

Abstract
Real-time monitoring of human health can be significantly improved by designing novel electronic skin (E-skin) platforms that mimic the characteristics and sensitivity of human skin. A high-quality E-skin platform that can simultaneously monitor multiple physiological and metabolic biomarkers without introducing skin irritation is an unmet medical need. Conventional E-skins are either monofunctional or made from elastomeric films that do not include key synergistic features of natural skin, such as multi-sensing, breathability, and thermal management capabilities in a single patch. Herein, we engineered and demonstrated a biocompatible and biodegradable E-skin patch based on flexible gelatin methacryloyl aerogel (FGA) for non-invasive and continuous monitoring of multiple biomarkers of interest. Taking advantage of cryogenic temperature treatment and slow polymerization, we fabricated FGA with a highly interconnected porous structure that displays good flexibility, passive-cooling capabilities, and ultra-lightweight properties that make it comfortable to wear for long periods of time. It also provides numerous permeable capillary channels for thermal-moisture transfer, ensuring its excellent breathability. Therefore, the engineered FGA-based E-skin can simultaneously monitor body temperature, hydration, and biopotentials via electrophysiological sensors and detect glucose, lactate, and alcohol levels via electrochemical sensors. This work offers a previously unexplored materials strategy for next-generation E-skin platforms with superior practicality.
Keywords: electronic skin, wearable electronics, flexible aerogel, biosensors, breathable electronics
Graphical Abstract
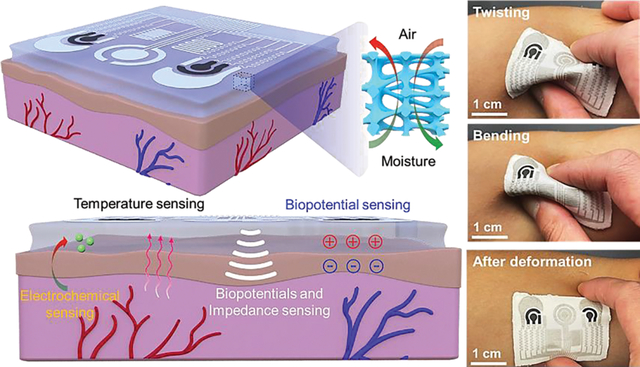
A breathable, passive-cooling, non-inflammatory, biodegradable, and flexible aerogel electronic skin is developed to monitor physical, electrophysiological, and metabolic biomarkers from the human body continuously and simultaneously.
1. Introduction
Human skin is the largest organ of the body, providing an essential somatosensory ecosystem for humans to perceive, and communicate with the physical world. Developing electronic skins (E-skins) that mimic features and functionalities of natural skin hold promise in widespread applications, including healthcare monitoring and management, intelligent human-machine interfaces, and precision medicine.[1–6] The E-skin typically consists of a sensor layer and a substrate.[7] The sensor transduces biomarker information into electrical signals.[8, 9] Whereas the substrate offers flexibility, stretchability, and tolerance to conditions such as high temperature, humidity, and mechanical deformation.[7] In particular, ideal E-skins should integrate multiplexed sensing capabilities on a single wearable device to provide greater insight into the users’ physiological status without hampering their routine or introducing discomfort.[10–14]
To date, various E-skins have been reported for unobtrusive monitoring of single or multiple physical/physiological parameters, including biopotentials (i.e., electrocardiogram, ECG; electroencephalogram, EEG; or electromyography, EMG), body temperature, human motion, skin hydration as well as a library of metabolic markers (such as glucose, insulin, lactate, and cortisol).[15–23] However, most E-skins are limited to conventional elastomeric substrates (e.g., polydimethylsiloxane, PDMS;[18] polyimide;[24] polyethylene terephthalate;[25] and Ecoflex[26]) which suffer from one or more drawbacks, including mechanical mismatch, poor biocompatibility, and low permeability. Such limitations prevent sweat evaporation off the skin and limit the emission of volatile organic components (VOCs), which lead to skin irritation and inflammation.[27–29] Also, typical elastomers are neither disposable nor recyclable, leading to more plastic waste and increased environmental burden.[30] The next generation of high-quality E-skin devices with enhanced comfort and practicality should therefore be breathable, biodegradable, biocompatible, and withstand mechanical deformation especially for long-term on-body use.[30–35]
To address these challenges comprehensively, materials designs for novel E-skin substrates are required. One of the leading candidates is gelatin methacryloyl (GelMA) hydrogel, which is a cost-effective, naturally derived polymer from skin collagen.[36] GelMA hydrogels have been widely used in tissue engineering,[37] bioprinting,[38] and implantations[39] because of their similarities to extracellular matrix (ECM) microenvironments and superior biocompatibility, biodegradability, bioadhesion, and tissue-like mechanical properties.[40–45] However, GelMA can offer additional features compared to its hydrogel forms, such as high porosity, anti-swelling, and ultra-lightweight properties, if used in an aerogel form. Despite some applications of GelMA hydrogel in wearable electronic devices,[36, 46] GelMA aerogel has not previously been explored in E-skin applications, to our knowledge, due to its natural brittleness when in a dry state. Therefore, to use GelMA aerogel for wearable E-Skin applications, it is essential to develop a strategy to prepare flexible GelMA aerogel (FGA) without sacrificing the advantageous features of GelMA.
Here, we explore a novel fabrication strategy by cryofreezing GelMA pre-polymer in liquid nitrogen followed by slow chemical polymerization at a low temperature (−80 °C) to provide flexibility to the GelMA aerogel. Furthermore, numerous three-dimensional (3D) capillary porous channels in FGA substrate offer efficient thermal-moisture transfer that ensures breathable capabilities for dynamic and comprehensive health monitoring. We demonstrate that soft sensor arrays can be integrated into the wearable FGA via scalable screen-printing techniques. Electrophysiological sensors were screen-printed on FGA substrate to monitor skin temperature, skin impedance/hydration levels, and ECG patterns continuously, while electrochemical sensors were integrated to characterize the levels of metabolic biomarkers (i.e., lactate, glucose, and alcohol). The engineered FGA-based E-skin can be in direct contact with the skin and simultaneously sense while stimulating sweat excretion and extracting interstitial fluid (ISF) through reverse iontophoresis. This FGA-based E-skin can detect dynamic physical, physiological, and metabolic responses to daily activities, including food/alcohol intake and exercise, as well as facilitate the early prediction of abnormal vital-sign changes. To the best of our knowledge, FGA with passive-cooling and E-skin functionality for multiplexed chemical-electrophysiological-physical analyses has not been reported previously.
2. Results and Discussion
2.1. Electronic design, device fabrication, and in vitro validation
Fig. 1a schematically shows an overview of the FGA-based E-skin patch worn on the skin. A 3D porous structure of FGA-based E-skin enables its outstanding breathability performance. Air and moisture can easily pass through the interconnected capillary microchannels to balance the thermal-moisture equilibrium of the microenvironment between human skin and the outer environment (Fig. 1b). The sensor arrays and serpentine interconnections were screen-printed onto the flexible and conformal aerogel substrate to construct an FGA-based E-skin. (Supplementary Figs. 1 and 2). Fig. 1c briefly introduces the multimodal sensing functionality of the FGA-based E-skin, including the skin impedance/hydration sensor, temperature sensor, biopotential, and electrochemical sensors. A digital photograph of the prepared FGA-based E-skin resting on a flower is exhibited in Fig. 1d, indicating its ultra-lightweight (approximately 200 mg) and extremely low densities (about 0.03 g/cm3). Fig. 1e displays the conformal capability of FGA-based E-skin on the skin.
Fig. 1. Schematic illustration of the flexible GelMA aerogel-based electronic skin (FGA-based E-skin).
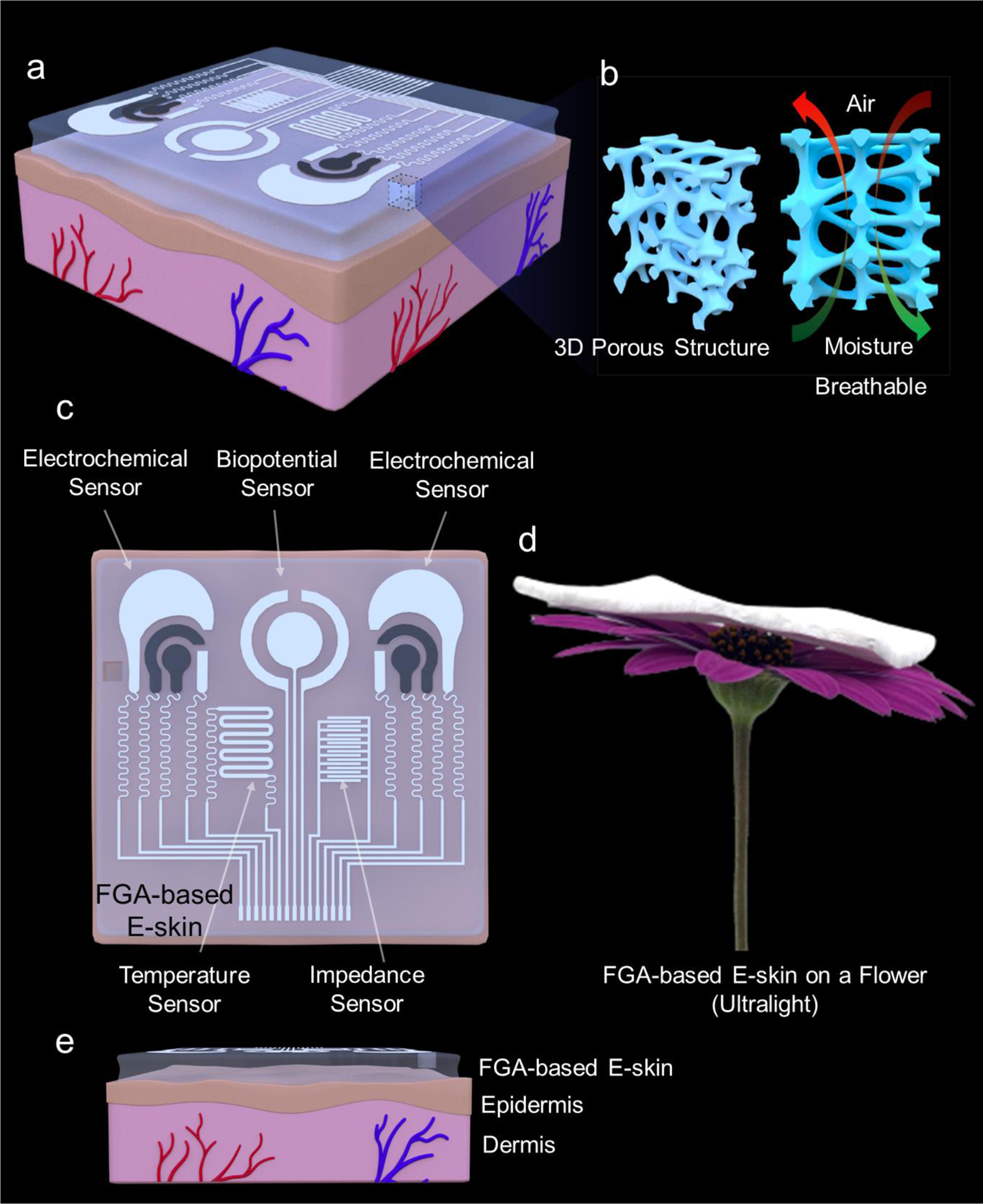
a, Application scenario of the integrated FGA-based E-skin that can be attached to the epidermis. b, Schematic illustration of the three-dimensional porous network structure of the FGA that makes it breathable. c, Top view of the multiple sensors integrated on the aerogel surface, including electrochemical, biopotential, temperature, and impedance sensors. d, FGA-based E-skin on a flower, indicating its ultra-lightweight. e, Side view of the FGA-based E-skin worn on the epidermal surface to suggest the conformal capability of the FGA-based E-skin.
We designed an impedance sensor for measuring the hydration level of the skin, as follows. We placed two interdigitated electrodes on the epidermis surface. Interdigitated patterns were chosen to maximize the interactions between the electrodes in a relatively small area. Fringing fields generated by the electrodes penetrate the upper skin layer, as shown conceptually in Supplementary Fig. 3 and Supplementary Note 1. The conductive ink was screen-printed in an interdigitated pattern with a finger spacing of 200 μm. In addition, the flexibility of the GelMA aerogel substrate enables conformal and soft contact with the skin surface. The impedance sensing capabilities of the FGA-based E-skin were characterized in vitro using a gelatin-based skin phantom. First, impedance frequency sweeps were carried out across various hydration states as determined by a commercial hydration meter (Supplementary Fig. 4). As expected, the measured impedance decreases as the skin phantom dries out because of its lower water content. Supplementary Fig. 5 demonstrates the average impedance at 100 kHz for each case plotted against the standard values of the commercial hydration meter.
