

Abstract
Caloric overconsumption in vertebrates promotes adipose and liver fat accumulation while perturbing the gut microbiome. This triad triggers pattern recognition receptor (PRR)-mediated immune cell signaling and sterile inflammation. Moreover, immune system activation perpetuates metabolic consequences, including the progression of nonalcoholic fatty liver disease (NAFLD) to nonalcoholic hepatic steatohepatitis (NASH). Recent findings show that nutrient overabundance sensing disrupts the activity and homeostasis of the cell’s central energy-generating organelle, the mitochondrion. In parallel, whether caloric excess-initiated PRR signaling and mitochondrial perturbations are coordinated to amplify this inflammatory process in NASH progression remains in question. Here, we hypothesize that altered mitochondrial function, classic PRR signaling, and complement activation in response to nutrient overload, play an integrated role across the immune cell landscape, leading to liver inflammation and NASH progression.
Keywords: cardiometabolic disease, liver, pattern recognition receptors, mitochondria, complement
NAFLD pathophysiology
Sustained caloric excess leads to metabolic syndrome, an umbrella term incorporating at least four pathophysiological conditions including obesity, insulin resistance, hypercholesterolemia, and hypertension. Metabolic syndrome, in turn, increases stroke, cardiovascular disease (CVD), and type 2 diabetes mellitus (T2DM) risk [1]. Nonalcoholic fatty liver disease (NAFLD) is a further manifestation of metabolic syndrome, encompassing initial excessive fat-infiltration into the liver (steatosis), which can progress to reversable nonalcoholic hepatic steatohepatitis (a cardinal feature of NASH), and the potential to progress towards non-reversable liver cirrhosis, increasing the risk of hepatocellular carcinoma (HCC) development [2]. The full spectrum of NAFLD constitutes the most common chronic liver disease and its prevalence is increasing in parallel with the global rise in obesity and metabolic syndrome [3]. Although genetic susceptibility is evident [4], the increasing prevalence implicates environmental triggers, with excess caloric load as a major driver. Under physiologic conditions, hepatocyte lipid deposition functions as a dynamic lipid storage site during eating and fasting, with a concomitant regulated machinery controlling lipid droplet (LD) synthesis and lipolysis [5]. Sustained nutritional overload associated with NAFLD overwhelms these regulatory programs leading to steatosis [6]. Subsequently, hepatocyte functional perturbations and cell death promote the development of systemic and local (chronic) inflammation; here, in parallel with induced liver fibrosis, changes in liver-resident and infiltrative immune cell activity contribute to the progression to NASH (Box 1)—a precursor to exacerbated hepatic and systemic consequences [7]. For the latter, major contributors include an elevated state of activation of the vascular endothelium with concomitant atherosclerosis risk [8] and an overconsumption-driven increase in intestinal permeability leading to gut microbe leakage [9] – both providing additional sources of inflammatory signals.
Box 1. Liver cells and hepatic immune cells.
The mammalian liver comprises multiple cell types that play distinct functional roles depending on their localization around either the portal (intestinal) venous and hepatic arterial vascular supplies to the liver and subsequent drainage through the hepatic venous and hepatobiliary sinusoidal systems [84]. The major hepatic parenchymal cell types linked to the pathophysiology of fatty liver include hepatocytes, the liver sinusoidal endothelial cells (LSECs) and stellate cells (SCs - mediators of hepatic fibrosis) [7]. The liver also harbors a large range of resident immune cells including myeloid cells (e.g. macrophages called Kupffer cells (KCs), and dendritic cells (DCs)), innate T cells (γδ T cells, natural killer T cells (NKT), and mucosal-associated invariant T cells (MAIT)) as well as adaptive αβ T cells and B cells [85]. In the healthy liver, the general immune cell landscape is sustained mostly in an anti-inflammatory and tolerogenic state [7,84]. During the progression to NASH, two principal events occur: first, liver cells and the resident hepatic immune cell repertoire are reshaped towards an activated state and, second, a marked influx of almost all subtypes of immune cells into the inflamed liver can be seen, with myeloid cells dominating recruited immune cells [86]. Together these events sustain the inflammatory environment that promotes liver injury. Although various immune cells affect different aspects of the pathogenesis of NAFLD, we currently know most about the contributions of myeloid cells (and specifically of neutrophils, monocytes, and macrophages) and conventional T cells to liver disease [7,84,86]. We therefore focus here with on potential novel nutrient-PRR-mitochondrial axes in these latter immune cell types.
In the healthy mammalian liver, immune cells generally have a ‘resting’ or tolerogenic phenotype [10]. However, during the transition to NASH, danger signals and proinflammatory mediators released by injured and/or dying hepatocytes, reshape the molecular signatures of resident immune and liver parenchymal cells via pattern recognition receptor (PRR) engagement towards proinflammatory phenotypes [11,12]. This, in combination with monocyte and neutrophil infiltration from the periphery, further augments liver dysfunction and fibrosis [13] (Figure 1). Most studies assess singular molecular pathways contributing to liver disease in isolation. In this opinion piece, we argue that there is a significant – and recently recognized – interplay between nutritional overload, PRR-mediated immune cell signaling, and the control of inflammatory cell programs that contribute to the progression of benign NAFLD to NASH. Further, we posit that as a canonical nutrient-sensing intracellular organelle, the mitochondrion constitutes a central node initiating these PRR-inflammatory programs. In addition, we suggest that the unexpected activities of intracellular complement may be an important constituent of metabolic and mitochondrial changes associated with NASH. Identifying novel mechanistic networks underlying NASH may uncover putative targets for urgently needed therapeutic interventions. Here, we provide a succinct overview of the known contributions of nutrient overload on mitochondria and subsequent engagement of classic PRRs such as toll-like receptors (TLRs), inflammasomes, and cyclic GMP-AMP synthase stimulator of interferon genes (cGAS-STING) pathways in NAFLD progression. We discuss recently discovered intracellular complement activities and how these may intersect with canonical hepatic PRR systems and mitochondrial perturbations during the progression to steatohepatitis. We also highlight potential new NASH/NAFLD pharmacological interventions targeting the proposed nutrient-PRR-mitochondrial axes, and assess limitations of this model as well as key outstanding questions.
Figure 1. Liver and immune cell contributions to NAFLD.
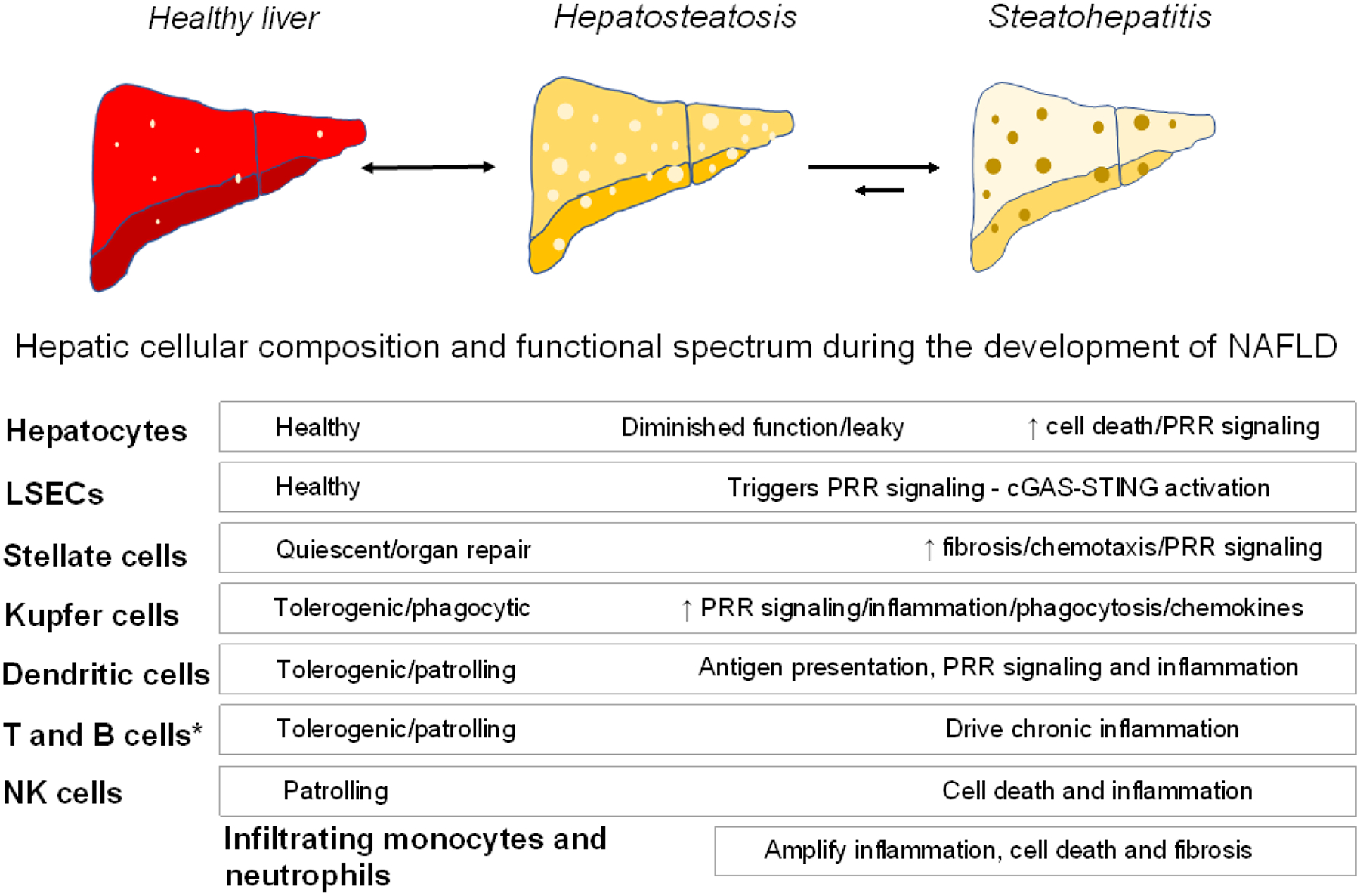
Shown is a schematic of various cell populations in the mammalian liver with their functional contributions to maintaining a healthy organ and their presumed role in NAFLD transitions from hepatosteatosis to steatohepatitis [7]. Hepatosteatosis has a high potential to revert to the healthy state with reversal of risk factors (indicated by a double-sided arrow). The capacity to reverse NASH is much more limited (indicated by a shorter arrow) [96]. The bulk of the pathophysiologic effects of the various cell types signal the transition from hepatosteatosis to steatohepatitis. LSECs, liver sinusoidal epithelial cells. *, includes αβ CD4+ and CD8+ T cells (resident memory T cell subset), γδ T cells, invariant natural killer T cells (iNKT cells), mucosal-associated invariant T cells (MAIT), innate lymphoid cells (ILCs), and B cell plasma cells [84].
