

Abstract
Background.
CD4+ T-cells temporally transition from protective to pathological during ischemic heart failure [HF; 8 weeks post-myocardial infarction (MI)]. Cellular mechanisms mediating this shift are unknown.
Methods.
RNA-sequencing of cardiac CD4+ T-cells and flow cytometric (FC) analysis of immune cells was conducted.
Results.
RNA sequencing of CD4+ T-cells from the failing hearts of male mice indicated activation of estrogen receptor (ER)-α signaling. FC analysis showed that ERα in CD4+ T-cells decreases significantly at 3 days post-MI but increases during HF. To antagonize ERα, we tested a novel ERβ agonist (OSU-ERb-012) to inhibit T-cells and blunt left-ventricular (LV) remodeling. Proliferation assays showed that OSU-ERb-012 dose-dependently inhibited proliferation and pro-inflammatory cytokine expression in anti-CD3/CD28 stimulated splenic T-cells isolated from both the sexes. For in-vivo efficacy, 10–12 week old male and ovariectomized female mice were randomized at 4 weeks post-MI and treated with either vehicle or drug (60 mg/kg/day; oral). While vehicle-treated HF mice displayed progressive LV dilatation with significantly increased end-systolic and end-diastolic volumes (ESV and EDV, respectively) from 4 to 8 weeks post-MI, treatment with OSU-ERb-012 significantly blunted these changes and stopped LV remodeling in both the sexes. Reduction in tibia-normalized heart and LV weights, cardiomyocyte hypertrophy and interstitial fibrosis further supported these results. Additionally, OSU-ERb-012 treatment selectively inhibited cardiac, splenic and circulating CD4+ T-cells without affecting other myeloid and lymphoid cells in the HF mice.
Conclusions.
Our studies indicate that ERβ agonists and OSU-ERb-012, in particular, could be used as selective immunomodulatory drugs to inhibit CD4+ T-cells during chronic HF.
Subject codes: Inflammation, Cell Biology, Heart Failure, Hypertrophy
Keywords: T-lymphocytes, Heart Failure, Myocardial Infarction, Left-Ventricular Remodeling, Estrogen Receptors
Introduction
Chronic heart failure (HF) is an inflammatory disease1, 2. Ischemic or non-ischemic cardiac injury, such as in myocardial infarction (MI) or transverse aortic constriction (TAC), respectively, initiate a cascade of rapid early innate immune responses followed by the activation of adaptive immune system3. Activation of both of these immune responses immediately after ischemic injury (1–14 days post-MI) is protective and essential for wound healing, scar formation, and to regain homeostasis4. However, progressive cardiac dysfunction and sustained low-grade inflammation initiate a 2nd phase of immune responses which are significantly slower, ranging from months to years, and coincide with the maladaptive left-ventricular (LV) remodeling. Previous studies have shown that this 2nd phase is also associated with activation and infiltration of monocytes, macrophages, and dendritic cells into the dysfunctional hearts5. Activation of innate immune responses, including antigen-presenting cells (APCs), would also suggest amplification of T-cell mediated adaptive immune responses. Accordingly, we have shown that CD4+ helper T-cells and its subsets, viz Th1, Th2, Th17 and regulatory T-cells (Tregs), are globally expanded during ischemic HF, and exert pathological effects, as antibody-mediated depletion of CD4+ T-cells blunts progressive cardiac dysfunction1. Moreover, the adoptive transfer of HF-activated splenic CD4+ T-cells induce substantial cardiac dysfunction in naïve mice1, suggesting that CD4+ T-cell activation during HF is antigenic and is mediated by T-cell receptor (TCR) stimulation. These studies also suggest that CD4+ T-cells transition from being protective during MI6 to pathological during ischemic HF1. However, the molecular switches that mediate this pathological temporal transition are unknown.
Besides antigen-specific CD4+ T-cell activation, autoimmune diseases also manifest significant gender bias7, such as in multiple sclerosis (MS) and experimental autoimmune encephalomyelitis (EAE). HF is no exception and mimic auto-immune diseases in both the characteristics. While premenopausal women are more protected from adverse myocardial events as compared to men, this protection is lost post-menopause8. Epidemiological and preclinical studies suggest that circulating estradiol (E2) concentrations mediate this protection8. E2 exerts its effects either via transcription factors, such as estrogen receptors (ERs)α and –β to regulate gene expression, or via membranous G–protein coupled estrogen receptor 1 (Gper1), and all of these receptors are expressed in the myocardium9 as well as in CD4+ T-cells10. Several studies using ERα−/− or ERβ−/− mice have shown either increased mortality or exacerbated LV remodeling following ischemic and non-ischemic injury11–13, however; cellular effectors of these responses have not been identified in any of these studies. It is, therefore, not known if the cardio-protective effects of E2 signaling are mediated directly through the myocardium or are indirectly mediated by altering CD4+ T-cell responses. Accordingly, we, for the first time, show that ERα signaling is upregulated in cardiac CD4+ T-cells and is a potential pathological switch that temporally promotes pathological transitioning of T-cells during chronic HF. We also identified a novel ERβ agonist to functionally antagonize ERα effects and demonstrate that it selectively inhibits TCR-mediated T-cell activation/proliferation, blunts progressive cardiac dysfunction, and could provide a new class of therapeutics for immune modulation of HF.
Methods
Animal studies were approved by the Institutional Animal Care and Use Committee at the Ohio State University and were done in accordance with the NIH Guide for the Care and Use of Laboratory Animals (DHHS publication No. 85–23, revised 1996). All mice had free access to food and water ad-libitum and 177 mice were used for all the studies. All raw data are available from the corresponding author upon reasonable request. OSU-ERb-012 is patented by the Ohio State University.
Mouse Model and Surgical Procedure
Male or ovariectomized female mice, 10 to 12-week-old C57BL/6 mice (Jackson Laboratories, stock# 000664) underwent left thoracotomy followed by permanent left coronary artery ligation to induce MI and ischemic HF (n=80 for males and 30 for females) or sham surgery (n=47), as described previously14. At 3 days and 8 weeks post MI, peripheral blood was collected from the facial vein and at both the time points, mice were euthanized by cervical dislocation. Hearts and spleens were collected, weighed and processed either for mononuclear cell isolation or histological analysis14. Tibia length was measured to normalize all gravimetric data.
Drug Treatment
All HF and sham mice underwent echocardiography at 25–26 days post-MI and were randomized according to their cardiac function to receive either the vehicle control or drug. All mice with akinetic area> 30% were included in the study following published guidelines15. A total of 25 males and 5 ovx females received vehicle whereas 33 males and 6 females received the drug. A 20-fold stock solution of OSU-ERb-012 was prepared in DMSO and stored at −20 °C. At the time of dosing, the drug was diluted using Tween 20 and saline maintaining a ratio of 5:5:90 (DMSO:Tween 20:saline). All mice were weighed every day and the drug was administered at 60 mg/kg dose via gavage.
