

Abstract
Motivation
Sequence motif discovery algorithms can identify novel sequence patterns that perform biological functions in DNA, RNA and protein sequences—for example, the binding site motifs of DNA- and RNA-binding proteins.
Results
The STREME algorithm presented here advances the state-of-the-art in ab initio motif discovery in terms of both accuracy and versatility. Using in vivo DNA (ChIP-seq) and RNA (CLIP-seq) data, and validating motifs with reference motifs derived from in vitro data, we show that STREME is more accurate, sensitive and thorough than several widely used algorithms (DREME, HOMER, MEME, Peak-motifs) and two other representative algorithms (ProSampler and Weeder). STREME’s capabilities include the ability to find motifs in datasets with hundreds of thousands of sequences, to find both short and long motifs (from 3 to 30 positions), to perform differential motif discovery in pairs of sequence datasets, and to find motifs in sequences over virtually any alphabet (DNA, RNA, protein and user-defined alphabets). Unlike most motif discovery algorithms, STREME reports a useful estimate of the statistical significance of each motif it discovers. STREME is easy to use individually via its web server or via the command line, and is completely integrated with the widely used MEME Suite of sequence analysis tools. The name STREME stands for ‘Simple, Thorough, Rapid, Enriched Motif Elicitation’.
Availability and implementation
The STREME web server and source code are provided freely for non-commercial use at http://meme-suite.org.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The ab initio discovery of biologically significant, fixed-length sequence patterns (ungapped motifs) in sets of unaligned DNA, RNA or protein sequences is an important and heavily studied problem. Motif discovery algorithms are especially useful in elucidating the DNA patterns bound by transcription factors (TFs), and the RNA patterns recognized by RNA-binding proteins (RBPs). In this work, we describe a novel motif discovery algorithm, STREME, and show its advantages over the most widely used existing algorithms in a careful and comprehensive evaluation using real DNA and RNA data. A key aspect of this work is that we use reference motifs created using in vitro data to judge the motifs found by the algorithms we evaluate here in in vivo data. This avoids the circularity found in some previous work, where the reference motifs were derived from the same type of data used to evaluate the algorithms.
As do several existing motif discovery algorithms [e.g. Weeder (Pavesi et al., 2004), HOMER (Heinz et al., 2010), STEME (Reid and Wernisch, 2011)], STREME makes use of a data structure called a generalized suffix tree (Weiner, 1973). It uses the generalized suffix tree to store the input sequences, and in the case of DNA, their reverse complements. Unlike Weeder and HOMER, however, which use the suffix tree to speed counting of approximate matches to individual words, STREME uses it to efficiently count matches to a position weight matrix (PWM) (Stormo, 2000) representing a candidate motif. Another innovation of STREME is that, unlike HOMER and Weeder, which use separate searches of a suffix tree to find motifs with different widths, the STREME algorithm scores PWMs of all widths in the user-specified range in a single depth-first traversal of the suffix tree. This makes STREME faster, especially for wide motifs (width up to 30 positions), and more thorough in its search for the ideal width for each motif.
Like DREME (Bailey, 2011) and HOMER, STREME evaluates motifs using a one-sided statistical test of the enrichment of matches to the motif in a primary set of sequences compared to a set of control sequences. STREME uses Fisher’s exact test (Fisher, 1922) if the primary and control sequences have the same length distribution, and the Binomial test otherwise. Like DREME, and unlike HOMER, STREME can also discover motifs given just a primary set of sequences. In that case, STREME will create a control set by shuffling the letters of the primary sequences, preserving certain (user-specified) lower-order statistics of the sequences. Preserving the lower-order sequence statistics helps STREME avoid discovering uninteresting motifs. In addition, STREME always creates a Markov model of a user-specified order from the control sequences. STREME uses the Markov model in conjunction with the PWM when counting matches to the motif to further bias the search away from motifs that are mere artifacts of the lower-order statistics of the input sequences.
STREME (like DREME) reports a useful estimate of the statistical significance of each motif that it discovers, in contrast to HOMER, Peak-motifs (Thomas-Chollier et al., 2011), ProSampler (Li et al., 2019) and Weeder, which do not report motif statistical significance, and MEME, whose motif significance estimates tend to be too conservative (Nagarajan et al., 2005). STREME estimates motif statistical significance by evaluating the ability of each motif it discovers to classify primary and control sequences that it holds out from the motif discovery process.
STREME assumes that each primary sequence may contain zero or one occurrences (sites) of the motif [the so-called ‘ZOOPS’ model (Bailey and Elkan, 1995)]. Motif discovery will not be negatively affected if a primary sequence contains more than one occurrence of a motif, but unlike MEME, STREME cannot discover motifs given only a single primary sequence.
