

Abstract
Background
Little is known about the relationship between lipoprotein (a) (Lp[a]) and high-sensitivity C-reactive protein (hsCRP) and their joint association with atherosclerotic cardiovascular disease (ASCVD).
Objective
To assess whether Lp(a)-associated ASCVD risk is modified by hsCRP in the context of primary prevention.
Methods
The current study included 4,679 participants from the Multi-Ethnic Study of Atherosclerosis (MESA) Apolipoprotein ancillary dataset. Cox proportional hazards models and Kaplan-Meier curves were used to assess the association between Lp(a), hsCRP, and time to CVD events.
Results
During a mean follow up of 13.6 years, 684 CVD events occurred. A significant interaction was observed between Lp(a) and hsCRP (P=0.04). With hsCRP < 2 mg/L, no significant CVD riskwas observed at any level of Lp(a) from < 50 mg/dL to above 100 mg/dL. However, with hsCRP ≥ 2 mg/L, a significant CVD risk was observed with Lp(a) of 50 to 99.9 mg/dL (HR, 1.36; 95%CI, 1.02–1.81) and Lp(a) ≥ 100 mg/dL (HR, 2.09; 95%CI, 1.40–3.13). Isolated elevations of either Lp(a) or hsCRP were not associated with increased CVD risk. In contrast, the combination of elevated Lp(a) (≥ 50 mg/dL) and hsCRP (≥ 2 mg/L) was independently associated with significant CVD risk (HR, 1.62; 95%CI, 1.25–2.10) and all-cause mortality (HR, 1.39; 95%CI, 1.12–1.72).
Conclusions
Lp(a)-associated ASCVD risk is observed only with concomitant elevation of hsCRP. Individuals with concomitant presence of elevated Lp(a) and systemic inflammation have greater ASCVD risk and all-cause mortality, thus may merit closer surveillance and more aggressive ASCVD risk management.
Keywords: ASCVD, Cardiovascular risk, high-sensitivity C-reactive protein (hsCRP), Inflammation, Lipoprotein(a), Multi-Ethic Study of Atherosclerosis (MESA)
CONDENSED ABSTRACT:
The current study tested the hypothesis that Lp(a)-associated ASCVD risk is modified by systemic inflammation in the general population. A mean of 13.6 years follow-up of 4,679 participants in the MESA cohort demonstrated that the risk of CVD was significantly associated with elevated Lp(a) only in the setting of hsCRP ≥ 2 mg/L. Individuals with concomitant increase in both Lp(a) and hsCRP had particularly higher ASCVD risk and all-cause mortality. These findings suggest that individuals with concomitant presence of elevated Lp(a) and systemic inflammation may merit closer surveillance and more aggressive ASCVD risk management.
Introduction
Significant declines in atherosclerotic cardiovascular disease (ASCVD) mortality have occurred over the last four decades (1). Despite this progress, cardiovascular disease remains the leading cause of death and disability globally accounting for about 17.6 million death worldwide in 2016 (1). Residual risk of atherosclerotic events remains high (2). Additional targets of therapy may be needed to further improve cardiovascular health and outcomes (3). Lipoprotein(a) [Lp(a)] is one such putative target (4). Lp(a) is an LDL-like particle with an additional glycoprotein, apolipoprotein(a), covalently bound to its apolipoprotein B moiety (5). Therefore, Lp(a) is an atherogenic, pro-inflammatory, and pro-thrombotic lipoprotein particle. Epidemiologic, experimental, and genetic studies suggest a causal relationship between elevated Lp(a) and risk for myocardial infarction (MI), stroke, and calcific aortic valve stenosis (6–10).
Inflammation is another emerging target of therapy to potentially reduce residual ASCVD risk. Results of recent large randomized controlled trials demonstrated improved cardiovascular outcomes with specific anti-inflammatory therapies (13,14). Among the numerous inflammatory biomarkers, high-sensitivity C-reactive protein (hsCRP) has the most extensive predictive validation with regard to ASCVD outcomes (15). Lp(a) and hsCRP are both listed as risk-enhancers in the 2018 American Heart Association (AHA) / American College of Cardiology (ACC) Cholesterol guideline (16) and the 2019 ACC/AHA guideline on primary prevention of cardiovascular disease (17). A recent post hoc analysis of the ACCELERATE trial (Assessment of Clinical Effects of Cholesteryl Ester Transfer Protein Inhibition with Evacetrapib in Patients at a High Risk for Vascular Outcomes) reported that Lp(a) was associated with greater risk of MACE (major adverse cardiovascular events) only when hsCRP was ≥ 2mg/L (18). It is not known whether the Lp(a)-associated ASCVD risk is modified by inflammation as measured by plasma hsCRP in primary prevention. The purpose of the current study was to examine the relationship between Lp(a), subclinical inflammation, and their joint association with ASCVD in a population free of clinical ASCVD at baseline.
Methods and Materials
Study Population
The study design of Multi-Ethnic Study of Atherosclerosis (MESA) has been previously described by Bild et al. (19). More protocol information about the MESA study can be found at www.mesa-nhlbi.org. Briefly, MESA recruited 6,814 asymptomatic men and women aged 45–84 years without overt clinical CVD from six communities in the United States. Informed consent and Institutional Review Board approval was completed at all MESA participating sites. A total of six exams have been completed since 2000.
The current study included 4,679 participants from the MESA Apolipoprotein ancillary dataset with the following race/ethnic distribution: Caucasian (n=1,709, 36.6%), Chinese-American (n=560, 12%), African-American (n=1,346, 28.7%), and Hispanic (n=1,064, 22.7%). Of this study population, 4654 participants had measurements of both Lp (a) and hsCRP and were included in the analysis for this paper.
Demographics and Baseline Characteristics
Age, gender, race/ethnicity, smoking status, education, medical history, family history, and medication use data were collected by standard questionnaires. Diabetes mellitus was defined as fasting glucose ≥ 126 mg/dL or hypoglycemic medication use. Body mass index (BMI) was calculated as weight (in kg) / height in meters-squared (m2). Resting blood pressure was repeated three times after five minutes in the seated position using a Dinamap automated oscillometric sphygmomanometer (Critikon, Tampa, Florida) and the average of the second and third readings was used in analyses.
Laboratory Measurements
Lp(a) mass was determined in plasma using a latex-enhanced turbidimetric immunoassay (Denka Seiken, Tokyo, Japan) at Health Diagnostics Laboratory (Richmond, Virginia). Total cholesterol (TC), high-density lipoprotein (HDL)-cholesterol (HDL-C), and triglyceride were measured using blood samples collected after overnight fasting. Low density lipoprotein (LDL)-cholesterol (LDL-C) was calculated using the Friedewald equation in specimens with triglycerides < 400 mg/dL (20), 43 participants with triglyceride > 400 mg/dl were assigned missing values. Serum creatinine was measured by rate reflectance spectrophotometry using thin film adaptation of the creatine amidinohydrolase method on the Vitros analyzer (Johnson & Johnson Clinical Diagnostics, Inc., Rochester, NY 14650) and estimated Glomerular Filtration Rate (eGFR) was calculated using the creatinine-based CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) equation. hsCRP was measured by a BNII nephelometer using a particle enhanced immunoelpolometric assay (N High Sensitivity CRP; Dade Behring Inc., Deerfield, IL).
