

Summary:
Pioneer transcription factors engage nucleosomal DNA in chromatin to initiate gene regulatory events that control cell fate1. To determine how different pioneer transcription factors initiate the formation of a locally accessible environment within silent, compacted chromatin and collaborate with an ATP-dependent chromatin remodeler, we generated nucleosome arrays in vitro with a central nucleosome that can be targeted by the hematopoietic ETS factor PU.1 and bZIP factors C/EBPα, and C/EBPβ. Each class of factor can expose target nucleosomes on linker histone-compacted arrays, but with different hypersensitivity patterns, as discerned by long-read sequencing. The DNA binding domain of PU.1 is sufficient for mononucleosome binding but requires an additional intrinsically disordered domain to bind and open compacted chromatin. The canonical mammalian SWI/SNF (BAF) complex, cBAF, was unable to act upon two forms of locally open chromatin, in the presence of linker histone, unless cBAF was enabled by the acidic- and glutamine-enriched transactivation domain of PU.1. However, cBAF complexes potentiate the nucleosome binding DBD of PU.1 to weakly open chromatin in the absence of the PU.1 unstructured domain. Together our findings provide a mechanism for how pioneer factors initially target chromatin structures to provide specificity for action by nucleosome remodelers that further open local domains.
Chromatin compaction by linker histone maintains and stabilizes cell fate by preventing access of chromatin regulators and the transcriptional machinery2–9. During cell fate changes, new genetic networks are initiated by regulatory proteins that access DNA control regions embedded in compacted, silent chromatin. Key among such proteins are pioneer transcription factors, which can bind nucleosomal DNA and locally engage other proteins to enable chromatin opening or further chromatin compaction3,4,10. Low-signal chromatin, which is silent and lacks enrichment for known histone modifications, is most frequently targeted by pioneer transcription factors during cellular reprogramming11. Thus, in vitro studies, which have identified diverse pioneer transcription factors that directly bind mononucleosomes, have utilized histones lacking modifications12,13. However, it is unclear how diverse pioneer factors engage and initiate the opening of compacted chromatin.
Time course studies of chromatin opening in vivo have reported that, shortly after being ectopically induced, various pioneer transcription factors bind closed chromatin rapidly, but require the recruitment of additional co-factors to induce wider changes to the underlying chromatin structure14,15. DNase I hypersensitivity and footprinting assays determined that the liver specific pioneer transcription factor FoxA1 not only binds H1-compacted nucleosome arrays derived from natural nucleosome sequences of the Alb1 gene, but also exposes the targeted nucleosome16,17. By contrast, linker histone H1 impedes the action of ATP-dependent, SWI/SNF nucleosome remodelers6–8. While diverse pioneer factors are now known to directly bind mononucleosomes12,13, it is not clear whether pioneer factors other than FoxA1 are sufficient to access their sites in H1-compacted chromatin and, if so, whether they expose a targeted nucleosome like FoxA1. Furthermore, it is unclear if locally opening the chromatin is sufficient to enable the action of a nucleosome remodeler, or whether direct recruitment by the pioneer factor is necessary.
In vivo studies have detailed reciprocity between pioneer factors and chromatin remodelers, where rapid depletion of Oct4 leads to loss of chromatin accessibility and BAF complex binding at some sites18,19, and rapid depletion of BAF complexes leads to loss of chromatin accessibility and reduction of transcription factor binding at other sites20–23. The pioneer transcription factor PU.113,24 physically interacts with components of the SWI/SNF chromatin remodeling complex25, and truncation or deletion of the non-DNA binding domains of PU.1 results in the loss of binding to closed chromatin sites, which is correlated with loss of chromatin accessibility and loss of SWI/SNF binding in vivo26,27. Such studies indicate that maintenance of an open chromatin environment is a dynamic process that requires the sustained action of regulatory proteins. Indeed, continuous binding of pioneer factors to nucleosomes within regulatory regions can be required to maintain an open chromatin environment28,29. While pioneer factor chromatin binding and BAF complex recruitment are both an integral part of tissue-specific gene expression, little is known about the individual steps in the process of chromatin opening and the mechanism by which pioneer factors may enable BAF complex activity, or vice-versa. Given redundancies and complexities in vivo, we took an in vitro approach with purified components previously studied in vivo to reveal the interplay between a pioneer factor and a chromatin remodeler.
PU.1 (SPI1) is a master regulator required for hematopoietic stem cell renewal and collaborates with other factors to give rise to myeloid and lymphoid lineages. Macrophage development from hematopoietic stem cells requires the coordinated expression of PU.1 with the myeloid transcription factors C/EBPα and C/EBPβ24, and ectopic expression of PU.1 and C/EBPα/β converts fibroblasts to the macrophage lineage30. Previously, we identified a nucleosome targeted by PU.1, C/EBPα, and C/EBPβ in vivo (Extended Data Fig. 1a) and reconstituted this “Cx3cr1” mononucleosome in vitro13. We found that all three transcription factors can directly bind Cx3cr1 mononucleosomes, with varying affinities in the nanomolar range13. Here, we inserted a Cx3cr1 mononucleosome sequence centrally within a 13X nucleosome array (Extended Data Fig. 1b) and 5’ end-labeled the DNA template with the fluorescent nucleotide, dCTP-Cy513. “Cx3cr1 nucleosome arrays” were assembled using human recombinant purified histones and the Cy5 DNA templates, and validated as previously described16 (Extended Data Figs. 1 and 2).
To map nucleosome positions along the 2733 bp Cx3cr1 nucleosome array, we performed partial MNase digests of free DNA and extended Cx3cr1 nucleosome arrays, purified the DNA, performed long-read Nanopore sequencing, then mapped the endpoints of each aligned fragment (Extended Data Fig. 2a–c). We subtracted the free DNA MNase cleavage signals from the nucleosome array MNase signals, to correct for MNase DNA sequence preferences (Extended Data Fig. 2c)31. The resulting plot yielded patterns of protection, indicating the positions of phased nucleosomes, and MNase hypersensitivity, indicating the positions of linker DNA (Extended Data Fig. 2d). To mimic a naïve, silent chromatin state32, we further compacted nucleosome arrays with linker histone (H1). As previously reported9,16, a 1:1 molar ratio of H1 to nucleosomes was sufficient to quantitatively compact the Cx3cr1 nucleosome arrays, as seen by a marked overall resistance to DNase I digestion and increased mobility on a native agarose, gel relative to the extended nucleosome arrays (Extended Data Fig. 1c and d).
Incubation of PU.1 and C/EBPs with the H1-compacted nucleosome arrays each resulted in induced DNase cleavages within the Cx3cr1 nucleosome (Fig. 1a–d, Extended data Figs. 3a–c). However, the factors’ patterns of chromatin opening differed markedly. PU.1 generated a strong DNase hypersensitive signal underlying its binding site within the Cx3cr1 nucleosome core and 3’ flanking linker DNA. By contrast, C/EBPα and C/EBPβ each elicited hypersensitivity, but less intensely than PU.1, within the entry/exit regions of the nucleosome as well as the 5’ linker region (Fig. 1b–d). IRF3, which specifically binds the Cx3cr1 free DNA, but fails to bind Cx3cr1 mononucleosomes13, did not change accessibility of the H1-compacted array (Extended Data Fig. 3d). We found that incubating PU.1 together with C/EBPα led to a synergistic, rather than additive, increase in the DNase I hypersensitive signal (Extended Data Fig. 4a–c), indicating that two pioneer factors can cooperate to open chromatin. Importantly, the observed transcription factor-induced hypersensitivity required the specific transcription factor binding motifs within the central Cx3cr1 nucleosome (Fig. 1e). We conclude that the different pioneer factors elicit different opening patterns on H1-compacted chromatin, in a target site-dependent manner.
Figure 1: Different patterns of chromatin opening by PU.1 and C/EBP.
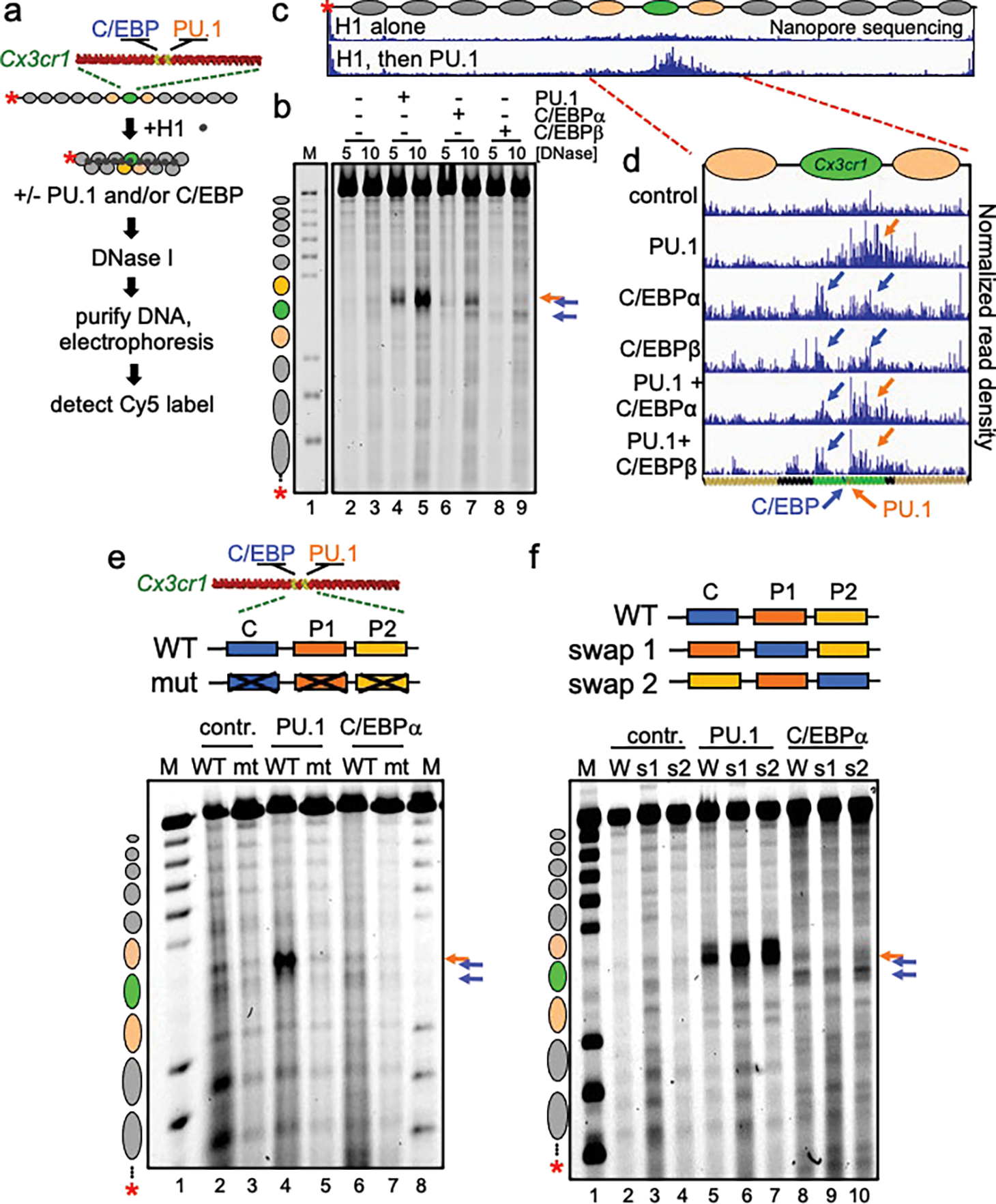
a, Schematic of chromatin assembly, compaction, transcription factor binding, and DNase I digestion assays. b, DNase I digestion analysis of TFs binding to H1-compacted nucleosome arrays visualized by gel electrophoresis at two DNase I concentrations (in ng/uL) Distinct hypersensitive sites are observed, relative to no TF control (lanes 2–3), upon PU.1 (lanes 4–5, orange arrow), C/EBPα (lanes 6–7, blue arrows), and C/EBPβ (lanes 8–9, blue arrows). Lane M, partial EcoRI digest of end-labeled array fragment. Positions of nucleosomes on reconstituted arrays are indicated. c and d, Nanopore sequencing endpoint analysis of DNase I digested (10 ng/uL) H1-compacted arrays. Ovals indicate the translational positions of nucleosomes determined in Extended Data Fig. 2d. Plots show normalized read density on the y axis. For each plot, the maximum value is set to 0.4% of reads. e, DNase I digestion (10 ng/uL) of H1-compacted wild-type Cx3cr1 nucleosome arrays (WT) or motif mutant Cx3cr1 nucleosome arrays (mt) incubated alone (contr., lanes 2–3), with PU.1 (lanes 4–5) or C/EBPα (lanes 6–7). f, DNase I digestion of H1-compacted wild-type Cx3cr1 nucleosome arrays (WT) or PU.1 and C/EBPα motif swap arrays (swap 1 or swap 2) incubated alone (contr., lanes 2–4), with PU.1 (lanes 5–7) or C/EBPα (lanes 8–10).
