). Together, these findings indicate that TBK1 LOF mutations can contribute to and modify ALS pathogenesis (Brenner et al., 2019; Gerbino et al., 2020; Xu et al., 2018).
The relationship between the two independent LOF ALS genes TBK1 and OPTN is one that warrants closer scrutiny. For instance, expression of the ALS-causing p.690–713del TBK1 mutant in human HEK293T cells abolishes its interaction with OPTN (Freischmidt et al., 2015). Moreover, this mutant TBK1 is still able to phosphorylate and interact with IRF3, suggesting that impairment of the interaction between TBK1 and OPTN may be sufficient to cause ALS (Freischmidt et al., 2015). This possibility is further strengthened by the finding that the p.E696K missense mutation in TBK1 abolishes TBK1’s interaction with OPTN in vitro and in vivo despite still being able to interact with other protein partners (Freischmidt et al., 2015; Li et al., 2018; Moore and Holzbaur, 2016; Richter et al., 2016), implying that the disrupted interaction with OPTN is specific. Since these initial studies, other missense mutations in TBK1 have been identified in ALS patients that also reduce levels of activated TBK1 and abolish its interactions with both IRF3 and OPTN (de Majo et al., 2018).
The overlapping functions of TBK1 and OPTN in autophagy and the innate immune system also suggests an impairment of common functions in the OPTN/TBK1 axis, such as RIPK1-dependent cell death, in the development of ALS. For instance, conditional knockout of Optn in microglia and oligodendrocytes from mice leads to axonal pathology, elevated production of proinflammatory cytokines, and accumulation of apoptosis/necroptosis markers that include phosphorylated RIPK1, which can be rescued by inhibition of RIPK1 or a kinase-dead mutant (Figure 4D) (Ito et al., 2016). Similarly, homozygous knockout of Tbk1 in mice is embryonic lethal owing to liver failure, which can be rescued by inactivating Ripk1 (Xu et al., 2018). Tbk1 has also been shown to inhibit Ripk1 together with Tak1 in mice; this is noteworthy given that Tak1 levels are upregulated in Tbk1 knockout cells and decline with aging in humans, more so in ALS patients compared to healthy individuals of the same age (Figure 4C) (Xu et al., 2018). Additionally, RIPK1 levels are elevated in the spinal cords of symptomatic SOD1G93A mice compared to age-matched controls (Dermentzaki et al., 2019). Together, this evidence suggests that perhaps OPTN and TBK1 LOF mechanisms in ALS may center on their role in the innate immune system, and that RIPK1-dependent signaling may play a key role in ALS pathogenesis.
In direct contrast to dysregulation of the OPTN/TBK1 axis via LOF mechanisms in ALS, GOF missense mutations in OPTN (Rezaie et al., 2002) and copy number variations in TBK1 (Fingert et al., 2011; Kawase et al., 2012; Ritch et al., 2014) have been identified in patients with some forms of glaucoma (Minegishi et al., 2016). In particular, the p.M98K missense mutation in OPTN induces RGC death and enhanced autophagosome formation (Sirohi et al., 2013). Moreover, the p.E50K mutation in OPTN leads to enhanced interaction of OPTN with TBK1 (Li et al., 2016; Minegishi et al., 2013), and its overexpression leads to RGC death (Chalasani et al., 2007) and accumulation of insoluble OPTN. These phenotypes of RGC death and OPTN accumulation can be reversed by treatment with the TBK1 inhibitor BX795 (Minegishi et al., 2013), while amlexanox, another TBK1 inhibitor, corrects the abnormal intracellular localization of OPTN E50K (Minegishi et al., 2016), suggesting potential therapeutic routes through inhibition of TBK1 in glaucoma. However, it is important to emphasize that complete or partial LOF of TBK1 affects cell survival and function differently depending on the cell type, indicating that TBK1-targeting therapeutic strategies should not be indiscriminately applied to all glaucoma patients.
6. GRN
Progranulin (PGRN), encoded by the GRN gene, is a secreted glycoprotein that regulates inflammatory and neuroprotective responses (Kao et al., 2017; Karamysheva et al., 2019). Since heterozygous null mutations in GRN were first described as causative for tau-negative FTD with TDP-43 pathology (Baker et al., 2006; Cruts et al., 2006), missense mutations have also been found in ALS patients with or without FTD across different cohorts (Schymick et al., 2006; Sleegers et al., 2008). Some of these mutations predict a LOF mechanism, although pathogenicity of these variants has not been investigated in much detail. Grn knockout mice show exacerbated neuronal damage after traumatic brain injury and lysosomal dysfunction (Tanaka et al., 2013), while knockdown of grn in zebrafish leads to impaired motor function (Chitramuthu et al., 2017). Knockdown of tardbp and fus in zebrafish causes similar motor neuron degeneration phenotypes which are rescued by the expression of human GRN (Chitramuthu et al., 2017). Overexpression of human GRN also seems to extend the survival and slow disease progression in a mouse model of TDP-43 proteinopathy (Beel et al., 2018), suggesting that boosting levels of PGRN could be beneficial for ameliorating ALS/FTD pathogenesis.
7. NEK1 and C21ORF2
NEK1 was first identified as an ALS gene through whole-exome sequencing of more than 2,000 ALS patients (Brenner et al., 2016; Cirulli et al., 2015; Gratten et al., 2017; Kenna et al., 2016; Nguyen et al., 2018). Rare variants in NEK1 are observed in 3–4% of ALS patients in European populations, among which 1% are confirmed heterozygous LOF variants (Brenner et al., 2016; Cirulli et al., 2015; Kenna et al., 2016; Nguyen et al., 2018).
As a member of the NIMA-related serine/threonine kinase protein family (NEK), NEK1 participates in controlling cell cycle, DNA damage repair, and ciliogenesis (Chen et al., 2011; Higelin et al., 2018; Pelegrini et al., 2010; Shalom et al., 2008). In fact, the first mechanistic characterization of an ALS-linked NEK1 mutation (p.R812X; heterozygous) in patient-derived motor neurons showed that upon DNA damage by irradiation, there was increased DNA damage as well as a compromised DNA damage response (Higelin et al., 2018). Moreover, knocking down Nek1 expression in 661W photoreceptor cells increases cell death by RIPK1-dependent apoptosis (Amin et al., 2018), supporting a LOF mechanism.
NEK1 has also been described to interact with the ALS-associated proteins ALS2, VAPB and C21orf2, sharing cellular functions with the latter in DNA damage response and ciliogenesis (Cirulli et al., 2015; Fang et al., 2015; Wheway et al., 2015). While the biological role of C21orf2 remains poorly characterized, it is known to participate in formation and maintenance of primary cilia (Khan et al., 2015; L. Wang et al., 2017; Wheway et al., 2015) and to directly interact with NEK1. Interestingly, mutations in both NEK1 and C21ORF2 are associated with Axial Spondylometaphyseal Dysplasia, an autosomal recessive disease characterized by dysplasia of axial skeleton and retinal dystrophy (Wang et al., 2016; Z. Wang et al., 2017). Given that C21ORF2, NEK1, and other novel ALS genes share common cellular functions/disease pathways, therapies targeting these pathways could be efficacious for a broad group of patients despite variations in genotype. However, it will be necessary to first better characterize the functions and mechanisms associated with these genes, their effects in animal models, and their roles in disease pathogenesis in ALS/FTD patients.
CURRENT CLINICAL EFFORTS
The success of targeting SOD1 mRNA for degradation in animal models of SOD1-ALS (McCampbell et al., 2018) led to the development and recent conclusion of a phase 1–2 clinical trial of the ASO tofersen (BIIB067), which binds to both mutant and WT SOD1 mRNA (Miller et al., 2020, 2013). 48 of the 50 enrolled SOD1-ALS patients received all five planned doses, of which those that received 100 mg of tofersen (the highest dose given in the trial) showed a ~36% decrease of SOD1 concentrations in the cerebrospinal fluid (CSF) compared to a ~3% decrease in the placebo group (Miller et al., 2020). Due to intrathecal injection of ASOs for wide distribution throughout the central nervous system (Schoch and Miller, 2017), most patients exhibited lumbar puncture-related adverse events (Miller et al., 2020). Although this trial was not powered to test clinical or biological efficacy beyond SOD1 levels in CSF, some exploratory outcomes showed that disease progression was slowed in the 100 mg dose group, with a particularly dramatic effect for the four fast-progressing patients (Miller et al., 2020). In this subgroup, the ALSFRS-R (ALS functional rating scale-revised) score at day 85 changed from baseline by 0.84 points in the treated group compared to −16.73 points in the placebo group. The treated group also showed better performance in two other clinical measures—slow vital capacity and hand-held dynamometry—compared to the placebo group (Miller et al., 2020). While this study was small and requires continued follow-up on the treated patients, the results so far are inspiring and augur well for future ASO-based therapeutic strategies for ALS. Further assessments of the safety and efficacy of tofersen are now being investigated in a phase 3 clinical trial in SOD1-ALS patients (ClinicalTrials.gov identifier: NCT02623699).
Non-ASO-based therapies are also in the works (Hester et al., 2009) . For example, an adeno-associated virus (AAV)-based gene therapy has been intrathecally administered to two SOD1-ALS patients to lower SOD1 levels using microRNAs (Mueller et al., 2020). One patient had lower SOD1 levels in spinal cord at autopsy and transiently lower SOD1 levels in CSF during life, while the other patient has shown no changes in SOD1 levels (Mueller et al., 2020). Moreover, the first patient developed meningoradiculitis (extreme nerve pain) after infusion, while the second patient did not have this complication perhaps due to pre-treatment with immuno-suppressive drugs (Mueller et al., 2020). Further investigations of AAV-based therapies and novel delivery routes (Bravo-Hernandez et al., 2020) will be necessary to assess their efficacy and side effects, given the small sample size to date. Regardless of whether the therapy is ASO- or AAV-based, achieving clinical benefit with only partial SOD1 suppression will help to mitigate potential problems associated with complete SOD1 LOF.
ASOs targeting C9orf72 sense strand hexanucleotide repeat-containing RNAs for degradation have also shown promising results in preclinical studies (Jiang et al., 2016; Lagier-Tourenne et al., 2013), leading to a current study of ASOs in human C9ALS patients. The ASO BIIB078 targets sense strand C9ORF72 transcripts that contain the hexanucleotide G4C2 repeat (Figure 1A), and the safety and tolerability of this ASO are currently being assessed in a phase 1 clinical trial for patients with C9ALS (ClinicalTrials.gov identifier: NCT03626012). If shown to be safe and efficacious, perhaps targeting both sense and antisense strand repeat-containing RNAs will be more efficacious than targeting the sense strand alone, as both strands contribute to RNA foci and DPR production (Gendron et al., 2013; Lagier-Tourenne et al., 2013; Mizielinska et al., 2013; Mori et al., 2013a).
Lastly, jacifusen—an experimental ASO (not tested extensively in preclinical studies) that targets mRNA encoding mutant FUSP525L (Figure 1A)—has been developed, as these mutations in FUS can cause juvenile onset and rapidly progressing forms of ALS. To date, the late Jaci Hermstad and two other FUS-ALS patients have received jacifusen, which is scheduled to be given to eight additional patients with FUS mutations (Figueiredo, Marta, 2020). With advancements in ASO-based therapies, we may expect personalized ASOs to become more common in treating ALS patients—even those with rare mutations.
A CALL FOR MULTI-TARGETED THERAPEUTIC APPROACHES
While mutations in many ALS genes have demonstrated evidence for toxic GOF mechanisms, such effects are often intrinsically linked to LOF effects that may also contribute to disease onset, progression, and/or severity. As such, we suspect that a multi-targeted therapeutic approach targeting both GOF and LOF mechanisms will be necessary to treat the majority of ALS patients.
For example, G4C2 repeat expansions in C9ORF72 result in RNA foci, DPRs, and reduced levels of C9ORF72, all of which may contribute to ALS pathogenesis. While G4C2 repeat-targeting ASOs would aim to mitigate RNA foci- and DPR-mediated toxicity, they would not affect the reduced levels of C9ORF72. Thus, a combination therapy that targets not only sense and antisense G4C2 repeat-containing RNAs but also increases levels of C9ORF72 whether that is via gene therapy, a small molecule, or CRISPR activation (CRISPRa) (Gilbert et al., 2014) may be more effective in ameliorating both the LOF and toxic GOF effects caused by this mutation (Figure 5A). For ALS genes that are thought to primarily be LOF, we should also consider the possibility of boosting their levels, such as for individuals harboring mutations in GRN, TBK1, OPTN, NEK1 or C21ORF2.
A multi-targeted therapeutic approach may also be beneficial in general when TDP-43 is depleted from the nucleus and forms cytoplasmic inclusions, which occurs in nearly all (~97%) ALS cases (Taylor et al., 2016). Cytoplasmic TDP-43 aggregates have probable toxic GOF properties, thus targeting them may be beneficial for ALS patients. Directly reducing levels of TDP-43 is not likely to be advantageous as TDP-43 plays a role in vital cellular processes, but targeting modifiers of TDP-43-mediated toxicity, such as Ataxin-2, may be a viable therapeutic strategy; reducing levels of Ataxin-2 with ASOs significantly extended lifespan and reduced pathology/ behavioral deficits in a mouse model of TDP-43 proteinopathy (Becker et al., 2017), and mutations in the Ataxin-2 gene (ATXN2) increase stability of the Ataxin-2 protein and risk for ALS in humans (Elden et al., 2010).
