

Abstract
BACKGROUND
Cerebral cavernous malformations (CCMs) are common sporadic and inherited vascular malformations of the central nervous system. Although familial CCMs are linked to loss-of-function mutations in KRIT1 (CCM1), CCM2, or PDCD10 (CCM3), the genetic cause of sporadic CCMs, representing 80% of cases, remains incompletely understood.
METHODS
We developed two mouse models harboring mutations identified in human meningiomas with the use of the prostaglandin D2 synthase (PGDS) promoter. We performed targeted DNA sequencing of surgically resected CCMs from patients and confirmed our findings by droplet digital polymerase-chain-reaction analysis.
RESULTS
We found that in mice expressing one of two common genetic drivers of meningioma — Pik3caH1047R or AKT1E17K — in PGDS-positive cells, a spectrum of typical CCMs develops (in 22% and 11% of the mice, respectively) instead of meningiomas, which prompted us to analyze tissue samples from sporadic CCMs from 88 patients. We detected somatic activating PIK3CA and AKT1 mutations in 39% and 1%, respectively, of lesion tissue from the patients. Only 10% of lesions harbored mutations in the CCM genes. We analyzed lesions induced by the activating mutations Pik3caH1074R and AKT1E17K in mice and identified the PGDS-expressing pericyte as the probable cell of origin.
CONCLUSIONS
In tissue samples from sporadic CCMs, mutations in PIK3CA were represented to a greater extent than mutations in any other gene. The contribution of somatic mutations in the genes that cause familial CCMs was comparatively small. (Funded by the Fondation ARC pour la Recherche contre le Cancer and others.)
CEREBRAL CAVERNOUS MALFORMATIONS (CCMs) are vascular lesions of the central nervous system that are composed of abnormally enlarged capillary cavities without intervening brain parenchyma; the condition affects 1 in 200 to 250 persons.1 Although they are primarily characterized by subclinical bleeding, CCMs can lead to seizures and hemorrhagic stroke with substantial neurologic complications, especially when localized in the brain stem. More than 80% of CCMs occur sporadically2; patients with familial CCMs harbor biallelic germline and somatic loss-of-function mutations in one of three “CCM genes,” which affect endothelium stabilization: Krev interaction trapped 1 (KRIT1, also called CCM1), cerebral cavernous malformation 2 (CCM2), and programmed cell death 10 (PDCD10, also called CCM3).3 At least some sporadic CCMs also involve somatic mutations in these genes,4–6 but their genetic architecture remains poorly understood.
Meningiomas, the most common primary tumors of the central nervous system,7 are caused by recurrent mutations in NF2, PIK3CA (encoding phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit A), and AKT1 (encoding serine threonine kinase 1).8 Activating mutations in PIK3CA, an oncogene that is mutated in 9% of human cancers (https://cancer.sanger.ac.uk/cosmic), are present in 4 to 7% of meningiomas, whereas another 10% of meningiomas (primarily meningothelial skull-base tumors) harbor the common AKT1E17K oncogenic mutation.9,10 In meningioma, PIK3CA as well as AKT1E17K mutations typically cooccur with mutations in other genes, most notably TRAF7,9,10 which has raised doubts as to whether PIK3CA or AKT1 mutations per se can drive tumorigenesis.
To address this question, we generated two mouse models to selectively express Pik3caH1047R, the most common meningioma-associated PIK3CA mutation, and AKT1E17K in prostaglandin D2 synthase (PGDS)–positive cells. The unexpected observation of typical CCMs in both models then motivated us to investigate the possible involvement of PIK3CA and AKT1 mutations in sporadic CCMs.
METHODS
MOUSE MODELS
We previously showed that meningeal precursor cells expressing PGDS are the cells of origin for diverse meningioma histologic subtypes in mice.11,12 To induce activation of Pik3ca in PGDS-expressing cells in mice, we bred R26-Pik3caH1047R mice with homozygous PGDSCre mice.11,13 We also forced the expression of AKT1E17K in cells expressing PGDS by injecting a FLAG-tagged RCAS-AKT1E17K vector — either subdurally at the convexity or intraorbitally at the skull base — into PGDStv-a neonates, as described previously.12,14 Additional details on PGDS expression and on both mouse models are provided in Supplementary Appendix 1, available with the full text of this article at NEJM.org.
