

Abstract
The COVID-19 pandemic has given a strong impetus to the search for antivirals active on SARS-associated coronaviruses. Over these years, numerous vaccines have been developed and many of these are effective and clinically available. Similarly, small molecules and monoclonal antibodies have also been approved by the FDA and EMA for the treatment of SARS-CoV-2 infection in patients who could develop the severe form of COVID-19. Among the available therapeutic tools, the small molecule nirmatrelvir was approved in 2021. It is a drug capable of binding to the Mpro protease, an enzyme encoded by the viral genome and essential for viral intracellular replication. In this work, by virtual screening of a focused library of β-amido boronic acids, we have designed and synthesized a focused library of compounds. All of them were biophysically tested by microscale thermophoresis, attaining encouraging results. Moreover, they also displayed Mpro protease inhibitory activity, as demonstrated by performing enzymatic assays. We are confident that this study will pave the way for the design of new drugs potentially useful for the treatment of SARS-CoV-2 viral infection.
Keywords: drug design, MM-GBSA, boronic acids, multicomponent reactions, protease, SARS-CoV-2, Mpro
1. Introduction
The COVID-19 pandemic caused by the SARS-CoV-2 virus continues to have a significant impact on the lives and economies of countries worldwide. To combat this pandemic, numerous pharmaceutical companies and academia have developed vaccines. The first vaccines were released at the end of 2020 by Pfizer, Moderna, and Astra Zeneca. Although these vaccines elicit an immune response to SARS-CoV-2 infection, unfortunately, they are ineffective against new and unpredicted mutations of the spike protein, which is the protein target of the triggered immune response. For this reason, in parallel with active research on new and more efficient vaccines, cheap and orally available drugs directed at molecular targets vital for the replication of the virus need to be developed. Among numerous viral molecular targets, the main protease (Mpro), also called 3CLpro, is considered the most therapeutically relevant target, as it is highly conserved among β-coronaviruses and possesses very few structural similarities to human proteases [1,2]. Due to the important role that Mpro plays in the replication of human coronaviruses, in particular, that of SARS-CoV-2, finding a specific inhibitor of this enzyme that could be clinically useful is the focus of current research. During 2022, the FDA approved the association of ritonavir (an anti-HIV drug) and nirmatrelvir (Paxlovid®, Pfizer Inc., New York, NY, USA) for the treatment of severe COVID-19, highlighting the importance of including Mpro inhibitors in the therapeutic armamentarium against SARS-CoV-2 (Figure 1).
Figure 1.

Chemical structure of Ritonavir (left) and Nirmatrelvir (right), both composing Paxlovid®, clinically employed for the treatment of COVID-19.
SARS-CoV-2 Mpro (MproCov-2) is a homodimeric chymotrypsin-like cysteine protease, with a non-canonical catalytic dyad composed of Cys145 and His41, which forms hydrogen bonds with the water molecule needed to hydrolyze the amide bond of the substrates. MproCov-2, together with SARS-CoV-2 papain-like protease (PLpro), contributes to viral polyprotein processing, acting in the early stages of viral replication inside infected cells. The inhibition of one or both of these enzymes constitutes a valuable therapeutic strategy to prevent SARS-CoV-2 proliferation in infected cells. Regarding the mechanism of action of nirmatrelvir, it covalently binds Mpro-Cys145, which is responsible for the catalytic cleavage of polypeptide amide bonds, by means of a nitrile warhead. In this way, nirmatrelvir blocks the production of functional viral proteins needed to build structural proteins, such as those that form the viral capsid.
Numerous compounds bearing different warheads that show non-covalent or covalent activity against Mpro have been reported in the literature [3,4,5,6,7]. Among these, research suggests that boron-containing compounds (BCCs) may have potential against SARS-CoV-2 infection [8], similar to that of boronic acid derivatives reported by Bacha et al., which are active against human β-coronaviruses other than SARS-CoV [9]. BCCs have attracted increasing attention in recent years, with five BCCs now used as therapeutic agents, three of which having been approved in the last 6 years [10]. In terms of the clinical applications of BCCs, tavaborole (Kerdyn®, Pfizer Inc., New York, NY, USA) is used as a treatment for fungal infections, and bortezomib (Velcade®, Takeda Pharmaceuticals, Lexington, MA, USA) is used as an anti-cancer agent. Another BCC, vaborbactam (Vabomere®, Melinta Therapeutics, Parsippany, NJ, USA), in association with meropenem, inhibits β-lactamases produced by antibiotic-resistant bacterial infections [11]. More recently, the boron neutron capture therapy, by the irradiation of nonradioactive boron-10, is an emerging treatment modality for tumors [12].
Based on the considerations of the importance of BCCs as covalent inhibitors and of MproCoV-2 as a therapeutic target for COVID-19 treatment, we performed a computational study, including covalent docking, molecular dynamics (MD) simulations, and Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) calculations, on a virtual library of compounds, all endowed with a boronic acid warhead. This study utilized a library of easily synthesizable β-amido boronic acids. Their chemical synthesis relies on multi-component methodologies, and they have advantages in terms of process speed, ease of work up, and use of commercial substrates compared to multi-step routes. By varying and modulating the chemical structure of the individual components, multi-component reactions allow access to peptidomimetic skeletons characterized by high levels of structural diversity. In this study, we synthesized a number of computationally optimized derivatives and then determined the affinities of these derivatives for recombinant MproCov-2 through biophysical experiments. We then evaluated the capability of the most promising compounds to inhibit the catalytic activity of MproCov-2.
2. Results and Discussion
2.1. Computational Design of New β-Amino Boronic Acids
Bacha et al. previously reported that bifunctional phenyl boronic acid compounds could bind to a cluster of serine residues (Ser139, Ser144, and Ser147) on MproCoV-2, the active sites of which are largely conserved among β-coronaviruses [9]. With the aim of designing new inhibitors targeting MproCov-2, we structurally aligned the X-ray structures of both Mpro (PDB 1P9U [13] for MproCoV and 7LKS [14] for MproCoV-2) and found that the majority of residues shaping the catalytic sites of both enzymes were largely conserved. Intriguingly, MproCoV-2 contains a peculiar Thr-rich cluster (Thr24-Thr25-Thr26) in place of the Ser-rich cluster found in MproCoV. Furthermore, among these three threonine residues, Thr25 projected its side chain in the catalytic site of the enzyme, and the Thr25 Cα atom was only 10.7 Å far from the one of Cys145, the residue targeted by most known Cys protease inhibitors, including nirmatrelvir (Figure 2).
Figure 2.

Three-dimensional depiction of the MproCoV (A) and MproCoV-2 (B) catalytic sites. The enzyme surfaces are represented as a “solvent accessible surface”, in which the partial charges of the residue atoms are colored in blue or in red, accordingly with their positive or negative charge. Residues are numbered as in the X-ray structures.
Accordingly, as proposed by Bacha et al., we used the Thr-rich cluster of MproCoV-2 as a target for the computational design of new boronic acids [9], creating a virtual library composed of β-amido boronic acids capable of covalently binding the side chain of Thr25. Relying on the Ugi reaction reported in Scheme 1, chemical diversity was introduced through 49 carboxylic acids, bearing different R1 groups, which were virtually combined with three isonitriles (benzyl, cyclohexyl, and t-butyl as R2, Table 1). Among the selected carboxylic acids, 29 were chosen among those already available in our organic synthesis laboratory, and the remaining 20 were natural amino acids. We then used the ‘enumeration’ tool of Maestro software (Schrödinger Inc., New York, NY, USA) to create a virtual library of 294 compounds containing both enantiomers of each compound. After performing covalent docking calculations, the three top-scoring binding poses of each ligand were simulated in complex with MproCoV-2 in 250 ns long MD simulations. The ligands displaying the lowest non-hydrogen atoms root mean square deviation (RMSD) over the simulation time were chosen for calculating ligand binding free energy (ΔG*) values, adopting the MM-GBSA single trajectory approach (see the Experimental section for details) [15,16,17]. The results obtained suggested that the compounds with the lowest ΔG* values (Table 1) possessed an R configuration at the chiral center and contained a 2-aminocarbonylethyl (3a, 3b, 3c) or 3-aminocarbonylpropyl (3f, 3ea, 3eb, 3g) chain as an R1 substituent. Among the compounds derived from the natural amino acids, only the compound retaining the L-tyrosine side chain (3h) appeared to be promising as potent MproCoV-2 inhibitors.
Scheme 1.

Synthesis of target β-amido boronic acids 3, overall two-steps yield in brackets.
Table 1.
Chemical structure and predicted binding free energy values (ΔG*) of the most promising β-amido boronic acids.
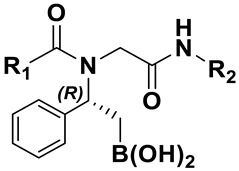
| |||
|---|---|---|---|
| Compound | R1 | R2 | ΔG* (Kcal/mol) |
| 3a | 2-aminocarbonyl-ethyl | -Cy | −64.9 ± 6.2 |
| 3b | 2-aminocarbonylethyl | -tBu | −42.7 ± 5.7 |
| 3c | 2-aminocarbonylethyl | -Bn | −70.5 ± 6.4 |
| 3d | phenyl | -Cy | −48.8 ± 8.0 |
| 3ea | 3-aminocarbonylpropyl | -Bn | −68.1 ± 7.4 |
| 3eb | 3-aminocarbonylpropyl | -Bn | −56.5 ± 8.1 |
| 3f | 3-aminocarbonylpropyl | -tBu | −66.0 ± 4.3 |
| 3g | 3-aminocarbonylpropyl | -Cy | −69.5 ± 7.7 |
| 3h | 4-[(2S)-2-aminopropyl]phenol | -Bn | −58.4 ± 5.0 |
As the most promising compounds contained 2-aminocarbonylethyl and 3-aminocarbonylpropyl as R1, we speculated that the number of methylene groups could be critical for the MproCoV-2 inhibitory activity. To establish whether this was the case, a compound derived using 3-amino-3-oxopropanoic acid as reactant, bearing only a methylene group between the amino boronic core and the primary amide group was designed. The ΔG* value of the resulting compound, simulated in complex with MproCoV-2, was higher than that of its homologs. Consequently, we can argue that effective (at least theoretically) inhibitors of MproCoV-2 depend critically on the full occupation of the MproCoV-2 pocket shaped by Leu141, Asn142, Gly143, Glu166, and His163 (Figure 3).