To measure skin temperature, we used the thermoresistive effect, which leads to changes in the resistance of conductive materials as temperature varies due to thermally enhanced charge transport (resistance decreases) or thermally introduced charge carrier scattering (resistance increases). Silver/silver chloride (Ag/AgCl) conductive ink was used to print the temperature sensor because Ag has a high thermal coefficient of resistance with a value of 3.8 ×10−3/°C.[47] The performance of the FGA-based E-skin was evaluated across a broad temperature range from 25 to 62 °C using a commercial hot plate as a temperature controller. As shown in Supplementary Fig. 6, the resistance changes were highly correlated with the corresponding temperature changes. Additionally, the time response of the FGA-based E-skin to the temperature was fast (a few seconds) upon heating and subsequent cooling (Supplementary Fig. 7). Note that the performance is limited by the ramp rate of the hot plate. These results indicate that FGA-based E-skin can reliably monitor skin temperature changes in response to environmental stimuli.
For ECG monitoring, we employed a concentric ring design, enabling Laplacian filtering to localize the potential bioelectric source with spatial resolution regulated by the ring distance.[48] The Ag/AgCl traces were printed to form robust conductive interconnections with a resistivity of a few Ω. When mounted on the forearm, continuous ECG measurement is shown in Supplementary Fig. 8, and the ECG is recorded with identified P, Q, R, and S waveforms depicted in Supplementary Fig. 9.
For metabolic biomarker analyses, electrochemical detection was used by implementing non-invasive sweat stimulation via transdermal pilocarpine delivery at the iontophoresis anode and ISF extraction via reverse iontophoresis at the cathode. Alcohol and lactate levels were tracked in sweat, whereas glucose levels were monitored in the ISF by modifying the working electrodes at their respective anodic/cathodic compartments. Chronoamperometry was applied for electrochemical measurements of the hydrogen peroxide generated from the alcohol oxidase (AOx), lactate oxidase (LOx), and glucose oxidase (GOx) enzymatic reactions. By contrast, in control experiments, no signal was captured or detected with the sensors without the surface modification with GOx, LOx, or AOx (Supplementary Figs. 10, 11, and 12). The enzymatic biochemical sensors for glucose, lactate, and alcohol were evaluated by making standard additions to their targets in 0.1 M phosphate-buffered saline (PBS). The amperometric response to glucose was investigated from 0–9 mM with 1 mM increments and showed a strong linear response induced by hydrogen peroxide reduction (Supplementary Fig. 13). This response was also robust in the presence of electroactive interferents (i.e., ascorbic acid and uric acid) and the other target biomarkers (lactate or alcohol) due to the selectivity of the enzyme-based biosensing (Supplementary Fig. 14). Similarly, the amperometric response of the alcohol sensor was evaluated over a broader range of 0–100 mM with 10 mM increments (Supplementary Fig. 15) and showed good selectivity in the presence of physiologically relevant interferents (Supplementary Fig. 16). Finally, the lactate sensor also showed a robust amperometric response in the tested range of 0–20 mM with 5 mM additions, even in the presence of interferents (Supplementary Figs. 17 and 18).
2.2. Flexible aerogel synthesis and characterization
The porous structures are essential to make the GelMA aerogel flexible. Currently, several attempts have been made to regulate the pore size and interconnectivity of the GelMA hydrogels by tuning GelMA concentration,[49] photoinitiator concentration,[50] degree of methacrylate substitution, and crosslinking time[51]. A few reports have also explored the effects of freezing temperatures and freezing time on their pore morphologies.[51, 52] It was found that such polymerized GelMA hydrogel with smaller pores could be obtained by lowering the freezing temperatures (i.e., −10 to −80 °C). However, GelMA molecules were covalently bonded together in this case, and the fixed GelMA polymer chain could prevent water molecules from rearranging and impede ice crystal nucleation, yielding low pore interconnectivity and a lack of flexibility in the structure. To this end, Mooney et al. studied the effect of the sub-zero temperature (−12 °C) regime during the polymerization of the GelMA to develop shape-recovery cryogels with highly interconnected porosity.[53] However, the group did not study the impact of their fabrication process on the flexibility of the dry cryogels. Indeed, their fabrication strategy could not maintain the shape recovery property of the cryogels when GelMA concentration increased above 1.5% w/v.
Motivated by this previously reported literature that validates the impact of low-temperature using liquid nitrogen on the flexibility of other polymers (i.e., poly(2-hydroxyethyl methacrylate) cryogels,[54] alginate (or cellulose)-based cryogels,[55, 56] and cellulose-chitosan aerogels[57]), we firstly employed such liquid nitrogen-assisted strategy to make GelMA aerogel flexible, which has not been studied before.
In addition, the freezing regime is critical in managing pore size and interconnectivity. At sub-zero temperatures, water solvent forms ice crystals, acting as inherent progens. Subsequent freeze-drying removes the ice crystals and maintains the pore structures where the ice crystals had formed. Utilizing these ice crystals as a temperature to regulate porosity, pore size, and interconnectivity, we first explored the effect of the cryogenic temperature regime on the flexibility of GelMA aerogels.
For typical fabrication, GelMA pre-polymer solution was cast in PDMS molds, followed by gelation at room temperature, storage at −80 °C and subsequent freeze-drying, which resulted in the formation of a brittle GelMA aerogel (BGA). Such brittleness could be attributed to the development of irregular ice crystals that randomly formed between the tightly bonded GelMA polymers, resulting in non-uniform pore structures with low interconnectivity (i.e., in the form of closed pores) as summarized in Supplementary Fig. 19, left panel.
One strategy to offer flexibility to cryostructures is applying freezing temperature before and/or during the polymerization of GelMA chains.[53] In this case, due to the mobility of the GelMA macromolecules, water molecules could freely contact each other and, therefore, form uniform interconnected ice crystals (Supplementary Fig. 19, right panel). GelMA would slowly self-organize and polymerize surrounding the ice crystals. Herein, we found that snap freezing of GelMA pre-polymer in liquid nitrogen (ca. −196 °C), followed by slow polymerization at low temperature (−80 °C), and subsequent freeze-drying, yielded FGA (Fig. 2a), which easily bent, fold, and roll without breakage (Supplementary Fig. 20).
Fig. 2. Mechanical and Electrical Characterization of the flexible GelMA aerogel (FGA).
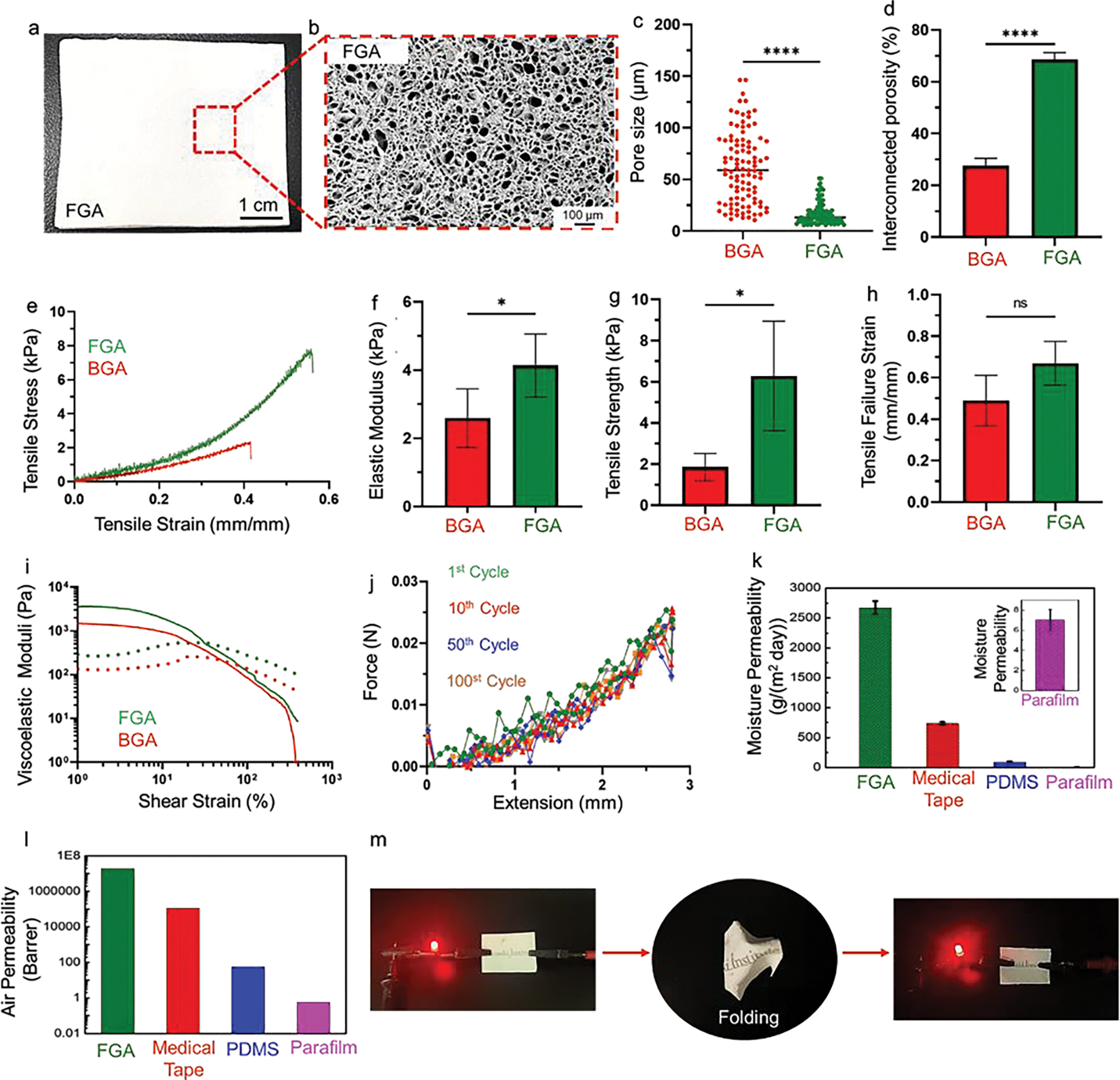
a, Digital image of the FGA. b, Scanning electron microscope image to show the microscale porous structure of FGA. c, Pore size distribution of conventional brittle GelMA aerogel (BGA) and FGA. d, Interconnected porosity of the FGA versus BGA. e, Representative tensile stress-strain curves for the FGA and BGA. f, Elastic moduli of the FGA as compared to BGA. g, Tensile strength of the FGA and BGA. h, Tensile failure strain of the FGA versus BGA. i, Viscoelastic moduli of the FGA as compared to BGA. j, Cyclic testing of the FGA for 1 cycle, 10 cycles, 50 cycles, and 100 cycles, separately. k, comparison of moisture permeability of the FGA, medical tape, polydimethylsiloxane (PDMS), and parafilm. Inset: moisture permeability of the parafilm. l, Air permeability measurement of the FGA. m, Photograph of the FGA with screen-printed “Terasaki Institute” letters through conductive silver ink. After cursing and unfolding, the FGA remained conducting.
As noted earlier, the freezing temperature has a significant effect on pore structure and morphology. For instance, lower freezing temperatures generate smaller pores with higher interconnectivity because the solvent freezes faster, preventing the further growth of ice crystals. To investigate the effects of our fabrication process on the pore size and morphology, scanning electron microscopy (SEM) was performed to show that FGAs have highly porous structures with smaller pore sizes and more uniform pore distributions than BGAs (Fig. 2b, Supplementary Fig. 21). The average pore size measured from the SEM images was approximately 17 μm and 61 μm for FGAs and BGAs, respectively (Fig. 2c). Since the formation of pores depends on the growth of ice crystals, uniformity in pore size of the FGAs is attributed to cryogenic temperature regime as compared to other sub-zero temperatures (−1 to −20 °C).[58–60] Furthermore, cryogenic temperatures significantly reduce chemical crosslinking (i.e., covalent interactions) of the methacrylate groups governed by ammonium persulfate (APS) and tetramethylethylenediamine (TEMED),[53] allowing the pronounced effect of physical crosslinking and easy molecular rearrangements that results in the formation of smaller and highly interconnected pores in FGAs. Incorporating inorganic salts such as PBS, APS, and TEMED in GelMA solutions further enhances physical crosslinking between the polymer chains due to ionic interactions and hydrogen bonding at cryogenic temperatures.[61, 62] Other factors that affect the pore structure and flexibility of the cryostructures are the concentration of the polymers and the thickness of the cryostructures. Higher concentrations of the polymer decrease the porosity, thus reducing flexibility.[56] The thinner aerogel reduces the heterogeneity of the temperature across its depth, resulting in uniform ice crystal formation and better flexibility after freeze-drying.[60]
We further investigated the interconnectivity of the micropores. The FGAs showed interconnective porosity of about 69%, 2.5 times higher than that for BGAs (Fig. 2d). Such highly uniform pore interconnectivity of FGAs supported complete shape recovery of the aerogels after deformation (Supplementary Mov. 1), whereas the BGAs broke under high strain and could not retain their original architectures (Supplementary Mov. 2). Retention of structural architecture for FGAs during mechanical deformation is also attributed to the small pore sizes, which could reinforce increased pore density throughout the aerogel and provide structural support to maintain the framework.[63]
Further characterization of FGAs by nitrogen adsorption analyses showed a pore volume of 0.02 cm3/g with a surface area of 40 m2/g (Supplementary Fig. 22). Fig. 2e shows representative tensile stress-strain curves of the FGAs and BGAs. The elastic moduli of FGAs calculated from the slope of the stress-strain curves were significantly higher than that of the BGAs (Fig. 2f). The tensile strength of FGAs is approximately 6.3 kPa, which is more than three-fold stronger than BGAs (Fig. 2g). The FGA displays over 70% stretchability compared with BGA (Fig. 2h). The advantageous mechanical properties of FGAs are the result of such highly porous structures and self-organization-mediated physical crosslinking, which provide increased crystallinity in the polymeric network (i.e., hydrogen bonding) at cryogenic temperatures.[64] Similarly, our rheology data demonstrated that FGAs have even higher viscoelastic moduli than BGAs (Fig. 2i), which suggests the presence of extended crosslinked polymer backbones. To characterize the strain recoverability of the FGAs, we performed cyclic tensile tests up to 100 loading and unloading cycles (Fig. 2j). The FGAs could recover almost entirely without significant hysteretic loss, indicating complete energy recovery during unloading. Such recovery during cyclic deformations is related to non-covalent interactions, which allow the reversible dynamic rearrangement of the physically crosslinked polymeric backbone.