Yet even if we target toxic GOF properties of cytoplasmic TDP-43, such a strategy may fall short in addressing negative consequences resulting from loss of TDP-43 function in the nucleus. One potential way to address this shortcoming is to target known processes closely affected by nuclear depletion of TDP-43, such as the decreased levels of STMN2 that may be contributing to ALS pathogenesis (Klim et al., 2019; Melamed et al., 2019). Perhaps combined therapies can aim to compensate for the negative effects of TDP-43 LOF by restoring STMN2 levels through gene therapy or an ASO that blocks cryptic exon inclusion (Figure 5B), to enhance the efficacy of approaches that target toxic cytoplasmic TDP-43.
CONCLUDING REMARKS
In just over 25 years, the field has shifted from identifying the first fALS gene to entering an exciting era of therapeutic development, ranging from the delivery of personalized ASOs for a single patient to finding druggable targets for common disease pathways in sALS (Becker et al., 2017; Klim et al., 2019; Melamed et al., 2019). As detailed above, the need to consider both loss and gain of function in developing new therapies is clear. While some LOF effects may not alone be enough to cause ALS phenotypes—such as reduced levels of C9ORF72 or TBK1—there is substantial evidence that they can significantly modify the toxic effects of the disease. Even for familial mutations that almost certainly work through GOF mechanisms like SOD1 mutations, complete inhibition must be carefully considered, as detrimental phenotypes have been observed in both humans and mice with loss of SOD1 activity.
Some potential ASO therapies, such as tofersen for SOD1-ALS, indiscriminately target both WT and mutant alleles—yet it is not known whether the incomplete silencing of the normal allele will produce unwanted side effects. To circumvent this caveat, other ASOs that target specifically the mutant allele are under consideration for C9ALS and ALS caused by FUSP525L mutations. Regardless, no current therapies for humans with ALS are aimed at mitigating deleterious LOF effects that may contribute to or modify disease pathogenesis, and these LOF effects should be considered.
This review showcases the importance and sometimes prevalence of the role of LOF in ALS, and the immense opportunity to target such functions. Whether LOF is the primary mechanism or a modifier of severity/susceptibility to ALS, it is clear that both GOF and LOF mechanisms deserve attention and careful consideration as the field moves forward to combat and prevent this devastating disease.
ACKNOWLEDGMENTS
This work was supported by NIH grants R35NS097263 and R01AG064690 (A.D.G.), the Robert Packard Center for ALS Research at Johns Hopkins (A.D.G.), and the Brain Rejuvenation Project of the Wu Tsai Neurosciences Institute (A.D.G.). G.K. is supported by a fellowship from the Stanford Knight-Hennessy Scholars Program. O.G. is supported by a National Science Foundation (NSF) Graduate Research Fellowship (DGE-1656518) and a Stanford Graduate Fellowship. X.R.M. is supported by the Stanford Genome Training Program (5T32HG000044-23). Figures were generated using biorender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
A.D.G. has served as a consultant for Aquinnah Pharmaceuticals, Prevail Therapeutics and Third Rock Ventures, and is a scientific founder of Maze Therapeutics.
REFERENCES
- Abo-Rady M, Kalmbach N, Pal A, Schludi C, Janosch A, Richter T, Freitag P, Bickle M, Kahlert A-K, Petri S, Stefanov S, Glass H, Staege S, Just W, Bhatnagar R, Edbauer D, Hermann A, Wegner F, Sterneckert JL, 2020. Knocking out C9ORF72 Exacerbates Axonal Trafficking Defects Associated with Hexanucleotide Repeat Expansion and Reduces Levels of Heat Shock Proteins. Stem Cell Rep. 14, 390–405. 10.1016/j.stemcr.2020.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad L, Zhang S-Y, Casanova J-L, Sancho-Shimizu V, 2016. Human TBK1: A Gatekeeper of Neuroinflammation. Trends Mol. Med 22, 511–527. 10.1016/j.molmed.2016.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akizuki M, Yamashita H, Uemura K, Maruyama H, Kawakami H, Ito H, Takahashi R, 2013. Optineurin suppression causes neuronal cell death via NF-κB pathway. J. Neurochem 126, 699–704. 10.1111/jnc.12326 [DOI] [PubMed] [Google Scholar]
- Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW, Kiskinis E, Winborn B, Freibaum BD, Kanagaraj A, Clare AJ, Badders NM, Bilican B, Chaum E, Chandran S, Shaw CE, Eggan KC, Maniatis T, Taylor JP, 2014. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–543. 10.1016/j.neuron.2013.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amick J, Roczniak-Ferguson A, Ferguson SM, 2016. C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol. Biol. Cell 27, 3040–3051. 10.1091/mbc.e16-01-0003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin P, Florez M, Najafov A, Pan H, Geng J, Ofengeim D, Dziedzic SA, Wang H, Barrett VJ, Ito Y, LaVoie MJ, Yuan J, 2018. Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFα-mediated apoptosis. Proc. Natl. Acad. Sci. U. S. A 115, E5944–E5953. 10.1073/pnas.1806973115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen PM, Forsgren L, Binzer M, Nilsson P, Ala-Hurula V, Keränen ML, Bergmark L, Saarinen A, Haltia T, Tarvainen I, Kinnunen E, Udd B, Marklund SL, 1996. Autosomal recessive adult-onset amyotrophic lateral sclerosis associated with homozygosity for Asp90Ala CuZn-superoxide dismutase mutation. A clinical and genealogical study of 36 patients. Brain J. Neurol 119 ( Pt 4), 1153–1172. 10.1093/brain/119.4.1153 [DOI] [PubMed] [Google Scholar]
- Andersen PM, Nilsson P, Ala-Hurula V, Keränen ML, Tarvainen I, Haltia T, Nilsson L, Binzer M, Forsgren L, Marklund SL, 1995. Amyotrophic lateral sclerosis associated with homozygosity for an Asp90Ala mutation in CuZn-superoxide dismutase. Nat. Genet 10, 61–66. 10.1038/ng0595-61 [DOI] [PubMed] [Google Scholar]
- Andersen PM, Nilsson P, Forsgren L, Marklund SL, 1998. CuZn-superoxide dismutase, extracellular superoxide dismutase, and glutathione peroxidase in blood from individuals homozygous for Asp90Ala CuZu-superoxide dismutase mutation. J. Neurochem 70, 715–720. 10.1046/j.1471-4159.1998.70020715.x [DOI] [PubMed] [Google Scholar]
- Andersen PM, Nordström U, Tsiakas K, Johannsen J, Volk AE, Bierhals T, Zetterström P, Marklund SL, Hempel M, Santer R, 2019. Phenotype in an Infant with SOD1 Homozygous Truncating Mutation. N. Engl. J. Med 381, 486–488. 10.1056/NEJMc1905039 [DOI] [PubMed] [Google Scholar]
- Aoki N, Higashi S, Kawakami I, Kobayashi Z, Hosokawa M, Katsuse O, Togo T, Hirayasu Y, Akiyama H, 2012. Localization of fused in sarcoma (FUS) protein to the post-synaptic density in the brain. Acta Neuropathol. (Berl.) 124, 383–394. 10.1007/s00401-012-0984-6 [DOI] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T, 2006. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun 10.1016/j.bbrc.2006.10.093 [DOI] [PubMed] [Google Scholar]
- Armstrong GAB, Drapeau P, 2013. Loss and gain of FUS function impair neuromuscular synaptic transmission in a genetic model of ALS. Hum. Mol. Genet 22, 4282–4292. 10.1093/hmg/ddt278 [DOI] [PubMed] [Google Scholar]
- Arnold C, 2019. Custom therapies pose huge financial burdens. Nat. Med. d41591-019-00021-w 10.1038/d41591-019-00021-w [DOI] [PubMed] [Google Scholar]
- Ash PEA, Bieniek KF, Gendron TF, Caulfield T, Lin W-L, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW, Rademakers R, Boylan KB, Dickson DW, Petrucelli L, 2013. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646. 10.1016/j.neuron.2013.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee H-C, Siao C-J, Brydges S, LaRosa E, Bai Y, Fury W, Burfeind P, Zamfirova R, Warshaw G, Orengo J, Oyejide A, Fralish M, Auerbach W, Poueymirou W, Freudenberg J, Gong G, Zambrowicz B, Valenzuela D, Yancopoulos G, Murphy A, Thurston G, Lai K-MV, 2016. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production and glomerulonephropathy in mice. Sci. Rep 6, 23204 10.1038/srep23204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala YM, Conti L, Eré Ndira Avendañ O-Vá Zquez S, Dhir A, Romano M, D’ambrogio A, Tollervey J, Ule J, Baralle M, Buratti E, Baralle FE, 2010. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 30, 277–288. 10.1038/emboj.2010.310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M, 2006. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919. 10.1038/nature05016 [DOI] [PubMed] [Google Scholar]
- Becker LA, Huang B, Bieri G, Ma R, Knowles DA, Jafar-Nejad P, Messing J, Kim HJ, Soriano A, Auburger G, Pulst SM, Taylor JP, Rigo F, Gitler AD, 2017. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 544, 367–371. 10.1038/nature22038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beel S, Herdewyn S, Fazal R, De Decker M, Moisse M, Robberecht W, Van Den Bosch L, Van Damme P, 2018. Progranulin reduces insoluble TDP-43 levels, slows down axonal degeneration and prolongs survival in mutant TDP-43 mice. Mol. Neurodegener 13, 55 10.1186/s13024-018-0288-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belancio VP, Roy-Engel AM, Pochampally RR, Deininger P, 2010. Somatic expression of LINE-1 elements in human tissues. Nucleic Acids Res. 38, 3909–3922. 10.1093/nar/gkq132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belly A, Moreau-Gachelin F, Sadoul R, Goldberg Y, 2005. Delocalization of the multifunctional RNA splicing factor TLS/FUS in hippocampal neurones: exclusion from the nucleus and accumulation in dendritic granules and spine heads. Neurosci. Lett 379, 152–157. 10.1016/j.neulet.2004.12.071 [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Lee MK, Slunt HS, Guarnieri M, Xu ZS, Wong PC, Brown RH, Price DL, Sisodia SS, Cleveland DW, 1994. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc. Natl. Acad. Sci. U. S. A 91, 8292–8296. 10.1073/pnas.91.17.8292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-Hernandez M, Tadokoro T, Navarro MR, Platoshyn O, Kobayashi Y, Marsala S, Miyanohara A, Juhas S, Juhasova J, Skalnikova H, Tomori Z, Vanicky I, Studenovska H, Proks V, Chen P, Govea-Perez N, Ditsworth D, Ciacci JD, Gao S, Zhu W, Ahrens ET, Driscoll SP, Glenn TD, McAlonis-Downes M, Da Cruz S, Pfaff SL, Kaspar BK, Cleveland DW, Marsala M, 2020. Spinal subpial delivery of AAV9 enables widespread gene silencing and blocks motoneuron degeneration in ALS. Nat. Med 26, 118–130. 10.1038/s41591-019-0674-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner D, Müller K, Wieland T, Weydt P, Böhm S, Lulé D, Hübers A, Neuwirth C, Weber M, Borck G, Wahlqvist M, Danzer KM, Volk AE, Meitinger T, Strom TM, Otto M, Kassubek J, Ludolph AC, Andersen PM, Weishaupt JH, 2016. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain J. Neurol 139, e28 10.1093/brain/aww033 [DOI] [PubMed] [Google Scholar]
- Brenner D, Sieverding K, Bruno C, Lüningschrör P, Buck E, Mungwa S, Fischer L, Brockmann SJ, Ulmer J, Bliederhäuser C, Philibert CE, Satoh T, Akira S, Boillée S, Mayer B, Sendtner M, Ludolph AC, Danzer KM, Lobsiger CS, Freischmidt A, Weishaupt JH, 2019. Heterozygous Tbk1 loss has opposing effects in early and late stages of ALS in mice. J. Exp. Med 216, 267–278. 10.1084/jem.20180729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broustal O, Camuzat A, Guillot-Noël L, Guy N, Millecamps S, Deffond D, Lacomblez L, Golfier V, Hannequin D, Salachas F, Camu W, Didic M, Dubois B, Meininger V, Le Ber I, Brice A, French clinical and genetic research network on FTD/FTD-MND, 2010. FUS mutations in frontotemporal lobar degeneration with amyotrophic lateral sclerosis. J. Alzheimers Dis. JAD 22, 765–769. 10.3233/JAD-2010-100837 [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW, 1997. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18, 327–338. 10.1016/s0896-6273(00)80272-x [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW, 1998. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281, 1851–1854. 10.1126/science.281.5384.1851 [DOI] [PubMed] [Google Scholar]