To address whether the relative positions of binding sites on the Cx3cr1 nucleosome determined the differences in DNase I cleavage patterns, we swapped the two PU.1 and one C/EBP binding motifs in two ways (Fig. 1f). Swapping motif positions had little effect on the patterns of nucleosome exposure (Fig. 1f). Thus, the transcription factor itself, rather than its precise binding position on the nucleosome, dictates a particular chromatin opening pattern. We note that a prior study using the 601 strong nucleosome positioning sequence only observed PU.1 binding at the entry and exit points of the nucleosome27. Such findings conflict with those observed with nucleosomes made with natural DNA13 and binding-selected sequences12 that allow factors to bind at multiple locations on the nucleosome.
We found that the DNA-binding domains of PU.1 and C/EBPα were sufficient to bind a Cx3cr1 mononucleosome (Fig. 2a, b, Extended Data Fig. 5a–d), yet were insufficient to efficiently elicit DNase I hypersensitivity on compacted nucleosome arrays (Fig. 2c, d). These in vitro data are concordant with previous reports that nucleosome binding is encoded within the DNA binding domains of pioneer factors12,29,33–35, but non-DNA binding domains are required for perturbations of higher-order chromatin structure in vivo17,26,36.
Figure 2: Domains outside of DBDs are needed to open chromatin.
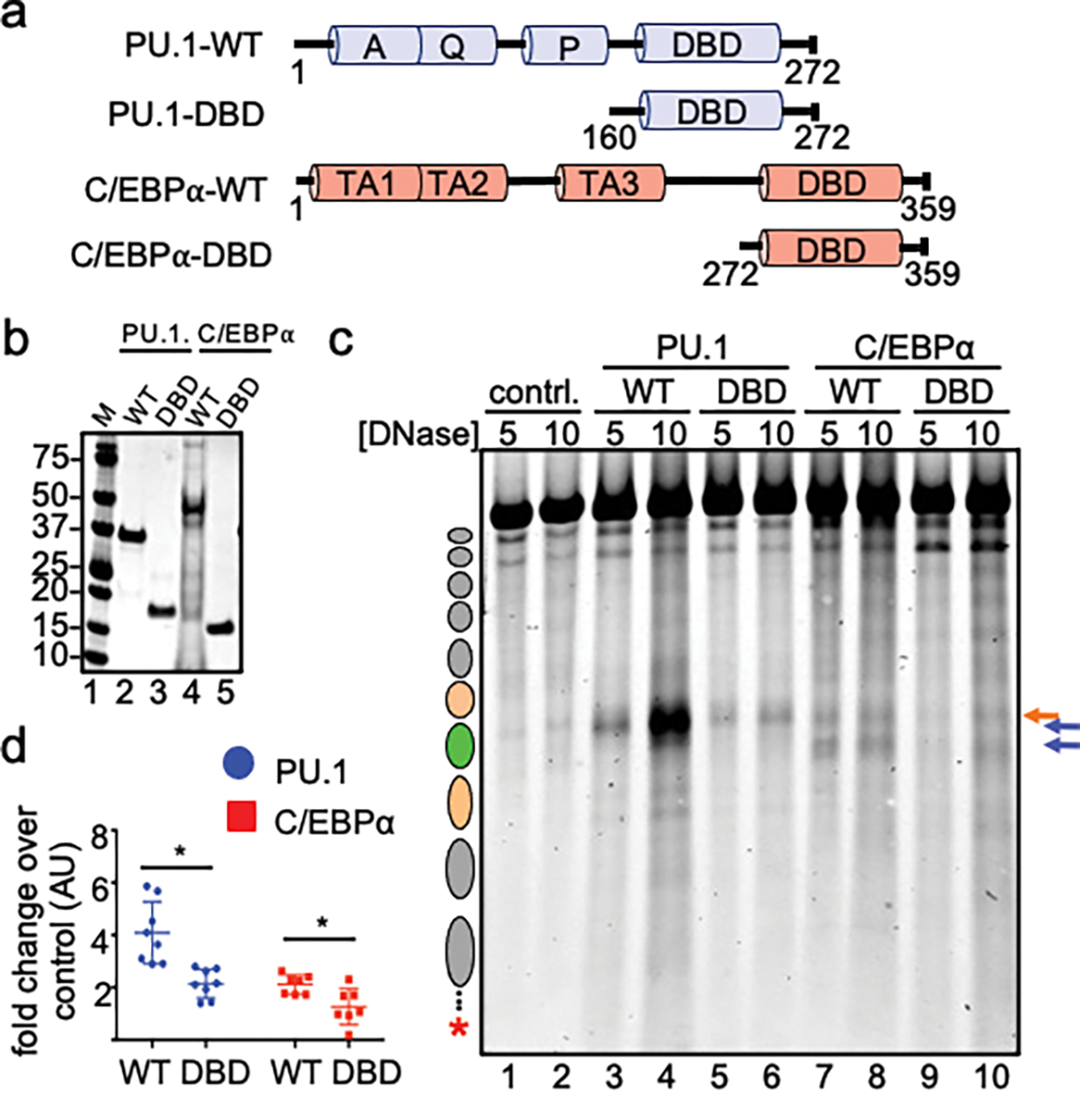
a, Illustration of wild-type (WT) and DNA-binding domain (DBD) recombinant PU.1 and C/EBPα. b, Coomassie stained SDS-PAGE of 1 ug of each indicated purified protein. c, DNase I digestion analysis of WT and DBD TF opening of H1-compacted chromatin. d, Quantified Cy5 signal in each lane normalized to no TF control (n=4, *p < 0.05). Data are mean ± SD and significance was determined by Student’s unpaired t-test.
We determined that purified cBAF37,38 (Extended Data Fig. 6a) plus ATP could move nucleosomes to expose a restriction site on extended Cx3cr1 nucleosome arrays, without H1, proximal to the Cx3cr1 nucleosome (Extended Data Fig. 6b, c). Consistent with previous studies6–8, H1-compacted arrays were refractory to nucleosome remodeling by cBAF in the presence of ATP (Fig. 3a, no change between lanes 2–3 vs 6–7). PU.1 incubated with cBAF complex, without the addition of ATP, showed no difference in eliciting hypersensitivity compared to PU.1 alone (Fig. 3a, no change between lanes 8–9 vs 10–11). However, when PU.1 was incubated with cBAF in the presence of ATP, we observed a 3’ expansion of hypersensitivity into the neighboring 3’ nucleosome (Fig, 3a, lanes 12–13, denoted by a red arrow), compared to PU.1 alone (lanes 8–9), as detailed by Nanopore sequencing (Fig. 3b and Extended Fig. 6d and e). Thus, PU.1 enabled cBAF remodeler action within the compacted chromatin in an ATP-dependent manner.
Figure 3: PU.1 Enables further chromatin opening by cBAF.
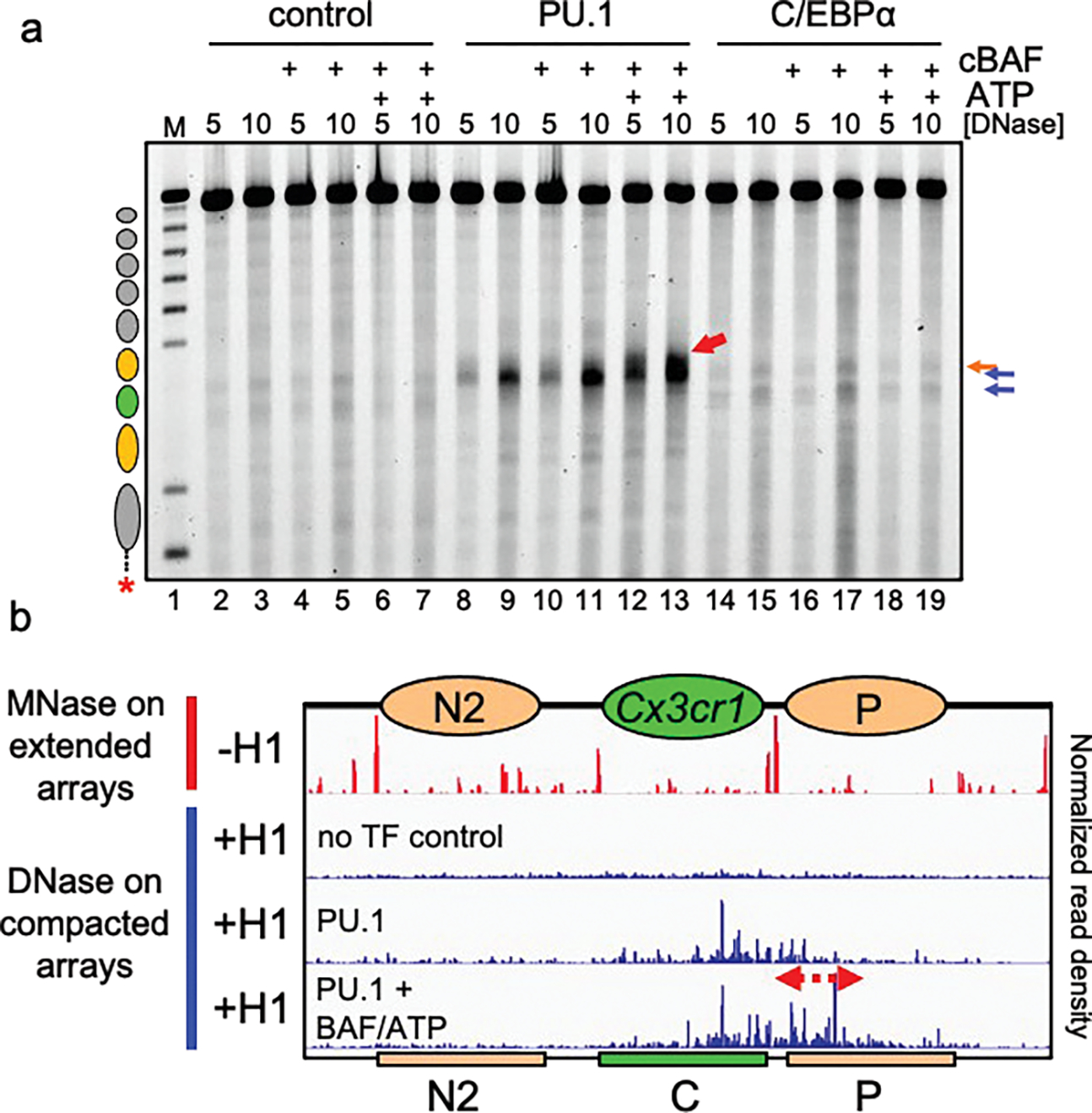
a, DNase I digestion analysis H1-compacted nucleosome arrays incubated with PU.1 (lanes 8–13) or C/EBPα (lanes 14–19), in the presence of cBAF alone (lanes 10–11 and lanes 16–17, respectively) or cBAF and ATP (lanes 12–13 and lanes 18–19, respectively). b, Nanopore analysis of MNase digested extended arrays (−H1) and DNase I digested compacted arrays (+H1) alone (no TF control), with PU.1 alone, or with PU.1, cBAF and ATP. Ovals indicate the translational positions of nucleosomes determined in Extended Data Fig. 2d. Plots show normalized read density on the y axis. For each plot, the maximum value is set to 0.4% of reads.
