

Abstract
In an effort to identify sets of yeast genes that are coregulated across various cellular transitions, gene expression data sets derived from yeast cells progressing through the cell cycle, sporulation, and diauxic shift were analyzed. A partitioning algorithm was used to divide each data set into 24 clusters of similar expression profiles, and the membership of the clusters was compared across the three experiments. A single cluster of 189 genes from the cell cycle experiment was found to share 65 genes with a cluster of 159 genes from the sporulation data set. Many of these genes were found to be clustered in the diauxic-shift experiment as well. The overlapping set was enriched for genes required for rRNA biosynthesis and included genes encoding RNA helicases, subunits of RNA polymerases I and III, and rRNA processing factors. A subset of the 65 genes was tested for expression by a quantitative-relative reverse transcriptase PCR technique, and they were found to be coregulated after release from alpha factor arrest, heat shock, and tunicamycin treatment. Promoter scanning analysis revealed that the 65 genes within this ribosome and rRNA biosynthesis (RRB) regulon were enriched for two motifs: the 13-base GCGATGAGATGAG and the 11-base TGAAAAATTTT consensus sequences. Both motifs were found to be important for promoting gene expression after release from alpha factor arrest in a test rRNA processing gene (EBP2), which suggests that these consensus sequences may function broadly in the regulation of a set of genes required for ribosome and rRNA biosynthesis.
Ribosome biosynthesis is a complex and demanding process that depends directly upon multiple metabolic pathways, including the activities of three different RNA polymerases (reviewed in references 21 and 27). In Saccharomyces cerevisiae there are 137 ribosomal protein genes (RP genes), and they are transcribed by RNA polymerase II to yield 78 ribosomal proteins. Because the RP genes are transcribed at such a high level, together they account for nearly 50% of the total RNA polymerase II-mediated transcription initiation events (18). The 25S, 18S, and 5.8S rRNAs are synthesized by RNA polymerase I, first as a large 35S transcript that subsequently gets processed into the three smaller, mature species. Synthesis of the 5S rRNA is distinct from the other rRNAs and is carried out by RNA polymerase III. In order to achieve the high levels of rRNA production that are required during rapid cell division, yeast cells contain roughly 150 repeats of the rRNA genes in a tandem array on chromosome XII. Together, these repeats represent 10% of the genome, and rRNA production alone accounts for some 60% of the total cellular transcription.
Ribosome biogenesis also depends upon the activities of a large number of protein and RNA molecules that are not themselves components of the final ribosome. The complex processing pathway that converts the 35S precursor rRNA into the mature 25S, 18S, and 5.8S rRNA species requires a multitude of factors, including RNA endonucleases, exonucleases, RNA helicases, base modification enzymes, and small nucleolar RNAs (24). Many of these processing factors are nucleolar proteins that were identified through the characterization of mutants that exhibit defects in ribosome biosynthesis. For example, Ebp2p is an essential, nucleolar protein that is required for processing of the 27S pre-rRNA (13). Temperature-sensitive ebp2-1 mutants become depleted of the mature 25S and 5.8S rRNAs at the restrictive temperature, and this diminution leads to a decline in ribosome production and the cessation of cell division. Similarly, there are dozens of other genes whose essential functions relate to the roles they play in rRNA biosynthesis.
Given the importance of ribosome biogenesis to the total economy of cellular metabolism, it is perhaps not surprising that cells have evolved mechanisms to regulate this process. Yeast cells can modulate ribosome production in response to nutrient availability, heat shock, and defects in the secretory pathway (27). The major mechanism whereby cells effect this regulation is through transcriptional control, and both heat shock and secretory defects cause a rapid repression of rRNA and RP gene transcription (18, 19). Most RP gene promoters contain two Rap1p binding sites (17) and Rap1p can act both as an activator and as a silencer of transcription (20). Although promoter swap experiments have demonstrated that the Rap1p binding sequences from the RPL30 promoter are sufficient to confer the repression response when placed upstream of the ACT1 gene, they are not the only cis-acting elements involved in this response (18).
The recent development of microarray hybridization technologies has provided the opportunity to investigate the regulation of gene expression on a genome-wide scale (10). By this approach researchers have been able to compile very large data sets that include transcription profiles for nearly all of the known yeast genes. These data sets include expression profiles derived from cells proceeding through the cell cycle, as well as expression responses to external stresses and drug treatments (5, 7, 11, 23). Analysis of these data sets has identified clusters of genes that exhibit similar expression profiles. The members of these clusters could potentially represent common targets of a particular transcriptional control pathway, such as genes containing the MCB element in their promoters. Alternatively, the clusters could represent sets of genes that are regulated by different mechanisms to achieve a coordinated response to a particular cellular requirement or stimulus. In either case, the identification and characterization of these clusters provide insights into how cells coordinate and regulate gene expression networks in response to diverse stimuli.
In this study we have used microarray expression data (7, 9, 23) to identify a novel cluster of coregulated genes. This regulon is enriched for genes that encode for proteins that have been implicated to play a role in RNA metabolism and rRNA processing. Promoter analysis reveals that this set of genes is highly enriched for two sequence motifs that are preferentially located between 50 and 200 bp upstream of their respective translation initiation sites. Site-directed mutagenesis was used to demonstrate that both promoter elements were important in vivo for regulating the expression of the rRNA processing-related EBP2 gene after release from alpha factor arrest.
MATERIALS AND METHODS
Strains and media.
The yeast strains and plasmids used in this study are described in Table 1. Standard yeast genetic and molecular biology techniques were used throughout (1, 12), and a list of the oligonucleotides used here can be found in Table 2.
TABLE 1.
S. cerevisiae strains used in this study
| Strain | Alias | Relevant genotype | Source |
|---|---|---|---|
| yMM13 | CH1584 | MATaleu2Δ1 trp1Δ63 ura3-52 | Connie Holm |
| yMM177 | CH355 | MATahis4-539 lys2-801 ura3-53 | Connie Holm |
| yMM354 | MATaleu2Δ1 trp1Δ63 ura3-52 pMM147 (EBP2ΔN62 LEU2) | This study | |
| yMM355 | MATaleu2Δ1 trp1Δ63 ura3-52 pMM322 (p-M1 EBP2ΔN62 LEU2) | This study | |
| yMM373 | MATaleu2Δ1 trp1Δ63 ura3-52 pMM341 (p-M2 EBP2ΔN62 LEU2) | This study |
TABLE 2.