Mitochondrial perturbations and NAFLD progression
One principal function of the liver, and of hepatocytes, is the synthesis and storage of triglycerides (TG) in LDs [6]. It is therefore not surprising that this process is augmented in response to chronic nutritional overload. The accumulation of LDs themselves do not induce injury or inflammation, as lipid encapsulation in the form of TGs neutralizes lipotoxic effects [6]. However, associated perturbations of mitochondria, as nutrient-sensing intracellular organelles, can trigger retrograde signaling, often via intracellular PRR engagement, to both initiate and amplify inflammation [14].
Mitochondria are highly abundant in hepatocytes and function in part, to supply energy for the liver, provide metabolic intermediates for cholesterol, steroid, bile acid, lipid, and glycogen synthesis, as well as for producing ketone bodies [15]. NAFLD and NASH show progressive histological and biochemical perturbations in mitochondria, as evidenced by mitochondrial swelling, cristae disruption, and impaired mitochondrial oxidative phosphorylation [16]. Of note, in obese human subjects, the loss of mitochondrial respiratory capacity and the increase in hepatic mitochondrial mass correlate with the transition from NAFLD to NASH [17]. The mechanism whereby perturbed mitochondrial fidelity promotes inflammation is driven in part by the evolutionary formation of these organelles as vestiges of their bacterial origins; indeed eukaryotic mitochondria retain numerous prokaryotic molecules, including hypomethylated mitochondrial DNA (mtDNA), formyl peptides, and cardiolipin [18]. Leakage or extrusion of these components through the outer mitochondrial membrane, are recognized as damage associated molecular patterns (DAMPs) [19]. Hence, disruption of mitochondrial integrity triggers numerous PRR immune surveillance programs, including the intracellular NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome and STING-interferon-regulatory factor 3 (IRF3) pathways, as well as extracellular TLR9-initiated signaling. Perturbed mitochondrial fidelity can also increase reactive oxygen species (ROS) production, another strong driver of inflammasome activation and cell injury and death [20,21]. We propose that the crosstalk between liver parenchymal cells and resident and infiltrating immune cells can contribute to the progression of NAFLD to steatohepatitis (NASH). Moreover, given emerging evidence on mitochondrial dysfunction-initiated pathways, and considerable evidence supporting PRR signaling triggered by nutrient overload in the contribution to NASH progression, we discuss such evidence and highlight areas that require further study to demonstrate our hypothesis of an integrated module of nutrient overload, mitochondrial dysfunction, and PRR activation driving liver inflammation.
Mitochondrial infidelity-driven PRR engagement and inflammation in NAFLD
One hypothetical potential consequence of NAFLD-associated enlarged mitochondria, termed megamitochondria, is that these become resistant to engulfment by autophagosomes, and thereby preclude recycling/repair via selective mitophagy [22]. This concept was explored in a methionine-choline-deficient dietary mouse model of NAFLD, where mitochondrial fusion was impaired by anti-sense knockdown of the fusion protein optic atrophy 1 (OPA1) [22]. Preventing megamitochondria formation, as assessed by confocal immunofluorescence of liver sections, inhibited the development of steatosis and reversed established steatosis [22], supporting the concept that disruption of mitochondrial quality control programs (mitochondrial fidelity) can contribute to disease progression. In parallel, attenuated mitochondrial fusion via OPA1 depletion blunted hepatic inflammation and fibrosis. Although mitochondrial integrity and specific inflammatory programs were not explored in this OPA1 knockdown model, the observed downregulation of the IL1B gene transcript encoding interleukin IL-1β following inhibition of mitochondrial fusion, implicates the NLRP3 inflammasome in this mechanism, given that IL-1β is a canonical inflammasome-induced cytokine [22]. Because autophagy processes (and their different specific iterations, e.g. mitophagy or lipophagy) are key mechanisms for removing intrinsic danger molecules across cells, it is not surprising that mitophagy impairment in the liver, in the context of NAFLD associated mitochondrial infidelity, might contribute to inflammation in the transition to NASH (Box 2).
Box 2. Autophagic pathways linked to NAFLD and inflammation.
Autophagy (self-digestion) is a general homeostatic catabolic process to recycle intracellular content and organelles that also removes intracellular danger components (pathogens, damaged organelles, unfolded proteins, etc.) targeting them to lysosomes for destruction. Autophagosome and lysosome biology in the liver can play myriad roles, including the quality control of mitochondria (mitophagy), the catabolism of lipid stores (autophagy, selective lipophagy, and lysosomal lipolysis). These processes provide essential building blocks for liver function and more globally promote, in a context-specific manner both pro- and anti-inflammatory effects [87]. During liver inflammation, the accumulation of lipids, associated lipotoxicity, and compromised mitochondria across parenchymal and immune cells engages autophagy in mammalian cells to combat these disturbances and reinstate homeostasis [88]. Although the underlying molecular events are not well defined, this beneficial autophagic flux is gradually reduced during the progression to NASH, consequently leading to accumulation of DAMPs with further mitochondrial disruption and reduced cell viability [89–91]. Similarly, DAMP-driven PRR activation can have a negative impact on autophagy, for example in mouse oxidant stressed cells, by reducing the expression of mitophagy activators [92]. Furthermore, NAFLD is generally associated with changes in autophagic, and specifically lipophagic machinery across the spectrum of parenchymal and immune cells in patients, and in several murine models of HFD-induced NAFLD [91]. Although targeting autophagy in NAFLD/NASH might be attractive (based on the central role of autophagy at the PRR-mitochondrial interface), this approach needs to be evaluated carefully because autophagy also serves important homeostatic and beneficial activities in the mammalian liver, such as the removal of compromised and non-functional mitochondria in the healthy liver and during distinct phases of NAFLD/NASH [93,94].
In primary hepatocytes and Kupffer cells (KCs), and in the intact murine liver, NLRP3 inflammasome activation is evident in response to elevated dietary fat and glucose induced NAFLD [23,24]. Further, genetic induction of murine hepatocyte pyroptosis and primary human NAFLD hepatocytes demonstrate inflammasome protein extrusion with subsequent internalization into stellate cells (SC), resulting in accentuated IL-1β driven inflammation and collagen formation— two cardinal features of NASH [25]. In a 60% high fat diet (HFD) mouse model, progression to NASH was linked to impaired liver mitochondrial turnover in parallel with primary hepatocyte NLRP3 activation [26]. We speculate that these mechanisms are likely operational in humans as the expression of NLRP3 markers such as caspase-1 were higher in serum, and the expression of transcripts encoding IL-1β were increased in frozen liver sections from patients with NASH, where the elevation of markers paralleled increased disease severity [25,27]. Similarly, primary human SCs showed internalization of extracellular NLRP3 inflammasome oligomers, further supporting the presumed role of the inflammasome in NASH [25]. Moreover, in experimental NAFLD mouse models, pharmacologic NLRP3 inhibition with small molecule inhibitors, sulforaphane and MCC950, alleviated NASH progression [28,29]; and germline genetic depletion of NLRP3 prevented the development of fibrosis (NASH) in an experimental mouse hepatic steatosis model [27]. Although, these studies do not directly link NLRP3-induction with mitochondrial dysfunction, NLRP3-inhibition in primary murine hepatocytes exposed to fatty acids (FAs), improved mitochondrial bioenergetics and reduced mitochondrial ROS production, supporting the integration of mitochondrial function with NRLP3 activity [28]. In parallel, blunting mitochondrial lipid metabolism via inhibition of the mitochondrial citrate carrier Slc25a1 prevented the evolution of steatohepatitis in a murine HFD model of NAFLD [30]. However, in this study, NLRP3 activity was not directly measured. Taken together, these studies appear to correlate mitochondrial dysfunction and NLRP3 inflammasome activation with the progression to NASH. However, the definitive link integrating mitochondrial dysfunction and the NLRP3 inflammasome in NASH remains to be established. In other tissue types, this link is stronger; for example, in severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) induced lung inflammation in wild type mice, mitochondrial stress in pulmonary macrophages promotes mtDNA oxidation, fragmentation, and cytoplasmic extrusion which, in turn, activates the NLRP3 inflammasome [31]. Furthermore, in human monocytes, mtDNA extrusion linked to NLRP3 inflammasome activation is evident in the pro-inflammatory milieu of refeeding following prolonged fasting [32]. We model how this might be operational in different cell types in the liver with NASH, as schematized in Figure 2.
