

Abstract
Background
The mutated α‐B‐Crystallin (CryABR120G) mouse model of desmin‐related myopathy (DRM) shows an age‐dependent onset of pathologic cardiac remodeling and progression of heart failure. CryABR120G expression in cardiomyocytes affects the mitochondrial spatial organization within the myofibrils, but the molecular perturbation within the mitochondria in the relation of the overall course of the proteotoxic disease remains unclear.
Methods and Results
CryABR120G mice show an accumulation of electron‐dense aggregates and myofibrillar degeneration associated with the development of cardiac dysfunction. Though extensive studies demonstrated that these altered ultrastructural changes cause cardiac contractility impairment, the molecular mechanism of cardiomyocyte death remains elusive. Here, we explore early pathological processes within the mitochondria contributing to the contractile dysfunction and determine the pathogenic basis for the heart failure observed in the CryABR120G mice. In the present study, we report that the CryABR120G mice transgenic hearts undergo altered mitochondrial dynamics associated with increased level of dynamin‐related protein 1 and decreased level of optic atrophy type 1 as well as mitofusin 1 over the disease process. In association with these changes, an altered level of the components of mitochondrial oxidative phosphorylation and pyruvate dehydrogenase complex regulatory proteins occurs before the manifestation of pathologic adverse remodeling in the CryABR120G hearts. Mitochondria isolated from CryABR120G transgenic hearts without visible pathology show decreased electron transport chain complex activities and mitochondrial respiration. Taken together, we demonstrated the involvement of mitochondria in the pathologic remodeling and progression of DRM‐associated cellular dysfunction.
Conclusions
Mitochondrial dysfunction in the form of altered mitochondrial dynamics, oxidative phosphorylation and pyruvate dehydrogenase complex proteins level, abnormal electron transport chain complex activities, and mitochondrial respiration are evident on the CryABR120G hearts before the onset of detectable pathologies and development of cardiac contractile dysfunction.
Keywords: desmin‐related myopathy, mitochondrial dynamics, mitochondrial respiration, oxidative phosphorylation, R120G‐αB‐crystallin
Subject Categories: Basic Science Research, Myocardial Biology, Pathophysiology, Mechanisms, Cell Biology/Structural Biology
Nonstandard Abbreviations and Acronyms
- BN‐PAGE
blue native polyacrylamide gel electrophoresis
- CryAB
α‐B‐crystallin
- CryABR120G
Arg120Gly missense mutation in CryAB
- CS
citrate synthase
- DRM
desmin‐related myopathy
- Drp1
dynamin‐related protein 1
- E1
pyruvate dehydrogenase
- E2
dihydrolipoamide transacetylase
- E3
dihydrolipoamide dehydrogenase
- ETC
electron transport chain
- FCCP
carbonyl cyanide‐p‐trifluoromethoxy‐phenylhydrazone
- HD
Huntington disease
- MFN1
mitofusin 1
- MFN2
mitofusin 2
- OCR
oxygen consumption rates
- OPA1
optic atrophy type 1
- OXPHOS
oxidative phosphorylation system
- PDH
pyruvate dehydrogenase complex
Clinical Perspective
What Is New?
A missense mutation in α‐B‐crystallin (R120G) causes desmin‐related cardiomyopathy, which is a myofibrillar protein aggregation disease characterized by abnormal accumulations of desmin and α‐B‐crystallin within muscle fibers; the proteotoxic sequelae leading to the development of cardiomyopathy in the CryABR120G model are unclear.
In the present study, we aim to determine the time point of the onset of impairment in mitochondrial dynamics and oxidative phosphorylation that ultimately contribute to the CryABR120G‐associated cardiomyopathy.
Here, we demonstrate that altered level of mitochondrial dynamics, oxidative phosphorylation system, and pyruvate dehydrogenase complex complex regulatory proteins and resultant mitochondrial respiratory dysfunction are the early events ultimately contributing to the development of cardiomyopathy.
What Are the Clinical Implications?
Desmin‐related myopathy pathogenesis is associated with the accumulation of electron‐dense aggregates affecting the derangement of the mitochondrial spatial organization, but the pathogenic molecular sequelae remain obscure.
Despite extensive studies, the relationship between maladaptive changes in mitochondrial structure and function to the development of desmin‐related myopathy associated cardiomyopathy remains unknown.
Our study showed that alterations in mitochondria dynamics and defects in oxidative phosphorylation appears in CryABR120G hearts before the onset of other detectable pathologies and compromised cardiac systolic contractile function; these findings suggesting a causal role for altered mitochondrial function in DRM pathogenesis and shed light on the molecular mechanism of desmin‐related myopathy‐associated cell death.
Cardiomyopathies are a heterogeneous group of progressive diseases of the myocardium, or heart muscle, often leading to progressive heart failure (HF) with significant morbidity and mortality. 1 Cardiomyopathies are classified according to their morphological and clinical symptoms into dilated cardiomyopathy, hypertrophic cardiomyopathy, restrictive cardiomyopathy, and arrhythmogenic right ventricular cardiomyopathy. Mutations in the cytoskeletal proteins and proteins associated with them can be causative in different forms of cardiomyopathies because these proteins function in structural, sensor, and signaling roles in the healthy and diseased cardiomyocyte. 2 One such genetically heterogeneous group of disorders is the desmin‐related myopathy (DRM), which is a rare heritable cardiac and skeletal muscle disease characterized by skeletal muscle weakness, conduction disturbance, and dilated cardiomyopathy. 3 Mutations in desmin, α‐B‐crystallin (CryAB), and other desmin‐related genes result in DRM leading to intracellular accumulation of misfolded protein and production of soluble pre‐amyloid oligomers, resulting in weakened skeletal and cardiac muscle. 4
CryAB, a member of the small heat shock protein family comprising 1% to 3% of total soluble cardiac protein, 5 , 6 , 7 binds desmin and cytoplasmic actin and acts as a chaperone for the protection of intermediate filaments in the heart. 5 An Arg120Gly missense mutation in CryAB (CryABR120G) was initially identified in a French family as causing DRM. 8 Patients with DRM with the CryABR120G mutation also develop hypertrophic, dilated, and restrictive cardiomyopathies. 9 The structure–function relationship of CryAB has been confirmed and further studied in cell culture and genetically modified animal models. 10 , 11 , 12 The cardiac‐specific transgenic mice expressing CryABR120G showed the accumulation of pre‐amyloid oligomer–positive aggregates, followed by progressive cardiomyocyte loss, cardiac dysfunction, and HF. Though a major histopathological feature of DRM is the accumulation of desmin‐positive aggregates caused by mutations in desmin or desmin‐interacting proteins, 13 , 14 DRM‐causing mutations are not the only cause of desmin aggregation in the heart. 13 Studies reported the accumulation of desmin‐positive pre‐amyloid oligomers and amyloid fibrils in small and large animal models of HF (HF) and in human tissue specimens in the absence of genetic mutations. 15 , 16 Moreover, studies also suggested the amyloidogenic properties of wild‐type desmin in skeletal muscle where desmin misfolding can propagate in a prion‐like fashion. 17 Overall, the pre‐amyloid oligomer accumulations are toxic for cells and membranous organelles, thus adversely affecting cellular energetics and leading to decreased cardiac contractile function observed with HF. 13 Increasing the clearance/degradation of these toxic protein aggregates by upregulation of small heat shock proteins, 18 activation of proteasomes, 19 stimulation of autophagy, 20 , 21 , 22 and exercise 20 are individually sufficient to attenuate and/or delayed the onset of the morphological and functional pathologies, increasing the probability of survival into midadulthood. Despite all these studies to treat DRM pathologies, studies were partially able to delay the progression of the disease, because the pathogenic molecular sequelae remain obscure.
In this study, we explored early pathological processes involved in contractile dysfunction and determined the pathogenic basis for the HF observed in the CryABR120G mice. The integrity of the cytoskeleton in both skeletal and cardiac muscles rigidly controls the mitochondrial organization, as DRM‐causing mutations result in early perturbations in mitochondrial‐sarcomere architecture and function. In DRM mice expressing either mutant desmin or CryABR120G, electron‐dense aggregates intersperse the myofibril, resulting in disruptions in mitochondrial spatial organization. Similarly, tissue biopsy samples from patients diagnosed with myofibrillar myopathies showed alterations in mitochondrial morphology and positioning, abnormal mitochondrial enzyme staining, and have reduced mitochondrial complex activities. 23 Despite extensive studies, the relationship between maladaptive changes in mitochondria to the development of cardiomyopathies in DRM remains unknown. In the present study, we aim to define the time point of the onset of impairment in mitochondrial dynamics and mitochondrial function that ultimately contribute to the CryABR120G‐associated cardiomyopathy. Here, we demonstrate that altered expression of regulatory proteins for mitochondrial dynamics and oxidative phosphorylation system (OXPHOS) and dysfunction of mitochondrial respiration are early events that ultimately contribute to the development of cardiomyopathy.
Methods
Data Availability Disclosure Statement
The authors declare that all supporting data and method descriptions are available within the article or from the corresponding author upon reasonable request.
Materials
Materials are as follows: Cell Lytic M (Sigma‐Aldrich), Protease Inhibitor Cocktail (Roche), pre‐cast 7.5% to 15% Criterion Gels (BioRad), Oligomycin (Sigma‐Aldrich), carbonyl cyanide‐p‐trifluoromethoxy‐phenylhydrazone (FCCP) (Sigma‐Aldrich), Rotenone (Sigma‐Aldrich), Antimycin A (Sigma‐Aldrich), and Ponceau S (Acros Organic) were used.
Animals
CryABR120G transgenic mice have been described previously and are in an FVB/N background. 20 All procedures for handling animals complied with the Guide for Care and Use of Laboratory Animals and were approved by the ACUC Committee of LSU Health Sciences Center‐Shreveport. All animals were cared for according to the National Institutes of Health guidelines for the care and use of laboratory animals.
Echocardiography
Echocardiograms were performed on isoflurane‐anesthetized mice with a VisualSonics Vevo 3100 Imaging System to assess cardiac functional parameters as described previously. 24 , 25 , 26 Briefly, we recorded the M‐mode echocardiographic images along the parasternal short axis by investigators blinded to genotype to determine left ventricular (LV) size and systolic function. M‐mode measurements included the LV internal dimensions in systole and diastole (LVID;s and LVID;d, respectively) as well as the diastolic thickness of LV posterior wall, and diastolic intraventricular septum thickness. Percent fractional shortening was calculated using: [(LVIDd‐LVIDs)/LVIDd] × 100. From these data, LV end‐systolic, LV end‐diastolic diameter, LV mass, and percent ejection fraction were calculated. Systolic shortening of the interventricular septum thickness and posterior wall was calculated using: [(systolic thickness‐diastolic thickness)/systolic thickness] × 100.
Mitochondria Isolation
Mitochondria from the hearts of CryABR120G transgenic and nontransgenic mice were isolated, as described previously. 24 , 25 , 26 Briefly, hearts were harvested, homogenized in MS‐EGTA buffer (225 mM mannitol, 75 mM sucrose, 5 mM HEPES, and 1 mM EGTA, pH 7.4) and subjected to differential centrifugation. Finally, mitochondria were lysed with 1x cell lytic M (Sigma‐Aldrich) containing protease and phosphatase inhibitors.
