

Abstract

The monoamine oxidase (MAO) enzyme class is a prevalent target for many neurodegenerative and depressive disorders. Even though scrutinization of many promising drugs for the treatment of MAO inhibition has been carried out in recent times, a conclusive structural requirement for potent activity needs to be developed. Numerous approaches have been examined for the identification of structural features for potent MAO inhibitors (MAOIs) that mainly involve an array of computational studies, synthetic approaches, and biological evaluation. In this paper, we have analyzed ∼2200 well-known MAOIs to expand perceptions in the chemical space of MAOIs. The physicochemical properties of the MAOIs disclosed a discernible hydrophobic feature making a bunch discrete from the central nervous system (CNS) acting drugs, as exposed using the principal component analysis (PCA). The Murcko scaffold structure study revealed unfavorable and favorable scaffold structures, in both data sets, with the highest biological activity shown by the 3-phenyl-2H-chromen-2-one scaffold. This scaffold showed a polypharmacological effect. R-group disintegration and automatic structure–activity relationship (SAR) study resulted in identification of substructures responsible for the inhibitory bioactivity of the MAO-A and MAO-B enzymes. Moreover, with activity cliff analysis, significant biological activity was detected by simple molecular conversion in the chemical compound structure. In addition, we used the machine learning tool to generate a hypothesis wherein pyrazole, benzene ring, and amide containing structural functionalities can exhibit potential biological activities. This hypothesis revealed that CNS target drugs, C4155, C13390, C21265, C43862, C31524, C24810, C37100, C42075, and C43644, could be repurposed as valuable candidates for the MAO-B enzyme. For researchers, this study will bring new perceptions in the discovery and development of MAOIs and direct lead and hit optimization for the progress of small molecules beneficial for MAO-targeting associated diseases.
Introduction
Monoamine oxidase (MAO) enzyme is one of the most important enzymes among the ones responsible for neurodegeneration.1 They are flavoproteins that contain a chemical bond with the cofactor flavin adenosine dinucleotide2 (FAD). MAOs are enzymes that catalyze the oxidation of biogenic monoamines.3 There are two isoforms of MAO enzymes, i.e., MAO-A and MAO-B. Both MAOs are needed for the inactivation of monoamine neurotransmitters, for which they have various peculiarities, such as the breakdown of serotonin, melatonin, norepinephrine, epinephrine, phenethylamine, benzylamine, dopamine, tyramine, and tryptamine. Dopamine to dihydroxyphenylacetic acid, 3-methoxytyramine to homovanillic acid, and metadrenaline or normetadrenaline to vanillylmandelic acid (VMA) are some of the reactions catalyzed by MAO.4 Owing to the crucial role that MAOs play in the deactivation of neurotransmitters, MAO-related disorder is noted to be responsible for a number of psychiatric and neurological issues.5 Schizophrenia, addiction, attention deficit disorder, alcohol misuse, and migraines have also been linked to abnormally increased or decreased levels of MAO enzymes in the body.6,7 Apart from neurological disorders, MAO enzymes are quite significant in cardiovascular and vascular diseases and so on.8,9
Both MAO enzymes have a substrate-binding cavity and an entrance cavity in which the substrate-binding sites are hydrophobic and are enclosed with aromatic and aliphatic residues.10 In human MAO-A, the substrate-binding site is a 400 Å3 cavity, but in MAO-B, a smaller lipophilic cavity, called the entry cavity, sits between the main substrate-binding site and the protein surface.11 Depending on the substrate or inhibitor present, the cavities remain separate or get interconnected. We identified that four critical amino acid residues in MAO-A, Lys305, Trp397, Tyr407, and Tyr444, and their equivalent amino acids in MAO-B, Lys296, Trp388, Tyr398, and Tyr435, play major roles in MAO catalytic activity. The noncovalent binding to FAD may be mediated by Lys305, Trp397, and Tyr407 in MAO-A and Lys296, Ile199, Trp388, and Tyr398 in MAO-B, according to the polyamine oxidase three-dimensional (3D) crystal structure. MAO-A (Tyr398 and Tyr435 in MAO-B) may form an aromatic sandwich that stabilizes substrate binding.12 Among them, Ile199, since it may exist in two distinct conformations, a closed conformation separating the substrate and entry cavities or an opened conformation merging the two cavities, was originally identified as the gate residue in human MAO-B13 (Figure 1). In addition, Ile335 in MAO-A vs Tyr326 in MAO-B are two further differences in the substrate-binding regions of the two enzymes where these residues contribute to the selectivity of the two enzymes.14,15
Figure 1.

Ligand binding active sites of MAO enzymes.
