





Official websites use .gov
A
.gov website belongs to an official
government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you've safely
connected to the .gov website. Share sensitive
information only on official, secure websites.

I miglioramenti registrati negli ultimi anni nella qualità del sequenziamento di nuova generazione, nella riduzione dei costi associati e in una complessiva evoluzione delle scienze omiche, hanno favorito lo sviluppo della medicina personalizzata o di precisione. Ad oggi, anche a livello di popolazione si possono ottenere dei benefici rilevanti attraverso tale approccio. La Sanità Pubblica di precisione consiste nel fornire il giusto intervento, alla popolazione che ne ha necessità, nel momento e con le modalità opportune. Significa, quindi, promuovere metodologie accurate per identificare e misurare le patologie ma anche le esposizioni, i comportamenti e la suscettibilità.
La Sanità Pubblica di precisione è in evoluzione e non è legata semplicemente a geni, trattamenti e malattia ma alla precisa identificazione e risposta ai bisogni di salute. È necessario, quindi, discutere dell’inclusione delle scienze omiche in Sanità Pubblica.
La medicina si è evoluta da un modello di diagnosi e trattamento basato essenzialmente sui sintomi ad uno sempre più dipendente dalla definizione bioinformatica di profili di rischio e/o patologici. Tali profili sono delineati mediante la produzione di informazioni attingendo a solide banche dati biologiche con il supporto dell’intelligenza artificiale. D’altra parte l’evoluzione nella pratica sanitaria è un processo complesso che include, tra l’altro, la sostenibilità dei costi sanitari, la valutazione dell’efficienza nella pratica clinica, l'integrazione dei nuovi progressi tecnologici e la rimodulazione dell'organizzazione dei servizi.
Nel Gruppo di Lavoro Genomica in Sanità Pubblica della SItI, attivo dal 2012, sono coinvolti prevalentemente docenti universitari ma anche operatori del Ministero della Salute e dei Dipartimenti di Prevenzione. In questo special issue illustriamo alcuni argomenti di ricerca trattati. Non stupirà l’eterogeneità dei temi proposti vista la trasversalità delle scienze omiche in molteplici aspetti della salute umana. In particolare sono illustrati esempi che vanno dalla prevenzione di tumori ad alta incidenza, alla prevenzione di patologie infettive, sia per gli aspetti acuti che cronici, tenendo conto di caratteristiche genetiche ed epigenetiche della popolazione. Inoltre, illustriamo le prospettive di integrazione offerte dallo studio del microbiota umano nella prevenzione. Procediamo con la discussione delle modalità di valutazione dei test genetici e genomici per la loro integrazione nell’offerta del Servizio Sanitario Nazionale. Infine, è illustrato il coinvolgimento della popolazione nell’impiego delle tecnologie omiche al fine di promuovere un cambiamento culturale nei confronti delle tecnologie disponibili e nella tutela della salute individuale e collettiva.
Gli agenti infettivi sono responsabili di circa il 15-20% dei tumori umani [1]. Tra questi, Human Papillomavirus (HPV) assume una posizione di rilievo causando il 30% di tutti i tumori correlati ad infezione [2]: a livello mondiale è il quarto tumore più frequente nelle donne, con oltre 600.000 nuovi casi nel 2021 [3], mentre in Italia rappresenta il quinto tumore per frequenza nelle donne sotto i 50 anni. Nel 2020 si sono registrate circa 3.500 nuove diagnosi di cancro cervicale e oltre 1.500 donne sono decedute a causa di questo tumore [4].
Gli HPV si differenziano a livello di genotipo, trofismo epiteliale e patogenicità [5]: a oggi, sono stati identificati oltre 200 genotipi, di cui 30 oncogeni per l’uomo e caratterizzati da un differente grado di cancerogenicità. Sono divisi in ceppi a basso rischio cancerogeno (LR-HPV) e ad alto rischio cancerogeno (HR-HPV), a seconda della loro capacità trasformante [6-9]. Gli HR-HPV sono responsabili di quasi tutti i tumori del collo dell’utero (> 99,7%) [8]. I dati epidemiologici affermano che il genotipo HPV16 è maggiormente identificato nei tumori cervicali, ma è anche il più comune in tutti i siti di cancro correlati all’HPV (vagina, vulva, ano, tratto orofaringeo e cute), seguito da HPV18 [10, 11]: insieme rappresentano oltre il 70% di tutti i casi di carcinoma a cellule squamose [12]. I ceppi LR-HPV sono invece associati allo sviluppo di lesioni benigne.
Ad oggi, l’HPV è l’infezione sessualmente trasmissibile più diffusa e nonostante la disponibilità di strategie preventive, i tumori associati sono una delle principali cause di morbilità e mortalità a livello globale [13].
La condizione necessaria, ma non sufficiente, per lo sviluppo del tumore cervicale è un’infezione persistente da HR-HPV con integrazione del genoma virale in quello della cellula ospite [14]. Altri fattori quindi sono implicati nella patogenesi della malattia, tra cui il fumo di sigaretta, la multiparità, l’utilizzo di contraccettivi per periodi prolungati, la co-infezione con HIV o con altre MST e altri fattori ancora oggi non ben chiariti [15-17].
Sebbene le infezioni da HR-HPV solitamente non causano sintomi, si risolvono spontaneamente [18] e sono generalmente eliminate dal sistema immunitario dell’ospite in 1-2 anni dall’esordio, il 10-20% possono persistere in modo latente, portando alla progressione verso lesioni precancerose (denominate CIN, cervical intraepithelial neoplasia) per poi evolvere, se non trattate, in cancro invasivo [19].
L’integrazione del genoma virale in quello della cellula ospite determina la sovraespressione delle oncoproteine virali E6 ed E7 [20, 21] che contribuisce al processo oncogenico cervicale inibendo le funzioni del soppressore tumorale p53 e promuovendo la degradazione della proteina retinoblastoma (pRB) [22-24]. L’interazione con p53 e pRB causa danni al DNA con conseguente alterazione del ciclo cellulare, trasformazione displastica e poi neoplastica [25].
A causa della genesi multifattoriale e della variabilità dei genotipi oncogeni, le lesioni cervicali HPV-relate non sono quindi tutte uguali: per riuscire a migliorare la diagnosi precoce, sta emergendo con forza l’esigenza di comprendere il processo infettivo di HPV e l’eventuale progressione neoplastica della lesione sia da un punto di vista molecolare, considerando aspetti legati alla biologia cellulare, trascrizionale e regolazione post-trascrizionale; e sia alcune caratteristiche proprie del virus infettante. Il fattore principale che determina il passaggio da lesione pre-maligna a cancro è la regolazione epigenetica esercitata dai microRNA. È stato dimostrato che i profili di espressione dei microRNA sono altamente alterati in diversi tumori rispetto ai controlli e i danni si accumulano durante la carcinogenesi. I microRNA sono stati correlati con i tumori, ed in particolare con la causa dei tumori, ma anche con la prognosi tumorale. Quando l’acido nucleico virale entra nella cellula ospite, il microRNA lo riconosce per complementarità attivando una serie di meccanismi che possono essere di difesa o patogenetici che attivano i toll like receptor producendo infiammazione cronica.
La vaccinazione anti-HPV rappresenta il principale e più efficace strumento di prevenzione primaria del cancro della cervice [26, 27], ma a oggi la copertura vaccinale è al di sotto degli obiettivi stabiliti [28], anche in Italia [29]. La strategia preventiva più efficace, ad oggi, rimane il programma di screening basato su Pap test e HPV-DNA test, che consentono di identificare e trattare precocemente le lesioni precancerose prima che si trasformino in cancro. Il Pap test è offerto ogni 3 anni alle donne di età compresa tra i 25 e i 30 anni; l’affidabilità è sensibilmente influenzata dal grado di esperienza dell’esaminatore e dal preparato sui vetrini.
Dal 2006 è stato introdotto l’HPV-DNA test per le pazienti al di sopra dei 30 anni, che ha dimostrato una maggiore efficacia e sensibilità rispetto allo screening citologico: il test viene offerto alle donne di età tra i 30 e i 64 anni, da ripetere ad intervalli di 5 anni. È poco utile, o addirittura dannoso, svolgerlo prima dei 30 anni, quando l’infezione da HPV è più frequente e nella maggior parte dei casi regredisce spontaneamente. Secondo le attuali linee guida, se l’esito dell’HPV-DNA test è positivo, dallo stesso prelievo si esegue anche un Pap test, con la finalità di evidenziare la presenza di anomalie cellulari.
Tuttavia, questi test presentano delle criticità non trascurabili che dipendono dal campionamento, dalla qualità del preparato citologico e ciò giustifica il 5-10% di risultati incerti o anomali [30]: solo associando la valutazione morfologica classica (Pap test e istologia) con l’analisi molecolare della lesione è possibile caratterizzarla in modo accurato, stabilire il reale rischio di progressione neoplastica ed eventualmente intervenire precocemente con programmi clinici e terapeutici personalizzati.
È necessario quindi prendere in considerazione fattori molecolari ed epigenetici a più livelli, come il profilo di espressione dei microRNA, lo status di integrazione virale, la genotipizzazione e la carica virale. I microRNA sono piccoli RNA non codificanti di circa 25 paia di basi che regolano negativamente l’espressione genica a livello post-trascrizionale. Sono nati per difendere le piante dai virus e questo meccanismo di difesa è così efficiente da funzionare anche nell’uomo. Sono 3000 nell’uomo, ognuno controlla 200 geni circa. Queste corte molecole di RNA riconoscono i loro messaggeri complementari neutralizzandoli tramite le RNAsi del complesso RISC (RNA inhibiting silecing complex). Per questo meccanismo il 95% dei messaggeri trascritti non raggiunge il ribosoma perché c’è un rigoroso controllo post-trascrizionale dell’espressione genica.
I fattori che determinano l’evoluzione dell’infezione da HR-HPV non sono ancora stati chiariti completamente, ma nell’ultimo decennio l’attenzione si sta concentrando sulle alterazioni epigenetiche (microRNA), che sono alla base della progressione verso il cancro, e su alcune caratteristiche dell’infezione stessa (integrazione virale, genotipizzazione e carica virale). Studi recenti dimostrano come il ruolo oncogenico o oncosoppressore dei microRNA possa influenzare la differenziazione cellulare, la regolazione del ciclo cellulare e l’apoptosi nella carcinogenesi [21] oltre a regolare l’espressione post-trascrizionale di alcuni geni.