Mitochondrial Respiration
Mitochondrial oxygen consumption rate was measured with an XF24 Extracellular Flux Analyzer (Seahorse Biosciences, North Billerica, MA) by methods as described previously. 24 , 25 , 26 Mitochondria were isolated from the mix‐sexed age‐matched CryABR120G and nontransgenic hearts using MS‐EGTA buffer (225 mM mannitol, 75 mM sucrose, 5 mM HEPES, and 1 mM EGTA, pH 7.4) by differential centrifugation as described above. Mitochondria (50 μg/well) were seeded in XF24 culture plates, and respiration was measured in mitochondrial assay buffer (220 mM mannitol, 7M mM sucrose, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA, 0.2% fatty acid‐free bovine serum albumin, pH 7.4) supplemented with 7 mM pyruvate and 1 mM malate. Mitochondrial oxygen consumption rate (OCR) was measured and plotted at basal conditions followed by sequential addition of 1 μg/mL oligomycin (ATP‐synthase inhibitor), 4 μM FCCP (a mitochondrial uncoupler), and 0.5 μM rotenone (complex I inhibitor) plus 0.5 μM antimycin A (complex III inhibitor). The OCR values were normalized to total protein content in the corresponding wells and expressed as pmol/min per µg protein.
electron transport chain complex activity assays
Mitochondrial electron transport chain complex activities were measured in isolated mitochondria spectrophotometrically as described previously. 25 , 26 Briefly, complex I activity was determined by measuring NADH (100 µmol/L) oxidation with and without the presence of complex I inhibitor (rotenone, 10 µmol/L) at 340 nm using a DS‐11 FX+spectrophotometer (DeNovix). Specific complex I activity was calculated as rotenone sensitive activity expressed as nmol min−1 mg−1 of mitochondrial protein. Complex II activity was determined by measuring the reduction of 2,6‐dichlorophenolindophenol (DCPIP) coupled to complex II catalyzed reduction of decylubiquinone with and without the presence of 10 µmol/L antimycin A, 10 µmol/L rotenone, 250 µmol/L KCN, 80 µmol/L DCPIP and 60 µmol/L decylqubiquinone. Complex III activity was determined by measuring the reduction of cytochrome C (75 µmol/L) with and without complex III inhibitor (antimycin A, 10 µmol/L). Specific complex III activity was calculated as antimycin A sensitive activity expressed as nmol min−1 mg−1 of mitochondrial protein. Extinction coefficient values for NADH (8 mM‐1 cm‐1), DCPIP (19.1 mM−1 cm−1), and reduced cytochrome C (18.5 mM‐1 cm‐1) were used to calculate complex I, II, and III activities, respectively.
Immunofluorescence microscopy
Paraffin‐embedded heart sections were used for immunofluorescence analyses as we described previously. 20 The following primary antibodies were used: mouse anti–troponin I (1:1000; Millipore), rabbit anti‐CryAB (1:400; Assay Designs). Alexa Fluor 488– or Alexa Fluor 568–conjugated secondary antibody (Molecular Probes) directed against mouse or rabbit IgG was used, and 4′,6‐diamidino‐2‐phenylindole (Invitrogen) was used to identify nuclei. Immunofluorescent staining for CryAB was used to quantitate the cytoplasmic area occupied by the aggregates, as described previously. 20
Cell fractionation, SDS‐PAGE, and Western blot analyses
Soluble and insoluble fractions of heart lysate were prepared by methods as we described previously. 20 , 26 Briefly, hearts were harvested in cold PBS (pH 7.4) containing 1% Triton‐X100, 2.5 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride, and a complete protease inhibitor mixture and then vortexed for 30 s. The heart extracts were centrifuged at 12 000g for 15 minutes, and the supernatants were collected (soluble fraction). The pellets were dissolved in DNAase (1 mg/kL in 10 mM Tris, 15 mM MgCl2; Roche) and sonicated on ice, and the protein was quantitated with a modified Bradford assay. The insoluble protein was then diluted in RIPA buffer, and 3 μg insoluble protein resuspended was used for subsequent immunoblotting with appropriate antibodies.
Whole‐cell lysate fractions were prepared from hearts as described previously. 20 The heart homogenates were centrifuged at 12 000g for 15 minutes and the supernatants were collected (soluble fraction). The protein content of the soluble lysates was measured using the modified Bradford protocol/reagent relative to a BSA standard curve (BioRad). Protein lysates (50 µg for the whole cell lysate and 30 µg for the mitochondrial fraction) were separated on SDS‐PAGE using pre‐cast 7.5% to 15% Criterion Gels (BioRad) and transferred to polyvinylidene difluoride membranes (BioRad). Membranes were blocked for 1 hour in 5% nonfat dried milk and exposed to primary antibodies overnight. The following primary antibodies were used for immunoblotting: anti‐Drp1 (1:1000, Cell Signaling Technology), anti‐pDrp1 Ser616 (1:1000, Cell Signaling Technology), anti‐mitofusin 1 (MFN1) (1:1000, Cell Signaling Technology), anti‐MFN2 (1:1000, Cell Signaling Technology), anti‐OPA1 (1:1000, Cell Signaling Technology), anti‐GAPDH (1:5000, EMD Millipore), anti‐β‐Actin (1:1000, Santa Cruz Biotechnology), anti‐α‐sarcomeric actinin (1:1000, Sigma‐Aldrich), anti‐OXPHOS (1:1000, Abcam), anti‐PDH (1:1000, Abcam), anti‐CryAB (1:10 000, Enzo Life Sciences), anti‐Periostin (1:500, Santa Cruz Biotechnology), and anti‐smooth‐muscle actin (1:500, Cell Signaling Technology). Membranes were then washed, incubated with alkaline‐phosphatase‐conjugated secondary antibodies (Santa Cruz Biotechnology), exposed with ECF reagent (Amersham), and finally, detected on a ChemiDoc Touch Imaging System (BioRad). Ponceau S protein stain of the transfer membrane was used as a loading control. Densitometry on scanned membranes was done using ImageJ software (National Institutes of Health, Bethesda, MD).
Nongradient blue native polyacrylamide gel electrophoresis
Mitochondrial extracts were prepared for blue native polyacrylamide gel electrophoresis (BN‐PAGE) analysis as described previously. 24 , 25 , 26 Briefly, fresh mitochondrial pellets were suspended in a solubilization buffer containing 50 mM Bis‐Tris (pH 7.0), 1% n‐dodecyl‐β‐d‐maltoside (vol/vol) and 750 mM ε‐amino‐N‐caproic acid. 27 The suspension was kept on ice for 1 hour with occasional vortexing followed by clarification with centrifugation at 8000g for 10 minutes. Following the centrifugation, the supernatant was quantified for protein concentration by the Bradford method and mixed with concentrated BN‐PAGE loading buffer (10×) containing 0.5 M ε‐amino‐N‐caproic acid and 3% Serva Blue G‐250 (wt/vol). The samples were run in an acrylamide/bisacrylamide gel containing 4% stacking gel, and 8% of the resolving gel. 28 Nongradient BN‐PAGE was performed at room temperature using gel buffer (500 mM aminocaproic acid and 50 mM Bis‐Tris, pH 7.0), cathode buffer (50 mM Tricine, 15 mM Bis‐Tris, pH 7.0), 0.02% Serva blue G‐250 (wt/vol), anode buffer (50 mM Bis‐Tris pH 7.0), and sample buffer (75 mM aminocaproic acid containing 0.3% Serva blue G‐250 [wt/vol]). 28 Proteins (30 μg) were loaded in the gel and run at 150 V until the front line had entered into two‐third of the gel. Next, the cathode buffer was replaced with buffer without Serva blue G‐250 (50 mM Tricine, 15 mM Bis‐Tris, pH 7.0) and gel running was completed at 250 V. Gel was stained with Coomassie blue (BioRad) using previously described methods. 29
Pyruvate dehydrogenase enzymatic activity
Pyruvate dehydrogenase (PDH) activity was measured in isolated mitochondria with the PDH Activity Assay Kit (MAK183, Sigma‐Aldrich) according to manufacturer’s instruction. 30 Mitochondria (150 µg) were homogenized in 50 µL of ice‐cold PDH assay buffer, and centrifuged at 10 000g for 5 minutes. Ten microliters of lysates was used for reactions. Reactions were carried out at 37°C, and A450 was recorded every minute for 30 minutes. PDH activity rates were measured in nmol/min per mg using the NADH standard curve.
Citrate synthase activity
Citrate synthase activity was measured in isolated mitochondria and whole heart homogenates as described previously. 31 Briefly, whole heart homogenates and isolated mitochondria were homogenized with Cell‐lytic‐M (Sigma Aldrich) supplemented with protease inhibitor. Mitochondrial lysate (10 µg) was used in reaction mixture containing 0.25% Triton X‐100, 0.31 mM acetyl CoA, and 0.1 mM 5,5´‐dithiobis(2‐nitrobenzoic acid). Finally 0.5 mM oxaloacetate was added in 1 mL reaction volume and citrate synthase activities were measured using a DS‐11 FX+spectrophotometer (DeNovix) at 412 nm at 37°C. Activity was expressed as nmol/min per mg of mitochondrial protein. Extinction coefficient values for TNB (13.6 mM‐1 cm‐1) were used to measure the citrate synthase enzyme activity.
Statistical Analysis
Data are expressed as mean±SEM. All statistical tests were done with GraphPad Prism software. All data were tested for normality using the Kolmogorov‐Smirnov test, and data that passed the normality assumption were analyzed using Student t test (P<0.05) for 2 groups and groups of 3 or more with 1‐way ANOVA, followed by the Tukey post hoc test. For certain data sets with smaller sample sizes (n=3–6), the Kruskal‐Wallis test was applied, and data were presented in graphs showing median and interquartile ranges. A value of P<0.05 was considered statistically significant.
Results
Age‐Dependent Cardiac Pathological Remodeling and Contractile Dysfunction in CryABR120G Transgenic Hearts
The earliest pathology manifested by the CryABR120G expression in cardiomyocytes is the accumulation of protein aggregates that is evident within days of birth. 12 Images of hearts from CryABR120G transgenic showed normal heart size at 2 months of age but showed development of hypertrophy at 6 months of age compared with littermate nontransgenic hearts (Figure 1A). Heart weight‐to‐body‐weight ratios were measured as an indicator of cardiac hypertrophy and were significantly increased in the CryABR120G transgenic mouse at 6 months of age (Figure 1B). Western blot analysis of proteins showed an age‐dependent increase in mutant CryAB protein content in the whole cell lysate (Figure 1C) and in the pellet (insoluble) fraction (Figure 1D) in CryABR120G hearts. We also used the immunofluorescent staining for CryAB to quantitate aggregate accumulation on a per‐cell‐area basis. Quantification of aggregate size per cell showed significantly increased aggregate size in 6 months compared with 2 months of age (Figure 1E). Previously, we extensively characterized the CryABR120G model where hearts from CryABR120G mice at 4 months displayed pathological remodeling, characterized by cardiomyocyte disarray and extensive interstitial fibrosis. 20 Therefore, we measured the periostin and α‐smooth muscle actin protein level to demonstrate the extent of fibrosis during 2 and 6 months of age. The CryABR120G transgenic hearts also showed an increased level of periostin and α‐smooth muscle actin, indicating increased fibrotic remodeling in these hearts at 6 months of age (Figure 1F).