Finally, STREME will work with user-specified (‘custom’) alphabets, as will MEME and DREME, but not the other algorithms studied here. This allows STREME to be applied to a much wider range of motif-discovery problems than HOMER, Peak-motifs, ProSampler or Weeder, including finding motifs in epigenetically modified DNA or post-translationally modified proteins.
In the remainder of this article, we describe the STREME algorithm in more detail and present experimental results comparing its performance with several widely used motif discovery algorithms. For the experimental comparisons, we consider motif discovery in TF ChIP-seq datasets and in RBP CLIP-seq datasets. In each case, we validate the predicted motifs using motifs derived using completely independent assays—high-throughput SELEX (Jolma et al., 2013) for TF motifs, and RNAcompete (Ray et al., 2013) for RBP motifs. In Supplementary Material, we present an example of using STREME to find motifs in sequences in a user-defined alphabet (English).
2 Materials and methods
2.1 The STREME algorithm
STREME searches for motifs by performing step 1 (below), and then iterating steps 2 through 6 until the stopping criterion is met. The stopping criterion, chosen by the user, can be either a maximum number of motifs, a minimum significance (maximum P-value) threshold or a maximum run-time. Details on each of the steps below are given in Supplementary Material.
-
Dataset preparation. STREME first reads the input sequence dataset(s) (primary, and optionally, control), converting to uppercase if the sequence alphabet is not case sensitive, and converting all ambiguous characters to a unique ‘separator’ character that is not present in the alphabet.
To ensure that STREME will give the same results regardless of the order of the sequences in the input dataset(s), it sorts the input dataset(s) alphabetically by sequence content, and then randomizes the order of the sequences in the dataset(s).
Next, if the user does not provide a set of control sequences, STREME creates one from the primary sequences. Each primary sequence is shuffled, preserving the frequencies of all words of length k (‘k-mers’) within it, where k can be specified by the user. The shuffling also preserves the positions of any separator characters. This prevents artifacts caused by the presence of ambiguous characters (such as the ‘N’ character used by DNA repeat-masking programs).
Based on the input sequences, STREME chooses the statistical test it will use. It will use Fisher’s exact test if the primary and control sequences have the same average length (within 0.01%), otherwise, it will use the Binomial test.
Then, STREME creates a Markov model of the control sequences of order. STREME uses this model in conjunction with the PWM to compute the likelihood ratio scores of words during all stages of motif discovery and evaluation.
Finally, STREME creates a ‘hold-out’ dataset for assigning statistical significance to each discovered motif. By default, the hold-out set consists of a random sample of 10% of the sequences in the primary and control datasets.
Suffix tree creation. STREME builds a generalized suffix tree that includes both the primary and control sequences (but not the hold-out set sequences). If the alphabet is complementable, STREME adds the reverse complement of each primary and control sequence to the tree as well. For the first round of STREME, the sequences are those created by Step 1, above. For subsequent rounds, the sequences from Step 6, with previous motifs erased, are used. STREME builds the suffix tree using code developed for MUMmer (Kurtz et al., 2004) based on the McCreight (1976) method.
Seed word evaluation. For each word of length 3 to the maximum motif width in the primary sequences, STREME uses the tree to efficiently count the numbers of exact matches to the word in the primary and control sequences, and computes its enrichment P-value. For each of the NEVAL (default = 25) most significant words of each width, STREME then uses the tree to count the number of approximate matches to it, and computes its enrichment P-value.
Motif refinement. STREME converts each of the NREF (default = 4) best seed words of each width in the user-specified range into a PWM (which we will refer to as the ‘motif’), and then iteratively refines each such motif. At each iteration of refinement, the current motif and the -order background are used with the suffix tree to efficiently find the best site in each sequence. The primary and control sequences are then sorted by the log-likelihood score of their best site, and the score threshold that optimizes the P-value of the statistical test is found. The iteration ends by using maximum likelihood estimation to estimate a new version of the motif from the single best site in each primary sequence whose score is above the optimal threshold. STREME uses this new motif in the next refinement iteration. Refinement stops when the P-value fails to improve, or NITER (default = 20) iterations have been performed. As the final motif for the round, STREME selects the motif that best discriminates the primary sequences from the control sequences.
Motif significance computation. STREME computes the statistical significance of the motif by using the motif and the optimal discriminative score threshold (based on the primary and control sequences) to classify the hold-out set sequences, and then applying the statistical test to the classification. Classification is based on the best match to the motif in each sequence (on either strand when the alphabet is complementable).
Motif erasing. STREME ‘erases’ every site matching the best motif in the primary, control and hold-out sequences (and their reverse complements, if the alphabet is complementable) by converting the positive-scoring letters in the sites to the separator character.