Il ruolo dei microRNA è stato ipotizzato anche nello sviluppo del cancro cervicale, sebbene la sua causa primaria resti la persistenza dell’infezione da HR-HPV [31, 32].
In generale, i microRNA possono regolare sia geni oncosoppressori che oncogeni e l’alterata espressione rappresenta un evento piuttosto precoce nell’induzione della carcinogenesi da parte dell’infezione da HPV [33]. La loro analisi, quindi, si dimostra essere molto utile nella diagnosi precoce dello sviluppo del tumore cervicale.
Sebbene siano necessari ulteriori studi per migliorare la comprensione dei profili di espressione dei microRNA, appare evidente il loro ruolo emergente nel meccanismo di cancerogenesi cervicale come mediatori chiave dell’espressione genica. I microRNA, dunque, possono essere considerati potenziali biomarcatori predittivi, utili anche nel campo della prevenzione delle malattie infettive e delle decisioni terapeutiche [34].
Ugualmente importante appare l’analisi molecolare di alcune caratteristiche dell’infezione da HPV a livello delle lesioni preneoplastiche. In particolare, la determinazione del genotipo infettante, la quantificazione della carica virale e la verifica dello status di integrazione del genoma virale nella cellula ospite.
Il cancro cervicale solitamente si sviluppa in un lasso di tempo piuttosto ampio. Le lesioni precancerose di alto grado (CIN2-CIN3) possono svilupparsi entro 3-5 anni a seguito di un’infezione da HR-HPV, mentre l’eventuale progressione verso il cancro può richiedere persino 20-30 anni [35]. Il lungo periodo permette di attuare strategie preventive per l’identificazione e il trattamento delle lesioni cervicali, prevenendone la progressione cancerosa [36]. Per questo motivo, l’analisi dei fattori molecolari che determinano lo sviluppo della lesione preneoplastica diventa fondamentale per capire la reale natura della CIN e il reale rischio di progressione neoplastica. In letteratura recenti studi mettono in relazione la carica virale di HPV con la presenza di infezioni cervicali persistenti e ne evidenziano l’associazione [36, 37]. I nostri precedenti studi hanno messo in evidenza che la sola analisi morfologica delle lesioni, soprattutto se a basso rischio (CIN1), tende a sovrastimare il numero delle lesioni HPV-relate, con notevoli ripercussioni per le donne: l’indagine molecolare, quindi, associata alle routinarie analisi morfologiche, migliorerebbe notevolmente la diagnosi precoce delle lesioni. La valutazione molecolare delle lesioni preneoplastiche potrebbe portare ad ampie prospettive traslazionali, consentendo una migliore diagnosi, prognosi e terapia e con preziose applicazioni nella prevenzione.
Infatti, il protocollo di screening non prevede mai un approfondimento molecolare delle lesioni, che vengono trattate a prescindere dal ceppo di HPV infettante e dalle caratteristiche molecolari ed epigenetiche.
Questo modus operandi, però, determina il considerare le lesioni come se fossero tutte uguali, ma in realtà non è così. Il ceppo infettante, la carica virale e gli aspetti epigenetici sono fattori che influenzano l’andamento della lesione nel tempo, definendo un quadro ad alto rischio di progressione, oppure con un rischio inferiore e per le quali basterebbe monitorarle nel tempo. Determinarli preventivamente consentirebbe di migliorare notevolmente la diagnosi precoce e di stabilire protocolli terapeutici personalizzati.
La recente disponibilità di sistemi per il sequenziamento ad alto rendimento, come le piattaforme di Next Generation Sequencing (NGS), che consentono la caratterizzazione di interi genomi microbici in pochi giorni e ad un costo contenuto, unitamente alla larga disponibilità di sequenze/genomi nelle banche dati pubbliche, si sono rivelati ottimi alleati per la sorveglianza delle epidemie emergenti e per la pianificazione e messa in atto di efficaci strategie di controllo.
La sorveglianza genomica è ampiamente riconosciuta come attività essenziale nella Sanità Pubblica, in particolare nella preparedness delle emergenze infettive, e lo è ancor di più oggi con il crescente problema della diffusione della multiresistenza, che conta quasi 700,000 casi e 33,000 morti all’anno in Europa [1].
Un esempio è dato dalla nascita dell’Antimicrobial Resistance Surveillance Program (ARSP) nelle Filippine, un sistema di sorveglianza che ogni anno pubblica un report sull’andamento delle resistenze, per sviluppare delle linee guida applicabili nelle procedure cliniche [2, 3].
Fin dagli inizi della pandemia COVID-19, il sequenziamento dei genomi virali di SARS-CoV-2, unitamente all’immediata disponibilità di dati genomici ed epidemiologici resi disponibili in database pubblici, quali GenBank o GISAID (Global Initiative on Sharing All Influenza Data), si è mostrato uno strumento chiave nella ricostruzione dei singoli eventi di trasmissione all’interno delle epidemie e nella comprensione delle dinamiche evolutive e della patogenesi del virus [3, 4] e delle varianti virali [5, 6].
SARS-CoV-2 (Severe Acute Respiratory Syndrome- Coronavirus 2) essendo un virus ad RNA, possiede un’elevata variabilità genetica, stimata tra 0,7 e 1,1 mutazioni ogni 1000 nucleotidi all’anno, che equivale, per un genoma della lunghezza di circa 29000 nucleotidi, a circa 2 mutazioni al mese, nonostante la presenza di un’esonucleasi con attività di “correzione di bozze” (proofreading), presente in tutti i coronavirus.
Dopo un periodo iniziale di limitata diversità genetica virale, conseguente al salto di specie dal serbatoio animale all’uomo, il successivo accumulo di mutazioni ha portato alla comparsa di numerosi lignaggi evolutivi susseguitisi nel corso del tempo con un meccanismo di sostituzione di una variante destinata all’estinzione con quella successiva. Dalla fine del 2020 sono emerse le varianti “di preoccupazione” (VOC, Variants of Concern), in quanto caratterizzate da mutazioni (in particolare nella proteina Spike) in grado di conferire importanti cambiamenti nel fenotipo virale, tra cui un’alterata infettività, trasmissibilità, patogenicità o antigenicità virale (capacità di sfuggire alle difese immuni indotte da precedenti infezioni o da vaccinazione-immune escape) [7].
Sono state anche definite le Varianti di Interesse (VOI) che possiedono alcune mutazioni delle VOC, ma il cui impatto sull’epidemiologia del virus e sulle sue caratteristiche fenotipiche sono ancora incerte, e le Varianti “sotto osservazione”, (VUM, Variants under monitoring) [8, 9] che si presentano come varianti minoritarie ma degne di attenzione.
Nel maggio 2021, L’Organizzazione Mondiale della Sanità (WHO, World Health Organization) ha proposto un sistema di nomenclatura delle Varianti di SARS-CoV-2 basato sulle lettere dell’alfabeto greco (Alpha, Beta, Gamma, ecc), da utilizzare accanto alle classificazioni filogenetiche più ampiamente utilizzate, basate sui lignaggi evolutivi (Pangolin) o sui clade filogenetici (Nexstrain).
L’impatto sull’epidemiologia e sulla Sanità Pubblica dell’emergenza di mutazioni nel genoma virale di SARS-CoV-2 è stata evidenziata già all’inizio del 2020, quando la sostituzione acido aspartico con glicina (D-> G) in posizione 614 nella proteina Spike del virus, aveva conferito un evidente vantaggio in termini di trasmissibilità al lignaggio B.1, che nell’arco di pochi mesi (da gennaio a maggio del 2020) aveva sostituito il wild type (i ceppi originari A e B) in tutto il mondo [10], divenendo il progenitore di tutte le successive varianti, comprese quelle attualmente circolanti.
La successiva variante Alpha, la prima definita VOC, in quanto dotata di elevata trasmissibilità e virulenza, identificata a dicembre 2020 nel Sudest del Regno Unito, si è diffusa in tutto il mondo divenendo la variante prevalente a livello globale nel marzo 2021 [11].
A maggio dello stesso anno, un’altra VOC, la Delta, identificata inizialmente (dicembre 2020) in India, dove ha causato la seconda (e più drammatica) ondata di infezioni aveva iniziato a diffondersi a livello globale, rimpiazzando la variante Alpha. Grazie alla sua elevata trasmissibilità e alla maggiore capacità di evadere le difese immuni indotte da infezione pregressa e vaccinazione [12], era diventata la variante prevalente (> 90% degli isolati) durante l’estate 2021. L’infezione causata dalla variante Delta è stata associata ad un livello di ospedalizzazioni e di infezioni severe più elevato della variante Alpha [13]. Durante questo periodo, la variante Delta si è differenziata accumulando una serie di mutazioni che hanno dato origine a più di 130 lignaggi (denominati con il prefisso AY nella classificazione Pangolin) di cui solo alcuni dotati di mutazioni amminoacidiche nella proteina Spike in grado di conferire resistenza al trattamento con alcuni anticorpi monoclonali (come AY.49 e AY.106) [14].
A partire da dicembre 2021 è iniziata la diffusione della VOC Omicron che, già nella seconda metà di gennaio 2022, rappresentava più del 90% degli isolati in tutto il mondo. Identificata per la prima volta in Sudafrica nel novembre 2021, mostra un numero elevato di mutazioni (più di 30 nella proteina Spike, di cui 15 nella RBD) che la rendono capace di evadere dall’azione degli anticorpi neutralizzanti indotti da infezione pregressa e/o vaccinazione, conferendo quindi al virus un vantaggio rispetto a tutti gli altri genotipi in termini di trasmissibilità. Gli studi in Sudafrica suggeriscono una ridotta patogenicità della variante Omicron, rispetto alle precedenti [15, 16], tuttavia non è chiaro se ciò sia direttamente attribuibile ad una ridotta virulenza o piuttosto ad un più elevato livello di immunità protettiva nei confronti dell’infezione severa, nella popolazione [17]. Omicron è attualmente caratterizzata da una elevata variabilità che ha dato origine ad un vero e proprio “sciame” di sottolignaggi, spesso caratterizzati da mutazioni identiche che compaiono ripetutamente in linee evolutive diverse, e che rappresentano probabilmente il prodotto di un’evoluzione convergente dovuta alla pressione selettiva esercitata dall’immune escape [18, 19].