Figure 3.

Supposed binding mode of 3ea (magenta sticks) in complex with MproCoV-2. The enzyme surface is represented as a “solvent accessible surface”, in which the partial charges of the residue atoms are colored in blue or in red, accordingly with their positive or negative charge. The hydrogen bonds are represented as dashed yellow lines. Residues are numbered as was found in the X-ray structure.
2.2. Chemistry
Based on the computational studies, the β-amido boronic acids reported in Table 1 were synthesized. Relying on our previous experience [18,19], the Ugi multi-component reaction (Ugi-4CR) was chosen as the key step for the synthesis of boron-enriched peptidomimetics. Starting from enantiopure β-amino boronic acid hydrochloride 1 and formaldehyde (fixed amino and carbonyl components, respectively), a series of selected acids and isocyanides were employed in the reaction. All compounds were easily obtained as pinacol esters 2, which were purified by direct flash chromatography and then subjected to mild boron deprotection using methylboronic acid, resulting in target-free boronic acids 3 (Scheme 1).
The synthesis protocol proved to be suitable with 5-amino-5-oxopentanoic acid, 4-amino-4-oxobutanoic acid, and benzoic acid as acid components, in combination with commercial isocyanides (3a–3g). Boc-l-tyrosine was suitable for the reaction, giving the desired product 3h, as a hydrochloride salt, with a good yield. Finally, as a control experiment in the biological evaluation, the S-enantiomer of compound 3ea, namely, 3eb, was also prepared.
All the obtained products were stable and were fully characterized by 1H, 11B, and 13C NMR and by high-resolution mass spectrometry. In most cases, the 1H and 13C spectra showed more rotamers, as expected in the presence of tertiary amide bonds in peptides and peptidomimetic compounds [20]. As such isomerism could not be solved by varying the polarity of deuterated solvents or running the experiments at higher temperature, the rotamers’ ratio was safely quantified by integration of the 1H NMR peaks assigned to each rotamer. As equilibrium between monomeric and trimeric forms of free boronic acids 3 is observed in deuterated solvents [21], one drop of water was added to the NMR solvent, in order to suppress intermolecular interactions (see Supplementary Materials).
2.3. Biophysical Assays by Microscale Thermophoresis (MST)
MST is a biophysical technique enabling the characterization and quantification of molecular interactions of two partners in liquid phase, without any sample immobilization. MST experiments aim at measuring ligand-dependent changes in the temperature-related intensity change (TRIC-Fnorm) as a function of the ligand concentration in a dose–response curve [22,23,24,25]. In the experiments conducted in this research, we used His-tagged recombinant MproCoV-2 provided by GeneTex. To determine the minimum concentration of ligand capable of covalently binding MproCoV-2, we used a standard protein labeling protocol, according to the manufacturer’s instructions (NanoTemper Technologies GmbH, München, Germany), using a constant concentration of fluorescent target molecule and fixed concentrations of ligands (Table 2).
Table 2.
Results of the MST binding check assays.
| Compound | Ligand Concentration Displaying: | |
|---|---|---|
| Binding | No Binding | |
| 3a | 500 nM | 250 nM |
| 3b | 5 µM | 1 µM |
| 3c | 4 µM | 1 µM |
| 3d | 20 µM | 5 µM |
| 3ea | 500 nM | 250 nM |
| 3eb | 3.1 µM | 1.2 µM |
| 3f | 1 µM | 500 nM |
| 3g | 5 µM | 1 µM |
| 3h | 100 µM | 20 µM |
| 3d-pin | / | 100 µM |
| 3f-pin | 20 µM | 4 µM |
Except for 3h, the obtained results suggested that all the synthesized compounds displayed significant affinity for the target, confirming the theoretical studies previously reported in the previous section. Moreover, as computationally predicted, the S enantiomer of compound 3ea (named 3eb) was less active than the R enantiomer (3ea). This β-amido boronic acid, together with 3a, displayed the highest affinity for MproCoV-2, as binding was detected at concentrations between 250 and 500 nM (Figure 4). The MST binding check plots of all the compounds are shown in Figure S1 (Supplementary Materials).
Figure 4.

MST binding check experiments of (A) 3ea and (B) 3a tested at the concentration of 500 and 250 nM, respectively. In red is highlighted the “no binding” Fnorm area due to an insufficient Response Amplitude (RA).
To investigate the role played by the boronic acid warhead in the creation of the complex with the target, MST experiments on β-amido boronic compounds protected by the pinacol group (3d-pin and 3f-pin) were also conducted (Table 2). Interestingly, although binding of 3d-pin was not observed at a concentration of 100 µM, binding of free boronic acid (3d) was observed at concentrations ranging between 20 and 5 µM. A comparison of the binding check experiments of both 3d and 3d-pin at a concentration of 100 µM is shown in Figure S2A (Supplementary Materials). Similarly, the binding affinity of the pinacol analogue 3f-pin was decreased compared to that of 3f (Table 2 and Figure S2B,C, Supplementary Materials). These data suggest that the hydroxyl groups of the boronic warhead can induce critical bonds with the Mpro binding pocket. In fact, increasing the size of the warhead through the pinacol-protecting group prevented the entry of the ligand into the catalytic site. The low affinity displayed by pinacol ester could also be due to the impossibility of establishing a covalent bond with the target.
To verify that the β-amido boronic acid compounds bind only to the MproCoV-2 protein, and that they do not interfere with His-labeling or the dye, we conducted a binding affinity assay, testing 3ea on the 6His peptide control provided by NanoTemper (NanoTemper Technologies GmbH, München, Germany), according to standard protocols suggested by MST developers (see Experimental Section). As shown in Figure S3 (Supplementary Materials), the MST traces do not display a binding curve, as was analyzed by “MO.Affinity Analysis” software provided by NanoTemper Technologies GmbH (München, Germany), and no significant fluorescence change is observed between the ligand samples in the capillaries, thus confirming the specific binding of compound 3ea (and consequently that of all the other analogues) to the MproCoV-2 protein.
2.4. Inhibition of MproCoV-2: Reversible or Irreversible
We used MST to determine whether the binding of the β-amido boronic acid compounds was reversible or irreversible. First, we pre-incubated the MproCoV-2/3ea complex with a ligand concentration (Figure S4A, Supplementary Materials) that was previously determined high enough to bind the target in solution (100% bound, stock). We then measured the Fnorm values of this and that of other solutions obtained progressively by diluting a MproCoV-2/3ea complex solution (stock). We then compared the Fnorm of all the dilutions to that of the stock dilution. If the ligand was reversible, diluting the stock solution would produce a change in the Fnorm. Conversely, if the ligand was irreversible, no Fnorm changes would be noted in the diluted solutions, as the ligand would be firmly bound to the target. In our experiments (Figure S4B, Supplementary Materials), the Fnorm values did not change with the dilution of the stock complex solution, indicating that the ligand, once bound to the target, was unable to rapidly return in the solvent. This points to ligand binding being irreversible. In addition, preliminary LC-MS/MS experiments showed that at least 6% of the ligand present in solution could bind in an irreversible manner to MproCoV-2 (see Experimental section for details).
2.5. In Vitro Enzymatic Activity against Recombinant SARS-CoV-2 Proteases
We evaluated the ability of boronic acid derivatives 3a and 3ea to inhibit in vitro the activity of recombinant MproCoV-2 in a FRET-based assay (Table 3). First, we incubated the compounds with the MproCoV-2 enzyme for 10 min at 25 °C, followed by the addition of fluorogenic substrate. All the boronic acids partially reduced enzyme activity, with inhibition of ≅23% at a concentration of 20 μM. In contrast, evaluation of the enzymatic activity of 3a and 3ae against recombinant SARS-CoV-2 PLpro revealed no inhibition, disclosing a noteworthy selectivity of the β-amido boronic acid compounds for MproCoV-2 with respect to PLpro. The selectivity of MproCoV-2 inhibitors against PLpro represents an interesting feature, avoiding a specific interaction with human DUBs (Deubiquitinases), which share similarity sequences at the C-terminal with viral PLpro [26]. Further extension of the incubation time to 30 min did not lead to an increase in percentage inhibition. Due to the low activity observed in the enzymatic inhibition assays, no further assays were conducted.
Table 3.
Evaluation of the inhibitory activity of 3a and 3ae against SARS-CoV-2 Proteases.
| Compound | Structure | SARS-CoV-2 Mpro | SARS-CoV-2 PLpro |
|---|---|---|---|
| 3a |
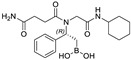
|
23.39% | n.i. |
| 3ea |
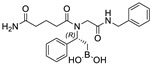
|
23.70% | n.i. |
| Nirmaltrevir |
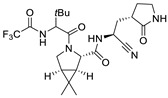
|
0.003 ± 0.0004 μM a | - |
Notes: % of inhibition of the enzymatic activity at 20 μM; a IC50 was determined for Nirmaltrevir; IC50 value includes standard deviation from two independent experiments, each performed in triplicate; n.i. = no inhibition.
