

Abstract
Metastasis is initiated and sustained through therapy by cancer cells with stem-like and immune evasive properties, termed metastasis initiating cells (MICs). Recent progress suggests that MICs result from the adoption of a normal regenerative progenitor phenotype by malignant cells, a phenotype with intrinsic programs to survive the stresses of the metastatic process, undergo epithelial-mesenchymal transitions, enter slow-cycling states for dormancy, evade immune surveillance, establish supportive interactions with organ-specific niches, and co-opt systemic factors for growth and recurrence after therapy. Mechanistic understanding of the molecular mediators of MIC phenotypes and host tissue ecosystems could yield cancer therapeutics to improve patient outcomes.
Significance
Understanding the origins, traits and vulnerabilities of progenitor cancer cells with the capacity to initiate metastasis in distant organs, and the host microenvironments that support the ability of these cells to evade immune surveillance and regenerate the tumor, is critical for developing strategies to improve the prevention and treatment of advanced cancer. Leveraging recent progress in our understanding of the metastatic process, here we review the nature of metastasis initiating cells and their ecosystems and offer a perspective on how this knowledge is informing innovative treatments of metastatic cancers.
Clinically evident metastasis remains largely incurable, but progress is being made in understanding its basic biological principles and laying the foundation for improving long-term outcomes in patients with advanced cancer(1,2). In spite of the aggressiveness of metastatic cancer, only a small proportion of cells released from a tumor end up forming a distant lesion. This is partly due to the extensive attrition that cancer cells suffer as they confront the stresses of cancer cell dissemination, dormancy, outgrowth, and resistance to therapy(2). Cancer cells must accumulate numerous phenotypic traits in order to survive these stresses and become proficient at initiating metastasis.
It is increasingly clear that metastasis is initiated by a subset of cancer cells with a stem cell-like phenotype that makes them more competent than the bulk cancer cell population at reinitiating tumor growth in distant sites(3–5). Cell “stemness” in mature tissues represents a phenotypically fluid condition rather than a developmentally imprinted and rigidly defined state. Stem cells in different tissues have distinct properties and developmental trajectories, as do stem cells that support tissue homeostasis versus those that are mobilized in response to injury to regenerate the disrupted tissue. Adult stem cells that maintain homeostasis may suffer oncogenic mutations and become tumor-initiating cancer stem cells (CSCs). However, the cells that initiate metastasis are not necessarily CSCs that acquired a few extra metastatic traits. Recent progress indicates that metastatic colonization of distant organs is initiated by stem-like cancer cells that are distinct not only in terms of expressing certain specialized pro-metastatic traits but also in terms of their overall phenotype, which is more akin to that of regenerative stem cells. Hence, we refer to these cells as “metastasis initiating cells” (MICs), a term which denotes what these cells can do and why they are of interest. Cells with MIC features are enriched in residual disease that survives cancer therapy, underscoring their importance not only in the original formation of distant metastases but also during relapse of increasingly lethal metastases after multiple treatment cycles in the clinic.
Understanding the origins, traits and vulnerabilities of MICs and their host ecosystems is critical for developing strategies to improve the prevention and treatment of metastasis. Here we leverage the conceptual framework and open questions from previous reviews on the metastatic process(1,2) and the cells and microenvironments that make it possible(3,5) to offer an update on recent progress towards the resolution of these questions. We present a summary of the metastatic process and the traits that cancer cells must possess in order to become MICs. We then address the question of how MICs acquire these traits. We consider the hypothesis that MICs emerge by the progressive accumulation of an array of pro-metastatic traits through evolution of highly heterogeneous cancer cell populations under the selective pressures of the multiple steps of the metastatic process. We also consider the alternative hypothesis that cancer cells acquire many of their critical traits at once by adopting a regenerative progenitor phenotype with intrinsic tumor regrowth capacity. Finally, we offer a perspective on the implications of the growing understanding for improving the treatment of patients with metastatic cancer.
Phases of the metastatic process
Metastasis starts with a phase of cancer cell dissemination from the primary tumor to regional and distal sites (Figure 1a). In certain cancers, such as breast cancer, dissemination may start during early stages of tumor progression while the tumor is still small and appears to be confined by basement membrane within a ductal structure – i.e. ductal carcinoma in situ(6,7). Dissemination can continue until the source tumor is removed, as demonstrated by the presence of circulating tumor cells (CTCs) in patients undergoing surgical resection of the tumor(8). CTCs culminate their journey by exiting from blood capillaries (a process called extravasation) and infiltrating the new host parenchyma.
Figure 1. Phases of Metastasis.

a. Metastasis proceeds through three distinct phases of dissemination, dormancy and colonization. Metastasis Initiating Cells (MICs) disseminate from the primary tumor and seed multiple organs, where they enter a subclinical state of dormancy. During dormancy, MICs may shuttle between quiescent and proliferative states, with proliferative cells being continually cleared by niche-specific or systemic immune defenses. MICs that acquire immune evasive and organ-specific growth adaptations are able to exit dormancy and generate clinically evident macrometastatic colonies. During dissemination and dormancy, MICs are in a dynamic equilibrium with host immunity, while failure of immune surveillance results in metastatic outbreaks and organ colonization. b. The three phases of metastasis can overlap with the growth of primary tumors and may co-exist in the same individual until removal of the primary tumor (Stage I-III). The latter two phases continue to co-exist during adjuvant therapy, with the eventual dominance of the colonization phase resulting in macrometastatic relapse (in cases that were originally diagnosed as Stage I-III), or in patients diagnosed with de novo Stage IV cancers.
The dissemination phase is followed by a phase of metastatic dormancy during which disseminated cancer cells do not yet grow to form a clinically manifest lesion because the host stroma presents physical, metabolic and immune barriers that prevent this growth(9). Disseminated cancer cells survive under these conditions by entering a state of proliferative quiescence. During dormancy, metastasis remains a cryptic process that may eventually progress towards overt tumor growth or disappear by exhaustion of the disseminated cancer cell population under these barriers or by the effect of therapy.
The final phase of the metastatic process involves overt outgrowth by colonization of the host organ (Figure 1a). Clinically detectable metastases may not become evident until months or decades after initial diagnosis and removal of a primary tumor. In some cancers overt metastasis occurs in multiple sites without marked preference for one or another organ. In other cancers, metastasis shows strong preference or “tropism” for certain organs. Notably, 2%-5% of metastatic cancers are diagnosed as carcinomas of unknown primary, in which the primary tumor is not detected either because of its small size or because it may have spontaneously regressed after seeding aggressive metastases(10).
The three phases of the metastatic process –dissemination, dormancy and outbreak– may coexist in time (Figure 1b). Cancer cell dissemination may persist as long as the source tumor remains in place, long after the first disseminated cells accumulated as metastatic seeds in distant organs. Precocious metastatic lesions may break out from dormancy while the primary tumor is still in place, as it occurs in patients whose cancer is first diagnosed as widely metastatic stage IV disease (Figure 1b). When distant relapse occurs in a particular site long after the surgical elimination of the primary tumor, other organs may still harbor dormant metastatic cells that can independently evolve and eventually initiate metastatic outgrowth with their own set of organ-specific metastatic traits. An example is provided by late brain recurrence in patients with HER2-positive breast cancer who were treated with anti-HER2 agents that suppressed visceral metastasis(11).
Metastatic dissemination
A combination of invasive cell migration, extracellular matrix remodeling, and the presence of leaky vasculature at the tumor invasion front enable the dispersion of cancer cells through the circulation, lymphatics, perineural and perivascular routes, or via direct infiltration of adjacent body cavities(12) (Figure 2). The hematogenous circulation is the principal route in most types of cancer, but lymphatic dissemination and forms of extravascular spread are prominent routes in various types of cancer as well.
Figure 2. Metastatic dissemination.

The most common and best characterized route of tumor dissemination is via the blood circulation (a-c). a. During hematogenous dissemination, cancer cells at the invasion front of primary tumors that undergo epithelial-mesenchymal transitions (EMT) lead intravasation into neoangiogenic capillaries in the tumor, and thus access the venous circulation for dissemination to multiple organs. Cancer cells may intravasate individually or in clusters during collective migration. Invasion and intravasation involve remodeling of the tumor extracellular matrix (ECM) and may be facilitated by tumor-resident fibroblasts and macrophages. b. In the circulation, cancer cells must rapidly adapt to overcome biomechanical, redox and immunological threats. Clustering of circulating tumor cells (CTCs) may enable paracrine growth factor signaling that promotes the niche-independent survival of disseminated epithelial cells. CTC clusters are enriched in stem-like cancer cells and can include other cell types including platelets and neutrophils, which in turn can protect CTCs from immune attack by secreting immunosuppressive factors. c. CTCs become trapped in the capillary beds of multiple organs and migrate into the organ parenchyma to seed nascent metastasis. Fenestrated capillary beds in the liver and bone marrow can facilitate extravasation. Non-tumor cells with CTC clusters, or in the parenchyma, including platelets and monocytes, can facilitate endothelial permeability and transmigration by secreting endothelial disjunction factors. Alternatively, CTCs can secrete factors that induce endothelial necroptosis. d. Beyond hematogenous dissemination, cancer cells originating in certain primary tumors can also reach distant organs via alternative routes.
Invasion and intravasation.
Malignant cells remodel the basement membrane to invade into tissue parenchyma, induce neoangiogenesis, move directionally towards blood vessels and undergo trans-endothelial migration to enter the portal or systemic venous circulations(13,14) (Figure 2a). These steps are enabled by dynamic changes in the repertoire and function of cell-cell adhesion molecules (cadherins, CAM family cell adhesion molecules), extracellular matrix remodeling enzymes (matrix metalloproteinases, ADAMTS endopeptidases), and cell-matrix adhesion molecules (integrins, syndecans)(15,16). Carcinoma cells undergo epithelial-to-mesenchymal transition (EMT), a phenotypic conversion process that enables cell migration and entry into the circulation (see below)(1,17). Stromal cells, including macrophages, fibroblasts and neutrophils, can further stimulate invasion and intravasation by direct interactions with carcinoma cells(18,19). Cancer cell-derived fibrogenic signals stimulate the deposition of desmoplastic matrix by mesenchymal cells in the tumor stroma(20,21), which in turn facilitates the invasion of cancer cells and their proliferation through mechanosignaling cues(15,16). The physical stress of migrating through tight interstitial spaces leads to nuclear envelope rupture and chromatin shearing, thus causing further genomic and phenotypic diversification of cancer cells that survive such stress(22,23).
Circulating tumor cells.
Unlike tumor tissues, which can only be accessed via invasive biopsies, CTCs derived from primary or metastatic tumors can be readily and repeatedly sampled via simple blood draws from cancer patients. This provides a unique window to study metastasis biology and for use as longitudinal clinical biomarkers of metastasis and therapy response(24),(25). For these reasons, CTCs have become a topic of intense study. Although, the sensitivity and specificity of current CTC detection assays remain limiting for some applications, the prospect of delineating CTC characteristics that discriminate between high and low metastatic capacity is of biological and clinical significance.
Cancer cells circulate singly and in clusters(26) (Figure 2b). CTC clusters are observed in the blood of patients and tumor-bearing mice and have a superior ability to seed metastasis in experimental models(27). CTC clusters may also include tumor-derived stromal cells(28). CTC clustering provides intercellular spaces that concentrate paracrine growth-promoting signals(29), and enable cooperation between different cancer cell states within a cluster(30). CTC clusters are enriched in genome methylation patterns that denote a stem-like cancer cell state(31), and therefore have features of MICs. MICs upregulate expression of certain cell-cell and cell-matrix adhesion genes(32), raising the question, are CTC clusters more metastatic because clustering augments the aggressiveness of cancer cells or because MICs are intrinsically prone to clustering?
Blood platelets coat CTCs providing protection from shear stress and immune attack, particularly attack by natural killer (NK) cells. Platelets can also facilitate cancer cell extravasation by mediating adhesion to selectins on the endothelium and by enforcing EMT in the cancer cells. Platelets release factors including transforming growth factor β (TGF-β) and platelet-derived growth factor (PDGF) that suppress NK cells and promote EMT of CTCs (14,33,34). A small proportion of CTCs in breast cancer patients are bound to neutrophils, an abundant white blood cell type; these CTCs are more proliferative and effective at forming metastases in mice than are neutrophil-free counterparts(35).
Extravasation.
Within seconds to minutes of leaving a primary tumor, CTCs may already become trapped in the capillaries of distant organs. In experimental systems, CTCs can remain lodged in capillaries for days until they either extravasate or disappear(36). The half-life of cancer cells in the circulation is estimated to be just a few hours, based on the rate of decline in the CTC numbers following surgical removal of primary tumors in patients(8).
Differences in the tightness of capillary endothelial walls determine the difficulty of seeding metastasis in different organs, contributing to organ-specific patterns of metastatic spread (Figure 2c). Liver and bone, the most common sites of metastasis from many cancers, contain sinusoid capillary beds with large intracellular fenestration gaps as well as gaps in the underlying basement membrane, which facilitate CTC extravasation. At the other end of the range, the blood-brain and blood-choroid plexus barriers restrict the movement of molecules and cells between the systemic circulation and the brain parenchyma and cerebrospinal fluid, respectively. Accordingly, metastasis to the brain and leptomeningeal spaces selects for MICs expressing factors that increase barrier permeability or disrupt the barrier altogether(37),(38,39).
Extravasation can be facilitated by platelets coating CTCs(34) and cytokines secreted by perivascular macrophages in target organs or by the cancer cells themselves(13) (Figure 2c). As an alternative to squeezing through tight interstitial spaces between endothelial cells, cancer cells may induce endothelial cell death. Human and mouse cancer cells have been shown to induce necroptosis of endothelial cells in co-culture and orthotopic lung metastasis models, while inhibition of RIPK1/3 dependent endothelial necroptosis reduced extravasation and metastasis in these models(40).
Following extravasation, metastatic cells remain situated near blood capillaries and migrate along the abluminal surface on the capillaries(36,41–44). Integrins mediate interactions between cancer cells and capillaries(45). This is most apparent in experimental models of metastasis to the brain where the elongated nature of capillaries facilitates imaging studies(41,43). However, perivascular residence of extravasated cancer cells is also observed in other sites including the liver, lungs and bone marrow(43).
Lymphatic dissemination.
Cancer cells disseminate via the lymphatic circulation, but whether lymph nodes are an obligatory staging post for metastasis remains controversial and likely depends on tumor type(46,47). Comparative genomics of regional lymph node and distant metastases from colorectal and breast cancers suggest that in the majority of cases, distinct clones within primary tumors give rise to nodal and distant metastases, with the latter undergoing more stringent clonal selection(48–50). In patients, highly aggressive triple negative breast cancers often metastasize without apparent regional lymph node involvement(47). Consistent with a correlative, not causal relationship between lymph node and distant metastasis, recent clinical trials have shown no survival benefit for regional lymph node dissection in patients with breast and ovarian cancers and melanoma(51–53). Even when injected directly into the lymph nodes of mice, cancer cells preferentially metastasize via the blood rather than through the lymphatics(54,55). However, transient passage through the lymphatics may confer long-term survival benefits to MICs as they join the blood circulation(56).
Melanomas implanted in mice lacking dermal lymphatic vessels exhibited lower cytokine expression and leukocyte infiltration than those implanted in wildtype controls, suggesting that cancer cell interactions with immune cells in lymphatic structures are important for anti-tumor immunity(57). The distinctive metabolic milieu of lymph may also enable cancer cells adaptation to the stresses of metastasis. For example, exposure to oleic acid in lymph protected melanoma cells from ferroptosis and increased their ability to initiate metastasis during subsequent hematogenous dissemination(58).
Extravascular spread.
The metastatic spread of some tumors occurs largely or at least partly through routes that obviate the need to enter and exit the circulation (Figure 2d). In ovarian cancer, cell clusters carried by the peritoneal fluid attach on the abdominal peritoneum where the cells grow(59). Peritoneal metastasis is also frequent in colorectal and pancreatic cancers. Melanomas and other cancer can spread without entering and exiting the circulation by engaging in extravascular migratory metastasis, a process of migration over the abluminal surface of vessels that is common during embryogenesis(60). Perineural spread is a long recognized, if under-appreciated form of metastatic spread in head and neck cancers and other squamous cell carcinomas(61). Regional spread of lung adenocarcinoma can occur by spread through air spaces where cancer cells detach from a primary tumor, migrate as clusters through air spaces, and reattach to the alveolar walls through vessel co-option to resume tumor growth, which is associated with tumor recurrence and poor survival of stage I lung adenocarcinomas(62,63).
Dormancy and immune evasion
Dormant metastasis and the ability to predict the likelihood of an eventual outbreak in patients diagnosed with early-stage cancer are major considerations in the clinic(64). For example, patients with estrogen receptor-positive early stage breast cancer who undergo complete surgical removal of the primary tumor continue to experience metastatic relapse as late as 20 years from their original diagnosis(65). Dormant metastasis is an understudied problem, owing to a dearth of appropriate experimental models. Models using aggressive metastatic cells sidestep the dormancy phase altogether. Some models of dormant metastasis rely on comparisons between non-isogenic cancer cell lines of varying propensity to develop metastatic colonies after inoculation in mice. Spontaneously arising models of dormancy have been characterized in detail(9). New experimental models have been developed by in vivo selection of early-stage tumor derived MICs that are competent to establish dormant metastasis and spontaneously generate late outbreaks, thus mimicking the course of the disease in patients(66). Studies on these various models have begun to shed light on the biology of MIC entry and exit from dormancy (Figure 3).
