

Abstract
Rhodopsin dephosphorylation in Drosophila is a calcium-dependent process that appears to be catalyzed by the protein product of the rdgC gene. Two vertebrate rdgC homologs, PPEF-1 and PPEF-2, have been identified. PPEF-1 transcripts are present at low levels in the retina, while PPEF-2 transcripts and PPEF-2 protein are abundant in photoreceptors. To determine if PPEF-2 alone or in combination with PPEF-1 plays a role in rhodopsin dephosphorylation and to determine if retinal degeneration accompanies mutation of PPEF-1 and/or PPEF-2, we have produced mice carrying targeted disruptions in the PPEF-1 and PPEF-2 genes. Loss of either or both PPEFs has little or no effect on rod function, as mice lacking both PPEF-1 and PPEF-2 show little or no changes in the electroretinogram and PPEF-2−/− mice show normal single-cell responses to light in suction pipette recordings. Light-dependent rhodopsin phosphorylation and dephosphorylation are also normal or nearly normal as determined by (i) immunostaining of PPEF-2−/− retinas with the phosphorhodopsin-specific antibody RT-97 and (ii) mass spectrometry of C-terminal rhodopsin peptides from mice lacking both PPEF-1 and PPEF-2. Finally, PPEF-2−/− retinas show normal histology at 1 year of age, and retinas from mice lacking both PPEF-1 and PPEF-2 show normal histology at 3 months of age, the latest time examined. These data indicate that, in contrast to loss of rdgC function in Drosophila, elimination of PPEF function does not cause retinal degeneration in vertebrates.
Vertebrate and invertebrate phototransduction is initiated after light absorption by 11-cis retinal bound to the G protein-coupled receptor rhodopsin, leading to photoisomerization of retinal to the all-trans form, conversion of rhodopsin to its active state, and G protein activation (35, 43). Downregulation of the light response in both systems involves phosphorylation of rhodopsin at its C terminus (8, 9, 25, 27) and subsequent binding of arrestin (4, 11, 13, 14, 16, 30, 50, 53). Regeneration of rhodopsin requires rhodopsin dephosphorylation, arrestin release, and return of the chromophore back to the 11-cis configuration (16, 35, 37).
Mutations that affect the rhodopsin cycle cause photoreceptor dysfunction and/or degeneration in both Drosophila and humans. For example, retinal degeneration in Drosophila can be caused by mutations in the ninaE gene (10, 19), which encodes the opsin in the photoreceptors R1 to R6, and defects in the light response are seen in arrestin mutants (11, 35). Similarly, in humans, defective rod function and, in many cases, retinal degeneration can be caused by mutations in the rhodopsin, arrestin, or rhodopsin kinase genes (reviewed in reference 36).
One part of the rhodopsin cycle that is still incompletely understood is rhodopsin dephosphorylation. In Drosophila, dephosphorylation is thought to be catalyzed by the protein product of the retinal degeneration C (rdgC) gene. rdgC mutants demonstrate abnormal responses to intense light flashes and a rapid, light-dependent photoreceptor degeneration (18, 41, 42, 49). In vertebrates, protein phosphatase 2A (PP2A) is thought to mediate this dephosphorylation. PP2A has been immunolocalized to rod outer segments (ROS) and shown to dephosphorylate rhodopsin in vitro (12, 29, 51). In addition, an enzymatic activity referred to as calcium-activated opsin phosphatase activity has been identified in extracts from bovine ROS (20), but the protein (or proteins) responsible for this activity has not been identified.
Two vertebrate homologs of rdgC have been discovered and named protein phosphatase with EF hands 1 and 2 (PPEF-1 and -2) for their most salient features—a serine/threonine-specific protein phosphatase catalytic domain and two consensus C-terminal EF hands (15, 26, 38). PPEF-1 transcripts have been localized by in situ hybridization to the inner ear, the dorsal root ganglia, and several brain stem nuclei in the developing mouse (26). PPEF-1 transcripts are also present at low levels in the retina and can be detected in retina RNA by reverse transcriptase PCR (RT-PCR) (15). PPEF-2 transcripts have been localized by in situ hybridization to photoreceptors and pinealocytes where they are present at high levels, and the PPEF-2 protein has been localized by immunostaining to photoreceptor inner segments (38).
To test whether vertebrate PPEF-1 and PPEF-2 contribute to rhodopsin dephosphorylation in vivo and to test whether, like rdgC, their loss causes retinal degeneration, we constructed mice carrying targeted disruptions of both the PPEF-1 and PPEF-2 genes. We show here that in PPEF mutant mice, rod light responses and rhodopsin dephosphorylation kinetics are normal and that there is no evidence of retinal degeneration. These data suggest that, despite their high degrees of homology, Drosophila rdgC and vertebrate PPEF-1 and PPEF-2 play distinct roles in photoreceptor cells.
MATERIALS AND METHODS
Construction of PPEF-1 targeting vector.
Lambda phage clones containing overlapping regions of the mouse PPEF-1 genomic locus were isolated after library screening with a human PPEF-1 cDNA probe. The 5′ homology region, consisting of a 3.5-kb DNA fragment extending from a StuI site to a point 62 bp from the 3′ end of exon 5, was constructed by a combination of subcloning and Bal 31 digestion. This 5′ homology region was cloned into pLacF (33) such that the lacZ sequence would be in frame with the truncated PPEF-1 open reading frame, and the 5′ homology region and lacZ were then cloned into pNeoTK just 5′ of the neomycin cassette. The 3′ homology region, consisting of a 2.4-kb EcoRI fragment from intron 5, was subsequently cloned into pNeoTK just 3′ of the neomycin cassette to produce the final knockout vector. Oligonucleotide linkers containing a HindIII site were ligated onto the 3′ homology fragment before cloning into pNeoTK to facilitate screening for targeting vector integration by Southern blotting.
Construction of PPEF-2 targeting vector.
Genomic DNA corresponding to the mouse PPEF-2 genomic locus was obtained by screening lambda phage and bacterial artificial chromosome libraries made from 129/Sv mouse genomic DNA (Genome Systems). The 5′ homology region, consisting of a 3.6-kb BamHI-BglII fragment ending 41 bp from the 3′ end of exon 6, was then inserted along with the lacZ gene into the region 5′ of the neo cassette of pNeoTK such that the lacZ sequences would be in frame with the truncated PPEF-2 open reading frame. A 3′ homology region consisting of a 5.0-kb StuI fragment from intron 9 was subsequently cloned into pNeoTK just 3′ of the neo cassette to produce the final knockout vector.
Targeting of PPEF-1 and PPEF-2 in ES cells.
