

Summary
Nutritional deficiency and genetic errors that impair the transport, absorption, and utilization of vitamin B12 (B12) lead to hematological and neurological manifestations. The cblC disease (cobalamin complementation type C) is an autosomal recessive disorder caused by mutations and epi-mutations in the MMACHC gene and the most common inborn error of B12 metabolism. Pathogenic mutations in MMACHC disrupt enzymatic processing of B12, an indispensable step before micronutrient utilization by the two B12-dependent enzymes methionine synthase (MS) and methylmalonyl-CoA mutase (MUT). As a result, patients with cblC disease exhibit plasma elevation of homocysteine (Hcy, substrate of MS) and methylmalonic acid (MMA, degradation product of methylmalonyl-CoA, substrate of MUT). The cblC disorder manifests early in childhood or in late adulthood with heterogeneous multi-organ involvement. This review covers current knowledge on the cblC disease, structure–function relationships of the MMACHC protein, the genotypic and phenotypic spectra in humans, experimental disease models, and promising therapies.
Subject areas: biochemistry, biological sciences, enzymology, health sciences
Graphical abstract
Biochemistry; Biological sciences; Enzymology; Health sciences
Introduction
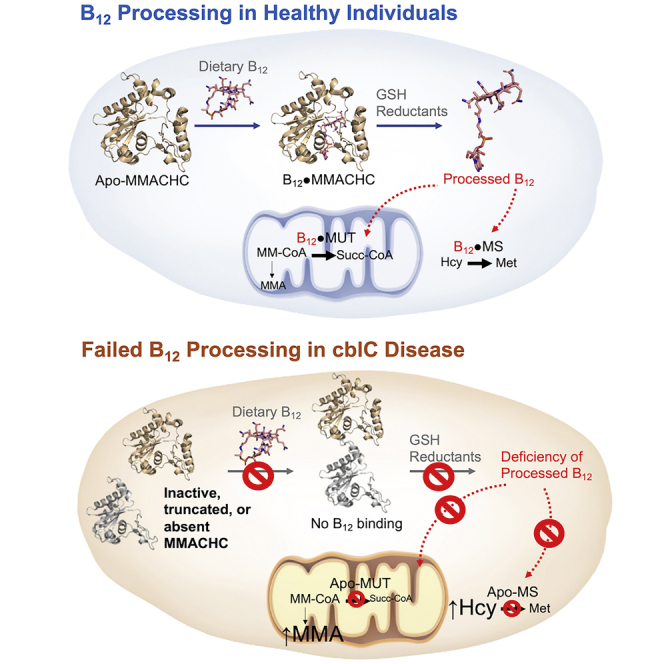
The cblC disease (cobalamin complementation type C) is the most common inborn error of vitamin B12 metabolism and presents combined homocystinuria and methylmalonic aciduria. This rare inherited disorder affects newborns, infants, and adults in a devastating manner. Its discovery goes back to S.H. Mudd and H.L. Levy, who described and investigated the first case in 1969 (Mudd et al., 1969). The infant was remitted to the Massachusetts General Hospital at four weeks of age with poor feeding, failure to thrive, lethargy, and anemia and died at 7 1/2 weeks of age. A previously unrecognized pattern of amino acid concentrations in plasma, urine, and tissue included homocystinemia(uria), cystathioninemia(uria), and hypomethioninemia(uria) in combination with methylmalonic aciduria and drew attention to vitamin B12 metabolism. A publication by McCully later in 1969 identified the same metabolic findings in a second patient with cblC disease (McCully, 1969a). McCully reported that the cblC patient exhibited certain clinical similarities to a patient with homocystinuria owing to cystathionine β-synthase (CBS) deficiency, and was first to propose that the vascular complications common to both patients may be driven or mediated by elevated plasma homocysteine (McCully, 1969a). This original proposal opened the field to the investigation of the role of homocysteine in vascular disease well beyond the scope of inborn errors of metabolism, stimulating a number of important epidemiological studies in the decades that followed (McCully, 2005).
Vitamin B12, also known as cobalamin, is synthesized by a limited number of archaeal and bacterial species (Roth et al., 1996). Yet, it is essential to all cells in humans and animals, who therefore rely on its dietary intake and a complex network of proteins for the transport, intestinal absorption, systemic distribution, and cellular utilization of the micronutrient (Figures 1A and 1B). Mammals possess two cobalamin-dependent enzymes, namely MS and MUT, localized in the cytosol and mitochondrion, respectively. Cytosolic MS utilizes MeCbl as a cofactor and N5-methyltetrahydrofolate Me-THF) as a substrate to catalyze the methylation of Hcy to form methionine (Met), thus connecting the methionine and folate metabolic pathways (Figure 1C). Mitochondrial MUT employs AdoCbl as a cofactor to catalyze the conversion of methylmalonyl-CoA into succinyl-CoA. Defective transformation of dietary cobalamin to the coenzymatically active forms MeCbl and AdoCbl, resulting in the loss of MS and MUT activities, provided the link between the biochemical abnormalities found in the first cblC patient (Levy et al., 1970; Mudd et al., 1970, 1969).
Figure 1.

Cellular processing and trafficking of dietary cobalamins
(A) Dietary intake and gastrointestinal absorption of cobalamin in humans. Dietary intake of B12 from animal foods like milk, eggs, and meet incorporates different cobalamin forms, such as MeCbl, AdoCbl, and H2OCbl. Commercially available supplements mainly contain CNCbl. Regardless of the specific chemical form, Cbl is released from dietary proteins in the stomach and then binds to haptocorrin, a transport protein that is produced by the salivary glands and protects cobalamin from gastric acid. Once in the duodenum, pancreatic proteases cleave haptocorrin, and Cbl binds to a second transport protein-intrinsic factor. The intrinsic factor is required for receptor-mediated endocytosis in the terminal ileum via the so-called cubam receptor, through which Cbl is taken up by the enterocytes. After lysosomal degradation of intrinsic factor, and Cbl efflux mediated by multidrug resistance protein 1 (MDR1) in the basolateral membrane of the enterocyte, the micronutrient reaches the bloodstream. Here cobalamin binds to its systemic transport apo-protein transcobalamin (apo-TC) and distributed to all cells (holo-TC).
(B) Cellular uptake, processing, and utilization of cobalamin. TC-receptor-mediated endocytosis (TCR) enables cellular uptake of circulating holoTC. Transcobalamin and its receptor undergo degradation in the lysosome (Gick et al., 2020), and free Cbl is released from the organelle via the transporters CblF and CblJ. In the cytosol, Cbl undergoes processing by MMACHC. The processing enzyme MMACHC catalyzes the removal of the upper axial ligand (β-ligand) of Cbls via a multitude of β-deligase mechanisms. MMACHC-MMADHC interactions direct the cofactor to either cytosolic methionine synthase (MS, CblG) or mitochondrial methylmalonyl-CoA mutase (MUT). Nutritional and functional cobalamin deficiencies impair the function of MS and MUT and lead to elevations of total homocysteine (Hcy) and methylmalonic acid (MMA).
(C) Role of Cbl in cellular metabolism. Cobalamin associated metabolic pathways include folate cycle, methionine cycle, S-adenosyl methionine (SAM)-dependent methylation, and catabolism of odd-chain fatty acids and branched-chain amino acids. Abbreviations: cobalamin derivatives (R-Cbl(III)), methylcobalamin (MeCbl), adenosylcobalamin (AdoCbl), dihydrofolate (DHF), tetrahydrofolate (THF), methionine (Met), S-adenosyl homocysteine (SAH); deoxyuridine monophosphate (dUMP); deoxythymidine monophosphate (dTMP), methylmalonyl-CoA (MMA-CoA), succinyl-CoA (SUC-CoA). Figures adapted from Hannibal et al. (2016) and Green et al. (2017).
In further studies, the failure to accumulate AdoCbl and MeCbl was confirmed (Mahoney et al., 1971). AdoCbl is formed by the reduction and adenosylation inside mitochondria near the target enzyme MUT (Mahoney et al., 1975). MeCbl was predicted to form directly near the cytosolic MS during the catalytic cycle (Quadros and Jacobsen, 1995). Mellman et al. stated that a previously unidentified enzyme with the activity of cob(III)alamin reductase affected the conversion of cyanocobalamin (CNCbl) and hydroxocobalamin (HOCbl) into a common coenzyme precursor (Mellman et al., 1979). Cofactor requirement synthesis appeared to be linked to β-ligand elimination and cobalt reduction. Studies on cell-extracts by Pezacka et al. confirmed the cob(III)alamin reductase and cyanocobalamin β-ligand transferase deficiency in cblC mutants and pointed to glutathionylcobalamin (GSCbl) as an intermediate in the synthesis of both AdoCbl and MeCbl (Pezacka, 1993; Pezacka and Jacobsen, 1992; Pezacka et al., 1990). Early treatment proposals based on the very first case included different cobalamin forms such as CNCbl and HOCbl, betaine, choline, and supplementation with methionine (Levy et al., 1970; Mudd et al., 1970, 1969). These are still the fundamental components of cblC disease treatment today (Huemer et al., 2017; Mudd et al., 1969).
Patients with disorders of isolated as well as combined versions of homocystinuria and methylmalonic aciduria present with clinical and biochemical heterogeneity. Mahoney et al. proposed their differentiation into complementation groups cblA, cblB, and cblC allocating the hypothesized underlying defects to different branches of cobalamin processing in cells (Mahoney et al., 1975). The simultaneous defect in AdoCbl and MeCbl synthesis to yield combined homocystinuria with methylmalonic aciduria was defined as the cblC type. They proposed that cblC was an intracellular defect occurring before MS and MUT activity. The complementation groups defined by their expression in fibroblast heterokaryons belong to separate gene loci and are inherited in an autosomal recessive manner (Gravel et al., 1975).
In 2006 Lerner-Ellis et al. identified the MMACHC gene encoding the cobalamin processing enzyme responsible for cblC disease (Lerner-Ellis et al., 2006). The MMACHC gene maps to chromosome region 1p34.1 and consists of 5 exons and 10,736 bp. The mRNA is 849 bp in length and encodes a protein of 282 amino acids with a predicted molecular weight of 31.7 kDa. MMACHC does not form part of any known gene family. It is well conserved among mammals, but no homologous gene or protein was found in other eukaryotes or prokaryotes. Still, its C-terminal domain presents similarities to cobalamin-binding enzymes in bacteria. A study examining 204 cblC individuals identified 42 mutations among which variant 271dupA is the most frequent, accounting for 40% of all pathogenic alleles. The transduction of wild-type MMACHC into 271dupA mutant fibroblasts restored cobalamin metabolism. The first genotype-phenotype correlations were established pointing to associations between combinations of specific mutations and early or late-onset disease. To date, approximately 1,000 patients have been identified worldwide, and the implementation of newborn screening allows an approximation of incidence ranging between ⅓,220 to ½00,000 in different populations (Wang et al., 2019). This review is focused on the MMACHC gene (OMIM ∗609831) and the functions of its protein product MMACHC.
Biochemical characterization of the MMACHC protein
Biological chemistry of cobalamins
Structure of cobalamins
The isolation (Barker et al., 1960) and structural elucidation of cyanocobalamin (Hodgkin et al., 1956), with the cyanide derivative being a vestige of the isolation process (Veer et al., 1950), and adenosylcobalamin (cofactor B12) (Galen Lenhert and Hodgkin, 1961) provided valuable insights into the chemistry of cobalamins. Cobalamins contain a corrin-ring core with seven side chains (Figures 2A–2F). The corrin core consists of four pyrrole rings joined by a C-CH3 methylene linker on opposite sides connecting the A, B, and C pyrrole rings (Figures 2A–2D). The A and D pyrrole rings are connected by a trans-C-H linker, leading to a helical geometry of the corrin ring. In comparison with the closely similar porphyrin ring, the corrin ring lacks a bridging C-atom, contains reduced pyrrole rings, and is not aromatic. These characteristics lend a degree of flexibility to the cobalamins. In natural cobalamins, the nitrogen atoms of the four pyrrole rings are coordinated to a central cobalt atom, which is coordinated 6-fold in total. The other two ligands that coordinate with the cobalt atom are the upper axial β-ligand and the lower axial nitrogen atom of a 5,6-dimethylbenzimidazole group (α-ligand). The 5,6-dimethylbenzimidazole (DMBI) group connects to a ribose moiety which phosphorylates with the C17 side chain of the corrin ring. The side chains of the corrin ring comprise a total of seven peripheral acetamide and propionamide side chains, which are important for anchoring the cofactor within protein skeletons.
Figure 2.

Structure of cobalamin
R = -CN (Cyanocobalamin), -Me (Methylcobalamin), -OH (Hydroxocobalamin), 5′deoxy-5′-adenosyl (Adenosylcobalamin), -OH2 (Aquacobalamin), -NO (Nitroxylcobalamin), L-Glutamyl-L-cysteinylglycine (Glutathionylcobalamin), L-Cys (Cysteinylcobalamin).
The biochemical reactions driven by cobalamins exploit the chemical versatility of three distinct structural motifs of the cobalamin skeleton:
-
1.
the equatorial corrin core, comprising the corrin ring, the cobalt ion, and the axial ligands, responsible for the reactivity and electronic interactions of cobalamins;
-
2.
the outer layer comprising of the nitrogen and oxygen atoms of the amide side chains and the nucleotide skeleton, which form inter and intramolecular hydrogen bonds influencing protein binding, enzymatic activity, and solubility; and
-
3.
an intermediate layer of aliphatic side chains affecting steric interactions.
Owing to the flexibility of the corrinoids that arises from a partially saturated corrin core and the side chains, the effects of interactions between the aforementioned motifs can be readily transmitted from one part of the cobalamin moiety to another (Pratt, 1972). The corrinoid ring folds upwards toward the upper axial β-ligand, like the wings of a butterfly. The fold line bisects the corrin ring through the center of the trans bridge joining the A and D rings on the western side, passing through the cobalt atom and onto the methyl bridge between the B and C rings on the eastern side. The fold angle is influenced by the nature of the α- and β-ligands. A shorter Co-Nax distance results in greater folding of the corrin ring (Pett et al., 2002).
Base-on/base-off configurations and redox states of cobalamins
At low pH, the DMBI base undergoes protonation to generate the base-off form of cobalamins. Competition with strongly coordinating ligands, for example, cyanide (Zelder, 2008), which displace the intra-molecularly coordinated DMBI, leads to the generation of the base-off form at neutral or alkaline pH. The base-on/base-off equilibria of cobalamins are often represented as in Figure 3A. The Co-β-ligand affects the base-on/base-off equilibria of the cobalamins through the trans and cis-effects (De March et al., 2012). The presence of a strong σ-donating β-ligand leads to stretching of the Co-Nax bond (structural trans influence). The Co-Nax bond lengths show an excellent correlation with the pKbase-off values, suggesting that the presence of a strong σ-donating β-ligand favors the base-off form (Figure 3B) (De March et al., 2012; Hassanin et al., 2009a). The Co-Nax bond stretching owing to the σ-donating β-ligand is accentuated in cobalamins in comparison with cobaloximes, octahedral cobalt complexes which are used as the functional model for cobalamins. This indicates a steric cis-effect, whereby the folding of the corrin ring decreases as the Co-Nax bond length increases. For bulkier β-ligands as in thiolatocobalamins, which feature a Co-S β-axial bond, Co-Nax bond lengths increase as the Co-S bond length decreases in the order SC(NH2)2 > S2O3 > SO3, resulting from the cis-effect (Randaccio et al., 2006). However, ground state cis- and trans-effects often do not translate to the kinetics of reactions involving axial ligand substitutions. An example is the formation of stable thiolatocob(III)alamins on reactions with reduced thiols. Cobinamides are corrinoids lacking the dimethylbenzyimidazole moiety of cobalamin. Aquahydroxocobin(III)amide undergoes reduction to cobin(II)amide in the presence of GSH (Dereven’kov et al., 2018). Glutathionylcobalamin, formed by the reaction of GSH with hydroxocobalamin, in spite of possessing a bulky β-ligand, does not undergo the aforementioned reduction.
Figure 3.

Base-on/base-off equilibria in cobalamins
(A) The base-on/base-off equilibria of cobalamins are governed by a combination of acid-base equilibria and coordination equilibria. The acid-base equilibria feature the protonation of the lower DMBI base. Coordination to the cobalt center by competing α-ligands determine the coordination equilibria of cobalamins. The overall equilibrium constant is given as KBase-Off = (1 + KCo) KBz (Brown et al., 1984).
(B) Correlation between the Co-Nax bond length and the pKbase-off of cobalamins with various β-ligands owing to the trans-effect (De March et al., 2012).
Cobalamins as well as cobinamides and cobaloximes react with hydrogen sulfide (Salnikov et al., 2014; Toohey, 2017), which is suggested to play a mechanistic role in cobalamin-dependent methyl group transfer as catalyzed by MS and radical S-adenosylmethionine methyl transfer (RSMT) (Toohey, 2017). Subsequent to complex formation, inner-sphere electron transfer from sulfur to the cobalt atom leads to the reduction of Co(III) to Co(II) and the formation of sulfur radicals (Salnikov et al., 2014). Dependent on acidic or alkaline pH, aquacob(III)alamin reacts to form six-coordinate cob(II)alamin with the anion-radical SSH2− or five-coordinate cob(II)alamin, respectively. As proposed by Toohey, the sulfur atom could be displaced by a methyl cation to form MeCbl or, alternatively, form part of the methyl transfer unit promoting the methylation of Hcy to Met on MS (Toohey, 2017). This may help to meet the requirements of strong reductants in the accepted mechanism of MS, which require anaerobic conditions and the two methyl donors, S-adenosylmethionine (SAM) and Me-THF, to form MeCbl from cob(II)alamin and cob(I)alamin, respectively.
Cobinamide, a drug in the advanced stages of development for cyanide poisoning, reduced sulfide toxicity and sulfide-induced oxidative stress in cells and whole animals, showing promise as a specific therapy for sulfide poisoning (Jiang et al., 2016). Boss et al. have shown that cobinamide was significantly more efficacious than hydroxocobalamin when compared at equal mg/kg dosages (Bebarta et al., 2017). Hydroxocobalamin or/and cobinamide are also the rational choices for treating methyl mercaptan (Hendry-Hofer et al., 2020) and azide (Tat et al., 2021) poisoning. Aquacobalamin forms a complex with isoniazid (isonicotinic acid hydrazide) (Tumakov et al., 2017), which is one of the most effective antituberculosis drugs. Many papers were also devoted to the influence of the other nitrogen-containing compound – N,N-dimethylbiguanide, known as the oral diabetes medicine metformin on cobalamin deficiency (Chapman et al., 2016). Although the exact mechanism is unknown, metformin’s ability to cause vitamin B12 deficiency is well established, and physicians should be cognizant of the increased incidence of vitamin B12 deficiency in long-term users of metformin (Green et al., 2017).
Coordination states and spectral properties of cobalamins in biological systems
Three coordination states of cobalamins exist under physiological conditions (cob(III)alamin, cob(II)alamin, and cob(I)alamin corrins). The number of axial ligands decreases with a decrease in coordination number with a few exceptions. Thus, recent spectroscopic studies revealed that cobalamins reduced by dithionite exhibited a unique property distinct from other cob(II)alamin (Mieda-Higa et al., 2020; Salnikov et al., 2011). It is shown that cob(II)alamin forms a relatively stable six-coordinated complex with sulfur dioxide anion radical SO2− having strong reducing properties. Indeed, dithionite (sulfur dioxide anion radical) produces the catalytically incompetent cob(II)alamin (Allen and Wang, 2014; Gagsteiger et al., 2022). Insights on the importance of the upper axial ligand in cobalamin structure and reactivity can be derived from a comparison of the structure of cob(II)alamin-SO2− with the other important complex – nitrosylcobalamin NOCbl (Hannibal et al., 2007; Hassanin et al., 2009b), which is readily formed when cob(II)alamin reacts with nitric oxide. Pallares and Brunold have shown (Pallares and Brunold, 2014) that NOCbl is best described as a hybrid of Co(III)−NO− and Co(II)−NO⋅ resonance structures. In contrast, the description of the structure of the complex of cobalamin with superoxide (superoxocobalamin) as Co(III)−O2− is adequate owing to the larger oxidizing power of O2 versus NO (Pallares and Brunold, 2014). Thus, the structure of cobalamins depends on the redox properties of the upper axial ligand.
Cyanocobalamin and other oxidized cobalamins (cob(III)alamins) are red in their base-on configuration, whereas cob(II)alamins are generally orange-brown in color. The base-on form of the paramagnetic free radical, cob(II)alamin, results from one-electron reduction of cob(III)alamin. Reduction leads to the formation of the diamagnetic cob(I)alamin form, which is emerald green in color. The colors of the three cobalamin coordination states also depend on the concentration and range, for example, for cob(I)alamin between black and light green. Thermodynamically, the cobalt center of cobalamins is stabilized against cob(III)alamin/cob(II)alamin reduction by strongly nucleophilic, coordinating, or basic ligands (Butler and Kräutler, 2006). Hence, the values for reduction potentials for cob(III)alamin/cob(II)alamin redox couples are greater for the base-on form in comparison with the base-off forms of cobalamins. The redox potentials for cob(II)alamin/cob(I)alamin are lower than those for cob(III)alamin/cob(II)alamin. The redox potential of cob(II)alaminbase-on/cob(I)alamin and cob(II)alaminbase-off/cob(I)alamin have been reported to be −0.61 V and −0.50 V (both values vs NHE, 22°C), respectively (Figure 4) (Lexa and Saveant, 1976).
Figure 4.

Shuttling of cobalamins between the cob(III)alamin, cob(II)alamin, and cob(I)alamin redox states (∗versus NHE, 22°C) Adapted from Butler and Kräutler (2006)
Thus, it would seem that the reduction cob(II)alamin/cob(I)alamin is thermodynamically uphill with physiological reducing agents. Yet, these reactions occur in the presence of the dual flavoenzymemethionine synthase reductase (Olteanu and Banerjee, 2003; Wolthers and Scrutton, 2004) and also by MMACHC (see next sections). One mechanism for the formation of cob(I)alamin involves the removal of nucleophiles from the coordination sphere of the cobalt atom, leading to the formation of the tetracoordinated cob(II)alamin form. This increases the redox potential of the cob(II)alamin/cob(I)alamin couple (Liptak and Brunold, 2006). Another mechanism based on DFT calculations postulates the formation of a pseudo pentacoordinated cob(I)alamin transition state involving one or two hydrogen bonds involving the cobalt ion. The pseudo-pentacoordinated cob(I)alamin transition state is more thermodynamically favorable than a tetracoordinated cob(I)alamin (Kumar et al., 2012; Kumar and Kozlowski, 2011). Trends in the redox transformation of cobalamins have been extensively studied and reviewed elsewhere (Butler and Kräutler, 2006; Dereven’kov et al., 2016; Johns et al., 2015; Lehene et al., 2021; Lexa and Saveant, 1976; Pugina et al., 2018; Salnikov et al., 2021).
Cobalamins have characteristic absorption spectra described as γ-, D/E−, and αβ-bands between 300 and 600 nm (Figure 5, panel a). The spectra arise owing to the π- π∗, d-d transitions of the cobalt ion, and charge transfer transitions of and between the β-ligand, corrin ring, and the equatorial ligands. High molar extinction coefficients of up to 3 × 104 M−1 cm−1 have been reported for the absorption spectra of various cobalamins (Beaven and Johnson, 1955; Chemaly, 2008). The absorption spectra of cobalamins, which is characteristic of the oxidation state of the cobalt atom and the β-ligand, have been used to study the reaction kinetics of various cobalamins in their free forms and when bound to cognate transport proteins, chaperones, and enzymes (Froese et al., 2012; Kim et al., 2009, 2008; Koutmos et al., 2011). Most cobalamin-dependent enzymes bind the cobalamins in the base-off configuration, where the lower α-ligand DMBI base is tucked away from the cobalt center of the corrin ring (Banerjee et al., 2021). Such is the case of MMACHC, whose base-off transitions have been confirmed via X-ray crystallography and UV-visible spectrometry (Kim et al., 2009; Koutmos et al., 2011) (Figure 5, panel b). Some proteins also bind to the free cobalt center arising from the protonation of the DMBI base via coordination to a histidine residue forming the base-off/His-on form of cobalamins. The coordination chemistry of the cobalt center and the functionality of the cobalamin-binding proteins often dictate the bound form of cobalamins. In plasma, cobalamins are associated with transcobalamin II, which selectively binds biologically relevant undegraded cobalamins in the base-on form (Wuerges et al., 2006). The cobalamin-dependent enzymes methionine synthase binds cob(II)alamin in the base-off/His-on form (Drennan et al., 1994). In the mitochondrial branch, where cob(II)alamin is converted into the cofactor form AdoCbl by the enzyme adenosyltransferase (ATR), the cob(II)alamin is bound in a unique tetra-coordinated base-off form (Stich et al., 2005). In contrast, the mitochondrial cobalamin-dependent enzyme MUT binds cobalamins in the base-off/His-on form (Padmakumar et al., 1995). The binding of cobalamins to apoenzymes is paramount to their cofactor activity and is dictated by the cleavage of the Co-C bond of the β-ligand. In AdoCbl-dependent enzymes, the cleavage of the Co-C bond occurs homolytically, generating a free radical cob(II)alamin and a radical form of the exiting β-ligand (Banerjee, 1997). The reaction is reversible and the cob(III)alamin form is regenerated via a free radical addition of the adenosyl moiety to the cob(II)alamin. This mechanism explains how enzyme-processed cobalamins are efficient free radical scavengers. In contrast, MeCbl-dependent enzymes require the Co-C bond to be cleaved heterolytically (Figure 6). The dealkylation by heterolytic cleavage of the Co-C bond occurs by a nucleophilic attack at the cobalt atom leading to the substitution of the β-ligand. The reaction proceeds via a two-step SN2 mechanism, where the immediate product generated is a base-off alkyl-substituted corrin, which subsequently changes to its base-on form (Banerjee, 1997). A host of alkyl substituted corrinoids have also been synthesized by SN2 reactions following heterolytic cleavage of cob(III)alamin corrins to the cob(I)alamin form electrochemically or by reducing agents (Kräutler, 2005; Kräutler and Puffer, 2012; Suarez-Moreira et al., 2006).
