

SUMMARY
An emerging principle of cell biology is the regulated conversion of macromolecules between soluble and condensed states. To screen for such regulation of the cyanobacterial proteome, we used quantitative mass spectrometry to identify proteins that change solubility during the day-night cycle. We find a set of night-insoluble proteins which includes many enzymes in essential metabolic pathways. Using time-lapse microscopy and isotope labeling, we show that these proteins reversibly transition between punctate structures at night and a soluble state during the day without substantial degradation. We find that the cyanobacterial circadian clock regulates the kinetics of puncta formation during the night, and that the appearance of puncta indicates the metabolic status of the cell. Reversible condensation of specific enzymes is thus a regulated response to the day-night cycle and may reflect a general bacterial strategy used in fluctuating growth conditions.
Keywords: Protein condensation, Metabolism, Circadian clock, Cyanobacteria
INTRODUCTION
Natural environments are characterized by fluctuations in conditions that restrict the growth of microorganisms. To cope with these challenges, cells regulate their physiology, gene expression, and metabolism to anticipate and respond to stressors. In eukaryotes, an emerging concept in cell biology is the dynamic organization of specific proteins and nucleic acids into condensates that have demixed from the cytosol. This biophysical mechanism can be used for multiple functional purposes, including activation or inactivation of enzymes, sequestration of components, and localization of molecules involved in a specific pathway. The resolution of these condensates then allows an appropriate return to the normal state when conditions improve (Franzmann and Alberti, 2019; Hyman et al., 2014; Riback et al., 2017; Wallace et al., 2015). There are now many examples of proteins and RNAs forming condensed phases in eukaryotic cells, including stress granules, processing bodies, and others (Greig et al., 2020; Hyman et al., 2014; Ivanov et al., 2019; Rhine et al., 2020; Snead and Gladfelter, 2019). These structures are likely diverse both in terms of physical state, with examples of both liquid phases and hydrogels (Shin and Brangwynne, 2017) and in terms of their regulation, including constitutive structures and stimulus-triggered phase separation (Brangwynne et al., 2009). Bacterial condensed phases have been reported (Al-Husini et al., 2018) as well as in vitro condensation of bacterial proteins (Monterroso et al., 2019; Wang et al., 2019). These studies reveal a general principle that the cytosol cannot be regarded as a well-mixed soup of soluble components; instead it contains biomolecular condensates that form dynamically in response to changing conditions.
Because bacterial cells lack internal membrane-bound compartments, a large set of enzymes and metabolites share a common cytosol. Do prokaryotes also use condensation to dynamically organize their cellular contents under typical growth conditions? We sought to define a model system where bacterial physiology could be studied under large but realistic changes in external conditions. Cyanobacteria are an ancient clade of photosynthetic prokaryotes that are responsible for a large fraction of primary production on Earth. Because of constraints imposed by the rotation of the Earth, cells are photosynthetically active and growth and division occur during the day; growth ceases, and energy reserves are consumed to ensure survival at night (Stanier and Cohen-Bazire, 1977). Many cyanobacteria are obligate phototrophs; little is known about how these bacteria regulate their metabolism in the dark and maintain viability during a prolonged night when photosynthesis is unavailable.
Here, using the model cyanobacterium Synechococcus elongatus, we report that dozens of proteins reversibly transition from a soluble state during the day to an insoluble state at night, associated with puncta that we visualize by fluorescence microscopy. In contrast to eukaryotic systems, where granules that form in response to stress tend to be enriched for RNA-binding proteins and in some cases form by liquid-liquid phase separation, these cyanobacterial protein condensates include many metabolic enzymes and lack obvious features associated with phase separation. Nevertheless, we show that the insolubility of these enzymes is regulated and reversible, as they are resolubilized in the next day.
Cyanobacteria anticipate the day-night cycle using a circadian clock which schedules gene expression and cell division throughout the day. We show that mutations which disrupt these rhythms coordinately alter the kinetics of punctum formation, indicating that the clock regulates changes in the physical status of the proteome during the night. Finally, by directly manipulating the metabolic status of the cell, both in light and in dark, we find evidence that changes in protein solubility closely reflect metabolic activity in the cell, suggesting that this phenomenon may be a general response to metabolic limitation.
RESULTS
Proteome-wide Profiling of Condensate Formation in a Day-Night Cycle
To characterize changes in the physical status of the proteome of the cyanobacterium Synechococcus elongatus (S. elongatus) during the daily light-dark cycle, we adapted an ultracentrifugation assay followed by quantitative mass spectrometry (Wallace et al., 2015). We synchronized S. elongatus cultures using a dark pulse and then subjected them to a 12h:12h light-dark cycle (Figure 1A), harvesting cells for analysis just before dark and again 8 hours into the night. This analysis revealed a set of proteins which are in the supernatant fraction, and are thus highly soluble during the day, but largely appear in the pellet fraction at night, reflecting their incorporation into condensates (Figure 1B). We identified a set of 197 proteins whose average supernatant fraction was >80% at the end of the day and <45% at night across two biological replicates (Figure S1). We refer to these proteins as “dark-demixing”, a generic term that is agnostic to the physical state of the sedimented proteins. They include proteins involved in photosynthesis, metabolism, DNA mismatch repair, proteases, and the circadian clock (Table S1). Many of the dark-demixing proteins are enzymes involved in amino acid biosynthesis, particularly pathways whose precursors are downstream of pyruvate (Figure 1C). Because of the importance of central carbohydrate metabolism to dark tolerance, we focused our attention on glycolysis, the TCA pathway, and the pentose phosphate pathway (Diamond et al., 2017; Puszynska and O’Shea, 2017).
Figure 1. Protein Condensation in the Cyanobacterial Proteome During the Day-Night Cycle.
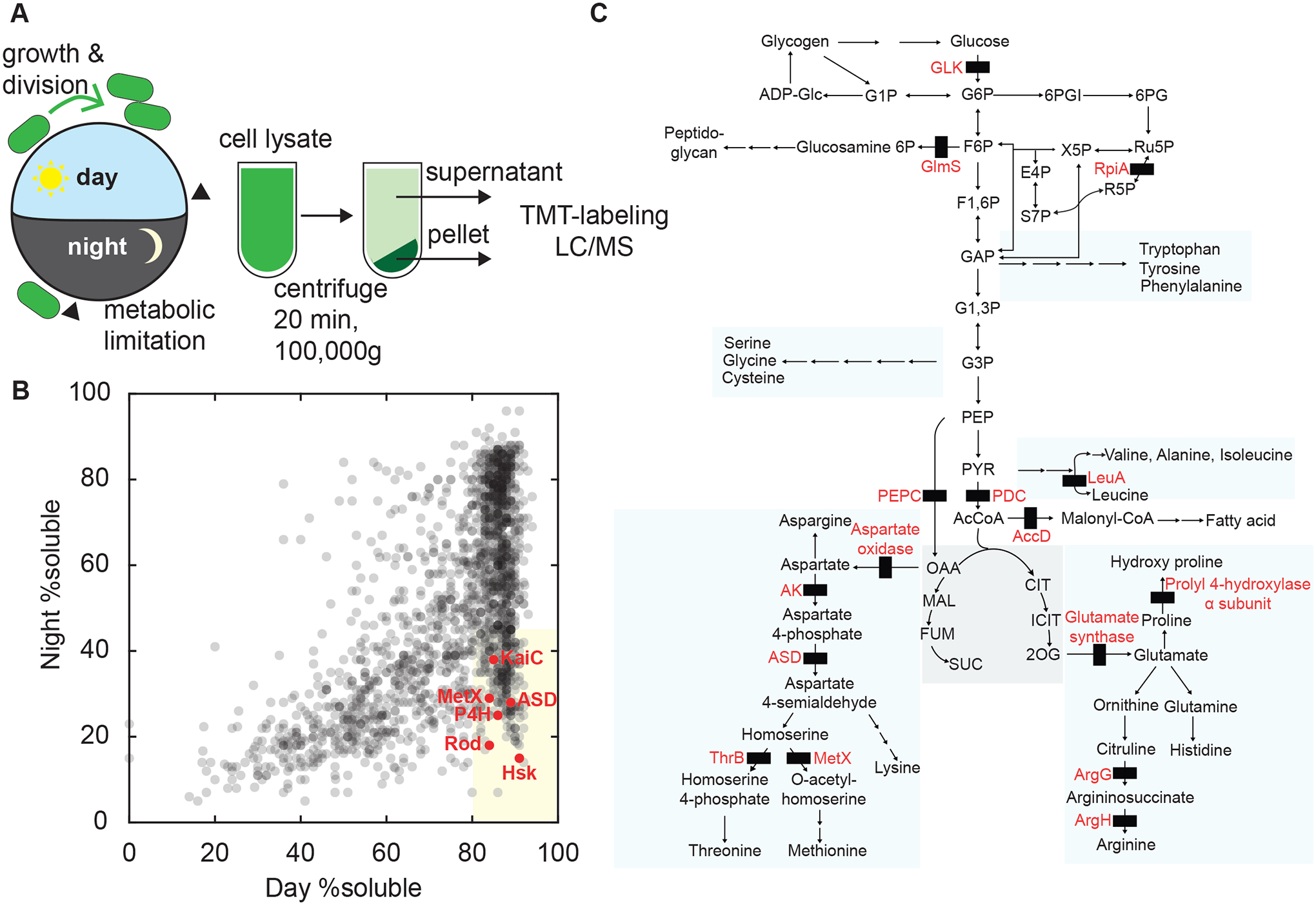
(A) Schematic of experimental design. Wild-type cell cultures were synchronized by one light-dark cycle. Cells were harvested at the end of the day and 8 h into the dark (arrowheads). Cells were lysed and fractionated by ultracentrifugation into soluble and pellet fractions. Fractions were trypsin digested, labeled with TMT tags and analyzed by tandem mass spectrometry.