Figure 3. Metastatic dormancy and outbreak.
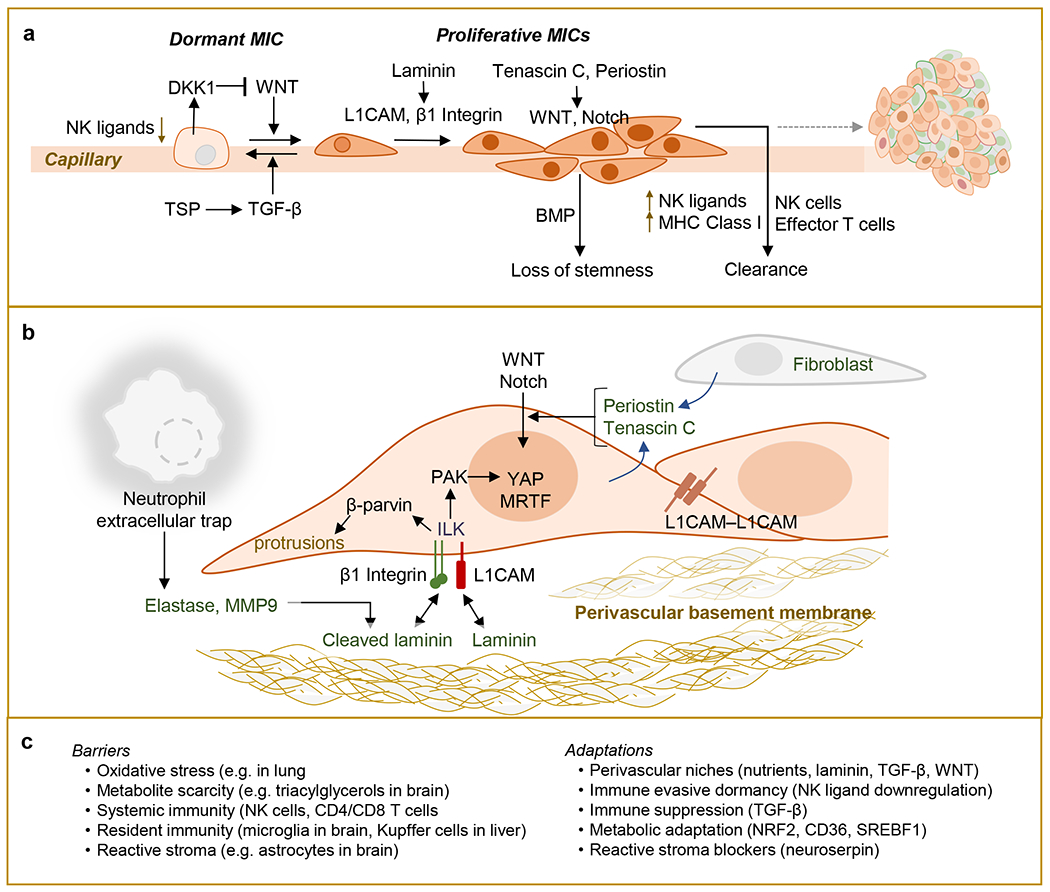
a. Metastasis initiating cells (MICs) seeding distant organs enter into a variable period of dormancy, when then cannot be detected by clinical imaging technologies. Clinical dormancy reflects an equilibrium incorporating cell-intrinsic growth arrest, and stochastic MIC proliferation events, which are countered by elimination of proliferating cells. Secreted factors in the perivascular microenvironment may either inhibit MIC proliferation (e.g. TGF-β), or promote it (e.g. WNT), which in turn can be countered by MIC secretion of WNT inhibitors (e.g. DKK1). MICs spread on the abluminal surface of capillaries by adhering to the perivascular basement membrane, which provides proliferative inputs. However, stromal factors can induce proliferating MICs to differentiate and lose stemness properties (e.g. BMP). In contrast to quiescent MICs, proliferating MICs upregulate cell surface expression of NK ligands and MHC Class I molecules, facilitating their detection and destruction by NK cells and adaptive immune cells. b. Exit from dormancy requires close interactions between MICs and extracellular matrix (ECM). MICs induce innate immune responses that include the recruitment of neutrophils and the formation of extracellular web-like chromatin decondensates harboring ECM remodeling enzymes, termed neutrophil extracellular traps (NETs). NET-dependent cleavage of laminin, a key component of the ECM, activates β1-integrin signaling in MICs. MICs can also activate signaling through integrin-like kinase (ILK) by upregulating L1CAM, a cell adhesion molecule that binds laminin on extracellular basement membranes, including that of blood capillaries. Integrin/ILK signaling enables cell stretching via the formation of actin-dependent protrusions, which in turn enables mechanosignaling to activate YAP-dependent transcriptional output and proliferation. Homophilic L1CAM interactions as cancer cells multiply can extend this signaling. Additional cues from the ECM, including the stem cell niche ECM components tenascin C and periostin, can potentiate growth factor signaling and enable MIC stemness and proliferation. c. Outgrowth of dormant metastasis might be prevented by bolstering barriers to MIC survival and proliferation or targeting the phenotypic adaptations required for escape from dormancy.
Causes of cancer cell elimination.
Clinical dormancy implies one of two complementary scenarios: either a cell-autonomous “temporary mitotic exit” as originally advanced by Geoffrey Hadfield in 1954(67), or alternatively the existence of persistent attempts at colonization that are aborted by cell-extrinsic limitations on metastatic outgrowth. While several studies have explored the niches, signals and mechanisms that maintain both modes of dormancy (reviewed in (9,68,69)), multiple lines of experimental and clinical data are converging on the view that dormancy reflects a dynamic equilibrium of MICs with antagonistic immune surveillance (Figure 3a).
Cancer cells suffer extensive attrition after infiltrating distant organs, as shown in mouse models of metastasis and as inferred from the low counts of disseminated cancer cells relative to the number of CTCs that a primary tumor likely shed while it was present in a patient. Elimination of cancer cells after extravasation is driven by passive factors such as high oxidative stress in the lungs(70,71), a dearth of growth factors and nutrients in the cerebrospinal fluid(38,72), or more generally, a lack of the appropriate microenvironment cues that had nursed the growth of MICs in the primary tumor. These passive mechanisms of disseminated cancer cell elimination are accompanied by the proactive role of tissue resident immunity and other defenses, such as astrocytes in the brain(36), against intruding cancer cells. Tissue resident macrophages, such as microglia in the brain and Kupffer cells in the liver, as well as NK cells attack infiltrated cancer cells(18,73,74). In experimental models, depletion of T cells and NK cells increases the survival and outgrowth of disseminated cancer cells(66,75,76). Tumor lymphocyte infiltration, indexed in the form of an “immunoscore” is inversely correlated with metastatic burden in patients(77), and metastatic clones can show branched evolution as a result of different immunoediting pressures(78). Dramatically, immunosuppressed recipients of transplant organs from donors deemed cured of localized melanoma or glioblastoma developed donor-derived metastasis(79,80). Underscoring the importance of immune surveillance of MICs, in patients with completely resected localized melanomas, post-surgical adjuvant therapy with immune checkpoint inhibitors to boost anti-tumor immunity reduced later metastatic relapse and prolonged survival(81–84).
Entering dormancy.
Disseminated MICs that enter proliferative quiescence may be particularly equipped to survive these threats. Quiescence allows cancer cells to evade the innate and adaptive arms of immunity mediated by NK and T cells respectively(66,85–88). As an added benefit, dormancy renders disseminated cancer cells resistant to anti-mitotic therapy. The ability to enter a slow-cycling state is a property of some adult stem cells. Thus, to the extent that MICs are primed to enter protective dormancy, dormancy will enrich for MICs in disseminated cancer cell populations.
Entry of MICs into quiescence is stimulated by growth inhibitory signals in the host microenvironment (Figure 3a). TGF-β has cytostatic effects on normal epithelial cells and carcinoma cells by inducing the expression of cyclin-dependent kinase inhibitors, while it triggers tumor suppressive apoptosis in premalignant epithelial cells. Carcinoma cells bypass the pro-apoptotic effect of TGF-β through various mechanisms while retaining the ability to respond to TGF-β with pro-metastatic effects, including entry into immunoprotective quiescence(89,90). TGF-β has been shown to stimulated dormancy in experimental models of prostate, breast, and head and neck carcinomas(9). The three isoforms, TGF-β1, -β2 and -β3, are known to be produced in tumor microenvironments by various stromal components and cancer cells. Latent TGF-β1 in particular is present in perivascular niches of the bone marrow and brain parenchyma of mice and is activated by thrombospondin (TSP) to drive extravasated breast cancer cells into proliferative quiescence(44)(Figure 3a). In this context TGF-β likely functions through canonical binding to the paired receptor kinases TGFBR1 and TGFBR2 with betaglycan, also known as type III TGF-β receptor, serving as a co-receptor that is particularly important for TGF-β2 binding(89).
Entry of disseminated MICs into dormancy is not merely a passive process resulting from the effect of local growth inhibitory signals from the tumor microenvironment. Entry into dormancy can also be initiated by MICs themselves such that MICs that actively avoid mitogenic stimulation are selected for because quiescence enables immune evasion(66). MICs derived from early-stage lung and breast tumors actively secrete DKK1, an inhibitor of WNT factors which are potent mitogens for adult stem cells (Figure 3a). This autocrine DKK1 enforces MICs dormancy in these models, and depleting DKK1 releases MICs from dormancy and delivers them to NK mediated clearance(66).
These insights suggest that dormant metastasis results from a dynamic equilibrium in which MICs enter proliferative quiescence and thus avoid immune-mediated killing or enter the cell cycle for tumor growth at the risk of being eliminated (Figure 3a). Another form of dormancy is tumor mass dormancy in which MICs start proliferating but are unable to satisfy the nutrient or oxygen requirements for growth beyond a small tumor mass due to lack of angiogenesis or other causes(9,91). However, large clinical trials have shown little success with antiangiogenic drugs in the adjuvant or neoadjuvant setting when targeting latent micrometatases, suggesting that (with the limitations of the drugs available) the angiogenic switch may not be a major limiting factor to metastatic outgrowth in patients, or that current therapies are inadequate to suppress angiogenesis(92).
Exiting from dormancy.
Little is known about the signals that lead disseminated cancer cells to exit from dormancy and develop clinically detectable metastases. It is possible that disseminated cancer cells are “awakened” from years of quiescence by signals that jolt them into entering the cell cycle. However, the cited studies on depleting mice of immunity suggest that there is always a fraction of disseminated cancer cells population attempting to proliferate perhaps due to a growth permitting balance between local mitogenic and antimitogenic signals (Figure 3a). In line with this idea, depletion of DKK1 in models of dormant lung adenocarcinoma and breast cancer metastasis enables exit from dormancy, implying that WNT ligands in the perivascular niche reactivate the proliferation of dormant cells(66). Bone morphogenetic proteins can trigger differentiation of disseminated breast cancer cells in mouse models, which is prevented by BMP sequestering factors(93). Cell metabolism may also play a role. Cell autophagy in response to metabolic stress promotes the survival of dormant breast cancer cells in mouse models(94), and depleting autophagy related 3 (ATG3) or p62/sequestosome-1 promotes cancer cell exit from dormancy with expression of 6-phosphofructo-2-kinase which supports glycolysis(95).
MICs have certain requirements for continued growth during exit from dormancy. MICs derived from breast, lung, colorectal and renal cell carcinomas express L1CAM and use this cell adhesion molecule to bind to laminin in the perivascular basement membranes of capillaries, spreading on the abluminal surface of capillaries (Figure 3b). Once engaged, L1CAM cooperates with β1 integrins, the integrin-actin bridging protein ILK, and p21-activating kinases (PAK) to activate transcription factors YAP and MRTF for the initiation of metastatic outgrowth(32),(43). ILK and β-parvin regulate actin-filaments supporting the formation of filopodia-like protrusions, which are required for metastatic outgrowth(96). The L1CAM-expressing MICs cells grow forming sheaths around the vessels, engulfing the capillary network before the micrometastatic mass acquires a spheroidal shape(32),(43). The requirement for L1CAM has been shown in experimental models of dormant MICs outgrowth following depletion of NK cells as well as models of aggressive metastatic outgrowth without an intervening dormancy phase(43).
Recent studies have revealed a role for neutrophil extracellular traps (NETs) in promoting YAP activation during cancer cell exit from domancy(97,98). NETs are protease-loaded chromatin webs that neutrophils release as an ultimate maneuver to entrap and clear pathogens(99). NETs are of interest for their roles in inflammation, autoimmune diseases, and cancer(100). Studies using intravital microscopy documented the presence of NETs around disseminated breast cancer cells in mouse models of metastasis and an inhibitory effect of DNAse I on metastatic growth, implying that cancer cell-triggered NETs promote metastasis(101). Indeed, NETs produced during inflammatory responses were shown to reactivate the growth of local dormant cancer cells through cleavage of basement membrane laminin by NET-associated elastase and matrix metalloproteinase 9 (MMP9) (Figure 3b). The resulting laminin fragment activates α3β1 integrin-mediated YAP and proliferation of the cancer cells(98).
Organ colonization and metastatic tropism
Colonization of distant organs is the most dramatic phase of the metastatic process clinically and the most complex biologically. The outgrowth of disseminated MICs into large metastatic colonies depends on the successful avoidance of organ-specific barriers and the productive interactions with specific components of the host tissue ecosystem. Many of these barriers and interactions vary with the site of metastasis. The lung, liver, or brain parenchyma, the lymph nodes, bone marrow, and leptomeninges present widely different tissue microenvironments. Not surprisingly, studies on metastatic colonization of these major target sites have uncovered different factors, metabolic changes, cell-cell interactions, and other determinants of metastatic colonization depending of the tumor type and the metastasis organ site. Detailed accounts of organ-specific metastatic colonization factors have been recently provided for metastasis to different organs(102,103), and specifically to bone(104), central nervous system(39,105), lung(106), and liver(107). Here we briefly summarize current concepts on general and organ-specific colonization processes.
Determinants of organ tropism.
Metastatic tropism is manifest in the tendency of tumors to preferentially relapse in certain organs. Some cancers (e.g. small cell lung carcinomas, metastatic melanomas) form metastasis in multiple sites without marked organ preference, whereas other cancers preferentially relapse in certain organs (e.g. the bones in prostate and hormone receptor-positive breast carcinomas, the liver in colorectal carcinomas and ocular melanomas, or the lungs in soft tissue sarcomas).
Metastatic tropism results from the combination of two distinct phenomena. One is the ability of cancer cells to reach particular organs through the circulation. Colon cancer is thought to predominantly metastasize to the liver because the mesenteric blood draining the intestines flows via the hepatic portal vein into the liver. The majority of CTCs derived from an intestinal tumor become trapped in the hepatic sinusoids and initiate metastases in the liver with far higher frequency than in the lungs, brain or bones. It has been proposed that cells derived from certain tumors use organ-specific endothelium adhesion molecules to concentrate in the capillaries of those organs and preferentially extravasate there(108). However, experimental(66,109,110) and clinical(111) evidence suggest that, in most types of cancer, disseminated cells reach all organs and leave dormant cells everywhere. This suggests that metastasis develops from latent surviving MICs in the least antagonistic site first, and in other sites later. Thus, the metastatic organ tropism of a particular type of cancer is a function of the organs that MICs can travel to and, most critically, the probability that MICs will growth in a particular organ site depending on their intrinsic or acquired ability to sort the barriers and utilize the supportive niches in that site.
Surpassing endothelial barriers.
As we noted earlier, differences in the permeability of capillary beds in different organs determine the ability of disseminated cancer cells to accumulate in these organs. Several mediators of cancer cells extravasation have been identified in experimental model systems, including cytokines (e.g. ANGPTL4, HBEGF), secreted enzymes (e.g. cathepsin S) and monocytes that facilitate extravasation in the lung capillary walls(1,2,13) or through the blood-brain barrier(37–39,112) (Figure 2c). Metastatic colon cancer cells that colonize the liver can gain lung metastatic ability by expressing the cytokine PTHLH, which mediates extravasation of these cells in the lungs after they reenter the circulation(113). In some instances, metastatic cells disrupt endothelial barriers to allow the influx of nutrients from the circulation. Breast and lung carcinoma cells that infiltrate the leptomeninges produce complement component 3 to activate the C3a receptor in the choroid plexus epithelium and disrupt the blood-CSF barrier, thereby allowing influx of growth factors and nutrients from blood into the nutritionally barren CSF(38).
Neutralizing hostilities.
The important role of immunity in suppressing the outgrowth of disseminated MICs can give rise to organ-specific differences depending on the ability of MICs from different types of cancer to evade tissue resident immunity(18,73,74). The ability of disseminated cancer cells to regulate their own exposure to immune attack by modifying the expression of MHC class I molecules(114), NK cell and effector T cell ligands(115), cytosolic DNA sensing pathways(116) and damage-associated RNAs as ligands for pattern recognition receptors(117) are under study given their likely importance in metastatic colonization. Moreover, the defenses that invading cancer cells must neutralize in order to seed distant organs are not limited to classical immunity. Breast cancer and lung adenocarcinoma cells that infiltrate the brain must contend with the release of pro-apoptotic Fas-ligand by reactive astrocytes. Fas-ligand is released from its membrane-bound precursor in astrocytes by the protease plasmin, and cancer cells can prevent this by expressing serpin inhibitors of plasminogen activator(36).