Figure 2. Model of mammalian cell mitochondrial-PRR crosstalk perturbations in NAFLD/NASH.

In mammals, high fat diet-induced mitochondrial perturbations include changes in morphology (fission/fusion), function (metabolism), fidelity and integrity (mtDNA release). These in turn, engage cell-intrinsic PRRs, which cumulatively induce pro-inflammatory cytokine production, release of DAMPs, and cell death (left side – see text for details) [7]. Nutrient overload–mediated mitochondrial dysfunction is most prominent in hepatocytes but is operational to various degrees across all liver cells [7]. The exogenous effects of cell-intrinsic PRR-driven cell activation/death (for example, in hepatocytes) on other cells is depicted on the right side in red underneath each cell type in a simplified fashion (e.g., IL-1β production and NLRP3 release by hepatocytes undergoing pyroptosis induce TGF-β in stellate cells, which supports fibrosis). Mitochondrial dysfunction and specific PRR engagement in specialized liver cells are listed within each cell type and known cross-communications between cells are indicated by red-arrows. The consequences of these inflammatory pathways emanating from different cell types comprise various components of inflammatory signaling during the progression to NASH [84]. *Of the broad range of resident and incoming immune cells with mitochondrial and PRR activation hallmarks, monocytes and macrophages are the best understood. cGAS-STING, cyclic GMP-AMP synthase and stimulator of interferon genes pathway; ECV, extracellular vesicle; FFAs, free fatty acids; Inflammas., inflammasomes; LDs, lipid droplets; LSEC; liver sinusoidal endothelial cell; Mito-Dysf., mitochondrial dysfunction; mtDNA, mitochondrial DNA; Oxphos; oxidative phosphorylation; ROS, reactive oxygen species; TLR9, toll-like receptor 9. Some icons/cell types were imported from BioRender (https://biorender.com/).
Like the NLRP3 inflammasome, the cGAS-STING pathway is an intracellular immune surveillance pathway that recognizes aberrant host DNA localized in the cytosol, including mtDNA (Figure 2). Immunohistochemical staining shows that STING expression is induced in non-parenchymal liver cells, including KCs, infiltrating macrophages, and liver sinusoidal endothelial cells (LSECs) from patients with NAFLD compared to controls [33]. In a murine HFD model, cGAS-STING was activated in adipose tissue in response to mtDNA release [34]. Furthermore, mtDNA extracted from HFD-exposed primary mouse hepatocytes activated STING in cultured KC, promoting nuclear factor κB (NF-κB)-mediated inflammation [35]. These data support the concept that STING functions as an mtDNA sensor in liver KC cells in response to lipid overload [35]. Consistent with in vitro culture-based studies, germline and myeloid cell-specific STING knockout (KO) mice (Tmem173 (STINGgt)) exposed to a HFD developed less severe hepatic steatosis, inflammation, and fibrosis when compared with wildtype controls [33]. In addition, the crosstalk between STING activation and hepatic function was reported in models where in HFD fed mouse livers, or upon free fatty acid (FFA) incubation of human hepatocyte-derived L-O2 cells, STING and IRF3 were activated, whereas genetic knockdown of these transcription factors in L-O2 cells reduced inflammation and apoptosis [36]. Of note, the integration of STING-IRF3 with hepatocyte metabolic remodeling was reinforced when concomitant knockdown of STING and IRF3 induced glycogen storage and alleviated lipid accumulation in L-O2 cells; this suggested a crosstalk between inflammatory signaling and glucose and lipid metabolism in hepatocytes [36]. However, similarly to the NLRP3 data, the direct link between mitochondrial DAMPs, cGAS-STING signaling, and NASH has not been definitively determined. Nevertheless, the data presented above suggest that the cGAS-STING immunosurveillance program is primarily activated in non-parenchymal and infiltrating macrophage cells and that the putative crosstalk between hepatocytes and non-parenchymal cells might exacerbate this inflammatory program [33]. Finally, the proposed role for cGAS-STING immunosurveillance in this context supports the notion that inflammatory signaling might exacerbate hepatocyte metabolic perturbations, and eventually contribute to the pathophysiology of NASH, although this still warrants robust investigation [36]. Thus, further research into targeting the inhibition of cGAS-STING might add to the armamentarium of approaches aiming to alleviate NASH progression.
From another angle, TLR9, an endosomal PRR for which CpG-rich bacterial DNA and mammalian self-DNA are ligands, is also operational in NASH [14]. Here, the release of intact hepatocyte derived microvesicles into the plasma in NASH patients and from HFD fed mice, have been shown to contain oxidized mtDNA that triggers TLR9 inflammation [14]. The removal of these microvesicles or TLR9 antagonists blocked murine NASH development [14]. In genetic mouse models, germline TLR9 knockout (KO) mice similarly resisted diet-induced NASH compared to wildtype controls. Here, global Tlr9–/– KO animals showed impaired Type I interferon (IFN) signaling in the liver, and hepatic KCs exhibited reduced TLR9 activation (with decreased IL-1β production) [37]. Moreover, genetic ablation of the IL-1 receptor Il1r–/– or MyD88 Myd88–/– (an adaptor molecule for TLR9 and IL-1R signaling, also showed reduced steatohepatitis and fibrosis in response to diet-induced NASH compared to wildtype control mice [37]. Moreover, recent experiments using macrophage-specific conditional MyD88 KO mice (Myd88f/f Lyz2-Cre) indicated that these mice were protected against HFD-mediated hepatic injury, lipid accumulation, and fibrosis [37b]. Together, these findings suggested mechanistic links between mitochondrial DAMPS and inflammation in hepatic injury and NASH.
We posit that collectively, such evidence supports a model where different PRRs –induced by nutrient overload with subsequent mitochondrial perturbations – can play a coordinated role in initiating and amplifying liver inflammation (Figure 2). Moreover, based on the current body of published data, se argue that the transfer of mitochondrial DAMPs (either via mitochondrial surface changes or leakage) or via PRR molecule transfer from hepatic parenchymal cells to resident immune cells, or between resident non-parenchymal cell types, might perpetuate inflammation and fibrosis - two key features of NAFLD to NASH progression. We suggest that the chronic activation of such PRR-initiated pathways might exacerbate the metabolic milieu linked to NAFLD and sustain a vicious cycle of positive feedback loops in the nutrient-overload-induced PRR-mitochondrial axis, although this remains to be rigorously demonstrated. Also, at the onset and progression of NASH, additionally/alternatively engaged and/or dysregulated physiological pathways, such as the endoplasmic reticulum (ER) stress response and DNA damage repair pathways could contribute to NAFLD pathology [38,39] and might also intersect at some point with such PRR-mitochondrial crosstalk, which also warrants further investigation.
Nutrient overload, the Complement system, and NAFLD
From another angle, there is an interesting association between nutrient overload, mitochondrial infidelity, and activation of one of the oldest PRRs, the complement system (Box 3), and NAFLD.
Box 3. Complement activation and classic key functions.
The complement system is evolutionary the oldest component of mammalian innate immunity [95]. It comprises over 50 different proteins operational in circulation, in extracellular spaces, within cells, organelles, and on cell membranes [44]. Complement components are categorized into four groups based on their activity: PRR proteins sensing pathogenic or self-derived noxious antigens, effector molecules (mostly activation fragments of the core complement components C3, C4, and C5) driving antigen opsonization and anaphylatoxin generation, activation fragment receptors recruiting and stimulating innate and adaptive immune cells, and regulators preventing unwanted or uncontrolled detrimental complement activation [95]. Danger sensing by a complement PRR component, such as C1q or ficolin, triggers the activation of one or several complement activation pathways, the lectin pathway (LP), the classical pathway (CP), and/or the alternative pathway (AP). This culminates in the proteolytic activation of C3 and C5, the induction of the general inflammatory reaction, and ultimately, the formation of the lytic membrane attack complex (MAC) to directly lyse pathogens [41,95]. The C3 and C5 fragments generated during complement activation (C3a and C3b/iC3b/C3dg, C5a and C5b) can engage respective receptors on immune and structural cells and induce signaling events that control cell activation states and (effector) functions in a cell-specific and often temporal manner [95]. Due to complement’s multifaceted roles in both the induction and resolution phases of the immune response, perturbations in its normal functions are associated with a wide range of human diseases [41].
An integral connection between the complement system and the liver has long been recognized because the liver secretes the majority of complement components circulating in the blood [40]. C3 and C5 are also considered acute-phase proteins and are produced and released in response to a wide array of liver insults [41]. Chronic nutrient overload similarly promotes hepatocyte C3 secretion in mice fed a HFD. Indeed, clinical and epidemiological studies have shown that patient C3 concentrations assessed in blood samples and liver biopsies were increased in NAFLD compared to healthy individuals, and correlated with NAFLD severity [42,43] (Figure 3).
Figure 3. Proposed model of human complosome-PRR signaling axes in mitochondrial function.
