

Abstract
Numerous biochemical experiments have invoked a model in which B-cell antigen receptor (BCR)-Fc receptor for immunoglobulin (Ig) G (FcγRII) coclustering provides a dominant negative signal that blocks B-cell activation. Here, we tested this model using quantitative confocal microscopic techniques applied to ex vivo splenic B cells. We found that FcγRII and BCR colocalized with intact anti-Ig and that the SH2 domain-containing inositol 5′-phosphatase (SHIP) was recruited to the same site. Colocalization of BCR and SHIP was inefficient in FcγRII−/− but not gamma chain−/− splenic B cells. We also examined the subcellular location of a variety of enzymes and adapter proteins involved in signal transduction. Several proteins (CD19, CD22, SHP-1, and Dok) and a lipid raft marker were corecruited to the BCR, regardless of the presence or absence of FcγRII and SHIP. Other proteins (Btk, Vav, Rac, and F-actin) displayed reduced colocalization with BCR in the presence of FcγRII and SHIP. Colocalization of BCR and F-actin required phosphatidylinositol (PtdIns) 3-kinase and was inhibited by SHIP, because the block in BCR/F-actin colocalization was not seen in B cells of SHIP−/− animals. Furthermore, BCR internalization was inhibited with intact anti-Ig stimulation or by expression of a dominant-negative mutant form of Rac. From these results, we propose that SHIP recruitment to BCR/FcγRII and the resulting hydrolysis of PtdIns-3,4,5-trisphosphate prevents the appropriate spatial redistribution and activation of enzymes distal to PtdIns 3-kinase, including those that promote Rac activation, actin polymerization, and receptor internalization.
B lymphocytes function to process and present foreign antigen to T lymphocytes and to proliferate and produce antigen-specific immunoglobulin (Ig). These responses are initiated by antigen binding to the B-cell antigen receptor (BCR) and by stimulation of the attending signal transduction process.
The BCR is composed of surface immunoglobulin associated with an immunoreceptor tyrosine-based activation motif (ITAM)-containing α/β heterodimer. BCR clustering is critical to B-cell activation and can be accomplished by antigen or anti-Ig to mimic antigen binding. Upon BCR clustering, tyrosine residues within ITAMs are phosphorylated by Src family protein tyrosine kinases (10a). The nascent phosphotyrosine residues become docking sites for the Src homology 2 (SH2) domains of various downstream signaling molecules, which activate additional signaling enzymes, including phosphatidylinositol (PtdIns) 3-kinase, Ras, and phospholipase Cγ.
Earlier studies established the ability of existing antibody to inhibit a humoral response to new epitopes on the same particle (12b–12d; reviewed in reference 22f). The inhibitory event, termed negative signaling, occurs through coclustering of the BCR and the Fc IgG receptor FcγRII (6a, 26a). This hypothesis was inferred from experimental evidence showing that a blocking monoclonal antibody directed against FcγRII reversed the inhibitory effect of intact anti-Ig on B-cell proliferation (22a, 22b) and antigen presentation to T cells (31). Furthermore, stimulation of B cells with Fc-bearing intact anti-Ig antibodies blocks activation events, such as Ca2+ influx (8a) and B-cell proliferation (15a). Although a BCR-FcγRII coclustering model of negative signaling is consistent with existing data, there is no direct evidence that these receptors actually cocluster.
FcγRII contains a 13-residue cytoplasmic immunoreceptor tyrosine-based inhibitory motif (ITIM) sequence common to other inhibitory receptors (reviewed in reference 22e). The tyrosine within the ITIM motif of FcγRII is phosphorylated upon BCR-FcγRII coclustering (19a). Studies using synthetic ITIM phosphopeptides (8, 9a, 27, 28a) or FcγRII coimmunoprecipitation (5, 28) identified the SH2 domain-containing inositol 5′-phosphatase (SHIP) and the protein phosphatase SHP-1 as capable of engaging the phosphorylated ITIM of FcγRIIb. B-cell lines deficient in SHIP expression lose the ability to undergo negative signaling, as defined by changes in cytoplasmic Ca2+ (12a, 19b, 21a, 27, 30). Thus, in vitro biochemical evidence supports a role for SHIP in negative signaling, but there is no direct evidence indicating that SHIP is recruited to the FcγRII. More importantly, specific biological events affected by SHIP recruitment to FcγRII are unclear.
Enzymes involved in signal transduction pathways are activated by a change in their subcellular location, providing new access to substrates or initiating noncovalent protein interactions that modify activity. Here, we have investigated the subcellular localization of several enzymes involved in B-cell signal transduction and of the two phosphatases thought to contribute to the negative signaling process. Using a novel quantitative method applied to confocal microscopic images of splenic B cells, we provide direct evidence that FcγRII and BCR are coclustered under negative signaling conditions. Likewise, SHIP was recruited to the site of colocalized BCR and FcγRII. Colocalization of BCR and SHIP was impaired in FcγRII−/− but not gamma chain−/− B cells.
We examined BCR recruitment of signaling proteins that might be affected by SHIP-mediated hydrolysis of PtdIns 3-kinase lipid products (phosphatidylinositol 3,4,5-trisphosphate [PtdIns-3,4,5-P3]), including Btk, Vav, Rac, and F-actin. Recruitment to the BCR of these proteins was reduced under conditions leading to BCR-FcγRII-SHIP colocalization, and their reduction was associated with a block in antigen receptor internalization. We hypothesize that hydrolysis of PtdIns-3,4,5-P3 by recruitment of the inositol 5′-phosphatase SHIP to the BCR/FcγRII cap site disturbs recruitment and activation of enzymes responding to PtdIns 3-kinase. Consistent with this hypothesis, we found that colocalization of BCR and F-actin was reduced by treatment with PtdIns 3-kinase inhibitor. Moreover, the reduction in colocalization of F-actin and BCR under intact anti-Ig was abolished in B cells from SHIP−/− animals. Together, our findings provide a spatial and temporal description of molecules involved in positive and negative signal transduction events in B cells.
MATERIALS AND METHODS
Animals and commercial antibodies.
Animals were purchased from Jackson Laboratories (Bar Harbor, Maine) or Taconic Farms (Germantown, N.Y.) and bred at our facility. Antibodies were purchased from commercial suppliers and labeled with N-hydroxysuccinimidyl biotin (Pierce Chemical Co., Rockford, Ill.). Nonlabeled and indodicarbocyanine (Cy5)- and fluorescein isothiocyanate (FITC)-labeled rabbit anti-mouse Ig were purchased from Pierce Biochemicals and Jackson Immunoresearch Laboratories (West Grove, Pa.). Antibodies to SHIP (27) were labeled with AlexaFluor 488 (Molecular Probes, Eugene, Oreg.). Monoclonal antibodies (MAbs) to mouse FcγRII/III, CD19, CD22, and B220 were purchased from Pharmingen (San Diego, Calif.). Antibodies to Vav, Dok, SHP-1, IκB, Btk, and Rac were purchased from Santa Cruz Biotechnology (Santa Cruz, Calif.). AlexaFluor 488-labeled phalloidin and anti-FITC antibody were purchased from Molecular Probes. FITC-cholera toxin B subunit and LY294002 were purchased from Sigma Chemical Co. (St. Louis, Mo.).
Splenic B-cell preparation, stimulation, and immunostaining.
B cells were prepared from splenocytes as previously described (22c) and stimulated with 10 or 15 μg of Cy5-conjugated F(ab′)2 fragment or intact rabbit anti-mouse Ig per ml at 37°C for 1 to 2 min. Stimulations were stopped by fixative solution (1% formaldehyde in phosphate-buffered saline [PBS] containing 0.1% NaN3) or by cold PBS and fixation as above. Unstimulated cells were incubated at 37°C in buffer only and fixed before staining. Fixed cells were washed in staining buffer (3% fetal bovine serum [FBS], 0.1% NaN3 in PBS), incubated with 1 μg of biotinylated MAb for 2 h at 4°C, washed, and stained with 1 μg of FITC-streptavidin or AlexaFluor 488-streptavidin.
The stained cells were added to poly-l-lysine (0.1%, wt/vol; Sigma)-coated cover slips and mounted to slides. For intracellular staining, cells were fixed and permeabilized in PBS with 2% bovine serum albumin (BSA), 2% FBS, 1 mM sodium vanadate, 1 mM EDTA, 0.2% Tween 20, and 0.01% sodium dodecyl sulfate (SDS), supplemented with the indicated biotinylated antibodies. The cells were incubated at 4°C for 4 to 16 h. After staining, cells were washed and incubated with FITC- or AlexaFluor 488-streptavidin.
A Leica TCS laser scanning confocal microscope was used for image acquisition. Fluorescein or AlexaFluor 488 was excited at 488 nm using an argon laser; Cy5 was excited using a helium-neon laser at 633 nm. Emission wavelengths between 530 and 560 nm were collected for FITC and AlexaFluor 488, and emission wavelengths greater than 645 nm were collected for Cy5. Image analysis of double-labeled cells was performed by determining the correlation coefficient, as previously described (22d). Briefly, correlation analysis of double-labeled cells was performed by determining the correlation coefficient (ρ):
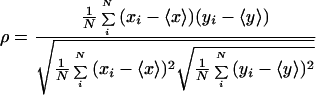 |
1 |
where xi and yi are the intensities of each point in equatorial images of double-labeled cells and 〈x〉 and 〈y〉 are the corresponding average values.