- Buratti E, Dörk T, Zuccato E, Pagani F, Romano M, Baralle FE, 2001. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 10.1093/emboj/20.7.1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burberry A, Suzuki N, Wang J-Y, Moccia R, Mordes DA, Stewart MH, Suzuki-Uematsu S, Ghosh S, Singh A, Merkle FT, Koszka K, Li Q-Z, Zon L, Rossi DJ, Trowbridge JJ, Notarangelo LD, Eggan K, 2016. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci. Transl. Med 8, 347ra93 10.1126/scitranslmed.aaf6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burberry A, Wells MF, Limone F, Couto A, Smith KS, Keaney J, Gillet G, van Gastel N, Wang J-Y, Pietilainen O, Qian M, Eggan P, Cantrell C, Mok J, Kadiu I, Scadden DT, Eggan K, 2020. C9orf72 suppresses systemic and neural inflammation induced by gut bacteria. Nature. 10.1038/s41586-020-2288-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Chiu Y-H, Chen ZJ, 2014. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 54, 289–296. 10.1016/j.molcel.2014.03.040 [DOI] [PubMed] [Google Scholar]
- Arnold Carrie, 2019. Tailored treatment for ALS poised to move ahead [WWW Document]. Nat. Med. News URL https://www.nature.com/articles/d41591-019-00013-w (accessed 6.9.20). [DOI] [PubMed] [Google Scholar]
- Chalasani ML, Radha V, Gupta V, Agarwal N, Balasubramanian D, Swarup G, 2007. A glaucoma-associated mutant of optineurin selectively induces death of retinal ganglion cells which is inhibited by antioxidants. Invest. Ophthalmol. Vis. Sci 48, 1607–1614. 10.1167/iovs.06-0834 [DOI] [PubMed] [Google Scholar]
- Chang LY, Slot JW, Geuze HJ, Crapo JD, 1988. Molecular immunocytochemistry of the CuZn superoxide dismutase in rat hepatocytes. J. Cell Biol 107, 2169–2179. 10.1083/jcb.107.6.2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Chen C-F, Chiang H-C, Pena M, Polci R, Wei RL, Edwards RA, Hansel DE, Chen P-L, Riley DJ, 2011. Mutation of NIMA-related kinase 1 (NEK1) leads to chromosome instability. Mol. Cancer 10, 5 10.1186/1476-4598-10-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia R, Chiò A, Traynor BJ, 2018. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 17, 94–102. 10.1016/S1474-4422(17)30401-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitramuthu BP, Kay DG, Bateman A, Bennett HPJ, 2017. Neurotrophic effects of progranulin in vivo in reversing motor neuron defects caused by over or under expression of TDP-43 or FUS. PLOS ONE 12, e0174784 10.1371/journal.pone.0174784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SY, Lopez-Gonzalez R, Krishnan G, Phillips HL, Li AN, Seeley WW, Yao W-D, Almeida S, Gao F-B, 2019. C9ORF72-ALS/FTD-associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo. Nat. Neurosci 22, 851–862. 10.1038/s41593-019-0397-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu Y-F, Wang Q, Krueger BJ, Ren Z, Keebler J, Han Y, Levy SE, Boone BE, Wimbish JR, Waite LL, Jones AL, Carulli JP, Day-Williams AG, Staropoli JF, Xin WW, Chesi A, Raphael AR, McKenna-Yasek D, Cady J, Vianney de Jong JMB, Kenna KP, Smith BN, Topp S, Miller J, Gkazi A, FALS Sequencing Consortium, Al-Chalabi A, van den Berg LH, Veldink J, Silani V, Ticozzi N, Shaw CE, Baloh RH, Appel S, Simpson E, Lagier-Tourenne C, Pulst SM, Gibson S, Trojanowski JQ, Elman L, McCluskey L, Grossman M, Shneider NA, Chung WK, Ravits JM, Glass JD, Sims KB, Van Deerlin VM, Maniatis T, Hayes SD, Ordureau A, Swarup S, Landers J, Baas F, Allen AS, Bedlack RS, Harper JW, Gitler AD, Rouleau GA, Brown R, Harms MB, Cooper GM, Harris T, Myers RM, Goldstein DB, 2015. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–1441. 10.1126/science.aaa3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland DW, Laing N, Hurse PV, Brown RH, 1995. Toxic mutants in Charcot’s sclerosis. Nature 378, 342–343. 10.1038/378342a0 [DOI] [PubMed] [Google Scholar]
- Conlon EG, Lu L, Sharma A, Yamazaki T, Tang T, Shneider NA, Manley JL, 2016. The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. eLife 5 10.7554/eLife.17820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crapo JD, Oury T, Rabouille C, Slot JW, Chang LY, 1992. Copper,zinc superoxide dismutase is primarily a cytosolic protein in human cells. Proc. Natl. Acad. Sci. U. S. A 89, 10405–10409. 10.1073/pnas.89.21.10405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin J-J, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C, 2006. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924. 10.1038/nature05017 [DOI] [PubMed] [Google Scholar]
- Daskalakis Stella, 2020. Jaci Hermstad receives eleventh treatment of potentially lifesaving drug [WWW Document]. KTIV. URL https://ktiv.com/2020/02/11/jaci-hermstad-receives-eleventh-treatment-of-potentially-lifesaving-drug/ (accessed 3.20.20). [Google Scholar]
- de Majo M, Topp SD, Smith BN, Nishimura AL, Chen H-J, Gkazi AS, Miller J, Wong CH, Vance C, Baas F, Ten Asbroek ALMA, Kenna KP, Ticozzi N, Redondo AG, Esteban-Pérez J, Tiloca C, Verde F, Duga S, Morrison KE, Shaw PJ, Kirby J, Turner MR, Talbot K, Hardiman O, Glass JD, de Belleroche J, Gellera C, Ratti A, Al-Chalabi A, Brown RH, Silani V, Landers JE, Shaw CE, 2018. ALS-associated missense and nonsense TBK1 mutations can both cause loss of kinase function. Neurobiol. Aging 71, 266.e1–266.e10. 10.1016/j.neurobiolaging.2018.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung G-YR, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R, 2011. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 72, 245–256. 10.1016/j.neuron.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bo R, Tiloca C, Pensato V, Corrado L, Ratti A, Ticozzi N, Corti S, Castellotti B, Mazzini L, Sorarù G, Cereda C, D’Alfonso S, Gellera C, Comi GP, Silani V, SLAGEN Consortium, 2011. Novel optineurin mutations in patients with familial and sporadic amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 82, 1239–1243. 10.1136/jnnp.2011.242313 [DOI] [PubMed] [Google Scholar]
- Deng H, Gao K, Jankovic J, 2014. The role of FUS gene variants in neurodegenerative diseases. Nat. Rev. Neurol 10, 337–348. 10.1038/nrneurol.2014.78 [DOI] [PubMed] [Google Scholar]
- Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP, 1993. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science 261, 1047–1051. 10.1126/science.8351519 [DOI] [PubMed] [Google Scholar]
- Dermentzaki G, Politi KA, Lu L, Mishra V, Pérez-Torres EJ, Sosunov AA, McKhann GM, Lotti F, Shneider NA, Przedborski S, 2019. Deletion of Ripk3 Prevents Motor Neuron Death In Vitro but not In Vivo. eneuro 6, ENEURO.0308–18.2018 10.1523/ENEURO.0308-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies J-E, Shkreta L, Moszczynski AJ, Sidibé H, Semmler S, Fouillen A, Bennett ER, Bekenstein U, Destroismaisons L, Toutant J, Delmotte Q, Volkening K, Stabile S, Aulas A, Khalfallah Y, Soreq H, Nanci A, Strong MJ, Chabot B, Velde C, 2018. TDP-43 regulates the alternative splicing of hnRNP A1 to yield an aggregation-prone variant in amyotrophic lateral sclerosis. Brain 141, 1320–1333. 10.1093/brain/awy062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donde A, Sun M, Ling JP, Braunstein KE, Pang B, Wen X, Cheng X, Chen L, Wong PC, 2019. Splicing repression is a major function of TDP-43 in motor neurons. Acta Neuropathol. (Berl.) 1–14. 10.1007/s00401-019-02042-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly CJ, Zhang P-W, Pham JT, Haeusler AR, Heusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, Maragakis N, Tienari PJ, Petrucelli L, Traynor BJ, Wang J, Rigo F, Bennett CF, Blackshaw S, Sattler R, Rothstein JD, 2013. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428. 10.1016/j.neuron.2013.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D, Haass C, 2011. TDP-43 and FUS: a nuclear affair. Trends Neurosci. 34, 339–348. 10.1016/j.tins.2011.05.002 [DOI] [PubMed] [Google Scholar]
- Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IRA, Capell A, Schmid B, Neumann M, Haass C, 2010. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 29, 2841–2857. 10.1038/emboj.2010.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elden AC, Kim H-J, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, Padmanabhan A, Clay-Falcone D, McCluskey L, Elman L, Juhr D, Gruber PJ, Rüb U, Auburger G, Trojanowski JQ, Lee VM-Y, Van Deerlin VM, Bonini NM, Gitler AD, 2010. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075. 10.1038/nature09320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Lin H, Wang X, Zuo Q, Qin J, Zhang P, 2015. The NEK1 interactor, C21ORF2, is required for efficient DNA damage repair. Acta Biochim. Biophys. Sin 47, 834–841. 10.1093/abbs/gmv076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feiguin F, Godena VK, Romano G, D’Ambrogio A, Klima R, Baralle FE, 2009. Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 583, 1586–1592. 10.1016/J.FEBSLET.2009.04.019 [DOI] [PubMed] [Google Scholar]
- Ferrari R, Kapogiannis D, Huey ED, Momeni P, 2011. FTD and ALS: a tale of two diseases. Curr. Alzheimer Res 8, 273–294. 10.2174/156720511795563700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Legati A, García-Monco JC, Gomez-Beldarrain M, Carmona M, Blanco R, Seeley WW, Coppola G, 2015. Familial behavioral variant frontotemporal dementia associated with astrocyte-predominant tauopathy. J. Neuropathol. Exp. Neurol 74, 370–379. 10.1097/NEN.0000000000000180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fifita JA, Williams KL, Sundaramoorthy V, Mccann EP, Nicholson GA, Atkin JD, Blair IP, 2017. A novel amyotrophic lateral sclerosis mutation in OPTN induces ER stress and Golgi fragmentation in vitro. Amyotroph. Lateral Scler. Front. Degener 18, 126–133. 10.1080/21678421.2016.1218517 [DOI] [PubMed] [Google Scholar]
- Figueiredo Marta, 2020. Collaboration Funds Experimental Therapy for Rare FUS-ALS [WWW Document]. ALS News Today. URL https://alsnewstoday.com/2020/03/16/jacifusen-collaboration-funds-experimental-therapy-for-patients-with-rare-fus-als/ (accessed 3.28.20). [Google Scholar]
- Fingert JH, Robin AL, Stone JL, Roos BR, Davis LK, Scheetz TE, Bennett SR, Wassink TH, Kwon YH, Alward WLM, Mullins RF, Sheffield VC, Stone EM, 2011. Copy number variations on chromosome 12q14 in patients with normal tension glaucoma. Hum. Mol. Genet 20, 2482–2494. 10.1093/hmg/ddr123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, De Vivo DC, Yamashita M, Rigo F, Hung G, Schneider E, Norris DA, Xia S, Bennett CF, Bishop KM, 2016. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet Lond. Engl 388, 3017–3026. 10.1016/S0140-6736(16)31408-8 [DOI] [PubMed] [Google Scholar]
- Fischer LR, Igoudjil A, Magrané J, Li Y, Hansen JM, Manfredi G, Glass JD, 2011. SOD1 targeted to the mitochondrial intermembrane space prevents motor neuropathy in the Sod1 knockout mouse. Brain J. Neurol 134, 196–209. 10.1093/brain/awq314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer LR, Li Y, Asress SA, Jones DP, Glass JD, 2012. Absence of SOD1 leads to oxidative stress in peripheral nerve and causes a progressive distal motor axonopathy. Exp. Neurol 233, 163–171. 10.1016/j.expneurol.2011.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao S-M, Maniatis T, 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol 4, 491–496. 10.1038/ni921 [DOI] [PubMed] [Google Scholar]
- Flood DG, Reaume AG, Gruner JA, Hoffman EK, Hirsch JD, Lin YG, Dorfman KS, Scott RW, 1999. Hindlimb motor neurons require Cu/Zn superoxide dismutase for maintenance of neuromuscular junctions. Am. J. Pathol 155, 663–672. 10.1016/S0002-9440(10)65162-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratta P, Poulter M, Lashley T, Rohrer JD, Polke JM, Beck J, Ryan N, Hensman D, Mizielinska S, Waite AJ, Lai M-C, Gendron TF, Petrucelli L, Fisher EMC, Revesz T, Warren JD, Collinge J, Isaacs AM, Mead S, 2013. Homozygosity for the C9orf72 GGGGCC repeat expansion in frontotemporal dementia. Acta Neuropathol. (Berl.) 126, 401–409. 10.1007/s00401-013-1147-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredi M, Cavazzana I, Biasiotto G, Filosto M, Padovani A, Monti E, Tincani A, Franceschini F, Zanella I, 2019. C9orf72 Intermediate Alleles in Patients with Amyotrophic Lateral Sclerosis, Systemic Lupus Erythematosus, and Rheumatoid Arthritis. Neuromolecular Med. 21, 150–159. 10.1007/s12017-019-08528-8 [DOI] [PubMed] [Google Scholar]
- Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee K-H, Badders N, Valentine M, Miller BL, Wong PC, Petrucelli L, Kim HJ, Gao F-B, Taylor JP, 2015. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525, 129–133. 10.1038/nature14974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibaum BD, Taylor JP, 2017. The Role of Dipeptide Repeats in C9ORF72-Related ALS-FTD. Front. Mol. Neurosci 10, 35 10.3389/fnmol.2017.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Müller K, Marroquin N, Nordin F, Hübers A, Weydt P, Pinto S, Press R, Millecamps S, Molko N, Bernard E, Desnuelle C, Soriani M-H, Dorst J, Graf E, Nordström U, Feiler MS, Putz S, Boeckers TM, Meyer T, Winkler AS, Winkelman J, de Carvalho M, Thal DR, Otto M, Brännström T, Volk AE, Kursula P, Danzer KM, Lichtner P, Dikic I, Meitinger T, Ludolph AC, Strom TM, Andersen PM, Weishaupt JH, 2015. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci 18, 631–636. 10.1038/nn.4000 [DOI] [PubMed] [Google Scholar]
- Fridovich I, 1995. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem 64, 97–112. 10.1146/annurev.bi.64.070195.000525 [DOI] [PubMed] [Google Scholar]
- Fridovich I, 1986. Biological effects of the superoxide radical. Arch. Biochem. Biophys 247, 1–11. 10.1016/0003-9861(86)90526-6 [DOI] [PubMed] [Google Scholar]
- Fujii R, Okabe S, Urushido T, Inoue K, Yoshimura A, Tachibana T, Nishikawa T, Hicks GG, Takumi T, 2005. The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr. Biol. CB 15, 587–593. 10.1016/j.cub.2005.01.058 [DOI] [PubMed] [Google Scholar]
- Fujii R, Takumi T, 2005. TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J. Cell Sci 118, 5755–5765. 10.1242/jcs.02692 [DOI] [PubMed] [Google Scholar]
- Fukushima M, Hosoda N, Chifu K, Hoshino S, 2019. TDP −43 accelerates deadenylation of target mRNA s by recruiting Caf1 deadenylase. FEBS Lett. 593, 277–287. 10.1002/1873-3468.13310 [DOI] [PubMed] [Google Scholar]
- Gendron TF, Bieniek KF, Zhang Y-J, Jansen-West K, Ash PEA, Caulfield T, Daughrity L, Dunmore JH, Castanedes-Casey M, Chew J, Cosio DM, van Blitterswijk M, Lee WC, Rademakers R, Boylan KB, Dickson DW, Petrucelli L, 2013. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. (Berl.) 126, 829–844. 10.1007/s00401-013-1192-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbino V, Kaunga E, Ye J, Canzio D, O’Keeffe S, Rudnick ND, Guarnieri P, Lutz CM, Maniatis T, 2020. The Loss of TBK1 Kinase Activity in Motor Neurons or in All Cell Types Differentially Impacts ALS Disease Progression in SOD1 Mice. Neuron S0896627320301914. 10.1016/j.neuron.2020.03.005 [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M, Weissman JS, 2014. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661. 10.1016/j.cell.2014.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordana MT, Piccinini M, Grifoni S, Marco G, Vercellino M, Magistrello M, Pellerino A, Buccinnà B, Lupino E, Rinaudo MT, 2010. TDP-43 redistribution is an early event in sporadic amyotrophic lateral sclerosis. Brain Pathol. 10.1111/j.1750-3639.2009.00284.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler AD, Tsuiji H, 2016. There has been an awakening: Emerging mechanisms of C9orf72 mutations in FTD/ALS. Brain Res. 1647, 19–29. 10.1016/j.brainres.2016.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier JL, 2016. Restricting retrotransposons: a review. Mob. DNA 7, 16 10.1186/s13100-016-0070-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratten J, Zhao Q, Benyamin B, Garton F, He J, Leo PJ, Mangelsdorf M, Anderson L, Zhang Z-H, Chen L, Chen X-D, Cremin K, Deng H-W, Edson J, Han Y-Y, Harris J, Henders AK, Jin Z-B, Li Z, Lin Y, Liu X, Marshall M, Mowry BJ, Ran S, Reutens DC, Song S, Tan L-J, Tang L, Wallace RH, Wheeler L, Wu J, Yang J, Xu H, Visscher PM, Bartlett PF, Brown MA, Wray NR, Fan D, 2017. Whole-exome sequencing in amyotrophic lateral sclerosis suggests NEK1 is a risk gene in Chinese. Genome Med. 9, 97 10.1186/s13073-017-0487-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory RI, Yan K-P, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R, 2004. The Microprocessor complex mediates the genesis of microRNAs. Nature 432, 235–240. 10.1038/nature03120 [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, 1994. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775. 10.1126/science.8209258 [DOI] [PubMed] [Google Scholar]
- Hamilton RT, Bhattacharya A, Walsh ME, Shi Y, Wei R, Zhang Y, Rodriguez KA, Buffenstein R, Chaudhuri AR, Van Remmen H, 2013. Elevated protein carbonylation, and misfolding in sciatic nerve from db/db and Sod1(−/−) mice: plausible link between oxidative stress and demyelination. PloS One 8, e65725 10.1371/journal.pone.0065725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Z, Liu L, Tao Z, Wang R, Ren H, Sun H, Lin Z, Zhang Z, Mu C, Zhou J, Wang G, 2019. Motor dysfunction and neurodegeneration in a C9orf72 mouse line expressing poly-PR. Nat. Commun 10, 2906 10.1038/s41467-019-10956-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes LR, Asress SA, Li Y, Galkin A, Stepanova A, Kawamata H, Manfredi G, Glass JD, 2019. Distal denervation in the SOD1 knockout mouse correlates with loss of mitochondria at the motor nerve terminal. Exp. Neurol 318, 251–257. 10.1016/j.expneurol.2019.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward LJ, Rodriguez JA, Kim JW, Tiwari A, Goto JJ, Cabelli DE, Valentine JS, Brown RH, 2002. Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J. Biol. Chem 277, 15923–15931. 10.1074/jbc.M112087200 [DOI] [PubMed] [Google Scholar]
- Heo J-M, Ordureau A, Paulo JA, Rinehart J, Harper JW, 2015. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 60, 7–20. 10.1016/j.molcel.2015.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog JJ, Deshpande M, Shapiro L, Rodal AA, Paradis S, 2017. TDP-43 misexpression causes defects in dendritic growth. Sci. Rep 7, 15656 10.1038/s41598-017-15914-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hester ME, Foust KD, Kaspar RW, Kaspar BK, 2009. AAV as a gene transfer vector for the treatment of neurological disorders: novel treatment thoughts for ALS. Curr. Gene Ther 9, 428–433. 10.2174/156652309789753383 [DOI] [PubMed] [Google Scholar]
- Hewitt C, Kirby J, Highley JR, Hartley JA, Hibberd R, Hollinger HC, Williams TL, Ince PG, McDermott CJ, Shaw PJ, 2010. Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol 67, 455–461. 10.1001/archneurol.2010.52 [DOI] [PubMed] [Google Scholar]
- Hicks GG, Singh N, Nashabi A, Mai S, Bozek G, Klewes L, Arapovic D, White EK, Koury MJ, Oltz EM, Van Kaer L, Ruley HE, 2000. Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat. Genet 24, 175–179. 10.1038/72842 [DOI] [PubMed] [Google Scholar]
- Higelin J, Catanese A, Semelink-Sedlacek LL, Oeztuerk S, Lutz A-K, Bausinger J, Barbi G, Speit G, Andersen PM, Ludolph AC, Demestre M, Boeckers TM, 2018. NEK1 loss-of-function mutation induces DNA damage accumulation in ALS patient-derived motoneurons. Stem Cell Res. 30, 150–162. 10.1016/j.scr.2018.06.005 [DOI] [PubMed] [Google Scholar]
- Ho WY, Tai YK, Chang J-C, Liang J, Tyan S-H, Chen S, Guan J-L, Zhou H, Shen H-M, Koo E, Ling S-C, 2019. The ALS-FTD-linked gene product, C9orf72, regulates neuronal morphogenesis via autophagy. Autophagy 15, 827–842. 10.1080/15548627.2019.1569441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoell JI, Larsson E, Runge S, Nusbaum JD, Duggimpudi S, Farazi TA, Hafner M, Borkhardt A, Sander C, Tuschl T, 2011. RNA targets of wild-type and mutant FET family proteins. Nat. Struct. Mol. Biol 18, 1428–1431. 10.1038/nsmb.2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Sahashi K, Rigo F, Hung G, Horev G, Bennett CF, Krainer AR, 2011. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 478, 123–126. 10.1038/nature10485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishigaki S, Fujioka Y, Okada Y, Riku Y, Udagawa T, Honda D, Yokoi S, Endo K, Ikenaka K, Takagi S, Iguchi Y, Sahara N, Takashima A, Okano H, Yoshida M, Warita H, Aoki M, Watanabe H, Okado H, Katsuno M, Sobue G, 2017. Altered Tau Isoform Ratio Caused by Loss of FUS and SFPQ Function Leads to FTLD-like Phenotypes. Cell Rep. 18, 1118–1131. 10.1016/j.celrep.2017.01.013 [DOI] [PubMed] [Google Scholar]
- Ishigaki S, Masuda A, Fujioka Y, Iguchi Y, Katsuno M, Shibata A, Urano F, Sobue G, Ohno K, 2012. Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci. Rep 2, 529 10.1038/srep00529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii KJ, Coban C, Kato H, Takahashi K, Torii Y, Takeshita F, Ludwig H, Sutter G, Suzuki K, Hemmi H, Sato S, Yamamoto M, Uematsu S, Kawai T, Takeuchi O, Akira S, 2006. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat. Immunol 7, 40–48. 10.1038/ni1282 [DOI] [PubMed] [Google Scholar]
- Ismail A, Cooper-Knock J, Highley JR, Milano A, Kirby J, Goodall E, Lowe J, Scott I, Constantinescu CS, Walters SJ, Price S, McDermott CJ, Sawcer S, Compston DAS, Sharrack B, Shaw PJ, 2013. Concurrence of multiple sclerosis and amyotrophic lateral sclerosis in patients with hexanucleotide repeat expansions of C9ORF72. J. Neurol. Neurosurg. Psychiatry 84, 79–87. 10.1136/jnnp-2012-303326 [DOI] [PubMed] [Google Scholar]
- Ito D, Seki M, Tsunoda Y, Uchiyama H, Suzuki N, 2011. Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann. Neurol 69, 152–162. 10.1002/ana.22246 [DOI] [PubMed] [Google Scholar]
- Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, Hitomi J, Zhu H, Chen H, Mayo L, Geng J, Amin P, DeWitt JP, Mookhtiar AK, Florez M, Ouchida AT, Fan J, Pasparakis M, Kelliher MA, Ravits J, Yuan J, 2016. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353, 603–608. 10.1126/science.aaf6803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivannikov MV, Van Remmen H, 2015. Sod1 gene ablation in adult mice leads to physiological changes at the neuromuscular junction similar to changes that occur in old wild-type mice. Free Radic. Biol. Med 84, 254–262. 10.1016/j.freeradbiomed.2015.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji YJ, Ugolino J, Brady NR, Hamacher-Brady A, Wang J, 2017. Systemic deregulation of autophagy upon loss of ALS- and FTD-linked C9orf72. Autophagy 13, 1254–1255. 10.1080/15548627.2017.1299312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji YJ, Ugolino J, Zhang T, Lu J, Kim D, Wang J, 2020. C9orf72/ALFA-1 controls TFEB/HLH-30-dependent metabolism through dynamic regulation of Rag GTPases. PLoS Genet. 16, e1008738 10.1371/journal.pgen.1008738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, Stauffer JE, Jafar-nejad P, Drenner K, Schulte D, Chun S, Sun S, Ling S-C, Myers B, Engelhardt J, Katz M, Baughn M, Platoshyn O, Marsala M, Watt A, Heyser CJ, Ard MC, De Muynck L, Daughrity LM, Swing DA, Tessarollo L, Jung CJ, Delpoux A, Utzschneider DT, Hedrick SM, de Jong PJ, Edbauer D, Van Damme P, Petrucelli L, Shaw CE, Bennett CF, Da Cruz S, Ravits J, Rigo F, Cleveland DW, Lagier-Tourenne C, 2016. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron 90, 535–550. 10.1016/j.neuron.2016.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Bercier V, Lissouba A, Liao M, Brustein E, Rouleau GA, Drapeau P, 2011. FUS and TARDBP but not SOD1 interact in genetic models of amyotrophic lateral sclerosis. PLoS Genet. 7, e1002214 10.1371/journal.pgen.1002214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao AW, McKay A, Singh PP, Brunet A, Huang EJ, 2017. Progranulin, lysosomal regulation and neurodegenerative disease. Nat. Rev. Neurosci 18, 325–333. 10.1038/nrn.2017.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapeli K, Pratt GA, Vu AQ, Hutt KR, Martinez FJ, Sundararaman B, Batra R, Freese P, Lambert NJ, Huelga SC, Chun SJ, Liang TY, Chang J, Donohue JP, Shiue L, Zhang J, Zhu H, Cambi F, Kasarskis E, Hoon S, Ares M, Burge CB, Ravits J, Rigo F, Yeo GW, 2016. Distinct and shared functions of ALS-associated proteins TDP-43, FUS and TAF15 revealed by multisystem analyses. Nat. Commun 7, 12143 10.1038/ncomms12143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamysheva ZN, Tikhonova EB, Karamyshev AL, 2019. Granulin in Frontotemporal Lobar Degeneration: Molecular Mechanisms of the Disease. Front. Neurosci 13, 395 10.3389/fnins.2019.00395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Mieda-Sato A, 2012. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. U. S. A 109, 3347–52. 10.1073/pnas.1112427109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawase K, Allingham RR, Meguro A, Mizuki N, Roos B, Solivan-Timpe FM, Robin AL, Ritch R, Fingert JH, 2012. Confirmation of TBK1 duplication in normal tension glaucoma. Exp. Eye Res 96, 178–180. 