Thirty micrograms of the NotI-linearized PPEF-1 and PPEF-2 targeting vectors were independently electroporated into R1 embryonic stem (ES) cells. After 9 days of selection on G418 and ganciclovir, ES colonies were picked, trypsinized, plated onto mouse embryo fibroblasts in 96-well trays, and amplified. Clones successfully incorporating the PPEF-1 targeting construct were identified by Southern blotting of HindIII-digested ES cell genomic DNA and by using a probe generated from a 1.0-kb EcoRI-XbaI fragment corresponding to the region just 3′ to the 3′ homology region. The wild-type PPEF-1 allele produces a 5.7-kb fragment, while the targeted allele produces a single 3.9-kb fragment, consistent with the male origin of R1 ES cells and PPEF-1 localization to the X chromosome (26). Putative mutant clones were confirmed for correct integration at the 5′ end by Southern blotting of KpnI-digested ES cell genomic DNA and using a probe generated from a 300-bp PCR fragment corresponding to a region 5′ of the 5′ homology region. Wild-type and PPEF-1-targeted alleles produce 18- and 23-kb fragments, respectively. One clone representing the correct homologous recombination event was identified among 700 colonies screened by Southern blotting.
Clones successfully incorporating the PPEF-2 construct were identified by Southern blotting of BglII-digested ES cell genomic DNA and by using a probe generated from a 250-bp SalI-BamHI fragment corresponding to the region 5′ of the 5′ homology region. Wild-type and targeted alleles produce 4.5- and 8.2-kb fragments, respectively. Putative mutant clones were then checked for correct integration at the 3′ end by Southern blotting of SpeI-digested ES cell genomic DNA by using a probe generated from a 200-bp PCR product corresponding to the region 3′ of the 3′ homology region. Wild-type and targeted alleles produce 10.5- and 5.5-kb fragments, respectively. Five clones representing the correct homologous recombination event were identified among 300 colonies screened by Southern blotting.
Generation of PPEF mutant mice.
Positive ES cell clones were injected into the blastocelic cavity of C57BL6 embryos, and the resulting embryos were implanted into a pseudopregnant females. Chimeric mice were bred to wild-type C57BL6 females. Genotyping was performed by Southern blotting of mouse tail DNA as described above for ES cell DNA, using the PPEF-1 3′ probe and the PPEF-2 5′ probe.
Suction pipette recordings from wild-type and PPEF-2−/− mice.
The procedure for recording from individual mouse rods with a suction pipette was similar to that described by Sung et al. (44). After dark adaptation overnight, a wild-type or PPEF-2−/− mouse was euthanatized by CO2 asphyxiation under dim red light. All subsequent procedures were performed under infrared light. The retina was isolated from an enucleated eye in chilled, oxygenated Leibovitz's L-15 medium (GIBCO) and cut into several pieces. Each piece of retina was placed photoreceptor side up on a glass capillary array (10-mm-diameter capillaries; Galileo Electro-Optics, Sturbridge, Mass.) on which the retina was held by suction, and the vitreous humor was removed by moving a razor blade between the retina and the array. The retinal pieces were stored in L-15 medium on ice until use. When needed, a piece of retina was chopped, under L-15 medium containing 8 mg of DNase I (Sigma)/ml, with a razor blade mounted on a lever arm and a suspension of small retinal fragments was transferred into the recording chamber. The chamber temperature was held at 36 to 38°C by continuous perfusion with heated solution buffered with bicarbonate and bubbled with 95% O2–5% CO2, pH 7.4. The outer segment of an isolated rod, or a rod projecting from a small fragment of retina, was drawn into a suction pipette connected to a current-to-voltage converter. The recorded membrane current was filtered with a low-pass, eight-pole Bessel filter at 30 Hz and was digitized with pCLAMP6.
The suction pipette was filled with a solution containing (in millimolars): 134.5 Na+, 3.6 K+, 2.4 Mg2+, 1.2 Ca2+, 136.3 Cl−, 3 succinate, 3 l-glutamate, 10 glucose, 10 HEPES, pH 7.4, and 0.02 EDTA plus basal medium Eagle amino acid supplement and basal medium Eagle vitamin supplement (GIBCO). The perfusion medium was the same except that 20 mM NaHCO3 replaced an equimolar amount of NaCl. The optical bench design was as previously described (3). Unpolarized, 8-ms flashes at 500 nm (10-nm bandwidth) were used for stimulation throughout.
Rhodopsin phosphorylation and dephospnorylation.
Dark-adapted mice were anesthetized with a xylazine/ketamine mix, and their pupils were dilated by topical application of tropicamide and phenylephrine before being exposed to an intense flash of light that bleached 20% of the rhodopsin in the eye. Mice were sacrificed at 3, 30, or 120 min following the bleach and were enucleated under infrared illumination. The dissected eyes were immediately homogenized in deionized 7 M urea using an Ultra-Turrax homogenizer. Membranes were harvested by centrifugation at 54,000 rpm in a Beckman Optima tabletop ultracentrifuge and were washed twice with sterile H2O to remove soluble proteins. The membranes were then incubated with Asp-N proteinase (2 μl of a 20-μg/ml solution) at room temperature for 15 to 17 h. Soluble peptides were collected in the supernatant after centrifugation at 54,000 rpm and were subjected to high-pressure liquid chromatography-mass spectrometry as previously described (17).
ERG recordings.
Mice were dark adapted overnight and anesthetized with a ketamine/xylazine mix (140 and 0.5 mg/kg of body weight, respectively). Mice were placed on a glass manifold connected to a circulating water bath set at 37°C. A gold ring electrode was placed on a drop of 2 to 3% methylcellulose on the cornea, and a copper reference electrode was placed in the mouth. The unattenuated energy of the flash at the surface of the cornea was 2.9 mJ/cm2. Electroretinogram (ERG) responses were amplified 10,000 times, filtered between 1 Hz and 3 kHz, and sampled at 5 kHz. Responses to a family of dim-to-moderate intensity test flashes causing 1,500, 4,400, 7,300, 15,000, 61,000 and 164,000 photoisomerizations/rod were evaluated based on comparisons with the amplification factor determined from mouse ERGs calibrated by Lyubarsky and Pugh (24).
Dark adaptation was monitored as described previously (17). Briefly, a family of test flashes was delivered at various time points following the initial conditioning bleach. Leading edges of the a-wave responses were fit with the following model for the activation of phototransduction: a(t) = amax[1 − e0.5AΦ(t − td)2]. a(t) is the a-wave amplitude at time t, amax is the maximum amplitude of the a-wave, A is a factor proportional to the gain of phototransduction, Φ is the number of photoisomerizations/rod elicited by the test flash, and td is an intrinsic delay time. Data were fit using a variation of this equation that takes into account the capacitive time constant of the photoreceptor (39). A corrected value for Φ was applied at each time point that took into account the reduced amount of rhodopsin present following the conditioning bleach. We assumed that rhodopsin was regenerated at a rate of 0.014/min (17). Data presented were obtained from fits to averages of six traces from different mice at each time point. The data in Fig. 4 were fit with a model that describes the decay of molecular species that can activate phototransduction in the absence of light stimulation; i.e., “equivalent background light” (45).
FIG. 4.

Dark adaptation in PPEF 2×KO and wild-type mice. Dark adaptation following an intense conditioning bleach was monitored using electroretinography. The parameters amax and A are proportional to the fraction of cation channels open at the time of the test flash and photoreceptor sensitivity, respectively. Wild-type amax (white squares), wild-type A (black squares), 2×KO amax (white circles), and 2×KO A (black circles) were fit with a model that describes the first-order decay of a desensitizing molecular species (solid lines).