The operation for calculating ρ was as follows. The mean fluorescence of the plasma membrane was measured and subtracted from each pixel associated with the plasma membrane. This was performed for each image to generate a pair of difference images defined by the terms
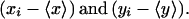 |
The corresponding difference images were multiplied and squared to generate images corresponding to
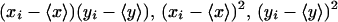 |
Next, the region corresponding to the plasma membrane in the last three images was summed and divided by the area (in pixels). Square root and division operations of the resulting integers were performed as defined by equation 1. All calculations were made using IP Lab Spectrum software (Signal Analytics Corp., Vienna, Va.).
B-cell receptor internalization assay.
B cells were incubated with FITC-labeled anti-Ig for 30 min at 4°C and washed to remove unbound antibody. The cells were then incubated at 37°C for the indicated time. The reaction was stopped and separated into two equal volumes. Mean fluorescence intensity from one set of the duplicate samples represented total internalized and surface-bound FITC-anti-Ig. The other set of samples were incubated for 1 h with anti-FITC antibody to quench noninternalized, surface-bound FITC-anti-Ig. Mean fluorescence intensity was measured and represented internalized fluorescence. Percent internalized BCR was obtained by dividing internalized mean fluorescence intensity by total mean fluorescence intensity. Values at the zero time points represent cells incubated with FITC-anti-Ig at 4°C for the entire experiment, followed by anti-FITC to quench.
Transfection of A20 B cells.
A20 B cells were transfected as described earlier (14a) with either green fluorescent protein (GFP) vector or a dominant negative mutant of Rac (N17Rac-GFP). Transfectants were incubated with Cy5-labeled F(ab′)2 anti-Ig for 30 min at 4°C and moved to 37°C for 10 min. The reaction was stopped with cold staining buffer and fixation. Cy5 staining of BCR at 4°C was used to indicate the noninternalized BCR from transfected cells.
Internalization measurements were modified from earlier studies (31a). Briefly, cells were stimulated with nonlabeled F(ab′)2 anti-Ig for the indicated times to permit receptor internalization. Reactions were stopped, and remaining noninternalized BCR was stained with Cy5-labeled F(ab′)2 anti-Ig. GFP-positive cells were gated, and mean fluorescence intensity was measured by flow cytometry. Mean fluorescence intensity at 4°C represented the total amount of BCR and mean fluorescence intensity at each time point represented by noninternalized BCR. Percent internalized BCR was measured as a loss of Cy5 fluorescence by subtracting noninternalized BCR from the total amount of BCR and divided by the total amount of BCR.
RESULTS
Coclustering of BCR-FcγRII with intact anti-Ig.
Biochemical studies examining inhibitory signaling events in B cells treated with F(ab′)2 or intact anti-Ig antibodies suggest that BCR and FcγRII colocalized with the latter reagent and that colocalization was essential to block B-cell activation. To obtain direct evidence that the two receptors are juxtaposed, splenic B cells were stimulated with Cy5-labeled anti-Ig, either as an F(ab′)2 fragment or as intact antibody. The cells were fixed and costained with FITC-labeled MAb directed against mouse FcγRII/III (Fig. 1A) or against CD45/B220 as a control (Fig. 1B). Images of the FITC label are shown in the left set of panels. Cy5 label data for the same microscopic field are shown are the middle set, and the merged images are shown on the right.
FIG. 1.
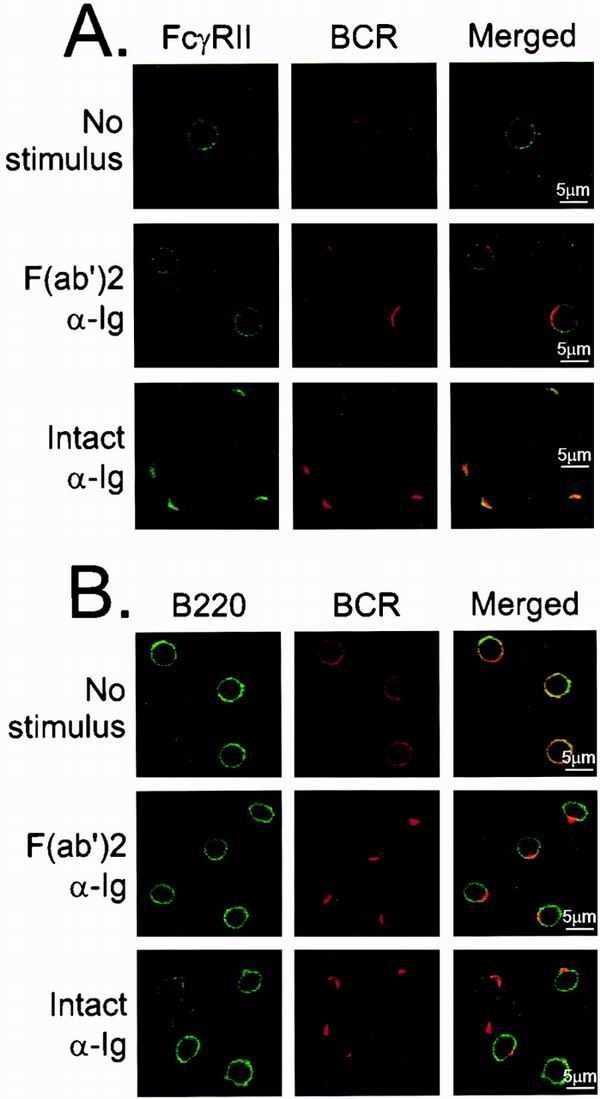
FcγRII and BCR colocalize under intact anti-Ig stimulation conditions. (A) Splenic B cells were stimulated with Cy5-labeled anti-Ig, either F(ab′)2 fragment [F(ab′)2 anti-Ig] or as intact antibody (intact anti-Ig). Cells were stimulated for 2 min and costained with FITC-labeled 2.4G2 to visualize FcγRII. FITC-labeled isotype antibody for 2.4G2 was used as a negative control for confocal microscopy (data not shown). Unstimulated samples (No stimulus) were fixed before staining with Cy5-labeled F(ab′)2 anti-Ig for 1 h at 4°C, washed twice, and stained with FITC-labeled 2.4G2. (B) B220 was visualized using FITC-conjugated anti-B220 antibodies. All cells were examined by confocal microscopy using argon and helium-neon lasers and visualized at mid-plane at ×100 magnification.
In resting cells, both the BCR and FcγRII receptors exhibited a random distribution. After stimulation with anti-Ig, the BCR rapidly formed a cap at one pole of the cell. Receptor capping occurred regardless of the nature of the anti-Ig reagent. B cells stimulated with F(ab′)2 fragments of anti-Ig maintained a random distribution of FcγRII identical to that in resting cells. Likewise, B220 remained randomly distributed on the surface of B cells before and after stimulation with F(ab′)2 fragments of anti-Ig. In contrast, FcγRII but not B220 cocapped with the BCR when the cells were stimulated with intact anti-Ig.
While such images are direct and therefore informative, they can be subjective and hence less instructive than other quantitative techniques. Earlier reports described a statistical means to quantify colocalization of molecules from confocal images (22d). Briefly, the image of each cell is scanned around the perimeter, and fluorescent signals from the two fluorochomes are quantitated at each position. The resulting histograms display overlapping peaks of high signal intensity if the two proteins colocalize. Fluorescent peaks can be statistically evaluated to derive correlation coefficients for each individual cell and each marker within a cell. The correlation coefficient rises to 1.0 when the two molecules are perfectly colocalized. Average correlation coefficients of fluorescent signals from cells stimulated in different ways and times can then be compared.
Fluorescent histograms of the BCR and FcγRII signals from a single cell, resting or stimulated with F(ab′)2 or intact anti-Ig, are shown in Fig. 2A to C. Panel D of Fig. 2 shows average correlation coefficients for 10 to 30 cells. BCR and FcγRII were randomly distributed in unstimulated B cells (Fig. 2A), with a correlation coefficient of ≈0.5 (Fig. 2D). After stimulation with F(ab′)2 or intact anti-Ig, the BCR cap showed a peak of fluorescence in the corresponding fluorescence histograms (Fig. 2B and C). However, FcγRII remained randomly distributed in B cells stimulated with F(ab′)2 anti-Ig. Hence, the correlation coefficient does not significantly change from the resting state. In contrast, B cells stimulated with intact anti-Ig showed a nearly perfect correlation of BCR and FcγRII, with a corresponding correlation coefficient of 0.91 (Fig. 2C and D). These findings confirm that the BCR and FcγRII colocalize when B cells are stimulated with intact anti-Ig antibodies.
FIG. 2.

