

Abstract
Maternal infection during pregnancy is a known risk factor for offspring mental health disorders. Animal models of maternal immune activation (MIA) have implicated specific cellular and molecular etiologies of psychiatric illness, but most rely on pathogen mimetics. Here, we developed a mouse model of live H3N2 influenza A virus (IAV) infection during pregnancy that induces a robust inflammatory response but is sublethal to both dams and offspring. We observed classic indicators of lung inflammation and severely diminished weight gain in IAV-infected dams. This was accompanied by immune cell infiltration in the placenta and partial breakdown of placental integrity. However, indications of fetal neuroinflammation were absent. Further hallmarks of mimetic-induced MIA, including enhanced circulating maternal IL-17A, were also absent. Respiratory IAV infection did result in an upregulation in intestinal expression of transcription factor RORγt, master regulator of a subset of T lymphocytes, TH17 cells, which are heavily implicated in MIA-induced etiologies. Nonetheless, subsequent augmentation in IL-17A production and concomitant overt intestinal injury was not evident. Our results suggest that mild or moderately pathogenic IAV infection during pregnancy does not inflame the developing fetal brain, and highlight the importance of live pathogen infection models for the study of MIA.
Keywords: maternal immune activation, influenza A virus, fetal neuroinflammation, gestational infection, influenza infection, maternal viral infection, fetal development, T helper 17 cells, maternal inflammation, infection during pregnancy
1.0. Introduction
Influenza A virus (IAV) infection during pregnancy poses a substantial risk for heightened morbidity and mortality in both mother and infant. Poor infant outcomes associated with maternal IAV infection, such as low birth weight, premature delivery, and stillbirth, occur in the absence of IAV vertical transmission, as the virus rarely breaches the maternal-fetal interface (Fatemi et al., 2012; Kanmaz et al., 2011; Rasmussen et al., 2012; Shi et al., 2005). Given a lack of direct fetal infection, prolonged health complications due to prenatal exposure to maternal IAV infection, including development of neuropsychiatric disorders (Brown et al., 2004; Mednick et al., 1988), have mostly been attributed to the maternal inflammatory response. While there is evidence in humans that elevated maternal cytokines during pregnancy contribute to poor offspring psychiatric outcomes (Zhang et al., 2018), much of the data on prenatal inflammation comes from animal models of maternal immune activation (MIA) (Estes and McAllister, 2016a). These models indicate that aberrant cytokine signaling can undermine placental integrity and disrupt homeostatic neurodevelopment prenatally (Choi et al., 2016; Meyer et al., 2009; Wu et al., 2017). However, it remains unclear whether there is a threshold of infection severity required for fetal neuroinflammation during MIA, particularly in IAV infections.
It is also important to note that the majority of MIA animal models utilize pathogen mimetics as immunostimulants, including bacterial endotoxin lipopolysaccharide (LPS) or synthetic viral dsRNA mimetic polyinosinic-polycytidylic acid (poly I:C). These non-replicating toll-like receptor (TLR) ligands trigger acute phase inflammatory responses that are short-lived (lasting 24–48 hours), and allow for the targeting of fetal developmental windows and precise control of cytokine responses through dose regulation (Meyer, 2014; Meyer and Feldon, 2012). While the enhanced control and reproducibility obtained with pathogen mimetics is advantageous (Meyer and Feldon, 2012), the immune response is difficult to translate to that of a live pathogen infection. For example, inhalation of live influenza virus into the respiratory tract is followed by continuous viral replication and spread over multiple days, triggering a prolonged and robust inflammatory response involving multiple concurrently activated intracellular detection and signaling pathways (Miao et al., 2010). Distinct inflammatory cascades, initiated by early innate responses (Iwasaki and Pillai, 2014) and then later adaptive responses to IAV (Braciale et al., 2012), generally occur over the span of one to two weeks until the virus is cleared. Overall, immune responses to IAV infection during pregnancy extend across a more prolonged period and involve a wider array of cellular inflammatory and regulatory pathways as compared to MIA induced by a bolus of LPS or poly I:C. To date, comparatively few studies have been conducted using live IAV infection in the MIA field (Kentner et al., 2019; Kneeland and Fatemi, 2013; Weber-Stadlbauer and Meyer, 2019).
While essential progress continues to be made in elucidating potential etiologies of disrupted fetal neurodevelopment during MIA, these mechanisms have yet to be confirmed in models that use live pathogen infections. In particular, recent evidence implicates maternal T helper 17 (TH17) cells and interleukin (IL)-17A as primary drivers of altered fetal brain development during poly I:C-induced MIA (Choi et al., 2016; Kalish et al., 2020; Kim et al., 2017; Shin Yim et al., 2017). Genetic knock out of transcription factor retinoic acid receptor-related orphan nuclear receptor gamma t (RORγt), expressed by IL-17-producing cells and a master regulator of TH17 cell development (Ivanov II et al., 2006), can rescue MIA-induced cortical malformations (Choi et al., 2016). Similarly, IL-17A blockade in pregnant dams protects the developing fetal brain from MIA-induced disruptions in morphology and proteostasis, and normalizes behavioral outcomes (Choi et al., 2016; Kalish et al., 2020; Lammert et al., 2018). The majority of TH17 cells reside in the intestine, and endogenous segmented filamentous intestinal bacteria (SFB), which promote TH17 cell development (Ivanov II et al., 2009), have been shown to be required for the emergence of MIA-associated offspring phenotypes (Kalish et al., 2020; Kim et al., 2017; Lammert et al., 2018). Notably, respiratory IAV infection also promotes TH17 cell development and IL-17A-dependent intestinal injury in non-pregnant male mice (Wang et al., 2014). Furthermore, shifts in the intestinal microbiome are evident during respiratory IAV infection despite the fact that the virus does not infect intestinal tissue (Hanada et al., 2018). Disrupted microbial composition is concomitant with intestinal inflammation (Groves et al., 2018) and altered production of antimicrobial peptides (Deriu et al., 2016; Yildiz et al., 2018), and microbe depletion prior to IAV inoculation has been shown to diminish IAV-induced IL-17A production and intestinal injury (Wang et al., 2014). Thus, there is mounting evidence that IAV infection during pregnancy may elicit heightened levels of maternal IL-17A through intestinal signaling mechanisms, similar to what is observed in poly I:C-induced MIA models, though this remains to be investigated.
Here, we sought to develop a maternal IAV infection mouse model using an H3N2 virus strain and dose that is well characterized to induce moderate pathogenicity in mice (Askovich et al., 2013; Hemann et al., 2019; Kenney et al., 2019; Pazos et al., 2012). A model of moderate infection severity was desirable as this represents the majority of sublethal seasonal influenza virus infections in humans (Hayward et al., 2014). Our results show that, despite lung and systemic inflammatory responses to this IAV infection, fetal brain inflammation was not observed. Consistent with the prevalence of mild-to-moderate infections during pregnancy, these data demonstrate that not all infection-induced MIA results in fetal neuroinflammation, and instead supports a model wherein a high severity threshold must be reached to induce detrimental effects on fetal neurodevelopment. Our study emphasizes the need for live pathogen-induced MIA models to further parse out discrete mechanisms that have the potential to drive aberrant offspring outcomes.
2.0. Materials and Methods
2.1. Animals.
Nulliparous adult C57BL/6NTac female mice were obtained from Taconic Biosciences (Cambridge City, IN), pair-housed, and allowed to acclimate to The Ohio State University Wexner Medical Center vivarium for one week. Two days prior to breeding, females were housed in the soiled cage of a male stud (to initiate estrous), and on the third day that male stud was added to the cage. Presence of a vaginal plug was designated as gestational day (GD) 1, and females were removed to clean cages after breeding. Animals were maintained under sterile conditions on a 12 h light-dark cycle and body weights were recorded daily. A total of 37 pregnant dams across four identical replicates were used in this study. All animal research was approved by and performed in accordance with The Institutional Animal Care and Use Committee at The Ohio State University.
2.2. Influenza A virus inoculation.
Mouse-adapted influenza A virus strain X31 (H3N2) was propagated and titered in 10-day embryonated chicken eggs as previously described (Kenney et al., 2019). On GD10, pregnant dams were randomly assigned to IAV or Con groups and anesthetized by inhalation of isoflurane (Henry Schein Animal Health, Dublin, OH) before intranasal inoculation with IAV strain X31 at a dose of 1000 TCID50 sterile saline or mock inoculated with sterile saline. This dose was selected to mimic the minimal infectious dose of IAV in humans(Edwards et al., 2004; Nikitin et al., 2014), and we have previously validated mild-to-moderate infection severity of this same dose and strain in non-pregnant male mice(Kenney et al., 2019). A total of 20 pregnant dams were inoculated with X31 (IAV group) and 17 pregnant dams were mock-inoculated (Con group) across 4 identical replicates. The MIA Model Reporting Guidelines Checklist (Kentner et al., 2019) is included as Supp. File A.