There were no differences in hypersensitivity patterns when the cBAF complex was added to C/EBPα and H1-compacted nucleosome arrays (Fig. 3a, lanes 14–19). Notably, the ability of C/EBPα to open the chromatin, but not enable BAF activity, indicated that the local chromatin opening pattern of C/EBP factors, per se, was insufficient to enable cBAF activity.
We performed a deletion analysis of PU.1 to determine the molecular features that enable initial nucleosome exposure and subsequent cBAF remodeling (Fig. 4a,b). Between the N-terminal acidic (AQ) domain and the DNA-binding domain of PU.1 is a highly anionic domain with similarity to a “PEST” domain. However, the PEST domain does not target PU.1 for metabolic turnover39, though there is evidence it controls the dynamics of PU.1 homodimerization40. Using two different methods to map protein structure41,42 (Fig. 4a), we found that the PEST domain is predicted to be highly disordered and thus we refer to it as an intrinsically disordered region (IDR).
Figure 4: cBAF action requires AQ and IDR domains of PU.1.
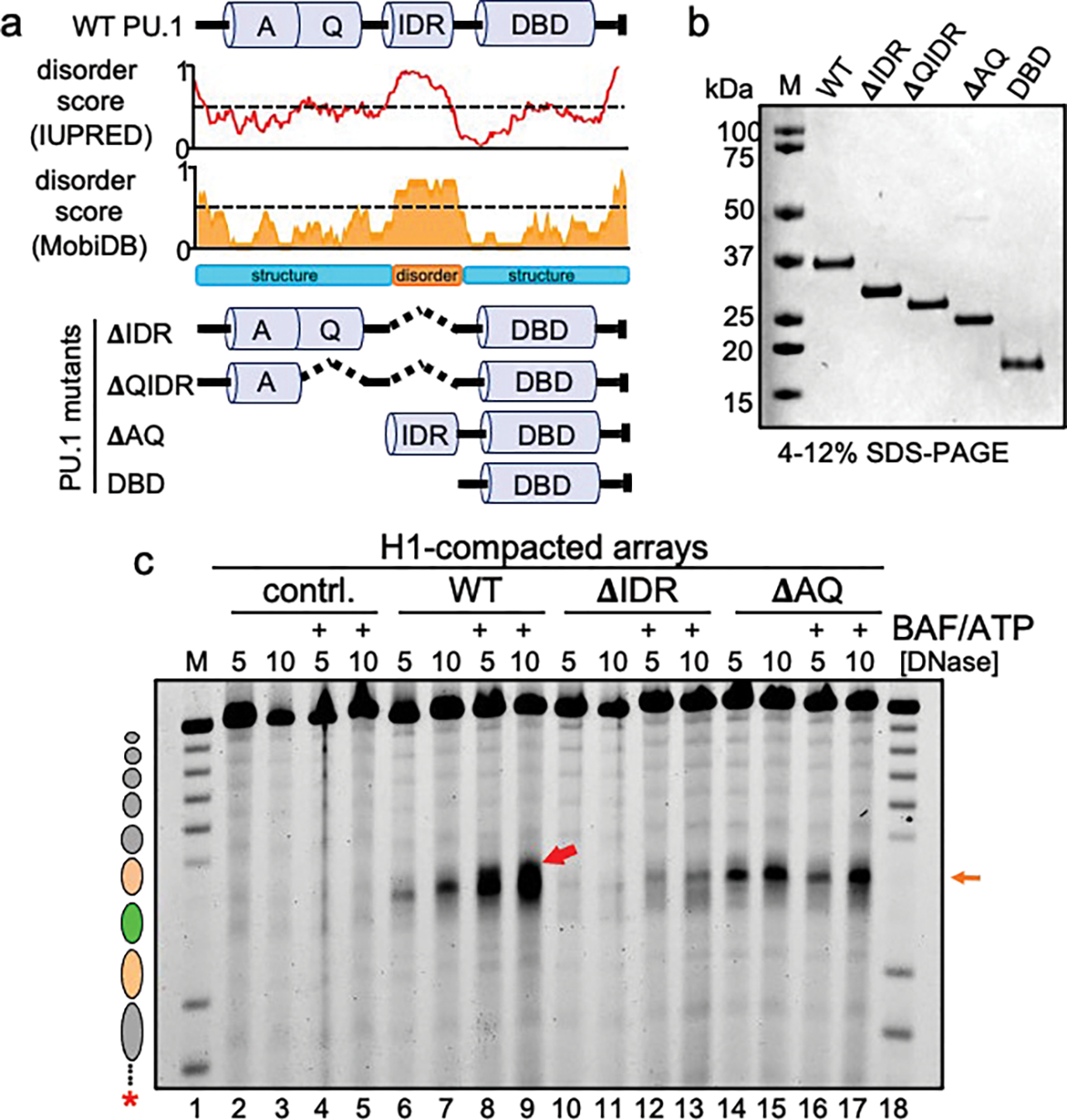
a, Predicted disorder tendency of WT PU.1. The red and orange lines indicate the predicted disorder tendency along the WT PU.1 protein (as calculated by IUPred and MobiDB, respectively) with values above 0.5 (indicated by the dotted line) considered disordered. Shown are the positions of the Acidic domain (A), Q-rich domain (Q), Intrinsically disordered region (IDR) and DNA-binding domain (DBD). The deletion mutant series of PU.1 are indicated below as ΔIDR, ΔQIDR, ΔAQ, and DBD. b, SDS-PAGE analysis of 1 μg of each protein, stained with Coomassie blue. c, DNase I digestion analysis H1-compacted nucleosome arrays incubated alone (contrl., lanes 2–5), WT PU.1 (lanes 6–9), ΔIDR deletion (lanes 10–13), and ΔAQ truncation (lanes 14–17) with or without the addition of BAF and ATP.
We interrogated DNA and chromatin binding by the PU.1 deletion mutants in a series of electromobility shift assays (Extended Data Fig. 7). The ΔAQ and ΔIDR mutants of PU.1 bound Cx3cr1 free DNA and mononucleosomes as well as WT PU.1 (Extended Data Fig. 7a–c). The ΔAQ truncation mutant bound extended arrays and H1-compacted arrays as well as WT PU.1 (Extended Data Fig. 7e, g. Compare lanes 2–3 to lanes 14–15, respectively). However, the ΔIDR mutant bound extended arrays less efficiently (Extended Data Fig. 7e, lanes 8–9) and failed to shift H1-compacted arrays (Extended Data Fig. 7g, lanes 8–9). Thus, the IDR domain of PU.1 is not needed for nucleosome binding but facilitates PU.1 engagement with chromatin.
The N-terminal ΔAQ truncation mutant, with the IDR of PU.1 tethered to the PU.1 DNA-binding domain, was sufficient to robustly expose the Cx3cr1 nucleosome within H1-compacted arrays (Extended Data Fig. 8a, b, lanes 11–12). By contrast, the ΔIDR mutant did not elicit chromatin opening (Extended Data Fig. 8a, b lanes 7–8; Extended Data Fig. 8c, lanes 14, 15), comparable to the low range observed with the DBD alone (Extended Data Fig. 8a, b lanes 5–6; Extended Data Fig. 8c, lanes 10–11). We conclude that the IDR domain is essential for PU.1 to interact with H1-compacted chromatin and for the intrinsic chromatin opening function of PU.1.
Strikingly, robust chromatin opening by the N-terminal ΔAQ truncation mutant (Fig. 4c, lanes 14–15) was insufficient to enable cBAF activity (Fig. 4c compare lanes 14–15 to lanes 16–17, respectively). Thus, the acidic domain interacts with27 or positions a unique orientation of cBAF complexes required for them to expand hypersensitivity within H1-compacted chromatin.
Interestingly, we found that incubation of cBAF and ATP with the ΔIDR deletion mutant, which can bind mononucleosomes (Extended Data Fig. 7b) yet is deficient for nucleosome exposure in compacted chromatin, resulted in a weak hypersensitive signal within the Cx3cr1 nucleosome of the compacted nucleosome array. Thus, residual nucleosome binding by the ΔIDR deletion mutant, which retains the N-terminal AQ region, enabled cBAF to elicit chromatin opening (Fig. 4c lanes 10–13). The DNA-binding domain alone, which like the ΔIDR deletion mutant can bind mononucleosomes (Extended Data Fig. 5b) and is deficient for nucleosome exposure in compacted chromatin (Extended Data Fig. 8c), did not enable cBAF activity (Extended Data Fig. 8c, compare lanes 10–11 to lanes 12–13). Together, these data indicate that the N-terminal AQ region is necessary for PU.1 to enable cBAF to engage and remodel H1-compacted chromatin in vitro.
In summary, to act on linker histone-compacted chromatin, cBAF chromatin remodeling complexes require initial chromatin opening by PU.1, mediated by the IDR and acidic domains. The lack of cBAF complex activity on the chromatin opened by C/EBPα and the PU.1 ΔAQ truncation, each of which elicit different chromatin opening patterns, demonstrate that mere local opening is not sufficient for cBAF activity and that cBAF complexes must also be stabilized by, positioned, or recruited to the compacted chromatin by the pioneer factor. Presumably, the necessity for the pioneer factor provides a specificity check for ATP-dependent remodeling at particular sites in chromatin. Furthermore, our finding that cBAF can weakly promote chromatin opening in cooperation with a PU.1 deletion mutant deficient for chromatin opening (Fig. 4c, lanes 12–13) indicates that the chromatin remodeler can “feed forward” weak interactions with chromatin by a nucleosome-binding transcription factor. These results can explain in vivo findings that the loss of SWI/SNF results in a subset of sites where transcription factor binding is lost22,23. However, our in vitro studies indicate that SWI/SNF activity is not required for the wild-type pioneer factors to initially access and locally open compacted chromatin. Our in vitro studies provide a mechanistic understanding of how pioneer factors and nucleosome remodeling complexes together play fundamental roles in enabling alterations of DNA-nucleosome contacts and chromatin accessibility, both necessary determinants of gene expression.
Methods
Protein Expression and Purification
The bacterial expression plasmids: pET-28b-PU.1, pET-28b-C/EBPα, pET-28b-C/EBPβ encode the mouse PU.1, C/EBPα, and C/EBPβ fused to an N-terminal 6X histidine tag, respectively13. pET-28b-PU.1-DBD (encodes PU.1-DBD, residues 160–272) and pET-28b-C/EBPα-DBD (encodes C/EBPα-DBD, residues 272–359) were generated by PCR from the respective full-length construct, introducing NdeI and XhoI restriction sites for insertion into the pET-28b plasmid. The remaining mutant PU.1 constructs, pET-28b-PU.1-ΔIDR (encodes residues 1–115, 161–272 of PU.1), pET-28b-PU.1-ΔQIDR (encodes residues 1–73, 101–272 of PU.1), pET-28b-PU.1-ΔAQ (encodes residues 116–272 of PU.1), were generated by infusion cloning of gene blocks (IDT) using XhoI NdeI digested pET-28b.