Oligonucleotides used in this study
| Name | Sequence (5′ to 3′) |
|---|---|
| EBP2-CP3 | GGAGGATCCTTATGCGGCTCTCTTTCCGTT |
| EBP2-CP6 | GATCGTCGACCCTTAGAATCTTCTGGCACGTCT |
| EBP2-CP12 | CAGACTCAGAGTCACTTTTGGCC |
| EBP2-CP14 | CGAGGACGTCGGAATGTATTTAGGCAGAAATA AAAAATTTTCAG |
| EBP2-CP15 | GGACGACGTCATGTCCCGATGAGTAGTTAT |
| EBP2-WP11 | GGCGCGATGCATAAAGATCTATACGATGATAC TGAAAGG |
| EBP2-WP14 | GTTGAACAACGGTATATAGAGAATG |
| EBP2-WP16 | GGACGACGTCACTTTATAGAATTTTACGCTGTT AAAAAG |
| EBP2-WP21 | GGACGACGTCAATTATTTCTGCCTAAATAC |
| HHT1&2-WP1 | CCACTGGTGGTAAAGCCCC |
| HHT1&2-CP1 | CGACGGATTCTTGCAAAGCACCG |
| NOP2-WP1 | GATGAAGATAATGACGCCCACCC |
| NOP2-CP1 | CTCCATCGCCTCTGCTGGTGAG |
| pMM147-WP1 | GGGAAGGGCGATCGGTGCGG |
| ROK1-WP1 | GGCATGTGGTCCCACAGGGTCTGG |
| ROK1-CP1 | CGATGGCTCTCTACAGGCGCTC |
| RRS1-WP1 | CGGAGTCTGTAGGTGGC |
| RRS1-CP1 | GCGCCCTTGTAGCCCCAC |
| SW14-WP1 | GACACCGTTCAGCGCAGG |
| SW14-CP1 | CGCCATAGCTGTAGCCC |
| SW16-WP1 | CGGAGACTGGCCATTTCCTCC |
| SW16-CP1 | CGGAGTGGAGTCACTCTCAGC |
| YML093W-WP1 | GCCACTGTCTCAAGTGTGGG |
| YML093W-CP1 | GGTAGCGGAACTTCATACGTCG |
Cluster analysis.
The microarray time-series data was analyzed by using the Partitioning Around Medoids (PAM) algorithm provided by the SPLUS statistical analysis software package (22). PAM is a variant of the well known “k-means” cluster algorithm for grouping multidimensional data. The relative expression measurements at T time points for each gene on the microarray correspond to a single point in a T-dimensional space. Nearby genes in this T-dimensional space will have similar expression profiles over time. The goal of the cluster algorithm is to identify the groups of points (genes) that are close together but far from other groups. The user specifies the number, k, of groups or clusters to be found and then the program automatically optimizes the membership of the clusters to minimize the distance between members of the cluster and to maximize the distance from other clusters (see reference 15 for details.).
Before application of the algorithm, the large group of genes that remain relatively constant across each experiment were removed. The cutoff level was chosen so that ca. one-half of the 6,000 genes would be included in the final cluster analysis. This cutoff varied from the requirement that the relative expression in at least one time point change by 1.65-fold for the alpha-pheromone experiments to a 2-fold change in the sporulation experiments.
The PAM algorithm was then applied to the (ca. 3,000) gene time-series. The “distance” between genes was determined by the Euclidean metric applied to the log-ratio time series. The log ratio was defined as the log2(gene expression value at a given time point/control gene expression value). We searched for k = 24 clusters so that each cluster would contain roughly 100 to 150 genes. For each experiment the clusters were graphically characterized by the time series of the most representative gene (the “medoid”) of the cluster, and a list of genes in each cluster was generated along with a quantitative measure of the “strength” of their membership. A complete list of the cluster membership can be obtained from the authors or at the ribosome and rRNA biosynthesis (RRB) regulon website (http://mmcalear.web.wesleyan.edu/rrb-regulon/).
Statistical analysis of clusters across experiments.
To estimate the numbers of overlapping genes from a purely random sampling of N ≅ 3,000 genes, we first computed the probability that any particular set of m genes found in a cluster in the first experiment would be found in a cluster of size n2 in the second experiment:
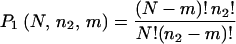 |
1 |
However, since there are
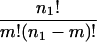 |
different ways of choosing a particular set of m genes from the n1 genes in the original cluster in the first experiment, the probability that a cluster with n1 genes in the first experiment shares m genes with a cluster of size n2 in the second experiment can be estimated by the equation:
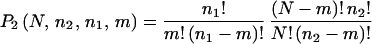 |
2 |
Note that equation 2 is an overestimate, since it double counts the occurrence of the same m + 1, m + 2, … genes in both clusters. As a consequence, this estimate erroneously gives a probability of >1 when m is small. Finally, since there are nc = 24 clusters in the first experiment that can be compared with any of the nc = 24 clusters in the second experiment, P2(N, n1, n2, m) should be multiplied by a factor of nc2 to estimate the odds of finding m genes in common between any two clusters in the two experiments by chance.
By using this very conservative estimate for the random probability that two clusters from two different experiments share m genes, we can assess whether an observed overlap could have occurred by chance or indicates a systematic relationship among the common genes. For example, equation 2 predicts that the random probability of the overlap of m = 65 genes between two clusters of size n1 ≅ n2 ≅ 125 sampled out of ca. 3,000 genes is 1.2 × 10−62 which is much, much smaller than 1/nc2 ≅ 2 × 10−3. These estimates indicate that only an overlap of ca. 16 genes could be attributed to chance.
Alpha factor arrest, heat shock, and tunicamycin treatment.
Yeast strains were grown to a density of 3 × 107 cells/ml in either yeast extract-peptone-dextrose (YPD; pH 5.5) (yMM13 and yMM177) or synthetic complete (SC)-Leu liquid medium (yMM354, yMM355, and yMM373) at 30°C. The cultures were arrested by the addition of alpha factor for 2 h at a final concentration of 5 μg/ml. The cells were washed free of the alpha factor and released into fresh medium at 30°C. At the indicated time intervals, 10-ml aliquots of the cultures were collected on ice, harvested with a table-top centrifuge, and flash-frozen in a dry ice-ethanol bath.
The heat shock experiment was performed by growing strains in either YPD (yMM13 and yMM177) or SC-Leu liquid medium (yMM354, yMM355, and yMM373) at 25°C to ca. 3 × 107 cells/ml. The cells were harvested by centrifugation and then resuspended in YPD or SC-Leu medium at 37°C. Samples were incubated at 37°C, and aliquots were collected every 10 min and then flash-frozen in dry ice and ethanol.
For the tunicamycin treatment, cells were grown in liquid YPD medium to a concentration of 3 × 107 cells/ml, and then tunicamycin was added to a concentration of 1 μg/ml. Cultures were incubated at 30°C, and samples were collected as outlined above.
RNA preparation and relative quantitative RT-PCR.
