

Abstract
The role of gene expression during learning and in short-term memories has been studied extensively1–3, but less is known about remote memories, which can persist for a lifetime4. We used long-term contextual fear memory as a paradigm to probe the single-cell gene expression landscape that underlies remote memory storage in the medial prefrontal cortex. Remarkably, we found persistent activity-specific transcriptional alterations in diverse populations of neurons that lasted for weeks after fear learning. Out of a vast plasticity-coding space, we identified membrane-fusion genes that could have important roles in the maintenance of remote memory. Unexpectedly, astrocytes and microglia also acquired persistent gene expression signatures that were associated with remote memory, suggesting that they actively contribute to memory circuits. The discovery of gene expression programmes associated with remote memory engrams adds an important dimension of activity-dependent cellular states to existing brain taxonomy atlases and sheds light on the elusive mechanisms of remote memory storage.
Long-term memories do not form immediately after learning, but are consolidated over time4. Previous studies have uncovered important contributions of molecular and cellular processes to learning and memory, such as gene expression changes, cAMP signalling and synaptic plasticity1, and identified a central role for RNA synthesis and protein translation in memory consolidation2. Despite these discoveries, the molecular underpinnings of memory consolidation remain elusive. For instance, while changes in gene expression are found in the first 24 h of learning, it is unclear whether these changes are maintained or whether new changes are acquired to consolidate a long-term memory trace3. Moreover, the dependence on the hippocampus for long-term memory is known to degrade over time, with cortical structures such as the medial prefrontal cortex (mPFC) becoming increasingly important5. Recently, the development of activity-dependent genetic labelling tools have allowed identification of sparsely activated neuronal ensembles, enabling access to the molecular mechanisms that underlie experience-dependent connectivity and plasticity6.
Neuron subtypes in remote memory engrams
To identify and study the transcriptional programmes of neurons involved in remote memory, we used TRAP2; Ai14 mice expressing Cre-ERT2 recombinase in an activity-dependent manner along with a Cre-dependent tdTomato (tdT) reporter allele (Extended Data Fig. 1a), enabling us to label memory recall-activated neurons. To genetically label the engrams that are associated with remote memory, we trained mice in a conditioning chamber with three pairs of tone–foot shocks on day 0, and induced fear memory recall (FR) on day 16 by returning mice to the conditioning chamber. Memory recall activates the consolidated memory engrams, thereby labelling the neuronal ensemble that encodes the consolidated remote memory7, while also inducing memory reconsolidation8. Because the molecular and cellular substrates of consolidation and reconsolidation were indistinguishable in this paradigm, we denote them here collectively as memory consolidation. We used control mice that were not fear conditioned but exposed to the recall context (no fear (NF)), fear conditioned but not subjected to recall (no recall (NR)), and neither fear conditioned nor exposed to the recall condition (homecage (HC)) (Fig. 1a–c). All mice were injected with 4-hydroxytamoxifen (4-OHT) before the FR procedure (or at the equivalent time) to allow activity-dependent production of tdT, thus enabling the distinction between the molecular programmes that are specific to remote memory versus background activation.
Fig. 1 |. Labelling and collection of single memory engram cells via the TRAP2; Ai14 line.

a, The experimental paradigm includes generating remote fear-memory traces via contextual fear conditioning, isolation of TRAP+-activated neurons via flow cytometry and plate-based single-cell RNA sequencing (scRNA-seq). b, Representative image of tdT+ TRAPed (red) cells in the anterior cingulate cortex (ACC) region of the mPFC 9 days after an injection of 4-OHT (at the time of remote memory recall). Scale bars, 1 mm (0.5 mm for zoomed in images). c, Degree of freezing upon return to the novel context 16 days after fear conditioning (FR) or no conditioning (NF) (n = 4 mice per condition, ***p= 4.89 × 10−9). d, Representative flow gating for tdT+ TRAPed cells (~1.5% of events in FR) in one mouse from the FR condition (left). Post-sequencing analysis of 722 example cells from a representative FR brain shows enrichment of tdT mRNA (pos) in the positive sort gate (orange violin) (middle). Enrichment of neuronal cells in the positive gate and neuronal and non-neuronal cell types in the negative gate in all brains (right). e, All sorted cells, with neuronal cells identified via the expression of Snap25 mRNA. All training and control conditions are represented in all cell clusters.
Nine days after FR9, single neuronal and non-neuronal cells were collected from the mPFC (Extended Data Fig. 1b) via fluorescence-activated cell sorting (FACS) and gating on the tdT signal, followed by plate-based single-cell mRNA sequencing (Fig. 1d). The percentage of TRAPed cells collected via FACS was significantly higher in FR (~1.5% of all cells) than in other conditions (Extended Data Fig. 2a), further confirming that the TRAP2 system captured increased neuronal activity during the FR process. We sequenced 3,691 neurons (Snap25+/tdT+ or tdT− mRNA) and 2,672 non-neuronal cells with high quality and depth (Extended Data Fig. 1c, d). Unbiased transcriptome clustering of cells from all four training conditions allowed the identification of major cell types and confirmed the dominance of neurons among tdT+ cells, whereas tdT− cells comprised both neurons and non-neuronal cells (Cldn5+ endothelial, Pdgfra+ oligodendrocyte progenitor cells (OPCs), Cx3cr1+ microglia and Aqp4+ astrocytes) (Fig. 1d, e, Extended Data Fig. 1e). Both tdT+ and tdT− cells from FR and control groups were represented in all clusters, which suggests that neither the neuronal activation state nor the training paradigm significantly altered fundamental cell-type identities (Fig. 1e).
Sub-clustering of 3,691 Snap25+ neurons using the top 2,000 highly variable genes revealed seven putative neuron sub-populations—four glutamatergic (C0, C1, C3 and C5) and three GABAergic neuron populations (C2, C4 and C6)—all of which were consistently observed throughout four biological replicates (Fig. 2a–c, Extended Data Fig. 2b). These are molecularly distinct populations (Fig. 2d), with each subtype expressing at least one distinctive marker gene (see Methods; Fig. 2e). All subtypes contained tdT+ cells, which indicated activation of all neuron subtypes regardless of the training state (Extended Data Fig. 2c). A comparison of key layer-specific marker genes (C0-Dkkl1, C1-Rprm, C2-Calb2, C3-Tesc, C4-Tnfaip8l3, C5-Tshz2 and C6-Lhx6) to existing cortical single-cell expression databases10 confirmed their presence in the mPFC (Extended Data Fig. 2d).
Fig. 2 |. Molecular identification of active neurons during (re)consolidation of a remote memory trace.