2.3. Tissue collection.
Tissues were collected and examined seven days post inoculation (7 dpi; GD17), at the expected peak of inflammatory responses to IAV (Kenney et al., 2019; Wang et al., 2014) and when possible repercussions of IAV-induced MIA would be detectable within the placenta and fetal brain (Liong et al., 2020). Pregnant dams were euthanized by CO2 inhalation, and tissues were excised under sterile conditions. Whole blood (obtained through cardiac puncture) was collected into EDTA-coated tubes and stored on ice until centrifugation at 13,300 rpm at 4°C for 15 min; plasma was aliquoted and stored at −80°C until analysis. The uterus was removed and individual fetuses and placentas were dissected out; any fetal resorptions were noted. Two placentas per litter were preserved in 10% Neutral Buffered Formalin for histology. Maternal intestinal tissue was removed, cleaned of mesenteric fat, and colon length obtained. Cross sections of the colon and ileum were preserved in methacarn solution (vol/vol: 60% methanol, 30% chloroform, 10% acetic acid) for histology. The small intestine was flushed with sterile PBS, sliced longitudinally, and cut into 1 cm pieces stored in ice cold HBSS with 5% FBS until further processing. All other tissues were snap frozen and stored at −80°C until analysis.
2.4. Intestinal epithelial cell (IEC) and intraepithelial lymphocyte (IEL) selection.
Sections of the small intestine were immediately transferred to a digestion solution (HBSS with 10mM HEPES, 5mM EDTA, 1mM DTT, 5% FBS) and incubated at 37°C for 20 min with slow rotation. Samples were then vortexed and supernatant (containing IECs and IELs) was passed through a 100 μm cell strainer and kept on ice. Intestinal sections were incubated and rotated in digestion solution two more times, vortexed, and supernatant passed through a 100 μm and then 70 μm cell strainer. The combined flow-through containing IECs and IELs was centrifuged at 400 x g for 10 min at 4°C, cells were washed in buffer (PBS with 2% FBS and 2 mM EDTA), passed through a 70 μm cell strainer, then centrifuged and resuspended in buffer containing CD45 MicroBeads (Miltenyi Biotec, Auburn, CA). Cells were incubated with microbeads for 15 min at 4°C, then washed with buffer and passed through LS columns for magnetic cell sorting, as per manufacturer’s protocol (Miltenyi Biotec). Both CD45 positive (IEL) and negative (IEC) cell fractions were collected and stored at −80°C until analysis.
2.5. Protein assays.
Protein was isolated from tissues by sonication (40% amp) in lysis buffer (100:1 T-PER Tissue Protein Extraction Reagent and Halt Protease and Phosphatase Inhibitor Cocktail; Thermo Scientific, Waltham, MA). Homogenized samples were then incubated for 30 min at 4°C, centrifuged at 13,300 rpm at 4°C for 15 min, and protein lysates were aliquoted. Protein concentration was determined using the Pierce BCA Protein Assay kit (Thermo Scientific) and normalized prior to each assay.
Protein lysates from fetal brains (one per litter, normalized to 4 mg/mL) and placentas (one per litter, normalized to 7.4 mg/mL) were pooled to obtain a total of 1 mL per treatment group per tissue. Each pooled sample was assayed for 40 different cytokines using the Proteome Profiler Mouse Cytokine Array Panel A kit according to manufacturer’s instructions (R&D Systems, Minneapolis, MN). Membranes were imaged using a LI-COR Odyssey Fc chemiluminescence imager (Lincoln, NE) and mean pixel density between duplicate spots was quantified using ImageJ software (NIH, Bethesda, MD).
Protein lysates from lung tissue (normalized to 6 mg/mL using Diluent 41) and plasma samples (diluted 1:1 in Diluent 41) were assayed in duplicate on a custom multiplex assay (U-PLEX Assay Platform, Meso Scale Discovery, Rockville, MD) containing 9 cytokines: IFN-α, IFN-β, IFN-γ, IL-1β, IL-6, TNFα, IL-17A, IL-17C, IL-17E/IL-25. The assay was performed according to manufacturer’s instructions using Alternate Protocol 1, Extended Incubation. Concentration of circulating LPS-binding protein (LBP) was determined in plasma samples (diluted 1:5) using Mouse LBP ELISA Kit (Abcam, Cambridge, MA) according to manufacturer’s instructions. Protein lysates from colon tissue (normalized to 3.5 mg/mL) were assayed in duplicate using the Mouse IL-17 DuoSet ELISA (R&D Systems) according to manufacturer’s instructions.
2.6. Quantitative real-time PCR.
RNA was isolated using TRIzol Reagent as per the TRI Reagent Protocol (Invitrogen, Carlsbad, CA). cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA), and PCR was performed on a QuantStudio 3 Real-Time PCR System machine using TaqMan Fast Advanced Master Mix (Applied Biosystems). Data were analyzed using the 2−ΔΔCt method against housekeeping gene Rpl19 and presented as relative expression compared to control, unless otherwise stated. Statistics were run on relative expression values. Primer information is listed in Supp. Table S7. Custom primers (Integrated DNA Technologies, Coralville, IA) against the IAV nucleoprotein (forward: 5’-CAGCCTAATCAGACCAAATG-3’; reverse: 5’-TACCTGCTTCTCAGTTCAAG-3’) were assayed using SYBR Green Master Mix (Applied Biosystems) to test for viral presence in the maternal lung, placenta, and fetal brain. cDNA from X31 viral stock was used as a positive control. For visualization purposes, samples with undetermined Ct values or no amplification were arbitrarily given a Ct value of 40.
2.7. NanoString Gene Expression Panel.
RNA was isolated from 12 fetal brains (one per litter) using TRIzol Reagent as per the TRI Reagent Protocol (Invitrogen), and delivered to the Genomics Shared Resource lab at The Ohio State University Comprehensive Cancer Center. RNA integrity was assessed using both BioAnalyzer and Qubit systems (mean RIN = 5.7), and the samples were assayed on the nCounter Neuroinflammation Panel (NanoString Technologies, Seattle, WA) according to manufacturer’s protocol. Raw data were analyzed using nSolver 4.0 Software (NanoString), applying the Advanced Analysis method with automated normalization gene selection. Automated probe exclusion using the “Omit Low Count Data” default selection resulted in omission of 166 probes. Based on quality control measures, two samples (one per treatment group) were identified as outliers and excluded from the analysis, resulting in 591 genes assayed across 10 samples. False discovery rate (FDR) correction for multiple comparisons was applied using Bonferroni correction.
2.8. Pathology.
Histology services were performed in the Histology Laboratory of the Comparative Pathology and Digital Imaging Shared Resource (CPDISR) of The Ohio State University Comprehensive Cancer Center. Tissues were routinely processed for histopathology on a Leica Peloris 3 Tissue Processor (Leica Biosystems, Buffalo Grove, IL), embedded in paraffin, sectioned at an approximate thickness of 4–5 micrometers, and batch stained with hematoxylin and eosin (H&E) on a Leica ST5020 autostainer (Leica Biosystems) using a routine and quality controlled protocol. Slides were then evaluated by a board-certified veterinary comparative pathologist (Kara N. Corps, who was blinded to treatment groups) using a Nikon Eclipse Ci-L Upright Microscope (Nikon Instruments, Inc., Melville, NY). Representative photomicrographs were taken using an 18 megapixel Olympus SC180 microscope-mounted digital camera and cellSens imaging software (Olympus Life Science, Center Valley, PA).
Semi-quantitative scoring of placental histopathology findings was performed using a scoring rubric modified from previously published placental scoring methods (Fricke et al., 2018; Ward et al., 2012). Mineralization was scored using the following criteria: 0 = no mineral present beyond scattered small foci; 1 = rare or mild, multifocal mineralization; 2 = multifocal scattered foci of mineralization; 3 = large multifocal zones of mineralization or diffuse mineralization within a zone. Infiltration of neutrophils and mononuclear cells were scored using the following criteria: 0 = normal; 1 = slight increase; 2 = moderate increase; 3 = marked increase. Necrosis was scored using the following criteria: 0 = no necrosis present; 1 = multifocal necrosis exclusively in the maternal placenta; 2 = multifocal necrosis throughout the placenta; 3 = multifocal to coalescing, large zones of necrosis throughout the placenta; 4 = diffuse placental necrosis. Two sections each from two representative placentas per litter were scored and then averaged for each litter.
Semi-quantitative scoring of intestinal histopathology findings was performed using a scoring rubric adapted from established scoring methods (Kennedy et al., 2000; Suzuki et al., 2006). Edema was scored using the following criteria: 0 = normal, 1 = mild/focal/single layer of ileum or colon, 2 = moderate/multifocal/multiple layers of ileum or colon, 3 = severe/widespread/transmural. Epithelial hyperplasia was scored using the following criteria: 0 = normal, 1 = slight/mild, 2 = moderate, 3 = marked: discrete nests of regenerated crypts delineated from adjacent mucosa with no obvious disruption of mucosal surface. Presence of neutrophils and lamina propria mononuclear cells were scored using the following criteria: 0 = normal, 1 = slight increase, 2 = moderate increase, 3 = marked increase. Crypt/gland inflammation was scored using the following criteria: 0 = normal mucosa, 1 = one-to-two inflammatory cells, 2 = cryptitis/significant gland inflammation affecting majority of structure, 3 = crypt/gland abscess/necrosis, 4 = crypt/gland absent due to mucosal destruction. Mucosal/crypt loss was scored using the following criteria: 0 = normal mucosa, 1 = shortening of basal one-third of crypts +/− slight inflammation and edema in the lamina propria, 2 = loss of basal two-thirds of crypts +/− moderate inflammation in the lamina propria, 3 = loss of all epithelium +/− severe inflammation in lamina propria or submucosa inflammation with surface epithelium intact/remaining, 4 = loss of all epithelium +/− severe inflammation in lamina propria +/− muscularis; exudate may cover damaged mucosa. Two sections of colon and ileum tissue per dam were scored and averaged.