Total RNA was prepared from the samples by using a hot acidic phenol extraction protocol (1). The concentrations of the RNA preparations were determined by spectrophotometry, and aliquots were tested by electrophoresis on 1.2% agarose–2.2 M formaldehyde gels. The samples were treated with DNase I (Ambion Co.) and tested by PCR to confirm that there was no contaminating DNA. Then, 2 μg of RNA from each sample was incubated with reverse transcriptase (RT) to yield cDNA as per the instructions in the Ambion RETROscript kit. PCRs were then performed as described by Ambion's QuantumRNA Universal 18S rRNA Internal Standards Kit. Briefly, for each gene, PCRs were performed to determine the linear range of amplification that would permit a quantitative assessment of expression levels (typically between 14 and 24 amplification cycles). We then empirically determined the 18S rRNA primer-to-competimer ratio that would allow for an appropriate level of amplification for the 18S rRNA internal control. PCRs containing two pairs of oligonucleotides (one for the 18S rRNA control and one for the gene of interest) were performed in the presence of [α-32P]dATP, and the fragments were separated by gel electrophoresis. The PCR products were quantified by PhosphorImager analysis and ImageQuant software. For each time point of each gene series the number of counts in the PCR product of interest were divided by the number of counts in the internal 18S control. Expression levels across a time course were then normalized to a mean of expression level of 1.
Construction of the motif 1 and motif 2 promoter element substitution strains.
The two promoter motif elements located upstream of the EBP2ΔN62 allele were altered by a PCR-based substitution strategy. Briefly, pairs of oligonucleotides were used to amplify separate DNA fragments that flanked the motif sequences within plasmid pMM147. Each of these fragments contained altered sequences including an AatII site at one end in place of the normal motif sequences. The two DNA fragments were digested with either AatII and NcoI or AatII and HindIII to produce compatible sticky ends. These two inserts were then ligated with an NcoI and HindIII fragment from plasmid pMM147 and transformed into Escherichia coli. The resulting motif 1 and motif 2 substitution plasmids (pMM322 and pMM341, respectively) were recovered, sequenced, and transformed into yeast strain yMM13.
RESULTS
Clustering of microarray expression profiles.
Genome-wide changes in gene expression have been monitored by microarray analysis as yeast cells progress through a variety of conditions, including traversal through the cell cycle (23), through sporulation (7), and through diauxic shift (9). Each of these studies produced large data sets containing time course expression profiles for more than 6,000 yeast genes. In the effort to identify groups of genes that share similar transcription patterns, these data sets were independently analyzed by using a partitioning cluster algorithm (described in Materials and Methods). This algorithm is designed to divide the data set into groups of genes that are similar to one another and yet distinct from other groups. As initial parameters for this partitioning, we limited the analysis to the expression profiles that varied the most within each data set (ca. 3,000 genes in each case), and we set the number of clusters to be 24. Although somewhat arbitrary, these parameters were chosen to exclude those genes that varied the least over the respective time courses and to allow for the potential discrimination of many different expression profiles. The resulting clusters typically contained 75 to 175 genes each, and the relative “strengths” of membership for each gene within a cluster could be assigned based on the similarity of their profiles to the most representative or medoid gene.
Analysis of the expression profiles and memberships of the clusters revealed that several sets overlapped with groups of genes that were previously identified to have distinct time courses in the transcriptional programs for the cell cycle, sporulation, and diauxic shift (Fig. 1). For example, the 24 clusters derived from the alpha factor arrest and release experiment (23) included seven clusters (totaling 523 genes) that clearly oscillated through two full cell cycles. With a few exceptions, the memberships of these clusters corresponded to those previously identified as being maximally expressed within the G1, S, G2, M, and M/G1 stages of the yeast cell cycle (23). One such cluster contained the histone genes, and they exhibited the characteristic expression profile of genes that peak in S phase. The remaining 17 clusters contained genes that varied by at least 1.65-fold at at least one time point but did not exhibit a strong cell cycle dependence.
FIG. 1.

Yeast gene expression profiles derived from microarray experiments were analyzed by a partitioning algorithm and grouped into 24 clusters. One gene expression data set came from cells after release from alpha factor arrest (23) (A), and another was derived from cells progressing through sporulation (7) (B). Expression profiles of two clusters from each set of 24 groups are depicted, including the number of genes within the cluster and the expression profiles of representative members. The gene expression profile corresponding to the medoid of each cluster is indicated by “M.”
Similarly, the grouping of the sporulation and diauxic-shift data sets identified clusters of genes that have previously been described as being important for the execution of these pathways. Our sporulation cluster number 24 contained 34 genes, and it included members of the Early I gene set recognized (7) as being important for chromsome synapsis or recombination (i.e., the ZIP1, DMC1, HOP1, and IME2 genes). In addition to these previously identified gene sets, there were numerous gene clusters that have not previously been characterized.
Progression through the cell cycle, sporulation, and diauxic shift all involve significant changes in the metabolic activities of the cell. In order to identify sets of gene that could potentially be important for more than one of these transitions, we investigated whether any of the clusters from one experiment shared an unusual number of genes with a cluster from another experiment. We reasoned that this approach could potentially identify gene sets whose regulation is important for general aspects of yeast cell metabolism. In the case where there was the largest degree of overlap, a single cell cycle cluster of 189 genes shared 65 genes with a sporulation cluster of 159 genes. In both cases, the clusters were characterized by expression profiles that varied transiently and did so at the beginning of their respective metabolic transitions. The large degree of overlap between these two gene sets was striking, since for all of the other cluster pairs, no two groups had more than 24 genes in common. The probability that these two clusters from two different experiments could have 65 genes in common by chance, estimated by using equation 2 (see Materials and Methods) is P(3,000, 189, 159, 65) ≅ 2 × 10−38.
In addition, many of these 65 genes were also found in two similar clusters of genes in the diauxic-shift experiments that showed a sharp decrease in expression at the start of the diauxic shift (9). The original cluster of 189 genes from the alpha factor release experiment shared 62 genes with the two diauxic-shift clusters with a combined total of 211 genes. The probability of this large overlap occurring by chance is estimated to be P(3,000, 189, 211, 62) ≅ 2 × 10−25. Therefore, the overlapping set of 65 genes appear to be coregulated across different growth conditions.
A set of rRNA biosynthesis genes are coregulated.
Since the theoretical calculations indicated that the set of 65 genes were highly unlikely to have been cogrouped into two distinct clusters by chance, we investigated this subset further to ascertain whether there was a physiologically relevant basis for their apparent coregulation. To do this, we first scanned the gene set to determine whether the members have been implicated to function in common areas of metabolism. There is little known about nearly half of the 65 genes in this set; however, for those genes that have reported activities, many encode proteins that function in aspects of rRNA metabolism (Table 3). There are six known or putative RNA helicases, three subunits of either RNA polymerase I or III, and twelve other proteins that have been indicated to function in rRNA processing or ribosome biogenesis. This observation is significant, since in a random selection, less than 10% of the characterized genes would be expected to have functions related to rRNA biosynthesis. Of particular interest to us was the coinclusion of the EBP2 and RRS1 genes in this gene set. Ebp2p and Rrs1p interact, and both are essential proteins that function in the rRNA processing pathway (13, 25). Because this set of 65 coregulated genes is enriched for genes that are required for ribosome and rRNA production, we suggest that they may represent members of an RRB regulon. It is worth noting that this set does not include any of the RP genes themselves.
