

Abstract
Background
Targeted RNA sequencing (RNA-seq) from FFPE specimens is used clinically in cancer for its ability to estimate gene expression and to detect fusions. Using a cohort of NSCLC patients, we sought to determine whether targeted RNA-seq could be used to measure tumour mutational burden (TMB) and the expression of immune-cell-restricted genes from FFPE specimens and whether these could predict response to immune checkpoint blockade.
Methods
Using The Cancer Genome Atlas LUAD dataset, we developed a method for determining TMB from tumour-only RNA-seq and showed a correlation with DNA sequencing derived TMB calculated from tumour/normal sample pairs (Spearman correlation = 0.79, 95% CI [0.73, 0.83]. We applied this method to targeted sequencing data from our patient cohort and validated these results against TMB estimates obtained using an orthogonal assay (Spearman correlation = 0.49, 95% CI [0.24, 0.68]).
Results
We observed that the RNA measure of TMB was significantly higher in responders to immune blockade treatment (P = 0.028) and that it was predictive of response (AUC = 0.640 with 95% CI [0.493, 0.786]). By contrast, the expression of immune-cell-restricted genes was uncorrelated with patient outcome.
Conclusion
TMB calculated from targeted RNA sequencing has a similar diagnostic ability to TMB generated from targeted DNA sequencing.
Subject terms: Cancer genomics, Tumour biomarkers, Molecular medicine, Predictive markers
Background
In the past decade, non-small cell lung cancer (NSCLC) patients have benefited from the discovery of biomarkers predicting response to targeted tyrosine kinase inhibitors (TKI), resulting in significantly improved outcomes in patients whose tumours harbour genomic alterations such as activating mutations in EGFR [1, 2], and cMET [3, 4] and in fusions involving ALK, ROS1, RET [5–7]. As the diversity of biomarkers has increased, diagnostic laboratories have increasingly used molecular methods with broader clinical utility and higher diagnostic accuracy to replace traditional in situ and immunohistochemical techniques for biomarker measurement. In this respect, RNA sequencing (RNA-seq) is becoming the method of choice for the detection of activating gene fusions and transcript variants in NSCLC due to the relative ease with which both recurrent and novel (but likely oncogenic) events may be detected [8–10].
In patients unsuitable for TKI therapy, immune checkpoint blockade (ICB) given alone or in combination with chemotherapy is approved in lung tumours expressing high levels of the negative immune modulator PD-L1 [11]. However, due to inherent issues of PD-L1 assays [12, 13], the measurement of biomarkers for ICB response is also evolving towards genomic alternatives [14]. Of these, tumour mutation burden (TMB) has received the most interest [15]. A high TMB (≥ 10 somatic mutations per megabase, as measured by a US Food & Drug Administration (FDA)-approved test) is now an FDA-approved companion diagnostic biomarker for treatment with the PD-1 inhibitor pembrolizumab in patients that have progressed following prior treatment and who have no satisfactory alternative treatment options, regardless of solid tumour type [16]. TMB in NSCLC might also be particularly relevant for patients receiving the anti-PD-1 antibody nivolumab plus low-dose ipilimumab (anti-CTLA-4) who have a better overall response and progression-free survival (but not overall survival) with high TMB, irrespective of PD-L1 expression [17, 18].
Formal TMB measurement requires whole-exome sequencing (WES) or whole genome sequencing (WGS) but these remain challenging clinical assays due to higher cost and relatively poorer performance on formalin-fixed paraffin-embedded (FFPE) material, and therefore TMB-capable targeted panels have been assessed [19]. Nonetheless, questions about TMB as a predictive biomarker remain, partly due to lack of standardisation but also with accumulating evidence suggesting TMB may be poorly predictive in some solid tumour histologies [20].
Given the utility of RNA-seq for the detection of other molecular biomarkers in NSCLC, we sought to determine whether RNA-seq could also form the basis for measuring TMB. We reasoned that an approach combining three important molecular biomarkers (fusions, splice variants and TMB) into a single assay could have cost-benefit advantages and, due to the inherent property of mRNA deriving only from expressed genes, might predict neoantigen burden, and therefore ICB response, more accurately than a DNA-based TMB. Finally, we sought to determine whether the expression of immune modulators could also be measured, and if so, could this act as an additional biomarker of ICB response.
Methods
RNA-seq library design
Since the primary goal was to develop a method with practical utility in the diagnostic setting, we evaluated RNA-seq methodologies for their suitability for use with formalin-fixed paraffin-embedded (FFPE) specimens. Our initial strategy involved the use of an rRNA-depleted, non-stranded cDNA library (NEBNext Ultra II RNA Library Prep, New England Biolabs Ltd.). Although this strategy theoretically maximised the sequenced region to effectively the transcriptome, we observed a high rate of sequence alignment to non-coding regions of the genome and unacceptably low coverage of coding regions (data not shown). We, therefore, evaluated a probe-based target-enrichment system, the TruSight RNA Pan-Cancer Panel (Illumina, San Diego, CA). Although the sequenced coding region obtainable using this product is limited to a maximum of 1385 targeted genes, this represents up to 3.7 MB of the coding region, over three times larger than popular targeted DNA sequencing panels such as TruSight Oncology 500 (Illumina, San Diego, CA) that are currently used to assess TMB.