Tremendous numbers of drugs have been marketed for the treatment of neurorelated diseases, among which MAO inhibitors (MAOIs) are the most often used drug types for the treatment of many neurodegenerative disorders, even if they are mostly used as a last resort due to the possibility of drug interactions with food or other medications.16 MAOIs are either reversible or irreversible. Phenelzine, isocarboxazid, and tranylcypromine are nonselective MAOIs that are approved by the U.S. Food and Drug Administration (USFDA).17 Orthostatic hypotension, dizziness, fatigue, sleeplessness, and nausea are the most prevalent early adverse effects of MAOIs.18 Weight gain, edema, muscular aches, myoclonus, paresthesias, and sexual dysfunction are among late adverse effects.19 MAOI has also been linked to erectile dysfunction. Although weight gain is common with all MAOIs, phenelzine may make it worse. Studies have shown that developing reversible and selective inhibitors of MAO isoenzymes can reduce the symptoms of many neurodegenerative diseases.20 Early development of irreversible MAOIs for treatment of depression (first-generation MAOI: phenelzine, tranylcypromine) caused unwanted side effects, which led to the development of second-generation MAOIs.21 But again, the irreversibility of inhibition was a major concern as it led to hypertensive crisis as potential side effects. There was need for a reversible inhibitor as most of the initially developed MAOI was irreversible.22 Later selective MAO-A (like moclobemide) and MAO-B inhibitors (selegiline) were developed, which showed lower toxicity and higher potency. With the therapeutic role of MAO in various neurodegenerative diseases, there was a keen interest in developing MAOI.23 A recently added FDA-approved MAO inhibitor is safinamide, which is used as an add-on therapeutic agent with levodopa for Parkinson’s disease.24 From the above studies, we realize that although many scaffolds have been brought to research, none of them are marketed due to failure in phase II trials, and there is a need for developing highly potent and isoform-specific inhibitors since currently used irreversible inhibitors are known to have significant adverse effects, such as tyramine-induced hypertensive crisis, while currently used reversible inhibitors are known to only block the MAO-B enzyme.25
A wide variety of computational, in vitro, and in vivo studies have been reported. Using X-ray diffraction methods, many PDB databases have been established in many studies like 2Z5X, 2Z5Y, etc. for MAO-A and 2BK5, 2BK4, etc. for MAO-B.26 A variety of in silico studies and molecular dynamics studies have been published on the MAO enzyme. Apart from the above-established computational methods, a new approach among computational techniques is cheminformatics. It is the utilization of computational and research-based knowledge to find a solution for a wide range of hypotheses in chemistry.27 In this study, our research focuses on organic substituents from a cheminformatics standpoint, with the goal of determining the total number of substituents from a known organic chemistry space and the optimal pharmacophore for MAO inhibitory action. Thus, there is a requirement of cheminformatics, quantitative structure–activity relationship (QSAR) analysis, pharmacophore analysis, and available structural information to guide scaffold optimization, to improve the pharmacodynamic properties of inhibitors of MAO. The current research employs a rigorous cheminformatic study with small compounds to identify the chemogenomic space of MAO-targeting compounds. The study involves the calculation of physicochemical properties, scaffold and fragment-based analysis, structural resemblance analysis of ligands, and activity cliff analysis and finally dissects the automated structure–activity relations. Based on the inductive learning technique, the DMax Chemistry Assistant (DCA) tool used for machine learning was used to discover the optimal chemical patterns that might play a key role in providing significant inhibitory action against the MAO enzyme.
Results and Discussion
Physicochemical Properties
The absorption, distribution, metabolism, excretion, and toxicity (ADMET) profile failure is the main factor that decides the pharmacological activity of drug molecules in later phases and is highly affected by their physicochemical properties. For this, it is necessary to analyze physicochemical properties for drug-likeness and bioavailability of drug molecules. Thus, we performed principal component analysis (PCA) for physicochemical properties,28i.e., total molecular weight, H-acceptor, H-donor, cLog P, cLog S, drug-likeness, rotatable bond (RB), total surface area (TSA), polar surface area (PSA), and relative polar surface area (RPSA) of the MAO inhibitor (n = 2177), the central nervous system (CNS) data set (n = 1017), and the FDA data set (n = 2309) to obtain a comparison of the physicochemical property space (Figure 2). The first three principal components (PCs) obtained 86.67% covariance. The concise information in Table 1 indicates that PC1 is primarily contributed by some H-acceptors (0.43) and PSA (0.43) and almost similar molecular weight (MW), RB, TSA and H-donors. PC2 has the highest loadings by cLog S (0.53) and RPSA (0.47), followed by aqueous solubility and relative PSA. The third PC has the highest loading by drug-likeness (0.97). Figure 2 illustrates a 3D PCA plot of the parameter scatter plot of CNS (blue), FDA (red), and MAO inhibitors (green). As observed in this figure, many of the MAO inhibitors are enlarged along both the PC2 and PC3 axes, showing that they vary ominously from CNS acting and FDA-approved. Estimation of the chemical variety of compounds focusing on MAO by PCA displayed that the MAO ligands vary drastically in physicochemical properties from the traditional medicinal property space. Thus, it is necessary to transform ligands selected from “MAO inhibitors” into potent molecules using scaffold hopping, 3D fragment, and computational chemistry.
Figure 2.

Three-dimensional plot of principal component analysis; red, blue, and green spheres denote FDA-approved drugs, CNS acting drugs, and MAO inhibitors, respectively.
Table 1. Principal Component Analysis of Physicochemical Properties with Loading Values for the First Three PCs.
| properties | PC1 | PC2 | PC3 |
|---|---|---|---|
| MW | 0.39 | –0.26 | 0.06 |
| cLog P | –0.18 | –0.51 | 0.03 |
| RB | 0.34 | –0.19 | –0.19 |
| drug-likeness | –0.02 | –0.01 | 0.97 |
| PSA | 0.43 | 0.13 | 0.02 |
| RPSA | 0.17 | 0.47 | –0.01 |
| TSA | 0.38 | –0.29 | 0.05 |
| H-donor | 0.37 | 0.16 | 0.04 |
| H-acceptor | 0.43 | 0.06 | 0.05 |
| cLog S | –0.07 | 0.53 | 0.01 |
| cumulative proportion (%) | 49.92 | 76.31 | 86.67 |
Scaffold Analysis
We analyzed the Murcko scaffold on MAO-A and MAO-B data sets. In this approach, the molecule is divided into a side-chain atom, a ring system, a framework, and a linker. We studied scaffold structures using bioactivity. The Murcko structure revealed 490 and 901 scaffolds from MAO-A and MAO-B data sets, respectively. The figure indicates the Murcko structure and their linked activities and colors depict frequencies. The Murcko structure analysis showed 68 and 91 frequencies to be the maximum in MAO-A and MAO-B, respectively. Afterward, the skeleton scaffold was built from the Murcko structure. The skeleton structure showed 327 and 440 frequencies for MAO-A and MAO-B data sets, respectively. The skeleton structure study showed 116 and 140 frequencies for the MAO-A and MAO-B data sets, respectively. The scaffold range is computed as a part of the number of scaffolds to the total number of compounds in the data set.29 The diversity of the Murcko, skeleton, and singleton scaffold structures of both data sets is shown in Table 2. This analysis suggests that scaffold diversity is low, and thus, we urgently need new scaffolds and diverse MAOIs.