La collaborazione fra i diversi centri internazionali e l’utilizzo di tutte le risorse necessarie risulta fondamentale per consentire a tutti i Paesi del mondo di avere un adeguato livello di caratterizzazione genomica di SARS-CoV-2 e, in prospettiva, anche di altri microrganismi emergenti.
Da gennaio 2020, nel corso della pandemia da COVID-19, a oggi (novembre 2022) con grande sforzo sono state caratterizzate e messe a disposizione sequenze di 13 milioni di genomi di SARS-CoV-2 depositati su banca dati GISAID.
Inizialmente, il livello di sorveglianza genomica considerato soddisfacente era stato fissato al sequenziamento di una proporzione di campioni positivi al SARS-CoV-2 intorno al 5-10% [20]. Tuttavia, dato il numero ridotto di Paesi in grado di produrre una quantità di sequenze genomiche così elevata, si è fissato un numero minimo di genomi da caratterizzare per avere la possibilità di individuare per tempo una variante minoritaria (≤ 5%) [21]. Più recentemente, è stato stimato che la caratterizzazione di una proporzione di casi intorno allo 0,5% in un tempo di 21 giorni sia sufficiente ad un buon livello di sorveglianza [22].
Sulla base di studi fin qui condotti, emerge una elevata eterogeneità nella capacità di sequenziamento tra i diversi paesi del mondo dimostrando come i paesi a più alto livello di reddito (HIC, High Income Countries) hanno avuto una capacità 10 volte maggiore rispetto agli altri paesi, caratterizzando in media il 4% di campioni positivi contro lo 0,3% dei paesi a basso o medio reddito (LMIC, Low Middle Income Countries) [22].
Bassi livelli di sequenziamento sono chiaramente emersi nella regione dell’Africa, Mediterraneo dell’Est e Sud-Est Asiatico. I livelli più alti di campioni positivi sequenziati si osservano invece in Europa (media 3,4%), Pacifico Occidentale (2,7%) e Americhe (2,0%). Tra i Paesi del mondo quelli con la più elevata proporzione di campioni positivi sequenziati sono stati Islanda, Danimarca, Nuova Zelanda, Australia, Regno Unito, Finlandia, Canada, Norvegia, Lussemburgo e Giappone. Le ragioni di un divario di questo tipo tra HIC e LMIC sono per lo più di natura socioeconomica. Naturalmente anche in regioni a basso rendimento medio di sequenze prodotte si possono osservare singoli Paesi in grado di coprire una proporzione elevata e soddisfacente di campioni positivi, come è il caso del Gambia (7%), Djbuti e Burkina Faso (intorno al 2%) [23].
Anche il tempo necessario per la pubblicazione delle sequenze genomiche sui principali database è risultata più breve negli HIC rispetto ai LMIC e si è ridotta a partire dalla individuazione delle VOC. In particolare, nel Nord Europa, si è passati da un tempo medio di 20 giorni a circa 10 giorni. Gli ECDC hanno stimato il numero di sequenze che devono essere caratterizzate settimanalmente, in base al numero di casi COVID-19 positivi, per poter individuare una variante [21]. Sulla base di questa stima, sono stati formulati 4 livelli di prestazioni in termini di volume di sequenze prodotte: il livello 1a per quei Paesi il cui volume di sequenziamento settimanale consente di individuare varianti con una prevalenza ≤ 1%; il livello 1b per quelli con una capacità di discriminazione superiore all’1% ma inferiore al 2,5%; il livello 1c, per quelli tra il 2,5% e il 5%; infine, il livello 2, per quelli che non hanno una capacità di stimare la presenza di una variante < 5% con sufficiente precisione.
Sulla base di questa classificazione, nella settimana 44 del 2022, 3 Paesi (Austria, Danimarca e Germania) risultano 1a; 4 Paesi (Estonia, Francia, Lettonia e Olanda) risultano 1b e 3 Paesi (Italia, Lussemburgo e Svezia) risultano 1c. Tutti gli altri (17) risultano L2, con l’eccezione di Cipro, Finlandia e Lituania, che non hanno fornito dati per le settimane 43-44 [24].
Per quanto riguarda l’Italia, l’ultimo rapporto disponibile dell’Istituto Superiore di Sanità relativo alla prevalenza e distribuzione delle varianti di SARS-CoV-2 [25], riporta che l’Italia sequenzia un numero di casi necessario ad individuare con sufficiente precisione le varianti virali con una prevalenza tra il 2,5 e il 5,0% (livello 1c). Alla fine di ottobre 2022, la piattaforma italiana I-Co-Gen (Italian COVID-19 Genomic), ha ricevuto circa 171.000 sequenze prodotte da circa 70 laboratori presenti in tutte le Regioni Italiane. In particolare, i dati ECDC ottenuti dal database GISAID, indicano per il periodo gennaio-inizio di novembre (settimana 45) 2022 una media di 1392 sequenze a settimana (escludendo le ultime 3 settimane) con un minimo di 461 (ad agosto) e un massimo di 3407 (a febbraio), mentre nelle ultime 3 settimane il numero di sequenze depositate scende da 473 (settimana 43) a 24 (settimana 45) (Fig. 1). Tale numerosità di sequenze corrisponde ad una proporzione di campioni positivi analizzati che varia tra l’1,1% e lo 0,2%, con una media che si attesta intorno allo 0,4% di campioni caratterizzati settimanalmente. A questa performance contribuiscono anche iniziative di gruppi di studio e di reti di expertise che includono centri clinici e laboratori specializzati in sequenziamento sparsi in tutto il Paese, come ad esempio il gruppo SCIRE (SARS-CoV-2 Italian Research Enterprise-Collaborative Group) [26].
Un caso particolare in Europa è rappresentato dal Regno Unito, che ha caratterizzato fino al 15% circa dei casi positivi alla PCR, con un numero di sequenze che ha superato anche le 10000 sequenze prodotte a settimana [27]. Questi numeri sono il risultato della creazione, nel marzo 2020, di un consorzio, denominato COG-UK (COVID-19 Genomics UK Consortium), una vera e propria rete di laboratori di genomica, comprendente le agenzie di Public Health, i laboratori ospedalieri, quelli di ricerca afferenti a più di 15 Università ed enti privati (tra cui Wellcome Sanger Institute) che è stato finanziato, già nell’aprile 2020, con 20 milioni di sterline provenienti da fondi sia pubblici che privati. Le finalità del network erano quelle di fornire dati, strumenti e metodi di analisi utili alle scelte di Sanità Pubblica durante la pandemia COVID-19, allo studio dei meccanismi alla base della variabilità genetica di SARS-CoV-2 e al supporto di studi di ricerca tra cui quelli farmacologici o volti alla prevenzione dell’infezione con vaccini o metodi non farmacologici [27]. Questa esperienza può rappresentare un valido modello a livello mondiale per il futuro sviluppo delle reti di genomica, elemento ormai centrale della preparedness nei confronti delle infezioni emergenti.
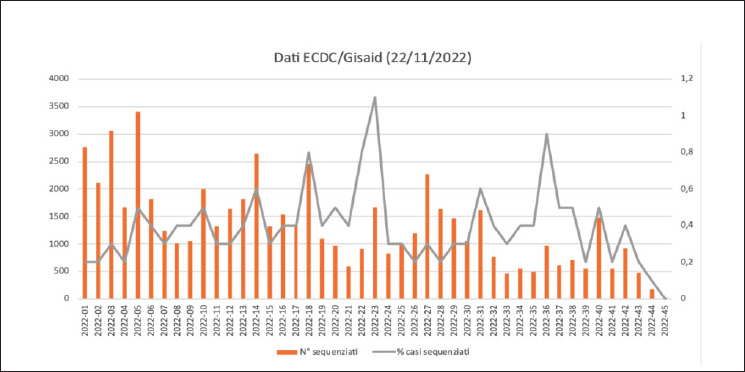
Distribuzione del numero di sequenziati in Italia dalla settimana 01 alla settimana 45 nel 2022.
COronaVIrus Disease 2019 (COVID-19) è definita come una patologia multisistemica con un’ampia varietà di manifestazioni cliniche [1] e la possibilità di una prognosi severa o fatale influenzata da diversi fattori [2]. Nonostante la vaccinazione rappresenti una strategia efficace e sicura per la prevenzione delle ospedalizzazioni e complicanze [3], i pazienti spesso riportano conseguenze a lungo termine dopo la risoluzione della fase acuta. Ciò ha portato alla definizione del “long-COVID” per indicare un insieme di sintomi che generalmente insorgono in individui tre mesi dopo l’insorgenza di COVID-19, permangono per almeno due mesi e non possono essere spiegati da una diagnosi alternativa [4]. I sintomi comuni includono affaticamento profondo e prolungato, dispnea, dolore muscolare, articolare e toracico, disfunzione cognitiva (brain fog), ma anche altri con impatto nella qualità di vita quotidiana [5]. Considerando la variabilità nell’entità, durata e tempo di insorgenza dei sintomi, il long-COVID è stato classificato in cinque categorie [6] (Tab. I).
Possibili cause del long-COVID potrebbero includere: il danno agli organi causato da un’eccessiva risposta infiammatoria attivata dal virus; serbatoi persistenti del virus in alcuni tessuti che potrebbero innescare una morbilità post-infezione; alterazioni del microbioma; problemi legati a coagulazione o produzione di autoanticorpi come risultato del mimetismo molecolare tra peptidi/metaboliti virali e quelli espressi dall’ospite generando una risposta infiammatoria intrinseca [7]. Sebbene casi di long-COVID siano sempre più descritti, sono ancora carenti le informazioni relative a prevalenza, determinanti e caratteristiche utili a predire un decorso prolungato della malattia, che rappresenterà un importante problema di Sanità Pubblica nei prossini anni. Questo lavoro riassume i dati a oggi disponibili sui fattori predisponenti il long-COVID, ponendo le basi per la definizione di un modello probabilistico.
È stata effettuata un’approfondita ricerca bibliografica nei principali database Medline/Pubmed, EMBASE e ISI Web of Science, utilizzando le parole chiavi “long-COVID” AND “genetic factors” OR “risk factors”.