Metabolic adaptations.
Metabolic adaptation is an important aspect of metastasis. Unlike glycolytic primary tumors, MICs in multiple cancer types undergo a bioenergetic shift into an oxidative phosphorylation state(70,118,119). Organ specific metabolic adaptations enable carcinoma MICs to reinitiate growth in tissues with variable bioavailability of oxygen, including in the lungs, where high oxygen levels cause oxidative stress that cancer cells can withstand by overexpressing NRF2(119–121) (Figure 3c). Metastatic melanoma cells also undergo metabolic adaptations in order to withstand high levels of oxidative stress in the blood and lungs(70). Adaptations to the bioavailability of amino acids(122,123), nucleotides(124) and other bioenergetic substrates(125,126) have also been noted in metastasis models and human tissue samples and are likely to vary depending on the host tissue. In turn, metabolic stresses may shape the genomic and epigenomic reprogramming of cancer cells to enable metastasis(127). Other cancer cell adaptations serve to source scarce nutrients. For example, metastatic cells secrete the iron chelating factor lipocalin A in order to scavenge scarce iron in the CSF(72).
Lipids are important for tumor growth. CD36, a scavenger receptor that can transport fatty acids and other molecules, is upregulated in MICs from oral carcinomas and other cancers and required for metastasis in various organs(128). Brain metastasis appears to place higher demands on MICs with regards to procuring their own lipids. The brain has a markedly lower level of triacylglycerol stores than other tissues. Breast cancer cell lines that are metastatic to the brain up-regulate sterol response element binding transcription factor 1 (SREBF1) to promote lipid synthesis and fatty acid metabolism or alternatively upregulate CD36 for metastasis growth(129).
Metastatic niches and stromal cooption.
MICs that survive the organ-specific stresses described above additionally require support from the host stroma. Stromal support is thought to be important for MICs immediately after their infiltration of new tissues and establishment as long-term seeds for metastases. This may depend on a combination of cellular and extracellular cues that fulfills the survival and self-renewal requirements of MICs by mimicking the native niches that these cells or their predecessors occupied in their tissue of origin. Various cellular and extracellular components of natural stem cell niches support the viability of disseminated MICs(3,5), including the extracellular matrix interacting components periostin and tenascin C, which MICs and fibroblasts produce to promote WNT and Notch signaling in MICs(130,131) (Figure 3b). Evidence that disseminated cancer cells intrude natural stem cell niches in the bone marrow has been provided in models of prostate(132) and breast cancer metastasis(133).
In addition to biochemical signals from the niche, biophysical and mechanical features of stromal niches can also promote metastasis(134). An expanding primary tumor mass exerts mechanical force on its tissue microenvironment, in turn triggering differentiation of myofibroblasts to produce extracellular proteins such as collagens and tenascin C(135), which further increase stiffness of the extracellular matrix. In turn, increased ECM stiffness can promote integrin-mediated focal adhesion assembly, RHOA-ROCK-mediated cell contractility, and mechanosignaling via TGF-β, Wnt and YAP pathways (Reviewed in (134)). Stiffness-mediated mechanosignaling may thus mediate MIC behaviors including motility, anoikis evasion and EMT. Consistently, in patients with breast and pancreatic cancer, tumors with increased ECM stiffness were associated with increased metastasis, therapy resistance and poor prognosis(136–138).
Support from the stroma is also essential during the expansion of macrometastatic colonies, maintenance or aggressive metastatic lesions, and resistance to therapy. Most parenchymal components in sites of metastasis have been implicated in the support of survival, outgrowth and/or chemoresistance of MICs through unidirectional or reciprocal signaling interactions with the cancer cells. This includes osteoblasts and osteoclasts in bone metastasis(102,104,139–141), astrocytes(142,143), microglia(144), and neurons(145) in brain metastasis(39,105), monocytes(146,147), neutrophils(98,130,148), fibroblasts(131,149), and epithelial cells(150) in lung metastasis(106), and mesenchymal, epithelial, macrophage and other cells in liver metastasis(107). Cytokines and enzymes released by a primary tumor in soluble or vesicular form can alter the parenchyma of distant organs and establish “pre-metastatic niches”, zones that favor the immediate growth of cancer cells reaching these sites(151). It is unclear if pre-metastatic niches established by a primary tumor could support MICs as dormant seeds for long periods after a primary tumor is surgically removed.
Most studies on stromal interactions with metastatic cells have utilized experimental models in which aggressive cancer cells proceed to form metastatic colonies without an intervening dormancy phase. Therefore, with few exceptions, these studies do not directly distinguish whether a supportive interaction benefits MICs prior to dormancy, during the incipient outgrowth of micrometastasis after dormancy, or during the continued expansion of macrometastases. In spite of this shortcoming, several organ-specific metastatic mediators identified to date are accompanied with evidence of clinical relevance, providing potential targets for therapy in the treatment of metastasis.
Epithelial-mesenchymal transitions
EMT, referred to in passing above, is a key process during tumor progression and metastasis(1). Epithelial cells undergoing an EMT lose polarity and downregulate key cell adhesion molecules, resulting in the loss of contacts with neighboring cells and an increased ability to migrate and invade adjacent tissue. The ability to undergo an EMT is a property of epithelial stem and progenitor cells, both normal and neoplastic, and reflects an intrinsic phenotypic plasticity of these cell states. EMTs play important roles in development and epithelial regeneration. During gastrulation, epiblast cells undergo an EMT in order to migrate to appropriate locations as they differentiate into mesoderm and endoderm progenitors. EMTs are also important in later morphogenic events and during wound healing in the adult (17,21,152). Cells that undergo an EMT return to an epithelial state through a mesenchymal-to-epithelial transition (MET)(17,21,152). The regained epithelial state after and EMT-MET sequence may be different from the starting one, as it occurs during gastrulation.
In normal and neoplastic cells EMTs are driven by SNAIL, SLUG, ZEB and TWIST and other transcription factors (referred to as EMT-TFs) which repress epithelial genes and stimulate the expression of mesenchymal components, together with micro-RNAs that balance this regulatory network(21). Stromal signals that trigger EMTs include TGF-β, WNT, NFkB activators(17,21,152). TGF-β induces the expression of EMT-TFs in cooperation with RAS-MAPK signaling; TGF-β-activated SMAD transcription factors and MAPK-activated RREB1 jointly drive expression of SNAIL and SLUG coupled to cell type-dependent differentiation or fibrogenesis gene sets, both in normal and neoplastic cells(153). WNT- activated TCF transcription factors, and NFkB transcription factor also converge on EMT-TF gene enhancers to regulate EMTs(21).
Carcinoma cells at the tumor invasion front undergoing an EMT can drive collective migrating by pulling cohesive ensembles of cancer cells that remain connected by cell-cell contacts(154). After efficiently disseminating and infiltrating distant organs using the mesenchymal properties acquired with the EMT, disseminated MICs undergo an MET in order to reinitiate tumor growth(155,156). In human and mouse carcinomas, loss of cell surface E-cadherin during EMT activates pro-survival signaling that is necessary in the detached state, but E-cadherin re-expression is required for the outgrowth of metastatic colonies(35). What signals trigger MET in disseminated MICs, and whether MET occurs before or after MICs pass through dormancy remain open questions.
Epithelial and mesenchymal states are now viewed not as discrete endpoints but a continuum of highly fluid, interconverting phenotypic states. Each particular epithelial progenitor cell state undergoes its own form of EMT, involving a distinct combination of EMT-TFs and different changes in cell morphology, adhesion and motility(17). The extent of mesenchymal conversion achieved by an epithelial progenitor during an EMT also depends on the developmental state of this cell and reflects the phenotypic space that this state is programed to sample. Many epithelial progenitors undergo what is referred to as a “partial” or “hybrid” EMT. However, the current trend is to consider every departure of epithelial cells from the polarized phenotype an EMT regardless of how far towards a stereotypical (“full”) mesenchymal phenotype this departure may reach(17). Although EMT is a common mechanism for metastatic dissemination, the cells that are most competent at forming metastasis are not necessarily those that adopt the most mesenchymal-like phenotype in their EMT(157–159). Some forms of dissemination, such as collective migration, may enable cancer cells to retain epithelial identity and intercellular contacts during migration and may not require an EMT.
Becoming a MIC
Cancer cells require a large number of traits in order to perform each step of the metastatic process and additionally survive therapy if present. Some of the traits required to become a MIC are imparted by the basic tumor-initiating phenotype resulting from genetic alterations that activate driver oncogenes and that disrupt tumor suppressor genes. In addition to uncontrolled survival and proliferation functions, these traits include the ability to migrate, invade, and remodel extracellular matrix, all of which are conducive to metastasis. The basic traits or “hallmarks” of cancer included with the neoplastic phenotype remain essential throughout the course of tumor progression(160). Moreover, growing evidence indicates that cancer is initiated by oncogenic mutations in stem cells whose normal function is to maintain tissue homeostasis. Turned into CSCs, these cells retain self-renewal capacity, or “stemness”, which is essential for long-term tumor growth(4).
But even with these powerful traits in place, a vast majority of cancer cells leaving a tumor fail to form distant metastasis, instead succumbing to the stresses of the metastatic process. The many obstacles that MICs must sort on their way to establishing metastasis imply a need for additional traits beyond those provided by the CSC phenotype alone. Indeed, work over the past two decades has identified a large number of “metastasis genes” whose expression (or repression) enhances the ability of cancer cells to enter the circulation, extravasate, avert stromal attack, resist metabolic stress, enter dormancy, evade immune surveillance, reinitiate tumor growth, secure a supportive niche, and coopt organ-specific stromal components(2,5,102).
Adaptability is another key trait of the MIC phenotype. Disseminated cancer cells must adapt to a succession of rapidly changing physical, metabolic and immune challenges. In order to survive these stresses, MICs adjust their phenotype accordingly. Phenotypic plasticity, the ability of cells to dynamically change their gene expression programs, is an overarching hallmark of tumor progression and metastasis, and a source of adaptability for MICs(161,162). Phenotypic plasticity enables cancer cells to undergo EMT, adapt to specific microenvironments, and resist therapy(163,164). Moreover, plasticity accentuates the phenotypic heterogeneity of genomically diverse cancer cell populations(165).
In sum, MICs acquire different classes of traits from different sources (Figure 4a): basic pro-metastatic traits acquired with the neoplastic phenotype, additional specialized traits acquired under selection for tumor regeneration in specific organs, and a plastic phenotype adept at deploying these traits under rapidly changing conditions and MICs attempt to regenerate the tumor in new locations.
Figure 4. Acquisition of MIC phenotypes.

a. MICs arise from primary tumors but must acquire distinct phenotypic traits in order to successfully disseminate, seed and colonize distant organs. MICs adopt key phenotypic traits of regenerative progenitors that respond to tissue injury, including phenotypic plasticity, the ability to restore heterogeneous and morphologically complex epithelial structures upon disruption of tissue structure, and the ability to evade killing by immune cells. In addition, MICs must acquire organ-specific adaptations that enable colonization of distinct microenvironments. b. Models of metastasis evolution. In the parallel evolution model, cancer cells disseminating from the primary tumor early during cancer progression seed distant organs and evolve genetically, independently from the primary tumor. Thus, mutations in the primary and metastatic tumors would be expected to be different. In contrast, in linear evolution models, MICs disseminate late during tumor evolution from the primary tumor, and thus are closely clonally related to the primary tumor. Linear evolution can be gradual, in which case tumors may consist of multiple distinct subclones with similar fitness and individual metastases may be derived from distinct subclones, or punctuated, in which case a dominant subclone arising in the primary tumor rapidly outcompetes and overtakes the entire population and seeds all metastases. Dash arrows represent the length of metastasis latency time periods.
Tumor evolution and metastatic progression
How do MICs acquire their unique adaptability and other specialized traits? Heterogeneous cancer cell populations evolve under continual selective pressures during tumor progression and metastasis. The identification of large numbers of tumor-associated mutations by large-scale genomic sequencing of matched normal and primary tumor tissues provides a basis for the notion that metastasis specific traits may emerge by selection from the genetic heterogeneity present in cancer cell populations. However, the search for mutations that specifically drive metastasis has met with few examples to date.
Genetic drivers of metastasis.
Studies seeking to identify metastasis-specific mutations pose several challenges(166,167). Metastasis typically evolves in patients over several years, and this temporal dimension cannot be adequately reproduced in mouse models. On the other hand, patient-based studies are logistically cumbersome, complicated by interpatient heterogeneity, and subject to sampling bias if only a small region of a large tumor is biopsied for genomic analysis(168). Therapy is another important confounder, since patients who experience metastatic relapse after initial removal of primary tumors will have typically received systemic anti-cancer therapies, and such metastases reflect therapy resistance and may not represent the natural history of metastasis per se. Despite these hurdles, several recent studies involving multi-region sampling of matched primary and metastatic cancers biopsied from large numbers of patients have illuminated the genetics of metastatic evolution(164,167,169–174). Notably, across tumors, there appear to be few recurrent metastasis-specific somatic mutations including single nucleotide variants (SNVs), insertions and deletions. Where such mutations have been identified, their functional significance is frequently linked to expanding the activity of the oncogenic driving pathway or conferring resistance to therapy(168,170,175,176). However, activation of certain general growth promoting pathways may translate into organ-specific metastatic advantage. For example, mutations in PIK3CA (phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit α) have been proposed to favor brain metastasis of breast cancer by driving lipid synthesis(129).
The association of genetic mutations with metastatic progression reflects a selection of pro-metastatic traits or an association of bystander mutational events with clones that gain a pro-metastatic phenotype by other means. A recent study of 1,421 samples from 394 tumors across 22 tumor types identified increased clonality of somatic copy number alterations (SCNAs) in metastases versus primary tumors, potentially indicating an as yet inadequately understood role for large scale genomic rearrangements in driving metastasis(177). In non-small cell-lung cancer, elevated copy-number heterogeneity across multiple sequenced regions of early stage primary tumors was associated with increased risk of relapse(178), suggesting that ongoing chromosomal instability is an important mechanism to generate evolutionary space for the emergence of pro-metastatic traits within tumors.
Modes of evolution.
Computational analyses of SCNAs and SNVs enable reconstruction of phylogenetic trees to determine the sequence and selection of mutations during metastasis(179). Two models for metastatic evolution have long been considered(180). The “linear evolution” model envisions that primary tumors gradually accumulate mutations, with a late subclone acquiring metastatic capacity (Figure 4b). The “parallel evolution” model, on the other hand, argues that cancer cells that disseminate early are more effective at forming metastasis because they evolve in the host microenvironment, unlike clones that remain in the primary tumor(181). In HER2+ mouse models of metastatic breast cancer, early disseminating cells were found to be more metastatically competent than late disseminators(182,183). Using sensitive cell-free DNA technology, circulating DNA with tumor-associated mutations can be detected in patients with subclinical primary tumors in a variety of cancers, suggesting that early dissemination is possible. However, the rarity of metastasis-specific SNVs in patients argues against the parallel evolution model(167,169). Other studies have identified both metastasis-specific recurrent alternations and recurrent alterations that are shared between primary and metastatic tumors, suggesting that both early and late metastatic dissemination can occur(164). Moreover, recent work suggests the presence of “punctuated evolution” in some tumors, with acquisition of mutations that rapidly clonally dominate the primary tumor, and then seed metastasis in an evolutionary “Big Bang”(171,184–187). Other tumors may evolve more gradually, with multiple clones of low metastatic fitness competing within the primary tumor.
Taken together, the evidence from clinical and murine studies suggest that metastasis imposes an evolutionary bottleneck that only select cells within primary cancer cell populations are able to overcome. Such metastases may be monoclonal or polyclonal(171,184–189), suggesting potentially distinct modes of seeding or selection. It is worth noting that the prevalent linear interpretation of the clonality and timing of metastases may be confounded by the possibility of ongoing “self-seeding” of metastases, wherein cells from metastatic tumors may recirculate and seed not only new metastatic lesions, but may also infiltrate pre-existing primary and metastatic tumors(190,191).
Evolutionary timing and cancer therapy.
Intriguingly, in clear cell renal cell carcinoma, dominant clones resulting from punctuated evolution seed aggressive multiorgan metastasis with little intermetastatic heterogeneity, while oligometastases with indolent behavior arise from tumors displaying gradual evolution(186) (Figure 4b). Unlike multiorgan metastasis, oligometastasis in a single organ may be amenable to curative surgery or other local treatments, and thus provides an opportunity to use tumor clonality as a predictive biomarker for such therapies(192). In patients, there is a strong correlation between tumor size (a proxy for tumor age) and the probability of future metastatic relapse, which is inconsistent with the possibility of exclusive early metastatic seeding. However, in a large cohort of breast cancer patients treated with mastectomy and no systemic therapy, tumor size was associated with early local recurrence and distant metastasis, but not with late metastasis(193). This observation suggests the possibility that in some tumors both early and late dissemination may coexist, with late, more proliferative disseminating cells driving mostly rapid relapse. In contrast, early disseminating cells may be particularly prone to latency for extended periods of time, either because of a capacity to enter a deep quiescent state, or because those that grow are rapidly cleared by the immune system.