Figure 1. Pathological cardiac remodeling in CryABR120G Tg hearts.
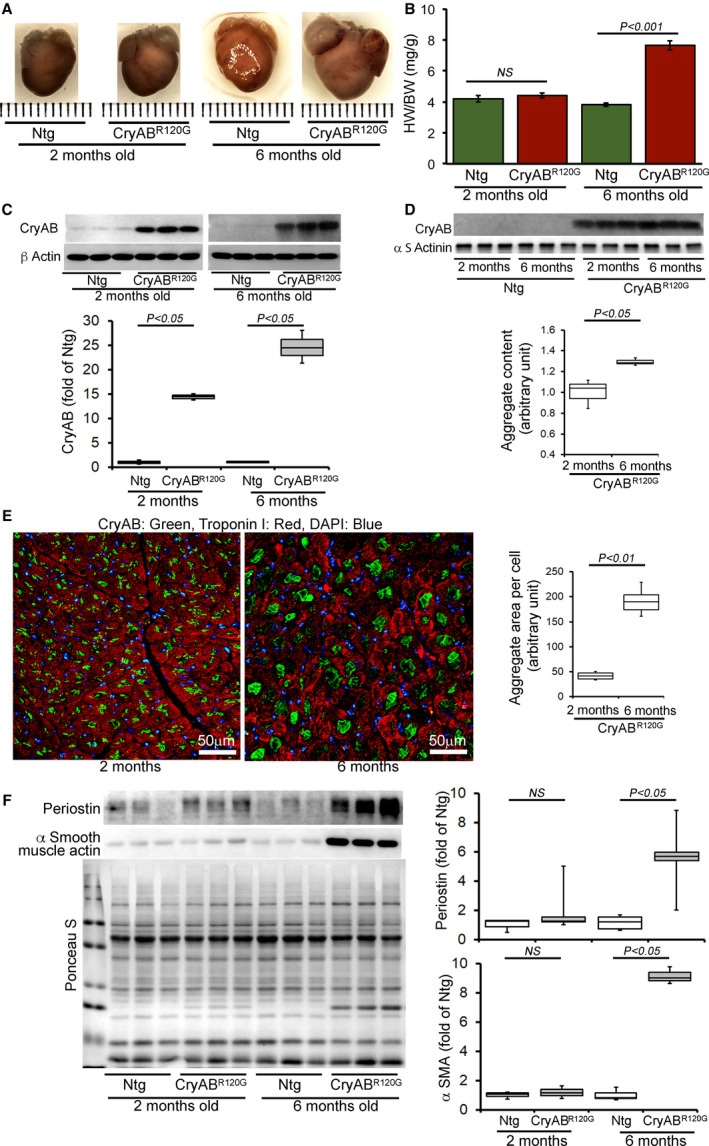
A, Images of hearts and B, quantification of the heart‐weight‐to‐body‐weight ratios (HW/BW) showing cardiac hypertrophy in the CryABR120G transgenic hearts (n ≥ 6 or more mice per group) at 6 months of age. C, Representative Western blot and densitometric quantification of αB‐crystallin (CryAB) protein level in the whole cell lysate derived from 2‐ and 6‐month‐old CryABR120G transgenic and nontransgenic hearts (n=3 mice per group). β actin was used as a loading control. D, Representative Western blot and densitometric quantification of aggregated CryAB protein level present in the insoluble fractions derived from 2‐ and 6‐month‐old CryABR120G transgenic and nontransgenic hearts (n=3 mice per group). α‐Sarcomeric actinin was used as a loading control. E, Immunofluorescence staining for CryAB (green) with troponin I counterstaining (red) showed aggregate levels in 2‐ and 6‐month‐old CryABR120G transgenic hearts. Quantification of CryAB aggregates showed age‐dependent increased aggregate size in the heart. F, Representative Western blot and densitometric quantification of periostin and (αSMA) proteins level in the whole cell lysate derived from 2‐ and 6‐month‐old CryABR120G transgenic and nontransgenic hearts (n=3 mice per group). Boxes represent interquartile ranges, lines represent medians, whiskers represent ranges, and P values were determined by Kruskal–Wallis test. CryABR120G Tg indicates mutant αB‐crystallin transgenic mouse; DAPI, 4′,6‐diamidino‐2‐phenylindole; NS, not significant; Ntg, nontransgenic; and αSMA, α‐smooth muscle actin. Scale bars: 50 μm.
In vivo LV function and chamber dimensions of CryABR120G transgenic and nontransgenic mice were assessed with echocardiography in mixed‐sex cohorts at 2 and 6 months of age (Figure 2). Similar to the cardiac morphometry, the CryABR120G transgenic hearts at 2 months age were indistinguishable from nontransgenic littermates with similar values for LV internal dimensions in diastole and systole (Figure 2A and 2B, respectively), LV fractional shortening (percent fractional shortening, Figure 2C), volumes in diastole and systole (LV Vol;d and LV Vol;s, respectively, Figure 2D and 2E), and LV ejection fraction (Figure 2F). However, with aging the CryABR120G transgenic hearts developed progressive systolic dysfunction, evidenced by an increase in LV internal dimensions (in systole and diastole) and volumes (LV Vol;s and LV Vol;s) resulting in decreased fractional shortening and ejection fraction compared with nontransgenic littermates (Figure 2A‐2F). These hemodynamic changes were also accompanied by an increase in the diastolic thickness of the interventricular septum, LV posterior wall, and LV mass index (Figure 2G‐2I) in CryABR120G transgenic compared with nontransgenic mice. Overall, the CryABR120G transgenic mice showed increased aggregate formation, adverse cardiac hypertrophic remodeling, and defective cardiac contractile function at 6 months of age.
Figure 2. Echocardiography indices of cardiac structure and function in CryABR120G Tg and Ntg hearts.
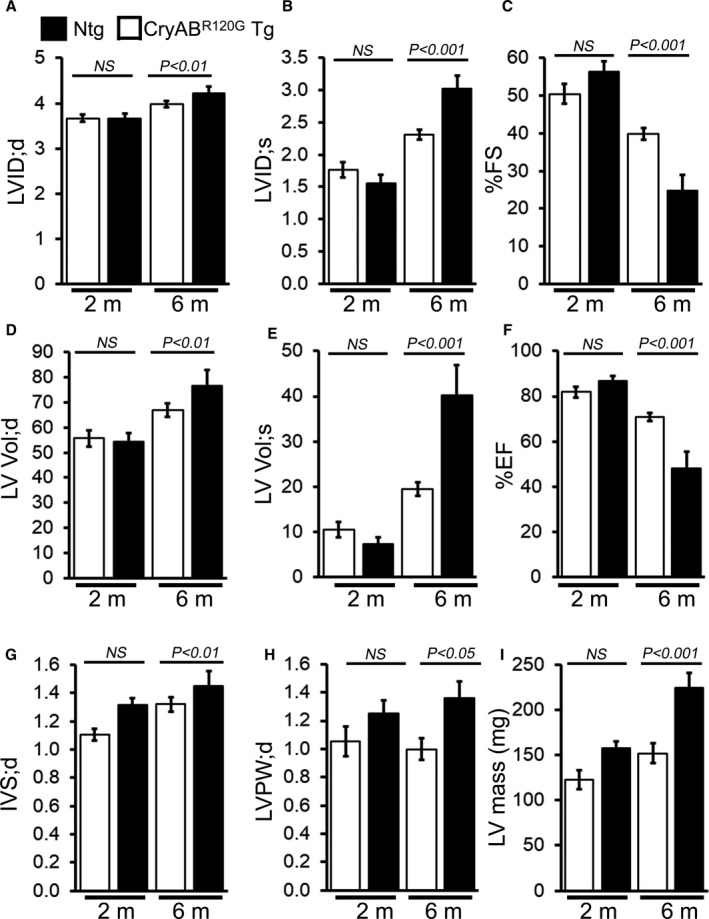
M‐mode echocardiography indices measured in 2‐ and 6‐month‐old CryABR120G transgenic mice with age‐matched littermate nontransgenic mice. A, Left ventricular (LV) diastolic internal dimension (LVID;d). B, LV systolic internal dimension (LVID;s). C, Percent fractional shortening (%FS). D, LV diastolic volume (LV Vol;d). E, LV systolic volume (LV Vol;s). F, Percent ejection fraction (%EF). G, LV diastolic interventricular septum thickness (IVS;d). H, LV diastolic posterior wall thickness (LVPW;d). I, LV mass (n=5–6 mice per group). Bars represent mean±SEM. P values were determined by Student t test. CryABR120G Tg indicates mutant αB‐crystallin transgenic mouse; NS, not significant; and Ntg, nontransgenic.
Altered Mitochondrial Dynamics Regulatory Protein Expression in CryABR120G Hearts
Ultrastructural analysis of CryABR120G hearts by electron microscopy showed perturbations in mitochondrial–sarcomere architecture resulting in alterations in mitochondrial morphology and positioning. 32 Therefore, we monitored whether the CryABR120G hearts show any imbalance in mitochondrial fission/fusion protein levels and investigated the levels of critical mitochondrial fission (ie, dynamin‐related protein 1 [Drp1]) and fusion (ie, optic atrophy type 1 [OPA1], mitofusin 2 [MFN2], and mitofusin 1 [MFN1]) regulatory proteins in the heart (Figure 3A through 3D). We observed the proteins level of the mitochondrial dynamics regulatory proteins in CryABR120G transgenic mice before the onset of cardiac hypertrophy (2 months of age), during the development of maladaptive concentric hypertrophy (4 months of age), and during the progression of HF (6 months of age) (Figure 3A through 3I). The level of mitochondrial fission regulatory protein DRP1 was significantly increased in whole‐cell lysates and the mitochondrial fraction isolated from CryABR120G transgenic hearts at 2, 4, and 6 months of age (Figure 3E). On the other hand, fusion regulatory protein OPA1 level was significantly decreased in whole‐cell lysates and the mitochondrial fraction isolated from CryABR120G transgenic hearts at 2, 4, and 6 months of age (Figure 3F). MFN2 levels were not altered in all these age groups (Figure 3G) but the MFN1 level was significantly decreased in the mitochondrial fraction (Figure 3H). We confirmed the purity of the mitochondrial fractionation by using GAPDH and β actin protein levels in the whole‐cell lysate fraction as a positive control and absence in the mitochondrial fraction as measures of purity. α‐Sarcomeric actinin was used to show the level of myofilament co‐enrichment in the mitochondrial fraction. Ponceau S protein staining of the transfer membrane confirmed approximately equal loading and transfer across the gel. The phosphorylation of Drp1 at Ser616 permits mitochondrial translocation of Drp1 facilitating the fission of mitochondria. 33 , 34 Consistent with increased Drp1 protein level in mitochondria, Drp1 phosphorylation at Ser616 was significantly increased in the CryABR120G transgenic hearts at 2, 4, and 6 months of age compared with nontransgenic hearts (Figure 3D and 3I). Therefore, the CryABR120G mouse hearts showed altered proteins level of mitochondrial dynamics regulatory proteins before the onset of pathological remodeling and development of systolic contractile dysfunction.
Figure 3. Mitochondrial dynamics regulatory proteins levels in CryABR120G Tg and Ntg hearts.