L’analisi ha evidenziato che fattori genetici, età, sesso femminile, ospedalizzazione durante la fase acuta di malattia, presenza di autoanticorpi e di RNA di SARS-CoV-2 e del virus Epstein-Barr (EBV), diabete ed elevato indice di massa corporea (IMC) contribuiscono a un rischio più elevato di sviluppo del long-COVID.
Mutazioni nei geni codificanti per i componenti immunitari innati come i recettori toll-like (TLR) e la lectina 2 legante il mannosio (MBL2) svolgono un ruolo critico nella capacità del sistema immunitario di riconoscere SARS-CoV-2, avviare una risposta precoce per la clearance virale e prevenire lo sviluppo di sintomi gravi [8]. Dal momento che tali mutazioni determinano alterazioni nella risposta all’infezione, potrebbero essere determinanti anche nel long-COVID [7]. È stato dimostrato che pazienti con COVID-19 grave presentano mutazioni nei geni coinvolti nella regolazione dell’immunità, quali interferoni (IFN) di tipo I e III, suggerendo la presenza di tali mutazioni anche nel long-COVID [9]. Considerando, inoltre, che problemi di coagulazione o coagulazione indotta da SARS-CoV-2 contribuiscono a sequele, varianti nei geni che hanno un impatto sulla coagulazione, come il fattore V di Leiden, potrebbero essere riscontrate in questi pazienti [7].
Una revisione sistematica della letteratura [8] condotta nel 2021 ha riportato che individui di età compresa tra 35 e 49 anni presentavano più frequentemente sintomi persistenti (27%) rispetto a quelli nel gruppo 50-69 anni (26%) e ≥ 70 anni (18%). Tuttavia, un recente studio multicentrico [10] che ha incluso 2333 individui provenienti da Italia, Israele, Svizzera e Spagna, ha evidenziato che la frequenza dei sintomi più comuni (affaticamento e dispnea) di long-COVID a 5 mesi dalla diagnosi di COVID-19, è stata significativamente più elevata (80 vs 64%) negli individui di età > 65 anni rispetto al gruppo 18-65 anni. Se nei bambini e adolescenti COVID-19 si è generalmente manifestato con sintomi lievi o addirittura assenti, uno studio su 129 pazienti di età ≤ 18 anni condotto nel centro Italia [11] ha riportato che il 67% e 27% rispettivamente manifestavano almeno un sintomo persistente a 60-120 giorni e dopo 120 giorni o più dalla diagnosi di COVID.
La prevalenza di almeno un sintomo di long-COVID è stata più elevata nelle donne rispetto agli uomini [8]. In uno studio svolto nel nord Italia su 377 adulti con diagnosi di COVID-19 [12] è stato osservato che, a un tempo mediano di 44 giorni dall’inizio della sintomatologia, il 70% presentava sintomi persistenti, in particolare l’82% delle donne rispetto al 62% dei maschi (p < 0,0001). Nelle donne, sono state anche osservate frequenze più elevate della maggior parte dei sintomi fisici e di tutti quelli psicologici.
In uno studio, 210 pazienti COVID, di cui il 70% era stato ricoverato in fase acuta, hanno fornito campioni di sangue nel corso di 2-3 mesi dalla diagnosi; il 24% e 37% riportavano rispettivamente uno-due e tre o più sintomi associati a long-COVID all’ultimo follow-up [13]. Sono state, inoltre, effettuate le misurazioni di autoanticorpi neutralizzanti IFN di tipo I, già associati precedentemente a disfunzioni immunitarie e morte da COVID-19, sia al momento della diagnosi di infezione, sia 2-3 mesi dopo l’insorgenza dei sintomi (T3); il 44% presentava autoanticorpi maturi a T3 e il 56% già alla diagnosi, indicando la loro possibile presenza anche prima dell’infezione. Inoltre, pazienti con problemi neurologici a T3 avevano elevati livelli di autoanticorpi IgG contro la proteina del nucleocapside di SARS-CoV-2, mentre autoanticorpi contro IFN-α2 erano associati in modo univoco a sequele di tipo respiratorio.
Frammenti di mRNA di SARS-CoV-2 (RNAemia), osservati nel 25% dei pazienti al momento della diagnosi dell’infezione, sono risultati significativamente correlati con sintomi relativi a difficoltà di concentrazione e di memoria dopo la fase acuta della malattia [13].
È stato riportato che i sintomi di long-COVID possano essere una conseguenza dell’infiammazione indotta dalla riattivazione di EBV, in quanto la viremia da EBV è stata osservata in pazienti testati a 2-3 mesi dalla diagnosi, in associazione con problemi persistenti di memoria e stanchezza [13]. In un altro studio, il 67% dei soggetti con long-COVID presentava titoli positivi di IgG per l’antigene precoce (EA-D) di EBV o di IgM per l’antigene del capside virale (VCA) di EBV [14].
Pazienti con T2D e infezione pregressa da SARS-CoV-2 presentano un rischio maggiore di sviluppo di long-COVID rispetto ai non diabetici [15]. COVID-19 può esacerbare T2D, in quanto le cellule β pancreatiche possono essere direttamente infettate da SARS-CoV-2, con conseguenti danni e incremento dell’insulino-resistenza. D’altra parte, se scarsamente controllato, T2D può causare danni agli organi e lesioni microvascolari, favorendo l’espressione di citochine pro-infiammatorie che inibiscono la via di segnalazione dell’insulina [13]. Uno studio ha evidenziato che il 53% dei pazienti con T2D e COVID-19 manifestava sintomi da affaticamento tipici del long-COVID rispetto al 37% di pazienti T2D non affetti da COVID-19 (p = 0,018) [16].
Uno studio di coorte condotto nel sud Italia ha riportato che circa il 51% dei pazienti con sintomi di long-COVID aveva IMC > 25 kg m-2 rispetto al 39% nel gruppo non long-COVID (p = 0,029) [17]. Anche il valore medio di IMC nei soggetti con long-COVID era significativamente più alto rispetto a quello osservato nel gruppo non-long COVID (25,9 kg m-2 vs 24,8 kg m-2; p = 0,020).
Il tipo di variante potrebbe essere associato a long-COVID. In uno studio condotto nel centro Italia sono stati confrontati i dati relativi a pazienti raccolti nel periodo marzo-dicembre 2020 in cui il ceppo wild-type era prevalente, rispetto ai casi diagnosticati nel periodo gennaio-aprile 2021 quando era predominante la variante B.1.1.7 o Alpha [18]. Sebbene la frequenza di sintomi persistenti e di quelli cronici più comuni (mancanza di respiro e stanchezza cronica) fosse simile, la prevalenza di mialgia, nebbia cerebrale e ansia/depressione aumentava significativamente in associazione al ceppo wild-type, mentre anosmia e disgeusia erano maggiormente riscontrate nei casi da variante Alpha.
Il long-COVID è una condizione complessa ed eterogenea con coinvolgimenti multisistemici. Sebbene i dati siano ancora contrastanti [8], l’età avanzata potrebbe contribuire allo sviluppo per la co-presenza di fattori di rischio come inattività fisica, comorbidità e uso di farmaci [10]. Meccanismi immunologici sarebbero invece responsabili di conseguenze dell’infezione a lungo termine nei bambini e adolescenti [11]. Il rischio di long-COVID, inoltre, aumenta di circa tre volte nella popolazione femminile rispetto a quella maschile per la risposta immunologica che si attiva dopo l’infezione o per la possibilità che SARS-CoV-2 generi uno stato infiammatorio cronico con livelli di citochine più elevati nelle donne [12]. La variabilità dei quadri clinici riscontrabile nei soggetti con long-COVID potrebbe essere associata al processo dell’autoimmunità, che si attiva dopo l’infezione ed è in grado di generare autoanticorpi diretti contro proteine coinvolte in un’ampia gamma di funzioni immunologiche [7, 13]. Molti sintomi nel long-COVID potrebbero essere una conseguenza della riattivazione di EBV indotta dallo stato infiammatorio da COVID-19 [14]. Anche il diabete è stato associato al long-COVID basandosi sull’osservazione che la dispnea persistente è più comune in individui con T2D scarsamente controllato [15, 16]. Tuttavia, se da un lato individui T2D presentano un maggior rischio di sviluppo di long-COVID, dall’altro soggetti con long-COVID hanno maggiori probabilità di sviluppare T2D. È possibile, infine, suggerire l’associazione tra elevato IMC e long-COVID, essendo stata osservata una maggiore predisposizione allo sviluppo nei pazienti in condizione di sovrappeso o obesità [17].
Considerando l’ampio spettro di fenotipi associati al long-COVID, ulteriori indagini sono necessarie per identificare i fattori di rischio, sebbene alcuni di essi, come T2D, autoanticorpi e RNA di SARS-CoV-2 e EBV, possano essere rilevati già alla diagnosi di COVID-19 e potrebbero essere validi nel predire l’insorgenza di long-COVID. La medicina personalizzata o di precisione rappresenta pertanto una valida strategia per comprendere aspetti associati allo sviluppo di long-COVID, al fine di implementare efficaci interventi di prevenzione e controllo adeguati alle caratteristiche e ai bisogni del singolo paziente.
Classificazione e caratteristiche del long-COVID [6].
| Tipo 1 | Sintomi con tempi variabili di recupero e riabilitazione direttamente correlati a gravità dell’infezione, danno dell’organo bersaglio e condizioni mediche preesistenti al momento dell’infezione iniziale |
| Tipo 2 | Sintomi persistenti per 6 settimane dal momento dell’infezione iniziale |
| Tipo 3 | Periodo di quiescenza o guarigione quasi completa dopo l’infezione iniziale, seguito da un ritorno dei sintomi che persistono per ≥ 3 mesi (Tipo 3A) o ≥ 6 mesi (Tipo 3B) |
| Tipo 4 | Inizialmente asintomatico al riscontro di un test positivo per SARS-CoV-2, con sintomi che insorgono dopo 1-3 mesi (Tipo 4A) o ≥ 3 mesi (Tipo 4B) e persistono per periodi variabili |
| Tipo 5 | Inizialmente asintomatico o paucisintomatico al momento di un test positivo per SARS-CoV-2 e morte improvvisa entro i successivi 12 mesi |
Ogni organismo superiore, pianta o animale, incluso l’uomo è il risultato di molteplici interazioni ospite-microrganismi, in cui almeno un membro (microrganismo e/o ospite) trae vantaggio. Tutte le superfici esposte dell’organismo umano, non solo la cute – dalla mucosa buccale all’intestino, dalle mucose nasali alla superficie alveolare, ecc. – ospitano eterogenee comunità di microrganismi [1]. I patogeni e i patogeni opportunisti possono far parte di tale comunità e sono meglio noti e conosciuti sia in termini morfologici che funzionali nell’indurre danni all’organismo. D’altra parte sono da molto tempo conosciuti anche numerosi microrganismi con attività tendenzialmente benefiche per l’organismo e comunemente offerti come probiotici, sebbene non siano sempre esaustive le evidenze scientifiche sul beneficio indotto [2].