2.9. Statistics.
Dam (or litter) was treated as the experimental unit for all outcomes, and one representative fetus per litter was used for each experimental outcome, unless otherwise stated. All data (except NanoString gene expression data, described above) were analyzed using GraphPad Prism 8 software (San Diego, CA), with significance set at α = 0.05. Dam body weights across time were analyzed using repeated measures two-way ANOVA (time x infection), with Geisser-Greenhouse correction for sphericity and Bonferroni correction for multiple comparisons. Unpaired parametric t tests were used to compare Con and IAV groups, with Welch’s correction for unequal variances when applicable. For data that were not normally distributed (multiplex cytokine concentrations and histopathology scores), non-parametric Mann-Whitney tests were used. Outliers were identified and removed using the ROUT method with Q = 1%.
3.0. Results
3.1. Respiratory IAV infection during gestation induces a robust maternal inflammatory response.
To determine whether infection with the moderately pathogenic H3N2 IAV strain X31 could induce MIA, we inoculated pregnant dams intranasally with a dose of 103 TCID50 on GD10. We have previously demonstrated that prenatal insults initiated at GD10 confer detrimental behavioral and neuroimmune outcomes in offspring (Chen et al., 2020; Gur et al., 2019, 2017). Furthermore, murine infection beginning at GD10 approximates early gestation in humans, a time when the risk of IAV infection in determining aberrant offspring mental health outcomes is especially high (Brown et al., 2004). We found that IAV infection severely blunted maternal body weight gain throughout the remainder of the gestational period, culminating in an average body mass that was 6.2 ± 1.2 g less than saline controls by GD17 (Fig. 1A). Indeed, 4 out of 20 IAV-infected dams lost weight across the 7-day period (Supp. Fig. S1A). IAV infection did not affect litter size or number of fetal resorptions, demonstrating that blunted body weight gain was not due to fetal loss (Supp. Table S1). Further, GD17 maternal body weight was still stunted when normalized to litter size (Supp. Fig. S1B). When corrected for body weight, maternal spleen mass at 7 dpi (GD17) was increased in the IAV group, suggesting systemic expansion of immune cells in response to the virus (Fig. 1B). We confirmed the presence of viral RNA in infected maternal lungs through quantitative PCR (Fig. 1C). Within infected lungs there was increased secretion of pro-inflammatory cytokines, including classical inflammatory mediators IL-1β, IL-6, TNFα, IFN-α, and IFN-γ, as well as TH17 cytokines IL-17A and IL-17C (Fig. 1D; IFN-β and IL-17E were below detectable levels). This coincided with upregulated immune cell marker genes (Cd4: T helper cells, Cd8a1: cytotoxic T cells, Itgam: neutrophils and monocytes, Ly6c1: inflammatory monocytes), indicating immune cell infiltration of infected lung tissue (Fig. 1E). Circulating levels of plasma IL-6, TNFα, and IFN-γ were also increased in response to IAV infection (Fig. 1F), but all other cytokines (IFN-α, IFN-β, IL-1β, IL-17A, IL-17C, IL-17E) were outside the detectable range. Overall, these results demonstrate that X31 IAV infection of pregnant dams induces a significant inflammatory response and prevents healthy maternal weight gain without loss of pups, establishing this infection system as a moderate MIA model.
Figure 1. Gestational respiratory IAV infection induces robust respiratory and systemic inflammation.

IAV inoculation at gestational day (GD)10 (A) suppressed body weight gain beginning three days post-inoculation (repeated measures ANOVA: time x infection, p < 0.001, Bonferroni multiple comparisons correction, *** = adj p < 0.001, ** = adj p < 0.01, * = adj p < 0.05), resulting in substantial reduction in maternal body weight compared to control dams at 7 dpi. (B) Relative to body weight, maternal spleen mass at 7 dpi was increased due to IAV infection. Con: n = 17, IAV: n = 20. (C) Presence of IAV in the lung was confirmed through PCR, using a cycle threshold of ≤ 32 cycles as a confirmed infection (dotted line). (D) Inflammatory cytokines associated with innate and adaptive anti-viral immune response were increased in the lungs of IAV infected dams at 7 dpi, concomitant with (E) upregulated immune cell marker genes (Cd4: T helper cells, Cd8a1: cytotoxic T cells, Itgam: neutrophils and monocytes, Ly6c1: inflammatory monocytes), indicating immune cell infiltration. (F) IAV infection increased concentrations of IL-6, TNFα, and IFN-γ in circulation. Lung cytokines: Con n = 8, IAV n = 6; lung PCR: Con n = 17, IAV n = 20; plasma cytokines: n = 13/group. IAV = influenza A virus; dpi = days post-inoculation; ND = not detectable; *** = p < 0.001, ** = p < 0.01, * = p < 0.05. Data are presented as mean ± SEM, with each individual dot representing one dam.
3.2. Inflammation and antimicrobial responses extend to the placenta during maternal IAV infection.
Given prior reports that placental disruptions can be induced by MIA (Fatemi et al., 2012; Liong et al., 2020), we next examined whether maternal respiratory IAV X31 infection had downstream effects on the placenta. Using PCR, we confirmed that the virus is not vertically transmitted to placental tissue (Supp. Fig. S2). Next, we scored H&E-stained placental sections for inflammatory lesions, and found increased incidence of necrosis, multifocal neutrophil infiltration, and mild-to-moderate mineralization in placentas from IAV-infected dams that was evident in both maternal and fetal layers (Fig. 2A–F). In order to broadly characterize the molecular immune profile of the placenta, we pooled samples of normalized protein extracts from placentas (one per litter) of control or IAV dams to obtain sufficient material to utilize a Proteome Profiler Cytokine Array Panel, which assessed 40 cytokines. Placentas from IAV infected dams showed modest increases in complement component C5/C5a, IL-1α, IL-1ra, and neutrophil chemoattractant KC/CXCL1 (Fig. 3A). Roughly 2-fold increases were observed for monocyte chemoattractant CCL2, T cell chemoattractant and CD4 ligand IL-16, and tissue inhibitor of metalloproteinases 1 (TIMP-1; Fig. 3A), consistent with the mild-to-moderate histological changes reported in Fig. 2. We next probed a select set of 26 genes of interest to examine transcription of classical inflammatory mediators implicated in MIA, as well as anti-viral and anti-bacterial response genes, and genes involved in placental barrier integrity (presented in Supp. Table S2). Of these, relative expression of three genes increased 2-fold (Ifng, bactericidal lectin Reg3b, and cytotoxic Gzmb; Fig. 3B), indicating a modest enhancement of antimicrobial responses. Expression of the remaining immune and barrier integrity genes were comparatively unchanged by maternal IAV infection (Supp. Table S2). Previous reports have suggested that type I IFN signaling during pregnancy can lead to disruptions of placental structural formation (Buchrieser et al., 2019; Yockey et al., 2018; Zani et al., 2019). Thus, we also included interferon-responsive genes, Ifitm3 and Irf7, but did not observe their placental upregulation upon IAV infection (Supp. Table S2), suggesting that the observed placental changes may not be driven by type I IFN signaling. Notably, relative expression of a subset of these same immune and antimicrobial genes was also largely unchanged in the uterus (Supp. Table S3). Together, our data suggest that maternal X31 infection leads to downstream immune and antimicrobial signaling and mild tissue breakdown in the placenta.
Figure 2. Maternal respiratory IAV-infection increases placental necrosis, mineralization and inflammation.

Representative photomicrographs of H&E-stained placental sections and lesion score quantification from Control and IAV-infected dams. While Con dams had small foci of necrosis only in the maternal portion of the placenta and lacked an overt inflammatory response (A), all IAV-infected dams had larger, multifocal to coalescing areas of necrosis in the maternal layers (B) and 2 of 6 had necrosis in the fetal layers. Necrosis in IAV dams was frequently accompanied by neutrophilic inflammation, and scattered neutrophils were present in all layers of the maternal tissue (C, arrows). Control dams had small foci of mineralization in the fetal vascular labyrinth, a frequent and expected finding (D, arrow). In contrast, IAV-infected dams had numerous scattered foci of mineral throughout the fetal vascular labyrinth that were occasionally accompanied by neutrophilic inflammation or focal necrosis (E, arrows). Increased overall basophilic staining was also evident (B, E) and is caused by a combination of increased circulating inflammatory cells, particularly mononuclear cells and multifocal neutrophils, and mild-to-moderate mineralization of capillary walls. Inflammatory lesion scores are quantified in (F); scoring criteria is listed in the Methods. Two sections each from two representative placentas per litter were scored and averaged; Con: n = 8 litters, IAV: n = 6 litters. A-B: 10X magnification, scale bar = 100 μm; C-E: 20X magnification, scale bar = 50 μm. IAV = influenza A virus; *** = p < 0.001, * = p < 0.05. Group scores are presented as mean ± SEM.
Figure 3. Maternal IAV infection increases placental inflammatory signaling and bactericidal responses.

(A) Pooled placental protein extracts from control (n = 8) and IAV-infected (n = 6) dams were assayed on the Proteome Profiler Cytokine Array, suggesting enhanced immune responses due to maternal IAV infection. (B) Upregulation of Ifng, bactericidal C-type lectin Reg3b, and Gzmb (Granzyme B) expression in placentas from IAV-infected dams (n = 9–13/group). IAV = influenza A virus; ** = p < 0.01, # = p ≤ 0.10. Proteome profiler data are presented as mean pixel density across the two spots (no error bars). Gene expression data are presented as mean ± SEM.