STUDY PATIENTS
Patients who had been treated for sporadic CCMs at Pitié-Salpêtrière Hospital, Paris, between 2010 and 2018 (82 patients) or at Caen Hospital, Caen, France, between 2008 and 2017 (6 patients) were included in the study. Patients with a family history of cavernomas or multiple lesions on preoperative T2-weighted magnetic resonance imaging (MRI) were excluded. Histologic diagnosis of the lesions was reviewed by the study pathologists and validated according to World Health Organization guidelines. Patients with cerebral arteriovenous malformations were included as controls. Written informed consent was provided by all patients who were 18 years of age or older and by both parents of the one patient who was younger than 18 years of age. Additional details are provided in Supplementary Appendix 1. The authors vouch for the accuracy and completeness of the data in this report.
GENOMIC ANALYSES
For formalin-fixed, paraffin-embedded samples, all slides were reviewed by a pathologist, and punches in the tissue block were performed to enrich each sample with lesion cells. Targeted deep sequencing of genomic DNA extracted from the lesions was performed. Molecular inversion probes were designed with the use of MIPgen to capture the exonic bases and exon–intron boundaries of CCM1, CCM2, and CCM3 and the recurrent variants of AKT1 (p.E17K) and PIK3CA (p.E542K, p.H1047R, and p.H1047L) on the canonical transcripts. Droplet digital PCR (ddPCR) was performed to orthogonally validate the PIK3CA and AKT1 mutations. Additional details of the genomic analyses are provided in Supplementary Appendix 1.
RESULTS
MICE WITH SOMATIC PIK3CA AND AKT1 MUTATIONS
We analyzed a cohort of 37 PGDSCre;R26-Pik3caH1047R mice (referred to as Pik3caH1047R mice hereafter; mean follow-up, 7.9 months) (Fig. S1 and Table S1 in Supplementary Appendix 1). On autopsy, only 1 mouse (3%) was found to have a grade I meningothelial meningioma (Fig. S2). To our surprise, 8 mice with weight loss or hemiplegia (22%; mean age at diagnosis, 2.4 months) were found to have intraparenchymal CCM lesions, most of which were located in the brain stem (Fig. 1A–1F). Histologically, the lesions ranged from intraparenchymal vessel dilatations to capillary telangiectasia and clusters of capillary telangiectasia forming “young” cavernomatous lesions, thus retracing the natural history of CCM formation in mice (Fig. 1G–1L). Of the 37 mice, 10 (27%) had meningothelial proliferations (Fig. S2). We then generated a second cohort of 21 Pik3caH1047R mice, a subset of which we analyzed at the onset of clinical symptoms (3 mice; onset at 3, 4, and 5 weeks of age): these mice had intraparenchymal CCMs in the brain stem.
Figure 1. Characterization of Mouse Pik3caH1047R CCMs.

Panel A is a whole-mount view of a dissected brain showing a cerebral cavernous malformation (CCM) in the brain stem of a 1-month-old PGDSCre;Pik3caH1047R mouse (scale bar, 1 mm). Panel B shows a section through the lesion in which blood-filled caverns are visible (scale bar, 200 μm). Panel C shows in situ hybridization, with prostaglandin D2 synthase (Pgds) messenger RNA expression in pericavernomatous cells (scale bar, 50 μm). Panels D, E, and F show representative examples of CCMs located in the brain stem of 3.2-month-old (Panel D), 1-month-old (Panel E), and 2.8-month-old (Panel F) PGDSCre;Pik3caH1047R mice (scale bar, 2 mm). Panel G shows dilated capillaries in normal brain parenchyma (asterisks) (scale bar, 100 μm). Panel H shows the formation of noncoalescent intraparenchymal telangiectasias with preserved spherical structure (asterisks) (scale bar, 100 μm). Panel I shows fusion of adjacent telangiectasias with a thin band of residual brain parenchyma between the two caverns (arrowheads) (scale bar, 100 μm). Panel J shows immunohistochemical detection of PGDS-positive cells around dilated blood vessels (scale bar, 50 μm). The inset shows an example of a PGDS-positive cell bordering a cavern (arrow). Panel K shows Masson’s trichrome staining of a brain-stem lesion with thin borders separating the caverns (scale bar, 2 mm). The inset shows an example of a more mature human cavernoma, with thick blue collagen fibers separating the caverns. Panel L shows an example of intraluminal thrombi found in some CCMs (scale bar, 100 μm).
We also analyzed a cohort of 35 asymptomatic PGDStv-a;RCAS-AKT1E17K mice (Fig. S1 and Table S1); on autopsy, none were found to have meningioma (mean follow-up, 14.5 months); 2 mice (6%) had convexity osteomas with adjacent meningothelial proliferation, the first step in meningioma tumorigenesis14,15 (Fig. S3). It was notable that 4 mice (11%) had large intraparenchymal CCMs (Fig. 2A and 2B), which, in contrast to findings in the first model, were histologically mature and developed later (mean age at diagnosis, 13.5 months). In all cases, the intraparenchymal lesions were superficial, located near the injection site in the forebrain (Fig. 2C and 2D), and associated with AKT1E17K expression (Fig. 2E).