a, Dimensional reduction of all Snap25+ neurons (n = 3,691 cells) reveals seven distinct neuronal subtypes (C0–C6). b, Neuronal subtypes fall into two distinct categories: excitatory (glutamatergic) and inhibitory (GABAergic). The Glut–GABA signature is calculated based on the difference of the scaled expression level of Gad1 and Slc17a7. c, Expression levels of the top marker gene for each neuron subtype (C0–C6). d, Differences in the composition of neuron subtypes of TRAPed populations in FR and NF conditions, as well as inactive populations in FR mice (1 mouse per condition per replicate, n = 5 replicates, two-sided t-test) in the composition of TRAPed populations between FR and NF conditions are found. FR TRAPed populations are composed of significantly more C2 (GABAergic) neurons and less C3 (glutamatergic) neurons than the inactive population (error bars indicate SEM).
Surprisingly, no significant differences were found in the neuron subtype composition of TRAPed populations between the FR and NF groups (Fig. 2f, Extended Data Fig. 2e), which suggests a lack of training-dependent recruitment of neuron types during consolidation compared to baseline active populations in a NF memory scenario. Both excitatory and inhibitory neuron types were found in active FR populations, with glutamatergic cells comprising ~60–70%. Within the same FR brains, active and inactive populations had roughly similar compositions of neuron subtypes, with the exception of C2-Calb2 and C3-Tesc, suggesting only slight shifts in the recruitment or retirement of neuron subtypes due to activity.
Memory-associated gene expression
To determine whether remote-memory-associated transcriptional changes occur in recall-activated neurons, we looked for differentially expressed genes (DEGs; log2 fold change (log2FC) > 0.2 and false discovery rate (FDR) < 0.05) in TRAPed FR versus NF cells (Fig. 3a). Single-cell resolution enables a comparison of neurons within the same subtype and the identification of genes that are specifically associated with memory consolidation and recall. Of 23,355 genes, 1,292 were found to be consolidation-dependent. Expression patterns indicated an overall transcriptional activation, with more genes upregulated than downregulated. Interestingly, DEGs were heterogenous across neuron subtypes, which suggests that remote memory consolidation involves subtype-specific transcriptional programmes (Fig. 3b).
Fig. 3 |. Transcriptional programmes activated by (re)consolidation of remote memories are distinct per neuron subtype.

a, DEGs in FR versus NF in the C0 neuron subtype (n = 126 cells (NF), n = 289 cells (FR)) with all replicated pooled. Differential expression is defined by FDR < 0.01 and abs(log2FC) > 0.3 (red points) via a two-sided Mann–Whitney test. Remote-memory-associated DEGs (that is, DEGs that remain differentially expressed in three-quarters or more of replicates and when FR is compared to the NR and HC groups (see Extended Data Fig. 3)) are labelled in black. b, The number of DEGs per neuron subtype (left) and the number of shared DEGs between each neuron subtype (using pooled cells) (right). c, log2FC of remote-memory-associated DEGs (FR versus NF) per neuron subtype. Each gene is further annotated with potential functions. DE, differentially expressed; EXH, excitatory; GO, Gene Ontology; INH, inhibitory; NS, not significant. d, The percentile in which a TRAPed neuron (from C0) lies in the distribution of expression of a C0 DEG for all TRAPed C0 cells. The box plots (median ± s.d.) show the log2cpm distribution for each C0 DEG. Hierarchical clustering reveals one common C0 transcriptional programme that is concertedly upregulated. e, The fraction of cells in each neuron subtype that is activated with the transcriptional programme (that is, DEGs) from C0.
We applied a set of strict criteria to identify possible effector genes. First, each DEG had to be differentially expressed in at least three-quarters of biological replicates, enforcing reproducibility. The removal of DEGs that are also differentially expressed between the inactive populations in FR versus NF mice allowed the identification of changes that were specific to active populations (Extended Data Fig. 3a). Next, DEGs must be differentially expressed when FR cells are compared to NR and HC controls, ensuring that DEGs are not just a consequence of a fear experience. Last, DEGs had to pass a permutation test with shuffled labels (Extended Data Fig. 3b). These criteria produced a set of 99 ‘remote-memory-associated DEGs’ (Fig. 3c; see Methods). Several genes encoded proteins with regulatory roles, including known regulators of transcription (Hmg20a, Hnrnpk and Zfp706) and translation (Nck2, Alpl1 and Eif2ak1). Interestingly, even among the condensed list of remote-memory-associated DEGs, we found strong enrichments in genes encoding proteins involved in vesicle exocytosis (Vamp2, Gdi2, Rab15, Rab5a, Rab24, Atp6v0c, Syt13, Stx1b and Nsf), transmembrane transport (Slc30a9, Slc25a46, Mfsd14a, Tmem50a, Gpm6a, Mfsd14b and Abcf3), dendritic spine density (Strip1, Pls3 and Gsk3b) and long-range intracellular transport (Timm29, Atad1, Pak1, Plehkb2, Sarnp, Rtn3, Dmtn, Sar1a and Hid1) (Fig. 3c, Extended Data Fig. 3b, c). More than half of the remote-memory-associated DEGs are associated with neuronal diseases, suggesting links between the functional role of these genes to various memory-affecting neuronal disorders, in addition to the regulation of remote memory.
We further investigated the specificity of our findings by analysing TRAPed neurons that were activated by a salient experience unrelated to fear memory (Extended Data Fig. 4a). Using food deprivation as a salience signal, we identified TRAPed neuronal ensembles that contained the same neuronal subtypes as in TRAPed FR ensembles, but with differences in the subtype composition ratios (Extended Data Fig. 4b–d). While we found a total of 143 DEGs (FDR < 0.01) when comparing salience to no salience groups, there was almost no intersection of these DEGs with those found in FR mice (Extended Data Figs. 4e, 5). Thus, while new transcriptional programmes are activated in salience ensembles, the nature of these molecular changes is experience-specific and probably modulated by the particular valence of and/or functional requirements arising from the experience.
Hierarchical clustering of TRAPed FR neurons by the expression levels for each remote-memory-associated DEG allowed distinct populations of ‘highly activated’ and ‘lowly activated’ cells to emerge, suggesting that different transcriptional modules are concertedly regulated during memory consolidation in each neuronal subtype (Fig. 3d). To determine the subtype specificity of these modules, we found that the fraction of cells activated with the subtype-specific DEGs was generally highest in the corresponding subtype when compared to the activation levels in other subtypes or in the inactive populations (Fig. 3e, Extended Data Fig. 6a). Together, this could indicate the presence of subtype-specific common regulatory elements.
To address this possibility, we analysed our DEGs using hypergeometric optimization of motif enrichment (HOMER) to search for common regulatory motifs in an unbiased manner (see Methods; Extended Data Fig. 6b). We found 12 putative de novo and 2 known motifs enriched within our target DEG set (P > 0.01). While we did not find significant enrichment of motifs within subtype-specific DEGs, the Hif1b binding motif was found in >40% of total DEGs, including the synaptic transmission and plasticity-related genes Rab5a, Rab24, Vamp2, Gdi2, Gpm6a, Strip1, Ptp4a1, Trim32, Mfsd14a, Mfsd14b and Slc25a46. Interestingly, these findings agree with recent studies indicating a potential dual role for HIF1 transcription factors during hippocampal-dependent spatial learning and consolidation under normoxic conditions11. Interestingly, motifs associated with CREB, NFKB, CBP and C/EBP—canonical transcriptional regulators of neuronal activity and plasticity12,13—were absent near the transcription start site (−400 to +100 bp).
Vesicle exocytosis signatures in memory
To further elucidate the significance of these remote-memory-dependent transcriptional programmes, we used STRING to look for known and predicted protein–protein interactions. K-means clustering of the gene nodes revealed a significantly connected network (P = 1.75 × 10−6) that was centred around a large cluster of genes related to vesicle-mediated transport, exocytosis and neurotransmitter secretion, all of which were highly connected (confidence = 0.4; Extended Data Fig. 6c). Remarkably, 20 out of 99 remote-memory-associated DEGs fell within these functional categories, including Stx1b, Syt13, Vamp2, the SNAP receptor (SNARE) ATPase (Nsf) and the GTPase Rab5a, all of which are functionally linked to the SNARE complex and to vesicle exocytosis (Fig. 4a). Interestingly, the two most highly and ubiquitously upregulated genes across subtypes were Serinc1 and Serinc3, which are thought to be serine incorporators14. Notably, phosphatidylserine phospholipids are calcium-dependent binding partners for synaptotagmins15, suggesting that Serinc1 and Serinc3 may have important roles in enhancing phosphatidylserine levels and vesicle membrane fusion during memory consolidation. Finally, in situ hybridization confirmed the endogenous proportions of neuronal subtypes in TRAPed populations (Extended Data Fig. 7a, b), as well as the upregulated expression of key remote-memory-associated DEGs, including Serinc3, Syt13, Vamp2 and Stx1b in respective neuronal subtypes (Fig. 4b, c, Extended Data Fig. 7c).
Fig. 4 |. Remote memory (re)consolidation is associated with specific markers for vesicle exocytosis.