3.3. Fetal neuroinflammation is not overtly apparent during maternal IAV infection.
Given that we observed a partial breakdown of placental integrity during maternal IAV infection, we further investigated whether immune homeostasis in the fetal brain was also affected. IAV RNA was not detectable in fetal brains (Supp. Fig. S2), confirming that the virus does not breach the fetal compartment. Cytokine array profiling of pooled fetal brain protein extracts revealed only two secreted proteins within detectable range, lymphocyte chemoattractant CXCL12 and soluble intercellular adhesion molecule-1 (sICAM-1), and these were at similar levels between groups (Fig. 4A). To more thoroughly examine the impacts of maternal IAV infection on neuroinflammation of the developing fetal brain, we investigated an extensive set of genes of interest across ten fetal brain samples using the NanoString nCounter Mouse Neuroinflammation Panel (a heatmap of the normalized data is presented in Supp. Fig. S3; log2 fold change and statistical results are presented in Supp. Table S4). However, out of 591 genes, we identified none that were differentially expressed in the fetal brain due to maternal IAV infection. To follow up on these findings, we conducted independent qPCR experiments to investigate relative expression of 23 genes implicated during prenatal insults and involved in neuroimmune responses, blood brain barrier integrity, and neurodevelopment (Fig. 4B and Supp. Table S5). We confirmed an overall lack of gene expression changes in the fetal brain upon maternal IAV infection, with only a modest but significant downregulation in Il17ra (encoding IL-17 Receptor A) and Il1b; Il17a mRNA was not detectable. In all, the notable absence of fetal neuroinflammation during moderately severe maternal IAV infection suggests that an infection severity threshold must be reached in order to induce immune imbalance in the developing fetal brain.
Figure 4. Fetal neuroinflammation is not overtly apparent during mild maternal IAV infection.
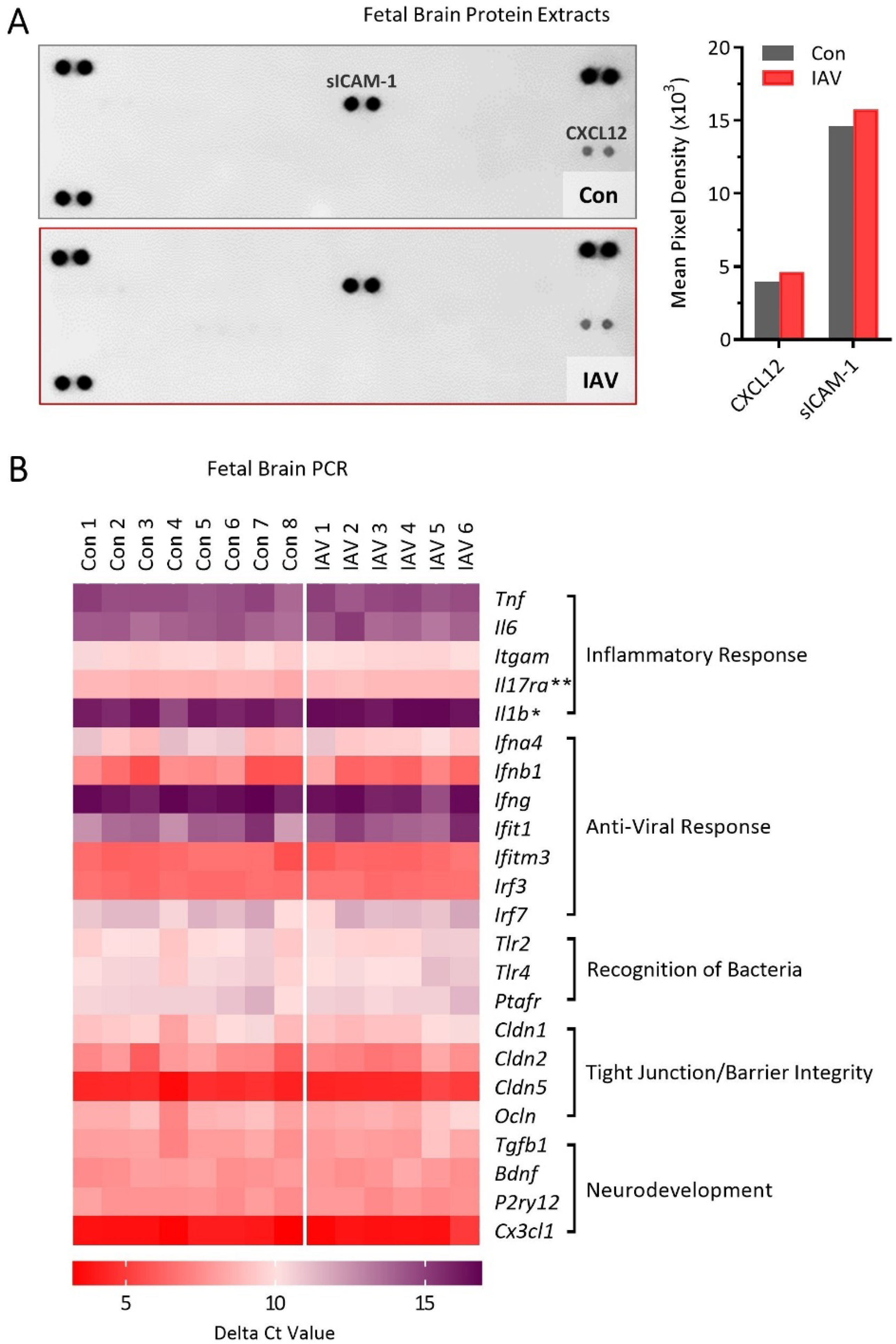
(A) Pooled fetal brain protein extracts from litters of control (n = 7) and IAV-infected (n = 4) dams assayed on the Proteome Profiler Cytokine Array revealed only two detectable signaling molecules, suggesting limited inflammatory response to maternal IAV infection in the fetal brain. Proteome profiler data are presented as mean pixel density across the two spots (no error bars). (B) Independent qPCR was carried out on fetal brain tissue to confirm and extend upon the NanoString Neuroinflammation Panel data. PCR data are represented as a heatmap of delta Ct values (compared to housekeeping control gene = Rpl19) of select genes involved in neuroimmune responses, blood brain barrier integrity, and neurodevelopment within the fetal brain. Purple indicates low expression compared to Rpl19 (i.e. higher cycle threshold); red indicates high expression compared to Rpl19 (i.e. lower cycle threshold). A modest but significant downregulation in Il17ra and Il1b expression was observed due to maternal IAV-infection, but no other genes were differentially expressed. Mean ± SEM and p-values for all genes are listed in Supp. Table S5. IAV = influenza A virus. ** = p < 0.01, * = p < 0.05.
3.4. Respiratory IAV infection has downstream consequences for maternal intestinal health.
Mimetic-induced MIA models have demonstrated that maternal IL-17A, derived from intestinal TH17 cells, is a crucial component in mediating detrimental offspring outcomes (Choi et al., 2016; Kalish et al., 2020; Kim et al., 2017). To investigate whether live IAV infection might also activate intestinal TH17 cells and propagate downstream IL-17A signaling, we examined the maternal intestine for indicators of immune activation and resultant tissue breakdown. As colon shortening is a hallmark indication of colitis (Chassaing et al., 2014), we first measured colon length, which was indeed reduced in IAV-infected pregnant dams compared to control (Fig. 5A). Relative expression of colonic Ifng and Rorc (which encodes T-cell dependent transcription factor RORγt) was upregulated by respiratory IAV infection, concurrent with a tendency for upregulated tight junction protein transcripts Cldn1 and Ocln (p < 0.10), but not Cldn2 or Cldn5 (Fig. 5A). IL-17 protein within colon protein extracts did not differ (Fig. 5A). In order to follow up on the potential incidence of colitis, sections of colon and small intestine (ileum) tissue were H&E stained and examined for inflammatory lesions. However, we observed no differences in intestinal pathology between control and IAV-infected dams, with almost all mean lesion scores falling at or below 1 (i.e., normal-to-mild pathology; Supp. Fig. S4).
Figure 5. Downstream inflammatory impacts of gestational IAV infection on the maternal intestine.

IAV-infected dams displayed (A) reduced colon lengths (Con: n = 17, IAV: n = 20) concurrent with upregulation in colonic expression of transcription factor Rorc, Ifng, and tight junction proteins Cldn1 and Ocln (n = 6–13/group), while there were no changes in Cldn2 and Cldn5 expression or IL-17 protein within colon protein extracts (n = 9/group). (B) Intraepithelial lymphocytes (IEL; CD45+ fraction) isolated from the small intestine upregulated Rorc expression (n = 5–8/group). (C) Relative expression of most antimicrobial response genes was dysregulated in intestinal epithelial cells (IEC; CD45− fraction) of IAV-infected dam small intestine, while TH17-related genes were unchanged (n = 5–8/group). (D) The plasma concentration of LPS-binding protein, a proxy for disrupted intestinal barrier integrity, was not impacted by IAV infection (p = 0.52; Con: n = 12, IAV: n = 14). IAV = influenza A virus; *** = p < 0.001, ** = p < 0.01, * = p < 0.05, # = p ≤ 0.10. Lines indicate mean ± SEM, with each individual dot representing one dam. Gene expression data are presented as mean ± SEM.