Clinical Endpoints
Incident cardiovascular events were recorded from baseline (2000) through calendar year 2017. Participants were followed for a mean of 13.6 years (standard deviation of 4.43 years). The cardiovascular events analyzed in this study include myocardial infarction, fatal and nonfatal coronary heart disease (CHD), definite angina, and probable angina if followed by revascularization, resuscitated cardiac arrest, fatal and nonfatal stroke, and other atherosclerotic or CVD death. Telephone interviews were conducted at 9–12 month-intervals for each participant to obtain data regarding interim hospitalizations, outpatient cardiovascular procedures and diagnoses, and deaths. Additional medical encounters were identified through cohort clinic visits, call-ins, medical record abstractions or obituaries. Copies of all death certificates and medical records of hospitalizations and selected outpatient cardiovascular procedures and diagnoses were collected to verify self-reported diagnoses (19).
Statistical Analysis
Continuous normally distributed variables were reported as mean and standard deviation (SD). Variables with skewed distributions were reported as median and interquartile range (IQR). Categorical variables were reported as frequency and percentage (%). Log transformed values of Lp(a) and hsCRP were used when they were included as continuous variables in statistical models (18,21). Elevated hsCRP was defined as ≥ 2 mg/L, consistent with the current ACC/AHA Cholesterol guideline (17). Lp(a) was also examined as a categorical variable by commonly used clinical cut points (50 mg/dL and 100 mg/dL) (22) or quartiles. Risk of CVD was also compared among four risk groups classified by elevation of none, one or both of risk factors (Lp[a] and hsCRP). Differences in baseline characteristics were compared using the Chi-square test for categorical variables and an unpaired Student’s t-test or Mann-Whitney U test for continuous variables. The time-to-ASCVD across Lp(a) categories by hsCRP threshold was evaluated using Kaplan Meier curves. The cumulative incidence of events during the follow-up period was computed. Multivariable Cox proportional hazards models were used to assess the relationship between Lp(a) and endpoints. Formal interaction test between Lp(a) and hsCRP was performed. Model adjustments were made for age, gender, ethnicity, hypertension, use of antihypertensive medication, diabetes, smoking status (never; former; current), HDL-C, triglycerides, total cholesterol and renal function as assessed by eGFR. Sensitivity analysis was performed by adjusting to corrected LDL-cholesterol (total LDL – 30% Lp(a)). The proportional hazard assumption was assessed by the ASSESS statement with the Ph option and also by the Schoenfeld residuals method.
Given the observation that median levels and distributions of Lp(a) differ significantly between African-American and other ethnic groups, a subgroup analysis was performed to compare the relationship of Lp(a) and hsCRP to CVD in African-Americans and non-African-American. A subgroup analysis was also conducted to explore this relationship across sex. Statistical analysis was performed using SAS, version 9.4 (SAS Institute Inc). Statistical significance was defined as two-tailed P value < 0 .05, except for interaction testing where a P value less than 0.2 was considered significant. We pre-specified 0.2 as the cut-off to be conservative in terms of determining if there was an interaction between Lp(a) and hsCRP and justifying more detailed inspection on its potentially important impact on clinical outcomes.
Results
Baseline Characteristics of Study Population
Baseline characteristics were presented by hsCRP thresholds (Table 1). The mean age of participants in this analysis was 62 years (52.5% females). Of the study population, 36.6% were Caucasians, 28.7% were African American, 22.7% were Hispanic, and 12% were Asian. Median Lp(a) and hsCRP were 17.85 mg/dl and 1.94 mg/L, respectively. Elevated hsCRP was associated with higher levels of Lp(a), triglycerides, total cholesterol and BMI. Additionally, there was a higher prevalence of female gender, hypertension, and use of antihypertensive medications, diabetes and current smoking among those with hsCRP ≥ 2 mg/L. African-American participants manifested a significantly higher median Lp(a) level of 35mg/dL compared with median values of approximately 13mg/dL in other ethnicities, consistent with prior reports (21) (Online Figure 1).
Table 1.
Baseline Characteristics of the Study Population Classified by high-sensitivity C - reactive protein
| hsCRP<2mg/L (n=2,388) | hsCRP≥2mg/L (n=2,273) | Total (n=4,661) | P value | |
|---|---|---|---|---|
| Age, mean (SD),y | 61.7 (10.7) | 62.2 (10.0) | 62 (10.4) | 0.1 |
| Female gender (%) | 43.2 | 62.4 | 52.5 | <0.001 |
| BMI, mean (SD) | 26.3 (4.4) | 30.2 (5.8) | 28.2 (5.5) | <0.001 |
| Ethnicity (%) | <0.001 | |||
| Caucasian | 38.8 | 34.2 | 36.6 | |
| Chinese American | 18.1 | 5.6 | 12.0 | |
| African-American | 23.7 | 34.0 | 28.7 | |
| Hispanic | 19.4 | 26.3 | 22.7 | |
| hsCRP, median (IQR), mg/L | 0.88 (0.53–1.33) | 4.44 (3.02–7.93) | 1.94 (0.86–4.32) | <0.001 |
| Lipids, mg/dL | ||||
| Lp(a), median (IQR) | 16 (7.7,37.2) | 20 (8.5,42.5) | 17.85 (8.2–40) | 0.001 |
| LogLp(a), mean (SD) | 1.19 (0.50) | 1.25 (0.53) | 1.22 (0.52) | <0.001 |
| LDL-C, mean (SD) | 119 (31) | 120 (32) | 120 (31) | 0.2 |
| HDL-C, mean (SD) | 52 (15.2) | 50 (14.9) | 51 (15.1) | 0.33 |
| Triglycerides, median (IQR) | 101 (17,147) | 116 (82,169) | 109 (76,158) | <0.001 |
| Total cholesterol, mean (SD) | 195 (34) | 198 (36) | 196 (35.5) | 0.01 |
| HTN (%) | 34.9 | 48.8 | 41.7 | <0.001 |
| HTN therapy (%) | 27.4 | 39.1 | 33.1 | <0.001 |
| Lipid lowering therapy (%) | 0.13 | 0.09 | 0.1 | 0.69 |
| Diabetes (%) | 9.8 | 14.0 | 11.9 | <0.001 |
| Current smoker (%) | 10.9 | 15.2 | 13.0 | <0.001 |
| eGFR, mean(SD), mL/min/1.73m2 | 75.6 (15.1) | 74.7 (16.6) | 75 (15.8) | 0.08 |
SD, standard deviation; IQR, interquartile range; BMI, body mass index; hsCRP, high-sensitivity C-reactive protein; Lp(a), lipoprotein(a); LogLp(a), log transformed lipoprotein(a); LDL-C, low-density lipoprotein; HDL-C, high-density lipoprotein cholesterol; HTN, hypertension; eGFR, estimated glomerular filtration rate.