Patient cohort
We requested archival FFPE material from the Melbourne Thoracic Malignancies Prospective Cohort Study, a prospective collection of clinical data from patients with lung cancer and other thoracic malignancies consented for access to tissue specimens and treatment data (Supplementary Table 2). All patients gave written informed consent to participate in this research which was approved by the Peter MacCallum Cancer Centre Human Research Ethics Committee 03/90. The initial cohort included 170 patients with advanced NSCLC, treated with first or second-line Anti PD-1-PD-L1 agents, starting between February 2013 and December 2017 at the Peter MacCallum Cancer Centre and the Royal Melbourne Hospital. The clinical information was collected retrospectively. Enough tissue and data for analysis were available for 77 patients, all of whom were treated with second-line nivolumab via a pharma-sponsored access programme. PD-L1 expression levels were not originally measured in these patients as this was not needed for the access programme and unfortunately most cases cannot now be stained due to tissue exhaustion. Responders were patients with a complete or partial response, or stable disease, as assessed retrospectively by the independent review of the radiologic response at the first clinical follow-up. Non-responders were patients who had progressed. A qualified pathologist reviewed an H&E-stained section from all received specimens, and those with at least 1 mm3 of tissue at ≥20 tumour cell nuclei per 100 nuclei and a non-small cell adenocarcinoma or squamous carcinoma lung histology were submitted for processing (n = 93). Ten patients were removed after a systematic review of case notes due to incomplete clinical data. RNA-seq analysis was successful according to standard sequencing metrics on 77 samples (83%). The clinical features of the analysed cohort are summarised in Table 1.
Table 1.
Clinical characteristics of the analysed cohort.
| Characteristic | Value |
|---|---|
| Total no. | 77 |
| Age at diagnosis (years) | |
| Median | 63 |
| Range | 30–83 |
| Gender | |
| Male | 44 |
| Female | 33 |
| Tumour morphology | |
| Adenocarcinoma | 51 |
| Squamous cell carcinoma | 23 |
| Neuroendocrine carcinoma | 2 |
| NSCLC NOS | 2 |
| Agent | |
| Nivolumab | 77 |
| Responder | |
| Yes | 33 |
| No | 44 |
NGS sequencing methods
DNA and RNA were extracted from methyl green stained formalin-fixed paraffin-embedded (FFPE) sections with the AllPrep DNA/RNA FFPE kit (Qiagen, Hilden, Germany) using a QIACube (Qiagen, Hilden, Germany) according to manufacturer’s instructions. Elution volumes of 40 µl for DNA and 30 µl of RNA were used. DNA samples were quantified using the BR Qubit dsDNA assay (Life Technologies, Waltham, MA) and RNA samples using the NanoDrop (Thermo Fisher, Waltham, MA).
For TruSight RNA Pan-Cancer Panel, 100 ng of RNA was synthesised into cDNA, end-repaired and ligated to Illumina adaptors, and sequence indexes added. Libraries were then checked and quantified using D1000 ScreenTapes on a 4200 TapeStation instrument (Agilent Technologies, Santa Clara, CA) all according to the manufacturer’s instructions. Libraries were then normalised to a consistent concentration, hybridised with the TruSight RNA Pan-Cancer probes, and checked and quantified using High Sensitivity D1000 ScreenTapes on a 4200 TapeStation instrument (Agilent Technologies, Santa Clara, CA) all according to the manufacturer’s instructions. Libraries were sequenced on a NextSeq 550DX in RUO mode using NextSeq 500/550 High Output Kit v2.5 (150 Cycles) (Illumina, San Diego, CA) according to the manufacturer’s instructions.
For TruSight Oncology 500, 100 ng of DNA was end-repaired and ligated to Illumina adaptors, and sequence indexes were added. Libraries were then hybridised using Oncology Probes DNA 2 (Illumina, San Diego, CA), and checked and quantified using High Sensitivity D1000 ScreenTapes on a 4200 TapeStation instrument (Agilent Technologies, Santa Clara, CA) all according to the manufacturer’s instructions. Libraries were sequenced on a NextSeq 550DX in RUO mode using NextSeq 500/550 High Output Kit v2.5 (300 Cycles) (Illumina, San Diego, CA) according to the manufacturer’s instructions.