Table 2. Diversity Calculation of MAO-A and MAO-B Inhibitor Scaffoldsa.
| data set (N) | Murcko scaffold structure (Nm) | singleton Murcko scaffolds (Nsm) | skeleton scaffold structure (Nsc) | Nm/N | Nsc/N | Nsm/N | Nsm/Nm | |
|---|---|---|---|---|---|---|---|---|
| MAO-A | 1546 | 490 | 259 | 327 | 0.32 | 0.21 | 0.17 | 0.53 |
| MAO-B | 2177 | 701 | 382 | 440 | 0.32 | 0.20 | 0.17 | 0.54 |
(Nsm/N) indicates the ratio of singleton Murcko scaffolds and the data set (N). Nm/N shows the ratio of Murcko scaffolds (Nm) and the data set (N). Nsc/N represents the ratio of skeleton scaffolds (Nsc) and the data set (N). (Nsm/Nm) signifies the quotient of a singleton Murcko scaffold (Nsm) and the Murcko scaffold structure (Nm).
The Murcko scaffold structure and the associated bioactivity (Figure S1A,B) show a change in biological properties with scaffold variety. This study uncovered favorable effective scaffolds and avoidable scaffolds (Table 3). For example, the Murcko scaffold, 3-phenyl-2H-chromen-2-one, shows the highest biological activities for both data sets. This analysis suggested that the chromenone-based compound shows a polypharmacological effect.30 This scaffold can be modified by scaffold hopping and exploring the SAR information, classifying the compound cycle, or creating persistent molecule libraries. The favorable scaffolds recognized in the MAO-A and MAO-B data sets from this study could be utilized as preliminary materials for the coherent designing of novel MAOIs by the implementation of in silico and in vivo studies.
Table 3. Some Illustrative Examples of Unfavorable and Favorable Scaffold Structures among the MAO-A and MAO-B Targeting Inhibitors.
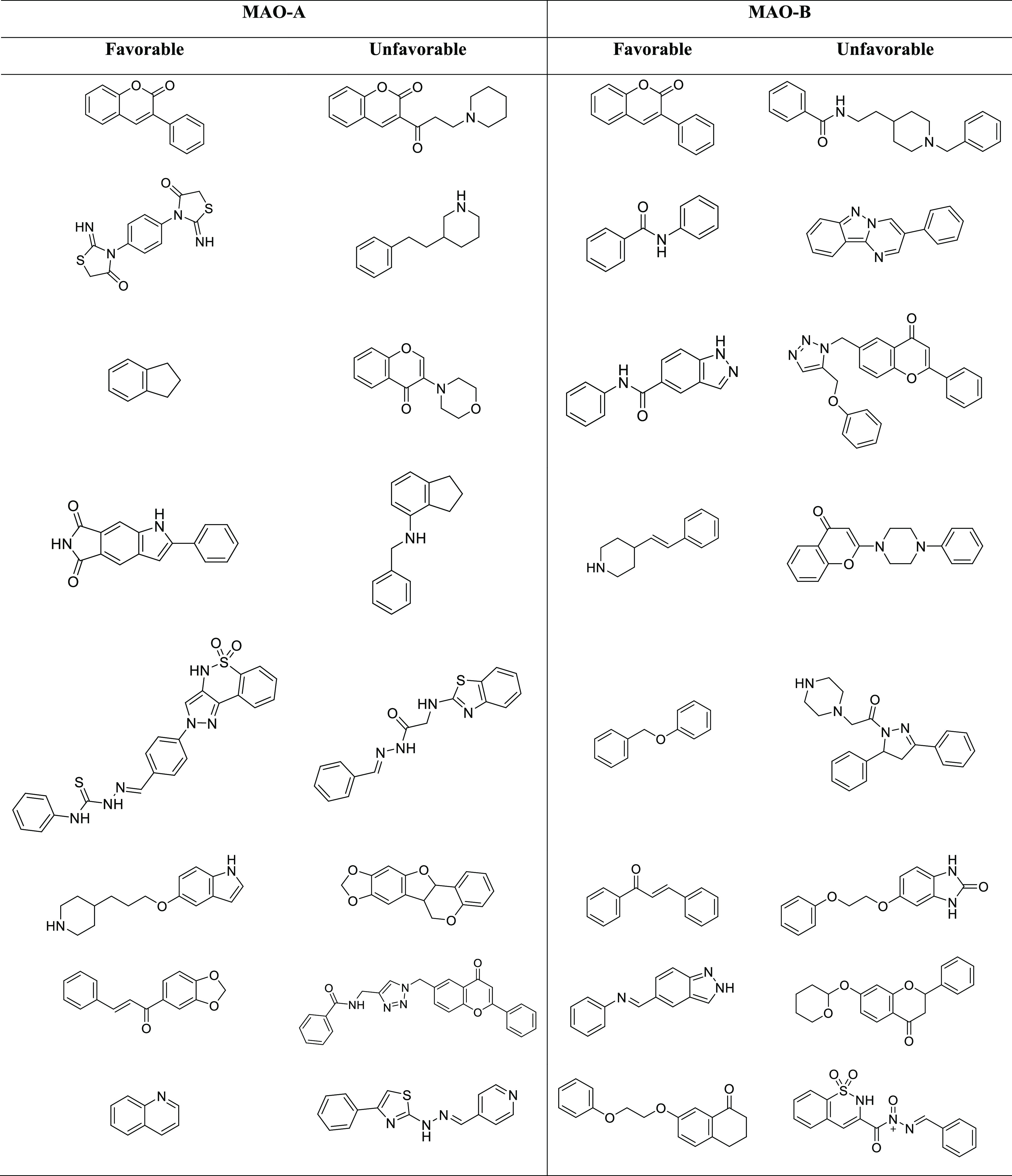
Using Scaffold Hunter, some frequent fragments such as chromenone, benzoquinone isoindoline-dione, pyrrole, indenone, naphthalene-dione, morpholine, thiophene, benzo-dioxole, imino-methylenethiazolidinone, cyclopropane, cyclobut-dione, furan, azetidine, indole, and piperidine (Figure 3), which represent latent scaffolds or fragments for creating the fragment library, were revealed. Furthermore, it will be beneficial to discover the synthetic possibility of the compounds. Hence, we need some 3D fragments to increase the pharmacophore efficiency as well as the solubility. Thus, we suggest screening of a 3D-based fragment library against MAO.