Association between Cardiovascular Events and Lp(a) according to hsCRP thresholds
Table 2 displays the results of the Cox proportional hazard models evaluating the association between risk of CVD events and Lp(a) by log-transformed (LogLp[a]) or 50mg/dL threshold. In the total study population, Lp(a) was significantly associated with CVD assessed by either per unit increase of LogLp(a) (hazard ratio[HR]=1.18; 95% confidence interval[CI]=1.00–1.41; P=0.05), or categorical threshold of 50 mg/dL (HR, 1.36; 95%CI, 1.10–1.65; P=0.001). A significant interaction was observed between Lp(a) and hsCRP tested as log-transformed (LogCRP)(P for interaction =0.04, and 0.08 when hsCRP was tested by 2mg/L threshold ) (Table 2). In those with hsCRP < 2 mg/L, there was not a significant association between risk of CVD events and Lp(a) when evaluated either by LogLp(a) (HR, 1.02; 95%CI, 0.81–1.27) or categorical threshold of 50mg/dL (HR, 1.19; 95%CI, 0.89–1.58)]. On the other hand, in those with hsCRP ≥ 2 mg/L, elevation of Lp(a), either by log unit increase (HR, 1.32; 95%CI, 1.05–1.65; P=0.016) or categorical threshold of 50mg/dL (HR, 1.52; 95%CI, 1.18–1.95; P=0.001)] was significantly associated with risk of CVD events.
Table 2.
Lipoprotein(a)-associated Cardiovascular Risk according to high-sensitivity C-Reactive Protein Threshold
| Groups | Events/Total (%) | HR(95%CI) | P value* |
|---|---|---|---|
| In entire cohort | |||
| LogLp(a), per unit | 676/4611(14.7) | 1.18(1.00–1.41) | 0.05 |
| Lp(a)<50mg/dL | 528/3764(14) | 1[Reference] | NA |
| Lp(a)≥50mg/dL | 156/889(17.5) | 1.36(1.10–1.65) | 0.001 |
|
| |||
| hsCRP<2mg/L | |||
| LogLp(a), per unit | 335/2379(14) | 1.02(0.81–1.27) | 0.88 |
| Lp(a)<50mg/dL | 268/1960(13.7) | 1[Reference] | NA |
| Lp(a)≥50mg/dL | 66/417(15.8) | 1.19(0.89–1.58) | 0.23 |
|
| |||
| hsCRP≥2mg/L | |||
| LogLp(a), per unit | 348/2263(15.4) | 1.32(1.05–1.65) | 0.016 |
| Lp(a)<50mg/dL | 256/1788(14.2) | 1[Reference] | NA |
| Lp(a)≥50mg/dL | 90/470(19.1) | 1.52(1.18–1.95) | 0.001 |
Elevation of Lp(a), either by log unit increase or above the cut-off of 50mg/dL was associated with significant CVD risk only in the setting of hsCRP≥2mg/L. HR, hazard ratio; CI, Confidence Interval; NA, not applicable; Lp(a), lipoprotein(a); LogLp(a), log transformed Lp(a); Multivariable Cox proportional hazards regression model was adjusted for age, gender, ethnicity, hypertension, use of hypertension medications, diabetes, smoking status, HDL-C, triglycerides, total cholesterol and renal function (eGFR).
P-value for interaction: LogLp(a)*LogCRP= 0.04; LogLp(a)*hsCRP(<2 or ≥2) =0.08.
Risk of Cardiovascular Events across Lp(a) Strata according to hsCRP thresholds
Figure 1A shows the results of Cox proportional hazard model exploring the risk of CVD events across Lp(a) strata (< 50 mg/dL, 50–99.9 mg/dL and ≥ 100 mg/dL). Again, in the setting of hsCRP < 2 mg/L, no significant association between Lp(a) and CVD risk was observed in any of the Lp(a) categories. However, in the setting of elevated hsCRP (≥ 2 mg/L), a significant risk of CVD events was observed with increased levels of Lp(a) 50 to 99.9mg/dL (HR, 1.36; 95%CI, 1.02–1.81; P=0.03) and Lp(a) ≥ 100 mg/dL (HR, 2.09; 95%CI, 1.40–3.13; P<0.001).
Figure 1. Cardiovascular Risk across Clinical Strata of Lipoprotein(a).
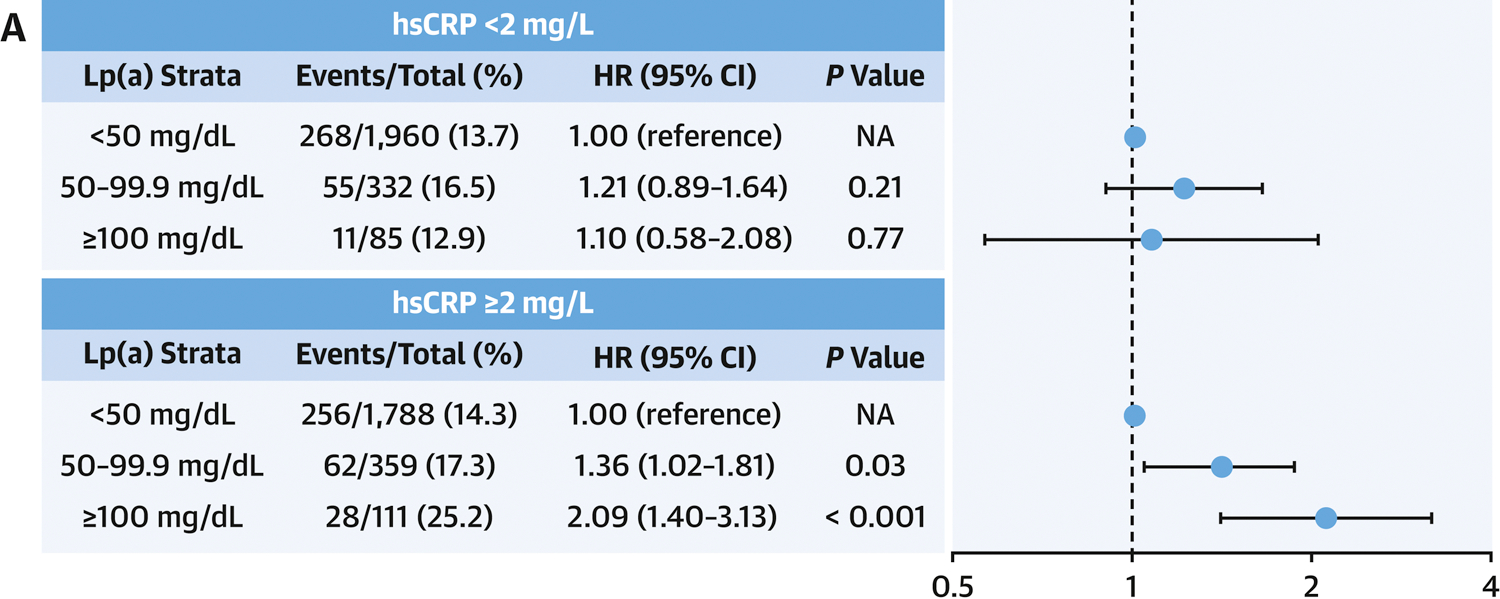

A: Hazard Ratios for Cardiovascular Events. Cox proportional hazards model was adjusted for age, gender, race/ethnicity, hypertension, use of hypertension medications, diabetes, smoking status, HDL-C, triglycerides, total cholesterol and renal function (eGFR). Point estimates of hazard ratios are represented by the black dot and 95%CI by the horizontal lines. B: Kaplan-Meier curves of Cumulative Incidence of Cardiovascular Events. Lp(a)<50mg/L was used as the reference group. Lp(a), lipoprotein(a); hsCRP, high-sensitivity C-reactive protein; HR, hazard ratio; CI, Confidence Interval; NA, not applicable.