Bioinformatic methods
TCGA lung adenocarcinoma data
Due to the necessity of working with somatic tissue, a critical issue requiring careful optimisation was the filtering strategy employed to remove germline variants from our sequenced reads. In order to validate this approach, we obtained The Cancer Genome Atlas Lung Adenocarcinoma (TCGA-LUAD) dataset as raw RNA-seq reads (fastq files) and DNA variant calls (vcf files) from the Genomic Data Commons (GDC) Data Portal (https://portal.gdc.cancer.gov/). An in-house bioinformatics pipeline was used to process the RNA-seq data as follows: Raw sequencing reads were quality-checked using FastQC v0.11.6 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc), trimmed using cutadapt v1.9.1 [21] then aligned to the GRCh37/hg19 human reference genome using hisat2 v2.0.4 [22]. Duplicate reads were filtered using Picard MarkDuplicates v2.17.3 (http://broadinstitute.github.io/picard). Base quality score recalibration, indel realignment and variants calling were then performed on the filtered reads using the Genome Analysis Toolkit (GATK) v3.8.0 [23].
TruSight RNA pan-cancer panel
Sequencer output was processed and all RNA variants and gene fusions were called using the BaseSpace RNA-Seq Alignment App V2 from Illumina (https://support.illumina.com/help/BS_App_RNASeq_Alignment_v2_OLH_1000000006112/Content/Source/Informatics/Apps/Versions_swBS_appRNASeqA.htm).
TruSight oncology 500
Sequencer output was processed and all DNA variants were called using the TSO500 Local App Software 2.02 from Illumina (https://sapac.support.illumina.com/downloads/trusight-oncology-500-local-app-user-guide-1000000067616.html).
Variant filtering
Estimation of TMB in the absence of the availability of a normal sample requires the removal of variants that are not of somatic origin. These unwanted variants are due to a variety of processes and are filtered using algorithms that take into account their corresponding patterns of occurrence across the population and within samples [24–26]. The most common of these are germline variants which occur in the order of 1000 variants per MB [27] compared with the relevant clinical threshold of 10 somatic variants per MB [16]. Filtering using databases removes most but not all germline variants and so heuristics based on variant allele fraction (VAF) can be used to identify rare variants not in the databases [25, 26], a technique that we also employed (Fig. 1c).
Fig. 1. Filtering variants from tumour-only RNA-seq to compute RNA TMB.

a Flowchart illustrating the sequence of filtering steps used to remove unwanted variants. b All variants in the cohort are aggregated into a table containing loci as rows and samples as columns. This enables the identification of loci-containing multiple variants. These commonly occurring variants (with loci marked by a red cross in the figure) are unlikely to be due to randomly distributed mutations and hence can be removed from TMB calculations based on their frequency within the cohort. c Variants are accepted or rejected based on their allele frequency in each sample. VAFs close to 0 or 1 are removed as are those in a range around 0.5. d RNA TMB versus DNA TMB for the TCGA-LUAD dataset with a linear regression fitted through the origin.
Variants due to post-transcriptional modification of RNA represent another possible source of error. While methods using prior knowledge of biology already exist to identify these [28, 29], we found that it was sufficient for our purposes to remove loci with multiple variants from consideration (Fig. 1b). This strategy has the added advantage of removing both technical artefacts specific to the assay as well as variants at mutational hotspots driven by selective pressure rather than by the (presumed) random processes that operate genome-wide.
The steps used to filter detected variants of all origins are summarised in Fig. 1a and can be described as follows:
Low-quality variants with less than 20 supporting reads were discarded as were coding regions with less than an average supporting read depth of 20.
Potential germline variants were removed by excluding those loci that occurred in the gnomAD database v2.0.1 [30] at a population frequency of more than one in 100,000.
Loci-containing variants in more than one sample were excluded on the basis that these variants could be due to either post-transcriptional modifications (in the case of RNA-seq) or technical/bioinformatic artefacts.
For RNA-seq, variants with allele frequencies less than 0.05 or greater than 0.95 were excluded in order to reject potential germline variants. Variants with allele frequencies between 0.3 and 0.6 were also excluded for the same reason.
Tuning of bioinformatic settings
Filtering as described above results in most unwanted variants and many wanted variants being discarded, affecting the sensitivity and specificity of the assay. Inadequate filtering can lead to the inclusion of unwanted variants leading to an inflated TMB estimate and a dependence on ethnicity [31], while overly stringent filtering removes wanted variants and inflates noise for the resulting TMB correlate. As has been discussed elsewhere [31–33], the selection and tuning of the various bioinformatic parameters used for filtering has a significant impact on TMB estimates. Our own choices were determined empirically using an optimisation procedure to maximise concordance between RNA-derived TMB (RNA TMB) and DNA-derived TMB (DNA TMB) across a patient cohort. In the case of the DNA TMB, somatic variants were removed using either a matched normal (TCGA LUAD) or bioinformatically using the Illumina-supplied TruSight Oncology 500 Local App [26]. The assumption is that the parameters that maximise the correlation of the RNA TMB correlate with the “true” DNA TMB will also provide optimal filtering of RNA variants. Application of this assumption is explored in the next section, but we note here that the parameters eventually chosen (Supplementary Table 1) were found to be applicable to both datasets despite the significant technical differences between them.