Figure 3.

Illustrations of frequent fragments recognized from MAO inhibitors using Scaffold Hunter.
Activity Cliff Analysis
The bioactivity scatter plot was generated for the activity cliff analysis of both data sets (Figures S2 and S3). This analysis of 4059 pairs was generated using a cutoff similarity threshold of 86% from the MAO-A data set. Similarly, 5567 pairs were generated for the MAO-B data set from the same similarity threshold. The pairwise relationships explained the structural similarity of effective molecules with surprising potential variations. A slight molecular modification entirely reverses the bioactivity in a drastic manner (Tables S1 and S2). In Figures S1 and S2 (Supporting Information), scatters represent molecules that are connected based on similarity, and the structure–activity landscape index (SALI) value signifies size. It is possible to identify compounds with significant activity in terms of the SALI value but with a similar scaffold structure (high molecular similarity). A greater percentage of pairs of molecules has shown bioactivity alteration around 2 and 4 log units of determination (Tables S1 and S2). For illustration, on matching between the pairs A1462 and A1468 of the MAO-A data set, the similarity value was 0.98, the determined SALI value was 254.56, and bioactivity values were 8.52 and 4.52, respectively. On matching between the pairs B1639 and B302 of the MAO-B data set, the similarity value was 0.99, the calculated SALI value was 445.82, and the bioactivity values were 5.01 and 7.39, respectively. These studies imply that these pairs of molecules could be investigated for the structure–activity relationship (SAR) to design more potent derivatives. In Figure 4, an informative case of SAR created for chromenone-based compounds in the MAO-B inhibitor is presented. These studies show that the major difference in effectiveness among the derivatives of several scaffolds targeting MAO-A and MAO-B results primarily from single replacements and offer valuable suggestions for molecule optimization attempts. The occurrence of the well-known activity cliffs in the MAO-A and MAO-B data sets can contribute as a key constraining factor in quantitative structure–activity relationship (QSAR) extrapolations, which will be reasonable to direct bioactivity estimates regarding attentive provinces of constant SAR in the MAO chemical space.
Figure 4.

Chromenone derivative activity cliff and SAR study. The colors signify the pIC50 value, where red color indicates the highest and blue color indicates the lowest. The tinted red color indicates structural deviation.
Similarity Analysis
A similarity measure between compounds plays a vital role where molecular similarity ranges from a chemical resemblance based on substructure fragments to biological similarities that take into account binding and 3D geometry.31 For a better interpretation of the chemical space of MAOIs, use of the rubberbanding scaling force (RSF) field method32 for structural similarity computation was beneficial. This procedure includes twisting, snapping, and then stretching the molecular bonds to the original positions. When compared to the regular PCA-based approach, the RSF method advances the similarity analysis. The similarity of compounds is calculated using structural descriptors obtained from the 3D structure of compounds. In this study, we developed a flexophore descriptor for investigating the 3D pharmacophore to discover compounds with related binding activities. Compounds with the same shape, pharmacophore, and size will have elevated indexes of flexophore similarity. A similarity threshold of 97% for 4155 pairs was generated from the MAO-A data set; a similar threshold was created for 5930 pairs from the MAO-B data set.
In Figure 5, the structure similarity of the flexophore is shown, where compounds with increased similarity are linked with lines and color predicting their similar pharmacophore. Based on the pharmacophoric features (Figure 5) from both data sets, the bulk of chemical scaffolds was clustered together, suggesting that the pharmacophoric structural diversity of MAOIs is low in the MAO chemical space. A comprehensive pharmacophore space study of structurally similar molecules may indicate that there is an important redundancy in chemical compound sets with respect to MAO. Thus, the current chemical space of compounds in relation to MAO must be varied by including illustrative compounds with diverse pharmacophores.
Figure 5.

Structure-similarity flexophore plot and neighbor tree visualization. Similarity is indicated by colors; the highest and lowest similarities are denoted by red and blue, respectively. (A) MAO-A data set. (B) MAO-B data set.
Structure–Activity Relationship (SAR)
Automatic SAR was used on the MAO data set (MAO-A and MAO-B), which resulted in the creation of core fragments and several linked R-groups. The most central ring system scaffolds were used for the automatic SAR, and two different SARs were created based on the MAO-A and MAO-B data sets. For the MAO-A data set, 103 core fragments linked to six R-groups were created (Figure 6). Similarly, for the MAO-B data set, 121 core fragments connected to six R-groups were created (Figure 6). In this analysis, it was detected that compounds with a similar core fragment show a large deviation in bioactivity, suggesting that different R-groups change the bioactivity idiosyncratically. As in Table 4, the incorporation of various R-groups on the core structure can significantly be responsible for high bioactivity profiles against both MAO-A and B. For example, chromen-2-one at the R-6 group shows the highest biological activity. The pyarazolo-thiazine-dioxide derivative at the R-1 group of the core fragment benzene results in high bioactivity, but substitution at any other R-2 and R-4 groups reduces the bioactivity in the MAO-A data set. In the MAO-B data set, an aniline at the R-2 group of the core fragment chromenone showed the highest biological activity. At R-2 or R-4 positions, the different moieties influence the biological activity. This analysis implies that chromenone shows the highest activity for MAO-A and MAO-B; although both proteins are identical with 70% similarity,33 the ligand–protein interaction did not seem to be much similar.