The results obtained by enzymatic assays, considered in conjunction with the results of the experiments using MST, in which some compounds displayed binding at a concentration of 500 nM, suggest that the studied compounds could bind to MproCoV-2 on a site different from the catalytic one. Varying the substrate concentration resulted in no significant changes in percentage inhibition, suggesting a non-competitive mode of target binding. Consequently, the existence and involvement of allosteric or multiple sites in the reaction with the studied compounds cannot be excluded. It is possible that due to its high reactivity, the boronic warhead could be attached to nucleophilic residues different from those in the Thr cluster. Alternatively, the ligands may not fully occupy the catalytic site of the enzyme, partially allowing the physiological activity of the MproCoV-2.
3. Experimental Section
3.1. Simulating System Setup and MD Simulations
The “protein preparation wizard” was used to build the MproCoV-2 computational model to be used as target, retrieving the coordinates from the Protein Data Bank (accession code 7LKS [14]). The virtual library of the amino boronic acids series was created by means of the “Reaction Based Enumeration” tool, implemented in Maestro (Schrödinger Inc., New York, NY, USA, release 2021-2). Then, the “ligprep” tool was used to generate the virtual library of compounds, containing both enantiomers of each compound, and to assign the OPLS4 force field. Docking calculations were accomplished by the GLIDE algorithm [27], supposing that a covalent bond was created between the side chain of Thr25 and the boronic warhead of the ligands composing the library. Subsequently, the ligand poses acquiring the lowest Gscore were simulated in complex with MproCoV-2 by 250 ns-long MD, evaluating also the RMSD/time plot (Figure S5, Supplementary Materials) to establish if the ligands remained anchored in the catalytic site of the enzyme. Finally, the ligands acquiring the most promising Gscore, and displaying the highest stability in the catalytic site, were analyzed in order to predict their ∆G values, by means of the PRIME tool of Maestro (Schrödinger Inc., New York, NY, USA). To this purpose, by a script developed by Schrodinger, the Thr25 was mutated into a Gly residue, leaving the ligand free in the MproCoV-2 catalytic site. Finally, the ligands binding free energy values (ΔG) were computed as the mean of the values acquired by the ligands in the trajectory frames in which they displayed the highest geometrical stability, as indicated by the RMSD/time plot. In these calculations, the single trajectory approach was applied, and the entropy contributions to the binding free energy, coming from the normal mode analysis, were neglected, due to the computational costs of the calculations and the inaccuracy in the estimation. For this reason, the calculated ΔG values are termed ΔG* throughout the text.
3.2. Chemistry, General Information
All hydrochlorides 1 were prepared according to the method reported in the literature [28]. All employed reagents, including aldehydes, carboxylic acids, and isocyanides, are commercially available or were synthesized according to the literature procedures. Solvents were purchased as “anhydrous” and used without further purification. 1H NMR, 13C NMR, and 11B NMR spectra were recorded using a Bruker AV 400 Ultrashield spectrometer. 1H NMR and 13C NMR chemical shifts were reported in parts per million (ppm) downfield from tetramethylsilane,11B NMR chemical shifts were determined relative to BF3·Et2O, and spectra were recorded using quartz NMR tubes. Coupling constants (J) were reported in Hertz (Hz). The residual solvent peaks were used as internal references: 1H NMR (CDCl3 7.26 ppm, CD3CN 1.94 ppm) and 13C NMR (CDCl3 77.0 ppm, CD3CN 1.32 and 118.26 ppm). The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, m = multiplet, br = broad, app = apparent. Reactions involving boron-containing compounds were followed by TLC using a curcumin solution, which was prepared as reported in the literature [29]. Chromatographic purifications were performed by Flash Chromatography (FC), using Merck Silica gel 60.
3.3. General Procedure (A) for the Synthesis of β-Amido Boronic Esters 2
In a flame-dried round-bottom flask, (R)-phenyl-β-amino boronic hydrochloride 1 (0.345 mmol, 1 eq) was suspended in dry dichloromethane (0.70 mL, 0.5M), then freshly distilled triethylamine (48 μL, 0.345 mmol, 1 eq) was added dropwise and the reaction stirred at room temperature for 5 min. Paraformaldehyde (0.345 mmol, 1 eq), carboxylic acid (0.380 mmol, 1.1 eq), and isocyanide (0.449 mmol, 1.3 eq) were added sequentially and the reaction was stirred at room temperature for 72 h (reaction changes from turbid pale-yellow to clear dark-yellow). The solvent was removed under reduced pressure and the crude product was purified by FC, to afford pure β-amido boronic esters 2.
3.3.1. (R)-N1-(2-(Cyclohexylamino)-2-oxoethyl)-N1-(1-phenyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethyl)succinamide (2a)
Synthesized according to the General Procedure (A), using succinamic acid and cyclohexyl isocyanide. Purified by FC (dichloromethane/methanol 96:4) to afford compound 2a as a pale-yellow foam (yield 35%). [α]20D = +63.5 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3, 55:45 rotameric mixture) δ 7.37–7.25 (m, 5H), 6.31 (br d, 1.45H), 6.16 (app t, 0.45H), 6.04 (br d, 0.45H), 5.62 (br d, 0.55H), 5.52 (br s, 0.55H), 5.45 (app t, 0.55H), 3.80–3.65 (m, 2H), 3.60–3.49 (m, 1H), 3.07–2.96 (m, 1H), 2.80–2.58 (m, 3H), 2.52–2.43 (m, 1H), 1.76–1.27 (m, 10H), 1.15 and 1.13 (s, 6H), 1.09 and 1.05 (s, 6H), 0.96–0.88 (m, 1H); 13C NMR (101 MHz, CDCl3, 55:45 rotameric mixture) δ 175.5 and 175.3 (Cq), 173.6 and 173.5 (Cq), 168.6 and 168.3 (Cq), 140.9 and 140.30 (Cq), 129.4, 129.3, 128.7, 128.6, 128.2, 84.3 and 84.1 (2 Cq), 57.7 and 53.8, 49.2 and 48.7, 47.9 and 47.0, 33.4 and 33.2 (2C), 33.0 and 32.8, 31.3 and 31.1, 29.6 and 29.1, 26.2 and 26.0, 25.8 and 25.7, 25.5 and 25.4 (2C), 25.3 and 25.2 (2C), 16.2 and 14.4 (CH2-B); 11B NMR (128 MHz, CDCl3) δ 33.6; HRMS (ESI) calcd for C26H40BN3NaO5+ [M + Na]+ 508.2959, found 508.2966.
3.3.2. (R)-N1-(2-(Tert-butylamino)-2-oxoethyl)-N1-(1-phenyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethyl)succinamide (2b)
Synthesized according to the General Procedure (A), using succinamic acid and tert-butyl isocyanide. Purified by FC (dichloromethane/methanol 96:4) to afford compound 2b as a pale-yellow foam (yield 29%). [α]20D = +40.3 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3, 50:50 rotameric mixture) δ 7.39–7.28 (m, 5H), 6.22–6.11 (m, 1.5H), 5.77 (br s, 0.5H), 5.58 (br s, 0.5H), 5.50–5.42 (m, 1.5H), 3.74–3.61 (m, 2H), 3.16–3.02 (m, 1H), 2.80–2.60 (m, 2H), 2.55–2.48 (m, 1H), 1.69 (dd, J2 = 15.5 Hz, J3 = 8.9 Hz, 1H), 1.55–1.53 (m, 1H), 1.26 (s, 3H), 1.17 (s, 6H), 1.12 (s, 6H), 1.11 (s, 3H), 1.05 (s, 3H); 13C NMR (101 MHz, CDCl3, 50:50 rotameric mixture) δ 175.3 and 175.0 (Cq), 173.5 and 173.2 (Cq), 168.8 and 168.2 (Cq), 141.1 and 140.4 (Cq), 129.5 (2C), 128.7, 128.6, 128.2, 84.3 and 84.0 (2 Cq), 57.7 and 53.7, 51.9 and 51.4 (Cq), 48.2 and 47.9, 31.3 and 31.1, 29.9 and 29.4, 25.5, 25.4 (2C), 25.3 (2C), 25.22, 25.16, 16.2 and 14.5 (CH2-B); 11B NMR (128 MHz, CDCl3) δ 34.4; HRMS (ESI) calcd for C24H38BN3NaO5+ [M + Na]+ 482.2802, found 482.2799.
3.3.3. (R)-N1-(2-(Benzylamino)-2-oxoethyl)-N1-(1-phenyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethyl)succinamide (2c)
Synthesized according to the General Procedure (A), using succinamic acid and benzyl isocyanide. Purified by FC (dichloromethane/methanol 96:4) to afford compound 2c as a pale-yellow foam (yield 31%). [α]20D = +54.2 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3, 65:35 rotameric mixture) δ 7.41–7.17 (m, 10H), 6.72 (br t, 0.65H), 6.13 (app t, 0.35H), 5.96–5.90 (m, 1.35H), 5.45 (app t, 0.65H), 5.40 (br s, 0.35H), 5.11 (br s, 0.65H), 4.37–4.23 (m, 2H), 3.91 (d, J2 = 16.0 Hz, 0.65H), 3.80 (s, 0.70H), 3.72 (d, J2 = 16.0 Hz, 0.65H), 3.09–2.99 (m, 1.30H), 2.75–2.61 (m, 2H), 2.57–2.52 (m, 0.70H), 1.69 (dd, J2 = 15.6 Hz, J3 = 8.4 Hz, 1H), 1.55 (dd, J2 = 15.6 Hz, J3 = 8.4 Hz, 1H), 1.18 (s, 3.90H), 1.14 (s, 2.10H), 1.13 (s, 3.90H), 1.07 (s, 2.10); 13C NMR (101 MHz, CDCl3, 65:35 rotameric mixture) δ 174.8 and 174.5 (Cq), 173.1 and 172.9 (Cq), 169.1 and 168.9 (Cq), 139.6 (Cq), 138.5 and 138.1 (Cq), 127.9, 128.8, 128.6, 128.5, 128.4, 128.1, 127.8, 127.6, 127.4, 127.0, 83.7 and 83.6 (2 Cq), 57.3 and 53.5, 47.6 and 46.5, 43.3 and 43.1, 30.8 and 30.6, 29.0 and 28.6, 24.8 (2C), 24.5 (2C), 15.5 and 15.2 (CH2-B); 11B NMR (128 MHz, CDCl3) δ 33.3; HRMS (ESI) calcd for C27H36BN3NaO5+ [M + Na]+ 516.2646, found 516.2650.