RNA expression profiling for targeted genes
Differential expression analysis was also performed on the 1412 targeted regions on the panel in order to identify differentially expressed genes between responders and non-responders. For RNA expression, counts were taken from BaseSpace RNA-Seq Alignment App V2 from Illumina and those regions not significantly expressed in more than five of the samples were discarded. The remaining 1353 genes were normalised using EDASeq [34], and differential expression was subsequently tested using edgeR [35] using as a significance threshold an adjusted FDR of 0.01. In addition, for four known immune-cell lineage-restricted genes, the correlation of log normalised expression with both response and each other was computed.
Statistical methods
Statistical analysis was performed using R4.0.2 [36] and standard packages therein. ROC curves and their uncertainties were computed using the R package pROC [37] while pairwise comparisons and correlations were plotted using the packages GGally [38] and corrplot [39], respectively.
Results
Establishing concordance between RNA TMB and DNA TMB
We applied the filtering strategy shown in Fig. 1a to the TCGA-LUAD dataset in order to test whether removing recurrent variants provided benefits over and above using the gnomAD database. This was done by adjusting both the minimum population frequency of the gnomAD variants used for filtering and the maximum number of times variants could occur at a given loci across all samples, and then examining how this affected correlation between estimates of DNA TMB with matching germline and RNA TMB without matching germline. Supplementary Fig. 2 shows that the optimal choice is to use the most stringent filtering in both cases. In order to assess whether germline variants were escaping filtering we stratified RNA variants by allele frequency and investigated their correlation with total DNA TMB across samples (Supplementary Fig. 3A). The dependence of the correlation on allele frequency supports the use of VAF-based filtering similar to Jessen et al. [25] but with more stringent cut offs around the expected germline VAF of 0.5. We optimised the filter settings as above (Supplementary Fig. 3B) to produce our final RNA TMB estimate (Fig. 1d), which was significantly correlated with DNA TMB (Spearman correlation = 0.79; with 95% CI [0.73, 0.83]).
TMB from RNA-seq and DNA-seq are correlated for the patient cohort
Our patient cohort has significant technical differences to the TCGA-LUAD dataset i.e., all the samples were sequenced from FFPE tissue and the target region for the TruSight RNA Pan-Cancer Panel produces far fewer bases with sufficient read depth for variant calling (median of 2.2 MB with read depth >20 compared with 27.9 MB for exome). Since these two differences act to increase the number of “false” variants and decrease the absolute number of “true” variants, respectively, we sought to determine if the filtering method established for the TCGA-LUAD dataset would be applicable to our independent patient cohort. In fact, these settings appeared to be overly stringent (Supplementary Fig. 4A), resulting in a correlation of 0.49 between the two TMB estimates (Fig. 2). Some improvement was obtained by relaxation of filtering based on allele frequencies (Supplementary Fig. 4B) to the point where TMB estimates from DNA and RNA had a Spearman correlation of 0.66 (Supplementary Fig. 5A). Nevertheless, although we note this observation for completeness, the original filtering settings derived using the TCGA-LUAD dataset were used in the following analyses.
Fig. 2. RNA TMB versus DNA TMB for the lung cancer cohort.
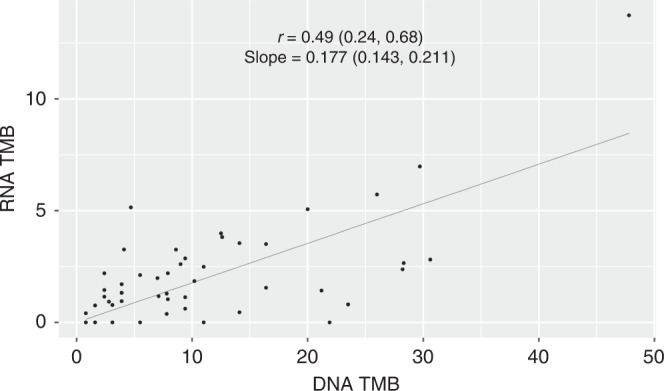
A linear regression line is fitted through the origin.
TMB from RNA-seq predicts response to checkpoint blockade for the patient cohort
Satisfied that our RNA-derived TMB correlate corresponded closely to a clinically validated DNA-based measure of TMB, we sought to test its suitability for use as a biomarker that predicts response to checkpoint inhibitors. RNA-derived TMB was significantly higher for responders (P = 0.028) than non-responders (Fig. 3a), although this was not associated with prolonged survival (Fig. 3b). Further, patient classification using a basic linear model yielded an AUC estimate of 0.640 (with 95% CI [0.493, 0.786], Fig. 3c). The size of the confidence intervals generated by the bootstrapping procedure in Fig. 3c shows the limited power of this particular study and suggests that a larger study is needed to further explore the predictive ability of RNA TMB for predicting response to therapy. As mentioned, this was done using the same settings as was used for Fig. 2 and detailed in Supplementary Table 1. The more permissive VAF filtering settings used in Supplementary Fig. 5A did not result in any greater separation of RNA TMB between groups (Supplementary Fig. 5B) and gave an inferior result for classification.
Fig. 3. Predictive ability of TMB derived from RNA-seq.