Details for i) Echocardiography, ii) Immune Cell Isolation and Fixation, iii) T-cell Proliferation Assays, iv) Flow Cytometric Staining, v) Limited-Cell RNA Sequencing (Lc-RNA seq) and Bioinformatics Analysis, and vi) Fibrosis and Hypertrophy Staining are given in supplemental materials.
Statistical Analysis
All data are shown as mean±SD. Statistical tests used for each data-set and, number of biological and experimental replicates for each ex-vivo assay are given in the Figures/Figure legends. Each data-point represent a separate biological replicate (mouse/experiment). GraphPad Prism version 9.0 was used for all statistical analyses.
Results
ERα and ERβ are Temporally and Spatially Altered in CD4+ T-cells during Ischemic HF.
CD4+ T-cells undergo a pathological phenotypic shift during chronic HF, and accentuate LV remodeling and cardiac dysfunction1. Therefore, to identify potential phenotypic switches, 150–300 CD4+ T-cells were flow-sorted from the failing hearts, and the mediastinal lymph nodes of male mice at 8 weeks post-MI and limited cell RNA-sequencing was conducted. IPA analysis of the differentially expressed genes showed that ESR1 (ERα) signaling was significantly activated (Figure 1A) with downregulation of ESR2 (ERβ) pathway (Figure S1A) in cardiac CD4+ T-cells and several genes downstream of ESR1 were altered when compared with the CD4+ T-cells isolated from the mediastinal lymph nodes of HF mice. This was interesting as CD4+ T-cells were sorted from failing male hearts. To further validate these findings, we measured gene expression of ERα and ERβ in the remote zones of LV tissues isolated from the male HF mice and found them to be significantly increased relative to sham controls (Figure S1B). Similar increase in the ERα expression has also been reported in the LV of male and female HF patients16. Studies in other autoimmune diseases have shown the role of ERs in modulating T-cell activity7 but these studies are mostly done using ovariectomized female mice and very limited work has been done in male mice. Therefore, we compared the steady-state expression of ERα and ERβ in the ovaries with the hearts of male and female mice, and spleens of naïve male mice. As shown in Figure S1C, while ERα expression in the hearts and spleens was comparable, ERβ expression was ~11-fold higher in spleens as compared to the hearts. We then magnetically sorted splenic CD4+ T-cells from the naïve male mice and measured the expression of ERs. Interestingly, ERβ expression in splenic CD4+ T-cells was 10-fold lower as compared to ERα (Figure S1D) suggesting that ERα signaling is predominant in CD4+ T-cells and, other splenic immune cells of myeloid origin express significantly higher levels of ERβ as compared to CD4+ T-cells (supplemental material and Figure S2A and S2B). These data are consistent with other studies showing that the highest expression of ERα is observed in T-cells obtained from human PBMCs10.
Figure 1:

A) At 8 weeks post-myocardial infarction (MI), CD4+ T-cells from the failing hearts (150 cells) and mediastinal lymph-nodes (300 cells) were flow sorted and limited-cell RNA sequencing (lcRNA seq) was conducted to identify differential gene expression changes. IPA analysis identified ESR1 activation as one of the upregulated nodes in cardiac CD4+ T-cells and ESR1 dependent gene expression changes in the dataset are indicated. Upregulated genes are shown in red while downregulated genes are shown in green. Orange color represents ‘predicted activation’ whereas blue represents ‘predicted inhibition’. Lines in orange and blue represent ‘leads to activation or inhibition’ respectively, whereas yellow lines show ‘findings inconsistent with state of downstream molecule’ and grey lines show effects that could not be predicted. (B) Representative flow histograms showing ERα and ERβ expression in cardiac CD4+ T-cells at 3 days post-MI. Group quantitation for ERα (C) and ERβ (D) expression in cardiac CD4+ T-cells at 3 days and at 8 weeks post-MI. (E) Representative flow histograms showing ERβ expression in splenic (left) and cardiac (right) immune cells of myeloid and lymphoid lineages at 3 days post-MI. Group quantitation for mean fluorescence intensity (MFI) of ERβ in CD19+ B-cells, CD4+ T-cells, CD11b+Ly6G+ neutrophils and CD11b+Ly6G−Ly6C+ monocytes in the spleens (F) and the hearts (G) at 3 days (left) and at 8 weeks post-MI (right). Group quantitation for mean fluorescence intensity (MFI) of ERβ in CD4+ T-cells in the spleen, blood and the hearts at 3 days (H) and at 8 weeks post-MI (I). Data in C and D was analyzed using two-tailed unpaired students T-test whereas 1-way ANOVA with Tukey’s post-hoc test was used to analyze data in F, G, H and I.
It is not known what role, if any, ER signaling plays in modulating CD4+ T-cell activation in the context of MI and HF. Since 3d post-MI marks the peak protective inflammatory response17 and 8w post-MI exhibits significant LV remodeling with pathological transitioning of CD4+ T-cells1, we measured ERα and ERβ expression in CD4+ T-cells at these time-points. As shown in Figure 1B/C, as compared to sham mice, ERα expression in CD4+ T-cells was significantly decreased at 3d but was significantly increased at 8w post-MI, consistent with our lc-RNA seq data showing the activation of ERα pathway at this time-point. ERβ expression, on the other hand, did not change at 3d but decreased significantly at 8w post-MI when compared with the sham mice (Figure 1D). Upon further analysis of spatial changes in ERβ expression, we found that despite low ERβ expression in spleen (Figure 1E and 1F), CD4+ T-cells infiltrating into the failing hearts (Figure 1G) had higher ERβ expression as compared to other cells of either myeloid origin (Ly6G+ neutrophils or Ly6C+ monocytes/macrophages) or lymphoid origin, (CD19+ B-cells). Representative gating strategy for measuring all of these cells in the ischemic myocardium is shown in Figure S3. Moreover, both at 3d and 8 wks post-MI, ERβ expression in cardiac CD4+ T-cells was much lower as compared to circulating or splenic T-cells (Figure 1H and 1I). These changes were not specific to all cells of lymphocytic lineage or were not due to differential non-specific binding of antibodies between splenic and cardiac single cell preparations, as B-lymphocytes (comparable in size and ERβ expression in the naive mice; Figure S2B) did not exhibit this change at 3d post-MI (Figure S4), the peak of acute inflammatory response post-MI. These data show that (1) ERα expression is temporally regulated and its high levels coincide with pathological transitioning of T-cells, 2) ERβ expression is significantly and sustainably increased in cardiac CD4+ T-cells when compared with other immune cells post-ischemic injury, 3) ERβ is spatially modulated, and its expression in CD4+ T-cells significantly declines upon infiltration into the ischemic hearts at 3d and 8w post-MI when compared with other tissues, and importantly, 4) ERβ activation to constrain ERα signaling could be a potential target to blunt pathological transitioning of CD4+ T-cells.