a, A subset of remote-memory-associated DEGs (FDR < 0.05 and log2FC > 0.2) that are known to regulate vesicle membrane fusion and exocytosis at the presynaptic terminal. The violin plots are overlayed with a drumstick plot indicating the average expression per mouse. The red points represent the median. The bubble plots depict the log2FC and the degree of significance (FDR) for each replicate and are coloured by such. In addition to the FR/NF log2FC (first column), DEGs were also confirmed to be differentially expressed when compared to NR (second column), and their activation is specific only to the active (inactive (In)) (last column). The red dots indicate which neuronal subtype these particular genes are upregulated in. b, Representative in situ images of Serinc3 expression (purple) in Dkkl1+/tdT+/DAPI+ cells in the mPFC. Scale bars, 100 μm. c, In situ validation of key genes involved in vesicle exocytosis in various neuron subtypes, including Serinc3 (in the Dkkl1+ subtype, n = 268 (FR), n = 62 (NF) cells), Stx1b (in the Rprm+ subtype, n = 342 (FR), n = 144 (NF) cells), Syt13 (in the Tnfai8lp+ subtype, n = 244 (FR), n = 44 (NF) cells) and Vamp2 (in the Tesc+ subtype, n = 326 (FR), n = 292 (NF) cells). Each point represents the normalized integrated intensity of the probe per cell analysed.
Non-neuronal gene expression changes
Remarkably, we discovered that non-neuronal cells also exhibited transcriptional changes associated with remote memory consolidation (FR compared to NF mice; Fig. 5a, b, Extended Data Fig. 8a, b). These signatures were distinct from those of neurons, indicating that non-neuronal programmes may support maintenance of the remote fear-memory trace. Surprisingly, >95% of these DEGs were upregulated, which suggests an overall transcriptional activation during consolidation. Not only was this response detectable weeks after the initial learning, it was observed even without enrichment of the non-neuronal cells directly associated with the TRAPed engram cells (i.e. the TRAP method is neuron-specific).
Fig. 5 |. Transcriptomic changes in non-neuronal cells associated with remote memory (re)consolidation.