Figure 5.

UV-visible spectra of cobalamins and structure of MMACHC-bound MeCbl
(A) UV-visible spectra of the predominant cobalamin forms in the cell – H2OCbl, AdoCbl, and MeCbl. Base-on spectra were taken in water, base-off spectra in 0.1 N HCl.
(B) Structure of MeCbl bound to human MMACHC (PDB ID 3SC0) and UV-visible spectra of MeCbl in the base-on form in buffer compared with the base-off form when bound to recombinant human MMACHC.
Figure 6.

Mechanism of heterolytic cleavage of the Co-C bond in cobalamins
The cob(I)alamin generated by reduction acts as a strong nucleophile that can be used to synthesize a wide array of cobalamins. Adapted from Butler and Kräutler (2006)
Likewise, the formation of base-off Cbl species constitutes an important step in CNCbl reductive decyanation. CNCbl reacts with reducing agents via slow DMBI dissociation followed by fast binding of reductant and further electron transfer (Salnikov et al., 2013). Generated cob(II)alamin binds CN- relatively weakly (Lexa et al., 1980), which results in CN- dissociation.
Enzymatic activity of MMACHC
The identification of the MMACHC gene in 2006 (Lerner-Ellis et al., 2006) enabled functional studies of its encoded protein. Banerjee and coworkers pioneered the expression and purification of human recombinant MMACHC protein and confirmed one of the originally proposed enzymatic activities, namely, CNCbl β-decyanase (Kim et al., 2008). While this work failed to identify β-dealkylase activity, follow-up in vitro studies with purified protein and cultured fibroblasts from cblC patients demonstrated that the two most abundant dietary forms of cobalamin, namely, MeCbl and AdoCbl were also processed by the MMACHC protein to yield reduced cobalamin under anaerobic conditions and aquacobalamin in the presence of oxygen (Hannibal et al., 2009; Kim et al., 2009). The dealkylation of dietary MeCbl and AdoCbl and the decyanation of supplemental CNCbl in the cytoplasm lead to the formation of vital base-off reduced cobalamin that can readily enter the catalytic cycles of recipient cobalamin-dependent enzymes MS and MUT (Figure 7).
Figure 7.

β-deligation mechanism of MMACHC
Cyanocobalamin and alkylcobalamins undergo reduction to cob(II)alamin, which is channeled to methionine synthase (MS) or via adenosylcobalamin transferase (ATR) to methylmalonyl-CoA mutase (MUT) to yield the respective biologically active forms, MeCbl and AdoCbl, respectively. Decyanation requires flavin coenzymes FMN or FAD and NADPH. Dealkylation proceeds in the presence of glutathione (GSH) to generate a cob(I)alamin thatundergoes oxidation to the cob(II)alamin cofactor form. The MMACHC bound cob(II)alamin binds to cysteine-261 of the adaptor protein MMADHC forming a protein-linked thiolatocobalamin (Li et al., 2020b).
Initial studies using recombinant MMACHC showed that MMACHC-bound CNCbl undergoes reductive decyanation that proceeds in the presence of NADPH yielding cob(II)alamin (Kim et al., 2008). The one-electron reduction is catalyzed by dual flavoprotein oxidoreductases, human methionine synthase reductase, or novel reductase 1. Crystallographic studies on the structure of MMACHC revealed that the protein contains an N-terminal flavin binding domain. Follow-up in vitro studies with recombinant MMACHC demonstrated decyanase activity in the presence of the reductants, NADPH and FMN/FAD (Koutmos et al., 2011). It had been previously reported that reduced GSH was required for decyanation of CNCbl by cell extracts (Pezacka et al., 1990). In vitro experiments using GSH under aerobic conditions could not demonstrate thiol-catalyzed decyanase activity of MMACHC. However, under anaerobic conditions and at high, albeit physiologically relevant concentrations of GSH, one-electron reductive decyanation activity of the MMACHC was also demonstrated (Li et al., 2014a).
The elucidation of the decyanase activity of MMACHC illustrated how the dietary supplement CNCbl is processed toward the cofactor form to supply MUT and MS. However, the mechanism for the utilization of the alkylcobalamins MeCbl and AdoCbl, the dietary sources of cobalamins, remained elusive. In early 2009, Hannibal et al. showed that cultured cells could utilize a series of n-alkylcobalamin derivatives to form the biologically viable cobalamin cofactors, MeCbl and AdoCbl (Hannibal et al., 2009). cblC cell lines however lacked the ability for biotransformation of alkylcobalamins to MeCbl and AdoCbl (Hannibal et al., 2009). Studies using recombinant MMACHC provided further evidence that the MMACHC possesses cobalamin dealkylase activity coupled to a thiol oxidase activity (Kim et al., 2009). Alkylcobalamins bound to MMACHC undergo reduction to cob(I)alamin in a dealkylation reaction involving GSH. The reaction proceeds via a nucleophilic displacement of the alkyl moiety by the thiolate group, resulting in the formation of the corresponding alkylated GSH (Figure 7).
The catalytic versatility of MMACHC is illustrated by the discovery of additional substrates and electron sources that generate the essential bioactive intermediate base-off cob(II)alamin or its precursor cob(I)alamin. Thiolatocobalamins including glutathionylcobalamin have been shown to be more viable substrates for several cobalamin-dependent enzymes (Pezacka et al., 1990). In this study, glutathionylcobalamin showed faster rates of β-deligation than other cobalamin substrates. In vitro studies showed that thiolatocobalamins are antioxidant/anti-inflammatory agents that inhibit manifestations of oxidative stress (Birch et al., 2009). Thiolatocobalamins exhibited superior inhibition of intracellular peroxide production, supported the maintenance of the intracellular GSH pool, and prevented cell death from necrosis and apoptosis in cell lines exposed to homocysteine and hydrogen peroxide. Further, the thiolatocobalamins used in the study were non-toxic even at supraphysiological concentrations. In enzymatic studies involving recombinant bovine MMACHC protein with glutathionylcobalamin as substrate, higher β-dethiolation rates in comparison with other cobalamin congeners, have been reported (Jeong et al., 2014). These findings have generated renewed interest in the use of thiolatocobalamins as possible drug candidates in diseases involving a defective cobalamin metabolism. In recent studies on the enzymatic activity of MMACHC and pathogenic mutants of MMACHC using synthetic thiolatocobalamins as substrates, a 4-fold increase in rates for β-deligation in comparison with MeCbl has been reported (Wingert et al., 2021). Additionally, these synthetic thiolatocobalamins featuring a reductant β-ligand joined to the cobalamin scaffold, undergo spontaneous β-deligation when MMACHC-bound in the absence of GSH. Thiolatocob(III)alamins have been also shown to be generated via protein–protein interactions between the MMACHC and MMADHC proteins (Li et al., 2020b). The MMADHC protein is a partitioning or adaptor protein that directs the delivery of the active cofactor cob(II)alamin to MS and MUT. A cysteine β--ligand supplied by the MMADHC protein to MMACHCbound cob(II)alamin is responsible for MMACHC-MMADHC interactions (Li et al., 2020b). This protein–protein interaction results in the channeling of the cofactor form of cobalamins to the mitochondrial MUT and the cytosolic MS compartments, respectively (Figure 7).
MMACHC also performs reductive β-deligation of substrates not featuring a cobalt-carbon or cobalt-sulfur bond. For example, MMACHC possesses nitrite reductase activity (Mascarenhas et al., 2020). It catalyzes the denitration of nitrocobalamin leading to the formation of intermediate cob(II)alamin, which further oxidizes under aerobic conditions to form aquacobalamin. MMACHC bound cob(I)alamin catalyses the reduction of nitrite to nitric oxide with the formation of cob(II)alamin. This reaction is then followed by the rapid formation of nitrosylcobalamin by the combination of the cob(II)alamin and nitrosyl free radical species. A summary of the reactions catalyzed by MMACHC is provided in Figure 8.
Figure 8.

A summary of the reactions catalyzed by mammalian MMACHC (citations are provided in the text)
Another salient aspect of the β-deligation chemistry of cobalamins is the generation of the physiologically active cob(II)alamin form. As evidenced from in vitro studies, the β-deligated cobalamins undergo transformation into aquacobalamin and remain bound to MMACHC. MMACHC-bound aquacobalamin undergoes reduction to cob(II)alamin in the presence of NADH and riboflavin (Dereven’kov et al., 2020). The reduction involves formation of a NADH⋅Co(III) complex which further decomposes to cob(II)alamin and NADH+. This study provided mechanistic and kinetic insights into the transformation of MMACHC-bound aquacobalamin to the cob(II)alamin form that can be readily utilized by MUT and MS.
The bovine homolog of MMACHC has been expressed and purified (Jeong et al., 2011). Bovine MMACHC shares 88% sequence identity with human MMACHC. Bovine recombinant MMACHC binds to cobalamins in the base-off configuration and is stabilized thereby. Studies with purified recombinant bovine MMACHC demonstrated that binding of GSH to MMACHC increases the protein’s affinity for cobalamins (Jeong and Kim, 2011). Furthermore, bound GSH protects base-off aquacobalamin from catalyzing the oxidation of GSH to form GSSG (Jeong et al., 2011). Altogether, the binding of GSH to MMACHC is vital to its enzymatic function. Also, it enhances MMACHC thermal stability and protects the enzyme from deleterious electron-transfer uncoupling. This suggests that redox control could play a role in the regulation of MMACHC longevity in the cell (Jeong et al., 2011; Jeong and Kim, 2011; Park et al., 2012).
Early crystallographic studies on the structure of the MMACHC suggested that Arg230, Arg161, and Arg206 as putative amino acid residues in the GSH binding domain of MMACHC (Froese et al., 2012). Missense mutations at the Arg161 residue are the most common cause of MMACHC disease (Froese et al., 2009). The Arg161 residue acts as an anchor for GSH binding by the interaction between the guanidinium group of arginine and the carboxamide group of the cysteine residue in GSH (Ruetz et al., 2017). However, these studies lacked the analysis of the structure of GSH bound to MMACHC protein.
Recent crystallographic studies with the anti-vitamin B12 2,4-diflurophenylethylcobalamin has provided newer insights into the differences between the holo-form and the GSH-bound form of MMACHC. Binding of GSH to MMACHC increases the binding affinity toward the anti-vitamin and induces a complete shift to its base-off form (Ruetz et al., 2017). The crystal structure reveals a network of H-bond interactions between all three peptide moieties of GSH (Glu, Gly, and Cys) and hydrophobic stacking interactions with the Gly moiety in the GSH binding pocket of MMACHC. The study also identified additional residues Tyr215, Asp77, and Asp80 that form H-bonds with GSH.
Structural characterization of the MMACHC protein
High-resolution X-ray crystal structures of apo-MMACHC, holo-MMACHC, and the MMACHC-bound anti-vitamin B12 derivative are available (Froese et al., 2012; Koutmos et al., 2011; Ruetz et al., 2017). The first structure of MMACHC published by Koutmos et al. (2011) showed that MMACHC comprises an N-terminal flavodoxin nitro-reductase domain, which can use FMN or FAD to catalyze the reductive decyanation of CNCbl (Koutmos et al., 2011) and a large cavity suitable for cobalamin binding in its base-off configuration (Figure 9, panel a). Studies in solution demonstrated dimerization of the MMACHC protein in the presence of FMN and to a lesser extent Cbl (Froese et al., 2012). MMACHC does not harbor a canonical Cbl-binding site as described in MS or MUT. The base-off configuration of MMACHC⋅Cbl leaves the fifth-coordination position unoccupied (Koutmos et al., 2011), which also differs from the base-off/His-on mode observed in Cbl-dependent enzymes. A cblC patient carrying a deletion in amino acid residue Gln131 exhibited nearly zero MMACHC enzymatic activity (Backe et al., 2013). Analysis of the crystal structure shows that Gln131 hydrogen bonds with the Cbl moiety. Deletion of Gln131 is predictably disruptive toward adequate Cbl positioning to drive catalysis (Backe et al., 2013). Likewise, cobalamin binding influences MMACHC conformation. Binding of MeCbl to MMACHC modifies the conformation of the protein in three different loops around the B12 cavity (Figure 9, panel b). The structure of MMACHC bound to AdoCbl showed that the overall fold of MMACHC⋅AdoCbl is similar to that of MMACHC⋅MeCbl (Froese et al., 2012) (Figure 9, panels c and d). Results from the study also revealed a highly conserved dimerization cap for the β-axial 5′-adenosyl ligand and an GSH-binding pocket featuring a triad of Arg residues up above the β-axial ligand position(Froese et al., 2012). The study elucidated an important structure-function relationship in residues proposed to be critical for anchoring GSH in catalytically relevant positions, namely, Arg161, Arg206, and Arg230 (Figure 9, panel c, yellow sticks) are sites for point mutations that cause cblC disease.
Figure 9.

The structures of apo-MMACHC, MMACHC•MeCbl, and MMACHC•AdoCbl
(A) Structure of MMACHC⋅MeCbl (3SC0, teal/dark red (Koutmos et al., 2011) in its base-off configuration. A surface representation illustrating the vast solvent accessibility of the Cbl moiety is provided in the inset
(B) Overlay of the structure of MMACHC⋅MeCbl and apo-MMACHC (3SBZ, gray; Koutmos et al., 2011). MeCbl binding influences the conformation of three loop regions, namely 103–115, 196–203, and 231–238 (black for apo-MMACHC and pink for MMACHC⋅MeCbl)
(C) Structure of MMACHC⋅AdoCbl (3SOM, yellow/orange; Froese et al., 2012). Residues Arg161, Arg206, and Arg230 necessary for GSH binding are shown as pink sticks. The citrate molecule within the GSH binding site is also shown in pink
(D) Overlay of the structures of MMACHC⋅MeCbl and MMACHC⋅AdoCbl. The overall fold of MMACHC is very similar regardless of the distinct β-ligand of MeCbl and AdoCbl. The three flexible loops are sensitive to the absence or presence of the cobalamin moiety but not to the chemical structure of its β-axial ligand. Illustrations adapted from Hannibal and Jacobsen (2017)
The crystal structure showed that the predicted region for GSH binding held a citrate molecule from the solvent (Figure 9, panel c, green) (Froese et al., 2012). Functional studies showed poor solubility of variant Arg206Gln indicative of a structural role for this residue, and reduced GSH binding and dealkylase activity of mutants Arg161Gln and Arg230Gln (Froese et al., 2012). In-depth biophysical characterization of MMACHC Arg161G and Arg161Q showed that Arg161 is essential to maintain the enzyme productively coupled toward Cbl processing and preventing futile redox cycling (Gherasim et al., 2015).
Residues Arg161 and Arg206 also appear to play important roles in supporting the interaction of MMACHC with MS (Fofou-Caillierez et al., 2013). Molecular modeling and docking studies predict that a loop of MS interacts with residues Arg161 and Arg206 of MMACHC (Fofou-Caillierez et al., 2013). Because Arg161 and Arg206 are vastly accessible to the solvent (Figure 9, panel c), they could engage in protein–protein interactions. The structural analysis also revealed a partially disordered C-terminus in MMACHC (Koutmos et al., 2011).
Crystallization of human MMACHC bound to the catalytically inactive anti-vitamin cobalamin derivative permitted investigations on the active site of human MMACHC with bound GSH (Ruetz et al., 2017). Besides the amino acid residues identified earlier, the study revealed that residues Tyr215, Asp77, and Asp80 form H-bonds with GSH (Ruetz et al., 2017). In terms of solvent accessibility, 70% of the GSH molecule is enclosed within the protein structure and another 20% hindered by the cobalamin molecule. While the sulfur center of GSH is positioned near the bound Cbl, the calculated Co-S atom distance (6.3 Å) as well as the CH3-S atom distance of bound MeCbl modeled as a substrate (4.9 Å) do not align with the geometric constraints for the currently proposed SN2 mechanism of GSH-driven dealkylation of Co-C cobalamins (Ruetz et al., 2017). The authors also observed that bound GSH does not display additional bonding contacts that would induce SH group deprotonation, thereby increasing its nucleophilic character (Ruetz et al., 2017). From the above observations, one possibility is that different β-axial ligands of the cobalamin molecule itself could influence Co-β-ligand-S proximity as well as the overall electronic properties of the bound GSH.
An isoform of MMACHC truncated at its C-terminus is predominantly expressed in mice (Koutmos et al., 2011). Unlike the murine counterpart, analysis of MMACHC expression in human material, namely, HEK93 cells and MCH46 fibroblasts, showed that MMACHC has an apparent molecular weight of 32 kDa, very close to the predicted value of its full-length size (31.9 kDa, 282 amino acid residues) (Deme et al., 2012). The role of the C-terminus in human MMACHC remains unsolved but its dispensable nature in mice suggests species-specific differences in structure-function relationships.
Pathogenic mutations of the MMACHC gene
Mutation spectrum and genotype-phenotype correlations
The NCBI ClinVar database reports 286 genetic variants in the MMACHC gene, of which 91 are classified as pathogenic or likely pathogenic. The predominant mutations are single nucleotides (62%) followed by short duplications (19%) and deletions (18%) (Landrum et al., 2018). Yet, for a substantial number of mutations reported in studies and case reports their listing in NCBI ClinVar is still pending. An overview of the currently known disease-associated mutations is provided in Table 1.
Table 1.