Additionally, the FGAs absorbed almost 1000% of their volume of water within the first 10 min, which is 13 times their original weight, and subsequently reached equilibrium. By contrast, the BGAs absorbed approximately 50% less water than the FGAs and reached equilibrium in 30 min, slower than FGAs (Supplementary Fig. 23). Note that no obvious changes in the original dimensions and structures of the FGAs were observed after water uptake, indicating excellent anti-swelling behavior, which is essential for FGA-based E-skins.
As validated in Fig. 2k,l and Supplementary Fig. 24, such porous FGAs provide excellent breathability. The moisture permeability of FGAs (2675 g m−2 day) was much larger than that of PDMS (91 g m−2 day), Parafilm (7 g m−2 day), and commercial medical tape (734 g m−2 day). By contrast, the moisture permeability of PDMS (300 μm), a widely used polymer elastomer in flexible electronics (e.g., electronic skin), was negligible compared to the FGA. At this point, FGAs are hydrophobic. That is, they are permeable to moisture but waterproof. The hydrated FGAs rendered the sample hydrophilic, where the water contact angle of the FGAs was decreased from 100.6° to 50.3° after hydration (Supplementary Fig. 25), which facilitates efficient contact of the sweat with the sensors. Furthermore, the air permeability of the FGAs was much higher than that of commercial medical tape (over 100 times), which is compatible with the perspiration of the human body under different scenarios such as exercise. In contrast, the air permeability of PDMS and parafilm were below 60 Barrer.
We screen-printed electrodes onto the FGAs to test their mechanical properties under extreme conditions. After crushing and then unfolding the electrodes on FGA, the electrodes remained conductive, as shown in Fig. 2m. The mechanistic explanation is that soft conductive ink printed on this FGA substrate remains conductive during folding and unfolding, undergoing deformation to tolerate the applied stress. To test the robustness of the printed electrodes on the FGA, we investigated cyclic bending and twisting deformation while monitoring the resistance changes across the electrodes. As shown in Supplementary Fig. 26, the relative resistance varied less than 20% (with initial resistance values ~50 Ω), even after 100 cycles of deformation. Such results confirm that the mechanical flexibility of the FGAs, in combination with stretchable and screen-printable inks, yields robust electrical contacts.
Further, we investigated the direct and conformal contact of the FGA to the skin using a standard wound closure test setup (Supplementary Fig. 27a). The FGA patch (100 mm2) was applied on porcine skin, where they were incubated for varying amounts of time. The skins were then axially displaced while the adhesion force was recorded. As shown in Supplementary Fig. 27b, the adhesion force steadily increased by displacement for all the samples until the FGA patch was detached from either side of the skin, corresponding to the abrupt decline in force. At the early stages of the test, the interfacial adhesion was strong enough to attach the FGA patch to the dynamic skin tissue (Supplementary Fig. 27cI) until the excess load forcefully delaminated the FGA (Supplementary Fig. 27cII). Independent of the time attached to the skin, all the FGA patches showed adhesive strengths over a range of 1.5 to 2.5 kPa (Supplementary Fig. 27d), which is consistent with the reported values for adhesive patches.[65] Such adhesive strength can be maintained for at least 72 h. We attribute the self-adhesive property to the rapid absorption of body fluids by dry FGA, facilitating electrostatic forces and hydrogen bonding with the tissue surface. These results make the FGA an attractive candidate for E-skin devices that adhere to the skin. However, to enhance the adhesive strength and to maintain the long-term stability and durability of the FGA patch on the skin, a small amount of commercial medical tape was applied (only) on the boundaries of the FGA-based E-skin, ensuring that the breathability of the device was not significantly affected by the tape. The twisting and bending test of the patch on the skin were shown in Supplementary Fig. 28, indicating its on-body conformability and mechanical integrity during and after deformation.
Furthermore, direct and close contact of the patch with the skin is of great importance to ensure high-quality sensing performance, especially for long-term monitoring of ECG. Herein, we demonstrated the effectiveness of the patch adhesion by continuously monitoring ECG for three hours. Recordings during the rest appear in Supplementary Fig. 29. The graphs present typical ECG signals with sharp features. The overall data reveal no significant difference from 0 h to 3 h, indicating the excellent self-adhesion property of the E-skin patch. In addition, we have also provided videos to show the real-time ECG monitoring by attaching the patch to the skin without adding external medical tape, as shown in Supplementary Mov. 3 and Supplementary Mov. 4.
2.3. Biocompatibility, non-inflammatory, and biodegradability of the flexible aerogel
To assess the biocompatibility of the aerogel on human skin, in vitro cytocompatibility assay against human dermal fibroblast (HDF) cells was performed. Direct incubation of the cells on the surface of the FGA-based E-skin did not induce a negative effect on the morphology of the cells for up to 7 days. Staining live cells with green-fluorescent dye (Calcein AM) suggested excellent cell viability, as a majority of the cells stained green (Fig. 3a, I). Likewise, when the FGA-based E-skin was not directly in touch with the cells and placed inside the transwells (indirect method), the cells maintained their elongated morphology with a slightly smaller number of dead cells (red spots in Fig. 3a, I) compared to the direct method. The overall viabilities of the cells in the direct and indirect methods were 91% and 97%, respectively, compared to the non-treated control (Fig. 3a, II). Moreover, extended incubation of the cells with the FGA-based E-skin for up to seven days did not affect the metabolic activity and proliferation of the cells, as indicated by the PrestoBlue assay. The cells in direct and indirect contact with the FGA-based E-skin showed higher fluorescence intensity by day 7 than by day 1, indicating no adverse effects on the metabolic activity of the cells (Fig. 3a, III).
Fig. 3. Biocompatibility and biodegradability of the flexible GelMA aerogel (FGA).
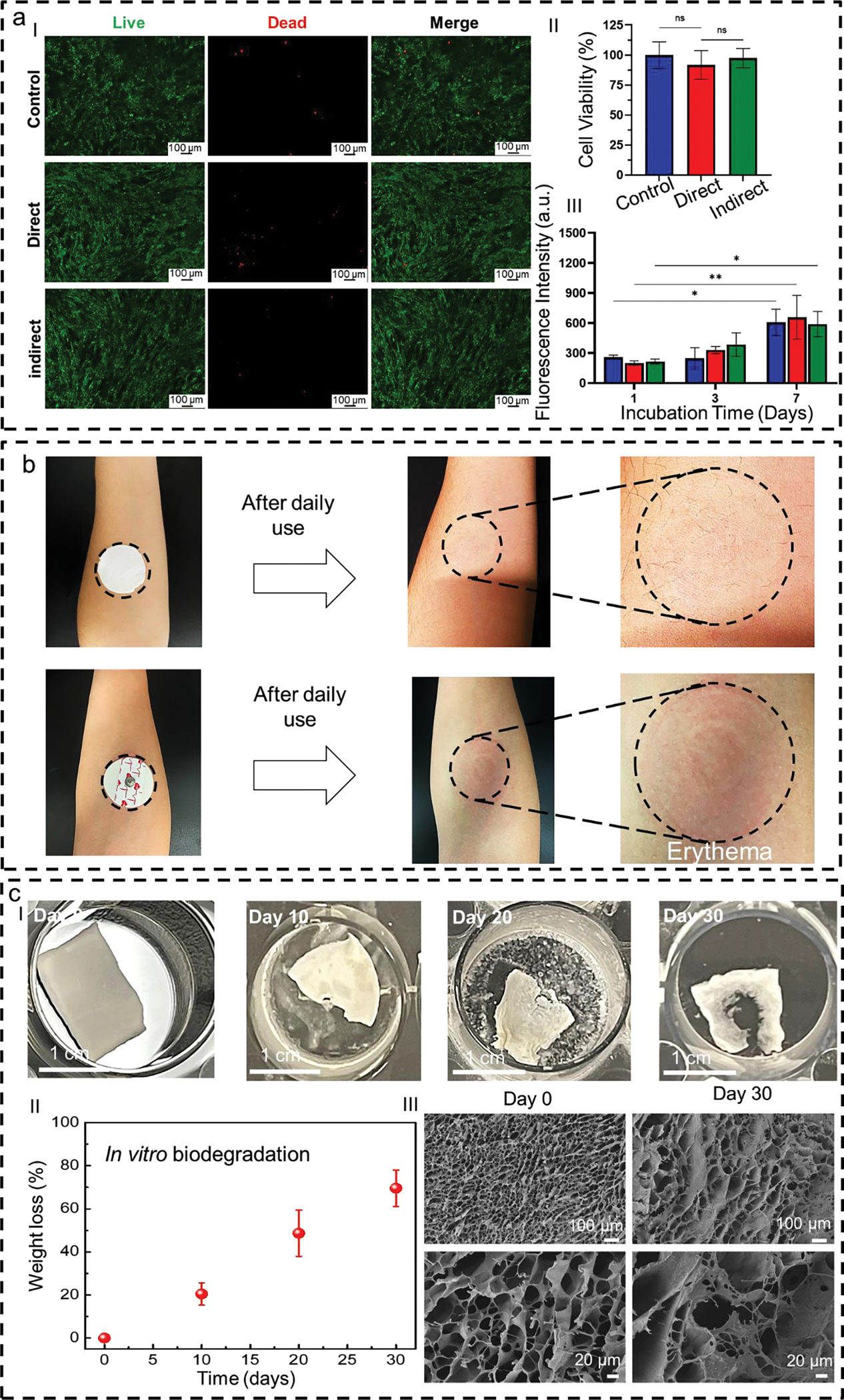
a, Fluorescent images of cells cultured in the incubation medium with the FGA (I). Quantification of human dermal fibroblast cell viability (II) and fluorescence intensity obtained by PrestoBlue assay in the presence of FGA after 1, 3, and 7 days of incubation (III). Error bars show a standard deviation of 4 replicates. b, Images of an FGA mounted on the forearm. It does not induce significant adverse effects after one day of wearing. By contrast, the skin has obvious erythema after 30 min of wearing gel electrodes. c, Sequential photograph of in vitro biodegradation of FGA in phosphate-buffered saline (PBS) solution containing enzyme (I). Weight loss rates of the FGAs within the degradation period of 30 days (II). Scanning electron microscope images to show the surface morphology of the FGAs before (Day 0) and after the 30-day degradation (III).
We also conducted on-skin tests to validate the importance of breathability for long-term wearability. For comparison, two patches (the FGA-based E-skin and the commercial gel electrode) were attached to the forearms of one adult subject for one day of use. As illustrated in Fig. 3b, the breathable FGA-based E-skin did not cause any adverse effects on the skin after daily use. In contrast, the commercial gel electrode induced obvious signs of skin erythema because of its poor permeability.
Biodegradation represents a significant feature of the E-skin, enabling it to operate over the desired time frame while still physically degrading into nonharmful constituents after use. GelMA is well-known to be biodegradable and can successfully break down over a controlled period.[41] We carried out in vitro biodegradation investigations of FGA-based E-skin over 30 days. The variations of their morphology and weight loss after corresponding degradation periods were recorded (Fig. 3cI). The results revealed bulk degradation and autocatalytic hydrolysis after 20 days of incubation, with weight loss of almost 60% (Fig. 3cII). Moreover, it almost reduced by at least 80% of its original weight after a 30-day degradation period. Concurrently, cracks appear on the FGA-based E-skin surface, and its density increases as the degradation progress (Fig. 3cIII). Additionally, the degradation time of the FGA-based E-skin is tunable based on the thickness of the GelMA aerogel and the concentration of GelMA. It can also be tailored depending on the intended wearable lifetime of the device.