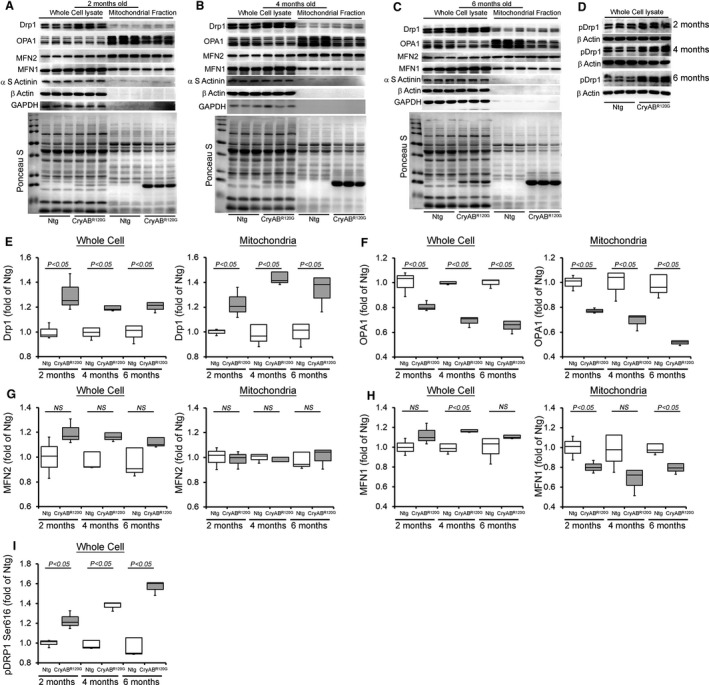
Representative Western blot showing the mitochondrial dynamic regulatory proteins level in the whole cell lysate and the mitochondrial fraction of CryABR120G transgenic and nontransgenic hearts at (A) 2 months, (B) 4 months, and (C) 6 months of age: Drp1, OPA1, MFN2, and MFN1. (D) pDrp1 Ser616 protein level in the whole cell lysate of CryABR120G transgenic and nontransgenic hearts at 2, 4, and 6 months of age. GAPDH and β actin were used as a control to show the purity of the mitochondrial fraction. α‐Sarcomeric actinin was used to show the level of myofilament co‐enrichment in the mitochondrial fraction. Ponceau S protein staining of the transfer membrane confirmed approximately equal loading across the gel (n=3 mice per group). Densitometric quantification of the protein levels of (E) Drp1, (F) OPA1, (G) MFN2, (H) MFN1, and (I) pDrp1 Ser616 in the CryABR120G transgenic and nontransgenic hearts at 2, 4, and 6 months of age. Boxes represent interquartile ranges, lines represent medians, whiskers represent ranges, and P values were determined by Kruskal–Wallis test. CryABR120G Tg indicates mutant αB‐crystallin transgenic mouse; Drp1, dynamin‐related protein 1; MFN1, mitofusin 1; MFN1, mitofusin 2; Ntg, nontransgenic; OPA1, optic atrophy type 1; and NS, not significant.
Altered OXPHOS Complex Regulatory Proteins Level in CryABR120G Hearts
The OXPHOS of the mitochondrial inner membrane is composed of 5 enzymes (complexes I‐V), which produce the majority of cellular energy in the form of ATP. Altered protein content of OXPHOS complex proteins was found in the brain of patients with proteotoxic neurodegenerative diseases. 35 , 36 , 37 Moreover, decreased protein level of complex I and II of OXPHOS have been reported in the in vivo and in vitro models of proteinopathy. 36 , 38 , 39 As these mice develop age‐dependent progression of cardiac contractile dysfunction, 12 we observed the level of the OXPHOS complex proteins in CryABR120G transgenic mice before the onset of cardiac systolic contractile dysfunction (2 months of age), during the development of maladaptive concentric hypertrophy (4 months of age), and during the progression of HF (6 months of age) (Figure 4A through 4G). We compared OXPHOS complex proteins level in whole‐cell lysates and the mitochondrial fractions using 2‐, 4‐, and 6‐month‐old CryABR120G transgenic and nontransgenic mice hearts of mixed sex (Figure 4A through 4C). We observed a significantly decreased proteins level of Complex I and Complex II of OXPHOS complex in the mitochondrial fraction isolated from the CryABR120G transgenic hearts of 2, 4, and 6 months of age (Figure 4F through 4G). The level of Complex III protein was significantly decreased in 2 months of age heart mitochondria (Figure 4E). Interestingly, the levels of Complex I, Complex II, Complex III, and Complex V proteins were significantly decreased in the mitochondrial fraction of 6 months of age when these mice develop the signs of HF (Figure 4D through 4G). The whole‐cell fraction did not show any significant difference in the levels of OXPHOS in all these age groups. Ponceau S staining of proteins was used to confirm equal loading (Figure 4A through 4C). Because many of these OXPHOS proteins are organized into complexes in the mitochondrial inner membrane, we observed the level of the mitochondrial oxidative phosphorylation complexes by BN‐PAGE (Figure 5). The assembly and stability of Complex I, Complex II, Complex III, Complex IV, and Complex V were reduced in the CryABR120G hearts compared with nontransgenic hearts at 2, 4, and 6 months of age (Figure 5). Collectively, we found an altered proteins level and alterations in native complexes of the oxidative phosphorylation system in the CryABR120G transgenic hearts before the onset of cardiac contractile dysfunction.
Figure 4. OXPHOS complex protein levels in CryABR120G Tg and Ntg hearts.
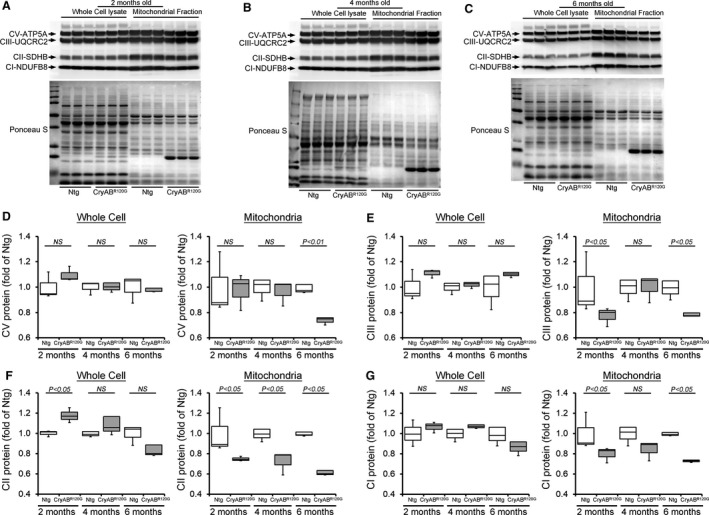
Representative Western blot showing the OXPHOS complex regulatory proteins level in the whole cell lysate and the mitochondrial fraction of CryABR120G transgenic and nontransgenic hearts at (A) 2 months, (B) 4 months, and (C) 6 months of age: Complex I, Complex II, Complex III, and Complex V protein. Ponceau S protein stain of the transfer membrane was used to confirm approximately equal loading (n=3 mice per group). Densitometric quantification of the protein levels of (D) Complex V (CV), (E) Complex III (CIII), (F) Complex II (CII), and (G) Complex I (CI) in the whole cell lysate and the mitochondrial fraction of CryABR120G transgenic and nontransgenic hearts at 2, 4, and 6 months of age. Boxes represent interquartile ranges, lines represent medians, whiskers represent ranges, and P values were determined by Kruskal–Wallis test. CryABR120G Tg indicates mutant αB‐crystallin transgenic mouse; Ntg, nontransgenic; NS, not significant; and OXPHOS, oxidative phosphorylation system.
Figure 5. Representative gel image showing mitochondrial protein complexes resolved by nongradient BN‐PAGE in CryABR120G Tg and Ntg hearts.
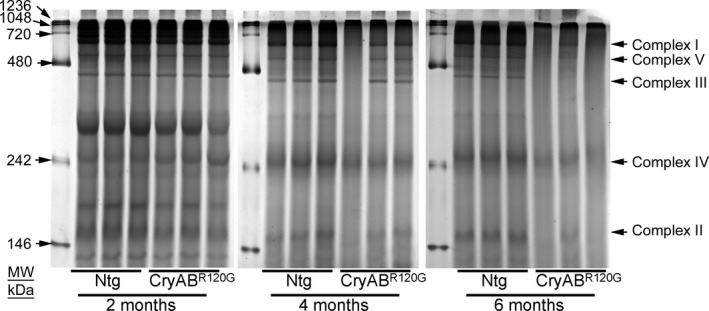
Mitochondria isolated from CryABR120G transgenic and nontransgenic hearts at 2, 4, and 6 months of age were analyzed, with each lane containing mitochondria prepared from independent mice. An 8% separating gel was used and the gel was stained by Coomassie blue after gel electrophoresis. Arrows indicate mitochondrial electron transport chain complexes visualized following Coomassie blue staining. BN‐PAGE indicates blue native polyacrylamide gel electrophoresis; and CryABR120G, mutant αB‐crystallin transgenic mouse.
Altered PDH Regulatory Protein Level in CryABR120G Hearts
PDH complex is a large multisubunit enzyme complex within the mitochondrial matrix, consisting of 3 proteins: pyruvate dehydrogenase (E1), dihydrolipoamide transacetylase (E2), and dihydrolipoamide dehydrogenase (E3). 40 PDH functions to connect the citric acid cycle and subsequent oxidative phosphorylation to the glycolysis, gluconeogenesis, and lipid and amino acid metabolism pathways. LV myocardium from adult patients with end‐stage systolic HF exhibits a higher expression of PDH complex subunits and higher rates of PDH activity. 41 Therefore, we observed the protein level of E1α/β, E2, and E3bp components of the PDH complex in the CryABR120G and nontransgenic mouse hearts at 2, 4, and 6 months of age (Figure 6A through 6F). CryABR120G and nontransgenic hearts at 2 months of age showed a similar level of E1α/β, E2, and E3bp proteins (Figure 6D through 6F). CryABR120G hearts at 4 months of age showed a significantly increased level of E1α/β and E3bp protein in the mitochondrial fraction (Figure 6D through 6F). At 6 months of age, CryABR120G hearts showed a significantly increased level of E1α/β, E2, and E3bp protein compared with nontransgenic hearts (Figure 6D through 6F). The whole‐cell fraction did not show any significant difference in the proteins level of components of the PDH complex in all these age groups except E1α/β in 6‐month‐old mice (Figure 6F). Ponceau S staining of proteins was used to confirm equal loading (Figure 6A through 6C). We also measured the PDH enzyme activity in the CryABR120G and nontransgenic mouse hearts at 2, 4, and 6 months of age. PDH enzymatic activity was significantly higher in the CryABR120G hearts at 4 and 6 months of age compared with nontransgenic hearts (Figure 7A). Collectively, we found that the altered PDH complex proteins level and PDH enzymatic activities in the CryABR120G transgenic hearts start during the maladaptive cardiac remodeling. Citrate synthase (CS) activity was used to assess mitochondrial content and purity of the isolated mitochondria in the CryABR120G and nontransgenic mouse hearts. 31 CS activities did not show any significant difference in the isolated mitochondrial fraction from CryABR120G transgenic hearts compared with nontransgenic hearts at 2, 4, and 6 months of age (Figure 7B), indicating similar mitochondrial purity among groups. We also measured CS activity in the whole heart homogenates to provide further insight into the mitochondrial changes and found significantly decreased CS activity in the CryABR120G hearts at 4 and 6 months of age compared with nontransgenic hearts (Figure 7C). Therefore, these key adaptations may afford the failing hearts crucial capacity to utilize glucose‐dependent energy production in the face of dwindling energy options.