2.2 Evaluating motif discovery algorithms with ChIP-seq and CLIP-seq datasets
In order to evaluate the accuracy, sensitivity, thoroughness and speed of motif discovery algorithms on ChIP-seq data, we identify 40 TF ChIP-seq experiments in K562 cells (from ENCODE) for which there is a motif derived from high-throughput SELEX data for the same TF from Jolma et al. (2013), or if not, for a member of the same TF family. To evaluate the accuracy of the algorithms on CLIP-seq data, we determine the set of 20 RBPs with ENCODE enhanced CLIP-seq (eCLIP) datasets from K562 cells that also have at least one motif in the RNAcompete database (Ray et al., 2013) for Homo sapiens. Complete details on how we identify the reference motif for each ChIP-seq and eCLIP dataset, how we create the primary and control sequence sets from each dataset, as well as details on how we compute the accuracy, sensitivity and thoroughness of each motif discovery algorithm, are given in Supplementary Material.
3 Results
3.1 Comparison of motif discovery algorithms on TF ChIP-seq data
3.1.1. Accuracy
The accuracy of a motif discovery algorithm on a TF ChIP-seq dataset is its ability to discover an accurate version of the binding motif of the ChIP-ed TF in the set of DNA sequences identified as bound by the TF. We evaluate the accuracy of motif discovery algorithms by measuring the similarity of each of the discovered motifs to the known motif for the TF, using the Tomtom motif comparison algorithm (Gupta et al., 2007). In all experiments in this article, we run Tomtom using its command-line defaults. So that higher scores will correspond to more accurate motifs, we use minus the base-10 logarithm of the Tomtom P-value of the similarity between the discovered motif and the reference motif as our motif similarity score. To comprehensively understand the relative quality of different algorithms, rather than picking a single similarity score, we plot their success rates across the whole gamut of similarity scores. To avoid circularity, we use motifs derived using SELEX—an in vitro assay—as our reference motifs. For each of 40 ENCODE TF ChIP-seq experiments in K562 cells for which there is a known TF motif in the Jolma et al. (2013) compendium of SELEX-derived motifs, we prepare primary and control datasets, and assign a single SELEX motif to be the reference motif for that experiment (see Supplementary Material). The primary sequence dataset for an experiment consists of the 100 bp sequence regions centered on each of the ChIP-seq peaks; the control dataset comprises a shuffled version of each of the primary sequences, with the frequency of 3-mers in each sequence preserved. (Weeder does not use a control dataset, so we present it with only the primary dataset.) Because ChIP-seq peak regions often contain enriched motifs besides that of the ChIP-ed TF (e.g. binding motifs of cofactors), the motif discovery algorithms were each allowed to report five motifs when presented with the primary and control datasets. We run each algorithm with its default settings, except we set the minimum and maximum motif widths to 8 and 12, respectively. For the three algorithms that construct a background model from their input sequences (STREME, MEME and Peak-motifs), we specify that a second-order Markov model be used.
As shown inFigure 1a, the accuracy of STREME is as good or better than that of the other algorithms tested here. The best motif found by STREME in ChIP-seq data is more similar to the reference motif derived from SELEX data, on average, than the best motif found by the other algorithms, across a wide range of similarity score thresholds. STREME, MEME and HOMER all discover a motif with a similarity score of at least 5 (Tomtom P-value ) in at least 70% of the ChIP-seq datasets. STREME actually finds such a motif 82.5% of the time. Additionally, STREME discovers about twice as many highly accurate motifs as the other two algorithms at the more stringent motif similarity score threshold of 9 (Tomtom P-value ). At this motif similarity score threshold, STREME is successful on 32.5% of the ChIP-seq datasets, whereas HOMER is only successful on 12.5% of the datasets.
Fig. 1.

Accuracy of motif discovery algorithms on ENCODE TF ChIP-seq datasets. The curves in (a) show the percentage of ChIP-seq datasets (Y) where the best motif found by the named algorithm has motif similarity score , averaged over 40 ChIP-seq datasets. (b) The sequence logos and accuracies of the best motifs found by STREME (top) and HOMER (bottom) in an ENCODE ChIP-seq dataset for the CTCF TF (UtaK562Ctcf), aligned to the SELEX reference motif (center) from the Jolma et al. (2013) compendium (CTCF_full). Similar alignments are given for all 40 ChIP-seq datasets in Supplementary Material
Figure 1b gives an example of a type of motif where the HOMER algorithm is sometimes less accurate than STREME because, unlike STREME, HOMER does not keep track of strand information for individual motif sites. This causes HOMER to sometimes combine overlapping sites on the two DNA strands into a motif that is a perfect palindrome, when, in fact the motif is quite asymmetrical (e.g. Fig. 1b). STREME avoids this problem by keeping track of strand information, and by never allowing more than one site in a sequence (on either strand) to contribute to a motif. Sequence logos for the best STREME and HOMER motifs are shown, along with their motif accuracies (Tomtom P-values), for all 40 TF ChIP-seq datasets studied here in Supplementary Table S6. As seen in that table, the STREME motif is more accurate than the HOMER motif for 29 of the 40 TF ChIP-seq datasets.