2.4. Passive-cooling capabilities of the flexible aerogel
Point-of-care wearable thermal management strategies to address Joule heating and personal cooling can help regulate human body temperature, significantly improving user comfort and energy saving.[66, 67] Although E-skins with personalized heating capabilities have been widely investigated,[68, 69] Developing E-skin that can passively cool human bodies without external energy consumption remains less explored. Porous materials, such as polyethylene, poly(vinylidene fluoride-co-hexafluoropropene), and polystyrene-blockpoly(ethylene-ran-butylene)-block-polystyrene, have been reported with passive-cooling features.[70–72] However, the balance between pore-size control and skin-similar mechanical properties impedes their ability to be integrated with E-skin platforms. Considering the importance of thermal management for E-skin applications, which could counteract deleterious phenomena, such as Joule heating, to improve human comfort and reduce energy consumption,[71] we also investigated the potential passive-cooling effects of the FGA-based E-skin. Fig. 4a and b depict the Fourier-transform infrared (FTIR) and ultraviolet-visible (UV-vis) spectra of the FGA-based E-skin, respectively. Due to the multiscale porous nature of the flexible GelMA aerogel, it can effectively reflect sunlight while allowing human-body mid-infrared to be mainly transmitted unimpeded. The spectrum of the GelMA can be characterized by comparing the characteristic peaks at 1532 cm−1 and 1643 cm−1 in Supplementary Fig. 30, which are attributed to the C-N stretch of amide groups and C=O stretch of the gelatin peptide groups, respectively. Additional peaks for GelMA were reported by Arica et al. and included 3255 cm−1, 1451 cm−1, and 1245 cm−1, which were attributed to N-H stretch, and in-plane stretching of -CH2 groups, respectively.[48, 73] Given these promising results, we conducted an outdoor on-body test by wearing the FGA on the skin (Fig. 4c). The inset reveals its highly porous structure. A thermal camera was employed to compare skin temperature with/without the FGA-based E-skin. These results show that the temperature of the skin covered with the FGA was maintained at temperatures ca. 5 °C lower than that of uncovered skin after 30 min of sunlight radiation exposure (Fig. 4d).
Fig. 4. Passive-cooling feature of the Flexible GelMA aerogel (FGA).
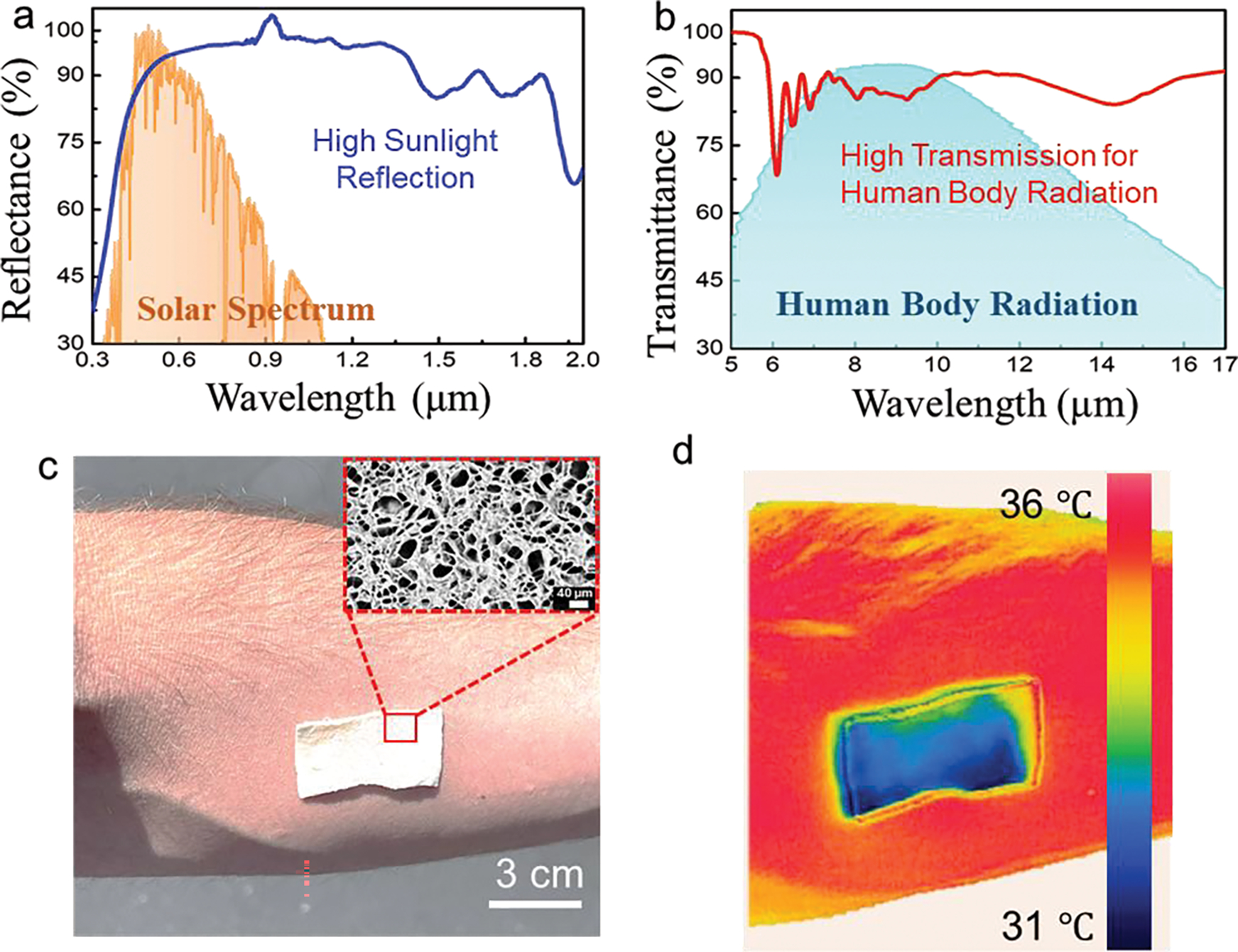
a, The ultraviolet-visible (UV-Vis) reflectance spectrum of the FGA is between 0.3 and 2.0 μm. The normalized ASTM G173 Global solar spectrum is shaded as orange as a reference. b, Fourier-transform infrared (FTIR) spectrum of the FGA between 5 and 17 μm. The human-body mid-IR radiation is shaded as the blue regions as a reference. All suggest that the FGA has a high sunlight reflectance and excellent thermal emission. c, Camera image of the FGA attached to human skin. The inset shows the porous structures of the FGA. d, Thermal mapping of the FGA on human skin, recorded using an infrared (IR) thermal camera. Photo credit: Martin C. Hartel.
2.5. On-body validation of the flexible aerogel-based E-skin with multiple stimuli
We then validated the E-skin’s performance in realistic scenarios in which subjects underwent various activities or were exposed to multiple stimuli that may have competing effects on the physical, physiological, and/or metabolic responses of the body. The E-skin was first investigated for use in tracking physical and physiological signals in addition to multiplexed biochemical monitoring on subjects exposed to different stimuli. A typical paradigm to demonstrate a direct effect on glucose and alcohol levels is consuming sugar- and alcohol-containing aliments or participating in an exercise routine. The reasoning is that glucose levels can be quickly depleted during exercise to produce energy, while glucose/alcohol can be rapidly metabolized while eating/drinking. To study these effects, the subject was asked to have a sugar-rich meal with alcohol-containing red wine while the ISF glucose and sweat alcohol levels were tracked before and after each step. Normal glucose and alcohol levels were observed before eating/drinking (Fig. 5aI,II). Afterward, the glucose and alcohol increased accordingly (Fig. 5aIII,IV). For comparison, a commercial fingerpick blood glucose tester and breathalyzer were applied to measure the blood glucose and blood alcohol levels separately, which have similar trends as the FGA-based E-skin (Fig. 5aV,VI). These results indicate that the FGA-based E-skin is able to detect and track daily activities, including simultaneous food and wine intake, the digestion of food and wine that produces glucose as an energy reservoir, and the accumulation of alcohol in the blood. Excessive alcohol consumption can cause severe hypoglycemia and hypotension, even when administered simultaneously with glucose intake, especially for insulin-dependent diabetics.[74] Therefore, concurrent monitoring of alcohol and glucose is helpful in differentiating between moderate and excessive drinking and can help prevent drinking-related incidents, particularly for those with underlying health issues.
Fig. 5. On-body validation of flexible gelatin methacryloyl aerogel-based electronic skin (FGA-based E-skin).
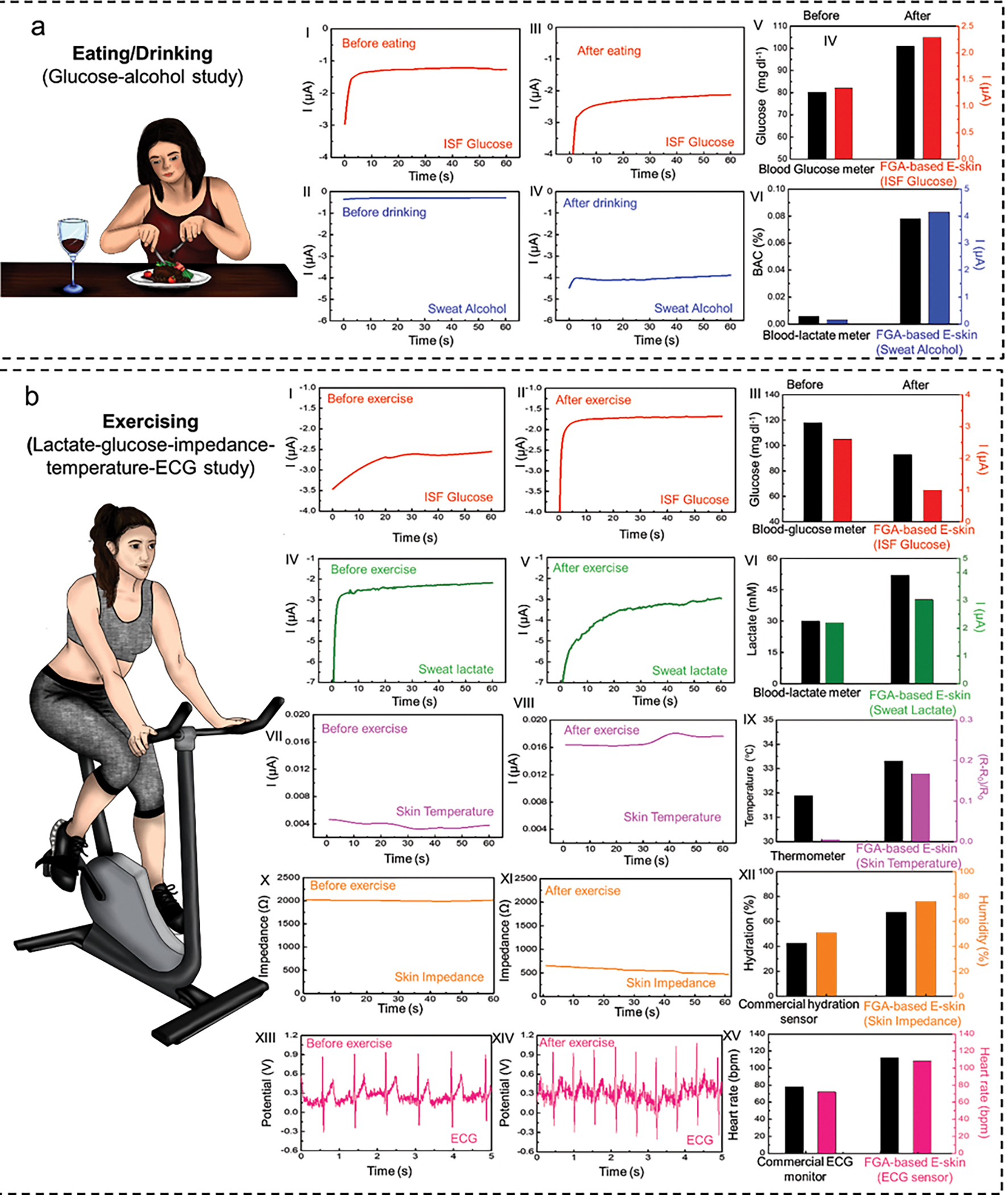
a, Multi-event study of the FGA-based E-skin involving food and alcohol consumption events. (I-II), Electrochemical sensor signal recordings for ISF glucose before and after food intake. (III) Comparison between the glucose levels in interstitial fluid (ISF) measured using the electrochemical sensor and in blood using a blood glucose meter. (IV-V) Electrochemical sensor signal recordings for sweat alcohol levels before and after wine intake. (VI) Comparison between the alcohol levels in sweat measured using the FGA-based E-skin and in blood using a commercial blood-lactate meter. b, Multiplexed chemical-electrophysiological analysis during exercise. (I-II) Electrochemical sensor signal recordings for ISF glucose levels before and after high-intensity exercise. (III) Comparison between the glucose levels in ISF measured using the FGA-based E-skin and in blood using a commercial blood-glucose meter. (IV-V) Electrochemical sensor recordings for sweat lactate levels before and after exercise. (VI) Comparison between the alcohol levels in sweat using the FGA-based E-skin and blood using a commercial blood-alcohol meter. (VII-VIII) Physiological signal recordings for skin temperature variations before and after exercise. (IX) Comparison between skin temperature measured using the FGA-based E-skin and a commercial infrared thermometer. (X-XI) Physiological signal recordings for skin impedance/hydration levels before and after exercise. (XII) Comparison between the skin impedance/hydration levels measured using the FGA-based E-skin and commercial hydration sensor. (XIII-XIV) Physiological signal recordings for electrocardiogram (ECG) before and after exercise. (XV) Comparison between the heart rate measured using the FGA-based E-skin and commercial ECG monitor.