The histidine-tagged proteins were expressed in E. Coli Rosetta (DE3) pLysS (Novagen # 70956–3). Transformed cells were grown at 37°C to a density of 0.5–0.7 at an absorbance of 600 nm and protein expression was induced with 1 mM IPTG at 16°C for 16 hr for PU.1 and PU.1 mutant constructs; 1 mM IPTG at 37°C for 4 hr CEBPβ; 2 mM IPTG at 37°C for 4 hr CEBPα and C/EBPα-DBD.
The recombinant proteins were purified over Ni-NTA resin under denaturing conditions (20 mM Tris-HCl pH 8.0, 0.5M NaCl, 6M Urea) with 5 mM imidazole and 20–300 mM single step imidazole changes follow by 4M and 2M urea step dialysis. The recombinant human full length histones H2A, H2B, H3, and H4 were expressed and purified as described previously43. Recombinant human IRF3 was purchased (Active Motif 31544).
Human cBAF Complex Purification
Human cBAF complexes were purified as described previously37,38, with several modifications. Briefly, HEK-293T cells were lentivirally transduced with HA-DPF2 for the specific capture of cBAF complexes. Pellets were washed in cold phosphate-buffered saline (PBS) (GIBCO). Cell suspension was then centrifuged for 5 minutes at 4 degrees C at 4000 g. Cell pellets were resuspended in HB (10 mM Tris-HCl, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM DTT, 1 mM PMSF) and homogenized in a glass Dounce homogenizer. The suspension was pelleted for 30 minutes at 4 degrees C at 4000 g and nuclear pellets were resuspended in pre-extraction buffer (50 mM Tris-HCl, pH 7.5, 100 mM KCl, 1 mM MgCl2, 1 mM EDTA, 1 mM, 0.1% (v/v) NP40, 1 mM DTT, 1 mM PMSF and protease inhibitor cocktail. After pelleting for 10 minutes at 4 degrees C at 4000 g, chromatin was resuspended in high-salt buffer (HSB; 50 mM Tris-HCl, pH 7.5, 300 mM KCl, 1 mM MgCl2, 1 mM EDTA, 1 mM, 1% NP40, 1 mM DTT, 1 mM PMSF and protease inhibitor cocktail) and incubated with rotation for one hour at 4 degrees C. Homogenates were centrifuged at 20,000 rpm in a SW32Ti rotor (Beckman Coulter) for 1 hour at 4 degrees C and the supernatant collected. Nuclear extracts were filtered with a 0.45-mm filter and rotated overnight at 4 degrees C with HA magnetic resin (Thermo Fisher Scientific). The beads were washed in HSB and proteins were eluted by incubating the beads 4 times for 1.5 hours with 1 mg/mL of HA peptide in HSB. Eluted protein complexes were separated by density using 10%–30% glycerol gradients and fractions with confirmed purified expression of full cBAF complex were pooled and concentrated using the appropriate molecular weight cutoff protein concentrator (Pierce). Samples were snap frozen and kept at −80°C until needed.
Generation of Nucleosome Array Templates
The 162 bp Cx3cr1 mononucleosome sequence that corresponds to the genomic location: mm9 chr9:119946611–11994676213 was extended on either side by 35 bp to incorporate linker DNA and BfuAI and XbaI flanking sequences: ACCTGCGTTACAGCATCCACTCAGTATCCCTTGAGCCCCGCGTGCAAAGCCCAGGAGCCCTTGCTTAGGTGCAGGGCCTCTCGGCTGCTGATCTTCAGCTGGTTGCTGAGAGTTGCAGCATTGCTGAGTCTTAGCAATGGATACTTCCCGATTCCCCTCACAAAAATAGGTCAGTCTGTCTGGCTAGTTCTGTACTTGCAGACACAGGGCATGTGGGGTTCCTATTTTTCTAGCTCCCAGGCTTCTGTCTGCTTCCTTCGTTTAGTATGTCTA
The DNA sequence was generated by PCR of genomic mouse DNA with BfuAI and XbaI restriction sites cloned into the p208–10.N1N216 to generate p208–10-Cx3cr1. Mutant Cx3cr1 arrays were generated by infusion cloning using linearized BfuAI XbaI digested p208–10-Cx3cr1 plasmids. Geneblocks for cloning were obtained from IDT:
TF mutant motif Cx3cr1:
AAAATAACCTGCGTTACAGCATCCACTCAGTATCCCTTGAGCCCCGCGTGCAAAGCCCAGGAGCCCTTGCTTAGGTGCAGGGCCTCTCGGCTGCTGATCTTCAGCTGGTTGCTGAGAGTTGCAGCATTGCTGAGTCCCGTATAGAGATCGTATAGCGTATAGAGCACAAAAATAGGTCAGTCTGTCTGGCTAGTTCTGTACTTGCAGACACAGGGCATGTGGGGTTCCTATTTTTCTAGCTCCCAGGCTTCTGTCTGCTTCCTTCGTTTAGTATGTCTAGACTACAGTTATTGGTT
Swap1 Cx3cr1:
AAAATAACCTGCGTTACAGCATCCACTCAGTATCCCTTGAGCCCCGCGTGCAAAGCCCAGGAGCCCTTGCTTAGGTGCAGGGCCTCTCGGCTGCTGATCTTCAGCTGGTTGCTGAGAGTTGCAGCATTGCTGAGTCTACTTCCCGAATTAGCAATGGTTCCCCTCACAAAAATAGGTCAGTCTGTCTGGCTAGTTCTGTACTTGCAGACACAGGGCATGTGGGGTTCCTATTTTTCTAGCTCCCAGGCTTCTGTCTGCTTCCTTCGTTTAGTATGTCTAGACTACAGTTATTGGTT
Swap2 Cx3cr1:
AAAATAACCTGCGTTACAGCATCCACTCAGTATCCCTTGAGCCCCGCGTGCAAAGCCCAGGAGCCCTTGCTTAGGTGCAGGGCCTCTCGGCTGCTGATCTTCAGCTGGTTGCTGAGAGTTGCAGCATTGCTGAGTCTACTTCCCGAATTAGCAATGGTTCCCCTCACAAAAATAGGTCAGTCTGTCTGGCTAGTTCTGTACTTGCAGACACAGGGCATGTGGGGTTCCTATTTTTCTAGCTCCCAGGCTTCTGTCTGCTTCCTTCGTTTAGTATGTCTAGACTACAGTTATTGGTT
Plasmids were amplified in E.coli, purified, and digested with MluI, SmaI and HhaI to release array fragments. Fragments were purified by ethanol precipitation then labeled with dCTP-Cy5 (Cytiva PA55021) by end-repair with Klenow fragment DNA polymeraase (3’-5’ exo-) (NEB). Cy5-DNA labeling reactions were carried out at final concentrations of 1 μM DNA, 4 mM Cy5-dCTP and 0.5 U/μL Klenow fragment in the presence of excess 4mM dATP, 4 mM dTTP, and 4 mM dGTP in NEB buffer 2 (50mMNaCl, 10mM Tris-HCl, 10 mM MgCl2, 1 mM DTT). The reaction was incubated at 37C for 1 hr. Labeling reactions were purified by phenol:chloroform extraction followed by FPLC purification of the 2.7 kb MluI-SmaI fragment using a Capto HiRes Q anion exchange column (Cytiva 29275878).
Nucleosome Array and Mononucleosome Reconstitution
Nucleosome arrays were assembled as previously described16. Assembly reactions containing 2 μg of end-labeled DNA fragments and core histones at a 1:1.0 molar ratio of octamers to nucleosomal sites, and 2 M NaCl in a total of 10 μl were incubated at 37°C for 15 min, serially diluted by adding 3.3, 6.7, 5, 3.6, 4.7, 6.7, 10, 30, and 20 μl of 50 mM HEPES (pH 7.5), 1 mM EDTA, 5 mM DTT, and 0.5 mM PMSF in 15 min incubation steps at 30°C, and brought to 0.1 M NaCl by adding 100 μl of 10 mM Tris·HCl (pH 7.5), 1 mM EDTA, IGEPAL, 5 mM DTT, 0.5 mM PMSF, and 20% glycerol, followed by incubation at 30°C for 15 min. Array saturation was determined by EcoRI digestion, performed with a concentration of 2 nM nucleosome array or free DNA in 20 mM HEPES (pH 7.5), 50 mM KCl, 1% glycerol, 5 mM DTT, 100 μg/ml BSA, and 2 U/μl EcoRI. Following a 2 hr incubation at 37°C, digestion products were resolved on 4% polyacrylamide 0.5X TBE gels and analyzed as described16.
Cx3cr1 mononucleosomes were reconstituted by dialysis with the Cy5 end-labeled 162 bp Cx3cr1 DNA fragment and recombinant human histones as described previously13.
TF Binding Reactions and Enzymatic Assays
Binding reactions for the DNase I digestion, MNase, and Restriction enzyme assays were carried out with 1 nM (1.5 ng/μl) of nucleosome array (13 nM of nucleosomes) in 6 mM HEPES (pH 7.5), 35 mM KCl, 1.5% glycerol, 2 mM DTT, 250 μg/ml BSA. In experiments with compacted nucleosome arrays, 13 nM purified histone H1 (Calbiochem) was incubated with the nucleosome arrays at room temperature for 1 hr. Purified transcription factors were then incubated with the nucleosome arrays at room temperature for 1 hr. For cBAF remodeling assays 10nM of indicated transcription factors were incubated with H1-compacted arrays at room temperature followed by incubation with cBAF (6 nM) and ATP (0.5mM) for 30 minutes at 30°C.
DNase digestions were carried out by 2.5–10 ug/mL of DNase I diluted in 50 mM MgCl2, followed by incubation at room temperature for 1 min. MNase digestion was performed by addition of .075–0.6 U/mL MNase diluted in 30 mM CaCl2, followed by incubation at room temperature for 1 min. XbaI restriction enzyme digestion was performed by incubation with 0.25–1 U/mL XbaI for 45 minutes at 30°C. Reactions were terminated by addition of 1 volume of stop buffer (20 mM Tris pH 7.5, 50 mM EDTA, 1% SDS, 0.5 mg/mL tRNA, and 0.2 mg/mL proteinase K) and incubated at 50C for 15 min. Digestion products are purified by ethanol precipitation and electrophoresed on 1% Agarose 1X TBE gels.
Electromobility shift assays with end-labeled 162 bp Cx3cr1-DNA or mononucleosomes incubated with recombinant proteins were performed in DNA-binding buffer (10 mM Tris-HCl pH7.5, 1 mM MgCl2, 10 mM ZnCl2, 1 mM DTT, 50 mM KCl, 3 mg/ml BSA, 5% Glycerol) at room temperature for 30 min. Free and bound DNA were separated on 4% non-denaturing polyacrylamide gels run in 0.5X Tris–borate–EDTA. Electromobility shift assays with extended or H1-compacted nucleosome arrays incubated with recombinant proteins were performed in 6 mM HEPES (pH 7.5), 35 mM KCl, 1.5% glycerol, 2 mM DTT for 1 hour, fixed on ice with 0.1% glutaraldehyde for 30 minutes, then electrophoresed on 1% agarose gel run in 0.5X Tris–borate–EDTA.
Gels were visualized using with an Amersham Typhoon RGB Biomolecular Imager using Cy5 fluorescence setting (excitation at 633 nm and emission filter 670 BP 30). All experiments were performed at least twice.