(B) Scatter plot shows supernatant-pellet ratio changes in light vs dark samples for one biological replicate (gray dots, N =1777). The proportion of protein in each fraction was estimated from ratios of spectral counts. The shaded region indicates proteins with supernatant fraction >80% in the light and <45% in the dark (dark-demixing). Selected proteins marked in red.
(C) Schematic showing steps catalyzed by dark-demixing enzymes (black rectangles) in central metabolism, including reactions from glycolysis and TCA cycle, pentose phosphate pathway, amino acid biosynthesis, lipid and cell wall synthesis.
Dark-Demixing Proteins Are Reversibly Resolubilized in the Day
To visualize the kinetics of protein localization for candidates we identified by mass spectrometry, we fused enzymes to a fluorescent protein (EYFP), then analyzed their localization in living cyanobacteria. Because the fluorescent protein fusion may be perturbative, and because not all condensates will be detectable as fluorescent puncta (Wallace et al., 2015), we screened these strains for a microscopy signal reflecting the mass spectrometry results. We identified strains without detectable fluorescent puncta during the day and reliable formation of puncta at night. Out of 12 candidates, 4 constructs passed these tests: homoserine O-acetyltransferase (MetX; Synpcc_1714), aspartate-semialdehyde dehydrogenase, (ASD; Synpcc_1848), prolyl 4-hydroxylase α subunit (Synpcc_2480), and a tyrosine phosphatase (Synpcc_0463) (Movies S1–S4). Fusing these proteins to EYFP does not prevent sedimentation at night, but can perturb it quantitatively (Figure S2). Thus, our finding that these proteins sediment by ultracentrifugation likely reflects assembly into discrete puncta, typically distributed throughout the cytosol.
Particle identification and kinetic analysis of these movies indicate a diversity of kinetic behavior (Figure 2). A particularly striking example is prolyl hydroxylase which shows two distinct peaks of puncta formation early in the night and then again late at night. We asked whether the proteins in these fluorescent puncta could reversibly return to a soluble state, consistent with a scenario where protein condensation is being used as a reversible regulatory mechanism. An alternative is that nighttime puncta are irreversible aggregates destined for proteolysis and that soluble protein in the daytime reflects new protein synthesis. We addressed this question in two ways; first we quantified the total fluorescence intensity following the night-to-day transition and the intensity in discrete puncta in the reporter strains. While the intensity of fluorescent puncta in strains expressing the MetX-EYFP or ASD-EYFP drops by 90% in the first hour after dawn, the total fluorescence intensity does not decrease, but instead increases slightly (Figure 2B). The decrease in fluorescence signal in puncta following dawn is balanced by a strongly anticorrelated increase in the diffuse fluorescent background, consistent with dispersal of enzymes from puncta into their soluble form (Figure 2C).
Figure 2. Live Cell Imaging of Reversible Protein Condensation.
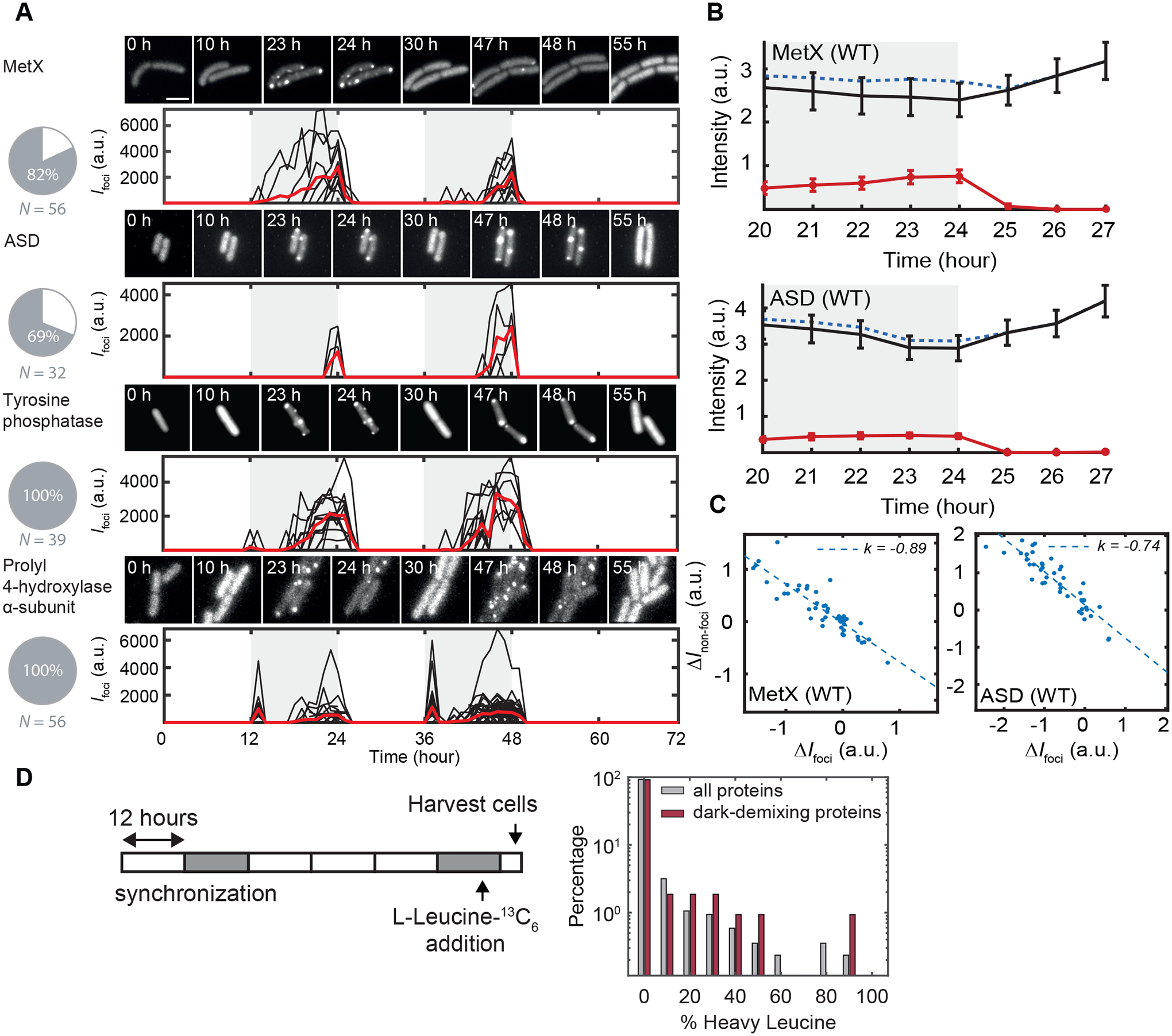
(A) Microscopy of dark-demixing proteins fused with EYFP. Cultures were grown under agar pads for live cell imaging during light-dark cycles. Micrograph filmstrips show the formation of EYFP puncta in dark and their disappearance following dawn (upper subpanels). Quantification of the total intensity of puncta during the experiment (lower subpanels). Black traces represent puncta fluorescence intensity from single cells. The red trace is the average of at least 10 cells. Pie charts show the percentage of cells with EYFP puncta. N = the number of cells analyzed. Scale bar = 3 μm.
(B) Average time trace of fluorescence intensity in the cytosol (black line), puncta (red line) and total (blue dashed line) in EYFP-MetX (top) or EYFP-ASD (bottom) expressing cells before and after the end of night (shaded region). N=108 cells, bars show 95% CI.
(C) Scatter plot showing change of cytosol fluorescence intensity (vertical axis) versus change in punctate fluorescence intensity (horizontal axis) over the 3 h window following lights-on. Dashed line shows linear regression with best-fit slope.
(D) Pulse-chase labeling experiment. At end of the night, cultures were labeled with L-Leucine 13C6 for 1 h before being transferred to light, harvested, and analyzed by tandem mass spectrometry. Normalized log-histogram in grey shows distribution of isotope labeling percentage in the soluble fraction after 4 h of light exposure for all detected proteins (gray, N = 864) and detected dark-demixing proteins (red, N=107).