Epigenetic basis for metastatic adaptability
Although genomic alterations generate selectable survival traits in cancer cells, mutations alone cannot account for the fundamentally distinct biology of MICs. Unlike primary tumors, which are thought to largely arise through oncogenic transformation of homeostatic tissue stem and progenitor cells in their native niche(194–198), MICs must rapidly change their phenotypes to detach from the primary tumor, survive mechanical, metabolic and immunological stresses during dissemination to distant organs, adapt to the foreign microenvironments, access proliferative cues to reinitiate tumor growth in distant organs, and resist therapy. Dynamic adaptation to changing circumstances during the metastatic process requires faster change than a purely genetic mechanism, which allows for selection of variants only once per cell cycle, might support.
While the dynamic expression of specific genes has long been associated with metastatic phenotypes, the master transcription factors and epigenetic remodeling mechanisms required for the different steps of the metastatic process in various tumors are only beginning to emerge(199). Genes required for chromatin modification (e.g. ARID1A/B, KMT2C/D) and DNA methylation (DNMTs) are frequently mutated in advanced tumors, underscoring the importance of epigenetic modulation in metastasis(173). A fundamental question is whether metastatic adaptability involves greater overall chromatin accessibility, and hence the potential to activate the expression of multiple genes, as implied by studies in genetically engineered mouse models of lung cancer(200–202), or whether the ability to transition to one or more pro-metastatic phenotypic states by activation of specific transcription factors is sufficient, as shown in models of pancreatic ductal adenocarcinoma(203) and renal cell carcinoma(204). Recent work illuminated the role of histone H3 variants(205), and specific histone marks, such as H3K36me2(206), as critical requirements for epigenomic reprogramming during metastasis. The question remains whether epigenetic alterations favor metastasis by randomly activating selectable genes or by enabling cancer cells to enter progenitor states that have a superior fitness to initiate metastasis.
Regenerative origins of metastatic traits
Several key traits of the MIC phenotype are normally present in regenerative progenitor cells. In wound-healing processes (Figure 5a), epithelial regenerative progenitors show intense phenotypic plasticity, undergo EMT, display migratory activity, interact with and remodel the extracellular matrix, and secrete signals that coopt mesenchymal, endothelial and immune stromal components to support the restoration of epithelial barrier integrity(207,208). Recent work raises the possibility that carcinoma MICs may capture these traits all at once by adopting a regenerative phenotype.
Figure 5. Model of the regenerative progenitor origin of MICs.

a. In the intestine, proliferative LGR5+ crypt base cells maintain intestinal homeostasis. Injury disrupts intercellular adherens junctions and induces the emergence of an L1CAM+ regenerative progenitor phenotype, which is required for epithelial repair, regeneration of heterogeneous cell types, and resolution of the wound. b. Oncogenic driver mutations in homeostatic stem and progenitor cells create tumor initiating cells (or “cancer stem cells”, CSCs), capable of adenoma formation within an intact epithelial niche. Additional mutations give rise to an invasive carcinoma, which further breaches the integrity of the epithelium and triggers the emergence of an L1CAM+ regenerative progenitor state in cancer cells. As regenerative progenitors, these malignant cells have the capacity to survive the loss of the epithelial niche during tumor dissemination, are competent to enter quiescence, evade immune surveillance and therapy, and upon the receipt of favorable growth signals, regenerate heterogeneous tumors in metastatic sites and after therapy.
Phenotypic plasticity in regenerative progenitors.
Phenotypic plasticity, a key feature of tumor progression and metastasis(161,162), is also a feature of normal adult stem cells, including those of the gut, lung and skin. These cells are not locked in a fixed pre-existing cell fate but rather enter and exit “stem-like” states in response to demands to preserve the structural and functional integrity of the tissue(4,162,209–211). In the intestinal mucosa, cells expressing the WNT signaling pathway component LGR5 function as stem cells in the maintenance of epithelial homeostasis(212). However, when LGR5+ cells are lost by genetic ablation or injury, LGR5− progenitor cells can become LGR5+ cells(208,211). Moreover, unique stem cell states are mobilized by damage to revive the epithelium through transcription factor YAP (213).
Cells from the mouse small intestine can grow as organoids, three-dimensional structures that form in laminin-rich basement membrane matrix and mimic the cellular composition and architecture of the intestinal epithelium(214). Both LGR5+ and LGR5− cells sorted from normal mouse intestinal epithelium can form organoids(211,215). Moreover, during the initial stages of organoid formation LGR5+ cells convert to a LGR5− state that includes YAP activation, and later re-express LGR5 and downregulate YAP to establish a differentiated epithelium(215). Thus, intestinal organoid formation appears to start as a process of epithelium recovery driven by LGR5− progenitors followed by restoration of differentiated structures driven by reemerging LGR5+ progenitors.
Dynamic phenotypic changes are also observed in cancer metastasis. In colorectal cancer (CRC), intestinal LGR5+ stem cells harboring constitutively activating mutations in the WNT signaling pathway act as tumor-initiating CSCs(197). However, intravital microscopy of experimental CRC metastasis in mice revealed that MICs disseminating from the primary tumor and seeding metastasis in the liver are predominantly LGR5− (216). To initiate metastatic tumor growth in the liver, some progeny of LGR5− cells reacquire LGR5 expression(216), while other cancers may acquire LGR5-independent regenerative capacity(217,218).
Adopting a regenerative phenotype.
Recent work has shown that MICs in CRC and other epithelial cancers dynamically express L1CAM(32). Originally identified as a neuronal cell adhesion molecule, L1CAM is expressed in carcinomas at the primary tumor invasion front and is associated with poor prognosis in multiple tumor types, including those of the breast, lung, colon, ovary and kidney(219). L1CAM is not expressed in intact normal epithelia or required for epithelial homeostasis, but its expression is induced by the breach of epithelial intercellular adherens junctions during colitis and tissue injury, and is required for the survival of detached epithelial progenitors and for tissue repair(32). L1CAM expression in dissociated CRC cells is required for the initiation of organoid formation, where most L1CAM+ cells are LGR5−. L1CAM+ cells are highly competent to initiate growth of transplanted CRC tumors and liver metastases. As MICs initiate tumor growth and regain epithelial structures, L1CAM expression is downregulated, but can be reactivated again when tumor architecture is disrupted by therapy. Repeated cycles of therapy select for cells with the capacity to upregulate L1CAM-expression, which can more efficiently reinitiate tumor growth(32). Thus, L1CAM seems to mark certain regenerative progenitor states and its expression in these cells mediates the regrowth of intestinal epithelium after injury and tumor regrowth during metastasis.
Several studies highlight a biphasic process of epithelial repair that is coopted during metastasis. Upon wounding, an immediate emergency response is activated in which epithelial progenitors enter distinct migratory and proliferate states to seal the breach in the mucosa(220,221) (Figure 5a). This “bandaid” phase is followed by a slower regenerative phase in which normal tissue stem/progenitor hierarchies are recreated. Borrowing from repair, dissociation of epithelial structures in invasive CRC induces malignant progenitors to adopt a highly plastic regenerative phenotype endowed with capacity to undergo EMT, migrate, avert anoikis, and drive L1CAM-dependent regrowth(32,43) (Figure 5b).
Further evidence for the regenerative nature of metastatic progression comes from single-cell RNA sequencing studies on tumor tissue from lung adenocarcinoma patients as well as lesions from a mouse model of metastatic latency and outbreak(222). Whereas primary tumors showed developmental lineage promiscuity and cell states that were intermediate between the various cell states of the regenerative lung epithelium, metastatic cells reverted to earlier states and recapitulated lung organogenesis and regeneration phenotypes with striking fidelity.
The adoption of a regenerative phenotype by MICs has several implications. Instead of having to select a pre-determined rigid MIC fate or randomly acquiring a multitude of independent metastatic traits, CRC cells seem to sample among a range of phenotypically transient states, some of which, such as the L1CAM+ state, are adept at supporting survival of disseminated cells and the initiation of metastatic growth and are hence selected for during the metastatic process(32,43).
Acquiring organ-specific colonization traits.
The highly specialized nature of most organ-specific metastatic traits suggest that these are acquired through selection from heterogeneous MIC populations that complete their evolution after seeding distant organs, rather than being intrinsic properties of the stem cell states that give rise to MICs. However, not all organ-specific traits result from clonal evolution after dissemination. The microenvironment of a primary tumor can pre-select clones with properties that are compatible with the environment of a particular distant organ, thus influencing the metastatic tropism of the disseminating cancer cells. For example, primary breast tumors with high TGF-β activity are associated with lung relapse, and this is linked to the ability of TGF-β to induce the expression of the cytokine ANGPTL4 in the cancer cells. ANGPTL4 causes endothelium disjunction to promote extravasation of circulating breast cancer cells that lodge in lung capillaries(223). Primary tumors may also pre-select for MIC clones that thrive in distant organs whose stroma the primary tumor mimics. For example, breast carcinomas rich in mesenchymal cells that produce CXCL12 select for MIC clones that thrive in a CXCL12-rich microenvironment, and thus are predestined to colonize the CXCL12-rich stem cell niches of the bone marrow(224).
Metastasis as a systemic disease
Metastasis is subject to important organ-specific barriers and adaptation, as in previous sections and recapitulated in Figure 6a. However, metastasis is by definition a systemic disease(225). Despite the growing understanding of MIC-intrinsic and metastatic niche dependent mechanisms, relatively little is known about the complex ways in which systemic physiological factors influence MIC plasticity and selection, and vice versa. However, recent studies have provided evidence for the potential role of various systemic sources in the development of metastasis (Figure 6b). While the molecular basis for a majority of these effects remains to be determined, and their causal relationship with metastatic progression remains to be firmly established, this is an area of rising interest.
Figure 6. Metastasis as a systemic disease with organ specific features.

a. A recap of organ-specific determinants of metastatic colonization. b. Systemic determinants of metastasis. During metastasis, cancer cells from primary tumors disseminate throughout the body via the blood and lymphatic circulations, among other routes. Tumor cells establish a dynamic equilibrium with both systemic immunity and organ-specific immune infiltrates. Systemic factors can modulate both primary and metastatic tumor growth as well as the anti-tumor immune responses. While the catalog of systemic factors that can modulate metastasis is incomplete, current evidence implicates circulating chemokines and cytokines and signals transmitted via nerves and hormones in controlling tumor growth and metastasis. In addition, intratumoral and gut microbiomes can influence cancer progression either through direct interactions with cancer cells and immune cells, or indirectly, via metabolites released into the systemic circulation. The dynamic equilibrium between tumors, metastases and their niches is influenced by systemic factors that impact MIC growth and therapy resistance.
Systemic niche components.
Both primary and metastatic tumors can release cytokines, chemokines and hormones either directly into the systemic circulation or packaged into exosomes, which in turn can modulate distant niches to favor or resist metastatic colonization, including organ-specific metastatic dissemination(147,151,226,227). While many critical mediators and mechanisms of pre-metastatic niche formation remain to be uncovered, studies to date suggest that organ specific stromal cell reprogramming induces changes in extracellular matrix and matrix-associated proteins, which in turn enable recruitment of specific myeloid populations that support future MIC growth(228). Indeed, bone marrow derived myeloid cells can play critical roles in either limiting or promoting MIC survival and growth, and the mechanisms enabling their context-specific recruitment, differentiation and activity at various steps in the metastatic process have been a focus of growing interest(18,98,130).
Systemic immunity, inflammation and metabolism.
The critical role of systemic immunity in controlling metastasis is now firmly established. Physiological and pathological phenomena that modulate systemic immunity indirectly control metastasis, in addition to having direct effects on MICs (Figure 6). Stress and depression have been associated with early metastasis and poor prognosis in patients with breast, lung and hepatobiliary carcinomas(229). In cell line xenograft models of metastatic breast cancer, increased corticosteroid stress hormone levels generated during tumor progression resulted in increased glucocorticoid receptor activation, downstream ROR1 upregulation, metastatic growth and chemoresistance(230). In addition to direct effects on MICs, glucocorticoids have widespread immunosuppressive effects, which in turn can promote MIC outgrowth(231,232). In addition to steroids, increased levels of the stress-induced catecholamine norepinephrine induced changes in neutrophils that enabled MIC escape from dormancy in syngeneic lung cancer models(233).
Systemic inflammation and immunosuppression resulting from a range of physiological and pathological conditions, including ageing, post-partum breast involution, asthma and osteoarthritis have been associated with increased metastasis risk and reduced survival from a range of epithelial cancers(234–238). In a recent study, inflammation induced by surgical wounding was shown to promote MIC outgrowth from dormancy in breast cancer mouse models(239), suggesting a potential explanation for the early peak in metastatic recurrence within one year of curative breast surgery in patients(240). Based on these observations, several groups have proposed the prophylactic use of non-steroidal anti-inflammatory agents and beta-blockers in the peri-operative setting, although clear clinical evidence for a significant anti-metastatic effect is lacking(239,241).
Tumors in turn may alter host metabolism, immunity and physiology locally as well as systemically (Figure 6). Circulating cytokines and metabolites derived from metastatic tumors can have a dramatic impact on tissues such as skeletal muscle and adipose. Indeed, many patients with metastatic cancer develop cachexia, a debilitating muscle-wasting syndrome that is associated poor prognosis and accelerated death(242). A better understanding of the molecular basis for how metastatic disease promotes cachexia is important for improving the care of patients with metastatic cancer.
Nerves and metastasis.
The nervous system is emerging as a potential systemic modulator of metastasis(243,244). Innervation is essential for physiological regeneration(245), but the role of nerves in metastasis had long been surmised to be only as passive conduits for tumor dissemination in cases of perineural invasion(61). Growing evidence, however, suggests that neurons can be active determinants of cancer progression and outcome(243,244) (Figure 6). Direct innervation of tumors can contribute to tumor progression at both the primary and metastatic sites. In mouse cancer models, autonomic denervation of the prostate, stomach and pancreas inhibited tumor progression and cell dissemination from these sites(246). In addition to effects mediated by direct tumor innervation, increasing understanding of neuronal regulation of immunity suggests a systemic means of neuronal metastatic regulation, although little mechanistic insight is currently available(247). For example, in a syngeneic metastatic mammary carcinoma model, denervation of sensory neurons suppressed NK cell activity resulting in increased lung and cardiac metastases(248).
Diet and exercise.
Epidemiological and experimental studies have identified correlations between lifestyle factors such as diet and exercise with cancer incidence, therapy resistance and survival(249–251). Proposed mechanisms of tumor modulation include direct effects on cancer cells (for example, by altering insulin signaling or lipid metabolism), indirect effects on tumor inflammation and immune surveillance, or especially for diet, tertiary effects mediated by the gut microbiome(128,250–255). Obesity, which is associated with both poor diet and low exercise, is an independent risk factor for the future development of distant metastasis and death in patients with surgically resected primary breast cancer(256). With some notable exceptions (e.g. (128,257)), the majority of animal studies of diet, exercise and microbiome modulation to date have been performed in models of tumor initiation or subcutaneous tumor cell transplantation, and the functional and mechanistic significance of these studies to the critical steps of MIC dissemination, dormancy and outbreak, as well as therapy resistance in patients with advanced cancer remain unresolved.
It is important to discern whether lifestyle factors that may influence tumor progression represent modifiable, and potentially therapeutically targetable, risk factors in patients who have already been diagnosed with primary tumors. A meta-analysis of 49,095 early stage breast and colon cancer survivors suggested that survivors who increased their physical activity from before diagnosis and surgery showed a statistically significant decreased total mortality risk in comparison with those who did not change their activity level(258). Prospective clinical studies randomizing newly diagnosed cancer patients to specific dietary or exercise interventions will help clarify the actionability of these factors in preventing and treating metastasis(259).
Microbiome.
The role of the microbiome as a player in influencing cancer biology is becoming increasingly evident(260,261) (Figure 6). The mucosal microbiome can interact directly with epithelial cells in niches such as the luminal gastrointestinal tract, respiratory tract and skin to influence tumor progression through local inflammation or immunosuppression(260,261). Gut pathobiotic strains of fusobacterium have been shown to colonize colorectal tumors and drive metastasis to the liver(262). Mouse xenografts derived from such human liver metastases continue to be colonized by fusobacterium, and antibiotics that target fusobacterium can inhibit metastasis in mouse models(262). Intriguingly, many cancers that are not directly exposed to environmental bacteria, including breast and bone cancers, have recently been shown to also harbor intratumoral microbiota, although the functional significance of such colonization remains unclear(263). In addition to local effects, metabolites secreted by microbiota may have diverse systemic effects on immunity, hormones and organ function, most of which are currently poorly understood(264,265). Other systemic factors, such as diet, can rapidly alter microbiome composition(266,267), and patient-specific microbiota can in turn influence responses to therapy(268,269). Given this complexity, there is an increasingly evident need to characterize and control for the microbiome composition as an independent variable in animal and human studies of metastasis.
Aging.
In some cancers, such as melanoma, age at cancer diagnosis is associated with increased tumor aggression and metastasis(270). Three broad mechanisms have been proposed for age-related local and systemic acceleration of metastasis(271,272). First, age-dependent accumulation of senescent cells, and their associated secretomes, in the tumor microenvironment may promote tumor invasion, pre-metastatic niche formation and metastatic outgrowth(273–275). Co-injection of senescent fibroblasts, but not normal fibroblasts, promoted metastatic progression in mouse models(276). Second, age-related deterioration of extracellular collagen matrices may facilitate tumor cell invasion and promote transendothelial migration(277,278). Third, age-associated systemic low-grade chronic inflammation, termed “inflammaging”(279), can disrupt tissue architecture and promote tumor dissemination(277,280), while immunosenescence may prevent effective immune editing of disseminated MICs emerging from dormancy(272,281). Further delineation of the mechanisms by which aging intersects with metastatic progression is needed to define therapeutic strategies to prevent and treat cancer in aging populations.