a TMB derived from RNA-seq is significantly different between responders and non-responders but is not predictive of overall patient survival time. b The threshold of RNA TMB is set to be equivalent to DNA TMB of 10 mutations/MB and 15 of 63 samples are TMB high. c AUC with confidence intervals derived from a bootstrap analysis of the patient response.
RNA expression profiling for targeted restricted genes
Differential expression analysis was performed in order to identify genes that might be significantly differentially expressed between responders and non-responders. Supplementary Table 2 contains the top 20 hits obtained from this analysis. None of these candidates were judged to be significant using a threshold set at an adjusted FDR of 0.01. Log of normalised expression for four immune-cell lineage-restricted genes was stratified according to patient response in order to further explore if there might be any relationship. Supplementary Fig. 6 shows that while the expression of these genes was uncorrelated with patient outcome, they were significantly correlated with each other.
Discussion
TMB is an emerging biomarker predicting benefit from ICB therapy in NSCLC [40]. A variety of assays have been proposed to measure TMB, with those methods using a larger (>1 MB) survey of the genome more accurately predicting TMB derived from the ‘gold standard’ whole exome [19]. Nonetheless, apart from those assays required to accurately report TMB, molecular profiling of NSCLC for treatment selection is achieved today using relatively small oncogene-targeted sequencing panels of only a few 10’s of kilobases [41]. Additionally, RNA-seq used as a sequential adjunct to a small targeted panel, has gained favour for fusion detection in lung cancer [42], with both sequencing modes able to be performed economically on a benchtop instrument. The goal of this study was to assess whether RNA-seq for the detection of TMB could add to established molecular biomarkers in NSCLC thereby providing a single cost-effective clinical assay. The underlying hypothesis of this approach is that RNA-based assays will accurately predict neoantigen burden, and therefore ICB response. Our results showing that a panel-derived RNA TMB is only 3–4% of the corresponding DNA TMB suggest that RNA TMB does not reflect the actual DNA TMB but is a strong correlate. Possible explanations for this are (i) the corresponding DNA TMB is an over-estimate of the actual TMB, (ii) the size of the candidate region used to normalise for RNA TMB is an over-estimate, (iii) not all the DNA variants are expressed or captured by RNA-seq or (iv) the filtering used to remove germline and low-quality variants also removes most of the actual variants. We cannot discount (i) for the case of the TCGA LUAD but think it most unlikely since our methodology is similar to that used by others [24, 25], while the TMB estimates for the TruSight Oncology 500 assay have been validated elsewhere [43]. With respect to (ii) the normalisation for RNA TMB is computed as the total number of bases in exons with an average coverage greater than 20 since these are the only regions from which variants are counted. However, there is an extra requirement of 20 supporting reads for a variant to be counted which means that the actual number of bases at which variants might be found is somewhat lower and hence our estimate for the denominator used to normalise RNA TMB will be high. In principle, failure to take into account VAF for the purposes of normalisation may lead to aneuploidy or tumour purity impacting on TMB estimates derived from both RNA and DNA. In practice correcting for this is difficult to do, especially for RNA, and so we assume that noise from this effect will be small compared to other sources of error, and therefore ignore it. In relation to (iii), there may well be technical or biological reasons that cause some variants detected with DNA-seq not to show up in RNA-seq however, to the extent that this is an unbiased process, it should not affect the proportionality between the corresponding TMB estimates. This suggests that filtering (iv) is the largest cause of the differential between RNA TMB and DNA TMB. We therefore argue that if we treat the RNA TMB as a correlate to DNA TMB and use a constant of proportionality to convert between them, we can use RNA TMB as a surrogate for DNA TMB. A similar concession was made by Sorokin et al. who used algorithmic filtering of ribosomal RNA-depleted, FFPE-derived, RNA-seq data to generate RNA-seq-TMB scores with good correlation to WES-derived DNA TMB. RNA-seq-TMB scores were ~25% of the corresponding DNA-TMB score when optimal filtering was applied [44], When compared to commercial panels generating DNA TMB at similar per sample cost, targeted RNA TMB has equivalent ability to discriminate ICB responders from non-responders (AUC 0.796 for Illumina TruSight RNA Pan-Cancer Panel v0.65 for Thermo Fisher Oncomine Tumor Mutational Load) [45] in addition to being able to identify more actionable mutations [46]. Nevertheless, validation against a DNA TMB assay using a single cohort, similar to a method comparison for two different DNA-TMB assays, would provide more substantial support for this conclusion.