a, tSNE of all non-neuronal cells reveals five non-neuronal cell types (astrocyte, endothelial (EC), microglia, OPC and oligodendrocyte (oligo)) that were collected in an unbiased manner through tdT-negative flow cytometry gates. b, DEGs in non-neuronal cells (FR versus NF) (left). DEGs are defined as log2FC > 1 and FDR < 0.01. The number of DEGs that satisfy these criteria in each non-neuronal cell type is also shown (right). The top DEGs (FR versus NF) for glial cells (astrocytes and microglia) that are also differentially expressed in FR versus NR are labelled. c, DEGs (determined by two-sided Mann–Whitney test) that are upregulated and downregulated in astrocytes (left) and microglia (right) in FR versus NF mice. DEGs (FDR < 0.01, log2FC > 1) are indicated by red dots, and the top DEGs are labelled in black. d, Pathway analysis of the DEGs (FR versus NF) in microglia and astrocytes. The score is defined as the –log2(P value) using the GeneAnalytics software.
Astrocytes and microglia showed the greatest number of transcriptional changes, with 181 and 308 genes perturbed, respectively (log2FC > 1 and FDR < 0.01) (Fig. 5c). Most of these DEGs represent largely diverging pathways (Fig. 5d). In particular, upregulated astrocytic genes were enriched in lipid, cholesterol and steroid metabolic functions (Gja1, Hmgcr, Dhcr7, Insig1, Acsl3, Idi1, Acsbg1, 10Asah1 and Hacd3) as well as glucose transport (Abcc5, Slc39a1, Slc6a1, Slc27a1, Slco1c1, Gnb1 and Ttyh1). Enhanced metabolic support from astrocytes may be required during memory consolidation since astrocyte–neuron metabolic coupling is elevated during neuronal activity16. Moreover, 95 out of 181 astrocyte DEGs were reproduced when comparing FR to NR mice, suggesting that a large portion of DEGs is specific to the recall experience itself and not merely a remnant of the fear experience.
By contrast, DEGs from microglial cells were enriched in innate immunity (Il6r, Stat6, Csf3r, Il1a, Irf5, Cd86, Tnfrsf1b, Ywhaz, Litaf, Ptgs1, Gdi2 and Rnf13) and cytoskeletal reorganization/focal adhesion maintenance pathways (Cdc42, Rhoa, Rhoh, Prkcd, Vasp, Arf6, Vav1 and Actr2), suggesting that upregulation of specific inflammatory molecules and enhancement of cell migration may be involved in the maintenance of memory. While less is known regarding the immunomodulatory roles of microglia in memory and learning, a previous study has shown that low levels of inflammatory cytokines (such as IL-1, IL-6 and tumour necrosis factor) can regulate neuronal circuit remodelling and long-term potentiation17.
In addition to neuron–neuron coupling, communication programmes between neurons and non-neuronal cells may support the memory trace over long periods. We looked for the expression of receptors or ligands in non-neuronal cells whose known binding partner18 is perturbed in TRAPed FR neurons (Extended Fig. 8c–e). We focused on genes that were differentially expressed in both the ligand-bound and the receptor-bound cell type (Extended Data Fig. 8c). In FR mice, we found upregulation of neuronal neuroligin-1 and neuroligin-3 (encoded by Nlgn1 and Nlgn3, respectively) and its binding partner neurexin-1 (encoded by Nrxn1) on astrocytes, complexes that may enhance neuron–glia adhesions and modulate synaptic function19. Thus, the concerted upregulation of these binding pairs in FR mice strongly suggests a role for astrocyte–neurexin–neuroligin interactions in the maintenance of synaptic strength during fear memory storage.
Discussion
While high-resolution gene expression atlases of the brain have provided invaluable information about cellular taxonomy10,20, characterization of activity-dependent states within these cell types is necessary to understand how experience modulates gene expression, synaptic plasticity and neuronal circuitry. The ability to form and maintain unique synaptic connections that encode a particular memory out of a vast pool of other experiences requires a complex coding space. By using a combination of activity-dependent labelling of neurons and single-cell transcriptomics, we discovered that (1) all mPFC neuron types can be activated during consolidation of remote memory via heterogenous transcriptional programmes; (2) enhanced membrane fusion and vesicle exocytosis may be a critical mode of synaptic strengthening during memory consolidation; (3) a specific set of exocytosis-related genes out of a vast coding space may be involved in allowing highly unique, experience-specific connections to be made; (4) these particular transcriptional programmes are detectable at remote time points and thus are probably involved in maintaining the memory trace weeks after learning; and (5) consolidation of remote memory also induces a persistent transcriptional programme in astrocytes and microglia.
Deciphering the temporal evolution of engram populations and their associated gene programmes through the various stages of initial learning, recent memory and remote memory is crucial for understanding the basis of conversion of short-term memories to long-term memories. We found that the majority of gene programmes affected in activated neurons during early stages of learning21–23 and recent memory24 do not intersect with our remote-memory-associated DEGs (Extended Data Fig. 9a), nor with genes enriched in TRAPed FR populations over inactive ones (Extended Data Fig. 9b). This suggests that remote memory could be governed by temporally unique transcriptional programmes. However, future experiments using unified technologies to deconvolve the neuronal compositions of recent and remote engrams and identify the immediate transcriptional changes in recent memory will be of great importance. Relevant to this point, TRAPed neurons in NR mice also exhibited continuous transcriptional changes at moderate levels (when compared to NF mice) (Extended Data Fig. 10). However, these DEGs are largely non-intersecting with remote-memory-associated DEGs, which suggests that the experience of fear itself can induce long-lasting changes in gene expression programmes and that the process of recall induces new transcriptional programmes in a different set of neurons. The current data therefore provide the first step towards deciphering the transcriptional coding landscape that is specifically associated with remote memory consolidation.
METHODS
Mice
All animal experiments were conducted following protocols approved by the Administrative Panel on Laboratory Animal Care at Stanford University. The TRAP2; Ai14 mouse line was kindly gifted by the Luo lab at Stanford. TRAP2 mice were heterozygous for the Fos2A-iCreER allele, and homozygous for Ai14, and were bred with Ai14 homozygous mice in the C57BL/6 background. Mice were group-housed (maximum five mice per cage) on a 12 h light–dark cycle (07:00 to 19:00, light) with food and water freely available. Male mice 42–49 days of age were used for all the experiments. Mice were handled daily for 3 days before their first behavioural experiment.
Genotyping
The following primers: GAG GGA CTA CCT CCT GTA CC (forward) and TGC CCA GAG TCA TCC TTG GC (reverse) were used for genotyping of the Fos2A-iCreER allele.
Fear conditioning
The fear conditioning training was performed as previously described25. Briefly, mice were individually placed in the fear conditioning chamber (Coulbourn Instruments) located in the centre of a sound attenuating cubicle, which was cleaned with 10% ethanol to provide a background odour. A ventilation fan provided a background noise at ~55 dB. After a 2-min exploration period, three tone–foot shock pairings separated by 1-min intervals were provided. The 85 dB 2 kHz tone lasted for 30 s, and the foot shocks were at 0.75 mA and lasted for 2 s. The foot shocks were co-terminated with the tone. The mice remained in the training chamber for another 60 s before being returned to the home cages. For the context recall, mice were placed back into the original conditioning chamber for 5 min 16 days after the training. 4-OHT injections were performed immediately (within 30 min) before the recall experiments. For the HC and the NR groups, 4-OHT was injected at a similar time when the other two groups were subjected to recall. The behaviour of the mice was recorded and analysed with the FreezeFrame software (version 4; Coulbourn Instruments). Motionless bouts that lasted more than 1 s were considered as freeze. Data were analysed with the tracking software Viewer III (Biobserve).
Food deprivation
Mice were deprived of food for 16 h, and then 4-OHT was injected to the animals and food was returned to one group (salience group) immediately afterwards (within 30 min), while no food was returned to the other group (no salience group) until 10 h later.
tdT florescence examination
Mice were deep anaesthetized with tribromoethanol and perfused with PBS followed by fixative (4% paraformaldehyde diluted in PBS). The brains were then removed and postfixed in 4 °C overnight and immersed in 30% sucrose solution for 2 days before being sectioned at a thickness of 50 μm on a cryostat (CM3050 S, Leica Biosystems). Imaging was performed with a scanning microscope (BX61VS, Olympus).
Single-cell dissociation and flow cytometry