MIC-intrinsic and acquired drug resistance mechanisms
A cardinal feature of metastatic disease is its ability to resist therapy. We recently reviewed the clinical contexts and approaches for targeting metastasis(282). Here we focus on the properties of MICs that enable therapy resistance, and potential approaches to overcome these.
Since tumors comprise numerous genetically distinct subclones, cells harboring therapy resistance mutations can be selected for during metastasis as the patient undergoes treatment for initially localized disease. Genetic analyses of matched primary tumors and metastases show that metastases from patients who had received adjuvant therapy for treatment of initially localized cancer had reduced clonal heterogeneity in comparison to metastases from previously untreated patients, suggesting that treatment imposes an evolutionary bottleneck for MIC emergence(173). This is particularly evident in the case of treatments that target specific oncogenic signaling pathways, where therapy with inhibitors of these pathways selects for the emergence of cancer cells with specific recurrent resistance mutations. Examples include EGFR T790M mutations in patients treated with first generation tyrosine kinase inhibitors gefitinib and erlotinib(283), and ESR1 mutations in estrogen receptor-positive breast cancer patients treated with anti-estrogen therapy(175). Clinical and experimental studies characterizing such targeted therapy-specific resistance mutations have enabled the rapid development of improved drugs that anticipate and target resistance mutations, with improved therapeutic results(284).
In addition, these genetically acquired mechanisms of drug resistance, MIC phenotypic properties may confer intrinsic therapy resistance(154,282,285). In most cancers, the mainstay of treatment of metastasis remains cytotoxic chemotherapy, which preferentially targets rapidly proliferating cells. Thus, MICs that are capable of entering quiescence may more readily evade chemotherapy than do proliferative cancer cells. When therapies result in partial destruction of glandular metastatic tumors, they may in turn disrupt epithelial adherens junctions and induce MICs to enter a regenerative progenitor state, as observed in colorectal cancer(32). Successive rounds of therapy may further select for residual cells that are epigenetically primed to reenter regenerative states(286), suggesting an explanation for the successively restricted duration of clinical response typically observed with sequential lines of therapy in patients with advanced cancer. In addition, the stem-like properties of MICs may include the expression of ATP-binding cassette drug efflux transporters that could confer multidrug resistance(287). Dying or senescent cancer cells induced by therapy that partially debulks metastatic tumors can release cytokines as secretomes, to which MICs can respond by activating growth factor signaling programs to regenerate tumors(288–290).
The immune evasive properties of MICs are likely also of importance in resisting both tumor cell-directed therapy and systemic immunotherapy. In addition to MIC-intrinsic mechanisms of immune escape, desmoplasia in tumors is an important mechanism of therapy resistance. In pancreatic ductal adenocarcinoma, Hedgehog signaling induces fibrosis, which in turn poses a physical barrier for drug delivery to tumor cells that are surrounded by fibroblasts(20,291,292). In many cancer types, immune cells are recruited to tumors from the systemic circulation but are spatially segregated from tumor cells by fibroblast barriers, resulting in immunologically “cold” tumors that do not respond to PD-1 checkpoint immunotherapy(293,294). Targeting these fibroblast barriers using hedgehog inhibitors disrupts desmoplasia in mouse models of pancreatic ductal adenocarcinoma (292). TGF-β is strongly immunosuppressive and fibrogenic(89,90); the use of TGF-β inhibitors in colorectal and breast cancer increase drug and immune cell infiltration into the tumor, reversing immunotherapy resistance in preclinical models and correlating with superior immunotherapy responses in patients(295,296). However, the translation of these approaches to patients has been challenging to date due to complex effects of drugs that target developmental signaling pathways on the tumor microenvironment, as well as broader systemic toxicities(297–299). Improved understanding of the mechanisms of tumor fibrogenesis and immune cell communication may yield more specific targets for therapeutic intervention.
The plasticity of MICs arguably poses the greatest bottleneck to therapeutic success, since MICs that are capable of sampling among numerous transcriptional and epigenetic states may be surmised to rapidly select for gene expression programs that enable therapeutic escape. However, understanding the molecular mechanisms of MIC plasticity may yield improved therapeutics(162,300). For example, epigenetic modulators could be used to inhibit or reverse MIC plasticity or to force MIC differentiation into terminal, non-proliferative states. Targeting genotype-agnostic conserved regenerative states may block the relapse of metastatic disease that is being debulked by antineoplastic chemotherapy. Characterizing the endpoints of plasticity, such as neuroendocrine lineage plasticity in prostate cancer, may enable the development of new drugs that target the emergent neuroendocrine state instead of or in addition to the pre-existing androgen-driven epithelial state(163).
Summary and Perspectives
In summary, recent work is illuminating the fundamental principles of metastasis initiating cells and their ecosystems, and importantly shedding light on the mechanisms by which metastatic tumors emerge and develop as phenotypically distinct, therapy resistant entities in comparison to primary tumors. In brief:
Metastasis develops through three distinct phases, namely dissemination, dormancy and outbreak, which can co-exist until the removal of the primary tumor.
Each phase of metastasis reflects a dynamic equilibrium of MICs with host immunity, with the eventual outbreak of MICs as macrometastatic colonies that can be detected using standard imaging reflecting a failure of immune surveillance.
While pre-existing genetically distinct tumor subclones with high metastatic propensity are selected for in primary tumors, phenotypic plasticity, enabled by epigenetic and metabolic adaptations, is an overarching hallmark of MICs.
Phenotypic plasticity endows MICs with the ability to sample multiple transcriptional states and dynamically adapt to diverse stresses during each phase of metastasis.
MICs can adopt epithelial regenerative progenitor states that possess stem-like, immune evasive, tissue regenerative, and therapy resistance capacities.
The dynamic equilibrium between MICs and their organ-specific microenvironmental niches can be modulated by systemic factors that impact either or both components.
Identifying the vulnerabilities of conserved regenerative MIC states, and the molecular determinants of communication between MICs and their ecosystems could yield improved therapeutics to prevent and treat metastasis, and thus improve outcomes for patients with advanced cancer.
With the advent of immunotherapy and continued advances in targeted therapy, the last two decades have witnessed considerable improvements in outcomes of patients with some types of metastatic cancers(270). These advances are driven by increasingly closer collaborative bidirectional interactions between clinical and bench scientists, and improved, more representative patient-derived and animal models of metastatic cancer. Significant therapeutic advances are resulting from the rapid recognition, molecular characterization and clinical translation of biologically discrete phenotypes of metastatic disease. Examples include curative local therapy for oligometastatic disease, immune activation phenotypes in extreme outlier patients with exceptional response to therapy, and mechanism-based therapies for metastases in specific organs, such as the brain, where even small improvements in tumor shrinkage can yield significant improvements in patient survival(282).
Further improvements in patient outcomes will emerge from a deeper understanding of the mechanisms that underlie the plasticity and dormancy of MICs as well as the complex interactions between the MICs, their niches and systemic factors. To do so, preclinical models used to study metastasis and test therapies for metastatic cancer must recapitulate the genomic complexity, plasticity and niche biology of advanced human cancers, rather than relying exclusively on simpler systems that may fail to capture these critical features.
In 1889, Steven Paget first hypothesized that metastasis might result from a fertile “seed” landing on congenial niche “soil”(301). While investigations in the subsequent 111 years have vindicated Paget’s view, a third dimension of the metastasis ecosystem – the “forest” of systemic factors, is emerging. By understanding and targeting the interactions between the seeds, soil and forest, we may be able to convert metastasis, which is today still typically a death sentence, into a manageable illness.
Acknowledgments
Financial support: The author’s work in the subject area of this review is supported by NIH grants R35CA252978 (JM), K08CA23021 (KG), P30-CA008748 (MSKCC), and the Alan and Sandra Gerry Metastasis and Tumor Ecosystems Center at MSKCC (JM).
Footnotes
Conflict of Interest Disclosure: JM owns stock of Scholar Rock Inc. One or both authors are inventors in the following patents and patent applications: US Patent 7829066 (JM); EU Patent 2831593 (JM); EU Patent 3047039 (JM). US Patent 10413522 (JM); Patent Application PCT/US2016/062880 (JM); and, Patent Application PCT/US2017/045145 (JM and KG).
References
- 1.Lambert AW, Pattabiraman DR, Weinberg RA. Emerging Biological Principles of Metastasis. Cell 2017;168(4):670–91 doi 10.1016/j.cell.2016.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Massagué J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature 2016;529(7586):298–306 doi 10.1038/nature17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oskarsson T, Batlle E, Massagué J. Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell 2014;14(3):306–21 doi 10.1016/j.stem.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Batlle E, Clevers H. Cancer stem cells revisited. Nat Med 2017;23(10):1124–34 doi 10.1038/nm.4409. [DOI] [PubMed] [Google Scholar]
- 5.Celià-Terrassa T, Kang Y. Metastatic niche functions and therapeutic opportunities. Nature Cell Biology 2018;20(8):868–77 doi 10.1038/s41556-018-0145-9. [DOI] [PubMed] [Google Scholar]
- 6.Hüsemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, et al. Systemic spread is an early step in breast cancer. Cancer Cell 2008;13(1):58–68 doi 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 7.Sänger N, Effenberger KE, Riethdorf S, Van Haasteren V, Gauwerky J, Wiegratz I, et al. Disseminated tumor cells in the bone marrow of patients with ductal carcinoma in situ. Int J Cancer 2011;129(10):2522–6 doi 10.1002/ijc.25895. [DOI] [PubMed] [Google Scholar]
- 8.Stott SL, Lee RJ, Nagrath S, Yu M, Miyamoto DT, Ulkus L, et al. Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Sci Transl Med 2010;2(25):25ra3 doi 10.1126/scitranslmed.3000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Risson E, Nobre AR, Maguer-Satta V, Aguirre-Ghiso JA. The current paradigm and challenges ahead for the dormancy of disseminated tumor cells. Nature Cancer 2020;1(7):672–80 doi 10.1038/s43018-020-0088-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Varadhachary GR. Carcinoma of unknown primary origin. Gastrointest Cancer Res 2007;1(6):229–35. [PMC free article] [PubMed] [Google Scholar]
- 11.Lin NU, Winer EP. Brain metastases: the HER2 paradigm. Clin Cancer Res 2007;13(6):1648–55 doi 10.1158/1078-0432.Ccr-06-2478. [DOI] [PubMed] [Google Scholar]
- 12.Sleeman JP, Nazarenko I, Thiele W. Do all roads lead to Rome? Routes to metastasis development. Int J Cancer 2011;128(11):2511–26 doi 10.1002/ijc.26027. [DOI] [PubMed] [Google Scholar]
- 13.Reymond N, d’Agua BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer 2013;13(12):858–70 doi 10.1038/nrc3628. [DOI] [PubMed] [Google Scholar]
- 14.Labelle M, Hynes RO. The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov 2012;2(12):1091–9 doi 10.1158/2159-8290.CD-12-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kai F, Drain AP, Weaver VM. The Extracellular Matrix Modulates the Metastatic Journey. Dev Cell 2019;49(3):332–46 doi 10.1016/j.devcel.2019.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamidi H, Ivaska J. Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer 2018;18(9):533–48 doi 10.1038/s41568-018-0038-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2020;21(6):341–52 doi 10.1038/s41580-020-0237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013;19(11):1423–37 doi 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pignatelli J, Bravo-Cordero JJ, Roh-Johnson M, Gandhi SJ, Wang Y, Chen X, et al. Macrophage-dependent tumor cell transendothelial migration is mediated by Notch1/Mena(INV)-initiated invadopodium formation. Sci Rep 2016;6:37874 doi 10.1038/srep37874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008;455(7211):406–10 doi 10.1038/nature07275. [DOI] [PubMed] [Google Scholar]
- 21.Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell 2016;166(1):21–45 doi 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 22.Denais CM, Gilbert RM, Isermann P, McGregor AL, te Lindert M, Weigelin B, et al. Nuclear envelope rupture and repair during cancer cell migration. Science 2016;352(6283):353–8 doi 10.1126/science.aad7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Helvert S, Storm C, Friedl P. Mechanoreciprocity in cell migration. Nat Cell Biol 2018;20(1):8–20 doi 10.1038/s41556-017-0012-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alix-Panabieres C, Schwarzenbach H, Pantel K. Circulating tumor cells and circulating tumor DNA. Annu Rev Med 2012;63:199–215 doi 10.1146/annurev-med-062310-094219. [DOI] [PubMed] [Google Scholar]
- 25.Pantel K, Alix-Panabières C. Liquid biopsy and minimal residual disease - latest advances and implications for cure. Nat Rev Clin Oncol 2019;16(7):409–24 doi 10.1038/s41571-019-0187-3. [DOI] [PubMed] [Google Scholar]
- 26.Fidler IJ. The relationship of embolic homogeneity, number, size and viability to the incidence of experimental metastasis. Eur J Cancer 1973;9(3):223–7 doi 10.1016/s0014-2964(73)80022-2. [DOI] [PubMed] [Google Scholar]
- 27.Aceto N, Toner M, Maheswaran S, Haber DA. En Route to Metastasis: Circulating Tumor Cell Clusters and Epithelial-to-Mesenchymal Transition. Trends Cancer 2015;1(1):44–52 doi 10.1016/j.trecan.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 28.Duda DG, Duyverman AM, Kohno M, Snuderl M, Steller EJ, Fukumura D, et al. Malignant cells facilitate lung metastasis by bringing their own soil. Proc Natl Acad Sci U S A 2010;107(50):21677–82 doi 10.1073/pnas.1016234107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wrenn ED, Yamamoto A, Moore BM, Huang Y, McBirney M, Thomas AJ, et al. Regulation of Collective Metastasis by Nanolumenal Signaling. Cell 2020;183(2):395–410 e19 doi 10.1016/j.cell.2020.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campbell NR, Rao A, Zhang M, Baron M, Heilmann S, Deforet M, et al. Cell state diversity promotes metastasis through heterotypic cluster formation in melanoma. bioRxiv 2020:2020.08.24.265140 doi 10.1101/2020.08.24.265140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gkountela S, Castro-Giner F, Szczerba BM, Vetter M, Landin J, Scherrer R, et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019;176(1-2):98–112 e14 doi 10.1016/j.cell.2018.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ganesh K, Basnet H, Kaygusuz Y, Laughney AM, He L, Sharma R, et al. L1CAM defines the regenerative origin of metastasis-initiating cells in colorectal cancer. Nature Cancer 2020;1(1):28–45 doi 10.1038/s43018-019-0006-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haemmerle M, Stone RL, Menter DG, Afshar-Kharghan V, Sood AK. The Platelet Lifeline to Cancer: Challenges and Opportunities. Cancer Cell 2018;33(6):965–83 doi 10.1016/j.ccell.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011;20(5):576–90 doi 10.1016/j.ccr.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Padmanaban V, Krol I, Suhail Y, Szczerba BM, Aceto N, Bader JS, et al. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019;573(7774):439–44 doi 10.1038/s41586-019-1526-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH, Lee DJ, et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 2014;156(5):1002–16 doi 10.1016/j.cell.2014.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009;459(7249):1005–9 doi 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boire A, Zou Y, Shieh J, Macalinao DG, Pentsova E, Massagué J. Complement Component 3 Adapts the Cerebrospinal Fluid for Leptomeningeal Metastasis. Cell 2017;168(6):1101–13 e13 doi 10.1016/j.cell.2017.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Achrol AS, Rennert RC, Anders C, Soffietti R, Ahluwalia MS, Nayak L, et al. Brain metastases. Nat Rev Dis Primers 2019;5(1):5 doi 10.1038/s41572-018-0055-y. [DOI] [PubMed] [Google Scholar]
- 40.Strilic B, Yang L, Albarran-Juarez J, Wachsmuth L, Han K, Muller UC, et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 2016;536(7615):215–8 doi 10.1038/nature19076. [DOI] [PubMed] [Google Scholar]
- 41.Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, Herms J, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med 2010;16(1):116–22 doi 10.1038/nm.2072. [DOI] [PubMed] [Google Scholar]
- 42.Im JH, Fu W, Wang H, Bhatia SK, Hammer DA, Kowalska MA, et al. Coagulation Facilitates Tumor Cell Spreading in the Pulmonary Vasculature during Early Metastatic Colony Formation. Cancer Research 2004;64(23):8613–9 doi 10.1158/0008-5472.Can-04-2078. [DOI] [PubMed] [Google Scholar]