We then isolated the CD45+ cell fraction (intraepithelial lymphocytes, IELs) of the small intestine to examine TH17-related genes. Again, while Rorc expression was increased in IELs of IAV-infected dams (Fig. 5B), there were no significant changes in expression of Il17a or Il17ra, nor interferons (Ifna4, Ifnb1, Ifng) or Il6 (Supp. Table S6). As TH17 cell differentiation is induced by shifts in commensal microbe populations (Ivanov II et al., 2008; Wang et al., 2014), and intestinal epithelial cells (IECs, CD45− fraction) mediate immune responses to endogenous microbes, we examined antimicrobial gene transcription within IECs as an indication of microbial dysbiosis. Expression of antimicrobial peptides alpha defensin 1 (Defa1), C-type lectins Reg3b and Reg3g, and dual oxidase Duoxa2 were differentially impacted by IAV infection (Fig. 5C), indicating a disruption in IEC-controlled bacterial segregation. IEC expression of toll-like receptor 4 (Tlr4) and LPS-binding protein (Lbp), which detect bacterial LPS and help regulate antimicrobial molecule production, were downregulated while TLR4 signal transducer Myd88 was unchanged (Fig. 5C). Relative expression of Il17ra, which binds IL-17A from TH17 cells and also aids in antimicrobial molecule production, was unchanged, as were Tgfb1 and Il23a, which promote TH17 cell polarization (Fig. 5C). As a proxy for intestinal breakdown leading to release of bacteria into circulation, we measured LPS-binding protein in maternal plasma but found no differences between groups (Fig. 5D), consistent with maintained or enhanced tight junction protein transcripts within the colon (Fig. 5A). Overall, respiratory IAV X31 infection appears to drive mild downstream intestinal immune and microbial dysregulation, and while we observed consistently increased expression of transcription factor RORγt, indications of augmented IL-17 signaling were markedly absent. Given the known role of IL-17A in development of fetal neuroinflammation in MIA, the lack of intestinal IL-17 signaling in our IAV X31 infection experiments is consistent with the absence of fetal brain inflammation.
4.0. Discussion
To address a major limitation of mimetic-induced MIA—the inability to capture a full pathogen-induced spectrum of inflammatory and regulatory immune responses—we developed a clinically relevant IAV infection model to probe the sequelae of MIA. Utilization of a moderately pathogenic IAV strain and dose, which is pertinent to the human condition, helps advance our understanding of the mechanisms mediating adverse fetal outcomes during seasonal IAV infection. Our data reveal that moderately severe IAV infection leads to maternal inflammation that is not restricted to the respiratory tract but also elicits off-target effects on the intestine and placenta. The notable absence of IL-17 signaling and of overt fetal brain inflammation supports the theory that an infection severity threshold likely exists for some MIA-mediated outcomes.
The clinically relevant moderate dose of X31 we selected is approximately 10 times lower than comparable studies (Liong et al., 2020), yet our IAV infection model still blunted maternal weight gain without impacting litter size. Others have shown that maternal IAV infection can slow placental and fetal growth, but does not increase fetal resorption (Harding et al., 2021; Liong et al., 2020; Pazos et al., 2012), in agreement with what we report here. While we did not record placental and fetal weights within each litter, it is likely that a similar growth retardation is present in our model considering the substantial body weight reduction in pregnant dams after correcting for litter size. Indeed, rates of intrauterine growth restriction leading to lower birth weight infants are increased following gestational IAV infection in humans (Newsome et al., 2019).
Although viral transmission to placental tissue during murine IAV infection is rare (Fatemi et al., 2012; Harding et al., 2021; Liong et al., 2020; Shi et al., 2005), and did not occur in our model, we observed mild-to-moderate placental pathology within all IAV litters, similar to a previous report (Fatemi et al., 2012). Infiltration of neutrophils, but not mononuclear cells, was especially apparent and is common to both bacterial-induced intrauterine inflammation (Kothary et al., 2017; Presicce et al., 2018) and preeclampsia (Cadden and Walsh, 2008). Recent evidence suggests that maternal IAV infection can result in a vascular signature that mimics preeclampsia, albeit this study used a much higher dose of X31 (Liong et al., 2020). In our model, changes in placental transcription of matrix metalloproteinases 2 and 9, implicated in preeclampsia (Espino et al., 2017), did not reach significance, although augmented secretion of metalloproteinase inhibitor TIMP-1 (Rahat et al., 2016) was detectable in placental lysates. Additionally, increased placental mineralization, as observed here, can be an indication of placental vascular malperfusion (Stanek, 2019). Thus, it is possible that a mild preeclampsia-like phenotype also exists in our maternal IAV infection model, but comprehensive measurements of vascular alterations are still required.
On the other hand, upregulation of antimicrobial transcripts within the placenta also suggest that neutrophil trafficking might correspond to infiltration of endogenous bacteria, although the concept of in utero bacterial colonization is extremely controversial (Chen and Gur, 2019; Walker et al., 2017). While vertical transmission of bacteria or their products during pregnancy has been demonstrated (Humann et al., 2016; Tan et al., 2013), the notion that maternal IAV infection may lead to bacterial translocation to the placenta requires extensive follow-up. Interestingly, while there is increased necrotic tissue breakdown in placentas of IAV litters, barrier integrity may still be intact at 7 dpi considering that transcripts of tight junction proteins were unchanged. Further indication of a relatively resilient placental barrier system may be inferred from the lack of inflammatory signaling in the fetal brain, despite significant maternal inflammation in the lung and systemically. Indeed, recent evidence has identified nonclassical estrogen receptor GPER1, which is constitutively expressed in all major zones of the placenta, as a primary regulator of IFN signaling that protects fetal health during maternal IAV infection (Harding et al., 2021). The lack of placental upregulation of interferon-response genes in our model indicates that GPER1 activation is likely suppressing maternally-derived IFN signaling within fetal tissues. Outside of IFNs, previous work has demonstrated the essential involvement of cytokine IL-6 in evoking neurodevelopmental abnormalities during mimetic-induced MIA (Garbett et al., 2012; Hameete et al., 2021; Smith et al., 2007), which revealed the necessity of transplacental IL-6 signaling (Wu et al., 2017). While IL-6 was increased approximately 3-fold in maternal circulation by IAV infection, IL-6 protein was not detectable from placental lysates and placental and fetal brain Il6 transcripts were not differentially regulated. It is possible that the pooling of placental and fetal brain lysates prior to profiling could have masked some variability in cytokine production in our model, and more sensitive methods may reveal subtle yet significant differences within and across litters. Further investigation is needed to discern the impact of heightened maternal IL-6 on fetal development within discrete model systems of MIA, especially when comparing live pathogen infections with bolus administration of LPS, poly I:C, or recombinant IL-6. Overall, it appears the placenta is able to maintain a functional barrier system during moderately severe IAV infection despite indications of mild pathology, proffering protection to the developing fetus. It should be noted, however, that further placental breakdown or inflammatory signaling may be occurring at time points other than 7 dpi.
Only one group, Fatemi and colleagues, has done extensive work in establishing an MIA mouse model of IAV infection by inoculating pregnant mice with human Influenza A/NWS/33 (H1N1) at various gestational time points (Fatemi et al., 2012, 2008, 2005, 2002a, 2002b, 1999; Shi et al., 2003). There are several important factors that differ between this model and the one we present here: (a) IAV strain and dose (Fatemi et al. used the H1N1 NWS strain, which possesses the unusual property of being neurotropic in mice, which is not generally a common feature of IAVs; dosing was reported as a dilution of an in-house stock virus and cannot be compared with our study); (b) gestational timing of IAV infection (Fatemi et al. use GD7, 9, 16, and 18); (c) mouse strain (Fatemi et al. use both C57BL/6 and Balb/c); and (d) Fatemi et al.’s use of a broad-spectrum antibiotic (oxytetracycline) in the drinking water to prevent secondary infection (Fatemi et al., 2012, 2008, 2005, 2002a, 2002b, 1999). Overall, this group’s pioneering work revealed the various adverse effects that maternal infection with IAV can have on postnatal mouse offspring brain morphology, chemistry, and transcription, despite the lack of H1N1 viral presence in offspring tissues ((Fatemi et al., 2012) and reviewed in (Kneeland and Fatemi, 2013)). In particular, inoculation of pregnant mice at GD9 resulted in 38 dysregulated genes in the brains of neonatal (P0) progeny (Fatemi et al., 2005). Another group, using the same H1N1 NWS infection model, reported 256 differentially expressed genes in the E12.5 embryonic brain, 3 days after IAV inoculation (Garbett et al., 2012). Here, we found almost no differentially expressed genes in the E17 fetal brain due to maternal IAV X31 infection at GD10. Further investigation into comparable neurodevelopmental and neurocognitive parameters, pre- and postnatally, is needed in order to determine if additional differences exist between the two models. Moreover, investigation at multiple time points following infection (in addition to E17 presented here) is needed in order to capture the full spectrum of fetal responses to a dynamic maternal immune activation profile. Nevertheless, the lack of transcriptional changes in immune, barrier integrity, and neurodevelopmental genes here suggests that offspring neurological outcomes, if present, are likely to be more mild than what has previously been described in the H1N1 A/NWS/33 infection model (Fatemi et al., 2002b, 2002a, 1999; Garbett et al., 2012). Indeed, recent epidemiological data indicates that severe IAV infection during pregnancy significantly elevated the risk of adverse infant outcomes, while less severe infection did not (Newsome et al., 2019). Our data support a model in which a moderately pathogenic IAV infection is below the severity threshold necessary to induce detrimental effects on fetal brain immune equilibrium.