TABLE 3.
Genes of the RRB cluster
| Gene | Pathwaya | Activity and/or function | Motifb
|
|
|---|---|---|---|---|
| 1 | 2 | |||
| YGL171W ROK1 | rRNA processing | RNA helicase | −107 W | −120 W |
| YJL033W HCA4 | rRNA processing | RNA helicase | −94 C, −79 C | −125 C |
| YMR290C HAS1 | RNA processing | RNA helicase (nucleolar) | −108 W | −168 W |
| YGL120C PRP43 | mRNA splicing | RNA helicase | −122 W | −140 W |
| YKR024C DBP7 | rRNA processing | RNA helicase (putative) | −141 W, −345 C | |
| YLR276C DBP9 | Unknown | RNA helicase (putative) | −155 C | |
| YJL148W RPA34 | Transcription | RNA polymerase I subunit | −124 W, −222 C | |
| YNL248C RPA49 | Transcription | RNA polymerase I subunit | −132 W | −147 W |
| YNL113W RPC19 | Transcription | RNA polymerase I and III subunit | −129 W | |
| YNR054C | Transcription | Nucleolar protein | −88 W | |
| YKL172W EBP2 | rRNA processing | Unknown | −87 W | −114 W |
| YOR294W RRS1 | rRNA processing | Unknown | −123 C | |
| YLR197W SIK1 | rRNA processing | U3 snoRNP complex | −129 W | −113 W, −174 W |
| YNL002C RLP7 | rRNA processing | Nucleolar protein | −102 W | −141 W |
| YLR175W CBF5 | rRNA processing | Pseudouridine synthase | −147 W, −203 W | |
| YGR159C NSR1 | rRNA processing | Nucleolar protein | −192 C | −173 C |
| YNL061W NOP2 | rRNA processing | Nucleolar protein | −136 C, −112 W | −193 C |
| YHR088W RPF1 | rRNA processing | Unknown | −101 W | |
| YMR131C RSA2 | Ribosome biogenesis | Nucleolar protein | −105 W | −161 W |
| YOL077C BRX1 | Ribosome biogenesis | Nucleolar protein | −162 W | |
| YMR049C ERB1 | Ribosome biogenesis | Unknown | −95 W | −120 W |
| YGL029W CGR1 | Ribosome biogenesis | Nucleolar protein | −133 W | |
| YNL175C NOP13 | RNA processing | Nucleolar protein | −101 W | −129 C |
| YPR112C MRD1 | RNA processing | Unknown | −96 C | |
| YAL025C MAK16 | RNA processing | Unknown | −137 W | |
| YOR361C PRT1 | Protein synthesis | Translation initiation factor eIF3 subunit | ||
| YBR079C RPG1 | Protein synthesis | Translation initiation factor eIF3 subunit | −99 W, −330 W | |
| YNL062C GCD10 | Protein synthesis | Translation initiation factor eIF3 subunit | −204 C, −228 C | −143 W |
| YHR196W | Unknown | Nucleolar protein | −132 W | |
| YGR103W | Unknown | Nucleolar protein | −131 W | −107 W, −143 W |
| YPL093W NOG1 | Unknown | Nucleolar protein | −148 C | −131 C |
| YBR084W MIS1 | Folate metabolism | C1-tetrahydrofolate synthase | ||
| YDR465C RMT2 | Arginine metabolism | Arginine methyltransferase | −107 C | −121 C |
| YIR026C YVH1 | Sporulation | Protein phosphatase | −75 W | |
| YOR272W YTM1 | Cytoskeleton (putative) | Microtubule-associated protein | −142 C | −169 C |
| YDR299W BFR2 | Protein translocation | Unknown | −111 W | −126 C |
| YGR245C SDA1 | Cell cycle control | Unknown | −150 C | |
| YCR055C | Cell polarity | Unknown | ||
| YAL036C FUN11 | Unknown | Unknown | −170 W | |
| YBL054W | Unknown | Unknown | −237 W, −252 W | |
| YBR172C SMY2 | Unknown | Unknown | −40 C | |
| YBR247C ENP1 | Unknown | Unknown | −138 W | |
| YCR072C | Unknown | Unknown | −94 W | −129 W |
| YDL167C | Unknown | Unknown | −116 W | |
| YDR101C | Unknown | Unknown | −82 W | |
| YDR365C | Unknown | Unknown | −124 C | −154 C |
| YGL099W | Unknown | Unknown | −135 C | |
| YGR145W | Unknown | Unknown | −89 C, −253 W | |
| YGR160W | Unknown | Unknown | ||
| YJL109C | Unknown | Unknown | −102 W | −117 C |
| YJR070C | Unknown | Unknown | −144 W | −156 W |
| YKL082C | Unknown | Unknown | −97 W | −166 W, −430 C |
| YKL099C | Unknown | Unknown | ||
| YKL143W LTV1 | Unknown | Unknown | −113 W | |
| YLR002C | Unknown | Unknown | −122 C | |
| YLR401C | Unknown | Unknown | −73 W | |
| YML093W | Unknown | Unknown | −95 W | −116 C |
| YNL110C | Unknown | Unknown | −115 C | −155 W |
| YNL132W | Unknown | Unknown | −148 W | −190 W, −358 W |
| YNL174W | Unknown | Unknown | ||
| YNR053C | Unknown | Unknown | −186 C | −153 C |
| YOL124C | Unknown | Unknown | ||
| YOR309C | Unknown | Unknown | ||
| YPL012W | Unknown | Unknown | −169 C | −217 W |
| YPR143W | Unknown | Unknown | ||
Abbreviated pathway and activity information as retrieved from the YPD database (http://www.proteome.com) as of 15 June 2001.
The values indicate motif positions in relation to the initiator ATG codons (W, coding strand; C, noncoding strand).