3.1.2 Sensitivity
The sensitivity of a motif discovery algorithm is its ability to discover motifs that are present in only a small fraction of the primary input sequences. To study this aspect of performance, we construct a series of progressively more difficult primary datasets from each of the original 40 primary TF ChIP-seq datasets by shuffling the letters of up to 99% of the sequences in the dataset, preserving the frequency of 3-mers, which removes any motif occurrences from the shuffled sequences. As before, for each such ‘diluted’ primary dataset, we create a control dataset by shuffling the letters of all the sequences in it, preserving the frequency of 3-mers. We let each motif discovery algorithm report five motifs, allowing the algorithm to choose the optimal motif width in the range 8 to 12.
To analyze the results, we must choose a motif similarity score threshold for deciding if the algorithm has found a motif matching the reference motif for the dataset. Figure 2 compares the algorithms using a motif similarity score threshold of 5 (Tomtom P-value ); using higher or lower thresholds gives similar results (data not shown). Across the range of dataset ‘purity’ from 100% (the original ChIP-seq peak sequences) to 1% (99% of sequences are shuffled), STREME is at least as likely as HOMER to discover the ChIP-ed TF’s motif, and more likely than the other algorithms. We conclude that the sensitivity of STREME is as good or better than that of the algorithms we examine here.
Fig. 2.

Sensitivity of motif discovery algorithms on ENCODE TF ChIP-seq datasets. Each point shows the percentage of times (Y) that the best motif found by the named algorithm has motif similarity score at least five (Tomtom P-value ) when run on a primary dataset that has been diluted to a given purity (X), averaged over 40 ChIP-seq datasets. Note that the lowest dataset purity tested is 1%
3.1.3. Thoroughness
The thoroughness of a motif discovery algorithm is the degree to which it can discover many distinct motifs in a given primary set of sequences. This is a particularly important aspect of motif discovery in TF ChIP-seq datasets, where multiple TFs often bind in close proximity to regulate transcriptional expression. To measure the ability of STREME and other algorithms to discover multiple motifs, we construct an artificial ChIP-seq dataset by combining 100 randomly selected sequences from each of 21 of our 40 ENCODE TF ChIP-seq primary datasets. (See Supplementary Material for how the 21 datasets were selected.) This creates a set of primary sequences containing bound regions for all 21 ChIP-ed TFs. We repeat the random sampling process 20 times to create 20 distinct primary datasets. Using these 20 primary datasets and shuffled versions of them as control datasets, we allow each motif discovery algorithm to report 25 motifs with widths in the range 8–12, and we measure each algorithm’s average success rate at finding all 21 reference motifs at different motif similarity score thresholds.
Figure 3 shows that STREME is generally more thorough in this setting than the other algorithms tested here. At motif accuracies less stringent than (similarity score < 7), STREME finds substantially more motifs than any of the other algorithms. At stricter accuracy thresholds, only MEME is as thorough as STREME.
Fig. 3.
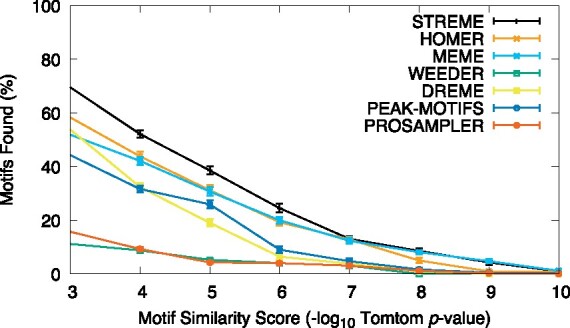
Thoroughness of motif discovery algorithms on combined ENCODE TF ChIP-seq datasets. The curves show the percentage of 21 reference motifs (Y) for which the named algorithm finds a motif matching it with given motif similarity score (X) or better, averaged over 20 combined datasets
3.1.4. Speed
Figure 4 shows the running time of the motif discovery algorithms on each of the 40 experiments described above in the Accuracy section. We run the algorithms using a single thread on a 4.0 GHz Intel Core i7 processor with 16GB of memory. The running time of STREME compares favorably with that of the other algorithms we test with the exception of ProSampler, which is about 100 times faster than STREME. Peak-motifs is slightly faster than STREME, but only when the primary sequence dataset contains more than 20 000 sequences of 100 bp. With fewer than 10 000 sequences, STREME is up to an order of magnitude faster than Peak-motifs. STREME is slightly faster in this setting than HOMER. As we shall show below (see Fig. 6), STREME is much faster than HOMER for finding motifs wider than 12 positions. At all dataset sizes, STREME is approximately an order of magnitude faster than DREME, the algorithm it replaces within the MEME Suite. In Supplementary Material, we show detailed results on the running time of STREME with DNA, RNA or Protein sequence datasets of different sizes and sequence lengths (Supplementary Fig. S1).