ERβ agonist Dose Dependently Inhibits Anti-CD3/CD28 Mediated T-cell Proliferation
ERα signaling is functionally antagonized by ERβ agonism18. Thus, we tested a novel ERβ agonist, OSU-ERb-012, a lipophilic and orally bioavailable compound with ~200-fold selectivity for ERβ as compared to ERα and ~300-fold selectivity against other nuclear hormone receptors (synthesis, ligand-binding studies, pharmacology and biopharmaceutical characterization is published elsewhere19). We evaluated this compound for its ability to inhibit TCR (anti-CD3/CD28) mediated proliferation of splenic CD4+ T-cells isolated from the naive male mice. As shown in Figure 2A, OSU-ERb-012 dose-dependently inhibited TCR mediated T-cell proliferation with an IC50 of 3.4 μM. Importantly, while the drug did not affect non-stimulated CD4+ T-cells (Figure S5A), it significantly inhibited the proliferation of TCR activated CD4+ T-cells even in the presence of low and high estradiol concentrations (Figure 2B) without affecting cell-survival (supplemental material and Figure S5B). Importantly, these effects were ERβ specific as OSU-ERb-012 failed to inhibit CD4+ T-cells isolated from ERβ−/− mice (Figure S5C). Using 5 μM concentration, we also tested the effects of OSU-ERb-012 on the expression of pro-inflammatory cytokines, such as TNFα and IFNγ, and activation marker CD69. As compared to non-stimulated T-cells, TCR stimulation significantly increased TNFα (Figure 2C and 2D) IFNγ (Figure 2E) and CD69 expression (Figure S5D) both in the absence as well as in the presence of estradiol, and was significantly inhibited by the drug. There was no change in either the TNFα or IFNγ expression in the non-stimulated cells treated with the drug in the presence or absence of estradiol (Figures S5E and S5F) suggesting that the drug did not affect homeostatic expression of these cytokines. Also, OSU-ERb-012 did not affect PMA/Ionomycin mediated T-cells activation that bypasses TCR (supplemental material and Figure S6). We also tested CD4+ T-cells isolated from the female mice and observed similar effects (Figure S7), suggesting that although the drug was specific for ERβ, the effects were not sex-specific, and T-cells from both male and female mice were equally inhibited. Interestingly, the effects of OSU-ERb-012 were drastically amplified in the presence of E2 and a more drastic reduction of TNFα and IFNγ expressing cells was observed with T-cells isolated from female mice, especially at 5 nM E2 concentration. Since, we did not track the estrous cycle of female mice before harvesting splenic T-cells, this could be due to increased susceptibility of CD4+ T-cells during different phases of estrous cycle. Nonetheless, these findings indicate an important role of estradiol, and ERs in mediating TCR activation and T-cell proliferation in both the sexes.
Figure 2:

(A) Representative flow cytometric histograms for Tag-it violet™ (TIV) labeled CD4+ T-cells either non-stimulated or stimulated with anti-CD3 and anti-CD28 antibodies in the absence and presence of different concentrations of OSU-ERb-012. Peak patterns from high to low fluorescence intensity in stimulated cells represent halving of dye concentration in the daughter cells with every successive cell division. Cell proliferation (%) in the presence of different concentrations of the drug is used to derive dose-response curve (lower panel, right). (B) Representative flow cytometric histograms for TIV labeled CD4+ T-cells either non-stimulated or stimulated with anti-CD3 and anti-CD28 antibodies and treated with either Estradiol (E2; 5 and 50 nM) or OSU-ERb-012 (5 μM) or both, and group quantitation for % cell proliferation (right). (C) Representative flow histograms showing TNFα expression in non-stimulated and stimulated CD4+ T-cells treated either with estradiol or OSU-ERb-012 or both. Group quantitation for the frequency of CD4+TNFα+ and CD4+IFNγ+ cells is shown in (D) and (E), respectively. Mean values from 3–4 separate experiments (conducted by isolating splenic CD4+ T-cells from 3–4 male mice) done in triplicate are reported. One way Anova with Tukey’s post-hoc test was used for the data analysis. *p represent significance with respect to non-stimulated cells whereas $p represent significance with respect to vehicle treated stimulated cells.
OSU-ERb-012 Activates ERβ Signaling
To investigate the specificity of the drug, we conducted RNA sequencing of anti-CD3/CD28 stimulated CD4+ T-cells cultured with and without 5 μM drug. Principal component analysis (Figure 3A) showed that the RNA transcriptome of TCR-activated CD4+ T-cells was significantly altered in the presence of 5 μM OSU-ERb-012 as compared to the stimulated cells treated with the vehicle. We identified several genes that were either downregulated or upregulated (log2 fold change>2 and p<0.01) in the presence of drug as shown in the volcano plot (Figure 3B). As expected, pathway analysis of differentially expressed genes in the dataset indicates activation of ERβ (ESR2) and concomitant functional suppression of ERα (ESR1) (Figure 3C and Figure S8). Of the ERβ dependent genes identified in the dataset, the majority were upregulated upon OSU-ERb-012 treatment (Figure 3D). We also observed downregulation of several genes involved in TCR activation, further supporting TCR specificity of OSU-ERb-012 (Figure 3E). We further assessed the effects of ERβ agonism on other pathways required for immune activation. Analysis of upregulated and downregulated gene transcripts showed that several pathways involved in inflammation, immune activation and metabolism were either inhibited or negatively-affected by OSU-ERb-012 (Figure 3F) leading to an overall reduction in antigenic CD4+ T-cell activation.