Although microarray analysis is a powerful technique for measuring gene expression levels of large sets of genes, this approach is subject to experimental variations within and between individual hybridizations. In order to independently measure gene expression levels, we used a relative-quantitative RT-PCR technique (see Materials and Methods). Briefly, in this approach RNAs are first converted to cDNAs, and then gene-specific oligonucleotides are used to quantitatively amplify the cDNA fragments (reviewed in reference 4). When care is taken to ensure that the genomic DNA is removed prior to the RT step and that the amplification reactions fall within a linear range, one can obtain a representative expression profile (Fig. 2). To account for differences between samples in a time course experiment, an internal 18S rRNA-derived fragment is coamplified for each reaction. Because the consistently high levels of rRNA within the RNA preparations would overwhelm the amplification reactions, nonamplifying rRNA competimer oligonucleotides are used to dampen the rRNA signal to a level that is comparable to the mRNA of interest. Once the appropriate competimer ratio and amplification cycles are determined for a given gene, the relative transcript abundance can be calculated in relation to the 18S rRNA levels. It should be noted that the relative mRNA expression levels, as detected by this analysis, are influenced both by changes in transcription, as well as mRNA turnover rates.
FIG. 2.

Gene expression levels can be assessed by a relative-quantitative RT-PCR technique. (A) RT-PCRs with primers specific to the HHT2 gene were performed on a total RNA sample derived from a logarithmically growing culture of strain yMM177. The products were labeled with [α-32P]dATP, separated by gel electrophoresis, and quantified by phosphorimaging analysis. In order to determine the linear range of the assay, levels of [α-32P]dATP incorporation were measured for up to 26 amplification cycles. (B) RT-PCR assays were performed simultaneously with primers sets specific to both the HHT2 and 18S rRNA sequences. Increasing amounts of nonamplifying competimer 18S primers were added to dampen the 18S amplification reactions down to a level comparable to the HHT2 reaction.
The RT-PCR technique was used to assess the expression profiles of a subset of genes from within and outside of the RRB cluster. Cells were first arrested with alpha factor, and then samples were collected every 10 min after release into the cell cycle. The efficiency of the arrest and release protocol was confirmed by monitoring the cells for the appearance of schmoos and new buds, respectively (data not shown). In order to validate the RT-PCR technique for obtaining relative mRNA transcript profiles, we used it to monitor histone (HHT2) gene expression levels in three independent experiments (Fig. 3). In each case, the profiles matched the expected pattern for an S-phase-expressed gene, and they correlated very well with the HHT2 microarray derived expression profiles. Similarly, we observed that our measurements of the SWI4 transcript levels were consistent with microarray data, although, by comparison, our profiles appeared to be delayed by 5 to 10 min.
FIG. 3.

Gene expression levels change as cells progress through the cell cycle. RT-PCR assays were performed on RNA samples derived from yMM13 cells after release from alpha factor arrest. Relative gene expression levels were determined as a ratio of the expression values for the gene of interest divided by the 18S values. Expression profiles for the HHT2 (A), SWI4 (B), and the EBP2, RRS1, ROK1, NOP2, and YML093W genes (C) are indicated. Expression profiles as determined by microarray hybridization studies were converted to a similar scale and are depicted as indicated for the HHT2 and SWI4 genes (array).
Having determined that the RT-PCR technique could reproducibly corroborate the microarray profiles, we then investigated a sampling of genes from the RRB cluster. Five genes were chosen for this analysis (EBP2, RRS1, ROK1, NOP1, and YML093W), and as expected from this cluster analysis, they exhibited similar expression profiles (Fig. 3C). Although the five genes exhibited a transient peak of expression 10 min after release from arrest, the RT-PCR expression profile was unlike the microarray data. The microarray profiles indicated that, rather than increase, the expression levels of these genes transiently declined after release from arrest. Differences in strain handling procedures could potentially be responsible for this discrepancy, particularly if one set of cell harvesting conditions triggers a stress response (see Discussion). In this regard, the microarray profiles for the RRB genes in the alpha factor release experiment (23) were similar to profiles observed when cells respond to stressful conditions (11). In any case, the sample of RRB cluster members did exhibit similar profiles, and they were unlike the expression patterns for the nonmember gene controls.
If the genes of the RRB cluster are members of a regulon, one might expect them to be coregulated under a variety of conditions. To test this hypothesis, we monitored gene expression levels following a shift from 25 to 37°C. Since heat shock has previously been shown to temporarily repress transcription of the rRNA and RP genes, we reasoned that it could also impact upon the transcription of the rRNA processing factors. RNA samples were collected from cells after a shift from 25 to 37°C, and transcript levels were determined by the RT-PCR technique (Fig. 4A). As a group, the RRB cluster subset exhibited similar profiles, with a transient drop in expression that recovered by 30 min after the heat shock. These profiles were similar to those that have been reported for these genes in microarray experiments (11) and were unlike the expression profile determined for the HHT2 gene.
FIG. 4.

RRB cluster members are coregulated following heat shock and tunicamycin treatment. RT-PCR assays were performed on RNA samples derived from yMM13 cells after a shift from 25 to 37°C (A) or after treatment with 1 μg of the drug tunicamycin/ml (B). Heat shock expression profiles of the HHT2 and EBP2 genes as determined by microarray hybridizations (11) (array) are also indicated.
We also tested whether the RRB sample subset was affected by the drug tunicamycin. Tunicamycin is known to interfere with the yeast secretory pathway, and this stress subsequently leads to an inhibition of rRNA biogenesis (19, 25, 26). Gene expression levels were monitored after cells were treated with tunicamycin, and again the RRB cluster subset of genes yielded similar profiles. After an initial rise, there was a gradual decline in expression levels that continued for at least two more hours (Fig. 4B and data not shown). Since this expression pattern was not observed for either the SWI6 or HHT2 genes, it does not represent a general transcriptional response to tunicamycin treatment.
RRB gene promoters share common sequence motifs.
One mechanism by which the RRB cluster of genes could be regulated is through their sharing of common regulatory sequences within their promoters. To identify any such sequences, we used the MEME program (2) to scan through the 500 bp of sequence that are immediately upstream of the coding sequences for each gene member. This program identified two enriched sequence motifs, each of which was present in at least 41 of the 65 RRB cluster promoters (Fig. 5 and Table 3). Motif 1 is a 13-base G(C/A)GATGAG(A/C)TGA(G/A) consensus sequence that is found 50 times (upstream of 44 genes) within the RRB cluster promoters. The corresponding P values for each of these occurrences range from 2.3 × 10−6 to 3 × 10−9. Within motif 1 one can recognize the 11-base GATGAGATGAG tandem repeat sequence. There are 67 perfect matches to this 11-base sequence within the entire yeast genome, and remarkably, 21 of these sequences can be found within the 65 RRB cluster promoters (i.e., upstream of only 1% of the total number of yeast genes). Typically, motif 1 sequences are located between 50 and 150 bases upstream and on the same strand (60%) of their respective initiator codons. Motif 2 is the 11-base TGAAAA(A/T)TTTT sequence. It is present 48 times (upstream of 41 genes) within the RRB promoters, and it is usually found on the same strand as the ATG codon (67% of the time). Of the 65 RRB promoters, 29 contain both motif 1 and motif 2 sequences, with a typical arrangement of motif 2 occurring some 15 to 50 bp upstream of motif 1.