3.3.4. (R)-N-(2-(Cyclohexylamino)-2-oxoethyl)-N-(1-phenyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethyl)benzamide (2d)
Synthesized according to the General Procedure (A), using benzoic acid and cyclohexyl isocyanide. Purified by FC (hexane/ethyl acetate 6:4) to afford compound 2d as a white foam (yield 55%). [α]20D = +56.5 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3, complex rotameric mixture: the section by section integration proves the overall number of protons) δ 7.65–7.11 (m, 10H), 6.36–5.30 (m, 1H), 4.61–3.15 (m, 4H), 1.98–1.88 (m, 2H), 1.69–1.38 (m, 10H), 1.15–1.09 (m, 12H); 13C NMR (101 MHz, CDCl3, complex rotameric mixture) δ 173.1 (1C), 170.1 and 168.7 (1C), 136.6 (1C), 134.1 (1C), 130.1–126.4 (10C), 84.0 (2C), 63.9 and 61.6 and 54.8 (1C), 63.2 and 59.0 and 58.5 (1C), 49.4–47.1 (1C), 33.2–33.0 (2C), 31.5 and 30.1 (1C), 25.9 (2C), 25.2–25.1 (4C), (CH2-B missing due to boron-quadrupole-induced relaxation); 11B NMR (128 MHz, CDCl3) δ 34.0; HRMS (ESI) calcd for C29H39BN2NaO4+ [M + Na]+ 513.2895, found 513.2891.
3.3.5. (R)-N1-(2-(Benzylamino)-2-oxoethyl)-N1-(1-phenyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethyl)glutaramide (2ea)
Synthesized according to the General Procedure (A), using 5-amino-5-oxopentanoic acid and benzyl isocyanide. Purified by FC (dichloromethane/methanol 96:4) to afford compound 2ea as a yellow foam (yield 37%). [α]20D = +40.1 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3, 60:40 rotameric mixture) δ 7.36–7.15 (m, 10H), 0.46 (br t, 0.60H), 6.40 (br s, 1H), 6.09 (app t, 0.40H), 5.97 (br s, 0.40H), 5.73 (br s, 0.40H), 5.34 (app t, 0.60H), 4.77–4.68 (m, 0.60H), 4.38–4.22 (m, 2H), 3.92 (d, J2 = 16.0 Hz, 0.60H), 3.73 (s, 0.80H), 3.65 (d, J2 = 16.0 Hz, 0.60H), 2.89–2.70 (m, 1H), 2.37–2.23 (m, 3H), 2.08–1.95 (m, 2H), 1.68 (dd, J2 = 15.7 Hz, J3 = 8.3 Hz, 1H), 1.52 (dd, J2 = 15.7 Hz, J3 = 8.3 Hz, 1H), 1.16–1.09 (m, 12H); 13C NMR (101 MHz, CDCl3, 60:40 rotameric mixture) δ 176.5 and 176.4 (Cq), 174.2 and 174.1 (Cq), 170.1 and 169.8 (Cq), 141.3 and 140.6 (Cq), 139.0 and 138.7 (Cq), 129.5, 129.4, 129.3, 129.2, 128.7, 128.6, 128.3, 128.2, 128.1, 127.8, 84.4 and 84.3 (2Cq), 57.7 and 54.0, 48.3 and 47.3, 43.9, 35.3 and 35.2, 33.5 and 32.7, 25.34, 25.30, 25.22 (2C), 21.6 and 21.5, 16.4 and 14.5 (CH2-B); 11B NMR (128 MHz, CDCl3) δ 33.2; HRMS (ESI) calcd for C28H38BN3NaO5+ [M + Na]+ 530.2802, found 530.2807.
3.3.6. (R)-N1-(2-(Tert-butylamino)-2-oxoethyl)-N1-(1-phenyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethyl)glutaramide (2f)
Synthesized according to the General Procedure (A), using 5-amino-5-oxopentanoic acid and tert-butyl isocyanide. Purified by FC (dichloromethane/methanol 96:4) to afford compound 2f as a pale-yellow foam (yield 29%). [α]20D = +76.7 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3, 55:45 rotameric mixture) δ 7.40–7.29 (m, 5H), 6.35 (br s, 0.45H), 6.23 (app t, 0.55H), 6.05 (br s, 0.45H), 5.83 (br s, 0.55H), 5.54–5.49 (m, 1H), 5.33 (app t, 0.45H), 5.19 (br s, 0.55H), 3.77 (d, J2 = 15.4 Hz, 0.55H), 3.71 (s, 0.90H), 3.56 (d, J2 = 15.4 Hz, 0.55H), 2.90–2.73 (m, 1H), 2.40–2.24 (m, 3H), 2.11–2.07 (m, 1H), 2.06–2.00 (m, 1H), 1.68 (dd, J2 = 15.8 Hz, J3 = 9.4 Hz, 1H), 1.49–1.44 (m, 1H), 1.21 (s, 4.70H), 1.18 (s, 3H), 1.15 (s, 3H), 1.12 (s, 3H), 1.07 (s, 7.30); 13C NMR (101 MHz, CDCl3, 55:45 rotameric mixture) δ 176.1 and 175.7 (Cq), 174.0 and 173.8 (Cq), 169.1 and 168.5 (Cq), 141.4 and 140.8 (Cq), 129.7, 129.4, 128.7, 128.6, 127.9, 84.3 and 84.1 (2Cq), 57.9 and 53.4, 51.8 and 51.5 (Cq), 48.2 and 48.0, 35.7 and 35.5, 33.4 and 32.6, 29.2 (2C), 28.9 (2C), 25.4, 25.32, 25.26, 21.9 and 21.6, 16.8 and 14.5 (CH2-B); 11B NMR (128 MHz, CDCl3) δ 33.0; HRMS (ESI) calcd for C25H40BN3NaO5+ [M + Na]+ 496.2959, found 496.2964.
3.3.7. (R)-N1-(2-(Cyclohexylamino)-2-oxoethyl)-N1-(1-phenyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethyl)glutaramide (2g)
Synthesized according to the General Procedure (A), using 5-amino-5-oxopentanoic acid and cyclohexyl isocyanide. Purified by FC (Dichloromethane/Methanol 96:4) to afford compound 2g as a pale-yellow foam (yield 27%). [α]20D = +51.0 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3, 65:35 rotameric mixture) δ 7.37–7.26 (m, 5H), 6.20–6.14 (m, 0.70H), 6.03–5.91 (m, 1H), 5.44 (app t, 0.65H), 5.38–5.32 (m, 0.65H), 3.79 (d, J2 = 15.8 Hz, 0.65H), 3.75 (s, 0.70H), 3.68 (d, J2 = 15.8 Hz, 0.65H), 3.61–3.49 (m, 1H), 3.06–3.03 (m, 1H), 2.80–2.44 (m, 3H), 1.79–1.72 (m, 2H), 1.71–1.24 (m, 12H), 1.16–1.14 (m, 6H), 1.10–1.07 (m, 6H), (1 exchangeable NH proton missing); 13C NMR (101 MHz, CDCl3, 65:35 rotameric mixture) δ 175.3 and 175.0 (Cq), 173.4 and 173.2 (Cq), 168.3 and 167.9 (Cq), 140.7 and 140.1 (Cq), 128.9, 128.7, 128.01 and 127.96, 127.8, 127.2, 83.7 and 83.6 (2Cq), 57.2 and 52.8, 48.2 and 48.0, 47.4 and 46.8, 35.1 and 34.8, 32.8 and 32.72, 32.68 and 32.5, 32.1 and 32.0, 25.5 and 25.4, 24.9 and 24.83, 24.75 (2C), 24.68 (2C), 24.6, 21.1 and 20.9, 15.9 and 13.7 (CH2-B); 11B NMR (128 MHz, CDCl3) δ 34.3; HRMS (ESI) calcd for C27H42BN3NaO5+ [M + Na]+ 522.3115, found 522.3111.
3.3.8. Tert-Butyl ((S)-1-((2-(Benzylamino)-2-oxoethyl)((R)-1-phenyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethyl)amino)-3-(4-hydroxyphenyl)-1-oxopropan-2-yl)carbamate (2h)
Synthesized according to the General Procedure (A), using (tert-butoxycarbonyl)-L-tyrosine, and benzyl isocyanide. Purified by FC (hexane/ethyl acetate 6:4) to afford compound 2h as a white foam (yield 37%). [α]20D = +42.9 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3, 50:50 rotameric mixture) δ 7.31–7.00 (m, 12H), 6.87 (br s, 1H), 6.75–6.67 (m, 2H), 6.03–5.99 (m, 0.50H), 5.49 (br. s, 0.50H), 5.14–5.13 (m, 1H), 5.02–4.97 (m, 0.50H), 4.38–4.28 (m, 1.50H), 4.12–4.10 (m, 1H), 3.76–3.74 (m, 2H), 3.03–2.96 (m, 1H), 2.85–2.75 (m, 1H), 1.74 (dd, J2 = 15.3 Hz, J3 = 9.9 Hz, 1H), 1.55 (dd, J2 = 15.3 Hz, J3 = 9.9 Hz, 1H), 1.39–1.37 (m, 9H), 1.16 (s, 3H), 1.13 (s, 3H), 1.09 (s, 3H), 1.04 (s, 3H), (1 exchangeable NH proton missing); 13C NMR (101 MHz, CDCl3, 50:50 rotameric mixture) δ 173.2 and 173.1 (Cq), 169.2 (Cq) 168.7 (Cq), 155.9 and 155.6 (Cq), 139.6 and 139.3 (Cq), 138.3 (Cq) 137.6 (Cq), 130.5, 130.3, 128.8–126.9 (11C), 116.8 and 116.7, 83.7 and 83.5 (2C), 80.3 and 80.2 (Cq), 57.9 and 52.4, 54.5 and 53.7, 47.1 and 46.7, 43.5 and 43.1, 37.8 and 37.6, 28.3 and 28.2 (3C), 24.7 (2C), 24.5 (2C), (CH2-B missing due to boron-quadrupole-induced relaxation); 11B NMR (128 MHz, CDCl3) δ 32.9; HRMS (ESI) calcd for C37H49BN3O7+ [M + H]+ 658.3658, found 658.3670.