Given the potential contribution of TH17 cell responses in MIA outcomes (Estes and McAllister, 2016b), one of our major goals was to investigate IL-17 signaling pathways during IAV infection. While respiratory IAV infection increased IL-17A and IL-17C in the lung, IL-17 cytokines were below detectable levels within circulation and did not differ in the intestine. It is possible that this is due to IAV strain differences (Bouvier and Lowen, 2010), as enhanced intestinal TH17 cell polarization and IL-17A signaling has been documented in male mice infected with a more pathogenic IAV strain (Wang et al., 2014). Even so, this more severe IAV infection did not alter circulating levels of IL-17A (Wang et al., 2014), similar to what we report here. This is in stark contrast to poly I:C-induced MIA, wherein increased levels of IL-17A in maternal circulation are readily apparent two days after poly I:C injection (Choi et al., 2016). In this model, increased maternal IL-17A augments IL-17RA expression in the fetal brain (Choi et al., 2016), whereas our maternal IAV infection model resulted in a minimal but significant downregulation of fetal brain Il17ra expression at 7 dpi. This suggests that either IL-17A signaling is not pervasive during moderate IAV-induced MIA, or we were unable to fully capture the maternal IL-17A signaling cascade during a single study end point.
Within the aforementioned poly I:C MIA model, intestinal dendritic cells detect poly I:C directly through TLR3 and induce IL-17A production from pre-existing intestinal TH17 cells (Kim et al., 2017). Conversely, IAV infection has been shown to indirectly disrupt intestinal microbial communities through lung-derived adaptive responses, which then lead to antimicrobial signaling by intestinal epithelial cells, ultimately promoting TH17 cell polarization from resident naive T cells (Wang et al., 2014). This is in line with our observation of upregulated Rorc within the CD45+ intestinal lymphocyte population, suggesting a similar induction of TH17 polarization. Importantly, this occurs much later (7 days post IAV inoculation) compared to 12–24 hours post poly I:C injection (Choi et al., 2016). Critically, however, it appears that IL-17 cytokine production is restricted to the lung at the time point examined in our model, with no detectable changes in IL-17 transcripts or protein in the intestine or in circulation at 7 dpi. Likewise, TH17-related gene transcripts (IL-17 receptor Il17ra and TH17-polarizing Tgfb1 and Il23a) were not differentially regulated within intestinal epithelial cells. While it is possible that changes in IL-17 signaling occurs at time points outside of 7 dpi, these findings bolster the theory that moderate IAV infection may be below severity threshold for eliciting IL-17A responses comparable to levels achieved by poly I:C bolus, which is administered directly into the peritoneum. It remains to be seen whether a more severe respiratory IAV infection may elicit appreciable IL-17 production within the intestine (and systemically), propagating an inflammatory signaling cascade that could negatively impact fetal brain development.
It has previously been shown that a maternal intestinal microbiome that contains segmented filamentous bacteria (SFB) is required for poly I:C-induced offspring abnormalities (Kim et al., 2017; Lammert et al., 2018), in line with the particular ability of SFB to promote TH17 cell development (Ivanov II et al., 2009). Yet IAV infection has been documented to substantially reduce SFB numbers, despite concomitant increases in TH17 cell polarization and in IL-17A production (Wang et al., 2014). Although microbial communities were not investigated here, we did observe a dysregulation in expression of antimicrobial response genes within the intestine, suggesting that endogenous microbes likely play a role in our IAV-induced MIA model. Overall, a marked decrease in colon length of IAV-infected dams (similar to prior observations (Wang et al., 2014)), in conjunction with differential expression of immune and antimicrobial genes, indicates that even moderately severe IAV infection during pregnancy can disrupt certain components of maternal intestinal health. However, trending increases or stable expression of colonic tight junction proteins, in addition to equivalent circulating levels of LPS-binding protein between IAV-infected and control dams, supports an intact intestinal barrier at this time point. Furthermore, the lack of intestinal pathology at 7 dpi across both colonic and ileal H&E-stained sections confirms that overt intestinal injury, outside of colonic shortening, is mostly absent at this time point. Further investigation encompassing multiple time points post-inoculation is needed. Overall, however, these conflicting reports between poly I:C- and IAV-induced intestinal responses epitomize the need for live pathogen MIA models that can further elucidate the influential potential of maternal microbes and adaptive immune responses on offspring outcomes. Indeed, the expansion of biologically relevant MIA models is needed not only to appreciate the full extent of disparate immune mechanisms and pathophysiologies elicited by distinct pathogens (virulent and nonvirulent), but also to enable more direct translation to humans (as reviewed in (Brown and Meyer, 2018)).
Taken together, our data indicate that a clinically relevant and moderately pathogenic IAV infection during pregnancy results in downstream inflammatory sequelea in the placenta but not the fetal brain. While live IAV infection is also characterized by indirect effects within maternal intestinal tissue, IL-17A signaling appears not to play a significant role if the infection is less severe. Critically, our IAV-induced MIA model mirrors a distinct component of epidemiological data wherein only a fraction of infections during pregnancy result in aberrant offspring outcomes, and that offspring psychiatric risk intensifies with increasing infection severity. Overall, our data emphasize the importance of conducting experiments using biologically relevant and translatable MIA models in order to better understand the mechanisms underlying the etiology of neurodevelopmental disorders.
Supplementary Material
Highlights.
A mouse model of influenza A virus (IAV) infection during pregnancy was established
Moderate IAV infection induced lung inflammation and blunted weight gain in dams
Maternal IAV infection caused mild pathology in the placenta without pup loss
Moderate gestational IAV infection did not induce fetal brain inflammation
Enhanced maternal IL-17A was not observed within the intestine nor in circulation
An IAV infection severity threshold may exist for inducing fetal neuroinflammation
Acknowledgements.
We would like to thank Sydney Schiff, Therese Rajasekera, Zachary Waite, and Hannah Rashidi for their assistance with animal care and tissue processing.
Funding.
This work was supported by the National Institutes of Health [T32 DE014320 to A.M.A. and K08 MH112892 to T.L.G.], The Ohio State University Infectious Disease Institute [idi.osu.edu; Interdisciplinary Research Seed Grant to T.L.G., A.M.A., J.S.Y. and Trainee Transformative Research Grant to A.M.A.], and The Ohio State University Institute for Behavioral Medicine Research [Pilot Grant to T.L.G., A.M.A]. The CPDISR is supported by The Ohio State University Comprehensive Cancer Center grant number P30CA016058. The funding sources had no role in study design; collection, analysis and interpretation of data; writing the report; nor the decision to submit the article for publication.