- 43.Er EE, Valiente M, Ganesh K, Zou Y, Agrawal S, Hu J, et al. Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat Cell Biol 2018;20(8):966–78 doi 10.1038/s41556-018-0138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol 2013;15(7):807–17 doi 10.1038/ncb2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang H, Fu W, Im JH, Zhou Z, Santoro SA, Iyer V, et al. Tumor cell alpha3beta1 integrin and vascular laminin-5 mediate pulmonary arrest and metastasis. J Cell Biol 2004;164(6):935–41 doi 10.1083/jcb.200309112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Naxerova K Defining the role of lymph node metastasis in systemic breast cancer evolution. EBioMedicine 2020;57:102852 doi 10.1016/j.ebiom.2020.102852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tjan-Heijnen V, Viale G. The Lymph Node and the Metastasis. N Engl J Med 2018;378(21):2045–6 doi 10.1056/NEJMcibr1803854. [DOI] [PubMed] [Google Scholar]
- 48.Reiter JG, Hung WT, Lee IH, Nagpal S, Giunta P, Degner S, et al. Lymph node metastases develop through a wider evolutionary bottleneck than distant metastases. Nat Genet 2020;52(7):692–700 doi 10.1038/s41588-020-0633-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Naxerova K, Reiter JG, Brachtel E, Lennerz JK, van de Wetering M, Rowan A, et al. Origins of lymphatic and distant metastases in human colorectal cancer. Science 2017;357(6346):55–60 doi 10.1126/science.aai8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Venet D, Fimereli D, Rothe F, Boeckx B, Maetens M, Majjaj S, et al. Phylogenetic reconstruction of breast cancer reveals two routes of metastatic dissemination associated with distinct clinical outcome. EBioMedicine 2020;56:102793 doi 10.1016/j.ebiom.2020.102793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Giuliano AE, Hunt KK, Ballman KV, Beitsch PD, Whitworth PW, Blumencranz PW, et al. Axillary dissection vs no axillary dissection in women with invasive breast cancer and sentinel node metastasis: a randomized clinical trial. JAMA 2011;305(6):569–75 doi 10.1001/jama.2011.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Faries MB, Thompson JF, Cochran AJ, Andtbacka RH, Mozzillo N, Zager JS, et al. Completion Dissection or Observation for Sentinel-Node Metastasis in Melanoma. N Engl J Med 2017;376(23):2211–22 doi 10.1056/NEJMoa1613210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harter P, Sehouli J, Lorusso D, Reuss A, Vergote I, Marth C, et al. A Randomized Trial of Lymphadenectomy in Patients with Advanced Ovarian Neoplasms. N Engl J Med 2019;380(9):822–32 doi 10.1056/NEJMoa1808424. [DOI] [PubMed] [Google Scholar]
- 54.Pereira ER, Kedrin D, Seano G, Gautier O, Meijer EFJ, Jones D, et al. Lymph node metastases can invade local blood vessels, exit the node, and colonize distant organs in mice. Science 2018;359(6382):1403–7 doi 10.1126/science.aal3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brown M, Assen FP, Leithner A, Abe J, Schachner H, Asfour G, et al. Lymph node blood vessels provide exit routes for metastatic tumor cell dissemination in mice. Science 2018;359(6382):1408–11 doi 10.1126/science.aal3662. [DOI] [PubMed] [Google Scholar]
- 56.Jones D, Pereira ER, Padera TP. Growth and Immune Evasion of Lymph Node Metastasis. Front Oncol 2018;8:36 doi 10.3389/fonc.2018.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lund AW, Wagner M, Fankhauser M, Steinskog ES, Broggi MA, Spranger S, et al. Lymphatic vessels regulate immune microenvironments in human and murine melanoma. J Clin Invest 2016;126(9):3389–402 doi 10.1172/JCI79434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ubellacker JM, Tasdogan A, Ramesh V, Shen B, Mitchell EC, Martin-Sandoval MS, et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 2020;585(7823):113–8 doi 10.1038/s41586-020-2623-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lengyel E Ovarian cancer development and metastasis. Am J Pathol 2010;177(3):1053–64 doi 10.2353/ajpath.2010.100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lugassy C, Kleinman HK, Vermeulen PB, Barnhill RL. Angiotropism, pericytic mimicry and extravascular migratory metastasis: an embryogenesis-derived program of tumor spread. Angiogenesis 2020;23(1):27–41 doi 10.1007/s10456-019-09695-9. [DOI] [PubMed] [Google Scholar]
- 61.Liebig C, Ayala G, Wilks JA, Berger DH, Albo D. Perineural invasion in cancer: a review of the literature. Cancer 2009;115(15):3379–91 doi 10.1002/cncr.24396. [DOI] [PubMed] [Google Scholar]
- 62.Kadota K, Nitadori JI, Sima CS, Ujiie H, Rizk NP, Jones DR, et al. Tumor Spread through Air Spaces is an Important Pattern of Invasion and Impacts the Frequency and Location of Recurrences after Limited Resection for Small Stage I Lung Adenocarcinomas. J Thorac Oncol 2015;10(5):806–14 doi 10.1097/JTO.0000000000000486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yagi Y, Aly RG, Tabata K, Barlas A, Rekhtman N, Eguchi T, et al. Three-Dimensional Histologic, Immunohistochemical, and Multiplex Immunofluorescence Analyses of Dynamic Vessel Co-Option of Spread Through Air Spaces in Lung Adenocarcinoma. J Thorac Oncol 2020;15(4):589–600 doi 10.1016/j.jtho.2019.12.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pan H, Gray R, Braybrooke J, Davies C, Taylor C, McGale P, et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N Engl J Med 2017;377(19):1836–46 doi 10.1056/NEJMoa1701830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pan H, Gray R, Braybrooke J, Davies C, Taylor C, McGale P, et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. New England. Journal of Medicine 2017;377(19):1836–46 doi 10.1056/NEJMoa1701830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, et al. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 2016;165(1):45–60 doi 10.1016/j.cell.2016.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hadfield G The dormant cancer cell. Br Med J 1954;2(4888):607–10 doi 10.1136/bmj.2.4888.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Phan TG, Croucher PI. The dormant cancer cell life cycle. Nat Rev Cancer 2020;20(7):398–411 doi 10.1038/s41568-020-0263-0. [DOI] [PubMed] [Google Scholar]
- 69.Shen S, Vagner S, Robert C. Persistent Cancer Cells: The Deadly Survivors. Cell 2020;183(4):860–74 doi 10.1016/j.cell.2020.10.027. [DOI] [PubMed] [Google Scholar]
- 70.Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015;527(7577):186–91 doi 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Le Gal K, Ibrahim MX, Wiel C, Sayin VI, Akula MK, Karlsson C, et al. Antioxidants can increase melanoma metastasis in mice. Science Translational Medicine 2015;7(308):308re8–re8 doi 10.1126/scitranslmed.aad3740. [DOI] [PubMed] [Google Scholar]
- 72.Chi Y, Remsik J, Kiseliovas V, Derderian C, Sener U, Alghader M, et al. Cancer cells deploy lipocalin-2 to collect limiting iron in leptomeningeal metastasis. Science 2020;369(6501):276–82 doi 10.1126/science.aaz2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mohme M, Riethdorf S, Pantel K. Circulating and disseminated tumour cells - mechanisms of immune surveillance and escape. Nat Rev Clin Oncol 2017;14(3):155–67 doi 10.1038/nrclinonc.2016.144. [DOI] [PubMed] [Google Scholar]
- 74.Garner H, de Visser KE. Immune crosstalk in cancer progression and metastatic spread: a complex conversation. Nat Rev Immunol 2020;20(8):483–97 doi 10.1038/s41577-019-0271-z. [DOI] [PubMed] [Google Scholar]
- 75.Eyles J, Puaux AL, Wang X, Toh B, Prakash C, Hong M, et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest 2010;120(6):2030–9 doi 10.1172/JCI42002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bidwell BN, Slaney CY, Withana NP, Forster S, Cao Y, Loi S, et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat Med 2012;18(8):1224–31 doi 10.1038/nm.2830. [DOI] [PubMed] [Google Scholar]
- 77.Van den Eynde M, Mlecnik B, Bindea G, Fredriksen T, Church SE, Lafontaine L, et al. The Link between the Multiverse of Immune Microenvironments in Metastases and the Survival of Colorectal Cancer Patients. Cancer Cell 2018;34(6):1012–26 e3 doi 10.1016/j.ccell.2018.11.003. [DOI] [PubMed] [Google Scholar]
- 78.Angelova M, Mlecnik B, Vasaturo A, Bindea G, Fredriksen T, Lafontaine L, et al. Evolution of Metastases in Space and Time under Immune Selection. Cell 2018;175(3):751–65 e16 doi 10.1016/j.cell.2018.09.018. [DOI] [PubMed] [Google Scholar]
- 79.MacKie RM, Reid R, Junor B. Fatal melanoma transferred in a donated kidney 16 years after melanoma surgery. N Engl J Med 2003;348(6):567–8 doi 10.1056/nejm200302063480620. [DOI] [PubMed] [Google Scholar]
- 80.Xiao D, Craig JC, Chapman JR, Dominguez-Gil B, Tong A, Wong G. Donor cancer transmission in kidney transplantation: a systematic review. Am J Transplant 2013;13(10):2645–52 doi 10.1111/ajt.12430. [DOI] [PubMed] [Google Scholar]
- 81.Eggermont AMM, Blank CU, Mandala M, Long GV, Atkinson V, Dalle S, et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N Engl J Med 2018;378(19):1789–801 doi 10.1056/NEJMoa1802357. [DOI] [PubMed] [Google Scholar]
- 82.Weber J, Mandala M, Del Vecchio M, Gogas HJ, Arance AM, Cowey CL, et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N Engl J Med 2017;377(19):1824–35 doi 10.1056/NEJMoa1709030. [DOI] [PubMed] [Google Scholar]
- 83.Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, et al. Prolonged Survival in Stage III Melanoma with Ipilimumab Adjuvant Therapy. N Engl J Med 2016;375(19):1845–55 doi 10.1056/NEJMoa1611299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zimmer L, Livingstone E, Hassel JC, Fluck M, Eigentler T, Loquai C, et al. Adjuvant nivolumab plus ipilimumab or nivolumab monotherapy versus placebo in patients with resected stage IV melanoma with no evidence of disease (IMMUNED): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2020;395(10236):1558–68 doi 10.1016/S0140-6736(20)30417-7. [DOI] [PubMed] [Google Scholar]
- 85.Pommier A, Anaparthy N, Memos N, Kelley ZL, Gouronnec A, Yan R, et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science 2018;360(6394) doi 10.1126/science.aao4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pantel K, Schlimok G, Kutter D, Schaller G, Genz T, Wiebecke B, et al. Frequent down-regulation of major histocompatibility class I antigen expression on individual micrometastatic carcinoma cells. Cancer Res 1991;51(17):4712–5. [PubMed] [Google Scholar]
- 87.Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007;450(7171):903–7 doi 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- 88.Eyob H, Ekiz HA, Derose YS, Waltz SE, Williams MA, Welm AL. Inhibition of ron kinase blocks conversion of micrometastases to overt metastases by boosting antitumor immunity. Cancer Discov 2013;3(7):751–60 doi 10.1158/2159-8290.CD-12-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.David CJ, Massagué J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat Rev Mol Cell Biol 2018;19(7):419–35 doi 10.1038/s41580-018-0007-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Batlle E, Massagué J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019;50(4):924–40 doi 10.1016/j.immuni.2019.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Naumov GN, Akslen LA, Folkman J. Role of angiogenesis in human tumor dormancy: animal models of the angiogenic switch. Cell Cycle 2006;5(16):1779–87 doi 10.4161/cc.5.16.3018. [DOI] [PubMed] [Google Scholar]
- 92.Ye W The Complexity of Translating Anti-angiogenesis Therapy from Basic Science to the Clinic. Dev Cell 2016;37(2):114–25 doi 10.1016/j.devcel.2016.03.015. [DOI] [PubMed] [Google Scholar]
- 93.Gao H, Chakraborty G, Lee-Lim AP, Mo Q, Decker M, Vonica A, et al. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 2012;150(4):764–79 doi 10.1016/j.cell.2012.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vera-Ramirez L, Vodnala SK, Nini R, Hunter KW, Green JE. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat Commun 2018;9(1):1944 doi 10.1038/s41467-018-04070-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.La Belle Flynn A, Calhoun BC, Sharma A, Chang JC, Almasan A, Schiemann WP. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat Commun 2019;10(1):3668 doi 10.1038/s41467-019-11640-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shibue T, Brooks MW, Weinberg RA. An integrin-linked machinery of cytoskeletal regulation that enables experimental tumor initiation and metastatic colonization. Cancer Cell 2013;24(4):481–98 doi 10.1016/j.ccr.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.De Cock JM, Shibue T, Dongre A, Keckesova Z, Reinhardt F, Weinberg RA. Inflammation Triggers Zeb1-Dependent Escape from Tumor Latency. Cancer Res 2016;76(23):6778–84 doi 10.1158/0008-5472.CAN-16-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018;361(6409) doi 10.1126/science.aao4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science 2004;303(5663):1532–5 doi 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 100.Branzk N, Papayannopoulos V. Molecular mechanisms regulating NETosis in infection and disease. Semin Immunopathol 2013;35(4):513–30 doi 10.1007/s00281-013-0384-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med 2016;8(361):361ra138 doi 10.1126/scitranslmed.aag1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Obenauf AC, Massagué J. Surviving at a Distance: Organ-Specific Metastasis. Trends Cancer 2015;1(1):76–91 doi 10.1016/j.trecan.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gao Y, Bado I, Wang H, Zhang W, Rosen JM, Zhang XH. Metastasis Organotropism: Redefining the Congenial Soil. Dev Cell 2019;49(3):375–91 doi 10.1016/j.devcel.2019.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Esposito M, Guise T, Kang Y. The Biology of Bone Metastasis. Cold Spring Harb Perspect Med 2018;8(6) doi 10.1101/cshperspect.a031252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Quail DF, Joyce JA. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017;31(3):326–41 doi 10.1016/j.ccell.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Altorki NK, Markowitz GJ, Gao D, Port JL, Saxena A, Stiles B, et al. The lung microenvironment: an important regulator of tumour growth and metastasis. Nat Rev Cancer 2019;19(1):9–31 doi 10.1038/s41568-018-0081-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Brodt P Role of the Microenvironment in Liver Metastasis: From Pre- to Prometastatic Niches. Clin Cancer Res 2016;22(24):5971–82 doi 10.1158/1078-0432.CCR-16-0460. [DOI] [PubMed] [Google Scholar]
- 108.Ruoslahti E Vascular zip codes in angiogenesis and metastasis. Biochem Soc Trans 2004;32(Pt3):397–402 doi 10.1042/BST0320397. [DOI] [PubMed] [Google Scholar]
- 109.Minn AJ, Kang Y, Serganova I, Gupta GP, Giri DD, Doubrovin M, et al. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest 2005;115(1):44–55 doi 10.1172/JCI22320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2002;2(8):563–72 doi 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 111.Iacobuzio-Donahue CA, Michael C, Baez P, Kappagantula R, Hooper JE, Hollman TJ. Cancer biology as revealed by the research autopsy. Nat Rev Cancer 2019;19(12):686–97 doi 10.1038/s41568-019-0199-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sevenich L, Bowman RL, Mason SD, Quail DF, Rapaport F, Elie BT, et al. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat Cell Biol 2014;16(9):876–88 doi 10.1038/ncb3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Urosevic J, Garcia-Albeniz X, Planet E, Real S, Cespedes MV, Guiu M, et al. Colon cancer cells colonize the lung from established liver metastases through p38 MAPK signalling and PTHLH. Nat Cell Biol 2014;16(7):685–94 doi 10.1038/ncb2977. [DOI] [PubMed] [Google Scholar]
- 114.Romero I, Garrido F, Garcia-Lora AM. Metastases in immune-mediated dormancy: a new opportunity for targeting cancer. Cancer Res 2014;74(23):6750–7 doi 10.1158/0008-5472.CAN-14-2406. [DOI] [PubMed] [Google Scholar]
- 115.Raulet DH, Marcus A, Coscoy L. Dysregulated cellular functions and cell stress pathways provide critical cues for activating and targeting natural killer cells to transformed and infected cells. Immunol Rev 2017;280(1):93–101 doi 10.1111/imr.12600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kwon J, Bakhoum SF. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov 2020;10(1):26–39 doi 10.1158/2159-8290.CD-19-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nabet BY, Qiu Y, Shabason JE, Wu TJ, Yoon T, Kim BC, et al. Exosome RNA Unshielding Couples Stromal Activation to Pattern Recognition Receptor Signaling in Cancer. Cell 2017;170(2):352–66 e13 doi 10.1016/j.cell.2017.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Davis RT, Blake K, Ma D, Gabra MBI, Hernandez GA, Phung AT, et al. Transcriptional diversity and bioenergetic shift in human breast cancer metastasis revealed by single-cell RNA sequencing. Nat Cell Biol 2020;22(3):310–20 doi 10.1038/s41556-020-0477-0. [DOI] [PubMed] [Google Scholar]
- 119.Basnet H, Tian L, Ganesh K, Huang YH, Macalinao DG, Brogi E, et al. Flura-seq identifies organ-specific metabolic adaptations during early metastatic colonization. Elife 2019;8 doi 10.7554/eLife.43627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dupuy F, Tabaries S, Andrzejewski S, Dong Z, Blagih J, Annis MG, et al. PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab 2015;22(4):577–89 doi 10.1016/j.cmet.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 121.Nguyen A, Loo JM, Mital R, Weinberg EM, Man FY, Zeng Z, et al. PKLR promotes colorectal cancer liver colonization through induction of glutathione synthesis. J Clin Invest 2016;126(2):681–94 doi 10.1172/JCI83587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Knott SRV, Wagenblast E, Khan S, Kim SY, Soto M, Wagner M, et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018;554(7692):378–81 doi 10.1038/nature25465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chen J, Lee HJ, Wu X, Huo L, Kim SJ, Xu L, et al. Gain of glucose-independent growth upon metastasis of breast cancer cells to the brain. Cancer Res 2015;75(3):554–65 doi 10.1158/0008-5472.CAN-14-2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yamaguchi N, Weinberg EM, Nguyen A, Liberti MV, Goodarzi H, Janjigian YY, et al. PCK1 and DHODH drive colorectal cancer liver metastatic colonization and hypoxic growth by promoting nucleotide synthesis. Elife 2019;8 doi 10.7554/eLife.52135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Loo JM, Scherl A, Nguyen A, Man FY, Weinberg E, Zeng Z, et al. Extracellular metabolic energetics can promote cancer progression. Cell 2015;160(3):393–406 doi 10.1016/j.cell.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 2014;159(7):1603–14 doi 10.1016/j.cell.2014.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lehuede C, Dupuy F, Rabinovitch R, Jones RG, Siegel PM. Metabolic Plasticity as a Determinant of Tumor Growth and Metastasis. Cancer Res 2016;76(18):5201–8 doi 10.1158/0008-5472.CAN-16-0266. [DOI] [PubMed] [Google Scholar]