3.3.9. (S)-N1-(2-(Benzylamino)-2-oxoethyl)-N1-(1-phenyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethyl)glutaramide (2eb)
Synthesized according to the General Procedure (A), with the exception of using (S)-phenyl-β-amino boronic hydrochloride 1, as well as paraformaldehyde, 5-amino-5-oxopentanoic acid, and benzyl isocyanide. Purified by FC (dichloromethane/methanol 96:4) to afford compound 2eb as a yellow foam (yield 36%). [α]20D = −39.8 (c 1.0, CHCl3); 1H, 13C, and 11B NMR data are identical to those of compound 2ea, previously reported. HRMS (ESI) calcd for C28H38BN3NaO5+ [M + Na]+ 530.2802, found 530.2799.
3.4. General Procedure (B) for the Synthesis of β-Amido Boronic Acids 3
The reaction was performed following a modified literature procedure [30]. In a round-bottom flask, the desired β-amido boronic ester 2 (0.20 mmol, 1 eq) and methylboronic acid (2.00 mmol, 10 eq) were dissolved in an acetone/0.2 N HClaq (1:1 v/v) solution (5 mL, 0.04 M) and stirred at room temperature for 4 h (the reaction changes from yellow to pale-yellow). The solvent was evaporated under reduced pressure using a 50 °C bath, then the reaction was diluted with 0.2 NHClaq (1.0 mL) and evaporated again under reduced pressure. The crude was then dissolved in MeCN/H2O 1:1 (4 mL) and freeze-dried to afford pure β-amido boronic acids 3.
3.4.1. (R)-(2-(4-Amino-N-(2-(cyclohexylamino)-2-oxoethyl)-4-oxobutanamido)-2-phenylethyl)boronic Acid (3a)
Synthesized according to the General Procedure (B) starting from compound 2a. Product 3a was obtained as a yellow powder (yield 99%). [α]20D = +33.4 (c 1.0, MeCN/H2O 1:1); 1H NMR (400 MHz, CD3CN + 1 drop of H2O, 60:40 rotameric mixture) δ 7.43–7.29 (m, 5H), 7.09–6.80 (m, 3H), 5.32 (dd, J3 = 9.7 Hz, J3 = 4.8 Hz 0.60H), 5.46 (dd, J3 = 9.7 Hz, J3 = 4.8 Hz 0.40H), 3.69–3.40 (m, 3H), 2.67–2.58 (m, 2H), 1.84–1.55 (m, 6H), 1.44–1.10 (m, 8H), (two exchangeable B(OH)2 protons missing); 13C NMR (101 MHz, CD3CN + 1 drop of H2O, 60:40 rotameric mixture) δ 176.4 and 175.1 (Cq), 174.3 and 173.8 (Cq), 170.2 and 169.3 (Cq), 141.7 and 141.6 (Cq), 129.3, 129.2, 128.6, 128.4 and 128.3, 128.1, 57.7 and 54.9, 49.3 and 49.1, 47.4 and 46.7, 33.0, 32.8, 32.7, 29.3 and 28.7, 25.9 and 25.4 (2C), 25.3, 18.5 and 18.0 (CH2-B); 11B NMR (128 MHz, CD3CN + 1 drop of H2O) δ 32.3 (-B(OH)2), 19.8 (-B(OH)2·H2O); HRMS (ESI) calcd for C22H34BNaN3O5+ [MB(OMe)2 + Na]+ 454.2489, found 454.2495.
3.4.2. (R)-(2-(4-Amino-N-(2-(tert-butylamino)-2-oxoethyl)-4-oxobutanamido)-2-phenylethyl)boronic Acid (3b)
Synthesized according to the General Procedure (B) starting from compound 2b. Product 3b was obtained as a yellow powder (yield >99%). [α]20D = +27.0 (c 1.0, MeCN/H2O 1:1); 1H NMR (400 MHz, CD3CN + 1 drop of H2O, 70:30 rotameric mixture) δ 7.42–7.28 (m, 5H), 7.02–6.59 (m, 2H), 5.57–5.54 (m, 0.70H), 5.46 (dd, J3 = 9.3 Hz, J3 = 5.6 Hz, 0.30H), 3.90–3.68 (m, 2H), 3.17–3.08 (m, 1H), 2.76–2.64 (m, 3H), 1.33–1.31 (m, 2.70H), 1.30–1.25 (m, 2H), 1.24–1.22 (m, 6.30H), (two exchangeable B(OH)2 and one NH protons missing); 13C NMR (101 MHz, CD3CN + 1 drop of H2O, 70:30 rotameric mixture) δ 178.1 and 177.8 (Cq), 176.5 and 175.1 (cq), 169.5 and 167.9 (Cq), 141.2 and 140.8 (Cq), 129.4 (2C), 128.7 (2C), 128.2, 58.8 and 58.2, 55.0 (Cq), 49.6, 47.1, 43.9, 28.5, 28.4 (2C), 19.5 and 18.5 (CH2-B); 11B NMR (128 MHz, CD3CN + 1 drop of H2O) δ 32.3 (-B(OH)2), 19.9 (-B(OH)2·H2O); HRMS (ESI) calcd for C20H32BNaN3O5+ [MB(OMe)2 + Na]+ 428.2333, found 428.2329.
3.4.3. (R)-(2-(4-Amino-N-(2-(benzylamino)-2-oxoethyl)-4-oxobutanamido)-2-phenylethyl)boronic Acid (3c)
Synthesized according to the General Procedure (B) starting from compound 2c. Product 3c was obtained as a dark-yellow powder (yield >99%). [α]20D = +26.8 (c 1.0, MeCN/H2O 1:1); 1H NMR (400 MHz, CD3CN + 1 drop of H2O, 55:45 rotameric mixture) δ 7.67–7.07 (m, 12H), 5.67–5.57 (m, 0.55H), 5.48 (app t, 0.45H), 4.44–4.24 (m, 2H), 4.03–3.74 (m, 2H), 3.17–2.93 (m, 1H), 2.84–2.53 (m, 3H), 1.53–1.21 (m, 2H), (two exchangeable B(OH)2 and one NH protons missing); 13C NMR (101 MHz, CD3CN + 1 drop of H2O, 55:45 rotameric mixture) δ 178.1 and 177.8 (Cq), 176.3 and 174.1 (Cq), 171.1 and 169.1 (Cq), 141.2 and 140.7 (Cq), 139.2 and 138.9 (Cq), 129.3, 129.2, 129.1, 128.8, 128.7, 128.5, 128.2, 128.0, 127.9, 127.7, 58.6 and 58.1, 49.1 and 46.7, 43.7 and 43.4, 30.7, 29.6 and 28.9, 19.5 and 18.7 (CH2-B); 11B NMR (128 MHz, CD3CN + 1 drop of H2O) δ 32.3 (-B(OH)2), 19.9 (-B(OH)2·H2O); HRMS (ESI) calcd for C23H30BNaN3O5+ [MB(OMe)2 + Na]+ 426.2176, found 426.2173.
3.4.4. (R)-(2-(N-(2-(Cyclohexylamino)-2-oxoethyl)benzamido)-2-phenylethyl)boronic Acid (3d)
Synthesized according to the General Procedure (B) starting from compound 2d. Product 3d was obtained as a white powder (yield 98%). [α]20D = +57.4 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3, complex rotameric mixture: the section by section integration proves the overall number of protons) δ 7.58–7.13 (m, 10H), 6.14–6.05 (m, 1H), 5.31–5.18 (m, 1H), 4.13–3.68 (m, 2H), 3.61–3.32 (m, 1H), 1.78–1.10 (m, 12H), (two exchangeable B(OH)2 protons missing); 13C NMR (101 MHz, CDCl3, complex rotameric mixture) δ 173.4 and 172.7 (Cq), 169.6 and 168.4 (Cq), 141.0 (Cq), 136.1 and 135.8 (Cq), 129.7, 128.7 (2C), 128.6 (2C), 127.7 (2C), 126.9, 126.7 (2C), 58.9 and 58.7, 48.9 and 48.8, 47.3, 32.63, 32.58, 25.4, 24.7, 24.6, 17.8 and 17.3 (CH2-B); 11B NMR (128 MHz, CDCl3) δ 32.5; HRMS (ESI) calcd for C25H33BNaN2O4+ [MB(OMe)2 + Na]+ 459.2431, found 459.2434.