Fig. 4.

Speed of motif discovery algorithms on ENCODE TF ChIP-seq datasets. Each point represents the running time (Y) of the named algorithm on one of 40 ENCODE TF ChIP-seq datasets containing the given number of sequences (X). For ease of interpretation, the points for each algorithm have been fit with a smooth Bezier curve
Fig. 6.

Running time as a function of maximum motif width. Each point shows the running time in seconds per motif found (Y) when the named motif finder is run with the given maximum motif width (X) set, averaged over 25 ChIP-seq primary sequence datasets each containing 10 000 sequences of length 100 bp. Error bars show standard error. The points for a given motif discovery algorithm are connected with straight lines for ease of interpretation. The algorithms were run on a 3.2 GHz Intel Core i7 processor with 16 GB of memory
3.1.5. Choosing the background model
In the preceding sections, we use a value of k = 3 when creating sets of control sequences from each primary set by shuffling each sequence while preserving the frequencies of all k-mers within it. For STREME, MEME and Peak-motifs, we also instruct them to build an internal Markov model of order .
This choice of k is justified by the results shown in Supplementary Figure S10, which shows how each of the algorithms studied here (except Weeder) performs in terms of accuracy, sensitivity and thoroughness using values of k from 1 to 4. (Weeder is not included in the figure as it does not use a set of control sequences.) For all algorithms tested here, using a value of k = 3 provides (near) optimum results. Supplementary Figure S10 shows that, with the TF ChIP-seq datasets we use here, the effect of k is particularly large for STREME and HOMER.
3.1.6. Accuracy on ‘small’ datasets
We also study the accuracy of motif discovery algorithms on datasets with from 10 to 1000 sequences, using randomly chosen samples of increasing size from each of the 40 ENCODE TF ChIP-seq datasets. From each of these primary sequence datasets, we create a control dataset by randomly shuffling each sequence while preserving the frequencies of 3-mers. We run the motif discovery algorithms on each of the resulting pairs of datasets, and measure the accuracy of the best motif out of five, as before (see the subsection on ‘Accuracy’, above).
Figure 5 shows, that with small datasets, the MEME algorithm, which is based on maximizing motif information content, more frequently finds a motif with similarity score at least 5 (Tomtom P-value ) than the other algorithms, all of which are based on maximizing the motif’s ability to classify sequences. In this setting, STREME performs the best of the classification-based algorithms, substantially better than HOMER. These observations remain true using motif similarity score thresholds from 4 to 7, inclusive (data not shown).
Fig. 5.

Accuracy of motif discovery algorithms on ‘small’ datasets. Each point shows the percentage of times (Y) the best motif found by the named algorithm in a (sampled) dataset with the given number of sequences (X) has motif similarity score at least 5 (Tomtom P-value ), averaged over each of 40 TF ChIP-seq datasets
With primary datasets containing more than 1000 sequences, MEME bases its search on only a random subsample of 1000 sequences. (It does this because the running time of MEME’s underlying algorithm increases at least quadratically with the size of the sequence dataset.) With full-size ChIP-seq datasets, which typically contain many thousands of sequences, the classification-based algorithms (especially STREME) find more accurate motifs (Fig. 1), find fainter motifs (Fig. 2) and find more co-factor motifs (Fig. 3). We have incorporated STREME into the MEME-ChIP tool (Machanick and Bailey, 2011), which now combines the motifs discovered by MEME and STREME into a single comprehensive analysis of ChIP-seq and similar data.
3.1.7. Performance finding ‘wide’ motifs
Many TF binding motifs are wider than 12 positions, the maximum motif width we set for the motif discovery algorithms in the results presented thus far. The C2H2 zinc finger proteins comprise the largest family of eukaryotic TFs, and many have motifs much wider than 12 positions (Fedotova et al., 2017), as do twelve of the TFs studied here (see Supplementary Table S6). Three of the motif discovery algorithms studied here (STREME, HOMER and MEME) allow the user to specify the range of motif widths that the algorithm may search for. We conduct additional experiments to determine how running time scales with maximum motif width (up to a maximum of 30), and how the accuracy, sensitivity and thoroughness of the algorithms on TF ChIP-seq datasets is affected when we set the maximum motif width to 18 (instead of 12).