L’introduzione di metodiche biomolecolari avanzate (in particolare del sequenziamento di nuova generazione, NGS), lo sviluppo della bioinformatica e l’introduzione di un approccio ecosistemico hanno fornito degli strumenti utili alla nascita e rapida crescita della microbiomica, intesa quale disciplina volta allo studio del microbioma con metodiche biomolecolari avanzate [3].
Il microbiota è definito come l’insieme di microrganismi (comunità microbica) che esistono in uno stesso spazio e nello stesso tempo mentre il microbioma è definito come l’insieme del complemento genico della stessa comunità [4]. Il microbiota umano è coinvolto in vari processi fisiologici: metabolizzazione e digestione dei nutrienti, stimolo delle risposte immunitarie della mucosa, produzione delle vitamine essenziali, stimolo dello sviluppo fisio-anatomico e supporto nella resistenza alla colonizzazione da parte dei patogeni.
Studi meccanicistici condotti in vivo su animali gnotobiotici hanno descritto il ruolo del microbiota nel determinare differenze anatomiche e funzionali coinvolgendo per esempio intestino, fegato, polmoni, sistema nervoso, immunità e funzioni endocrine [5]. Tali studi hanno descritto alcune interazioni microbo-microbo e ospite-microbo con un ruolo determinante nel modulare il rischio di eventi morbosi acuti e cronici [6]. Le priorità in termini di conoscenza sin dal 2010, a seguito dell’introduzione dei metodi di NGS, sono state: determinare le caratteristiche del microbiota di soggetti in salute, riconoscere le perturbazioni del microbiota in condizioni patologiche conclamate, individuare le interazioni microbiota-ospite attraverso l’integrazione di diversi approcci omici per condizioni patologiche o di fragilità specifiche, in particolare nel caso della nascita pretermine, della sindrome dell’intestino irritabile e del diabete di tipo 2 [7].
Il microbiota costituisce la prima interfaccia tra esposizione ambientale e ospite e agisce sia come filtro che come modulatore degli effetti indotti dalla stessa esposizione. Per tale ragione, a oggi, lo studio del microbiota fornisce uno strumento aggiuntivo della Genomica in Sanità Pubblica, proprio per il ruolo centrale del microbiota, riconosciuto in molteplici aspetti della salute umana suggerendo delle opportunità preventive di sicuro interesse in tutte le aree della prevenzione. D’altra parte, la modulazione del microbiota è soggetta a molteplici fattori influenti che sono ampiamente sovrapposti ai determinanti di salute umana e per tale ragione è necessario applicare un approccio epidemiologico rigoroso, che integri adeguate informazioni esposomiche al fine di individuare proxy o driver di malattia depurati dall’effetto di fattori confondenti.
Scopo del presente lavoro è illustrare alcune opportunità preventive fornite dall’analisi del microbiota, supportate da evidenze scientifiche, in riferimento alle aree della prevenzione. La figura 1 mostra a quali livelli della transizione da salute a malattia il microbiota può fornire biomarker utili nell’anticipare la patogenesi, nel condurre ad una diagnosi precoce, nell’individuare risposte personalizzate ai trattamenti somministrati e nello stabilire il ripristino delle condizioni di salute.
I microrganismi che popolano il corpo umano sono in rapporto circa 10:1 con le cellule umane, pertanto non stupisce che il loro ruolo sia fondamentale nel mantenimento di una condizione di equilibrio delle funzioni fisiologiche umane. Solo una ridotta percentuale di microrganismi è patogena per l’uomo ma una interazione uomo-microbo è sempre presente pur prescindendo dal percorso patologico. Lo studio di queste interazioni conduce a nuove classificazioni dei microrganismi: probiotici [2] e/o patobionti [8]; arrivando sino a microbi e/o consorzi microbici terapeutici [9]. Negli ultimi anni la ricerca ha cercato di definire quale composizione del microbiota sia ottimale per garantire la salute dell’organismo (eubiosi). Sebbene alcune evidenze siano presenti per alcuni siti anatomici caratterizzati da una più bassa biodiversità (es. microbiota vaginale) le ampie differenze interpersonali e di popolazione rendono tale stato ancora non definibile in termini di composizione. Per contro si può invece definire con maggiore chiarezza una condizione di disbiosi, in quanto associata alla perdita di microrganismi health-associated (principalmente produttori di acidi grassi a catena corta), l’aumento di patogeni opportunisti (come batteri mucolitici, produttori di idrogeno, metano, acido solfidrico), aumento dei livelli di lipopolisaccaride con un’ampia destrutturazione dell’ecosistema microbico e compromissione dell’integrità della mucosa intestinale, infiammazione acuta della mucosa stessa con traslocazione dei batteri e dei loro frammenti, effetti tossici sugli enterociti e danno ossidativo, alterazione del pattern di citochine prodotte ed altri effetti a livello sistemico [9, 10].
I fattori influenti sin dalla nascita sull’equilibrio del microbiota sono molteplici e la loro azione può modulare i livelli di salute dell’individuo per tutta la durata della vita. Sebbene recenti evidenze mostrino un’elevata resistenza e resilienza del microbiota soprattutto nel medio e lungo periodo [11]. Ad esempio, il parto vaginale è un processo essenziale per la semina di specie microbiche sulla pelle e nell’intestino dei neonati, mentre i parti cesarei precludono il trasferimento di tali microbi madre-figlio e la prima colonizzazione è operata da microbi acquisiti dall’ambiente determinando delle differenze nel successivo sviluppo del microbiota. Altra variabile influente è rappresentata dall’uso di antibiotici ad ampio spettro che influisce drasticamente sulla composizione del microbiota. A questo proposito recenti evidenze mostrano un chiaro bioaccumulo intracellulare di questi farmaci che non viene rilevato dalle normali indagini di persistenza sierica dei principi attivi somministrati [12].
Durante il corso della vita, l’assunzione di farmaci, la modalità di allattamento e la successiva alimentazione, gli stili di vita ed i fattori ambientali modulano la composizione del microbiota. Riconoscere delle perturbazioni che anticipino uno stato di disbiosi potrebbe condurre ad una riduzione importante dell’incidenza di patologie ad elevato impatto sulla popolazione come il diabete ma anche di patologie cardiovascolari [13] e neurodegenerative [14].
Una prima indicazione di perdita dell’eubiosi è la diminuzione della biodiversità microbica [15], in termini di alfa-diversità (diversità di specie all’interno del microbiota), beta-diversità (variazione delle specie tra diversi microbioti) e gamma-diversità (la somma del numero di specie caratterizzata attraverso la lettura di diverse unità spaziali, es microbiota intestinale considerando diverse porzioni dell’intestino) [4].
Tale perdita di biodiversità sembra essere il frutto di una carente interazione uomo-ambiente dovuta alla antropizzazione che avrebbe comportato un indebolimento immunitario e una maggiore suscettibilità alle patologie autoimmuni [16] e neurodegenerative [17]. A titolo di esempio citiamo indicatori precoci di perdita di eubiosi nei bambini, nei soggetti adulti e negli anziani.
Nei bambini è stato proposto un percorso per il ciclo transgenerazionale dell’obesità [18]. L’elevato BMI pre-gravidanza, l’elevato aumento del peso durante la gravidanza così come la presenza di diabete gestazionale ed una dieta in stile occidentale (ricca di grassi) possono provocare disbiosi intestinale materna. Questa condizione può essere trasmessa direttamente al bambino e può causare disbiosi nell’intestino del bambino provocando alterazioni nella produzione di acidi grassi a catena corta, uno stato pro-infiammatorio, alterazioni epigenetiche ed un maggiore assorbimento energetico dai nutrienti ingeriti. Tali cambiamenti possono comportare la nascita di bambini di età gestazionale elevata o con un eccesso di adiposità, entrambi fattori che pongono il bambino in una condizione di aumentato rischio per obesità, disfunzioni immunologiche e steatosi epatica non alcolica più avanti nel corso della vita [18]. Seguire appropriatamente la gravidanza e intervenire precocemente mantenendo una eubiosi intestinale sia materna che del neonato interromperebbe il ciclo perpetuo dell’obesità e parte della predisposizione familiare allo sviluppo di diabete di tipo 1 e 2 [19].
Analogamente, un secondo esempio, riconoscibile in età adulta, riguarda la sindrome metabolica dove perturbazioni del microbiota intestinale anticipano ed accompagnano il cambiamento delle variabili fisiologiche utilizzate per la diagnosi: alterata glicemia, obesità, ipertensione [20]. Individuare precocemente nel microbiota una riduzione dello strato mucoso intestinale, la diminuzione dell’acetato e del butirrato prodotti ad opera dei microrganismi consentirebbe di intervenire precocemente, evitando la conseguente riduzione della produzione di sostanze antimicrobiche che regolano la presenza di profili molecolari associati ai patogeni e l’espressione del PPAR-gamma recettore nucleare coinvolto nell’omeostasi del glucosio e dei lipidi [10].
Infine, delle prospettive di prevenzione primaria sono individuabili anche durante l’anzianità dove il microbiota intestinale assume un ruolo determinante nella modulazione dell’inflammosoma. In questo caso commensali che hanno generalmente un ruolo benefico per l’eubiosi intestinale quali Akkermansia muciniphila e i produttori di acidi grassi a catena corta diminuiscono mentre le specie appartenenti al phylum Firmicutes aumentano influenzando il processo infiammatorio che coinvolge monociti e macrofagi nella mucosa e sub-mucosa intestinale [21]. Recenti ricerche hanno consentito di riconoscere generi e specie associati ad un invecchiamento in salute (es. Roseburia, Bifidobacterium, Coprococcus, Prevotella, Akkermansia muciniphila, Barnesiella) così come microrganismi con una azione opposta (es. Bacteroides fragilis, Actinomyces, Desulfovibrio) [16]. L’attenzione al mantenimento dello stato eubiotico intestinale, pur tenendo conto delle normali modificazioni dovute all’anzianità, anticiperebbe problemi di salute importanti ed eviterebbe successive co-morbosità che rendono sempre più fragile il soggetto anziano.