3.4.5. (R)-(2-(5-Amino-N-(2-(benzylamino)-2-oxoethyl)-5-oxopentanamido)-2-phenylethyl)boronic Acid (3ea)
Synthesized according to the General Procedure (B) starting from compound 2ea. Product 3ea was obtained as a dark-yellow powder (yield >99%). [α]20D = +39.2 (c 1.0, MeCN/H2O 1:1); 1H NMR (400 MHz, CD3CN + 1 drop of H2O, 55:45 rotameric mixture) δ 8.19–6.79 (m, 12H), 5.84 (dd, J3 = 9.4 Hz, J3 = 7.0 Hz, 0.45H), 5.42 (dd, J3 = 9.4 Hz, J3 = 7.0 Hz, 0.55H), 4.40–4.36 (m, 1H), 4.31–4.26 (m, 1H), 3.85 (d, J2 = 18.1 Hz, 0.45H), 3.74 (d, J2 = 18.1 Hz, 0.45H), 3.72 (d, J2 = 16.3 Hz, 0.55H), 3.45 (d, J2 = 16.3 Hz, 0.55H), 2.77–2.66 (m, 1H), 2.38–2.21 (m, 2H), 1.96–1.83 (m, 2H), 1.49–1.39 (m, 1H), 1.34–1.29 (m, 2H), (two exchangeable B(OH)2 and one NH protons missing); 13C NMR (101 MHz, CD3CN + 1 drop of H2O, 55:45 rotameric mixture) δ 178.7 and 178.2 (Cq), 176.7 and 174.4 (Cq), 171.3 and 169.3 (Cq), 141.5 and 141.1 (Cq), 139.2 and 139.0 (Cq), 129.4 (2C), 129.2, 129.1, 128.7, 128.6 and 128.5, 128.3, 128.1, 128.0 and 127.9, 127.7, 58.3 and 57.9, 49.1 and 46.8, 43.7 and 43.4, 35.3 and 35.1, 33.4 and 32.6, 21.7 and 21.5, 19.5 and 18.5 (CH2-B); 11B NMR (128 MHz, CD3CN + 1 drop of H2O) δ 32.4 (-B(OH)2), 19.9 (-B(OH)2·H2O); HRMS (ESI) calcd for C24H32BNaN3O5+ [MB(OMe)2 + Na]+ 476.2333, found 476.2337.
3.4.6. (R)-(2-(5-Amino-N-(2-(tert-butylamino)-2-oxoethyl)-5-oxopentanamido)-2-phenylethyl)boronic Acid (3f)
Synthesized according to the General Procedure (B) starting from compound 2f. Product 3f was obtained as a pale-yellow powder (yield 99%). [α]20D = +51.8 (c 1.0, MeCN/H2O 1:1); 1H NMR (400 MHz, CD3CN + 1 drop of H2O, 65:35 rotameric mixture) δ 7.47–7.32 (m, 5H), 7.26–6.56 (m, 2H), 5.61 (app t, 0.65H), 5.41 (dd, J3 = 9.8 Hz, J3 = 5.6 Hz, 0.35H), 3.76 (br s, 1.30H), 3.66 (d, J2 = 15.8 Hz, 0.35H), 3.27 (d, J2 = 15.8 Hz, 0.35H), 2.80–2.71 (m, 1H), 2.51–2.31 (m, 4H), 1.96–1.89 (m, 1H), 1.46–1.40 (m, 1H), 1.36–1.29 (m, 3.15H), 1.25 (br s, 6.85H), (two exchangeable B(OH)2 and one NH protons missing); 13C NMR (101 MHz, CD3CN + 1 drop of H2O, 65:35 rotameric mixture) δ 178.6 and 178.3 (Cq), 176.6 and 174.3 (Cq), 170.5 and 168.3 (Cq), 141.6 and 141.3 (Cq), 129.4 (2C), 128.6, 128.5, 128.1, 58.1 and 57.8, 52.0 and 51.8 (Cq), 49.3 and 47.1, 34.8, 33.2 and 32.5, 28.5 and 28.4 (3C), 21.7 and 21.5, 19.5 and 18.2 (CH2-B); 11B NMR (128 MHz, CD3CN + 1 drop of H2O) δ 32.3 (-B(OH)2), 20.0 (-B(OH)2·H2O); HRMS (ESI) calcd for C21H34BNaN3O5+ [MB(OMe)2 + Na]+ 442.2489, found 442.2492.
3.4.7. (R)-(2-(5-Amino-N-(2-(cyclohexylamino)-2-oxoethyl)-5-oxopentanamido)-2-phenylethyl)boronic acid (3g)
Synthesized according to the General Procedure (B) starting from compound 2g. Product 3g was obtained as a fluoro-yellow powder (yield >99%). [α]20D = +27.3 (c 1.0, MeCN/H2O 1:1); 1H NMR (400 MHz, CD3CN + 1 drop of H2O, 60:40 rotameric mixture) δ 7.44–7.24 (m, 5H), 7.18–6.75 (m, 1H), 5. 53 (app, t, 0.60H), 5.42 (dd, J3 = 9.6 Hz, J3 = 6.4 Hz, 0.40H), 3.88–3.78 (m, 1.20H), 3.68 (d, J2 = 15.7 Hz, 0.40H), 3.61–3.52 (m, 1H), 3.36 (d, J2 = 15.7 Hz, 0.40H), 2.76 (t, J3 = 6.7 Hz, 0.60H), 2.52–2.35 (m, 3H), 1.96–1.91 (m, 1H), 1.85–1.60 (m, 5.40H), 1.45–1.40 (m, 1H), 1.37–1.11 (7H), (two exchangeable B(OH)2 and two NH protons missing); 13C NMR (101 MHz, CD3CN + 1 drop of H2O, 60:40 rotameric mixture) δ 179.1 and 178.7 (Cq), 176.8 and 174.5 (Cq), 170.3 and 168.1 (Cq), 141.5 and 141.1 (Cq), 129.4 and 129.2 (2C), 128.6 and 128.4 (2C), 128.1, 58.7 and 57.9, 49.4 and 49.3, 46.7 and 43.3, 34.9, 33.4 and 32.5, 32.8 (2C), 25.9, 25.3, 25.2, 21.7 and 21.5, 19.8 and 18.4; 11B NMR (128 MHz, CD3CN + 1 drop of H2O) δ 32.3 (-B(OH)2), 19.4 (-B(OH)2·H2O); HRMS (ESI) calcd for C23H36BNaN3O5+ [MB(OMe)2 + Na]+ 468.2646, found 468.2649.
3.4.8. (S)-1-((2-(Benzylamino)-2-oxoethyl)((R)-2-borono-1-phenylethyl)amino)-3-(4-hydroxyphenyl)-1-oxopropan-2-aminium Chloride (3h)
In a round-bottom flask, compound 2h (0.20 mmol, 1 eq) was dissolved in dry DCM (0.7 mL, 0.30 M), then TFA (0.3 mL, 1.76 mmol, 9 eq) was added dropwise and the reaction stirred at room temperature for 10 min. The solvent was removed under reduced pressure to afford the N-Boc-deprotected intermediate as a white powder (yield >99%). [α]20D = +25.1 (c 1.0, CHCl3); HRMS (ESI) calcd for C32H41BN3O5+ [M]+ 558.3134, found 558.3130. The obtained N-Boc-deprotected intermediate was treated according to the General Procedure (B), to obtain pure compound 3h as a dark-yellow powder (yield 99%). [α]20D = +44.7 (c 1.0, MeCN/H2O 1:1); 1H NMR (400 MHz, CD3CN + 1 drop of H2O, 60:40 rotameric mixture) δ 8.00 (br s, 3H), 7.51 (br dd, amide NH, 0.40H), 7.42 (br dd, amide NH, 0.60H), 7.35–7.22 (m, 8H), 7.12–7.10 (m, 2H), 7.00 and 6.96 (d, J3 = 7.4 Hz, 2H), 6.83 and 6.79 (d, J3 = 8.5 Hz, 2H), 5.99 (app t, 0.60H), 5.40 (app t, 0.40H), 5.00 (br s, 0.40H), 4.33–4.27 (m, 1.60H), 4.00 (br d, 1H), 3.71 (d, J2 = 17.8 Hz, 0.60H), 3.58 (hidden by the H2O signal, detected by 1H/13C HSQC, 0.80H), 3.44 (d, J2 = 17.8 Hz, 0.60H), 3.13–3.09 (m, 2H), 1.55 (d, J3 = 8.0 Hz, 0.80H), 1.47 (dd, J2 = 15.4 Hz, J3 = 9.7 Hz, 0.60H), 1.17 (dd, J2 = 15.4 Hz, J3 = 9.7 Hz, 0.60H), (two exchangeable B(OH)2 and one NH protons missing); 13C NMR (101 MHz, CD3CN + 1 drop of H2O, 60:40 rotameric mixture) δ 170.4 and 170.08 (Cq), 170.05 and 169.3 (Cq), 157.5 and 157.4 (Cq), 140.6 and 139.3 (Cq), 139.2 (Cq), 138.5 (Cq), 131.8, 131.6, 129.5, 129.4, 129.1 (2C), 128.9, 128.6 and 128.5, 128.2, 128.1, 127.9, 127.8, 125.5 and 125.3, 116.6 and 116.5, 58.7 and 55.5, 53.8 and 52.4, 47.3 and 45.9, 43.6 and 43.5, 36.9 and 36.7, 20.2 and 18.1 (CH2-B); 11B NMR (128 MHz, CD3CN + 1 drop of H2O) δ 32.4 (-B(OH)2), 19.9 (-B(OH)2·H2O); HRMS (ESI) calcd for C28H35BN3O5+ [MB(OMe)2]+ 504.2604, found 504.2609.
3.4.9. (S)-(2-(5-Amino-N-(2-(benzylamino)-2-oxoethyl)-5-oxopentanamido)-2-phenylethyl)boronic Acid (3eb)
Synthesized according to the General Procedure (B) starting from compound 2eb. Product 3eb was obtained as a dark-yellow powder (yield >99%). [α]20D = −40.1 (c 1.0, MeCN/H2O 1:1); 1H, 13C, and 11B NMR data are identical to those of compound 3ea, previously reported. HRMS (ESI) calcd for C24H32BNaN3O5+ [MB(OMe)2 + Na]+ 476.2333, found 476.2330.