Figure 2. Characterization of Mouse AKT1E17K CCMs.
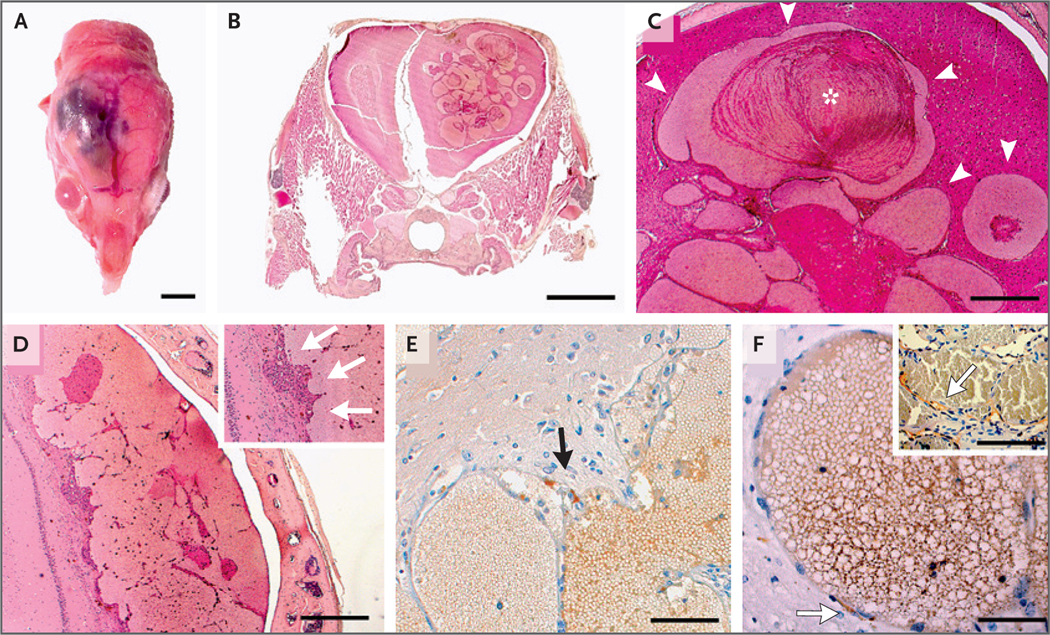
Panel A shows the macroscopic appearance of a right cerebral cortical lesion in a 13-month-old PGDStv-a;RCAS-AKT1E17K mouse (scale bar, 5 mm). Panel B shows an example of a frontal lobe CCM in a 13-month-old PGDStv-a;RCAS-AKT1E17K mouse (scale bar, 2 mm). Panel C shows the lesions, which consist of tortuous dilated vascular channels lined with a single layer of endothelium without intervening parenchyma (arrowheads), similar to human CCMs. Cavities are filled with erythrocytes along with intraluminal and parietal thrombi (asterisk); also evident are clusters of siderophages, lymphocyte infiltrates, and reactive gliosis, indicating past hemorrhages (scale bar, 500 μm). Panel D shows a cerebral cortical lesion formed by the coalescence of several small cavities with intraluminal thrombi (scale bar, 500 μm). The inset shows endothelial cells of neovessels (arrows) that line the cavities. Panel E shows FLAG immunohistochemical staining, which detected AKT1E17K expression in pericavernous cells of a mouse CCM, in the arachnoid cells of meningothelial proliferations, and in cells bordering CCMs (arrow), suggesting that the lesions are associated with AKT1E17K expression (scale bar, 100 μm). Panel F shows PGDS immunohistochemical staining, which indicates the presence of scattered PGDS-positive cells (arrow) boarding the intraparenchymal caverns without any contact with the meninges (scale bar, 50 μm). The inset shows a human CCM lesion with PGDS-positive cells bordering vascular channels (arrow).