Mutations in the MMACHC genea
| Mutation | Protein change | Mutation type | Phenotype/population | Frequencyb | References |
|---|---|---|---|---|---|
| c.1A>G | p.Met1Val | Missense Initiation codon change |
South Asian, Latino | 0.4–3.6% | (Lerner-Ellis et al., 2009; Liu et al., 2010; Wang et al., 2019) |
| c.1A>T | p.Met1? | Missense Initiation codon change |
0.1–0.2% | (Lerner-Ellis et al., 2009, 2006), not in ClinVar | |
| c.2T>G | p.Met1Arg | Missense Initiation codon change |
|||
| c.2T>C | p.Met1Thr | Missense Initiation codon change |
|||
| c.3G>A | p.Met1Ile | Missense Initiation codon change |
South Asian, European | 2.5% | (Lerner-Ellis et al., 2009, 2006) |
| c.14_24del | p.Val5fs | Deletion Frameshift |
0.1% | (Lerner-Ellis et al., 2009), not in ClinVar | |
| c.48_49del | p.Cys17fs | Deletion Frameshift |
|||
| c.72C>A | p.Tyr24Ter | Nonsense Premature stop |
|||
| c.80A>G | p.Gln27Arg | Missense Exon-intron boundary Splice variant? |
East Asian, founder effect | 0.3–9.1% | (Lerner-Ellis et al., 2009, 2006; Liu et al., 2010; Wang et al., 2019) |
| c.81G>A | p.Gln27 = | Synonymous Exon-intron boundary Splice variant? |
(Guéant et al., 2018) | ||
| c.81 + 1G>A | ? | Missense Splice donor |
0.1–1.8% | (Lerner-Ellis et al., 2009, 2006; Wang et al., 2019) | |
| c.81 + 2T>G | ? | Missense Splice donor |
(Richard et al., 2009) | ||
| c.82-1G>A | ? | Missense Splice acceptor |
0.3% | (Lerner-Ellis et al., 2009) | |
| c.82-9_12del | ? | Deletion Splice variant? |
0.3–0.5% | (Lerner-Ellis et al., 2009, 2006), not in ClinVar | |
| c.89G>A | p.Trp30Ter | Nonsense Premature stop |
|||
| c.90G>A | p.Trp30Ter | Nonsense Premature stop |
(Richard et al., 2009) | ||
| c.99del | p.Glu33fs | Deletion Frameshift, premature stop |
0.6% | (Liu et al., 2010), not in ClinVar | |
| c.122dup | p.Pro42fs | Duplication Frameshift |
0.3% | (Lerner-Ellis et al., 2009), not in ClinVar | |
| c.122del | p.Leu41fs | Deletion Frameshift |
|||
| c.123dup | p.Pro42fs | Duplication Frameshift |
|||
| c.126_141del | p.Leu43fs | Deletion Frameshift |
0.1% | (Lerner-Ellis et al., 2009), not in ClinVar | |
| c.145G>C | p.Ala49Pro | Missense | 0.1% | (Lerner-Ellis et al., 2009) | |
| c.146_154del | p.Ala49_Val52delinsVal | Indel | 0.6% | (Liu et al., 2010), not in ClinVar | |
| c.158T>C | p.Leu53Pro | Missense | |||
| c.178dup | p.Asp60fs | Duplication Frameshift |
|||
| c.182G>C | p.Arg61Pro | Missense | |||
| c.202C>T | p.Gln68Ter | Nonsense Premature stop |
|||
| c.217C>T | p.Arg73Ter | Nonsense Premature stop CpG hotspot |
Early onset? | 0.6–2.2% | (Lerner-Ellis et al., 2009, 2006; Liu et al., 2010) |
| c.248dup | p.Ala84fs | Duplication Frameshift, premature stop |
0.6% | (Liu et al., 2010), not in ClinVar | |
| c.270dup | p.Arg91Ter | Nonsense Premature stop |
|||
| c.271dup | p.Arg91fs | Duplication Frameshift, premature stop |
Early onset; Different ethnic groups, primarily European |
0,6–48% | (Lerner-Ellis et al., 2009, 2006; Liu et al., 2010; Wang et al., 2019) |
| c.271_272AG[2] | p.Glu92fs | Insertion Frameshift |
|||
| c.276G>A | p.Glu92= | Silent Splice variant |
Reno-pulmonary phenotype | (Kömhoff et al., 2013) | |
| c.276G>T | p.Glu92Asp | Missense Splice variant |
Reno-pulmonary phenotype; Dutch/German, founder effect |
(Kömhoff et al., 2013) | |
| c.277-3_303del | p.Ser93_Gln143del | Deletion Splice variant? |
0.6% | (Liu et al., 2010), not in ClinVar | |
| c.285dup | p.Glu96fs | Duplication Frameshift |
|||
| c.292C>T | p.Gln98Ter | Nonsense Premature stop |
|||
| c.310_313del | p.Asp104fs | Deletion Frameshift |
|||
| c.315C>G | p.Tyr105Ter | Nonsense Premature stop |
1.3–3.6% | (Liu et al., 2010; Wang et al., 2019) | |
| c.328_331del | p.Asn110fs | Deletion Frameshift, premature stop |
Hispanic/Latino | 1.9–2.0% | (Lerner-Ellis et al., 2009, 2006) |
| c.331C>T | p.Arg111Ter | Nonsense Premature stop CpG hotspot |
Early onset French Canadian, Acadian, Cajun, European, Palestinian |
7,4–8.8% | (Lerner-Ellis et al., 2009, 2006) |
| c.347T>C | p.Leu116Pro | Missense | 0.5–0.6% | (Lerner-Ellis et al., 2009, 2006; Liu et al., 2010) | |
| c.352del | p.Gln118fs | Deletion Frameshift, premature stop |
0.5–1% | (Lerner-Ellis et al., 2009, 2006) | |
| c.364dup | p.His122fs | Duplication Frameshift |
1.8% | (Wang et al., 2019) | |
| c.365A>G | p.His122Arg | Missense | 0.2–0.3% | (Lerner-Ellis et al., 2009, 2006), not in ClinVar | |
| c.365A>T | p.His112Leu | Missense | 1.3% | (Liu et al., 2010), not in ClinVar | |
| c.382_384TAC[2] | p.Tyr130del | Deletion In-frame |
|||
| c.384del | p.Tyr129fs | Deletion Frameshift |
|||
| c.388T>C | p.Tyr130His | Missense | 0.4–0.5% | (Lerner-Ellis et al., 2009, 2006) | |
| c.388_390del | p.Tyr130del | Deletion In-frame |
0.8–1.5% | (Lerner-Ellis et al., 2009, 2006), not in ClinVar | |
| c.389A>G | p.Tyr130Cys | Missense | 0.3% | (Lerner-Ellis et al., 2009) | |
| c.391C>T | p.Gln131Ter | Nonsense Premature stop |
0.1–0.2% | (Lerner-Ellis et al., 2009, 2006) | |
| c.392_394del | p.Gln131del | Deletion In-frame |
One late-onset case | (Backe et al., 2013) | |
| c.394C>T | p.Arg132Ter | Nonsense Premature stop CpG hotspot |
Late onset, good response to treatment; Indian, Pakistani, Middle Eastern, European |
5,5–13,7% | (Lerner-Ellis et al., 2009, 2006; Liu et al., 2010; Wang et al., 2019) |
| c.398_399del | p.Gln133fs | Deletion Frameshift, premature stop |
0.1–0.6% | (Lerner-Ellis et al., 2009, 2006; Liu et al., 2010) | |
| c.415_416delinsTA | p.Pro139Ter | Nonsense Premature stop |
|||
| c.420G>A | p.Trp140Ter | Nonsense Premature stop |
0.3–0.5% | (Lerner-Ellis et al., 2009, 2006) | |
| c.427C>T | p.Gln143Ter | Nonsense Premature stop |
(He et al., 2020) | ||
| c.429 + 1G>C | ? | Missense Splice variant |
|||
| c.435_436del | p.Ser146fs | Deletion Frameshift, premature stop |
0.3–0.5% | (Lerner-Ellis et al., 2009, 2006), not in ClinVar | |
| c.440G>A | p.Gly147Asp | Missense | Early onset | 1.4–2.2% | (Lerner-Ellis et al., 2009, 2006) |
| c.440G>C | p.Gly147Ala | Missense | Native American | 0.5–0.7% | (Lerner-Ellis et al., 2009, 2006) |
| c.441_442TG[2] | p.Cys149fs | Insertion Frameshift |
|||
| c.445_446insA | p.Cys149Ter | Insertion Premature stop |
0.6–3.6% | (Liu et al., 2010; Wang et al., 2019), not in ClinVar | |
| c.448_449delinsCC | p. Ile150Pro | Indel | (Guan et al., 2020), not in ClinVar | ||
| c.449T>A | p.Ile150Lys | Missense | |||
| c.450_479dup | p.Ile150_Ala159dup/p.His151_Ile160dup | Duplication In-frame |
0.1–0.2% | (Lerner-Ellis et al., 2009, 2006), not in ClinVar | |
| c.452A>G | p.His151Arg | Missense | Late onset, mild phenotype | 0.2% | (Liu et al., 2010), not in ClinVar |
| c.457C>T | p.Arg153Ter | Nonsense Premature stop CpG hotspot |
0.6–1.5% | (Lerner-Ellis et al., 2009, 2006; Liu et al., 2010) | |
| c.464G>A | p.Gly155Glu | Missense | |||
| c.467G>A | p.Gly156Asp | Missense | 0.1–0.2% | (Lerner-Ellis et al., 2009, 2006) | |
| c.468_469del | p.Trp157fs | Deletion Frameshift, premature stop |
0.1–0.5% | (Lerner-Ellis et al., 2006), not in ClinVar | |
| c.471G>A | p.Trp157Ter | Nonsense Premature stop |
0.1% | (Lerner-Ellis et al., 2009) | |
| c.471G>C | p.Trp157Cys | Missense | 0.1–0.2% | (Lerner-Ellis et al., 2009, 2006) | |
| c.481C>G | p.Arg161Gly | Missense | Early onset | 0.1–0.2% | (Lerner-Ellis et al., 2009, 2006) |
| c.481C>T | p.Arg161Ter | Nonsense Premature stop CpG hotspot |
0.6–1.6% | (Lerner-Ellis et al., 2009, 2006; Liu et al., 2010) | |
| c.482G>A | p.Arg161Gln | Missense CpG hotspot |
Late onset, mild phenotype; Multi-ethnic, East Asian, founder effect |
1,8%7.3% | (Lerner-Ellis et al., 2009, 2006; Liu et al., 2010; Wang et al., 2019) |
| c.484G>T | p.Gly162Trp | Missense | Pulmonary hypertension | (Gündüz et al., 2014), not in ClinVar | |
| c.487_489del | p.Val164del | Deletion In-frame |
0.3% | (Lerner-Ellis et al., 2009), not in ClinVar | |
| c.497dup | p.Pro167fs | Duplication Frameshift |
|||
| c.500del | p.Pro167fs | Deletion Frameshift, premature stop |
0.1–0.2% | (Lerner-Ellis et al., 2009, 2006) | |
| c.507_519del | p.Glu170fs | Deletion Frameshift, premature stop |
0.1% | (Lerner-Ellis et al., 2009) | |
| c. 536_537insAT | p.His180fs | Insertion Frameshift, premature stop |
East Asian | 0.1% | (Lerner-Ellis et al., 2009), not in ClinVar |
| c.541_548del | p.Asp181fs | Deletion Frameshift |
|||
| c.544T>C | p.Cys182Arg | Missense | 0.1% | (Lerner-Ellis et al., 2009) | |
| c.545_546GT[1] | p.Val183fs | Insertion Frameshift |
|||
| c.547_548del | p.Val183fs | Deletion Frameshift, premature stop |
Early onset? Middle Eastern |
1.4–2.0% | (Lerner-Ellis et al., 2009, 2006), not in ClinVar |
| c.551_554dup | p.Arg186fs | Frameshift, premature stop | (Bourque et al., 2021) | ||
| c.565del | p.Arg189fs | Deletion Frameshift, premature stop |
0.2–0.4% | (Lerner-Ellis et al., 2009, 2006) | |
| c.565C>A | p.Arg189Ser | Missense | 0.2–0.4% | (Lerner-Ellis et al., 2009, 2006) | |
| c.565C>T | p.Arg189Cys | Missense | 1.8% | (Wang et al., 2019) | |
| c.567dup | p.Ile190fs | Duplication Frameshift, premature stop |
0,1–3.8% | (Lerner-Ellis et al., 2009; Liu et al., 2010) | |
| c.568insT | Insertion Frameshift |
(Weisfeld-Adams et al., 2010), not in ClinVar | |||
| c.574del | p.Leu192fs | Deletion Frameshift |
|||
| c.578T>C | p.Leu193Pro | Missense | 0.4–0.7% | (Lerner-Ellis et al., 2009, 2006) | |
| c.599G>A | p.Trp200Ter | Nonsense | |||
| c.600G>A | p.Trp200Ter | Nonsense Premature stop |
0.2–0.3% | (Lerner-Ellis et al., 2009, 2006), not in ClinVar | |
| c.603_604del | p.Asp202fs | Deletion Frameshift |
|||
| c.608G>A | p.Trp203Ter | Nonsense Premature stop |
Hispanic | 1,0–1.5% | (Lerner-Ellis et al., 2009, 2006) |
| c.609G>A | p.Trp203Ter | Nonsense Premature stop |
East Asian, founder effect | 1,8–48,1% | (Lerner-Ellis et al., 2009, 2006; Liu et al., 2010; Wang et al., 2019) |
| c.615C>A | p.Tyr205Ter | Nonsense Premature stop |
0.6% | (Liu et al., 2010) | |
| c.615C>G | p.Tyr205Ter | Nonsense Premature stop |
1.6–2.7% | (Lerner-Ellis et al., 2009, 2006) | |
| c.616C>T | p.Arg206Trp | Missense CpG hotspot |
0.5–0.6% | (Lerner-Ellis et al., 2009, 2006; Liu et al., 2010) | |
| c.616del | p.Arg206fs | Deletion Frameshift |
|||
| c.617G>A | p.Arg206Gln | Missense | |||
| c.617G>C | p.Arg206Pro | Missense | 0.3–0.5% | (Lerner-Ellis et al., 2009, 2006) | |
| c.619dup | p.Asp207fs | Duplication Frameshift |
|||
| c.624_625TG[1] | p.Val209fs | Duplication Frameshift |
|||
| c.626dup | p.Thr210fs | Duplication Frameshift, premature stop |
0.6–3.6% | (Liu et al., 2010; Wang et al., 2019) | |
| c.626_627del | Deletion Frameshift, premature stop |
1.8% | (Wang et al., 2019), not in ClinVar | ||
| c.649_650del | p.Glu217fs | Deletion Frameshift |
|||
| c.658_659del | p.Lys200del | Deletion Frameshift, premature stop |
0.2% | (Lerner-Ellis et al., 2006) | |
| c.658_660del | p.Lys220del | Deletion In-frame |
East Asian, founder effect | 0.3–13.9% | (Lerner-Ellis et al., 2009; Liu et al., 2010; Wang et al., 2019) |
| c.666C>A | p.Tyr222Ter | Nonsense Premature stop |
0.7–1.8% | (Lerner-Ellis et al., 2009, 2006) | |
| c.688C>T | p.Arg230Ter | Nonsense Premature stop |
|||
| c.792_818del | p.Ser264_Pro272del | Deletion In-frame |
0.1% | (Lerner-Ellis et al., 2009) |
Mutations in the MMACHC gene listed in ClinVar as pathogenic or likely pathogenic (May 12, 2021) or reported in the following publications: Lerner-Ellis et al. (2006, 2009), Liu et al. (2010), Wang et al. (2019), Guéant et al. (2018), Richard et al. (2009), Kömhoff et al. (2013), Hoff Backe et al. (2013), Guan et al. (2020), Gündüz et al. (2014), and Weisfeld-Adams et al. (2010).
Frequency among pathogenic alleles found in cohorts studied by Lerner-Ellis et al. (2006, 2009), Liu et al. (2010), Guéant et al. (2018,) and Wang et al. (2019).
In 2006, Lerner-Ellis et al. identified 42 different mutations among 204 cblC individuals (Lerner-Ellis et al., 2006). In 2009, a study with 118 cblC patients revealed 34 different mutations among which 7 were new variants (Lerner-Ellis et al., 2009). Since then, research groups worldwide have identified additional mutations and contributed to investigating genotype-phenotype associations and distinct population frequencies.
The genotypes and disease frequency differ significantly between populations (Lerner-Ellis et al., 2006). French Canadian, Cajun, Indian, Chinese, Middle Eastern, and European populations appear to be affected with higher frequency, still for many regions, representative data are limited. The most common mutation is c.271dupA (p.Arg91LysfsTer14). It accounts for more than 40% of pathogenic alleles in Caucasian patients and is prevalent throughout different ethnic groups. The duplication of a single nucleotide in exon 2 predicts a change in the amino acid sequence from arginine to lysine at position 91 followed by a premature termination codon 14 sites downstream owing to a frameshift and thus to a truncated protein lacking the cobalamin binding site. In homozygous states and in several compound heterozygous combinations, c.271dupA is associated with early onset disease. c.394C>T (p.Arg132Ter) is the second most prevalent mutation in Caucasian patients and results in a premature stop codon at position 132 (Lerner-Ellis et al., 2009). Still the partially mild late-onset phenotype and observation of relatively increased mRNA levels may indicate rescue from nonsense-mediated decay and thus a residual function of the protein. Likewise, c.331C>T (p.Arg111Ter) is a nonsense mutation within exon 3 and located on a CpG hotspot. Hypermutability owing to CpG site methylation explain their relatively high frequency as well as their recurrence in different populations. In contrast to nearby mutation c.394C>T, variant c.331C>T is associated with early onset.
The mutation spectrum in East Asian cohorts differs fundamentally from Caucasians (Wang et al., 2019). Liu et al. discovered c.609G>A (p.Trp203Ter) as the most common pathogenic mutation in cblC patients from China. c.609G>A accounts for 47% of pathogenic alleles in Northern and up to 75% in southern China and is associated with early onset disease (Liu et al., 2010). Haplotype analysis suggests a non-recent founder effect for c.609G>A as well as for c.658_660del and c.80A>G. The in-frame mutation c.658_660del (p.Lys220del) accounts for >13%, the two missense mutations c.482G>A (p.Arg161Gln) and c.80A>G (p.Gln27Arg) account for >5% of pathogenic alleles in Chinese patients (Wang et al., 2019). c.452A>G has been associated with late onset and a relatively mild phenotype. Also, c.482G>A (p.Arg161Gln) is mainly but not exclusively found in late-onset patients.
Correlation to the early or late onset and disease severity could be established for some of the most common mutations. Meanwhile, associations to clinical features are more complex and small numbers of highly heterogeneous patients make it difficult to draw conclusions. Among the remarkable features of cblC disease, severe reno-pulmonary phenotypes were reported (Kömhoff et al., 2013). c.276G>A (p.Glu92 = ) and c.276G>T (p.Glu92Asp) affect the last nucleotide of exon 2 on the exon-intron boundary and likely result in aberrant splicing. They were solely discovered in Dutch and German patients with combined renal thrombotic microangiopathy and pulmonary hypertension suggesting a founder effect on one hand and a specific vascular pathogenicity on the other hand, which remains to be elucidated in further detail. Besides, in Chinese patients, the mutation c.80A>G was associated with diffuse lung disease (Liu et al., 2020). Also, several case reports revealed a high prevalence of renal impairment in patients compound heterozygous for c.271dupA (early termination) and c.389A>G (p.Tyr130Cys; missense mutation within the cobalamin binding site) (Cornec-Le Gall et al., 2014; Higashimoto et al., 2020; Lemoine et al., 2018; Lerner-Ellis et al., 2009). Patients differed in the age of onset from infancy to presentation during adult life. Even among three siblings with the same mutations, clinical manifestations were highly heterogeneous (Higashimoto et al., 2020). One presented with severe neurological deterioration, one with a moderate renal phenotype and the third was clinically asymptomatic but presented with biochemical alterations (Higashimoto et al., 2020). Good responsiveness to treatment with HOCbl appeared to be a common feature of this genotype (Cornec-Le Gall et al., 2014; Higashimoto et al., 2020; Lemoine et al., 2018). Altogether, clinical heterogeneity among cblC patients is not fully explained by specific mutations. Different haplotypes, polymorphisms, trans-acting factors and environmental factors such as diet that influences epigenetics may contribute to phenotype complexity in ways that are yet to be identified (Lerner-Ellis et al., 2009).
Pathogenic mutations in other genes: cblC-like disorders
During the past few years, pathogenic mutations in genes functionally unrelated to MMACHC such as PRDX1 (peroxiredoxin 1, an enzyme), HCFC1 (host cell factor 1, a transcriptional regulator), THAP11 (THAP Domain-Containing Protein 11, a transcriptional regulator) and ZNF143 (Zinc finger protein 143, a transcriptional regulator) were found to cause combined methylmalonic acidemia and homocystinemia mimicking classic cblC disease (Guéant et al., 2018; Pupavac et al., 2016; Quintana et al., 2017; Yu et al., 2013). MMACHC expression depends on versatile transcription factors such as THAP11, which forms a complex with the co-regulator HCFC1. Yu et al. discovered mutations in the HCFC1 gene (OMIM ∗300019, Xq28) in 14 of 17 male patients who were initially diagnosed with cblC deficiency without presenting MMACHC mutations. This specific set of mutations in HCFC1 was designated as “cblX” disease (Yu et al., 2013). One of the remaining patients was later diagnosed with a homozygous mutation in the THAP11 gene (OMIM ∗609119, 16q22.1) and designated as “cblX-like.” Mutations within the highly conserved kelch domain of HCFC1 mediating protein–protein interactions or within the DNA-binding THAP-domain of THAP11 are suggested to cause cblX and cblX-like phenotypes owing to the downregulation of MMACHC expression (Quintana et al., 2017; Yu et al., 2013). The structural domain affected by HCFC1 mutations is relevant in determining whether or not the patient may present with disrupted cobalamin metabolism. A study with five patients identified exon 10 skipping mutations in the basic domain of HCFC1 (Wongkittichote et al., 2021). The basic domain of HCFC1 (amino acids 478–875) is located downstream of the Kelch domain and is important for proteolytic maturation of HCFC1. The mutation was predicted to yield a truncated protein with in-frame deletion of amino acids 536–601 (Wongkittichote et al., 2021). These patients exhibited characteristic features of X-linked intellectual developmental disorder, without metabolic disturbances of cobalamin metabolism (Wongkittichote et al., 2021).
Studies performed on a mouse model of HCFC1 and THAP11 deficiency (Hcfc1 A115V/Y and Thap11 F80L/F80L) revealed a dual phenotype that recapitulated characteristics of cblC disease and ribosomopathies (Chern et al., 2022). ZNF143 is another wide-ranging transcriptional activator known to interact with HCFC1 and THAP11. To date, one patient compound heterozygous for two variants in ZNF143 (OMIM ∗603433, 11p15.4) was reported with a slightly different cellular phenotype to cblC, yet with typical clinical manifestations (Pupavac et al., 2016). In 2018, Guéant et al. described the first epimutation resulting in cblC disease in three patients. A primary mutation located in the PRDX1 gene (OMIM ∗176763, 1p34.1) causes antisense transcription of MMACHC resulting in secondary hypermethylation of the promotor region and subsequent silencing of MMACHC (Guéant et al., 2018). Since then, a number of works identified epi-mutations in MMACHC (Cavicchi et al., 2021; Guéant et al., 2022; Oussalah et al., 2022; Zhang et al., 2021), including in compound heterozygous form, suggesting that their occurrence is probably more frequent than previously estimated. An important consideration is that transcriptional regulators modify the expression of multiple genes, not just MMACHC, and therefore, the clinical phenotype of patients with cblC-like disorders likely represents the compound effects of abnormal expression of multiple genes, as recently demonstrated in a rodent model of cblC-like disease (Chern et al., 2022).
Biochemical findings in pathogenic MMACHC variants: Enzymatic activity, thermal stability, redox cycling, and protein–protein interactions
Enzymatic activity and loss of function
As described in earlier sections of this review, the MMACHC protein is strikingly versatile regarding its substrates and catalyzed reactions. MMACHC binds to different naturally occurring cobalamins such as AdoCbl, MeCbl, HOCbl, and CNCbl as well as synthetic straight-chain alkylcobalamins (Hannibal et al., 2009). However, certain synthetic variants of cobalamin with highly stable Co-β-ligand bonds are not substrates for catalysis by MMACHC and have been denominated anti-vitamin B12 (Ruetz et al., 2017). MMACHC catalyzes the removal of the upper axial ligand by reductive decyanation or nucleophilic dealkylation. Reductive decyanation requires NADPH and FMN/FAD as cofactors and produces cob(II)alamin and cyanide. Nucleophilic dealkylation depends on GSH, which binds near the upper-axial ligand of cobalamin within an arginine-rich pocket of MMACHC. Products are the respective GSH thioether and cob(I)alamin, which rapidly oxidizes to cob(II)alamin and serves as intermediate for the further synthesis of the active coenzymes AdoCbl and MeCbl (Kim et al., 2009, 2008). Additionally, GSH-dependent decyanation and reduction of H2OCbl occur under oxygen-depleted conditions (Li et al., 2014a).
Aside from its catalytic function, MMACHC is described as a “trafficking chaperone” in reference to its role in intracellular cofactor trafficking and the stabilization of the cobalamin molecule in its base-off configuration (Kim et al., 2008). The DMBI of the corrin ring is coordinated to the central cobalt in the base-on conformation or un-coordinated in the base-off coordination. The base-off configuration of cobalamins does not occur under neutral or near-neutral physiological conditions of pH. Yet, the two Cbl-dependent enzymes MS and MUT utilize base-off, reduced cobalamin as the preferred cofactor state to enter their respective catalytic cycles. The known reactivity of cobalamin in its three oxidation states further suggests the need for protective targeted transport before delivery to MS and MUT. MMACHC stabilizes the base-off conformation of different cobalamin derivatives. It is predicted to interact with the lysosomal transporters LMBRD1 and ABCD4 to receive the cobalamin cargo at its passage from lysosome to cytosol (Banerjee, 2006; Banerjee et al., 2009; Coelho et al., 2012; Hannibal et al., 2013; Kim et al., 2008). Downstream, the interaction of MMACHC with MMADHC (the cobalamin complementation group D, cblD, gene ID MMADHC) facilitates the allocation of cobalamin to cytosolic MS or mitochondrial MUT (Froese et al., 2015; Gherasim et al., 2013; Plesa et al., 2011). Thus, current experimental evidence suggests that MMACHC plays a central role in chaperoning the cobalamin molecule from its exit from the lysosome to its delivery to recipient enzymes MS and MUT.
Pathogenic mutations affect the expression and function of the MMACHC protein in different ways. Depending on the location and type of mutation, impairments include defective binding to cobalamin and GSH, reduced thermal stability, increased redox cycling (thiol oxidase activity), and impaired protein–protein interactions with adaptor protein MMADHC.
Defective substrate binding
Several missense mutations are located within the cobalamin binding site of MMACHC and are predicted to compromise the binding and metabolism of cobalamin (Gherasim et al., 2015). Earlier studies demonstrated that fibroblasts from cblC patients lack the capacity to dealkylate any alkylcobalamins (Hannibal et al., 2009). In vitro studies with human recombinant MMACHC showed that mutant p.Gly147Asp (c.440G>A), which causes a severe early-onset phenotype non-responsive to supplementation therapy, does not bind CNCbl or HOCbl (Froese et al., 2009). In the same study, p.Arg161Gln was shown to impair CNCbl binding and decyanation, but not HOCbl binding. Subsequent studies revealed that the loss of arginine within the GSH binding pocket decreases GSH-binding by 25 to 40-fold and impairs dealkylation, but not decyanation (Froese et al., 2012; Gherasim et al., 2015). Rates of MeCbl dealkylation diminished by 6- and 10-fold in the late-onset mutation p.Arg161Gln (c.482G>A) and the early-onset mutation p.Arg161Gly (c.481C>G), respectively.
Thermal lability
Certain mutations such as those occurring at residue Arg161 render the MMACHC protein less stable to temperature (Froese et al., 2010). Reduced thermal stability is partially counteracted by cobalamin binding (Froese et al., 2010). In vitro, wild-type MMACHC denatured at a surprisingly low, nearly physiological temperature (Thermal melting point, Tm = 39°C), but stabilized when binding to cobalamins, especially with AdoCbl and MeCbl. In comparison, p.Arg161Gln mutant MMACHC exhibited a slight, but significant decrease in its melting temperature with half of the protein unfolding at 37°C. Similar to wild-type MMACHC, the mutant protein underwent stabilization upon binding to AdoCbl, MeCbl, and HOCbl (and less so by CNCbl). Yet, Arg161 variants required several-fold higher concentrations of cobalamins to counter thermal destabilization. Gherasim et al. extended these observations and revealed even lower denaturation temperature in the early-onset mutation p.Arg161Gly (Tm = 38.1°C) than in late-onset mutation p.Arg161Gln (Tm = 43.4°C) and wild-type protein (Tm = 46.6°C) (Gherasim et al., 2015). Thus, mutant MMACHC proteins that may exhibit normal expression, may have however a significantly reduced half-life in cells, thereby giving rise to a state of MMACHC deficiency. These findings together with the fact that the concentration of free cobalamin in cells is predicted to be negligible may explain the responsiveness of such cblC patients (c.482G>A, p.Arg161Gln) to high-dose HOCbl treatment. Yet, a benefit of treatment with AdoCbl and MeCbl based on more effective stabilization compared with HOCbl did not bear out in the clinical context (Froese et al., 2010; Gherasim et al., 2015). Park et al. described stabilization of bovine MMACHC not only by cobalamin, but also by GSH (Park and Kim, 2012). This observation may provide a putative link between the mutational changes of Arg161 and thermal instability by impaired GSH binding.
Oxidative stress and increased futile redox cycling
Oxidative stress is considered an important element in the pathophysiology of several metabolic diseases (Jacobsen and Hannibal, 2019). Evidence of oxidative stress as part of the pathomechanism of cblC exists from in vitro studies with purified recombinant proteins, cells, and by examination of urinary markers of oxidative damage.
In vitro studies with recombinant MMACHC from Caenorhabditis elegans (ceMMACHC) by Li et al. revealed a catalytically competent decyanase and dealkylase enzyme with the surprising finding of a robust thiol oxidase activity (Li et al., 2017, 2014b). In human MMACHC, the seemingly latent thiol oxidase activity is suppressed in wild-type protein but uncovered by some mutations affecting the GSH binding site (e.g. p.Arg161Gln and p.Arg161Gly). Thiol oxidation is coupled to dealkylation or decyanation of cobalamins and leads to futile redox cycling with consumption of GSH and O2, to form GSSG, and stabilization of reduced cobalamin. The striking stabilization of highly reactive cob(I)alamin and cob(II)alamin in human MMACHC has been seen under strict anaerobic conditions. In the presence of oxygen, catalysis by human wild-type MMACHC produces H2Ocbl as the predominant species (Kim et al., 2009).
Mechanistic studies performed with ceMMACHC showed that cob(I)alamin or cob(II)alamin initiate the futile redox cycle at the expense of reduced GSH and oxygen (Gherasim et al., 2015; Li et al., 2017). GSCbl, a proposed intermediate, might react with a second GSH molecule to form GSSG and set free cob(I)alamin to reenter the redox cycle. Human wild-type MMACHC is suggested to favor cob(III)alamin α-ligated to H2O, and thus, withdraw it from reentering the redox cycle. Conversely, ceMMACHC exhibits a more compact active site than human MMACHC and might favor GSH to locate closer to the β-face of Cbl. In pathogenic mutations p.Arg161Gln and p.Arg161Gly, the exchange of the positively charged residues in the arginine-rich binding pocket by the smaller glutamine or glycine residue may destabilize O2.-, hinder further oxidation and thereby stabilize cob(II)alamin (Gherasim et al., 2015; Li et al., 2017). Simultaneously, it may enhance binding of a second GSH molecule adjacent to the proposed GSCbl intermediate, resulting in GSSG formation. The confirmation of the reaction mechanism requires further investigation. For example, Li et al. recently discovered chlorocobalamin (ClCbl) as a new intermediate of the thiol oxidase activity of ceMMACHC (Li et al., 2020a). In the presence of KCl and GSH, an unexpected ceMMACHC-bound cob(II)alamin form was captured and then identified as Cl-cob(II)alamin by spectroscopic and computational analyses. ClCbl might facilitate GSH-dependent ligand switching for the formation of GSCbl. GSSG formation increased drastically in a biphasic manner dependent on the concentration of KCl as well as KBr, but not so with KI and KF. Yet, KCl concentration in commonly used buffers is significantly higher than in the cellular environment. Whether this reactivity also occurs with wild-type human MMACHC and especially in pathogenic mutants known to engage in thiol oxidase reactions remains to be elucidated.
