

Version Changes
Revised. Amendments from Version 1
The following author was added: Robert A Scott. This author was omitted in the initial submission as an accident. All co-authors are aware (from previous drafts of the paper) that this author should be included. A conflict of interest statement was also added.
Abstract
Drugs whose targets have genetic evidence to support efficacy and safety are more likely to be approved after clinical development. In this paper, we provide an overview of how natural sequence variation in the genes that encode drug targets can be used in Mendelian randomization analyses to offer insight into mechanism-based efficacy and adverse effects. Large databases of summary level genetic association data are increasingly available and can be leveraged to identify and validate variants that serve as proxies for drug target perturbation. As with all empirical research, Mendelian randomization has limitations including genetic confounding, its consideration of lifelong effects, and issues related to heterogeneity across different tissues and populations. When appropriately applied, Mendelian randomization provides a useful empirical framework for using population level data to improve the success rates of the drug development pipeline.
Keywords: Drugs, Genetics, Mendelian randomization
Introduction
The majority of small molecule and biologic drugs exert their effects by perturbing protein targets 1. The identification of such targets is therefore central to drug discovery. Despite increasing investment in research and development within the pharmaceutical industry 2, overall drug development failure rates remain high 3– 8, most notably for targets that represent novel mechanisms. Such failures result in increased costs and reduced availability of novel agents 9.
With the recent growth in genetic data 10, there has been substantial progress in the identification of genes that are linked to human health and disease. Genetic data can potentially be used for identifying and prioritizing novel drug targets and indications 2. For example, genome-wide association studies (GWAS) have corroborated approximately 70 of the 670 known effects of licensed drugs through associations at the loci of the genes coding for their corresponding target proteins 11. Studies of drug development programs have also shown that targets with genomic support have a higher rate of success 2, 12– 15.
Mendelian randomization
Through the random allocation of genetic variants at conception, genetic studies in human populations can imitate the design of randomized controlled trials (RCT) 16, 17. Such investigation uses genetic variants as instrumental variables for studying the effect of an exposure on an outcome, and has been referred to as Mendelian randomization (MR) 18. Phenotypic observational studies are limited in their ability to draw causal inferences due to bias from confounding and reverse causation 18. In contrast, MR uses the random allocation of genetic variants from parents to offspring during conception to guard against these biases.
MR requires the following instrumental variable assumptions: the genetic variant i) is associated with the exposure (relevance), ii) has no common cause with the outcome (independence), and iii) only affects the outcome via the exposure (exclusion restriction) 19, 20. The first of these is testable; the remaining assumptions are untestable but falsifiable. Assumption iii) the exclusion restriction, assumes that the genetic variant affects the outcome through the exposure and not any other horizontally pleiotropic pathways 18, 21. Further assumptions are also required to obtain valid point estimates, for instance, that the influence of the exposure on the outcome is the same for all individuals (effect homogeneity) or that the exposure is a monotonic (always increasing or always decreasing) function of the instrument for all individuals in the population (monotonicity) 19. In addition, the interpretation of MR findings can have particular nuances, as previously described 22.
Where the exposure under study is perturbation of a drug target, MR can be used to explore drug effects ( Figure 1) 23, 24. For drug target MR specifically, genetic variants such as single-nucleotide polymorphisms (SNPs) related to the function or expression of the drug target protein can be used as instrumental variables to study the effect of perturbing that drug target 25, 26. These variants are typically in or near the gene that encodes the drug target ( cis-variants). Such MR can be used in drug development to investigate the likely efficacy and safety of perturbing novel drug targets 27, 28, as well as explore the repurposing potential and adverse effects of existing drugs 25.
Figure 1. Principles of Mendelian randomization studies (MR) studying drug effects.

MR makes use of genetic variants located within or close to a gene encoding a drug target (e.g. at HMGCR encoding the drug target of statins) that lead to downstream effects similar to the desired drug response (e.g. lowering of low-density lipoprotein [LDL] cholesterol) in order to explore effects on clinical outcomes (e.g. risk of coronary artery disease). SNP: single-nucleotide polymorphism.
The identification and validation of appropriate genetic variants as instrumental variables for an exposure is critical for the design and interpretation of all MR analyses 29. While previous work has offered practical advice on selecting instruments for MR studies considering disease biomarkers 24, the field is continuing to evolve rapidly 30. The growth in genetic association study data that extends to tissue-specific gene expression 31, circulating proteins 32, metabolites 33, 34 and cytokines 35, has been coupled with increased efficiency of MR studies using automated software, databases, statistical packages and readily available code 30, 36– 38. However, there is still no consensus on the strategy for identifying genetic instruments and exploring potential drug effects with MR. Here we discuss practical considerations while also offering illustrative examples for the most relevant points. We describe issues relating to selection of genetic variants as proxies for drug target perturbation, evaluation of the plausibility of genetic variants as proxies for drug target perturbation, generation and interpretation of MR estimates, and limitations of MR for investigating drug target perturbation. Finally, we offer a step-by-step framework for how to conduct a drug target MR study ( Box 1).
Box 1. Step-by-step guide for conducting Mendelian randomization (MR) analyses of drug target perturbation.