10.1016/j.exer.2011.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keithley EM, Canto C, Zheng QY, Wang X, Fischel-Ghodsian N, Johnson KR, 2005. Cu/Zn superoxide dismutase and age-related hearing loss. Hear. Res 209, 76–85. 10.1016/j.heares.2005.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller GA, Warner TG, Steimer KS, Hallewell RA, 1991. Cu,Zn superoxide dismutase is a peroxisomal enzyme in human fibroblasts and hepatoma cells. Proc. Natl. Acad. Sci. U. S. A 88, 7381–7385. 10.1073/pnas.88.16.7381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenna KP, van Doormaal PTC, Dekker AM, Ticozzi N, Kenna BJ, Diekstra FP, van Rheenen W, van Eijk KR, Jones AR, Keagle P, Shatunov A, Sproviero W, Smith BN, van Es MA, Topp SD, Kenna A, Miller JW, Fallini C, Tiloca C, McLaughlin RL, Vance C, Troakes C, Colombrita C, Mora G, Calvo A, Verde F, Al-Sarraj S, King A, Calini D, de Belleroche J, Baas F, van der Kooi AJ, de Visser M, Ten Asbroek ALMA, Sapp PC, McKenna-Yasek D, Polak M, Asress S, Muñoz-Blanco JL, Strom TM, Meitinger T, Morrison KE, SLAGEN Consortium, Lauria G, Williams KL, Leigh PN, Nicholson GA, Blair IP, Leblond CS, Dion PA, Rouleau GA, Pall H, Shaw PJ, Turner MR, Talbot K, Taroni F, Boylan KB, Van Blitterswijk M, Rademakers R, Esteban-Pérez J, García-Redondo A, Van Damme P, Robberecht W, Chio A, Gellera C, Drepper C, Sendtner M, Ratti A, Glass JD, Mora JS, Basak NA, Hardiman O, Ludolph AC, Andersen PM, Weishaupt JH, Brown RH, Al-Chalabi A, Silani V, Shaw CE, van den Berg LH, Veldink JH, Landers JE, 2016. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat. Genet 48, 1037–1042. 10.1038/ng.3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AO, Eisenberger T, Nagel-Wolfrum K, Wolfrum U, Bolz HJ, 2015. C21orf2 is mutated in recessive early-onset retinal dystrophy with macular staphyloma and encodes a protein that localises to the photoreceptor primary cilium. Br. J. Ophthalmol 99, 1725–1731. 10.1136/bjophthalmol-2015-307277 [DOI] [PubMed] [Google Scholar]
- Kino Y, Washizu C, Kurosawa M, Yamada M, Miyazaki H, Akagi T, Hashikawa T, Doi H, Takumi T, Hicks GG, Hattori N, Shimogori T, Nukina N, 2015. FUS/TLS deficiency causes behavioral and pathological abnormalities distinct from amyotrophic lateral sclerosis. Acta Neuropathol. Commun 3, 24 10.1186/s40478-015-0202-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R, Moccia R, Cassel SH, Chen K, Wainger BJ, Woolf CJ, Eggan K, 2019. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci 22, 167–179. 10.1038/s41593-018-0300-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppers M, Blokhuis AM, Westeneng H-J, Terpstra ML, Zundel CAC, Vieira de Sá R, Schellevis RD, Waite AJ, Blake DJ, Veldink JH, van den Berg LH, Pasterkamp RJ, 2015. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits: C9orf72 Ablation. Ann. Neurol 78, 426–438. 10.1002/ana.24453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korac J, Schaeffer V, Kovacevic I, Clement AM, Jungblut B, Behl C, Terzic J, Dikic I, 2013. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J. Cell Sci 126, 580–592. 10.1242/jcs.114926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer BC, Schuck T, Wheeler JM, Robinson LC, Trojanowski JQ, Lee VMY, Schellenberg GD, 2010. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. (Berl.) 119, 409–419. 10.1007/s00401-010-0659-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug L, Chatterjee N, Borges-Monroy R, Hearn S, Liao WW, Morrill K, Prazak L, Rozhkov N, Theodorou D, Hammell M, Dubnau J, 2017. Retrotransposon activation contributes to neurodegeneration in a Drosophila TDP-43 model of ALS. PLoS Genet. 10.1371/journal.pgen.1006635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Hasan GM, Hassan MI, 2017. Unraveling the Role of RNA Mediated Toxicity of C9orf72 Repeats in C9-FTD/ALS. Front. Neurosci 11, 711 10.3389/fnins.2017.00711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda M, Sok J, Webb L, Baechtold H, Urano F, Yin Y, Chung P, de Rooij DG, Akhmedov A, Ashley T, Ron D, 2000. Male sterility and enhanced radiation sensitivity in TLS(−/−) mice. EMBO J. 19, 453–462. 10.1093/emboj/19.3.453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH, 2009. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. 10.1126/science.1166066 [DOI] [PubMed] [Google Scholar]
- Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T, Kim J, Yun J, Xie Y, McKnight SL, 2014. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 345, 1139–1145. 10.1126/science.1254917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li H-R, Jiang J, Watt AT, Chun S, Katz M, Qiu J, Sun Y, Ling S-C, Zhu Q, Polymenidou M, Drenner K, Artates JW, McAlonis-Downes M, Markmiller S, Hutt KR, Pizzo DP, Cady J, Harms MB, Baloh RH, Vandenberg SR, Yeo GW, Fu X-D, Bennett CF, Cleveland DW, Ravits J, 2013. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. U. S. A 110, E4530–4539. 10.1073/pnas.1318835110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Cleveland DW, 2010. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet 19, R46–64. 10.1093/hmg/ddq137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling S-C, Liang TY, Mazur C, Wancewicz E, Kim AS, Watt A, Freier S, Hicks GG, Donohue JP, Shiue L, Bennett CF, Ravits J, Cleveland DW, Yeo GW, 2012. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci 15, 1488–1497. 10.1038/nn.3230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y, Sullivan PM, Hu F, 2019. SMCR8 negatively regulates AKT and MTORC1 signaling to modulate lysosome biogenesis and tissue homeostasis. Autophagy 15, 871–885. 10.1080/15548627.2019.1569914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin LM, Davis CS, Sims-Robinson C, Kostrominova TY, Van Remmen H, Richardson A, Feldman EL, Brooks SV, 2011. Skeletal muscle weakness due to deficiency of CuZn-superoxide dismutase is associated with loss of functional innervation. Am. J. Physiol. Regul. Integr. Comp. Physiol 301, R1400–1407. 10.1152/ajpregu.00093.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ, 2015. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. 10.1038/nature14893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K-H, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, Cika J, Coughlin M, Messing J, Molliex A, Maxwell BA, Kim NC, Temirov J, Moore J, Kolaitis R-M, Shaw TI, Bai B, Peng J, Kriwacki RW, Taylor JP, 2016. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 167, 774–788.e17. 10.1016/j.cell.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Ji Y, Tang L, Zhang N, He J, Ye S, Liu X, Fan D, 2015. Optineurin mutations in patients with sporadic amyotrophic lateral sclerosis in China. Amyotroph. Lateral Scler. Front. Degener 16, 485–489. 10.3109/21678421.2015.1089909 [DOI] [PubMed] [Google Scholar]
- Li F, Xie X, Wang Y, Liu J, Cheng X, Guo Y, Gong Y, Hu S, Pan L, 2016. Structural insights into the interaction and disease mechanism of neurodegenerative disease-associated optineurin and TBK1 proteins. Nat. Commun 7, 12708 10.1038/ncomms12708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Xu D, Wang Y, Zhou Z, Liu J, Hu S, Gong Y, Yuan J, Pan L, 2018. Structural insights into the ubiquitin recognition by OPTN (optineurin) and its regulation by TBK1-mediated phosphorylation. Autophagy 14, 66–79. 10.1080/15548627.2017.1391970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Jin Y, Prazak L, Hammell M, Dubnau J, 2012. Transposable Elements in TDP-43-Mediated Neurodegenerative Disorders. PLoS ONE. 10.1371/journal.pone.0044099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling JP, Pletnikova O, Troncoso JC, Wong PC, 2015. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349, 650–5. 10.1126/science.aab0983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling S-C, Dastidar SG, Tokunaga S, Ho WY, Lim K, Ilieva H, Parone PA, Tyan S-H, Tse TM, Chang J-C, Platoshyn O, Bui NB, Bui A, Vetto A, Sun S, McAlonis-Downes M, Han JS, Swing D, Kapeli K, Yeo GW, Tessarollo L, Marsala M, Shaw CE, Tucker-Kellogg G, La Spada AR, Lagier-Tourenne C, Da Cruz S, Cleveland DW, 2019. Overriding FUS autoregulation in mice triggers gain-of-toxic dysfunctions in RNA metabolism and autophagy-lysosome axis. eLife 8 10.7554/eLife.40811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liscic RM, Grinberg LT, Zidar J, Gitcho MA, Cairns NJ, 2008. ALS and FTLD: two faces of TDP-43 proteinopathy. Eur. J. Neurol 15, 772–780. 10.1111/j.1468-1331.2008.02195.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu EY, Russ J, Cali CP, Phan JM, Amlie-Wolf A, Lee EB, 2019. Loss of Nuclear TDP-43 Is Associated with Decondensation of LINE Retrotransposons. Cell Rep. 10.1016/j.celrep.2019.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu Y-T, Grishin NV, Chen ZJ, 2015. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630 10.1126/science.aaa2630 [DOI] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderwyde T, Citro A, Mehta T, Zaarur N, McKee A, Bowser R, Sherman M, Petrucelli L, Wolozin B, 2010. Tar DNA binding protein-43 (TDP-43) associates with stress granules: Analysis of cultured cells and pathological brain tissue. PLoS ONE. 10.1371/journal.pone.0013250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Erauskin J, Tadokoro T, Baughn MW, Myers B, McAlonis-Downes M, Chillon-Marinas C, Asiaban JN, Artates J, Bui AT, Vetto AP, Lee SK, Le AV, Sun Y, Jambeau M, Boubaker J, Swing D, Qiu J, Hicks GG, Ouyang Z, Fu X-D, Tessarollo L, Ling S-C, Parone PA, Shaw CE, Marsala M, Lagier-Tourenne C, Cleveland DW, Da Cruz S, 2018. ALS/FTD-Linked Mutation in FUS Suppresses Intra-axonal Protein Synthesis and Drives Disease Without Nuclear Loss-of-Function of FUS. Neuron 100, 816–830.e7. 10.1016/j.neuron.2018.09.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovinovic A, Cimbro R, Ljutic T, Kriz J, Rogelj B, Munitic I, 2017. Optineurin in amyotrophic lateral sclerosis: Multifunctional adaptor protein at the crossroads of different neuroprotective mechanisms. Prog. Neurobiol 154, 1–20. 10.1016/j.pneurobio.2017.04.005 [DOI] [PubMed] [Google Scholar]
- Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, Kinoshita Y, Kamada M, Nodera H, Suzuki H, Komure O, Matsuura S, Kobatake K, Morimoto N, Abe K, Suzuki N, Aoki M, Kawata A, Hirai T, Kato T, Ogasawara K, Hirano A, Takumi T, Kusaka H, Hagiwara K, Kaji R, Kawakami H, 2010. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226. 10.1038/nature08971 [DOI] [PubMed] [Google Scholar]
- Masuda A, Takeda J, Okuno T, Okamoto T, Ohkawara B, Ito M, Ishigaki S, Sobue G, Ohno K, 2015. Position-specific binding of FUS to nascent RNA regulates mRNA length. Genes Dev. 29, 1045–1057. 10.1101/gad.255737.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM, Grässer FA, Mori K, Kremmer E, Banzhaf-Strathmann J, Mann M, Meissner F, Edbauer D, 2014. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. (Berl.) 128, 485–503. 10.1007/s00401-014-1329-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCampbell A, Cole T, Wegener AJ, Tomassy GS, Setnicka A, Farley BJ, Schoch KM, Hoye ML, Shabsovich M, Sun L, Luo Y, Zhang M, Comfort N, Wang B, Amacker J, Thankamony S, Salzman DW, Cudkowicz M, Graham DL, Bennett CF, Kordasiewicz HB, Swayze EE, Miller TM, 2018. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J. Clin. Invest 128, 3558–3567. 10.1172/JCI99081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauley ME, Baloh RH, 2019. Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. (Berl.) 137, 715–730. 10.1007/s00401-018-1933-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauley ME, O’Rourke JG, Yáñez A, Markman JL, Ho R, Wang X, Chen S, Lall D, Jin M, Muhammad AKMG, Bell S, Landeros J, Valencia V, Harms M, Arditi M, Jefferies C, Baloh RH, 2020. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature. 10.1038/s41586-020-2625-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald KK, Aulas A, Destroismaisons L, Pickles S, Beleac E, Camu W, Rouleau GA, Velde C, 2011. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet 10.1093/hmg/ddr021 [DOI] [PubMed] [Google Scholar]