At the cellular level, cblC mutant fibroblasts suffer from increased apoptosis likely owing to the overproduction of reactive oxygen species (ROS) (Richard et al., 2009). Pastore et al. demonstrated an imbalance of reduced and oxidized GSH represented by the GSH/GSSG ratio in cblC lymphocytes (Pastore et al., 2014). In contrast to its elevated precursor molecule Hcy, GSH was shown to be decreased by 82%, likely by increased consumption. GSH is one of the principal cellular non-protein antioxidants and cofactor to several proteins involved in detoxification. Thus, its deficiency may lead to the incapacity of direct and indirect detoxification and oxidative damage. Within the scope of a global analysis of cblC cell proteome, Hannibal et al. revealed a deficient detoxification of ROS and xenobiotics by GSH-dependent proteins (Hannibal et al., 2011). Among various downregulated proteins were three isoforms of glutathione-S-transferases (GST) that catalyze GSH-dependent conversion of xenobiotics and protects against oxidative stress. Downregulation of GST could mainly be restored by HOCbl supplementation. In contrast, peroxiredoxins PRDX1, 2, and 6 were downregulated in cobalamin supplemented fibroblasts losing their protective function against oxidative stress by detoxification of peroxides. Consequently, dysregulation in the proteome may also contribute to oxidative damage and aggravation of cblC manifestations (Hannibal et al., 2011).
In comparison with other metabolic disorders, cblC patients exhibit especially high readouts of biomarkers of oxidative damage in urine (McGuire et al., 2009). In a study by McGuire et al., F2-isoprostanes, di-tyrosine, and antioxidant activity representing the overall redox state by lipid peroxidation, protein oxidation, and antioxidant capacity, respectively, were measured in urine samples of various inborn errors of metabolism and controls. Urinary isoprostanes were proposed as readily available and stable non-invasive biomarkers of oxidative stress in vivo (Kadiiska et al., 2005). All three parameters were significantly altered in the patient cohort and consistently showed the highest alterations in cblC patients (McGuire et al., 2009). In a longitudinal dataset, oxidative stress markers showed good concordance with biochemical control and acute decompensation in patients with maple syrup urine disease. Monitoring oxidative stress markers in urine may be a promising tool for disease monitoring in inborn errors of metabolism.
Protein–protein interactions with MMADHC and formation of a multiprotein complex with MS
MMACHC interacts with other proteins to ensure the protected and targeted transport of the scarce and reactive cobalamin within the cell. It appears to accept newly internalized cobalamins as they exit the lysosome via the transporter LMBRD1 and the ATP-binding cassette (ABC) transporter ABCD4 (Banerjee, 2006; Banerjee et al., 2009; Coelho et al., 2012; Hannibal et al., 2013; Kim et al., 2008). After processing of the cobalamins by removal of the upper ligand, the interaction of MMACHC with MMADHC allows delivery of the coenzymes to their target enzymes MS in cytosol and MUT in mitochondria. MMADHC is the main interaction partner of MMACHC. Mutations in the MMADHC gene lead to the rare, but strikingly heterogeneous cblD disease presenting with either isolated methylmalonic aciduria (cblD-MMA) or homocystinuria (cblD-HC), or a combination of both (cblD-MMA/HC) (Coelho et al., 2008; Froese et al., 2015). Genotype-phenotype correlations revealed that the N-terminal domain of MMADHC is required for the mitochondrial route, whereas domains within the C-terminus contribute to the cytosolic route. Consistently, a putative mitochondrial targeting sequence was identified within the N-terminal domain (Coelho et al., 2008). Then, adenosyltransferase (cblB, MMAB), methylmalonyl-CoA mutase (MUT) and presumably its chaperone (CblA) may function as a transfer complex. Yet, the details of cobalamin delivery to and into the mitochondria remain unknown. The MMADHC protein possesses motifs with homology to the bacterial and the MMACHC nitroreductase fold and to bacterial ABC-transporter, yet it does not directly process cobalamins and lacks ATPase activity or any other known enzymatic activity (Gherasim et al., 2013). Likewise, the presence of MMADHC does not modify β-deligation of cobalamins by MMACHC. According to Gherasim et al. MMADHC serves an adapter role that orchestrates cobalamin-loaded MMACHC toward delivery of the micronutrient to the cytosolic or mitochondrial routes that ultimately produce the cofactors MeCbl and AdoCbl, respectively (Gherasim et al., 2013). Experiments with cultured cblC fibroblasts of the isolated as well as the combined forms of the MMADHC defect showed a clear impairment of cobalamin partitioning in the cell leading to the imbalanced partition of MeCbl and AdoCbl with respect to the pattern of cobalamin forms observed in cells from healthy controls (Gherasim et al., 2013). The defect in one pathway, either the mitochondrial for N-terminal mutations or the cytosolic for C-terminal mutations, led to overproduction of the cofactor of the alternative pathway. In cells from healthy controls, MeCbl synthesis and MS function seem to be prioritized reflecting in 42% of total cellular cobalamin presenting as MeCbl. In cells from MMADHC patients with isolated homocystinuria up to 76% of the total intracellular cobalamin pool presented as AdoCbl. Concurrently, studies with isotopically labeled [57Co]-MeCbl confirmed that MMACHC-dependent dealkylation is not affected by MMADHC mutations, but instead, major effects are observed downstream in cofactor partition (Gherasim et al., 2013).
Plesa et al. demonstrated the complexation of MMACHC and MMADHC in vitro and vivo by surface plasmon resonance and a bacterial two-hybrid system, respectively (Plesa et al., 2011). The interface involved the two modified nitroreductase folds, the cobalamin binding site of MMACHC and the homologue within the C-terminal domain of MMADHC. In a similar manner, but with less affinity, MMACHC can form homodimers when binding cobalamins, which might provide the protection of the active center for proper cobalamin processing. Several pathogenic mutations on both sites have been shown to impair the interaction of MMACHC and MMADHC. Froese et al. demonstrated that the common MMACHC mutation p.Arg161Gln (c.482G>A) diminished complex formation, whereas the artificial deletion of residues 109–111 completely inhibited complexation (Froese et al., 2015). Meanwhile, recombinant MMADHC mutants lacking different portions of the unstructured N-terminus, were not impaired in complex formation with MMACHC as shown in native electrophoresis (Gherasim et al., 2013). The finding that the N-terminal 115 residues are not necessary for MMACHC-MMADHC-complex formation goes along with the identification of putatively involved short peptides between residues 142 and 290 in the C-terminal domain of MMADHC (Gherasim et al., 2013; Plesa et al., 2011). The MMACHC-MMADHC heterodimer may form before dealkylation or decyanation and is favored by the presence of bound cobalamins and excess GSH, i.e. catalytic conditions (Froese et al., 2015; Gherasim et al., 2013).
Work by Li et al. uncovered a previously unrecognized feature of MMADHC: direct binding to free H2Ocbl and to MMACHC-bound cob(II)alamin (Li et al., 2020b). Binding of H2Ocbl by MMADHC occurs via residues Cys261/Cys262 that serve as sulfur ligand donor for the formation of a transient thiolato-cob(III)alamin complex with a rarely seen Co−S bond anchored on the protein. In the case of the interaction between MMADHC and cob(II)alamin⋅MMACHC, a cob(II)alamin thiolate complex is formed. Although rare examples of labile Co(II)-corrinoid-thiolate complexes are known (Cockle et al., 1972; Dereven’kov et al., 2018, 2017), intraprotein Co(II)-S interaction is significantly tighter. Both MMACHC-bound cob(II)alamin or MMADHC-bound cob(III)alamin could initiate the formation of the interprotein complex between MMACHC and MMADHC, yet kinetics favor the former in line with the established cobalamin processing sequence. These new insights may give an explanation to the mechanism of cobalamin translocation between the active sites of the enzymes along its metabolic pathway.
MMACHC also interacts with cobalamin acceptor protein MS. Residues in different regions of MMACHC including the disease-relevant R161 have been predicted to be involved in this interaction (Fofou-Caillierez et al., 2013). The complexation of MMACHC with truncated MS was found to affect upstream decyanation of CNCbl and HOCbl production by MMACHC, whereas AdoCbl production was increased. Therefore, MS is proposed as one more central player in the regulation of cobalamin distribution between the MeCbl and AdoCbl branch. Bassila et al. further investigated the formation of a multiprotein interactome involving MS, MMACHC, MMADHC, and methionine synthetase reductase (MSR; Bassila et al., 2017). Studies in patient fibroblasts and siRNA-transfected HepG2 cells confirmed interactions of MS-MSR, MS-MMACHC and MMACHC-MMADHC and revealed interactions of MSR-MMACHC, MSR-MMADHC, and MS-MMADHC. All interactions were severely affected by the absence of either MS, MMACHC, or MSR, but not so by MMADHC mutations. These experimental findings suggest the formation of a tripartite complex between MMACHC, MS, and MSR, in which each protein would enhance the interactions between the others for optimal MeCbl synthesis. This may add to the explanation of shared biochemical features and pathophysiologic basis between the respective genetic defects of MS (cblG), MSR (cblE), MMACHC, and MMADHC.
Subcellular localization of the MMACHC protein
Considering the biochemical phenotype of cblC and the known localization of MS and MUT, MMACHC was predicted to localize in the cytoplasm, whereas MMADHC should co-localize in cytoplasm and mitochondria enabling fractionated distribution of cobalamin. Mah et al. demonstrated the dispersed presence of MMACHC throughout cytoplasm and its absence in mitochondria by immunofluorescence, confirming the findings by subcellular fractionation and immunoblotting of GFP (green fluorescent protein)-fused MMACHC as well as by the presence of endogenous MMACHC protein in the cytosol but not in the mitochondria of human fibroblasts (Mah et al., 2013). These results were in contrast with previous studies showing MMACHC localization to the mitochondrion, namely, mitochondrial expression of MMACHC in mice and HeLa cells (Deme et al., 2012; Pagliarini et al., 2008). Yet, these earlier findings involved the use of a truncated variant of MMACHC lacking the first N-terminal 57 amino acids. This lab-made truncation artificially generated a probable mitochondrial targeting sequence that is otherwise lacking in the full-length, native wild-type MMACHC protein. Thus, MMACHC is unambiguously localized to the cytosol.
Clinical overview
Clinical findings
Patients with cblC disease present with a variety of clinical symptoms with different ages of onset and severity (Huemer et al., 2017; Rosenblatt et al., 1997). The most severe cases are acutely ill in the first months of life, and most patients are diagnosed within the first year of life. Some already present with intrauterine growth restriction (IUGR) or nonimmune hydrops fetalis. The first symptoms are generally feeding difficulties and lethargy followed by progressive neurological deterioration, including seizures. Also, severe pancytopenia or a non-regenerative megaloblastic anemia may be present. cblC disease is a multisystem disease progressing to renal failure, hepatic dysfunction, cardiomyopathy, pulmonary hypertension, and other symptoms owing to widespread microangiopathy including retinopathy. Overall, the combination of neurologic and/or hematologic and/or ophthalmologic symptoms should always raise the suspicion of a remethylation disorder as stated by Huemer et al. (2017). While most patients present early with this severe multisystem phenotype, a small number of cblC patients were not diagnosed until after the first year of life and some as late as the end of the fourth decade of life (Huemer et al., 2017; Lerner-Ellis et al., 2006). In late-onset patients, neurologic abnormalities prevail, major clinical findings include confusion, disorientation, and gait abnormalities as well as incontinence. Macrocytic anemia was seen in only about a third of the older patients. Mild forms of the disease presenting with cardiorenal and cardiopulmonary symptoms and more generally, those with late-onset cblC, are ascertained by clinical symptoms (Kalantari et al., 2022) (i.e. not by laboratory markers during newborn screening, and/or genetic testing) and thus, many years may transcur without diagnosis and treatment, and in some cases, undertreatment. It is unknown whether these symptoms would ever manifest in patients with adequate treatment, but based on published information, this appears unlikely. Age- and system-specific manifestations of cblC disease are listed in Tables 2 and 3, respectively.
Table 2.
Age-specific manifestations of cblC diseasea
| In utero/Neonatal (0–28 days) | Infants (1–12 months) | Children (1–12 years) | Adolescents and adults |
|---|---|---|---|
| Intrauterine growth restriction (IUGR) Fetal hydrops Feeding difficulties Encephalopathy: Apnea Lethargy Seizures Muscular hypotonia Microcephaly Hydrocephalus Nystagmus Anemia: Thrombocytopenia Pancytopenia Megaloblastic anemia Renal impairment: Hemolytic uremic syndrome (HUS) with hematuria and proteinuria Cardiomyopathy Congenital heart disease Pulmonary hypertension Facial dysmorphia |
Feeding difficulties Failure to thrive: poor growth and/or weight gain Acute encephalopathy: Apnea Impaired consciousness Chronic encephalopathy: Lethargy Seizures Muscular hypotonia Acute behavior changes Developmental retardation, regression Maculopathy (“bull's eye”), retinopathy and/or optic atrophy: Nystagmus Visual inattention Anemia Renal impairment Pulmonary hypertension Metabolic decompensation |
Acute encephalopathy Chronic encephalopathy: Lethargy Seizures Muscular hypotonia or spasticity Acute behavior changes Personality changes Developmental retardation, regression or dementia Subacute degeneration of the spinal cord (SCD): Paresthesia, incontinence ataxia/spasticity, limb weakness (legs > arms) Visual loss, blindness Anemia Thromboembolic events: Recurrent venous thrombosis Pulmonary embolism Cerebrovascular events Renal impairment Pulmonary hypertension |
Acute encephalopathy Chronic encephalopathy: Lethargy Neuropsychiatric disturbances Acute behavior changes Personality changes Developmental retardation, regression or dementia SCD Anemia Thromboembolic events Renal impairment Pulmonary hypertension Marfanoid features |
According to Huemer et al. (2017), Carrillo-Carrasco et al. (2012), and Sloan et al. (2008).
Table 3.
Symptoms and signs of cblC disease according to the organ system and frequencya
| General symptoms | |
| Feeding difficulties, failure to thrive | +++ |
| Small for gestational age | + |
| Intrauterine growth restriction (IUGR) | + |
| Temperature instability/hypothermia | + |
| Metabolic acidosis and/or hyperammonemia | + |
| Neurologic | |
| Developmental disorder/cognitive impairment | +++ |
| Encephalopathy, acute or chronic | |
| Seizures | +++ |
| Movement disorder and/or muscular hypotonia | +++ |
| Neuropsychiatric symptoms including behavioral disorders, personality changes, speech difficulties, psychosis, and/or dementia | ++ |
| Decreased consciousness, lethargy and/or apnea | |
| Subacute degeneration of spinal cord (SCD) | ++ |
| presenting as paresthesia, incontinence, ataxia, spasticity, and/or progressive limb weakness (legs>arms) | ++ |
| Hydrocephalus | ++ |
| Microcephaly | ++ |
| Hematologic | |
| Megaloblastic anemia | ++ |
| Pancytopenia/thrombocytopenia/neutropenia | ++ |
| Recurrent severe infections | (+) |
| Ophthalmologic | +++ |
| Maculopathy, retinopathy, and/or optic atrophy | |
| Inability to fixate, nystagmus, and/or vision loss up to blindness | |
| Renal | |
| Haemolytic uraemic syndrome (HUS) | ++ |
| Glomerulopathy | + |
| Tubulointerstitial nephropathy | + |
| Thrombotic microangiopathyb | + |
| Cardiopulmonary | |
| Cardiomyopathy | ++ |
| Cardiac malformation | + |
| Interstitial pneumonia | + |
| Pulmonary hypertension | + |
| Vascular | |
| Thromboembolic events: deep venous thrombosis, pulmonary thrombosis, cerebrovascular events | + |
| Gastrointestinal | |
| Liver steatosis | + |
| Vomiting | ? |
| Diarrhea | ? |
| Stomatitis/glossitis | ? |
| Gastritis/enteropathy | ? |
| Others | |
| Facial dysmorphism | + |
| Skeletal deformity | (+) |
| Marfanoid features | ? |
| Dermatitis/rash/hyperpigmentation | + |
| Hydrops fetalis | + |
| Metabolic decompensation | ? |
+++ frequent (25–50%of cases), ++ infrequent (10–25% of cases), + occasional (<10% of cases), (+) single case reports, adapted from Huemer et al. (2017).
An increasing number of case reports suggest that this presentation is sporadic but certainly not infrequent.
Central and peripheral neurologic signs include decreased consciousness and apnea, seizures, microcephalia and hydrocephalus, late developmental disorders, cognitive impairment, personality changes, psychosis as well as abnormal muscle tone, movement disorders, and subacute combined degeneration of the spinal cord (SCD) (Carrillo-Carrasco and Venditti, 2012; Huemer et al., 2017; Sloan et al., 2008). SCD owing to vitamin B12 deficiency affects motor functions, continence, as well as deep and surface sensibility. SCD owing to B12 deficiency typically presents paresthesia, decreased proprioception, hypo-/hyperreflexia, and limb weakness, which can progress to spasticity.
Megaloblastic anemia, a classic feature of nutritional vitamin B12 deficiency, together with pancytopenia or even agranulocytosis is reported in 10–25% of cblC cases (Huemer et al., 2017). Megaloblastosis of the bone marrow may present with hypersegmented peripheral neutrophils, macrocytes, and thrombocytopenia (Panchabhai et al., 2016; Rao et al., 2020; Routh and Koenig, 2014). Recurrent infections owing to an impaired immune system have been reported, albeit as a rare presentation (Huemer et al., 2017).
Ophthalmologic manifestations occur with high incidence in the cblC disease (Carrillo-Carrasco and Venditti, 2012; Huemer et al., 2017) and visual impairment affects up to 50% of infant patients. Yet, it is unknown why some but not all cblC patients present with or develop ophthalmological problems. The phenotype is an unusual retinopathy consisting of perimacular hypopigmentation surrounded by a hyperpigmented ring and a more peripheral salt- and pepper retinopathy sometimes accompanied by nystagmus, microcephaly, and hydrocephalus. Vision loss shows a limited response to current treatment and may progress to complete blindness. Meanwhile, ophthalmological complications are rarely seen in late-onset cblC disease.
Microangiopathy and vascular complications such as recurrent venous thrombosis, pulmonary thrombosis, and cerebrovascular events play a substantial role in the morbidity and mortality in cblC patients. Hyperhomocysteinemia is generally considered as a risk factor for arteriosclerosis and established factor for morbidity and mortality in patients with classic homocystinuria (cystathionine beta-synthase (CBS) deficiency, OMIM #236200) (McCully, 1969b; Morris et al., 2017). High levels of total plasma Hcy (tHcy, > 45 μM) have been associated with the occurrence of vascular complications in cblC disease as well (Carrillo-Carrasco and Venditti, 2012; Huemer et al., 2017). Normal tHcy level range between 5 and 15 μmol/L, whereas untreated cblC patients frequently present with elevations >50–100 μmol/L. Therapies aimed at lowering elevated tHcy in patients at risk for or with manifest cardiovascular disease as a means to prevent further cardiovascular events have been largely unsuccessful (Armitage et al., 2010; Løland et al., 2010; Martí-Carvajal et al., 2009). However, the lack of evidence in support of the effectiveness of Hcy reduction for the prevention of cardiovascular events in healthy patients with mildly elevated tHcy cannot be directly extrapolated to cohorts highly affected by vascular events owing to inborn hyperhomocysteinemia wherein plasma Hcy frequently reaches concentrations >100 μM. Successful treatment and prevention of CBS deficiency (Morris et al., 2017; Yap, 2003) such as case reports documenting the resolution of microangiopathy under treatment endorse homocysteine lowering interventions in cblC disease (Huemer et al., 2014; Profitlich et al., 2009b; Van Hove et al., 2002). Adequate treatment to reduce elevated tHcy remains a key approach to reducing and preventing vascular complications in cblC patients (Carrillo-Carrasco and Venditti, 2012; Huemer et al., 2017, 2014). However elevated plasma metabolite concentrations usually do not completely normalize.
While patients with cblC disease are predisposed to renal dysfunction, its occurrence is overall rare. Renal involvement in cblC commonly presents intravascular hemolysis, hematuria, proteinuria, oliguria, and hypertension (Beck et al., 2017). In a study that reviewed an aggregate cohort of 36 cblC patients with renal involvement, the authors identified that about two-thirds of affected cblC patients exhibited atypical hemolytic uremic syndrome (aHUS) based on thrombotic microangiopathy (TMA) (Beck et al., 2017). HUS, characterized by the triad of hemolytic anemia, uremia, and thrombopenia, may lead to renal failure and require dialysis. Mortality is high among patients with renal manifestations (44%) and aggravates with neurologic (56%) or cardiopulmonary (79%) co-involvement. Furthermore, pulmonary hypertension has been described as an isolated manifestation as well as in combination with renal disease in several cblC patients of different ages (Iodice et al., 2013; Kömhoff et al., 2013). Right ventricular failure is a severe complication of pulmonary hypertension with poor prognosis. The report of four new cases of chronic lung disease presenting with diffuse alveolar hemorrhage and pulmonary microangiopathy further amplifies the range of pulmonary complications in cblC disease (Liu et al., 2020). Aside from secondary cardiac decompensation, congenital cardiac malformations and cardiomyopathy are also relevant features of cblC disease and demand cardiac evaluation on a regular basis (Carrillo-Carrasco and Venditti, 2012). Structural heart defects were seen in 5 of a cohort of 10 cblC patients (Profitlich et al., 2009b). The anomalies included left ventricular non-compaction, atrial and ventricular septal defects, dysplastic pulmonary valve, and mitral valve prolapse. In addition, pulmonary embolism led to cor pulmonale in one patient. Even in the presence of normal ventricular function, cardiac screening is particularly important regarding the high risk for thromboembolic events accompanying cblC disease. While the pathogenic mechanisms of many of those symptoms are not fully understood, some can be clearly attributed to systemic microangiopathy.
Disease progression and prognosis are as heterogeneous as their manifestations. Historically, the cblC disease is perceived to have poor long-term outcomes and a reported overall mortality of 30% (Rosenblatt et al., 1997). Prompt diagnosis and treatment can improve mortality and morbidity outcomes, especially if treatment starts before the establishment of chronic neurological impairment. More recent estimates suggest a decreased mortality rate of 11.4% (Fischer et al., 2014), however long-term outcome remains uncertain. Treatment significantly improves hematologic abnormalities and may prevent renal impairment and hydrocephalus (He et al., 2020). Meanwhile, the impact on the neurological outcome varies and is very poor concerning eye disease (Huemer et al., 2017).
Diagnostic methods
Biomarkers of cblC disease
Timely diagnosis is crucial to initiate adequate treatment and prevent complications. Diagnostics should proceed promptly upon clinical suspicion of a remethylation disorder, elevated tHcy, or an abnormal newborn screening (NBS) (Carrillo-Carrasco et al., 2012; Huemer et al., 2017). Treatment should not be delayed after diagnostic confirmation. The biochemical hallmarks of the cblC disease are elevated concentrations of tHcy and urinary/plasma MMA that are brought about by impaired biosynthesis of MeCbl and AdoCbl, respectively. Additionally, serum vitamin B12 and folate, plasma methionine, and the blood acylcarnitine profile should be measured to delineate the differential diagnoses. Typical laboratory findings are summarized in Table 4 tHcy is the preferred biomarker for a follow-up.
Table 4.
Key laboratory parametersa
| Parameter | Normal rangeb | Untreated cblC |
|---|---|---|
| tHcy (plasma) | 5–15 μM | ↑↑ |
| MMA (plasma) | <0.27 μM | ↑↑ |
| MMA (urine) | <3 mg/gCr | ↑↑ |
| Methionine (plasma) | 13–42 μM | ≈/↓ |
| Vitamin B12 (plasma) | 170–800 pg/mL | ≈/↑ |
Adapted by Carrillo-Carrasco et al. (2012) and Beck et al. (2017).
Varies across laboratories and age.
Confirmation of the diagnosis is primarily done by molecular genetic analysis and coenzyme formation analysis in dermal fibroblasts obtained from skin biopsy. Coenzyme formation analysis by incorporation of [14C]-propionate and [14C]-N5-methyltetrahydrofolate into amino acids and then cellular macromolecules, give valuable information on cofactor synthesis in the absence of a known pathogenic mutation and on vitamin responsiveness in vitro (Huemer et al., 2017; Watkins and Rosenblatt, 2013).