Nanopore Sequencing Library Prep
Partially MNase-digested chromatin retaining contiguous oligonucleosomes was used for generation of libraries for nanopore sequencing. Purified DNA was end-repaired and A-tailed with the NEBnext Ultra II end repair module (New England Biolabs), purified with Ampure XP beads (Beckman Coulter) and ligated to AMX adapter from the Oxford Nanopore 1D sequencing Kit (SQK-LSK108, Oxford Nanopore Technologies) using NEB Blunt/TA ligase master mix. Excess adapters were removed by adding 0.5 volumes of Ampure XP beads and washing with ABB buffer from the 1D sequencing kit. Purified library was eluted and loaded to a MinION R9.4 flowcell according to the manufacturer’s instructions and run for 24 h.
Nanopore Sequencing Analysis
The fast5 raw data files from the Nanopore sequencer were basecalled and converted to fastq format with Guppy Software (Oxford Nanopore Technologies). Reads were mapped to the Cx3cr1 Array reference with minimap2, a mapping algorithm specifically designed to analyze nanopore sequencing reads. Bam files produced by minimap2 were imported into R44 then filtered and normalized using tidyverse packages45.Specifically, reads were excluded if they were identified as multimappers by minimap2 or were located entirely within the non-unique regions of the nucleosome array DNA sequence. The ends of each read from an array digestion identify the precise nucleotide at which a nuclease was able to access and cleave the DNA in the array. We quantified the endpoints of each read using the hash package and then normalized each dataset to the total number of mapped reads. To determine nucleosome translational positions, the data was visualized using the ggplot2 package and the smoothed cleavage traces were produced with the kernel smoothing function in R.
Protein disorder prediction
Disorder tendency of PU.1 was calculated using the IUPRED tool46 and MobiDB41, as recommended by the programs.
Statistics
Unpaired, two-tailed, Student’s t-tests were used to test differences in chromatin opening efficiencies between full-length transcription factors and their DNA-binding domains.
Extended Data
Extended Data Figure 1: Generation of H1-compacted Cx3cr1 nucleosome arrays.
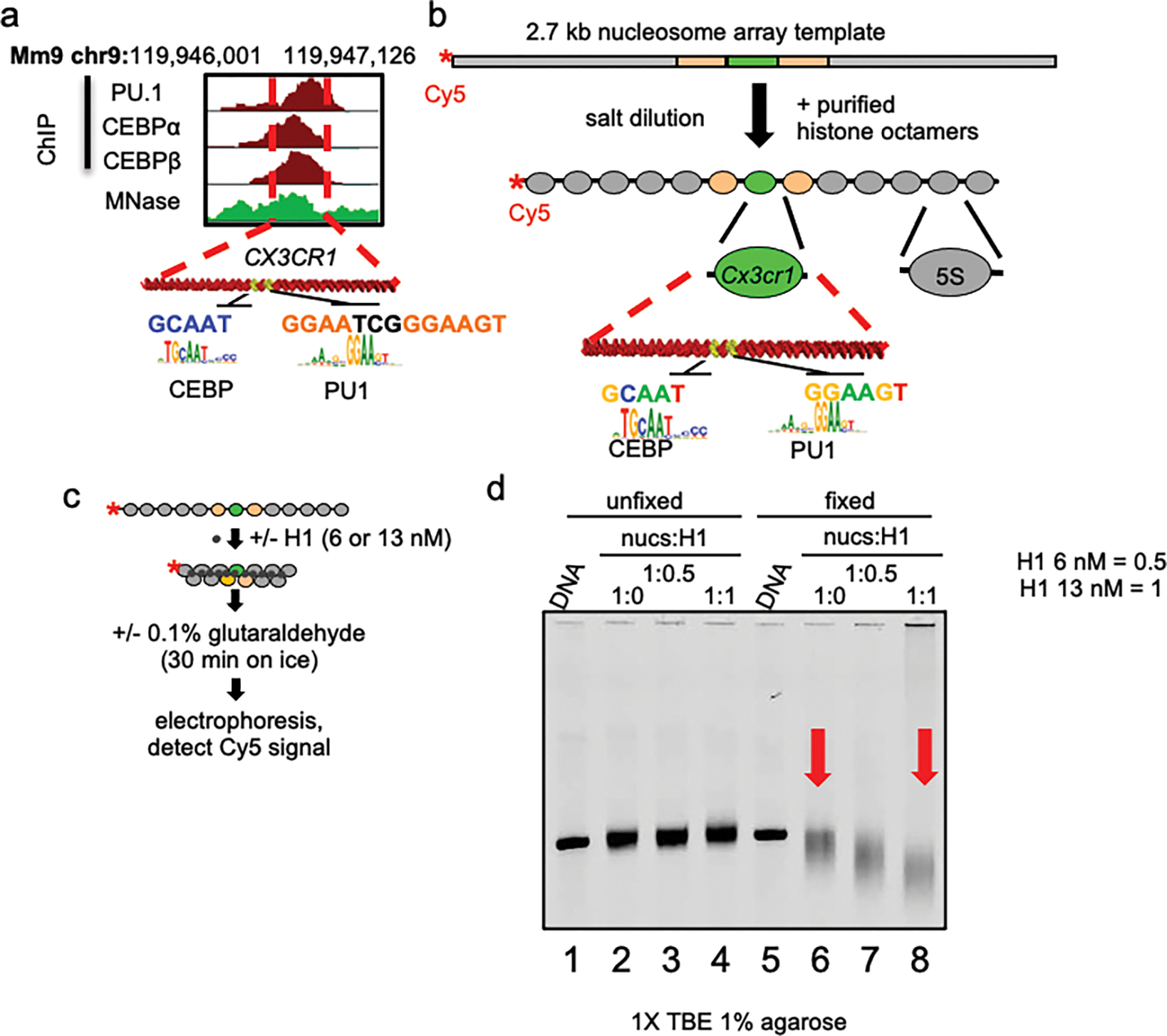
a, PU.1 and C/EBP ChIP-seq profiles (red) in macrophages and MNase-seq profile (green) in fibroblasts near the Cx3cr1 gene within the displayed region of the mouse genome with TF motifs indicated. b, Diagram of the dCTP-Cy5 end-labeled Cx3cr1 nucleosome array. c, Schematic of linker histone-mediated chromatin compaction. d, Analysis of linker histone-mediated chromatin compaction by native agarose gel electrophoresis. Free DNA (lanes 1 and 5) and nucleosome arrays with nucleosome:linker histone ratios of 0 (lanes 2 and 6), 0.5 (lanes 3 and 7) and 1.0 (lanes 4 and 8) are shown.
Extended Data Figure 2: Nanopore sequencing to determine translational position of nucleosomes in the Cx3cr1 array.
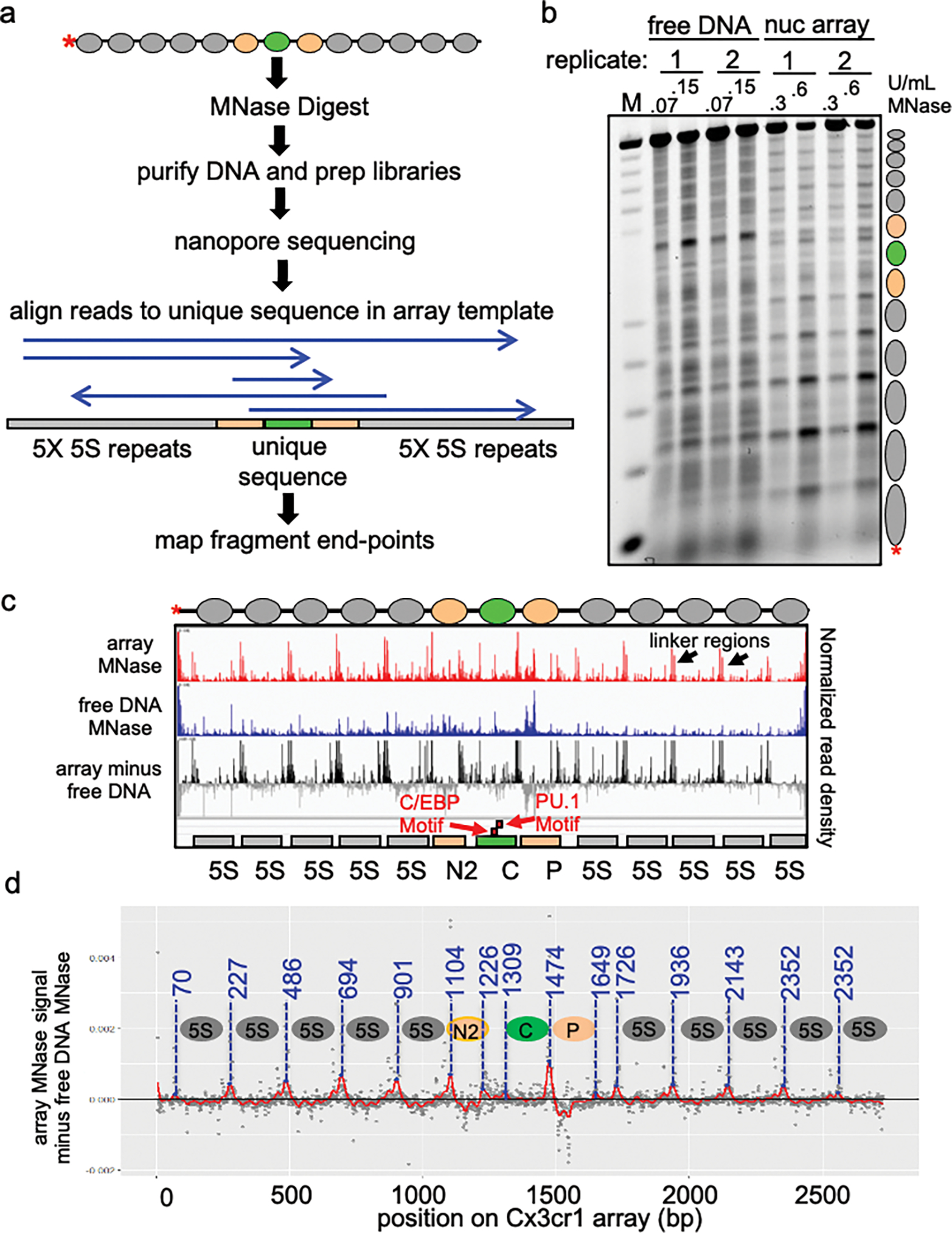
a, Schematic of Nanopore sequencing and endpoint analysis pipeline. b, MNase digestion analysis of free DNA and extended nucleosome arrays at two MNase concentrations (U/mL) visualized by gel electrophoresis. Reactions shown in this gel are the same ones analyzed by nanopore sequencing. c, IGV visualization of Nanopore sequencing endpoint analysis of MNase digested free DNA (0.07 U/mL MNase), extended nucleosome arrays (0.3 U/mL) and free DNA signal subtracted from array signal to account for MNase site bias. Plots show normalized read density on the y axis. For each plot, the maximum value is set to 0.4% of reads. d, Determination of nucleosome translational positions.
Extended Data Figure 3: Nanopore sequencing of TFs incubated with H1-compacted arrays.