Figure 1B displays the cumulative incidence of CVD events over a mean follow-up of 13.6 years across Lp(a) strata according to hsCRP thresholds. In those with hsCRP < 2 mg/L, increasing Lp(a) was not associated with a higher cumulative incidence of CVD (P =0.41). However, in those with hsCRP ≥ 2 mg/L, cumulative incidence of CVD was significantly higher in the Lp(a) 50–99.9 mg/dL group, and highest in those with Lp(a) ≥ 100 mg/dL (P =0.001). Similar results were found while Lp(a) was assessed by quartiles (Online Figure 2).
Risk of Cardiovascular Events and All-cause Mortality in Different Risk Groups Classified by Lp(a) and hsCRP
Risk of CVD was then compared by classifying participants to four risk groups: elevation of none, one or both risk factors (Lp(a) and hsCRP). Figure 2 displays the results of Cox proportional hazard models and Kaplan-Meier curves of the cumulative incidence of CVD events over a mean of 13.6 years follow up, grouped by Lp(a) and hsCRP thresholds. Compared with the reference group (Lp(a) < 50 mg/dL and hsCRP < 2 mg/L), an isolated elevation of either Lp(a) ( ≥ 50 mg/dL) or hsCRP ( ≥ 2 mg/L) was not associated with a higher risk of CVD events. However, concomitant elevation in both Lp(a) (≥ 50 mg/dL) and hsCRP (≥ 2 mg/L) was associated with a significantly increased risk of CVD events (HR, 1.62; 95%CI, 1.25–2.10; P<0.001). Given the inherent relationship between Lp(a), LDL-cholesterol and CVD risk, a sensitivity analysis was performed by adjusting to corrected LDL-cholesterol (total LDL – 30% Lp(a)) in the Cox proportional hazard model. And the results were consistent as shown in Online Table 1. When coronary heart disease and stroke were analyzed as separate endpoints, greater hazard ratios were consistently observed in the group with Lp(a)≥50mg/dL and hsCRP≥2mg/L (Online Table 2A, 2B and Figure 3). Additionally, dual-elevation of Lp(a) and hsCRP was associated with significantly higher all-cause mortality (HR, 1.39; 95%CI, 1.12–1.72; P<0.01) (Online Table 2C).
Figure 2. Cardiovascular Risk Classified by Lipoprotein(a) and high-sensitivity C-Reactive Protein.
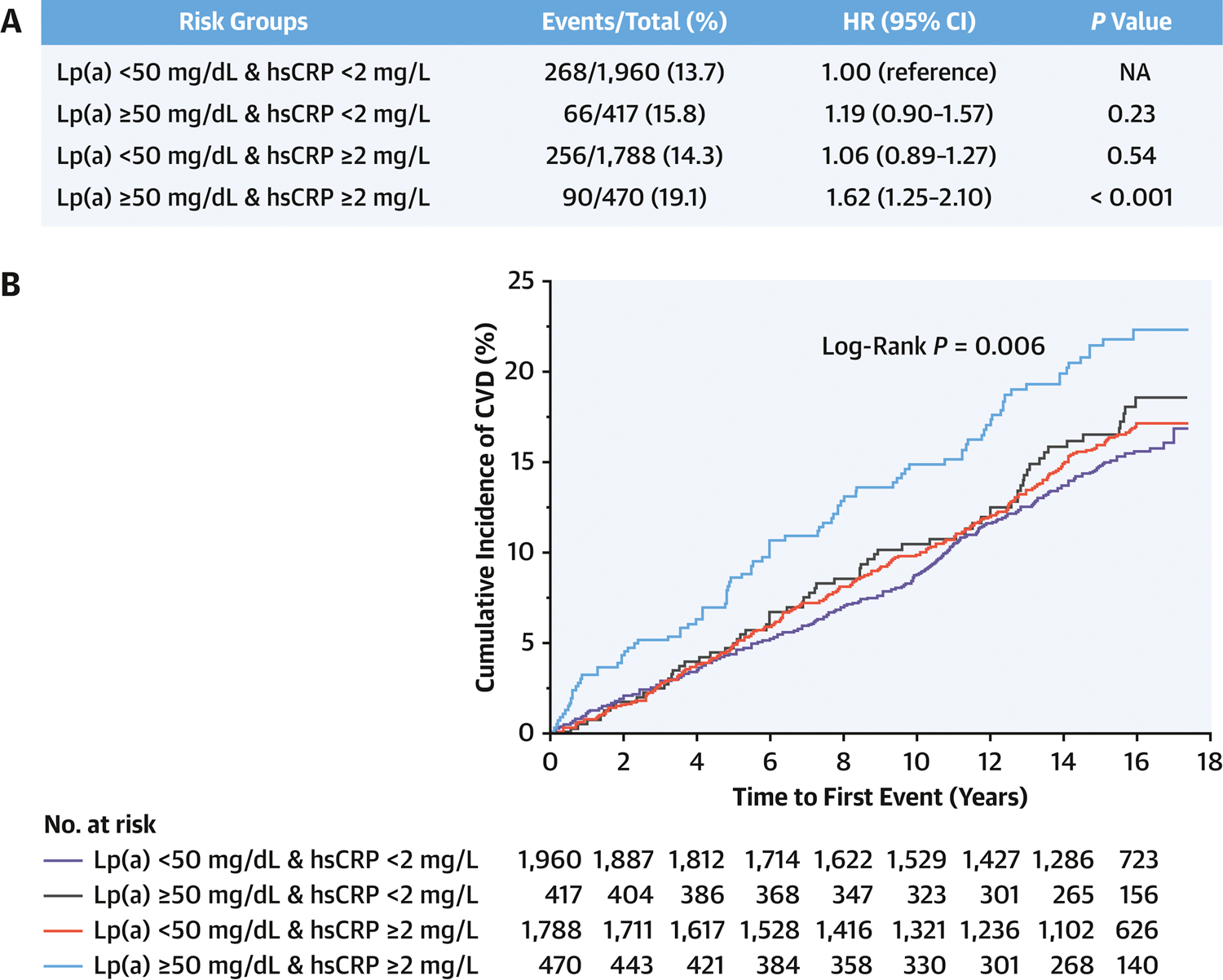
A: Hazard Ratios for Cardiovascular Events. Cox proportional hazards model was adjusted for age, gender, race/ethnicity, hypertension, use of hypertension medications, diabetes, smoking status, HDL-C, triglycerides, total cholesterol and renal function (eGFR). B: Kaplan-Meier curves of Cumulative Incidence of Cardiovascular Events. Lp(a), lipoprotein(a); hsCRP, high-sensitivity C-reactive protein; HR, hazard ratio; CI, Confidence Interval; NA, not applicable; Lp(a)<50mg/dL & hsCRP<2mg/L was used as the reference group. P-value for interaction between Lp(a) and hsCRP =0.04 (as continuous variable) or 0.17 (as a dichotomous variable).