FIG. 5.

RRB cluster promoters are enriched for two sequence motifs that cluster to positions between 50 to 200 bp upstream of their respective genes. (A) The MEME program was used to identify sequence motifs that are highly represented within the 500 bp of sequence immediately upstream of the RRB cluster genes. The consensus sequences of the two identified motifs are indicated, along with the two examples of the promoter occurrence set that correspond to the highest and lowest P values, as defined by the MEME program. (B) The relative positions of the two motif sequences are represented with respect to the initiator ATG codons of their respective genes.
Motifs 1 and 2 are important for the regulation of EBP2 gene expression in vivo
Given the high representation of sequence motifs 1 and 2 within the RRB cluster gene promoters, we sought to determine whether they played a role in transcriptional regulation in vivo. Since the promoter of the EBP2 gene contains both of these consensus sequences, we chose it as a reporter gene. To test the elements, we constructed a yeast strain that would allow us to simultaneously monitor expression levels in EBP2 alleles that both contained and that lacked the consensus sequences (Fig. 6). In this way, our experiments would contain an internal control consisting of the integrated wild-type EBP2 gene and promoter. To do this, we constructed a CEN ARS LEU2 plasmid that contained a 62-codon N-terminal EBP2 deletion construct that was driven by the natural EBP2 promoter. This allele produced a truncated but functional Ebp2 protein. By choosing an appropriate single pair of oligonucleotides, we could amplify and distinguish RT-PCR fragments derived from transcripts of the normal, integrated EBP2 allele and from the truncated, plasmid-borne EBP2ΔN62 construct (443 and 257 bp, respectively).
FIG. 6.

A plasmid-borne EBP2 construct can be used to simultaneously monitor gene expression levels in two EBP2 alleles. A plasmid-borne EBP2ΔN62 allele was created that lacked the first 62 codons of the EBP2 gene but retained the natural EBP2 promoter (pEBP2, represented by a boldface line). The EBP2 primer set used for the RT-PCRs could simultaneously amplify fragments of different sizes (dashed lines) that were derived from both the integrated EBP2 and the plasmid-derived EBP2ΔN62 transcripts. This plasmid was subsequently modified, replacing the EBP2 promoter sequences within motifs 1 and 2 (boxed) with heterologous sequences containing an AatII restriction site (underlined).
To test whether the plasmid-borne EBP2ΔN62 allele exhibited a transcription profile that was similar to the wild-type EBP2 allele, we arrested yeast strain yMM354 with alpha factor and released it into fresh medium. The transcription profile of the EBP2ΔN62 construct did very closely match the profile of the wild-type allele, although the overall normalized levels of the plasmid-derived transcripts were consistently higher (by ca. 60%) than those of the integrated allele (Fig. 7A). This elevated expression of plasmid versus integrated EBP2 alleles could be due to the plasmid being present on average in more than one copy per cell.
FIG. 7.

Motifs 1 and 2 are important for the regulated expression of the EBP2 gene after release from alpha factor arrest. Yeast strains containing either the EBP2ΔN62, the p-M1 EBP2ΔN62, or the p-M2 EBP2ΔN62 plasmid-borne alleles were arrested with alpha factor and released into the cell cycle (A) or else subjected to heat shock (B). RNA samples were prepared for RT-PCR analysis and the amplification products derived from the 18S rRNA, the integrated EBP2, and the EBP2ΔN62 alleles were separated and quantitated. Each expression time point value represents the mean expression level derived from at least two experiments.
Next, we mutated the deletion construct promoter, replacing the motif 1 element with a heterologous sequence that included an AatII restriction site. This plasmid was reintroduced into yeast creating strain yMM355, and expression levels after release from alpha factor arrest were monitored again. In contrast to what was observed for the natural EBP2 promoter plasmid, the p-M1 EBP2ΔN62 plasmid allele did not exhibit the characteristic EBP2 expression profile. We observed in three separate experiments that the transient peak in expression that occurs 10 min after release from alpha factor arrest was much less pronounced with this allele. We also altered motif 2 sequences and tested that allele (p-M2 EBP2ΔN62) for expression levels in strain yMM373. Again, motif 2 sequences were found to be important for the characteristic peak of expression that normally occurs soon after release from alpha factor arrest. Also, the overall levels of expression were lower for the p-M2 EBP2ΔN62 allele. The transcription profiles of the integrated, wild-type EBP2 control allele remained consistent across the three different yeast strains (i.e., compare the EBP2 profiles across the three panels of Fig. 7A). Thus, motif 1 and motif 2 sequences are highly enriched, positionally biased, and functionally important for the regulated expression of at least one member of the RRB regulon (EBP2).
In order to investigate the extent to which motif 1 and motif 2 sequences contribute to the regulated expression of the EBP2 gene, the same set of alleles were monitored for expression levels after heat shock treatment (Fig. 7B). In the case of heat shock, the transcription profile of the EBP2ΔN62 allele did not match as closely the expression profile of the integrated EBP2 allele. The decline in expression after heat shock was less pronounced for the plasmid-borne allele, and the same general expression profiles were observed for the plasmid alleles lacking motif 1 and 2 sequences. However, as seen for the alpha factor release experiment, the allele that lacked motif 2 sequences exhibited a lower overall level of expression. Therefore, within the context of the alleles tested here, motif 1 and 2 sequences were found to be most important for promoting EBP2 expression soon after release from alpha factor arrest.
It is worth considering how the RRB regulon, as defined by this study, relates to other groups of genes that are coordinately regulated after changes in environmental conditions. The environmental stress response group of genes includes subsets of genes that are either induced or repressed after exposure to a wide variety of conditions, including osmotic shock, heat shock, nutrient deprivation, hydrogen peroxide, and diamide or dithiothreitol treatment (11). The large set of 600 genes that are repressed after these stresses include genes involved in growth related processes, such as nucleotide biosynthesis, RNA metabolism, protein synthesis, and secretion. The RRB regulon appears to be a subset of the environmental stress response (ESR) repression group. Interestingly, most of the RP genes are also found within the ESR set, although as a group, they exhibit an expression profile that is distinct from the RRB set under a number of conditions, including heat shock, diauxic shift, and diamide or dithiothreitol treatment (Fig. 8). The fact that the clustering strategy that we used did not result in any RP genes being included in the RRB gene set also suggests that the regulation of the RP and RRB genes is distinguishable. However, the overall transcriptional patterns are consistent between these two sets of genes and can be understood in terms of strategies that cells use to regulate cell growth and division in response to varied conditions. It would be appropriate for cells to increase rRNA and RP synthesis at the onset of progression through the cell division cycle (i.e., after alpha factor arrest), as it would be to scale back these processes at the onset of sporulation or diauxic shift. It will be of interest to determine what factors regulate this set of genes and how the respective signaling pathways are coordinated to effect these rapid and genome-wide changes in gene expression.