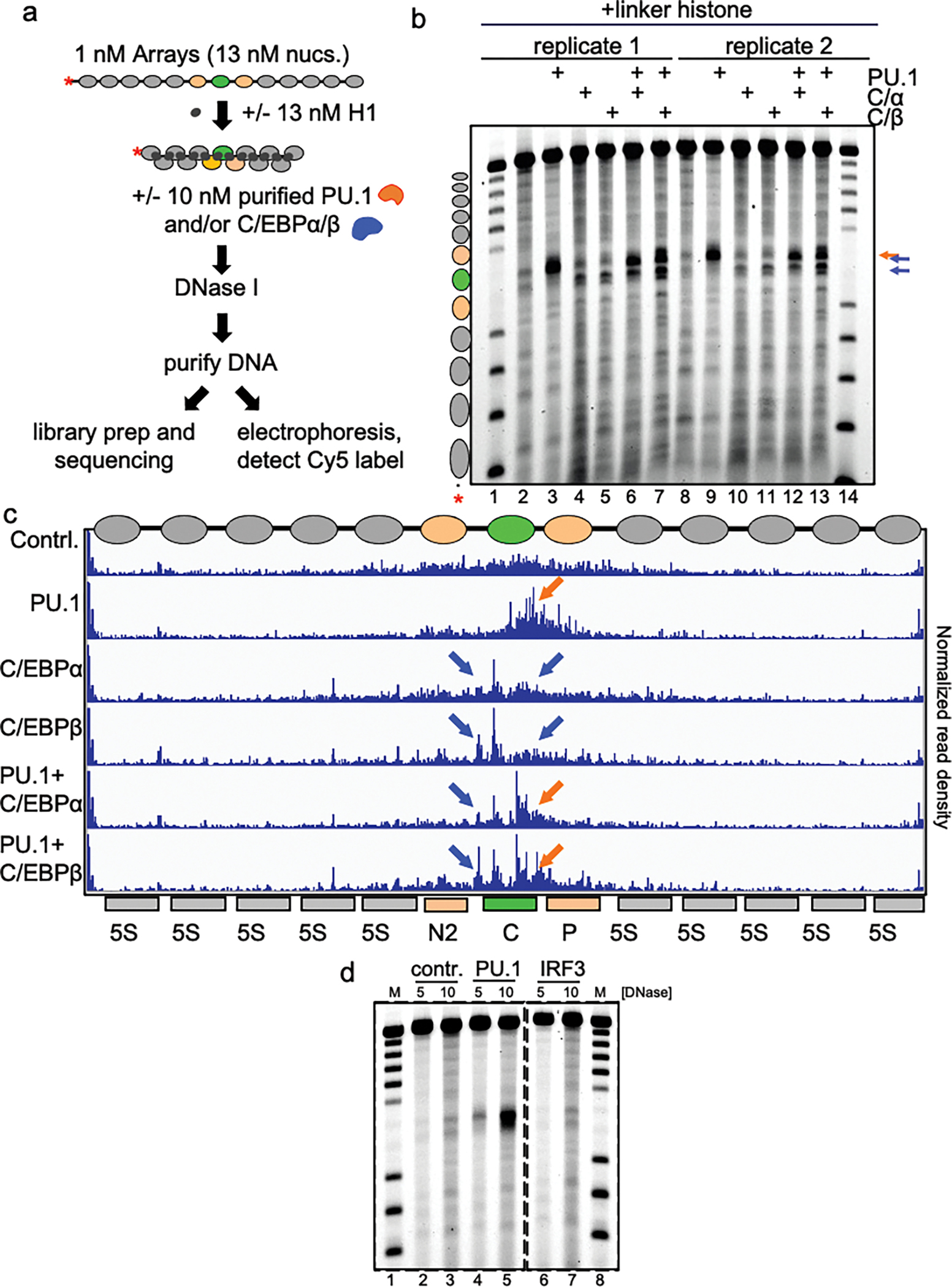
a, Schematic of DNase I digestion analysis. b, DNase I digestion analysis of TFs binding to H1-compacted nucleosome arrays visualized by gel electrophoresis (10 ng/uL DNase I). The same samples shown on the gel were used for nanopore sequencing shown in panel c. c, IGV visualization of Nanopore endpoint analysis of DNase I digested H1-compacted nucleosome arrays incubated with indicated transcription factor(s). Plots show normalized read density on the y axis. For each plot, the maximum value is set to 0.4% of reads. d, DNase I digestion analysis of indicated TFs binding to H1-compacted nucleosome arrays visualized by gel electrophoresis.
Extended Data Figure 4: PU.1 and C/EBPa open chromatin cooperatively.
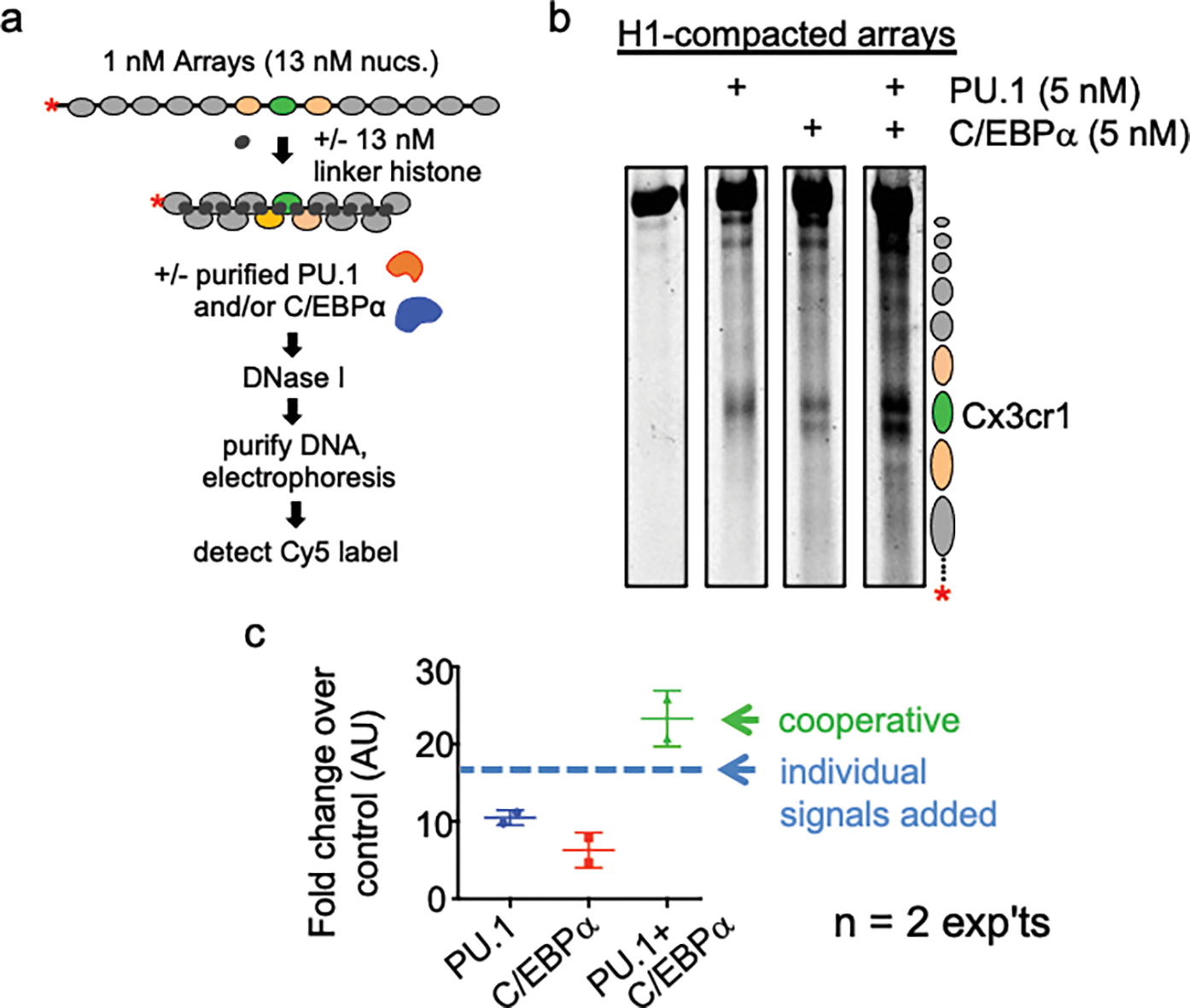
a, Schematic of DNase I digestion analysis. b, DNase I digestion analysis of TFs binding to H1-compacted nucleosome arrays visualized by gel electrophoresis. c, Quantified Cy5 signal in each lane normalized to no TF control (n=2).
Extended Data Figure 5: DNA-binding domains of PU.1 and C/EBPα are sufficient for nucleosome binding.
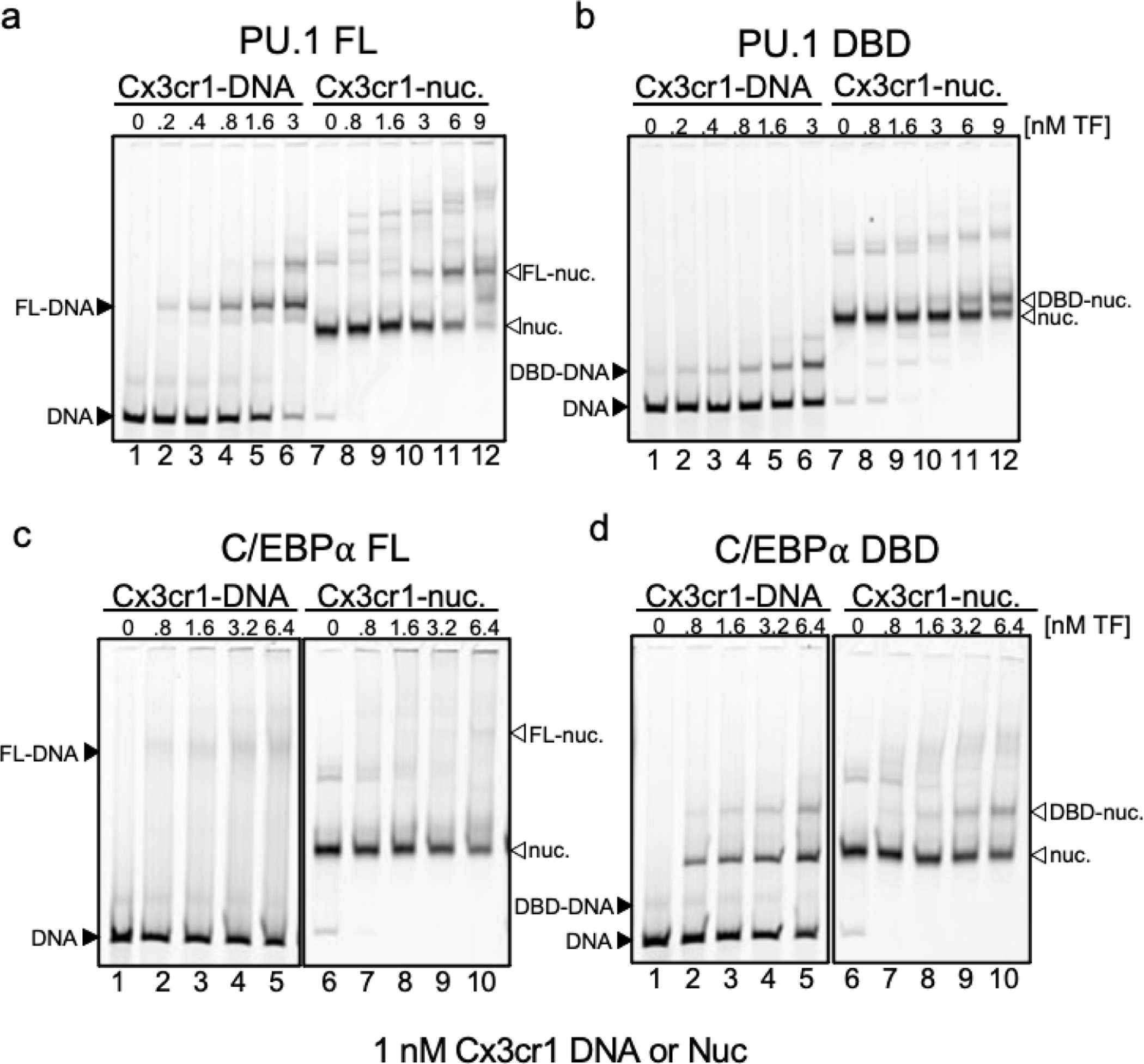
a-d, Representative EMSAs showing the affinity of increasing amounts of a, Full-length PU.1 (PU.1 FL), b, PU.1-DBD, c, Full-length C/EBPα (C/EBPα FL), d, C/EBP-DBD to Cy5-labeled Cx3cr1 free DNA (black arrows) or mononucleosomes (white arrows).