Figure 3. Cumulative Incidence of Cardiovascular Events in African-Americans and Non-African-Americans.
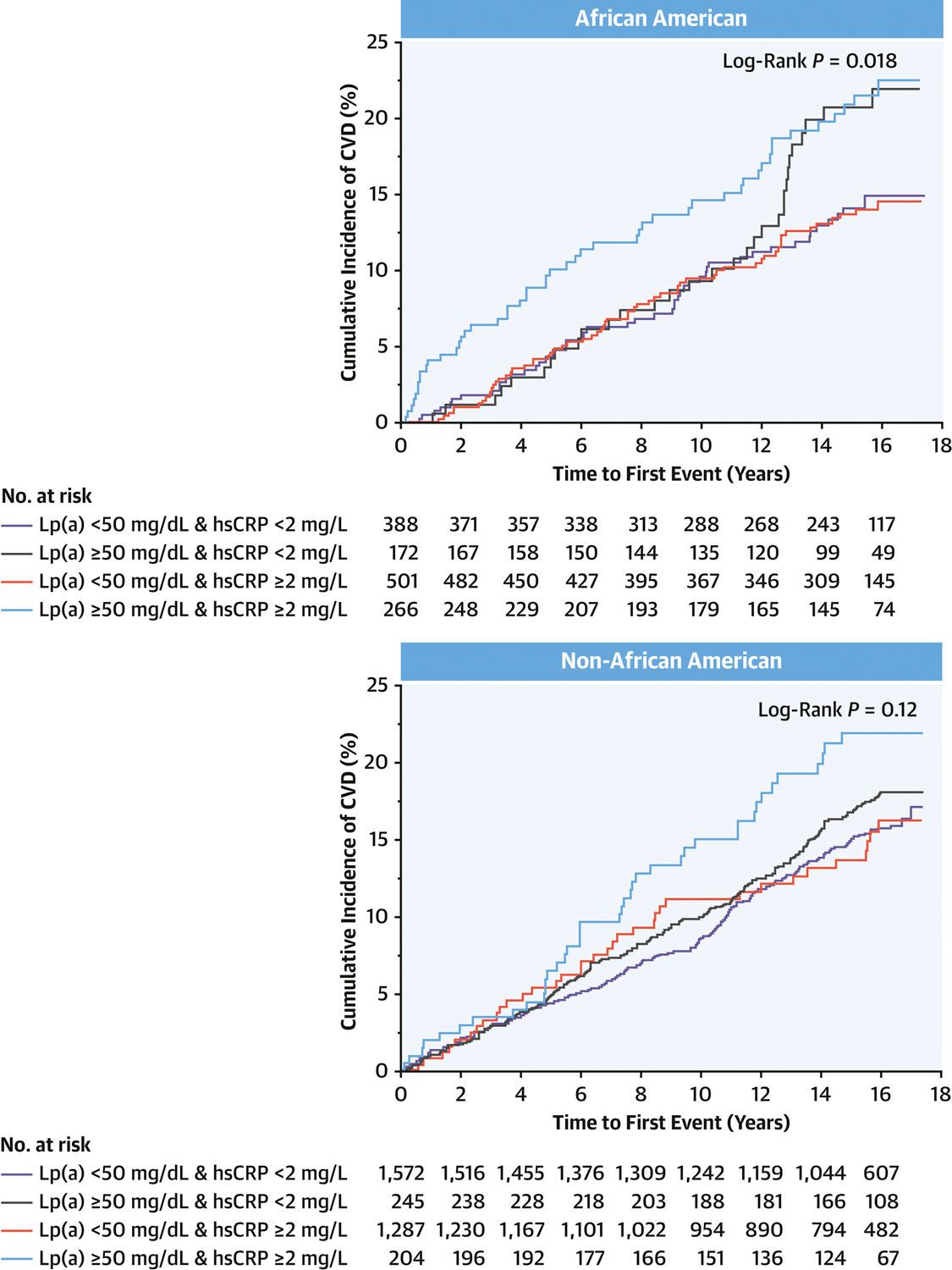
Kaplan-Meier curves of the cumulative incidence of CVD events are shown. In both African-American and Non-African-American, greater cumulative risk of CVD was observed only in individuals with concomitant elevation of Lp(a) (≥ 50 mg/dL) and hsCRP (≥ 2 mg/L).
Association between Cardiovascular Events and Lp(a) According to hsCRP in African-Americans and Non-African-Americans
Given the observation that median levels and distributions of Lp(a) differ significantly between African-Americans and other ethnic groups (Online Table 3 and Online Figure 1), a subgroup analysis was conducted by categorizing the study population to African-American (AA) and Non-African-American (Table 3 and Figure 3). P value for interaction was 0.03 and 0.2 in AA and non-AA, respectively. P value for interaction was 0.42 and 0.37 when Lp(a) was tested by 50mg/dL threshold and hsCRP by 2mg/L threshold in AA and non-AA, respectively. In both African-Americans and non-African-Americans, elevation of either Lp(a) ( ≥ 50 mg/dL) or hsCRP ( ≥ 2 mg/L) alone was not associated with a higher risk of CVD events compared with the reference group (Lp(a) < 50 mg/dL and hsCRP < 2 mg/L). The greatest risk of CVD events was observed in those with concomitant elevation in Lp(a) (≥ 50 mg/dL) and hsCRP (≥ 2 mg/L) (HR, 1.63; 95%CI, 1.09–2.44; P=0.016 and HR, 1.66; 95%CI, 1.17–2.35; P=0.005 for AA and Non-AA, respectively).
Table 3.
Hazard Ratio for Cardiovascular Events By Race
| African-American | Non-African-American | |||||
|---|---|---|---|---|---|---|
|
|
||||||
| Risk groups | Events/Total (%) | HR(95%CI) | P-value | Events/Total (%) | HR(95%CI) | P-value |
| Lp(a)<50mg/dL & hsCRP<2mg/L | 49/388(12.6) | 1[Reference] | NA | 219/1572(13.9) | 1[Reference] | NA |
| Lp(a)≥50mg/dL & hsCRP<2mg/L | 31/172(19) | 1.26(0.80–2.00) | 0.32 | 35/245(14.3) | 1.15(0.80–1.66) | 0.44 |
| Lp(a)<50mg/dL & hsCRP≥2mg/L | 61/501(12.2) | 0.99(0.68–1.45) | 0.97 | 195/1287(15.2) | 1.13(0.92–1.37) | 0.24 |
| Lp(a)≥50mg/dL & hsCRP≥2mg/L | 50/266(18.8) | 1.63(1.09–2.44) | 0.016 | 40/204(19.6) | 1.66(1.17–2.35) | 0.005 |
In both African-American and Non-African-American, elevated Lp(a) ≥50mg/dL was associated with greater cardiovascular risk only with concomitant elevation of hsCRP ≥2mg/L. Lp(a), lipoprotein(a); hsCRP, high-sensitivity C-reactive protein; HR, hazard ratio; CI, Confidence Interval; NA, not applicable; Cox proportional hazards model was adjusted for age, gender, hypertension, use of hypertension medications, diabetes, smoking status, HDL-C, triglycerides, total cholesterol and renal function (eGFR). P for interaction between Lp(a) and hsCRP =0.03 in African-American and 0.2 in Non-African-American.