Light adaptation was measured by exposing the animal to steady background illumination for 2 min before recording ERG responses to a family of dim-to-moderate intensity flashes superimposed on the background illumination. The intensity of the unattenuated background light source measured 5.9 mW/cm2; neutral density filters were used to vary the intensity of background light over 4 orders of magnitude.
PPEF-2 immunoblotting.
Two retinas from PPEF-2−/−, PPEF-2+/− and PPEF-2+/+ mice were solubilized in 100 μl of 1×phosphate-buffered saline (PBS), and 1% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), and insoluble material was removed by microcentrifugation. Protein concentrations were measured using the Bradford assay (Bio-Rad), sodium dodecyl sulfate-polyacrylamide gel electrophoresis sample buffer was added to each detergent extract, and 20 μg of each sample was then separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a nitrocellulose filter. PPEF-2 was visualized with the Supersignal West Pico system (Pierce) after sequential incubation with anti-PPEF-2 C-terminal antibodies and horseradish peroxidase-conjugated goat anti-rabbit antibodies (Vector).
Histology and immunostaining of retinas.
Eyes were rapidly removed from mice killed by cervical dislocation and fixed for 1 h in 4% paraformaldehyde in PBS. For staining with anti-PPEF-2 C-terminal antibodies, an eyecup was prepared by removing the cornea, iris, and lens. For staining with the phosphorhodopsin-specific monoclonal antibody RT-97, a retina whole mount was prepared by peeling away the sclera, cornea, and iris beginning at the optic nerve head. Eyecups or retina whole mounts were further fixed for 4 h in 4% paraformaldehyde in PBS, kept in PBS with 30% sucrose overnight, embedded in OCT (TissueTek), and cryosectioned at 10 μM. For whole mounts prepared from dark-adapted retinas, all steps up to the 30% sucrose incubation were performed in darkness or under a 15-W red light passed through a Kodak Safelight filter.
For immunostaining, sections were blocked for 1 h in PBS with 5% normal goat serum and were incubated overnight in PBS with 5% normal goat serum containing a 1:200 dilution of affinity-purified anti-PPEF-2 C-terminal antibodies or a 1:500 dilution of RT-97 hybridoma supernatant. PPEF-2 or phosphorhodopsin immunoreactivity was visualized using horse radish peroxidase-conjugated goat anti-rabbit or biotinylated rabbit anti-mouse secondary antibodies and the DAB Peroxidase Substrate kit (Vector) or the Vectastain ABC kit (Vector), respectively.
For toluidine blue staining, eyecups were additionally fixed overnight in 0.5% glutaraldehyde before equilibration in PBS with 30% sucrose. Sections were stained for 2 min in 0.1% sodium borate and 0.05% toluidine blue, rinsed in distilled water, and mounted in Aqua Polymount (Polysciences, Inc.).
RT-PCR analysis.
Total RNA was harvested from wild-type and PPEF-2−/−; PPEF-1−/− female or PPEF-2−/−; PPEF-1−/− male [both are referred to as (2×KO)] eyes (six each) and from wild-type and 2×KO brain stems (two each) using the STAT-60 reagent (Tel-Test, Inc.). First-strand cDNA was synthesized from 5 μg of total RNA using hexamer primers and Superscript RT (GIBCO-BRL) for 1 h at 37°C. Amplification of PCR products from PPEF-1 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) cDNA was performed with primers in different exons for 35 cycles.
Primer sequences.
For identification by PCR of a bacterial artificial chromosome carrying the PPEF-2 locus, 5′GGTCTACCATCACCAGAGAGAGC3′ and 5′TAGGTTCACTAGATGGTCCTCAT3′ were used. For PCR amplification of the PPEF-1 3′ probe, 5′TTTCAATTTGCAGTTGGCAGG3′ and 5′GCAAGCAAGAAAGAAAAGGGA3′ were used. For PCR amplification of the PPEF-2 3′ probe, 5′CCCATAAAAATTTGAGCGGGAAA3′ and 5′CCTACAAAGCCGGCAGGTCC3′ were used. For PPEF-1 RT-PCR, 5′ATACTTCATGCTCACTACGTC and 5′ATAGAAAATCAGCATAAGATC3′ were used. For GAPDH RT-PCR, 5′ACCACAGTCCATGCCATCAC3′ and 5′TCCACCACCCTGTTGCTGTA3′ were used.
RESULTS
Targeted deletion of mouse PPEF-1 and PPEF-2.
To produce targeted disruptions of the mouse PPEF-1 and PPEF-2 genes, critical regions of each PPEF phosphatase catalytic domain, determined by comparison to the minimal catalytic domain defined for bacteriophage lambda phosphatase (1), were deleted such that the targeted allele would be incapable of producing a catalytically activated protein. The regions deleted include the last 21 codons of exon 5 in PPEF-1 and the last 14 codons of exon 6 and all of exons 7, 8, and 9 in PPEF-2 (Fig. 1A and B). Additionally, proper targeting resulted in the fusion of lacZ in frame to PPEF-1 exon 5 and PPEF-2 exon 6. Germ line transmission of the targeted alleles yielded male and female PPEF-2+/− mice but yielded only female PPEF-1+/− mice, consistent with the autosomal location of PPEF-2, the X-chromosomal location of PPEF-1, and the male origin of the ES cells. Further breeding generated PPEF-1−/− females, PPEF-1−/Y males, PPEF-2−/− mice of both sexes, PPEF-2−/−; PPEF-1−/− females, and PPEF-2−/−; PPEF-1−/Y males (Fig. 1C and D). As stated above, the latter two classes, in which both PPEF genes are disrupted, are referred to as 2×KO mice.
FIG. 1.

PPEF-1 and PPEF-2 targeting constructs and genotyping of PPEF-1 and PPEF-2 mutant mice. (A and B) Targeting vectors for PPEF-1 (A) and PPEF-2 (B). Numbers shown below the wild-type allele and the targeted allele indicate the positions of mapped exons. An asterisk next to an exon number indicates that the exon has been partially removed by the homologous recombination event. Homologous recombination mediates removal of part of exon 5 for PPEF-1 and part of exon 6 and all of exons 7 through 9 in PPEF-2. The boxes shown in the targeting vector represent, from left to right, the 5′ homology arm (5′ arm), the lacZ gene (lacZ), the neomycin resistance gene (N), the 3′ homology arm (3′ arm), and the herpes simplex virus thymidine kinase gene (TK). The DNA fragments used as Southern blot probes are shown as shaded boxes underneath the targeted alleles. B, BamHI; E, EcoRI; G, BglII; H, HindIII; P, SpeI; S, SalI; and T, StuI. (C) Genotyping of PPEF-1 mutant mice (male progeny from crosses between a wild-type male and PPEF-1+/− females) by Southern blotting of HindIII-digested DNA probed with a 3′ flanking probe. (D) Genotyping of PPEF-2 mutant mice (progeny from a cross between male and female PPEF-2+/− mice) by Southern blotting of BglII-digested DNA probed with a 5′ flanking probe. (E) Genotyping of 2×KO mutant mice (progeny from a cross between a PPEF-1−/Y; PPEF-2+/− male and a PPEF-1+/−; PPEF-2+/− female) by Southern blotting as in panels C and D. In panel E the first part of the genotype refers to the PPEF-1 genotype, while the second part refers to the PPEF-2 genotype. In panels C to E WT refers to the wild-type allele, while Mut refers to the mutant or targeted allele.