Extended Data Figure 6: cBAF readily remodels extended nucleosome arrays but requires PU.1 to access H1-compacted arrays.
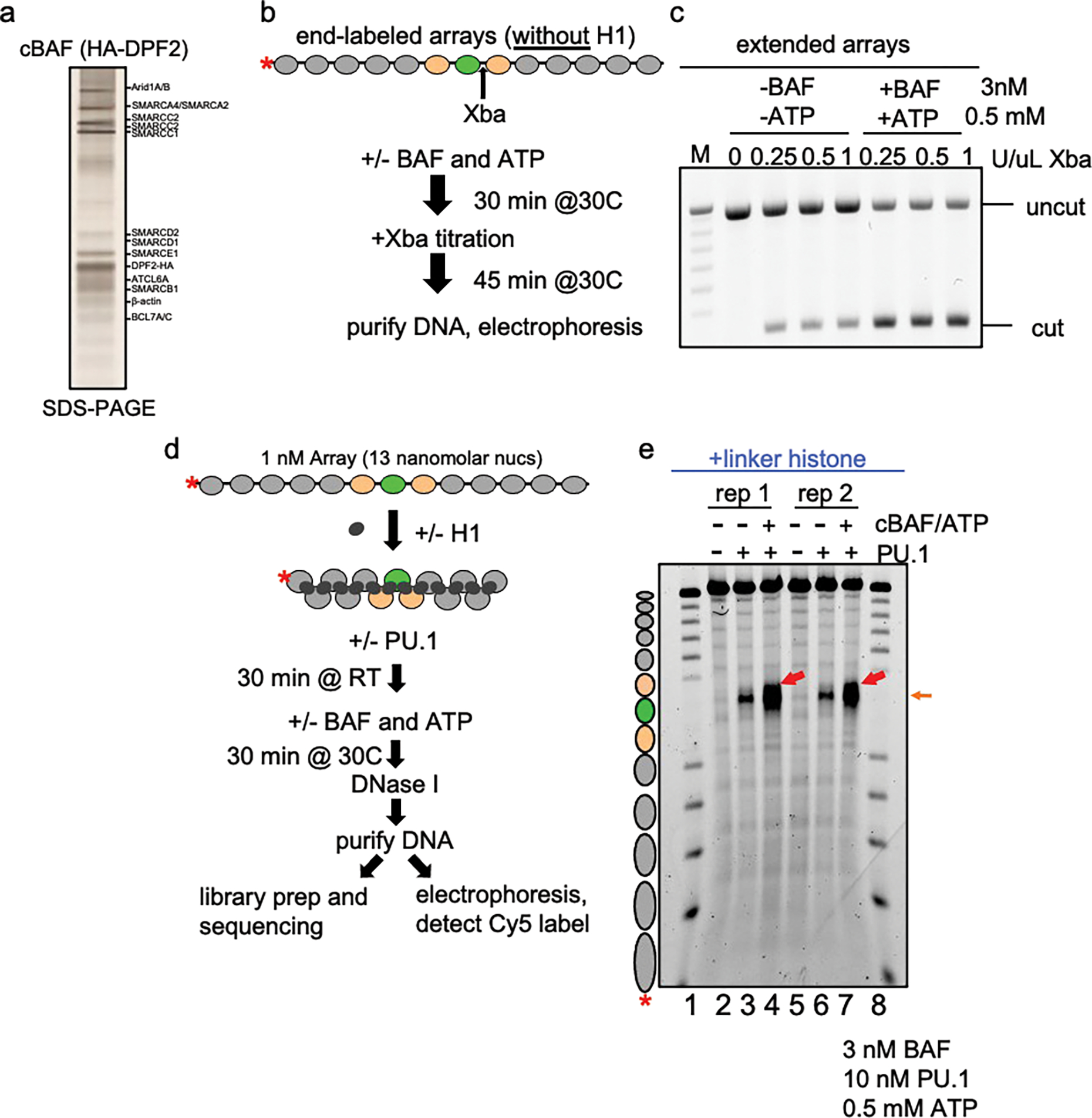
a, silver-stain of affinity-purified cBAF complexes from mammalian HEK293T cells expressing HA-DPF2. b, Schematic of XbaI accessibility assay with extended arrays and cBAF remodeling complex. c, Agarose gel visualization of XbaI accessibility assay of extended nucleosome arrays (no linker histone) incubated without and with cBAF plus ATP. d, Schematic of DNase I digestion analysis of transcription factors and cBAF with H1-compacted arrays. e, DNase I digestion analysis H1-compacted nucleosome arrays incubated alone (no TF, lanes 2 and 5), PU.1 alone (lanes 3 and 6), and PU.1 with cBAF and ATP (lanes 4 and 7). The same samples were used to perform nanopore sequencing shown in Fig. 3b.
Extended Data Figure 7: Hierarchy of chromatin binding by PU.1 WT and mutants.
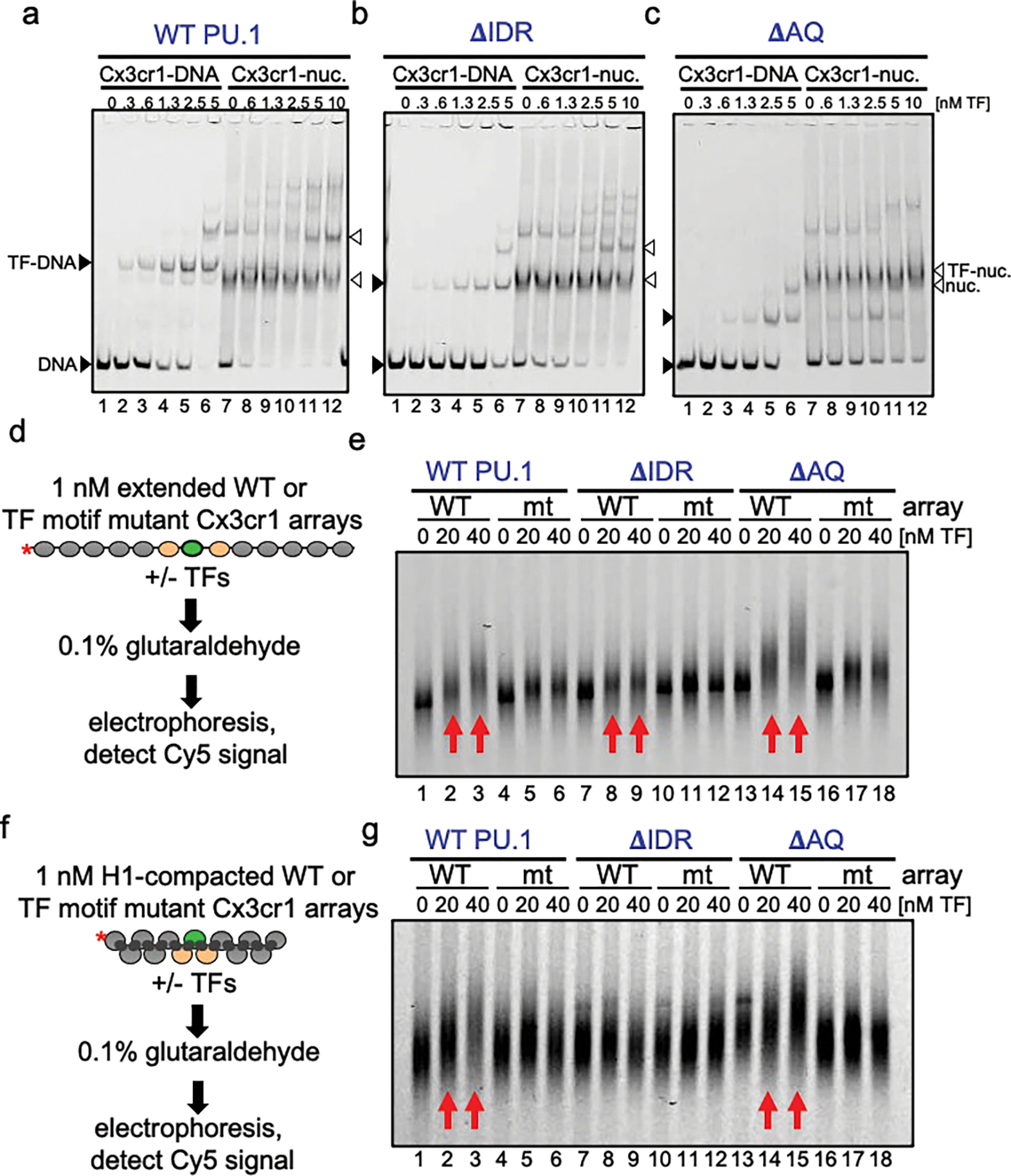
a-c, Representative EMSAs showing the affinity of increasing amounts of a, WT PU.1, b, ΔIDR, c, ΔAQ to Cy5-labeled Cx3cr1 free DNA (black arrows) or mononucleosomes (white arrows). d, Schematic of EMSA performed with transcription factors and extended nucleosome arrays e, EMSA showing the affinity of increasing amounts of WT PU.1 (lanes 1–6), ΔIDR (lanes 7–12), and ΔAQ (lanes 13–18) to Cy5-labeled extended WT or TF motif mutant Cx3cr1 nucleosome arrays. f, Schematic of EMSA performed with transcription factors and H1-compacted nucleosome arrays. g, Representative EMSA showing the affinity of increasing amounts of WT PU.1 (lanes 1–6), ΔIDR (lanes 7–12), and ΔAQ (lanes 13–18) to Cy5-labeled H1-compacted WT or TF motif mutant Cx3cr1 nucleosome arrays.
Extended Data Figure 8: The IDR domain of PU.1 is most crucial for chromatin opening.