SOMATIC PIK3CA MUTATIONS IN HUMAN SPORADIC CCMS
We then performed targeted DNA sequencing in a series of 88 sporadic CCMs (Table S2 [note that Tables S2 through S7 are provided in Supplementary Appendix 2]) to investigate the involvement of activating PIK3CA and AKT1 mutations. Given the small number of endothelial cells lining the caverns, we conducted targeted sequencing of the three CCM genes and the hotspot mutations in PIK3CA (E542K, H1047R, and H1047L) and AKT1 (E17K). We found that 28 CCMs (32%) had a PIK3CA mutation (PIK3CAE542K in 16 cases, PIK3CAH1047R in 10 cases, and PIK3CAH1047L in 2 cases). There was no predominant anatomical location for PIK3CA-mutant CCMs (Fig. 3). Four PIK3CAmutant samples also had mutations in CCM2 (Patient 1), CCM1 and AKT1 (Patient 5), or CCM1 (Patients 7 and 9); co-occurrence of mutations in PIK3CA with mutations in other genes is frequently seen in tumors. We also found one CCM2 mutation (Patient 4) and 2 CCM1 mutations (Patients 6 and 8). Finally, two samples harbored multiple mutations in CCM genes: one (Patient 2) had two loss-of-function CCM2 mutations16 and a CCM1 substitution of unknown pathologic significance, and the other (Patient 3) had two mutations (one in CCM1 and one in CCM3) with similar variantallele frequencies, both of which are described in the COSMIC database (Fig. 3 and Tables S3 and S4).
Figure 3. Detection of Mutations in Human Sporadic CCMs.

Panel A shows the allele frequencies of variants in PIK3CA, CCM1, CCM2, CCM3, and AKT1, determined on the basis of either the percentage of sequence reads that contained variants on next-generation sequencing (NGS) or the fractional abundance of variants on digital droplet polymerase-chain-reaction (ddPCR) analysis, in human sporadic CCM samples. Panel B shows clinical characteristics, including sample type (fresh-frozen or formalin-fixed, paraffin-embedded [FFPE] tissue), the sex of the patient, CCM location, and the specific activating mutation detected. For some samples, insufficient tissue remained for ddPCR analysis. Panel C shows examples of PIK3CA- and AKT1-mutant human CCMs. Axial three-dimensional T1-weighted MRI of the insular cavernoma harboring PIK3CA, CCM1, and AKT1 mutations (Patient 5) is shown, followed by axial MRIs of the PIK3CA-mutant CCMs with the highest fractional abundance of mutant reads. Patient numbers are shown beneath the scans.
A total of 78 of the CCM samples, including 3 for which paired blood samples were available, had remaining DNA that we used for orthogonal validation by ddPCR analysis. To assay sequence artifacts in DNA samples isolated from formalin-fixed, paraffin-embedded tissue, we genotyped, in parallel, AKT1E17K, PIK3CAE542K, PIK3CAH1047L, and PIK3CAH1047R in 11 formalin-fixed, paraffin-embedded samples of arteriovenous malformations. The false positives in these samples did not exceed a fractional abundance of 0.1%. We therefore selected a threshold for fractional abundance of 0.5% for both PIK3CA and AKT1E17K mutations, with a requirement of at least five positive droplets, as described previously.17 We confirmed all PIK3CA mutations and the single AKT1 mutation detected by next-generation sequencing (fractional abundance, 1.55 to 15% and 1.6%, respectively) and identified PIK3CA variants in 6 additional samples (fractional abundance, 1.95 to 4.25%) (Figs. 3 and S4 and Table S3). We found no PIK3CA or AKT1 mutations in the control series of arteriovenous malformations.
In total, 39 sporadic CCMs in our cohort were found to harbor somatic variants in PIK3CA, one of the three CCM genes, or AKT1; variants in PIK3CA were most prevalent (39% of all samples). CCM genes were found to be mutated in 10% of samples and AKT1 in 1%.
We then evaluated the histologic characteristics of PIK3CA-mutant and AKT1-mutant CCMs in mice as compared with those in humans and observed that the former recapitulated all the key features of human CCMs (Fig. S5A–S5J). Similar to CCM-mutant lesions in humans, PIK3CA-mutant CCMs in humans and mice showed increased phosphorylation of myosin light chain (Fig. S5K and S5L). We confirmed activation of the PI3K–AKT–mTOR pathway in PIK3CA-mutant CCMs in humans and mice by means of phospho-S6 ribosomal protein immunohistochemical analysis (Fig. S5M–S5R).
CELLULAR ORIGIN OF MOUSE LESIONS
Our mouse models harbored activating mutations in Pik3ca or AKT1 in PGDS-expressing cells. Although PGDS is expressed in meningeal pre-cursor cells during embryogenesis and its expression is sustained in the meninges throughout life, it is also expressed in perivascular cells in intraparenchymal vessels, which coexpress the pericyte marker platelet-derived growth factor beta (PDGFRβ) (Fig. S6). We infer from these data that PGDS is expressed in pericytes surrounding intraparenchymal vessels, a finding consistent with those of another study.18 PGDS immunostaining identified scattered PGDS-positive cells lining the caverns outside the endothelial border in lesions in both models (Figs. 1J and 2F), and we detected a few PGDS-positive periendothelial cells around the caverns of human CCMs (Fig. 2F, inset).