Having shown that RNA TMB is predictive of patient outcome, we sought to confirm whether additional prognostic biomarkers from the TruSight RNA Pan-Cancer Panel could be leveraged for our NSCLC patient cohort. In our thoracic disease cohort, 22 patients (25.6%) receiving first-line ICB therapy harboured Tier I or II [47] actionable variants detectable by targeted DNA or RNA sequencing (Table 2), confirming the clinical use of these approaches. Given the importance of tumour-infiltrating lymphocytes (TILs) for response to immunotherapy [48], we also explored the expression of immune-cell-restricted genes across the patient cohort. While the genes CD8A, IFNG and CD274 (PD-L1) were not significantly correlated to either RNA TMB or patient outcome, they were all significantly correlated with each other (Supplementary Fig. 6). Differential expression analysis on the 1412 targeted regions on the panel also failed to identify differentially expressed genes between responders and non-responders (threshold set at an adjusted FDR of 0.01). These observations are likely to be due to the presence of a non-neoplastic cell component in the tumour and suggest that unmixing of gene expression might be applied to remove contamination by both TILs and stromal cells. However, unlike an exome, the small size and the selection of genes on the TruSight RNA Pan-Cancer Panel mean that this is not an appropriate strategy here. The small size of the TruSight RNA Pan-Cancer Panel also makes it impractical to derive mutational signatures from the RNA-seq data in tumours with sufficiently high TMB [24] since there are insufficient filtered variants per sample. We do note, however, the possibility of using both FFPE and 'germline' signatures as an overall quality measure for artefact removal across a sample cohort.
Table 2.
Tier I or II driver events discovered in the cohort.
| Gene | Tier | No. of cases |
|---|---|---|
| BRAF | I | 3 |
| EGFR | I | 2 |
| KRAS | I | 4 |
| PIK3CA | II | 1 |
| RET fusion | I | 2 |
| ROS1 fusion | I | 2 |
| Other | II | 8 |
| Total | 22 (28.6%) |
In summary, we have shown that an inexpensive targeted RNA-seq assay may, in addition to revealing actionable gene fusions, generate a TMB correlate that is predictive of IO response in a NSCLC cohort. This finding now needs confirmation on a larger prospective cohort of patients. The reproducibility of this RNA-seq TMB approach across other laboratories should also be investigated.
Although we were unable to detect prognostic gene expression patterns from this data, it is possible that purposeful redesign of the targeted RNA-seq panel may allow effective quantitation of additional independent biomarkers of IO response, such as lymphocytic infiltration within the tumour.
Supplementary information
Acknowledgements
We thank all the patients who participated in this study. We acknowledge the cooperation of the Melbourne Health Shared Pathology Service. We thank Ann Officer and Marliese Alexander for the study coordination. We thank Dr Michael Christie for the pathology review. Some results shown here are based on data generated and made publicly available by the TCGA Research Network (http://cancergenome.nih.gov/).
Author contributions
JFM and APF designed the work, played important roles in interpreting the results, and drafted and revised the manuscript. RL and DC played important roles in interpreting the results and revising the manuscript. TG acquired the laboratory data and drafted the manuscript. TM acquired the clinical data. JLL, TJ, BS and SBF played important roles in interpreting the results and revising the manuscript. All authors approved the final version of this manuscript and are accountable for all aspects of the work.
Funding
This work was funded by Bristol Myers Squibb.
Data availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
Code availability
R code that implements the variant filtering strategies, performs differential expression analysis and generates all figures contained in this manuscript, along with all relevant data are available on Figshare at 10.6084/m9.figshare.21332646.
Competing interests
SF is on advisory boards for AstraZeneca, Pfizer, Merck, Bayer, GSK, Roche, Janssen, Novartis, and Thermo Fisher. Honoraria are paid to Peter MacCallum Cancer Centre. BS is on advisory boards/receives honoraria for Roche/Genentech, Pfizer, Novartis, AstraZeneca, Merck, Bristol Myers Squibb, Amgen, BeiGene, Janssen and Lilly.
Ethics approval and consent to participate
All patients gave written informed consent for their tissue samples and medical history to be used in this research. This research was approved by the Peter MacCallum Cancer Centre Human Research Ethics Committee (HREC ref HREC/17/PMCC/42). This study was performed in accordance with the Declaration of Helsinki.
Consent to publish
All patients gave consent for the publication of anonymised information.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: John F. Markham, Andrew P. Fellowes.
Supplementary information
The online version contains supplementary material available at 10.1038/s41416-022-02105-w.