Association between Cardiovascular Events and Lp(a) According to hsCRP by Gender
Online Table 4 demonstrates that males had relatively lower median levels of Lp(a) and hsCRP than females. Table 4 and Central Illustration display the results of the Cox proportional hazard models and Kaplan-Meier curves of the cumulative incidence of CVD events by gender. P value for interaction was 0.04 and 0.47 in female and male, respectively. P value for interaction was 0.09 and 0.85when Lp(a) was tested by 50mg/dL threshold and hsCRP by 2mg/L threshold , respectively. Compared with the reference group (Lp(a) < 50 mg/dL and hsCRP < 2 mg/L), the only significantly increased risk of CVD events was observed amongst participants with dual elevations of Lp(a) and hsCRP in both females and males (HR, 1.52; 95%CI, 1.06–2.18; P=0.02 and HR, 1.61; 95%CI, 1.12–2.31; P=0.01, respectively). Isolated elevation of either Lp(a) or hsCRP had higher cumulative incidence of CVD events in males, but this association was not statistically significant.
Table 4.
Hazard Ratio for Cardiovascular Events By Gender
| Female | Male | |||||
|---|---|---|---|---|---|---|
|
|
||||||
| Risk groups | Events/Total(%) | HR(95%CI) | P-value | Events/Total(%) | HR(95%CI) | P-value |
| Lp(a)<50mg/dL & hsCRP<2mg/L | 81/820(9.9) | 1[Reference] | NA | 187/1140(16.4) | 1[Reference] | NA |
| Lp(a)≥50mg/dL & hsCRP<2mg/L | 21/204(10.3) | 0.91(0.55–1.50) | 0.71 | 45/213(21.1) | 1.35(0.97–1.87) | 0.08 |
| Lp(a)<50mg/dL & hsCRP≥2mg/L | 115/1078(10.7) | 0. 95(0.71–1.27) | 0.71 | 141/710(19.9) | 1.12(0.90–1.40) | 0.31 |
| Lp(a)≥50mg/dL & hsCRP≥2mg/L | 53/333(15.9) | 1.52(1.06–2.18) | 0.02 | 37/137(27) | 1.61(1.12–2.31) | 0.01 |
In both female and male, elevated Lp(a) ≥50mg/dL was associated with greater cardiovascular risk only with concomitant elevation of hsCRP ≥2mg/L. Lp(a), lipoprotein(a); hsCRP, high-sensitivity C-reactive protein; HR, hazard ratio; CI, confidence interval; NA, not applicable; Cox proportional hazards model was adjusted for age, race/ethnicity, hypertension, use of hypertension medications, diabetes, smoking status, HDL-C, triglycerides, total cholesterol and renal function (eGFR).
P-value for interaction between Lp(a) and hsCRP =0.04 and 0.47 in female and male, respectively.
Central Illustration. Cumulative Incidence of Cardiovascular Events in Female and Male.
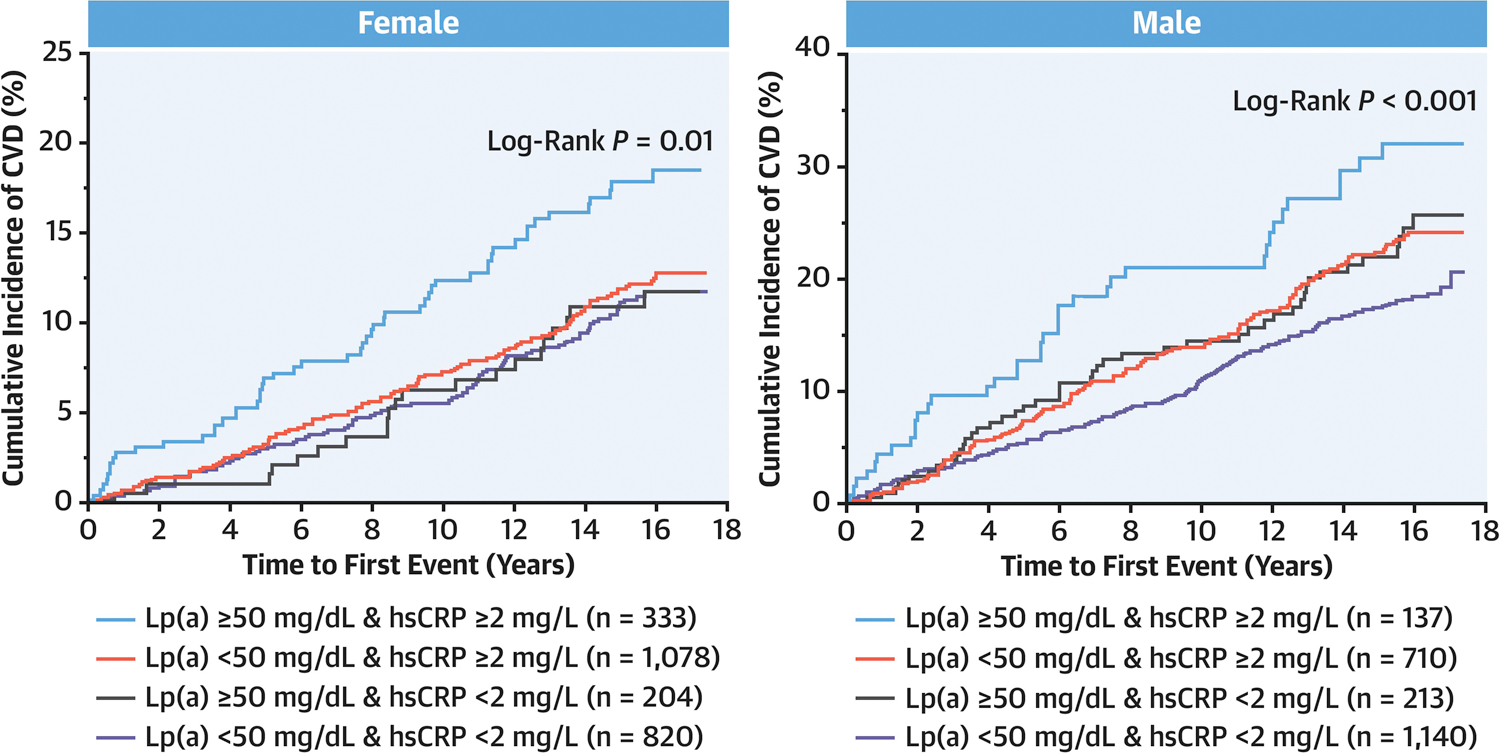
Kaplan-Meier curves of the cumulative incidence of CVD events are shown. In both female and male, greater cumulative risk of CVD was observed only in individuals with concomitant elevation of Lp(a) (≥ 50 mg/dL) and hsCRP (≥ 2 mg/L). Lp(a), lipoprotein(a); hsCRP, high-sensitivity C-reactive protein.