Exercise, which includes any physically demanding action, has a significant impact on the physical, physiological, and metabolic response of the body, including changes in body temperature, skin hydration, ECG patterns, heart rate, and glucose/lactate levels. During high-intensity exercise, the lactate levels in the blood and sweat increase because of the anaerobic metabolic pathway, whereas blood glucose levels decrease due to glucose consumption during exercise to produce energy via aerobic metabolism. Meanwhile, the heart rate rises to meet the muscles’ demand for oxygen, and the average body temperature increases according to sharply increased muscle metabolism. Concurrently, the skin hydration level also increases due to sweat generation. To study these effects, the subjects were asked to have a sugar-rich meal and then to perform high-intensity stationary cycling for 30 min, followed by 5 min of rest.
The ISF glucose, sweat lactate, skin temperature, skin impedance/hydration levels, and ECG patterns were monitored by FGA-based E-skin before and after exercise. As shown in Fig. 5b, high glucose levels (Fig. 5bI), low lactate levels (Fig. 5bII), normal skin temperature (Fig. 5bIII), impedance/hydration (Fig. 5bIV), and ECG patterns (Fig. 5bV) were measured before the cycling activity. The glucose level (Fig. 5bVI) and skin impedance decreased after exercise (Fig. 5bIX), accompanied by an increase in sweat lactate levels (Fig. 5bVII), skin temperature (Fig. 5bVIII), and heart rate (Fig. 5bX). The findings are consistent with a previously reported study using a similar electrochemical monitoring system.[75] Note that all the sensor performances were validated by commercial devices, as shown in Fig. 5bXI,XII,XIII,XIV,XV.
2.6. Continuous on-body validation of flexible aerogel-based E-skin
In addition to the previously demonstrated end-point measurements, the FGA-based E-skin is also capable of continuously capturing dynamic biosignals, and physiological signal fluctuations in real time, which was demonstrated in Fig. 6 and Supplementary Fig. 31. Physically active individuals are expected to have lower resting heart rates, indicating efficient heart function and good cardiovascular fitness. The lower resting heart rate can be characterized by smaller heart rate increases, as higher cardiac output is required. Smaller increases in lactate levels, temperature, and skin hydration are also expected. The heart rate of an individual is expected to increase following an intense workout, eventually returning to normal baseline levels. Such complex dynamic processes require hybrid sensors (such as electrochemical and physiological sensors) to continuously capture these real-time fluctuations through the activity. In order to validate the ability of the FGA-based E-skin to capture and detect such changes, subjects were asked to perform high-intensity exercises. The continuous sweat lactate profile was measured by FGA-based E-skin, indicating higher sweat lactate levels during the workout (Fig. 6a,b). Additionally, higher hydration levels (Fig. 6c), skin temperature (Fig. 6d), and heart rate (Fig. 6e) were recorded before, during, and after exercise, which had consistent trends, as expected. Therefore, we conclude that our durable and reliable FGAs hold promise in personalized healthcare applications, rehabilitation of patients, and athletic performance optimization.
Fig. 6. Continuous on-body validation of flexible gelatin methacryloyl aerogel-based electronic skin (FGA-based E-skin).
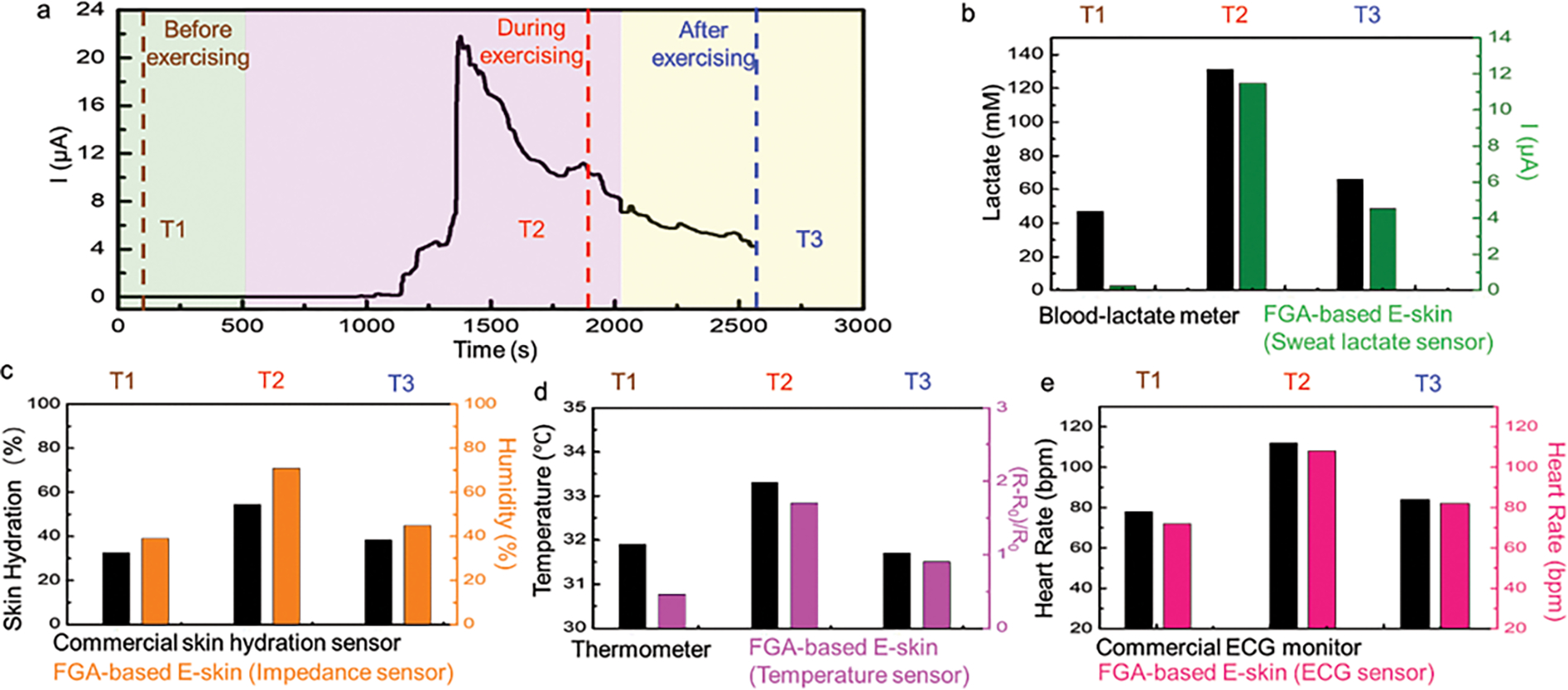
a, Continuous signal recording showing the sweat-lactate profile during high-intensity stationary cycling. Dashed lines mark the time corresponding to the plotted physiological signals recordings before, during, and after exercise. b, validation using a commercial blood-lactate meter and FGA-based E-skin readings. Multiplexed signal readings before, during, and after stationary cycling. c, comparison between skin hydration signal measured by a commercial skin hydration level and FGA-based E-skin before, during, and after cycling. d, comparison between skin temperature signal measured by a commercial thermometer and FGA-based E-skin before, during, and after cycling. e, comparison between heart rate signal measured by a commercial electrocardiogram (ECG) monitor and FGA-based E-skin before, during, and after cycling. For an example, an ECG trace, see Supplementary Fig. 9.
3. Conclusions and Outlook
The seamless integration of biomaterials with diversified functions is a significant breakthrough in developing next-generation flexible electronics, including wearable and implantable electronics. There are two main aspects to these advances: 1) Although current flexible electronics with high sensitivity and high functional integration have been developed, conventional polymeric substrates with mechanical mismatches and limited moisture/gas permeability can cause skin inflammation and irritation and reduce comfort, especially for the long-term on-body use. However, such comfort, safety, and inflammation concerns are often ignored, impeding practical applications to a large extent. GelMA is a promising candidate to address these long-standing issues due to its tunable mechanical properties, excellent biocompatibility, and breathable porous network. 2) The majority of electronic substrate materials are not degradable or recyclable, which introduces plastic waste once they reach their end-of-life period and potentially pollute the environment downstream. GelMA-based biodegradable green electronics can function over the prescribed time frames and then degrade completely into nonharmful products without any adverse effects, alleviating the environmental burden of wearable electronics.
Continuous monitoring of physical, physiological, and metabolic biomarkers can potentially offer new insight into the pathophysiology of the body. This multifunctional GelMA aerogel E-skin platform has thus been validated to support the possibility of developing hybrid wearable sensors with complex integration of physical, physiological, and chemical sensing functionalities on a single flexible and conformal patch to record multiple vital signs in real time.
We have demonstrated a breathable, passive-cooling, biocompatible, biodegradable, and flexible FGA-based E-skin for non-invasive, real-time, and simultaneous monitoring of hybrid chemical-electrophysiological-physical signals. We validated the performance of this aerogel E-skin by monitoring the ISF glucose, lactate, and alcohol levels from sweat, skin temperature, impedance/hydration, and ECG patterns as model analytes. We investigated this multiplexed configuration in response to stimuli associated with regular daily activities, such as fitness, exercise, and food and wine consumption. Although the integrated aerogel E-skin platform suggests promising functionalities, plenty of opportunities remain to improve such devices: (1) full miniaturization of the system through the development of integrated electronics capable of processing and wirelessly transmitting data from the various sensor modalities;[15] (2) integrating other modules (for example, drug-delivery systems) to form a closed-loop theranostic platform; (3) expanding the multiplexed sensing capability to additional analytes (e.g., growth factors, insulin, or cortisol); and (4) conducting comprehensive validation involving more participants with different health conditions, including patients with cardiovascular disease, diabetes, and/or alcohol use disorder. By addressing these opportunities, we envision an integrated multiplexed wearable healthcare system that can offer insights into the physiological and overall health status of individuals while empowering them to self-regulate and manage various chronic diseases. This work paves the way for a multifunctional aerogel-based E-skin capable of providing informative data regarding human healthcare and lays the foundation for next-generation, patient-centered diagnostic and therapeutic tools.
4. Experimental Section
Materials:
Porcine gelatin, Dulbecco’s phosphate-buffered saline (DPBS), Dulbecco’s modified eagle medium (DMEM), methacrylic anhydride, dialysis tubing (12–14 kD), Milli-Q water, tetramethylethylenediamine (TEMED), ammonium persulfate (APS) were purchased from Thermo Fisher Scientific (Waltham, MA). SYLGARD silicone elastomer kit was obtained from Dow (Midland, MI). Glucose oxidase (GOx), lactate oxidase (LOx), chitosan, bovine serum albumin (BSA), agarose, pilocarpine nitrate, acetic acid, ascorbic acid (AA), uric acid, ethanol, lactate, glucose, 0.5% trypsin-EDTA, fetal bovine serum, and penicillin/streptomycin (Pen/Strep, 10000 U/mL), kerosene, safflower oil, propanol, and Triton-X-100 were obtained from Sigma-Aldrich (St. Louis, MO). Prussian blue (soluble) and carbon ink were purchased from Sun Chemical Ltd (Parsippany-Troy Hills, NJ). The Ag/AgCl conductive ink was obtained from Creative Materials, Inc (Ayer, MA). The human dermal fibroblast was purchased from American Type Culture Collection (ATCC), USA. All reagents were used without further purification.
Fabrication of gelatin methacryloyl:
Gelatin methacryloyl (GelMA) was prepared according to our previously published procedure.[76] The reaction started by adding 10 g of gelatin to 100 mL of pre-heated DPBS on a magnetic stirring hotplate (240 rpm) at ~50 °C. Methacrylic anhydride (MA, 8 mL) was then added dropwise to the dissolved gelatin under continuous stirring at dark and allowed to react for ~2 h at 50 °C and 240 rpm. Next, 100 mL of pre-heated (~40 °C) DPBS was added to the solution and magnetically stirred for ~10 min at 50 °C to stop the reaction. The solutions were transferred to dialysis tubing and dialyzed against Milli-Q water at 40 °C and 400 rpm for 5–7 days to remove unreacted MA. After completing the dialysis process, the solution was transferred to the 50 mL centrifuge tubes, placed at −80 °C overnight, and freeze-dried (Free zone, 2.5 L, Labconco, USA) for 7 days to form white GelMA foams. The freeze-dried GelMA foams were placed in moist-free condition at room temperature until use.
Fabrication of conventional (brittle) gelatin methacryloyl aerogel:
Conventional brittle GelMA aerogel (BGA) was fabricated by dissolving freeze-dried GelMA (6% w/v) in DPBS at ~60 °C for ~20 min. Equal volumes of APS (1% w/v in DPBS) and an additional 0.25% v/v TEMED were added to the GelMA mixture to yield the GelMA pre-polymer. The pre-polymer mixture was transferred to a PDMS mold (6 mm × 5 mm × 2 mm) and allowed to crosslink for ~1 h at room temperature to form GelMA hydrogels with a final concentration of 3% w/v. The hydrogels were frozen at −80 °C for 24 h, followed by freeze-drying for 1 day to form 2 mm-thick BGAs. The samples were stored in a dry cabinet at room temperature before use in the experiments.