Single and double PPEF mutant mice are viable and fertile and show no overt abnormalities. Neither PPEF-1−/Y nor PPEF-2−/− mice show evidence of β-galactosidase activity, suggesting that either the mRNAs expressed from the targeted loci are unstable or poorly expressed or that the PPEF–β-galactosidase fusion proteins are unstable or enzymatically inactive. Analogous fusions between the Caenorhabditis elegans PPEF and green fluorescent protein are also unstable and/or inactive in transgenic nematodes (P. Ramulu and J. Nathans, unpublished).
To determine whether the PPEF-2 protein is eliminated in PPEF-2−/− mice, retina lysates from wild-type, PPEF-2+/−, and PPEF-2−/− mice were first analyzed by immunoblotting (Fig. 2A). Antibodies specific for the C terminus of PPEF-2 recognize a protein of ∼80 kDa from both wild-type and PPEF-2+/− retinas which is missing from PPEF-2−/− retinas. Consistent with the immunoblotting data, immunostaining of wild-type and PPEF-2−/− retinas with affinity-purified anti-PPEF-2 C terminus-specific antibodies (38) revealed the expected immunoreactivity of rod inner segments and synaptic termini in wild-type retinas but no immunoreactivity in PPEF-2−/− retinas (Fig. 2C and D). The loss of PPEF-1 transcripts in PPEF-1−/− mice was demonstrated by RT-PCR using total RNA from eye and brain stem (Fig. 2B).
FIG. 2.

Absence of PPEF-2 protein in PPEF-2−/− mice and PPEF-1 RNA in 2×KO mice. (A) Immunoblot analysis of 20 μg of detergent-solubilized retina protein from wild-type, PPEF-2+/−, and PPEF-2−/− mice. Molecular mass markers are indicated in kilodaltons. (B) RT-PCR analysis of brain stem and eye RNA from wild-type (WT) and 2×KO (KO) mice using PPEF-1 specific primers in exons 5 and 6. G3PDH, GAPDH; neg, negative control. (C and D) PPEF-2 immunoreactivity in photoreceptor inner segments and synaptic terminals of wild-type mice (C) and lack of PPEF-2 immunoreactivity in PPEF-2−/− mice (D). OS, photoreceptor outer segments; IS, photoreceptor inner segments; ONL, outer nuclear layer; and OPL, outer plexiform layer. Scale bars in panels C and D correspond to 25 μm.
Normal rod function in PPEF-2−/− and 2×KO mice.
To test rod function in PPEF mutant mice, dark-adapted and light-adapted ERGs were recorded from 2×KO mice (Fig. 3 and 4) and the light responses of single-rod cells from PPEF-2−/− mice were measured by suction pipette recordings (Fig. 5 and Table 1). ERG responses to a moderate intensity flash (causing ∼61,000 photoisomerizations in a dark-adapted rod) were measured in the presence of background illumination of various intensities. The effects of background illumination on the ERG a-wave response were similar in 2×KO and wild-type mice over a range of intensities (Fig. 3). The a-wave was completely suppressed in the presence of the brightest background in both wild-type and 2×KO mice.
FIG. 3.

Light adaptation in 2×KO and wild-type mice. ERG responses to a moderate intensity flash (eliciting approximately 61,000 photoisomerizations/rod) were recorded in the presence of background illumination using wild-type (A) and 2×KO (B) mice. The intensity of background illumination was varied over 4 orders of magnitude using neutral density filters. The value of the filter is given next to each trace (e.g., OD5 represents background light that was attenuated by a factor of 105 compared to unfiltered background). DA, dark adapted.
FIG. 5.

Suction pipette recording from single rods in wild-type and PPEF-2−/− mice. (A and B) Normalized response intensity families from a wild-type rod (A) and a PPEF-2−/− rod (B). Five-hundred-nanometer flashes are shown. Each trace is the averaged response from multiple flash trials. The records were low-pass-filtered at 30 Hz. Flash monitor output is shown by the bottom trace in each panel. The maximal response for panel A was 10.1 pA and for panel B was 11.0 pA. (C and D) Relations between the peak amplitude of flash response and the flash intensity for the two rods are shown in panels A and B, respectively. Curves were fit with the exponential saturation function r/rmax = 1 − exp(−ki), where k is a constant inversely proportional to the sensitivity of the cell and i is the flash strength. Half-maximal responses occurred at 46.9 photons/μm2 for the wild-type rod and 46.2 photons/μm2 for the PPEF-2−/− rod.
TABLE 1.
Parameters of flash responses in PPEF-2+/+ and PPEF-2−/− rodsa
| Rod type | n | tp (ms) | ti (ms) | a (pA) | io (photons/ μm2) | rMAX |
|---|---|---|---|---|---|---|
| PPEF-2+/+ | 20 | 124. ± 8.6 | 368.8 ± 44.7 | 0.36 ± 0.03 | ||
| 21 | 47.1 ± 2.3 | |||||
| 22 | 11.1 ± 0.7 | |||||
| PPEF-2−/− | 22 | 135.2 ± 3.5 | 359.5 ± 28.0 | 0.43 ± 0.03 | ||
| 21 | 41.5 ± 2.3 | |||||
| 23 | 9.7 ± 0.6 |
Values are means ± standard errors of the means; tp is the time to peak of dim flash response; ti is the integration time of the dim flash response; a is the estimated amplitude of the single photon response derived from the variance/mean rates in experiments with multiple dim flash trials; io is the flash strength at 500 nm that gave rise to a half-maximal response; and rmax is the saturating photoresponse amplitude.
Rod photoreceptor dark adaptation was monitored using methods previously described (17, 45). Dark-adapted mice were exposed to a bright conditioning flash that bleached approximately 20% of the rhodopsin in the eye. The time course of dark adaptation was monitored by recording ERG responses to a family of dim-to-moderate intensity flashes at various times following the conditioning bleach. The leading edge of the a-waves was fit as an ensemble with a model describing the activation of phototransduction (5) corrected for photoreceptor capacitance (39). The parameter amax is proportional to the fraction of outer segment cation channels open; the recovery of amax therefore represents the inactivation of phototransduction. The amplification parameter (A) reflects the efficiency with which a photoisomerized rhodopsin stimulates phototransduction; the recovery of A therefore represents resensitization of the photoreceptor. The rate of recovery of A has been shown to correlate with the rate of rhodopsin regeneration and complete dephosphorylation of rhodopsin (17). The rates of recovery of both amax and A are shown in Fig. 4 for 2×KO mice and wild-type littermate controls. The data are fit with a model describing the decay of residual phototransduction (45). The kinetics of dark adaptation in the 2×KO mice were similar to those for wild-type controls.
In single-rod recordings with a suction pipette, a single ROS either from an isolated cell or projecting from a retinal fragment was drawn into the suction pipette (3). Figure 5A and B show flash response intensity families from a wild-type rod and a PPEF-2−/− rod. The kinetics of the two sets of responses were very similar. The relation between peak response amplitude and flash intensity is also comparable (Fig. 5C and D). The curve fits are saturating exponential functions, with half-maximal intensities of 46.9 photons/μm2 (Fig. 5C, wild type) and 46.2 photons/μm2 (Fig. 5D, PPEF-2−/−), respectively. Collected results are given in Table 1.