Differential diagnosis of cblC disease
As clinical manifestations are unspecific and nutritional deficiencies as well as other rare inborn errors of metabolism may present with similar features, differential diagnoses are of central importance. In neonates and breast-fed infants, availability of cobalamin or folate may be impaired by maternal deficiency of these micronutrients owing to inadequate intake or malabsorption (Fowler et al., 2008; Huemer et al., 2017; Scolamiero et al., 2014). Disorders of extracellular cobalamin and folate metabolism include transcobalamin (TC)-deficiency (Trakadis et al., 2014) and autoimmune and genetic disorders such as pernicious anemia or hereditary folate malabsorption (Huemer et al., 2017; Watkins and Rosenblatt, 2012; Whitehead, 2006). Intracellular cobalamin and folate metabolism may also be affected by drugs such as nitrous oxide or antifolate agents (Hannibal et al., 2016; Huemer et al., 2017). In nutritional cobalamin deficiency, vitamin B12 is decreased, whereas cblC disease vitamin B12 levels are normal or elevated. Low to normal plasma methionine concentrations indicate remethylation disorders, whereas elevated Met is seen in classic homocystinuria owing to CBS deficiency. Hematologic manifestations can resemble severe iron deficiency, some infectious diseases, or myeloproliferative disorders (Huemer et al., 2017). Furthermore, several rare metabolic disorders can also present with hematologic and/or neurologic symptoms, for instance, mitochondrial respiratory chain disorders, orotic aciduria, and branched chain organic acidemias among others (Huemer et al., 2017). In general, the assessment of a combination of biomarkers, as described in Table 4, leads to the accurate and sensitive identification of cblC disease.
Prenatal diagnosis
Prenatal diagnostics may be carried out in individual high-risk cases (Carrillo-Carrasco et al., 2012; Huemer et al., 2017). Molecular genetic analysis then is performed on amniotic cells or chorionic villi samples. A recent study proposed haplotype-based non-invasive prenatal diagnosis (NIPD) as a promising alternative (Han et al., 2019). Metabolites in maternal urine or amniotic fluid may be measured as follow-up during pregnancy. Early treatment was found to improve long-term outcomes in cblC patients, as the identification of the MMACHC gene molecular genetic analysis offers reliable and feasible confirmation diagnosis. Currently, newborn screening (NBS) for cblC is performed, for instance, in the USA, Portugal, some regions of China (Zhou et al., 2019) and in a prospective pilot project in Germany (Gramer et al., 2017; Nogueira et al., 2017; Weisfeld-Adams et al., 2010; Zhou et al., 2018). Still, NBS remains a subject of discussion as the sensitivity of available markers is not optimal and small cohorts impede the supply of strong evidence required for nationwide interventions. C3 acylcarnitine together with the C3/C2 ratio are considered feasible primary markers and C17 might be a promising alternative (Malvagia et al., 2015). Methionine is less sensitive, but frequently decreased and might be included in diagnostic algorithm (Gramer et al., 2017; Weisfeld-Adams et al., 2010). In confirmation diagnostics, second-tier biomarkers tHcy and methylmalonic acid (MMA) should be measured to increase the specificity and differentiate from other inborn errors of metabolism (Huemer et al., 2017, 2015). The unequivocal distinction of nutritional cobalamin deficiency versus genetic disorders that yield combined Hcy and MMA elevation requires the measurement of additional biomarkers after NBS analysis.
cblC disease monitoring
Regular follow-up is required over the course of disease to monitor and adapt the treatment and ensure early recognition of possible complications. Sloan et al. proposed evaluation once or twice a month in infancy and to lengthen the intervals to half a year during childhood and to a yearly basis later on (Sloan et al., 2008). Screening for ophthalmologic, neurologic, cardiopulmonary, and renal manifestations should be performed at the time of diagnosis and in regular intervals depending on the phenotype.
Therapeutic strategies
Treatments are designed to improve both clinical and biochemical abnormalities. Parental HOCbl has shown better responses than CNCbl in vitro and in vivo. Still, some patients present with partially or non-responsive phenotypes. Further investigation on different cobalamin derivatives, such as thiolatocobalamins (Wingert et al., 2021), and their interaction with specific mutants of MMACHC show promise to improve therapeutic strategies which have changed very little during the last decades. Moreover, experiences and data for treatment in asymptomatic patients diagnosed by NBS to date are very limited, especially for late-onset genotypes. A single-center study from Philadelphia revealed decreased mortality, but constantly poor long-term outcomes, especially for neuropsychological and ocular impairment in patients diagnosed by NBS (Ahrens-Nicklas et al., 2017). The severity of impairment was correlated more closely to initial metabolic alterations reflecting in utero and pretreatment damage than to long-term metabolic control. Prospective investigations on larger cohorts, late-onset phenotypes, and prenatal treatment may help to prevent manifestations and complications of cblC in the future. A summary of current therapeutic strategies is given below and in Table 5.
Table 5.
Treatment recommendationsa
| Drug | Dose | Target parameter |
|---|---|---|
| Hydroxocobalamin (i.v., i.m., s.c.) | 0.3 mg/kg/day (standard for infants) 1 mg/day (standard for infants) 25 mg/day (high-dose for adults) Titrate, consider less frequent administration after stabilization |
Reduce MMA and tHcy, normalize Met |
| Betaine (p.o.) | 250 mg/day | Reduce tHcy, normalize Met |
| Folinic acid (p.o.) | Consider 5–15 mg/day | |
| L-Carnitine (p.o.) | Consider 50–200 mg/kg/day | |
| Methionine (p.o.) | If required | Normalize Met |
Avoid: nitrous oxide, protein restriction.
Adapted by Carrillo-Carrasco et al. (2012) and Beck et al. (2017).
Hydroxocobalamin
Treatment with parenteral HOCbl should be initiated as soon as a remethylation disorder is suspected. Guidelines provided by Huemer et al. (2017) and Carrillo-Carrasco et al. (2012) established a recommended initial dose of 1 mg HOCbl daily via IV, IM, or SQ injections. For long-term treatment, the dose and frequency should be individually titrated and IM injections provide an effective as well as a feasible way of administration. If patients require anticoagulation, low-weight heparin has been reported as an effective and safe treatment concomitant to HOCbl IM injections. More generally, it has been suggested that high-dose regimens that raise plasma HOCbl concentrations far above normal plasma levels are effective in achieving metabolic control, i.e. reduction of Hcy and MMA. Carrillo-Carrasco et al. could show a dose-dependent improvement of the biochemical markers, particularly plasma MMA, via dose escalation from 1 mg to 20 mg HOCbl/day (Carrillo-Carrasco et al., 2009). Results from an intervention by Scalais et al. using formulations of HOCbl of 5, 25, or 50 mg/mL, given daily, also showed the reduction of biomarker metabolites Hcy and MMA and improvement of neurocognitive and motor symptoms in cblC patients (Scalais et al., 2019). In three infants homozygous for c.271dupA dose intensification allowed biochemical control with a sensitive decrease of tHcy and better neurological and ophthamological outcomes compared with cblC patients on standard treatment (Scalais et al., 2019). An investigation by Higashimoto et al. on the optimal dosage of HOCbl for adult cblC patients demonstrated metabolic control and enhanced improvement of neuropsychological symptoms with 25 mg HOCbl/day and was able to maintain clinical and metabolic control with weekly administration (Higashimoto et al., 2020). Clinical response to therapy remains highly variable in terms of organ damage and genotypes and thus requires further investigation. Although rare, side-effects of HOCbl treatment include reversible discoloration of skin and urine, hypersensitivity and photosensitivity, nausea, headache, and infusion-site reactions (Carrillo-Carrasco et al., 2012). Overall, published studies on dose-escalation with HOCbl suggest biochemical and clinical improvements, yet, the use of doses in the upper limits of the above-mentioned studies should be considered carefully in the context of life-long therapy, as is the case of cblC patients. Human beings compete with their own gut microbiome for the use of vitamin B12. Thus, the gut microbiome is thought to play an important role in B12 deficiency (Rowley and Kendall, 2019; Sokolovskaya et al., 2020). Bacterial overgrowth in the small intestine can deplete dietary B12 levels and B12 deficiency is known to occur in patients with inflammatory bowel disease (IBD), including Crohn disease and ulcerative colitis (Allen and Stabler, 2008; Ward et al., 2015). Some intestinal bacteria can strip B12 from transport protein intrinsic factor, a property only recently reported (Wexler et al., 2018). The colon houses the greatest abundance of bacteria and archaea in humans, with cobalamin and non-cobalamin analogs themselves modulating the bacterial growth; hence, large oral doses of B12 will alter the gut microbial composition. Colonic bacteria produce structural analogs of vitamin B12 (Allen and Stabler, 2008; Sokolovskaya et al., 2020) that have been reported to inhibit the two B12-dependent enzymes in rodents (Stabler et al., 1991) and humans (Bito et al., 2020; Sokolovskaya et al., 2021, 2019). These B12 analogs traverse the gut epithelium and their presence in blood has, in a few cases, been correlated with severe neurological impairment (Carmel et al., 1988). An examination of microbiome composition and the presence of B12 and B12 analogs in the feces, cells, and plasma of cblC patients under different dose regimens of HOCbl administration is necessary to evaluate the long-term impact of ultra-high doses of HOCbl.
Betaine, folate, and carnitine
Oral betaine is recommended to reduce Hcy under conditions when Met is low (Carrillo-Carrasco et al., 2012; Huemer et al., 2017). Betaine enables the hepatic MeCbl-independent remethylation of Hcy to Met via betaine homocysteine-methyltransferase and thus reduces hyperhomocysteinemia. All cblC patients are recommended to receive betaine, usually doses of 250 mg/kg/day are prescribed but may be titrated higher according to Hcy and Met levels. It is well tolerated and normally does not cause side effects, yet, one case of epileptic seizures and two of cerebral edema in the association of highly increased Met were reported (Devlin et al., 2004; Enns et al., 1999; Yaghmai et al., 2002). Supplementation with oral folic or folinic acid is commonly used in cblC disease (Ahrens-Nicklas et al., 2017; Bourque et al., 2021; Carrillo-Carrasco et al., 2012). It is thought to bypass the folate trap owing to decreased MS activity. In practice, folic acid is used more frequently, although folinic acid provides the benefit of passing the blood-brain barrier more efficiently. Alternatively, methylfolate might also be administered as used in methylenetetrahydrofolate reductase deficiency (MTHFR deficiency, OMIM #236250) (Huemer et al., 2017). As cblC patients may present a low level of Met and carnitine synthesis depends on Met, supplementation with Met and L-carnitine may be considered. L-carnitine promotes the excretion of propionyl groups and hence MMA reduction. Folate compounds as well as L-carnitine, Met and other supplements such as valine are generally very well tolerated, yet clinical benefit remains unclear as case numbers are low and long-term data is missing.
Treatment of complications
While treatment of the underlying biochemical abnormalities in cblC disease is crucial to prevent and/or manage severe complications, certain complications may require close monitoring and additional treatment. Cardiovascular risk factors such as dyslipidemias should be evaluated and treated (Carrillo-Carrasco et al., 2012). Resolution of cor pulmonale secondary to pulmonary embolism after acute treatment with heparin, milrinone, furosemide, dioxin, and sildenafil has been described (Profitlich et al., 2009a). Aspirin is not included in common treatment but has been used safely and may be considered when the risk for thromboembolic events is increased. Pulmonary hypertension in cblC patients has been addressed with targeted drugs and symptomatic treatment, yet improvement was mainly seen after initiation and intensification of causal treatment with HOCbl (Gündüz et al., 2014; Iodice et al., 2013; Kömhoff et al., 2013). In acute kidney disease renal replacement therapy has to be discussed (Beck et al., 2017; Morath et al., 2013). Metabolic decompensations are relatively rare and usually improve under the administration of dextrose-containing fluids and standard medication (Ahrens-Nicklas et al., 2017). Seizures, which does not respond to HOCbl treatment, may be complex to treat. In patients presenting with hydrocephalus, chirurgic intervention, i.e. paraventricular shunts, should be considered if neurological development is impaired and may also improve epileptic symptoms (He et al., 2020).
Dietary management
Protein-restricted diets are a fundamental pillar of treatment in many metabolic diseases. In cblC disease, special diets (also known as “medical foods”) have been prescribed to improve weight gain and lower MMA concentrations. However, incomplete protein diets failed to prove better long-term outcomes and can cause iatrogenic amino acid imbalance, especially hypomethioninemia (Ahrens-Nicklas et al., 2017; Manoli et al., 2016). Of concern, the study identified a correlation between the intake of medical food and impaired head growth, even though a causal link cannot be drawn based on the available data. In comparison with severe isolated methylmalonic acidurias, the cblC disease presents lower MMA levels and rare acute metabolic decompensation. Thus, dietary restriction of propionate precursors may have limited benefits and possibly aggravate Met deficiency. Protein restriction and specifically Met-free formulas designed for isolated methylmalonic aciduria should therefore be avoided as stated by Huemer et al. (Ahrens-Nicklas et al., 2017; Huemer et al., 2017). Prolonged fasting and dehydration may trigger metabolic decompensation and should be avoided as well (Sloan et al., 2008). Concerning general anesthesia, nitrous oxide (N2O), a known MS inhibitor, must be avoided as a fatal adverse effect was reported in a patient with MTHFR deficiency (Selzer et al., 2003). When needed, propofol is considered a safe alternative (Ktena et al., 2015).
Prenatal treatment
Prenatal treatment attempts with high-dose HOCbl and folic acid administered to pregnant women with an affected fetus are reported in only a few cases with divergent results (Huemer et al., 2005; Trefz et al., 2016). So far, neither HOCbl nor betaine have been associated with any fetal or neonatal adverse effects (Huemer et al., 2017).
Advances in therapy
As our understanding of the intricate molecular mechanisms of disease progression deepens, new therapies may emerge. Different cobalamin derivatives are under investigation to improve intracellular cofactor supply. Thiolatocobalamins as promising drug candidates have been shown to undergo facilitated β-deligation and serve as antioxidant agents in vitro (Birch et al., 2009; Wingert et al., 2021). Another interesting example is the investigation of the SIRT1 agonist SRT1720. Ghemrawi et al. suggested that decreased expression of SIRT1 in cblC and cblG cells leads to ER stress. In addition, decreased methylation and phosphorylation alter RNA trafficking and splicing of mainly neurologically relevant gene products (Ghemrawi et al., 2019). Methylation, phosphorylation, and localization of RNA binding proteins (RBPs) were measured in treated and untreated patient and control cells. SRT1720 as well as cobalamin and SAM restored multiple parameters of ER stress and mislocalization of RBPs and mRNA. In the mouse model of cblG disease, SRT1720 treatment restored SIRT1 expression in the hippocampus and demonstrated to improve learning ability. Thus, SIRT1 agonists might provide an alternative treatment for non-responsive neuropsychiatric symptoms in cobalamin deficiencies. Wang et al. proposed to use stop codon readthrough as a therapeutic option in cblC patients with mutations leading to premature stop to codons and nonsense-mediated decay (Wang et al., 2019). Small molecules, for example, aminoglycoside antibiotics, were found to enhance readthrough of stop codons during translation and enable the production of untruncated proteins with a normal or residual function (Dabrowski et al., 2018). Different substances such as chloramphenicol and gentamicin were investigated in cellular models of lysosomal storage diseases and methylmalonic aciduria but their use has not yet been extended to cblC disease (Buck et al., 2009; Quoos Mayer et al., 2013). Mutations c.609G > A (p.W203Ter) and c.394C > T (p.Arg132Ter) with high prevalence in Chinese and Indian-Pakistani populations, respectively, cause stop codons within exon 4 and could be targets for stop codon readthrough approaches (Kaur et al., 2021; Wang et al., 2019). Enzyme replacement therapy is not available for disorders of intracellular cobalamin metabolism so far. An et al. investigated systemic mRNA as alternative replacement therapy in a mouse model for methylmalonic aciduria owing to MUT deficiency (An et al., 2017). Codon-optimized hMMUT mRNA was encapsulated in bio-degradable lipid nanoparticles (LNP) and administered in increasing doses and repetitions. MUT was successfully expressed and localized in mitochondria of hepatocytes and showed to improve metabolite levels, growth, and survival in mice. The effect persisted for over a week and even at repeated doses, no signs of hepatotoxicity, immune response, or inflammation were noted. mRNA replacement might be a promising approach for various metabolic disorders, although there are still challenges to overcome, among others the blood brain barrier for CNS delivery in the case of cblC. The discovery of CRISPR/Cas9 genome editing is a promising advance toward curative therapies for inborn errors of metabolism including various entities with devastating manifestations where current treatments are ineffective (Doudna and Charpentier, 2014; Schneller et al., 2017). Adeno-associated virus (AAV) is used as a vector for gene transfer and site-specific endonucleases as zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), or CRISPR/Cas9 enable targeted editing. First in vivo studies were performed in mouse models of hemophilia B (Barzel et al., 2015; Li et al., 2011; Sharma et al., 2015), hereditary tyrosinemia type I (Nygaard et al., 2016; Pankowicz et al., 2016; Yin et al., 2016, 2014), ornithine transcarbamylase deficiency (Yang et al., 2016), lysosomal storage disorders (Sharma et al., 2015), and glycogen storage disease (Landau et al., 2016) showing successful expression of the inserted genes and improved biochemical and clinical parameters as outlined in the review by Schneller et al. (2017). Nonetheless, there is still a long way from bench to bedside. In several of the studies, the efficacy was still low and safety will have to be proven by ruling out risks of genotoxicity by insertional mutagenesis and immune activation.
Cellular and animal models of cblC disease
Induced pluripotent stem cells (iPSCs)
Specific and disease-relevant cellular assays are a key instrument for in vitro modeling of pathogenesis as well as therapeutic response. Recently, Guan et al. created the first iPSC line from a cblC patient that was compound heterozygous for MMACHC mutations (Guan et al., 2020). C.482G>A (p.Arg161Gln) is one of the most common mutations in East Asian patients and associated with late-onset and mild phenotype, whereas the second mutation, c.448_449delinsCC, to date is not reported elsewhere. Peripheral blood mononuclear cells were reprogrammed by transfer and transient expression of non-integrated episomal plasmids carrying the factors OCT4, SOX2, KLF4, C-MYC, and BCL-XL. The iPSCs obtained by this method showed pluripotency and differentiation potential for all three germ layers. As of now, more cellular models of different pathogenic mutations will be of interest to elucidate the basis of clinical heterogeneity among cblC patients and to test new therapeutic drug candidates.
Zebrafish
Work by Sloan et al. identified the first fully viable and stable animal model of cblC disease with zebrafish (Sloan et al., 2020). Knockdown of the Mmachc gene by morpholino antisense oligonucleotides resulted in severe and lethal embryonic phenotype. Germline mutants with two differently modified alleles were then created by using zinc finger nucleases (ZNFs) targeting exon 2, the location of the most common pathogenic mutation c.271dup1 in humans. Mmachc mutant zebrafish showed survival of the embryonic stage but died in an early juvenile stage after severe growth retardation and developmental delay. Analysis of Mmachc expression in wild-type fish revealed the highest expression in early embryogenesis with a rapid decrease thereafter, followed by a return to increased expression during the juvenile period. This time course of gene expression coincides with the death of cblC mutant fish in the early juvenile period. A protective effect of maternal MMACHC is discussed for early development (Chern et al., 2020; Sloan et al., 2020). In adult zebrafish, Mmachc expression is highest in the liver, muscle, kidney, and spinal cord, the latter ones reflecting organs typically affected by cblC disease. Biochemical parameters demonstrated a similarity of the animal model to findings in human cblC patients with increased MMA levels, decrease in Met, and abnormal GSH/GSSG ratio as an indicator of oxidative stress. Investigation of ophthalmologic features, characteristic of cblC disease, revealed further analogies rendering the mutants as suitable models to study the pathogenesis of visual impairment in the cblC disorder. After a normal embryonic period, retinopathy with a thin outer nuclear layer, shortened outer segment of cones, decreased number of rods, and thin optic nerves were observed consistent with findings in cblC patients (Traboulsi et al., 1992). mRNA analysis unveiled the dysregulation of multiple genes involved in photoreception and cholesterol metabolism. Thus, further questions were raised concerning the underlaying mechanism and the suggestion of possibly altered RNA trafficking (Ghemrawi et al., 2019). Similar to humans, cblC zebrafish showed very limited response to CNCbl therapy, whereas standard treatment with HOCbl improved biochemical markers, despite the less successful reversal of growth anomalies. Notably, MeCbl had little effect on biochemical markers but was most effective to restore growth. Despite limitations owing to possibly quite different uptake, transport, and metabolism of cobalamin in fish, responses were comparable with trends seen in cblC patients (Sloan et al., 2020). Quintana et al. performed experiments on hcfc1b and thap11 morphant zebrafish to elucidate the principles of cblX and cblX-like disease confirming relevant downregulation of Mmachc expression in both (Quintana et al., 2014). Hcfc1, thap11, and Mmachc genes were shown to play a crucial role in craniofacial and brain development. Craniofacial dysmorphia is not a cardinal manifestation of neither cblC, cblX, nor cblX-like, but was described in all three entities and could be reproduced by knockdown of each of the three genes in zebrafish. In addition, human MMACHC mRNA was able to restore the craniofacial phenotype in hcfc1b morphant zebrafish. Yet, many known mutations in humans do not cause craniofacial defects, which is suggested to correlate with the level of MMACHC expression and function (Quintana et al., 2017, 2014).
Lethality in mice
Mmachc is conserved in mice and is known to express in a tissue- and developmental stage-specific manner (Lerner-Ellis et al., 2006; Pupavac et al., 2011; Smith et al., 2019). Using in situ hybridization in mouse embryos, Pupavac et al. discovered cell-type specific expression of Mmachc in cardiovascular, respiratory, renal, and nervous systems (Pupavac et al., 2011). Contrarily, the interaction partner Mmachc was uniformly expressed throughout all tissues. Mainly, Mmachc expression was stronger in endoderm than in mesenchyme except for a strong presence in head mesenchyme. These patterns correlate with typical organ manifestations of cblC disease, for example, the high prevalence of structural heart defects including atrial and ventricular septum defects based on altered endocardial cushion development (Profitlich et al., 2009b). Reporting on left ventricular non-compaction (LNVC), Tanpaiboon et al. stressed the role of Mmachc in cardiac pathogenesis and suggested the contribution of mitochondrial dysfunction and global dysregulation of cytoskeletal proteins, two phenomena independently associated with both cblC and LNVC (Hannibal et al., 2011; Richard et al., 2009; Tanpaiboon et al., 2012). Besides, expression in head mesenchyme supports a special role of Mmachc in cranial development reflected in microcephaly and facial dysmorphia of some cblC patients and also in recent findings in mutant zebrafish and mouse embryos (Chern et al., 2020; Sloan et al., 2020). The observed malformations in combination with the reported expression pattern indicate a still unclear, but a unique role of Mmachc during embryogenesis.
Overall, the generation of a viable and clinically relevant mammalian model of cblC disease has remained a challenge. Moreno-Garcia et al. created mice heterozygous and homozygous for a loss-of-function allele by gene trapping (Moreno-Garcia et al., 2014). Heterozygous mice were viable and fertile, presented reduced MMACHC protein expression compared with wild-type animals, and increased plasma concentrations of MMA and Hcy, thus confirming a significant disruption of MMACHC function. In contrast, homozygous mutants were not viable and vanished in an early embryonic stage after impaired transformation from morula to blastocyst, with the disabled formation of the trophectoderm needed for hatching and implantation. Thus, Mmachc plays a crucial role in early embryonic development and mutations may lead to even more severe phenotypes in mice than in humans. These observations also pose the question of comparability and fidelity of animal models in representing cblC disease in humans. Nevertheless, during the last two years, four promising new mouse models were developed. In 2019, Ma et al. documented the first murine model of cblC disease created by CRISPR/Cas9 (Ma et al., 2019). The mutant mice carried homozygous mutation c.609G>A encoding p.Trp203Ter, which is a common variant among East Asian patients. The cblC mutant mice were transiently viable and presented with a typical increase of propionyl carnitine. Further assessment of the phenotype of this cblC animal model is awaited with great interest. Chern et al. created two mouse models, a conditional null as well as a gain-of-function model (Chern et al., 2020). A floxed Mmachc allele was generated by CRISPR/Cas9 to enable temporal and tissue-specific knockout. Mmachc flox/flox mice presented viable, fertile, and with unaffected phenotypes. MmachcΔ/Δ were created by Cre mediated deletion and crossbreeding but died in the embryonic stage (Chern et al., 2020) as also seen in the Mmachc GT/GT model of Moreno-Garcia et al. (2014). One Mmachc Δ/Δ embryo recovered at E15.5 exhibited growth restriction, anophthalmia, cranial dysmorphia, and right ventricular hypoplasia (Chern et al., 2020) reiterating features reported in the zebrafish model as well as in singular human patients (Sloan et al., 2020). In the second model, overexpression was gained by a CAG (cytomegalovirus/β-actin) enhancer-promoter followed by an IRES-GFP-pA cassette and verified by elevated mRNA expression and protein levels in GFP fluorescence and Western blot analysis, respectively (Chern et al., 2020). Not only were Mmachc-OE+/tg mice viable, fertile and showed no abnormalities, but also transgenic Mmachctm1.1/tm1.1; Mmachc-OE+/tg mice were rescued from embryonic lethality. Mmachctm1.1/tm1.1; Mmachc-OE+/tg mice were smaller and presented with elevated MMA and Hcy levels and though may serve as a disease model. Interestingly, two viable Mmachctm1.1/tm1.1 weanlings were rescued from mothers carrying the overexpressed allele suggesting a maternal-effect rescue as also discussed for the zebrafish model. To address the pathogenesis of the striking ocular phenotypes in cblC patients, Kiessling et al. generated the first organ- and tissue-specific mouse model (Kiessling et al., 2021). Mmachcflox/flox; Pax6Cre mice, named retinaΔMmachc mice, are depleted of Mmachc expression in the peripheral, but not in the central retina, as of embryonic day E10.5. Analysis of retinal samples showed that this functional knockdown presented typical metabolic alterations such as a significant increase in MMA and folate-dependent purine synthesis intermediates AICA-riboside and SAICA-riboside. Surprisingly, no ocular defects were found in the transgenic mice, suggesting that peripheral murine retinal neurons do not depend on local expression of Mmachc for survival, tissue-integrity, and function. One interpretation of this outcome is that local deficiency may be rescued by the bordering cells, which could be controlled by additional knowdown as intended in follow-up by the research team. If rather caused by systemic than by local effects, non-responsiveness of ocular defects to treatment remains unclear. Also, ocular development may arise earlier than Cre expression initiates Mmachc depletion. Further investigations are awaited as the peripheral retina is just one first piece affected in the puzzle of ocular phenotypes including the central macula, optical nerve, and even lense and iris.