Second, we directly interrogated whether the increase in soluble protein after dark-to-light transition is the product of new protein synthesis using an isotopic labeling strategy. We introduced 13C6-leucine to the medium one hour before the end of night and then measured the appearance of heavy leucine, reflecting new protein synthesis, in the pool of soluble proteins after 4 hours of light exposure (Figure 2D; Table S2). Most of the dark-demixing proteins have no detectable incorporation of heavy leucine in the soluble fraction. The average isotopic labeling of the dark-demixing proteins (2.9% labeling) is not noticeably different from the entire proteome (2.2% labeling). Highly labeled (>80% heavy leucine) peptides act as an internal control indicating that the isotope will be extensively incorporated into de novo synthesized proteins. Taken together, these results lead us to conclude that condensates formed by the dark-demixing proteins are generally reversible, and these proteins are returned to a soluble state for use during the day.
The Circadian Clock Regulates the Condensation Kinetics of Specific Metabolic Enzymes
One possibility is that the proteins we identified form condensates as an inevitable physical response to nightfall. Another is that this process is subject to regulatory systems, indicating its active management by the cell. Cyanobacteria have a robust circadian clock based on the Kai proteins whose function is to temporally organize gene expression, metabolism, and cell division in anticipation of the light-dark cycle (Ito et al., 2009; Mori et al., 1996a; Pattanayak et al., 2014). We first tested whether the internal clock state at lights-off influenced the appearance of fluorescent puncta. We observed a clear timing difference in both the MetX-EYFP and ASD-EYFP reporters: when cells were transferred to dark in sync with a previous light-dark cycle, i.e. at subjective dusk, puncta formed slowly over the course of the night. However, when cells were transferred to dark in the subjective morning, puncta appeared rapidly, within an hour after nightfall (Figure 3A), suggesting that the clock plays a role in the dynamics of puncta formation.
Figure 3. The Circadian Clock Regulates the Kinetics of Puncta Formation.
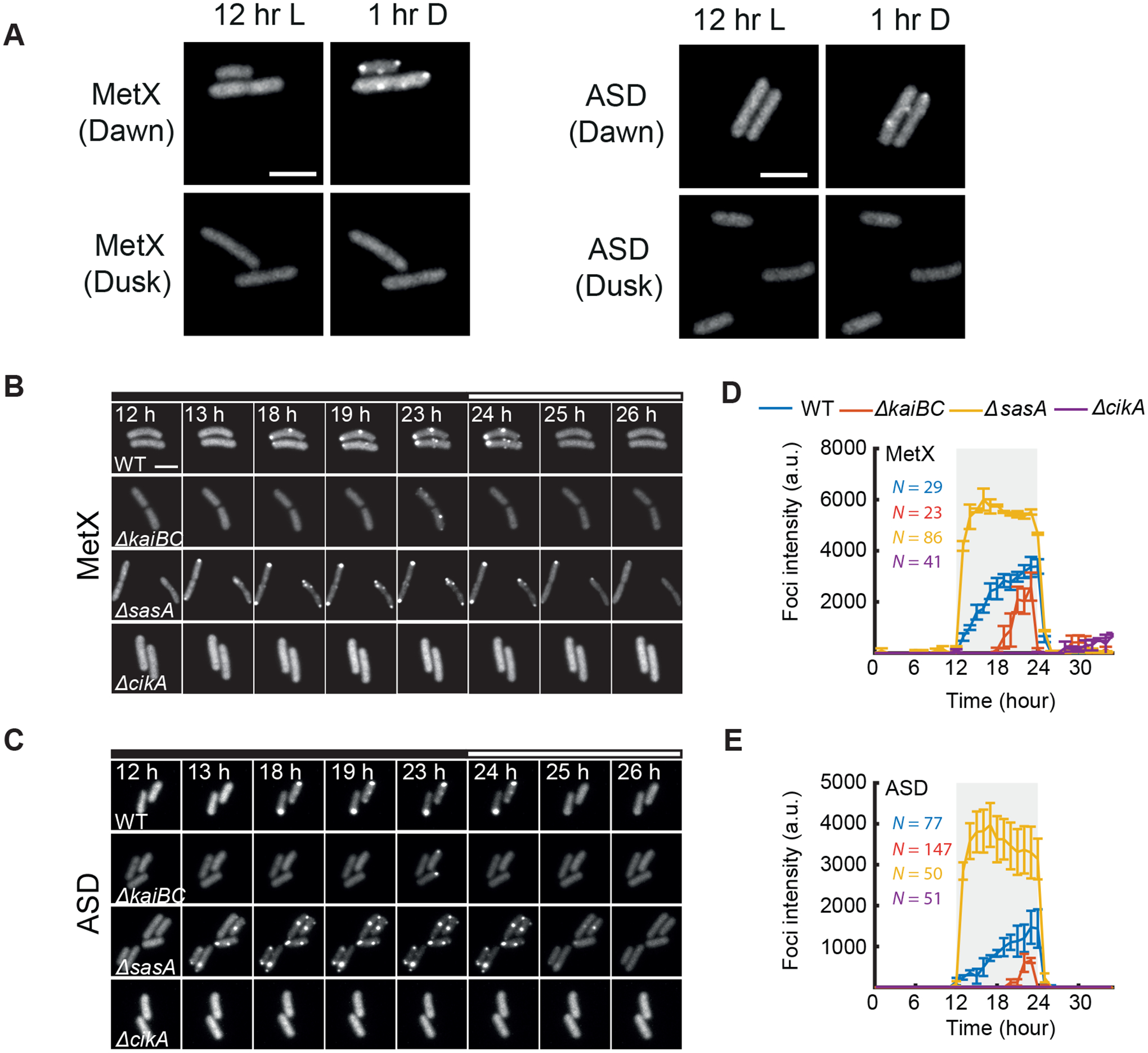
(A) Imaging of dark-demixing proteins (MetX and ASD) follow transition to dark at either subjective dawn (upper) or subjective dusk (lower).
(B-C) Imaging of MetX-EYFP and ASD-EYFP in circadian clock mutants. Micrograph filmstrips show the formation of EYFP puncta following the transfer of cells to darkness. Black bars indicates times when cells are not illuminated.
(C-D) Puncta fluorescence intensity during a light-dark cycle in circadian clock mutants. Error bars show standard deviation from 2 independent experiments. All scale bars = 3 μm.
The core circadian oscillator sends opposing output signals through the histidine kinases SasA and CikA needed to engage dusk and dawn transcriptional programs, respectively (Gutu and O’Shea, 2013; Takai et al., 2006). To test the role of the circadian clock mechanism in regulating the physical state of the proteome, we monitored puncta formation in the MetX-EYFP and ASD-EYFP strains when these clock components were mutated (Figures 3B, C). We find that these clock mutations cause marked differences in the kinetics of the formation of fluorescent puncta. The sasA-null mutant, which mimics a dawn-like transcriptional state, causes puncta to form much more rapidly following darkness. In contrast, loss of the circadian clock genes kaiB and kaiC, which instead mimics a dusk-like state, causes a substantial delay in puncta formation. cikA-null, which results in an exaggerated dusk-like state (Pattanayak et al., 2014), largely prevents the appearance of puncta (Figures 3C, D). From this, we conclude that the circadian clock system regulates the night-time condensation of these enzymes.
Protein Condensation Reflects Metabolic Limitation
The metabolic state of photosynthetic microbes is dramatically affected by the transition from light to dark. Photosynthetic energy production and carbon fixation are unavailable at night, resulting in depressed energy status, lack of biomass incorporation, and inhibited gene expression. Further, it is known that the circadian clock controls the timing of these metabolic changes (Diamond et al., 2015; Pattanayak et al., 2014). To test whether metabolic effects are an important determinant underlying protein condensation, we used genetic and pharmacological manipulations to alter metabolism and monitored puncta formation of MetX-EYFP.
Metabolism at night is supported by breakdown of glycogen reserves that accumulate during the day. To impair this metabolic pathway, we deleted the glycogen phosphorylase gene (glgP) and found that detectable puncta appeared rapidly (< 1 hr) after nightfall compared to wildtype (~7 hr average delay time before fluorescent spots appear), consistent with the interpretation that MetX-EYFP puncta formation reflects the metabolic status of the cell (Figure 4A).
Figure 4. Metabolic Limitation Is a Key Driver of Protein Condensation.