References
- 1.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 2.Recondo G, Facchinetti F, Olaussen KA, Besse B, Friboulet L. Making the first move in EGFR-driven or ALK-driven NSCLC: first-generation or next-generation TKI? Nat Rev Clin Oncol. 2018;15:694–708. doi: 10.1038/s41571-018-0081-4. [DOI] [PubMed] [Google Scholar]
- 3.Paik PK, Drilon A, Fan PD, Yu H, Rekhtman N, Ginsberg MS, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015;5:842–9. doi: 10.1158/2159-8290.CD-14-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drilon A, Clark JW, Weiss J, Ou SI, Camidge DR, Solomon BJ, et al. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat Med. 2020;26:47–51. doi: 10.1038/s41591-019-0716-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drilon A, Hu ZI, Lai GGY, Tan DSW. Targeting RET-driven cancers: lessons from evolving preclinical and clinical landscapes. Nat Rev Clin Oncol. 2018;15:151–67. doi: 10.1038/nrclinonc.2017.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18:378–81. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- 7.Remon J, Pignataro D, Novello S, Passiglia F. Current treatment and future challenges in ROS1- and ALK-rearranged advanced non-small cell lung cancer. Cancer Treat Rev. 2021;95:102178. doi: 10.1016/j.ctrv.2021.102178. [DOI] [PubMed] [Google Scholar]
- 8.Majewski IJ, Mittempergher L, Davidson NM, Bosma A, Willems SM, Horlings HM, et al. Identification of recurrent FGFR3 fusion genes in lung cancer through kinome-centred RNA sequencing. J Pathol. 2013;230:270–6. doi: 10.1002/path.4209. [DOI] [PubMed] [Google Scholar]
- 9.Reguart N, Teixido C, Gimenez-Capitan A, Pare L, Galvan P, Viteri S, et al. Identification of ALK, ROS1, and RET fusions by a multiplexed mRNA-based assay in formalin-fixed, paraffin-embedded samples from advanced non-small-cell lung cancer patients. Clin Chem. 2017;63:751–60. doi: 10.1373/clinchem.2016.265314. [DOI] [PubMed] [Google Scholar]
- 10.McLeer-Florin A, Duruisseaux M, Pinsolle J, Dubourd S, Mondet J, Phillips Houlbracq M, et al. ALK fusion variants detection by targeted RNA-next generation sequencing and clinical responses to crizotinib in ALK-positive non-small cell lung cancer. Lung cancer. 2018;116:15–24. doi: 10.1016/j.lungcan.2017.12.004. [DOI] [PubMed] [Google Scholar]
- 11.Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375:1823–33. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 12.Hendry S, Byrne DJ, Wright GM, Young RJ, Sturrock S, Cooper WA, et al. Comparison of four PD-L1 immunohistochemical assays in lung cancer. J Thorac Oncol. 2018;13:367–76. doi: 10.1016/j.jtho.2017.11.112. [DOI] [PubMed] [Google Scholar]
- 13.Cooper WA, Russell PA, Cherian M, Duhig EE, Godbolt D, Jessup PJ, et al. Intra- and interobserver reproducibility assessment of PD-L1 biomarker in non-small cell lung cancer. Clin Cancer Res. 2017;23:4569–77. doi: 10.1158/1078-0432.CCR-17-0151. [DOI] [PubMed] [Google Scholar]
- 14.Butter R, t Hart NA, Hooijer GKJ, Monkhorst K, Speel EJ, Theunissen P, et al. Multicentre study on the consistency of PD-L1 immunohistochemistry as predictive test for immunotherapy in non-small cell lung cancer. J Clin Pathol. 2020;73:423–30. doi: 10.1136/jclinpath-2019-205993. [DOI] [PubMed] [Google Scholar]
- 15.Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377:2500–1. doi: 10.1056/NEJMc1713444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21:1353–65. doi: 10.1016/S1470-2045(20)30445-9. [DOI] [PubMed] [Google Scholar]
- 17.Ready N, Hellmann MD, Awad MM, Otterson GA, Gutierrez M, Gainor JF, et al. First-line nivolumab plus ipilimumab in advanced non-small-cell lung cancer (CheckMate 568): outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. J Clin Oncol. 2019;37:992–1000. doi: 10.1200/JCO.18.01042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N Engl J Med. 2019;381:2020–31. doi: 10.1056/NEJMoa1910231. [DOI] [PubMed] [Google Scholar]
- 19.Stenzinger A, Endris V, Budczies J, Merkelbach-Bruse S, Kazdal D, Dietmaier W, et al. Harmonization and standardization of panel-based tumor mutational burden measurement: real-world results and recommendations of the quality in pathology study. J Thorac Oncol. 2020;15:1177–89. doi: 10.1016/j.jtho.2020.01.023. [DOI] [PubMed] [Google Scholar]
- 20.Strickler JH, Hanks BA, Khasraw M. Tumor mutational burden as a predictor of immunotherapy response: is more always better? Clin Cancer Res. 2021;27:1236–41.. doi: 10.1158/1078-0432.CCR-20-3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:3. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 22.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Current protocols in bioinformatics/editoral board. Andreas D Baxevanis [et al.] 2013;43:11 0 1–0 33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DiGuardo MA, Davila JI, Jackson RA, Nair AA, Fadra N, Minn KT, et al. RNA-seq reveals differences in expressed tumor mutation burden in colorectal and endometrial cancers with and without defective DNA-mismatch repair. J Mol Diagn. 2021;23:555–64. doi: 10.1016/j.jmoldx.2021.01.008. [DOI] [PubMed] [Google Scholar]
- 25.Jessen E, Liu Y, Davila J, Kocher JP, Wang C. Determining mutational burden and signature using RNA-seq from tumor-only samples. BMC Med Genomics. 2021;14:65. doi: 10.1186/s12920-021-00898-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao C, Jiang T, Hyun Ju J, Zhang S, Tao J, Fu Y, et al. TruSight oncology 500: enabling comprehensive genomic profiling and biomarker reporting with targeted sequencing. bioRxiv. [Preprint] 2020. https://www.biorxiv.org/content/10.1101/2020.10.21.349100v1.