Figure 3:
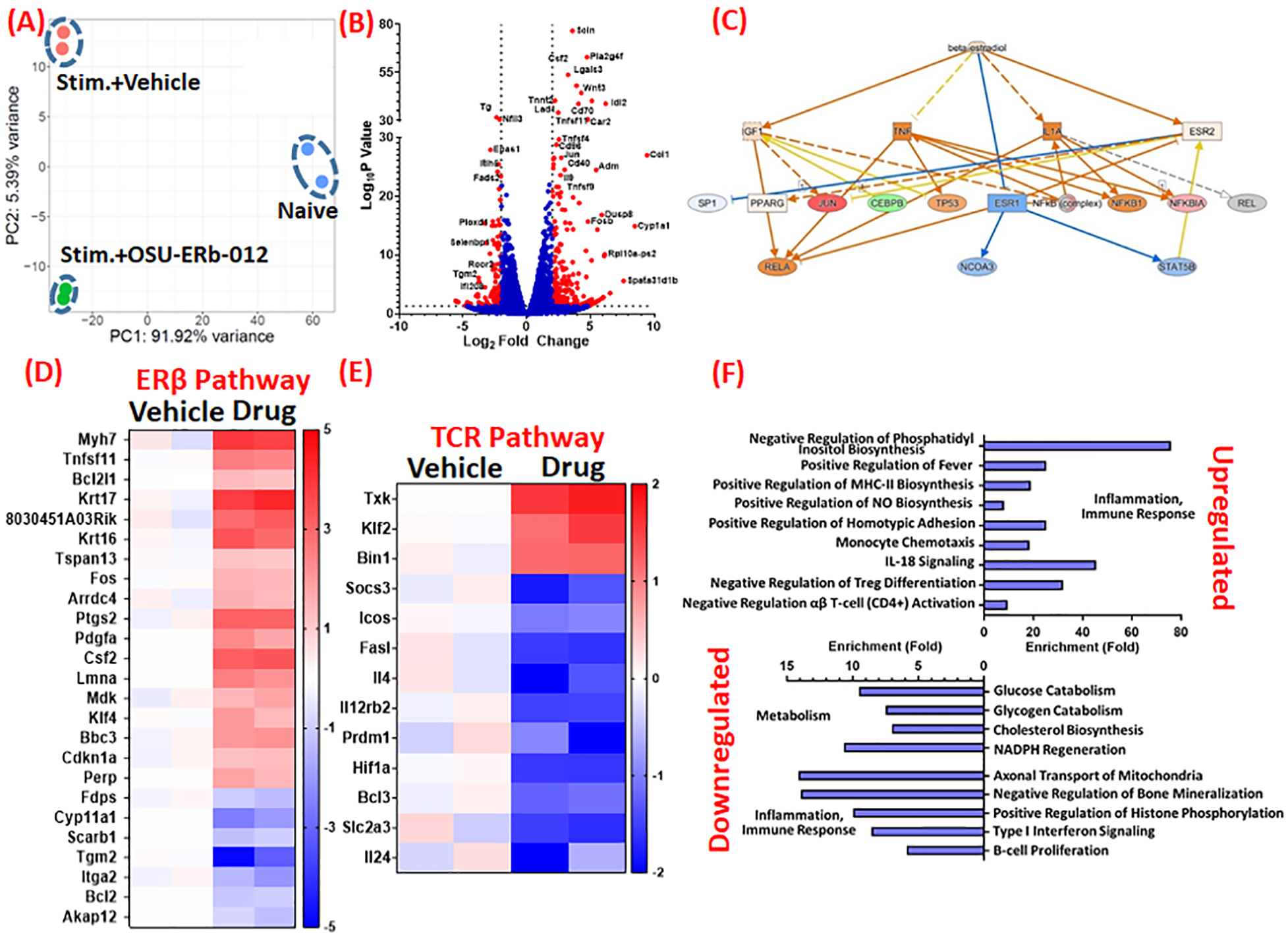
(A) Principal Component Analysis of RNA transcriptomes of naïve and stimulated (stim) CD4+ T-cells treated either with the vehicle (DMSO) or OSU-ERb-012 (5 μM). (B) Volcano plot showing several genes (marked as red) are either upregulated or downregulated by more than 2-fold in stimulated CD4+ T-cells treated with OSU-ERb-012 (5 μM) as compared to the vehicle treatment. Some of the representative genes that showed either very high LogP values or very high fold-changes are labeled. (C) Ingenuity Pathway analysis of RNA transcriptomes of stimulated CD4+ T-cells treated either with the vehicle control (DMSO) or OSU-ERb-012 (5 μM). Heat maps depicting genes in the Estrogen Receptor (ER) β (D) and T-cell receptor (TCR) (E) pathway that are either significantly up- or down-regulated in stimulated CD4+ T-cells upon treatment with OSU-ERb-012 (5 μM). (F) Pathways that are most affected in stimulated CD4+ T-cells upon treatment with OSU-ERb-012. N=2/gp.
OSU-ERb-012 Treatment Specifically during Chronic HF is Protective and Ameliorates Cardiac Remodeling
Considering that CD4+ T-cells are critical for wound healing during the initial 14 days post-MI6 but are pathological and dispensable during chronic HF1, we decided to test the efficacy of the drug at both the phases. For acute-MI, using echocardiography we measured akinetic area at 7d post-MI and randomized mice to either receive vehicle or 60 mg/kg OSU-ERb-012 (summarized in Figure 4A). Daily administrations of the drug via gavage did not affect the body weight of infarcted or sham mice (Figure 4B), suggesting that the drug did not have any overt side-effects on rodent physiology. However, we observed significantly increased mortality (Figure 4C) with the drug, as several mice (~65%) died during the first week of treatment when compared with the vehicle control (20–25%). This observation is consistent with other studies6 and underscores the vital role of CD4+ T-cells for healing post-MI.
Figure 4:

(A) Schematic for the experimental plan to test the efficacy of OSU-ERb-012 during acute phase of MI and during chronic HF. Random: Randomization; Echo: echocardiography. (B) Body weight (g) of mice that either underwent sham-operation or myocardial infarction (MI) followed by treatment either with the vehicle or OSU-ERb-012 (60 mg/kg/day; gavage). For clarity of data, SD is shown only for HF group treated with vehicle and was comparable in all groups. N=4–7 in each group (C) Kaplan-Meier curve showing mortality rate in MI mice treated either with the vehicle or drug. N=6–10 in each group (D) Representative B-mode tracings depicting systole and diastole of failing hearts at 4 w (at the time of randomization) and 8 weeks post-MI after treatment either with the vehicle or the drug. (E) Group quantitation for the change in end-systolic and end-diastolic volumes (ESV and EV), and the ejection fraction (EF) from 4 to 8 weeks post-MI after treatment either with the vehicle or the drug. Unpaired students two-tailed T-test was used for analyzing data in (E).
For chronic HF, cardiac function was evaluated at 4w post-MI. All animals were randomized according to the degree of cardiac dysfunction (Figure S9A), measured by end-systolic and end-diastolic volumes (ESV and EDV), and ejection fraction (EF), to either receive vehicle or 60 mg/kg drug, daily, via gavage, for 4 weeks. Interestingly, we did not observe any drug related mortality or any changes in body weight (Figure S9B) when OSU-ERb-012 was administered during the chronic phase. Echocardiography measurements at 8w post-MI (4w post-treatment) showed that the cardiac dysfunction in vehicle-treated HF mice progressed during this time with increased ESV and EDV, and reduced ejection fraction (Figures 4D and 4E, and Figure S10A). In contrast, the cardiac function of OSU-ERb-012 treated HF mice did not worsen, and the ESV, EDV and EF did not change over 4 weeks of drug treatment (Figure S10A). The cardiac function in drug-treated HF mice was significantly better than the vehicle-treated mice at 8w post-MI (Figures 4D and 4E). We observed similar effects in ovx female mice treated with OSU-ERb-012 from 4 to 8 weeks post-MI (Figure S10B). This improvement in cardiac function was not due to differences in heart rate as the average heart rate was comparable between both the groups and was more than 480 BPM at both time-points (Figure S10C).