Figure 6.

Scatter plot of activity (pIC50) and core fragment. The pIC50 is indicated by colors: red indicates the highest and blue indicates the lowest activity. (A) MAO-A data set. (B) MAO-B data set.
Table 4. SAR of Benzene and Chromenone Scaffolds from MAO-A and MAO-B Data Sets, Respectively.

QSAR Modeling by Inductive Logical Programing (ILP)
To reveal and associate the mutual chemical forms of the MAO-B inhibitors and their biological activities, we applied the artificial intelligence tool DCA. DMax Chemistry Assistant is an ILP-based method that utilizes the existing information, e.g., moiety, element, substructure relationship, and electron flow, to create hypotheses that are best validated with the known information. It starts by reading the functional groups and rings followed by the construction of hypothesis. To obtain a valid result, we require a logical number of inhibitors with high biological activity parameters. Thus, we chose the MAO-B inhibitors having high biological activities for this study. In this study, we generated the phenotype inhibitory hypothesis successfully. The hypothesis implies that inhibitors with precise arrangements comprising a five-membered heteroaromatic ring such as a pyrazole ring, a six-membered aromatic ring such as a benzene ring, and a functional group such as an amide may retain the p-value (7 × 10–2). Figure 7 displays an informative example of molecules with high activity (pIC50). The developed model was acceptable with a root-mean-square error (RMSE) of 0.35, correlation 0.44, and a “rank high” cutoff of 8.55. The inhibitor exhibiting a cumulative response plot indicates the 60th percentile on ∼80% of the hit coverage, and the lift curve implies that the top 40% of information will outdo a random model by ∼2.60 times. A receiver operating characteristic curve (ROC curve) openly differentiates the false alarms (x-axis) from hits, and an area under curve (AUC) value of 0.77 indicates a good measure of parting between hits and false alarm (nonhits). In Figure 7D, the plot shows the predicted vs actual activity of the inhibitory model.
Figure 7.

(A) Inhibitor model of the cumulative response plot, demonstrating the relation between the percentiles (x-axis) and percentage of hits (y-axis). (B) Inhibitor model of the lift plot, to validate the presentation of the model. (C) Inhibitor model of the ROC plot openly differentiates the percentage of false alarms (nonhits) (x-axis) from percentage hits. The AUC of 0.77 indicates a significant measure of parting between hits and nonhits. (D) Inhibitor model of the actual-predicted scatter plot. (E) Illustrations of the high biological activity (pIC50) of MAO-B inhibitors, emphasizing the typical outlines of hypothesis. The hypothesis states that inhibitors with a high activity (pIC50) may have a five-membered heteroaromatic ring-A (yellow) with a fused benzene ring (green) and the general functional group (red), which are at distance 2. The benzene ring (green) is linked to the general functional group (red) with a single bond. On the benzene ring (green), the substituted general function group (red) and the unsubstituted atoms are at distances of 1 and 2.
To confirm the study, we utilized the hypothesis to screen the CNS target drug library34 (n = 44 085). In this study with the created hypothesis model for the MAO-B inhibitor, the outcome revealed a five-membered heteroaromatic ring, a benzene ring, and a functional group. Figure 8 displays representative examples: C4155, C13390, C21265, C43862, C31524, C24810, C37100, C42075, and C43644 of the CNS target drug library with high predicted pIC50 values (Table S3). We repurposed compounds showing high predicted pIC50 values against MAO-B. It may be showing excellent binding affinity toward the MAO-B enzyme.
Figure 8.

Few representative examples of the CNS target drug library that displayed a high predicted biological activity (pIC50).
Conclusions
Many virtual screenings have been performed for MAO, but no inhibitors showed any polypharmacological effects on MAO-A and MAO-B proteins, whereas the existing inhibitors showed major side effects. Although it has been several decades since the drug discovery, the success rate in developing this type of inhibitor is less. Thus, in this study, we focused on chemical space and AI analysis to study in depth the target MAO, contributing a novel perception to drug discovery development. Evaluation of chemical space including study of molecular similarity, scaffold analysis, activity cliff, physicochemical parameters, and SAR for optimizing the scaffold analogues can help in designing novel compounds. Furthermore, a slight change in the molecule leads to major fluctuations in the bioactivity. The automatic SAR analysis disclosed varied core fragment structures representing the highest bioactivity in MAO-A and MAO-B inhibitor data sets. Scaffold Hunter used for fragment-based analysis exposed some fragments and a few of the 3D fragments in the chemical space of compounds against MAO, which may help in characterizing an exciting framework that might prove useful in designing novel compounds. The structure–activity landscape index (SALI) plot presented a perception of the relation between the structure and bioactivities. The proportional physicochemical parameter study of MAOIs, FDA-approved drugs, and CNS acting drugs showed that CNS acting drugs occupied a wide range in chemical space, with a hydrophobic character of cLog P = ∼2.06. Similarly, MAOIs showed a more hydrophobic (cLog P = ∼3.55) nature. Thus, this perception can be greatly beneficial in the coherent designing of a molecule library against MAO. Additionally, we used the ILP method to generate a hypothesis on MAO-B inhibitors and applied this hypothesis to the CNS target drug library and found several highly predicted activity ligands. Artificial intelligence techniques clubbed with molecular modeling approaches such as scaffold hopping and fragment-based drug design might be helpful in the design and identification of a potential class of MAO inhibitors. Further, experimental validation of these identified classes of compounds, viz., synthetic methodologies, followed by their biological evaluation may result in promising candidates for neurodegenerative disorders.