- Melamed Z, López-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, Freyermuth F, McMahon MA, Beccari MS, Artates JW, Ohkubo T, Rodriguez M, Lin N, Wu D, Bennett CF, Rigo F, Cruz S, Ravits J, Lagier-Tourenne C, Cleveland DW, 2019. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci 22, 180–190. 10.1038/s41593-018-0293-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L, Ward AJ, Chun S, Bennett CF, Beaudet AL, Rigo F, 2015. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 518, 409–412. 10.1038/nature13975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merner ND, Girard SL, Catoire H, Bourassa CV, Belzil VV, Rivière J-B, Hince P, Levert A, Dionne-Laporte A, Spiegelman D, Noreau A, Diab S, Szuto A, Fournier H, Raelson J, Belouchi M, Panisset M, Cossette P, Dupré N, Bernard G, Chouinard S, Dion PA, Rouleau GA, 2012. Exome sequencing identifies FUS mutations as a cause of essential tremor. Am. J. Hum. Genet 91, 313–319. 10.1016/j.ajhg.2012.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller T, Cudkowicz M, Shaw PJ, Andersen PM, Atassi N, Bucelli RC, Genge A, Glass J, Ladha S, Ludolph AL, Maragakis NJ, McDermott CJ, Pestronk A, Ravits J, Salachas F, Trudell R, Van Damme P, Zinman L, Bennett CF, Lane R, Sandrock A, Runz H, Graham D, Houshyar H, McCampbell A, Nestorov I, Chang I, McNeill M, Fanning L, Fradette S, Ferguson TA, 2020. Phase 1–2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med 383, 109–119. 10.1056/NEJMoa2003715 [DOI] [PubMed] [Google Scholar]
- Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, Andres PL, Mahoney K, Allred P, Alexander K, Ostrow LW, Schoenfeld D, Macklin EA, Norris DA, Manousakis G, Crisp M, Smith R, Bennett CF, Bishop KM, Cudkowicz ME, 2013. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 12, 435–442. 10.1016/S1474-4422(13)70061-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller ZA, Sturm VE, Camsari GB, Karydas A, Yokoyama JS, Grinberg LT, Boxer AL, Rosen HJ, Rankin KP, Gorno-Tempini ML, Coppola G, Geschwind DH, Rademakers R, Seeley WW, Graff-Radford NR, Miller BL, 2016. Increased prevalence of autoimmune disease within C9 and FTD/MND cohorts: Completing the picture. Neurol. - Neuroimmunol. Neuroinflammation 3, e301 10.1212/NXI.0000000000000301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minegishi Y, Iejima D, Kobayashi H, Chi Z-L, Kawase K, Yamamoto T, Seki T, Yuasa S, Fukuda K, Iwata T, 2013. Enhanced optineurin E50K-TBK1 interaction evokes protein insolubility and initiates familial primary open-angle glaucoma. Hum. Mol. Genet 22, 3559–3567. 10.1093/hmg/ddt210 [DOI] [PubMed] [Google Scholar]
- Minegishi Y, Nakayama M, Iejima D, Kawase K, Iwata T, 2016. Significance of optineurin mutations in glaucoma and other diseases. Prog. Retin. Eye Res 55, 149–181. 10.1016/j.preteyeres.2016.08.002 [DOI] [PubMed] [Google Scholar]
- Mitchell JC, McGoldrick P, Vance C, Hortobagyi T, Sreedharan J, Rogelj B, Tudor EL, Smith BN, Klasen C, Miller CCJ, Cooper JD, Greensmith L, Shaw CE, 2013. Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol. (Berl.) 125, 273–288. 10.1007/s00401-012-1043-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizielinska S, Grönke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IOC, Pietrzyk J, Cleverley K, Nicoll AJ, Pickering-Brown S, Dols J, Cabecinha M, Hendrich O, Fratta P, Fisher EMC, Partridge L, Isaacs AM, 2014. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345, 1192–1194. 10.1126/science.1256800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizielinska S, Lashley T, Norona FE, Clayton EL, Ridler CE, Fratta P, Isaacs AM, 2013. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. (Berl.) 126, 845–857. 10.1007/s00401-013-1200-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AS, Holzbaur ELF, 2016. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. U. S. A 113, E3349–3358. 10.1073/pnas.1523810113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Arzberger T, Grässer FA, Gijselinck I, May S, Rentzsch K, Weng S-M, Schludi MH, van der Zee J, Cruts M, Van Broeckhoven C, Kremmer E, Kretzschmar HA, Haass C, Edbauer D, 2013a. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. (Berl.) 126, 881–893. 10.1007/s00401-013-1189-3 [DOI] [PubMed] [Google Scholar]
- Mori K, Lammich S, Mackenzie IRA, Forné I, Zilow S, Kretzschmar H, Edbauer D, Janssens J, Kleinberger G, Cruts M, Herms J, Neumann M, Van Broeckhoven C, Arzberger T, Haass C, 2013b. hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. (Berl.) 125, 413–423. 10.1007/s00401-013-1088-7 [DOI] [PubMed] [Google Scholar]
- Mori K, Weng S-M, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C, Edbauer D, 2013c. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. 10.1126/science.1232927 [DOI] [PubMed] [Google Scholar]
- Morlando M, Dini Modigliani S, Torrelli G, Rosa A, Di Carlo V, Caffarelli E, Bozzoni I, 2012. FUS stimulates microRNA biogenesis by facilitating co-transcriptional Drosha recruitment. EMBO J. 31, 4502–4510. 10.1038/emboj.2012.319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris HR, Waite AJ, Williams NM, Neal JW, Blake DJ, 2012. Recent advances in the genetics of the ALS-FTLD complex. Curr. Neurol. Neurosci. Rep 12, 243–250. 10.1007/s11910-012-0268-5 [DOI] [PubMed] [Google Scholar]
- Mueller C, Berry JD, McKenna-Yasek DM, Gernoux G, Owegi MA, Pothier LM, Douthwright CL, Gelevski D, Luppino SD, Blackwood M, Wightman NS, Oakley DH, Frosch MP, Flotte TR, Cudkowicz ME, Brown RH, 2020. SOD1 Suppression with Adeno-Associated Virus and MicroRNA in Familial ALS. N. Engl. J. Med 383, 151–158. 10.1056/NEJMoa2005056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FL, Song W, Liu Y, Chaudhuri A, Pieke-Dahl S, Strong R, Huang T-T, Epstein CJ, Roberts LJ, Csete M, Faulkner JA, Van Remmen H, 2006. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic. Biol. Med 40, 1993–2004. 10.1016/j.freeradbiomed.2006.01.036 [DOI] [PubMed] [Google Scholar]
- Neelagandan N, Gonnella G, Dang S, Janiesch PC, Miller KK, Küchler K, Marques RF, Indenbirken D, Alawi M, Grundhoff A, Kurtz S, Duncan KE, 2019. TDP-43 enhances translation of specific mRNAs linked to neurodegenerative disease. Nucleic Acids Res. 47, 341–361. 10.1093/nar/gky972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IRA, 2009. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain J. Neurol 132, 2922–2931. 10.1093/brain/awp214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VMY, 2006. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- Nguyen HP, Van Mossevelde S, Dillen L, De Bleecker JL, Moisse M, Van Damme P, Van Broeckhoven C, van der Zee J, BELNEU Consortium, 2018. NEK1 genetic variability in a Belgian cohort of ALS and ALS-FTD patients. Neurobiol. Aging 61, 255.e1–255.e7. 10.1016/j.neurobiolaging.2017.08.021 [DOI] [PubMed] [Google Scholar]
- Niblock M, Smith BN, Lee Y-B, Sardone V, Topp S, Troakes C, Al-Sarraj S, Leblond CS, Dion PA, Rouleau GA, Shaw CE, Gallo J-M, 2016. Retention of hexanucleotide repeat-containing intron in C9orf72 mRNA: implications for the pathogenesis of ALS/FTD. Acta Neuropathol. Commun 4, 18 10.1186/s40478-016-0289-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noad J, von der Malsburg A, Pathe C, Michel MA, Komander D, Randow F, 2017. LUBAC-synthesized linear ubiquitin chains restrict cytosol-invading bacteria by activating autophagy and NF-κB. Nat. Microbiol 2, 17063 10.1038/nmicrobiol.2017.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke JG, Bogdanik L, Yanez A, Lall D, Wolf AJ, Muhammad AKMG, Ho R, Carmona S, Vit JP, Zarrow J, Kim KJ, Bell S, Harms MB, Miller TM, Dangler CA, Underhill DM, Goodridge HS, Lutz CM, Baloh RH, 2016. C9orf72 is required for proper macrophage and microglial function in mice. Science 351, 1324–1329. 10.1126/science.aaf1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco D, Tahirovic S, Rentzsch K, Schwenk BM, Haass C, Edbauer D, 2012. Loss of fused in sarcoma (FUS) promotes pathological Tau splicing. EMBO Rep. 13, 759–764. 10.1038/embor.2012.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HT, 2011. FTD and ALS: genetic ties that bind. Neuron 72, 189–190. 10.1016/j.neuron.2011.10.001 [DOI] [PubMed] [Google Scholar]
- Padman BS, Nguyen TN, Uoselis L, Skulsuppaisarn M, Nguyen LK, Lazarou M, 2019. LC3/GABARAPs drive ubiquitin-independent recruitment of Optineurin and NDP52 to amplify mitophagy. Nat. Commun 10, 408 10.1038/s41467-019-08335-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Elpers C, Reunert J, McCormick ML, Mohr J, Biskup S, Schwartz O, Rust S, Grüneberg M, Seelhöfer A, Schara U, Boltshauser E, Spitz DR, Marquardt T, 2019. SOD1 deficiency: a novel syndrome distinct from amyotrophic lateral sclerosis. Brain J. Neurol 142, 2230–2237. 10.1093/brain/awz182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus JD, Link BA, 2014. Loss of optineurin in vivo results in elevated cell death and alters axonal trafficking dynamics. PloS One 9, e109922 10.1371/journal.pone.0109922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelegrini AL, Moura DJ, Brenner BL, Ledur PF, Maques GP, Henriques JAP, Saffi J, Lenz G, 2010. Nek1 silencing slows down DNA repair and blocks DNA damage-induced cell cycle arrest. Mutagenesis 25, 447–454. 10.1093/mutage/geq026 [DOI] [PubMed] [Google Scholar]
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling S-C, Sun E, Wancewicz E, Mazur C, Kordasiewicz H, Sedaghat Y, Donohue JP, Shiue L, Bennett CF, Yeo GW, Cleveland DW, 2011. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci 14, 459–468. 10.1038/nn.2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh N, Pandey UB, 2017. Autophagy Dysregulation in ALS: When Protein Aggregates Get Out of Hand. Front. Mol. Neurosci 10, 263 10.3389/fnmol.2017.00263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, Scott RW, Snider WD, 1996. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat. Genet 13, 43–47. 10.1038/ng0596-43 [DOI] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita V-M, Kaivorinne A-L, Hölttä-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chiò A, Restagno G, Borghero G, Sabatelli M, ITALSGEN Consortium, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ, 2011. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. 10.1016/j.neuron.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezaie T, Child A, Hitchings R, Brice G, Miller L, Coca-Prados M, Héon E, Krupin T, Ritch R, Kreutzer D, Crick RP, Sarfarazi M, 2002. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science 295, 1077–1079. 10.1126/science.1066901 [DOI] [PubMed] [Google Scholar]
- Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, Youle RJ, Dikic I, 2016. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. U. S. A 113, 4039–4044. 10.1073/pnas.1523926113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritch R, Darbro B, Menon G, Khanna CL, Solivan-Timpe F, Roos BR, Sarfarzi M, Kawase K, Yamamoto T, Robin AL, Lotery AJ, Fingert JH, 2014. TBK1 gene duplication and normal-tension glaucoma. JAMA Ophthalmol. 132, 544–548. 10.1001/jamaophthalmol.2014.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogelj B, Easton LE, Bogu GK, Stanton LW, Rot G, Curk T, Zupan B, Sugimoto Y, Modic M, Haberman N, Tollervey J, Fujii R, Takumi T, Shaw CE, Ule J, 2012. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci. Rep 2, 603 10.1038/srep00603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer JD, Isaacs AM, Mizielinska S, Mead S, Lashley T, Wray S, Sidle K, Fratta P, Orrell RW, Hardy J, Holton J, Revesz T, Rossor MN, Warren JD, 2015. C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. Lancet Neurol. 14, 291–301. 10.1016/S1474-4422(14)70233-9 [DOI] [PubMed] [Google Scholar]
- Romano G, Klima R, Feiguin F, 2020. TDP-43 prevents retrotransposon activation in the Drosophila motor system through regulation of Dicer-2 activity. BMC Biol. 18, 82 10.1186/s12915-020-00816-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, 1993. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. 10.1038/362059a0 [DOI] [PubMed] [Google Scholar]