OSU-ERb-012 Treatment Ameliorates Cardiac Hypertrophy and Fibrosis
Gravimetric analysis of heart and LV weights showed that vehicle treated HF mice exhibited significantly increased tibia-normalized heart and LV weights, as compared to the sham mice, which was significantly ameliorated with drug treatment from 4 to 8 weeks post-MI (Figure 5A). To further validate these results, we conducted WGA staining of LVs and measured cardiomyocyte area. A shown in Figure 5B, drug treatment significantly decreased cardiomyocyte area and cellular hypertrophy, when compared with the vehicle-treated HF mice. We also observed a significant decrease in the gene expression of several hypertrophy markers such as Gja1, Gja5, MyH7, and MyH6 while some others such as BNP and RyR2 showed a decreasing trend (Figure 5C and Table S1). We also measured fibrosis in the LVs of drug-treated HF male mice and observed a significant reduction in interstitial fibrosis in the remote-zone upon treatment with OSU-ERb-012 (Figure 5D). Similar amelioration of hypertrophy and fibrosis was also observed in ovx HF females (Figure S10D).
Figure 5:

(A) Gravimetric data for tibia normalized heart and left-ventricular (LV) weights of sham and heart failure (HF) mice treated either with the vehicle or OSU-ERb-012. N=9–12 in each group. (B) Representative images of LV sections stained with FITC conjugated Wheat-germ agglutinin to show cardiac hypertrophy. Boxed area in the upper panel is shown in the lower panel, and the group quantitation for cardiomyocyte area is shown in right. N=4 in each group. (C) Gene expression of cardiac hypertrophy markers in the remote-zone LV of HF mice treated either with the vehicle or OSU-ERb-012 from 4 to 8 weeks post-MI. N=4–8 in each group. (D) Representative images showing interstitial fibrosis in the LVs of HF mice treated either with the vehicle or the drug and their group quantitation. N=4–5 in each group. Two way Anova with Tukey’s post-hoc test was used for data analysis in (A), while two-tailed students T-test was used for (B), (C) and (D).
OSU-ERb-012 Treatment Selectively Depletes CD4+ Helper T-cells in HF mice
Using flow cytometry, we measured CD4+ T-cells in the circulation, hearts and the spleens of vehicle- and drug-treated HF mice at 8w post-MI (4w post-treatment). Daily treatments with the OSU-ERb-012 significantly decreased circulating CD4+ T-cells at 8 wks in HF male as well as ovx female mice (Figures S11 and S12). Reduction in T-cell numbers was not limited to a particular helper T-cell subset as both pro- and anti-inflammatory cells, viz CD4+TNFα+, CD4+FoxP3+ Tregs, CD4+IFNγ+ Th1 T-cells, CD4+IL-4+ Th2 T-cells, and CD4+IL-17+ Th17 T-cells were significantly decreased (Figure S11). Similarly, we also observed a significant decrease in cardiac CD4+ T-cells in OSU-ERb-012-treated HF mice when compared with the vehicle-treated mice (Figure 6A and 6B). This decrease was reflected in a significant decline of both TNFα+ and IFNγ+ cells, suggesting inhibition of pro-inflammatory cytokine producing CD4+ T-cells, consistent with our data from ex-vivo T-cell inhibition assays. A similar decrease in CD4+ T-cell numbers (and frequency) was also observed in splenic CD4+ T-cells (Figure 6C and 6D). Interestingly, while cell counts for FoxP3+ Tregs were also decreased in spleens (Figures S13A and S13B), the overall frequency of Tregs (Figure S13C) as well as expression of FoxP3 (Figure S13D) was increased suggesting that the other Th subsets were decreased more than FoxP3+ Tregs and Tregs remaining after the drug treatment were significantly potent and immune-suppressive20. Importantly, OSU-ERb-012 did not affect other immune cells either in the heart, blood or spleens (supplemental material and Figure S14) of HF mice.
Figure 6:

(A) Representative flow scatter plots depicting CD4+ and CD8+ T-cells in total CD45+ cells. HF: heart failure. (B) Levels of CD4+ Helper T-cells and its pro-inflammatory subsets viz CD4+TNFα+ cells and CD4+IFNγ+ (Th1), T-cells at 8 weeks post-MI in mice treated either with the vehicle or OSU-ERb-012 from 4 to 8 weeks post-MI. (C) Representative flow scatter plots for splenic CD4+ and CD8+ T-cells in CD45+ leukocytes, and (D) group quantitation for splenic CD4+ Helper T-cells (total cell numbers and frequency) at 8 weeks post-MI in mice treated either with the vehicle or OSU-ERb-012 from 4 to 8 weeks post-surgery. Unpaired Students 2-tailed T-test was used for the data analysis in (B) and (D).
Discussion
ERα expression is known to increase in the LVs of both male and female HF patients16. Studies have also shown that systemic estrogen levels are strong predictors of survival in male HF patients with both high and low concentrations being associated with increased mortality21. Complementing these clinical observations, we demonstrate several novel key findings in this study. First, we show that during chronic HF ERα signaling is significantly upregulated in cardiac CD4+ T-cells also. While ERα expression is temporally regulated to significantly decrease during MI and increase during chronic HF, ERβ expression is spatially, and temporally regulated in a cell- and tissue-dependent context. Second, ERβ is selectively increased in cardiac CD4+ T-cells as compared to other innate and adaptive immune cells infiltrated into the myocardium both at 3 days and 8 weeks post-MI. However, spatial comparison in different tissues showed that ERβ expression is significantly decreased in cardiac CD4+ T-cells as compared to splenic or circulating CD4+ T-cells at both the time-points. Third, OSU-ERb-012, a novel ERβ agonist, dose dependently inhibited CD4+ T-cell activation, proliferation and the expression of pro-inflammatory cytokines in both sexes. Fourth, ERβ agonists, if given early after MI, result in increased mortality, but improve cardiac function and reduce tissue hypertrophy if given during chronic HF, in both the sexes. This underscores significant immunological differences between MI and chronic HF, at least from the perspective of adaptive immunity. Fifth, OSU-ERb-012 specifically blunts CD4+ T-cells in the failing hearts, lymphoid tissues and circulation without affecting other immune cells, and can be developed as a selective therapeutic for temporal modulation of CD4+ T-cells during ischemic HF.