On a more positive note, an important breakthrough in our understanding of cblC-like disorders emerged from the characterization of a mouse model of HCFC1/THAP11 deficiency (Chern et al., 2022). The mouse model was created to carry pathogenic human mutations, namely, Hcfc1A115V/Y and Thap11F80L/F80L) (Chern et al., 2022). The HCFC1/THAP11 mutant animals exhibited elevations of plasma Hcy and MMA, greatly reduced biosynthesis of cofactors MeCbl and AdoCbl, as well as defective development as previously noted in cblC disease (Chern et al., 2022). In addition, the study uncovered a previously unrecognized impairment in the expression of genes that encode ribosome protein subunits with important roles during embryonic development (Chern et al., 2022). In sum, this study was the first to demonstrate that this cblC-like disorder (for these specific genes, also known as cblX disease) presents a dual phenotype consistent with hallmarks of cblC disease and ribosomopathies.
While recapitulation of the homozygous phenotypes of human disease has proven unattainable in mice, further studies on these animal models will permit valuable insights into the role of Mmachc in early life, especially during early development and organ-specific pathogenesis. These studies may guide therapeutic options that could be applicable in utero and perhaps in the pre-conceptional phase in cases where parents attempting a new pregnancy are known carriers of a defective Mmachc gene.
Current limitations and future studies
The complex molecular consequences of MMACHC mutations that lead to cblC disease remain enigmatic. Huemer et al. proposed five main hypotheses for the pathogenesis of cobalamin-related remethylation disorders: direct toxicity of metabolites, missing products, impaired methylation capacity, oxidative stress, and impaired non-enzymatic protein functions (Huemer et al., 2017). Whether metabolites in plasma such as Hcy and MMA are disease-causing mediators, or bystander biomarkers remains unclarified (Hannibal and Blom, 2017). For instance, Hcy is a known risk factor for arteriosclerosis and thrombotic events and Hcy lowering treatment can prevent vascular complications in cblC patients. However, other manifestations do not respond to Hcy normalization and are not sufficiently explained by the direct toxicity of metabolites. Research over the last decade uncovered significant functional versatility of the MMACHC protein, including the biochemical characteristics of its mutants, in vitro oxidative stress owing to futile redox cycling, dysregulation of gene expression, and proteomic changes in seemingly unrelated biochemical pathways. iPSCs that could be differentiated into organ-relevant cell types promise to give insights into tissue-specific alterations in cellular cobalamin trafficking, signaling, proliferation, and differentiation, and biochemical response to drugs. Refined animal models may help to further understand the role of the MMACHC gene during organogenesis and the fundaments of the striking clinical heterogeneity. Pathogenesis and treatment response may be investigated in the scope of specific organs as well as their systemic interactions for specific pathogenic mutations. Approximately 1,000 patients worldwide have been diagnosed with cblC disease in the last 50 years (Wang et al., 2019). International cooperation is essential to obtain representative and adaptable insights in light of the limited case numbers. Guidelines provide best-practice orientation for a more standardized and comparable management. Despite improvements regarding timely diagnosis, early treatment and overall survival, neurological, and ophthalmological dysfunctions remain unsolved. The incorporation of cblC disease biomarkers into newborn screening will support the earliest possible interventions and facilitate the initiation of prospective studies. Prenatal diagnosis and prenatal treatment trials may play a central role in learning how to prevent in utero damage and long-term sequelae. Synthetic cobalamin derivatives that can bypass MMACHC-driven processing may be useful to treat genotypes where residual MMACHC protein expression and/or enzymatic activity are nearly zero.
Acknowledgments
The article processing charge was funded by the Baden-Wuerttemberg Ministry of Science, Research and Art and the University of Freiburg funding program Open Access Publishing. The authors are grateful to the Federal Ministry of Economics and Technology (BMWi), Germany, for grant Nr. 03THWBW002 to L.H., and to the anonymous reviewers for their highly constructive insights.
Author contributions
A.J.E and L.H. conceived and outlined the work and worked jointly on cataloging relevant literature. A.J.E. prepared the first complete draft of the manuscript. S.M., I.A.D., and S.V.M. contributed to the sections covering chemistry and biochemistry of cobalamins. A.J.E., D.W.J., and L.H. contributed to the sections covering enzymology, and cellular and subcellular studies of MMACHC expression. A.J.E and U.S. contributed to sections covering animal models and clinical overview of the cblC disease. All authors edited all sections of the manuscript and agreed to the final version submitted for publication.
Declaration of interests
The authors declare no competing interests.
References
- Ahrens-Nicklas R.C., Whitaker A.M., Kaplan P., Cuddapah S., Burfield J., Blair J., Brochi L., Yudkoff M., Ficicioglu C. Efficacy of early treatment in patients with cobalamin C disease identified by newborn screening: a 16-year experience. Genet. Med. 2017;19:926–935. doi: 10.1038/gim.2016.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen K.D., Wang S.C. Spectroscopic characterization and mechanistic investigation of P-methyl transfer by a radical SAM enzyme from the marine bacterium Shewanella denitrificans OS217. Biochim. Biophys. Acta. 2014;1844:2135–2144. doi: 10.1016/j.bbapap.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen R.H., Stabler S.P. Identification and quantitation of cobalamin and cobalamin analogues in human feces. Am. J. Clin. Nutr. 2008;87:1324–1335. doi: 10.1093/ajcn/87.5.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An D., Schneller J.L., Frassetto A., Liang S., Zhu X., Park J.S., Theisen M., Hong S.J., Zhou J., Rajendran R., et al. Systemic messenger RNA therapy as a treatment for methylmalonic acidemia. Cell Rep. 2017;21:3548–3558. doi: 10.1016/j.celrep.2017.11.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine SEARCH Collaborative Group. Armitage J.M., Bowman L., Clarke R.J., Wallendszus K., Bulbulia R., Rahimi K., Haynes R., Parish S., Sleight P., Peto R., Collins R. Effects of homocysteine-lowering with folic acid plus vitamin B 12 vs placebo on mortality and major morbidity in myocardial infarction survivors: a randomized trial. JAMA. 2010;303:2486–2494. doi: 10.1001/jama.2010.840. [DOI] [PubMed] [Google Scholar]
- Backe P.H., Ytre-Arne M., Røhr A.K., Brodtkorb E., Fowler B., Rootwelt H., Bjørås M., Mørkrid L. Novel deletion mutation identified in a patient with late-onset combined methylmalonic acidemia and homocystinuria, cblC type. JIMD Rep. 2013;11:79–85. doi: 10.1007/8904_2013_225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R. The Yin-Yang of cobalamin biochemistry. Chem. Biol. 1997;4:175–186. doi: 10.1016/S1074-5521(97)90286-6. [DOI] [PubMed] [Google Scholar]
- Banerjee R. B12 trafficking in mammals: a for coenzyme escort service. ACS Chem. Biol. 2006;1:149–159. doi: 10.1021/cb6001174. [DOI] [PubMed] [Google Scholar]
- Banerjee R., Gherasim C., Padovani D. The tinker, tailor, soldier in intracellular B12 trafficking. Curr. Opin. Chem. Biol. 2009;13:484–491. doi: 10.1016/j.cbpa.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R., Gouda H., Pillay S. Redox-linked coordination chemistry directs vitamin B12 trafficking. Acc. Chem. Res. 2021;54:2003–2013. doi: 10.1021/acs.accounts.1c00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker H.A., Smyth R.D., Weissbach H., Toohey J.I., Ladd J.N., Volcani B.E. Isolation and properties of crystalline cobamide coenzymes containing benzimidazole or 5, 6-dimethylbenzimidazole. J. Biol. Chem. 1960;235:480–488. [PubMed] [Google Scholar]
- Barzel A., Paulk N.K., Shi Y., Huang Y., Chu K., Zhang F., Valdmanis P.N., Spector L.P., Porteus M.H., Gaensler K.M., Kay M.A. Promoterless gene targeting without nucleases ameliorates haemophilia B in mice. Nature. 2015;517:360–364. doi: 10.1038/nature13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassila C., Ghemrawi R., Flayac J., Froese D.S., Baumgartner M.R., Guéant J.L., Coelho D. Methionine synthase and methionine synthase reductase interact with MMACHC and with MMADHC. Biochim. Biophys. Acta, Mol. Basis Dis. 2017;1863:103–112. doi: 10.1016/j.bbadis.2016.10.016. [DOI] [PubMed] [Google Scholar]
- Beaven G.H., Johnson E.A. The reduction of vitamin B12. Nature. 1955;176:1264–1265. doi: 10.1038/1761264b0. [DOI] [Google Scholar]
- Bebarta V.S., Garrett N., Brenner M., Mahon S.B., Maddry J.K., Boudreau S., Castaneda M., Boss G.R. Efficacy of intravenous cobinamide versus hydroxocobalamin or saline for treatment of severe hydrogen sulfide toxicity in a swine (Sus scrofa) model. Acad. Emerg. Med. 2017;24:1088–1098. doi: 10.1111/acem.13213. [DOI] [PubMed] [Google Scholar]
- Beck B.B., van Spronsen F., Diepstra A., Berger R.M.F., Kömhoff M. Renal thrombotic microangiopathy in patients with cblC defect: review of an under-recognized entity. Pediatr. Nephrol. 2017;32:733–741. doi: 10.1007/s00467-016-3399-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birch C.S., Brasch N.E., McCaddon A., Williams J.H.H. A novel role for vitamin B12: cobalamins are intracellular antioxidants in vitro. Free Radic. Biol. Med. 2009;47:184–188. doi: 10.1016/j.freeradbiomed.2009.04.023. [DOI] [PubMed] [Google Scholar]
- Bito T., Bito M., Hirooka T., Okamoto N., Harada N., Yamaji R., Nakano Y., Inui H., Watanabe F. Biological activity of pseudovitamin B12on cobalamin-dependent methylmalonyl-CoA mutase and methionine synthase in mammalian cultured COS-7 cells. Molecules. 2020;25:E3268. doi: 10.3390/molecules25143268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourque D.K., Mellin-Sanchez L.E., Bullivant G., Cruz V., Feigenbaum A., Hewson S., Raiman J., Schulze A., Siriwardena K., Mercimek-Andrews S. Outcomes of patients with cobalamin c deficiency: a single center experience. JIMD Rep. 2021;57:102–114. doi: 10.1002/jmd2.12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown K.L., Hakimi J.M., Nuss D.M., Montejano Y.D., Jacobsen D.W. Acid-base properties of α-ribazole and the thermodynamics of dimethylbenzimidazole association in alkylcobalamins. Inorg. Chem. 1984;23:1463–1471. doi: 10.1021/ic00178a032. [DOI] [Google Scholar]
- Buck N.E., Wood L., Hu R., Peters H.L. Stop codon read-through of a Methylmalonic aciduria mutation. Mol. Genet. Metab. 2009;97:244–249. doi: 10.1016/j.ymgme.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Butler P.A., Kräutler B. Biological organometallic chemistry of B12. Top. Organomet. Chem. 2006;17:1–55. doi: 10.1007/3418_004. [DOI] [Google Scholar]
- Carmel R., Karnaze D.S., Weiner J.M. Neurologic abnormalities in cobalamin deficiency are associated with higher cobalamin “analogue” values than are hematologic abnormalities. J. Lab. Clin. Med. 1988;111:57–62. [PubMed] [Google Scholar]
- Carrillo-Carrasco N., Sloan J., Valle D., Hamosh A., Venditti C.P. Hydroxocobalamin dose escalation improves metabolic control in cblC. J. Inherit. Metab. Dis. 2009;32:728–731. doi: 10.1007/s10545-009-1257-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo-Carrasco N., Chandler R.J., Venditti C.P. Combined methylmalonic acidemia and homocystinuria, cblC type. I. Clinical presentations, diagnosis and management. J. Inherit. Metab. Dis. 2012;35:91–102. doi: 10.1007/s10545-011-9364-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo-Carrasco N., Venditti C.P. Combined methylmalonic acidemia and homocystinuria, cblC type. II. Complications, pathophysiology, and outcomes. J. Inherit. Metab. Dis. 2012;35:103–114. doi: 10.1007/s10545-011-9365-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavicchi C., Oussalah A., Falliano S., Ferri L., Gozzini A., Gasperini S., Motta S., Rigoldi M., Parenti G., Tummolo A., et al. PRDX1 gene-related epi-cblC disease is a common type of inborn error of cobalamin metabolism with mono- or bi-allelic MMACHC epimutations. Clin. Epigenetics. 2021;13:137. doi: 10.1186/s13148-021-01117-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman L.E., Darling A.L., Brown J.E. Association between metformin and vitamin B12 deficiency in patients with type 2 diabetes: a systematic review and meta-analysis. Diabetes Metab. 2016;42:316–327. doi: 10.1016/J.DIABET.2016.03.008. [DOI] [PubMed] [Google Scholar]
- Chemaly S.M. Cobalamins and the spectrochemical series. Dalton Trans. 2008:5766–5773. doi: 10.1039/b806927a. [DOI] [PubMed] [Google Scholar]
- Chern T., Achilleos A., Tong X., Hsu C.W., Wong L., Poché R.A. Mouse models to study the pathophysiology of combined methylmalonic acidemia and homocystinuria, cblC type. Dev. Biol. 2020;468:1–13. doi: 10.1016/j.ydbio.2020.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chern T., Achilleos A., Tong X., Hill M.C., Saltzman A.B., Reineke L.C., Chaudhury A., Dasgupta S.K., Redhead Y., Watkins D., et al. Mutations in Hcfc1 and Ronin result in an inborn error of cobalamin metabolism and ribosomopathy. Nat. Commun. 2022;13:134. doi: 10.1038/s41467-021-27759-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockle S., Hill H.A.O., Ridsdale S., Williams R.J.P. The chemistry of vitamin B12. Part XVI. Binding of thiols to the cobalt(II) corrins. J. Chem. Soc. Dalton Trans. 1972:297–302. doi: 10.1039/DT9720000297. [DOI] [Google Scholar]
- Coelho D., Suormala T., Stucki M., Lerner-Ellis J.P., Rosenblatt D.S., Newbold R.F., Baumgartner M.R., Fowler B. Gene identification for the cblD defect of vitamin B12 metabolism. N. Engl. J. Med. 2008;358:1454–1464. doi: 10.1056/NEJMoa072200. [DOI] [PubMed] [Google Scholar]
- Coelho D., Kim J.C., Miousse I.R., Fung S., Du Moulin M., Buers I., Suormala T., Burda P., Frapolli M., Stucki M., et al. Mutations in ABCD4 cause a new inborn error of vitamin B12 metabolism. Nat. Genet. 2012;44:1152–1155. doi: 10.1038/ng.2386. [DOI] [PubMed] [Google Scholar]
- Cornec-Le Gall E., Delmas Y., De Parscau L., Doucet L., Ogier H., Benoist J.F., Fremeaux-Bacchi V., Le Meur Y. Adult-onset eculizumab-resistant hemolytic uremic syndrome associated with cobalamin C deficiency. Am. J. Kidney Dis. 2014;63:119–123. doi: 10.1053/j.ajkd.2013.08.031. [DOI] [PubMed] [Google Scholar]
- Dabrowski M., Bukowy-Bieryllo Z., Zietkiewicz E. Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol. Med. 2018;24:25. doi: 10.1186/s10020-018-0024-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De March M., Demitri N., Geremia S., Hickey N., Randaccio L. Trans and cis influences and effects in cobalamins and in their simple models. J. Inorg. Biochem. 2012;116:215–227. doi: 10.1016/j.jinorgbio.2012.07.018. [DOI] [PubMed] [Google Scholar]
- Deme J.C., Miousse I.R., Plesa M., Kim J.C., Hancock M.A., Mah W., Rosenblatt D.S., Coulton J.W. Structural features of recombinant MMADHC isoforms and their interactions with MMACHC, proteins of mammalian vitamin B12 metabolism. Mol. Genet. Metab. 2012;107:352–362. doi: 10.1016/j.ymgme.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Dereven’kov I.A., Salnikov D.S., Silaghi-Dumitrescu R., Makarov S.V., Koifman O.I. Redox chemistry of cobalamin and its derivatives. Coord. Chem. Rev. 2016;309:68–83. doi: 10.1016/j.ccr.2015.11.001. [DOI] [Google Scholar]
- Dereven’kov I.A., Hannibal L., Dürr M., Salnikov D.S., Bui Thi T.T., Makarov S.V., Koifman O.I., Ivanović-Burmazović I. Redox turnover of organometallic B12 cofactors recycles vitamin C: sulfur assisted reduction of dehydroascorbic acid by cob(II)alamin. J. Org. Chem. 2017;839:53–59. doi: 10.1016/j.jorganchem.2017.01.002. [DOI] [Google Scholar]
- Dereven’kov I.A., Makarov S.V., Bui Thi T.T., Makarova A.S., Koifman O.I. Studies on the reduction of dehydroascorbic acid by glutathione in the presence of aquahydroxocobinamide. Eur. J. Inorg. Chem. 2018;2018:2987–2992. doi: 10.1002/ejic.201800066. [DOI] [Google Scholar]
- Dereven’kov I.A., Hannibal L., Makarov S.V., Molodtsov P.A. Catalytic effect of riboflavin on electron transfer from NADH to aquacobalamin. J. Biol. Inorg. Chem. 2020;25:125–133. doi: 10.1007/s00775-019-01745-3. [DOI] [PubMed] [Google Scholar]
- Devlin A.M., Hajipour L., Gholkar A., Fernandes H., Ramesh V., Morris A.A.M. Cerebral edema associated with betaine treatment in classical homocystinuria. J. Pediatr. 2004;144:545–548. doi: 10.1016/j.jpeds.2003.12.041. [DOI] [PubMed] [Google Scholar]
- Doudna J.A., Charpentier E. The new Frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Drennan C.L., Huang S., Drummond J.T., Matthews R.G., Ludwig M.L. How a protein binds B12: a 3.0 A X-ray structure of B12-binding domains of methionine synthase. Science. 1994;266:1669–1674. doi: 10.1126/science.7992050. [DOI] [PubMed] [Google Scholar]
- Enns G.M., Barkovich A.J., Rosenblatt D.S., Fredrick D.R., Weisiger K., Ohnstad C., Packman S. Progressive neurological deterioration and MRI changes in cblC methylmalonic acidaemia treated with hydroxocobalamin. J. Inherit. Metab. Dis. 1999;22:599–607. doi: 10.1023/A:1005517727451. [DOI] [PubMed] [Google Scholar]
- Fischer S., Huemer M., Baumgartner M., Deodato F., Ballhausen D., Boneh A., Burlina A.B., Cerone R., Garcia P., Gökçay G., et al. Clinical presentation and outcome in a series of 88 patients with the cblC defect. J. Inherit. Metab. Dis. 2014;37:831–840. doi: 10.1007/s10545-014-9687-6. [DOI] [PubMed] [Google Scholar]
- Fofou-Caillierez M.B., Mrabet N.T., Chéry C., Dreumont N., Flayac J., Pupavac M., Paoli J., Alberto J.M., Coelho D., Camadro J.M., et al. Interaction between methionine synthase isoforms and MMACHC: characterization in cblG-variant, cblG and cblC inherited causes of megaloblastic anaemia. Hum. Mol. Genet. 2013;22:4591–4601. doi: 10.1093/hmg/ddt308. [DOI] [PubMed] [Google Scholar]
- Fowler B., Leonard J.V., Baumgartner M.R. Causes of and diagnostic approach to methylmalonic acidurias. J. Inherit. Metab. Dis. 2008;31:350–360. doi: 10.1007/s10545-008-0839-4. [DOI] [PubMed] [Google Scholar]
- Froese D.S., Zhang J., Healy S., Gravel R.A. Mechanism of vitamin B12-responsiveness in cblC methylmalonic aciduria with homocystinuria. Mol. Genet. Metab. 2009;98:338–343. doi: 10.1016/j.ymgme.2009.07.014. [DOI] [PubMed] [Google Scholar]
- Froese D.S., Healy S., McDonald M., Kochan G., Uppermann U., Niesen F.H., Gravel R.A. Thermolability of mutant MMACHC protein in the vitamin B12- responsive cblC disorder 100. 2010. [DOI] [PMC free article] [PubMed]
- Froese D.S., Krojer T., Wu X., Shrestha R., Kiyani W., Von Delft F., Gravel R.A., Oppermann U., Yue W.W. Structure of MMACHC reveals an arginine-rich pocket and a domain-swapped dimer for its B12 processing function. Biochemistry. 2012;51:5083–5090. doi: 10.1021/bi300150y. [DOI] [PubMed] [Google Scholar]
- Froese D.S., Kopec J., Fitzpatrick F., Schuller M., McCorvie T.J., Chalk R., Plessl T., Fettelschoss V., Fowler B., Baumgartner M.R., Yue W.W. Structural insights into the MMACHC-MMADHC protein complex involved in vitamin B12 trafficking. J. Biol. Chem. 2015;290:29167–29177. doi: 10.1074/jbc.M115.683268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagsteiger J., Jahn S., Heidinger L., Gericke L., Andexer J.N., Friedrich T., Loenarz C., Layer G. A cobalamin-dependent radical SAM enzyme catalyzes the unique Cα-methylation of glutamine in methyl-coenzyme M reductase. Angew. Chem. Int. Ed. Engl. 2022:e202204198. doi: 10.1002/anie.202204198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galen Lenhert P., Hodgkin D.C. Structure of the 5, 6-dimethylbenzimidazolylcobamide coenzyme. Nature. 1961;192:937–938. doi: 10.1038/192937a0. [DOI] [PubMed] [Google Scholar]
- Ghemrawi R., Arnold C., Battaglia-Hsu S.-F., Pourié G., Trinh I., Bassila C., Rashka C., Wiedemann A., Flayac J., Robert A., et al. SIRT1 activation rescues the mislocalization of RNA-binding proteins and cognitive defects induced by inherited cobalamin disorders. Metabolism. 2019;101:153992. doi: 10.1016/J.METABOL.2019.153992. [DOI] [PubMed] [Google Scholar]
- Gherasim C., Hannibal L., Rajagopalan D., Jacobsen D.W., Banerjee R. The C-terminal domain of CblD interacts with CblC and influences intracellular cobalamin partitioning. Biochimie. 2013;95:1023–1032. doi: 10.1016/j.biochi.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherasim C., Ruetz M., Li Z., Hudolin S., Banerjee R. Pathogenic mutations differentially affect the catalytic activities of the human B12-processing chaperone CblC and increase futile redox cycling. J. Biol. Chem. 2015;290:11393–11402. doi: 10.1074/jbc.M115.637132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gick G.G., Arora K., Sequeira J.M., Nakayama Y., Lai S.-C., Quadros E. v. Cellular uptake of vitamin B12: role and fate of TCblR/CD320, the transcobalamin receptor. Exp. Cell Res. 2020;396:112256. doi: 10.1016/j.yexcr.2020.112256. [DOI] [PubMed] [Google Scholar]
- Gramer G., Abdoh G., Ben-Omran T., Shahbeck N., Ali R., Mahmoud L., Fang-Hoffmann J., Hoffmann G.F., Al Rifai H., Okun J.G. Newborn screening for remethylation disorders and vitamin B12 deficiency-evaluation of new strategies in cohorts from Qatar and Germany. World J. Pediatr. 2017;13:136–143. doi: 10.1007/s12519-017-0003-z. [DOI] [PubMed] [Google Scholar]
- Gravel R.A., Mahoney M.J., Ruddle F.H., Rosenberg L.E. Genetic complementation in heterokaryons of human fibroblasts defective in cobalamin metabolism. Proc. Natl. Acad. Sci. USA. 1975;72:3181–3185. doi: 10.1073/pnas.72.8.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green R., Allen L.H., Bjørke-Monsen A.L., Brito A., Guéant J.L., Miller J.W., Molloy A.M., Nexo E., Stabler S., Toh B.H., et al. Vitamin B12 deficiency. Nat. Rev. Dis. Primers. 2017;3:17040. doi: 10.1038/nrdp.2017.40. [DOI] [PubMed] [Google Scholar]