-
1.
Determine the drug targets of interest
-
2.
Identify the gene(s) encoding the relevant protein(s)
-
3.
Choose data source for identifying instruments
-
4.
Select genetic variants as instruments based on:
-
a.
Strength of associations with downstream effects of drug target perturbation
-
b.
Linkage disequilibrium structure
-
c.
Distance from gene(s) encoding the drug target
-
a.
-
5.
Validate genetic variants for use as instruments by confirming that they recapitulate known on-target drug effects
-
6.
Estimate effects of drug target perturbation on outcome(s) of interest using MR
-
a.
Use appropriate method to account for linkage disequilibrium structure between variants
-
b.
Scale estimates appropriately
-
c.
Interpret MR as representing effects of lifelong drug target perturbation
-
a.
-
7.
Investigate potential adverse effects and repurposing opportunity using phenome-wide association study
-
8.
Triangulate using other interventional, observational and experimental data
Instrument selection
MR investigations of drug effects have mainly studied small molecule, peptide and biotherapeutic drugs 39, 40, where genetic instruments are selected as variants that mimic perturbation of their protein targets. Instrument selection can be considered in two parts: i) identifying the gene or group of genes corresponding to the drug target proteins and ii) selecting genetic variants to proxy perturbation of the drug targets. These steps are discussed in detail below, followed by consideration of drugs that have targets made up of multiple proteins.
Identifying genes corresponding to drug target proteins
The key difference between conventional MR for an exposure and MR for the investigation of drug effects is that for the latter the instrument can be constructed in relation to the gene corresponding to the drug target, rather than genetic variants from across the genome ( Table 1). The first step of this process is therefore to identify the drug target of interest and its corresponding gene. Resources such as DrugBank (which is freely available for non-commercial purposes) provide information about existing drugs, including their mechanism of action, targets and their corresponding gene(s), and indications 41. Where the target of a drug is known, information regarding the corresponding gene can also be obtained from other databases such as Ensembl and UniProt 42, 43.
Table 1. Differences between conventional Mendelian randomization (MR) and MR specifically exploring drug target perturbation.
| Conventional MR | MR investigating drug effects | |
|---|---|---|
| Aim of the analysis | To investigate the effect of an exposure on an
outcome |
To investigate the effect of perturbing a drug target on an
outcome |
|
Genomic location of
instruments |
Genome-wide | Often restricted to the locus of the gene encoding the drug
target under study |
|
Selection of genetic
instruments |
Variants associated with the exposure under
study |
Variants associated with perturbation of the drug target under
study |
| Statistical analysis | Typically uses uncorrelated variants; higher
risk of pleiotropic effects on the outcome through pathways unrelated to the exposure |
More frequent use of methods to account for correlation
between instrument variants; lower risk of pleiotropic effects on the outcome through pathways unrelated to the drug target |
Selecting genetic variants to proxy drug targets
Several factors need to be considered when selecting genetic variants to proxy the effects of drug target perturbation. If MR is being used to investigate effects of perturbing the target of a drug with an existing indication, then instruments can be selected based on their location at the corresponding gene and association with that indication. For instance, Gill et al. selected genetic variants to proxy antihypertensive drug class effects as those located at the gene corresponding to the drug target that also related to systolic blood pressure in a GWAS 44. If the indication is not known, one possible approach is to use quantitative trait loci for expression of the gene encoding the drug target of interest (in relevant tissues or cell contexts) as instruments for drug target perturbation. An important limitation of gene expression is that variants affecting gene expression may not necessarily also affect protein expression, and vice versa ( Figure 2) 45. Furthermore, gene expression quantitative loci have been reported to account for little of the heritability of complex diseases 46. Therefore, protein expression quantitative loci may make better instruments for proxying drug effects than gene expression data, if they are available in relevant tissues and contexts.
Figure 2. Potential strategies for selecting genetic variants as instruments for a protein drug target.
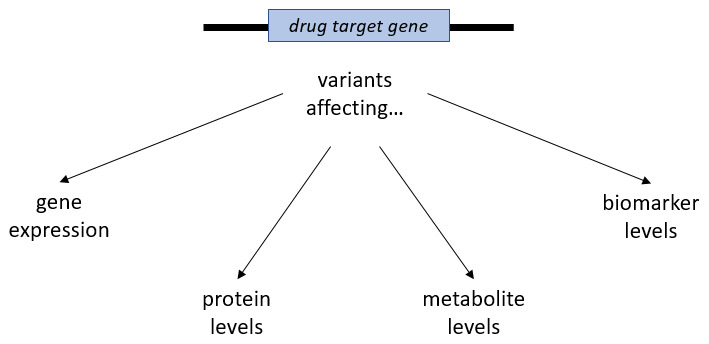
Variants within or close to the drug target gene might be selected on the basis of their associations with gene expression and levels of the target protein or known downstream functions such as effects on metabolite levels or biomarkers. Notably, variants influencing gene expression and protein levels do not always influence the function of the derived protein product and might not translate to downstream effects comparable to those achieved by the pharmacological modulation of the drug target.