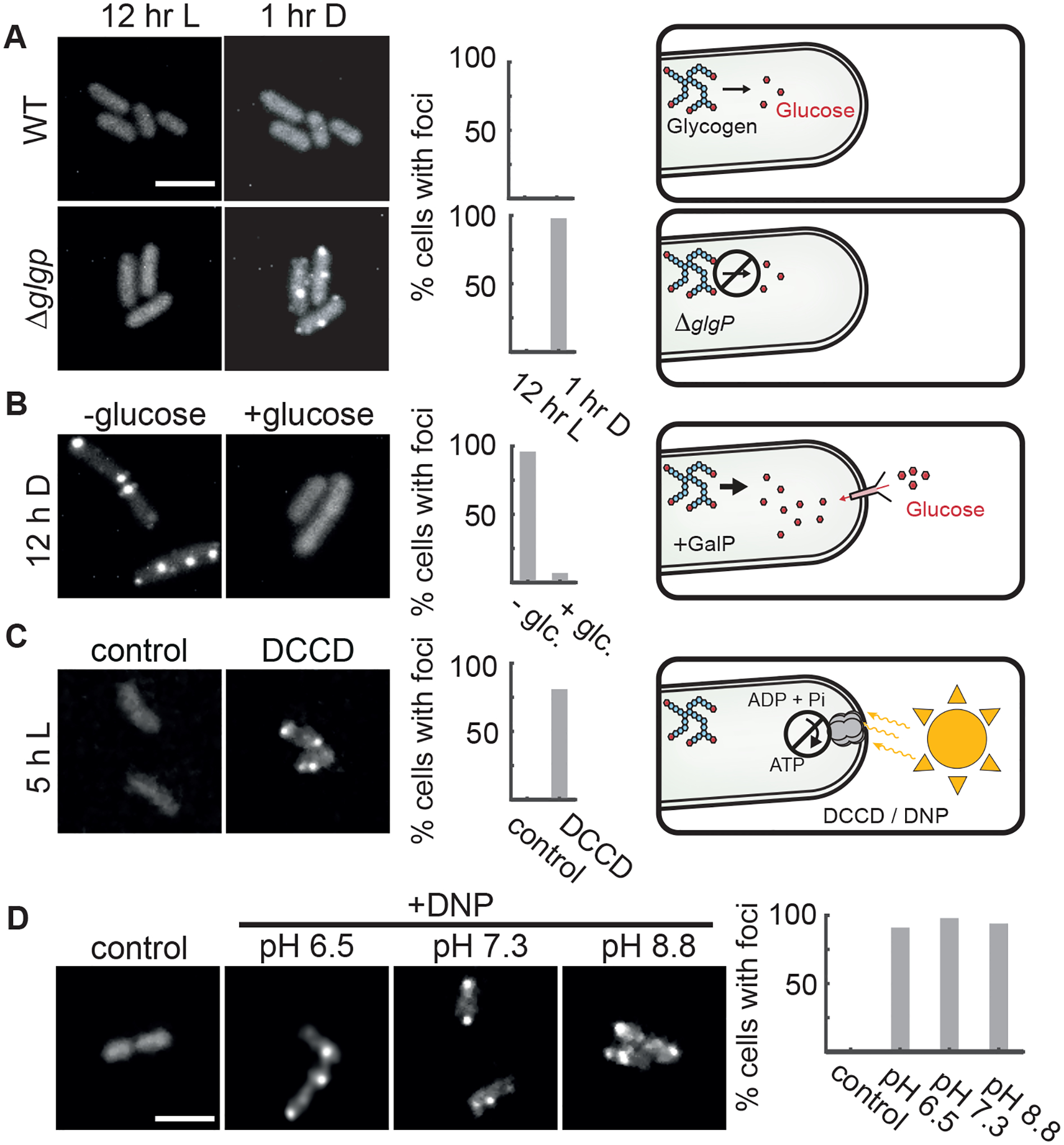
(A) Live cell imaging of MetX-EYFP in the light (left) or after 1 h in darkness (right) in either wildtype (top) or in a glycogen breakdown-deficient mutant (ΔglgP). Scale bar: 3μm.
(B) MetX-EYFP fluorescence in a strain expressing the GalP sugar transporter and capable of glucose-dependent growth in darkness. Images were captured after 12 h of dark treatment, with or without glucose.
(C) MetX-EYFP fluorescence in cells incubated in light when ATP synthesis was blocked (DCCD, 15 μM). Control shows the effect of 0.1% methanol vehicle. Images were taken 5 h after treatment.
(D) Representative micrographs of DNP-treated MetX-EYFP cells showing fluorescent puncta in light. The pH of the media was adjusted with Bis-Tris or Tris buffer. Scale bar is 3μm. Images were taken 5–8 h after treatment. Histograms show percentages of cells with foci, N = 50–100 cells.
S. elongatus grows autotrophically making it difficult to separate possible light-dependent signaling from light-driven effects on metabolism. To isolate the effect of growth, we expressed the E. coli sugar transporter GalP and added glucose to the culture medium, previously shown to raise ATP levels and allow slow growth in the dark (Pattanayak et al., 2015). Under these conditions, fluorescent EYFP-MetX puncta do not form (Figure 4B), suggesting that this protein is responding to the metabolic effects of darkness rather than darkness itself.
Finally, we sought to test whether interruption of energy production, even during the day, would be sufficient to induce protein condensation. We inhibited ATP production using either dicyclohexylcarbodiimide (DCCD), an inhibitor of F1FO-ATP synthase (Figure 4C), or 2,4-dinitrophenol (DNP), which allows protons to cross the membrane and prevents proton gradient formation (Figure 4D). In both conditions, EYFP-MetX puncta formed even in the light, indicating that darkness per se is not required, and metabolic limitation alone can cause this protein to condense from solution. Notably, the effect of DNP did not depend on the pH of the culture medium (Figure 4D), suggesting that intracellular pH itself is not the main causal factor. Taking these data together, we conclude that metabolic limitation is both necessary and sufficient to induce protein condensation in cyanobacteria.
DISCUSSION
Cyanobacteria present a unique opportunity to uncover general principles of how prokaryotic cells respond to fluctuating environments, both because it is experimentally straightforward to drive cells through repeated light-dark cycles, and because the regularity of the day-night cycle allows for regulatory mechanisms that anticipate metabolic changes. During the day, cells are metabolically active, grow and divide. At nightfall, the stringent response is activated, the rates of transcription and translation are markedly reduced (Hood et al., 2016), metabolism slows, and cell elongation ceases (Mori et al., 1996b). These phenomena are presumably linked to a need for the cell to restrict metabolic consumption to remain viable until the next sunrise (Grundel et al., 2012). This study reveals that entry into this restricted metabolic state is coupled with the transition of a specific set of proteins from a soluble cytosolic state into condensed, localized puncta. These protein molecules are largely preserved so that they can be reversibly dispersed back into the cytosol in the day. Interestingly, previous studies have shown that proteins involved in the circadian oscillator switch to an insoluble state on a daily schedule even in constant light. These proteins were also found in our mass spectrometry screen and likely represent a specialized metabolism-independent mechanism (Cohen et al., 2014; Kitayama et al., 2003).
This finding opens many questions, including the regulatory roles of the nighttime puncta and the underlying biophysical mechanisms controlling the physical state of these proteins. Many of the enzymes identified in our data divert precursors from glycolysis and the TCA pathway to produce amino acids. Flux through glycolysis and the oxidative pentose phosphate pathway has been shown to be necessary for cyanobacteria to tolerate light-dark cycles (Diamond et al., 2017; Puszynska and O’Shea, 2017) and, because protein synthesis is inhibited at night (Hood et al., 2016), the demand for amino acids is presumably low. Thus, we speculate that enzyme condensation at night may function to temporarily inactivate these enzymes until they are needed in the next day. Notably, the E. coli MetA protein, which has a homologous role in methionine biosynthesis to MetX, is known to be inactivated by aggregation in response to increased temperature (Gur et al., 2002). Future in-depth analysis of particular bacterial metabolic pathways will determine whether condensates are composed of heterogeneous assemblies of many proteins or whether these structures are composed of single protein species.
These bacterial results suggest a link to findings in yeast and animal cells where metabolic enzymes have be shown to dynamically condense in response to metabolic shifts (Narayanaswamy et al., 2009; O’Connell et al., 2012). In existing eukaryotic examples, protein phase separation can be achieved through diverse biophysical mechanisms. Proteins with self-interacting domains or proteins having intrinsically disordered regions are prone to condensation (Li et al., 2012; Molliex et al., 2015; Patel et al., 2015). Similarly, the propensity of proteins to form condensates can be altered by the charge status of amino acids which can be altered by post-translational modification (Monahan et al., 2017; Nott et al., 2015), and the ionic strength of the cellular environment (Petrovska et al., 2014) alter the propensity of proteins to form condensates. Low complexity and disordered regions are not obviously present in the bacterial proteins we identify here. However, such regions are neither required nor sufficient for condensate formation (Kroschwald et al., 2018; Riback et al., 2017; Wang et al., 2019). The precise physical state of the protein assemblies we detect at night is not clear and may include droplets of liquid-like phases as well as protein polymers or gel-like aggregates.
The circadian clock generates metabolic rhythms that partitions some metabolic pathways important for growth in light-dark cycles, such as glycogen storage, into the latter part of the day (Lambert et al., 2016; Pattanayak et al., 2014). Previous work has shown that the metabolic status of cells after nightfall depends on the circadian clock. For example, the cikA-null mutant which markedly delays puncta formation has elevated energy charge in the dark. Further, ATP levels in the dark depend on clock-environment alignment: ATP falls faster when darkness occurs near subjective dawn (Pattanayak et al., 2014). This effect may explain why fluorescent puncta appear rapidly in MetX-EYFP and ASD-EYFP strains when cells were subjected to darkness after subjective dawn. In other words, the clock may be programming certain metabolic pathways to shut down partway through the night.