Footnotes
Declarations of Interest. The authors declare no interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Askovich PS, Sanders CJ, Rosenberger CM, Diercks AH, Dash P, Navarro G, Vogel P, Doherty PC, Thomas PG, Aderem A, 2013. Differential host response, rather than early viral replication efficiency, correlates with pathogenicity caused by influenza viruses. PLoS One 8, e74863. 10.1371/journal.pone.0074863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvier NM, Lowen AC, 2010. Animal Models for Influenza Virus Pathogenesis and Transmission. Viruses 2, 1530–1563. 10.3390/v20801530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braciale TJ, Sun J, Kim TS, 2012. Regulating the adaptive immune response to respiratory virus infection. Nat Rev Immunol 12, 295–305. 10.1038/nri3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M, Babulas VP, Susser ES, 2004. Serologic-evidence of prenatal influenza in the etiology of schizophrenia. Arch Gen Psychiat 61, 774–780. 10.1001/archpsyc.61.8.774 [DOI] [PubMed] [Google Scholar]
- Brown AS, Meyer U, 2018. Maternal immune activation and neuropsychiatric illness: A translational research perspective. Am. J. Psychiatry 10.1176/appi.ajp.2018.17121311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchrieser J, Degrelle SA, Couderc T, Nevers Q, Disson O, Manet C, Donahue DA, Porrot F, Hillion KH, Perthame E, Arroyo MV, Souquere S, Ruigrok K, Dupressoir A, Heidmann T, Montagutelli X, Fournier T, Lecuit M, Schwartz O, 2019. IFITM proteins inhibit placental syncytiotrophoblast formation and promote fetal demise. Science (80-. ). 365, 176–180. 10.1126/science.aaw7733 [DOI] [PubMed] [Google Scholar]
- Cadden KA, Walsh SW, 2008. Neutrophils, but not lymphocytes or monocytes, infiltrate maternal systemic vasculature in women with preeclampsia. Hypertens Pregnancy 27, 396–405. 10.1080/10641950801958067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M, 2014. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr Protoc Immunol 104, 15 25 1–15 25 14. 10.1002/0471142735.im1525s104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HJ, Antonson AM, Rajasekera TA, Patterson JM, Bailey MT, Gur TL, 2020. Prenatal stress causes intrauterine inflammation and serotonergic dysfunction, and long-term behavioral deficits through microbe- and CCL2-dependent mechanisms. Transl Psychiatry 10, 191. 10.1038/s41398-020-00876-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HJ, Gur TL, 2019. Intrauterine Microbiota: Missing, or the Missing Link? Trends Neurosci 42, 402–413. 10.1016/j.tins.2019.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, Hoeffer CA, Littman DR, Huh JR, 2016. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science (80-. ). 351, 933–939. 10.1126/science.aad0314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deriu E, Boxx GM, He X, Pan C, Benavidez SD, Cen L, Rozengurt N, Shi W, Cheng G, 2016. Influenza Virus Affects Intestinal Microbiota and Secondary Salmonella Infection in the Gut through Type I Interferons. PLoS Pathog 12, e1005572. 10.1371/journal.ppat.1005572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DA, Man JC, Brand P, Katstra JP, Sommerer K, Stone HA, Nardell E, Scheuch G, 2004. Inhaling to mitigate exhaled bioaerosols. Proc Natl Acad Sci U S A 101, 17383–17388. 10.1073/pnas.0408159101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espino YSS, Flores-Pliego A, Espejel-Nunez A, Medina-Bastidas D, Vadillo-Ortega F, Zaga-Clavellina V, Estrada-Gutierrez G, 2017. New Insights into the Role of Matrix Metalloproteinases in Preeclampsia. Int J Mol Sci 18. 10.3390/ijms18071448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes ML, McAllister AK, 2016a. Maternal immune activation: Implications for neuropsychiatric disorders. Science (80-. ). 353, 772–777. 10.1126/science.aag3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes ML, McAllister AK, 2016b. IMMUNOLOGY. Maternal TH17 cells take a toll on baby’s brain. Science (80-. ). 351, 919–920. 10.1126/science.aaf2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Earle J, Kanodia R, Kist D, Emamian ES, Patterson PH, Shi L, Sidwell R, 2002a. Prenatal viral infection leads to pyramidal cell atrophy and macrocephaly in adulthood: implications for genesis of autism and schizophrenia. Cell Mol Neurobiol 22, 25–33. 10.1023/a:1015337611258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Emamian ES, Kist D, Sidwell RW, Nakajima K, Akhter P, Shier A, Sheikh S, Bailey K, 1999. Defective corticogenesis and reduction in Reelin immunoreactivity in cortex and hippocampus of prenatally infected neonatal mice. Mol Psychiatry 4, 145–154. 10.1038/sj.mp.4000520 [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Emamian ES, Sidwell RW, Kist DA, Stary JM, Earle JA, Thuras P, 2002b. Human influenza viral infection in utero alters glial fibrillary acidic protein immunoreactivity in the developing brains of neonatal mice. Mol Psychiatry 7, 633–640. 10.1038/sj.mp.4001046 [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Folsom TD, Rooney RJ, Mori S, Kornfield TE, Reutiman TJ, Kneeland RE, Liesch SB, Hua K, Hsu J, Patel DH, 2012. The viral theory of schizophrenia revisited: abnormal placental gene expression and structural changes with lack of evidence for H1N1 viral presence in placentae of infected mice or brains of exposed offspring. Neuropharmacology 62, 1290–1298. 10.1016/j.neuropharm.2011.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Pearce DA, Brooks AI, Sidwell RW, 2005. Prenatal viral infection in mouse causes differential expression of genes in brains of mouse progeny: a potential animal model for schizophrenia and autism. Synapse 57, 91–99. 10.1002/syn.20162 [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Reutiman TJ, Folsom TD, Huang H, Oishi K, Mori S, Smee DF, Pearce DA, Winter C, Sohr R, Juckel G, 2008. Maternal infection leads to abnormal gene regulation and brain atrophy in mouse offspring: implications for genesis of neurodevelopmental disorders. Schizophr Res 99, 56–70. 10.1016/j.schres.2007.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke EM, Elgin TG, Gong H, Reese J, Gibson-Corley KN, Weiss RM, Zimmerman K, Bowdler NC, Kalantera KM, Mills DA, Underwood MA, McElroy SJ, 2018. Lipopolysaccharide-induced maternal inflammation induces direct placental injury without alteration in placental blood flow and induces a secondary fetal intestinal injury that persists into adulthood. Am J Reprod Immunol 79, e12816. 10.1111/aji.12816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbett KA, Hsiao EY, Kalman S, Patterson PH, Mirnics K, 2012. Effects of maternal immune activation on gene expression patterns in the fetal brain. Transl Psychiatry 2, e98. 10.1038/tp.2012.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves HT, Cuthbertson L, James P, Moffatt MF, Cox MJ, Tregoning JS, 2018. Respiratory Disease following Viral Lung Infection Alters the Murine Gut Microbiota. Front. Immunol. 9, 182. 10.3389/fimmu.2018.00182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur TL, Palkar AV, Rajasekera T, Allen J, Niraula A, Godbout J, Bailey MT, 2019. Prenatal stress disrupts social behavior, cortical neurobiology and commensal microbes in adult male offspring. Behav Brain Res 359, 886–894. 10.1016/j.bbr.2018.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur TL, Shay L, Palkar AV, Fisher S, Varaljay VA, Dowd S, Bailey MT, 2017. Prenatal stress affects placental cytokines and neurotrophins, commensal microbes, and anxiety-like behavior in adult female offspring. Brain Behav Immun 64, 50–58. 10.1016/j.bbi.2016.12.021 [DOI] [PubMed] [Google Scholar]
- Hameete BC, Fernández-Calleja JMS, de Groot MWGDM, Oppewal TR, Tiemessen MM, Hogenkamp A, de Vries RBM, Groenink L, 2021. The poly(I:C)-induced maternal immune activation model; a systematic review and meta-analysis of cytokine levels in the offspring. Brain, Behav. Immun. - Heal. 11, 100192. 10.1016/j.bbih.2020.100192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada S, Pirzadeh M, Carver KY, Deng JC, 2018. Respiratory Viral Infection-Induced Microbiome Alterations and Secondary Bacterial Pneumonia. Front. Immunol. 9, 2640. 10.3389/fimmu.2018.02640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding AT, Goff MA, Froggatt HM, Lim JK, Heaton NS, 2021. GPER1 is required to protect fetal health from maternal inflammation. Science (80-. ). 371, 271–276. 10.1126/science.aba9001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward AC, Fragaszy EB, Bermingham A, Wang L, Copas A, Edmunds WJ, Ferguson N, Goonetilleke N, Harvey G, Kovar J, Lim MS, McMichael A, Millett ER, Nguyen-Van-Tam JS, Nazareth I, Pebody R, Tabassum F, Watson JM, Wurie FB, Johnson AM, Zambon M, Flu Watch G, 2014. Comparative community burden and severity of seasonal and pandemic influenza: results of the Flu Watch cohort study. Lancet Respir Med 2, 445–454. 10.1016/S2213-2600(14)70034-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemann EA, Green R, Turnbull JB, Langlois RA, Savan R, Gale M Jr., 2019. Interferon-lambda modulates dendritic cells to facilitate T cell immunity during infection with influenza A virus. Nat Immunol 20, 1035–1045. 10.1038/s41590-019-0408-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humann J, Mann B, Gao G, Moresco P, Ramahi J, Loh LN, Farr A, Hu Y, Durick-Eder K, Fillon SA, Smeyne RJ, Tuomanen EI, 2016. Bacterial Peptidoglycan Traverses the Placenta to Induce Fetal Neuroproliferation and Aberrant Postnatal Behavior. Cell Host Microbe 19, 388–399. 10.1016/j.chom.2016.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K, Littman DR, 2009. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498. 10.1016/j.cell.2009.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, Littman DR, 2008. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4, 337–349. 10.1016/j.chom.2008.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR, 2006. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133. 10.1016/j.cell.2006.07.035 [DOI] [PubMed] [Google Scholar]