Quantitative analysis of confocal microscopy image. Confocal microscopic images were obtained from resting cells or cells stimulated with either F(ab′)2 or intact anti-Ig. Fluorescence intensities of positions around the perimeter of a single cell stained for BCR and FcγRII were determined. Fluorescence intensities were plotted for a single cell without stimulus (A), stimulated with F(ab′)2 anti-Ig (B), or stimulated with intact anti-Ig (C). (D) The degree of codistribution of two fluorochrome signals (FITC-labeled 2.4G2 and Cy5-labeled BCR) measured from single cells was assessed with correlation coefficients, as described in the text. The results shown are averages of correlation coefficients and standard errors from cells with no stimulus or F(ab′)2 or intact anti-Ig stimulus for 1 or 2 min. F(ab′)2 and intact anti-Ig treatments at each time point were statistically analyzed for significance of differences using the paired t test. For both cases, the P value was less than 0.0001.
Differential recruitment of SHIP but not SHP-1 to FcγRII.
Earlier experiments by us and others demonstrated that the phosphatases SHIP and SHP-1 can engage a synthetic phosphorylated ITIM peptide of FcγRII. These observations predicted that SHIP would associate with FcγRII in B cells stimulated by intact but not F(ab′)2 fragments of anti-Ig antibodies, since the ITIM is phosphorylated only in the former case.
To address this issue, B cells were stimulated with Cy5-labeled F(ab′)2 or intact anti-Ig, then fixed and permeabilized before staining with rabbit polyclonal anti-SHIP antiserum directly conjugated with AlexaFluor-488. Antibodies to IκB were used as a negative control because IκB is not associated with receptors upon stimulation. In resting and F(ab′)2 anti-Ig stimulation conditions, SHIP showed a diffuse cytoplasmic staining pattern with a corresponding low degree of colocalization with BCR (Fig. 3A). However, consistent with the earlier biochemical data using synthetic phosphopeptides, BCR and SHIP showed a high degree of colocalization when the cells were stimulated with intact anti-Ig. B cells stained for IκB showed little colocalization with the BCR regardless of the form of anti-Ig stimulation (Fig. 3B).
FIG. 3.

SHIP recruitment to the BCR-FcγRII coclustering site, (A) Colocalization of SHIP and BCR was examined by cytoplasmic staining of SHIP. Cells were unstimulated or stimulated for 2 min with F(ab′)2 or intact anti-Ig antibodies. The location of SHIP was examined by AlexaFluor 488-labeled anti-SHIP antibody. (B) Cytoplasmic staining of IκB was performed using biotinylated anti-IκB followed by streptavidin-AlexaFluor 488. (C) Average correlation coefficient for SHIP and BCR from cells unstimulated or stimulated with F(ab′)2 or intact anti-Ig stimulus for 1 or 2 min. N is number of cells examined for each experimental condition, and standard error is indicated as a bar. F(ab′)2 and intact anti-Ig treatment at each time point was analyzed for significance of difference as P value. For both cases the P value was less than 0.0001.
The extent of BCR-SHIP colocalization was quantitated by determining the correlation coefficient in a number of identically stimulated cells. The results (Fig. 3C) showed a correlation coefficient of ≈0.4 for unstimulated B cells, which increased to 0.55 in F(ab′)2 anti-Ig-stimulated B cells. The average correlation coefficient after stimulation with intact anti-Ig antibodies increased to 0.9, confirming the high degree of colocalization of SHIP and BCR under these conditions. However, the correlation coefficient of BCR and IκB was less than 0.36 in resting, F(ab′)2, or intact anti-Ig stimulation conditions (Table 1). Thus, as indicated by in vitro experiments using phosphopeptides, BCR-SHIP colocalization was inefficient when the cells were stimulated with F(ab′)2. In contrast, SHIP (Fig. 3), FcγRII, and BCR (Fig. 1 and 2) are efficiently colocalized when B cells are stimulated with intact anti-Ig.
TABLE 1.
Association of BCR with molecules implicated in B-cell signal transductiona
| Marker | Avg correlation coefficient ± SE (no. of cells)
|
P value | ||
|---|---|---|---|---|
| Resting | F(ab′)2 α-Ig | Intact α-Ig | ||
| Btk | 0.13 ± 0.05 (8) | 0.80 ± 0.05 (10) | 0.50 ± 0.08 (9) | =0.0045 |
| Vav | 0.39 ± 0.06 (8) | 0.87 ± 0.02 (24) | 0.59 ± 0.04 (41) | ≤0.0001 |
| Rac | 0.024 ± 0.04 (10) | 0.83 ± 0.03 (46) | 0.61 ± 0.07 (48) | ≤0.0001 |
| F-actin | 0.28 ± 0.03 (10) | 0.55 ± 0.04 (21) | 0.38 ± 0.06 (23) | =0.0226 |
| FcγRII | 0.49 ± 0.03 (21) | 0.42 ± 0.06 (11) | 0.87 ± 0.01 (10) | ≤0.0001 |
| CD19 | 0.76 ± 0.03 (5) | 0.84 ± 0.03 (7) | 0.86 ± 0.04 (8) | NS |
| CD22 | 0.63 ± 0.04 (8) | 0.77 ± 0.02 (19) | 0.85 ± 0.02 (15) | =0.0105 |
| Dok | 0.30 ± 0.05 (10) | 0.77 ± 0.05 (10) | 0.71 ± 0.02 (12) | NS |
| pTyr | 0.42 ± 0.08 (9) | 0.82 ± 0.03 (12) | 0.80 ± 0.04 (9) | NS |
| SHIP | 0.41 ± 0.04 (12) | 0.55 ± 0.06 (17) | 0.91 ± 0.01 (12) | ≤0.0001 |
| SHP-1 | 0.23 ± 0.03 (10) | 0.70 ± 0.04 (28) | 0.77 ± 0.03 (28) | NS |
| GM1 | 0.21 ± 0.04 (11) | 0.68 ± 0.04 (12) | 0.67 ± 0.05 (11) | NS |
| IκB | 0.23 ± 0.05 (5) | 0.36 ± 0.05 (10) | 0.27 ± 0.05 (9) | NS |
B cells from BALB/c mouse spleens were stimulated with Cy5-labeled F(ab′)2 or intact anti-Ig for 2 min. The cells were fixed, permeabilized, and counterstained with biotinylated antibodies against the various intracellular proteins listed in the table, followed by FITC-labeled streptavidin. The cells were visualized by confocal microscopy, and colocalization of the BCR was quantitated as described in the text. The data are average correlation coefficients ± standard errors for N cells for the BCR and the molecules listed. The last column shows the P values obtained by paired t tests, representing the significance of difference between the correlation coefficient obtained using F(ab′)2 and intact anti-Ig reagents. NS, not significant (P > 0.05). pTyr, phosphotyrosine.
Of the murine IgG receptors, only FcγRII bears an ITIM motif capable of engaging inhibitory phosphatases like SHIP and SHP-1. The other IgG receptors, FcγRI and FcγRIII, are associated with an ITAM-containing gamma chain, and hematopoietic cells from gamma chain−/− animals do not express these receptors (24). Thus, B cells lacking FcγRII but not gamma chain should display deficient BCR-SHIP colocalization.
To test this prediction, we isolated B cells from wild-type animals and from animals deficient in FcγRII, the gamma chain, or both. B cells were stimulated with F(ab′)2 or intact anti-Ig, stained with antibodies to SHIP, and examined as above to determine the degree of colocalization of the BCR with SHIP (Fig. 4A). We found that B cells from gamma chain−/− and wild-type mice displayed a higher correlation coefficient when the cells were stimulated under negative signaling conditions. In contrast, B cells from FcγRII-deficient or gamma chain/FcγRII doubly deficient mice showed a significantly lower correlation coefficient of SHIP and BCR upon intact anti-Ig stimulation. Thus, consistent with earlier experiments using phosphopeptides (27), SHIP colocalization with the BCR under negative signaling conditions is optimal in cells expressing FcγRII.
FIG. 4.

Differential recruitment of SHIP but not SHP-1 to FcγRII. (A) Splenic B cells were derived from wild-type animals or mice deficient in FcγRII, gamma chain, or both receptors. Subcellular locations of SHIP and BCR were visualized after stimulation with Cy5-labeled F(ab′)2 or intact anti-Ig. Confocal microscopic analysis was done as described for Fig. 2. Numbers of cells analyzed after F(ab′)2 or intact anti-Ig treatment are shown below the label. The asterisks indicate a significant difference with a P value of ≤0.0003. (B) Splenic B cells of wild-type or FcγRII−/− mice were stimulated with Cy5-labeled F(ab′)2 or intact anti-Ig, fixed, permeabilized, and stained with biotinylated SHP-1 antibody followed by streptavidin-FITC. The stimulated samples showed no significant difference.
