

Abstract
Corticobasal degeneration (CBD) is a rare, progressive neurodegenerative disorder with onset in the 5th to 7th decade of life. It is associated with heterogeneous motor, sensory, behavioral and cognitive symptoms, which make its diagnosis difficult in a living patient. The etiology of CBD is unknown; however, neuropathological and genetic evidence supports a pathogenetic role for microtubule-associated protein tau. CBD pathology is characterized by circumscribed cortical atrophy with spongiosis and ballooned neurons; the distribution of these changes dictates the patient’s clinical presentation. Neuronal and glial tau pathology is extensive in gray and white matter of the cortex, basal ganglia, diencephalon and rostral brainstem. Abnormal tau accumulation within astrocytes forms pathognomonic astrocytic plaques. The classic clinical presentation, termed corticobasal syndrome (CBS), comprises asymmetric progressive rigidity and apraxia with limb dystonia and myoclonus. CBS also occurs in conjunction with other diseases, including Alzheimer disease and progressive supranuclear palsy. Moreover, the pathology of CBD is associated with clinical presentations other than CBS, including Richardson syndrome, behavioral variant frontotemporal dementia, primary progressive aphasia and posterior cortical syndrome. Progress in biomarker development to differentiate CBD from other disorders has been slow, but is essential in improving diagnosis and in development of disease-modifying therapies.
Introduction
Corticobasal degeneration (CBD) is a rare, progressive neurodegenerative 4-repeat (4R) tauopathy (Box 1) associated with a wide variety of motor, sensory, behavioral and cognitive symptoms that can also occur in other conditions. This clinical heterogeneity and lack of specific features make CBD difficult to diagnose before the patient’s death.1 Owing to the wide spectrum of clinical presentations of CBD, neuropathological examination is required for a definitive diagnosis. Little is known about the epidemiology of CBD, and the available studies offer only crude estimates of its incidence. CBD has an estimated prevalence of 4.9–7.3 cases per 100,000 individuals2 and an annual incidence rate of 0.02 cases per 100,000 individuals on the basis of an Eastern European and Asian population study.3 Disease onset is typically in the 5th to 7th decades of life, with an average disease duration of 7 years. No sex bias has been observed. The pathogenesis of CBD is unknown, and no effective therapy is currently available to treat this disease or even to slow its progression.
Box 1 |. Tauopathies.
When tau pathology is considered to be the main contributing factor to neurodegeneration, the disorder is classified as a tauopathy. Tauopathies are characterized by their distinct biochemical protein isoform profiles, with regard to the presence of either three or four repeats (3R or 4R, respectively) in the tau microtubule-binding domain.
In Alzheimer disease, tau tangles are composed of equimolar 3R and 4R isoforms
Corticobasal degeneration, progressive supranuclear palsy and many variants of frontotemporal dementia and parkinsonism linked to chromosome 17 (and associated with MAPT mutations) have predominantly 4R tau pathology105
Pick disease has predominantly 3R tau pathology,106 and alterations from normal tau isoform levels contribute to its pathogenesis107
CBD was originally described as a movement disorder, but over the past few decades, as more clinical and pathological studies have been performed, the disease is now considered a disorder of both movement and cognition. CBD was first reported in 1968 by Rebeiz et al., who described clinical and neuropathological features of three patients in whom the disorder was termed corticodentatonigral degeneration with neuronal achromasia.4 Gibb and Marsden coined the term CBD in 1989,5 and this pathology has also been referred to as corticobasal ganglionic degeneration by some authors.6–8 The initial descriptions of CBD emphasized progressive movement abnormalities, which began as a unilateral slowing of voluntary movements with concurrent involuntary movements that eventually became generalized but remained asymmetric. In their paper, Rebeiz et al. noted, “…[the patients’] intellect was relatively preserved until the end.”4 However, their well-documented report describes cognitive as well as behavioral dysfunction in all three patients. On neuropathological examination, the researchers found distinct features common among the three patients, which included frontoparietal atrophy with neuronal loss, gliosis, ‘achromatic’ or ballooned neurons, and pigment loss in the substantia nigra.