- 128.Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017;541(7635):41–5 doi 10.1038/nature20791. [DOI] [PubMed] [Google Scholar]
- 129.Jin X, Demere Z, Nair K, Ali A, Ferraro GB, Natoli T, et al. A metastasis map of human cancer cell lines. Nature 2020;588(7837):331–6 doi 10.1038/s41586-020-2969-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wculek SK, Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 2015;528(7582):413–7 doi 10.1038/nature16140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG, et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med 2011;17(7):867–74 doi 10.1038/nm.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest 2011;121(4):1298–312 doi 10.1172/JCI43414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang H, Tian L, Liu J, Goldstein A, Bado I, Zhang W, et al. The Osteogenic Niche Is a Calcium Reservoir of Bone Micrometastases and Confers Unexpected Therapeutic Vulnerability. Cancer Cell 2018;34(5):823–39 e7 doi 10.1016/j.ccell.2018.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Northey JJ, Przybyla L, Weaver VM. Tissue Force Programs Cell Fate and Tumor Aggression. Cancer Discov 2017;7(11):1224–37 doi 10.1158/2159-8290.CD-16-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 2002;3(5):349–63 doi 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 136.Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nature Medicine 2016;22(5):497–505 doi 10.1038/nm.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, et al. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integrative Biology 2015;7(10):1120–34 doi 10.1039/c5ib00040h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Maller O, Drain AP, Barrett AS, Borgquist S, Ruffell B, Zakharevich I, et al. Tumour-associated macrophages drive stromal cell-dependent collagen crosslinking and stiffening to promote breast cancer aggression. Nat Mater 2020. doi 10.1038/s41563-020-00849-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, et al. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest 1999;103(2):197–206 doi 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zheng H, Bae Y, Kasimir-Bauer S, Tang R, Chen J, Ren G, et al. Therapeutic Antibody Targeting Tumor- and Osteoblastic Niche-Derived Jagged1 Sensitizes Bone Metastasis to Chemotherapy. Cancer Cell 2017;32(6):731–47 e6 doi 10.1016/j.ccell.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhang J, Dai J, Qi Y, Lin DL, Smith P, Strayhorn C, et al. Osteoprotegerin inhibits prostate cancer-induced osteoclastogenesis and prevents prostate tumor growth in the bone. J Clin Invest 2001;107(10):1235–44 doi 10.1172/JCI11685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016;533(7604):493–8 doi 10.1038/nature18268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Priego N, Zhu L, Monteiro C, Mulders M, Wasilewski D, Bindeman W, et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat Med 2018;24(7):1024–35 doi 10.1038/s41591-018-0044-4. [DOI] [PubMed] [Google Scholar]
- 144.Guldner IH, Wang Q, Yang L, Golomb SM, Zhao Z, Lopez JA, et al. CNS-Native Myeloid Cells Drive Immune Suppression in the Brain Metastatic Niche through Cxcl10. Cell 2020;183(5):1234–48 e25 doi 10.1016/j.cell.2020.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zeng Q, Michael IP, Zhang P, Saghafinia S, Knott G, Jiao W, et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 2019;573(7775):526–31 doi 10.1038/s41586-019-1576-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen Q, Zhang XH, Massagué J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell 2011;20(4):538–49 doi 10.1016/j.ccr.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012;150(1):165–78 doi 10.1016/j.cell.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Teijeira A, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, et al. CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps that Interfere with Immune Cytotoxicity. Immunity 2020;52(5):856–71 e8 doi 10.1016/j.immuni.2020.03.001. [DOI] [PubMed] [Google Scholar]
- 149.Malanchi I, Santamaria-Martinez A, Susanto E, Peng H, Lehr HA, Delaloye JF, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2011;481(7379):85–9 doi 10.1038/nature10694. [DOI] [PubMed] [Google Scholar]
- 150.Ombrato L, Nolan E, Kurelac I, Mavousian A, Bridgeman VL, Heinze I, et al. Metastatic-niche labelling reveals parenchymal cells with stem features. Nature 2019;572(7771):603–8 doi 10.1038/s41586-019-1487-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer 2017;17(5):302–17 doi 10.1038/nrc.2017.6. [DOI] [PubMed] [Google Scholar]
- 152.Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, et al. Identification of the tumour transition states occurring during EMT. Nature 2018;556(7702):463–8 doi 10.1038/s41586-018-0040-3. [DOI] [PubMed] [Google Scholar]
- 153.Su J, Morgani SM, David CJ, Wang Q, Er EE, Huang YH, et al. TGF-beta orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature 2020;577(7791):566–71 doi 10.1038/s41586-019-1897-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol 2017;14(10):611–29 doi 10.1038/nrclinonc.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012;22(6):725–36 doi 10.1016/j.ccr.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 2012;22(6):709–24 doi 10.1016/j.ccr.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 157.Pastushenko I, Blanpain C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol 2019;29(3):212–26 doi 10.1016/j.tcb.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 158.Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, et al. Identification of the tumour transition states occurring during EMT. Nature 2018;556(7702):463–8 doi 10.1038/s41586-018-0040-3. [DOI] [PubMed] [Google Scholar]
- 159.Pastushenko I, Mauri F, Song Y, de Cock F, Meeusen B, Swedlund B, et al. Fat1 deletion promotes hybrid EMT state, tumour stemness and metastasis. Nature 2021;589(7842):448–55 doi 10.1038/s41586-020-03046-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144(5):646–74 doi 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 161.Gupta PB, Pastushenko I, Skibinski A, Blanpain C, Kuperwasser C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019;24(1):65–78 doi 10.1016/j.stem.2018.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yuan S, Norgard RJ, Stanger BZ. Cellular Plasticity in Cancer. Cancer Discov 2019;9(7):837–51 doi 10.1158/2159-8290.CD-19-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Quintanal-Villalonga A, Chan JM, Yu HA, Pe’er D, Sawyers CL, Sen T, et al. Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat Rev Clin Oncol 2020;17(6):360–71 doi 10.1038/s41571-020-0340-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Priestley P, Baber J, Lolkema MP, Steeghs N, de Bruijn E, Shale C, et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019;575(7781):210–6 doi 10.1038/s41586-019-1689-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Biswas D, Birkbak NJ, Rosenthal R, Hiley CT, Lim EL, Papp K, et al. A clonal expression biomarker associates with lung cancer mortality. Nat Med 2019;25(10):1540–8 doi 10.1038/s41591-019-0595-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hunter KW, Amin R, Deasy S, Ha NH, Wakefield L. Genetic insights into the morass of metastatic heterogeneity. Nat Rev Cancer 2018;18(4):211–23 doi 10.1038/nrc.2017.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Reiter JG, Baretti M, Gerold JM, Makohon-Moore AP, Daud A, Iacobuzio-Donahue CA, et al. An analysis of genetic heterogeneity in untreated cancers. Nat Rev Cancer 2019;19(11):639–50 doi 10.1038/s41568-019-0185-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Turajlic S, Swanton C. Metastasis as an evolutionary process. Science 2016;352(6282):169–75 doi 10.1126/science.aaf2784. [DOI] [PubMed] [Google Scholar]
- 169.Makohon-Moore AP, Zhang M, Reiter JG, Bozic I, Allen B, Kundu D, et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat Genet 2017;49(3):358–66 doi 10.1038/ng.3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Reiter JG, Makohon-Moore AP, Gerold JM, Heyde A, Attiyeh MA, Kohutek ZA, et al. Minimal functional driver gene heterogeneity among untreated metastases. Science 2018;361(6406):1033–7 doi 10.1126/science.aat7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Notta F, Chan-Seng-Yue M, Lemire M, Li Y, Wilson GW, Connor AA, et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016;538(7625):378–82 doi 10.1038/nature19823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yates LR, Knappskog S, Wedge D, Farmery JHR, Gonzalez S, Martincorena I, et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 2017;32(2):169–84 e7 doi 10.1016/j.ccell.2017.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hu Z, Li Z, Ma Z, Curtis C. Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases. Nature Genetics 2020;52(7):701–8 doi 10.1038/s41588-020-0628-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010;467(7319):1114–7 doi 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018;34(3):427–38 e6 doi 10.1016/j.ccell.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018;173(2):321–37 e10 doi 10.1016/j.cell.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Watkins TBK, Lim EL, Petkovic M, Elizalde S, Birkbak NJ, Wilson GA, et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 2020;587(7832):126–32 doi 10.1038/s41586-020-2698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N Engl J Med 2017;376(22):2109–21 doi 10.1056/NEJMoa1616288. [DOI] [PubMed] [Google Scholar]
- 179.Turajlic S, Sottoriva A, Graham T, Swanton C. Resolving genetic heterogeneity in cancer. Nat Rev Genet 2019;20(7):404–16 doi 10.1038/s41576-019-0114-6. [DOI] [PubMed] [Google Scholar]
- 180.Hu Z, Curtis C. Looking backward in time to define the chronology of metastasis. Nat Commun 2020;11(1):3213 doi 10.1038/s41467-020-16995-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Klein CA. Parallel progression of primary tumours and metastases. Nat Rev Cancer 2009;9(4):302–12 doi 10.1038/nrc2627. [DOI] [PubMed] [Google Scholar]
- 182.Hosseini H, Obradovic MMS, Hoffmann M, Harper KL, Sosa MS, Werner-Klein M, et al. Early dissemination seeds metastasis in breast cancer. Nature 2016;540(7634):552–8 doi 10.1038/nature20785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, Nobre R, et al. Mechanism of early dissemination and metastasis in Her2(+) mammary cancer. Nature 2016;540(7634):588–92 doi 10.1038/nature20609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Turajlic S, Xu H, Litchfield K, Rowan A, Horswell S, Chambers T, et al. Deterministic Evolutionary Trajectories Influence Primary Tumor Growth: TRACERx Renal. Cell 2018;173(3):595–610 e11 doi 10.1016/j.cell.2018.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sottoriva A, Kang H, Ma Z, Graham TA, Salomon MP, Zhao J, et al. A Big Bang model of human colorectal tumor growth. Nat Genet 2015;47(3):209–16 doi 10.1038/ng.3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Turajlic S, Xu H, Litchfield K, Rowan A, Chambers T, Lopez JI, et al. Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell 2018;173(3):581–94 e12 doi 10.1016/j.cell.2018.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Noorani A, Li X, Goddard M, Crawte J, Alexandrov LB, Secrier M, et al. Genomic evidence supports a clonal diaspora model for metastases of esophageal adenocarcinoma. Nat Genet 2020;52(1):74–83 doi 10.1038/s41588-019-0551-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Sanborn JZ, Chung J, Purdom E, Wang NJ, Kakavand H, Wilmott JS, et al. Phylogenetic analyses of melanoma reveal complex patterns of metastatic dissemination. Proc Natl Acad Sci U S A 2015;112(35):10995–1000 doi 10.1073/pnas.1508074112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Maddipati R, Stanger BZ. Pancreatic Cancer Metastases Harbor Evidence of Polyclonality. Cancer Discov 2015;5(10):1086–97 doi 10.1158/2159-8290.CD-15-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kim MY, Oskarsson T, Acharyya S, Nguyen DX, Zhang XH, Norton L, et al. Tumor self-seeding by circulating cancer cells. Cell 2009;139(7):1315–26 doi 10.1016/j.cell.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Comen E, Norton L, Massagué J. Clinical implications of cancer self-seeding. Nat Rev Clin Oncol 2011;8(6):369–77 doi 10.1038/nrclinonc.2011.64. [DOI] [PubMed] [Google Scholar]
- 192.Weichselbaum RR, Hellman S. Oligometastases revisited. Nat Rev Clin Oncol 2011;8(6):378–82 doi 10.1038/nrclinonc.2011.44. [DOI] [PubMed] [Google Scholar]
- 193.Demicheli R, Abbattista A, Miceli R, Valagussa P, Bonadonna G. Time distribution of the recurrence risk for breast cancer patients undergoing mastectomy: further support about the concept of tumor dormancy. Breast Cancer Res Treat 1996;41(2):177–85 doi 10.1007/BF01807163. [DOI] [PubMed] [Google Scholar]
- 194.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994;367(6464):645–8 doi 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 195.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997;3(7):730–7 doi 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 196.Shimokawa M, Ohta Y, Nishikori S, Matano M, Takano A, Fujii M, et al. Visualization and targeting of LGR5(+) human colon cancer stem cells. Nature 2017;545(7653):187–92 doi 10.1038/nature22081. [DOI] [PubMed] [Google Scholar]
- 197.Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009;457(7229):608–11 doi 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 198.Visvader JE. Cells of origin in cancer. Nature 2011;469(7330):314–22 doi 10.1038/nature09781. [DOI] [PubMed] [Google Scholar]
- 199.Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science 2017;357(6348) doi 10.1126/science.aal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Denny SK, Yang D, Chuang CH, Brady JJ, Lim JS, Gruner BM, et al. Nfib Promotes Metastasis through a Widespread Increase in Chromatin Accessibility. Cell 2016;166(2):328–42 doi 10.1016/j.cell.2016.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Marjanovic ND, Hofree M, Chan JE, Canner D, Wu K, Trakala M, et al. Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell 2020;38(2):229–46 e13 doi 10.1016/j.ccell.2020.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.LaFave LM, Kartha VK, Ma S, Meli K, Del Priore I, Lareau C, et al. Epigenomic State Transitions Characterize Tumor Progression in Mouse Lung Adenocarcinoma. Cancer Cell 2020;38(2):212–28 e13 doi 10.1016/j.ccell.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Roe JS, Hwang CI, Somerville TDD, Milazzo JP, Lee EJ, Da Silva B, et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 2017;170(5):875–88 e20 doi 10.1016/j.cell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Vanharanta S, Shu W, Brenet F, Hakimi AA, Heguy A, Viale A, et al. Epigenetic expansion of VHL-HIF signal output drives multiorgan metastasis in renal cancer. Nat Med 2013;19(1):50–6 doi 10.1038/nm.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Gomes AP, Ilter D, Low V, Rosenzweig A, Shen ZJ, Schild T, et al. Dynamic Incorporation of Histone H3 Variants into Chromatin Is Essential for Acquisition of Aggressive Traits and Metastatic Colonization. Cancer Cell 2019;36(4):402–17 e13 doi 10.1016/j.ccell.2019.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Yuan S, Natesan R, Sanchez-Rivera FJ, Li J, Bhanu NV, Yamazoe T, et al. Global Regulation of the Histone Mark H3K36me2 Underlies Epithelial Plasticity and Metastatic Progression. Cancer Discov 2020;10(6):854–71 doi 10.1158/2159-8290.CD-19-1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Dekoninck S, Blanpain C. Stem cell dynamics, migration and plasticity during wound healing. Nature Cell Biology 2019;21(1):18–24 doi 10.1038/s41556-018-0237-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.de Sousa EMF, de Sauvage FJ. Cellular Plasticity in Intestinal Homeostasis and Disease. Cell Stem Cell 2019;24(1):54–64 doi 10.1016/j.stem.2018.11.019. [DOI] [PubMed] [Google Scholar]
- 209.Rompolas P, Deschene ER, Zito G, Gonzalez DG, Saotome I, Haberman AM, et al. Live imaging of stem cell and progeny behaviour in physiological hair-follicle regeneration. Nature 2012;487(7408):496–9 doi 10.1038/nature11218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Tata PR, Mou H, Pardo-Saganta A, Zhao R, Prabhu M, Law BM, et al. Dedifferentiation of committed epithelial cells into stem cells in vivo. Nature 2013;503(7475):218–23 doi 10.1038/nature12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Tian H, Biehs B, Warming S, Leong KG, Rangell L, Klein OD, et al. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature 2011;478(7368):255–9 doi 10.1038/nature10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007;449(7165):1003–7 doi 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 213.Ayyaz A, Kumar S, Sangiorgi B, Ghoshal B, Gosio J, Ouladan S, et al. Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature 2019;569(7754):121–5 doi 10.1038/s41586-019-1154-y. [DOI] [PubMed] [Google Scholar]