How does metabolic status regulate the condensation of enzymes? One possibility is that metabolite binding may alter protein conformation to favor either solubility or condensation. The role of energy metabolism in the protein condensation phenomena we report here suggest that an energy-dependent process may be involved in maintenance of protein solubility, possibly including ATP-dependent disaggregation enzymes or a direct biophysical role for the ATP molecule itself (Patel et al., 2017). Multiple lines of evidence now suggest that the physical state of a bacterial cell is profoundly different under conditions that restrict growth. The cytosol enters a glass-like state where mobility of macromolecular diffusion probes is extremely limited (Parry et al., 2014). Bacterial protein condensation phenomenon have been observed in C. crescentus (Al-Husini et al., 2018), in M. tuberculosis (Heinkel et al., 2019), in cyanobacteria (Bar Eyal et al., 2017; Wang et al., 2019), during cell division and during antibiotic treatment in E. coli (Kwiatkowska et al., 2008; Laskowska et al., 2003; Monterroso et al., 2019; Pu et al., 2019). Along with our results showing that reversible protein condensation is a highly regulated aspect of the normal growth cycle of cyanobacteria, this suggests that dynamic remodeling of the physical state of the proteome is a general principle of bacterial physiology.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further request for resources should be directed to the lead contact, Michael J. Rust (mrust@uchicago.edu)
Materials Availability
Plasmids generated in this study will be made available on request through the lead author but may require a shipping payment and/or a completed Material Transfer Agreement.
Data and Code Availability
The published article includes all analyzed data. Raw mass spectrometry data are available on Chorus (Project 1682) and the codes used for microscope data analysis is available upon request from the lead contact.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The photosynthetic cyanobacterium Synechococcus elongatus PCC 7942 wildtype (WT) cells were kindly provided by Dr. Susan S. Golden (University of California, San Diego). All the EYFP reporter strains were generated in the S. elongatus PCC 7942 wildtype cells. Different mutants used in this study were generated in the EYFP reporter background cells.
METHOD DETAILS
Cloning and Strain Construction
All the EYFP (enhanced yellow fluorescence protein) reporter strains used in this study are derivatives of Synechococcus elongatus PCC 7942 wildtype (WT) cells. Description of molecular cloning, construction of individual plasmids, description of EYFP reporter strains, and different metabolic and clock mutant strains are detailed below.
The MetX (Synpcc_1714), ASD (Synpcc_1848), tyrosine phosphatase (Synpcc_0463) and prolyl 4-hydroxylase α subunit (Synpcc_2480) genes were PCR amplified from Synechococccus genomic DNA using unique pairs of oligonucleotide primers (Supplementary Table 3; Related to Figure 2 and 4) specific for the respective genes. Similarly, the EYFP DNA fragment was PCR amplified from pEYFP plasmid (Clontech) by EYFP_linker forward and EYFP reverse primers (Supplementary Table 3; Related to Figure 2 and 4). The neutral site 1 (NSI) plasmid pAM2991 (Ivleva et al., 2005) was linearized with EcoRI restriction enzyme. The PCR amplified gene fragments, EYFP DNA fragment and EcoRI digested pAM2991 plasmid DNA products were gel purified and Gibson assembled to create the individual plasmids; pAM2991-MetX-EYFP (pMR0202), pAM2991-ASD-EYFP (pMR0203), pAM2991-tyrosine-phosphatase-EYFP (pMR0204) and pAM2991- prolyl 4-hydroxylase α subunit-EYFP (pMR0205), respectively. In the above plasmids, each individual EYFP-fused gene was under the control of Ptrc promoter (IPTG-inducible). Plasmids used to generate circadian clock mutants in EYFP reporter backgrounds were described elsewhere (Pattanayak et al., 2014).
To disrupt the glgP gene from the Synechococcus genome, DNA fragments were PCR amplified from the genomic DNA using two pairs of oligonucleotide primers (Supplementary Table 3), glgPko1 and glgPko2; and glgPko3 and glgPko4, respectively. The DNA fragment (352 bp) amplified by glgPko1/glgPko2 primers was digested with Hind III and EcoRI and inserted into pMR0089 plasmid at the respective restriction enzyme sites, to create the pMR0089A plasmid. The DNA fragment (307 bp) amplified by glgPko3/glgPko4 primers was digested with XbaI and SacI and cloned into the pMR0089A plasmid at the respective restriction sites, resulting in the pΔglgP::Gmr plasmid (pMR0206).
The plasmid pMR0094 (Pattanayak et al., 2015) was used as a template and two primers i.e., GalP-NS3 Gib forward and GalP-NS3 Gib reverse were used to amplify the GalP DNA fragment. The neutral site III (NS3) plasmid pAM5433 was digested with SwaI restriction enzymes. The PCR amplified GalP fragment and the linearized pAM5433 plasmid DNA products were gel purified and Gibson assembled to create the individual plasmid; pAM5433-GalP (pMR0207).
Transformation of S. elongatus
S. elongatus transformations were performed as previously described (Golden et al., 1986). pMR0202, pMR0203, pMR0204 and pMR0205 plasmids were used to transform wild-type S. elongatus cells to generate the MRC1054, MRC1055, MRC1057 and MRC1058 reporter strains, respectively. Similarly, pMR0090, pMR0091 and pMR0092 plasmids (Pattanayak et al., 2014) were used to make cikA null, kaiBC null and sasA null strains, respectively either in MRC1054 background or in MRC1055 background strain. The resulting strains were named as MRC1105 (ΔcikA/MRC1054), MRC 1115 (ΔkaiBC/MRC1054), MRC1106 (ΔsasA/MRC1054) and MRC1125 (ΔcikA/MRC1055), MRC1123 (ΔkaiBC/MRC1055) and MRC1124 (ΔsasA/MRC1055). The pMR0206 and pMR0207 plasmids were transformed in MRC1054 background strain to either delete glgP or to overexpress galP, respectively. The respective strains are named MRC1145 and MRC1121. The transformed colonies were grown repeatedly in BG11 liquid medium supplemented with appropriate antibiotics followed by genomic DNA extraction and confirmation of the presence of the heterologous genes at the targeted neutral sites. Similarly, all the mutant strains were allowed to segregate completely by growing them repeatedly in BG11 liquid medium supplemented with appropriate antibiotics followed by genomic DNA extraction and confirmation of the absence of the wild-type allele or presence of the mutant allele by PCR analysis.
Culture Growth Conditions
All cyanobacterial strains were grown in BG11 liquid medium with appropriate combinations of antibiotics at 30°C under continuous illumination (LL) ~75 μmol photons m−2 s−1; cool white fluorescent light (Phillips, USA) with shaking at 170 rpm. Cell densities were monitored by measuring the optical density at 750 nm (OD750). For cell fractionation experiments, cultures (300 mL each and OD750 ~ 0.3) synchronized with a light-dark (12 h:12 h) cycles were released into LL for 36 hours (subjective dusk) followed by 8 hrs of dark incubation. Cultures were collected just before transferred to dark and 8 hrs into the night. For 13C6-leucine incubation experiment, wild-type cultures were grown as mentioned above, but instead of 8 hrs of darkness cells were incubated for 12 hrs in dark. After 11 hrs into dark, cells were fed with 13C6-leucine, (25μg/mL) incubated in darkness for another 1 hr, and then exposed to light for 4 hrs. Cell pellets were harvested by vacuum filtration, snap frozen in liquid nitrogen, and stored at −80°C until further processing.
For microscope experiments, starter cultures were grown in 5 mL of BG11 (in a 30 mL tube) till the OD750 reaches between 0.2–0.3. For entrainment, the cultures were then diluted to OD750 = 0.1 using fresh BG11 medium and 250 μL of the diluted cell cultures were pipetted into different wells of a black (opaque) 96-well plate. The plate was sealed with adhesive film and a hole was punched for aeration over each well. A custom-made Arduino driven LED array (628 nm peak emission, Super Bright LEDs, Inc.) was used to illuminate each well (Lambert et al., 2016). The LED array provided the equivalent of ~75 μmol photons m−2 s−1 of red light to each well. The Arduino was programmed to generate 2 cycles of light/dark (12 h: 12h) conditions (light conditions: ~75 μmol photons m−2s−1 (23 mA); Dark conditions: 0 mA) followed by continuous light. For glucose feeding experiments, MetX-EYFP cells expressing galP were grown in liquid BG11 supplemented with 20 mM HEPES (pH 7) in presence and absence of glucose (1%) under red LEDs as described above.