Materials and Methods
Data Collection
A database of MAOIs with their biological activities (IC50) and molecular structures was constructed by revising the journals from PubMed, Google Scholar, and Web of Science since the years 2015–2021. We investigated and collected information on around ∼2200 compound inhibitors of MAO, for which the data set is divided into two subset MAO-A and MAO-B inhibitors. The database comprising “MAO inhibitors” had MAO-A (n = 1546) and MAO-B (n = 2177) inhibitor values in the micromolar range, while compounds with IC50 values of more than 100 μM were omitted from our databank. We utilized the data of “CNS acting drug molecules”35 (n = 1017) and “FDA-approved drug molecules”35 (n = 2309) for principal component analysis36 (PCA). The MAOI chemical space evaluation included scaffold analysis,29 examination of the structure–activity relationship (SAR), similarity, and various physicochemical properties, and activity cliff analysis,37 which were implemented by DataWarrior38 (V5.5.0), and using ECFP4 fingerprint39 in Scaffold Hunter40 on a DESKTOP-9KJUAO8 Intel core i5-72000CPU@2.50GHz.
Physicochemical Properties
The physicochemical parameters important for drug optimization, like molecular weight (MW), cLog S, total surface area (TSA), H-acceptor, polar surface area (PSA), cLog P, rotatable bond (RB), H-donor, relative polar surface area (RPSA), and drug-likeness, were calculated for the data sets, “MAO inhibitors,” “FDA-approved drugs” (FDA data set), and “CNS acting drugs” (CNS data set). The graphical representation of the 3D PCA plot was developed by operating R studio with the rgl package.41
Scaffold Analysis
Each molecule having an accompanying inimitable scaffold and the core structures of each molecule were evaluated. The removal of all of the side chains connected via the terminal to the ring resulted in the scaffold structure. The study was implemented by the Murcko structure and the Skeleton structure. The Murcko structures were created by removing the α attached atom and the exocyclic double bonds.29 Additionally, a skeleton structure was generated by the Murcko structure. The skeleton investigation includes only the ring, and any other heteroatom (S, N, and O) is replaced by the carbon atom. Additionally, using the ECFP4 fingerprint in Scaffold Hunter, and the data set was decomposed into parental and child structure.
Activity Cliff Analysis
The challenge in drug innovation and development is understanding the relation between molecular descriptions and the associated biological activity. The small variations in the structure of a compound change the associated bioactivity. The SALI was revealed using SkeleSphere descriptors. The structure–activity landscape index42 values were estimated on the basis of activity cliff analysis, associating the activity (pIC50) with the chemical variety of MAOIs.
where A1 is the activity of the first compound and A2 is the activity of the second compound of the MAO data set, and sim(1,2) is the similarity proportion between the pair of compounds.
Similarity Analysis
Using the identical flexophore43 descriptors, we calculated the similarity between two compounds from the molecular structure. This included generating an illustrative variety of conformers.
QSAR Modeling by Inductive Logical Programing
In this analysis, we used DMax Chemistry Assistant44 to obtain the suppositions and establish the correlation between the structural and morphological features of MAO-B inhibitors and their activities. Inductive logical programing (ILP)45 leads to a hierarchical approach by including the related data of molecular structure, type of elements (e.g., N, C, O, S), electrostatics, functional groups, and linkage (the location of topology in the ring, fused, linked, how different rings and functional groups are connected) of the compound. DMax Chemistry Assistant (DCA) can correlate past data of compounds with their bioactivity (pIC50), e.g., inhibition and stimulation. In this analysis, we applied DCA to develop the hypothesis to associate the traditional molecular characteristics of the MAO-B inhibitors with the associated biological activity.
Acknowledgments
The authors acknowledge Taif University Researchers Supporting Project number (TURSP-2020/68), Taif University, Taif, Saudi Arabia.
Glossary
Abbreviations
- FAD
flavin adenine dinucleotide
- MAO
monoamine oxidase
- PCA
principal component analysis
- ILP
inductive logical programing
- DCA
DMax Chemistry Assistant
- RSF
rubberbanding scaling force
- SALI
structure–activity landscape index
- SAR
structure–activity relationship
- MW
molecular weight
- TSA
total surface area
- PSA
polar surface area
- RB
rotatable bond
- RPSA
relative polar surface area
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c03250.
This study received a grant from Amrita Vishwa Vidyapeetham University (Seed Grant Number K-PHAR-20-628 to B.M.).
The authors declare no competing financial interest.
Notes
Any data generated and analyzed for this study that are included in this article and its Supporting Information are available. DataWarrior (V5.5.0) tool is publicly available free of charge at https://openmolecules.org/datawarrior. R studio is freely available at https://www.rstudio.com/products/rstudio/download/. DMax Chemistry Assistant is also freely available at https://dtai.cs.kuleuven.be/dmax/download/ChemistryAssistant.html. FDA-approved drugs and the CNS acting drug library are from https://www.selleckchem.com/. CNS target drug library is from https://www.chemdiv.com/cns-targets-library.