- Saccon RA, Bunton-Stasyshyn RKA, Fisher EMC, Fratta P, 2013. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain J. Neurol 136, 2342–2358. 10.1093/brain/awt097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakellariou GK, Davis CS, Shi Y, Ivannikov MV, Zhang Y, Vasilaki A, Macleod GT, Richardson A, Van Remmen H, Jackson MJ, McArdle A, Brooks SV, 2014. Neuron-specific expression of CuZnSOD prevents the loss of muscle mass and function that occurs in homozygous CuZnSOD-knockout mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol 28, 1666–1681. 10.1096/fj.13-240390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sareen D, O’Rourke JG, Meera P, Muhammad AKMG, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, Gendron T, Petrucelli L, Baughn M, Ravits J, Harms MB, Rigo F, Bennett CF, Otis TS, Svendsen CN, Baloh RH, 2013. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med 5, 208ra149 10.1126/scitranslmed.3007529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasayama H, Shimamura M, Tokuda T, Azuma Y, Yoshida T, Mizuno T, Nakagawa M, Fujikake N, Nagai Y, Yamaguchi M, 2012. Knockdown of the Drosophila fused in sarcoma (FUS) homologue causes deficient locomotive behavior and shortening of motoneuron terminal branches. PloS One 7, e39483 10.1371/journal.pone.0039483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage AL, Schumann GG, Breen G, Bubb VJ, Al-Chalabi A, Quinn JP, 2019. Retrotransposons in the development and progression of amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 90, 284–293. 10.1136/jnnp-2018-319210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch KM, Miller TM, 2017. Antisense Oligonucleotides: Translation from Mouse Models to Human Neurodegenerative Diseases. Neuron 94, 1056–1070. 10.1016/j.neuron.2017.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoen M, Reichel JM, Demestre M, Putz S, Deshpande D, Proepper C, Liebau S, Schmeisser MJ, Ludolph AC, Michaelis J, Boeckers TM, 2015. Super-Resolution Microscopy Reveals Presynaptic Localization of the ALS/FTD Related Protein FUS in Hippocampal Neurons. Front. Cell. Neurosci 9, 496 10.3389/fncel.2015.00496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz PJ, Reaume A, Scott R, Coyle JT, 1998. Effects of over- and under-expression of Cu,Zn-superoxide dismutase on the toxicity of glutamate analogs in transgenic mouse striatum. Brain Res. 789, 32–39. 10.1016/s0006-8993(97)01469-8 [DOI] [PubMed] [Google Scholar]
- Schymick JC, Yang Y, Andersen PM, Vonsattel JP, Greenway M, Momeni P, Elder J, Chio A, Restagno G, Robberecht W, Dahlberg C, Mukherjee O, Goate A, Graff-Radford N, Caselli RJ, Hutton M, Gass J, Cannon A, Rademakers R, Singleton AB, Hardiman O, Rothstein J, Hardy J, Traynor BJ, 2006. Progranulin mutations and amyotrophic lateral sclerosis or amyotrophic lateral sclerosis-frontotemporal dementia phenotypes. J. Neurol. Neurosurg. Psychiatry 78, 754–756. 10.1136/jnnp.2006.109553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoles DR, Meera P, Schneider MD, Paul S, Dansithong W, Figueroa KP, Hung G, Rigo F, Bennett CF, Otis TS, Pulst SM, 2017. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 544, 362–366. 10.1038/nature22044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, Campanari M-L, Julie Corbier C, Gaucherot A, Kolb-Cheynel I, Oulad-Abdelghani M, Ruffenach F, Page A, Ciura S, Kabashi E, Charlet-Berguerand N, 2016. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 35, 1276–1297. 10.15252/embj.201593350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj BT, Livesey MR, Zhao C, Gregory JM, James OT, Cleary EM, Chouhan AK, Gane AB, Perkins EM, Dando O, Lillico SG, Lee Y-B, Nishimura AL, Poreci U, Thankamony S, Pray M, Vasistha NA, Magnani D, Borooah S, Burr K, Story D, McCampbell A, Shaw CE, Kind PC, Aitman TJ, Whitelaw CBA, Wilmut I, Smith C, Miles GB, Hardingham GE, Wyllie DJA, Chandran S, 2018. C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca2+-permeable AMPA receptor-mediated excitotoxicity. Nat. Commun 9, 347 10.1038/s41467-017-02729-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalom O, Shalva N, Altschuler Y, Motro B, 2008. The mammalian Nek1 kinase is involved in primary cilium formation. FEBS Lett. 582, 1465–1470. 10.1016/j.febslet.2008.03.036 [DOI] [PubMed] [Google Scholar]
- Shao Q, Liang C, Chang Q, Zhang W, Yang M, Chen J-F, 2019. C9orf72 deficiency promotes motor deficits of a C9ALS/FTD mouse model in a dose-dependent manner. Acta Neuropathol. Commun 7, 32 10.1186/s40478-019-0685-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Lyashchenko AK, Lu L, Nasrabady SE, Elmaleh M, Mendelsohn M, Nemes A, Tapia JC, Mentis GZ, Shneider NA, 2016. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun 7, 10465 10.1038/ncomms10465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W-C, Li H-Y, Chen G-C, Chern Y, Tu P-H, 2015. Mutations in the ubiquitin-binding domain of OPTN/optineurin interfere with autophagy-mediated degradation of misfolded proteins by a dominant-negative mechanism. Autophagy 11, 685–700. 10.4161/auto.36098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Hung S-T, Rocha G, Lin S, Linares GR, Staats KA, Seah C, Wang Y, Chickering M, Lai J, Sugawara T, Sagare AP, Zlokovic BV, Ichida JK, 2019. Identification and therapeutic rescue of autophagosome and glutamate receptor defects in C9ORF72 and sporadic ALS neurons. JCI Insight 5 10.1172/jci.insight.127736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Ivannikov MV, Walsh ME, Liu Y, Zhang Y, Jaramillo CA, Macleod GT, Van Remmen H, 2014. The lack of CuZnSOD leads to impaired neurotransmitter release, neuromuscular junction destabilization and reduced muscle strength in mice. PloS One 9, e100834 10.1371/journal.pone.0100834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lin S, Staats KA, Li Y, Chang W-H, Hung S-T, Hendricks E, Linares GR, Wang Y, Son EY, Wen X, Kisler K, Wilkinson B, Menendez L, Sugawara T, Woolwine P, Huang M, Cowan MJ, Ge B, Koutsodendris N, Sandor KP, Komberg J, Vangoor VR, Senthilkumar K, Hennes V, Seah C, Nelson AR, Cheng T-Y, Lee S-JJ, August PR, Chen JA, Wisniewski N, Hanson-Smith V, Belgard TG, Zhang A, Coba M, Grunseich C, Ward ME, van den Berg LH, Pasterkamp RJ, Trotti D, Zlokovic BV, Ichida JK, 2018. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med 24, 313–325. 10.1038/nm.4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiihashi G, Ito D, Yagi T, Nihei Y, Ebine T, Suzuki N, 2016. Mislocated FUS is sufficient for gain-of-toxic-function amyotrophic lateral sclerosis phenotypes in mice. Brain J. Neurol 139, 2380–2394. 10.1093/brain/aww161 [DOI] [PubMed] [Google Scholar]
- Sippl C, Bosserhoff AK, Fischer D, Tamm ER, 2011. Depletion of optineurin in RGC-5 cells derived from retinal neurons causes apoptosis and reduces the secretion of neurotrophins. Exp. Eye Res 93, 669–680. 10.1016/j.exer.2011.08.011 [DOI] [PubMed] [Google Scholar]
- Sirohi K, Chalasani MLS, Sudhakar C, Kumari A, Radha V, Swarup G, 2013. M98K-OPTN induces transferrin receptor degradation and RAB12-mediated autophagic death in retinal ganglion cells. Autophagy 9, 510–527. 10.4161/auto.23458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleegers K, Brouwers N, Maurer-Stroh S, van Es MA, Damme PV, van Vught PWJ, van der Zee J, Serneels S, Pooter TD, Van den Broeck M, Cruts M, Schymkowitz J, Jonghe PD, Rousseau F, van den Berg LH, Robberecht W, Broeckhoven CV, 2008. Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology 71, 253–259. 10.1212/01.wnl.0000289191.54852.75 [DOI] [PubMed] [Google Scholar]
- Slowicka K, Vereecke L, van Loo G, 2016. Cellular Functions of Optineurin in Health and Disease. Trends Immunol. 37, 621–633. 10.1016/j.it.2016.07.002 [DOI] [PubMed] [Google Scholar]
- Smith RA, Miller TM, Yamanaka K, Monia BP, Condon TP, Hung G, Lobsiger CS, Ward CM, McAlonis-Downes M, Wei H, Wancewicz EV, Bennett CF, Cleveland DW, 2006. Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Invest 116, 2290–2296. 10.1172/JCI25424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- So E, Mitchell JC, Memmi C, Chennell G, Vizcay-Barrena G, Allison L, Shaw CE, Vance C, 2018. Mitochondrial abnormalities and disruption of the neuromuscular junction precede the clinical phenotype and motor neuron loss in hFUSWT transgenic mice. Hum. Mol. Genet 27, 463–474. 10.1093/hmg/ddx415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC, 2001. A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J. Biol. Chem 276, 38084–38089. 10.1074/jbc.M105296200 [DOI] [PubMed] [Google Scholar]
- Sudria-Lopez E, Koppers M, de Wit M, van der Meer C, Westeneng H-J, Zundel CAC, Youssef SA, Harkema L, de Bruin A, Veldink JH, van den Berg LH, Pasterkamp RJ, 2016. Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta Neuropathol. (Berl.) 132, 145–147. 10.1007/s00401-016-1581-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PM, Zhou X, Robins AM, Paushter DH, Kim D, Smolka MB, Hu F, 2016. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun 4, 51 10.1186/s40478-016-0324-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N, Maroof AM, Merkle FT, Koszka K, Intoh A, Armstrong I, Moccia R, Davis-Dusenbery BN, Eggan K, 2013. The mouse C9ORF72 ortholog is enriched in neurons known to degenerate in ALS and FTD. Nat. Neurosci 16, 1725–1727. 10.1038/nn.3566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen B, Bento-Abreu A, Gendron TF, Boeynaems S, Bogaert E, Nuyts R, Timmers M, Scheveneels W, Hersmus N, Wang J, Mizielinska S, Isaacs AM, Petrucelli L, Lemmens R, Van Damme P, Van Den Bosch L, Robberecht W, 2018. A zebrafish model for C9orf72 ALS reveals RNA toxicity as a pathogenic mechanism. Acta Neuropathol. (Berl.) 135, 427–443. 10.1007/s00401-017-1796-5 [DOI] [PubMed] [Google Scholar]
- Sztainberg Y, Chen H, Swann JW, Hao S, Tang B, Wu Z, Tang J, Wan Y-W, Liu Z, Rigo F, Zoghbi HY, 2015. Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides. Nature 528, 123–126. 10.1038/nature16159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan AY, Manley JL, 2010. TLS inhibits RNA polymerase III transcription. Mol. Cell. Biol 30, 186–196. 10.1128/MCB.00884-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Q, Yalamanchili HK, Park J, Maio A, Lu HC, Wan YW, White JJ, Bondar V, Sayegh LS, Liu X, Gao Y, Sillitoe R, Orr HT, Liu Z, Zoghbi HY, 2016. Extensive cryptic splicing upon loss of RBM17 and TDP43 in neurodegeneration models. Hum. Mol. Genet 10.1093/hmg/ddw337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Chen ZJ, 2012. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal 5, ra20 10.1126/scisignal.2002521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Matsuwaki T, Yamanouchi K, Nishihara M, 2013. Increased lysosomal biogenesis in activated microglia and exacerbated neuronal damage after traumatic brain injury in progranulin-deficient mice. Neuroscience 250, 8–19. 10.1016/j.neuroscience.2013.06.049 [DOI] [PubMed] [Google Scholar]
- Tao Z, Wang H, Xia Q, Li Ke, Li Kai, Jiang X, Xu G, Wang G, Ying Z, 2015. Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum. Mol. Genet 24, 2426–2441. 10.1093/hmg/ddv005 [DOI] [PubMed] [Google Scholar]
- Taylor JP, Brown RH, Cleveland DW, 2016. Decoding ALS: from genes to mechanism. Nature 539, 197–206. 10.1038/nature20413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston TL, Boyle KB, Allen M, Ravenhill BJ, Karpiyevich M, Bloor S, Kaul A, Noad J, Foeglein A, Matthews SA, Komander D, Bycroft M, Randow F, 2016. Recruitment of TBK1 to cytosol-invading Salmonella induces WIPI2-dependent antibacterial autophagy. EMBO J. 35, 1779–1792. 10.15252/embj.201694491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, König J, Hortobágyi T, Nishimura AL, Župunski V, Patani R, Chandran S, Rot G, Zupan B, Shaw CE, Ule J, 2011. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Publ. Group 14 10.1038/nn.2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth RP, Atkin JD, 2018. Dysfunction of Optineurin in Amyotrophic Lateral Sclerosis and Glaucoma. Front. Immunol 9, 1017 10.3389/fimmu.2018.01017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tümer Z, Bertelsen B, Gredal O, Magyari M, Nielsen KC, Lucamp null, Grønskov K, Brøndum-Nielsen K, 2012. Novel heterozygous nonsense mutation of the OPTN gene segregating in a Danish family with ALS. Neurobiol. Aging 33, 208.e1–5. 10.1016/j.neurobiolaging.2011.07.001 [DOI] [PubMed] [Google Scholar]