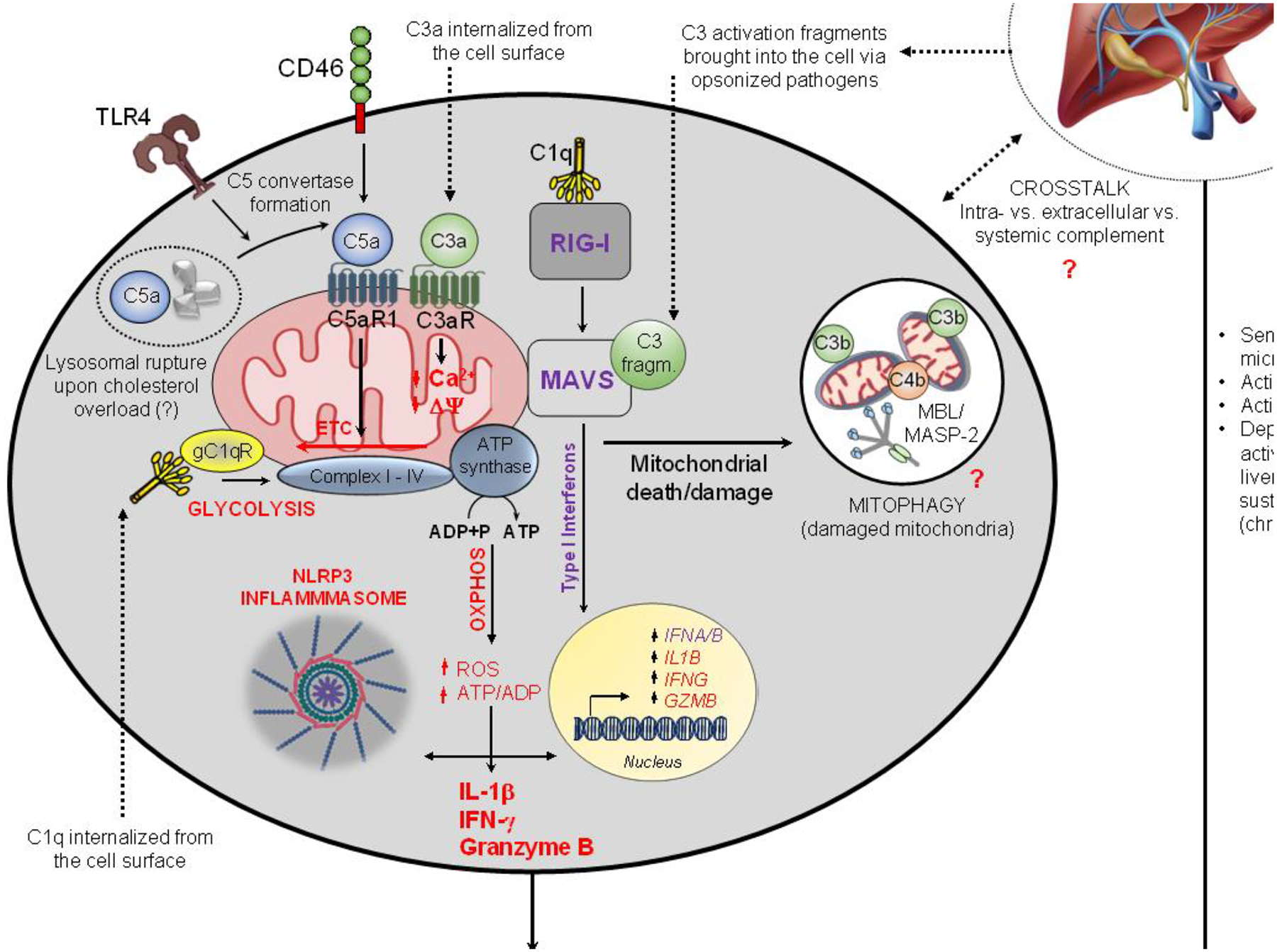
In this model, circulating and intracellular complement contribute to the amplification of inflammation seen in NAFLD [95]. Liver-derived complement, activated by microbial components released from compromised intestinal barriers, can aggravate human disease by sustaining vasculature and hepatocyte activation [97]. Intracellularly active complement can engage the mitochondrial C5aR1 which increases ROS production, reverses direction of the ETC and induces NLRP3 inflammasome assembly and IL-1β production [65,71]. TLR4 fosters cell-autonomous C5a generation via increased C3/C5 convertase formation and human monocytes can also activate intracellular C4 [65]. The RIG-I/MAVS axis is activated by cell-intrinsic C1q and C3 activation fragments introduced via opsonized pathogens and triggers type I IFN production [68]. Internalized C1q and C3a impact on ROS production and mitochondrial membrane potential, respectively, which compromise mitochondrial integrity and overall sustain cell-intrinsic DAMP presence [70,72]. Complosome-mediated mitophagy may mitigate this process by removal of injured mitochondria. The shown complosome perturbations can contribute to pathological cell changes in infection and sterile inflammation [65,98]; whether they also operate in liver cells and/or contribute to NASH remains to be assessed. ADP, adenosine diphosphate; ATP, adenosine triphosphate; CTL, cytotoxic T lymphocyte; gC1qR, globular-head C1q receptor; MAVS, mitochondrial anti-viral signaling protein; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3 inflammasome; Oxphos, oxidative phosphorylation; RIG-I, retinoic-inducible gene I; ROS, reactive oxygen species; Th1, T helper type 1 cell; TLR4, toll-like receptor 4. Some icons were imported from BioRender (https://biorender.com/).
The potential contributions of complement in the progression to NASH are rooted in the ability of complement to detect microbes released by overnutrition-induced gut microbiome dysbiosis and impaired intestinal integrity, subsequently contributing to a general state of (hepatic) inflammation [44]. For example, increased anaphylatoxin C3a and C5a production engages the respective C3aR and C5aRs expressed on the liver vasculature, promoting pro-inflammatory and pro-coagulant signals in mice and humans [45,46]. The complement-stimulated vascular endothelium also facilitates immune cell extravasation into an inflamed liver which, in turn, further aggravates disease progression [44]. The latter is aided by the formation of the membrane attack complex (MAC) (Box 3) on steatotic hepatocytes, which display complement-activating DAMPs [47]. Importantly, C3 plays a central, albeit not yet fully understood, role in the control of lipid metabolism [48,49]. For example, in mice, circulating C3 has negatively controlled serum triglyceride concentrations and counteracted Western diet-induced hyperlipidemia and atherosclerosis [50]. In contrast, inhibition of C3aR via a specific antagonist in a diet-induced obesity male Wistar rat model, attenuated metabolic dysfunction by normalizing FA uptake vs. lipolysis, compared with vehicle treated controls [51]. These data implicate complement proteins in lipid metabolism pathways, which might also contribute to NAFLD [7].
At the same time, suggesting that complement components increase NAFLD via liver local tissue inflammation is probably too simplistic given that complement also contributes to liver repair [52]. In fact, complement has emerged as a central player in general tissue homeostasis and regeneration [41,53]. Thus, augmented hepatic complement production during NAFLD could also reflect repair and rescue pathways [54]. These potential divergent spatial and temporal aspects of complement function in NAFLD therefore require further investigation.
An additional layer of complexity exists with the discovery of the complosome, which refers to the intracellular cell-autonomous and and/or cell-intrinsic complement system [55]. Functionally, the complosome can modulate immune cell functions (Figure 3), as epitomized by intracellular human CD4+ T cell-generated C3a, which supports tonic mammalian target of rapamycin (mTOR) activation to sustain T cell survival [56]. Furthermore, T cell receptor (TCR) stimulation promotes intracellular C3b translocation to the cell surface to engage the C3b receptor, CD46, which in turn can stimulate the expression of genes encoding nutrient transporters, foster mTORC1 assembly on lysosomes, and increase glycolysis and oxidative phosphorylation [56]. These events drive IFN-γ secretion and T helper cell type 1 (Th1) induction [57]. Also, in isolated, activated, human CD8+ T cells in vitro, the complosome/CD46 increased FA metabolism and amplified cytolytic protective immunity, as evidenced from augmented FA synthase activity and granzyme B production [58]. In line with an important role of complement in tissue homeostasis (see above), the complosome can also control timely Th1 cell number contraction (which prevents tissue pathology) by mediating the co-expression of Th1-derived anti-inflammatory IL-10; this is achieved by complosome-instructed metabolic reprogramming [59], in particular by inducing Th1 cell cholesterol efflux [60]. Moreover, augmented expression and activity of C3 and C5 in human CD4+ T cells can contribute to Th1 hyper-responses in patients suffering from autoimmune manifestations, increasing the production of pro-inflammatory IFNγ and IL-1β [61,62]. Further, dysregulation of the complosome is being increasingly linked to other human diseases such as atherosclerosis, rheumatoid arthritis, and clear cell renal cell carcinoma [63–65]. Although NAFLD pathology is exacerbated by infiltrating/circulating CD4+ Th1 and CD8+ T cells [66], whether intracellular complosome signaling plays a role in hepatosteatosis remains uncharacterized.