To characterize the PGDS-positive cell of origin in the murine lesions, we used the AKT1E17K mouse, which, by virtue of the FLAG epitope present in the RCAS-AKT1E17K vector, allows unequivocal identification of cells expressing AKT1E17K. We performed double immunolabeling with FLAG and cell-type–specific markers (CD31 for endothelial cells, glial fibrillary acidic protein [GFAP] for astrocytes, and oligodendrocyte transcription factor 2 [OLIG2] for oligodendrocytes) and then counted immunolabeled cells within the region between the injection site and the lesion. A mean of 25% of pericavernomatous cells were FLAG-positive, and no pericavernomatous cells were CD31-positive (Fig. S7A–S7C and S7E) or GFAP-positive (Fig. S7D and S7F); some cells, primarily along the tissue scar, were positive for both FLAG and OLIG2 (Fig. S7G–S7I). The cytoplasmic rather than nuclear staining of OLIG2, however, suggested that these cells were reactive astrocytes.19 The vast majority (98%) of observed FLAG-positive pericavernomatous cells also expressed PDGFRβ (Fig. S7J–S7L). Taken together, these observations are consistent with PGDS-positive pericytes being the origin of the CCM lesions in our mouse models.
DISCUSSION
Somatic activating mutations in oncogenes of the RAS–RAF–MAPK and PI3K–AKT–mTOR pathways — for example, KRAS,17BRAF,20 and MAP2K121 — have been reported in sporadic vascular malformations, including high-flow and low-flow limb malformations and high-flow brain arteriovenous malformations. Our data show that sporadic CCMs (low-flow brain vascular malformations) also result from activating mutations in the PI3K–AKT–mTOR pathway, mainly in PIK3CA. In sporadic CCMs, the incidence of activating mutations in PIK3CA far exceeds that of activating mutations in CCM1, CCM2, and CCM3, the genes that cause familial (and some sporadic) CCMs.6 These findings are in line with the occurrence of CCMs and other vascular malformations in rare overgrowth disorders, including CLOVES (congenital lipomatous overgrowth, vascular malformations, epidermal nevi, and spinal or skeletal anomalies) syndrome, which is associated with activating mutations in PIK3CA22; the Klippel–Trenaunay syndrome,23,24 which is also PIK3CA-related25; and the Proteus syndrome, which is caused by the AKT1E17K mutation26 and may also involve multiple meningiomas.27,28 Somatic activating mutations in PIK3CA have also been reported in sporadic venous malformations.29
The causative nature of both PIK3CA and AKT1 mutations in sporadic CCMs was observed in two mouse models that had initially been engineered for the purpose of studying meningeal tumorigenesis. Both models differed from previous models generated by means of inducible, endothelium-specific deletion of Ccm1, Ccm2, or Ccm3 in neonatal mice, in all of which lesions develop exclusively in the cerebellum.30 In our models, we found CCM lesions mostly in the brain stem (Pik3caH1047R-related) or, in older mice, in the convexity (AKT1E17K-related), similar to models generated with neural progenitor–specific promoters.31 It is not possible to determine whether these differences are linked to the causative mutation (Pik3caH1047R vs. AKT1E17K) or to the method used to generate the two mouse models (genetic engineering vs. direct intracranial injection).
In sporadic CCMs in humans, we found that the frequency of PIK3CA mutations was high (39%), albeit lower than that of KRAS mutations (62%) in brain arteriovenous malformations.17 However, although the fractional abundance of KRAS mutations varied between 0.5% and 6%,17 we found 14 CCMs with a fractional abundance of PIK3CA variant surpassing 8%, despite the apparently small number of mutant cells surrounding the caverns. In a previous study,20 the prevalence of somatic mutations in genes in the RAS–MAPK pathway in low-flow vascular malformations was 3.7% (5 of 135), and the fractional abundance of mutant alleles varied between 2% and 7%.
Studies in mice have shown that CCMs might originate from clonal expansion of a few mutant endothelial cells that express stem-cell markers and attract surrounding wild-type endothelial cells to contribute to cavernoma growth,32,33 which would account for the very small number of mutant cells around the caverns. Indeed, it was previously reported that, depending of the proportion of mutant cells within the lesion, direct sequencing of DNA extracted from CCMs does not always detect the mutation. In early studies in which repeated cycles of amplification and subcloning were used, somatic mutations were identified in all three forms of familial CCM,34,35 whereas in a more recent analysis, in which targeted next-generation sequencing of sporadic CCMs has been performed, somatic mutations in CCM genes were detected in 4 of 11 samples (36%).6 The number of clones harboring the somatic mutation varied between 0.4% and 7.2%, a finding similar to ours.