Figure 6. PDH complex protein levels in CryABR120G Tg and Ntg hearts.
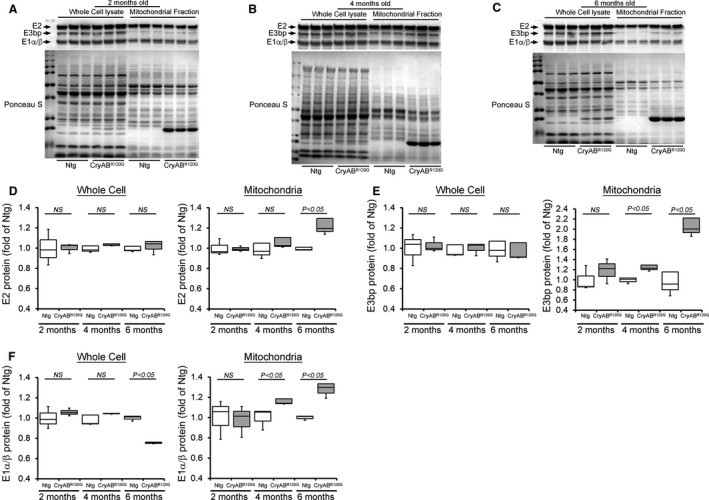
Representative Western blot showing the PDH complex regulatory proteins level in the whole cell lysate and the mitochondrial fraction of CryABR120G transgenic and nontransgenic hearts at (A) 2 months, (B) 4 months, and (C) 6 months of age: E2, E3bp, and E1α/β protein. Ponceau S protein stain of the transfer membrane was used to confirm approximately equal loading (n=3 mice per group). Densitometric quantification of the protein levels of (D) E2, (E) E3bp, and (F) E1α/β component of the PDH complex in the whole cell lysate and the mitochondrial fraction of CryABR120G transgenic and nontransgenic hearts at 2, 4, and 6 months of age. Boxes represent interquartile ranges, lines represent medians, whiskers represent ranges, and P values were determined by Kruskal–Wallis test. CryABR120G Tg indicates mutant αB‐crystallin transgenic mouse; E1, pyruvate dehydrogenase; E2, dihydrolipoamide transacetylase; E3, dihydrolipoamide dehydrogenase; Ntg, nontransgenic; NS, not significant; and PDH, pyruvate dehydrogenase complex.
Figure 7. Mitochondrial PDH and CS activities in CryABR120G Tg and Ntg hearts.
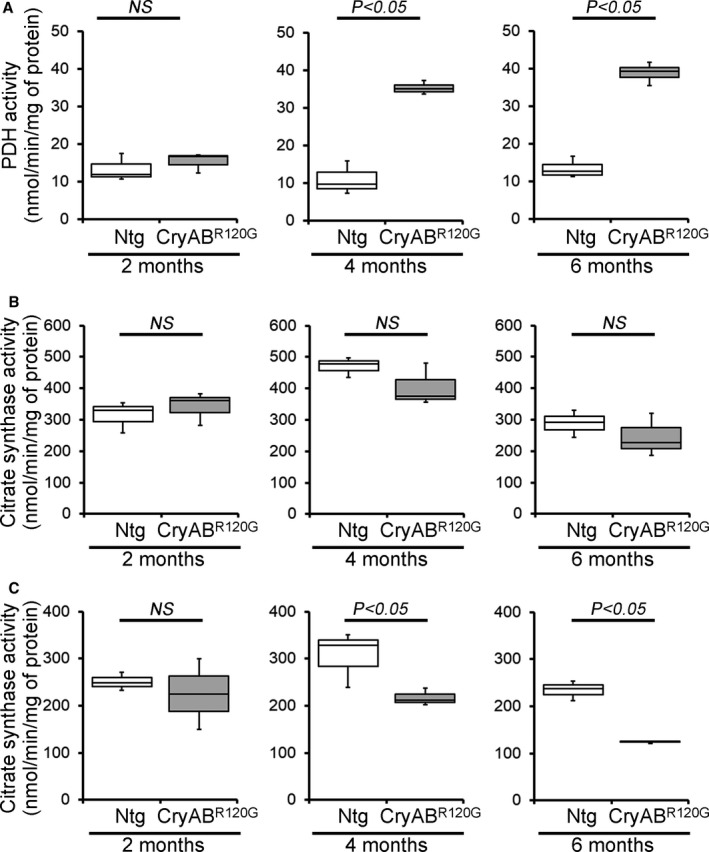
A, PDH enzymatic activity in 2‐, 4‐, and 6‐month‐old CryABR120G transgenic and nontransgenic hearts (n=3 mice per group). B and C, CS activity in the mitochondrial fraction (B) and the whole heart homogenates (C) isolated from 2‐, 4‐, and 6‐month‐old CryABR120G transgenic and nontransgenic hearts (n=3 mice per group). Boxes represent interquartile ranges, lines represent medians, whiskers represent ranges, and P values were determined by Kruskal–Wallis test. CryABR120G Tg indicates mutant αB‐crystallin transgenic mouse; CS, citrate synthase; NS, not significant; Ntg, nontransgenic; and Tg, transgenic.
Mitochondrial Respiratory Dysfunction in CryABR120G Transgenic Hearts
The abnormal mitochondrial dynamics, OXPHOS complex, and PDH complex regulatory protein level in CryABR120G mice heart at an early age with preserved systolic function pointed to potential mitochondrial dysfunction contribution to the cardiac dysfunction. To ascertain this, we isolated mitochondria from both CryABR120G and nontransgenic mouse hearts of 2 months age and measured mitochondrial electron transport chain complex activities (Figure 8) as well as real‐time OCRs (Figure 9). Mitochondrial Complex I, II, and III activities were significantly decreased in the CryABR120G transgenic heart at 2 months of age compared with age‐matched littermate nontransgenic hearts (Figure 8). Real‐time monitoring of OCR showed significantly decreased basal respiration in the CryABR120G mitochondria, indicating a lower respiratory function compared with nontransgenic mitochondria as the basal respiration representing the sum of all physiological mitochondrial oxygen consumption (Figure 9A and 9B). The ATP‐linked OCR was similar in both of the groups, determined by the addition of an ATP synthase inhibitor (oligomycin) that leads to a decrease in basal respiration (Figure 9C). The addition of FCCP uncouples respiration from oxidative phosphorylation and allows for the measurement of maximal OCR, which was lower in CryABR120G mitochondria, indicating lower overall mitochondrial activity (Figure 9D). The extent of nonmitochondrial oxygen‐consuming processes was similar in both groups of mice, estimated by inhibiting the respiratory chain with rotenone (complex I inhibitor) and antimycin A (complex III inhibitor) (Figure 9E). The respiratory reserve capacity, calculated by subtracting basal OCR from FCCP‐stimulated OCR, was significantly lower in CryABR120G mitochondria (Figure 9F), and ATP turnover measured by ATP‐linked respiration subtracted from the basal OCR was significantly decreased in CryABR120G mitochondria (Figure 9G). The maximum respiration calculated by nonmitochondrial respiration subtracted from FCCP‐stimulated OCR was also significantly lower in CryABR120G mitochondria (Figure 9H). We conclude that mitochondria isolated from young (2 months old) CryABR120G hearts with normal systolic contractile function and cardiac morphometry are functionally compromised.
Figure 8. Mitochondrial electron transport chain complex activities in CryABR120G Tg and Ntg hearts.
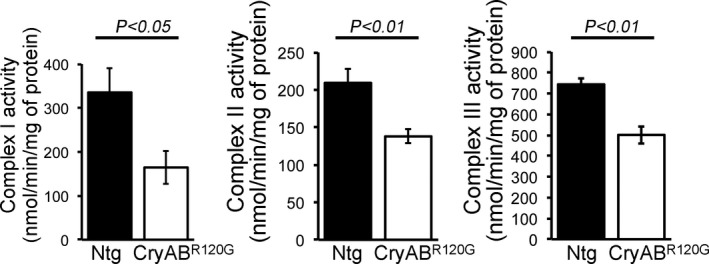
Mitochondrial Complex I, II, and III activities in 2‐month‐old CryABR120G transgenic and nontransgenic hearts (n=6 mice per group). Bars represent mean±SEM. P values were determined by Student t test. CryABR120G Tg indicates mutant αB‐crystallin transgenic mouse; NS, not significant; and Ntg, nontransgenic.
Figure 9. Mitochondrial respiration in isolated mitochondria from CryABR120G Tg and Ntg mice hearts.
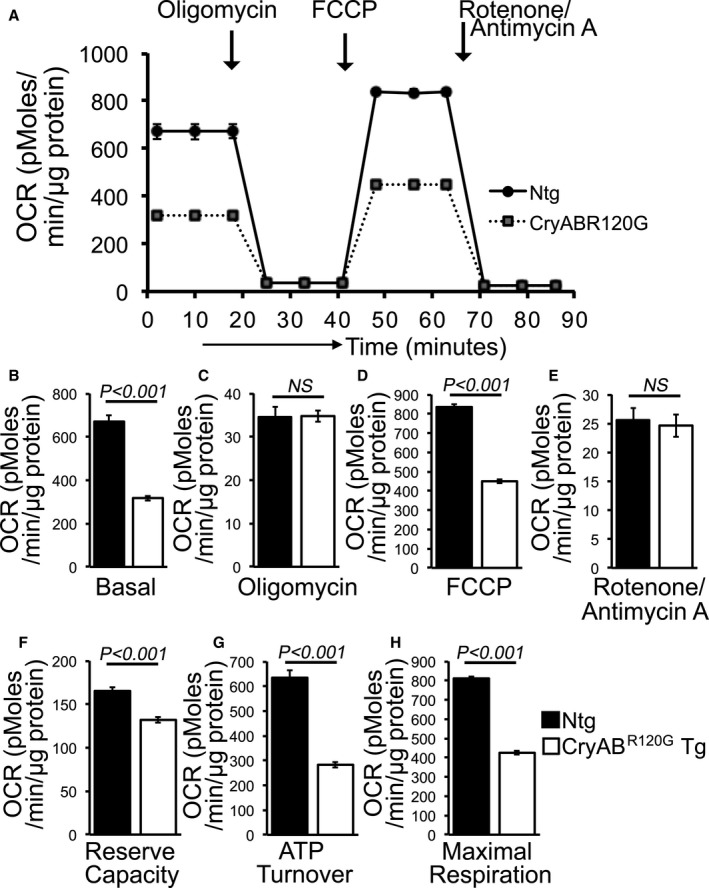
A, Mitochondrial oxygen consumption rate (OCR) profiles in isolated mitochondria from 2 months of age littermate CryABR120G Tg and Ntg hearts. Arrow indicates the sequential addition of oligomycin (1 µM), FCCP (4 µM), and rotenone (0.5 µM) plus antimycin A (0.5 µM). OCR profile is expressed as pmol O2/min per µg of protein, and each point represents average OCR values for 5 mice. Graphs showing OCR under (B) baseline as well as with the addition of (C) oligomycin, (D) FCCP, and (E) rotenone plus antimycin A. Key parameters of mitochondrial respiration, including (F) reserve capacity, (G) ATP turnover, and (H) maximal respiration were significantly decreased in CryABR120G Tg mitochondria. Bars represent mean±SEM. P values were determined by Student t test. CryABR120G Tg indicates mutant αB‐crystallin transgenic mouse; FCCP, carbonyl cyanide‐p‐trifluoromethoxy‐phenylhydrazone; NS, not significant; and Ntg, nontransgenic.