- 214.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009;459(7244):262–5 doi 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 215.Serra D, Mayr U, Boni A, Lukonin I, Rempfler M, Challet Meylan L, et al. Self-organization and symmetry breaking in intestinal organoid development. Nature 2019;569(7754):66–72 doi 10.1038/s41586-019-1146-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Fumagalli A, Oost KC, Kester L, Morgner J, Bornes L, Bruens L, et al. Plasticity of Lgr5-Negative Cancer Cells Drives Metastasis in Colorectal Cancer. Cell Stem Cell 2020;26(4):569–78.e7 doi 10.1016/j.stem.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Han T, Goswami S, Hu Y, Tang F, Zafra MP, Murphy C, et al. Lineage Reversion Drives WNT Independence in Intestinal Cancer. Cancer Discov 2020;10(10):1590–609 doi 10.1158/2159-8290.Cd-19-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Morral C, Stanisavljevic J, Hernando-Momblona X, Mereu E, Alvarez-Varela A, Cortina C, et al. Zonation of Ribosomal DNA Transcription Defines a Stem Cell Hierarchy in Colorectal Cancer. Cell Stem Cell 2020;26(6):845–61 e12 doi 10.1016/j.stem.2020.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Altevogt P, Doberstein K, Fogel M. L1CAM in human cancer. Int J Cancer 2016;138(7):1565–76 doi 10.1002/ijc.29658. [DOI] [PubMed] [Google Scholar]
- 220.Miyoshi H, Ajima R, Luo CT, Yamaguchi TP, Stappenbeck TS. Wnt5a potentiates TGF-beta signaling to promote colonic crypt regeneration after tissue injury. Science 2012;338(6103):108–13 doi 10.1126/science.1223821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wang Y, Chiang IL, Ohara TE, Fujii S, Cheng J, Muegge BD, et al. Long-Term Culture Captures Injury-Repair Cycles of Colonic Stem Cells. Cell 2019;179(5):1144–59 e15 doi 10.1016/j.cell.2019.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Laughney AM, Hu J, Campbell NR, Bakhoum SF, Setty M, Lavallee VP, et al. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat Med 2020;26(2):259–69 doi 10.1038/s41591-019-0750-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR, et al. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 2008;133(1):66–77 doi 10.1016/j.cell.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zhang XH, Jin X, Malladi S, Zou Y, Wen YH, Brogi E, et al. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell 2013;154(5):1060–73 doi 10.1016/j.cell.2013.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Alečković M, McAllister SS, Polyak K. Metastasis as a systemic disease: molecular insights and clinical implications. Biochim Biophys Acta Rev Cancer 2019;1872(1):89–102 doi 10.1016/j.bbcan.2019.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005;438(7069):820–7 doi 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Hoshino A, Kim HS, Bojmar L, Gyan KE, Cioffi M, Hernandez J, et al. Extracellular Vesicle and Particle Biomarkers Define Multiple Human Cancers. Cell 2020;182(4):1044–61.e18 doi 10.1016/j.cell.2020.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Kaczanowska S, Kaplan RN. Mapping the switch that drives the pre-metastatic niche. Nature Cancer 2020;1(6):577–9 doi 10.1038/s43018-020-0076-9. [DOI] [PubMed] [Google Scholar]
- 229.Moreno-Smith M, Lutgendorf SK, Sood AK. Impact of stress on cancer metastasis. Future Oncol 2010;6(12):1863–81 doi 10.2217/fon.10.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Obradovic MMS, Hamelin B, Manevski N, Couto JP, Sethi A, Coissieux MM, et al. Glucocorticoids promote breast cancer metastasis. Nature 2019;567(7749):540–4 doi 10.1038/s41586-019-1019-4. [DOI] [PubMed] [Google Scholar]
- 231.Flint TR, Janowitz T, Connell CM, Roberts EW, Denton AE, Coll AP, et al. Tumor-Induced IL-6 Reprograms Host Metabolism to Suppress Anti-tumor Immunity. Cell Metab 2016;24(5):672–84 doi 10.1016/j.cmet.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Webster JI, Tonelli L, Sternberg EM. Neuroendocrine regulation of immunity. Annu Rev Immunol 2002;20:125–63 doi 10.1146/annurev.immunol.20.082401.104914. [DOI] [PubMed] [Google Scholar]
- 233.Perego M, Tyurin VA, Tyurina YY, Yellets J, Nacarelli T, Lin C, et al. Reactivation of dormant tumor cells by modified lipids derived from stress-activated neutrophils. Science Translational Medicine 2020;12(572):eabb5817 doi 10.1126/scitranslmed.abb5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Harper EI, Sheedy EF, Stack MS. With Great Age Comes Great Metastatic Ability: Ovarian Cancer and the Appeal of the Aging Peritoneal Microenvironment. Cancers (Basel) 2018;10(7) doi 10.3390/cancers10070230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Amant F, von Minckwitz G, Han SN, Bontenbal M, Ring AE, Giermek J, et al. Prognosis of women with primary breast cancer diagnosed during pregnancy: results from an international collaborative study. J Clin Oncol 2013;31(20):2532–9 doi 10.1200/jco.2012.45.6335. [DOI] [PubMed] [Google Scholar]
- 236.Pennock ND, Martinson HA, Guo Q, Betts CB, Jindal S, Tsujikawa T, et al. Ibuprofen supports macrophage differentiation, T cell recruitment, and tumor suppression in a model of postpartum breast cancer. Journal for ImmunoTherapy of Cancer 2018;6(1):98 doi 10.1186/s40425-018-0406-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Taranova AG, Maldonado D 3rd, Vachon CM, Jacobsen EA, Abdala-Valencia H, McGarry MP, et al. Allergic pulmonary inflammation promotes the recruitment of circulating tumor cells to the lung. Cancer Res 2008;68(20):8582–9 doi 10.1158/0008-5472.Can-08-1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Roy LD, Ghosh S, Pathangey LB, Tinder TL, Gruber HE, Mukherjee P. Collagen induced arthritis increases secondary metastasis in MMTV-PyV MT mouse model of mammary cancer. BMC Cancer 2011;11(1):365 doi 10.1186/1471-2407-11-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Krall JA, Reinhardt F, Mercury OA, Pattabiraman DR, Brooks MW, Dougan M, et al. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Science Translational Medicine 2018;10(436):eaan3464 doi 10.1126/scitranslmed.aan3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Demicheli R, Retsky MW, Hrushesky WJ, Baum M. Tumor dormancy and surgery-driven interruption of dormancy in breast cancer: learning from failures. Nat Clin Pract Oncol 2007;4(12):699–710 doi 10.1038/ncponc0999. [DOI] [PubMed] [Google Scholar]
- 241.Benish M, Bartal I, Goldfarb Y, Levi B, Avraham R, Raz A, et al. Perioperative use of beta-blockers and COX-2 inhibitors may improve immune competence and reduce the risk of tumor metastasis. Ann Surg Oncol 2008;15(7):2042–52 doi 10.1245/s10434-008-9890-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Biswas AK, Acharyya S. Understanding cachexia in the context of metastatic progression. Nat Rev Cancer 2020;20(5):274–84 doi 10.1038/s41568-020-0251-4. [DOI] [PubMed] [Google Scholar]
- 243.Faulkner S, Jobling P, March B, Jiang CC, Hondermarck H. Tumor Neurobiology and the War of Nerves in Cancer. Cancer Discov 2019;9(6):702–10 doi 10.1158/2159-8290.CD-18-1398. [DOI] [PubMed] [Google Scholar]
- 244.Kuol N, Stojanovska L, Apostolopoulos V, Nurgali K. Role of the nervous system in cancer metastasis. J Exp Clin Cancer Res 2018;37(1):5 doi 10.1186/s13046-018-0674-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Kumar A, Brockes JP. Nerve dependence in tissue, organ, and appendage regeneration. Trends Neurosci 2012;35(11):691–9 doi 10.1016/j.tins.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 246.Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, et al. Autonomic nerve development contributes to prostate cancer progression. Science 2013;341(6142):1236361 doi 10.1126/science.1236361. [DOI] [PubMed] [Google Scholar]
- 247.Schiller M, Ben-Shaanan TL, Rolls A. Neuronal regulation of immunity: why, how and where? Nat Rev Immunol 2021;21(1):20–36 doi 10.1038/s41577-020-0387-1. [DOI] [PubMed] [Google Scholar]
- 248.Erin N, Boyer PJ, Bonneau RH, Clawson GA, Welch DR. Capsaicin-mediated denervation of sensory neurons promotes mammary tumor metastasis to lung and heart. Anticancer Res 2004;24(2b):1003–9. [PubMed] [Google Scholar]
- 249.Cannioto RA, Hutson A, Dighe S, McCann W, McCann SE, Zirpoli GR, et al. Physical Activity Before, During, and After Chemotherapy for High-Risk Breast Cancer: Relationships With Survival. J Natl Cancer Inst 2021;113(1):54–63 doi 10.1093/jnci/djaa046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Koelwyn GJ, Quail DF, Zhang X, White RM, Jones LW. Exercise-dependent regulation of the tumour microenvironment. Nat Rev Cancer 2017;17(10):620–32 doi 10.1038/nrc.2017.78. [DOI] [PubMed] [Google Scholar]
- 251.Chambers AF. Influence of diet on metastasis and tumor dormancy. Clin Exp Metastasis 2009;26(1):61–6 doi 10.1007/s10585-008-9164-4. [DOI] [PubMed] [Google Scholar]
- 252.Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D, et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018;560(7719):499–503 doi 10.1038/s41586-018-0343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Klil-Drori AJ, Azoulay L, Pollak MN. Cancer, obesity, diabetes, and antidiabetic drugs: is the fog clearing? Nat Rev Clin Oncol 2017;14(2):85–99 doi 10.1038/nrclinonc.2016.120. [DOI] [PubMed] [Google Scholar]
- 254.Lien EC, Vander Heiden MG. A framework for examining how diet impacts tumour metabolism. Nat Rev Cancer 2019;19(11):651–61 doi 10.1038/s41568-019-0198-5. [DOI] [PubMed] [Google Scholar]
- 255.Christ A, Lauterbach M, Latz E. Western Diet and the Immune System: An Inflammatory Connection. Immunity 2019;51(5):794–811 doi 10.1016/j.immuni.2019.09.020. [DOI] [PubMed] [Google Scholar]
- 256.Ewertz M, Jensen MB, Gunnarsdóttir K, Højris I, Jakobsen EH, Nielsen D, et al. Effect of obesity on prognosis after early-stage breast cancer. J Clin Oncol 2011;29(1):25–31 doi 10.1200/jco.2010.29.7614. [DOI] [PubMed] [Google Scholar]
- 257.Rubio-Patiño C, Bossowski JP, De Donatis GM, Mondragón L, Villa E, Aira LE, et al. Low-Protein Diet Induces IRE1α-Dependent Anticancer Immunosurveillance. Cell Metabolism 2018;27(4):828–42.e7 doi 10.1016/j.cmet.2018.02.009. [DOI] [PubMed] [Google Scholar]
- 258.Schmid D, Leitzmann MF. Association between physical activity and mortality among breast cancer and colorectal cancer survivors: a systematic review and meta-analysis. Annals of Oncology 2014;25(7):1293–311 doi 10.1093/annonc/mdu012. [DOI] [PubMed] [Google Scholar]
- 259.Kanarek N, Petrova B, Sabatini DM. Dietary modifications for enhanced cancer therapy. Nature 2020;579(7800):507–17 doi 10.1038/s41586-020-2124-0. [DOI] [PubMed] [Google Scholar]
- 260.Helmink BA, Khan MAW, Hermann A, Gopalakrishnan V, Wargo JA. The microbiome, cancer, and cancer therapy. Nat Med 2019;25(3):377–88 doi 10.1038/s41591-019-0377-7. [DOI] [PubMed] [Google Scholar]
- 261.Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017;357(6356):1156–60 doi 10.1126/science.aah5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, Cai D, et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017;358(6369):1443–8 doi 10.1126/science.aal5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020;368(6494):973–80 doi 10.1126/science.aay9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Chen YE, Fischbach MA, Belkaid Y. Skin microbiota-host interactions. Nature 2018;553(7689):427–36 doi 10.1038/nature25177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Fischbach MA. Microbiome: Focus on Causation and Mechanism. Cell 2018;174(4):785–90 doi 10.1016/j.cell.2018.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Cotillard A, Kennedy SP, Kong LC, Prifti E, Pons N, Le Chatelier E, et al. Dietary intervention impact on gut microbial gene richness. Nature 2013;500(7464):585–8 doi 10.1038/nature12480. [DOI] [PubMed] [Google Scholar]
- 267.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014;505(7484):559–63 doi 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018;359(6371):91–7 doi 10.1126/science.aan3706. [DOI] [PubMed] [Google Scholar]
- 269.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018;359(6371):97–103 doi 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70(1):7–30 doi 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 271.Fane M, Weeraratna AT. Normal Aging and Its Role in Cancer Metastasis. Cold Spring Harb Perspect Med 2020;10(9) doi 10.1101/cshperspect.a037341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Fane M, Weeraratna AT. How the ageing microenvironment influences tumour progression. Nature Reviews Cancer 2020;20(2):89–106 doi 10.1038/s41568-019-0222-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010;5:99–118 doi 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Aging Campisi J., cellular senescence, and cancer. Annu Rev Physiol 2013;75:685–705 doi 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Kaur A, Webster MR, Marchbank K, Behera R, Ndoye A, Kugel CH 3rd, et al. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature 2016;532(7598):250–4 doi 10.1038/nature17392. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 276.Krtolica A, Parrinello S, Lockett S, Desprez P-Y, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proceedings of the National Academy of Sciences 2001;98(21):12072–7 doi 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Kaur A, Ecker BL, Douglass SM, Kugel CH 3rd, Webster MR, Almeida FV, et al. Remodeling of the Collagen Matrix in Aging Skin Promotes Melanoma Metastasis and Affects Immune Cell Motility. Cancer Discov 2019;9(1):64–81 doi 10.1158/2159-8290.Cd-18-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Ecker BL, Kaur A, Douglass SM, Webster MR, Almeida FV, Marino GE, et al. Age-Related Changes in HAPLN1 Increase Lymphatic Permeability and Affect Routes of Melanoma Metastasis. Cancer Discov 2019;9(1):82–95 doi 10.1158/2159-8290.Cd-18-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013;153(6):1194–217 doi 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Leonardi GC, Accardi G, Monastero R, Nicoletti F, Libra M. Ageing: from inflammation to cancer. Immun Ageing 2018;15:1 doi 10.1186/s12979-017-0112-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Kugel CH 3rd, Douglass SM, Webster MR, Kaur A, Liu Q, Yin X, et al. Age Correlates with Response to Anti-PD1, Reflecting Age-Related Differences in Intratumoral Effector and Regulatory T-Cell Populations. Clin Cancer Res 2018;24(21):5347–56 doi 10.1158/1078-0432.Ccr-18-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Ganesh K, Massagué J. Targeting metastatic cancer. Nature Medicine 2021;27(1):34–44 doi 10.1038/s41591-020-01195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Balak MN, Gong Y, Riely GJ, Somwar R, Li AR, Zakowski MF, et al. Novel D761Y and Common Secondary T790M Mutations in Epidermal Growth Factor Receptor–Mutant Lung Adenocarcinomas with Acquired Resistance to Kinase Inhibitors. Clinical Cancer Research 2006;12(21):6494–501 doi 10.1158/1078-0432.Ccr-06-1570. [DOI] [PubMed] [Google Scholar]
- 284.Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature 2019;575(7782):299–309 doi 10.1038/s41586-019-1730-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Al-Hajj M, Becker MW, Wicha M, Weissman I, Clarke MF. Therapeutic implications of cancer stem cells. Curr Opin Genet Dev 2004;14(1):43–7 doi 10.1016/j.gde.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 286.Naik S, Larsen SB, Gomez NC, Alaverdyan K, Sendoel A, Yuan S, et al. Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature 2017;550(7677):475–80 doi 10.1038/nature24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med 2001;7(9):1028–34 doi 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]
- 288.Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015;520(7547):368–72 doi 10.1038/nature14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008;6(12):2853–68 doi 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Faget DV, Ren Q, Stewart SA. Unmasking senescence: context-dependent effects of SASP in cancer. Nature Reviews Cancer 2019;19(8):439–53 doi 10.1038/s41568-019-0156-2. [DOI] [PubMed] [Google Scholar]
- 291.Bailey JM, Swanson BJ, Hamada T, Eggers JP, Singh PK, Caffery T, et al. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res 2008;14(19):5995–6004 doi 10.1158/1078-0432.Ccr-08-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science (New York, NY) 2009;324(5933):1457–61 doi 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Gajewski TF, Meng Y, Blank C, Brown I, Kacha A, Kline J, et al. Immune resistance orchestrated by the tumor microenvironment. Immunol Rev 2006;213:131–45 doi 10.1111/j.1600-065X.2006.00442.x. [DOI] [PubMed] [Google Scholar]
- 294.Gajewski TF, Woo SR, Zha Y, Spaapen R, Zheng Y, Corrales L, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol 2013;25(2):268–76 doi 10.1016/j.coi.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 295.Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018;554(7693):538–43 doi 10.1038/nature25492. [DOI] [PubMed] [Google Scholar]
- 296.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554(7693):544–8 doi 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Catenacci DVT, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. Journal of Clinical Oncology 2015;33(36):4284–92 doi 10.1200/jco.2015.62.8719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Ciardiello D, Elez E, Tabernero J, Seoane J. Clinical development of therapies targeting TGFβ: current knowledge and future perspectives. Ann Oncol 2020;31(10):1336–49 doi 10.1016/j.annonc.2020.07.009. [DOI] [PubMed] [Google Scholar]
- 299.Lee JJ, Perera RM, Wang H, Wu D-C, Liu XS, Han S, et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proceedings of the National Academy of Sciences 2014;111(30):E3091–E100 doi 10.1073/pnas.1411679111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Boumahdi S, de Sauvage FJ. The great escape: tumour cell plasticity in resistance to targeted therapy. Nat Rev Drug Discov 2020;19(1):39–56 doi 10.1038/s41573-019-0044-1. [DOI] [PubMed] [Google Scholar]
- 301.Paget S The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev 1989;8(2):98–101. [PubMed] [Google Scholar]