Time-lapse Microscopy and EYFP Fluorescence Intensity Quantification
Microscope experiments were set up following procedures detailed in (Lambert et al., 2016) with minor modifications. Briefly, 1 μL of S. elongatus was taken out from cell cultures grown under red LEDs as described above and was pipetted onto a 6-well glass bottom plate (MatTek Corp., Part Number: P06G-1.5–20-F). A small 2% low melting point agarose (LMPA, Thermo Fisher Scientific Inc., Part Number: 16520–050) pad containing 1x BG11 was placed atop the cell suspension and the 6-well plate was transferred to a motorized microscope (IX71, Olympus Inc.). Then, 10 mL of liquid BG11 + 2% LMPA supplemented with 200 μM IPTG (BG11-LMPA) was poured inside the well to cover the LMPA pad. When required, glucose (1%) and 20 mM HEPES (pH 7) was added to BG11-LMPA medium to cover the cells expressing the glucose transporter. When we inhibited ATP synthesis during the light cycle, we added 15 μM of DCCD (N,N-dicyclohexylcarbodiimide) and 1 mM of DNP (2,4-Dinitrophenol) independently to the BG11-LMPA medium. DCCD was dissolved in methanol and DNP was dissolved in dimethyl sulfoxide (DMSO), respectively. The final concentration of methanol in the culture medium was 0.01% and that of DMSO was 0.5%.
On the microscope, cells were usually grown for 12–15 hrs before the 12 hr of dark pulse was administered. Two 12:12 hr light/dark cycles were given, during which time fluorescence and bright field images were recorded. Every 60 minutes, a motorized microscope stage (Prior Scientific Inc.) moved a total of 24 pre-assigned locations containing S. elongatus into the field of view (FOV). Bright-field (exposure: 100ms), chlorophyll (exposure: 200ms; excitation: 501 nm; emission: 590 nm) and YFP signals (exposure: 2s; excitation: 501nm; emission: 550 nm) were then captured with a 100x Olympus oil immersion objective and a Luca EMCCD camera (Andor). The cells were exposed to a continuous light source of 7 μmol photons m−2 s−1 of light (660 nm wavelength), and the illumination condenser was removed in order to widen the light beam to sufficiently illuminate the well evenly. The “SimpleAutofocus” program provided by the Micro-manager suite used the chlorophyll autofluorescence of the cells within each FOV to identify the optimal focal plane before each bright-field and fluorescence images were recorded.
To quantify the EYFP fluorescence intensity, fluorescence images were first subject to a median filter to eliminate salt-and-pepper noises. Bright-field images of cells were segmented using the watershed algorithm, which produced a mask for each cell, allowing quantifications of fluorescence signals to be carried out at the single-cell level. A bandpass filter was applied to YFP fluorescence images to identify pixels corresponding to locations of fluorescence foci. The intensity of each focus was calculated as Ifoci = 2πIpeakσ2, where Ipeak and σ are respectively the amplitude and the width of the 2D Gaussian fit of the intensity profile. The cytosol intensity of each cell was calculated by adding fluorescence intensities of all none-foci pixels within the corresponding cell mask.
Cell Fractionation and Sample Preparation for Mass Spectrometry
For cell fractionation experiments, cultures (300 mL each and OD750 ~ 0.3) synchronized with a light-dark (12 h:12 h) cycle were released into LL for 36 hours (dusk) followed by 8 hrs of dark incubation. Cultures were collected just before transferred to dark and 8 hrs into the night by vacuum filtration, snap frozen in liquid nitrogen, and stored at −80°C until further processing. Cell pellets from −80°C were lysed and fractionated according to (Wallace et al., 2015) with minor modifications The cell pellets were placed in a 2 mL Eppendorf Safe-Lok tube containing a 7 mM stainless steel ball (Retsch) pre-chilled in liquid nitrogen (LN). Cells were lysed with five 90 sec 30Hz pulses in a Retsch MM100 mixer mill, chilling in LN between pulses. 500 μL ice-cold buffer S (20 mM HEPES-NaOH, pH7.4, 120mM KCl, 2 mM EDTA, 2 mM DTT, 1 mM PMSF, 1:1000 serine and cysteine protease inhibitor (cOmplete, EDTA free cocktail tablets, Sigma-Aldrich) was added, then the thawed lysate was clarified by centrifugation at 3, 000g for 30 seconds at 4°C. The clarified supernatant was transferred to a 1.5mL ultracentrifuge tube, and centrifuged at 100, 000g for 20 minutes at 4°C. The supernatant was taken carefully and this is the soluble (S) sample. The pellet was washed in 500 μL buffer S, and centrifuged again at 100,000g for 20 min at 4°C. The remaining pellet was mixed with 500 μL buffer P [8 M urea, 20 mM HEPES-NaOH, pH7.4, 150mM NaCl, 2% SDS, 2 mM EDTA, 2 mM DTT, 1 mM PMSF, 1:1000 cOmplete, EDTA free protease inhibitor cocktail), by vortexing vigorously for 15–20 seconds for three to four times. The resuspended pellet was centrifuged at 20, 000g at RT for 5 minutes, and the aqueous phase was designated as the pellet (P) fraction. Protein from supernatant and pellet fractions was precipitated by chloroform/methanol extraction(Wessel and Flugge, 1984).
Mass Spectrometry, Data Extraction and Analysis
Protein from supernatant and pellet fractions was chloroform methanol extracted, trypsin digested and TMT labelled. The TMT-labelled samples were fractionated and fractions were submitted for LC-MS/MS analysis on Fusion Tribrid Lumos Orbitrap (Thermo Fisher). Each sample was submitted for single LC-MS/MS experiment that was performed on a Fusion Lumos Tribrid Instrument (Thermo Fischer) equipped with EASY1000 HPLC pump Peptides were separated onto a 100 μm inner diameter microcapillary trapping column packed first with approximately 5 cm of C18 Reprosil resin (5 μm, 100 Å, Dr. Maisch GmbH, Germany) followed by analytical column ~20 cm of Reprosil resin (1.8 μm, 200 Å, Dr. Maisch GmbH, Germany). Separation was achieved through applying a gradient from 5–27% ACN in 0.1% formic acid over 180 min at 200 nl min−1. Electrospray ionization was enabled through applying a voltage of 3 kV using a home-made electrode junction at the end of the microcapillary picofrit column (New Objective, MA). The Lumos was operated in data-dependent mode for the mass spectrometry methods. The mass spectrometry survey scan was performed in the Orbitrap in the range of 395–1,800 m/z at a resolution of 6 × 104, followed by the selection of the twenty most intense ions (TOP20) were subjected to HCD MS2 event in Orbitrap part of the instrument. The fragment ion isolation width was set to 0.7 m/z, AGC was set to 50,000, the maximum ion time was 100 ms, normalized collision energy was set to 37V and an activation time of 1 ms for each HCD MS2 scan.
Raw data were submitted for analysis in Proteome Discoverer 2.1.0.83 (Thermo Scientific) software. Assignment of MS/MS spectra were performed using the Sequest HT algorithm by searching the data against a protein sequence database including all entries from the Uniprot S. Elongates database (SwissProt 1,257 and TrEMBL7, 367; 2015) and other known contaminants such as human keratins and common lab contaminants. Sequest HT searches were performed using a 20 ppm precursor ion tolerance and requiring each peptides N-/C termini to adhere with Trypsin protease specificity, while allowing up to two missed cleavages. 6-plex TMT tags on peptide N termini and lysine residues (+229.162932 Da) was set as static modifications while methionine oxidation (+15.99492 Da) and heavy Leucine (+6.0200 Da) was set as variable modification. A MS2 spectra assignment false discovery rate (FDR) of 1% on protein level was achieved by applying the target-decoy database search. Filtering was performed using a Percolator (64bit version, reference 1). For quantification, a 0.02 m/z window centered on the theoretical m/z value of each the six reporter ions and the intensity of the signal closest to the theoretical m/z value was recorded. Reporter ion intensities were exported in result file of Proteome Discoverer 2.1 search engine as an excel tables for TMT6plex outputs and MS1 based signal intensities were exported for SILAC type heavy Leucine quantitation searches. The total signal intensity across all peptides quantified was summed for each TMT channel, and all intensity values were adjusted to account for potentially uneven TMT labeling and/or sample handling variance for each labeled channel.
Each TMT labeled set of samples was submitted for ERLIC separation. After digestion peptides were separated on Agilent (Santa Clara, CA) 1200 HPLC system using PolyWAX LP column (PolyLC, Columbia MD) 200×2.1 mm, 5μm, 300A for ERLIC (Electrostatic Repulsion Hydrophilic Interaction Chromatography) chromatography. Peptides were separated across 70 min gradient from 0% buffer A (90% acetonitrile 0.1% acetic acid) to 75 % buffer B (30% acetonitrile, 0.1% formic acid) with 20 fractions been collected by time. Each fraction was dried in SpeedVac (Eppendorf, Germany) and re-suspended in 0.1% formic acid solution before injection to mass spectrometry. Single LC-MS/MS experiment that was performed on Orbitrap Q Exactive Plus (Thermo, CA) equipped with 1200 Agilent (Santa Clara, CA) Nano HPLC pump. Peptides were separated onto a 150 μm inner diameter microcapillary trapping column packed first with approximately 3 cm of C18 Reprosil resin (5 μm, 100 Å, Dr. Maisch GmbH, Germany) followed by analytical column ~20 cm of Reprosil resin (1.8 μm, 200 Å, Dr. Maisch GmbH, Germany). Separation was achieved through applying a gradient from 5–27% ACN in 0.1% formic acid over 90 min at 200 nl min−1. Electrospray ionization was enabled through applying a voltage of 2 kV using a home-made electrode junction at the end of the microcapillary column and sprayed from fused silica pico tips (New Objective, MA). The Q Exactive Plus was operated in data-dependent mode for the mass spectrometry methods. The mass spectrometry survey scan was performed in the Orbitrap in the range of 395–1,800 m/z at a resolution of 6 × 104, followed by the selection of the twenty most intense ions (TOP20) for HCD-MS2 fragmentation in the Orbitrap using a precursor isolation width window of 2 m/z, AGC setting of 10,000, and a maximum ion accumulation of 200 ms. Singly charged ion species were not subjected to HCD fragmentation. Normalized collision energy was set to 37 V and an activation time of 1 ms. Ions in a 10 ppm m/z window around ions selected for MS2 were excluded from further selection for fragmentation for 60s.