La fase sub-clinica è a oggi riconoscibile con maggiore affidabilità, in quanto prevede delle modificazioni del microbiota intestinale decisamente marcate e che si discostano chiaramente da uno stato eubiotico. Al fine di discutere le prospettive di sviluppo che conducano a un anticipo della diagnosi, illustriamo due esempi oggi discussi e a carico di microbioti decisamente differenti tra di loro: il microbiota vaginale e le conseguenze della disbiosi vaginale nel modulare il rischio di tumore alla cervice uterina e la modulazione del microbiota intestinale in relazione all’insorgenza del tumore del colon.
Il microbiota vaginale disbiotico conduce a vaginosi e complicanze infettive successive come le infezioni da HPV che possono condurre a lesioni neoplastiche e successiva diagnosi di tumore della cervice uterina.
Le infezioni da HPV, sebbene con genotipi ad alto rischio, non sono sufficienti a produrre lesioni significative, diviene quindi importante il coinvolgimento di diversi cofattori in grado di modularne la persistenza o l’eliminazione. In relazione a questo aspetto, la disbiosi vaginale è stata proposta come un fattore in grado di modulare significativamente l’insorgenza di stati patologici dovuti all’infezione da parte di HPV. In diversi studi è stato osservato che nella maggior parte dei casi di cancro cervicale è presente un microbiota vaginale alterato, suggerendo il potenziale coinvolgimento della disbiosi vaginale nel tumore cervicale [22-24]. Una meta-analisi, recentemente pubblicata si è focalizzata sul rapporto tra i lattobacilli vaginali e diversi esiti clinici come lo stato di infezione da HPV, neoplasia intraepiteliale cervicale ed il cancro cervicale. L’odds ratio di metanalisi valutato per i microbioti vaginali dominati da Lactobacillus crispatus rispetto a microbioti vaginali dominati da altre specie è di 0,49 (95% IC 0,31-0,79); 0,50 (95% IC 0,29-0,88), 0,17 (95% IC 0,03-1,05) rispettivamente per infezione con genotipi HPV ad alto rischio, sviluppo di neoplasie, sviluppo di cancro alla cervice uterina [25]. Il microbiota vaginale prende parte nei processi che coinvolgono l’infezione virale da HPV con un aumento della diversità microbica; fattore che, in modo opposto a quanto accade per il microbiota intestinale, si associa ad un microambiente vaginale alterato che si discosta dallo stato di salute. Risulta pertanto evidente come poter intercettare i soggetti con microbioti vaginali non dominati da Lactobacillus spp. e una più elevata biodiversità possa presentare utili prospettive per una intensificazione dei controlli, una rapida diagnosi e un approccio terapeutico precoce.
Tra le patologie tumorali a più alta incidenza, il carcinoma del colon-retto è una delle più studiate in relazione alla possibilità di integrare lo screening con una indicazione di disbiosi fecale associabile alla disbiosi generata dal tumore. In particolare è stato dimostrato che alcuni generi e specie vengano significativamente modulati durante la progressione della massa tumorale in particolare Fusobacterium nucleatum ma anche Streptococcus spp., Alistipes spp. e Prevotella spp. Inoltre, vengono significativamente modulati metaboliti fecali come l’acido solfidrico (aumenta in presenza di massa) e il butirrato (diminuisce in presenza di tumore) [5, 26]. Anche in questo caso, integrare lo screening con una valutazione della composizione del microbiota, potrebbe migliorare la sensibilità dello screening ad oggi ancora non ottimale [27].
Le prospettive più interessanti in questo ambito riguardano la modulazione dell’azione dei farmaci mediata dal microbiota e le strategie di intervento terapeutico basate sulla modulazione del microbiota.
La modulazione dell’azione dei farmaci, soprattutto immunoterapici, da parte del microbiota è un aspetto di medicina personalizzata estremamente promettente al fine di migliorare l’efficacia dell’intervento terapeutico. Infatti alla composizione del microbioma è associata la risposta antitumorale e all’efficacia clinica dei trattamenti. Ciò è stato, per esempio, dimostrato a partire da modelli animali gnotobiotici dove man mano che la colonizzazione intestinale da parte di specifici generi veniva promossa risultava aumentata l’efficacia terapeutica degli anticorpi monoclonali somministrati [28]. Inoltre, il microbiota intestinale può contribuire all’efficacia e sicurezza del farmaco trasformando per via enzimatica la sua struttura e alterandone la biodisponibilità, bioattività o tossicità. La levodopa, usata per il trattamento del morbo di Parkinson, è un discusso esempio dell’impatto microbico sull’efficacia del farmaco. Dopo la somministrazione orale, la levodopa deve essere assorbita attraverso il piccolo intestino, in modo che possa attraversare la barriera emato-encefalica ed arrivare al cervello, dove una decarbossilasi converte la levodopa nella dopamina terapeuticamente attiva. Per tale ragione la biodisponibilità della levodopa al cervello è un fattore chiave per l’efficacia del farmaco e la levodopa è spesso co-somministrata con un inibitore al fine di preservare la possibilità della levodopa di divenire attiva nel sito bersaglio. Recentemente anche diversi microrganismi del microbiota hanno mostrato sia capacità di attivazione della levodopa in dopamina, sia capacità inibenti [11]. La composizione del microbiota risulta pertanto un elemento da non sottovalutare nella somministrazione del farmaco al fine di ottenerne il massimo vantaggio terapeutico. Numerose evidenze sono inoltre disponibili sul ruolo del microbiota nella metabolizzazione non solo dei farmaci ma di tutti gli xenobiotici ai quali l’organismo è esposto, modulandone anche in questo caso biodisponibilità, bioattività e tossicità [11].
Visto il ruolo fin qua descritto del microbiota è conseguenza logica ragionare su interventi terapeutici basati sulla modulazione del microbiota stesso che ovviamente prescindono dalla semplice somministrazione di prebiotici (substrati che favoriscano alcuni gruppi microbici) o probiotici (microrganismi vivi e attivi noti per una attività benefica nei confronti dell’organismo). Per pre e probiotici la somministrazione sinergica e regolare può condurre ad un beneficio per l’organismo sebbene le evidenze scientifiche prodotte con metodologia affidabile siano spesso limitate [2].
L’intervento eticamente più accettabile e proponibile è un intervento sugli stili di vita e soprattutto sull’alimentazione. In questo modo si possono ottenere delle modulazioni interessanti del microbiota sebbene recenti evidenze mostrino come nel medio e lungo periodo il microbiota tenda a riassumere la configurazione pre intervento [11]. In particolare, è in studio la formulazione di alimenti diretti al microbiota, che possano operare alterando le configurazioni funzionali della comunità microbica intestinale di un consumatore, sia fornendo substrati per la trasformazione microbica sia promuovendo la produzione di biomolecole necessarie per uno stato di salute o ancora agendo attraverso una combinazione di questi meccanismi [6, 29]. Altra strategia di intervento è la terapia fagica che ha l’obiettivo di ridurre le popolazioni di microrganismi con effetti negativi per la salute favorendo nella competizione microrganismi con effetti positivi. D’altra parte a oggi non sono chiaramente definiti né i bersagli né le conseguenze che si potrebbero ottenere da trattamenti di questo tipo [30]. Infine, tra gli interventi basati sulla modulazione del microbiota che sono già disponibili e validati secondo una valutazione costo/efficacia troviamo sia il trapianto di microbiota fecale per il trattamento delle infezioni persistenti da Clostridium difficile vancomicina resistenti in soggetti adulti [31] che la somministrazione di consorzi microbici approvati recentemente dalla FDA a seguito di studi di fase III [32]. Per il trapianto di microbiota fecale accanto ad efficacia in diversi quadri patologici sono presenti delle controindicazioni dovute alla selezione dei donatori ed alla presenza potenziale di rischi non solo di natura infettiva per il ricevente. Il trapianto di microbiota è anche in studio per molteplici patologie sebbene l’ipotesi di trattare anche soggetti giovani comporti maggiori perplessità in termini di eventuali effetti collaterali difficilmente prevedibili sul medio e lungo termine [31].
I consorzi microbici purificati, consistono in frazioni purificate di microbioma umano, tipo il SER-109, contenente dozzine di specie microbiche selezionate per la cura delle infezioni persistenti da C. difficile, dovrebbe essere più sicuro del trapianto di microbiota fecale e somministrabile per via orale, rendendo questo farmaco commercialmente attraente. Diversi altri farmaci basati sul microbiota sono in fase I di valutazione clinica rispettivamente per melanoma metastatico, sindrome dell’intestino irritabile, sindrome metabolica, allergia alimentare; fase II per la colite ulcerosa [32].
Per quanto fino a ora descritto, in ottica di Sanità Pubblica, non possiamo prescindere dal discutere due aspetti: da una parte il possibile sinergico effetto del mantenimento dello stato di eubiosi ed una modulazione salutistica degli stili di vita attraverso interventi di promozione della salute, dall’altro la necessità di valutare l’efficacia degli interventi e di evitare l’erogazione di prestazioni ed interventi inappropriati attraverso la prevenzione quaternaria. Sia in un caso che nell’altro la valutazione dell’efficacia degli interventi attraverso metodologia epidemiologica e valutazione delle tecnologie utilizzabili in sanità costituiscono dei punti di riferimento imprescindibili. D’altra parte, rinunciare a vantaggi di salute per la popolazione dovuto alla rigidità e lentezza procedurale dei processi di valutazione comparata è elevato, pertanto è auspicabile che la ricerca possa procedere celermente appoggiata e seguita da valutazioni esaustive ed appropriate al fine di rafforzare la prevenzione delle malattie sin dai primi anni di vita attraverso strumenti basati sul microbiota umano e nel prossimo futuro sul microbiota ambientale.