Complosome activity also directly engages other intracellular PRRs, such as TLRs, the NLRP3 inflammasome, the retinoic acid-inducible gene I (RIG-I) system, and the mitochondrial antiviral-signaling protein (MAVS) (Figure 3) [67–69]. A more direct impact of intracellular complement activation on mitochondrial activity has recently been uncovered where not only is C3aR and C5aR expression evident on the outer mitochondrial membrane, but there is also intra-organelle signaling upon engagement with respective anaphylatoxin ligands [61,65]. Here, in oxidatively-stressed human retinal epithelial cells (RPEs), surface C3aR translocates into mitochondria, induces mitochondrial Ca2+ uptake, and reduces mitochondrial respiration and bioenergetics, resulting in RPE cell death [70]. Similarly, intracellular engagement of C5aR1 in human CD4+ T cells drives mitochondrial ROS production [71]. The mechanism of C5aR1 action on mitochondria was recently delineated in human monocytes and macrophages in vitro : the pertussis toxin could block these events, implicating Gαi-dependent signaling [65]. Moreover, mitochondrial C5aR1 (mtC5aR1) reduced phosphorylation of mitochondrial extracellular signal-regulated kinase (ERK) 1/2, reversed electron transport chain (ETC) flow, and redirected mitochondria away from ATP and towards ROS production (Figure 3) [65]. In these cells, surface TLR4 binding to oxidized low-density lipoprotein (oxLDL) or lysosomal rupture resulting from cholesterol overload (visualized cholesterol crystal accumulation), increased intracellular C5a activation which led to mtC5aR1 engagement and IL-1β release [65]. Heightened mitochondrial C5aR1 activity accompanied atherosclerosis progression, showing immune cell activation in arterial plaques isolated from patients with cardiovascular disease [65]. These observations demonstrate that cholesterol/nutrient overload can activate mtC5aR1 and presumably, this pathway might also be engaged in NASH. Additionally, intracellular C1q has been reported to engage the globular head C1qR (gC1qR) expressed on mitochondria in isolated mouse and human CD8+ T cells, also impacting glycolysis vs. ATP balance (via Seahorse); overall, C1q restrained CD8+ T cell activation and was protective in a mouse model of systemic lupus erythematosus [72]. Further, C1q drove mitochondrial ROS production in neurons of neonatal mice exposed to hypoxic-ischemic brain injury [73]; also, gC1qR stimulation engaged cell-preserving mitochondrial responses, such as inhibition of the mitochondrial permeability transition pore - a central controller of mitochondrial fitness [74]. Given the direct impact of complosome components on various aspects of mitochondrial function and fidelity, the complosome has emerged as an important upstream activator of canonical PRRs (Figure 3) [75]. For example, ROS-driven NLRP3 assembly and subsequent IL-1β productìon by human CD4+ T cells and macrophages is dependent on mtC5aR1 activity [65]. This was demonstrated using in vitro activation of purified human CD4+ T cells and macrophages in which C5aR1 activity was prevented either via a cell-permeable C5aR1 inhibitor, by knockdown of C5aR1 expression via RNA silencing, or by using myeloid-specific C5aR1-deficient mice (LyzMcre-C5ar1fl/fl) [65]. Human and mouse T cells or macrophages that could not engage C5aR1 normally, exhibited reduced NLRP3 activation and intrinsic IL-1β production compared to wildtype C5aR1 cells. This translated into reduced Th1 cell activity in a mouse model of T cell transfer-induced colitis and reduced the disease score in an HFD-fed atherosclerosis mouse model [65,71]. Similarly, the α-chain of the C1q molecule directly interacts with members of the RIG-1 pathway in co-immunoprecipitation experiments using human kidney HEK293T cells and enhances RIG-I and TANK-binding kinase 1 (TBK1)-driven activation of the IFNB gene promoter in these cells when probed via in vitro luciferase reporter assays [76]. Further, C3 activation fragments introduced into cultured HEK293T cells via C3-opsonized adenovirus in vitro, directly activate an important RIG-I system downstream component, MAVS; intracellular C3-driven MAVS activation induced subsequent type I IFN signaling and proinflammatory cytokine secretion, restricting virus titers in in vitro infected HEK293T cells (Figure 3) [68]. Of note, and in line with these dichotomous roles, the complosome can contribute to cell homeostasis by playing a central role in autophagy and possibly, mitophagy (Box 2) (Figure 3).
Thus, the newly described complosome-metabolism-PRR axis is emerging as an integral part of normal cellular responses to infection or changes in cell homeostasis, but which can also contribute to inflammatory diseases when perturbed or sustained. Because mitochondria and PRRs can contribute to the transition to NASH, we argue that the complosome may likely be involved in this process, although this remains to be tested [77]. Specifically, we hypothesize that the involvement of cell-autonomous complement in the control of FA synthesis and cholesterol flux, in combination with the accumulation of cholesterol in cells can change structural liver cells and/or liver immune cell activities. Of note, there is emerging support for our hypothesis as a recent study found that global C3–/– mice displayed spontaneous accelerated TG accumulation in hepatocytes when compared to wild type mice [78]. C3-deficient mouse hepatocytes exhibited reduced lipophagy compared to controls, given that intracellular C3 normally controls autolysosome assembly via its interaction with autophagy-related 16 like 1 (ATG16L1) [78]. However, it is clear that a potential role for the complosome in NAFLD needs to be formally explored and – given the broad presence of the complosome across cell types – it will be important to assess complosome activity in liver parenchymal cells as well as in resident and incoming immune cells, and probing for any putative contributions to NASH pathology.
Targeting the PRR-mitochondrial dysfunction axis to ameliorate NAFLD
Current therapeutics options for NAFLD in general, and for preventing NASH progression, remain limited. Major interventions include lifestyle modifications to lose weight and/or bariatric surgery. In patients with diabetes, anti-diabetic agents may have some benefit, especially those that promote weight loss and/or rely on the peroxisome proliferator activated receptor γ (PPARγ agonist pioglitazone to reduce adipogenesis. However, once inflammation and fibrosis set in, only vitamin E, with potential antioxidant effects has been considered to provide modest benefit [79]. Based on our model of PRR-signaling and mitochondrial perturbation crosstalk, targeting mitochondrial fidelity and/or function, or PRR pathways directly, might represent an additional putative therapeutic approach to investigate. For example, an emerging strategy to improve mitochondrial fidelity and function, linked to blunting inflammation is to boost intracellular nicotinamide adenine dinucleotide (NAD)+ pools. This is currently being investigated for various pathologies where NAD+ concentrations are raised via provision of oral nicotinamide riboside, a NAD+ precursor [32,80]. NAD+ is a coenzyme for numerous redox enzymes and a cofactor in sirtuin activation, and has been widely linked to improved mitochondrial integrity and function, as well as reduced myeloid cell inflammation; NAD+ has also been associated with negative control of inflammatory programs (including those associated with NASH), such as NLRP3 inflammasome activation [32], and type I IFN signaling [81]. Recent studies also show that NASH mice harbor lower hepatocyte NAD+ concentrations due to NAD kinase mediated NAD+ consumption, and NAD-boosting using nicotinamide mononucleotide supplementation has repressed NASH-associated lipogenesis in murine hepatocytes and fibrosis in human and murine hepatic stellate cells [82]. However, the role of PRRs or inflammatory signaling in response to NAD+ boosting was not explored in this study. Other approaches to improve NAFLD outcomes shave been considered, such as targeting PRR pathways directly. For example, when compared to wildtype mice, the NLRP3 inhibitor MCC950 reduces liver inflammation and NASH pathology in foz/foz mice (animals bearing an Alms1 mutation eliciting hyperphagia with NASH on a Western diet) [29,83] (Figure 4). Of note, TLR4 induces increased cell-autonomous C5 activation in human monocytes and macrophages via induction of Factor B – a key component of the C5 activating C5 convertase (Figure 3) [65]. Further, a cell-permeable, Factor B inhibitor reduces cholesterol crystal-driven human macrophage activation and IL-1β production in vitro [65], suggesting another potential novel approach to treating NAFLD. Overall, targeting PRR signaling is emerging as a strategy to investigate the management of NAFLD. Conversely, complexities in the treatment of NAFLD and NASH include environmental triggers and concomitant systemic comorbidities (e.g. insulin resistance, metabolic syndrome, and HCC risk) which might add putative unexpected adverse effects with targeting PRR-immune cell pathways.
Figure 4. Potential novel therapeutic intervention points for mitochondrial-PRR axis perturbations in NAFLD/NASH.

Current NAFLD/NASH interventions aim to reduce cellular lipid accumulation by means of diet, bariatric surgery, and pharmacological inhibition of adipogenesis [99]. Increasing cellular NAD+ concentrations via oral provision of the NAD precursor nicotinamide riboside is currently being explored in the clinic (Clinicaltrials.gov NCT02835664). Targeting PRRs associated with mitochondrial dysfunction, induction of cytokines/chemokines, and/or cell death may be an additional means to reduce or revert the general pro-inflammatory state of liver cells in NAFLD/NASH (see text). However, such approaches need to be carefully explored because the nutrient-mitochondria-PRRs pathways are likely operative across cell subpopulations and may serve distinct functions within specialized cells, including the control of pathways contributing to cell survival, homeostasis, and regeneration. cGAS-STING, cyclic GMP-AMP synthase, and stimulator of interferon genes pathway; LDs, lipid droplets; NAD+, nicotinamide adenine dinucleotide; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3 inflammasome; PPAR γ, peroxisome proliferator activated receptor γ; RIG-I, retinoic-inducible gene I; ROS, reactive oxygen species; TLR, toll-like receptor. Some icons/cell types were imported from BioRender (https://biorender.com/).
Concluding remarks
In this opinion piece, we have proposed that mitochondrial infidelity, PRR signaling, and metabolic perturbations cooperatively drive inflammation in response to caloric overload, and further, that these may represent important contributors to hepatosteatosis progression to NASH. The bi-directional control of mitochondrial metabolism and PRR signaling has been substantially advanced by the recognition of the role of the complosome in regulating mitochondrial quality control programs. Furthermore, as therapeutic advances have begun to directly target components of ‘classic’ TLR and inflammasome PRR signaling, we propose that targeting the complosome should also be investigated. Overall, this strategy may not be simple due to several substantial limitations and caveats. For example, we must develop a better understanding of the molecular mechanisms underlying non-canonical PRR activities, as well as the complex systemic, mitochondrial, and parenchymal-immune cell crosstalk that may be implicated in the pathophysiology of NAFLD (see Outstanding questions). Further, the nutrient-mitochondrial-PRR axes discussed here might operate within most organ systems and perhaps also function in homeostasis and repair pathways. Thus, it will be important to understand when during NAFLD pathogenesis one might intervene, which cell type(s) to target, and if possible, determine which subcellular compartments are implicated; this may be key to preventing possible detrimental side effects, such as uncontrolled infections. Ideally, this should accompany a better stratification of patients and the development of improved pre-clinical disease models. Advancing experimental systems and generating new tools to gain insight into inflammatory- and immune-related molecular mechanisms in NAFLD will not be trivial but well worth the efforts to combat one of the most prevalent disease conditions in the Western world.