mPFC regions were microdissected from live vibratome sections (300 μm thick) of the prefrontal cortex. Tissue pieces were enzymatically dissociated via a papain-based digestion system (LK003150, Worthington). Briefly, tissue chunks were incubated in 1 ml of papain (containing l-cysteine and EDTA), DNase and kynurenic acid for 1 h at 37 °C and 5% CO2. After 10 min of incubation, tissues were triturated briefly with a P1000 wide bore pipette tip and returned. Cells were triturated another four times (~30 each) with a P200 pipette tip over the rest of the remaining incubation time. At room temperature, cell suspensions were centrifuged at 350g for 10 min, resuspended in 1 ml of EBSS with 10% v/v ovomucoid inhibitor, 4.5% v/v DNase and 0.1% v/v kynurenic acid, and centrifuged again. Supernatant was removed and 1 ml ACSF was added to cells. ACSF was composed of: 1 mM KCl, 7 mM MgCl2, 0.5 mM CaCl2, 1.3 mM NaH2PO4, 110 mM choline chloride, 24 mM NaHCO3, 1.3 mM Na ascorbate, 20 mM glucose and 0.6 mM sodium pyruvate. Cells were passed through a 70-μm cell strainer to remove debris. Hoechst stain was added (1:2,000; H3570, Life Technologies) and incubated in the dark at 4 °C for 10 min. Samples were centrifuged (350g for 8 min at 4 °C) and resuspended in 0.5 ml of ACSF and kept on ice for flow cytometry.
Cells were sorted via the Sony SH800 into 96-well or 384-well plates (Bio-Rad) directly into lysis buffer26 with oligodT, and immediately snap frozen until processing. A positive ‘TRAP’ gate was set for cells that were both Hoechst+ and tdT+. A negative ‘TRAP’ gate was set for all Hoechst+ and tdT− cells in general. No gating on forward or backscatter was used to avoid size biases that might be present in a heterogenous neuronal population. Each plate was kept on the sorter for <25 min to prevent evaporation.
Sequencing
Whole-cell lysis, first-strand synthesis and cDNA synthesis were performed using the Smart-seq-2 protocol as described previously26 in both 96-well and 384-well formats, with some modifications. After cDNA amplification (23 cycles), cDNA concentrations were determined via capillary electrophoresis (96-well format) or the PicoGreen quantitation assay (384-well format) and wells were cherry-picked to improve quality and cost of sequencing. This was done by selecting only wells with > 0.2 ng/ μl of cDNA and subsequently normalizing cDNA concentrations to ~0.2 ng/μl per sample, using the TPPLabtech Mosquito HTS and Mantis (Formulatrix) robotic platforms. Libraries were prepared, pooled and cleaned using the Illumina Nextera XT kits or in-house Tn5, following the manufacturer’s instructions. Libraries were then sequenced on Nextseq or Novaseq (Illumina) using 2 × 75 bp paired-end reads and 2 × 8 bp index reads with a 200 cycle kit (20012861, Illumina). Samples were sequenced at an average of 1.5 million reads per cell.
RNAscope
The RNAscope experiment was performed following the manufacturer’s instructions using the RNAscope multiplex fluorescent reagent kit v2 (323100, ACD). All probes were purchased from existing stocks of custom design from ACD.
Bioinformatics and data analysis
Mapping to the genome
Sequences from Nextseq or Novaseq were demultiplexed using bcl2fastq, and reads were aligned to the mm10 genome augmented with ERCC (External RNA Controls Consortium) [Author: Please define ERCC.] sequences, using STAR version 2.5.2b. Gene counts were made using HTSEQ version 0.6.1p1. All packages were called and run through a custom Snakemake pipeline. We applied standard algorithms for cell filtration, feature selection and dimensionality reduction. Briefly, genes that appeared in fewer than five cells, samples with fewer than 100 genes and samples with less than 50,000 reads were excluded from the analysis. Out of these cells, those with more than 30% of reads as ERCC, and more than 10% mitochondrial or 10% ribosomal were also excluded from analysis. Counts were log-normalized and then scaled where appropriate.
Next, the ‘canonical correlation analysis’ function from the Seurat package27 was used to align raw data from multiple experiments. Only the first ten canonical components were used. After alignment, relevant features were selected by filtering expressed genes to a set of ~2,500 with the highest positive and negative pairwise correlations. Genes were then projected into principal component space using the robust principal component analysis. Single-cell principal component scores and gene loadings for the first 20 principal components were used as inputs into Seurat’s (v2) FindClusters and RunTsne functions to calculate 2D tSNE coordinates and search for distinct cell populations. Briefly, a shared-nearest-neighbour graph was constructed based on the Euclidean distance metric in the principal component space, and cells were clustered using the Louvain method. Cells and clusters were then visualized using 3D tSNE embedding on the same distance metric. A neuron was characterized as ‘TRAPed’ trapped if it satisfied two conditions: (1) from the tdT+ sort gate (tdT protein positive) and (2) tdT mRNA raw count > 0. Neuron subtype marker genes were found by using the FindAllMarkers function in Seurat (min.pct = 0.3, thresh.use = 0.25, min.diff.pct = 0.2). DEG analysis was done by applying the Mann–Whitney U-test on various cell population. Raw P values were adjusted via the FDR. Permutation tests were then performed on all genes of interest. All graphs and analyses were generated and performed in R. GeneAnalytics and GeneCards packages offered by the gene set enrichment analysis tool were used for GO/KEGG/REACTOME pathway analysis and classification of enriched genes in each subpopulation.
Finding remote-memory-associated DEGs
To reduce our list of DEGs (FR TRAP versus NF TRAP results in 1,291 DEGs, cells from 4 biological replicates pooled, logFC > 0.3, FDR < 0.01) to only the most recall-specific, 4 steps were taken. Analysis was limited to C0–C4 neuron subtypes due to insufficient numbers of cells in C5 and C6 across all experimental conditions to make meaningful comparisons. First, DEGs are recalculated by assessing each experiment individually using the whole transcriptome, and only DEGs (via the same criteria as pooled) that intersect in three-quarters of replicates are kept. While this decreases the statistical power, it ensures biological reproductivity. Three out of four criteria were chosen as a compromise due to the high strictness of four out of four, which yielded only a maximum of seven DEGs (for a neuron subtype). All resulting DEGs are found in the initial DEG list (all replicates pooled), indicating that no additional DEGs were found as a result of analysing replicates separately. Second, ‘inactive’ (tdT-negative) populations were also compared (FR inactive versus NF inactive) and any DEGs that intersected with the DEGs left after the first criteria, were removed. This ensures that DEGs are activity-dependent, and not merely an overall upregulation in all cells due to the experience. This routinely removed genes such as Hsp90aa1 and Pcna-ps2. Third, the remaining DEGs had to be differentially expressed when FR TRAP was compared to either NR TRAP or HC TRAP. This ensures that the DEGs are specific to only neuronal ensembles that labelled by memory recall, and not due to forms of baseline activity (HC) or activity that remained from the initial fear learning (NR). Last, the remaining DEGs must pass a permutation test in which the training labels are shuffled and a distribution of log2FC is computed based on these labels. The true observed logFC must be above the 95th percentile of the distribution of the shuffled distribution. After placing these constraints, 99 genes remain from the original list of 1,291.
Assessment of activation score
A TRAPed (or inactive) cell is considered to be ‘activated’ by the remote-memory DEG programme if 25%, 50% or 75% of the subtype-specific DEGs (remote-memory-associated DEGs only) is expressed above the 90th percentile of the distribution of that gene in NF TRAP controls from the same subtype. This calculation is then repeated with DEG programmes that are specific to each neuronal subtype. The fraction of cells activated with the subtype-specific signature is calculated as the number of activated cells divided by all cells in the subtype/activity group.
Regulatory motif analysis
Enrichment of known and de novo motifs was found using HOMER by inputting the list of 99 remote-memory-associated DEGs and using the function findMotifs.pl and the criteria ‘–start −400 -end 100 -len 8,10 -p 2’. The locations of the motifs in specific DEGs were found using the -find < motif file > option of findMotifs.pl.
RNAscope image analysis
Images were taken using a Nikon Confocal Microscope (at ×10 or ×20, NA = 0.45) and images were processed in ImageJ to only obtain the mPFC regions. The resulting images were fed into a custom image analysis pipeline and were developed on CellProfiler (using a combination of the functions IdentifyPrimaryObjects, RelateObjects, FilterObjects, MeasureObjectIntensity, ClassifyObjects and CalculateMath. The custom pipeline can be found in Supplementary Methods). Briefly, images were corrected with control slides (unstained sample and negative control probes) and cells were segmented using the DAPI signal. Those harbouring a signal (above a set threshold level) for both the subtype marker and the tdT probe were retained. The integrated fluorescence intensity of the DEG probe was calculated for each DAPI+/subtype+/tdT+ cell. Cells that were not double-positive were not considered. The integrated fluorescent intensity was then normalized to the integrated DAPI signal per cell and results were plotted with custom scripts in R.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The accession number for the single-cell RNA sequencing data reported in this paper is GSE152632.
Code availability
Custom scripts can be found at (https://github.com/mbchen-424/memory-sc-rnaseq).
Extended Data
Extended Data Fig. 1 |. Fidelity of the TRAP2; Ai14 line and sequencing quality metrics.