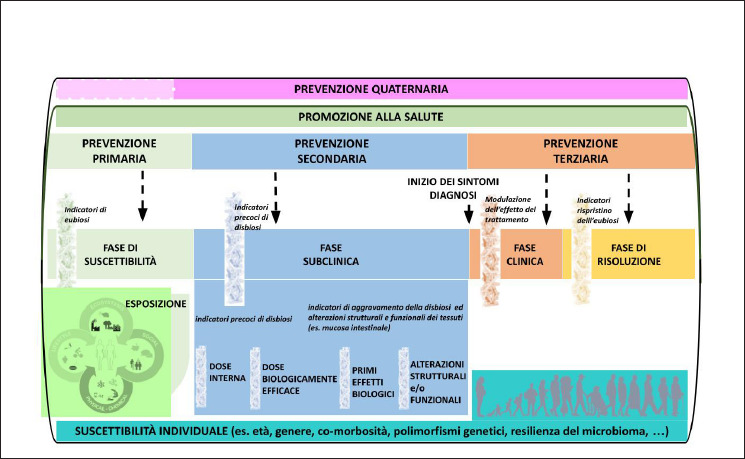
Individuazione di vari livelli di inclusione delle indagini sul microbiota umano a supporto della prevenzione nelle sue varie aeree: dal mantenimento (primaria) dalla promozione della salute (promozione), alla diagnosi precoce (secondaria), alla mitigazione degli effetti di una condizione morbosa (terziaria). Lo studio del microbiota umano potrebbe fornire strumenti per riconoscere e limitare l’uso improprio o eccessivo dei servizi sanitari (quaternaria), fornendo informazioni personalizzate che consentano di ottimizzare diagnosi e trattamenti.
I progressi scientifici nel campo della genetica e della biologia molecolare, come il sequenziamento del DNA e dell’RNA, la proteomica, la metabolomica, la biologia computazionale e le relative piattaforme di condivisione dei dati, hanno comportato nel tempo avanzamenti sempre più concreti nello sviluppo di tecnologie finalizzate ad una migliore gestione della salute dei pazienti e della popolazione [1].
Ciononostante, l’iniziale entusiasmo circa l’impatto della genomica sulla salute e sulla prevenzione delle malattie è stato smorzato dalla difficoltà di tradurre la ricerca di laboratorio nella pratica clinica e di Sanità Pubblica [2]. Infatti, se da un lato l’utilizzo incontrollato delle tecnologie genomiche può portare ad effetti dannosi sulla gestione e sulla salute dei pazienti, oltre a sottrarre risorse sanitarie già limitate [3], dall’altro lato l’identificazione di quelle applicazioni di comprovata efficacia e sostenibilità, che possono o devono essere considerate un “diritto” dei cittadini, è resa difficile dalla mancanza di una solida base di evidenze scientifiche riguardo ai rischi e ai benefici connessi all’utilizzo di queste tecnologie [3]. In questo contesto, alla luce della scarsità di risorse che caratterizza i sistemi sanitari di molti Paesi, risulta fondamentale definire un quadro metododologico di riferimento per la valutazione dell’efficacia e dei costi rispetto ai potenziali benefici a breve e a lungo termine che tali tecnologie comportano, in primis per i pazienti ma anche per i loro familiari [4].
Il processo di valutazione formale delle tecnologie in ambito sanitario è l’HTA (Health Technology Assessment) [5]. Si tratta di un approccio multidisciplinare che da una parte si concentra sulla valutazione di una o più proprietà tecniche della nuova tecnologia, dall’altra esamina l’impatto socioeconomico che l’introduzione della stessa può comportare. Quest’ultimo aspetto dovrà essere valutato congiuntamente sul piano dell’efficacia (capacità di migliorare lo stato di salute del paziente rispetto ad una precisa problematica), della sicurezza (considerazione dei rischi nell’utilizzo di una nuova tecnologia e valutazione della loro accettabilità), del rapporto tra costi ed efficacia, degli aspetti organizzativi, nonché di quelli etici, sociali e legali [6].
L’HTA è in grado di supportare scientificamente il processo decisionale a diversi livelli, a partire dagli enti regolatori che si occupano dell’introduzione nella pratica clinica di un farmaco o di un’altra tecnologia sanitaria, fino alle organizzazioni sanitarie che spesso si trovano a doverne gestire le modalità di acquisto e di erogazione [7]. L’approccio HTA integra quindi le informazioni disponibili sulle questioni cliniche, economiche, organizzative, sociali ed etiche connesse all’uso di una tecnologia sanitaria, in modo sistematico e trasparente, al fine di supportare politiche sanitarie sicure, efficaci, ed incentrate sui pazienti [6].
Tuttavia, proprio la valutazione dei rischi e dei benefici connessi all’utilizzo dei test genetici e genomici si è rivelato un processo non privo di difficoltà. Data la loro eterogeneità, il rapido sviluppo, l’impatto sui familiari e sulla società, e la mancanza di risultati comparabili, una delle principali sfide per la loro valutazione è la mancanza di modelli appropriati in grado di cogliere la complessità del valore di queste tecnologie [8]. In passato sono stati proposti diversi metodi di valutazione ad hoc per i test genetici e genomici, per lo più basati sul modello ACCE, un modello di valutazione che prende il nome dalle dimensioni incluse (vale a dire, validità analitica, validità clinica, utilità clinica, implicazioni etiche, legali e sociali), mentre solo una minoranza si è basata su rivisitazioni dell’approccio HTA [3]. Tuttavia, se da un lato i modelli basati sull’ACCE sono focalizzati prevalentemente sugli aspetti tecnici dei test genetici e genomici [3,4], dall’altra parte i modelli basati sull’HTA hanno il vantaggio di espandere la valutazione all’analisi degli aspetti economici e organizzativi legati al programma di offerta del test genetico, ma tali riadattamenti risultano spesso carenti in termini di validazione esterna e generalizzabilità [4, 9].
A livello europeo, riconoscendo l’importanza di uniformare la valutazione delle tecnologie sanitarie [10], la direttiva 2011/24/UE ha posto le basi per la formazione di una rete volontaria di istituzioni che cooperassero tra loro tramite sviluppo di metodologie di valutazione comuni e condivisione dei risultati [11]. Tale rete HTA è stata formalmente istituita nel 2013 [12], sostenuta dal punto di vista tecnico-scientifico dall’azione dell’EUnetHTA (European Network for Health Technology Assessment) tramite tre joint action susseguitesi tra il 2010 e il 2021 [13]. L’HTA Core Model è il manuale metodologico per la produzione e condivisione delle evidenze sulla valutazione delle tecnologie sanitarie sviluppato nell’ambito di queste joint action [14]. Esso permette la valutazione di una tecnologia sanitaria tramite l’analisi di nove domini: ogni dominio è suddiviso in argomenti, a loro volta declinati in quesiti, infine organizzati in quattro application che definiscono le tecnologie sanitarie cui si rivolge il modello (tecnologie diagnostiche, interventi medici e chirurgici, prodotti farmaceutici, e tecnologie di screening) [15].
Nel 2018, per rafforzare ulteriormente la cooperazione tra gli Stati Membri dell’Unione Europea nella valutazione delle tecnologie sanitarie, la Commissione Europea ha pubblicato una proposta legislativa, approvata formalmente dal Parlamento Europeo nel 2021 [16], che istituisce un quadro giuridico e procedurale per la cooperazione degli Stati Membri. Questo nuovo Regolamento Europeo di HTA, per cui è prevista una implementazione progressiva sino al 2030, cambierà sostanzialmente lo scenario europeo dell’HTA, dal momento che l’azione europea di valutazione diventerà centrale e tutti i Paesi dovranno adeguarsi [16].
In linea con le indicazioni europee, in Italia il Piano Nazionale HTA per i Dispositivi Medici (PNHTADM) ha adottato l’HTA Core Model come base metodologica [17]. Il piano è coordinato da una Cabina di Regia, costituita da Ministero della Salute, Agenzie nazionali (AGENAS e AIFA) e rappresentanti delle Regioni, che ha il compito di individuare le priorità valutative di interesse nazionale, promuovere e coordinare le attività di HTA sviluppate a livello nazionale e locale, validare indirizzi metodologici condivisi tra tutti gli attori e promuovere l’utilizzo e la diffusione delle valutazioni effettuate [17]. La piena attuazione del piano è stata però ostacolata principalmente da tre fattori, vale a dire la pandemia di COVID-19 (poiché la definizione del programma è stata completata nel 2019); la frammentazione operativa e la sovrapposizione tra le istituzioni centrali coinvolte nel programma (cioè Ministero della Salute, ISS, AGENAS, AIFA); e la mancanza di accordo sui meccanismi di finanziamento [8]. Tuttavia, in accordo con le iniziative e spinte europee dell’ultimo decennio, la recente legislazione ha sottolineato la necessità di migliorare l’efficienza del programma, ridefinendo le responsabilità e i compiti delle istituzioni centrali coinvolte, e coinvolgendo produttori e distributori nel suo finanziamento [18]. Questi aspetti sono particolarmente rilevanti nello scenario attuale, in cui è presente il rischio che le Regioni, che si trovano in fasi diverse di adozione del piano e sviluppo delle capacità di valutazione HTA, si organizzino autonomamente e promuovano iniziative disparate per l’HTA dei DM, aumentando pertanto la frammentazione [8]. In questo contesto, il ricorso ad uno dei modelli di valutazione specificamente dedicato alle applicazioni genomiche potrebbe creare confusione riguardo alle procedure metodologiche già stabilite dal PNHTADM, la cui centralità e importanza sono state recentemente ribadite in due Decreti Legislativi di agosto 2022 [19, 20]. Inoltre, la natura pubblica del sistema sanitario italiano, in cui l’equità e i vincoli di risorse sono fra le principali preoccupazioni, rende l’uso di standard comuni per la valutazione di tutti gli interventi medici il modo migliore di sostenere i decisori nell’identificazione degli interventi con il maggior potenziale di salute per la popolazione, siano essi basati sulla genomica o meno [8]. Un altro aspetto da considerare è che il nuovo Regolamento HTA prevede valutazioni congiunte a livello europeo attraverso la metodologia delineata dall’EUnetHTA Core Model, le quali dovranno poi essere riprese e contestualizzate nei diversi ambiti nazionali [16].