ERα signaling play a pivotal role in CD4+ T-cells by mediating i) activation against exogenous antigens22, ii) polarization into IFNγ producing Th1 T-cells22, and iii) trafficking and migration into tissues by regulating the expression of chemokine receptors such as CCR1, CCR2, CCR3, CCR4 and CCR523. Together, these studies suggest a potential pathological role of ERα signaling in T-cell biology by promoting excessive polarization into pro-inflammatory subtypes. Our findings are consistent with these studies and underscore the importance of decreased ERα signaling in CD4+ T-cells at 3d post-MI to tone down non-antigenic T-cell activation and fine-tune Th1 polarization to promote repair processes. Upregulation of ERα signaling at 8w post-MI compromises these protective responses and may, partly, mediate pathological transitioning and antigenic activation of CD4+ T-cells during HF.
Previous studies have shown that hearts from ERα−/− mice are more prone to ventricular fibrillations and tachycardia, and exhibit increased cell death and reduced contractility11, 13. Interestingly, ERβ loss in male and female mice also exacerbate LV remodeling24 and increase mortality post-MI12, and treatment only with ERβ, and not ERα, agonists improve cardiac function in TAC-induced HF mice25. These suggest that while both ERα and ERβ are cardio-protective, ERβ agonists alone can ameliorate LV remodeling during non-ischemic HF. However, the cellular responders of protective ERβ signaling have not been identified in any of these studies. Other studies conducted using isolated hearts have further shown that cardiac ERα or ERβ expression is not required for estradiol-mediated cardio-protection9. This suggests that perhaps cells other than cardiomyocytes play a critical role in dictating ERβ mediated protection during CVDs. Our studies suggest CD4+ T-cells to be one of the key players in this regard.
Our data also suggest a cell- and tissue-specific response for ERβ expression. Although, there was significantly higher ERβ expression in cardiac CD4+ T-cells as compared to other immune cells, we observed an overall reduction in ERβ expression upon infiltration into the ischemic hearts when compared with circulating and splenic CD4+ T-cells. These findings are significant considering the fact that HIF1α activation during ischemic injury is known to increase ERβ signaling to auto-regulate its own transcriptional activity26. ERβ, in turn, enhances integrin α1 and β1 expression to augment vinculin-mediated adhesive potential, cellular motility and transmigration of cells27. This increase in ERβ expression could, therefore, either be a direct result of hypoxic milieu encountered by infiltrating T-cells, or a mechanism to amplify integrin-mediated trafficking and transmigration of T-cells into the tissues. Reduced ERβ expression and enhanced ERα signaling at 8w post-MI, thus, indicate an imbalance in ER signaling with amplification of Th1/Th17 mediated pro-inflammatory responses during HF.
CD4+ T-cells during HF, (ischemic or non-ischemic), exhibit TCR-mediated activation and proliferation against cardiac neo-antigens1, 2. Hence, for HF immune modulation, it is critical that strategies targeting CD4+ T-cells specifically diminish TCR activation to avoid general immune suppression and infections by opportunistic pathogens. The ER pathway is, thus, of significant therapeutic potential as CD4+ T-cells isolated from ERα−/− mice exhibit decreased NFAT1, Zap70 and STAT5 levels, and diminished TCR activation28. It is important to note that a decrease in these transcription factors could either be a direct consequence of lost ERα signaling or could be indirect due to amplified ERβ signaling (in the absence of ERα). Our studies show latter to be the case, as our selective ERβ agonist dose-dependently inhibited the proliferation of TCR activated CD4+ T-cells in both sexes. ERα also regulates the gene expression of several T-cell specific cytokines (such as IFNγ, TNFα, IL-4, and IL-17), chemokine receptors (such as CCR1, CCR2, CCR3, CCR4 and CCR5)23, and other transcription factors (such as NF-kB and NFAT)29, all of which have been implicated in auto-immune diseases as well as in HF30. Although, in our RNA sequencing data we did not observe any significant changes in chemokines, OSU-ERb-012 significantly inhibited the expression of pro-inflammatory cytokines. The fact that these effects were limited only to the TCR pathway, as OSU-ERb-012 did not inhibit T-cells stimulated with PMA/Ionomycin, suggest a unique advantage of using this drug as other immune activation pathways are spared. As of now it is not clear if increased ER signaling in HF patients (and in our rodent studies) is ligand-dependent or independent as increased estrogen levels have also been reported in male HF patients21. However, our ex-vivo T-cell proliferation assays and studies from others show that E2 is not critical for T-cell activation, and absence of either ERα28 or ERβ (Figure S5C) does not completely abolish T-cell activation/proliferation. These suggest that ERs are not necessary for T-cell activation and are accessory pathways that regulate transcription and expression of pro-inflammatory cytokines and activation signals in T-cells downstream of TCR. This regulation could also be due to other parallel signaling mechanisms such as EGFR31, neuregulin31, IGF-132 or cAMP/PKA33 which are all known to enhance ER signaling in a ligand-independent manner. However, if any or all of these pathways are mediating these effects is not known and needs further exploration using specific inhibitors and activators of these pathways.
CD4+ T-cells mediate wound-healing and fibrotic scar formation post-MI6. In contrast, we have previously shown that ischemic HF is associated with a 2nd wave of CD4+ T-cell activation (reflected as a global increase in both pro- and anti-inflammatory helper T-cell subsets that exhibit a pathological phenotype), and promote LV remodeling and progressive cardiac dysfunction1. Together, these studies suggest that CD4+ T-cells activated immediately after ischemic injury, such as in MI, are immunologically and phenotypically different than those activated during chronic HF. Our studies showing increased mortality with OSU-ERb-012 if given early after-MI, but improved function and blunted LV remodeling if given during HF further bolster these findings. Our studies also show that ERβ agonists can potentially promote Foxp3 expression as well as frequency of Tregs, and could be a great tool to fine-tune immune activation by promoting immuno–suppressive mechanisms34. In-vitro experiments showing significant inhibition of CD3/CD28 mediated T-cell proliferation in the absence of APCs also indicate that the anti-proliferative effects of OSU-ERb-012 during in-vivo studies were probably not mediated through effects on APCs. This is also supported by the fact that OSU-ERb-012 did not change other innate and adaptive immune cells.
Conclusions
We show that similar to other autoimmune diseases, ER signaling in CD4+ T-cells is temporally and spatially altered during MI and HF in a context-dependent manner. ERs play an essential role in TCR-mediated antigen-specific T-cell activation, and are downstream regulators of amplified gene expression required to effect activation, proliferation and cytokine expression in CD4+ T-cells. Due to this central role, ERs are highly suitable drug targets for immunomodulation to blunt CD4+ T-cells during HF. However, caution is warranted as the optimal timing of such therapies needs to be carefully ascertained to refrain from inhibiting protective wound-healing responses required during MI.