FIG. 8.

The RRB and RP gene set expression profiles are similar and yet distinguishable. The average expression profiles for a set of 59 RRB and 128 RP genes are depicted as determined by microarray hybridization analysis (11) after heat shock (A) or diamide treatment (B).
DISCUSSION
In this study we have used computational analysis of gene expression profiles to identify a set of coregulated genes that function in various aspects of rRNA metabolism. However, because the strategy used to define this gene set relied upon both experimental data and computational assumptions, it is likely that additional RRB genes exist and that some of the listed 65 genes may not have rRNA-related functions. For example, the original expression profile data sets were compiled from single microarray hybridizations on over 6,000 genes for each time point. The inevitable experimental variations that arise within or between hybridizations could significantly affect the apparent expression profile for a given gene. Because the RRB cluster profiles were typified by early and transient gene expression changes, measurement errors in one or two early expression time points would be particularly significant. Additionally, by changing the parameters of the clustering algorithm, one would tend to either merge (for low k values) or split (for larger k values) sets of dissimilar expression profiles. The choice to divide the expression data sets into k = 24 clusters was somewhat arbitrary, and it did produce clusters that contained similar expression profiles. Therefore, rather than being a definitive membership list for the RRB regulon, the identified set of 65 coregulated genes from the overlap of a single cell cycle cluster and a single sporulation cluster is perhaps best considered as representatives of the RRB regulon.
Even though the exact membership of the RRB regulon as defined in this study is likely to be incomplete, the identification of this class of genes is useful. One prediction is that the large fraction of uncharacterized genes within the RRB cluster may also have rRNA-related functions. Likewise, it is possible that even the genes reported to have functions in other aspects of cellular metabolism could also play a role in rRNA metabolism. For example, the Cbf5 protein, originally identified as a centromere-binding factor, is now believed to be involved in rRNA pseudouridylation (16). Similarly, since translation initiation factor 6 (eIF6) has recently been shown to be involved in pre-rRNA processing (3), eIF3 could have a similar role.
One can also investigate other known ribosome and rRNA metabolism-related genes and determine whether they, too, exhibit expression profiles characteristic of the RRB cluster members. An investigation of 24 ribosome and rRNA-related genes that were not among the original 65 RRB genes revealed that several (i.e., DBP8, DIM1, NOP8, SPB1, NIP7, MTR4, SQT1, and NMD3) exhibited alpha factor release and sporulation microarray expression profiles that were similar to the RRB cluster members. Two genes (XRN1 and RSA1) exhibited profiles that were unlike the RRB members, and the remaining 14 genes (RNT1, FAL1, RRP3, DBP6, NOP4, MAK5, DRS1, DBP3, RRP5, RAT1, DBP10, SPB4, RRP7, and NOP3) had expression profiles that were somewhat similar to the RRB genes. One must be careful with these categorizations, however, because the microarray profile for any given gene may contain either widely varying or missing data points that could significantly alter the shape of the expression profile. Although these individual variations may not be as problematic when one considers multiple, large data sets covering thousands of genes, they are relevant to single gene investigations. In this regard, the RT-PCR approach can be a useful, independent method for monitoring changes in mRNA levels.
Although for the most part, we observed that the gene expression profiles as determined by RT-PCR assays were very similar to those reported in microarray studies, there was one notable exception. We consistently found that the expression profiles of the RRB cluster members peaked 10 min after release from alpha factor arrest. In contrast, the expression profiles for the same gene set, as determined by a microarray hybridization method (23), exhibited a transient decline in expression levels. This discrepancy could reflect the underlying fundamental differences in the way that the relative mRNA levels are measured in the two different methodologies, or it could be due to variations in strains or experimental conditions. Support for the latter explanation comes from the fact that 59 of the 65 RRB cluster members were identified as being part of the set of 600 genes known collectively as the ESR repression subgroup (11). This group of genes covers a range of biological pathways and exhibits expression profiles that declined transiently upon stressful environmental conditions (see below). Therefore, depending on the methods used to release cells from alpha factor arrest (i.e., sample harvesting, medium changes, etc.), one could potentially trigger a stress response. We suggest that it would be reasonable to expect cells to increase expression of rRNA metabolism related genes as they entered the cell cycle. In either case, it is significant that both the microarray and RT-PCR assays indicated that the expression profiles of the RRB cluster gene members were similar to one another.
Our results indicate that motifs 1 and 2 are strong candidates for promoting expression of members of the RRB regulon after release from alpha factor arrest. We also noted that these two motif sequences are frequently located adjacent to one another and within 200 bp upstream of their respective start codons. Sequences related to these two motifs have been described previously. The G(C/A)GATGAGAT sequence was recognized as being one of 17 positionally biased and over-represented sequences in a study designed to identify DNA-binding motifs for novel yeast transcription factors (14). That same study identified a subsequence of motif 2, TGAAAA(A/T)TTT (named RRPE), that was preferentially associated with genes involved in rRNA processing. Likewise, similar sequences were recognized as potential regulatory sequences within a group of genes that share a common response to environmental changes (5), as well as within the ESR gene set (11). The motif 1 G(C/A)GATGAG(A/C)TGA(G/A) consensus contains a tandem overlapping repeat of the GATGAG sequence. The related TG(C/A)GATGAG sequence was previously named the PAC element because it was frequently found upstream of subunits of RNA polymerase A (I) and C (III) (8). However, the specific functions of these motifs were not previously defined, nor have their presumptive binding factors been identified.
In the effort to determine whether motif 1 could stimulate gene expression within the context of a heterologous promoter, the corresponding sequences were inserted into a reporter construct bearing the lacZ gene under control of the minimal CYC1 promoter (6). lacZ expression levels were monitored by liquid β-galactosidase assays (1), and it was observed that motif 1 sequences did not stimulate expression either in logarithmically growing cells or in cells released from alpha factor arrest (data not shown). While this result may not provide much insight into the function of motif 1 (since there are many reasons why motif 1 might not be active in this artificial context), it does suggest that the one-hybrid approach could potentially be used to identify the putative factor that binds to motif 1 sequences. If this putative factor could bind to motif 1 and act as a transcriptional activator on its own, one would not be able to select for such an activity from a one-hybrid fusion library.
The dual-allele relative quantitative RT-PCR strategy that we have developed should be generally useful for testing putative promoter elements for their activities under a variety of conditions. The advantage of this system is that it allows for the simultaneous assessment of expression levels for both a wild-type control and a mutated allele of a given gene at the same time. The mutated allele could be integrated instead of plasmid-borne and could contain deletions, insertions, or even novel restriction sites that would allow its RT-PCR product to be distinguished from the wild-type product. Once the appropriate alleles and strains were generated, one could test expression levels under a variety of conditions. Given the increasing potential for mapping out genetic networks based on genome-wide expression and promoter sequence analysis data (5, 10, 28, 29), it will be useful to have a reliable, internally controlled, and flexible experimental approach to test putative promoter elements for their functions in individual test genes in vivo.