a, Fidelity with which the tdTomato reporter in TRAP2; Ai14 mice captures endogenous cFos expression during fear memory encoding by in situ hybridization of cFos and iCre in the mPFC directly following fear conditioning. (Left) Percentage of cFos+ cells in either fear conditioned (FC) or homecage (HC) mice as seen by in situ staining. Percentage of cFos+ cells that are also iCre+. (Right) Representative in situ hybridization images of FC mice (mPFC). b, Representative images of regions of the mPFC analysed in this study (anterior cingulate cortex (ACC), the prelimbic (PrL) and the infralimbic cortex (IL)). Scale bars are 1mm (and 0.5 mm in the insets). c, Violin plots of the number of reads and number of genes per biological replicates (m1-m4), per cell. d, Scatterplot depicting the lack of strong relationship between number of genes detected and number of reads obtained per cell. e, Scaled expression of canonical markers in non-neuronal cells (Cldn5-BECs, Pdgfra-OPCs, Cx3cr1-microglia, Aqp4-astrocytes)
Extended Data Fig. 2 |. Distribution of cell numbers and neuronal subtypes across various training conditions.

a, Representative flow cytometry plot of the amount of tdT+ events per training condition. In scatter plot, each point represents one mouse. b, Number of cells from each biological replicate that were annotated as one of 7 defined neuron subtypes (C0-C6). c, Number of TRAPed and Inactive cells (as defined by non-zero expression of tdT mRNA) collected per neuron subtype, in either fear-recall (FR) or no-fear (NF) mice. d, Representative images of subtype marker genes (C0 to C6) in the Allen Brain ISH atlas. e, Number of TRAPed (tdT mRNA+) cells collected in each experimental condition that fall in one of 7 neuronal subtype categories.
Extended Data Fig. 3 |. Differential gene expression in distinct neuronal subtypes (FR over NF TRAPed populations).

a, DEGs in each neuron subtype when inactive (tdT-) neurons are compared between FR and NF mice. b, (Left) Volcano plots of DEGs in FR vs NF mice for each neuron subtype (C0 to C4). DEGs found when all replicates are pooled analysed in a combined manner are shown in red. Recall-dependent DEGs (defined as being differentially expressed in at least ¾ replicates, when analysed replicates are analysed individually) are labelled in black. (Right) Permutations are performed for every recall-dependent DEG for each neuronal population. Upregulated DEGs lying above the y = x line (red) and downregulated DEGs lying below the y = x line (blue) are considered to be above the 99th percentile of the permuted distribution. c, GO enrichment analysis of all up- and downregulated DEGs (941 DEGs up, 384 DEGs down, all neuron subtypes combined) when all replicates are pooled. Bars show the enrichment scores (GeneAnalytics) for the GO pathway and dot indicates the number of DEGs involved in that pathway. d, GO enrichment analysis of only the upregulated remote-memory-specific DEGs (from Fig. 3c) from all neuron subtypes combined. Bar indicates enrichment score (Gene Analytics) and number indicates number of recall-dependent DEGs involved in that pathway.
Extended Data Fig. 4 |. Analysis of TRAPed ensembles in food salience (S) versus no salience (NS) mice.