Fabrication of flexible aerogel:
To fabricate FGAs, freeze-dried GelMA (6% w/v) was allowed to dissolve completely in DPBS at 60 °C for ~20 min. Then, an equal volume of refrigerated-cold APS (1% w/v in DPBS) was added to the GelMA solution as an initiator to adjust the final concentrations of GelMA and APS to 3% and 0.5% w/v, respectively. Next, TEMED (0.25% v/v) as a catalyst was added to the mixture and snapped vortexed for 3 s. The solution was immediately poured into pre-cooled PDMS molds (6 mm × 5 mm × 2 mm) and quickly transferred to liquid nitrogen to slow down the gelation process and to form small and uniform interconnected ice crystals. The liquid nitrogen-frozen samples were placed at −80 °C for 24 h for slow completion of the crosslinking process. Finally, the samples were freeze-dried for 1 day to form an aerogel. The aerogels were placed in a tightly closed container and stored in a moist-free environment (i.e., dry cabinet) at ambient temperature until use.
Fabrication of the flexible aerogel-based electronic skin:
The screen-printing was carried out with customized-built stainless steel stencils with 8-inch by 10-inch in width and length, which were designed using AutoCAD software (Autodesk) and fabricated by Metal Etch Services (San Marcos, CA). The sensors were then fabricated via stencil masks to pattern two sets of iontophoresis (IP) electrodes, two reference electrodes, and other physiological electrodes. All the sensors and interconnections were printed with flexible Ag/AgCl inks. The electrochemical sensors were additionally patterned with carbon working and counter electrodes. After each printing step, the resulting device was baked at 60 °C in a convection oven for ca. 30 min to cure the conductive ink thoroughly. The biosensor electrodes were selectively modified by drop-casting their respective enzyme and polymer layers.
Lactate biosensor:
Chitosan solution was mixed with LOx (40 mg/mL) in BSA solution at a ratio of 1:1 (v/v), followed by drop-casting the mixture (4 μL) onto the working electrode surface. All BSA solutions used in biosensor fabrications were prepared in PBS at a concentration of 10 mg/mL. The PBS used in the electrode modifications for all the biosensors was prepared in 0.1 M with a pH of 7.4. The chitosan solution was prepared by dissolving 0.5 wt% of chitosan in 0.1 M acetic acid.
Alcohol biosensor:
AOx (10–40 units/mg in BSA solution) was mixed with the chitosan solution at a ratio of 8:1 (v/v), and 4 μL of the mixture was dropped cast onto the working electrode surface.
Glucose biosensor:
Equal volume of GOx (40 mg/mL) in BSA solution was mixed with the chitosan solution, and 4 μL of the mixture was dropped and cast onto the working electrode surface. After drying at room temperature for 1 h, another 2 μL of the chitosan solution was dropped cast to all previously enzyme-modified surfaces, followed by storing at 4 °C overnight to ensure complete drying before the test.
For the dual IP process, epoxy molds of 1.5 mm thickness were printed via MONO resin 3D printer (Boston, MA). The cathode hydrogel solution was prepared by dissolving 4% w/v agarose in 0.1 M PBS solution at 150 °C while stirring until all agarose was visibly dissolved. Under the same temperature and stirring conditions, the anode hydrogel solution was prepared by dissolving a 4% w/v agarose solution in DI water. For the anode hydrogel, the temperature was immediately decreased to 60 °C after agarose dissolution, and pilocarpine was added at 2% w/v under continuous stirring. Either 200 μL (for the anode) or 300 μL (for the cathode) aliquots of the warm solutions were added into each circular mold and allowed to solidify. After the solution cooled down and completely solidified in the mold, the gels were removed and stored in a wet chamber at 4 °C until further use.
In vitro calibration of the sensors:
The printed sensors (glucose, lactate, alcohol biosensors, temperature, skin hydration, and ECG sensors) were calibrated separately in in vitro settings. The biosensors were calibrated using 0.1 M PBS (pH 7.4) and by spiking the corresponding analytes while recording the amperometric responses for glucose, lactate, and alcohol. Note that all amperometric responses were recorded after 1 min incubation in the sample solutions by applying a potential of −0.2 V (vs. Ag/AgCl) over 1 min. The selectivity of all sensors was evaluated by comparing their response to the target in the presence of common electroactive interferences, specifically 10 μM UA and 10 μM AA, in addition to the other biomarkers of interest (10 mM lactate and 1 mM glucose). Finally, one final standard addition of the target biomarker was added to verify that the sensors could accurately track concentration changes in such complex environments.
Glucose sensor:
The calibration curve of the glucose sensor was obtained using an initial 100 μL PBS drop on the sensor surface. The solution was spiked with 4 μL of 0.1 M glucose solution to increase the concentration of glucose incrementally from 0 to 9 mM after each spiking. The selectivity of the glucose sensor was evaluated by performing amperometry while spiking the PBS successively with glucose. The glucose sensor was examined by performing 10 repetitive measurements of 2 mM glucose and calculating its relative response changes in %.
Alcohol sensor:
The calibration curve of the alcohol sensor was obtained using an initial 100 μL PBS droplet on the sensor surface. The solution was spiked with 1 μL of 0.8 M ethanol solution to increase the concentration of alcohol incrementally from 0 to 100 mM after each spiking. The selectivity of the alcohol sensor was evaluated by performing CA while spiking the PBS measured successively with ethanol (20 mM), lactate (10 mM), glucose (0.2 mM), and ascorbic acid (10 μM).
Lactate sensor:
The calibration curve of the lactate sensor was obtained using an initial PBS droplet (100 μL volume) on the sensor surface. The solution was spiked with 1 μL of 0.5 M lactate solution to increase the lactate concentration incrementally from 0 to 30 mM with CA at −0.2 V for 60 s after each spiking.
Temperature sensor:
The calibration curve of the temperature sensor was characterized on a hot plate with gradually increasing temperature from 24 to 64 °C. A thermocouple thermometer (Fluke 80PK-1) was used to obtain the local temperature on the temperature sensor, and a precision source meter (B2902A, Keysight) was used to monitor the resistance of the sensor by four-probe measurements.
Impedance sensor:
The calibration curve of the impedance sensor was obtained over the 0–100 kHz range using a gelatin-based skin-mimicking phantom prepared according to Sunny et al.[77] Briefly, a 1:1 mixture of kerosene and safflower oil was brought to 70 °C. Meanwhile, 4 g of gelatin was added to 21.5 mL of deionized water and sonicated at a temperature of 80 °C for 15 min. 1.5 mL of propanol was then added to eliminate leftover bubbles. The oil mixture and gelatin solution were then mixed at a temperature of 46 °C and combined with 1.5 mL of Triton-X-100. The mixture was then poured, covered, and cooled at room temperature until testing. To drive water evaporation and mimic different hydration states, the gel was placed on a hotplate at 45 °C in between measurements and allowed to cool completely before repeating measurements.
ECG sensor:
The volunteer was either at rest or exercising. Two FGA-based E-skin patches containing the abovementioned concentric ECG electrodes were directly placed on the volunteer’s left and right forearms. A commercially available Ag/AgCl electrode was attached to the volunteer’s left elbow as a reference electrode. The ECG signals were collected by a commercial ECG board (Sparkfun AD8232 ECG monitor) and recorded by an Oscilloscope (Tektronix DPO3014 digital phosphor oscilloscope). This electronic circuit board was powered at approximately 3.3V by a portable power source (Dr.Meter PS-305DM power supply). Note that this circuit board relied on an AD8232 chip to amplify and filter the raw ECG signals. In this work, the amplification gain was 100 (V/V). Hence, a peak-to-peak ECG signal of 10 mV would be measured to 1 V. The established setup can be found in Supplementary Mov.3 and Mov.4.
On-body validation of flexible aerogel-based electronic skin:
Epidermal evaluation of the FGA-based E-skin was performed on healthy adult individuals with no heart conditions, diabetes, or chronic pain and in strict compliance with the protocol approved by the institutional review board at the University of California, Los Angeles (IRB #17-000170). Further, informed written consent of all participants was also obtained.
The FGA-based E-skin was placed on the forearm for all on-body validations. Volunteers were instructed to clean their forearm with soap and water and thoroughly dry it before the sensor was placed. An additional sensor fabrication step for on-body tests involved laser cutting double-sided tape (3M, United States) to provide better body adhesion and form fluid chambers for the sensors. The glucose, lactate, alcohol, temperature, skin hydration, and ECG signals were validated with a commercial glucometer (CareTouch) (Brooklyn, NY), blood-lactate meter (the EDGE, USA), infrared thermometer (ETEKCITY) (Anaheim, CA), skin analyzer (SK-8, FANTEXY) (Shenzhen, China), and ECG monitor (Prince 180B, Heal Force) (Shanghai, China), respectively, before every set of measurements using the integrated sensor. Sweat stimulation and ISF extraction were realized simultaneously using an Agilent B2902A source-measure unit (Santa Clara, CA) to apply a current density of 0.3 mA cm2 between the cathode and anode IP electrodes for 10 min. In all cases, a pre-conditioning step was carried out on the skin by first applying the same current density using agarose gels for 10 min, followed by immediate placement of the enzyme-modified FGA-based E-skin device with the anode pilocarpine gel on the conditioned area. All amperometric measurements were read at −0.2 V for 60 s immediately after 10 min of iontophoresis at 0.3 mA cm2 with the pilocarpine-loaded gel. For all measurements, a single device was used for each volunteer to perform the “before” and “after” tests. The device was kept on the forearm of the volunteer throughout the entire experiment unless otherwise specified.
Exercise:
Volunteers were instructed to mount an exercise bike for 25 min, followed by a 5 min cool-down period. Herein, the FGA-based E-skin was applied immediately before exercise to acquire the baseline values and subsequently removed and placed in a humid chamber so that exercise-induced sweat would not confound the glucose ISF measurements. Glucose and lactate levels, temperature, skin hydration, and ECG signals were acquired for the healthy volunteers before and after exercise. The FGA-based E-skin was removed from their skin during the 30 min workout on a stationary bike and kept for further use.
Alcohol intake:
Alcohol levels were measured before and 20 min after alcohol consumption (250 mL red wine, alcohol 17% vol). The FGA-based E-skin was kept on the forearm of the volunteers during the entire experiment.
Food intake:
Signals for ISF glucose were acquired for the healthy volunteers in the fasting state (12 h) and 20 min after consuming sugar-rich food. The FGA-based E-skin was kept on the forearm of the volunteer throughout the entire experiment.
Continuous on-body validation during exercise:
The FGA-based E-skin was further tested by monitoring dynamic changes in sweat lactate during ongoing physical activity. Individuals were asked to perform 30-min cycling activities at constant intensity activity. The skin temperature, skin hydration, ECG, and lactate levels were measured just before the start of the exercise, and the sweat was first generated. These signals were recorded within approximately 20 min of the exercise. These selected signals were also recorded on completion of the exercise for validation. Note that no IP electrodes/gels were needed for this protocol since the exercise-induced sufficient sweating.
Characterization and measurements:
The air permeability was measured by using a customized setup based on the ASTM E96 standard; the testing process is as follows: a 20 mL sized glass bottle was filled with 15 mL of the distilled water, then sealed with a sample using double-sided tape. The bottle was placed in a chamber with a temperature of 35 °C and relative humidity of 40%. The mass of the bottle was measured every two days. The water vapor transmission was calculated based on the mass change. Electrical characterization was performed at room temperature using a CHI660E potentiostat (Englewood, CO) with its accompanying software. Temperature analysis was performed with an Agilent B2902A source-meter unit under an applied bias of 100 mV controlled by Keysight quick IV measurement curve software. Spectral reflectance and transmittance of the flexible GelMA aerogel in the sunlight and mid-IR range were obtained using a UV/Vis spectrophotometer (Shimadzu UV-3101 PC; Kyoto, Japan) with an integrating sphere and a Fourier transform IR spectrophotometer (Agilent 620/660; Santa Clara, CA), respectively.
Imaging with the scanning electron microscope (SEM):
The surface morphology and porous interior structure of the FGA-based E-skin were characterized by a field-emission SEM (Supra 40 VP, Zeiss, Germany). Specifically, the GelMA aerogels (i.e., flexible and brittle) were adhered to double-sided carbon tape and placed on SEM holders. The samples’ surfaces were coated with iridium using a sputter coater (South Bay Technology, USA). The images were captured at an accelerating voltage of 12 kV. ImageJ software (Version 1.52e, USA) was used to analyze pore size with a two-dimensional (2D) aspect.
Adhesion test:
A standard wound closure test was conducted to assess the adhesive strength of FGA to porcine skin following the ASTM F2458-05 protocol with slight modifications.[78] The porcine skin was purchased from a slaughterhouse and cut into pieces with dimensions of 20 mm × 10 mm × 2 mm. The skins were utilized in the experiments within 6 h of sacrificing the animals. Before the experiment, skin moisture was maintained by a wet towel in a humidifying chamber. Two pieces of the cut skin were put together such that their ends touched. The FGA (20 mm × 5 mm × 2 mm) was placed on top of the touching edges, and the adhesion test was performed immediately (0 h) or 8 h and 72 h after incubating the FGA on top of the skin in a humidifier chamber. Force-displacement curves were plotted at a rate of 4 mm/min, using Bluehill software (Version 3, USA). The adhesive strength was calculated by dividing the force at the point of delamination by the cross-sectional area of the FGA under the force.
Assessment of pore interconnectivity:
To analyze the interconnectivity of the aerogel’s pores, dry samples were immersed in DPBS for 30 min to hydrate fully and to reach equilibrium liquid uptake. The weight of the hydrated samples was recorded. Next, the excess DPBS was lightly blotted with a Kimwipe for ~30 s, and the samples were weighed again. The interconnected pore volume was measured as the weight of the blotted DPBS divided by the total weight of the hydrated samples.