Supplementary Material
What is New?
Preclinical studies suggest protective roles of Estrogen Receptor (ER) β activation in cardiovascular diseases (CVDs). However, cellular mediators of these protective effects are unknown.
We show that increased ERα signaling in CD4+ T-cells is protective during myocardial infarction (MI) and its sustained activation could be one of the molecular switches that lead to pathological transitioning of CD4+ T-cells during chronic HF.
Activation of ERβ (to functionally antagonize ERα) using a novel drug inhibits T-cell receptor mediated T-cell activation, ameliorate left-ventricular remodeling, and blunt progressive cardiac dysfunction in HF mice.
ERβ agonists selectively inhibit CD4+ T-cells without affecting other immune cells.
What are the Clinical Implications?
Epidemiological studies show protective effects of estrogen (E2) and its receptors (ERs) in CVDs, and clinical studies show upregulated E2 levels and ERα signaling in male HF patients.
We show that ERα during MI is protective but chronic ERα activation is involved in the pathological transitioning of CD4+ T-cells. Importantly, therapy using ERβ agonists, to functionally antagonize ERα, could be a potential immune-modulatory therapy for ischemic HF.
Time-dependent effects are important for therapeutic immuno-modulation and temporal pathological immune activation needs to be identified to ascertain optimal therapeutic window for immune modulation in patients.
Acknowledgements:
Authors thank Drs. Chris Coss and Chad Bennett from the Drug Development Institute for helpful discussions.
Funding:
This work was supported by the National Institutes of Health (NIH) grants to SSB (R00 HL132123), RJG (R01 HL127442), and MSS (AG056848). This work was also supported by the Drug Development Institute within The Ohio State University Comprehensive Cancer Center and Pelotonia. Medicinal Chemistry and Flow Cytometry Shared Resources are supported by NCI/NIH Grants P30CA01605 and P30CA016058, respectively. Funding sources did not influence any part of this work.
Non-standard Abbreviations and Acronyms
- MI
Myocardial infarction
- HF
Heart failure
- ER
Estrogen receptor
- FC
Flow Cytometry
- LV
Left ventricle
- APCs
Antigen-presenting cells
- TCR
T-cell receptor
- ESV
End-systolic volume
- EDV
End-diastolic volume
- EF
Ejection fraction
- TAC
Transverse aortic constriction
- Treg
regulatory T-cell
- MS
Multiple sclerosis
- EAE
Experimental autoimmune encephalomyelitis
- E2
Estradiol
- GPER
G-protein coupled estrogen receptor
- LcRNA seq
Limited-cell RNA sequencing
- PKC
Protein kinase C
- MFI
Mean fluorescence intensity
- CVD
Cardiovascular disease
- RBC
Red blood cell
- DEGs
Differentially expressed genes
Footnotes
Disclosures: OSU-ERb-012 is an investigational drug designed and supplied by the Medicinal Chemistry Shared Resource, which is a part of The Ohio State University Comprehensive Cancer Center.
Declarations of Interest: None
Supplemental Materials:
References
- 1.Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G and Prabhu SD. Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circ Heart Fail. 2017;10:e003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ngwenyama N, Kirabo A, Aronovitz M, Velazquez F, Carrillo-Salinas F, Salvador AM, Nevers T, Amarnath V, Tai A, Blanton RM et al. Isolevuglandin-Modified Cardiac Proteins Drive CD4+ T-Cell Activation in the Heart and Promote Cardiac Dysfunction. Circulation. 2021;143:1242–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carrillo-Salinas FJ, Ngwenyama N, Anastasiou M, Kaur K and Alcaide P. Heart Inflammation: Immune Cell Roles and Roads to the Heart. Am J Pathol. 2019;189:1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nehra S, Gumina RJ and Bansal SS. Immune cell Dilemma in Ischemic Cardiomyopathy: To Heal or Not to Heal. Curr Opin Physiol. 2021;19:39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR and Prabhu SD. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ Res. 2014;114:266–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, Ertl G, Kerkau T and Frantz S. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation. 2012;125:1652–63. [DOI] [PubMed] [Google Scholar]
- 7.Rubtsova K, Marrack P and Rubtsov AV. Sexual dimorphism in autoimmunity. J Clin Invest. 2015;125:2187–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lerner DJ and Kannel WB. Patterns of coronary heart disease morbidity and mortality in the sexes: a 26-year follow-up of the Framingham population. Am Heart J. 1986;111:383–90. [DOI] [PubMed] [Google Scholar]
- 9.Kabir ME, Singh H, Lu R, Olde B, Leeb-Lundberg LM and Bopassa JC. G Protein-Coupled Estrogen Receptor 1 Mediates Acute Estrogen-Induced Cardioprotection via MEK/ERK/GSK-3beta Pathway after Ischemia/Reperfusion. PLoS One. 2015;10:e0135988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Phiel KL, Henderson RA, Adelman SJ and Elloso MM. Differential estrogen receptor gene expression in human peripheral blood mononuclear cell populations. Immunol Lett. 2005;97:107–13. [DOI] [PubMed] [Google Scholar]
- 11.Wang M, Crisostomo P, Wairiuko GM and Meldrum DR. Estrogen receptor-alpha mediates acute myocardial protection in females. Am J Physiol Heart Circ Physiol. 2006;290:H2204–9. [DOI] [PubMed] [Google Scholar]
- 12.Pelzer T, Loza PA, Hu K, Bayer B, Dienesch C, Calvillo L, Couse JF, Korach KS, Neyses L and Ertl G. Increased mortality and aggravation of heart failure in estrogen receptor-beta knockout mice after myocardial infarction. Circulation. 2005;111:1492–8. [DOI] [PubMed] [Google Scholar]
- 13.Zhai P, Eurell TE, Cooke PS, Lubahn DB and Gross DR. Myocardial ischemia-reperfusion injury in estrogen receptor-alpha knockout and wild-type mice. Am J Physiol Heart Circ Physiol. 2000;278:H1640–7. [DOI] [PubMed] [Google Scholar]