Six sporadic CCMs harbored multiple mutations, including two with mutations affecting two different CCM genes and four with co-occurrence of CCM gene and PIK3CA variants. Cases of somatic double CCM-gene mutants are rare if not absent from the literature, and we know of one report of concomitant germline pathogenic mutations in a patient with multiple CCMs.36 Because the two mutations (one in CCM1 and one in CCM3) in the CCM of one patient had similar allele frequencies and are known to be pathogenic, it is difficult to determine whether one is a driver of CCM formation and the other is a passenger. The other patient had a CCM with concomitant CCM2 and CCM1 variants, but the latter is of unknown pathologic significance and may be irrelevant to CCM formation. The occurrence of CCM and PIK3CA mutations in the same lesion is less surprising, considering that in meningioma, PIK3CA mutations may occur alone or in association with TRAF7 mutations.9,10 These results add new evidence regarding the similarities between cavernomas and meningiomas, in light of the fact that some persons with constitutive CCM3 mutations can have not only multiple CCMs but also dura-based lesions that have the typical MRI and histopathological features of meningioma.37
Our results shed light on the cell of origin of CCMs, which is generally considered to be of endothelial lineage on the basis of genetically engineered mouse models.38 CCM lesions also form after deletion of Ccm3 in neural progenitor cells,31 leading to the hypothesis that CCMs form as a result of altered interactions between the components of the neurovascular unit. However, a recent study suggested that increased transforming growth factor β signaling in brain pericytes triggers changes in endothelial behavior and acquisition of pathologic landmarks associated with CCMs,39 and mural cell-specific deletion of Ccm3 induces formation of CCMs in mice.40 Our findings support the hypothesis that aberrant signaling in the pericyte causes CCMs. PGDS is not expressed in endothelial cells but is expressed in perivascular cells lying outside the basement membrane of blood vessels in the pia–arachnoid and subpial cortex,41 and we were able to confirm the existence of perivascular PGDS-expressing and PDGFRβ-expressing cells in our mouse model. These PGDS-positive pericytes may have a direct role in the neurovascular unit, which would support a role in CCM formation.
Our findings may provide a new understanding of the biology of sporadic CCMs. Rather than somatic mutations in the CCM genes playing a major causative role,4–6 somatic mutations in the PI3K–AKT–mTOR pathway predominated in our study. This result, which was supported by findings in a preclinical model, offers potential for the development of targeted therapies for the treatment of sporadic human PIK3CA-mutated CCMs that are refractory to surgery and radiotherapy or radiosurgery and cause frequent complications, especially given that PIK3CA inhibitors have shown promising results in patients with CLOVES syndrome42 as well as in patients with a wide range of tumors.43
Supplementary Material
Acknowledgments
Supported by grants from the Fondation ARC pour la Recherche contre le Cancer (PJA 20131200431, to Dr. Kalamarides, and project PJA 20171206327, to Dr. Peyre), the Fondation pour la Recherche sur les AVC (AVC 201908009719, to Dr. Peyre), and the National Institutes of Health (NIH R21NS1055001, to Dr. Louvi). The PHENO-ICMice core facility, which housed the mice used in this study, is supported by Investissements d’Avenir grants (ANR-10- IAIHU-06 and ANR-11-INBS-0011-NeurATRIS).