ACKNOWLEDGMENTS
We thank Susan Baserga, David Stillman, and members of the McAlear lab for helpful discussions.
This study was supported by a National Science Foundation (NSF) Career Grant (MCB-9875283) to M.A.M. and by NSF grant PHY-9900746 to R.V.J. C.W. was supported in part by a Research Experiences for Undergraduates Grant from the NSF.
REFERENCES
- 1.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. New York, N.Y: Wiley Interscience; 1993. [Google Scholar]
- 2.Bailey T L, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- 3.Basu U, Si K, Warner J R, Maitra U. The Saccharomyces cerevisiae TIF6 gene encoding translation initiation factor 6 is required for 60S ribosomal subunit biogenesis. Mol Cell Biol. 2001;21:1453–1462. doi: 10.1128/MCB.21.5.1453-1462.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bustin S A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol. 2000;25:169–193. doi: 10.1677/jme.0.0250169. [DOI] [PubMed] [Google Scholar]
- 5.Causton H C, Ren B, Koh S S, Harbison C T, Kanin E, Jennings E G, Lee T I, True H L, Lander E S, Young R A. Remodeling of yeast genome expression in response to environmental changes. Mol Biol Cell. 2001;12:323–337. doi: 10.1091/mbc.12.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang Y C, Timberlake W E. Identification of Aspergillus brlA response elements (BREs) by genetic selection in yeast. Genetics. 1993;133:29–38. doi: 10.1093/genetics/133.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown P O, Herskowitz I. The transcriptional program of sporulation in budding yeast. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- 8.Dequard-Chablat M, Riva M, Carles C, Sentenac A. RPC19, the gene for a subunit common to yeast RNA polymerases A (I) and C (III) J Biol Chem. 1991;266:15300–15307. [PubMed] [Google Scholar]
- 9.DeRisi J L, Iyer V R, Brown P O. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- 10.Futcher B. Microarrays and cell cycle transcription in yeast. Curr Opin Cell Biol. 2000;12:710–715. doi: 10.1016/s0955-0674(00)00156-3. [DOI] [PubMed] [Google Scholar]
- 11.Gasch A P, Spellman P T, Kao C M, Carmel-Harel O, Eisen M B, Storz G, Botstein D, Brown P O. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guthrie C, Fink G R, editors. Methods in enzymology. Vol. 194. 1991. p. 305. . Guide to yeast genetics and molecular biology. Academic Press, Inc., San Diego, Calif. [DOI] [PubMed] [Google Scholar]
- 13.Huber M D, Dworet J H, Shire K, Frappier L, McAlear M A. The budding yeast homolog of the human EBNA1-binding protein 2 (Ebp2p) is an essential nucleolar protein required for pre-rRNA processing. J Biol Chem. 2000;275:28764–28773. doi: 10.1074/jbc.M000594200. [DOI] [PubMed] [Google Scholar]
- 14.Hughes J D, Estep P W, Tavazoie S, Church G M. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J Mol Biol. 2000;296:1205–1214. doi: 10.1006/jmbi.2000.3519. [DOI] [PubMed] [Google Scholar]
- 15.Kaufman L, Rousseauw P J. Finding groups in data: an introduction to cluster analysis. New York, N.Y: Wiley Interscience; 1990. [Google Scholar]
- 16.Kendall A, Hull M W, Bertrand E, Good P D, Singer R H, Engelke D R. A CBF5 mutation that disrupts nucleolar localization of early tRNA biosynthesis in yeast also suppresses tRNA gene-mediated transcriptional silencing. Proc Natl Acad Sci USA. 2000;97:13108–13113. doi: 10.1073/pnas.240454997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lascaris R F, Mager W H, Planta R J. DNA-binding requirements of the yeast protein Rap1p as selected in silico from ribosomal protein gene promoter sequences. Bioinformatics. 1999;15:267–277. doi: 10.1093/bioinformatics/15.4.267. [DOI] [PubMed] [Google Scholar]
- 18.Li B, Nierras C R, Warner J R. Transcriptional elements involved in the repression of ribosomal protein synthesis. Mol Cell Biol. 1999;19:5393–5404. doi: 10.1128/mcb.19.8.5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mizuta K, Warner J R. Continued functioning of the secretory pathway is essential for ribosome synthesis. Mol Cell Biol. 1994;14:2493–2502. doi: 10.1128/mcb.14.4.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moehle C M, Hinnebusch A G. Association of RAP1 binding sites with stringent control of ribosomal protein gene transcription in Saccharomyces cerevisiae. Mol Cell Biol. 1991;11:2723–2735. doi: 10.1128/mcb.11.5.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Planta R J. Regulation of ribosome synthesis in yeast. Yeast. 1997;13:1505–1518. doi: 10.1002/(SICI)1097-0061(199712)13:16<1505::AID-YEA229>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 22.Ripley B D, Venables W N. Applied statistics with S-Plus. New York, N.Y: Springer-Verlag; 1997. [Google Scholar]
- 23.Spellman P T, Sherlock G, Zhang M Q, Iyer V R, Anders K, Eisen M B, Brown P O, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tollervey D. trans-Acting factors in ribosome synthesis. Exp Cell Res. 1996;229:226–232. doi: 10.1006/excr.1996.0364. [DOI] [PubMed] [Google Scholar]
- 25.Tsuno A, Miyoshi K, Tsujii R, Miyakawa T, Mizuta K. RRS1, a conserved essential gene, encodes a novel regulatory protein required for ribosome biogenesis in Saccharomyces cerevisiae. Mol Cell Biol. 2000;20:2066–2074. doi: 10.1128/mcb.20.6.2066-2074.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vai M, Popolo L, Alberghina L. Effect of tunicamycin on cell cycle progression in budding yeast. Exp Cell Res. 1987;171:448–459. doi: 10.1016/0014-4827(87)90176-5. [DOI] [PubMed] [Google Scholar]
- 27.Warner J R. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci. 1999;24:437–440. doi: 10.1016/s0968-0004(99)01460-7. [DOI] [PubMed] [Google Scholar]
- 28.Wolfsberg T G, Gabrielian A E, Campbell M J, Cho R J, Spouge J L, Landsman D. Candidate regulatory sequence elements for cell cycle-dependent transcription in Saccharomyces cerevisiae. Genome Res. 1999;9:775–792. [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang M Q. Promoter analysis of coregulated genes in the yeast genome. Comput Chem. 1999;23:233–250. doi: 10.1016/s0097-8485(99)00020-0. [DOI] [PubMed] [Google Scholar]