Discussion
CryABR120G transgenic hearts begin accumulating protein aggregates within days of birth, 12 and the aggregate size as well as number increased over aging in CryABR120G hearts with the gradual progression of HF. 12 , 42 , 43 Earlier studies on CryABR120G transgenic mice showed they develop concentric hypertrophy by 4 to 5 months of age and HF by 6 to 8 months of age. 12 Mutant CryABR120G transgenic mouse hearts display electron‐dense aggregates (composed of CryAB, desmin, ubiquitin, and p62), myofibrillar disarray, and abnormal mitochondria, recapitulating the human pathology. In the present study, we found an altered level of mitochondrial dynamics regulatory proteins, decreased level of OXPHOS proteins, and increased level of PDH complex protein results in defects in mitochondrial complex activities and respiration at 2 months of age. At this age, CryABR120G hearts show normal cardiac morphometry and systolic contractile function even though the cardiomyocytes showed the accumulation of protein aggregates. These findings suggest that mitochondrial dysfunction occurs in CryABR120G hearts before the manifestation of pathological adverse remodeling and resultant cardiac systolic contractile dysfunction.
Extensive studies demonstrated several causative factors linking to the onset and development of pathologic cardiac remodeling and progression of HF in the CryABR120G mice. These include displacement of mitochondrial positioning, myofilament disarray, reduction in contractile function because of accumulation of insoluble aggregates, and apoptotic cell loss. The involvement of mitochondria in the pathogenic processes in CryABR120G transgenic hearts is evident by the findings showing CryABR120G can associate with the mitochondrial voltage‐dependent anion channel and activate apoptosis by opening the mitochondrial permeability transition. 43 However, the genetic approach to inhibit cardiomyocyte apoptosis by overexpression of anti‐apoptotic B‐cell lymphoma 2 in the CryABR120G hearts slowed the pathogenic progression but eventually leads to the development of HF. Overexpression of B‐cell lymphoma 2 leads to a reversal of calcium sensitivity of the mitochondria but resulted in the accumulation of an increased number of lysosomes and autophagosome. 44 In fact, activation of autophagy in CryABR120G hearts by genetic autophagy‐related protein 7 (Atg7) overexpression, 20 voluntary exercise, 20 , 45 ubiquitin‐conjugating enzyme 9–mediated SUMOylation, 46 and transcription factor EB overexpression 21 , 22 resulted in attenuated and/or delayed the onset of the morphological and functional pathologies. Similarly, activation of the ubiquitin–proteasome pathway in CryABR120G hearts by overexpression of proteasome 28 subunit α19, constitutive photomorphogenesis 9 signalosome, 47 ubiquitin‐conjugating enzyme 9, 48 and interferon‐inducible protein NEDD8 ultimate buster one long 49 resulted in reduced intracellular aggregates and partially rescued/delayed the cardiomyopathy. All these prior studies were only able to delay and/or partially rescue the accumulation of amyloid oligomers and progression of HF. Therefore, we wanted to determine the factors involved in the progression of this multifocal disease process and found the altered mitochondrial dynamics, and oxidative phosphorylation protein early in the disease process in these mice.
The metabolic requirement of the heart is maintained by functionally active mitochondria ensured through a balanced mitochondrial fission and fusion events that are indispensable to maintain the normal mitochondrial network. During mitochondrial fission, the dynamin‐related GTPase Drp1 is normally recruited from the cytosol into the mitochondrial outer membrane through interaction with the mitochondrial outer membrane in which the adapter proteins reside. Mitochondrial translocation of Drp1 is facilitated by the phosphorylation of Drp1 at Ser616, activating mitochondrial fission. 33 , 34 Moreover, studies also showed increased Drp1 phosphorylation at Ser616 in failing human hearts. 33 We found significantly increased Drp1 protein level, Drp1 Ser616 phosphorylation, and increased protein level in the mitochondrial fraction in the CryABR120G heart over the age ranges of this study. Conversely, mitochondrial fusion is regulated by OPA1 proteins, 50 and we observed a decreased protein level of mitochondrial OPA1, suggesting a decreased mitochondrial fusion in the CryABR120G hearts. Similar to the CryABR120G models of DRM, proteotoxic neurodegenerative diseases such as Huntington disease (HD) pathogenesis showed altered mitochondrial dynamics, fragmentation of mitochondria, subsequent mitochondrial dysfunction, and cellular death in HD‐affected neurons. 51 Studies also demonstrated that Huntingtin interacts with the mitochondrial fission protein Drp1, and elevates levels of GTPase Drp1, resulting in increased fission and reduced fusion in HD‐affected neurons. 51 However, the direct role of Drp1 and/or OPA1 on CryABR120G aggregate formation and the subsequent development of cardiac contractile dysfunction remains unknown. Along with the altered mitochondrial dynamics, CS activity in whole heart homogenate was significantly decreased in the CryABR120G hearts, indicating decreased mitochondrial content with the progression of cardiac contractile dysfunction. Future studies are needed to define whether CryAB directly interacts with Drp1 and/or OPA1 regulating their GTPase enzymatic activities, which in turn causes imbalances of mitochondrial fission/fusion events, resulting in impaired mitochondrial dynamics as well as alterations in mitochondrial content.
Proteotoxic neurodegenerative diseases have been shown to alter the expression of OXPHOS complex proteins in the brain of patients with Alzheimer disease, 35 Huntington disease, 36 and Parkinson disease. 37 For instance, decreased expression of Complex II of OXPHOS complex has been reported in the striatum of patients with HD, 36 in vitro primary striatal neurons expressing the N‐terminal fragment of huntingtin, 38 and genetic models of HD expressing N‐terminal fragments of mutant huntingtin. 39 As these mice develop age‐dependent progression of cardiac contractile dysfunction, 12 we observed the level of the OXPHOS complex proteins in CryABR120G transgenic mice along with the progression of cardiac contractile dysfunction. In line with these protein aggregation–based diseases, there was significantly decreased protein level of Complex I and Complex II of OXPHOS complex in the mitochondrial fraction isolated from the CryABR120G transgenic 2, 4, and 6 months age hearts. Moreover, CryABR120G hearts showed significantly increased levels of E1α/β, E2, and E3bp protein as well as PDH enzymatic activities during the progression of pathologic cardiac remodeling and development of contractile dysfunction similar to that observed in patients with end‐stage systolic HF. 41 CS activity in the mitochondrial fraction was similar among all groups that served as an important mitochondrial quality control and an index of mitochondrial purity among groups. Mitochondrial complex activities and mitochondrial respiratory parameters were significantly decreased in the CryABR120G transgenic heart at an early stage of the disease process in these mice when they have normal systolic contractile function and cardiac morphometry. Reduction in mitochondrial OCR in the mitochondria of CryABR120G hearts could result from defects in mitochondrial substrate uptake, PDH activity, the activity of the entire TCA cycle, or the flux through the electron transport chain. However, further studies are needed to define the role of CryABR120G proteotoxicity on cardiac mitochondrial substrate preference.
Overall, CryABR120G hearts showed mitochondrial dysfunction in the form of altered mitochondrial dynamics, modified OXPHOS and PDH complex protein expression, abnormal electron transport chain complex activities, and dysfunctional mitochondrial respiration before the onset of detectable pathologies and development of cardiac contractile dysfunction. Earlier studies of DRM animal models suggested that ultrastructural changes resulting from the protein aggregates as well as sarcomere disarray are partially responsible for impairment of cardiomyocyte contractility, 32 , 52 and do not lead to cardiomyocyte cell death by themselves. 52 Therefore, the accumulation of the protein aggregates early in the disease process may perturb the mitochondrial orientation and function, activating a cascade of events leading to cellular dysfunction and cell death.
Source of Funding
This work was supported by The National Institutes of Health grants: HL122354, HL145753, and HL145753‐01S1 to M.S.B; P20GM121307 to C.G.K.; HL098435, HL133497, and HL141155 to A.W.O; HL141998 to S.M.; AA025744 and AA026708 to M.P.; AHA Postdoctoral Fellowship to S.A. and C.S.A.; and LSUHSC‐S Malcolm Feist Pre‐doctoral Fellowship to R.A.
Disclosures
None.
Acknowledgments
The authors acknowledge the COBRE Center for Redox Biology and Cardiovascular Disease and the Animal Models and Histology Core at LSU Health Sciences Center‐Shreveport.
Author contributions: S.A., C.S.A., R.A., M.M., S.N., S.M., and M.S.B. performed experiments. M.P., C.G.K., and A.W.O. contributed to analytic tools. S.A. and M.S.B. designed experiments. S.A. and M.S.B. wrote the manuscript, and all authors contributed to the preparation of the manuscript.
(J Am Heart Assoc. 2020;9:e017195 DOI: 10.1161/JAHA.120.017195.)
For Source of Funding and Disclosures, see page 7.