- Guan J., Li Z., Zhang H., Yang X., Ma Y., Li Y., Dong R., Gai Z., Liu Y. Generation of a Human iPSC line (SDQLCHi021-A) from a patient with methylmalonic acidemia cblC type carrying compound heterozygous mutations in MMAHC gene. Stem Cell Res. 2020;43:101709. doi: 10.1016/j.scr.2020.101709. [DOI] [PubMed] [Google Scholar]
- Guéant J.L., Chéry C., Oussalah A., Nadaf J., Coelho D., Josse T., Flayac J., Robert A., Koscinski I., Gastin I., et al. A PRDX1 mutant allele causes a MMACHC secondary epimutation in cblC patients. Nat. Commun. 2018;9:67. doi: 10.1038/s41467-017-02306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guéant J.L., Siblini Y., Chéry C., Schmitt G., Guéant-Rodriguez R.M., Coelho D., Watkins D., Rosenblatt D.S., Oussalah A. Epimutation in inherited metabolic disorders: the influence of aberrant transcription in adjacent genes. Hum. Genet. 2022;141:1309–1325. doi: 10.1007/s00439-021-02414-9. [DOI] [PubMed] [Google Scholar]
- Gündüz M., Ekici F., Özaydın E., Ceylaner S., Perez B. Reversible pulmonary arterial hypertension in cobalamin-dependent cobalamin C disease due to a novel mutation in the MMACHC gene. Eur. J. Pediatr. 2014;173:1707–1710. doi: 10.1007/s00431-014-2330-6. [DOI] [PubMed] [Google Scholar]
- Han L., Chen C., Guo F., Ye J., Peng Z., Qiu W., Wang Y., Li W., Zhang H., Liang L., et al. Noninvasive prenatal diagnosis of cobalamin C (cblC) deficiency through target region sequencing of cell-free DNA in maternal plasma. Prenat. Diagn. 2019;40:324–332. doi: 10.1002/pd.5601. [DOI] [PubMed] [Google Scholar]
- Hannibal L., Blom H.J. Homocysteine and disease: causal associations or epiphenomenons? Mol. Aspects Med. 2017;53:36–42. doi: 10.1016/j.mam.2016.11.003. [DOI] [PubMed] [Google Scholar]
- Hannibal L., DiBello P.M., Yu M., Miller A., Wang S., Willard B., Rosenblatt D.S., Jacobsen D.W. The MMACHC proteome: hallmarks of functional cobalamin deficiency in humans. Mol. Genet. Metab. 2011;103:226–239. doi: 10.1016/j.ymgme.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannibal L., DiBello P.M., Jacobsen D.W. Proteomics of vitamin B 12 processing. Clin. Chem. Lab. Med. 2013;51:477–488. doi: 10.1515/cclm-2012-0568. [DOI] [PubMed] [Google Scholar]
- Hannibal L., Jacobsen D. 3. Intracellular processing and utilization of cobalamins: advances and insights. Vitamin B12. 2017:46–93. doi: 10.1201/9781315119540-4. [DOI] [Google Scholar]
- Hannibal L., Kim J., Brasch N.E., Wang S., Rosenblatt D.S., Banerjee R., Jacobsen D.W. Processing of alkylcobalamins in mammalian cells: a role for the MMACHC (cblC) gene product. Mol. Genet. Metab. 2009;97:260–266. doi: 10.1016/j.ymgme.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannibal L., Smith C.A., Jacobsen D.W., Brasch N.E. Nitroxylcob(III)alamin: synthesis and X-ray structural characterization. Angew. Chem. Int. Ed. Engl. 2007;46:5140–5143. doi: 10.1002/anie.200701131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassanin H.A., Hannibal L., Jacobsen D.W., Brown K.L., Marques H.M., Brasch N.E. NMR spectroscopy and molecular modelling studies of nitrosylcobalamin: further evidence that the deprotonated, base-off form is important for nitrosylcobalamin in solution. Dalton Trans. 2009:424–433. doi: 10.1039/b810895a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassanin H.A., Hannibal L., Jacobsen D.W., El-Shahat M.F., Hamza M.S.A., Brasch N.E. Mechanistic studies on the reaction between R2N-NONOates and aquacobalamin: evidence for direct transfer of a nitroxyl group from R 2N-NONOates to cobalt(III) centers. Angew. Chem. Int. Ed. Engl. 2009;48:8909–8913. doi: 10.1002/anie.200904360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannibal L., Lysne V., Bjørke-Monsen A.L., Behringer S., Grünert S.C., Spiekerkoetter U., Jacobsen D.W., Blom H.J. Biomarkers and algorithms for the diagnosis of vitamin B 12 deficiency. Front. Mol. Biosci. 2016 doi: 10.3389/fmolb.2016.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R., Zhang H., Kang L., Li H., Shen M., Zhang Y., Mo R., Liu Y., Song J., Chen Z., et al. Analysis of 70 patients with hydrocephalus due to cobalamin C deficiency. Neurology. 2020;95:e3129–e3137. doi: 10.1212/WNL.0000000000010912. [DOI] [PubMed] [Google Scholar]
- Hendry-Hofer T.B., Ng P.C., McGrath A.M., Soules K., Mukai D.S., Chan A., Maddry J.K., White C.W., Lee J., Mahon S.B., et al. Intramuscular cobinamide as an antidote to methyl mercaptan poisoning. Inhal. Toxicol. 2020;33:25–32. doi: 10.1080/08958378.2020.1866123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashimoto T., Kim A.Y., Ogawa J.T., Sloan J.L., Almuqbil M.A., Carlson J.M., Manoli I., Venditti C.P., Gunay-Aygun M., Wang T. High-dose hydroxocobalamin achieves biochemical correction and improvement of neuropsychiatric deficits in adults with late onset cobalamin C deficiency. JIMD Rep. 2020;51:17–24. doi: 10.1002/jmd2.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin D.C., Kamper J., Mackay M., Pickworth J., Trueblood K.N., White J.G. Structure of vitamin B12. Nature. 1956;178:64–66. doi: 10.1038/178064a0. [DOI] [PubMed] [Google Scholar]
- Huemer M., Simma B., Fowler B., Suormala T., Bodamer O.A., Sass J.O. Prenatal and postnatal treatment in cobalamin C defect. J. Pediatr. 2005;147:469–472. doi: 10.1016/j.jpeds.2005.04.040. [DOI] [PubMed] [Google Scholar]
- Huemer M., Scholl-Bürgi S., Hadaya K., Kern I., Beer R., Seppi K., Fowler B., Baumgartner M.R., Karall D. Three new cases of late-onset cblC defect and review of the literature illustrating when to consider inborn errors of metabolism beyond infancy. Orphanet J. Rare Dis. 2014;9:161. doi: 10.1186/s13023-014-0161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huemer M., Kožich V., Rinaldo P., Baumgartner M.R., Merinero B., Pasquini E., Ribes A., Blom H.J. Newborn screening for homocystinurias and methylation disorders: systematic review and proposed guidelines. J. Inherit. Metab. Dis. 2015;38:1007–1019. doi: 10.1007/s10545-015-9830-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huemer M., Diodato D., Schwahn B., Schiff M., Bandeira A., Benoist J.F., Burlina A., Cerone R., Couce M.L., Garcia-Cazorla A., et al. Guidelines for diagnosis and management of the cobalamin-related remethylation disorders cblC, cblD, cblE, cblF, cblG, cblJ and MTHFR deficiency. J. Inherit. Metab. Dis. 2017;40:21–48. doi: 10.1007/s10545-016-9991-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iodice F.G., Di Chiara L., Boenzi S., Aiello C., Monti L., Cogo P., Dionisi-Vici C. Cobalamin c defect presenting with isolated pulmonary hypertension. Pediatrics. 2013;132 doi: 10.1542/peds.2012-1945. e248-251. [DOI] [PubMed] [Google Scholar]
- Jacobsen D.W., Hannibal L. Redox signaling in inherited diseases of metabolism. Current Opin. Physiol. 2019;9:48–55. doi: 10.1016/j.cophys.2019.04.024. [DOI] [Google Scholar]
- Jeong J., Kim J. Glutathione increases the binding affinity of a bovine B12 trafficking chaperone bCblC for vitamin B12. Biochem. Biophys. Res. Commun. 2011;412:360–365. doi: 10.1016/j.bbrc.2011.07.103. [DOI] [PubMed] [Google Scholar]
- Jeong J., Ha T.S., Kim J. Protection of aquo/hydroxocobalamin from reduced glutathione by a B12 trafficking chaperone. BMB Rep. 2011;44:170–175. doi: 10.5483/BMBRep.2011.44.3.170. [DOI] [PubMed] [Google Scholar]
- Jeong J., Park J., Park J., Kim J. Processing of glutathionylcobalamin by a bovine B12 trafficking chaperone bCblC involved in intracellular B12 metabolism. Biochem. Biophys. Res. Commun. 2014;443:173–178. doi: 10.1016/j.bbrc.2013.11.075. [DOI] [PubMed] [Google Scholar]
- Jiang J., Chan A., Ali S., Saha A., Haushalter K.J., Lam W.L.M., Glasheen M., Parker J., Brenner M., Mahon S.B., et al. Hydrogen sulfide-mechanisms of toxicity and development of an antidote. Sci. Rep. 2016;6:20831. doi: 10.1038/srep20831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns P.W., Das A., Kuil E.M., Jacobs W.A., Schimpf K.J., Schmitz D.J. Cocoa polyphenols accelerate vitamin B12 degradation in heated chocolate milk. Int. J. Food Sci. Technol. 2015;50:421–430. doi: 10.1111/IJFS.12632. [DOI] [Google Scholar]
- Kadiiska M.B., Gladen B.C., Baird D.D., Germolec D., Graham L.B., Parker C.E., Nyska A., Wachsman J.T., Ames B.N., Basu S., et al. Biomarkers of Oxidative Stress Study II. Are oxidation products of lipids, proteins, and DNA markers of CCl4 poisoning? Free Radic. Biol. Med. 2005;38:698–710. doi: 10.1016/j.freeradbiomed.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Kalantari S., Brezzi B., Bracciamà V., Barreca A., Nozza P., Vaisitti T., Amoroso A., Deaglio S., Manganaro M., Porta F., Spada M. Adult-onset CblC deficiency: a challenging diagnosis involving different adult clinical specialists. Orphanet J. Rare Dis. 2022;17:33. doi: 10.1186/s13023-022-02179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur R., Attri S.V., Saini A.G., Sankhyan N. A high frequency and geographical distribution of MMACHC R132∗ mutation in children with cobalamin C defect. Amino Acids. 2021;53:253–264. doi: 10.1007/s00726-021-02942-8. [DOI] [PubMed] [Google Scholar]
- Kiessling E., Nötzli S., Todorova V., Forny M., Baumgartner M.R., Samardzija M., Krijt J., Kožich V., Grimm C., Froese D.S. Absence of MMACHC in peripheral retinal cells does not lead to an ocular phenotype in mice. Biochim. Biophys. Acta, Mol. Basis Dis. 2021;1867:166201. doi: 10.1016/j.bbadis.2021.166201. [DOI] [PubMed] [Google Scholar]
- Kim J., Gherasim C., Banerjee R. Decyanation of vitamin B12 by a trafficking chaperone. Proc. Natl. Acad. Sci. USA. 2008;105:14551–14554. doi: 10.1073/pnas.0805989105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Hannibal L., Gherasim C., Jacobsen D.W., Banerjee R. A human vitamin B12 trafficking protein uses glutathione transferase activity for processing alkylcobalamins. J. Biol. Chem. 2009;284:33418–33424. doi: 10.1074/jbc.M109.057877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kömhoff M., Roofthooft M.T., Westra D., Teertstra T.K., Losito A., van de Kar N.C.A.J., Berger R.M.F. Combined pulmonary hypertension and renal thrombotic microangiopathy in cobalamin C deficiency. Pediatrics. 2013;132 doi: 10.1542/peds.2012-2581. e540-544. [DOI] [PubMed] [Google Scholar]
- Koutmos M., Gherasim C., Smith J.L., Banerjee R. Structural basis of multifunctionality in a vitamin B 12- processing enzyme. J. Biol. Chem. 2011;286:29780–29787. doi: 10.1074/jbc.M111.261370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kräutler B. Biochemical Society Transactions. 2005. Vitamin B12: chemistry and biochemistry; pp. 806–810. [DOI] [PubMed] [Google Scholar]
- Kräutler B., Puffer B. 2012. Vitamin B 12 -Derivatives: Organometallic Catalysts, Cofactors and Ligands of Bio-Macromolecules; pp. 131–263. [Google Scholar]
- Ktena Y.P., Ramstad T., Baker E.H., Sloan J.L., Mannes A.J., Manoli I., Venditti C.P. Propofol administration in patients with methylmalonic acidemia and intracellular cobalamin metabolism disorders: a review of theoretical concerns and clinical experiences in 28 patients. J. Inherit. Metab. Dis. 2015;38:847–853. doi: 10.1007/s10545-015-9816-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M., Hirao H., Kozlowski P.M. Co+-H interaction inspired alternate coordination geometries of biologically important cob(I)alamin: possible structural and mechanistic consequences for methyltransferases. J. Biol. Inorg. Chem. 2012;17:1107–1121. doi: 10.1007/s00775-012-0924-x. [DOI] [PubMed] [Google Scholar]
- Kumar M., Kozlowski P.M. A biologically relevant Co1+⋯H bond: possible implications in the protein-induced redox tuning of Co2+/Co 1+ reduction. Angew. Chem. Int. Ed. Engl. 2011;50:8702–8705. doi: 10.1002/anie.201100469. [DOI] [PubMed] [Google Scholar]
- Landau D.J., Brooks E.D., Perez-Pinera P., Amarasekara H., Mefferd A., Li S., Bird A., Gersbach C.A., Koeberl D.D. In vivo zinc finger nuclease-mediated targeted integration of a glucose-6-phosphatase transgene promotes survival in mice with glycogen storage disease Type IA. Mol. Ther. 2016;24:697–706. doi: 10.1038/mt.2016.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W., et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D1067. doi: 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehene M., Plesa D., Ionescu-Zinca S., Iancu S.D., Leopold N., Makarov S.V., Brânzanic A.M.V., Silaghi-Dumitrescu R. Adduct of aquacobalamin with hydrogen peroxide. Inorg. Chem. 2021;60:12681–12684. doi: 10.1021/acs.inorgchem.1c01483. [DOI] [PubMed] [Google Scholar]
- Lemoine M., François A., Grangé S., Rabant M., Châtelet V., Cassiman D., Cornec-Le Gall E., Ambrosetti D., Deschênes G., Benoist J.F., Guerrot D. Cobalamin C deficiency induces a typical histopathological pattern of renal arteriolar and glomerular thrombotic microangiopathy. Kidney Int. Rep. 2018;3:1153–1162. doi: 10.1016/j.ekir.2018.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner-Ellis J.P., Tirone J.C., Pawelek P.D., Doré C., Atkinson J.L., Watkins D., Morel C.F., Fujiwara T.M., Moras E., Hosack A.R., et al. Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type. Nat. Genet. 2006;38:93–100. doi: 10.1038/ng1683. [DOI] [PubMed] [Google Scholar]
- Lerner-Ellis J.P., Anastasio N., Liu J., Coelho D., Suormala T., Stucki M., Loewy A.D., Gurd S., Grundberg E., Morel C.F., et al. Spectrum of mutations in MMACHC, allelic expression, and evidence for genotype-phenotype correlations. Hum. Mutat. 2009;30:1072–1081. doi: 10.1002/humu.21001. [DOI] [PubMed] [Google Scholar]
- Levy H.L., Mudd S.H., Schulman J.D., Dreyfus P.M., Abeles R.H. A derangement in B12 metabolism associated with homocystinemia, cystathioninemia, hypomethioninemia and methylmalonic aciduria. Am. J. Med. 1970;48:390–397. doi: 10.1016/0002-9343(70)90070-7. [DOI] [PubMed] [Google Scholar]
- Lexa D., Saveant J.M. Electrochemistry of vitamin B12. I. Role of the base-on/base-off reaction in the oxidoreduction mechanism of the B12r-B12s system. J. Am. Chem. Soc. 1976;98:2652–2658. doi: 10.1021/ja00425a039. [DOI] [PubMed] [Google Scholar]
- Lexa D., Sayeant J.M., Zickler J. Electrochemistry of vitamin B12. 5. Cyanocobalamins. J. Am. Chem. Soc. 1980;102:2654–2663. doi: 10.1021/ja00528a023. [DOI] [Google Scholar]
- Li H., Haurigot V., Doyon Y., Li T., Wong S.Y., Bhagwat A.S., Malani N., Anguela X.M., Sharma R., Ivanciu L., et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217–221. doi: 10.1038/nature10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Gherasim C., Lesniak N.A., Banerjee R. Glutathione-dependent one-electron transfer reactions catalyzed by a B 12 trafficking protein. J. Biol. Chem. 2014;289:16487–16497. doi: 10.1074/jbc.M114.567339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Lesniak N.A., Banerjee R. Unusual aerobic stabilization of Cob(I)alamin by a B12-trafficking protein allows chemoenzymatic synthesis of organocobalamins. J. Am. Chem. Soc. 2014;136:16108–16111. doi: 10.1021/ja5077316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Shanmuganathan A., Ruetz M., Yamada K., Lesniak N.A., Kräutler B., Brunold T.C., Koutmos M., Banerjee R. Coordination chemistry controls the thiol oxidase activity of the B12-trafficking protein CblC. J. Biol. Chem. 2017;292:9733–9744. doi: 10.1074/jbc.M117.788554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Greenhalgh E.D., Twahir U.T., Kallon A., Ruetz M., Warncke K., Brunold T.C., Banerjee R. Chlorocob(II)alamin formation which enhances the thiol oxidase activity of the B 12 -trafficking protein CblC. Inorg. Chem. 2020;59:16065–16072. doi: 10.1021/acs.inorgchem.0c02653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Mascarenhas R., Twahir U.T., Kallon A., Deb A., Yaw M., Penner-Hahn J., Koutmos M., Warncke K., Banerjee R. An interprotein Co-S coordination complex in the B12-trafficking pathway. J. Am. Chem. Soc. 2020;142:16334–16345. doi: 10.1021/jacs.0c06590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liptak M.D., Brunold T.C. Spectroscopic and computational studies of Co1+cobalamin: spectral and electronic properties of the “superreduced” B 12 cofactor. J. Am. Chem. Soc. 2006;128:9144–9156. doi: 10.1021/ja061433q. [DOI] [PubMed] [Google Scholar]
- Liu M.Y., Yang Y.L., Chang Y.C., Chiang S.H., Lin S.P., Han L.S., Qi Y., Hsiao K.J., Liu T.T. Mutation spectrum of MMACHC in Chinese patients with combined methylmalonic aciduria and homocystinuria. J. Hum. Genet. 2010;55:621–626. doi: 10.1038/jhg.2010.81. [DOI] [PubMed] [Google Scholar]
- Liu J., Tang X., Zhou C., Xu H., Yang H., He R., Li H., Zhao S. Cobalamin C deficiency presenting with diffuse alveolar hemorrhage and pulmonary microangiopathy. Pediatr. Pulmonol. 2020;55:1481–1486. doi: 10.1002/ppul.24781. [DOI] [PubMed] [Google Scholar]
- Løland K.H., Bleie Ø., Blix A.J., Strand E., Ueland P.M., Refsum H., Ebbing M., Nordrehaug J.E., Nygård O. Effect of homocysteine-lowering B vitamin treatment on angiographic progression of Coronary artery disease: a western Norway B vitamin intervention trial (WENBIT) substudy. Am. J. Cardiol. 2010;105:1577–1584. doi: 10.1016/j.amjcard.2010.01.019. [DOI] [PubMed] [Google Scholar]
- Ma F., Shi C.C., Liang P.P., Li S.T., Gu X., Xiao X., Hao H. Construction of a mouse model of cblC type methylmalonic acidemia with W203X mutation based on the CRISPR/Cas9 technology. Chin. J. Contemp. Pediatr. 2019;21:824–829. doi: 10.7499/j.issn.1008-8830.2019.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah W., Deme J.C., Watkins D., Fung S., Janer A., Shoubridge E.A., Rosenblatt D.S., Coulton J.W. Subcellular location of MMACHC and MMADHC, two human proteins central to intracellular vitamin B12 metabolism. Mol. Genet. Metab. 2013;108:112–118. doi: 10.1016/j.ymgme.2012.11.284. [DOI] [PubMed] [Google Scholar]
- Mahoney M.J., Rosenberg L.E., Mudd S.H., Uhlendorf B.W. Defective metabolism of vitamin B12 in fibroblasts from children with methylmalonicaciduria. Biochem. Biophys. Res. Commun. 1971;44:375–381. doi: 10.1016/0006-291x(71)90610-3. [DOI] [PubMed] [Google Scholar]
- Mahoney M.J., Hart A.C., Steen V.D., Rosenberg L.E. Methylmalonicacidemia: biochemical heterogeneity in defects of 5’ deoxyadenosylcobalamin synthesis. Proc. Natl. Acad. Sci. USA. 1975;72:2799–2803. doi: 10.1073/pnas.72.7.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvagia S., Haynes C.A., Grisotto L., Ombrone D., Funghini S., Moretti E., McGreevy K.S., Biggeri A., Guerrini R., Yahyaoui R., et al. Heptadecanoylcarnitine (C17) a novel candidate biomarker for propionic and methylmalonic acidemias during expanded newborn screening. Clin. Chim. Acta. 2015;450:342–348. doi: 10.1016/j.cca.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoli I., Myles J.G., Sloan J.L., Carrillo-Carrasco N., Morava E., Strauss K.A., Morton H., Venditti C.P. A critical reappraisal of dietary practices in methylmalonic acidemia raises concerns about the safety of medical foods. Part 2: cobalamin C deficiency. Genet. Med. 2016;18:396–404. doi: 10.1038/gim.2015.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martí-Carvajal A.J., Solà I., Lathyris D., Salanti G. Homocysteine lowering interventions for preventing cardiovascular events. Cochrane Database Syst. Rev. 2009:CD006612. doi: 10.1002/14651858.CD006612.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascarenhas R., Li Z., Gherasim C., Ruetz M., Banerjee R. The human B12 trafficking protein CblC processes nitrocobalamin. J. Biol. Chem. 2020;295:9630–9640. doi: 10.1074/jbc.ra120.014094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCully K.S. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am. J. Pathol. 1969;56:111–128. [PMC free article] [PubMed] [Google Scholar]
- McCully K.S. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am. J. Pathol. 1969;56:111–128. [PMC free article] [PubMed] [Google Scholar]
- McCully K.S. Hyperhomocysteinemia and arteriosclerosis: historical perspectives. Clin. Chem. Lab. Med. 2005 doi: 10.1515/CCLM.2005.172. [DOI] [PubMed] [Google Scholar]
- McGuire P.J., Parikh A., Diaz G.A. Profiling of oxidative stress in patients with inborn errors of metabolism. Mol. Genet. Metab. 2009;98:173–180. doi: 10.1016/j.ymgme.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellman I., Willard H., Youngdahl-Turner P., Rosenberg L.E. Cobalamin coenzyme synthesis in normal and mutant human fibroblasts. J. Biol. Chem. 1979;254:11847–11853. [PubMed] [Google Scholar]
- Mieda-Higa K., Mamun A.A., Ogura T., Kitagawa T., Kozlowski P.M. Resonance Raman investigation of dithionite-reduced cobalamin. J. Raman Spectrosc. 2020;51:1331–1342. doi: 10.1002/jrs.5909. [DOI] [Google Scholar]
- Morath M.A., Hörster F., Sauer S.W. Renal dysfunction in methylmalonic acidurias: review for the pediatric nephrologist. Pediatr. Nephrol. 2013;28:227–235. doi: 10.1007/s00467-012-2245-2. [DOI] [PubMed] [Google Scholar]
- Moreno-Garcia M.A., Pupavac M., Rosenblatt D.S., Tremblay M.L., Jerome-Majewska L.A. The Mmachc gene is required for pre-implantation embryogenesis in the mouse. Mol. Genet. Metab. 2014;112:198–204. doi: 10.1016/j.ymgme.2014.05.002. [DOI] [PubMed] [Google Scholar]
- Morris A.A.M., Kožich V., Santra S., Andria G., Ben-Omran T.I.M., Chakrapani A.B., Crushell E., Henderson M.J., Hochuli M., Huemer M., et al. Guidelines for the diagnosis and management of cystathionine beta-synthase deficiency. J. Inherit. Metab. Dis. 2017;40:49–74. doi: 10.1007/s10545-016-9979-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudd S.H., Levy H.L., Abeles R.H., Jennedy J.P., Jr. A derangement in B12 metabolism leading to homocystinemia, cystathioninemia and methylmalonic aciduria. Biochem. Biophys. Res. Commun. 1969;35:121–126. doi: 10.1016/0006-291X(69)90491-4. [DOI] [PubMed] [Google Scholar]
- Mudd S.H., Uhlendorf B.W., Hinds K.R., Levy H.L. Deranged B12 metabolism: studies of fibroblasts grown in tissue culture. Biochem. Med. 1970;4:215–239. doi: 10.1016/0006-2944(70)90050-5. [DOI] [PubMed] [Google Scholar]
- Nogueira C., Marcão A., Rocha H., Sousa C., Fonseca H., Valongo C., Vilarinho L. Molecular picture of cobalamin C/D defects before and after newborn screening era. J. Med. Screen. 2017;24:6–11. doi: 10.1177/0969141316641149. [DOI] [PubMed] [Google Scholar]