We thank the PHENO-ICMice core facility; the Fondation pour la Recherche Médicale; Yannick Marie (Molecular Biology Core, Brain Institute, Paris); Véronique Parietti (Histology Core, Brain Institute, Paris); Onconeurotek (Brain Institute, Paris) and Tumorothèque Caen Basse-Normandie (Caen, France) for management of surgical samples; and Piotr Topilko, Sayoko Nishimura, Francesco Lopez-Giraldez, Jungmin Choi, Tanyeri Barak, Zeynep Erson-Omay, and Kaya Bilguvar for discussion and advice.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
REFERENCES
- 1.Awad IA, Polster SP. Cavernous angiomas: deconstructing a neurosurgical disease. J Neurosurg 2019; 131: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rigamonti D, Hadley MN, Drayer BP, et al. Cerebral cavernous malformations: incidence and familial occurrence. N Engl J Med 1988; 319: 343–7. [DOI] [PubMed] [Google Scholar]
- 3.Riant F, Bergametti F, Ayrignac X, Boulday G, Tournier-Lasserve E. Recent insights into cerebral cavernous malformations: the molecular genetics of CCM. FEBS J 2010; 277: 1070–5. [DOI] [PubMed] [Google Scholar]
- 4.D’Angelo R, Marini V, Rinaldi C, et al. Mutation analysis of CCM1, CCM2 and CCM3 genes in a cohort of Italian patients with cerebral cavernous malformation. Brain Pathol 2011; 21:215–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.D’Angelo R, Alafaci C, Scimone C, et al. Sporadic cerebral cavernous malformations: report of further mutations of CCM genes in 40 Italian patients. Biomed Res Int 2013; 2013: 459253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McDonald DA, Shi C, Shenkar R, et al. Lesions from patients with sporadic cerebral cavernous malformations harbor somatic mutations in the CCM genes: evidence for a common biochemical pathway for CCM pathogenesis. Hum Mol Genet 2014;23: 4357–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro Oncol 2017; 19:Suppl_5: v1–v88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Preusser M, Brastianos PK, Mawrin C. Advances in meningioma genetics: novel therapeutic opportunities. Nat Rev Neurol 2018; 14: 106–15. [DOI] [PubMed] [Google Scholar]
- 9.Clark VE, Erson-Omay EZ, Serin A, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 2013; 339: 1077–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clark VE, Harmancı AS, Bai H, et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat Genet 2016; 48: 1253–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalamarides M, Stemmer-Rachamimov AO, Niwa-Kawakita M, et al. Identification of a progenitor cell of origin capable of generating diverse meningioma histological subtypes. Oncogene 2011; 30: 2333–44. [DOI] [PubMed] [Google Scholar]
- 12.Peyre M, Salaud C, Clermont-Taranchon E, et al. PDGF activation in PGDS-positive arachnoid cells induces meningioma formation in mice promoting tumor progression in combination with Nf2 and Cdkn2ab loss. Oncotarget 2015; 6: 32713–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boetto J, Apra C, Bielle F, Peyre M, Kalamarides M. Selective vulnerability of the primitive meningeal layer to prenatal Smo activation for skull base meningothelial meningioma formation. Oncogene 2018; 37:4955–63. [DOI] [PubMed] [Google Scholar]
- 14.Kalamarides M, Niwa-Kawakita M, Leblois H, et al. Nf2 gene inactivation in arachnoidal cells is rate-limiting for meningioma development in the mouse. Genes Dev 2002; 16: 1060–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peyre M, Stemmer-Rachamimov A, Clermont-Taranchon E, et al. Meningioma progression in mice triggered by Nf2 and Cdkn2ab inactivation. Oncogene 2013; 32: 4264–72. [DOI] [PubMed] [Google Scholar]
- 16.Riant F, Cecillon M, Saugier-Veber P, Tournier-Lasserve E. CCM molecular screening in a diagnosis context: novel unclassified variants leading to abnormal splicing and importance of large deletions. Neurogenetics 2013;14: 133–41. [DOI] [PubMed] [Google Scholar]
- 17.Nikolaev SI, Vetiska S, Bonilla X, et al. Somatic activating KRAS mutations in arteriovenous malformations of the brain. N Engl J Med 2018; 378: 250–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ganfornina MD, Sánchez D, Pagano A, Tonachini L, Descalzi-Cancedda F, Martínez S. Molecular characterization and developmental expression pattern of the chicken apolipoprotein D gene: implications for the evolution of vertebrate lipocalins. Dev Dyn 2005; 232: 191–9. [DOI] [PubMed] [Google Scholar]
- 19.Cassiani-Ingoni R, Coksaygan T, Xue H, et al. Cytoplasmic translocation of Olig2 in adult glial progenitors marks the generation of reactive astrocytes following autoimmune inflammation. Exp Neurol 2006; 201: 349–58. [DOI] [PubMed] [Google Scholar]