Experiments using the phosphorylated ITIM motif of FcγRII in peptide pull-down experiments identified the protein phosphatase SHP-1 as being able to engage this motif (8). If SHP-1 is specifically recruited to FcγRII, the correlation coefficient of SHP-1 and BCR should be higher under negative signaling conditions. We applied correlation coefficient analysis of cells stained with anti-SHP-1 and -BCR antibodies to address this issue. The results (Fig. 4B) revealed a low correlation coefficient (0.23) of SHP-1 and the BCR in resting cells.
Interestingly, SHP-1 was recruited to BCR upon stimulation with F(ab′)2 anti-Ig as well as intact anti-Ig, with correlation coefficients of 0.70 and 0.77, respectively. The difference in the correlation coefficients was not significant (Table 1). Hence, despite the increased presence of the ITIM-bearing FcγRII in the BCR cap, SHP-1 recruitment to the BCR was not improved under negative signaling conditions. Furthermore, recruitment of SHP-1 to BCR upon stimulation with any form of anti-Ig still occurred in FcγRII−/− B cells (Fig. 4B). These results indicate that SHP-1 recruitment to the BCR is not mediated by FcγRII.
Differential recruitment of other signaling molecules to FcγRII.
We applied this technique to analyze intracellular effector molecules involved in positive or negative signal transduction pathways. These molecules are listed in Table 1. The data shown are the average correlation coefficients of the BCR with each molecule in resting B cells and activated B cells. P values have been calculated to determine whether the correlation coefficients are significantly different. Several molecules, including Btk, Vav, Rac, and F-actin, showed low values of BCR colocalization in the resting state, increased values upon activation by F(ab′)2 anti-Ig, but significantly reduced values upon activation by intact anti-Ig. CD19 and CD22 both displayed high degrees of colocalization with BCR in resting cells, as observed previously (4, 16), and the association increased slightly upon stimulation. The adapter protein Dok and tyrosine-phosphorylated proteins showed a random pattern of BCR colocalization in resting cells, which increased upon BCR stimulation. Increased colocalization of BCR and Dok was not different under positive and negative signaling conditions, suggesting that Dok recruitment to the BCR is not affected by the presence of SHIP or FcγRII.
The BCR showed a stimulation-induced increase in colocalization with lipid rafts, marked by the cholera toxin subunit B, which binds gangliosides present in rafts (6). BCR migration into lipid rafts was not affected by stimulation conditions that led to colocalization with FcγRII and the inositol phosphatase SHIP.
SHIP prevents PtdIns 3-kinase-dependent colocalization of F-actin to BCR cap.
The observation that association of Vav, Rac, and F-actin with the BCR is reduced under negative signaling suggests an effect on the actin cytoskeleton, which might alter antigen receptor internalization. Colocalization of F-actin and BCR might be a crucial prerequisite for functional BCR signaling and internalization, since perturbing actin filaments with cytochalasin D reduced the rate of BCR internalization and blocked the movement of the BCR to late endosomes (2). Moreover, it has been reported that engagement of FcγRII with BCR negatively regulated antigen internalization, processing, and presentation (19, 31).
To explore the effect of the presence of SHIP in the BCR cap on cytoskeletal rearrangement, we measured the kinetics of BCR colocalization with Vav, Rac, and F-actin when B cells were stimulated with F(ab′)2 or intact anti-Ig. Figure 5 shows the averages of three independent experiments measuring colocalization of BCR with Vav (Fig. 5A), Rac (Fig. 5B), and F-actin (Fig. 5C). In all cases, the data show that BCR colocalization with these molecules was reduced in B cells stimulated under conditions leading to SHIP recruitment.
FIG. 5.

Differential recruitment of Vav, Rac, and F-actin to BCR and reduced BCR internalization under intact anti-Ig stimulation conditions. (A) Splenic B cells stimulated with Cy5-labeled F(ab′)2 or intact anti-Ig and stained with biotinylated anti-Vav antiserum were visualized by confocal microscopy and correlation coefficients were calculated. The inset shows the percentage of B cells exhibiting BCR cocapping with Vav, assessed visually by counting >100 cells. Correlation coefficient values of intact anti-Ig-stimulated samples at all time points showed a significant difference with P < 0.0001. (B) B cells were stimulated as in panel A, stained with anti-Rac antiserum, and analyzed for average correlation coefficients of the BCR and Rac. The inset shows the percentage of B cells exhibiting BCR cocapping with Rac, assessed visually by counting >100 cells. Correlation coefficient values of intact anti-Ig-stimulated samples at all time points showed a significant difference, with P < 0.0001. (C) B cells were stimulated as in panel A, stained with phalloidin, and analyzed for the average correlation coefficient of the BCR and F-actin. Correlation coefficient values of intact anti-Ig-stimulated samples at all time points showed a significant difference, with P < 0.0226. (D) A20 B cells were labeled with FITC-conjugated F(ab′)2 or intact anti-Ig and incubated at 37°C for the indicated times. The percent internalized BCR was derived as described in the text. (A to D) Open circles, F(ab′)2 stimulation; solid circles, intact anti-Ig stimulation.
Interestingly, although colocalization of these molecules and BCR was reduced under conditions leading to SHIP recruitment, it was not completely abrogated. Percent cocapping between BCR and Vav or BCR and Rac was measured by visually comparing the staining pattern of more than 100 cells at each time point (insets in Fig. 5A and 5B). Both the correlation coefficient and the percent cocapping under negative signaling conditions were significantly lower than those under positive signaling conditions. We also measured BCR internalization by staining cells with FITC-labeled anti-Ig reagents and quenching extracellular fluorescence with anti-FITC antiserum after incubation for the indicated times. The results (Fig. 5D) likewise showed a reduction of internalization at all time points in B cells stimulated with intact anti-Ig.
The data indicate that recruitment of SHIP to BCR reduces the level of the PtdIns 3-kinase product, PtdInse-3,4,5,-P3, which is needed for Rac activation and thus contributes to the initiation of actin polymerization and antigen receptor internalization.
To directly test the role of PtdIns 3-kinase in actin reorganization, we examined BCR-F-actin colocalization in B cells from wild-type animals untreated or treated with LY294002, a PtdIns 3-kinase inhibitor. Although F-actin distribution was not exclusively present on the BCR cap, the accumulation of F-actin with the BCR cap region was apparent upon treatment with F(ab′)2 anti-Ig. Beginning at 2 min after stimulation, the BCR formed a condensed protrusion that was highly enriched with F-actin. B cells from wild-type mice showed reduced colocalization of BCR and F-actin (Fig. 6A) after stimulation with intact anti-Ig, as described above.
FIG. 6.
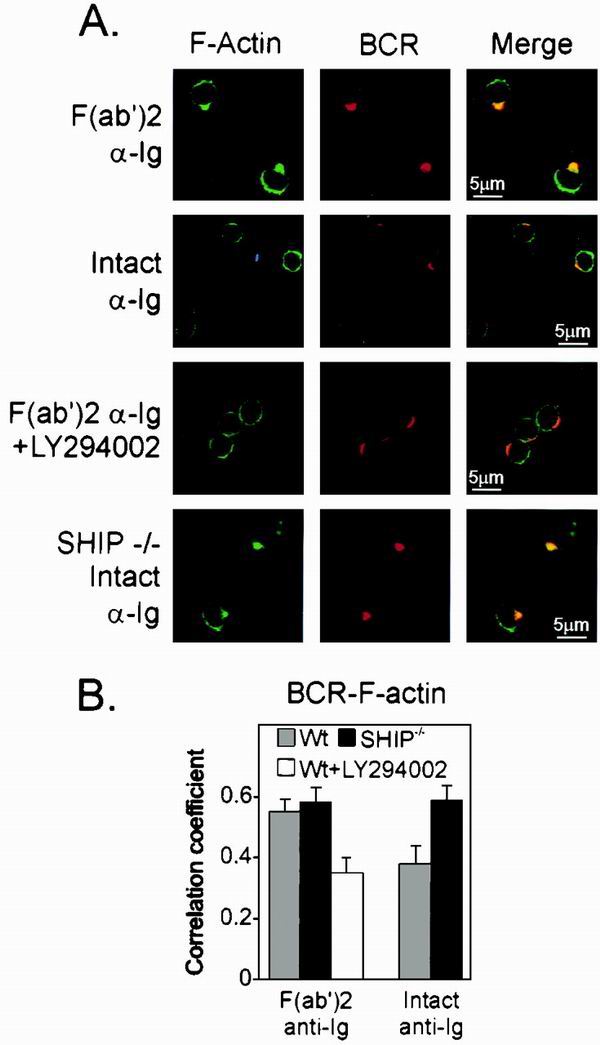
SHIP recruitment reduces PtdIns-3 kinase dependent colocalization of F-actin to BCR cap. (A) B cells from wild-type or SHIP−/− mice were stimulated with F(ab′)2 or intact anti-Ig in the presence or absence of 25 μM LY294002, as indicated. Cells were stained for BCR and F-actin as described for Fig. 5. (B) Codistribution of F-actin and BCR was examined as described in the text by correlation coefficient. Gray bar, wild-type B cells; black bars, SHIP−/− B cells. The white bar represents the average correlation coefficient of BCR and F-actin of splenic B cells treated with the PtdIns-3 kinase inhibitor LY294002.