Rheological characterization:
The rheological properties of FGA and BGA were characterized by a rheometer (Anton Paar, MCR 302, USA) using a sandblasted parallel-plate geometry (8 mm) with a gap of 1 mm at 25 °C. Oscillatory strain sweep experiments were conducted over a range of 1–1000% strain at 1 Hz. The storage and loss moduli were plotted versus strain by Anton Paar Rheocompass software.
Contact angle characterization:
The surface energy of the FGA-based E-skin was characterized by measuring the water contact angles at room temperature after depositing 20 μL of Milli-Q water on the flat surface of the aerogels and subsequent imaging. Images were analyzed by image-J software (version 1.53a, National Institute of Health, USA), and the water contact angle was reported as an average of three measurements for each sample.
Measurement of pore volume and surface area:
The aerogels’ pore volume and surface area were calculated by nitrogen adsorption-desorption isotherms using a volumetric instrument (Quadrasorb by Quantachrome) (Graz, Austria) at 77 K. The samples were degassed at 150 °C for 24 h in a vacuum before measurements. The surface area was measured in a range of 0.007 < P/P0 < 0.035 using the Brunauer–Emme–Teller (BET) method.[79]
Mechanical tests:
Tensile tests were performed using an Instron instrument (model 5943, USA) and a load cell of 100 N. Rectangular-shaped (20 mm × 5 mm × 2 mm) hydrated samples were glued on the paper from the top and bottom corners and pulled away at a constant strain rate of 2 mm/min until failure. Stress-strain curves were recorded, and the slope of the linear range for the first 10% of the graphs was used to calculate the elastic modulus for five samples. The maximum stress before the abrupt drop in the graph was reported as tensile strength. Similarly, a cyclic tensile test was performed for 10 cycles when the samples linearly stretched and relaxed by 50% to analyze the hysteresis behavior of the hydrated samples. In addition, a precision source meter (B2902A, Keysight) was used to monitor the resistance of the printed sensors every 10 cycles after 100 cyclic bendings (bending curvature of 2 mm) and twisting (180°) deformations.
Degradation tests:
The degradation behavior was monitored in the presence of 5 mL collagenase type II (2.5 U/mL in DPBS) at 37 °C. The initial dry weight and dry weight of the degraded samples at each time point were measured after freeze-drying. Weight loss of the aerogels was calculated using the following equation:
Where Wd is the initial dry sample weight, and Wf is the freeze-dried weight of the degraded aerogels at each time point. The weight loss was calculated for n=4 replicates.
Air permeability measurements:
A commercial test chamber consisting of a feed and permeate chamber was constructed to measure the transient air permeation through the different sample films. Before loading, the film was mounted on a 47-mm nonporous brass sample disc with a hole (6.5 or 10 mm in diameter) in the center. The film covered the hole, and the edges were sealed by epoxy resin (Gorilla). The film was then carefully placed on a round filter paper with a diameter of 47 mm. The filter paper acted as a support with mechanical stability while having negligible resistance to the air. After sample loading, the chamber was fully degassed with a mechanical pump for over 4 h to a base pressure of 10−4 kPa.
The flux of the leak (dPd/dt, leak) was tested before introducing the air to the chamber. After that, the air was introduced to the chamber, and the flux of downstream air (dPd/dt, air) and upstream pressure of the air (Pup) were recorded. With known downstream volume (Vd), gas constant R, measured sample thickness (l) and surface area (A), and temperature (T), the permeability is calculated by the equation:
Where Vd is the downstream volume, l is the sample thickness, A is the surface area, T is the temperature, Pup is the upstream pressure of the air, Pd is the downstream pressure, and R is the gas constant.
Moisture permeability measurements:
The moisture permeability of each film was measured on a customized-built test system based on the ASTM E96 standard. The testing process is summarized below: a 20 mL plastic bottle was filled with 17 mL of distilled water, then sealed with a sample using double-sided tape. The bottle was placed in a chamber with a constant temperature of 36 °C and relative humidity of 45%. The mass of the bottle was measured every 24 h. The moisture permeability performance was calculated based on the mass change.
Cell viability assay:
Human dermal fibroblasts were cultured in DMEM supplemented with 10% FBS and 1% Pen-Strep until ~90% confluency. The confluent cells were trypsinized and seeded either directly on an FGA-based E-skin (10 mm × 10 mm ×1 mm) or in a 12-well plate at a density of 5000 cells/well for the direct and indirect cell viability assessments, respectively. For the indirect method, 24 h after cell seeding, the FGA-based E-skin (10 mm × 10 mm × 1 mm) was placed in the transwells (4 μm mesh) on the upper compartments of the wells and kept in a 5% CO2 incubator (ThermoFisher Scientific, United States). PrestoBlue cell viability assay in a ratio of 1:9 in DMEM was conducted according to the manufacturer’s instructions on days 1, 3, and 7 of the experiment. Fluorescence intensity corresponding to the metabolic activity of the cells was measured after 1.5 h incubation of the cells with PrestoBlue using a microplate reader (VARIOSKAN LUX, Thermo Scientific, United States) at excitation/emission of 530/590 nm. A live/dead assay was performed on day 7 of incubation after staining the cells with calcein-AM (0.5 μL) and ethidium homodimer-1 (2 μL) in DPBS (1 mL). After 15 min incubation, the staining solution was discarded, and the cells were washed twice with DPBS. Fluorescence images were captured by Keyence fluorescence microscope (BZ-X700 Series) using red and green channels for ethidium homodimer-1 and calcein-AM, respectively.
Statistical analysis:
Quantitative data were statistically analyzed using GraphPad Prism software (version 9.3.0). One-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test or Two-way ANOVA tests were applied, where appropriate. Statistically significant differences were presented as * (p< 0.05), ** (p < 0.01), *** (p < 0.001), and **** (p < 0.0001). Statistically non-significant is shown by ns.
Supplementary Material
Acknowledgments
Funding:
This work is supported by the National Institutes of Health (AR074234, UG3TR003148, GM126571, GM126831). We also acknowledge the support of the Natural Sciences and Engineering Research Council of Canada (NSERC), [funding reference number PDF 546263-2020]. Cette recherche a été financée par le Conseil de recherches en sciences naturelles et en génie du Canada (CRSNG), [numéro de référence PDF 546263-2020].
Footnotes
Competing Interests: All the authors declare that they have no conflict of interest.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the authors.
Contributor Information
Yangzhi Zhu, Terasaki Institute for Biomedical Innovation, Los Angeles, California 90064, United States.
Reihaneh Haghniaz, Terasaki Institute for Biomedical Innovation, Los Angeles, California 90064, United States.
Martin C. Hartel, Terasaki Institute for Biomedical Innovation, Los Angeles, California 90064, United States Department of Bioengineering, University of California, Los Angeles, Los Angeles, California 90095, United States.
Shenghan Guan, Terasaki Institute for Biomedical Innovation, Los Angeles, California 90064, United States; Mork Family Department of Chemical Engineering & Materials Science, Viterbi School of Engineering, University of Southern California, Los Angeles, California 90007, United States.
Jamal Bahari, Terasaki Institute for Biomedical Innovation, Los Angeles, California 90064, United States.
Zijie Li, Terasaki Institute for Biomedical Innovation, Los Angeles, California 90064, United States; Mork Family Department of Chemical Engineering & Materials Science, Viterbi School of Engineering, University of Southern California, Los Angeles, California 90007, United States.
Avijit Baidya, Department of Chemical and Biomolecular Engineering, University of California, Los Angeles, Los Angeles, California 90095, United States.
Ke Cao, Chemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37830, United States.
Xiaoxiang Gao, Department of Nanoengineering, University of California, San Diego, La Jolla, California 92093, United States.
Jinghang Li, Terasaki Institute for Biomedical Innovation, Los Angeles, California 90064, United States.
Zhuohong Wu, Department of Nanoengineering, University of California, San Diego, La Jolla, California 92093, United States.
Xuanbing Cheng, Department of Bioengineering, University of California, Los Angeles, Los Angeles, California 90095, United States.
Bingbing Li, Terasaki Institute for Biomedical Innovation, Los Angeles, California 90064, United States; Department of Manufacturing Systems Engineering and Management, California State University, Northridge, California 91330, United States.
Sam Emaminejad, Department of Bioengineering, University of California, Los Angeles, Los Angeles, California 90095, United States.
Paul S. Weiss, Department of Bioengineering, University of California, Los Angeles, Los Angeles, California 90095, United States Department of Chemistry and Biochemistry, Department of Materials Science and Engineering and California NanoSystems Institute, University of California Los Angeles, Los Angeles, California 90095, United States.
Ali Khademhosseini, Terasaki Institute for Biomedical Innovation, Los Angeles, California 90064, United States.
Data and material availability:
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the corresponding authors.
Reference
- [1].Wang S, Oh JY, Xu J, Tran H, Bao Z, Acc. Chem. Res. 2018, 51, 1033. [DOI] [PubMed] [Google Scholar]
- [2].Jung D, Lim C, Shim HJ, Kim Y, Park C, Jung J, Han SI, Sunwoo S-H, Cho KW, Cha GD, Science 2021, 373, 1022. [DOI] [PubMed] [Google Scholar]
- [3].Wang C, Yokota T, Someya T, Chem. Rev. 2021, 121, 2109. [DOI] [PubMed] [Google Scholar]
- [4].Someya T, Amagai M, Nat. Biotechnol. 2019, 37, 382. [DOI] [PubMed] [Google Scholar]
- [5].Jiang Y, Zhang Z, Wang Y-X, Li D, Coen C-T, Hwaun E, Chen G, Wu H-C, Zhong D, Niu S, Wang W, Saberi A, Lai J-C, Wu Y, Wang Y, Trotsyuk AA, Loh KY, Shih C-C, Xu W, Liang K, Zhang K, Bai Y, Gurusankar G, Hu W, Jia W, Cheng Z, Dauskardt RH, Gurtner GC, Tok JB-H, Deisseroth K, Soltesz I, Bao Z, Science 2022, 375, 1411. [DOI] [PubMed] [Google Scholar]
- [6].Wang Y, Haick H, Guo S, Wang C, Lee S, Yokota T, Someya T, Chem. Soc. Rev. 2022, 51, 3759. [DOI] [PubMed] [Google Scholar]
- [7].Oh HS, Lee CH, Kim NK, An T, Kim GH, Polymers (Basel) 2021, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Liu S, Wang S, Xuan S, Zhang S, Fan X, Jiang H, Song P, Gong X, ACS Applied Materials & Interfaces 2020, 12, 15675. [DOI] [PubMed] [Google Scholar]
- [9].Jian W, Hengyi L, Junjing M, Zhiqin P, Bing W, Junmin W, Sensors and Actuators B: Chemical 2020, 305, 127529. [Google Scholar]