Supplementary Material
References
- Riederer P.; Müller T. Use of monoamine oxidase inhibitors in chronic neurodegeneration. Expert Opin. Drug Metab. Toxicol. 2017, 13, 233–240. 10.1080/17425255.2017.1273901. [DOI] [PubMed] [Google Scholar]
- Yeung A. W. K.; Georgieva M. G.; Atanasov A. G.; Tzvetkov N. T. Monoamine Oxidases (MAOs) as Privileged Molecular Targets in Neuroscience: Research Literature Analysis. Front. Mol. Neurosci. 2019, 12, 143 10.3389/fnmol.2019.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar B.; Prakash Gupta V.; Kumar V. Send Orders for Reprints to Reprints@benthamscience.Ae A Perspective on Monoamine Oxidase Enzyme as Drug Target: Challenges and Opportunities. Curr. Drug Targets 2017, 18, 87–97. 10.2174/1389450117666151209123402. [DOI] [PubMed] [Google Scholar]
- Kalgutkar A. S.; Dalvie D. K.; Castagnoli N.; Taylor T. J. Interactions of nitrogen-containing xenobiotics with monoamine oxidase (MAO) isozymes A and B: SAR studies on MAO substrates and inhibitors. Chem. Res. Toxicol. 2001, 14, 1139–1162. 10.1021/tx010073b. [DOI] [PubMed] [Google Scholar]
- Domino E. F.; Khanna S. S. Decreased blood platelet MAO activity in unmedicated chronic schizophrenic patients. Am. J. Psychiatry 1976, 10.1176/ajp.133.3.323. [DOI] [PubMed] [Google Scholar]
- Sharma A.; Kumar A.; Kaswan V.; Kaushik N.; Kaushik A. Role of monoamine Oxidase A (MAO-A) in drug addiction. J. Pharmacogn. Phytochem. 2018, 7, 727–729. [Google Scholar]
- Sanlier N.; Bektesoglu M. Migraine and Biogenic Amines. Ann. Med. Health Sci. Res. 2021, 11, 1362. [Google Scholar]
- Kaludercic N.; Carpi A.; Menabò R.; Di Lisa F.; Paolocci N. Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim. Biophys. Acta, Mol. Cell Res. 2011, 1813, 1323–1332. 10.1016/j.bbamcr.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ra̧iu C.; U̧u D.; Petruş A.; Norbert P.; Olariu S.; Duicu O.; Sturza A.; Muntean D. M. Monoamine oxidase inhibition improves vascular function and reduces oxidative stress in rats with lipopolysaccharide-induced inflammation. Gen. Physiol. Biophys. 2018, 37, 687–694. 10.4149/gpb_2018014. [DOI] [PubMed] [Google Scholar]
- Mathew B.; Carradori S.; Guglielmi P.; Uddin M. S.; Kim H. New Aspects of Monoamine Oxidase B Inhibitors: The Key Role of Halogens to Open the Golden Door. Curr. Med. Chem. 2020, 28, 266–283. 10.2174/0929867327666200121165931. [DOI] [PubMed] [Google Scholar]
- Tripathi A. C.; Upadhyay S.; Paliwal S.; Saraf S. K. Privileged Scaffolds as MAO Inhibitors: Retrospect and Prospects. Eur. J. Med. Chem. 2018, 145, 445–497. 10.1016/j.ejmech.2018.01.003. [DOI] [PubMed] [Google Scholar]
- Geha R. M.; Chen K.; Wouters J.; Ooms F.; Shih J. C. Analysis of conserved active site residues in monoamine oxidase A and B and their three-dimensional molecular modeling. J. Biol. Chem. 2002, 277, 17209–17216. 10.1074/jbc.M110920200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubálek F.; Binda C.; Khalil A.; Li M.; Mattevi A.; Castagnoli N.; Edmondson D. E. Demonstration of isoleucine 199 as a structural determinant for the selective inhibition of human monoamine oxidase B by specific reversible inhibitors. J. Biol. Chem. 2005, 280, 15761–15766. 10.1074/jbc.M500949200. [DOI] [PubMed] [Google Scholar]
- Son S. Y.; Ma J.; Kondou Y.; Yoshimura M.; Yamashita E.; Tsukihara T. Structure of Human Monoamine Oxidase A at 2.2-Å Resolution: The Control of Opening the Entry for Substrates/Inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 5739–5744. 10.1073/pnas.0710626105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaweska H.; Fitzpatrick P. F. Structures and Mechanism of the Monoamine Oxidase Family. Biomol. Concepts 2011, 2, 365–377. 10.1515/BMC.2011.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thase M. E. The role of monoamine oxidase inhibitors in depression treatment guidelines. J. Clin. Psychiatry 2012, 73, 10–16. 10.4088/JCP.11096su1c.02. [DOI] [PubMed] [Google Scholar]
- Gross M. E.; Agus D. B.; Dorff T. B.; Pinski J. K.; Quinn D. I.; Castellanos O.; Gilmore P.; Shih J. C. Phase 2 trial of monoamine oxidase inhibitor phenelzine in biochemical recurrent prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 61–68. 10.1038/s41391-020-0211-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hroudová J.; Nováková T.; Korábečný J.; Maliňák D.; Górecki L.; Fišar Z. Effects of Novel Tacrine Derivatives on Mitochondrial Energy Metabolism and Monoamine Oxidase Activity—In Vitro Study. Mol. Neurobiol. 2021, 58, 1102–1113. 10.1007/s12035-020-02172-1. [DOI] [PubMed] [Google Scholar]
- Jones D. N.; Raghanti M. A. The role of monoamine oxidase enzymes in the pathophysiology of neurological disorders. J. Chem. Neuroanat. 2021, 101957 10.1016/j.jchemneu.2021.101957. [DOI] [PubMed] [Google Scholar]
- Mathew B.; Parambi D. G.; Mathew G. E.; Uddin M. S.; Inasu S. T.; Kim H.; Marathakam A.; Unnikrishnan M. K.; Carradori S. Emerging therapeutic potentials of dual-acting MAO and AChE inhibitors in Alzheimer’s and Parkinson’s diseases. Arch. Pharm. 2019, 352, 1900177 10.1002/ardp.201900177. [DOI] [PubMed] [Google Scholar]
- López-Muñoz F.; Álamo C.; Juckel G.; Assion H. J. Half a Century of Antidepressant Drugs - On the Clinical Introduction of Monoamine Oxidase Inhibitors, Tricyclics, and Tetracyclics. Part I: Monoamine Oxidase Inhibitors. J. Clin. Psychopharmacol. 2007, 27, 555–559. 10.1097/jcp.0b013e3181bb617. [DOI] [PubMed] [Google Scholar]