Figure 6 shows how the running times of the three motif discovery algorithms scale with maximum motif width using randomly chosen DNA sequences. We run each of the algorithms on 25 (artificial) primary sequence datasets, with the maximum motif width specified as a number from 10 to 30, and the minimum motif width always set to 8. We create each primary sequence dataset by sampling one of our 40 ENCODE TF ChIP-seq datasets. Each such artificial sequence dataset contains 10 000 length 100 bp DNA sequences, and we create a matching control dataset by shuffling the primary dataset while preserving the frequencies of 3-mers.
STREME is much faster than either HOMER or MEME for finding motifs wider than 12, as seen in Figure 6. For finding motifs up to 30 positions wide, STREME is almost an order of magnitude faster than the other two algorithms. The improved speed relative to HOMER is due to the way STREME searches its suffix tree, computing scores for motifs from the minimum to the maximum specified width in a single, depth-first traversal of the tree. By contrast, HOMER repeats its traversal of the tree for each width in the width range. The running time of MEME with large datasets such as used here (10 000 sequences) is generally much higher than that of HOMER, except for the widest motifs.
Next, we study how the accuracy, sensitivity, thoroughness and speed of the motif discovery algorithms compare when they search for wider motifs in the same 40 ENCODE TF ChIP-seq datasets we used in the previous sections. The only change to the experiments from those sections is that we ran STREME, HOMER and MEME with the maximum motif width parameter of each set to 18, rather than 12.
Supplementary Figure S7a shows that STREME finds more motifs across all motif similarity score thresholds than either HOMER or MEME even with the wider maximum motif setting. Similarly, Supplementary Figure S7b shows that STREME is still at least as sensitive as HOMER, and considerably more so than MEME, even with the maximum motif width set to 18 instead of 12. STREME is also still more thorough than HOMER and MEME when we set the motif similarity score threshold below 7 (Supplementary Fig. S7c). When searching for motifs with widths up to 18, STREME is about an order of magnitude faster than HOMER (Supplementary Fig. S7d).
3.2 Comparison of motif discovery algorithms on CLIP-seq datasets
The accuracy of a motif discovery algorithm on an RBP CLIP-seq dataset is its ability to discover an accurate version of the binding motif of the RBP in the set of RNA sequences identified as bound by the RBP. As with the DNA (ChIP-seq) experiments above, we compare motif discovery algorithms run on data from an in vivo assay—eCLIP data from ENCODE (Van Nostrand et al., 2016)—using reference motifs for the same RBPs derived using an in vitro assay—RNAcompete (Ray et al., 2013). As before, we use the Tomtom motif comparison algorithm to estimate the accuracy of the discovered motifs. We identify twenty eCLIP datasets for which an RNAcompete motif exists for the same RBP. As the primary sequence dataset, we use the full-length RNA sequences identified as bound by the ENCODE eCLIP experiment. We find that using shuffled sequences as the control dataset gives poorer results (data not shown) than using a random sample of 10 000 full-length bound sequences identified in the 120 ENCODE RNAcompete experiments, and that is what we use in the following experiments. We let each motif discovery algorithm discover five motifs, and allow the width to range from 7 to 8, since that is the range of widths in the RNAcompete RBP motif compendium. As with the DNA (ChIP-seq) experiments above, for the three algorithms that construct a background model from their input sequences (STREME, MEME and Peak-motifs), we instruct them to build a second order Markov model.
Figure 7 shows that none of the motif discovery algorithms finds a motif that matches the reference motif more than 50% of the time even at a very permissive motif similarity score threshold of 3 (Tomtom P-value ). STREME, ProSampler and Weeder are perhaps slightly more accurate than the others, but the sample size is too small to declare that the difference is significant. Supplementary Table S7 shows sequence logos (Schneider and Stephens, 1990) for the best STREME and Weeder motifs, and their accuracies (Tomtom P-values), aligned to the logo of the reference motif, for each of the 20 eCLIP experiments.
Fig. 7.