3.5. MST Experiments
The ligand capacity to bind MproCoV-2 was measured by Monolith NT.115 instrument (NanoTemper Technologies GmbH, München, Germany). Briefly, histidine-tagged MproCoV-2 was labeled by using a non-covalent His-tag dye for 30 min at room temperature, using the His-Tag Labeling Kit RED-tris-NTA 2nd Generation (MO-L018), purchased from NanoTemper Technologies (GmbH, München, Germany). A fixed concentration of the labeled MproCoV-2 enzyme (50 nM) was mixed with different dilutions of the synthesized β-amido boronic compounds, and “binding check” experiments were conducted, using the “expert mode” of the Monolith software MO.Control v1.6 (München, Germany). At least two concentration points for each compound were performed for the Fnorm evaluation. The enzyme and the ligands were incubated for 60–90 min at room temperature. The MST measurements were accomplished using standard capillaries, using a medium MST power (40%), in order to create the temperature gradient, and an excitation power of 60% at the temperature of 25 °C. The ligand capability to bind the enzyme (Table 2) was calculated from compound concentration-dependent changes in normalized fluorescence (Fnorm). In all the experiments, both interacting species were dissolved in PBS-T buffer (phosphate-buffered saline + 0.05% Tween ™ 20) of NanoTemper Technologies (GmbH, München, Germany) and 2.5% DMSO. The auto-fluorescence of each ligand was assessed before proceeding to the evaluation of the Fnorm. In all the “binding check” assays performed, only the time of 1.5 s was considered for the evaluation of the Fnorm. Consequently, the Fnorm difference between the one observed for the ligand and the MproCoV-2 protein (namely, the Response Amplitude, RA), necessary to consider the ligand binding, must be greater than 1.
The capability of 3ea compound to specifically bind the MproCoV-2 protein was assessed through the “binding affinity” experiment on the 6His Peptide Control provided by NanoTemper Technologies (GmbH, München, Germany), applying the standard suggested protocol. Initially, the protein was labeled using the same protocol previously adopted, then a fixed concentration of the labeled MproCoV-2 enzyme (50 nM) was mixed with sixteen 1:1 serial dilutions of the 3ea compound (ranging from 100 to 3 nM). The protein and the molecule were incubated for at least 15 min at room temperature. The MST measurements were accomplished using a medium MST power, and an excitation power of 40% at the temperature of 25 °C, using standard capillaries. The full analysis report was generated using the Monolith software MO.Affinity Analysis v2.3 (München, Germany) (Figure S3, Supporting Information).
3.6. MST Experiments to Ascertain the Reversible or Irreversible Inhibition of Mpro
We conducted these experiments by pre-incubating the MproCov-2/3ea complex with a protein concentration of 400 nM, in order to obtain a sufficient fluorescence value for the subsequent dilution of the ligand/enzyme complex. At that target concentration, we verified the concentration of 3ea, in which no binding was observed, obtaining a ligand concentration of 2 µM (Figure S4A, Supplementary Materials). Then, we measured the Fnorm of the other solutions obtained progressively by diluting a MproCov-2/3ea complex solution to 6.25, 3.125, and 1.56 µM.
3.7. LC-MS/MS Experiment to Verify the Ligand Binding of Mpro
The 3ea sample (75 µL, 20 µM) was incubated with purified Mpro (75 µL, 400 nM) for 16 h at 4 °C, in the dark. The sample was concentrated and filtered through Amicon Centrifugal Filter Devices with a 3 kDa cut-off (Merck Millipore, Milan, Italy) for 30 min at 3030× g. Then, the eluent solution (5 µL) was analyzed for protein-free ligands using LC-MS/MS. The supernatant solution (ligand bound to Mpro + free ligand, 20 µL) was deproteinized by ACN (20 µL) and centrifuged (10 min at 3030× g) to displace the binding between the protease and β-amido boronic acid. Then, an aliquot (5 µL) was injected in LC-MS/MS. UHPLC–MS/MS analyses were performed on a 1290 Infinity ultra-high-performance liquid chromatography system (Agilent Technologies, Palo Alto, CA, USA) coupled to a Q Trap 5500 linear ion trap triple quadrupole mass spectrometer (Sciex, Darmstadt, Germany) and equipped with an electrospray ionization (ESI) source. Chromatographic separation was achieved on a reversed-phase Zorbax SB-C18 column 3.5 μm, 2.1 × 150 mm (Agilent Technologies, Palo Alto, CA, USA) equipped with pre-column using as mobile phases (A) water + 0.2 mM ammonium acetate and (B) acetonitrile. The flow rate was 0.5 mL/min and the column temperature was set to 40 °C. The elution gradient (%B) was set as follows: 0–1 min (1%), 1–4 min (1–95%), 4–8 min (95%), 8–8.1 (95–1%), held until 2 min. Analyses were performed by multiple reaction monitoring (MRM) in negative mode according to the transitions corresponding to 3ea: m/z 424.6 > 92.3 (target ion, DP −37, CE −47 eV); m/z 424.6 > 208.0 (DP −37, CE −20 eV); m/z 424.6 > 268.0 (target ion, DP −37, CE −20 eV); m/z 424.6 > 326.0 (DP −37, CE −16 eV); m/z 210.8 > 152.8 (DP −37, CE −18 eV); m/z 210.8 > 95 (DP −37, CE −18 eV) (Figure S6, Supplementary Materials).
3.8. Enzymatic Assays
The inhibitory activity of the compounds was evaluated by a Förster resonance energy transfer (FRET)-based enzymatic cleavage assay on a TECAN Infinite F2000 PRO plate reader (Agilent Technologies, Santa Clara, CA, USA) using white flat-bottom 96-well microtiter plates (Greiner bio-one, Kremsmünster, Austria) [31]. Nirmatrelvir was purchased from AOBIUS (Gloucester, MA, USA) and used as positive control. Recombinant MproCoV-2 was expressed and purified as previously described [32], whereas the peptidic substrate Dabcyl-KTSAVLQ↓SGFRKME-Edans (TFA salt) was obtained by commercial source (Genescript, NJ, USA). The arrow indicates the cleavage position. The proteolytic activity of the MproCoV-2 was measured by monitoring the increasing fluorescence of SGFRKME-Edans upon hydrolytic shedding of the quencher Dabcyl-KTSAVLQ, at 25 °C with a 335 nm excitation filter and a 493 nm emission filter. Each well contained 200 µL composed of 185 µL reaction buffer (20 mM Tris pH 7.5, 0.1 mM EDTA, 1 mM DTT, and 200 mM NaCl), 5 µL MproCoV-2 in enzyme buffer at a final concentration of 50 nM together with 5 µL of the fluorogenic substrate (final concentration 25 μM) and 10 µL of the compounds present at a final concentration of 20 μM (screening assay). DMSO was used as a negative control. Inhibitors and substrate were dissolved and diluted in DMSO, leading to a final DMSO concentration of 7.5% (v/v). The compounds and enzyme were incubated for 10 min at 25 °C prior to substrate addition. Product release from substrate hydrolysis was monitored in 30 s increments over a period of 10 min. The related KM value was determined in a separate experiment (33 µM). IC50 value was determined as previously described by us [33].
4. Conclusions
In this study, to investigate whether SARS-CoV-2 Mpro Thr25 could be targeted by boron-containing compounds, we designed and synthesized eight compounds displaying affinity to the target in the low micromolar range, as suggested by MST experiments. Enzymatic assays suggested that the most promising of the eight compounds slightly inhibited the catalytic activity of the target, permitting us to suppose that an allosteric site of the enzyme is covalently targeted by the compounds. However, our results provide experimental evidence that BCCs represent new and promising MproCoV-2 inhibitors. We are confident that this research can pave the way toward the design of new boron-containing compounds with antiviral activity against SARS-CoV-2, potentially useful for the treatment of diseases caused by coronaviruses.
Acknowledgments
G.G. dedicates this paper to the memory of his grandmother A.E.d.S., a rare example of family dedication. We would like to thank INDACO for providing high-performance computing resources and support. G.G. gratefully acknowledges the support of NVIDIA Corporation for donating the Titan Xp GPU, which was utilized to perform part of the MD simulations. G.G. and EMAF thank the Vincenzo Pisapia and NanoTemper team for the suggestions and the kind help for the MST assay development. G.G. and A.S. thank Nicola Sormani for his contribution during the preparation of his master’s thesis.
Abbreviations
MD: Molecular dynamics; RMSD root mean square deviation; MM-GBSA, Molecular Mechanics-Generalized Born Surface Area.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/molecules28052356/s1, Figure S1, MST binding check experiment of β-amido boronic compounds reported in Table 2. Figure S2, MST binding check experiment of β-amido boronic compounds protected by the pinacol group. Figure S3, MST analysis reporting the binding affinity assay of 3ea on 6His Control Peptide. Figure S4, MST binding check experiments to determine whether the 3ea compound binds reversibly or irreversibly to the MproCov-2. Figure S5, RMSD/time plots of β-amido boronic compounds reported in Table 1. Figure S6, LC-MS/MS experiment on a β-amido boronic acid. Figure S7, HPLC chromatograms for compounds 3a and 3ea. Copies of 1H, 13C, and 11B NMR spectra.