OUTSTANDING QUESTIONS.
Does intracellular complement operate in hepatocytes and what is its function in liver homeostasis and/or NAFLD/NASH? Although initial emerging evidence indicating that the complosome is operative within hepatocytes and within resident liver immune cells in mammals, its presence and function remains to be formally explored across cells in healthy and diseased livers, before we can consider it further as a putative therapeutic target for NAFLD.
Does the maladaptive PRR-mitochondrial axis also contribute to non-myeloid immune cell dysfunction in NAFLD/NASH? Most of our understanding of the pathologic contributions of immune cells to NAFLD/NASH are based on explorations in human and mouse myeloid cells. However, other immune cells (particularly conventional and non-conventional T cells and NK cells) are also clear drivers of disease. It will be important to assess if the nutrient-mitochondrial-PRR pathways and networks discussed here are functional in these latter cells and if they contribute to normal effector functions in health and/or liver disease.
Can PRR-mediated signaling orchestrate programs to counteract the progression to NASH e.g. by efferocytosis? Addressing this question is particularly important because inhibiting the potential beneficial contributions of these programs -- e.g. liver regeneration during therapeutic targeting – could potentially aggravate certain aspects of NAFLD. By contrast, harnessing any disease ameliorating properties from these networks via targeted activation, might provide a future alternative approach.
What are the exact lines of cross-communication between hepatocytes (liver injury), hepatic stellate cells (fibrosis), and resident and infiltrating immune cells in humans? Most studies explore phenotypical changes associated with NAFLD in a cell-specific manner and often in isolation. However, local pathophysiological paracrine signaling between cell types within the liver are central drivers of disease. The application of experimental techniques to provide a more holistic vantage point – e.g. single-cell RNA sequencing of tissue biopsies and spatial transcriptomics – may help define certain (mal)adaptive cellular network responses in NAFLD and NASH.
What are the molecular tipping points leading to cirrhosis and HCC? Despite considerable evidence for the contributions of nutrient overload, mitochondrial dysfunction, and PRR engagement in NASH, it is currently unclear what the relative contributions of these pathways towards non-reversible end-stage liver disease and cancer are. An initial question to address could be whether this detrimental transition involves a chronic gradual increase in a specific PRR-mitochondrial axis, eventually triggering a threshold-guarded permanent change, or whether qualitative changes in mitochondrial dysfunction and PRR activation are the culprit – or a combination of both.
Do early biomarkers of maladaptive PRR-mitochondrial axis exist and how can we identify those? Early intervention in (chronic) inflammatory diseases can slow disease progression and improve long-term outcomes. Measuring plasma proteins, metabolites, and/or gut/microbiome components via ‘omic’ approaches are currently being explored as promising early diagnostics in NAFLD. Although downstream outcomes of mitochondrial perturbation and PRR activation such mtDNA release and IL-1β and IFN secretion are likely suitable markers per se, it is possible that these most consistently occur locally in the liver, rather than in circulation – a notion which should be explored.
What is the suitability of currently tested mouse models? Small animal models of NAFLD/NASH (particularly mice) have been significantly helpful in understanding the pathology of liver disease. However, none of these fully recapitulate the complete human NAFLD spectrum, including related metabolic features associated with this chronic liver condition. Further, there are important speciesspecific differences in the cellular composition and function of aspects of the immune response and complosome between mice and humans which hamper the straightforward exploration of such systems in NAFLD. Thus, efforts to transition existing models towards humanized tissues/cells in combination with ablating genes of interest in a time- and cell-specific manner may be useful.
Can better immunophenotyping risk stratify patients with NAFLD and/or NASH, without the need for a liver biopsy? There is an unmet need for accurate i) diagnostics to differentiate steatosis from steatohepatitis, ii) fibrosis stage identification to better predict progression, iii) risk-stratification for candidate treatment strategies that can avoid complications. A more refined examination of the role of immunity in NAFLD can provide us a wider window of understanding of this disease.
Highlights.
NAFLD and NASH are on the rise and therapeutics to successfully target these detrimental conditions are very limited
Nutrient-induced hepatic lipid overload across cells in the mammalian liver contributes to mitochondrial dysfunction, producing DAMPs that initiate hepatosteatosis
Pattern recognition receptors integrate mitochondrial perturbations into proinflammatory cellular behavior in mammalian liver parenchymal cells and immune cells, supporting NAFLD pathogenesis and progression to NASH
Intracellular components of the canonical PRR system, complement (the complosome), operate across a range of cells in humans and mice, and may contribute to NAFLD/NASH pathogenesis.
Pharmacologically targeting nutrient-mitochondrial-PRR pathways, including intracellularly active complement, might lead to new putative NAFLD/NASH treatments
Significance.
The mechanistic understanding of the recently identified functional interplay between nutrient sensing, mitochondrial activity, classic PRR and intracellular complement activation across cell populations may help uncover novel therapeutic intervention targets to treat nonalcoholic fatty liver disease.
ACKNOWLEDGMENTS
This work was supported in part by the Intramural Research Program of the NIH, the National Heart, Lung, and Blood Institute (1ZIAHL006222-03 to C.K.), and (ZIA-HL005102 to M.N.S.).
GLOSSARY
- Adipose tissue
constitutes connective tissue composed mostly of adipocytes, which can store energy in the form of lipids (fat)
- Autophagy
cell-intrinsic machinery mediating the controlled degradation and recycling of a range of cellular components to maintain cellular health
- Autophagosome
double-membraned structure formed around cytoplasmic content that requires removal (e.g. damaged mitochondria and organelles, and invading pathogens); content is delivered to lysosomes for destruction
- cGAS-STING pathway
cytosolic PRR system consisting of the synthase for the second messenger cyclic GMP-AMP (cGAS) and the cGAS receptor stimulator of interferon genes (STING). Detects pathogenic DNA, triggering protective type I IFN responses (IFNα and -β)
- Cirrhosis
late stage of liver disease where healthy liver tissue is replaced by scar tissue and the liver is permanently damaged
- Complosome
cell-intrinsically expressed complement functioning in an intracellular and/or cell-autonomous fashion
- Damage-associated molecular pattern (DAMP)
endogenous molecules released from or displayed on damaged, stressed, or dying cells; can activate PRRs
- Fibrosis
development of fibrous connective tissue (thickening and scarring) as a repair response to injury and damage
- Hepatosteatosis
excess lipid droplet deposits present in the liver during NAFLD; precedes the development of inflammation (NASH)
- Inflammasomes
cytosolic PRRs; regulate the activation of caspases and generation of pro-inflammatory cytokines such as IL-1β and IL-18. There are several subtypes of inflammasomes (NLRP3, NLRP4, AIM2, etc.)