References
- 1. Wexler RK, Elton T, Pleister A, Feldman D. Cardiomyopathy: an overview. Am Fam Physician. 2009;79:778–784. [PMC free article] [PubMed] [Google Scholar]
- 2. Knoll R, Hoshijima M, Hoffman HM, Person V, Lorenzen‐Schmidt I, Bang ML, Hayashi T, Shiga N, Yasukawa H, Schaper W, et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002;111:943–955. [DOI] [PubMed] [Google Scholar]
- 3. Goldfarb LG, Dalakas MC. Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest. 2009;119:1806–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McLendon PM, Robbins J. Desmin‐related cardiomyopathy: an unfolding story. Am J Physiol Heart Circ Physiol. 2011;301:H1220–H1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bennardini F, Wrzosek A, Chiesi M. Alpha B‐crystallin in cardiac tissue. Association with actin and desmin filaments. Circ Res. 1992;71:288–294. [DOI] [PubMed] [Google Scholar]
- 6. Kato K, Shinohara H, Kurobe N, Inaguma Y, Shimizu K, Ohshima K. Tissue distribution and developmental profiles of immunoreactive alpha B crystallin in the rat determined with a sensitive immunoassay system. Biochim Biophys Acta. 1991;1074:201–208. [DOI] [PubMed] [Google Scholar]
- 7. Benjamin IJ, McMillan DR. Stress (heat shock) proteins: molecular chaperones in cardiovascular biology and disease. Circ Res. 1998;83:117–132. [DOI] [PubMed] [Google Scholar]
- 8. Vicart P, Caron A, Guicheney P, Li Z, Prevost MC, Faure A, Chateau D, Chapon F, Tome F, Dupret JM, et al. A missense mutation in the alphaB‐crystallin chaperone gene causes a desmin‐related myopathy. Nat Genet. 1998;20:92–95. [DOI] [PubMed] [Google Scholar]
- 9. Dalakas MC, Park KY, Semino‐Mora C, Lee HS, Sivakumar K, Goldfarb LG. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med. 2000;342:770–780. [DOI] [PubMed] [Google Scholar]
- 10. Bova MP, Yaron O, Huang Q, Ding L, Haley DA, Stewart PL, Horwitz J. Mutation R120G in alphaB‐crystallin, which is linked to a desmin‐related myopathy, results in an irregular structure and defective chaperone‐like function. Proc Natl Acad Sci USA. 1999;96:6137–6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang X, Klevitsky R, Huang W, Glasford J, Li F, Robbins J. AlphaB‐crystallin modulates protein aggregation of abnormal desmin. Circ Res. 2003;93:998–1005. [DOI] [PubMed] [Google Scholar]
- 12. Wang X, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, Hewett T, Robbins J. Expression of R120G‐alphaB‐crystallin causes aberrant desmin and alphaB‐crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89:84–91. [DOI] [PubMed] [Google Scholar]
- 13. Singh SR, Kadioglu H, Patel K, Carrier L, Agnetti G. Is desmin propensity to aggregate part of its protective function? Cells. 2020;9:491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brodehl A, Gaertner‐Rommel A, Milting H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys Rev. 2018;10:983–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Agnetti G, Bezstarosti K, Dekkers DH, Verhoeven AJ, Giordano E, Guarnieri C, Caldarera CM, Van Eyk JE, Lamers JM. Proteomic profiling of endothelin‐1‐stimulated hypertrophic cardiomyocytes reveals the increase of four different desmin species and alpha‐B‐crystallin. Biochim Biophys Acta. 2008;1784:1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Agnetti G, Halperin VL, Kirk JA, Chakir K, Guo Y, Lund L, Nicolini F, Gherli T, Guarnieri C, Caldarera CM, et al. Desmin modifications associate with amyloid‐like oligomers deposition in heart failure. Cardiovasc Res. 2014;102:24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kedia N, Arhzaouy K, Pittman SK, Sun Y, Batchelor M, Weihl CC, Bieschke J. Desmin forms toxic, seeding‐competent amyloid aggregates that persist in muscle fibers. Proc Natl Acad Sci USA. 2019;116:16835–16840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sanbe A, Yamauchi J, Miyamoto Y, Fujiwara Y, Murabe M, Tanoue A. Interruption of CryAB‐amyloid oligomer formation by HSP22. J Biol Chem. 2007;282:555–563. [DOI] [PubMed] [Google Scholar]
- 19. Li J, Horak KM, Su H, Sanbe A, Robbins J, Wang X. Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J Clin Invest. 2011;121:3689–3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, Hill JA, Sadoshima J, Robbins J. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123:5284–5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ma X, Mani K, Liu H, Kovacs A, Murphy JT, Foroughi L, French BA, Weinheimer CJ, Kraja A, Benjamin IJ, et al. Transcription factor EB activation rescues advanced alphaB‐crystallin mutation‐induced cardiomyopathy by normalizing desmin localization. J Am Heart Assoc. 2019;8:e010866 10.1161/JAHA.118.010866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pan B, Zhang H, Cui T, Wang X. TFEB activation protects against cardiac proteotoxicity via increasing autophagic flux. J Mol Cell Cardiol. 2017;113:51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Reimann J, Kunz WS, Vielhaber S, Kappes‐Horn K, Schroder R. Mitochondrial dysfunction in myofibrillar myopathy. Neuropathol Appl Neurobiol. 2003;29:45–51. [DOI] [PubMed] [Google Scholar]
- 24. Abdullah CS, Alam S, Aishwarya R, Miriyala S, Bhuiyan MAN, Panchatcharam M, Pattillo CB, Orr AW, Sadoshima J, Hill JA, et al. Doxorubicin‐induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respiration. Sci Rep. 2019;9:2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Abdullah CS, Alam S, Aishwarya R, Miriyala S, Panchatcharam M, Bhuiyan MAN, Peretik JM, Orr AW, James J, Osinska H, et al. Cardiac dysfunction in the sigma 1 receptor knockout mouse associated with impaired mitochondrial dynamics and bioenergetics. J Am Heart Assoc. 2018;7:e009775 DOI: 10.1161/JAHA.118.009775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alam S, Abdullah CS, Aishwarya R, Miriyala S, Panchatcharam M, Peretik JM, Orr AW, James J, Robbins J, Bhuiyan MS. Aberrant mitochondrial fission is maladaptive in desmin mutation‐induced cardiac proteotoxicity. J Am Heart Assoc. 2018;7:009289 DOI: 10.1161/JAHA.118.009289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jha P, Wang X, Auwerx J. Analysis of mitochondrial respiratory chain supercomplexes using blue native polyacrylamide gel electrophoresis (BN‐PAGE). Curr Protoc Mouse Biol. 2016;6:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yan LJ, Forster MJ. Resolving mitochondrial protein complexes using nongradient blue native polyacrylamide gel electrophoresis. Anal Biochem. 2009;389:143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luo X, Wu J, Jin Z, Yan LJ. Non‐gradient blue native polyacrylamide gel electrophoresis. Curr Protocols Protein Sci. 2017;87(1):1–19. 29 1–19 29 12. [DOI] [PubMed] [Google Scholar]
- 30. Pendleton AL, Humphreys LR, Davis MA, Camacho LE, Anderson MJ, Limesand SW. Increased pyruvate dehydrogenase activity in skeletal muscle of growth‐restricted ovine fetuses. Am J Physiol Regul Integr Comp Physiol. 2019;317:R513–R520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lai N, Fealy CE, Kummitha CM, Cabras S, Kirwan JP, Hoppel CL. Mitochondrial utilization of competing fuels is altered in insulin resistant skeletal muscle of non‐obese rats (Goto‐Kakizaki). Front Physiol. 2020;11:677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maloyan A, Osinska H, Lammerding J, Lee RT, Cingolani OH, Kass DA, Lorenz JN, Robbins J. Biochemical and mechanical dysfunction in a mouse model of desmin‐related myopathy. Circ Res. 2009;104:1021–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu S, Wang P, Zhang H, Gong G, Gutierrez Cortes N, Zhu W, Yoon Y, Tian R, Wang W. CaMKII induces permeability transition through Drp1 phosphorylation during chronic beta‐AR stimulation. Nat Commun. 2016;7:13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin‐related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–11529. [DOI] [PubMed] [Google Scholar]
- 35. Manczak M, Park BS, Jung Y, Reddy PH. Differential expression of oxidative phosphorylation genes in patients with Alzheimer's disease: implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med. 2004;5:147–162. [DOI] [PubMed] [Google Scholar]
- 36. Benchoua A, Trioulier Y, Zala D, Gaillard MC, Lefort N, Dufour N, Saudou F, Elalouf JM, Hirsch E, Hantraye P, et al. Involvement of mitochondrial complex II defects in neuronal death produced by N‐terminus fragment of mutated huntingtin. Mol Biol Cell. 2006;17:1652–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Perier C, Vila M. Mitochondrial biology and Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2:a009332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Benchoua A, Trioulier Y, Diguet E, Malgorn C, Gaillard MC, Dufour N, Elalouf JM, Krajewski S, Hantraye P, Deglon N, et al. Dopamine determines the vulnerability of striatal neurons to the N‐terminal fragment of mutant huntingtin through the regulation of mitochondrial complex II. Hum Mol Genet. 2008;17:1446–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Damiano M, Diguet E, Malgorn C, D'Aurelio M, Galvan L, Petit F, Benhaim L, Guillermier M, Houitte D, Dufour N, et al. A role of mitochondrial complex II defects in genetic models of Huntington's disease expressing N‐terminal fragments of mutant huntingtin. Hum Mol Genet. 2013;22:3869–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Patel MS, Nemeria NS, Furey W, Jordan F. The pyruvate dehydrogenase complexes: structure‐based function and regulation. J Biol Chem. 2014;289:16615–16623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sheeran FL, Angerosa J, Liaw NY, Cheung MM, Pepe S. Adaptations in protein expression and regulated activity of pyruvate dehydrogenase multienzyme complex in human systolic heart failure. Oxid Med Cell Longev. 2019;2019:4532592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sanbe A, Osinska H, Saffitz JE, Glabe CG, Kayed R, Maloyan A, Robbins J. Desmin‐related cardiomyopathy in transgenic mice: a cardiac amyloidosis. Proc Natl Acad Sci USA. 2004;101:10132–10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Maloyan A, Sanbe A, Osinska H, Westfall M, Robinson D, Imahashi K, Murphy E, Robbins J. Mitochondrial dysfunction and apoptosis underlie the pathogenic process in alpha‐B‐crystallin desmin‐related cardiomyopathy. Circulation. 2005;112:3451–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maloyan A, Sayegh J, Osinska H, Chua BH, Robbins J. Manipulation of death pathways in desmin‐related cardiomyopathy. Circ Res. 2010;106:1524–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maloyan A, Gulick J, Glabe CG, Kayed R, Robbins J. Exercise reverses preamyloid oligomer and prolongs survival in alphaB‐crystallin‐based desmin‐related cardiomyopathy. Proc Natl Acad Sci USA. 2007;104:5995–6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gupta MK, McLendon PM, Gulick J, James J, Khalili K, Robbins J. UBC9‐mediated sumoylation favorably impacts cardiac function in compromised hearts. Circ Res. 2016;118:1894–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Su H, Li J, Zhang H, Ma W, Wei N, Liu J, Wang X. COP9 signalosome controls the degradation of cytosolic misfolded proteins and protects against cardiac proteotoxicity. Circ Res. 2015;117:956–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gupta MK, Gulick J, Liu R, Wang X, Molkentin JD, Robbins J. Sumo E2 enzyme UBC9 is required for efficient protein quality control in cardiomyocytes. Circ Res. 2014;115:721–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li J, Ma W, Li H, Hou N, Wang X, Kim IM, Li F, Su H. NEDD8 Ultimate buster 1 Long (NUB1L) protein suppresses atypical Neddylation and promotes the Proteasomal degradation of misfolded proteins. J Biol Chem. 2015;290:23850–23862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. 2016;27:105–117. [DOI] [PubMed] [Google Scholar]
- 51. Joshi AU, Saw NL, Vogel H, Cunnigham AD, Shamloo M, Mochly‐Rosen D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol Med. 2018;10:e8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sanbe A, Osinska H, Villa C, Gulick J, Klevitsky R, Glabe CG, Kayed R, Robbins J. Reversal of amyloid‐induced heart disease in desmin‐related cardiomyopathy. Proc Natl Acad Sci USA. 2005;102:13592–13597. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors declare that all supporting data and method descriptions are available within the article or from the corresponding author upon reasonable request.
Materials
Materials are as follows: Cell Lytic M (Sigma‐Aldrich), Protease Inhibitor Cocktail (Roche), pre‐cast 7.5% to 15% Criterion Gels (BioRad), Oligomycin (Sigma‐Aldrich), carbonyl cyanide‐p‐trifluoromethoxy‐phenylhydrazone (FCCP) (Sigma‐Aldrich), Rotenone (Sigma‐Aldrich), Antimycin A (Sigma‐Aldrich), and Ponceau S (Acros Organic) were used.
Animals
CryABR120G transgenic mice have been described previously and are in an FVB/N background. 20 All procedures for handling animals complied with the Guide for Care and Use of Laboratory Animals and were approved by the ACUC Committee of LSU Health Sciences Center‐Shreveport. All animals were cared for according to the National Institutes of Health guidelines for the care and use of laboratory animals.