- [10].Chung HU, Kim BH, Lee JY, Lee J, Xie Z, Ibler EM, Lee K, Banks A, Jeong JY, Kim J, Ogle C, Grande D, Yu Y, Jang H, Assem P, Ryu D, Kwak JW, Namkoong M, Park JB, Lee Y, Kim DH, Ryu A, Jeong J, You K, Ji B, Liu Z, Huo Q, Feng X, Deng Y, Xu Y, Jang K-I, Kim J, Zhang Y, Ghaffari R, Rand CM, Schau M, Hamvas A, Weese-Mayer DE, Huang Y, Lee SM, Lee CH, Shanbhag NR, Paller AS, Xu S, Rogers JA, Science 2019, 363, eaau0780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kim K, Kim B, Lee CH, Adv. Mater. 2020, 32, 1902051. [DOI] [PubMed] [Google Scholar]
- [12].Imani S, Bandodkar AJ, Mohan A, Kumar R, Yu S, Wang J, Mercier PP, Nat. Commun. 2016, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gao W, Emaminejad S, Nyein HYY, Challa S, Chen K, Peck A, Fahad HM, Ota H, Shiraki H, Kiriya D, Lien D-H, Brooks GA, Davis RW, Javey A, Nature 2016, 529, 509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yu Y, Nassar J, Xu C, Min J, Yang Y, Dai A, Doshi R, Huang A, Song Y, Gehlhar R, Ames AD, Gao W, Sci. Robot. 2020, 5, eaaz7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang B, Zhao C, Wang Z, Yang K-A, Cheng X, Liu W, Yu W, Lin S, Zhao Y, Cheung KM, Sci. Adv. 2022, 8, eabk0967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lim C, Hong YJ, Jung J, Shin Y, Sunwoo S-H, Baik S, Park OK, Choi SH, Hyeon T, Kim JH, Sci. Adv. 2021, 7, eabd3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Huang Z, Hao Y, Li Y, Hu H, Wang C, Nomoto A, Pan T, Gu Y, Chen Y, Zhang T, Li W, Lei Y, Kim N, Wang C, Zhang L, Ward JW, Maralani A, Li X, Durstock MF, Pisano A, Lin Y, Xu S, Nat. Electron. 2018, 1, 473. [Google Scholar]
- [18].Zhu Y, Hartel MC, Yu N, Garrido PR, Kim S, Lee J, Bandaru P, Guan S, Lin H, Emaminejad S, de Barros NR, Ahadian S, Kim H-J, Sun W, Jucaud V, Dokmeci MR, Weiss PS, Yan R, Khademhosseini A, Small Methods 2022, 6, 2100900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhu Y, Haghniaz R, Hartel MC, Mou L, Tian X, Garrido PR, Wu Z, Hao T, Guan S, Ahadian S, Kim H-J, Jucaud V, Dokmeci MR, Khademhosseini A, ACS Biomater. Sci. Eng. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang X, Meng X, Zhu Y, Ling H, Chen Y, Li Z, Hartel MC, Dokmeci MR, Zhang S, Khademhosseini A, IEEE Electron Device Lett. 2021, 42, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yu Y, Li J, Solomon SA, Min J, Tu J, Guo W, Xu C, Song Y, Gao W, Sci. Robot. 2022, 7, eabn0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cheng J, Shang J, Yang S, Dou J, Shi X, Jiang X, Adv. Funct. Mater. 2022, 32, 2200444. [Google Scholar]
- [23].Yang B, Jiang X, Fang X, Kong J, Lab Chip 2021, 21, 4285. [DOI] [PubMed] [Google Scholar]
- [24].Chen X, Luo F, Yuan M, Xie D, Shen L, Zheng K, Wang Z, Li X, Tao L-Q, Adv. Funct. Mater. 2019, 29, 1904706. [Google Scholar]
- [25].Zhu Y, Kim S, Ma X, Byrley P, Yu N, Liu Q, Sun X, Xu D, Peng S, Hartel MC, Zhang S, Jucaud V, Dokmeci MR, Khademhosseini A, Yan R, Nano Research 2021. [Google Scholar]
- [26].Gao X, Chen X, Hu H, Wang X, Mu J, Lou Z, Zhang R, Shi K, Yue W, Chen X, Res. Sq. 2021. [Google Scholar]
- [27].Zhou W, Yao S, Wang H, Du Q, Ma Y, Zhu Y, ACS Nano 2020, 14, 5798. [DOI] [PubMed] [Google Scholar]
- [28].Sharifuzzaman M, Zahed MA, Sharma S, Rana SMS, Chhetry A, Shin YD, Asaduzzaman M, Zhang S, Yoon S, Hui X, Yoon H, Park JY, Adv. Funct. Mater. 2022, 32, 2107969. [Google Scholar]
- [29].Li H, Wang Z, Sun M, Zhu H, Liu H, Tang CY, Xu L, Adv. Funct. Mater. 2022, 32, 2202792. [Google Scholar]
- [30].Peng X, Dong K, Ye C, Jiang Y, Zhai S, Cheng R, Liu D, Gao X, Wang J, Wang ZL, Sci. Adv. 2020, 6, eaba9624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Miyamoto A, Lee S, Cooray NF, Lee S, Mori M, Matsuhisa N, Jin H, Yoda L, Yokota T, Itoh A, Sekino M, Kawasaki H, Ebihara T, Amagai M, Someya T, Nat. Nanotechnol. 2017, 12, 907. [DOI] [PubMed] [Google Scholar]
- [32].Yan Z, Xu D, Lin Z, Wang P, Cao B, Ren H, Song F, Wan C, Wang L, Zhou J, Zhao X, Chen J, Huang Y, Duan X, Science 2022, 375, 852. [DOI] [PubMed] [Google Scholar]
- [33].Matsuhisa N, Niu S, O’Neill SJK, Kang J, Ochiai Y, Katsumata T, Wu H-C, Ashizawa M, Wang G-JN, Zhong D, Wang X, Gong X, Ning R, Gong H, You I, Zheng Y, Zhang Z, Tok JBH, Chen X, Bao Z, Nature 2021, 600, 246. [DOI] [PubMed] [Google Scholar]
- [34].Ding L, Hang C, Yang S, Qi J, Dong R, Zhang Y, Sun H, Jiang X, Nano Lett. 2022, 22, 4482. [DOI] [PubMed] [Google Scholar]
- [35].Wang J, Lee S, Yokota T, Someya T, Adv. Funct. Mater. 2022, 32, 2200458. [Google Scholar]
- [36].Li Z, Zhang S, Chen Y, Ling H, Zhao L, Luo G, Wang X, Hartel MC, Liu H, Xue Y, Haghniaz R, Lee K, Sun W, Kim H, Lee J, Zhao Y, Zhao Y, Emaminejad S, Ahadian S, Ashammakhi N, Dokmeci MR, Jiang Z, Khademhosseini A, Adv. Funct. Mater. 2020, 30, 2003601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zoratto N, Di Lisa D, de Rutte J, Sakib MN, Alves e Silva AR, Tamayol A, Di Carlo D, Khademhosseini A, Sheikhi A, Bioengineering & Translational Medicine 2020, 5, e10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Erdem A, Darabi MA, Nasiri R, Sangabathuni S, Ertas YN, Alem H, Hosseini V, Shamloo A, Nasr AS, Ahadian S, Dokmeci MR, Khademhosseini A, Ashammakhi N, Adv. Healthc. Mater. 2020, 9, 2070047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Davoodi E, Montazerian H, Esmaeilizadeh R, Darabi AC, Rashidi A, Kadkhodapour J, Jahed H, Hoorfar M, Milani AS, Weiss PS, Khademhosseini A, Toyserkani E, ACS Applied Materials & Interfaces 2021, 13, 22110. [DOI] [PubMed] [Google Scholar]
- [40].Nasrollahi F, Haghniaz R, Hosseini V, Davoodi E, Mahmoodi M, Karamikamkar S, Darabi MA, Zhu Y, Lee J, Diltemiz SE, Montazerian H, Sangabathuni S, Tavafoghi M, Jucaud V, Sun W, Kim H-J, Ahadian S, Khademhosseini A, Small 2021, 17, 2100692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yue K, Trujillo-de Santiago G, Alvarez MM, Tamayol A, Annabi N, Khademhosseini A, Biomaterials 2015, 73, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Liu W, Wang Y, Wang J, Lanier OL, Wechsler ME, Peppas NA, Gu Z, Engineering 2022, 13, 53. [Google Scholar]
- [43].Annabi N, Tamayol A, Uquillas JA, Akbari M, Bertassoni LE, Cha C, Camci-Unal G, Dokmeci MR, Peppas NA, Khademhosseini A, Adv. Mater. 2014, 26, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Richbourg NR, Wancura M, Gilchrist AE, Toubbeh S, Harley BAC, Cosgriff-Hernandez E, Peppas NA, Sci. Adv. 2021, 7, eabe3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Tavafoghi M, Sheikhi A, Tutar R, Jahangiry J, Baidya A, Haghniaz R, Khademhosseini A, Adv. Healthc. Mater. 2020, 9, 1901722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Xuechuan W, Zhongxue B, Manhui Z, Ouyang Y, Mengdi H, Boqiang C, Rongrong S, Chao W, Xinhua L, Sci J.: Adv. Mater. Devices. 2022, 7, 100451. [Google Scholar]
- [47].Cui Z, Poblete FR, Zhu Y, ACS Appl. Mater. Interfaces 2019, 11, 17836. [DOI] [PubMed] [Google Scholar]
- [48].Arica TA, Guzelgulgen M, Yildiz AA, Demir MM, Mater. Sci. Eng. C 2021, 120, 111720. [DOI] [PubMed] [Google Scholar]
- [49].Celikkin N, Mastrogiacomo S, Jaroszewicz J, Walboomers XF, Swieszkowski W, J Biomed Mater Res A 2018, 106, 201. [DOI] [PubMed] [Google Scholar]
- [50].Benton JA, DeForest CA, Vivekanandan V, Anseth KS, Tissue Eng Part A 2009, 15, 3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Van Vlierberghe S, Cnudde V, Dubruel P, Masschaele B, Cosijns A, De Paepe I, Jacobs PJS, Van Hoorebeke L, Remon JP, Schacht E, Biomacromolecules 2007, 8, 331. [DOI] [PubMed] [Google Scholar]
- [52].Liu T, Zhang Y, Sun M, Jin M, Xia W, Yang H, Wang T, Front Bioeng Biotechnol 2021, 9, 810155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Koshy ST, Ferrante TC, Lewin SA, Mooney DJ, Biomaterials 2014, 35, 2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Takase H, Shiomori K, Okamoto Y, Watanabe N, Matsune H, Umakoshi H, ACS Applied Polymer Materials 2022, 4, 7081. [Google Scholar]
- [55].Barros A, Quraishi S, Martins M, Gurikov P, Subrahmanyam R, Smirnova I, Duarte ARC, Reis RL, Chemie Ingenieur Technik 2016, 88, 1770. [Google Scholar]
- [56].Tyshkunova IV, Poshina DN, Skorik YA, Int J Mol Sci 2022, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Li G, Nandgaonkar AG, Habibi Y, Krause WE, Wei Q, Lucia LA, RSC Advances 2017, 7, 13678. [Google Scholar]
- [58].Zhang X, Sun P, Yan T, Huang Y, Ma Z, Zou B, Zheng W, Zhou J, Gong Y, Sun CQ, Prog. Solid. State Ch. 2015, 43, 71. [Google Scholar]
- [59].Chang K-H, Liao H-T, Chen J-P, Acta Biomaterialia 2013, 9, 9012. [DOI] [PubMed] [Google Scholar]
- [60].Bruns J, McBride-Gagyi S, Zustiak SP, Macromol. Meter. Eng. 2018, 303, 1800298. [Google Scholar]
- [61].Li L, Ren L, Wang L, Liu S, Zhang Y, Tang L, Wang Y, RSC Advances 2015, 5, 25525. [Google Scholar]
- [62].Li W, Wang D, Yang W, Song Y, RSC Advances 2016, 6, 20166. [Google Scholar]
- [63].Kim S, Seo M, J. Polym. Sci. A. Polym. Chem. 2018, 56, 900. [Google Scholar]
- [64].Tyshkunova IV, Poshina DN, Skorik YA, Int. J. Mol. Sci. 2022, 23, 2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Montazerian H, Baidya A, Haghniaz R, Davoodi E, Ahadian S, Annabi N, Khademhosseini A, Weiss PS, ACS Appl. Mater. Interfaces 2021, 13, 40290. [DOI] [PubMed] [Google Scholar]
- [66].Mandal J, Fu Y, Overvig AC, Jia M, Sun K, Shi NN, Zhou H, Xiao X, Yu N, Yang Y, Science 2018, 362, 315. [DOI] [PubMed] [Google Scholar]
- [67].Dai B, Li K, Shi L, Wan X, Liu X, Zhang F, Jiang L, Wang S, Adv. Mater. 2019, 31, 1904113. [DOI] [PubMed] [Google Scholar]
- [68].Ma Z, Xiang X, Shao L, Zhang Y, Gu J, Angew. Chem. 2022, 61, e202200705. [DOI] [PubMed] [Google Scholar]
- [69].Yu H, Yang X, Lian Y, Wang M, Liu Y, Li Z, Jiang Y, Gou J, Sens. and Actuators A: Phys. 2021, 318, 112514. [Google Scholar]
- [70].Hsu PC, Song AY, Catrysse PB, Liu C, Peng Y, Xie J, Fan S, Cui Y, Science 2016, 353, 1019. [DOI] [PubMed] [Google Scholar]
- [71].Xu Y, Sun B, Ling Y, Fei Q, Chen Z, Li X, Guo P, Jeon N, Goswami S, Liao Y, Ding S, Yu Q, Lin J, Huang G, Yan Z, Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Leroy A, Bhatia B, Kelsall CC, Castillejo-Cuberos A, Di Capua H M, Zhao L, Zhang L, Guzman A, Wang E, Sci. Adv. 2019, 5, eaat9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Sreekumaran S, Radhakrishnan A, Rauf AA, Kurup GM, Prog. Biomater. 2021, 10, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Emanuele NV, Swade TF, Emanuele MA, Alcohol Health Res World 1998, 22, 211. [PMC free article] [PubMed] [Google Scholar]
- [75].Kim J, Sempionatto JR, Imani S, Hartel MC, Barfidokht A, Tang G, Campbell AS, Mercier PP, Wang J, Adv. Sci. 2018, 5, 1800880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Liu H, Zhang S, Li Z, Lu TJ, Lin H, Zhu Y, Ahadian S, Emaminejad S, Dokmeci MR, Xu F, Khademhosseini A, Matter 2021, 4, 2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Sunny AI, Rahman M, Koutsoupidou M, Cano-Garcia H, Thanou M, Rafique W, Lipscombe O, Kassanos P, Triantis I, Kallos E, Kosmas P, “Feasibility Experiments to Detect Skin Hydration Using a Bio-Impedance Sensor”, presented at 2019 41st Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), 23–27 July 2019, 2019. [DOI] [PubMed] [Google Scholar]
- [78].Standard Test Method for Wound Closure Strength of Tissue Adhesive and Sealants, ASTM F2458-05(2015), ASTM F2458-05(2015), ASTM International, West Conshohocken, PA, 2015, www.astm.org. [Google Scholar]
- [79].Nguyen BN, Meador MAB, Scheiman D, McCorkle L, ACS Appl. Mater. Interfaces 2017, 9, 27313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the corresponding authors.