- 27.Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang C, Davila JI, Baheti S, Bhagwate AV, Wang X, Kocher JP, et al. RVboost: RNA-seq variants prioritization using a boosting method. Bioinformatics. 2014;30:3414–6. doi: 10.1093/bioinformatics/btu577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neums L, Suenaga S, Beyerlein P, Anders S, Koestler D, Mariani A, et al. VaDiR: an integrated approach to variant detection in RNA. Gigascience. 2018;7:gix122. [DOI] [PMC free article] [PubMed]
- 30.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang H, Sasson A, Srinivasan S, Golhar R, Greenawalt DM, Geese WJ, et al. Bioinformatic methods and bridging of assay results for reliable tumor mutational burden assessment in non-small-cell lung cancer. Mol diagnosis Ther. 2019;23:507–20. doi: 10.1007/s40291-019-00408-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merino DM, McShane LM, Fabrizio D, Funari V, Chen SJ, White JR, et al. Establishing guidelines to harmonize tumor mutational burden (TMB): in silico assessment of variation in TMB quantification across diagnostic platforms: phase I of the Friends of Cancer Research TMB Harmonization Project. J Immunother Cancer. 2020;8:e000147. doi: 10.1136/jitc-2019-000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fancello L, Gandini S, Pelicci PG, Mazzarella L. Tumor mutational burden quantification from targeted gene panels: major advancements and challenges. J Immunother Cancer. 2019;7:183. doi: 10.1186/s40425-019-0647-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Risso D, Schwartz K, Sherlock G, Dudoit S. GC-content normalization for RNA-Seq data. BMC Bioinforma. 2011;12:480. doi: 10.1186/1471-2105-12-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Team RC. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2020.
- 37.Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinforma. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schloerke B, Cook D, Larmarange J, Briatte F, Marbach M, Thoen E, et al. GGally: Extension to ‘ggplot2’. 2021; R package version; 2.
- 39.Simko TWaV. R package “corrplot”: visualization of a correlation matrix. 2017. https://bmjopenrespres.bmj.com/content/4/1/e000250#ref-22andelsewhere.
- 40.Galvano A, Gristina V, Malapelle U, Pisapia P, Pepe F, Barraco N, et al. The prognostic impact of tumor mutational burden (TMB) in the first-line management of advanced non-oncogene addicted non-small-cell lung cancer (NSCLC): a systematic review and meta-analysis of randomized controlled trials. ESMO Open. 2021;6:100124. doi: 10.1016/j.esmoop.2021.100124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mosele F, Remon J, Mateo J, Westphalen CB, Barlesi F, Lolkema MP, et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: a report from the ESMO Precision Medicine Working Group. Annal Oncol. 2020;31:1491–505. [DOI] [PubMed]
- 42.Cohen D, Hondelink LM, Solleveld-Westerink N, Uljee SM, Ruano D, Cleton-Jansen AM, et al. Optimizing mutation and fusion detection in NSCLC by sequential DNA and RNA sequencing. J Thorac Oncol. 2020;15:1000–14. doi: 10.1016/j.jtho.2020.01.019. [DOI] [PubMed] [Google Scholar]
- 43.Pestinger V, Smith M, Sillo T, Findlay JM, Laes JF, Martin G, et al. Use of an integrated pan-cancer oncology enrichment next-generation sequencing assay to measure tumour mutational burden and detect clinically actionable variants. Mol diagnosis Ther. 2020;24:339–49. doi: 10.1007/s40291-020-00462-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sorokin M, Gorelyshev A, Efimov V, Zotova E, Zolotovskaia M, Rabushko E, et al. RNA sequencing data for FFPE tumor blocks can be used for robust estimation of tumor mutation burden in individual biosamples. Front Oncol. 2021;11:732644. doi: 10.3389/fonc.2021.732644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alborelli I, Leonards K, Rothschild SI, Leuenberger LP, Savic Prince S, Mertz KD, et al. Tumor mutational burden assessed by targeted NGS predicts clinical benefit from immune checkpoint inhibitors in non-small cell lung cancer. J Pathol. 2020;250:19–29. doi: 10.1002/path.5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benayed R, Offin M, Mullaney K, Sukhadia P, Rios K, Desmeules P, et al. High yield of RNA sequencing for targetable kinase fusions in lung adenocarcinomas with no mitogenic driver alteration detected by DNA sequencing and low tumor mutation burden. Clin Cancer Res. 2019;25:4712–22. doi: 10.1158/1078-0432.CCR-19-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn: JMD. 2017;19:4–23. doi: 10.1016/j.jmoldx.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Linette GP, Carreno BM. Tumor-infiltrating lymphocytes in the checkpoint inhibitor era. Curr Hematol Malig Rep. 2019;14:286–91. doi: 10.1007/s11899-019-00523-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
R code that implements the variant filtering strategies, performs differential expression analysis and generates all figures contained in this manuscript, along with all relevant data are available on Figshare at 10.6084/m9.figshare.21332646.