Normal rhodopsin dephosphorylation in PPEF-2−/− and 2×KO mice.
As a preliminary test of the ability of PPEF-2−/− mice to dephosphorylate rhodopsin, retinas from mice that were either dark adapted overnight or kept in ambient light were sectioned and stained with an antibody specific for the phosphorylated C terminus of rhodopsin, RT-97 (2). Sections from both light-adapted wild-type and PPEF-2−/− retinas show high levels of phosphorhodopsin in ROS (Fig. 6A and B), while sections from both dark-adapted wild-type and PPEF-2−/− retinas show undetectable levels of phosphorhodopsin (Fig. 6C and D). These data suggest that PPEF-2 is either not involved in rhodopsin dephosphorylation or is not the only rhodopsin phosphatase in vertebrate photoreceptors.
FIG. 6.
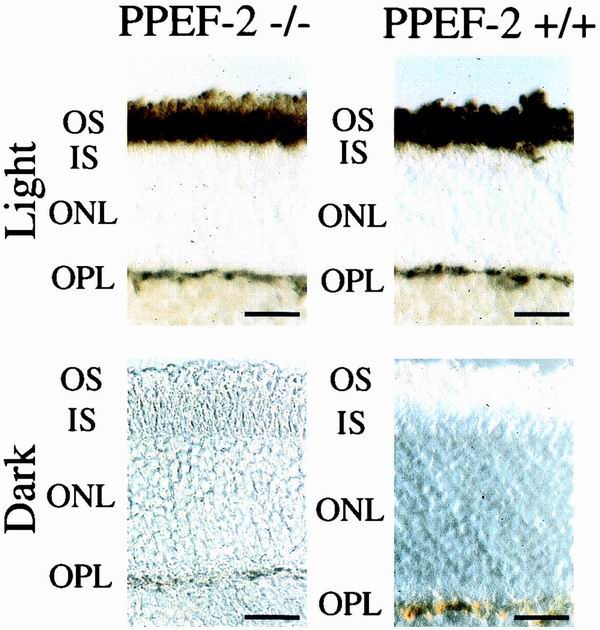
Antiphosphorhodopsin immunostaining of dark- and light-adapted retinas from wild-type and PPEF-2−/− retinas. Dark-adapted mice were kept in complete darkness for 12 h, and light-adapted mice were exposed to ambient light for 1 h. Whole-mount retinas from light-adapted mice were prepared in ambient light, and whole-mount retinas from dark-adapted mice were prepared under a 15-W light passed through a Kodak Safelight filter. Retinas were fixed, embedded in OCT, cut into 10-μm-thick sections, and immunostained with RT-97 hybridoma supernatant. The bar in the lower right-hand corner of each panel corresponds to 25 μm.
To directly assess the contribution of PPEF-1 and PPEF-2 to rhodopsin dephosphorylation, the C-terminal phosphopeptide from rhodopsin was analyzed from ROS membranes isolated from normal mice and 2×KO mice at different times following an intense flash that bleached ∼20% of the rhodopsin in the eye (17). Figure 7 shows the distribution of rhodopsin C termini modified with zero, one, two, three, or four phosphates in dark-adapted animals and at times 3, 30, and 120 min following the 20% photobleach. The amounts of phosphorylation at 3 min following the bleach were not significantly different for 2×KO mice and wild-type controls, indicating that the absence of PPEF-1 and PPEF-2 does not affect rhodopsin phosphorylation. More important, rhodopsin dephosphorylation in 2×KO mice appeared to be only mildly delayed relative to wild-type control mice.
FIG. 7.

Phosphorylation state of rhodopsin in PPEF 2×KO and wild-type mice in vivo. The phosphorylation state of rhodopsin was measured 3, 30, and 120 min following a flash of light that bleached roughly 20% of the rhodopsin in the eye in wild-type (left side) and 2×KO mice (right side). Dark-adapted values of phosphorylation are given by the 0-min time point. Values are reported as a fraction of the total amount of rhodopsin.
Absence of retinal degeneration in PPEF-2−/− and 2×KO mice.
To determine whether mutation of the vertebrate PPEFs leads to retinal degeneration, as observed in Drosophila rdgC mutants, retinas from 1-year-old PPEF-2−/− and 3-month-old 2×KO mice were sectioned and stained with toluidine blue (Fig. 8). PPEF-2−/− mice at 1 year of age show no evidence of photoreceptor loss, as judged by the thickness of the outer nuclear layer and no evidence of inner or outer segment disruption. Moreover, in PPEF-2−/− mice, dark-adapted ERG a-wave and b-wave amplitudes are stable over at least 10 months (data not shown), indicative of stable rod function. In 2×KO mice at 3 months of age, the latest time point analyzed, retinal histology is indistinguishable from that of normal age-matched controls, indicating that the absence of retinal degeneration observed in PPEF-2−/− mice is not due to the compensatory action of PPEF-1.
FIG. 8.

Normal histology of PPEF-2−/− mice at 1 year and 2×KO mice at 3 months. Sections (10 μm) of eyes from 12-month-old PPEF-2−/− mice and 3-month-old 2×KO mice were stained with toluidine blue. OS, photoreceptor outer segments; IS, photoreceptor inner segments; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; and GCL, ganglion cell layer. Scale bar in lower right of each panel corresponds to 25 μm.
DISCUSSION
This work represents the first functional characterization of PPEF-1 and PPEF-2, the vertebrate homologs of Drosophila rdgC. The results reported here show that, in contrast to Drosophila rdgC mutants, mice lacking both PPEF-1 and PPEF-2 demonstrate normal rod function, show no major changes in rhodopsin dephosphorylation, and do not exhibit retinal degeneration. Each of the above findings is discussed in more detail below.
Drosophila rdgC represents the most extensively characterized member of the PPEF family. rdgC mutant flies accumulate hyperphosphorylated rhodopsin in response to blue light and cannot dephosphorylate rhodopsin in response to orange light, suggesting that the rdgC protein dephosphorylates rhodopsin (6, 49). Furthermore, rhodopsin phosphatase activity in extracts of fly heads is stimulated by calcium (6). Like other PPEFs, rdgC has at least two C-terminal EF hands, which in the C. elegans PPEF have been shown to bind calcium (34). As a consequence of their inability to dephosphorylate rhodopsin, rdgC flies demonstrate prolonged ERGs and lower light intensity thresholds for prolonged depolarization afterpotentials. The latter reflects an increased photosensitivity due to titration of the available arrestin (49).