B cells treated with LY294002 and stimulated with F(ab′)2 fragments of anti-Ig appeared like those stimulated with intact anti-Ig, showing reduced BCR-F-actin colocalization. These findings show that receptor-initiated actin reorganization requires PtdIns 3-kinase and hence might be subject to inhibition by SHIP.
To directly test the influence of SHIP in BCR-initiated actin reorganization, we measured BCR-F-actin colocalization in B cells derived from SHIP−/− animals. SHIP−/− B cells showed a pronounced colocalization of F-actin and BCR upon stimulation with intact anti-Ig. The correlation coefficient data for both experiments are shown in Fig. 6B. Since B cells of SHIP−/− mice have FcγRII and SHP-1, these observations indicate that SHIP is required for the FcγRII-mediated inhibition of F-actin redistribution. The data are consistent with our central hypothesis that SHIP reduces F-actin colocalization with BCR by hydrolysis of PtdIns-3,4,5-P3.
Rac is essential for antigen receptor internalization.
Vav promotes nucleotide exchange and activation of Rac, and the exchange activity of Vav is dependent on PtdIns 3-kinase (12). G protein-coupled receptors reorganize the cytoskeleton through the activation of PtdIns 3-kinase, Vav, and Rac (18). In conjunction with these earlier findings, our data suggest that SHIP-mediated hydrolysis of PtdIns 3-kinase products may prevent activation of Vav and Rac, resulting in reduced colocalization of BCR and F-actin and reduced antigen receptor internalization.
To explore the role of Rac in BCR internalization, we transfected B cells with a dominant-negative form of Rac (N17Rac) fused to the GFP sequence or with GFP alone. The cells were incubated with F(ab′)2 fragments of Cy5-labeled anti-Ig and transferred to 37°C to induce receptor internalization. Midpoint confocal images of the transfectants held at 4°C and those incubated at 37°C are shown in Fig. 7. The GFP-only transfectants (Fig. 7A) showed surface labeling of the BCR in cells held at 4°C, with no attending receptor internalization, as indicated by a lack of Cy5 fluorescence in the cytoplasm. Transfer of the cells to 37°C initiated receptor internalization. Internalized BCR was apparent in the images of both the untransfected cells and the GFP+-transfected subpopulation. Cy5-anti-Ig labeling of B cells transfected with N17Rac (Fig. 7B) and held at 4°C likewise showed a surface staining pattern. When the temperature was raised to 37°C, the GFP− cells in this population, which do not express N17Rac, displayed internalized BCR. In contrast, the GFP+ cells expressing the transfected dominant-negative N17Rac showed Cy5-labeled BCR on the surface only.
FIG. 7.
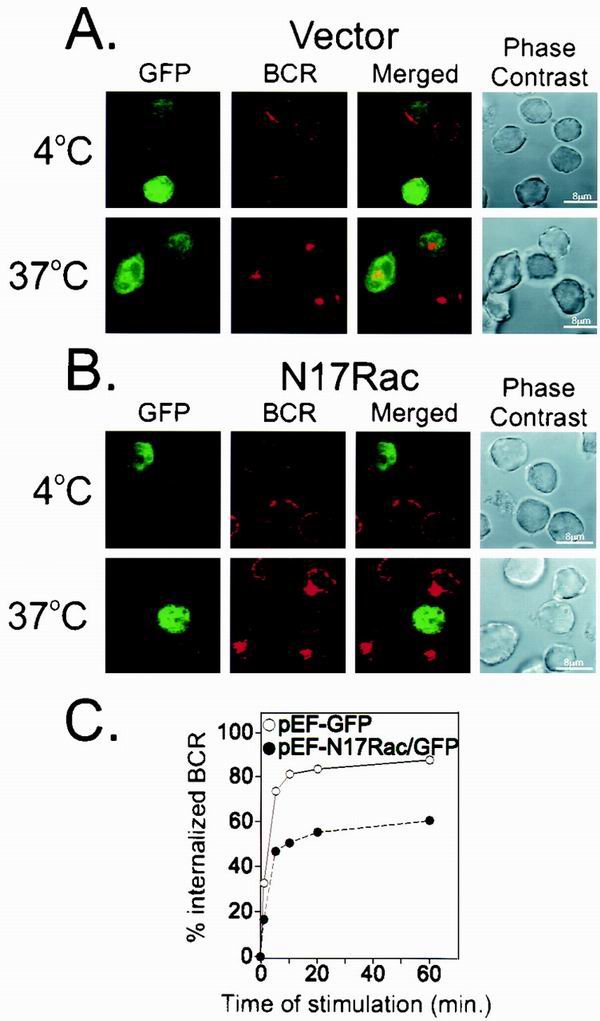
Rac is required for BCR internalization. (A) A20 cells were transfected with either GFP vector control (A) or dominant negative form of Rac (N17Rac-GFP) (B). After 24 h, Cy5-labeled F(ab′)2 anti-Ig was added for 30 min at 4°C. The anti-Ig-treated cells were held at 4°C or moved to 37°C for 10 min to permit receptor internalization. Internalized BCR was visualized using confocal microscopy. Cells appearing green represent those transfected with the GFP vector (A) or GFP-N17Rac (B). (C) To quantify receptor internalization from GFP vector- and N17Rac-GFP-transfected cells, cells were stimulated with unlabeled F(ab′)2 anti-Ig for the indicated times to allow internalization. The reactions were stopped using cold staining buffer, and remaining uninternalized BCR was labeled with Cy5-labeled F(ab′)2 anti-Ig and quantitated by flow cytometry. GFP-positive cells were gated, and mean fluorescence intensity of Cy5-labeled F(ab′)2 anti-Ig was measured at each time point. This value represented noninternalized BCR. Mean fluorescence intensity at 4°C was used to represent the total amount of BCR. Percent internalized BCR was measured as a loss of Cy5 fluorescence by subtracting noninternalized BCR from the total amount of BCR and divided by the total amount of BCR.
We quantified the effect of N17Rac expression on BCR internalization by analyzing the GFP-labeled cells using flow cytometry. The data in Fig. 7C represent the loss of surface BCR relative to the amount present on B cells held at 4°C before initiation of receptor internalization. Using this assay, we found that cells expressing GFP alone internalized the BCR rapidly, so that approximately 80% of Cy5-anti-Ig binding activity was lost after 10 min. B cells expressing N17Rac showed a similar rate of receptor internalization but failed to achieve the same extent of internalization as the GFP-only control. These observations show that Rac is essential in BCR internalization following its engagement by ligand.
DISCUSSION
Enzymes involved in a signal transduction process are primarily regulated by subcellular location. Alteration in enzyme location is a function of protein interaction domains and the ligand-induced generation of specific binding elements. Thus, tyrosine-phosphorylated proteins are generated upon receptor clustering to recruit SH2 domain-containing enzymes. Likewise, 3-phosphoinositides are generated by PtdIns 3-kinase to recruit proteins containing a pleckstrin homology (PH) domain. Consistent with this paradigm, our earlier studies (5, 22, 27, 28) and those of others (8, 21) using a variety of biochemical techniques reported findings consistent with the notion that the FcγRII provokes an inhibitory signal by recruitment of the SH2 domain-containing inositol phosphatase SHIP to FcγRII and the BCR.
Here, we have explored the subcellular localization of several enzymes and regulatory proteins involved in B-cell signal transduction. By quantitative confocal microscopic methods, we identified colocalized complexes composed of the BCR, FcγRII, and SHIP when B cells were stimulated with intact anti-Ig. Formation of these complexes required that responding B cells express the ITIM-bearing FcγRII. Conditions leading to SHIP-containing receptor complexes were accompanied by decreased recruitment of PH domain-containing proteins Vav and Btk, reduced recruitment of Rac and F-actin, as well as decreased receptor internalization.
Consistent with a role for the inositol phosphatase SHIP in reducing receptor recruitment of these molecules, the block in BCR-F-actin association was not seen in SHIP-deficient B cells. Likewise, inhibition of PtdIns 3-kinase or expression of dominant-negative Rac prevented F(ab′)2 anti-Ig-triggered BCR-F-actin association and receptor internalization, respectively. From these findings, we suggest the model shown in Fig. 8. Coclustering of BCR and FcγRII promotes the recruitment of SHIP, which consumes the PtdIns 3-kinase product, PtdIns-3,4,5-P3. PtdIns-3,4,5-P3 hydrolysis by SHIP inhibits the recruitment and activation of Vav and the subsequent activation of Rac. The resulting disruption of the actin cytoskeleton causes a defect in antigen receptor internalization.
FIG. 8.