- 20.Al-Olabi L, Polubothu S, Dowsett K, et al. Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted therapy. J Clin Invest 2018; 128: 1496–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Couto JA, Huang AY, Konczyk DJ, et al. Somatic MAP2K1 mutations are associated with extracranial arteriovenous malformation. Am J Hum Genet 2017; 100: 546–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindhurst MJ, Parker VER, Payne F, et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat Genet 2012; 44: 928–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oda K, Morimoto D, Kim K, Yui K, Kitamura T, Morita A. Spinal cavernous angioma associated with Klippel-Trenaunay-Weber syndrome: case report and literature review. World Neurosurg 2018; 109: 333–7. [DOI] [PubMed] [Google Scholar]
- 24.Südmeyer M, Maroof P, Saleh A, Hartmann C, Wojtecki L, Schnitzler A. Action tremor caused by olivary cavernoma in Klippel-Trénaunay syndrome mimicking asymmetric essential tremor. J Neurol 2011; 258: 140–2. [DOI] [PubMed] [Google Scholar]
- 25.Vahidnezhad H, Youssefian L, Uitto J. Klippel-Trenaunay syndrome belongs to the PIK3CA-related overgrowth spectrum (PROS). Exp Dermatol 2016; 25: 17–9. [DOI] [PubMed] [Google Scholar]
- 26.Lindhurst MJ, Sapp JC, Teer JK, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med 2011; 365: 611–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horie Y, Fujita H, Mano S, Kuwajima M, Ogawa K. Regional Proteus syndrome: report of an autopsy case. Pathol Int 1995; 45:530–5. [DOI] [PubMed] [Google Scholar]
- 28.Keppler-Noreuil KM, Baker EH, Sapp JC, Lindhurst MJ, Biesecker LG. Somatic AKT1 mutations cause meningiomas co-localizing with a characteristic pattern of cranial hyperostosis. Am J Med Genet A 2016; 170: 2605–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Castillo SD, Tzouanacou E, Zaw-Thin M, et al. Somatic activating mutations in Pik3ca cause sporadic venous malformations in mice and humans. Sci Transl Med 2016;8: 332ra43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boulday G, Rudini N, Maddaluno L, et al. Developmental timing of CCM2 loss influences cerebral cavernous malformations in mice. J Exp Med 2011; 208: 1835–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Louvi A, Chen L, Two AM, Zhang H, Min W, Günel M. Loss of cerebral cavernous malformation 3 (Ccm3) in neuroglia leads to CCM and vascular pathology. Proc Natl Acad Sci U S A 2011; 108: 3737–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Detter MR, Snellings DA, Marchuk DA. Cerebral cavernous malformations develop through clonal expansion of mutant endothelial cells. Circ Res 2018; 123: 1143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malinverno M, Maderna C, Abu Taha A, et al. Endothelial cell clonal expansion in the development of cerebral cavernous malformations. Nat Commun 2019; 10: 2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akers AL, Johnson E, Steinberg GK, Zabramski JM, Marchuk DA. Biallelic somatic and germline mutations in cerebral cavernous malformations (CCMs): evidence for a two-hit mechanism of CCM pathogenesis. Hum Mol Genet 2009; 18: 919–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gault J, Awad IA, Recksiek P, et al. Cerebral cavernous malformations: somatic mutations in vascular endothelial cells. Neurosurgery 2009;65: 138–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.da Fontoura Galvão G, Veloso da Silva E, Fontes-Dantas FL, Filho RC, Alves-Leon S, Marcondes de Souza J. First report of concomitant pathogenic mutations within MGC4607/CCM2 and KRIT1/CCM1 in a familial cerebral cavernous malformation patient. World Neurosurg 2020; 142: 481.e1–486.e1. [DOI] [PubMed] [Google Scholar]
- 37.Riant F, Bergametti F, Fournier HD, et al. CCM3 mutations are associated with early-onset cerebral hemorrhage and multiple meningiomas. Mol Syndromol 2013; 4: 165–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wetzel-Strong SE, Detter MR, Marchuk DA. The pathobiology of vascular malformations: insights from human and model organism genetics. J Pathol 2017; 241: 281–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diéguez-Hurtado R, Kato K, Giaimo BD, et al. Loss of the transcription factor RBPJ induces disease-promoting properties in brain pericytes. Nat Commun 2019; 10: 2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang K, Zhang H, He Y, et al. Mural cell-specific deletion of cerebral cavernous malformation 3 in the brain induces cerebral cavernous malformations. Arterioscler Thromb Vasc Biol 2020; 40: 2171–86. [DOI] [PubMed] [Google Scholar]
- 41.Nagasaka T, Hiraide M, Sugimoto T, Shindo K, Shiozawa Z, Yokota S. Localization of lipocaline-type prostaglandin D synthase in rat brain: immunoelectron microscopic study. Histochem Cell Biol 2004;121: 483–91. [DOI] [PubMed] [Google Scholar]
- 42.Venot Q, Blanc T, Rabia SH, et al. Targeted therapy in patients with PIK3CArelated overgrowth syndrome. Nature 2018; 558: 540–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.André F, Ciruelos E, Rubovszky G, et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N Engl J Med 2019; 380: 1929–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.