Author Contributions
Conceptualization and methodology, G.G. and A.S.; formal analysis, all authors; investigation, E.M.A.F., M.M., A.C., M.D.C. and S.C.; resources, G.R., N.M., T.S. and A.S.; writing—original draft preparation, all authors; writing—review and editing, G.G. and A.S; supervision, G.G., T.S., N.M. and A.S. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Supporting data including NMR spectral charts are available from the Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This research received no external funding.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Citarella A., Scala A., Piperno A., Micale N. SARS-CoV-2 Mpro: A Potential Target for Peptidomimetics and Small-Molecule Inhibitors. Biomolecules. 2021;11:607. doi: 10.3390/biom11040607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 3.Amin S.A., Banerjee S., Ghosh K., Gayen S., Jha T. Protease targeted COVID-19 drug discovery and its challenges: Insight into viral main protease (Mpro) and papain-like protease (PLpro) inhibitors. Bioorg. Med. Chem. 2021;29:115860. doi: 10.1016/j.bmc.2020.115860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Song S., Gao P., Sun L., Kang D., Kongsted J., Poongavanam V., Zhan P., Liu X. Recent developments in the medicinal chemistry of single boron atom-containing compounds. Acta Pharm. Sin. B. 2021;11:3035–3059. doi: 10.1016/j.apsb.2021.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elmaaty A.A., Eldehna W.M., Khattab M., Kutkat O., Alnajjar R., El-Taweel A.N., Al-Rashood S.T., Abourehab M.A.S., Binjubair F.A., Saleh M.A., et al. Anticoagulants as Potential SARS-CoV-2 Mpro Inhibitors for COVID-19 Patients: In Vitro, Molecular Docking, Molecular Dynamics, DFT, and SAR Studies. Int. J. Mol. Sci. 2022;23:12235. doi: 10.3390/ijms232012235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elagawany M., Elmaaty A.A., Mostafa A., Abo Shama N.M., Santali E.Y., Elgendy B., Al-Karmalawy A.A. Ligand-based design, synthesis, computational insights, and in vitro studies of novel N-(5-Nitrothiazol-2-yl)-carboxamido derivatives as potent inhibitors of SARS-CoV-2 main protease. J. Enz. Inhib. Med. Chem. 2022;37:2112–2132. doi: 10.1080/14756366.2022.2105322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abo Elmaaty A., Hamed M.I.A., Ismail M.I., Elkaeed E.B., Abulkhair H.S., Khattab M., Al-Karmalawy A.A. Computational Insights on the Potential of Some NSAIDs for Treating COVID-19: Priority Set and Lead Optimization. Molecules. 2021;26:3772. doi: 10.3390/molecules26123772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ataseven H., Sayin K., Tüzün B., Gedikli M.A. Could boron compounds be effective against SARS-CoV-2? Bratisl. Med. J. 2021;122:753–758. doi: 10.4149/BLL_2021_121. [DOI] [PubMed] [Google Scholar]
- 9.Bacha U., Barrila J., Velazquez-Campoy A., Leavitt S.A., Freire E. Identification of Novel Inhibitors of the SARS Coronavirus Main Protease 3CLpro. Biochemistry. 2004;43:4906–4912. doi: 10.1021/bi0361766. [DOI] [PubMed] [Google Scholar]
- 10.Plescia J., Moitessier N. Design and discovery of boronic acid drugs. Eur. J. Med. Chem. 2020;195:112270. doi: 10.1016/j.ejmech.2020.112270. [DOI] [PubMed] [Google Scholar]
- 11.Sgrignani J., Novati B., Colombo G., Grazioso G. Covalent docking of selected boron-based serine beta-lactamase inhibitors. J. Comput.-Aided Mol. Des. 2015;29:441–450. doi: 10.1007/s10822-015-9834-7. [DOI] [PubMed] [Google Scholar]
- 12.Malouff T.D., Seneviratne D.S., Ebner D.K., Stross W.C., Waddle M.R., Trifiletti D.M., Krishnan S. Boron Neutron Capture Therapy: A Review of Clinical Applications. Front. Oncol. 2021;11:601820. doi: 10.3389/fonc.2021.601820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus Main Proteinase (3CLpro) Structure: Basis for Design of Anti-SARS Drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 14.Dampalla C.S., Kim Y., Bickmeier N., Rathnayake A.D., Nguyen H.N., Zheng J., Kashipathy M.M., Baird M.A., Battaile K.P., Lovell S., et al. Structure-Guided Design of Conformationally Constrained Cyclohexane Inhibitors of Severe Acute Respiratory Syndrome Coronavirus-2 3CL Protease. J. Med. Chem. 2021;64:10047–10058. doi: 10.1021/acs.jmedchem.1c00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller B.R., 3rd, McGee T.D., Jr., Swails J.M., Homeyer N., Gohlke H., Roitberg A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 16.Hou T., Wang J., Li Y., Wang W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Mod. 2011;51:69–82. doi: 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lammi C., Sgrignani J., Arnoldi A., Lesma G., Spatti C., Silvani A., Grazioso G. Computationally Driven Structure Optimization, Synthesis, and Biological Evaluation of Imidazole-Based Proprotein Convertase Subtilisin/Kexin 9 (PCSK9) Inhibitors. J. Med. Chem. 2019;62:6163–6174. doi: 10.1021/acs.jmedchem.9b00402. [DOI] [PubMed] [Google Scholar]
- 18.Manenti M., Gusmini S., Lo Presti L., Silvani A. Exploiting Enantiopure β-Amino Boronic Acids in Isocyanide-Based Multicomponent Reactions. Eur. J. Org. Chem. 2022;2022:e202200435. doi: 10.1002/ejoc.202200435. [DOI] [Google Scholar]
- 19.Manenti M., Gusmini S., Lo Presti L., Molteni G., Silvani A. Enantiopure β-isocyano-boronic esters: Synthesis and exploitation in isocyanide-based multicomponent reactions. Mol. Div. 2022:1–8. doi: 10.1007/s11030-022-10549-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laursen J.S., Engel-Andreasen J., Fristrup P., Harris P., Olsen C.A. Cis–Trans Amide Bond Rotamers in β-Peptoids and Peptoids: Evaluation of Stereoelectronic Effects in Backbone and Side Chains. J. Am. Chem. Soc. 2013;135:2835–2844. doi: 10.1021/ja312532x. [DOI] [PubMed] [Google Scholar]
- 21.Yuji T., Hiroki U., Youji S., Toshihiro S. Formation of Boroxine: Its Stability and Thermodynamic Parameters in Solution. Heterocycles. 2002;57:787–790. doi: 10.3987/COM-02-9464. [DOI] [Google Scholar]
- 22.Rainard J.M., Pandarakalam G.C., McElroy S.P. Using Microscale Thermophoresis to Characterize Hits from High-Throughput Screening: A European Lead Factory Perspective. SLAS Discov. 2018;23:225–241. doi: 10.1177/2472555217744728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jerabek-Willemsen M., Wienken C.J., Braun D., Baaske P., Duhr S. Molecular Interaction Studies Using Microscale Thermophoresis. Assay Drug Dev. Technol. 2011;9:342–353. doi: 10.1089/adt.2011.0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jerabek-Willemsen M., André T., Wanner R., Roth H.M., Duhr S., Baaske P., Breitsprecher D. MicroScale Thermophoresis: Interaction analysis and beyond. J. Mol. Struct. 2014;1077:101–113. doi: 10.1016/j.molstruc.2014.03.009. [DOI] [Google Scholar]
- 25.Fassi E.M.A., Sgrignani J., D’Agostino G., Cecchinato V., Garofalo M., Grazioso G., Uguccioni M., Cavalli A. Oxidation State Dependent Conformational Changes of HMGB1 Regulate the Formation of the CXCL12/HMGB1 Heterocomplex. Comput. Struct. Biotechnol. J. 2019;17:886–894. doi: 10.1016/j.csbj.2019.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen Z., Ratia K., Cooper L., Kong D., Lee H., Kwon Y., Li Y., Alqarni S., Huang F., Dubrovskyi O., et al. Design of SARS-CoV-2 PLpro Inhibitors for COVID-19 Antiviral Therapy Leveraging Binding Cooperativity. J. Med. Chem. 2022;65:2940–2955. doi: 10.1021/acs.jmedchem.1c01307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friesner R.A., Murphy R.B., Repasky M.P., Frye L.L., Greenwood J.R., Halgren T.A., Sanschagrin P.C., Mainz D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 28.Park J., Lee Y., Kim J., Cho S.H. Copper-Catalyzed Diastereoselective Addition of Diborylmethane to N-tert-Butanesulfinyl Aldimines: Synthesis of β-Aminoboronates. Org. Lett. 2016;18:1210–1213. doi: 10.1021/acs.orglett.6b00376. [DOI] [PubMed] [Google Scholar]
- 29.Lawrence K., Flower S.E., Kociok-Kohn G., Frost C.G., James T.D. A simple and effective colorimetric technique for the detection of boronic acids and their derivatives. Anal. Methods. 2012;4:2215–2217. doi: 10.1039/c2ay25346a. [DOI] [Google Scholar]
- 30.Hinkes S.P.A., Klein C.D.P. Virtues of Volatility: A Facile Transesterification Approach to Boronic Acids. Org. Lett. 2019;21:3048–3052. doi: 10.1021/acs.orglett.9b00584. [DOI] [PubMed] [Google Scholar]
- 31.Citarella A., Moi D., Pedrini M., Pérez-Peña H., Pieraccini S., Stagno C., Micale N., Schirmeister T., Sibille G., Gribaudo G., et al. Discovery of a Novel Trifluoromethyl Diazirine Inhibitor of SARS-CoV-2 Mpro. Molecules. 2023;28:514. doi: 10.3390/molecules28020514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amendola G., Ettari R., Previti S., Di Chio C., Messere A., Di Maro S., Hammerschmidt S.J., Zimmer C., Zimmermann R.A., Schirmeister T., et al. Lead Discovery of SARS-CoV-2 Main Protease Inhibitors through Covalent Docking-Based Virtual Screening. J. Chem. Inf. Mod. 2021;61:2062–2073. doi: 10.1021/acs.jcim.1c00184. [DOI] [PubMed] [Google Scholar]
- 33.Previti S., Ettari R., Calcaterra E., Di Maro S., Hammerschmidt S.J., Müller C., Ziebuhr J., Schirmeister T., Cosconati S., Zappalà M. Structure-based lead optimization of peptide-based vinyl methyl ketones as SARS-CoV-2 main protease inhibitors. Eur. J. Med. Chem. 2023;247:115021. doi: 10.1016/j.ejmech.2022.115021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Supporting data including NMR spectral charts are available from the Supplementary Materials.