After extracting the TMT spectral counts from supernatant and pellet fractions of light and dark incubated sample, we defined the soluble fraction as the ratio of counts in the supernatant to the sum of both supernatant and pellet. To correct for possible systematic batch effects between the two replicates, we used linear regression to normalize data from the 2nd replicate to have the same intercept and slope as the 1st replicate. We defined two thresholds to consider a protein as a dark-demixing: its soluble fraction must be >80% at the end of the day and <45% during the night. If a protein was detected in both replicates, we required that it pass these criteria in both data sets.
Western Blotting Analysis
Equal amounts of fractionated samples (soluble and pellet) were added to 3x DTT-containing SDS-PAGE sample buffer. The samples were heated at 95°C for 5 min and were separated by SDS-PAGE gels (4–20% Criterion TGX Stain-Free protein gels, Biorad) and then transferred onto polyvinylidene fluoride membranes. The membrane filters were blocked for 2 hr at room temperature by incubating in 2% nonfat dry milk/Tris-buffered saline with 0.1% Tween 20 (TBST). Membranes were then incubated overnight at 4°C with a polyclonal anti-GFP antibody (Clontech; 1:1000 dilutions in 2% nonfat dry milk/TBST). After washing three to four times (15 min each), membranes were incubated for 2 hr at room temperature with 1: 1000 dilutions of secondary antibody (horseradish peroxidase-conjugated goat anti-rabbit, Thermo Fisher). The blots were developed using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) according to the manufacturer’s directions. The blots were photographed with a charge-coupled device camera using the Chemi- Doc MP Imaging System (Bio-Rad). The signal intensity was estimated using ImageJ.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical data analysis was performed using GraphPad Prism 7 (GraphPad Software Inc., CA, USA) on 2 biological replicates. Fluorescence intensity of the EYFP puncta was quantified using custom-made MATLAB programs.
Supplementary Material
Movies S1. Live-cell Imaging of MetX-EYFP, Related to Figure 2. S. elongatus cells expressing MetX fused with EYFP show fluorescent puncta in dark and their disappearance following light exposure. Frame rate = 1 hr/frame.
Movies S2. Live-cell Imaging of ASD-EYFP, Related to Figure 2. S. elongatus cells expressing ASD fused with EYFP show fluorescent puncta in dark and their disappearance following light exposure. Frame rate = 1 hr/frame.
Movies S3. Live-cell imaging of Tyrosine Phosphatase-EYFP, Related to Figure 2. S. elongatus cells expressing Tyrosine Phosphatase fused with EYFP show fluorescent puncta in dark and their disappearance following light exposure. Frame rate = 1 hr/frame.
Table S1. Protein Solubility in Light and Dark, Related to Figure 1. Proteins identified in the soluble and pellet fractions isolated from light and dark incubated wild-type Synechococcus elongatus cells, using TMT labeling. The proportion of protein in each fraction was estimated from ratios of spectral counts.
Movies S4. Live-cell imaging of Prolyl 4-hydroxylase α-subunit-EYFP, Related to Figure 2. S. elongatus cells expressing Prolyl 4-hydroxylase α-subunit fused with EYFP show fluorescent puncta in dark and their disappearance following light exposure. Frame rate = 1 hr/frame.
Table S2. Isotopic Labeling of Proteins Following a Dark-to-Light Transition, Related to Figure 2. Proteins labelled with heavy leucine (L-Leucine 13C6) identified in the soluble fraction isolated from light incubated cells. After 11 h in dark incubation, cultures were labeled with L-Leucine 13C6 for 1 h and then released to light. Cells were harvested 4 h after light exposure, fractionated and analyzed by tandem mass spectrometry.
ACKNOWLEDGMENTS
We thank Susan S. Golden (University of California, San Diego) and Carl H. Johnson (Vanderbilt University) for the generous gifts of plasmids. We thank members of the Rust lab, particularly Eugene Leypunskiy and Justin Chew for useful discussions. The work was supported by Pew Biomedical Scholarships (to M.J.R. and D.A.D.), the European Union’s Horizon 2020 programme (Marie Sklodowska-Curie grant 661179) to E.W., NIH R01 GM126547 and GM127406 to D.A.D., a Chicago Biomedical Consortium catalyst award to M.J.R., an HHMI-Simons Faculty Scholar award to M.J.R., and NIH R01 GM107369 to M.J.R.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Al-Husini N, Tomares DT, Bitar O, Childers WS, and Schrader JM (2018). alpha-Proteobacterial RNA Degradosomes Assemble Liquid-Liquid Phase-Separated RNP Bodies. Molecular cell 71, 1027–1039 e1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar Eyal L, Ranjbar Choubeh R, Cohen E, Eisenberg I, Tamburu C, Dorogi M, Unnep R, Appavou MS, Nevo R, Raviv U, et al. (2017). Changes in aggregation states of light-harvesting complexes as a mechanism for modulating energy transfer in desert crust cyanobacteria. Proceedings of the National Academy of Sciences of the United States of America 114, 9481–9486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Julicher F, and Hyman AA (2009). Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324, 1729–1732. [DOI] [PubMed] [Google Scholar]
- Cohen SE, Erb ML, Selimkhanov J, Dong G, Hasty J, Pogliano J, and Golden SS (2014). Dynamic localization of the cyanobacterial circadian clock proteins. Current biology : CB 24, 1836–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond S, Jun D, Rubin BE, and Golden SS (2015). The circadian oscillator in Synechococcus elongatus controls metabolite partitioning during diurnal growth. Proc Natl Acad Sci U S A 112, E1916–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond S, Rubin BE, Shultzaberger RK, Chen Y, Barber CD, and Golden SS (2017). Redox crisis underlies conditional light-dark lethality in cyanobacterial mutants that lack the circadian regulator, RpaA. Proc Natl Acad Sci U S A 114, E580–E589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzmann TM, and Alberti S (2019). Protein Phase Separation as a Stress Survival Strategy. Cold Spring Harbor perspectives in biology 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden SS, Brusslan J, and Haselkorn R (1986). Expression of a family of psbA genes encoding a photosystem II polypeptide in the cyanobacterium Anacystis nidulans R2. EMBO J 5, 2789–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig JA, Nguyen TA, Lee M, Holehouse AS, Posey AE, Pappu RV, and Jedd G (2020). Arginine-Enriched Mixed-Charge Domains Provide Cohesion for Nuclear Speckle Condensation. Molecular cell 77, 1237–1250 e1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundel M, Scheunemann R, Lockau W, and Zilliges Y (2012). Impaired glycogen synthesis causes metabolic overflow reactions and affects stress responses in the cyanobacterium Synechocystis sp. PCC 6803. Microbiology 158, 3032–3043. [DOI] [PubMed] [Google Scholar]
- Gur E, Biran D, Gazit E, and Ron EZ (2002). In vivo aggregation of a single enzyme limits growth of Escherichia coli at elevated temperatures. Molecular microbiology 46, 1391–1397. [DOI] [PubMed] [Google Scholar]
- Gutu A, and O’Shea EK (2013). Two antagonistic clock-regulated histidine kinases time the activation of circadian gene expression. Mol Cell 50, 288–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinkel F, Abraham L, Ko M, Chao J, Bach H, Hui LT, Li H, Zhu M, Ling YM, Rogalski JC, et al. (2019). Phase separation and clustering of an ABC transporter in Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences of the United States of America 116, 16326–16331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood RD, Higgins SA, Flamholz A, Nichols RJ, and Savage DF (2016). The stringent response regulates adaptation to darkness in the cyanobacterium Synechococcus elongatus. Proceedings of the National Academy of Sciences of the United States of America 113, E4867–4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman AA, Weber CA, and Julicher F (2014). Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol 30, 39–58. [DOI] [PubMed] [Google Scholar]
- Ito H, Mutsuda M, Murayama Y, Tomita J, Hosokawa N, Terauchi K, Sugita C, Sugita M, Kondo T, and Iwasaki H (2009). Cyanobacterial daily life with Kai-based circadian and diurnal genome-wide transcriptional control in Synechococcus elongatus. Proceedings of the National Academy of Sciences of the United States of America 106, 14168–14173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov P, Kedersha N, and Anderson P (2019). Stress Granules and Processing Bodies in Translational Control. Cold Spring Harbor perspectives in biology 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivleva NB, Bramlett MR, Lindahl PA, and Golden SS (2005). LdpA: a component of the circadian clock senses redox state of the cell. EMBO J 24, 1202–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitayama Y, Iwasaki H, Nishiwaki T, and Kondo T (2003). KaiB functions as an attenuator of KaiC phosphorylation in the cyanobacterial circadian clock system. EMBO J 22, 2127–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroschwald S, Munder MC, Maharana S, Franzmann TM, Richter D, Ruer M, Hyman AA, and Alberti S (2018). Different Material States of Pub1 Condensates Define Distinct Modes of Stress Adaptation and Recovery. Cell Rep 23, 3327–3339. [DOI] [PubMed] [Google Scholar]