Discussion
Despite optimal management of traditional risk factors, including aggressive LDL-C lowering therapy, significant residual cardiovascular risk remains. There are many drivers of residual risk, including inflammatory, pro-thrombotic and metabolic pathways (3). Lp(a) and hsCRP are established biomarkers that can facilitate ASCVD risk estimation in primary prevention according to the 2018 AHA/ACC Cholesterol guideline (16) and the 2019 ACC/AHA guideline on primary prevention of cardiovascular disease (17). A recent post hoc analysis suggested hsCRP may modulate Lp(a)-associated CVD risk in the ACCELERATE trial which is essentially in a secondary prevention population (18). However, it is not known whether the ASCVD risk associated with elevated Lp(a) is modified by the presence of inflammation as measured by plasma hsCRP in general population. The purpose of the current study was to determine whether subclinical inflammation modifies Lp(a)-associated cardiovascular risk in a population without established clinical ASCVD at baseline. Several key features of MESA, including large sample size, participation amongst multiple ethnic groups, and long follow-up duration allow this question to be addressed in a robust fashion.
In this analysis of over 4,600 MESA participants, a significant interaction was observed between Lp(a) and hsCRP. Lp(a) was associated with incident CVD events over a mean of 13.6 years follow-up only in those with elevated hsCRP (≥2mg/L). In participants without elevation in hsCRP (<2mg/L), no significant risk of CVD events was observed across any level of Lp(a), from below 50mg/dL to above 100mg/dL. Compared with those who had neither elevated Lp(a) nor elevated hsCRP, the presence of either elevated Lp(a) or hsCRP was not significantly associated with an increased risk of CVD events. However, concomitant elevation of both Lp(a) and hsCRP was associated with a significantly higher risk of CVD events. Similar impacts of dual elevations in hsCRP and Lp(a) were observed in both African-Americans and Non-African-Americans and in both genders. Finally, dual elevation of Lp(a) and hsCRP was associated with significantly higher risk of all-cause mortality.
Epidemiologic, genetic, and post-hoc analyses of clinical trials suggest that Lp(a) plays a causal role in the development of MI, stroke, and calcific aortic valve stenosis (6–9). A meta-analysis of multiple statin trials including 95,576 person-years at risk and 5,751 events revealed a linear association between elevated Lp(a) level and ASCVD, with risk being particularly high among patients with Lp(a) levels greater than 50 mg/dL (8). Plasma Lp(a) levels are largely determined by the LPA gene locus with minimal effects from lifestyle or environmental factors (25). Recently, an antisense oligonucleotide, pelacarsen, was developed to silence the expression of the apolipoprotein(a) gene, LPA (11). In a phase IIB trial, pelacarsen reduced plasma Lp(a) by up to 80% (12). Whether Lp(a) lowering with this strategy will improve cardiovascular outcomes in patients with established CVD and elevated Lp(a) is currently being evaluated in the Lp(a) HORIZON trial (Assessing the Impact of Lipoprotein(a) Lowering with TQJ230 on Major Cardiovascular Events in Patients with CVD) (https://clinicaltrials.gov/ct2/show/NCT04023552).
Systemic inflammation, frequently found as a pathophysiological feature of metabolic syndrome, has been recognized as a major component of residual cardiovascular risk (3,26). Recent trials on the Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) inhibitors also demonstrated persistent residual inflammatory CVD risk among patients with stable ASCVD despite treatment with both high intensity statin therapy and PCSK9 inhibition (23,24). Among the numerous inflammatory biomarkers, hsCRP has the greatest validation (15). In a review of multiple clinical trials and registry data, the proportion of atherosclerotic patients on statin therapy who have residual inflammatory risk (defined as an on-treatment LDL-C <70 mg/dL and hsCRP ≥2 mg/L) is more than twice the proportion of patients with residual cholesterol risk (defined as an on-treatment LDL-C ≥70 mg/dL and hsCRP <2 mg/L) (27). Results of recent large randomized controlled trials testing anti-inflammatory therapies targeting the NLRP3 inflammasome (canakinumab and colchicine) in participants with established ASCVD demonstrated improved cardiovascular outcomes (13,14). While the cardiovascular benefits of canakinumab were not observed in those who had on-treatment hsCRP ≥ 2mg/L (13). The connection between Lp(a) and inflammation was suggested by an early in vivo study by Dangas et al. where a strong correlation between plaque KP-1 and Lp(a) was found in atherectomy specimens from patients with unstable rest angina (28). Along those lines, a recent post hoc analysis of participants from the ACCELERATE trial, a secondary prevention study, reported that elevated Lp(a) was only significantly associated with MACE when hsCRP was ≥ 2mg/L (18). The results of our present analysis in primary prevention are consistent with the findings from ACCELERATE. Thus, data from both primary and secondary prevention studies suggest that elevated Lp(a) associates with ASCVD risk only in the presence of systemic inflammation, as measured by elevated hsCRP. Subgroups analysis further confirmed the presence of systemic inflammation potentiated Lp(a)-associated ASCVD risk in both African-Americans and Non-African-Americans as well as in both genders.
In the 2018 AHA/ACC Cholesterol guideline and the 2019 ACC/AHA primary prevention guideline, hsCRP and Lp(a) are both recommended as a risk-enhancers to help inform clinical decision-making about initiation of statin therapy in individuals with intermediate ASCVD risk (16). According to the current study, hsCRP levels may further refine Lp(a)-associated cardiovascular risk in the setting of primary prevention. Individuals with elevated levels of both Lp(a) and hsCRP appear to be at particularly higher ASCVD risk and all-cause mortality, thus may need to be more closely monitored and aggressively treated. A common dilemma in the assessment and management of individuals without known ASCVD but elevated Lp(a) is whether to obtain additional diagnostic tests and/or to start preventive medical therapies. Based on the data from this analysis, measurement of subclinical inflammation with hsCRP may help to guide further decisions, though this would need to be confirmed in trials designed to specifically answer this question.
Limitations
There are several limitations to this study. Baseline levels of Lp(a), hsCRP and LDL-C were used in the analysis, which precluded the ability to evaluate the impact of the dynamic changes of these markers over time. While very few participants were on lipid lowering therapy at baseline, use of lipid lowering medications during the follow up period may represent a source of confounding. We evaluated lipid lowering therapy at follow up visit 5. As summarized in Online Table 5, the group with Lp(a) ≥ 50mg/dL demonstrated a larger proportion of statin or any lipid lowering therapy compared with those with Lp(a) <50mg/dL. Additionally, the group with concomitant elevation of Lp(a) and hsCRP had the highest proportion on lipid-lowering therapy. These findings are consistent with the primary hypothesis that this is the group with highest CVD risk and thus had higher percentage on lipid therapy at follow up. Moreover, if there is any bias, it would be toward the null hypothesis and thus would not dramatically change the conclusion of the manuscript. Though we consistently observed significant interaction between Lp(a) and hsCRP with or without including BMI in the initial model, the final models did not include BMI as a covariate given that BMI is not included in the Pooled Cohort Equations and was not included in prior studies on Lp(a) (21). All ASCVD event was used as the primary outcome measure as pinpointing which specific cardiovascular disease was more or less impacted by the Lp(a)-hsCRP interaction was not the primary focus of the current study. Future studies with larger sample size may be needed to address this question. Finally, results from the study suggest an association between Lp(a), hsCRP and CVD events. However, given that this is an analysis of a population-based observational cohort, future experimental and randomized studies are needed to confirm whether the observed interaction between Lp(a) and hsCRP plays a causal role in ASCVD.