As mentioned above, selected instruments for drug target perturbation are often restricted to cis-acting genetic variants – those in or close to the gene of interest. In general, such variants are more likely to have effects specific to the protein of interest than genetic variants that are not located within the gene locus ( trans-acting). This point is well highlighted by the example of C-reactive protein, for which MR analyses investigating effects of its circulating levels produce very different results depending on whether the instruments are selected from the CRP locus or from throughout the genome 24. Related considerations include how proximal cis-variants should be to the corresponding gene, and whether enhancer or promoter regions for the gene should be counted as cis-acting 47. While some evidence currently supports that genetic variants affecting gene expression typically lie within 200kB of the gene locus 48, there is no established consensus on the issue of proximity. The pertinent requirement is that the variant be related to the function or expression of the drug target. In the past, single-region MR analyses have sometimes used only the top variant in the region as a genetic instrument 28, 49, 50. However, this approach can also be suboptimal if studying a region containing variants that have multiple conditionally independent associations with the exposure. For example, genetic association studies have suggested that the SHBG region encoding sex-hormone binding globulin (SHBG) may harbor up to nine variants independently associated with circulating SHBG concentration 51, and that using only the top variant may limit the statistical power of such MR analysis.
The degree to which variants at the same locus should be allowed to correlate with each other through linkage disequilibrium (LD) while still being modelled as independent also warrants attention. Unaccounted correlation between the variants used can result in underestimation of the standard error of MR estimates, yet there is no recommended LD threshold. To circumvent this issue, methods are available to adjust for LD between genetic variants used as instruments, which may help confirm the robustness of the findings and maximize statistical power 52– 54.
Investigating drugs with multiple targets
Many drugs do not have a target that is encoded by a single gene. For example, the calcium channel blocker class of antihypertensive drugs have targets that are made up of proteins coded by several different genes 44, 55. At present there is no consensus on the best way to combine data from multiple genes corresponding to a single target into an instrument. Previous studies have selected genetic variants related to the individual genes and combined their data to investigate the effect of perturbing the drug target, while applying clumping to ensure independence as described above 44, 55.
Instrument evaluation
Once the instrument has been selected, it can be evaluated to ascertain its validity for the analysis of interest. MR analyses exploring drug effects can be biased if the genetic variants incorporated as instruments have “horizontal” pleiotropic effects, where there are pathways from the variant to the specific outcome under consideration that do not pass through the exposure of interest 56. In contrast, “vertical” pleiotropy lies on the causal pathway between the pharmacological mechanism and outcome 56. Vertical pleiotropy does not bias MR estimates and is often of interest as it can provide insight into causal mechanisms and mediation. As with MR generally, one of the most useful approaches for evaluating instrument validity is to investigate its relation to a known effect of the exposure under consideration 57. This approach is feasible for MR used to predict the effect of perturbing targets for which there are drugs with established indications and known associations with biomarkers 58. For example, Walker et al. selected genetic variants to proxy antihypertensive drugs from gene expression data and validated these instruments through their associations with systolic blood pressure, prior to applying MR analyses investigating the outcome of interest, Alzheimer’s disease 55. An instrument may also be examined in relation to potential confounders, in order to investigate violations of the independence and exclusion restriction assumptions necessary for MR 56. Berry et al. illustrated such an approach during their evaluation of genetic proxies for vitamin D status 59. In this study, the association of variants with social, dietary and lifestyle factors was investigated, to identify potential sources of confounding.
Complementary data may also be used for instrument evaluation. For example, MR studies designed to investigate the effect of genetically predicted variations in interleukin-6 (IL6) signaling would be expected to show that the selected instruments associate with molecules that are downstream of the pathway 49. Genetic association estimates for the serum levels of several of these molecules are available, including IL6 and IL6 receptor (IL6R), C-reactive protein (CRP) and fibrinogen 60. Hence, if the selected genetic instruments are valid proxies for IL6 signaling, they may be expected to show consistent effects across these molecules. An alternative example is provided by Wurtz et al. who demonstrated consistency between the metabolic changes associated with starting statins and metabolomic associations of the HMGCR variant rs12916 that was used to proxy statin effect 61.
Analysis
Given a set of genetic instruments, the statistical methods used for MR investigation of drug target perturbation are similar to those used for MR more generally 62. Interpretability is often facilitated by scaling of genetic associations to unit change in a trait related to drug target perturbation. For example, for analyses considering associations of variants in the HMGCR gene that are used to proxy statin drug effects, estimates may be scaled to change in low-density lipoprotein cholesterol levels 50, 63– 65. As another example, for analyses investigating IL6R signaling using variants in the IL6R gene, effects may be scaled to downstream changes in CRP levels 49, 60. Care must be taken in the interpretation of such scaled estimates however, because although MR estimates may be directionally concordant to the effect of drug target perturbation on the biomarker, their magnitudes may not be comparable 66.
Statistical approaches used to evaluate potential bias from horizontal pleiotropy in MR analyses can also be used in MR investigating drug target perturbation 62. However, variants selected as instruments for drug target perturbation are often selected from within a specific locus rather than from throughout the genome, and may be limited in number. Statistical sensitivity analyses for investigating horizontal pleiotropy typically require large numbers of genetic variants, and so may not be suitable for many drug target MR analyses 29. Assessment of heterogeneity between MR estimates produced by variants in a single locus is still possible however, and can be used to inform on potential bias related to horizontal pleiotropy 53, 67.