- Kwiatkowska J, Matuszewska E, Kuczynska-Wisnik D, and Laskowska E (2008). Aggregation of Escherichia coli proteins during stationary phase depends on glucose and oxygen availability. Res Microbiol 159, 651–657. [DOI] [PubMed] [Google Scholar]
- Lambert G, Chew J, and Rust MJ (2016). Costs of Clock-Environment Misalignment in Individual Cyanobacterial Cells. Biophys J 111, 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowska E, Kuczynska-Wisnik D, Bak M, and Lipinska B (2003). Trimethoprim induces heat shock proteins and protein aggregation in E. coli cells. Curr Microbiol 47, 286–289. [DOI] [PubMed] [Google Scholar]
- Li P, Banjade S, Cheng HC, Kim S, Chen B, Guo L, Llaguno M, Hollingsworth JV, King DS, Banani SF, et al. (2012). Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, and Taylor JP (2015). Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan Z, Ryan VH, Janke AM, Burke KA, Rhoads SN, Zerze GH, O’Meally R, Dignon GL, Conicella AE, Zheng W, et al. (2017). Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J 36, 2951–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monterroso B, Zorrilla S, Sobrinos-Sanguino M, Robles-Ramos MA, Lopez-Alvarez M, Margolin W, Keating CD, and Rivas G (2019). Bacterial FtsZ protein forms phase-separated condensates with its nucleoid-associated inhibitor SlmA. EMBO reports 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T, Binder B, and Johnson CH (1996a). Circadian gating of cell division in cyanobacteria growing with average doubling times of less than 24 hours. Proc Natl Acad Sci U S A 93, 10183–10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T, Binder B, and Johnson CH (1996b). Circadian gating of cell division in cyanobacteria growing with average doubling times of less than 24 hours. Proceedings of the National Academy of Sciences of the United States of America 93, 10183–10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanaswamy R, Levy M, Tsechansky M, Stovall GM, O’Connell JD, Mirrielees J, Ellington AD, and Marcotte EM (2009). Widespread reorganization of metabolic enzymes into reversible assemblies upon nutrient starvation. Proceedings of the National Academy of Sciences of the United States of America 106, 10147–10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott TJ, Petsalaki E, Farber P, Jervis D, Fussner E, Plochowietz A, Craggs TD, Bazett-Jones DP, Pawson T, Forman-Kay JD, et al. (2015). Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Molecular cell 57, 936–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell JD, Zhao A, Ellington AD, and Marcotte EM (2012). Dynamic reorganization of metabolic enzymes into intracellular bodies. Annu Rev Cell Dev Biol 28, 89–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry BR, Surovtsev IV, Cabeen MT, O’Hern CS, Dufresne ER, and Jacobs-Wagner C (2014). The bacterial cytoplasm has glass-like properties and is fluidized by metabolic activity. Cell 156, 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, et al. (2015). A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 162, 1066–1077. [DOI] [PubMed] [Google Scholar]
- Patel A, Malinovska L, Saha S, Wang J, Alberti S, Krishnan Y, and Hyman AA (2017). ATP as a biological hydrotrope. Science 356, 753–756. [DOI] [PubMed] [Google Scholar]
- Pattanayak GK, Lambert G, Bernat K, and Rust MJ (2015). Controlling the Cyanobacterial Clock by Synthetically Rewiring Metabolism. Cell reports 13, 2362–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak GK, Phong C, and Rust MJ (2014). Rhythms in Energy Storage Control the Ability of the Cyanobacterial Circadian Clock to Reset. Current Biology 24, 1934–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovska I, Nuske E, Munder MC, Kulasegaran G, Malinovska L, Kroschwald S, Richter D, Fahmy K, Gibson K, Verbavatz JM, et al. (2014). Filament formation by metabolic enzymes is a specific adaptation to an advanced state of cellular starvation. Elife. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu Y, Li Y, Jin X, Tian T, Ma Q, Zhao Z, Lin SY, Chen Z, Li B, Yao G, et al. (2019). ATP-Dependent Dynamic Protein Aggregation Regulates Bacterial Dormancy Depth Critical for Antibiotic Tolerance. Molecular cell 73, 143–156 e144. [DOI] [PubMed] [Google Scholar]
- Puszynska AM, and O’Shea EK (2017). Switching of metabolic programs in response to light availability is an essential function of the cyanobacterial circadian output pathway. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhine K, Vidaurre V, and Myong S (2020). RNA Droplets. Annual review of biophysics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riback JA, Katanski CD, Kear-Scott JL, Pilipenko EV, Rojek AE, Sosnick TR, and Drummond DA (2017). Stress-Triggered Phase Separation Is an Adaptive, Evolutionarily Tuned Response. Cell 168, 1028–1040 e1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y, and Brangwynne CP (2017). Liquid phase condensation in cell physiology and disease. Science 357. [DOI] [PubMed] [Google Scholar]
- Snead WT, and Gladfelter AS (2019). The Control Centers of Biomolecular Phase Separation: How Membrane Surfaces, PTMs, and Active Processes Regulate Condensation. Molecular cell 76, 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanier RY, and Cohen-Bazire G (1977). Phototrophic prokaryotes: the cyanobacteria. Annu Rev Microbiol 31, 225–274. [DOI] [PubMed] [Google Scholar]
- Takai N, Nakajima M, Oyama T, Kito R, Sugita C, Sugita M, Kondo T, and Iwasaki H (2006). A KaiC-associating SasA-RpaA two-component regulatory system as a major circadian timing mediator in cyanobacteria. Proc Natl Acad Sci U S A 103, 12109–12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace EW, Kear-Scott JL, Pilipenko EV, Schwartz MH, Laskowski PR, Rojek AE, Katanski CD, Riback JA, Dion MF, Franks AM, et al. (2015). Reversible, Specific, Active Aggregates of Endogenous Proteins Assemble upon Heat Stress. Cell 162, 1286–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yan X, Aigner H, Bracher A, Nguyen ND, Hee WY, Long BM, Price GD, Hartl FU, and Hayer-Hartl M (2019). Rubisco condensate formation by CcmM in beta-carboxysome biogenesis. Nature 566, 131–135. [DOI] [PubMed] [Google Scholar]
- Wessel D, and Flugge UI (1984). A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Analytical biochemistry 138, 141–143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movies S1. Live-cell Imaging of MetX-EYFP, Related to Figure 2. S. elongatus cells expressing MetX fused with EYFP show fluorescent puncta in dark and their disappearance following light exposure. Frame rate = 1 hr/frame.
Movies S2. Live-cell Imaging of ASD-EYFP, Related to Figure 2. S. elongatus cells expressing ASD fused with EYFP show fluorescent puncta in dark and their disappearance following light exposure. Frame rate = 1 hr/frame.
Movies S3. Live-cell imaging of Tyrosine Phosphatase-EYFP, Related to Figure 2. S. elongatus cells expressing Tyrosine Phosphatase fused with EYFP show fluorescent puncta in dark and their disappearance following light exposure. Frame rate = 1 hr/frame.
Table S1. Protein Solubility in Light and Dark, Related to Figure 1. Proteins identified in the soluble and pellet fractions isolated from light and dark incubated wild-type Synechococcus elongatus cells, using TMT labeling. The proportion of protein in each fraction was estimated from ratios of spectral counts.
Movies S4. Live-cell imaging of Prolyl 4-hydroxylase α-subunit-EYFP, Related to Figure 2. S. elongatus cells expressing Prolyl 4-hydroxylase α-subunit fused with EYFP show fluorescent puncta in dark and their disappearance following light exposure. Frame rate = 1 hr/frame.
Table S2. Isotopic Labeling of Proteins Following a Dark-to-Light Transition, Related to Figure 2. Proteins labelled with heavy leucine (L-Leucine 13C6) identified in the soluble fraction isolated from light incubated cells. After 11 h in dark incubation, cultures were labeled with L-Leucine 13C6 for 1 h and then released to light. Cells were harvested 4 h after light exposure, fractionated and analyzed by tandem mass spectrometry.
Data Availability Statement
The published article includes all analyzed data. Raw mass spectrometry data are available on Chorus (Project 1682) and the codes used for microscope data analysis is available upon request from the lead contact.