Conclusion
In the setting of primary prevention, Lp(a)-associated ASCVD risk is observed only with concomitant elevation of hsCRP. Measurement of hsCRP may further refine Lp(a)-associated ASCVD risk. Individuals with elevated levels of both hsCRP and Lp(a) may merit closer surveillance and more aggressive ASCVD risk management strategies.
Supplementary Material
Clinical Perspectives.
Competency In Medical Knowledge:
Patients with elevated Lp(a) and systemic inflammation face greater risk of ischemic events and mortality.
Translational Outlook:
Future studies of Lp(a) lowering agents should assess risk reduction according to baseline levels of high-sensitivity C-reactive protein.
Acknowledgments
Funding statement: The MESA (Multi-Ethnic Study of Atherosclerosis) is supported by contracts HHSN268201500003I, N01-HC-95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168 and N01-HC-95169 from the National Heart, Lung, and Blood Institute, and by grants UL1-TR-000040, UL1-TR-001079, and UL1-TR-001420 from National Center for Advancing Translational Sciences. The MESA Air ancillary study was supported by a grant from the US Environmental Protection Agency’s (EPA’s) Science to Achieve Results (STAR) program - Assistance Agreement number RD831697 awarded by the EPA to the University of Washington (WA).
ABBREVIATIONS
- ACC
American College of Cardiology
- AHA
American Heart Association
- ASCVD
atherosclerotic cardiovascular disease
- CVD
cardiovascular disease
- hsCRP
high sensitivity C-reactive protein
- HTN
Hypertension
- IQR
interquartile range
- LDL-C
Low-density lipoprotein -cholesterol
- Lp(a)
Lipoprotein(a)
- MESA
Multi-Ethnic Study of Atherosclerosis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement: The authors have nothing to disclose.
References
- 1.Benjamin EJ, Muntner P, Alonso A et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019;139:e56–e528. [DOI] [PubMed] [Google Scholar]
- 2.Patel KV, Pandey A, de Lemos JA. Conceptual Framework for Addressing Residual Atherosclerotic Cardiovascular Disease Risk in the Era of Precision Medicine. Circulation 2018;137:2551–2553. [DOI] [PubMed] [Google Scholar]
- 3.Dhindsa DS, Sandesara PB, Shapiro MD, Wong ND. The Evolving Understanding and Approach to Residual Cardiovascular Risk Management. Front Cardiovasc Med 2020;7:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mayr M, Gerszten R, Kiechl S. Cardiovascular Risk Beyond Low-Density Lipoprotein Cholesterol. J Am Coll Cardiol 2018;71:633–635. [DOI] [PubMed] [Google Scholar]
- 5.Tsimikas S A Test in Context: Lipoprotein(a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. J Am Coll Cardiol 2017;69:692–711. [DOI] [PubMed] [Google Scholar]
- 6.Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA 2009;301:2331–9. [DOI] [PubMed] [Google Scholar]
- 7.Clarke R, Peden JF, Hopewell JC et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med 2009;361:2518–28. [DOI] [PubMed] [Google Scholar]
- 8.Willeit P, Ridker PM, Nestel PJ et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: individual patient-data meta-analysis of statin outcome trials. Lancet 2018;392:1311–1320. [DOI] [PubMed] [Google Scholar]
- 9.Emerging Risk Factors C, Erqou S, Kaptoge S et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA 2009;302:412–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsimikas S, Hall JL. Lipoprotein(a) as a potential causal genetic risk factor of cardiovascular disease: a rationale for increased efforts to understand its pathophysiology and develop targeted therapies. J Am Coll Cardiol 2012;60:716–21. [DOI] [PubMed] [Google Scholar]
- 11.Viney NJ, van Capelleveen JC, Geary RS et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016;388:2239–2253. [DOI] [PubMed] [Google Scholar]
- 12.Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N Engl J Med 2020;382:244–255. [DOI] [PubMed] [Google Scholar]
- 13.Ridker PM, MacFadyen JG, Everett BM et al. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet 2018;391:319–328. [DOI] [PubMed] [Google Scholar]
- 14.Tardif JC, Kouz S, Waters DD et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N Engl J Med 2019;381:2497–2505. [DOI] [PubMed] [Google Scholar]
- 15.Ridker PM. From C-Reactive Protein to Interleukin-6 to Interleukin-1: Moving Upstream To Identify Novel Targets for Atheroprotection. Circ Res 2016;118:145–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grundy SM, Stone NJ, Bailey AL et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 2019;73:e285–e350. [DOI] [PubMed] [Google Scholar]
- 17.Arnett DK, Blumenthal RS, Albert MA et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2019September10;74(10):e177–e232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Puri R, Nissen SE, Arsenault BJ et al. Effect of C-Reactive Protein on Lipoprotein(a)-Associated Cardiovascular Risk in Optimally Treated Patients With High-Risk Vascular Disease: A Prespecified Secondary Analysis of the ACCELERATE Trial. JAMA Cardiol 2020. [DOI] [PMC free article] [PubMed]
- 19.Bild DE, Bluemke DA, Burke GL et al. Multi-Ethnic Study of Atherosclerosis: objectives and design. Am J Epidemiol 2002;156:871–81. [DOI] [PubMed] [Google Scholar]
- 20.Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem 1972;18:499–502. [PubMed] [Google Scholar]
- 21.Guan W, Cao J, Steffen BT et al. Race is a key variable in assigning lipoprotein(a) cutoff values for coronary heart disease risk assessment: the Multi-Ethnic Study of Atherosclerosis. Arterioscler Thromb Vasc Biol 2015;35:996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verbeek R, Sandhu MS, Hovingh GK et al. Lipoprotein(a) Improves Cardiovascular Risk Prediction Based on Established Risk Algorithms. J Am Coll Cardiol 2017;69:1513–1515. [DOI] [PubMed] [Google Scholar]
- 23.Pradhan AD, Aday AW, Rose LM, Ridker PM. Residual Inflammatory Risk on Treatment With PCSK9 Inhibition and Statin Therapy. Circulation 2018;138:141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bohula EA, Giugliano RP, Leiter LA et al. Inflammatory and Cholesterol Risk in the FOURIER Trial. Circulation 2018;138:131–140. [DOI] [PubMed] [Google Scholar]
- 25.Kronenberg F, Utermann G. Lipoprotein(a): resurrected by genetics. J Intern Med 2013;273:6–30. [DOI] [PubMed] [Google Scholar]
- 26.Dandona P, Aljada A, Chaudhuri A, Mohanty P, Garg R. Metabolic syndrome: a comprehensive perspective based on interactions between obesity, diabetes, and inflammation. Circulation 2005;111:1448–54. [DOI] [PubMed] [Google Scholar]
- 27.Ridker PM. How Common Is Residual Inflammatory Risk? Circ Res 2017;120:617–619. [DOI] [PubMed] [Google Scholar]
- 28.Dangas G, Mehran R, Harpel PC et al. Lipoprotein(a) and inflammation in human coronary atheroma: association with the severity of clinical presentation. J Am Coll Cardiol 1998;32:2035–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.