In an effort to better explore the target region and increase statistical power, genetic variants that have weaker associations with perturbation of the drug target may be considered as instruments 27, 68. Despite the potential benefits of this approach 69, care must be taken to avoid weak instrument bias 54, 70. Under a two-sample design, weak instrument bias will attenuate MR estimates towards the null 71.
MR can be used to assess a wide range of outcome traits and thus investigate potential effects of perturbing the drug target on these traits 72. Such studies are often conducted as hypothesis-free, phenome-wide association analyses (PheWAS) 73, 74, and can be helpful for exploring potential adverse effects or identifying previously unknown re-purposing opportunities. For example, Schmidt et al. conducted a PheWAS of the PCSK9 locus to assess potential adverse effects of PCSK9 inhibitor drugs 75.
In addition to using MR, it is also possible to generate genetic evidence supporting a causal effect of drug target perturbation on an outcome by identifying proportionality of genetic associations with traits proxying drug target perturbation and the outcome, at the corresponding drug target gene locus. Such investigation is referred to as genetic colocalization, and can help distinguish causation from genetic confounding (such as may arise due to horizontal pleiotropy). Popular colocalization methods include coloc 76, moloc 77, eCAVIAR 78 and HEIDI 79. However, a limitation of many colocalization approaches is that they assume there is only a single causal variant at the considered locus.
Triangulation, the practice of integrating evidence from several different methodological approaches and data sources that each differ in their susceptibility to bias, is another important aspect of interpreting the analysis 80. MR evidence should be considered alongside other study designs to increase confidence in findings 58. For example, the European Atherosclerosis Society consensus statement on the role of low-density lipoproteins on atherosclerotic cardiovascular disease considers evidence from inherited disorders of lipid metabolism, prospective epidemiologic studies, MR investigations and RCTs 81. Moreover, comparing instruments between different MR studies of the same exposure can provide additional evidence. For example, both Gill et al. and Walker et al. independently derived instruments for antihypertensive drug effects that perform comparably when tested against a common outcome 44, 55. Although different MR studies may use similar or overlapping data sources, different instrument selection approaches can make analyses vulnerable to distinct biases and so also have a role in triangulation of evidence.
Limitations
As with all research methods, MR has limitations 82. RCTs remain the best source of evidence evaluating drug efficacy and guiding clinical practice 83. While MR and RCTs have the same aim – reliable evidence of causation – they estimate different treatment parameters, which are not directly comparable. Genetic variants typically have smaller effects which accumulate across the entire life-course, whereas pharmacological agents are often prescribed later in life and typically have larger effects. Therefore, MR estimates reflect the lifelong effects of perturbing a drug target, which may not be equivalent to interventions given at a specific point in time and for a shorter time period ( Figure 3). While these differences make it unlikely that MR estimates will accurately reflect the size of effect of a pharmacological intervention, they are still a useful indication of presence and direction of causal effects 58.
Figure 3. Comparison between Mendelian randomization (MR) study for drug effects and randomized clinical trial (RCT).

Similar to the randomization process of RCTs, the random allocation of alleles at a drug target gene in MR studies allows the distribution of individuals to groups that differ only regarding the downstream effects of the drug target and not other confounders. While the random allocation of alleles in MR studies happens at conception and leads to lifelong effects, the randomization in RCTs typically happens later in life and focuses on the effects of short-term interventions.
A further limitation of MR for studying the effects of drug target perturbation is that it may not account for post-transcriptional and post-translational modification in the pathway from a gene to a biologically functional protein. Well-conducted MR analyses may be able to inform broadly on drug class effects, but not necessarily provide information on the effects of a specific pharmacological agent. For example, dihydropyridine and non-dihydropyridine subclasses of calcium-channel blocker antihypertensive drugs have distinct pharmacological effects. Genetic variants that affect blood pressure via calcium-channel blockade can estimate the effects of calcium-channel targeting drugs in general, but cannot differentiate the relative effects of dihydropyridine versus non-dihydropyridine subclasses 44. Furthermore, MR in this context is applied to drug targets and not compounds – so it can be used to investigate the effects of perturbing a drug target, but is unlikely to be able to offer insight towards molecule specific effects 28.
Drug effects also vary in different tissues and populations, and similarly MR estimates for the effects of perturbing drug targets may only be valid if genetic association data from the relevant tissues or populations are used. This limitation can have implications for both identifying instruments and using MR to study drug effects, as highlighted in an example that used gene expression data to identify instruments for antihypertensive drug classes in the investigation of repurposing potential for the prevention of Alzheimer’s disease 55. Here, it is not clear whether the same genetic variants related to gene expression in vascular, cardiac and brain tissue. Furthermore, to date, most genotyped samples have been sampled from European ancestry populations. While this approach minimizes the risk of population stratification and false-positive GWAS signals, consideration of distinct ancestral groups is likely to offer novel insight. For example, genetic evidence on the effects of alcohol comes from variants in the ALDH2 gene, which are common in Asian, but not European populations 84.