- Santin Y.; Resta J.; Parini A.; Mialet-Perez J. Monoamine oxidases in age-associated diseases: New perspectives for old enzymes. Ageing Res. Rev. 2021, 101256 10.1016/j.arr.2021.101256. [DOI] [PubMed] [Google Scholar]
- Tripathi A. C.; Upadhyay S.; Paliwal S.; Saraf S. K. Privileged Scaffolds as MAO Inhibitors: Retrospect and Prospects. Eur. J. Med. Chem. 2018, 145, 445–497. 10.1016/j.ejmech.2018.01.003. [DOI] [PubMed] [Google Scholar]
- Joy M.; Mathew B.; Sudarsanakumar C. Structural Features of Safinamide: A Combined Hirshfeld Surface Analysis & Quantum Chemical Treatment. Chem. Data Collect. 2018, 17–18, 404–414. 10.1016/j.cdc.2018.10.009. [DOI] [Google Scholar]
- Van der Walt M. M.; Terre’Blanche G.; Petzer J. P.; Petzer A. Benzyloxynitrostyrene analogues–A novel class of selective and highly potent inhibitors of monoamine oxidase B. Eur. J. Med. Chem. 2017, 125, 1193–1199. 10.1016/j.ejmech.2016.11.016. [DOI] [PubMed] [Google Scholar]
- Binda C.; Aldeco M.; Mattevi A.; Edmondson D. E. Interactions of Monoamine Oxidases with the Antiepileptic Drug Zonisamide: Specificity of Inhibition and Structure of the Human Monoamine Oxidase B Complex. J. Med. Chem. 2011, 54, 909–912. 10.1021/jm101359c.Interactions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash N.; Gareja D. A. Cheminformatics. J. Proteomics Bioinf. 2010, 3, 249–252. 10.4172/jpb.1000147. [DOI] [Google Scholar]
- Rankovic Z. CNS drug design: balancing physicochemical properties for optimal brain exposure. J. Med. Chem. 2015, 58, 2584–2608. 10.1021/jm501535r. [DOI] [PubMed] [Google Scholar]
- Lipkus A. H.; Yuan Q.; Lucas K. A.; Funk S. A.; Bartelt W. F. III; Schenck R. J.; Trippe A. J. Structural diversity of organic chemistry. A scaffold analysis of the CAS Registry. J. Org. Chem. 2008, 73, 4443–4451. 10.1021/jo8001276. [DOI] [PubMed] [Google Scholar]
- Besnard J.; Ruda G. F.; Setola V.; Abecassis K.; Rodriguiz R. M.; Huang X. P.; Norval S.; Sassano M. F.; Shin A. I.; Webster L. A.; Simeons F. R.; et al. Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A.; Glen R. C. Molecular similarity: a key technique in molecular informatics. Org. Biomol. Chem. 2004, 2, 3204–3218. 10.1039/b409813g. [DOI] [PubMed] [Google Scholar]
- Chhabra S.; Kumar S.; Parkesh R. Chemical Space Exploration of DprE1 Inhibitors Using Chemoinformatics and Artificial Intelligence. ACS Omega 2021, 14430. 10.1021/acsomega.1c01314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro A.; Montecinos A.; Gómez-Molina C.; Núñez G.; Aldeco M.; Edmondson D. E.; Vilches-Herrera M.; Lühr S.; Iturriaga-Vásquez P.; Reyes-Parada M.. Similarities between the Binding Sites of Monoamine Oxidase (MAO) from Different Species. Is Zebrafish a Useful Model for the Discovery of Novel MAO Inhibitors. An Integrated View of the Molecular Recognition and Toxinology. From Analytical Procedures to Biomedical Applications; InTech: Rijeka, 2021; pp 405–431. [Google Scholar]
- CNS Target Drug Library. https://www.chemdiv.com/cns-targets-library (accessed May 10, 2021).
- FDA Approved Drugs and CNS Acting Drugs. https:www.selleckchem.comscreeningfda-approved-drug-library.html (accessed May 02, 2021).
- Abdi H.; Williams L. J. Principal component analysis. Wiley Interdiscip. Rev.: Comput. Stat. 2010, 2, 433–459. 10.1002/wics.101. [DOI] [Google Scholar]
- Bajorath J. Evolution of the activity cliff concept for structure–activity relationship analysis and drug discovery. Future Med. Chem. 2014, 6, 1545–1549. 10.4155/fmc.14.94. [DOI] [PubMed] [Google Scholar]
- Sander T.; Freyss J.; von Korff M.; Rufener C. DataWarrior: an open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. 10.1021/ci500588j. [DOI] [PubMed] [Google Scholar]
- Gardiner E. J.; Holliday J. D.; O’Dowd C.; Willett P. Effectiveness of 2D fingerprints for scaffold hopping. Future Med. Chem. 2011, 3, 405–414. 10.4155/fmc.11.4. [DOI] [PubMed] [Google Scholar]
- Schäfer T.; Kriege N.; Humbeck L.; Klein K.; Koch O.; Mutzel P. Scaffold Hunter: a comprehensive visual analytics framework for drug discovery. J. Cheminf. 2017, 9, 28 10.1186/s13321-017-0213-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tierney L.; Jarjour R.; Tierney M. L.. Package ‘Proftools’, 2016.
- Guha R.; Van Drie J. H. Structure–activity landscape index: identifying and quantifying activity cliffs. J. Chem. Inf. Model. 2008, 48, 646–658. 10.1021/ci7004093. [DOI] [PubMed] [Google Scholar]
- Von korff M.; Freyss J.; Sander T. Flexophore, a new versatile 3D pharmacophore descriptor that considers molecular flexibility. J. Chem. Inf. Model. 2008, 48, 797–810. 10.1021/ci700359j. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Loharch S.; Kumar S.; Ringe R. P.; Parkesh R. Exploiting cheminformatic and machine learning to navigate the available chemical space of potential small molecule inhibitors of SARS-CoV-2. Comput. Struct. Biotechnol. J. 2021, 19, 424–438. 10.1016/j.csbj.2020.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y.; Zhou H.; Zhu H.; Leung S. W. Ligand-based virtual screening and inductive learning for identification of SIRT1 inhibitors in natural products. Sci. Rep. 2016, 6, 19312 10.1038/srep19312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.