Accuracy of motif discovery algorithms on ENCODE RBP eCLIP datasets. The curves in the figure show the percentage of times (Y) the best motif found by the named algorithm has motif similarity score X or better, averaged over 20 eCLIP datasets
The lower motif accuracy we observe in this experiment is partly due to the fact that the RBP reference motifs are generally much shorter than TF binding motifs, which causes the Tomtom P-values of even very accurate motifs to be higher. In addition, in the three (out of 20) datasets where both STREME and Weeder fail to find a motif that is at all similar to the reference motif (FMR1, FUS, IGF2B2 in Supplementary Table S7), the problem might lie with the algorithms, with those particular eCLIP datasets, or with the RNAcompete reference motifs. To check the first possibility, we compared the statistical enrichment of the RNAcompete reference motif and the most similar STREME motif for each experiment using the ENR motif enrichment algorithm from the MEME Suite. The motifs found by STREME and Weeder in the FMR1, FUS and IGF2BP2 eCLIP datasets are much more enriched than the reference motifs in those datasets (data not shown). For only two of the 20 eCLIP experiments was the RNAcompete reference motif more enriched in the eCLIP sequences than the STREME motif. The same was true for the motifs found by Weeder—in only two out of 20 cases was the RNAcompete motif more enriched than the Weeder motif. These results suggest that there may be problems with either the in vitro RNAcompete reference motifs or with the in vivo eCLIP datasets for FMR1, FUS and IGF2BP2.
3.3 STREME provides useful estimates of motif statistical significance
Knowing the statistical significance of a discovered motif is a first step in deciding if it might be biologically significant. HOMER, Weeder, Peak-motifs and ProSampler produce no estimates of motif significance. The estimates produced by MEME can be extremely conservative, potentially causing biologically interesting motifs to be rejected by the user based on the estimate of statistical significance provided by MEME. DREME provides useful estimates of statistical significance, and we show here that STREME does, too.
For each motif STREME discovers, it reports a P-value that it estimates using a reserved portion of the sequences in its input. To verify the accuracy of these P-values, we run STREME on 10 000 randomly generated datasets, each containing 10 000 sequences, allowing STREME to find exactly one motif in each dataset. Since all 1000 motifs are ‘discovered’ in random sequences, the P-values should follow a uniform distribution. To check that they are uniformly distributed, we create a Q-Q plot (Wilk and Gnanadesikan, 1968), which plots the theoretical value of the nth largest P-value, , versus the P-value reported by STREME, y.
The Q–Q plots in Figure 8 and Supplementary Figure S8 show that, for motifs with at least 10 predicted sites in the hold-out sequences, the P-values reported by STREME are accurate to within factor of 2 for P-values greater than 0.001. Since the points in the Q–Q plots are above the line y = x, STREME’s P-values are conservative. The P-values of motifs with lower support (fewer than 10 sites in the hold-out sequences) are more conservative, skewing the distribution in the overall Q–Q plot (Supplementary Fig. S9). These results hold for sequences over the DNA, RNA or protein alphabets and regardless of whether the control sequences are the same length as the primary sequences (Fisher exact test) or not (Binomial test). They also hold when we do not provide a set of control sequences to STREME, causing it to create a control set by shuffling the primary sequences, and when the input sequences have variable lengths from 50 to 150 characters (data not shown).
Fig. 8.

Q–Q accuracy plot of the P-values reported by STREME for motifs with at least 10 sites in the hold-out set. Shown is the Q–Q plot for the P-values reported by STREME run on 10 000 datasets containing 10 000 random DNA sequences. Primary and control sequences are 100 characters long. Ideally, the points should lie along the line y = x
To use STREME P-values to determine if motifs are significant at the α level, set , where n is the number of motifs reported by STREME. Motifs with P-values at most P are significant at the α level. Since STREME P-values are conservative, the probability of a type I error will be at most α.
4 Discussion
The two major contributions of this work are the new STREME algorithm, and a comprehensive and rigorous evaluation of the performance of STREME and a number of other motif discovery algorithms on ChIP-seq data. Our evaluation includes the most widely used algorithms for ChIP-seq motif analysis—HOMER, MEME, DREME and Peak-motifs—as well as the recently developed, ultrafast, ProSampler. We call attention to the circularity of testing motif discovery algorithms using reference motifs derived from the same type of data, and use SELEX reference motifs when evaluating motif discovery in ChIP-seq data. An additional contribution of this work is that the P-values reported by STREME should reduce the number of biologically significant motifs missed, as well as the number of spurious motifs reported in the literature.
Our results uncover important differences in the performance of these algorithms that were not previously apparent. For example, although ProSampler has a comparable success rate to STREME, MEME and HOMER when the similarity threshold to the reference motif is lax, at a more stringent similarity threshold where STREME is successful on 45% of the ChIP-seq datasets, ProSampler succeeds on only 20% (Fig. 1a, Motif Similarity Score ). We also observe that HOMER, which is perhaps the most widely used algorithm for motif discovery in ChIP-seq data, has an algorithmic weakness that sometimes causes it to report erroneous motifs. HOMER allows overlapping sites on opposite DNA strands to contribute to the motif’s PWM, which can cause problems even with highly information-rich TF motifs such as that of CTCF (Fig. 1b).