Conclusion
Over the last decade, MR has become a widely used epidemiological tool for estimating the causal effects of risk factors on clinical outcomes. On top of this well-studied application, there are now multiple examples highlighting its power for investigating drug effects. Despite its explicit assumptions, modern developments in statistical methodology and the widespread availability of multiple levels of omics data have provided the necessary resources to more reliably and efficiently use MR in order to study drug effects. As such, it has found a growing niche within the broader framework for exploring therapeutic targets, efficacy, adverse effects and repurposing potential. Given the high failure rates of clinical trials and that drug targets with genetic support are more likely to make it through the development pipeline 13, 15, 23, MR can provide evidence for prioritizing agents to move forward in development.
Data availability
No data is associated with this article.
Funding Statement
DG is supported by the Wellcome Trust 4i Programme (203928/Z/16/Z) and British Heart Foundation Centre of Research Excellence (RE/18/4/34215) at Imperial College London, and by a National Institute for Health Research Clinical Lectureship at St. George's, University of London (CL-2020-16-001). MKG has received funding by the Onassis Foundation and the German Academic Exchange Service (DAAD). AG is funded by a Medical Research Council Methodology Research Panel Grant (RG88311). This project has received funding from the European Union’s Horizon 2020 research and innovation programme (No 666881, to MD,), SVDs@target (to MD) and No 667375, CoSTREAM (to MD); the DFG as part of the Munich Cluster for Systems Neurology (EXC 1010 SyNergy, to MD) and the CRC 1123 (B3, to MD); the Corona Foundation (to MD); the Fondation Leducq (Transatlantic Network of Excellence on the Pathogenesis of Small Vessel Disease of the Brain) (to MD). CF and ADH are supported by the UCL NIHR Biomedical Research Centre and UCL BHF Research Accelerator. ADH is an NIHR Senior Investigator. SB is supported by Sir Henry Dale Fellowship jointly funded by the Wellcome Trust and the Royal Society (204623/Z/16/Z). MVH works in a unit that receives funding from the UK Medical Research Council and is supported by a British Heart Foundation Intermediate Clinical Research Fellowship (FS/18/23/33512) and the National Institute for Health Research Oxford Biomedical Research Centre. JZ is funded by a Vice-Chancellor Fellowship from the University of Bristol. The Medical Research Council (MRC) and the University of Bristol support the MRC Integrative Epidemiology Unit (MC_UU_12013/1, MC_UU_12013/9, MC_UU_00011/1). The Norwegian Research Council support NMD grant number 295989. This work was supported by the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) National Heart, Lung, and Blood Institute (NHLBI) grant R01HL105756-09.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
[version 2; peer review: 3 approved, 1 approved with reservations]
References
- 1. Santos R, Ursu O, Gaulton A, et al. : A comprehensive map of molecular drug targets. Nat Rev Drug Discov. 2017;16(1):19–34. 10.1038/nrd.2016.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Plenge RM, Scolnick EM, Altshuler D: Validating therapeutic targets through human genetics. Nat Rev Drug Discov. 2013;12(8):581–94. 10.1038/nrd4051 [DOI] [PubMed] [Google Scholar]
- 3. Paul SM, Mytelka DS, Dunwiddie CT, et al. : How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nat Rev Drug Discov. 2010;9(3):203–14. 10.1038/nrd3078 [DOI] [PubMed] [Google Scholar]
- 4. Hay M, Thomas DW, Craighead JL, et al. : Clinical development success rates for investigational drugs. Nat Biotechnol. 2014;32(1):40–51. 10.1038/nbt.2786 [DOI] [PubMed] [Google Scholar]
- 5. Munos B: Lessons from 60 years of pharmaceutical innovation. Nat Rev Drug Discov. 2009;8(12):959–68. 10.1038/nrd2961 [DOI] [PubMed] [Google Scholar]
- 6. Pammolli F, Magazzini L, Riccaboni M: The productivity crisis in pharmaceutical R&D. Nat Rev Drug Discov. 2011;10(6):428–38. 10.1038/nrd3405 [DOI] [PubMed] [Google Scholar]
- 7. Scannell JW, Blanckley A, Boldon H, et al. : Diagnosing the decline in pharmaceutical R&D efficiency. Nat Rev Drug Discov. 2012;11(3):191–200. 10.1038/nrd3681 [DOI] [PubMed] [Google Scholar]
- 8. Kola I, Landis J: Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3(8):711–5. 10.1038/nrd1470 [DOI] [PubMed] [Google Scholar]
- 9. Wouters OJ, McKee M, Luyten J: Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018. JAMA. 2020;323(9):844–53. 10.1001/jama.2020.1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Welter D, MacArthur J, Morales J, et al. : The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42(Database issue):D1001–6. 10.1093/nar/gkt1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Finan C, Gaulton A, Kruger FA, et al. : The druggable genome and support for target identification and validation in drug development. Sci Transl Med. 2017;9(383):eaag1166. 10.1126/scitranslmed.aag1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cook D, Brown D, Alexander R, et al. : Lessons learned from the fate of AstraZeneca's drug pipeline: a five-dimensional framework. Nat Rev Drug Discov. 2014;13(6):419–31. 10.1038/nrd4309 [DOI] [PubMed] [Google Scholar]