- Turner MR, Goldacre R, Ramagopalan S, Talbot K, Goldacre MJ, 2013. Autoimmune disease preceding amyotrophic lateral sclerosis: an epidemiologic study. Neurology 81, 1222–1225. 10.1212/WNL.0b013e3182a6cc13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udagawa T, Fujioka Y, Tanaka M, Honda D, Yokoi S, Riku Y, Ibi D, Nagai T, Yamada K, Watanabe H, Katsuno M, Inada T, Ohno K, Sokabe M, Okado H, Ishigaki S, Sobue G, 2015. FUS regulates AMPA receptor function and FTLD/ALS-associated behaviour via GluA1 mRNA stabilization. Nat. Commun 6, 7098 10.1038/ncomms8098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Blitterswijk M, DeJesus-Hernandez M, Niemantsverdriet E, Murray ME, Heckman MG, Diehl NN, Brown PH, Baker MC, Finch NA, Bauer PO, Serrano G, Beach TG, Josephs KA, Knopman DS, Petersen RC, Boeve BF, Graff-Radford NR, Boylan KB, Petrucelli L, Dickson DW, Rademakers R, 2013. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol. 12, 978–988. 10.1016/S1474-4422(13)70210-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Blitterswijk M, van Vught PWJ, van Es MA, Schelhaas HJ, van der Kooi AJ, de Visser M, Veldink JH, van den Berg LH, 2012. Novel optineurin mutations in sporadic amyotrophic lateral sclerosis patients. Neurobiol. Aging 33, 1016.e1–7. 10.1016/j.neurobiolaging.2011.05.019 [DOI] [PubMed] [Google Scholar]
- van Doormaal PTC, Gallo A, van Rheenen W, Veldink JH, van Es MA, van den Berg LH, 2013. Amyotrophic lateral sclerosis is not linked to multiple sclerosis in a population based study. J. Neurol. Neurosurg. Psychiatry 84, 940–941. 10.1136/jnnp-2012-304864 [DOI] [PubMed] [Google Scholar]
- Van Langenhove T, van der Zee J, Sleegers K, Engelborghs S, Vandenberghe R, Gijselinck I, Van den Broeck M, Mattheijssens M, Peeters K, De Deyn PP, Cruts M, Van Broeckhoven C, 2010. Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology 74, 366–371. 10.1212/WNL.0b013e3181ccc732 [DOI] [PubMed] [Google Scholar]
- van Wijk SJL, Fricke F, Herhaus L, Gupta J, Hötte K, Pampaloni F, Grumati P, Kaulich M, Sou Y-S, Komatsu M, Greten FR, Fulda S, Heilemann M, Dikic I, 2017. Linear ubiquitination of cytosolic Salmonella Typhimurium activates NF-κB and restricts bacterial proliferation. Nat. Microbiol 2, 17066 10.1038/nmicrobiol.2017.66 [DOI] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo J-M, Miller CC, Shaw CE, 2009. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. 10.1126/science.1165942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas JNS, Wang C, Bunker E, Hao L, Maric D, Schiavo G, Randow F, Youle RJ, 2019. Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol. Cell 74, 347–362.e6. 10.1016/j.molcel.2019.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Dreyfuss G, 2017. Splicing-Correcting Therapy for SMA. Cell 170, 5 10.1016/j.cell.2017.06.028 [DOI] [PubMed] [Google Scholar]
- Wang J-W, Brent JR, Tomlinson A, Shneider NA, McCabe BD, 2011. The ALS-associated proteins FUS and TDP-43 function together to affect Drosophila locomotion and life span. J. Clin. Invest 121, 4118–4126. 10.1172/JCI57883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Gu L, Meng D, Wu Q, Deng H, Pan J, 2017. Comparative Proteomics Reveals Timely Transport into Cilia of Regulators or Effectors as a Mechanism Underlying Ciliary Disassembly. J. Proteome Res 16, 2410–2418. 10.1021/acs.jproteome.6b01048 [DOI] [PubMed] [Google Scholar]
- Wang X, Arai S, Song X, Reichart D, Du K, Pascual G, Tempst P, Rosenfeld MG, Glass CK, Kurokawa R, 2008. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 454, 126–130. 10.1038/nature06992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Horemuzova E, Iida A, Guo L, Liu Y, Matsumoto N, Nishimura G, Nordgren A, Miyake N, Tham E, Grigelioniene G, Ikegawa S, 2017. Axial spondylometaphyseal dysplasia is also caused by NEK1 mutations. J. Hum. Genet 62, 503–506. 10.1038/jhg.2016.157 [DOI] [PubMed] [Google Scholar]
- Wang Z, Iida A, Miyake N, Nishiguchi KM, Fujita K, Nakazawa T, Alswaid A, Albalwi MA, Kim O-H, Cho T-J, Lim G-Y, Isidor B, David A, Rustad CF, Merckoll E, Westvik J, Stattin E-L, Grigelioniene G, Kou I, Nakajima M, Ohashi H, Smithson S, Matsumoto N, Nishimura G, Ikegawa S, 2016. Axial Spondylometaphyseal Dysplasia Is Caused by C21orf2 Mutations. PloS One 11, e0150555 10.1371/journal.pone.0150555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster CP, Smith EF, Bauer CS, Moller A, Hautbergue GM, Ferraiuolo L, Myszczynska MA, Higginbottom A, Walsh MJ, Whitworth AJ, Kaspar BK, Meyer K, Shaw PJ, Grierson AJ, De Vos KJ, 2016. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 35, 1656–1676. 10.15252/embj.201694401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, Lin S, Shneider NA, Monaghan J, Pandey UB, Pasinelli P, Ichida JK, Trotti D, 2014. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84, 1213–1225. 10.1016/j.neuron.2014.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheway G, Schmidts M, Mans DA, Szymanska K, Nguyen T-MT, Racher H, Phelps IG, Toedt G, Kennedy J, Wunderlich KA, Sorusch N, Abdelhamed ZA, Natarajan S, Herridge W, van Reeuwijk J, Horn N, Boldt K, Parry DA, Letteboer SJF, Roosing S, Adams M, Bell SM, Bond J, Higgins J, Morrison EE, Tomlinson DC, Slaats GG, van Dam TJP, Huang L, Kessler K, Giessl A, Logan CV, Boyle EA, Shendure J, Anazi S, Aldahmesh M, Al Hazzaa S, Hegele RA, Ober C, Frosk P, Mhanni AA, Chodirker BN, Chudley AE, Lamont R, Bernier FP, Beaulieu CL, Gordon P, Pon RT, Donahue C, Barkovich AJ, Wolf L, Toomes C, Thiel CT, Boycott KM, McKibbin M, Inglehearn CF, UK10K Consortium, University of Washington Center for Mendelian Genomics, Stewart F, Omran H, Huynen MA, Sergouniotis PI, Alkuraya FS, Parboosingh JS, Innes AM, Willoughby CE, Giles RH, Webster AR, Ueffing M, Blacque O, Gleeson JG, Wolfrum U, Beales PL, Gibson T, Doherty D, Mitchison HM, Roepman R, Johnson CA, 2015. An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat. Cell Biol 17, 1074–1087. 10.1038/ncb3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dötsch V, Bumann D, Dikic I, 2011. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333, 228–233. 10.1126/science.1205405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL, 1995. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14, 1105–1116. 10.1016/0896-6273(95)90259-7 [DOI] [PubMed] [Google Scholar]
- Wong YC, Holzbaur ELF, 2014. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. U. S. A 111, E4439–4448. 10.1073/pnas.1405752111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MJA, Talbot K, Bowerman M, 2017. Spinal muscular atrophy: antisense oligonucleotide therapy opens the door to an integrated therapeutic landscape. Hum. Mol. Genet 26, R151–R159. 10.1093/hmg/ddx215 [DOI] [PubMed] [Google Scholar]
- Wu L-S, Cheng W-C, Chen C-Y, Wu M-C, Wang Y-C, Tseng Y-H, Chuang T-J, Shen C-KJ, 2019. Transcriptomopathies of pre- and post-symptomatic frontotemporal dementia-like mice with TDP-43 depletion in forebrain neurons. Acta Neuropathol. Commun 7, 50 10.1186/s40478-019-0674-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia R, Liu Y, Yang L, Gal J, Zhu H, Jia J, 2012. Motor neuron apoptosis and neuromuscular junction perturbation are prominent features in a Drosophila model of Fus-mediated ALS. Mol. Neurodegener 7, 10 10.1186/1750-1326-7-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Zou Q, Xie X, Liu T, Li HS, Jie Z, Jin J, Hu H, Manyam G, Zhang L, Cheng X, Wang H, Marie I, Levy DE, Watowich SS, Sun S-C, 2017. The kinase TBK1 functions in dendritic cells to regulate T cell homeostasis, autoimmunity, and antitumor immunity. J. Exp. Med 214, 1493–1507. 10.1084/jem.20161524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Jin T, Zhu H, Chen H, Ofengeim D, Zou C, Mifflin L, Pan L, Amin P, Li W, Shan B, Naito MG, Meng H, Li Y, Pan H, Aron L, Adiconis X, Levin JZ, Yankner BA, Yuan J, 2018. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell 174, 1477–1491.e19. 10.1016/j.cell.2018.07.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, Li H, Hales CM, Gearing M, Wingo TS, Jin P, 2013. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc. Natl. Acad. Sci. U. S. A 110, 7778–7783. 10.1073/pnas.1219643110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Wang H, Qiao T, Yang B, Aliaga L, Qiu L, Tan W, Salameh J, McKenna-Yasek DM, Smith T, Peng L, Moore MJ, Brown RH, Cai H, Xu Z, 2014. Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U. S. A 111, E1121–9. 10.1073/pnas.1322641111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Liang C, Swaminathan K, Herrlinger S, Lai F, Shiekhattar R, Chen J-F, 2016. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci. Adv 2, e1601167 10.1126/sciadv.1601167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda K, Zhang H, Loiselle D, Haystead T, Macara IG, Mili S, 2013. The RNA-inding protein Fus directs translation of localized mRNAs in APC-RNP granules. J. Cell Biol 203, 737–746. 10.1083/jcb.201306058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi S, Udagawa T, Fujioka Y, Honda D, Okado H, Watanabe H, Katsuno M, Ishigaki S, Sobue G, 2017. 3’UTR Length-Dependent Control of SynGAP Isoform α2 mRNA by FUS and ELAV-like Proteins Promotes Dendritic Spine Maturation and Cognitive Function. Cell Rep. 20, 3071–3084. 10.1016/j.celrep.2017.08.100 [DOI] [PubMed] [Google Scholar]
- Zachari M, Gudmundsson SR, Li Z, Manifava M, Shah R, Smith M, Stronge J, Karanasios E, Piunti C, Kishi-Itakura C, Vihinen H, Jokitalo E, Guan J-L, Buss F, Smith AM, Walker SA, Eskelinen E-L, Ktistakis NT, 2019. Selective Autophagy of Mitochondria on a Ubiquitin-Endoplasmic-Reticulum Platform. Dev. Cell 50, 627–643.e5. 10.1016/j.devcel.2019.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavolan M, Kondo S, Schönbach C, Adachi J, Hume DA, Arakawa T, Carninci P, Kawai J, Hayashizaki Y, Gaasterland T, 2003. Impact of alternative initiation, splicing, and termination on the diversity of the mRNA transcripts encoded by the mouse transcriptome. 10.1101/gr.1017303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Shang G, Gui X, Zhang X, Bai X-C, Chen ZJ, 2019. Structural basis of STING binding with and phosphorylation by TBK1. Nature 567, 394–398. 10.1038/s41586-019-1000-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y-J, Gendron TF, Ebbert MTW, O’Raw AD, Yue M, Jansen-West K, Zhang X, Prudencio M, Chew J, Cook CN, Daughrity LM, Tong J, Song Y, Pickles SR, Castanedes-Casey M, Kurti A, Rademakers R, Oskarsson B, Dickson DW, Hu W, Gitler AD, Fryer JD, Petrucelli L, 2018. Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat. Med 24, 1136–1142. 10.1038/s41591-018-0071-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Guo L, Gonzales PK, Gendron TF, Wu Y, Jansen-West K, O’Raw AD, Pickles SR, Prudencio M, Carlomagno Y, Gachechiladze MA, Ludwig C, Tian R, Chew J, DeTure M, Lin WL, Tong J, Daughrity LM, Yue M, Song Y, Andersen JW, Castanedes-Casey M, Kurti A, Datta A, Antognetti G, McCampbell A, Rademakers R, Oskarsson B, Dickson DW, Kampmann M, Ward ME, Fryer JD, Link CD, Shorter J, Petrucelli L, 2019. Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science. 10.1126/science.aav2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Liu S, Liu G, Oztürk A, Hicks GG, 2013. ALS-associated FUS mutations result in compromised FUS alternative splicing and autoregulation. PLoS Genet. 9, e1003895 10.1371/journal.pgen.1003895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Jiang J, Gendron TF, McAlonis-Downes M, Jiang L, Taylor A, Diaz Garcia S, Ghosh Dastidar S, Rodriguez MJ, King P, Zhang Y, La Spada AR, Xu H, Petrucelli L, Ravits J, Da Cruz S, Lagier-Tourenne C, Cleveland DW, 2020. Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat. Neurosci 23, 615–624. 10.1038/s41593-020-0619-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H, Sok J, Immanuel D, Yin Y, Ron D, 1997. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J. Cell Sci 110 (Pt 15), 1741–1750. [DOI] [PubMed] [Google Scholar]
- Zou Z-Y, Zhou Z-R, Che C-H, Liu C-Y, He R-L, Huang H-P, 2017. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 88, 540–549. 10.1136/jnnp-2016-315018 [DOI] [PubMed] [Google Scholar]
- Zu T, Liu Y, Bañez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH, Ostrow LW, Rothstein JD, Troncoso JC, Ranum LPW, 2013. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. U. S. A 110, E4968–4977. 10.1073/pnas.1315438110 [DOI] [PMC free article] [PubMed] [Google Scholar]