Other differences we uncover in the algorithms are their ability to handle noisy data, which we evaluate by injecting noise into real ChIP-seq datasets; their ability to find co-factor motifs, which we evaluate by combining sequences from multiple ChIP-seq datasets; and their ability to deal with datasets with fewer than 10 000 sequences, which we evaluate by subsampling from ChIP-seq datasets. In each of these three aspects, STREME is superior to the other algorithms, with the exception that MEME and Weeder perform better with very small (fewer than 100) sequence datasets. STREME is about 15% more successful than HOMER in the co-factor discovery evaluation, and significantly better than HOMER when the ChIP-seq dataset contains few than 10 000 sequences—a frequent occurrence in practice. These three evaluations also show that ProSampler rapidly loses sensitivity as the number of sequences containing the motif decreases. But because ProSampler is orders of magnitude faster than the other algorithms, it would be worthwhile to explore if its sensitivity problem could be corrected.
We also show that STREME performs as well as other motif discovery algorithms when applied to RNA from CLIP-seq experiments. In addition to DNA and RNA sequences, STREME can be applied to motif discovery in protein sequences, although we do not study that application here. STREME also allows the user to define their own sequence alphabet. In Supplementary Material, we illustrate this capability by using STREME to discover the key words and phrases in a highly corrupted version of an English text.
Supplementary Material
Acknowledgements
The author thank Charles E. Grant for proofreading the manuscript.
Funding
This work was supported by National Institutes of Health award [R01 GM103544].
Conflict of Interest: none declared.
References
- Bailey T.L. (2011) DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics, 27, 1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T.L., Elkan C. (1995) The value of prior knowledge in discovering motifs with MEME. In Proceedings of the Third International Conference on Intelligent Systems for Molecular Biology, July 16–19, 1995, Cambridge, UK, Vol. 3, pp. 21–29. [PubMed]
- Fedotova A.A. et al. (2017) C2h2 zinc finger proteins: the largest but poorly explored family of higher eukaryotic transcription factors. Acta Nat., 9, 47–58. [PMC free article] [PubMed] [Google Scholar]
- Fisher R.A. (1922) On the interpretation of from contingency tables, and the calculation of p. J. R. Stat. Soc., 85, 87–94. [Google Scholar]
- Gupta S. et al. (2007) Quantifying similarity between motifs. Genome Biol., 8, R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S. et al. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell, 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolma A. et al. (2013) DNA-binding specificities of human transcription factors. Cell, 152, 327–339. [DOI] [PubMed] [Google Scholar]
- Kurtz S. et al. (2004) Versatile and open software for comparing large genomes. Genome Biol., 5, R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. et al. (2019) ProSampler: an ultrafast and accurate motif finder in large ChIP-seq datasets for combinatory motif discovery. Bioinformatics (Oxford, England), 35, 4632–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machanick P., Bailey T.L. (2011) MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics, 27, 1696–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCreight E.M. (1976) A space-economical suffix tree construction algorithm. J. ACM, 23, 262–272. [Google Scholar]
- Nagarajan N. et al. (2005) Computing the P-value of the information content from an alignment of multiple sequences. Bioinformatics, 21, i311–i318. [DOI] [PubMed] [Google Scholar]
- Pavesi G. et al. (2004) Weeder Web: discovery of transcription factor binding sites in a set of sequences from co-regulated genes. Nucleic Acids Res., 32, W199–W203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray D. et al. (2013) A compendium of RNA-binding motifs for decoding gene regulation. Nature, 499, 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid J.E., Wernisch L. (2011) STEME: efficient EM to find motifs in large data sets. Nucleic Acids Res., 39, e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T.D., Stephens R.M. (1990) Sequence logos: a new way to display consensus sequences. Nucleic Acids Res., 18, 6097–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stormo G.D. (2000) DNA binding sites: representation and discovery. Bioinformatics, 16, 16–23. [DOI] [PubMed] [Google Scholar]
- Thomas-Chollier M. et al. (2011) RSAT 2011: regulatory sequence analysis tools. Nucleic Acids Res., 39, W86–W91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand E.L. et al. (2016) Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods, 13, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner P. (1973) Linear pattern matching algorithms. In: 14th Annual Symposium on Switching and Automata Theory. IEEE, pp. 1–11.
- Wilk M.B., Gnanadesikan R. (1968) Probability plotting methods for the analysis of data. Biometrika, 55, 1–17. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.