- 13. Nelson MR, Tipney H, Painter JL, et al. : The support of human genetic evidence for approved drug indications. Nat Genet. 2015;47(8):856–60. 10.1038/ng.3314 [DOI] [PubMed] [Google Scholar]
- 14. Sanseau P, Agarwal P, Barnes MR, et al. : Use of genome-wide association studies for drug repositioning. Nat Biotechnol. 2012;30(4):317–20. 10.1038/nbt.2151 [DOI] [PubMed] [Google Scholar]
- 15. King EA, Davis JW, Degner JF: Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. 2019;15(12):e1008489. 10.1371/journal.pgen.1008489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hingorani A, Humphries S: Nature's randomised trials. Lancet. 2005;366(9501):1906–8. 10.1016/S0140-6736(05)67767-7 [DOI] [PubMed] [Google Scholar]
- 17. Thanassoulis G, O'Donnell CJ: Mendelian randomization: nature's randomized trial in the post-genome era. JAMA. 2009;301(22):2386–8. 10.1001/jama.2009.812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Davey Smith G, Ebrahim S: Mendelian randomization: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32(1):1–22. 10.1093/ije/dyg070 [DOI] [PubMed] [Google Scholar]
- 19. Burgess S, Small DS, Thompson SG: A review of instrumental variable estimators for Mendelian randomization. Stat Methods Med Res. 2017;26(5):2333–55. 10.1177/0962280215597579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lawlor DA, Harbord RM, Sterne JAC, et al. : Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27(8):1133–63. 10.1002/sim.3034 [DOI] [PubMed] [Google Scholar]
- 21. Davey Smith G, Hemani G: Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89–98. 10.1093/hmg/ddu328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Holmes MV, Ala-Korpela M, Davey Smith G: Mendelian randomization in cardiometabolic disease: challenges in evaluating causality. Nat Rev Cardiol. 2017;14(10):577–90. 10.1038/nrcardio.2017.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hingorani AD, Kuan V, Finan C, et al. : Improving the odds of drug development success through human genomics: modelling study. Sci Rep. 2019;9(1):18911. 10.1038/s41598-019-54849-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Swerdlow DI, Kuchenbaecker KB, Shah S, et al. : Selecting instruments for Mendelian randomization in the wake of genome-wide association studies. Int J Epidemiol. 2016;45(5):1600–16. 10.1093/ije/dyw088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Walker VM, Davey Smith G, Davies NM, et al. : Mendelian randomization: a novel approach for the prediction of adverse drug events and drug repurposing opportunities. Int J Epidemiol. 2017;46(6):2078–89. 10.1093/ije/dyx207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schmidt AF, Finan C, Gordillo-Marañón M, et al. : Genetic drug target validation using Mendelian randomization. bioRxiv. 2019;781039. 10.1101/781039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ference BA, Ray KK, Catapano AL, et al. : Mendelian Randomization Study of ACLY and Cardiovascular Disease. N Engl J Med. 2019;380(11):1033–42. 10.1056/NEJMoa1806747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sofat R, Hingorani AD, Smeeth L, et al. : Separating the mechanism-based and off-target actions of cholesteryl ester transfer protein inhibitors with CETP gene polymorphisms. Circulation. 2010;121(1):52–62. 10.1161/CIRCULATIONAHA.109.865444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Burgess S, Davey Smith G, Davies NM, et al. : Guidelines for performing Mendelian randomization investigations [version 2; peer review: 2 approved]. Wellcome Open Res. 2019;4:186. 10.12688/wellcomeopenres.15555.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burgess S, Smith GD: How humans can contribute to Mendelian randomization analyses. Int J Epidemiol. 2019;48(3):661–4. 10.1093/ije/dyz152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. GTEx Consortium, Laboratory Data Analysis Coordinating Center Analysis Working Group, Statistical Methods Analysis Working Group, et al. : Genetic effects on gene expression across human tissues. Nature. 2017;550(7675):204–13. 10.1038/nature24277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sun BB, Maranville JC, Peters JE, et al. : Genomic atlas of the human plasma proteome. Nature. 2018;558(7708):73–9. 10.1038/s41586-018-0175-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kettunen J, Demirkan A, Wurtz P, et al. : Genome-wide study for circulating metabolites identifies 62 loci and reveals novel systemic effects of LPA. Nat Commun. 2016;7:11122. 10.1038/ncomms11122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shin SY, Fauman EB, Petersen AK, et al. : An atlas of genetic influences on human blood metabolites. Nat Genet. 2014;46(6):543–50. 10.1038/ng.2982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ahola-Olli AV, Wurtz P, Havulinna AS, et al. : Genome-wide Association Study Identifies 27 Loci Influencing Concentrations of Circulating Cytokines and Growth Factors. Am J Hum Genet. 2017;100(1):40–50. 10.1016/j.ajhg.2016.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zheng J, Baird D, Borges MC, et al. : Recent Developments in Mendelian Randomization Studies. Curr Epidemiol Rep. 2017;4(4):330–45. 10.1007/s40471-017-0128-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yavorska OO, Burgess S: MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol. 2017;46(6):1734–9. 10.1093/ije/dyx034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hemani G, Zheng J, Elsworth B, et al. : The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7:e34408. 10.7554/eLife.34408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Johnson DE: Biotherapeutics: Challenges and Opportunities for Predictive Toxicology of Monoclonal Antibodies. Int J Mol Sci. 2018;19(11):3685. 10.3390/ijms19113685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Scott DE, Bayly AR, Abell C, et al. : Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat Rev Drug Discov. 2016;15(8):533–50. 10.1038/nrd.2016.29 [DOI] [PubMed] [Google Scholar]