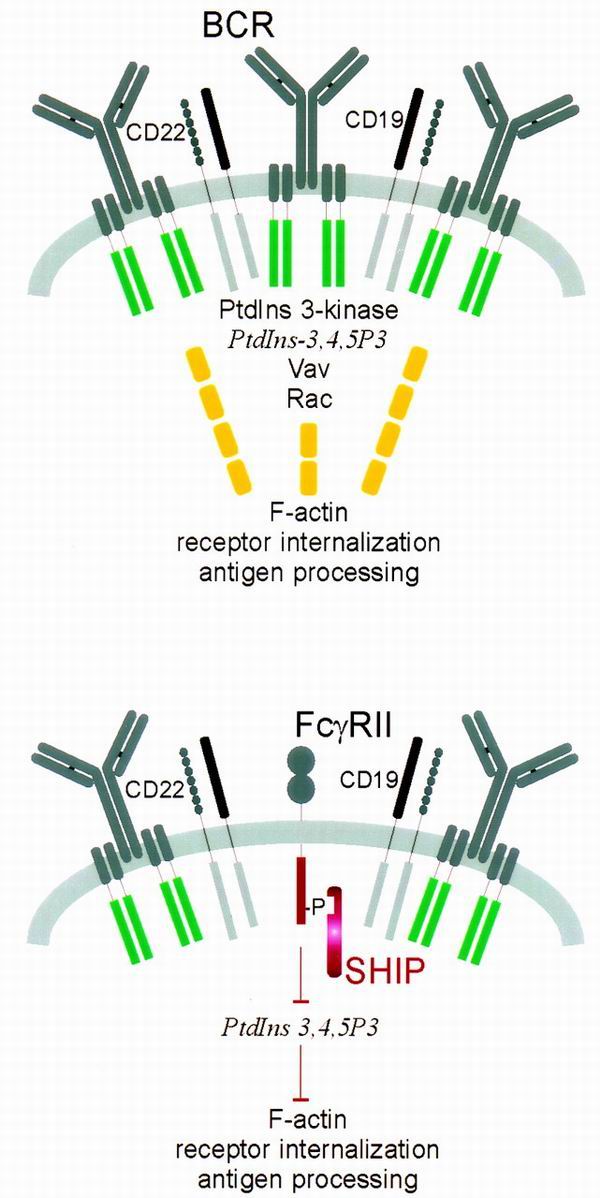
Model of inhibition of actin redistribution and receptor internalization by FcγRII-SHIP. Upon receptor clustering, the BCR forms a cap that includes CD22, CD19, and intracellular signaling molecules PtdIns 3-kinase, Vav, and Rac. BCR clustering activates PtdIns 3-kinase to form PtdIns-3,4,5-P3, which acts to recruit and stimulate Vav. Active Vav stimulates the small GTPase Rac to elicit actin reorganization. When FcγRII and SHIP are present with the BCR, formation of PtdIns-3,4,5-P3 is reduced, and hence all events distal to PtdIns-3,4,5-P3 are blocked.
When the BCR is clustered by antigen or anti-Ig, it becomes physically associated with the detergent-insoluble cytoskeletal components and undergoes patching, capping, and endocytosis. Actin polymerization and antigen receptor redistribution are critical for this process, since disturbing actin filaments with cytochalasin D, which blocks actin polymerization, inhibited BCR internalization as well as BCR targeting to late endosomes (2). Treatment of T cells with the same reagent was shown to inhibit capping of the T-cell antigen receptor and produce defects in the signaling pathways leading to interleukin-2 (IL-2) transcription (14). How the signals generated from BCR link to reorganizing actin cytoskeleton is not yet understood, but the importance of Vav and Rac in this process has been demonstrated (reviewed in reference 10).
Vav is a guanine nucleotide exchange factor for rho-family GTPases such as Rac, which are involved in reorganizing the actin cytoskeleton (3). Vav is essential for T-cell receptor capping, effective actin polymerization in response to antigen receptor activation (14, 29, 32), and actin-dependent receptor translocation to the interface between the lymphocyte and the antigen-presenting cell (32). Moreover, T and B cells of Vav-deficient mice show defects in development, antigen-induced proliferation, calcium influx, and IL-2 production (26, 34). Vav is regulated both by phosphorylation and by the binding of PtdIns 3-kinase products to its PH domain (12) and acts proximal to Rac activation (7). The hydrolysis of PtdIns-3,4,5-P3 by SHIP could therefore perturb actin cytoskeleton rearrangement by preventing Vav and Rac activation.
However, it is noteworthy that Btk, another PH domain-containing protein, was reported to be involved in actin rearrangement, acting as a link between PtdIns 3-kinase and Rac (20). These observations strongly suggest a role for PtdIns 3-kinase in regulating PH domain-containing molecules during cytoskeleton reorganization. We observed that the colocalization of polymerized actin and BCR was severely inhibited when SHIP was recruited to the BCR-FcγRII complex. Treatment with PtdIns 3-kinase inhibitor produced the same effect, and the inhibition was not seen in the SHIP−/− B cells. These findings demonstrate that SHIP reduces F-actin and BCR colocalization by consumption of PtdIns 3-kinase products.
SHIP has a documented role in the regulation of actin-dependent processes such as chemokine-dependent migration (15, 30). Splenocytes from SHIP−/− mice demonstrated enhanced F-actin levels and increased migration towards chemokines (15). SHIP inhibited insulin-induced GLUT4 translocation as well as membrane ruffling triggered by growth factor in adipocytes and fibroblasts (30). Interestingly, recent findings suggested that PTEN (phosphatase and tensin homolog deleted on chromosome 10), an inositol 3-phosphatase and negative regulator for PtdIns 3-kinase, suppresses cell migration by hydrolysis of PtdIns-3,4,5-P3 and consequently downregulating Rac and Cdc42 in fibroblasts (17).
The protein phosphatase SHP-1 is capable of binding the phosphorylated ITIM peptide corresponding to FcγRII (8). This in vitro binding activity was invoked to account for the reduction in CD19 phosphorylation upon stimulation of B cells with intact anti-Ig reagent (13). However, we demonstrate here that SHP-1 is not differentially recruited to receptor clusters containing BCR or BCR-FcγRII and that BCR colocalization of SHP-1 does not require FcγRII expression. Likewise, CD22 is coclustered with the BCR regardless of the form of the stimulating anti-Ig reagent. Phosphopeptide pull-down experiments indicated that SHP-1 can engage phosphotyrosines of CD22 (1, 9, 23, 33), a receptor bearing several cytoplasmic tyrosines in an ITIM configuration. Thus, a more likely scenario is that SHP-1 acts to negatively regulate BCR signal transduction independent of FcγRII and possibly through its interaction with other ITIM-containing receptors like CD22.
Stimulation of B cells with intact anti-Ig antibodies causes a block in stimulation of the Ras pathway. Dok is an adapter protein that recruits the Ras GTPase-activating protein RasGAP. Dok is more efficiently phosphorylated upon BCR-FcγRII coclustering relative to BCR clustering alone (25). It was proposed that SHIP acts to recruit a complex of proteins containing Dok and RasGAP upon BCR-FcγRII coclustering and that the Dok-associated RasGAP blocks GTP loading of Ras (25), which implies differential recruitment of Dok to SHIP or FcγRII. However, we found essentially identical BCR-Dok colocalization when B cells were stimulated with intact or F(ab′)2 fragments of anti-Ig. Other experiments in fibroblasts suggest that Dok is recruited to the plasma membrane and exerts its inhibitory effect on Ras by engaging 3-phosphoinositides via the Dok PH domain (35).
The Dok PH domain appeared to have equivalent affinity for PtdIns-3,4,5-P3, the SHIP substrate, and PtdIns-3,4-P2, the SHIP product (35). Neither our studies of BCR-Dok localization nor the recent experiments in fibroblasts on the role of the Dok PH domain can account for the reported differential tyrosine phosphorylation of Dok and its association with RasGAP (25). It is possible that these events occur at later times than used in this study. Indeed, Dok phosphorylation and association with RasGAP are submaximal 2 min after stimulation (25), the time used in our experiments.
The BCR cap shown here with its accumulation of various signaling molecules may describe the signalosome of BCR signal transduction (11). According to the signalosome model, a complex composed of phospholipase Cγ2, Syk, Btk, and B-cell linker protein (designated BLNK) is initiated by PtdIns-3,4,5-P3 formation and recruitment and phosphorylation of the adapter molecule BLNK. Thus, gene-targeted disruption of any of the components could result in destabilizing the complex. Accordingly, animals deficient in any single component of the signalosome exhibit a phenotype similar to that of xid mice, which are deficient in Btk (11). PtdIns-3,4,5-P3 produced by PtdIns 3-kinase in the BCR cap nucleates the signalosome by recruiting PH domain-containing enzymes. Polymerized actin in the signalosome may stabilize the complex, thereby generating prolonged and sustained mediators. Consumption of PtdIns-3,4,5-P3 by SHIP would attenuate the formation of signalosome and decrease the presence of polymerized actin in the BCR cap. This notion predicts that the BCR cap would be enriched with respect to PtdIns-3,4,5-P3 and the enrichment would be decreased upon stimulation under conditions leading to recruitment of SHIP.