- Mitochondrial anti-viral signaling protein (MAVS)
essential intermediate in the RIG-I response against invading viruses. Located in mitochondria; drives type I IFN production
- Microbiome
collection of bacteria, fungi and viruses that naturally associate and live symbiotically with the host, mostly, but not exclusively, at mucosal tissues
- Mitochondrion
double-membrane organelle present in most eukaryotes; mediates chemical cellular energy production via respiration
- Mitochondrial fusion
molecular driven program that combines mitochondria as a homeostatic process to optimize mitochondrial size to functional demand
- Mitophagy
specialized form of autophagy that specifically removes damaged or superfluous mitochondria
- Nonalcoholic fatty liver disease (NAFLD)
pathology characterized by excess fat accumulation and inflammation in the liver not caused by alcohol abuse; often reversible
- Nonalcoholic steatohepatitis (NASH)
advanced form of NAFLD; often not reversible as liver damage and fibrosis have progressed substantially
- Pattern-recognition receptors (PRRs)
range of proteina that can recognize specific molecular patterns displayed on pathogens or altered host cells (e.g. dying, malignantly transformed, and infected cells)
- Pyroptosis
i form of necrotic cell death; triggered by proinflammatory signals and associated with inflammation
- Retinoic acid-inducible gene I (RIG-I)
PRR; key sensor of viral infections mediating the transcriptional induction of type I IFNs
- Steatosis
abnormal retention of lipids within a cell or an organ (also known as fatty change)
- Steatohepatitis
activation of the immune system in the progression of hepatic steatosis to NASH
- Toll-like receptors (TLRs)
membrane-spanning PRRs (on cell surfaces and organelles); recognize a range of molecular patterns derived from pathogens and altered self (including oxidized lipids)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.O’Neill S and O’Driscoll L (2015) Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev 16, 1–12. 10.1111/obr.12229 [DOI] [PubMed] [Google Scholar]
- 2.Anstee QM et al. (2019) From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol 16, 411–428. 10.1038/s41575-019-0145-7 [DOI] [PubMed] [Google Scholar]
- 3.Estes C et al. (2018) Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 67, 123–133. 10.1002/hep.29466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker PR 2nd and Friedman JE (2018) Mitochondrial role in the neonatal predisposition to developing nonalcoholic fatty liver disease. The Journal of clinical investigation 128, 3692–3703. 10.1172/JCI120846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kramer DA et al. (2018) Fasting and refeeding induces changes in the mouse hepatic lipid droplet proteome. Journal of proteomics 181, 213–224. 10.1016/j.jprot.2018.04.024 [DOI] [PubMed] [Google Scholar]
- 6.Gluchowski NL et al. (2017) Lipid droplets and liver disease: from basic biology to clinical implications. Nat Rev Gastroenterol Hepatol 14, 343–355. 10.1038/nrgastro.2017.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schuster S et al. (2018) Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol 15, 349–364. 10.1038/s41575-018-0009-6 [DOI] [PubMed] [Google Scholar]
- 8.Fracanzani AL et al. (2008) Carotid artery intima-media thickness in nonalcoholic fatty liver disease. Am J Med 121, 72–78. 10.1016/j.amjmed.2007.08.041 [DOI] [PubMed] [Google Scholar]
- 9.Henao-Mejia J et al. (2012) Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185. 10.1038/nature10809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dou L et al. (2018) Hepatic Dendritic Cells, the Tolerogenic Liver Environment, and Liver Disease. Semin Liver Dis 38, 170–180. 10.1055/s-0038-1646949 [DOI] [PubMed] [Google Scholar]
- 11.Zhou Z et al. (2018) Neutrophil-Hepatic Stellate Cell Interactions Promote Fibrosis in Experimental Steatohepatitis. Cell Mol Gastroenterol Hepatol 5, 399–413. 10.1016/j.jcmgh.2018.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daemen S et al. (2021) Dynamic Shifts in the Composition of Resident and Recruited Macrophages Influence Tissue Remodeling in NASH. Cell reports 34, 108626. 10.1016/j.celrep.2020.108626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gadd VL et al. (2014) The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 59, 1393–1405. 10.1002/hep.26937 [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Martinez I et al. (2016) Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. The Journal of clinical investigation 126, 859–864. 10.1172/JCI83885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Legaki AI et al. (2022) Hepatocyte Mitochondrial Dynamics and Bioenergetics in Obesity-Related Non-Alcoholic Fatty Liver Disease. Curr Obes Rep 11, 126–143. 10.1007/s13679-022-00473-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei Y et al. (2008) Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J Gastroenterol 14, 193–199. 10.3748/wjg.14.193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koliaki C et al. (2015) Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell metabolism 21, 739–746. 10.1016/j.cmet.2015.04.004 [DOI] [PubMed] [Google Scholar]
- 18.Calfee CS and Matthay MA (2010) Clinical immunology: Culprits with evolutionary ties. Nature 464, 41–42. 10.1038/464041a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sack MN (2018) Mitochondrial fidelity and metabolic agility control immune cell fate and function. The Journal of clinical investigation 128, 3651–3661. 10.1172/JCI120845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sorbara MT and Girardin SE (2011) Mitochondrial ROS fuel the inflammasome. Cell Res 21, 558–560. 10.1038/cr.2011.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu T et al. (2022) Imbalanced GSH/ROS and sequential cell death. J Biochem Mol Toxicol 36, e22942. 10.1002/jbt.22942 [DOI] [PubMed] [Google Scholar]
- 22.Yamada T et al. (2022) Prevention and regression of megamitochondria and steatosis by blocking mitochondrial fusion in the liver. iScience 25, 103996. 10.1016/j.isci.2022.103996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ganz M et al. (2014) High fat diet feeding results in gender specific steatohepatitis and inflammasome activation. World J Gastroenterol 20, 8525–8534. 10.3748/wjg.v20.i26.8525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng T et al. (2020) High Glucose-Aggravated Hepatic Insulin Resistance: Role of the NLRP3 Inflammasome in Kupffer Cells. Obesity (Silver Spring) 28, 1270–1282. 10.1002/oby.22821 [DOI] [PubMed] [Google Scholar]
- 25.Gaul S et al. (2021) Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J Hepatol 74, 156–167. 10.1016/j.jhep.2020.07.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang NP et al. (2019) Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Laboratory investigation; a journal of technical methods and pathology 99, 749–763. 10.1038/s41374-018-0177-6 [DOI] [PubMed] [Google Scholar]
- 27.Wree A et al. (2014) NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J Mol Med (Berl) 92, 1069–1082. 10.1007/s00109-014-1170-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang G et al. (2016) A pharmacological inhibitor of NLRP3 inflammasome prevents non-alcoholic fatty liver disease in a mouse model induced by high fat diet. Sci Rep 6, 24399. 10.1038/srep24399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mridha AR et al. (2017) NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol 66, 1037–1046. 10.1016/j.jhep.2017.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tan M et al. (2020) Inhibition of the mitochondrial citrate carrier, Slc25a1, reverts steatosis, glucose intolerance, and inflammation in preclinical models of NAFLD/NASH. Cell death and differentiation 27, 2143–2157. 10.1038/s41418-020-0491-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xian H et al. (2021) Metformin inhibition of mitochondrial ATP and DNA synthesis abrogates NLRP3 inflammasome activation and pulmonary inflammation. Immunity 54, 1463–1477 e1411. 10.1016/j.immuni.2021.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Traba J et al. (2015) Fasting and refeeding differentially regulate NLRP3 inflammasome activation in human subjects. The Journal of clinical investigation 125, 4592–4600. 10.1172/JCI83260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo X et al. (2018) Expression of STING Is Increased in Liver Tissues From Patients With NAFLD and Promotes Macrophage-Mediated Hepatic Inflammation and Fibrosis in Mice. Gastroenterology 155, 1971–1984 e1974. 10.1053/j.gastro.2018.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bai J et al. (2017) DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS-cGAMP-STING pathway. Proceedings of the National Academy of Sciences of the United States of America 114, 12196–12201. 10.1073/pnas.1708744114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu Y et al. (2019) STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. The Journal of clinical investigation 129, 546–555. 10.1172/JCI121842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qiao JT et al. (2018) Activation of the STING-IRF3 pathway promotes hepatocyte inflammation, apoptosis and induces metabolic disorders in nonalcoholic fatty liver disease. Metabolism 81, 13–24. 10.1016/j.metabol.2017.09.010 [DOI] [PubMed] [Google Scholar]
- 37.Miura K et al. (2010) Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 139, 323–334 e327. 10.1053/j.gastro.2010.03.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37 b.Yang B et al. (2022). Macrophage-specific MyD88 deletion and pharmacological inhibition prevents liver damage in non-alcoholic fatty liver disease via reducing inflammatory response, Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, 1868:166480, 10.1016/j.bbadis.2022.166480. [DOI] [PubMed] [Google Scholar]
- 38.Kawamura S et al. (2022) Inhibiting SCAP/SREBP exacerbates liver injury and carcinogenesis in murine nonalcoholic steatohepatitis. The Journal of clinical investigation 132. 10.1172/JCI151895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu C et al. (2021) FOXA3 induction under endoplasmic reticulum stress contributes to non-alcoholic fatty liver disease. J Hepatol 75, 150–162. 10.1016/j.jhep.2021.01.042 [DOI] [PubMed] [Google Scholar]
- 40.Alper CA et al. (1969) Human C’3: evidence for the liver as the primary site of synthesis. Science 163, 286–288. 10.1126/science.163.3864.286 [DOI] [PubMed] [Google Scholar]
- 41.Hajishengallis G et al. (2017) Novel mechanisms and functions of complement. Nature immunology 18, 1288–1298. 10.1038/ni.3858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jia Q et al. (2015) Association between complement C3 and prevalence of fatty liver disease in an adult population: a cross-sectional study from the Tianjin Chronic Low-Grade Systemic Inflammation and Health (TCLSIHealth) cohort study. PloS one 10, e0122026. 10.1371/journal.pone.0122026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu C et al. (2016) Serum complement C3 levels are associated with nonalcoholic fatty liver disease independently of metabolic features in Chinese population. Sci Rep 6, 23279. 10.1038/srep23279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Merle NS et al. (2015) Complement System Part I - Molecular Mechanisms of Activation and Regulation. Front Immunol 6, 262. 10.3389/fimmu.2015.00262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Monsinjon T et al. (2003) Regulation by complement C3a and C5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. FASEB J 17, 1003–1014. 10.1096/fj.02-0737com [DOI] [PubMed] [Google Scholar]