a, Schematic of experimental paradigm for generating TRAPed neuronal ensembles as a result of food salience (food deprivation followed by food return (salience) or no food return (no salience)). b, Percentage of events in flow cytometry that were tdT+, by experience. c, tSNE of the merge of data from fear-recall experiments and food-salience experiments colored by neuron subtype (left) and experimental paradigm (right). d, Subtype composition differences between TRAPed fear-recalled ensembles and TRAPed food salience ensembles, as compared to background Inactive ensembles. e, Heatmap of the average log2 fold change of DEGs in each neuron subtype when comparing fear-recall vs no-fear, and salience vs no salience. Only DEGs with FDR <0.01 are shown.
Extended Data Fig. 5 |. DEGs when comparing ensembles from food salience (S) to no salience (NS) mice.
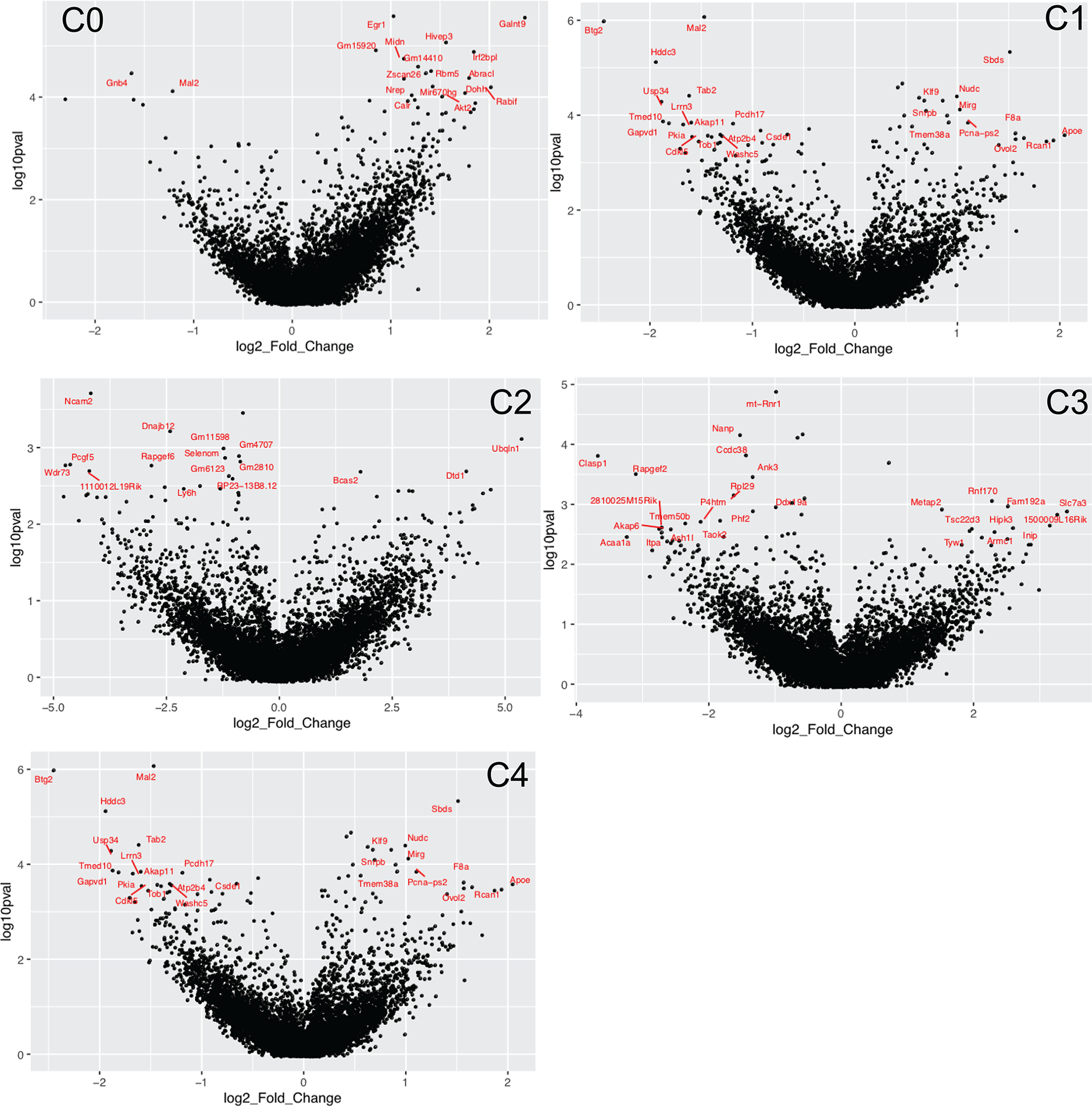
Volcano plots showing the log2 fold change and adjusted p-values (in log10 scale) of genes when comparing food salience over no salience groups within each neuron subtype (C0 through C4). Top DEGs per neuron subtype are labelled in red. Positive log2 fold change indicates upregulation in food salience (S) group.
Extended Data Fig. 6 |. Neuron subtype-specific activation programs, hypothesized protein–protein interactions and upstream regulatory motifs.

a, Fraction of cells in each neuron subtype that are induced with the transcriptional program (that is, DEGs) from a neuron subtype. Overall, the activation program of each TRAPed neuron subtype is found to be more specific to it than the inactive population, or other subtypes. b, (Left) De novo regulator motif discovery: analysis was performed using HOMER on the subset of 99 remote-memory-associated DEGs by looking at the sequences −400 to +100 bp from the TSS. 12 de novo and 2 known motifs were found (only motifs with an enrichment p-value <1e-2) were kept). Heatmap depicts the “motif score” of each DEG for each motif, and genes and motifs were clustered via the ward.D method. (Right) Bar graph depicting the % of the DEGs (target sequences) that possess a match for the motif within −400 to +100 bp from the TSS, vs the % of background sequences. For de novo motifs, the best match gene is listed on the right. HIF1b and HIF1a are matches to known motifs. c) (Left) Hypothesized protein–protein interactions of a subset of recall-dependent DEGs (TRAPed FR/NF) using the STRING database (https://string-db.org/). Only genes that are connected at a confidence level of 0.4 (medium) are shown. Connections indicate a possible existence of an interaction between two proteins. Genes are colored by up of downregulation in FR/NF. (Right) Same network plot, with nodes colored by the neuron subtype which differentially regulates the DEG.
Extended Data Fig. 7 |. In situ validation of tdT levels, neuronal subtype compositions and remote-memory-specific DEGs in the mPFC.

a, Ratio of Nuclei that are tdT+ (mRNA level) per training condition. Each data point represents one region of interest. b, Ratio of TRAPed cells that are positive for a neuronal subtype marker obtained either via the RNA-scope method, or by scRNA-seq (see Fig. 2). TRAPed cells are defined as DAPI+/tdT+ in RNAscope quantification, and as tdT mRNA count >1 in scRNA-seq (post-QC). No significant differences are found between FR and NF within either RNA-scope or scRNA-methods, indicating no major changes in neuronal subtype composition of active populations in different training conditions. c, in situ hybridization of key remote-memory specific DEGs including Stx1b in Rprm+/tdT+ cells, Syt13 in Tnfaip8l3+/tdT+ cells, Vamp2 in Tesc+/tdT+ cells. Scale bars = 100 μm.
Extended Data Fig. 8 |. DEGs and potential cell–cell interactions in non-neuronal cells during memory (re)consolidation.