- Iwasaki A, Pillai PS, 2014. Innate immunity to influenza virus infection. Nat Rev Immunol 14, 315–328. 10.1038/nri3665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalish BT, Kim E, Finander B, Duffy EE, Kim H, Gilman CK, Yim YS, Tong L, Kaufman RJ, Griffith EC, Choi GB, Greenberg ME, Huh JR, 2020. Maternal immune activation in mice disrupts proteostasis in the fetal brain. Nat Neurosci 10.1038/s41593-020-00762-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanmaz HG, Erdeve O, Ogz SS, Uras N, Celen S, Korukluoglu G, Zergeroglu S, Kara A, Dilmen U, 2011. Placental transmission of novel pandemic influenza a virus. Fetal Pediatr Pathol 30, 280–285. 10.3109/15513815.2011.572956 [DOI] [PubMed] [Google Scholar]
- Kennedy RJ, Hoper M, Deodhar K, Erwin PJ, Kirk SJ, Gardiner KR, 2000. Interleukin 10-deficient colitis: new similarities to human inflammatory bowel disease 87, 1346–1351. 10.1046/j.1365-2168.2000.01615.x [DOI] [PubMed] [Google Scholar]
- Kenney AD, McMichael TM, Imas A, Chesarino NM, Zhang L, Dorn LE, Wu Q, Alfaour O, Amari F, Chen M, Zani A, Chemudupati M, Accornero F, Coppola V, Rajaram MVS, Yount JS, 2019. IFITM3 protects the heart during influenza virus infection. Proc Natl Acad Sci U S A 116, 18607–18612. 10.1073/pnas.1900784116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kentner AC, Bilbo SD, Brown AS, Hsiao EY, McAllister AK, Meyer U, Pearce BD, Pletnikov MV, Yolken RH, Bauman MD, 2019. Maternal immune activation: reporting guidelines to improve the rigor, reproducibility, and transparency of the model. Neuropsychopharmacology 44, 245–258. 10.1038/s41386-018-0185-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim H, Yim YS, Ha S, Atarashi K, Tan TG, Longman RS, Honda K, Littman DR, Choi GB, Huh JR, 2017. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature 549, 528–532. 10.1038/nature23910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneeland RE, Fatemi SH, 2013. Viral infection, inflammation and schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 42, 35–48. 10.1016/j.pnpbp.2012.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothary V, Doster RS, Rogers LM, Kirk LA, Boyd KL, Romano-Keeler J, Haley KP, Manning SD, Aronoff DM, Gaddy JA, 2017. Group B Streptococcus Induces Neutrophil Recruitment to Gestational Tissues and Elaboration of Extracellular Traps and Nutritional Immunity. Front Cell Infect Microbiol 7, 19. 10.3389/fcimb.2017.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammert CR, Frost EL, Bolte AC, Paysour MJ, Shaw ME, Bellinger CE, Weigel TK, Zunder ER, Lukens JR, 2018. Cutting Edge: Critical Roles for Microbiota-Mediated Regulation of the Immune System in a Prenatal Immune Activation Model of Autism. J. Immunol. 201, 845–850. 10.4049/jimmunol.1701755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liong S, Oseghale O, To EE, Brassington K, Erlich JR, Luong R, Liong F, Brooks R, Martin C, O’Toole S, Vinh A, O’Neill LAJ, Bozinovski S, Vlahos R, Papagianis PC, O’Leary JJ, Brooks DA, Selemidis S, 2020. Influenza A virus causes maternal and fetal pathology via innate and adaptive vascular inflammation in mice. Proc Natl Acad Sci U S A 117, 24964–24973. 10.1073/pnas.2006905117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mednick SA, Machon RA, Huttunen MO, Bonett D, 1988. Adult Schizophrenia Following Prenatal Exposure to an Influenza Epidemic. Arch Gen Psychiat 45, 189–192. 10.1001/archpsyc.1988.01800260109013 [DOI] [PubMed] [Google Scholar]
- Meyer U, 2014. Prenatal poly(I:C) exposure and other developmental immune activation models in rodent systems. Biol Psychiatry 75, 307–315. 10.1016/j.biopsych.2013.07.011 [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, 2012. To poly(I:C) or not to poly(I:C): advancing preclinical schizophrenia research through the use of prenatal immune activation models. Neuropharmacology 62, 1308–1321. 10.1016/j.neuropharm.2011.01.009 [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Fatemi SH, 2009. In-vivo rodent models for the experimental investigation of prenatal immune activation effects in neurodevelopmental brain disorders. Neurosci Biobehav Rev 33, 1061–1079. 10.1016/j.neubiorev.2009.05.001 [DOI] [PubMed] [Google Scholar]
- Miao H, Hollenbaugh JA, Zand MS, Holden-Wiltse J, Mosmann TR, Perelson AS, Wu H, Topham DJ, 2010. Quantifying the early immune response and adaptive immune response kinetics in mice infected with influenza A virus. J Virol 84, 6687–6698. 10.1128/JVI.00266-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newsome K, Alverson CJ, Williams J, McIntyre AF, Fine AD, Wasserman C, Lofy KH, Acosta M, Louie JK, Jones-Vessey K, Stanfield V, Yeung A, Rasmussen SA, 2019. Outcomes of infants born to women with influenza A(H1N1)pdm09. Birth Defects Res 111, 88–95. 10.1002/bdr2.1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikitin N, Petrova E, Trifonova E, Karpova O, 2014. Influenza virus aerosols in the air and their infectiousness. Adv Virol 2014, 859090. 10.1155/2014/859090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazos MA, Kraus TA, Munoz-Fontela C, Moran TM, 2012. Estrogen mediates innate and adaptive immune alterations to influenza infection in pregnant mice. PLoS One 7, e40502. 10.1371/journal.pone.0040502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presicce P, Park CW, Senthamaraikannan P, Bhattacharyya S, Jackson C, Kong F, Rueda CM, DeFranco E, Miller LA, Hildeman DA, Salomonis N, Chougnet CA, Jobe AH, Kallapur SG, 2018. IL-1 signaling mediates intrauterine inflammation and chorio-decidua neutrophil recruitment and activation. JCI Insight 3, e98306. 10.1172/jci.insight.98306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahat B, Sharma R, Bagga R, Hamid A, Kaur J, 2016. Imbalance between matrix metalloproteinases and their tissue inhibitors in preeclampsia and gestational trophoblastic diseases. Reproduction 152, 11–22. 10.1530/REP-16-0060 [DOI] [PubMed] [Google Scholar]
- Rasmussen SA, Jamieson DJ, Uyeki TM, 2012. Effects of influenza on pregnant women and infants. Am J Obs. Gynecol 207, S3–8. 10.1016/j.ajog.2012.06.068 [DOI] [PubMed] [Google Scholar]
- Shi L, Fatemi SH, Sidwell RW, Patterson PH, 2003. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci 23, 297–302. 10.1523/JNEUROSCI.23-01-00297.2003 %J The Journal of Neuroscience [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Tu N, Patterson PH, 2005. Maternal influenza infection is likely to alter fetal brain development indirectly: the virus is not detected in the fetus. Int J Dev Neurosci 23, 299–305. 10.1016/j.ijdevneu.2004.05.005 [DOI] [PubMed] [Google Scholar]
- Shin Yim Y, Park A, Berrios J, Lafourcade M, Pascual LM, Soares N, Yeon Kim J, Kim S, Kim H, Waisman A, Littman DR, Wickersham IR, Harnett MT, Huh JR, Choi GB, 2017. Reversing behavioural abnormalities in mice exposed to maternal inflammation. Nature 549, 482–487. 10.1038/nature23909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH, 2007. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci 27, 10695–10702. 10.1523/JNEUROSCI.2178-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanek J, 2019. Segmental villous mineralization: A placental feature of fetal vascular malperfusion. Placenta 86, 20–27. 10.1016/j.placenta.2019.07.011 [DOI] [PubMed] [Google Scholar]
- Suzuki R, Kohno H, Sugie S, Nakagama H, Tanaka T, 2006. Strain differences in the susceptibility to azoxymethane and dextran sodium sulfate-induced colon carcinogenesis in mice. Carcinogenesis 27, 162–169. 10.1093/carcin/bgi205 [DOI] [PubMed] [Google Scholar]
- Tan Q, Xu H, Xu F, Aguilar ZP, Yang Y, Dong S, Chen T, Wei H, 2013. Survival, distribution, and translocation of Enterococcus faecalis and implications for pregnant mice. FEMS Microbiol Lett 349, 32–39. 10.1111/1574-6968.12280 [DOI] [PubMed] [Google Scholar]
- Walker RW, Clemente JC, Peter I, Loos RJF, 2017. The prenatal gut microbiome: are we colonized with bacteria in utero? Pediatr Obes 12 Suppl 1, 3–17. 10.1111/ijpo.12217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Li F, Wei H, Lian ZX, Sun R, Tian Z, 2014. Respiratory influenza virus infection induces intestinal immune injury via microbiota-mediated Th17 cell-dependent inflammation. J Exp Med 211, 2397–2410. 10.1084/jem.20140625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JM, Elmore SA, Foley JF, 2012. Pathology methods for the evaluation of embryonic and perinatal developmental defects and lethality in genetically engineered mice. Vet. Pathol 10.1177/0300985811429811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber-Stadlbauer U, Meyer U, 2019. Challenges and opportunities of a-priori and a-posteriori variability in maternal immune activation models. Curr. Opin. Behav. Sci. 28, 119–128. 10.1016/j.cobeha.2019.02.006 [DOI] [Google Scholar]
- Wu WL, Hsiao EY, Yan Z, Mazmanian SK, Patterson PH, 2017. The placental interleukin-6 signaling controls fetal brain development and behavior. Brain Behav Immun 62, 11–23. 10.1016/j.bbi.2016.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz S, Mazel-Sanchez B, Kandasamy M, Manicassamy B, Schmolke M, 2018. Influenza A virus infection impacts systemic microbiota dynamics and causes quantitative enteric dysbiosis. Microbiome 6, 9. 10.1186/s40168-017-0386-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yockey LJ, Jurado KA, Arora N, Millet A, Rakib T, Milano KM, Hastings AK, Fikrig E, Kong Y, Horvath TL, Weatherbee S, Kliman HJ, Coyne CB, Iwasaki A, 2018. Type I interferons instigate fetal demise after Zika virus infection. Sci Immunol 3, eaao1680. 10.1126/sciimmunol.aao1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zani A, Zhang L, McMichael TM, Kenney AD, Chemudupati M, Kwiek JJ, Liu SL, Yount JS, 2019. Interferon-induced transmembrane proteins inhibit cell fusion mediated by trophoblast syncytins. J Biol Chem 294, 19844–19851. 10.1074/jbc.AC119.010611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Luo W, Huang P, Peng L, Huang Q, 2018. Maternal C-reactive protein and cytokine levels during pregnancy and the risk of selected neuropsychiatric disorders in offspring: A systematic review and meta-analysis. J Psychiatr Res 105, 86–94. 10.1016/j.jpsychires.2018.09.002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.