These findings demonstrate the importance of the subcellular location of proteins involved in signal transduction pathways. Protein location depends on the receptor-induced production of intracellular mediators like 3-phosphoinositide lipids and tyrosine-phosphorylated proteins. The levels of these mediators are in turn regulated by phosphatases like SHIP and SHP-1. Our findings reported here provide direct visual evidence for such negative regulation. Furthermore, visualizing and quantitating movements of molecules provide a powerful technique to resolve mechanisms of signal transduction in lymphocytes and their regulation by phosphatases.
ACKNOWLEDGMENTS
This work was supported by NIH grants CA64268 and AI41447. K. M. Coggeshall is a Scholar of the Leukemia and Lymphoma Society.
REFERENCES
- 1.Blasioli J, Paust S, Thomas M L. Definition of the sites of interaction between the protein tyrosine phosphatase SHP-1 and CD22. J Biol Chem. 1999;274:2303–2307. doi: 10.1074/jbc.274.4.2303. [DOI] [PubMed] [Google Scholar]
- 2.Brown B K, Song W. The actin cytoskeleton is required for the trafficking of the B-cell antigen receptor to the late endosomes. Traffic. 2001;2:414–427. doi: 10.1034/j.1600-0854.2001.002006414.x. [DOI] [PubMed] [Google Scholar]
- 3.Bustelo X R. Regulatory and signaling properties of the Vav family. Mol Cell Biol. 2000;20:1461–1477. doi: 10.1128/mcb.20.5.1461-1477.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carter R H, Doody G M, Bolen J B, Fearon D T. Membrane IgM-induced tyrosine phosphorylation of CD19 requires a CD19 domain that mediates association with components of the B-cell antigen receptor complex. J Immunol. 1997;158:3062–3069. [PubMed] [Google Scholar]
- 5.Chacko G W, Tridandapani S, Damen J, Liu L, Krystal G, Coggeshall K M. Negative signaling in B-lymphocytes induces tyrosine phosphorylation of the 145 kDa inositol polyphosphate 5-phosphatase, SHIP. J Immunol. 1996;157:2234–2238. [PubMed] [Google Scholar]
- 6.Cheng P C, Dykstra M L, Mitchell R N, Pierce S K. A role for lipid rafts in B-cell antigen receptor signaling and antigen targeting. J Exp Med. 1999;190:1549–1560. doi: 10.1084/jem.190.11.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6a.Coggeshall K M. Inhibitory signaling by the B cell FcγRIIb. Curr Opin Immunol. 1998;10:306–312. doi: 10.1016/s0952-7915(98)80169-6. [DOI] [PubMed] [Google Scholar]
- 7.Crespo P, Schuebel K E, Ostrom A A, Gutkind J S, Bustelo X R. Phosphotyrosine-dependent activation of Rac-1 GDP/GTP exchange by the vav proto-oncogene product. Nature. 1997;385:169–172. doi: 10.1038/385169a0. [DOI] [PubMed] [Google Scholar]
- 8.D'Ambrosio D, Hippen K L, Minskoff S A, Mellman I, Pani G, Siminovitch K A, Cambier J C. Recruitment and activation of PTP1C in negative regulation of antigen receptor signaling by Fc gamma RIIB1. Science. 1995;268:293–297. doi: 10.1126/science.7716523. [DOI] [PubMed] [Google Scholar]
- 8a.Diegel M L, Rankin B M, Bolen J B, Dubois P M, Kiener P A. Cross-linking of Fc gamma receptor to surface immunoglobulin on B cells provides an inhibitory signal that closes the plasma membrane calcium channel. J Biol Chem. 1994;269:11409–11416. [PubMed] [Google Scholar]
- 9.Doody G M, Justement L B, Delibrias C C, Matthews R J, Lin J, Thomas M L, Fearon D T. A role in B-cell activation for CD22 and the protein tyrosine phosphatase SHP. Science. 1995;269:242–244. doi: 10.1126/science.7618087. [DOI] [PubMed] [Google Scholar]
- 9a.Famiglietti S J, Nakamura K, Cambier J C. Unique features of SHIP, SHP-1 and SHP-2 binding to FCγRIIb revealed by surface plasmon resonance analysis. Immunol Lett. 1999;68:35–40. doi: 10.1016/s0165-2478(99)00027-9. [DOI] [PubMed] [Google Scholar]
- 10.Fischer K D, Tedford K, Penninger J M. Vav links antigen-receptor signaling to the actin cytoskeleton. Semin Immunol. 1998;10:317–327. doi: 10.1006/smim.1998.0124. [DOI] [PubMed] [Google Scholar]
- 10a.Flaswinkel H, Reth M. Dual role of the tyrosine activation motif of the Ig-alpha protein during signal transduction via the B cell antigen receptor. EMBO J. 1994;13:83–89. doi: 10.1002/j.1460-2075.1994.tb06237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fruman D A, Satterthwaite A B, Witte O N. Xid-like phenotypes: a B-cell signalosome takes shape. Immunity. 2000;13:1–3. doi: 10.1016/s1074-7613(00)00002-9. [DOI] [PubMed] [Google Scholar]
- 12.Han J, Luby-Phelps K, Das B, Shu X, Xia Y, Mosteller R D, Krishna U M, Falck J R, White M A, Broek D. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science. 1998;279:558–560. doi: 10.1126/science.279.5350.558. [DOI] [PubMed] [Google Scholar]
- 12a.Hashimoto A, Hirose K, Okada H, Kurosaki T, Iino M. Inhibitory modulation of B cell receptor-mediated Ca2+ mobilization by Src homology 2 domain-containing inositol 5′-phosphatase (SHIP) J Biol Chem. 1999;274:11203–11208. doi: 10.1074/jbc.274.16.11203. [DOI] [PubMed] [Google Scholar]
- 12b.Herzenberg L A, Tokuhisa T. Carrier-priming leads to hapten-specific suppression. Nature. 1980;285:664–667. doi: 10.1038/285664a0. [DOI] [PubMed] [Google Scholar]
- 12c.Herzenberg L A, Tokuhisa T. Epitope-specific regulation. I. Carrier-specific induction of suppression for IgG anti-hapten antibody responses. J Exp Med. 1982;155:1730–1740. doi: 10.1084/jem.155.6.1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12d.Herzenberg L A, Tokuhisa T, Hayakawa K. Lack of immune response gene control for induction of epitope-specific suppression by TGAL antigen. Nature. 1982;295:329–331. doi: 10.1038/295329a0. [DOI] [PubMed] [Google Scholar]
- 13.Hippen K L, Buhl A M, D'Ambrosio D, Nakamura K, Persin C, Cambier J C. Fc gammaRIIB1 inhibition of BCR-mediated phosphoinositide hydrolysis and Ca2+ mobilization is integrated by CD19 dephosphorylation. Immunity. 1997;7:49–58. doi: 10.1016/s1074-7613(00)80509-9. [DOI] [PubMed] [Google Scholar]
- 14.Holsinger L J, Graef I A, Swat W, Chi T, Bautista D M, Davidson L, Lewis R S, Alt F W, Crabtree G R. Defects in actin-cap formation in Vav-deficient mice implicate an actin requirement for lymphocyte signal transduction. Curr Biol. 1998;8:563–572. doi: 10.1016/s0960-9822(98)70225-8. [DOI] [PubMed] [Google Scholar]
- 14a.Jacob A, Cooney D, Tridandapani S, Kelley T, Coggeshall K M. FcγRIIb modulation of surface immunogloblin-induced Akt activation in murine B cells. J Biol Chem. 1999;274:13704–13710. doi: 10.1074/jbc.274.19.13704. [DOI] [PubMed] [Google Scholar]
- 15.Kim C H, Hangoc G, Cooper S, Helgason C D, Yew S, Humphries R K, Krystal G, Broxmeyer H E. Altered responsiveness to chemokines due to targeted disruption of SHIP. J Clin Investig. 1999;104:1751–1759. doi: 10.1172/JCI7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15a.Klaus G G, Hawrylowicz C M, Holman M, Keeler K D. Activation and proliferation signals in mouse B cells. III. Intact (IGG) anti-immunoglobulin antibodies activate B cells but inhibit induction of DNA synthesis. Immunology. 1984;53:693–701. [PMC free article] [PubMed] [Google Scholar]
- 16.Leprince C, Draves K E, Geahlen R L, Ledbetter J A, Clark E A. CD22 associates with the human surface IgM-B-cell antigen receptor complex. Proc Natl Acad Sci USA. 1993;90:3236–3240. doi: 10.1073/pnas.90.8.3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liliental J, Moon S Y, Lesche R, Mamillapalli R, Li D, Zheng Y, Sun H, Wu H. Genetic deletion of the Pten tumor suppressor gene promotes cell motility by activation of Rac1 and Cdc42 GTPases. Curr Biol. 2000;10:401–404. doi: 10.1016/s0960-9822(00)00417-6. [DOI] [PubMed] [Google Scholar]