a, Volcano plots of non-neuronal cell types when comparing cells in FR over NF nice. DEGs (FDR >0.01, log2FC >1) are labelled in red, and exemplary DEGs (high log2FC and log10FDR) are labelled in black. b, Number of non-neuronal cells collected in this study, for each cell type and experimental condition. c, Heatmap of a subset of neuronal ligands and glial receptors that are found to be differentially perturbed upon memory consolidation. Only receptors and ligands which were found to be (differentially) expressed are shown. d, (Left) Heatmap of the log2FC of DEGs (FR over NF) in neurons that are classified as ligands. (Middle and right) Sankey plot of known ligand-receptor pairs and heatmap of the average scaled expression level of the corresponding receptors in each cell type. e, (Left) Heatmap of the log2FC of DEGs (FR over NF) in neurons that are classified as receptors. (Middle and right) Sankey plot of known ligand-receptor pairs and heatmap of the average scaled expression level of the corresponding ligands in each cell type.
Extended Data Fig. 9 |. Comparison of remote-memory DEGs with previously published data sets of experience-dependent transcriptional activity.

a, (Left) Heatmap of the log2FC of all 1292 DEGs (FDR <0.05, FR over NF, all cells pooled) in this manuscript, and their log2FC values in previously published data sets of experience-dependent DEGs in activated neurons during: recent fear memory retrieval (Rao-Ruiz, 2019), associative fear-learning (Cho, 2016), post-visual stimulus (Hrvatin, 2018), or novel environment exposure (Lacar, 2016). A value of zero log2FC indicates the gene was not differentially expressed in a data set. (Right) Same as (left), but now DEGs are filtered down to the “Recall-dependent DEG ” set derived from this manuscript. Only genes differentially expressed in ¾ replicates are remaining. b, (Left) Log2 fold change heatmap of the recall-dependent DEGs between tdT+ vs tdT- neurons in FR mice (genes are differentially expressed in >3/4 replicates) undergoing remote fear memory consolidation. (Right) The log2FC values of these genes if they are found in previously published data sets of experience-dependent DEGs (see (a)). A value of zero log2FC indicates the gene was not differentially expressed in that data set.
Extended Data Fig. 10 |. Comparison of remote-memory-specific DEGs and fear-experience-related DEGs.

(Left) Log2 fold change heatmap of the union of DEGs from comparing FR vs NF (remote-memory specific) and NR vs NF (fear-related). Sustained transcriptional changes from the fear-experience itself is shown in the yellow highlighted columns. (Right) Zoomed in view of the portion of the heatmap within the green boxes where most fear experience-related DEGs are located.
Acknowledgements
We thank L. Chen, M. Zhou and J. Li for discussion of the experimental design; S. Kolluru and D. Henderson for assistance in library preparation; N. Neff and J. Okamoto for assistance with sequencing; J. Lui for advice on brain dissociation; L. Denardo, J. Lui and L. Luo for the gift and help with the TRAP2 line; and W. Wang, G. Stanley and F. Horns for helpful discussions and computational assistance. S.R.Q. is a Chan Zuckerberg Investigator.
Footnotes
Competing interests The authors declare no competing interests.
Online content Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at
Additional information
Supplementary information is available for this paper at
Peer review information Nature thanks Steve Ramirez and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Kandel ER The molecular biology of memory storage : a dialogue between genes and synapses. Science 294, 1030–1038 (2001).</unknown> [DOI] [PubMed] [Google Scholar]
- 2.Flexner LB & Flexner JB Effect of acetoxycycloheximide and of an acetoxycycloheximide–puromycin mixture on cerebral protein synthesis and memory in mice. Proc. Natl Acad. Sci. USA 55, 369–374 (1966).</unknown> [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alberini CM & Kandel ER The regulation of transcription in memory consolidation. Cold Spring Harb. Perspect. Biol. 7, a021741 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Squire LR Mechanisms of memory. Science 232, 1612–1619 (1986).</unknown> [DOI] [PubMed] [Google Scholar]
- 5.Kitamura T et al. Engrams and circuits crucial for systems consolidation of a memory. Science 356, 73–78 (2017).</unknown> [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeNardo L & Luo L Genetic strategies to access activated neurons. Curr. Opin. Neurobiol. 45, 121–129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeNardo LA et al. Temporal evolution of cortical ensembles promoting remote memory retrieval. Nat. Neurosci. 22, 460–469 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKenzie S & Eichenbaum H Consolidation and reconsolidation: two lives of memories? Neuron 71, 224–233 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guenthner CJ, Miyamichi K, Yang HH, Heller HC & Luo L Permanent genetic access to transiently active neurons via TRAP: targeted recombination in active populations. Neuron 78, 773–784 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeisel A et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142 (2015).</unknown> [DOI] [PubMed] [Google Scholar]
- 11.O’Sullivan CN, Sheridan G & Murphy K in Transcription Factors CREB and NF-κB: Involvement in Synaptic Plasticity and Memory Formation 43–65 (Bentham Science, 2012). [Author: Please provide the editor(s) for this reference.] [Google Scholar]
- 12.Suzuki A et al. Upregulation of CREB-mediated transcription enhances both short- and long-term memory. J. Neurosci. 31, 8786–8802 (2011).</unknown> [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kida S et al. CREB required for the stability of new and reactivated fear memories. Nat. Neurosci. 5, 348–355 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Inuzuka M, Hayakawa M & Ingi T Serinc, an activity-regulated protein family, incorporates serine into membrane lipid synthesis. J. Biol. Chem. 280, 35776–35783 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Zhang X, Rizo J & Südhof TC Mechanism of phospholipid binding by the C2A-domain of synaptotagmin I. Biochemistry 37, 12395–12403 (1998). [DOI] [PubMed] [Google Scholar]
- 16.Bélanger M, Allaman I & Magistretti PJ Brain energy metabolism: focus on astrocyte–neuron metabolic cooperation. Cell Metab. 14, 724–738 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Williamson LL, Sholar PW, Mistry RS, Smith SH & Bilbo SD Microglia and memory: modulation by early-life infection. J. Neurosci. 31, 15511–15521 (2011).</unknown> [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramilowski JA et al. A draft network of ligand–receptor-mediated multicellular signalling in human. Nat. Commun. 6, 7866 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Südhof TC Synaptic neurexin complexes: a molecular code for the logic of neural circuits. Cell 171, 745–769 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saunders A et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lacar B et al. Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat. Commun. 7, 11022 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho J-H, Huang BS & Gray JM RNA sequencing from neural ensembles activated during fear conditioning in the mouse temporal association cortex. Sci. Rep. 6, 31753 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hrvatin S et al. Single-cell analysis of experience-dependent transcriptomic states in the mouse visual cortex. Nat. Neurosci. 21, 120–129 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rao-Ruiz P et al. Engram-specific transcriptome profiling of contextual memory consolidation. Nat. Commun. 10, 2232 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou M et al. A central amygdala to zona incerta projection is required for acquisition and remote recall of conditioned fear memory. Nat. Neurosci. 21, 1515–1519 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Picelli S et al. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protocols 9, 171–181 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The accession number for the single-cell RNA sequencing data reported in this paper is GSE152632.