- 46.Fischetti F and Tedesco F (2006) Cross-talk between the complement system and endothelial cells in physiologic conditions and in vascular diseases. Autoimmunity 39, 417–428. 10.1080/08916930600739712 [DOI] [PubMed] [Google Scholar]
- 47.Rensen SS et al. (2009) Activation of the complement system in human nonalcoholic fatty liver disease. Hepatology 50, 1809–1817. 10.1002/hep.23228 [DOI] [PubMed] [Google Scholar]
- 48.Xin Y et al. (2021) C3 and alternative pathway components are associated with an adverse lipoprotein subclass profile: The CODAM study. J Clin Lipidol 15, 311–319. 10.1016/j.jacl.2021.01.011 [DOI] [PubMed] [Google Scholar]
- 49.Merle NS et al. (2021) Integrins meet complement: The evolutionary tip of an iceberg orchestrating metabolism and immunity. Br J Pharmacol 178, 2754–2770. 10.1111/bph.15168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Persson L et al. (2004) Lack of complement factor C3, but not factor B, increases hyperlipidemia and atherosclerosis in apolipoprotein E−/− low-density lipoprotein receptor−/− mice. Arterioscler Thromb Vasc Biol 24, 1062–1067. 10.1161/01.ATV.0000127302.24266.40 [DOI] [PubMed] [Google Scholar]
- 51.Lim J et al. (2013) C5aR and C3aR antagonists each inhibit diet-induced obesity, metabolic dysfunction, and adipocyte and macrophage signaling. FASEB J 27, 822–831. 10.1096/fj.12-220582 [DOI] [PubMed] [Google Scholar]
- 52.Strey CW et al. (2003) The proinflammatory mediators C3a and C5a are essential for liver regeneration. The Journal of experimental medicine 198, 913–923. 10.1084/jem.20030374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reis ES et al. (2019) New insights into the immune functions of complement. Nature reviews. Immunology 19, 503–516. 10.1038/s41577-019-0168-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thorgersen EB et al. (2019) The Role of Complement in Liver Injury, Regeneration, and Transplantation. Hepatology 70, 725–736. 10.1002/hep.30508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.West EE et al. (2018) Complement and the Regulation of T Cell Responses. Annu Rev Immunol 36, 309–338. 10.1146/annurev-immunol-042617-053245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liszewski MK et al. (2013) Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 39, 1143–1157. 10.1016/j.immuni.2013.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kolev M et al. (2015) Complement Regulates Nutrient Influx and Metabolic Reprogramming during Th1 Cell Responses. Immunity 42, 1033–1047. 10.1016/j.immuni.2015.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arbore G et al. (2018) Complement receptor CD46 co-stimulates optimal human CD8(+) T cell effector function via fatty acid metabolism. Nature communications 9, 4186. 10.1038/s41467-018-06706-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cardone J et al. (2010) Complement regulator CD46 temporally regulates cytokine production by conventional and unconventional T cells. Nature immunology 11, 862–871. 10.1038/ni.1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perucha E et al. (2019) The cholesterol biosynthesis pathway regulates IL-10 expression in human Th1 cells. Nature communications 10, 498. 10.1038/s41467-019-08332-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ellinghaus U et al. (2017) Dysregulated CD46 shedding interferes with Th1-contraction in systemic lupus erythematosus. Eur J Immunol 47, 1200–1210. 10.1002/eji.201646822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arbore G et al. (2020) Deep phenotyping detects a pathological CD4(+) T-cell complosome signature in systemic sclerosis. Cell Mol Immunol 17, 1010–1013. 10.1038/s41423-019-0360-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Friscic J et al. (2021) The complement system drives local inflammatory tissue priming by metabolic reprogramming of synovial fibroblasts. Immunity 54, 1002–1021 e1010. 10.1016/j.immuni.2021.03.003 [DOI] [PubMed] [Google Scholar]
- 64.Daugan MV et al. (2021) Intracellular Factor H Drives Tumor Progression Independently of the Complement Cascade. Cancer Immunol Res 9, 909–925. 10.1158/2326-6066.CIR-20-0787 [DOI] [PubMed] [Google Scholar]
- 65.Niyonzima N et al. (2021) Mitochondrial C5aR1 activity in macrophages controls IL-1beta production underlying sterile inflammation. Sci Immunol 6, eabf2489. 10.1126/sciimmunol.abf2489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ferreyra Solari NE et al. (2012) The role of innate cells is coupled to a Th1-polarized immune response in pediatric nonalcoholic steatohepatitis. Journal of clinical immunology 32, 611–621. 10.1007/s10875-011-9635-2 [DOI] [PubMed] [Google Scholar]
- 67.Zhang X et al. (2007) Regulation of Toll-like receptor-mediated inflammatory response by complement in vivo. Blood 110, 228–236. 10.1182/blood-2006-12-063636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tam JC et al. (2014) Intracellular sensing of complement C3 activates cell autonomous immunity. Science 345, 1256070. 10.1126/science.1256070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Posch W et al. (2021) Complement Potentiates Immune Sensing of HIV-1 and Early Type I Interferon Responses. mBio 12, e0240821. 10.1128/mBio.02408-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ishii M et al. (2021) Mitochondrial C3a Receptor Activation in Oxidatively Stressed Epithelial Cells Reduces Mitochondrial Respiration and Metabolism. Front Immunol 12, 628062. 10.3389/fimmu.2021.628062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arbore G et al. (2016) T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4(+) T cells. Science 352, aad1210. 10.1126/science.aad1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ling GS et al. (2018) C1q restrains autoimmunity and viral infection by regulating CD8(+) T cell metabolism. Science 360, 558–563. 10.1126/science.aao4555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ten VS et al. (2010) Complement component c1q mediates mitochondria-driven oxidative stress in neonatal hypoxic-ischemic brain injury. The Journal of neuroscience : the official journal of the Society for Neuroscience 30, 2077–2087. 10.1523/JNEUROSCI.5249-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McGee AM and Baines CP (2011) Complement 1q-binding protein inhibits the mitochondrial permeability transition pore and protects against oxidative stress-induced death. The Biochemical journal 433, 119–125. 10.1042/BJ20101431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rahman J et al. (2021) Complement’s favourite organelle-Mitochondria? Br J Pharmacol 178, 2771–2785. 10.1111/bph.15238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang Y et al. (2012) The complement C1qA enhances retinoic acid-inducible gene-I-mediated immune signalling. Immunology 136, 78–85. 10.1111/j.1365-2567.2012.03561.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lung T et al. (2019) The complement system in liver diseases: Evidence-based approach and therapeutic options. J Transl Autoimmun 2, 100017. 10.1016/j.jtauto.2019.100017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li Y et al. (2021) Intracellular C3 prevents hepatic steatosis by promoting autophagy and very-low-density lipoprotein secretion. FASEB J 35, e22037. 10.1096/fj.202100856R [DOI] [PubMed] [Google Scholar]
- 79.Sato K et al. (2015) Vitamin E has a beneficial effect on nonalcoholic fatty liver disease: a meta-analysis of randomized controlled trials. Nutrition 31, 923–930. 10.1016/j.nut.2014.11.018 [DOI] [PubMed] [Google Scholar]
- 80.Brakedal B et al. (2022) The NADPARK study: A randomized phase I trial of nicotinamide riboside supplementation in Parkinson’s disease. Cell metabolism 34, 396–407 e396. 10.1016/j.cmet.2022.02.001 [DOI] [PubMed] [Google Scholar]
- 81.Wu J et al. (2022) Boosting NAD+ blunts TLR4-induced type I IFN in control and systemic lupus erythematosus monocytes. The Journal of clinical investigation 132. 10.1172/JCI139828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gao H et al. (2022) MiR-690 treatment causes decreased fibrosis and steatosis and restores specific Kupffer cell functions in NASH. Cell metabolism 34, 978–990 e974. 10.1016/j.cmet.2022.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang F et al. (2022) Oxy210, a Semi-Synthetic Oxysterol, Exerts Anti-Inflammatory Effects in Macrophages via Inhibition of Toll-like Receptor (TLR) 4 and TLR2 Signaling and Modulation of Macrophage Polarization. International journal of molecular sciences 23. 10.3390/ijms23105478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jenne CN and Kubes P (2013) Immune surveillance by the liver. Nature immunology 14, 996–1006. 10.1038/ni.2691 [DOI] [PubMed] [Google Scholar]
- 85.MacParland SA et al. (2018) Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nature communications 9, 4383. 10.1038/s41467-018-06318-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cai J et al. (2019) The Role of Innate Immune Cells in Nonalcoholic Steatohepatitis. Hepatology 70, 1026–1037. 10.1002/hep.30506 [DOI] [PubMed] [Google Scholar]
- 87.Das UN (2021) Essential Fatty Acids and Their Metabolites in the Pathobiology of Inflammation and Its Resolution. Biomolecules 11. 10.3390/biom11121873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Czaja MJ et al. (2013) Functions of autophagy in normal and diseased liver. Autophagy 9, 1131–1158. 10.4161/auto.25063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lavallard VJ and Gual P (2014) Autophagy and non-alcoholic fatty liver disease. Biomed Res Int 2014, 120179. 10.1155/2014/120179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ueno T and Komatsu M (2017) Autophagy in the liver: functions in health and disease. Nat Rev Gastroenterol Hepatol 14, 170–184. 10.1038/nrgastro.2016.185 [DOI] [PubMed] [Google Scholar]
- 91.Filali-Mouncef Y et al. (2022) The menage a trois of autophagy, lipid droplets and liver disease. Autophagy 18, 50–72. 10.1080/15548627.2021.1895658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sun Q et al. (2017) Inflammasome and autophagy regulation - a two-way street. Mol Med 23, 188–195. 10.2119/molmed.2017.00077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Onishi M et al. (2021) Molecular mechanisms and physiological functions of mitophagy. EMBO J 40, e104705. 10.15252/embj.2020104705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Saha S et al. (2018) Autophagy in health and disease: A comprehensive review. Biomed Pharmacother 104, 485–495. 10.1016/j.biopha.2018.05.007 [DOI] [PubMed] [Google Scholar]
- 95.Merle NS et al. (2015) Complement System Part II: Role in Immunity. Front Immunol 6, 257. 10.3389/fimmu.2015.00257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ebrahimi H et al. (2018) New Concepts on Reversibility and Targeting of Liver Fibrosis; A Review Article. Middle East J Dig Dis 10, 133–148. 10.15171/mejdd.2018.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xie CB et al. (2021) Complement-activated human endothelial cells stimulate increased polyfunctionality in alloreactive T cells. Am J Transplant 21, 1902–1909. 10.1111/ajt.16485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yan B et al. (2021) SARS-CoV-2 drives JAK1/2-dependent local complement hyperactivation. Sci Immunol 6. 10.1126/sciimmunol.abg0833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mundi MS et al. (2020) Evolution of NAFLD and Its Management. Nutr Clin Pract 35, 72–84. 10.1002/ncp.10449 [DOI] [PubMed] [Google Scholar]