Several observations are consistent with the conclusion from the present work that neither of the vertebrate rdgC homologs plays a significant role in rhodopsin dephosphorylation. First, no PPEF family member has yet been localized to the outer segments of vertebrate photoreceptors. Two alternatively spliced PPEF-2 transcripts are present in the retina, one encoding a short isoform ending just after the phosphatase domain and the second encoding a long isoform containing the C-terminal EF hands (38). To date, only the long isoform has been immunolocalized, and it was found principally in rod inner segments (38). As noted in the introduction, PPEF-1 transcripts are extremely rare in the retina and at present it is not known in which cell type(s) they reside. As most proteins involved in phototransduction are relatively abundant in the retina and are encoded by correspondingly abundant transcripts, the low abundance of PPEF-1 transcripts argues against a role for PPEF-1 in the outer segment and therefore in rhodopsin dephosphorylation. Second, as noted in the introduction, a number of lines of evidence suggest that PP2A is the rhodopsin phosphatase in vertebrates (12, 16, 29, 51). Third, present evidence suggests that the rise in intracellular calcium that follows light activation of invertebrate photoreceptors increases the enzymatic activity of rdgC. In contrast, light activation of vertebrate photoreceptors results in a lowering of intracellular calcium and would therefore be predicted to decrease the activity of vertebrate PPEF at precisely the time that efficient recycling of visual pigment would require enhanced dephosphorylation of rhodopsin. In the present work, we have used a combination of antiphosphorhodopsin immunostaining and mass spectrometry to demonstrate that the contribution of PPEF-1 and PPEF-2 to rhodopsin dephosphorylation is at most minimal, consistent with the idea that PP2A accounts for all or almost all rhodopsin phosphatase activity in vivo.
Phosphorylation has emerged as a general mechanism for modulating the activity of G protein-coupled receptors. Many G protein-coupled receptors, including vertebrate and invertebrate rhodopsin, contain conserved serine and threonine residues near their C termini that are phosphorylated. Phosphorylation of rhodopsin's C terminus has been shown to be required for effective inactivation of phototransduction (8, 9). Because phosphorylation limits the gain with which rhodopsin stimulates phototransduction, rhodopsin must be dephosphorylated and regenerated with 11-cis retinal in order for a photoreceptor to recover sensitivity.
Rod photoreceptors from 2×KO mice recover sensitivity at the same rate as those from wild-type controls (Fig. 4). This would not be expected if a significant portion of rhodopsin remained phosphorylated since rhodopsin cannot efficiently activate transducin in its phosphorylated state (14). More important, we show directly that rhodopsin is dephosphorylated with nearly normal kinetics in the 2×KO mice. At short times following a 20% conditioning bleach, approximately 30% of rhodopsin is modified with at least one phosphate in both the wild-type and 2×KO mice. After 120 min in the dark, rhodopsin is effectively dephosphorylated to nearly dark-adapted values in both wild type and 2×KO mice. We note that these data do not preclude the possibility that a different phosphatase might be able to fully compensate for the deletion of PPEF-1 or PPEF-2.
Current evidence indicates that the photoreceptor degeneration in rdgC flies occurs as a result of excessive arrestin-mediated phosphorhodopsin internalization. Flies carrying the rdgC mutation and expressing a rhodopsin mutant in which the phosphorylation sites are mutated or deleted in place of the wild-type rhodopsin do not exhibit a retinal degeneration, demonstrating that rdgC-dependent retinal degeneration requires rhodopsin phosphorylation (18, 49). Furthermore, shibere mutants, which show defects in endocytosis due to a temperature-sensitive dynamin allele, rescue rdgC-dependent photoreceptor degeneration, suggesting that internalization of phosphorhodopsin-arrestin complexes mediates degeneration (18).
If phosphorhodopsin were to accumulate in vertebrate photoreceptors, it is unlikely that it would mediate a retinal degeneration by the same mechanism observed in Drosophila. While arrestin-mediated internalization of phosphorylated G protein-coupled receptors has been observed in vertebrate systems (21, 22, 23), it seems unlikely that this mechanism for downregulation could operate for vertebrate rhodopsin in the outer segment, where the topological separation of outer segment disc membranes from the plasma membrane and from the inner segment would appear to be incompatible with recycling by internalization. Indeed, in vivo biosynthetic labeling of vertebrate outer segment proteins (principally rhodopsin) shows no evidence of protein turnover during the entire transit period from the base to the tip of the outer segment (52). Thus, the rhodopsin internalization phenomenon associated with the rdgC mutation is unlikely to apply to vertebrates.
The biological role of the vertebrate PPEFs remains an open question. Based on its subcellular localization, PPEF-2 may play a role in photoreceptor inner segment biology. Currently, the inner segment is recognized for its role in ATP generation (7, 48), in extruding sodium via the Na/K ATPase at the plasma membrane (40, 46, 47), and transporting rhodopsin and other molecules to the outer segments (31, 32). Interestingly, other regulatory proteins, such as guanylate cyclase activating protein 2, have been detected in the inner segment (28), suggesting that as-yet-uncharacterized signaling pathways may exist in this subcellular compartment. Based on the relative abundance of its transcripts in different tissues, the primary role of PPEF-1 is likelier to be in the inner ear or dorsal root ganglia than in the retina, although we cannot rule out the possibility that a relatively small amount of PPEF-1 in the retina may provide a degree of functional redundancy with PPEF-2.
The substrates for other members of the PPEF family of phosphatases, aside from rdgC, remain unidentified. Given that rdgC protein dephosphorylates the G protein-coupled receptor rhodopsin and that the C. elegans PPEF is localized to the cilia of sensory neurons, where G protein-coupled receptors and other components of G protein signaling are known to be sequestered (34), it is possible that all PPEFs function as G protein-coupled receptor phosphatases. Alternately, it is possible that PPEFs have evolved to act on a variety of targets within sensory neurons. In particular, PPEF-2 may either dephosphorylate an orthologue of a substrate other than rhodopsin that is also dephosphorylated by rdgC but which is not critically involved in retinal degeneration, or PPEF-2 may have acquired a substrate in vertebrate photoreceptors that does not exist in Drosophila photoreceptors. The PPEF mutant mice generated here will be a valuable resource for discovering the substrates of the vertebrate PPEFs and for elucidating their physiological functions.
ACKNOWLEDGMENTS
This work was supported by the Howard Hughes Medical Institute (K.-W.Y.). P.R. is a trainee of the Visual Neurosciences Training Program and the Medical Scientist Training Program.
We thank Ursula Drager for the gift of MAb RT-97 and Richard Behringer for the pNeo-TK plasmid.