- 41. Wishart DS, Feunang YD, Guo AC, et al. : DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46(D1):D1074–D82. 10.1093/nar/gkx1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. UniProt Consortium: UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47(D1):D506–D15. 10.1093/nar/gky1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hunt SE, McLaren W, Gil L, et al. : Ensembl variation resources. Database (Oxford). 2018;2018:bay119. 10.1093/database/bay119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gill D, Georgakis MK, Koskeridis F, et al. : Use of Genetic Variants Related to Antihypertensive Drugs to Inform on Efficacy and Side Effects. Circulation. 2019;140(4):270–9. 10.1161/CIRCULATIONAHA.118.038814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zheng J, Haberland V, Baird D, et al. : Phenome-wide Mendelian randomization mapping the influence of the plasma proteome on complex diseases. bioRxiv. 2019;627398. 10.1101/627398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yao DW, O’Connor LJ, Price AL, et al. : Quantifying genetic effects on disease mediated by assayed gene expression levels. bioRxiv. 2020;730549. 10.1101/730549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fishilevich S, Nudel R, Rappaport N, et al. : GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford). 2017;2017:bax028. 10.1093/database/bax028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Võsa U, Claringbould A, Westra HJ, et al. : Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv. 2018;447367. 10.1101/447367 [DOI] [Google Scholar]
- 49. Swerdlow DI, Holmes MV, Kuchenbaecker KB, et al. : The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet. 2012;379(9822):1214–24. 10.1016/S0140-6736(12)60110-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Swerdlow DI, Preiss D, Kuchenbaecker KB, et al. : HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet. 2015;385(9965):351–61. 10.1016/S0140-6736(14)61183-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Coviello AD, Haring R, Wellons M, et al. : A genome-wide association meta-analysis of circulating sex hormone-binding globulin reveals multiple Loci implicated in sex steroid hormone regulation. PLoS Genet. 2012;8(7):e1002805. 10.1371/journal.pgen.1002805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Burgess S, Zuber V, Valdes-Marquez E: Mendelian randomization with fine-mapped genetic data: Choosing from large numbers of correlated instrumental variables. Genet Epidemiol. 2017;41(8):714–25. 10.1002/gepi.22077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Burgess S, Butterworth A, Thompson SG: Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7):658–65. 10.1002/gepi.21758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Patel A, Burgess S, Gill D, et al. : Inference with many correlated weak instruments and summary statistics. arXiv: 2005.01765. 2020. Reference Source [Google Scholar]
- 55. Walker VM, Kehoe PG, Martin RM, et al. : Repurposing antihypertensive drugs for the prevention of Alzheimer’s disease: a Mendelian randomization study. Int J Epidemiol. 2019;49(4):1132–1140. 10.1093/ije/dyz155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Davies NM, Holmes MV, Davey Smith G: Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601. 10.1136/bmj.k601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Scott RA, Freitag DF, Li L, et al. : A genomic approach to therapeutic target validation identifies a glucose-lowering GLP1R variant protective for coronary heart disease. Sci Transl Med. 2016;8(341):341ra76. 10.1126/scitranslmed.aad3744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gill D, Walker VM, Martin RM, et al. : Comparison with randomized controlled trials as a strategy for evaluating instruments in Mendelian randomization. Int J Epidemiol. 2020;49(4):1404–1406. 10.1093/ije/dyz236 [DOI] [PubMed] [Google Scholar]