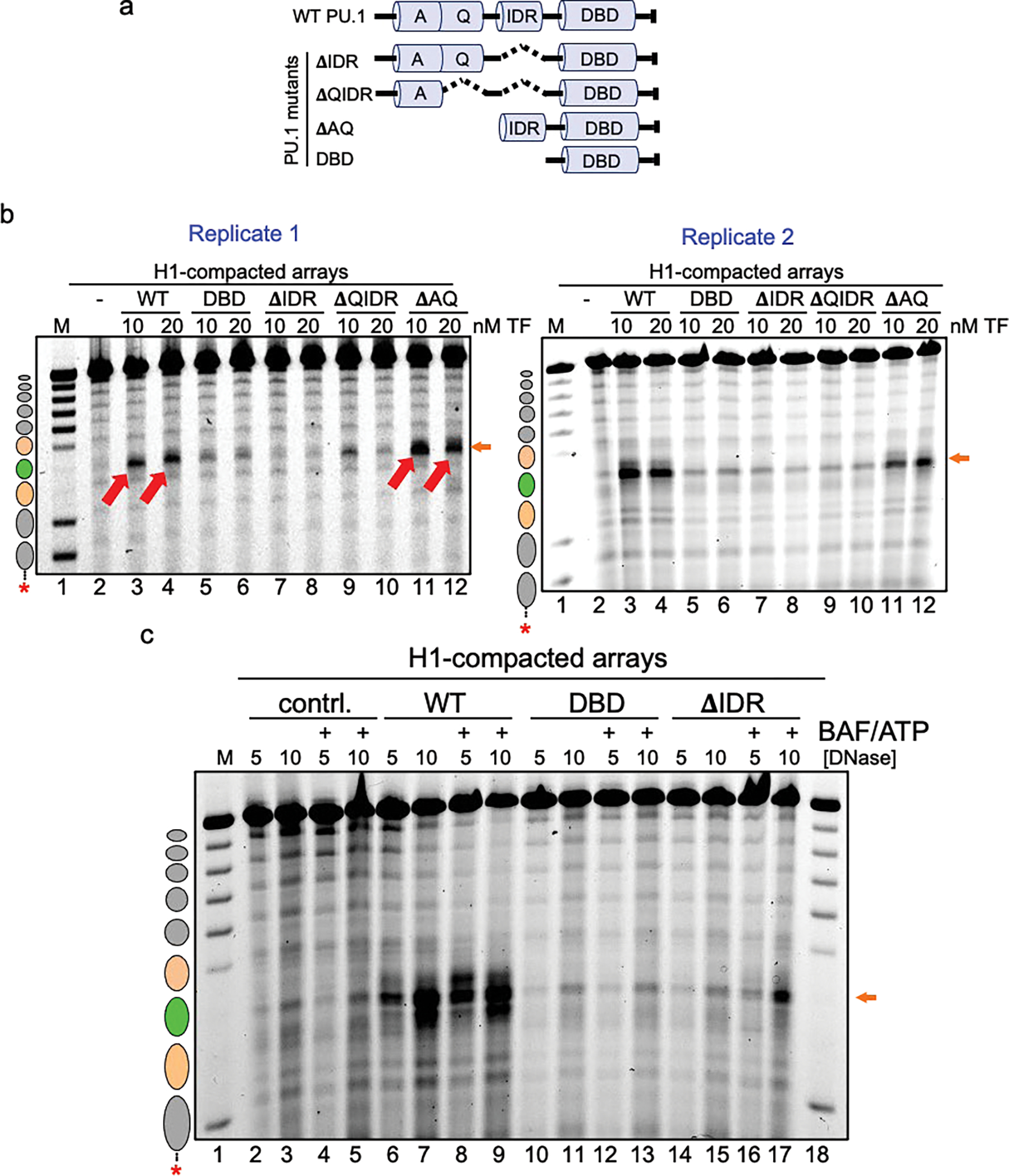
a, Illustration of the PU.1 deletion mutant series. Shown are the positions of the acidic domain (A), Q-rich domain (Q), intrinsically disordered region (IDR) and DNA-binding domain (DBD). b, DNase I digestion analysis of two transcription factor concentrations binding to H1-compacted nucleosome arrays visualized by gel electrophoresis (10 ng/uL DNase I). c, DNase I digestion analysis of H1-compacted nucleosome arrays incubated with no TFs (contrl., lanes 2–5), WT PU.1 (lanes 6–9), DBD (lanes 10–13), and ΔIDR deletion (lanes 14–17) with or without the addition of cBAF and ATP.
Acknowledgments
Funding: M.A.F. was supported by NIH training grant T32 DK07780 and NIH fellowship F31 DK123886. K.E.W. was supported by the National Science Foundation NSF Graduate Research Fellowship Program award and the Harvard Medical School Landry Cancer Biology Award. The authors are grateful to N. Mashtalir (Kadoch Lab) for guidance and technical support with respect to purification of cBAF complexes. The research was supported by NIH GM36477 to K.S.Z.
Footnotes
Competing interests: C.K. is the Scientific Founder, fiduciary Board of Directors member, Scientific Advisory Board member, shareholder, and consultant for Foghorn Therapeutics, Inc. C.K. also serves on the scientific advisory boards of Nereid Therapeutics, Nested Therapeutics, and is a consultant for Cell Signaling Technologies. K.E.W. is an employee and shareholder of Flare Therapeutics. The remaining authors declare no competing interest.
Code availability: All code for data analysis and visualization is available from the corresponding author on request.
Data and materials availability:
All cloned reagents described are available from the authors. Raw nanopore FAST5, fastq, and mapped bed files will be deposited to in the NCBI Trace Archive prior to publication of this manuscript and are available upon request.
References:
- 1.Iwafuchi-Doi M & Zaret KS Pioneer transcription factors in cell reprogramming. Genes and Development (2014) doi: 10.1101/gad.253443.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwarz PM & Hansen JC Formation and stability of higher order chromatin structures Contributions of the histone octamer. Journal of Biological Chemistry (1994). [PubMed] [Google Scholar]
- 3.Adams CC & Workman JL Binding of disparate transcriptional activators to nucleosomal DNA is inherently cooperative. Molecular and Cellular Biology (2015) doi: 10.1128/mcb.15.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mirny LA Nucleosome-mediated cooperativity between transcription factors. Proceedings of the National Academy of Sciences (2010) doi: 10.1073/pnas.0913805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bustin M, Catez F & Lim JH The dynamics of histone H1 function in chromatin. Molecular Cell (2005) doi: 10.1016/j.molcel.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 6.Hill DA & Imbalzano AN Human SWI/SNF nucleosome remodeling activity is partially inhibited by linker histone H1. Biochemistry (2000) doi: 10.1021/bi001330z. [DOI] [PubMed] [Google Scholar]
- 7.Horn PJ et al. Phosphorylation of linker histones regulates ATP-dependent chromatin remodeling enzymes. Nat Struct Mol Biol 9, 263–267 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Ramachandran A, Omar M, Cheslock P & Schnitzler GR Linker histone H1 modulates nucleosome remodeling by human SWI/SNF. J Biol Chem 278, 48590–48601 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Shimada M et al. Gene-Specific H1 Eviction through a Transcriptional Activator→p300→NAP1→H1 Pathway. Molecular Cell 74, 268–283.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sekiya T & Zaret KS Repression by Groucho/TLE/Grg Proteins: Genomic Site Recruitment Generates Compacted Chromatin In Vitro and Impairs Activator Binding In Vivo. Molecular Cell (2007) doi: 10.1016/j.molcel.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soufi A, Donahue G & Zaret KS Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 151, 994–1004 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu F et al. The interaction landscape between transcription factors and the nucleosome. Nature 562, 76–81 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernandez Garcia M et al. Structural Features of Transcription Factors Associating with Nucleosome Binding. Molecular Cell (2019) doi: 10.1016/j.molcel.2019.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mayran A et al. Pioneer and nonpioneer factor cooperation drives lineage specific chromatin opening. Nat Commun 10, 3807 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cernilogar FM et al. Pre-marked chromatin and transcription factor co-binding shape the pioneering activity of Foxa2. Nucleic Acids Research 47, 9069–9086 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cirillo LA et al. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Molecular Cell (2002) doi: 10.1016/S1097-2765(02)00459-8. [DOI] [PubMed] [Google Scholar]
- 17.Iwafuchi M et al. Gene network transitions in embryos depend upon interactions between a pioneer transcription factor and core histones. Nat Genet 52, 418–427 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Friman ET et al. Dynamic regulation of chromatin accessibility by pluripotency transcription factors across the cell cycle. eLife 8, e50087 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.King HW & Klose RJ The pioneer factor OCT4 requires the chromatin remodeller BRG1 to support gene regulatory element function in mouse embryonic stem cells. eLife 6, e22631 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boulay G et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 171, 163–178.e19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sandoval GJ et al. Binding of TMPRSS2-ERG to BAF Chromatin Remodeling Complexes Mediates Prostate Oncogenesis. Mol Cell 71, 554–566.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iurlaro M et al. Mammalian SWI/SNF continuously restores local accessibility to chromatin. Nature Genetics 1–9 (2021) doi: 10.1038/s41588-020-00768-w. [DOI] [PubMed] [Google Scholar]
- 23.Xiao L et al. Targeting SWI/SNF ATPases in enhancer-addicted prostate cancer. Nature 1–6 (2021) doi: 10.1038/s41586-021-04246-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heinz S et al. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Molecular Cell (2010) doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hosokawa H et al. Transcription Factor PU.1 Represses and Activates Gene Expression in Early T Cells by Redirecting Partner Transcription Factor Binding. Immunity 48, 1119–1134.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ungerbäck J et al. Pioneering, chromatin remodeling, and epigenetic constraint in early T-cell gene regulation by SPI1 (PU.1). Genome Research (2018) doi: 10.1101/gr.231423.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Minderjahn J et al. Mechanisms governing the pioneering and redistribution capabilities of the non-classical pioneer PU.1. Nat Commun 11, 402 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iwafuchi-Doi M et al. The Pioneer Transcription Factor FoxA Maintains an Accessible Nucleosome Configuration at Enhancers for Tissue-Specific Gene Activation. Molecular Cell (2016) doi: 10.1016/j.molcel.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts GA et al. Dissecting OCT4 defines the role of nucleosome binding in pluripotency. Nat Cell Biol 23, 834–845 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feng R et al. PU.1 and C/EBP alpha/beta convert fibroblasts into macrophage-like cells. Proceedings of the National Academy of Sciences (2008) doi: 10.1073/pnas.0711961105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drew HR & Travers AA DNA bending and its relation to nucleosome positioning. Journal of Molecular Biology 186, 773–790 (1985). [DOI] [PubMed] [Google Scholar]
- 32.Fyodorov DV, Zhou B-R, Skoultchi AI & Bai Y Emerging roles of linker histones in regulating chromatin structure and function. Nat Rev Mol Cell Biol 19, 192–206 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dodonova SO, Zhu F, Dienemann C, Taipale J & Cramer P Nucleosome-bound SOX2 and SOX11 structures elucidate pioneer factor function. Nature 580, 669–672 (2020). [DOI] [PubMed] [Google Scholar]
- 34.Cirillo LA et al. Binding of the winged-helix transcription factor HNF3 to a linker histone site on the nucleosome. EMBO Journal (1998) doi: 10.1093/emboj/17.1.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michael AK et al. Mechanisms of OCT4-SOX2 motif readout on nucleosomes. Science eabb0074 (2020) doi: 10.1126/science.abb0074. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y et al. A Prion-like Domain in Transcription Factor EBF1 Promotes Phase Separation and Enables B Cell Programming of Progenitor Chromatin. Immunity 53, 1151–1167.e6 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Mashtalir N et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 175, 1272–1288.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mashtalir N et al. A Structural Model of the Endogenous Human BAF Complex Informs Disease Mechanisms. Cell 183, 802–817.e24 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perkel JM & Atchison ML A Two-Step Mechanism for Recruitment of Pip by PU.1. The Journal of Immunology 160, 241–252 (1998). [PubMed] [Google Scholar]
- 40.Xhani S et al. Intrinsic disorder controls two functionally distinct dimers of the master transcription factor PU.1. Science Advances 6, eaay3178 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piovesan D et al. MobiDB: intrinsically disordered proteins in 2021. Nucleic Acids Research 49, D361–D367 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dosztányi Z, Csizmók V, Tompa P & Simon I The Pairwise Energy Content Estimated from Amino Acid Composition Discriminates between Folded and Intrinsically Unstructured Proteins. Journal of Molecular Biology 347, 827–839 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Tanaka Y et al. Expression and purification of recombinant human histones. Methods (2004) doi: 10.1016/j.ymeth.2003.10.024. [DOI] [PubMed] [Google Scholar]
- 44.R: The R Project for Statistical Computing. https://www.r-project.org/.
- 45.Wickham H et al. Welcome to the Tidyverse. Journal of Open Source Software (2019) doi: 10.21105/joss.01686. [DOI] [Google Scholar]
- 46.The Pairwise Energy Content Estimated from Amino Acid Composition Discriminates between Folded and Intrinsically Unstructured Proteins - ScienceDirect. https://www.sciencedirect.com/science/article/pii/S0022283605001294?via%3Dihub. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All cloned reagents described are available from the authors. Raw nanopore FAST5, fastq, and mapped bed files will be deposited to in the NCBI Trace Archive prior to publication of this manuscript and are available upon request.