REFERENCES
- 1.Ansai T, Dupuy L C, Barik S. Interactions between a minimal protein serine/threonine phosphatase and its phosphopeptide substrate sequence. J Bio Chem. 1996;271:24401–24407. doi: 10.1074/jbc.271.40.24401. [DOI] [PubMed] [Google Scholar]
- 2.Balkema G W, Drager U C. Light-dependent antibody labelling of photoreceptors. Nature. 1985;316:630–633. doi: 10.1038/316630a0. [DOI] [PubMed] [Google Scholar]
- 3.Baylor D A, Lamb T D, Yau K W. The membrane current of single rod outer segments. J Physiol (London) 1979;288:589–611. [PMC free article] [PubMed] [Google Scholar]
- 4.Bennett N, Sitaramayya A. Inactivation of photoexcited rhodopsin in retinal rods: the roles of rhodopsin kinase and 48-kDa protein (arrestin) Biochemistry. 1988;27:1710–1715. doi: 10.1021/bi00405a049. [DOI] [PubMed] [Google Scholar]
- 5.Breton M E, Schueller A W, Lamb T D, Pugh E N., Jr Analysis of ERG a-wave amplification and kinetics in terms of the G-protein cascade of phototransduction. Investig Ophthalmol Vis Sci. 1994;35:295–309. [PubMed] [Google Scholar]
- 6.Byk T, Bar-Yaacov M, Doza Y N, Minke B, Selinger Z. Regulatory arrestin cycle secures the fidelity and maintenance of the fly photoreceptor cell. Proc Natl Acad Sci USA. 1993;90:1907–1911. doi: 10.1073/pnas.90.5.1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carter-Dawson L D, LaVail M M. Rods and cones in the mouse retina. I. Structural analysis using light and electron microscopy. J Comp Neurol. 1979;188:245–262. doi: 10.1002/cne.901880204. [DOI] [PubMed] [Google Scholar]
- 8.Chen C K, Burns M E, Spencer M, Niemi G A, Chen J, Hurley J B, Baylor D A, Simon M I. Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase. Proc Natl Acad Sci USA. 1999;96:3718–3722. doi: 10.1073/pnas.96.7.3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J, Makino C L, Peachey N S, Baylor D A, Simon M I. Mechanisms of rhodopsin inactivation in vivo as revealed by a COOH-terminal truncation mutant. Science. 1995;267:374–377. doi: 10.1126/science.7824934. [DOI] [PubMed] [Google Scholar]
- 10.Colley N J, Cassill J A, Baker E K, Zuker C S. Defective intracellular transport is the molecular basis of rhodopsin-dependent dominant retinal degeneration. Proc Natl Acad Sci USA. 1995;92:3070–3077. doi: 10.1073/pnas.92.7.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dolph P J, Ranganathan R, Colley N J, Hardy R W, Socolich M, Zuker C S. Arrestin function in inactivation of G protein-coupled receptor rhodopsin in vivo. Science. 1993;260:1910–1916. doi: 10.1126/science.8316831. [DOI] [PubMed] [Google Scholar]
- 12.Fowles C, Akhtar M, Cohen P. Interplay of phosphorylation and dephosphorylation in vision: protein phosphatases of bovine rod outer segments. Biochemistry. 1989;28:9385–9391. doi: 10.1021/bi00450a020. [DOI] [PubMed] [Google Scholar]
- 13.Gibson S K, Parkes J H, Cohen P A. Phosphorylation modulates the affinity of light-activated rhodopsin for G protein and arrestin. Biochemistry. 2000;39:5738–5749. doi: 10.1021/bi991857f. [DOI] [PubMed] [Google Scholar]
- 14.Gurevich V V, Benovic J L. Visual arrestin interaction with rhodopsin. Sequential multisite binding ensures strict selectivity toward light-activated phosphorylated rhodopsin. J Biol Chem. 1993;268:11628–11638. [PubMed] [Google Scholar]
- 15.Huang X, Honkanen R E. Molecular cloning, expression, and characterization of a novel human serine/threonine protein phosphatase, PP7, that is homologous to Drosophila retinal degeneration C gene product (rdgC) J Biol Chem. 1998;273:1462–1468. doi: 10.1074/jbc.273.3.1462. [DOI] [PubMed] [Google Scholar]
- 16.Hurley J B, Spencer M, Niemi G A. Rhodopsin phosphorylation and its role in photoreceptor function. Vision Res. 1998;38:1341–1352. doi: 10.1016/s0042-6989(97)00459-8. [DOI] [PubMed] [Google Scholar]
- 17.Kennedy M J, Lee K A, Niemi G A, Craven K B, Garwin G G, Saari J C, Hurley J B. Multiple phosphorylation of rhodopsin and the in vivo chemistry underlying rod photoreceptor dark adaptation. Neuron. 2001;31:87–101. doi: 10.1016/s0896-6273(01)00340-3. [DOI] [PubMed] [Google Scholar]
- 18.Kiselev A, Socolich M, Vinos J, Hardy R W, Zuker C S, Ranganathan R. A molecular pathway for light-dependent photoreceptor apoptosis in Drosophila. Neuron. 2000;28:139–152. doi: 10.1016/s0896-6273(00)00092-1. [DOI] [PubMed] [Google Scholar]
- 19.Kurada P, O'Tousa J E. Retinal degeneration caused by dominant rhodopsin mutations in Drosophila. Neuron. 1995;14:571–579. doi: 10.1016/0896-6273(95)90313-5. [DOI] [PubMed] [Google Scholar]
- 20.Kutuzov M A, Bennett N. Calcium-activated opsin phosphatase activity in retinal rod outer segments. Eur J Biochem. 1996;238:613–622. doi: 10.1111/j.1432-1033.1996.0613w.x. [DOI] [PubMed] [Google Scholar]
- 21.Laporte S A, Oakley R H, Holt J A, Barak L S, Caron M G. The interaction of beta-arrestin with the AP-2 adaptor is required for the clustering of beta 2-adrenergic receptor into clathrin-coated pits. J Biol Chem. 2000;275:23120–23126. doi: 10.1074/jbc.M002581200. [DOI] [PubMed] [Google Scholar]
- 22.Lin F T, Krueger K M, Kendall H E, Daaka Y, Fredericks Z L, Pitcher J A, Lefkowitz R J. Clathrin-mediated endocytosis of the β-adrenergic receptor is regulated by phosphorylation/dephosphorylation of β-arrestinI. J Biol Chem. 1997;272:31051–31057. doi: 10.1074/jbc.272.49.31051. [DOI] [PubMed] [Google Scholar]
- 23.Luttrell L M, Ferguson S S, Daaka Y, Miller W E, Maudsley S, Della Rocca G J, Lin F, Kawakatsu H, Owada K, Luttrell D K, Caron M G, Lefkowitz R J. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 24.Lyubarsky A L, Pugh E N., Jr Recovery phase of the murine rod photoresponse reconstructed from electroretinographic recordings. J Neurosci. 1996;16:563–571. doi: 10.1523/JNEUROSCI.16-02-00563.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mendez A, Burns M E, Roca A, Lem J, Wu L W, Simon M I, Baylor D A, Chen J. Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron. 2000;28:153–164. doi: 10.1016/s0896-6273(00)00093-3. [DOI] [PubMed] [Google Scholar]
- 26.Montini E, Rugarli E I, Van de Vosse E, Andolfi G, Mariani M, Puca A A, Consalez G G, den Dunnen J T, Ballabio A, Franco B. A novel human serine-threonine phosphatase related to the Drosophila retinal degeneration C (rdgC) gene is selectively expressed in sensory neurons of neural crest origin. Hum Mol Genet. 1997;6:1137–1145. doi: 10.1093/hmg/6.7.1137. [DOI] [PubMed] [Google Scholar]