Per questi motivi, ricondurre formalmente la valutazione delle tecnologie genetiche o genomiche alla metodologia del sistema di HTA europeo potrebbe essere una valida opzione. A livello pratico, si potrebbe prendere come riferimento le application del modello EUnetHTA e valutare se queste possano essere utilizzate “come sono” o se debbano essere integrate con contenuti specifici quando la tecnologia in esame sia un test genetico o genomico. Questo approccio presenterebbe diversi vantaggi, tra cui la solidità della metodologia di riferimento, basata su robusti principi teorici e interamente (o quasi interamente) validata, la completezza della valutazione, che includerebbe tutti gli aspetti tecnici, clinici e di offerta del programma di test e, ultimo ma non meno importante, la possibilità di condivisione degli standard procedurali e dei risultati in tutta Europa [4]. In questo modo, la valutazione delle applicazioni genetiche e genomiche avverrebbe attraverso una metodologia HTA completa e condivisa ma al contempo capace di affrontare le caratteristiche peculiari di ciascuna tecnologia [4, 8].
A circa vent’anni dal completamento del progetto genoma umano, viviamo una fase di innovazioni tecnologiche in ambito genetico e “omico” senza precedenti. La medicina personalizzata è stata definita nel 2015 come “un modello medico che utilizza la caratterizzazione dei fenotipi e dei genotipi degli individui (ad esempio, profilo molecolare, imaging medico, dati sullo stile di vita) per personalizzare la strategia terapeutica giusta per la persona giusta al momento giusto, e/o per determinare la predisposizione alla malattia e/o per fornire una prevenzione tempestiva e mirata” [1].
Essa è dunque antitetica al paradigma “one-size-fits-all”, secondo il quale tutti i pazienti ricevono le stesse cure e secondo cui un trattamento efficace è quello di cui si conosce il beneficio per la maggior parte della popolazione target e che può essere valutato quantitativamente utilizzando la misura epidemiologica “number needed to treat” (NTT) [2]. La medicina personalizzata non si applica solo al settore diagnostico o terapeutico, ma anche a quello della prevenzione e pone quindi particolare attenzione al rischio di malattia conferito dalle caratteristiche individuali, dalla genetica e dall’ambiente, e alla sua “quantificazione” al fine di prevenire, ove possibile, la malattia stessa con una pianificazione strategica ed interventi mirati. Se lo sviluppo della malattia è inevitabile, invece, il rischio verrebbe comunque identificato in una fase più precoce, possibilmente riducendo l’impatto negativo in termini di esiti di salute e di costi. La sostenibilità dei sistemi sanitari, infatti, richiede un cambiamento radicale dell’assistenza sanitaria, passando dal trattamento delle malattie conclamate alla loro prevenzione e diagnosi precoce [3]. Oggi l’86% delle malattie si associa a patologie croniche, riconducibili principalmente agli stili di vita e, nonostante l’aumento dell’aspettativa di vita negli ultimi 50 anni, il numero medio di anni di vita vissuti con una malattia cronica in Europa è di 18 anni [4]. Questo contesto si associa ad un enorme onere economico per i sistemi sanitari europei, mettendone in discussione la sostenibilità e costringendo i decisori politici ad individuare nuovi modelli di prevenzione delle malattie, in un’ottica di assistenza sanitaria personalizzata [5]; pertanto, risulta prioritario innovare l’attuale modello di prevenzione. Gli ultimi dati disponibili suggeriscono che non esiste una relazione lineare tra gli investimenti in prevenzione, come quota della spesa sanitaria, e gli anni di vita in buona salute alla nascita; ad esempio, l’Estonia e la Finlandia, che spendono in prevenzione più della spesa media dell’Unione Europea (UE) per l’assistenza sanitaria, registrano la peggiore durata degli anni di vita in buona salute alla nascita [4]. La prevenzione personalizzata offre la potenzialità di accrescere l’efficacia e l’efficienza degli interventi preventivi e ruota attorno all’adozione di azioni mirate che combinano le informazioni biologiche (ad esempio, genetica e altri biomarcatori, dati demografici, condizioni di salute) e le caratteristiche ambientali e comportamentali e il contesto socioeconomico e culturale degli individui [6]. Per il cancro le previsioni di rischio basate sull’età e sul sesso sono molto utili in quanto entrambi sono fortemente associati al rischio di malattia. Tuttavia, con il miglioramento delle conoscenze sull’eziopatogenesi dei tumori, sono stati identificati ulteriori fattori ambientali e biologici associati all’insorgenza delle malattie oncologiche. Per molti dei tumori più comuni, i modelli di previsione del rischio, che combinano una serie di fattori diversi, possono fornire una stima del rischio stesso, ma l’impegno e i costi necessari per raccogliere tali informazioni, possono limitare l’inclusione e l’applicazione di tali modelli predittivi nell’ambito degli interventi di prevenzione e nella pratica clinica [7].
Se consideriamo la prevenzione primaria del cancro, dove gli stili di vita non salutari hanno un impatto maggiore in termini di morbilità evitabile, le evidenze suggeriscono di valutare il ruolo potenziale degli scores poligenici (PRS, varianti genetiche ereditarie comuni che contribuiscono al rischio di malattia) come misura del contributo genetico al rischio di sviluppare il cancro, da utilizzare indipendentemente o come parte dei modelli di predizione del rischio, per innescare cambiamenti nello stile di vita, indirizzando gli sforzi educativi personalizzati [8]. Lo stesso vale per la prevenzione secondaria. Ad esempio è stato dimostrato che l’uso del PRS ha un buon potere predittivo per identificare le donne a più alto e a più basso rischio di cancro al seno e potrebbe essere utilizzato per migliorare i programmi di screening della mammella [9].
A livello europeo sono ancora esigui gli esempi di medicina personalizzata applicata a percorsi strutturati in ambito di prevenzione, come lo screening del cancro al seno, mediante la ricerca di varianti patogenetiche dei geni BRCA [10], o lo screening del cancro al colon associato alla sindrome di Lynch [11, 12].
Pertanto, come possiamo accelerare l’implementazione di approcci di prevenzione personalizzata nell’assistenza sanitaria?
È certamente possibile potenziando le attuali conoscenze in ambito genetico/genomico di tutti gli attori coinvolti nel Sistema Salute, così come anche indicato nel “Piano Nazionale per l’innovazione del sistema sanitario basata sulle scienze omiche” del 2017 [13]. Sarà, quindi, essenziale implementare interventi di formazione specifica per gli operatori sanitari e i decisori politici [14, 15] nonché promuovere la valutazione dei test genetici/genomici mediante l’applicazione di metodologie evidence-based come l’Health Technology Assessment [16, 17].
Inoltre, fra i prerequisiti necessari per un cambiamento radicale nelle cure, fondato sulla medicina personalizzata, sono e saranno necessarie, azioni mirate al raggiungimento di una migliore “alfabetizzazione” in campo genetico/omico tra i cittadini e le loro famiglie [18-20], al fine di consentire decisioni appropriate sulla propria salute.
Nel corso degli ultimi anni, a livello internazionale, è notevolmente aumentato l’interesse di tutti gli stakeholder sull’importanza dell’alfabetizzazione dei cittadini sulle tematiche concernenti le scienze omiche e le tecnologie ad esse correlate, e numerosi consorzi e iniziative europee (come ad esempio ICPerMed [3, 21], 1+MG/B1MG [22], ExACT [23], PROPHET [24]) sottolineano oggi l’importanza di un maggiore coinvolgimento dei cittadini, proponendo l’implementazione di strategie mirate per la loro alfabetizzazione, affinché siano maggiormente consapevoli dei benefici e dei rischi della medicina personalizzata e siano in grado di orientare le decisioni sulla salute in base ai loro valori personali, impegnandosi in maniera consapevole e volontaria alla condivisione dei propri dati sanitari, ivi compresi quelli genetici e genomici.
A oggi, infatti, l’implementazione della medicina personalizzata è fortemente minata dallo scarso livello di conoscenze, sulla genomica e sulle altre scienze omiche, che si riscontra nella popolazione generale. I cittadini, infatti, non devono solo essere informati sui benefici della salute personalizzata, ma devono essere coinvolti attivamente nella definizione delle questioni che li riguardano e devono essere educati, incoraggiati e stimolati dai professionisti sanitari ad adottare stili di vita sani e a seguire trattamenti/interventi specifici nel medio e lungo termine.
Inoltre, il futuro successo della medicina personalizzata dipenderà dalla creazione di grandi database in grado di raccogliere diverse ed innumerevoli tipologie di dati, inclusi quelli clinici, genomici, farmacogenetici, quelli legati a fattori ambientali e agli stili di vita. La British UK Biobank, il Finland FinnGen Project e la Estonian Biobank sono alcuni esempi di queste banche dati già realizzate su larga scala.
Tuttavia, la realizzazione di tali infrastrutture dipenderà dalla partecipazione volontaria di popolazioni sane e su larga scala. La diversità biologica e la rappresentatività di tutte le etnie saranno, quindi, essenziali per applicare le nuove scoperte, nell’ambito della salute personalizzata, a tutte le popolazioni, anche al fine di non esacerbare le disparità sociali e di salute. Esistono, infatti, forti disuguaglianze nello stato di salute e nell’aspettativa di vita tra i diversi gruppi di popolazione, disuguaglianze che sono legate a molti fattori, tra cui l’eterogeneità nell’esposizione ai fattori di rischio e nell’accesso all’assistenza sanitaria [7].
L’accesso ai dati si baserà, in ultima analisi, sulla conoscenza e sulla fiducia che i pazienti e i cittadini manifesteranno nei confronti della scienza. Secondo diverse valutazioni condotte nell’UE, l’atteggiamento attuale dei cittadini, il loro interesse a ricevere ulteriori informazioni e la disponibilità a partecipare attivamente alle ricerche scientifiche in ambito di medicina personalizzata, sono generalmente molto soddisfacenti, con una preoccupazione comune su come i dati personali vengono utilizzati e da quale istituzione [25].
Risulta, quindi, evidente che la medicina personalizzata può davvero trasformare l’assistenza sanitaria nel prossimo decennio, rendendola più sostenibile nel lungo periodo. Per fare ciò però ci si dovrà concentrare su alcune aree chiave, quali: la creazione di enormi coorti longitudinali con l’applicazione di tecniche avanzate di elaborazione dei dati; l’introduzione della genomica nella pratica clinica insieme alla valutazione congiunta di caratteristiche fenotipiche, esposizioni ambientali e stili di vita; l’identificazione di priorità e azioni che consentano di garantire a tutti cittadini/pazienti l’opportunità di raccogliere i benefici per la salute derivanti dai progressi della medicina personalizzata.