- Nygaard S., Barzel A., Haft A., Major A., Finegold M., Kay M.A., Grompe M. A universal system to select gene-modified hepatocytes in vivo. Sci. Transl. Med. 2016;8:342ra79. doi: 10.1126/scitranslmed.aad8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olteanu H., Banerjee R. Redundancy in the pathway for redox regulation of mammalian methionine synthase. J. Biol. Chem. 2003;278:38310–38314. doi: 10.1074/jbc.M306282200. [DOI] [PubMed] [Google Scholar]
- Oussalah A., Siblini Y., Hergalant S., Chéry C., Rouyer P., Cavicchi C., Guerrini R., Morange P.-E., Trégouët D., Pupavac M., et al. Epimutations in both the TESK2 and MMACHC promoters in the Epi-cblC inherited disorder of intracellular metabolism of vitamin B12. Clin. Epigenetics. 2022;14:52. doi: 10.1186/s13148-022-01271-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmakumar R., Taoka S., Padmakumar R., Banerjee R. Coenzyme B12 is coordinated by histidine and not dimethylbenzimidazole on methylmalonyl-CoA mutase. J. Am. Chem. Soc. 1995;117:7033–7034. [Google Scholar]
- Pagliarini D.J., Calvo S.E., Chang B., Sheth S.A., Vafai S.B., Ong S.E., Walford G.A., Sugiana C., Boneh A., Chen W.K., et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallares I.G., Brunold T.C. Spectral and electronic properties of nitrosylcobalamin. Inorg. Chem. 2014;53:7676–7691. doi: 10.1021/ic500986x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchabhai T.S., Patil P.D., Riley E.C., Mitchell C.K. When the picture is fragmented: vitamin B12 deficiency masquerading as thrombotic thrombocytopenic purpura. Int. J. Crit. Illn. Inj. Sci. 2016;6:89–92. doi: 10.4103/2229-5151.183026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankowicz F.P., Barzi M., Legras X., Hubert L., Mi T., Tomolonis J.A., Ravishankar M., Sun Q., Yang D., Borowiak M., et al. Reprogramming metabolic pathways in vivo with CRISPR/Cas9 genome editing to treat hereditary tyrosinaemia. Nat. Commun. 2016;7:12642. doi: 10.1038/ncomms12642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J., Jeong J., Kim J. Destabilization of a bovine B 12 trafficking chaperone protein by oxidized form of glutathione. Biochem. Biophys. Res. Commun. 2012;420:547–551. doi: 10.1016/j.bbrc.2012.03.031. [DOI] [PubMed] [Google Scholar]
- Park J., Kim J. Glutathione and vitamin B 12 cooperate in stabilization of a B 12 trafficking chaperone protein. Protein J. 2012;31:158–165. doi: 10.1007/s10930-011-9385-2. [DOI] [PubMed] [Google Scholar]
- Pastore A., Martinelli D., Piemonte F., Tozzi G., Boenzi S., Di Giovamberardino G., Petrillo S., Bertini E., Dionisi-Vici C. Glutathione metabolism in cobalamin deficiency type C (cblC) J. Inherit. Metab. Dis. 2014;37:125–129. doi: 10.1007/s10545-013-9605-3. [DOI] [PubMed] [Google Scholar]
- Pett V.B., Fischer A.E., Dudley G.K., Zacharias D.E. Probing the mechanism of coenzyme B12: synthesis, crystal structures, and molecular modeling of coenzyme B12 model compounds. Comments Mod. Chem. 2002;23:385–400. doi: 10.1080/02603590216080. [DOI] [Google Scholar]
- Pezacka E., Green R., Jacobsen D.W. Glutathionylcobalamin as an intermediate in the formation of cobalamin coenzymes. Biochem. Biophys. Res. Commun. 1990;169:443–450. doi: 10.1016/0006-291X(90)90351-M. [DOI] [PubMed] [Google Scholar]
- Pezacka E.H. Identification and characterization of two enzymes involved in the intracellular metabolism of cobalamin. Cyanocobalamin β-ligand transferase and microsomal cob(III) alamin reductase. Biochim. Biophys. Acta. 1993;1157:167–177. doi: 10.1016/0304-4165(93)90061-C. [DOI] [PubMed] [Google Scholar]
- Pezacka E.H., Jacobsen D.W. Glial cells as a model for the role of cobalamin in the nervous system: impaired synthesis of cobalamin coenzymes in cultured human astrocytes following short-term cobalamin deprivation. Biochem. Biophys. Res. Commun. 1992;184 doi: 10.1016/0006-291x(92)90665-8. [DOI] [PubMed] [Google Scholar]
- Plesa M., Kim J., Paquette S.G., Gagnon H., Ng-Thow-Hing C., Gibbs B.F., Hancock M.A., Rosenblatt D.S., Coulton J.W. Interaction between MMACHC and MMADHC, two human proteins participating in intracellular vitamin B12 metabolism. Mol. Genet. Metab. 2011;102:139–148. doi: 10.1016/j.ymgme.2010.10.011. [DOI] [PubMed] [Google Scholar]
- Pratt J.M. Academic Press Inc. (London) Ltd; 1972. Inorganic Chemistry of Vitamin B12. [Google Scholar]
- Profitlich L., Kirmse B., Wasserstein M.P., Diaz G., Srivastava S. Resolution of cor pulmonale after medical management in a patient with cblC-type methylmalonic aciduria and homocystinuria: a case report. Cases J. 2009;2:8603. doi: 10.4076/1757-1626-2-8603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Profitlich L.E., Kirmse B., Wasserstein M.P., Diaz G.A., Srivastava S. High prevalence of structural heart disease in children with cblC-type methylmalonic aciduria and homocystinuria. Mol. Genet. Metab. 2009;98:344–348. doi: 10.1016/j.ymgme.2009.07.017. [DOI] [PubMed] [Google Scholar]
- Pugina R.A., Denisova E.A., Ivlev P.A., Salnikov D.S., Makarov S.V. Synthesis of vitamin B12 derivatives with sodium hydroxymethanesulfinate. J. Porphyr. Phthalocyanines. 2018;22:1092–1098. doi: 10.1142/S1088424618501092. [DOI] [Google Scholar]
- Pupavac M., Garcia M.A.M., Rosenblatt D.S., Jerome-Majewska L.A. Expression of mmachc and mmadhc during mouse organogenesis. Mol. Genet. Metab. 2011;103:401–405. doi: 10.1016/j.ymgme.2011.04.004. [DOI] [PubMed] [Google Scholar]
- Pupavac M., Watkins D., Petrella F., Fahiminiya S., Janer A., Cheung W., Gingras A.C., Pastinen T., Muenzer J., Majewski J., et al. Inborn error of cobalamin metabolism associated with the intracellular accumulation of transcobalamin-bound cobalamin and mutations in ZNF143, which Codes for a transcriptional activator. Hum. Mutat. 2016;37:976–982. doi: 10.1002/humu.23037. [DOI] [PubMed] [Google Scholar]
- Quadros E.V., Jacobsen D.W. The dynamics of cobalamin utilization in L-1210 mouse leukemia cells: a model of cellular cobalamin metabolism. Biochim. Biophys. Acta. 1995;1244:395–403. doi: 10.1016/0304-4165(95)00037-C. [DOI] [PubMed] [Google Scholar]
- Quintana A.M., Geiger E.A., Achilly N., Rosenblatt D.S., Maclean K.N., Stabler S.P., Artinger K.B., Appel B., Shaikh T.H. Hcfc1b, a zebrafish ortholog of HCFC1, regulates craniofacial development by modulating mmachc expression. Dev. Biol. 2014;396:94–106. doi: 10.1016/j.ydbio.2014.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana A.M., Yu H.C., Brebner A., Pupavac M., Geiger E.A., Watson A., Castro V.L., Cheung W., Chen S.H., Watkins D., et al. Mutations in THAP11 cause an inborn error of cobalamin metabolism and developmental abnormalities. Hum. Mol. Genet. 2017;26:2838–2849. doi: 10.1093/hmg/ddx157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quoos Mayer F., Alfonso Artigalas O., Lizzi Lagranha V., Baldo G., Vanessa Schwartz I., Matte U., Giugliani R. Chloramphenicol enhances IDUA activity on fibroblasts from mucopolysaccharidosis I patients. Curr. Pharm. Biotechnol. 2013;14:194–198. doi: 10.2174/138920113805219467. [DOI] [PubMed] [Google Scholar]
- Randaccio L., Geremia S., Nardin G., Wuerges J. X-ray structural chemistry of cobalamins. Coord. Chem. Rev. 2006;250:1332–1350. doi: 10.1016/j.ccr.2005.12.005. [DOI] [Google Scholar]
- Rao S., Colon Hidalgo D., Doria Medina Sanchez J.A., Navarrete D., Berg S. Et Tu, B12? Cobalamin deficiency masquerading as pseudo-thrombotic microangiopathy. Cureus. 2020;12:e9097. doi: 10.7759/cureus.9097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard E., Jorge-Finnigan A., Garcia-Villoria J., Merinero B., Desviat L.R., Gort L., Briones P., Leal F., Pérez-Cerdá C., Ribes A., et al. MMACHC Working Group Genetic and cellular studies of oxidative stress in methylmalonic aciduria (MMA) cobalamin deficiency type C (cblC) with homocystinuria (MMACHC) Hum. Mutat. 2009;30:1558–1566. doi: 10.1002/humu.21107. [DOI] [PubMed] [Google Scholar]
- Rosenblatt D.S., Aspler A.L., Shevell M.I., Pletcher B.A., Fenton W.A., Seashore M.R. Clinical heterogeneity and prognosis in combined methylmalonic aciduria and homocystinuria (cblC) J. Inherit. Metab. Dis. 1997;20:528–538. doi: 10.1023/A:1005353530303. [DOI] [PubMed] [Google Scholar]
- Roth J.R., Lawrence J.G., Bobik T.A. Cobalamin (coenzyme B12): synthesis and biological significance. Annu. Rev. Microbiol. 1996;50:137–181. doi: 10.1146/annurev.micro.50.1.137. [DOI] [PubMed] [Google Scholar]
- Routh J.K., Koenig S.C. Severe vitamin B12 deficiency mimicking thrombotic thrombocytopenic purpura. Blood. 2014;124:1844. doi: 10.1182/blood-2014-03-562488. [DOI] [PubMed] [Google Scholar]
- Rowley C.A., Kendall M.M. To B 12 or not to B 12 : five questions on the role of cobalamin in host-microbial interactions. PLoS Pathog. 2019;15:e1007479. doi: 10.1371/journal.ppat.1007479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruetz M., Shanmuganathan A., Gherasim C., Karasik A., Salchner R., Kieninger C., Wurst K., Banerjee R., Koutmos M., Kräutler B. Antivitamin B 12 inhibition of the human B 12 -processing enzyme CblC: crystal structure of an inactive ternary complex with glutathione as the Cosubstrate. Angew. Chem. Int. Ed. Engl. 2017;56:7387–7392. doi: 10.1002/anie.201701583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salnikov D.S., Dereven’kov I.A., Artyushina E.N., Makarov S.V. Interaction of cyanocobalamin with sulfur-containing reducing agents in aqueous solutions. Russ. J. Phys. Chem. 2013;87:44–48. doi: 10.1134/S0036024413010226. [DOI] [Google Scholar]
- Salnikov D.S., Makarov S.V., van Eldik R., Kucherenko P.N., Boss G.R. Kinetics and mechanism of the reaction of hydrogen sulfide with diaquacobinamide in aqueous solution. Eur. J. Inorg. Chem. 2014;2014:4123–4133. doi: 10.1002/ejic.201402082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salnikov D.S., Makarov S.V., Koifman O.I. The radical versus ionic mechanisms of reduced cobalamin inactivation by tert-butyl hydroperoxide and hydrogen peroxide in aqueous solution. New J. Chem. 2021;45:535–543. doi: 10.1039/D0NJ04231E. [DOI] [Google Scholar]
- Salnikov D.S., Silaghi-Dumitrescu R., Makarov S.V., van Eldik R., Boss G.R. Cobalamin reduction by dithionite. Evidence for the formation of a six-coordinate cobalamin(II) complex. Dalton Trans. 2011;40:9831–9834. doi: 10.1039/c1dt10219b. [DOI] [PubMed] [Google Scholar]
- Scalais E., Osterheld E., Geron C., Pierron C., Chafai R., Schlesser V., Borde P., Regal L., Laeremans H., van Gassen K.L.I., et al. Parenteral hydroxocobalamin dose intensification in five patients with different types of early onset intracellular cobalamin defects: clinical and biochemical responses. JIMD Rep. 2019;49:70–79. doi: 10.1002/jmd2.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneller J.L., Lee C.M., Bao G., Venditti C.P. Genome editing for inborn errors of metabolism: advancing towards the clinic. BMC Med. 2017;15:43. doi: 10.1186/s12916-017-0798-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scolamiero E., Villani G.R.D., Ingenito L., Pecce R., Albano L., Caterino M., di Girolamo M.G., Di Stefano C., Franzese I., Gallo G., Ruoppolo M. Maternal vitamin B12 deficiency detected in expanded newborn screening. Clin. Biochem. 2014;47:312–317. doi: 10.1016/j.clinbiochem.2014.08.020. [DOI] [PubMed] [Google Scholar]
- Selzer R.R., Rosenblatt D.S., Laxova R., Hogan K. Adverse effect of nitrous oxide in a child with 5, 10-methylenetetrahydrofolate reductase deficiency. N. Engl. J. Med. 2003;349:45–50. doi: 10.1056/NEJMoa021867. [DOI] [PubMed] [Google Scholar]
- Sharma R., Anguela X.M., Doyon Y., Wechsler T., DeKelver R.C., Sproul S., Paschon D.E., Miller J.C., Davidson R.J., Shivak D., et al. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood. 2015;126:1777–1784. doi: 10.1182/blood-2014-12-615492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan J.L., Carrillo N., Adams D., Venditti C.P. Disorders of intracellular cobalamin metabolism. GeneReviews. 2008 [Google Scholar]
- Sloan J.L., Achilly N.P., Arnold M.L., Catlett J.L., Blake T., Bishop K., Jones M., Harper U., English M.A., Anderson S., et al. The vitamin B12 processing enzyme, mmachc, is essential for zebrafish survival, growth and retinal morphology. Hum. Mol. Genet. 2020;29:2109–2123. doi: 10.1093/hmg/ddaa044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C.M., Hayamizu T.F., Finger J.H., Bello S.M., McCright I.J., Xu J., Baldarelli R.M., Beal J.S., Campbell J., Corbani L.E., et al. The mouse gene expression database (GXD): 2019 update. Nucleic Acids Res. 2019;47:D774–D779. doi: 10.1093/nar/gky922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolovskaya O.M., Mok K.C., Park J.D., Tran J.L.A., Quanstrom K.A., Taga M.E. Cofactor selectivity in methylmalonyl coenzyme a mutase, a model cobamide-dependent enzyme. mBio. 2019;10 doi: 10.1128/mBio.01303-19. e01303-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolovskaya O.M., Shelton A.N., Taga M.E. Sharing vitamins: Cobamides unveil microbial interactions. Science. 2020;369:eaba0165. doi: 10.1126/science.aba0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolovskaya O.M., Plessl T., Bailey H., Mackinnon S., Baumgartner M.R., Yue W.W., Froese D.S., Taga M.E. Naturally occurring cobalamin (B12) analogs can function as cofactors for human methylmalonyl-CoA mutase. Biochimie. 2021;183:35–43. doi: 10.1016/j.biochi.2020.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabler S.P., Brass E.P., Marcell P.D., Allen R.H. Inhibition of cobalamin-dependent enzymes by cobalamin analogues in rats. J. Clin. Invest. 1991;87:1422–1430. doi: 10.1172/JCI115148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stich T.A., Yamanishi M., Banerjee R., Brunold T.C. Spectroscopic evidence for the formation of a four-coordinate Co 2+cobalamin species upon binding to the human ATP:cobalamin adenosyltransferase. J. Am. Chem. Soc. 2005;127:7660–7661. doi: 10.1021/ja050546r. [DOI] [PubMed] [Google Scholar]
- Suarez-Moreira E., Hannibal L., Smith C.A., Chavez R.A., Jacobsen D.W., Brasch N.E. A simple, convenient method to synthesize cobalamins: synthesis of homocysteinylcobalamin, N-acetylcysteinylcobalamin, 2-N-acetylamino-2- carbomethoxyethanethiolatocobalamin, sulfitocobalamin and nitrocobalamin. Dalton Trans. 2006:5269–5277. doi: 10.1039/b610158e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanpaiboon P., Sloan J.L., Callahan P.F., McAreavey D., Hart P.S., Lichter-Konecki U., Zand D., Venditti C.P. Noncompaction of the ventricular myocardium and hydrops fetalis in cobalamin C disease. JIMD Rep. 2012;10:33–38. doi: 10.1007/8904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tat J., Heskett K., Satomi S., Pilz R.B., Golomb B.A., Boss G.R. Sodium azide poisoning: a narrative review. Clin. Toxicol. 2021;59:683–697. doi: 10.1080/15563650.2021.1906888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toohey J.I. Possible involvement of hydrosulfide in B12-dependent methyl group transfer. Molecules. 2017;22:E582. doi: 10.3390/molecules22040582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traboulsi E.I., Silva J.C., Geraghty M.T., Maumenee I.H., Valle D., Green W.R. Ocular histopathologic characteristics of cobalamin C type vitamin B12 defect with methylmalonic aciduria and homocystinuria. Am. J. Ophthalmol. 1992;113:269–280. doi: 10.1016/S0002-9394(14)71578-8. [DOI] [PubMed] [Google Scholar]
- Trakadis Y.J., Alfares A., Bodamer O.A., Buyukavci M., Christodoulou J., Connor P., Glamuzina E., Gonzalez-Fernandez F., Bibi H., Echenne B., et al. Update on transcobalamin deficiency: clinical presentation, treatment and outcome. J. Inherit. Metab. Dis. 2014;37:461–473. doi: 10.1007/s10545-013-9664-5. [DOI] [PubMed] [Google Scholar]
- Trefz F.K., Scheible D., Frauendienst-Egger G., Huemer M., Suomala T., Fowler B., Haas D., Baumgartner M.R. Successful intrauterine treatment of a patient with cobalamin C defect. Mol. Genet. Metab. Rep. 2016;6:55–59. doi: 10.1016/j.ymgmr.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumakov S.O., Dereven’kov I.A., Salnikov D.S., Makarov S.V. Kinetics and mechanism of the reaction between aquacobalamin and isoniazid. Russ. J. Phys. Chem. 2017;91:1839–1844. doi: 10.1134/S0036024417100405. [DOI] [Google Scholar]
- Van Hove J.L.K., Van Damme-Lombaerts R., Grünewald S., Peters H., Van Damme B., Fryns J.P., Arnout J., Wevers R., Baumgartner E.R., Fowler B. Cobalamin disorder Cbl-C presenting with late-onset thrombotic microangiopathy. Am. J. Med. Genet. 2002;111:195–201. doi: 10.1002/ajmg.10499. [DOI] [PubMed] [Google Scholar]
- Veer W.L.C., Edelhausen J.H., Wijmenga H.G., Lens J. Vitamin B12. 1. The relation between vitamins B12 and B12b. Biochim. Biophys. Acta. 1950;6:225–228. doi: 10.1016/0006-3002(50)90095-3. [DOI] [PubMed] [Google Scholar]
- Wang C., Li D., Cai F., Zhang X., Xu X., Liu X., Zhang C., Wang D., Liu X., Lin S., et al. Mutation spectrum of MMACHC in Chinese pediatric patients with cobalamin C disease : a case series and literature review. Eur. J. Med. Genet. 2019;62:103713. doi: 10.1016/j.ejmg.2019.103713. [DOI] [PubMed] [Google Scholar]
- Ward M.G., Kariyawasam V.C., Mogan S.B., Patel K.v., Pantelidou M., Sobczyńska-Malefora A., Porté F., Griffin N., Anderson S.H.C., Sanderson J.D., et al. Prevalence and risk factors for functional vitamin B 12 deficiency in patients with Crohn’s disease. Inflamm. Bowel Dis. 2015;21:2839–2847. doi: 10.1097/MIB.0000000000000559. [DOI] [PubMed] [Google Scholar]
- Watkins D., Rosenblatt D.S. Update and new concepts in vitamin responsive disorders of folate transport and metabolism. J. Inherit. Metab. Dis. 2012;35:665–670. doi: 10.1007/s10545-011-9418-1. [DOI] [PubMed] [Google Scholar]
- Watkins D., Rosenblatt D.S. Lessons in biology from patients with inherited disorders of Vitamin B12 and folate metabolism. Biochimie. 2013;126:3–5. doi: 10.1016/j.biochi.2016.05.001. [DOI] [PubMed] [Google Scholar]
- Weisfeld-Adams J.D., Morrissey M.A., Kirmse B.M. Newborn screening and early biochemical follow-up in combined methylmalonic aciduria and homocystinuria, cblC type, and utility of methionine as a secondary screening analyte. Mol. Genet. Metab. 2010;99:116–123. doi: 10.1016/j.ymgme.2009.09.008.Newborn. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler A.G., Schofield W.B., Degnan P.H., Folta-Stogniew E., Barry N.A., Goodman A.L. Human gut bacteroides capture vitamin B12 via cell surface-exposed lipoproteins. Elife. 2018;7:e37138. doi: 10.7554/eLife.37138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead V.M. Acquired and inherited disorders of cobalamin and folate in children. Br. J. Haematol. 2006;134:125–136. doi: 10.1111/j.1365-2141.2006.06133.x. [DOI] [PubMed] [Google Scholar]
- Wingert V., Mukherjee S., Esser A.J., Behringer S., Tanimowo S., Klenzendorf M., Derevenkov I.A., Makarov S.V., Jacobsen D.W., Spiekerkoetter U., Hannibal L. Thiolatocobalamins repair the activity of pathogenic variants of the human cobalamin processing enzyme CblC. Biochimie. 2021;183:108–125. doi: 10.1016/j.biochi.2020.10.006. [DOI] [PubMed] [Google Scholar]
- Wolthers K.R., Scrutton N.S. Electron transfer in human methionine synthase reductase studied by stopped-flow spectrophotometry. Biochemistry. 2004;43:490–500. doi: 10.1021/bi0356303. [DOI] [PubMed] [Google Scholar]
- Wongkittichote P., Wegner D.J., Shinawi M.S. Novel exon-skipping variant disrupting the basic domain of HCFC1 causes intellectual disability without metabolic abnormalities in both male and female patients. J. Hum. Genet. 2021;66:717–724. doi: 10.1038/s10038-020-00892-9. [DOI] [PubMed] [Google Scholar]
- Wuerges J., Garau G., Geremia S., Fedosov S.N., Petersen T.E., Randaccio L. Structural basis for mammalian vitamin B12 transport by transcobalamin. Proc. Natl. Acad. Sci. USA. 2006;103:4386–4391. doi: 10.1073/pnas.0509099103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaghmai R., Kashani A.H., Geraghty M.T., Okoh J., Pomper M., Tangerman A., Wagner C., Stabler S.P., Allen R.H., Mudd S.H., Braverman N. Progressive cerebral edema associated with high methionine levels and betaine therapy in a patient with cystathionine β-synthase (CBS) deficiency. Am. J. Med. Genet. 2002;108:57–63. doi: 10.1002/ajmg.10186. [DOI] [PubMed] [Google Scholar]
- Yang Y., Wang L., Bell P., McMenamin D., He Z., White J., Yu H., Xu C., Morizono H., Musunuru K., et al. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat. Biotechnol. 2016;34:334–338. doi: 10.1038/nbt.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap S. Classical homocystinuria: vascular risk and its prevention. J. Inherit. Metab. Dis. 2003;26:259–265. doi: 10.1023/A:1024497419821. [DOI] [PubMed] [Google Scholar]
- Yin H., Xue W., Chen S., Bogorad R.L., Benedetti E., Grompe M., Koteliansky V., Sharp P.A., Jacks T., Anderson D.G. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 2014;32:551–553. doi: 10.1038/nbt.2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H., Song C.Q., Dorkin J.R., Zhu L.J., Li Y., Wu Q., Park A., Yang J., Suresh S., Bizhanova A., et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol. 2016;34:328–333. doi: 10.1038/nbt.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H.C., Sloan J.L., Scharer G., Brebner A., Quintana A.M., Achilly N.P., Manoli I., Coughlin C.R., Geiger E.A., Schneck U., et al. An X-linked cobalamin disorder caused by mutations in transcriptional coregulator HCFC1. Am. J. Hum. Genet. 2013;93:506–514. doi: 10.1016/j.ajhg.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelder F.H. Specific colorimetric detection of cyanide triggered by a conformational switch in vitamin B12. Inorg. Chem. 2008;47:1264–1266. doi: 10.1021/ic702368b. [DOI] [PubMed] [Google Scholar]
- Zhang X., Chen Q., Song Y., Guo P., Wang Y., Luo S., Zhang Y., Zhou C., Li D., Chen Y. Epimutation of MMACHC compound to a genetic mutation in cblC cases. Mol. Genet. Genomic. Med. 2021:e1625. doi: 10.1002/mgg3.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Cui Y., Han J. Methylmalonic acidemia: current status and research priorities. Intractable and Rare Dis. Res. 2018;7:73–78. doi: 10.5582/irdr.2018.01026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W., Li H., Wang C., Wang X., Gu M. Newborn screening for methylmalonic acidemia in a Chinese population: molecular genetic confirmation and genotype phenotype correlations. Front. Genet. 2019;9:726. doi: 10.3389/fgene.2018.00726. [DOI] [PMC free article] [PubMed] [Google Scholar]