Echocardiography
Echocardiograms were performed on isoflurane‐anesthetized mice with a VisualSonics Vevo 3100 Imaging System to assess cardiac functional parameters as described previously. 24 , 25 , 26 Briefly, we recorded the M‐mode echocardiographic images along the parasternal short axis by investigators blinded to genotype to determine left ventricular (LV) size and systolic function. M‐mode measurements included the LV internal dimensions in systole and diastole (LVID;s and LVID;d, respectively) as well as the diastolic thickness of LV posterior wall, and diastolic intraventricular septum thickness. Percent fractional shortening was calculated using: [(LVIDd‐LVIDs)/LVIDd] × 100. From these data, LV end‐systolic, LV end‐diastolic diameter, LV mass, and percent ejection fraction were calculated. Systolic shortening of the interventricular septum thickness and posterior wall was calculated using: [(systolic thickness‐diastolic thickness)/systolic thickness] × 100.
Mitochondria Isolation
Mitochondria from the hearts of CryABR120G transgenic and nontransgenic mice were isolated, as described previously. 24 , 25 , 26 Briefly, hearts were harvested, homogenized in MS‐EGTA buffer (225 mM mannitol, 75 mM sucrose, 5 mM HEPES, and 1 mM EGTA, pH 7.4) and subjected to differential centrifugation. Finally, mitochondria were lysed with 1x cell lytic M (Sigma‐Aldrich) containing protease and phosphatase inhibitors.
Mitochondrial Respiration
Mitochondrial oxygen consumption rate was measured with an XF24 Extracellular Flux Analyzer (Seahorse Biosciences, North Billerica, MA) by methods as described previously. 24 , 25 , 26 Mitochondria were isolated from the mix‐sexed age‐matched CryABR120G and nontransgenic hearts using MS‐EGTA buffer (225 mM mannitol, 75 mM sucrose, 5 mM HEPES, and 1 mM EGTA, pH 7.4) by differential centrifugation as described above. Mitochondria (50 μg/well) were seeded in XF24 culture plates, and respiration was measured in mitochondrial assay buffer (220 mM mannitol, 7M mM sucrose, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA, 0.2% fatty acid‐free bovine serum albumin, pH 7.4) supplemented with 7 mM pyruvate and 1 mM malate. Mitochondrial oxygen consumption rate (OCR) was measured and plotted at basal conditions followed by sequential addition of 1 μg/mL oligomycin (ATP‐synthase inhibitor), 4 μM FCCP (a mitochondrial uncoupler), and 0.5 μM rotenone (complex I inhibitor) plus 0.5 μM antimycin A (complex III inhibitor). The OCR values were normalized to total protein content in the corresponding wells and expressed as pmol/min per µg protein.
electron transport chain complex activity assays
Mitochondrial electron transport chain complex activities were measured in isolated mitochondria spectrophotometrically as described previously. 25 , 26 Briefly, complex I activity was determined by measuring NADH (100 µmol/L) oxidation with and without the presence of complex I inhibitor (rotenone, 10 µmol/L) at 340 nm using a DS‐11 FX+spectrophotometer (DeNovix). Specific complex I activity was calculated as rotenone sensitive activity expressed as nmol min−1 mg−1 of mitochondrial protein. Complex II activity was determined by measuring the reduction of 2,6‐dichlorophenolindophenol (DCPIP) coupled to complex II catalyzed reduction of decylubiquinone with and without the presence of 10 µmol/L antimycin A, 10 µmol/L rotenone, 250 µmol/L KCN, 80 µmol/L DCPIP and 60 µmol/L decylqubiquinone. Complex III activity was determined by measuring the reduction of cytochrome C (75 µmol/L) with and without complex III inhibitor (antimycin A, 10 µmol/L). Specific complex III activity was calculated as antimycin A sensitive activity expressed as nmol min−1 mg−1 of mitochondrial protein. Extinction coefficient values for NADH (8 mM‐1 cm‐1), DCPIP (19.1 mM−1 cm−1), and reduced cytochrome C (18.5 mM‐1 cm‐1) were used to calculate complex I, II, and III activities, respectively.
Immunofluorescence microscopy
Paraffin‐embedded heart sections were used for immunofluorescence analyses as we described previously. 20 The following primary antibodies were used: mouse anti–troponin I (1:1000; Millipore), rabbit anti‐CryAB (1:400; Assay Designs). Alexa Fluor 488– or Alexa Fluor 568–conjugated secondary antibody (Molecular Probes) directed against mouse or rabbit IgG was used, and 4′,6‐diamidino‐2‐phenylindole (Invitrogen) was used to identify nuclei. Immunofluorescent staining for CryAB was used to quantitate the cytoplasmic area occupied by the aggregates, as described previously. 20
Cell fractionation, SDS‐PAGE, and Western blot analyses
Soluble and insoluble fractions of heart lysate were prepared by methods as we described previously. 20 , 26 Briefly, hearts were harvested in cold PBS (pH 7.4) containing 1% Triton‐X100, 2.5 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride, and a complete protease inhibitor mixture and then vortexed for 30 s. The heart extracts were centrifuged at 12 000g for 15 minutes, and the supernatants were collected (soluble fraction). The pellets were dissolved in DNAase (1 mg/kL in 10 mM Tris, 15 mM MgCl2; Roche) and sonicated on ice, and the protein was quantitated with a modified Bradford assay. The insoluble protein was then diluted in RIPA buffer, and 3 μg insoluble protein resuspended was used for subsequent immunoblotting with appropriate antibodies.
Whole‐cell lysate fractions were prepared from hearts as described previously. 20 The heart homogenates were centrifuged at 12 000g for 15 minutes and the supernatants were collected (soluble fraction). The protein content of the soluble lysates was measured using the modified Bradford protocol/reagent relative to a BSA standard curve (BioRad). Protein lysates (50 µg for the whole cell lysate and 30 µg for the mitochondrial fraction) were separated on SDS‐PAGE using pre‐cast 7.5% to 15% Criterion Gels (BioRad) and transferred to polyvinylidene difluoride membranes (BioRad). Membranes were blocked for 1 hour in 5% nonfat dried milk and exposed to primary antibodies overnight. The following primary antibodies were used for immunoblotting: anti‐Drp1 (1:1000, Cell Signaling Technology), anti‐pDrp1 Ser616 (1:1000, Cell Signaling Technology), anti‐mitofusin 1 (MFN1) (1:1000, Cell Signaling Technology), anti‐MFN2 (1:1000, Cell Signaling Technology), anti‐OPA1 (1:1000, Cell Signaling Technology), anti‐GAPDH (1:5000, EMD Millipore), anti‐β‐Actin (1:1000, Santa Cruz Biotechnology), anti‐α‐sarcomeric actinin (1:1000, Sigma‐Aldrich), anti‐OXPHOS (1:1000, Abcam), anti‐PDH (1:1000, Abcam), anti‐CryAB (1:10 000, Enzo Life Sciences), anti‐Periostin (1:500, Santa Cruz Biotechnology), and anti‐smooth‐muscle actin (1:500, Cell Signaling Technology). Membranes were then washed, incubated with alkaline‐phosphatase‐conjugated secondary antibodies (Santa Cruz Biotechnology), exposed with ECF reagent (Amersham), and finally, detected on a ChemiDoc Touch Imaging System (BioRad). Ponceau S protein stain of the transfer membrane was used as a loading control. Densitometry on scanned membranes was done using ImageJ software (National Institutes of Health, Bethesda, MD).
Nongradient blue native polyacrylamide gel electrophoresis
Mitochondrial extracts were prepared for blue native polyacrylamide gel electrophoresis (BN‐PAGE) analysis as described previously. 24 , 25 , 26 Briefly, fresh mitochondrial pellets were suspended in a solubilization buffer containing 50 mM Bis‐Tris (pH 7.0), 1% n‐dodecyl‐β‐d‐maltoside (vol/vol) and 750 mM ε‐amino‐N‐caproic acid. 27 The suspension was kept on ice for 1 hour with occasional vortexing followed by clarification with centrifugation at 8000g for 10 minutes. Following the centrifugation, the supernatant was quantified for protein concentration by the Bradford method and mixed with concentrated BN‐PAGE loading buffer (10×) containing 0.5 M ε‐amino‐N‐caproic acid and 3% Serva Blue G‐250 (wt/vol). The samples were run in an acrylamide/bisacrylamide gel containing 4% stacking gel, and 8% of the resolving gel. 28 Nongradient BN‐PAGE was performed at room temperature using gel buffer (500 mM aminocaproic acid and 50 mM Bis‐Tris, pH 7.0), cathode buffer (50 mM Tricine, 15 mM Bis‐Tris, pH 7.0), 0.02% Serva blue G‐250 (wt/vol), anode buffer (50 mM Bis‐Tris pH 7.0), and sample buffer (75 mM aminocaproic acid containing 0.3% Serva blue G‐250 [wt/vol]). 28 Proteins (30 μg) were loaded in the gel and run at 150 V until the front line had entered into two‐third of the gel. Next, the cathode buffer was replaced with buffer without Serva blue G‐250 (50 mM Tricine, 15 mM Bis‐Tris, pH 7.0) and gel running was completed at 250 V. Gel was stained with Coomassie blue (BioRad) using previously described methods. 29
Pyruvate dehydrogenase enzymatic activity
Pyruvate dehydrogenase (PDH) activity was measured in isolated mitochondria with the PDH Activity Assay Kit (MAK183, Sigma‐Aldrich) according to manufacturer’s instruction. 30 Mitochondria (150 µg) were homogenized in 50 µL of ice‐cold PDH assay buffer, and centrifuged at 10 000g for 5 minutes. Ten microliters of lysates was used for reactions. Reactions were carried out at 37°C, and A450 was recorded every minute for 30 minutes. PDH activity rates were measured in nmol/min per mg using the NADH standard curve.
Citrate synthase activity
Citrate synthase activity was measured in isolated mitochondria and whole heart homogenates as described previously. 31 Briefly, whole heart homogenates and isolated mitochondria were homogenized with Cell‐lytic‐M (Sigma Aldrich) supplemented with protease inhibitor. Mitochondrial lysate (10 µg) was used in reaction mixture containing 0.25% Triton X‐100, 0.31 mM acetyl CoA, and 0.1 mM 5,5´‐dithiobis(2‐nitrobenzoic acid). Finally 0.5 mM oxaloacetate was added in 1 mL reaction volume and citrate synthase activities were measured using a DS‐11 FX+spectrophotometer (DeNovix) at 412 nm at 37°C. Activity was expressed as nmol/min per mg of mitochondrial protein. Extinction coefficient values for TNB (13.6 mM‐1 cm‐1) were used to measure the citrate synthase enzyme activity.
Statistical Analysis
Data are expressed as mean±SEM. All statistical tests were done with GraphPad Prism software. All data were tested for normality using the Kolmogorov‐Smirnov test, and data that passed the normality assumption were analyzed using Student t test (P<0.05) for 2 groups and groups of 3 or more with 1‐way ANOVA, followed by the Tukey post hoc test. For certain data sets with smaller sample sizes (n=3–6), the Kruskal‐Wallis test was applied, and data were presented in graphs showing median and interquartile ranges. A value of P<0.05 was considered statistically significant.