- 14.Covarrubias R, Ismahil MA, Rokosh G, Hamid T, Accornero F, Singh H, Gumina RJ, Prabhu SD and Bansal SS. Optimized protocols for isolation, fixation, and flow cytometric characterization of leukocytes in ischemic hearts. Am J Physiol Heart Circ Physiol. 2019;317:H658–H666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lindsey ML, Brunt KR, Kirk JA, Kleinbongard P, Calvert JW, de Castro Bras LE, DeLeon-Pennell KY, Del Re DP, Frangogiannis NG, Frantz S, Gumina RJ, Halade GV, Jones SP, Ritchie RH, Spinale FG, Thorp EB, Ripplinger CM and Kassiri Z. Guidelines for in vivo mouse models of myocardial infarction. Am J Physiol Heart Circ Physiol. 2021;321:H1056–H1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahmoodzadeh S, Eder S, Nordmeyer J, Ehler E, Huber O, Martus P, Weiske J, Pregla R, Hetzer R and Regitz-Zagrosek V. Estrogen receptor alpha up-regulation and redistribution in human heart failure. FASEB J. 2006;20:926–34. [DOI] [PubMed] [Google Scholar]
- 17.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R and Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams C, Edvardsson K, Lewandowski SA, Strom A and Gustafsson JA. A genome-wide study of the repressive effects of estrogen receptor beta on estrogen receptor alpha signaling in breast cancer cells. Oncogene. 2008;27:1019–32. [DOI] [PubMed] [Google Scholar]
- 19.Sedlak D, Wilson TA, Tjarks W, Radomska HS, Wang H, Kolla JN, Lesnikowski ZJ, Spicakova A, Ali T, Ishita K, et al. Structure-Activity Relationship of para-Carborane Selective Estrogen Receptor beta Agonists. J Med Chem. 2021;64:9330–9353. [DOI] [PubMed] [Google Scholar]
- 20.van Loosdregt J, Fleskens V, Fu J, Brenkman AB, Bekker CP, Pals CE, Meerding J, Berkers CR, Barbi J, Grone A, et al. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg-cell-suppressive capacity. Immunity. 2013;39:259–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jankowska EA, Rozentryt P, Ponikowska B, Hartmann O, Kustrzycka-Kratochwil D, Reczuch K, Nowak J, Borodulin-Nadzieja L, Polonski L, Banasiak W, et al. Circulating estradiol and mortality in men with systolic chronic heart failure. JAMA. 2009;301:1892–901. [DOI] [PubMed] [Google Scholar]
- 22.Maret A, Coudert JD, Garidou L, Foucras G, Gourdy P, Krust A, Dupont S, Chambon P, Druet P, Bayard F and Guery JC. Estradiol enhances primary antigen-specific CD4 T cell responses and Th1 development in vivo. Essential role of estrogen receptor alpha expression in hematopoietic cells. Eur J Immunol. 2003;33:512–21. [DOI] [PubMed] [Google Scholar]
- 23.Mo R, Chen J, Grolleau-Julius A, Murphy HS, Richardson BC and Yung RL. Estrogen regulates CCR gene expression and function in T lymphocytes. J Immunol. 2005;174:6023–9. [DOI] [PubMed] [Google Scholar]
- 24.Fliegner D, Schubert C, Penkalla A, Witt H, Kararigas G, Dworatzek E, Staub E, Martus P, Ruiz Noppinger P, Kintscher U, et al. Female sex and estrogen receptor-beta attenuate cardiac remodeling and apoptosis in pressure overload. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1597–606. [DOI] [PubMed] [Google Scholar]
- 25.Iorga A, Umar S, Ruffenach G, Aryan L, Li J, Sharma S, Motayagheni N, Nadadur RD, Bopassa JC and Eghbali M. Estrogen rescues heart failure through estrogen receptor Beta activation. Biol Sex Differ. 2018;9:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim W, Cho J, Kwon HY, Park Y, Rhyu MR and Lee Y. Hypoxia-inducible factor 1 alpha activates and is inhibited by unoccupied estrogen receptor beta. FEBS Lett. 2009;583:1314–8. [DOI] [PubMed] [Google Scholar]
- 27.Lindberg K, Strom A, Lock JG, Gustafsson JA, Haldosen LA and Helguero LA. Expression of estrogen receptor beta increases integrin alpha1 and integrin beta1 levels and enhances adhesion of breast cancer cells. J Cell Physiol. 2010;222:156–67. [DOI] [PubMed] [Google Scholar]
- 28.Mohammad I, Starskaia I, Nagy T, Guo J, Yatkin E, Vaananen K, Watford WT and Chen Z. Estrogen receptor alpha contributes to T cell-mediated autoimmune inflammation by promoting T cell activation and proliferation. Sci Signal. 2018;11:eaap9415. [DOI] [PubMed] [Google Scholar]
- 29.Hirano S, Furutama D and Hanafusa T. Physiologically high concentrations of 17beta-estradiol enhance NF-kappaB activity in human T cells. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1465–71. [DOI] [PubMed] [Google Scholar]
- 30.Gordon JW, Shaw JA and Kirshenbaum LA. Multiple facets of NF-kappaB in the heart: to be or not to NF-kappaB. Circ Res. 2011;108:1122–32. [DOI] [PubMed] [Google Scholar]
- 31.Giuliano M, Trivedi MV and Schiff R. Bidirectional Crosstalk between the Estrogen Receptor and Human Epidermal Growth Factor Receptor 2 Signaling Pathways in Breast Cancer: Molecular Basis and Clinical Implications. Breast Care (Basel). 2013;8:256–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yee D and Lee AV. Crosstalk between the insulin-like growth factors and estrogens in breast cancer. J Mammary Gland Biol Neoplasia. 2000;5:107–15. [DOI] [PubMed] [Google Scholar]
- 33.Carascossa S, Dudek P, Cenni B, Briand PA and Picard D. CARM1 mediates the ligand-independent and tamoxifen-resistant activation of the estrogen receptor alpha by cAMP. Genes Dev. 2010;24:708–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bansal SS, Ismahil MA, Goel M, Zhou G, Rokosh G, Hamid T and Prabhu SD. Dysfunctional and Proinflammatory Regulatory T-Lymphocytes Are Essential for Adverse Cardiac Remodeling in Ischemic Cardiomyopathy. Circulation. 2019;139:206–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walker LA, Sovic MG, Chiang CL, Hu E, Denninger JK, Chen X, Kirby ED, Byrd JC, Muthusamy N, Bundschuh R and Yan P. CLEAR: coverage-based limiting-cell experiment analysis for RNA-seq. J Transl Med. 2020;18:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raju R, Bland KI and Chaudry IH. Estrogen: a novel therapeutic adjunct for the treatment of trauma-hemorrhage-induced immunological alterations. Mol Med. 2008;14:213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giaglis S, Stoikou M, Sur Chowdhury C, Schaefer G, Grimolizzi F, Rossi SW, Hoesli IM, Lapaire O, Hasler P and Hahn S. Multimodal Regulation of NET Formation in Pregnancy: Progesterone Antagonizes the Pro-NETotic Effect of Estrogen and G-CSF. Front Immunol. 2016;7:565. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.