- 27.Ohguro H, Van Hooser J P, Milam A H, Palezewski K. Rhodopsin phosphorylation and dephosphorylation in vivo. J Biol Chem. 1995;270:14259–14262. doi: 10.1074/jbc.270.24.14259. [DOI] [PubMed] [Google Scholar]
- 28.Otto-Bruc A, Fariss R N, Haeseleer F, Huang J, Buczylko J, Surgucheva I, Baehr W, Milam A H, Palczewski K. Localization of guanylate cyclase-activating protein 2 in mammalian retinas. Proc Nat Acad Sci USA. 1997;94:4727–4732. doi: 10.1073/pnas.94.9.4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palczewski K, Hargrave P A, McDowell J H, Ingebritsen T S. The catalytic subunit of phosphatase 2A dephosphorylates phosphoopsin. Biochemistry. 1989;28:415–419. doi: 10.1021/bi00428a001. [DOI] [PubMed] [Google Scholar]
- 30.Palczewski K, Rispoli G, Detwiler P B. The influence of arrestin (48K protein) and rhodopsin kinase on visual transduction. Neuron. 1992;8:117–126. doi: 10.1016/0896-6273(92)90113-r. [DOI] [PubMed] [Google Scholar]
- 31.Papermaster D S, Converse C A, Siuss J. Membrane biosynthesis in the frog retina: opsin transport in the photoreceptor cell. Biochemistry. 1975;14:1343–1352. doi: 10.1021/bi00678a001. [DOI] [PubMed] [Google Scholar]
- 32.Papermaster D S, Schneider B G, Besharse J C. Vesicular transport of newly synthesized opsin from the Golgi apparatus toward the rod outer segment. Ultrastructural immunocytochemical and autoradiographic evidence in Xenopus retinas. Investig Ophthalmol Vis Sci. 1985;26:1386–1404. [PubMed] [Google Scholar]
- 33.Peschon J J, Behringer R R, Brinster R L, Palmiter R D. Spermatid-specific expression of protamine 1 in transgenic mice. Proc Natl Acad Sci USA. 1987;84:5316–5319. doi: 10.1073/pnas.84.15.5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramulu P, Nathans J. Cellular and subcellular localization, N-terminal acylation, and calcium binding of Caenorhabditis elegans protein phosphatase with EF-hands. J Biol Chem. 2001;276:25127–25135. doi: 10.1074/jbc.M011712200. [DOI] [PubMed] [Google Scholar]
- 35.Ranganathan R, Malicki D M, Zuker C S. Signal transduction in Drosophila photoreceptors. Annu Rev Neurosci. 1995;18:283–317. doi: 10.1146/annurev.ne.18.030195.001435. [DOI] [PubMed] [Google Scholar]
- 36.Rattner A, Sun H, Nathans J. Molecular genetics of human retinal disease. Annu Rev Genet. 1999;33:89–131. doi: 10.1146/annurev.genet.33.1.89. [DOI] [PubMed] [Google Scholar]
- 37.Selinger Z, Doza Y N, Minke B. Mechanisms and genetics of photoreceptors desensitization in Drosophila flies. Biochim Biophys Acta. 1993;1179:283–299. doi: 10.1016/0167-4889(93)90084-3. [DOI] [PubMed] [Google Scholar]
- 38.Sherman P M, Sun H, Macke J P, Williams J, Smallwood P M, Nathans J. Identification and characterization of a conserved family of protein serine/threonine phosphatases homologous to Drosophila retinal degeneration C. Proc Natl Acad Sci USA. 1997;94:11639–11644. doi: 10.1073/pnas.94.21.11639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith N P, Lamb T D. The a-wave of the human electroretinogram recorded with a minimally invasive technique. Vision Res. 1997;37:2943–2952. doi: 10.1016/s0042-6989(97)00094-1. [DOI] [PubMed] [Google Scholar]
- 40.Spencer M, Detwiler P B, Bunt-Milam A H. Distribution of membrane proteins in mechanically dissociated retinal rods. Investig Ophthalmol Vis Sci. 1988;29:1012–1020. [PubMed] [Google Scholar]
- 41.Steele F, O'Tousa J E. Rhodopsin activation causes retinal degeneration in Drosophila rdgC mutant. Neuron. 1990;4:883–890. doi: 10.1016/0896-6273(90)90141-2. [DOI] [PubMed] [Google Scholar]
- 42.Steele F R, Washburn T, Rieger R, O'Tousa J E. Drosophila retinal degeneration C (rdgC) encodes a novel serine/threonine protein phosphatase. Cell. 1992;69:669–676. doi: 10.1016/0092-8674(92)90230-a. [DOI] [PubMed] [Google Scholar]
- 43.Stryer L. Cyclic GMP cascade of vision. Annu Rev Neurosci. 1986;9:87–119. doi: 10.1146/annurev.ne.09.030186.000511. [DOI] [PubMed] [Google Scholar]
- 44.Sung C-H, Makino C, Baylor D A, Nathans J. A rhodopsin gene mutation responsible for autosomal dominant retinitis pigmentosa results in a protein that is defective in localization to the photoreceptor outer segment. J Neurosci. 1994;14:5818–5833. doi: 10.1523/JNEUROSCI.14-10-05818.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomas M M, Lamb T D. Light adaptation and dark adaptation of human rod photoreceptors measured from the a-wave of the electroretinogram. J Physiol (London) 1999;518:479–496. doi: 10.1111/j.1469-7793.1999.0479p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ueno S, Bambauer H J, Umar H, Ueck M, Ogawa K. Ultracytochemical study of Ca++-ATPase and K+-NPPase activities in retinal photoreceptors of the guinea pig. Cell Tissue Res. 1984;237:479–489. doi: 10.1007/BF00228432. [DOI] [PubMed] [Google Scholar]
- 47.Ueno S, Umar H, Bambauer H J, Ueck M. Localization of ATPases in retinal receptor cells. Ophthalmic Res. 1984;16:15–20. doi: 10.1159/000265287. [DOI] [PubMed] [Google Scholar]
- 48.Vigh B, Vigh-Teichmann I. Three types of photoreceptors in the pineal and frontal organs of frogs: ultrastructure and opsin immunoreactivity. Arch Histol Jpn. 1986;49:495–518. doi: 10.1679/aohc.49.495. [DOI] [PubMed] [Google Scholar]
- 49.Vinos J, Jalink K, Hardy R W, Britt S G, Zuker C S. A G protein-coupled receptor phosphatase required for rhodopsin function. Science. 1997;277:687–690. doi: 10.1126/science.277.5326.687. [DOI] [PubMed] [Google Scholar]
- 50.Wilden U, Hall S W, Kuhn H. Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kDa protein of rod outer segments. Proc Natl Acad Sci USA. 1986;83:1174–1178. doi: 10.1073/pnas.83.5.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang S D, Fong Y L, Benovic J L, Sibley D R, Caron M G, Lefkowitz R J. Dephosphorylation of the beta 2-adrenergic receptor and rhodopsin by latent phosphatase 2. J Biol Chem. 1988;263:8856–8858. [PubMed] [Google Scholar]
- 52.Young R W. The renewal of the photoreceptor cell outer segments. J Cell Biol. 1967;33:61–72. doi: 10.1083/jcb.33.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang L, Sports C D, Osawa S, Weiss E R. Rhodopsin phosphorylation sites and their role in arrestin binding. J Biol Chem. 1997;272:14762–14768. doi: 10.1074/jbc.272.23.14762. [DOI] [PubMed] [Google Scholar]