- 59. Berry DJ, Vimaleswaran KS, Whittaker JC, et al. : Evaluation of genetic markers as instruments for Mendelian randomization studies on vitamin D. PLoS One. 2012;7(5):e37465. 10.1371/journal.pone.0037465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Georgakis MK, Malik R, Gill DK, et al. : Interleukin-6 signaling effects on ischemic stroke and other cardiovascular outcomes: a Mendelian Randomization study. medRxiv. 2019;19007682. 10.1101/19007682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wurtz P, Wang Q, Soininen P, et al. : Metabolomic Profiling of Statin Use and Genetic Inhibition of HMG-CoA Reductase. J Am Coll Cardiol. 2016;67(10):1200–10. 10.1016/j.jacc.2015.12.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Slob EAW, Burgess S: A Comparison Of Robust Mendelian Randomization Methods Using Summary Data. bioRxiv. 2019;577940. 10.1101/577940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Benn M, Nordestgaard BG, Frikke-Schmidt R, et al. : Low LDL cholesterol, PCSK9 and HMGCR genetic variation, and risk of Alzheimer's disease and Parkinson's disease: Mendelian randomisation study. BMJ. 2017;357:j1648. 10.1136/bmj.j1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ference BA, Majeed F, Penumetcha R, et al. : Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or both: a 2 x 2 factorial Mendelian randomization study. J Am Coll Cardiol. 2015;65(15):1552–61. 10.1016/j.jacc.2015.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ference BA, Robinson JG, Brook RD, et al. : Variation in PCSK9 and HMGCR and Risk of Cardiovascular Disease and Diabetes. N Engl J Med. 2016;375(22):2144–53. 10.1056/NEJMoa1604304 [DOI] [PubMed] [Google Scholar]
- 66. Ference BA: How to use Mendelian randomization to anticipate the results of randomized trials. Eur Heart J. 2018;39(5):360–2. 10.1093/eurheartj/ehx462 [DOI] [PubMed] [Google Scholar]
- 67. Del Greco MF, Minelli C, Sheehan NA, et al. : Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat Med. 2015;34(21):2926–40. 10.1002/sim.6522 [DOI] [PubMed] [Google Scholar]
- 68. Chong M, Sjaarda J, Pigeyre M, et al. : Novel Drug Targets for Ischemic Stroke Identified Through Mendelian Randomization Analysis of the Blood Proteome. Circulation. 2019;140(10):819–30. 10.1161/CIRCULATIONAHA.119.040180 [DOI] [PubMed] [Google Scholar]
- 69. Dudbridge F: Power and predictive accuracy of polygenic risk scores. PLoS Genet. 2013;9(3):e1003348. 10.1371/journal.pgen.1003348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Burgess S, Thompson SG, Collaboration CCG: Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. 2011;40(3):755–64. 10.1093/ije/dyr036 [DOI] [PubMed] [Google Scholar]
- 71. Burgess S, Davies NM, Thompson SG: Bias due to participant overlap in two-sample Mendelian randomization. Genet Epidemiol. 2016;40(7):597–608. 10.1002/gepi.21998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Allara E, Morani G, Carter P, et al. : Genetic Determinants of Lipids and Cardiovascular Disease Outcomes: A Wide-Angled Mendelian Randomization Investigation. Circ Genom Precis Med. 2019;12(12):e002711. 10.1161/CIRCGEN.119.002711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Denny JC, Ritchie MD, Basford MA, et al. : PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics. 2010;26(9):1205–10. 10.1093/bioinformatics/btq126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Pendergrass SA, Brown-Gentry K, Dudek SM, et al. : The use of phenome-wide association studies (PheWAS) for exploration of novel genotype-phenotype relationships and pleiotropy discovery. Genet Epidemiol. 2011;35(5):410–22. 10.1002/gepi.20589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Schmidt AF, Holmes MV, Preiss D, et al. : Phenome-wide association analysis of LDL-cholesterol lowering genetic variants in PCSK9. BMC Cardiovasc Disord. 2019;19(1):240. 10.1186/s12872-019-1187-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Giambartolomei C, Vukcevic D, Schadt EE, et al. : Bayesian Test for Colocalisation between Pairs of Genetic Association Studies Using Summary Statistics. PLoS Genet. 2014;10(5):e1004383. 10.1371/journal.pgen.1004383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Giambartolomei C, Liu JZ, Zhang W, et al. : A Bayesian framework for multiple trait colocalization from summary association statistics. Bioinformatics. 2018;34(15):2538–45. 10.1093/bioinformatics/bty147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hormozdiari F, van de Bunt M, Segrè AV, et al. : Colocalization of GWAS and eQTL Signals Detects Target Genes. Am J Hum Genet. 2016;99(6):1245–60. 10.1016/j.ajhg.2016.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhu Z, Zhang F, Hu H, et al. : Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48(5):481–7. 10.1038/ng.3538 [DOI] [PubMed] [Google Scholar]
- 80. Lawlor DA, Tilling K, Smith GD: Triangulation in aetiological epidemiology. Int J Epidemiol. 2016;45(6):1866–86. 10.1093/ije/dyw314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Borén J, Chapman MJ, Krauss RM, et al. : Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2020;41(24):2313–2330. 10.1093/eurheartj/ehz962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Haycock PC, Burgess S, Wade KH, et al. : Best (but oft-forgotten) practices: the design, analysis, and interpretation of Mendelian randomization studies. Am J Clin Nutr. 2016;103(4):965–78. 10.3945/ajcn.115.118216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bothwell LE, Greene JA, Podolsky SH, et al. : Assessing the Gold Standard--Lessons from the History of RCTs. N Engl J Med. 2016;374(22):2175–81. 10.1056/NEJMms1604593 [DOI] [PubMed] [Google Scholar]
- 84. Millwood IY, Walters RG, Mei XW, et al. : Conventional and genetic evidence on alcohol and vascular disease aetiology: a prospective study of 500 000 men and women in China. Lancet. 2019;393(10183):1831–42. 10.1016/S0140-6736(18)31772-0 [DOI] [PMC free article] [PubMed] [Google Scholar]