- 18.Ma A D, Metjian A, Bagrodia S, Taylor S, Abrams C S. Cytoskeletal reorganization by G protein-coupled receptors is dependent on phosphoinositide 3-kinase gamma, a Rac guanosine exchange factor, and Rac. Mol Cell Biol. 1998;18:4744–4751. doi: 10.1128/mcb.18.8.4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Minskoff S A, Matter K, Mellman I. Fc gamma RII-B1 regulates the presentation of B-cell receptor-bound antigens. J Immunol. 1998;161:2079–2083. [PubMed] [Google Scholar]
- 19a.Muta T, Kurosaki T, Misulovin Z, Sanchez M, Nussenzweig M C, Ravetch J V. A 13-amino-acid motif in the cytoplasmic domain of FcγRIIB modulates B-cell receptor signalling. Nature. 1994;368:70–73. doi: 10.1038/368070a0. [DOI] [PubMed] [Google Scholar]
- 19b.Nadler M J S, Chen B, Anderson J S, Wortis H H, Neel B G. Protein-tyrosine phosphatase SHP-1 is dispensable for FcγRIIB-mediated inhibition of B cell antigen receptor activation. J Biol Chem. 1997;272:20038–20043. doi: 10.1074/jbc.272.32.20038. [DOI] [PubMed] [Google Scholar]
- 20.Nore B F, Vargas L, Mohamed A J, Branden L J, Backesjo C M, Islam T C, Mattsson P T, Hultenby K, Christensson B, Smith C I. Redistribution of Bruton's tyrosine kinase by activation of phosphatidylinositol 3-kinase and Rho-family GTPases. Eur J Immunol. 2000;30:145–154. doi: 10.1002/1521-4141(200001)30:1<145::AID-IMMU145>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 21.Ono M, Bolland S, Tempst P, Ravetch J V. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fc(gamma)RIIB. Nature. 1996;383:263–266. doi: 10.1038/383263a0. [DOI] [PubMed] [Google Scholar]
- 21a.Ono M, Okada H, Bolland S, Yanagi S, Kurosaki T, Ravetch J V. Deletion of SHIP or SHP-1 reveals two distinct pathways for inhibitory signaling. Cell. 1997;90:293–301. doi: 10.1016/s0092-8674(00)80337-2. [DOI] [PubMed] [Google Scholar]
- 22.Phee H, Jacob A, Coggeshall K M. Enzymatic activity of the Src homology 2 domain-containing inositol phosphatase is regulated by a plasma membrane location. J Biol Chem. 2000;275:19090–19097. doi: 10.1074/jbc.M001093200. [DOI] [PubMed] [Google Scholar]
- 22a.Phillips N E, Parker D C. Cross-linking of B lymphocyte Fcγ receptors and membrane immunoglobulin inhibits anti-immunoglobulin-induced blastogenesis. J Immunol. 1984;132:627–632. [PubMed] [Google Scholar]
- 22b.Phillips N E, Parker D C. Fc-dependent inhibition of mouse B cell activation by whole anti-μ antibodies. J Immunol. 1983;130:602–606. [PubMed] [Google Scholar]
- 22c.Sakar S, Schlottmann K, Cooney D, Coggeshall K M. Negative signaling via FcγIIB1 in B cells blocks phospholipase Cγ2 phosphorylation but not Syk or Lyn activation. J Biol Chem. 1996;271:20182–20186. doi: 10.1074/jbc.271.33.20182. [DOI] [PubMed] [Google Scholar]
- 22d.Sheets E D, Holowka D, Baird B. Critical role for cholesterol in Lyn-mediated tyrosine phosphorylation of FcɛRI and their association with detergent-resistant membranes. J Cell Biol. 1999;145:877–887. doi: 10.1083/jcb.145.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22e.Sinclair N R. Immunoreceptor tyrosine-based inhibitory motifs on activating molecules. Crit Rev Immunol. 2000;20:89–102. [PubMed] [Google Scholar]
- 22f.Sinclair N R, Anderson C C. Co-stimulation and co-inhibition: equal partners in regulation. Scand J Immunol. 1996;43:597–603. doi: 10.1046/j.1365-3083.1996.d01-267.x. [DOI] [PubMed] [Google Scholar]
- 23.Smith K G C, Tarlinton D M, Doody G M, Hibbs M L, Fearon D T. Inhibition of the B-cell by CD22: a requirement for Lyn. J Exp Med. 1998;187:807–811. doi: 10.1084/jem.187.5.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takai T, Li M, Sylvestre D, Clynes R, Ravetch J V. FcR gamma chain deletion results in pleiotropic effector cell defects. Cell. 1994;76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 25.Tamir I, Stolpa J C, Helgason C D, Nakamura K, Bruhns P, Daeron M, Cambier J C. The RasGAP-binding protein p62dok is a mediator of inhibitory FcgammaRIIB signals in B cells. Immunity. 2000;12:347–358. doi: 10.1016/s1074-7613(00)80187-9. [DOI] [PubMed] [Google Scholar]
- 26.Tarakhovsky A, Turner M, Schaal S, Mee P J, Duddy L P, Rajewsky K, Tybulewicz V L. Defective antigen receptor-mediated proliferation of B and T cells in the absence of Vav. Nature. 1995;374:467–470. doi: 10.1038/374467a0. [DOI] [PubMed] [Google Scholar]
- 26a.Tridandapani S, Kelley T, Cooney D, Pradhan M, Coggeshall K M. Negative signaling in B cells: SHIP Grbs Shc. Immunol Today. 1997;18:424–427. doi: 10.1016/s0167-5699(97)01112-2. [DOI] [PubMed] [Google Scholar]
- 27.Tridandapani S, Kelley T, Pradhan M, Cooney D, Justement L B, Coggeshall K M. Recruitment and phosphorylation of SHIP and Shc to the B-cell Fcγ ITIM peptide motif. Mol Cell Biol. 1997;17:4305–4311. doi: 10.1128/mcb.17.8.4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tridandapani S, Phee H, Shivakumar L, Kelley T, Coggeshall K M. Role of SHIP in FcgammaRIIb-mediated inhibition of Ras activation in B cells. Mol Immunol. 1999;35:1135–1146. doi: 10.1016/s0161-5890(98)00097-2. [DOI] [PubMed] [Google Scholar]
- 28a.Tridandapani S, Pradhan M, LaDine J R, Garber S, Anderson C L, Coggeshall K M. Protein interactions of Src homology 2 (SH2) domain-containing inositol phosphatase (SHIP): association with Shc displaces SHIP from FcγRIIb in B cells. J Immonol. 1999;162:1408–1414. [PubMed] [Google Scholar]
- 29.Villalba M, Coudronniere N, Deckert M, Teixeiro E, Mas P, Altman A. A novel functional interaction between Vav and PKCtheta is required for TCR-induced T cell activation. Immunity. 2000;12:151–160. doi: 10.1016/s1074-7613(00)80168-5. [DOI] [PubMed] [Google Scholar]
- 30.Vollenweider P, Clodi M, Martin S S, Imamura T, Kavanaugh W M, Olefsky J M. An SH2 domain-containing 5′-inositolphosphatase inhibits insulin-induced GLUT4 translocation and growth factor-induced actin filament rearrangement. Mol Cell Biol. 1999;19:1081–1091. doi: 10.1128/mcb.19.2.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wagle N M, Faassen A E, Kim J H, Pierce S K. Regulation of B-cell receptor-mediated MHC class II antigen processing by FcgammaRIIB1. J Immunol. 1999;162:2732–2740. [PubMed] [Google Scholar]
- 31a.Walker J K, Premont R T, Barak L S, Caron M G, Shetzline M A. Properties of secretin receptor internalization differ from those of the beta(2)-adrenergic receptor. J Biol Chem. 1999;274:31515–31523. doi: 10.1074/jbc.274.44.31515. [DOI] [PubMed] [Google Scholar]
- 32.Wulfing C, Bauch A, Crabtree G R, Davis M M. The vav exchange factor is an essential regulator in actin-dependent receptor translocation to the lymphocyte-antigen-presenting cell interface. Proc Natl Acad Sci USA. 2000;97:10150–10155. doi: 10.1073/pnas.97.18.10150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yohannan J, Wienands J, Coggeshall K M, Justement L B. Analysis of tyrosine phosphorylation-dependent interactions between stimulatory effector proteins and the B-cell coreceptor CD22. J Biol Chem. 1999;274:18769–18776. doi: 10.1074/jbc.274.26.18769. [DOI] [PubMed] [Google Scholar]
- 34.Zhang R, Alt F W, Davidson L, Orkin S H, Swat W. Defective signalling through the T- and B-cell antigen receptors in lymphoid cells lacking the vav proto-oncogene. Nature. 1995;374:470–473. doi: 10.1038/374470a0. [DOI] [PubMed] [Google Scholar]
- 35.Zhao M, Schmitz A A, Qin Y, Di Cristofano A, Pandolfi P P, Van Aelst L. Phosphoinositide 3-kinase-dependent membrane recruitment of p62(dok) is essential for its negative effect on mitogen-activated protein (MAP) kinase activation. J Exp Med. 2001;194:265–274. doi: 10.1084/jem.194.3.265. [DOI] [PMC free article] [PubMed] [Google Scholar]