The classic clinical movement disorder described by Rebeiz et al. in 1968 is now referred to as corticobasal syndrome (CBS), and its cardinal features are progressive asymmetric rigidity and apraxia with additional signs of cortical and basal ganglia involvement, such as aphasia and dystonia.5,8 Although CBS has some predictive value for finding CBD pathology, autopsy studies demonstrate considerable heterogeneity in the pathology of patients who present with CBS.9 Over the years, clinical and pathological studies have highlighted several focal cortical syndromes associated with CBD pathology and, therefore, defining widely accepted clinical diagnostic criteria for research purposes is nearly impossible.7,10–12 Tau pathology is also found in other neurodegenerative disorders, such as Alzheimer disease (AD)13 and progressive supranuclear palsy (PSP), and as tau-directed therapies undergo clinical trials, determining the correct underlying pathology in patients with CBS has become of more than merely academic interest.
This Review focuses on the range of clinical presentations associated with CBD pathology and the latest advances in our understanding of its pathogenesis. We describe progress in the development of therapeutic agents for CBD and briefly outline efforts to develop biomarkers that can distinguish tauopathies from tau-unrelated degenerative disorders. The latter will become increasingly important for designing future pharmacological trials in this rare disorder.
Clinical syndromes associated with CBD
The clinical syndrome in patients with neurodegenerative diseases is a representation of the topography of the lesions but not necessarily the nature of the underlying pathology. Since CBD is a disease with prominent focal cortical atrophy and variable subcortical pathology, patients with CBD can exhibit a range of distinct clinical syndromes, including CBS, Richardson syndrome (the most common clinical presentation of PSP), progressive aphasia, frontotemporal dementia (FTD), posterior cortical atrophy (PCA), and dementia with features similar to AD (Table 1).6,14–23 Although diagnostically far from perfect, clinical features can help to distinguish CBD from its so-called mimickers in many patients (Box 2).
Table 1 |.
Clinical heterogeneity of pathologically confirmed CBD
| Reference | Female:male ratio | Age at onset/disease duration (years) | Common features at onset (% of patients) | Clinical diagnosis* (% of patients) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||||
| Limb apraxia | Memory impairment | Behavioral changes | Gait disturbance | Speech disorder | CBS | AD or PD | FTD | RS | |||
|
| |||||||||||
| Schneider et al. (1997)18 | 10:1 | 67/7.9 | 64 | 18 | 9 | 9 | 9 | 64 | 27 | 0 | 0 |
| Litvan et al. (2000)26 | 14:13 | 64/7.0 | 37 | 33 | 33 | 26 | 22 | 22 | 30 | 7 | 4 |
| Murray et al. (2007)16 | 8:7 | 61/5.4 | 33 | 33 | 40 | 27 | 40 | 40 | 13 | 33 | 13 |
| Ling et al. (2010)17 | 9:10 | 66/6.0 | 26 | 37 | 26 | 63 | 16 | 26 | 10 | 5 | 42 |
| Totals | 41:31 | 65/6.6 | 38 | 32 | 29 | 33 | 22 | 33 | 21 | 11 | 15 |
The total does not equal 100% because some series had final diagnoses other than the diseases listed here. Abbreviations: AD, Alzheimer disease; CBD, corticobasal degeneration; CBS, corticobasal syndrome; FTD, frontotemporal dementia; PD, Parkinson disease; RS, Richardson syndrome.
Box 2 |. Diagnosis of corticobasal degeneration.
Several clinical features can help to distinguish corticobasal degeneration from its mimickers.
Progressive supranuclear palsy
Postural instability with falls usually occurring within the 1st year of disease onset
Vertical (especially downward) supranuclear gaze palsy
Usually symmetric parkinsonism, although cases of asymmetric parkinsonism associated with progressive supranuclear palsy have been described
Absent cortical features, such as myoclonus and apraxia
Parkinson disease
Marked and sustained benefit from dopaminergic therapy
Resting tremor
Dementia with Lewy bodies
Visual hallucinations (not drug-related)
Rapid eye movement sleep behavior disorder
Fluctuating cognition
Multiple system atrophy
Pronounced dysautonomia
Cerebellar ataxia
Disruption of rapid eye movement sleep behavior
Alzheimer disease
Initial prominent short-term memory impairment
Creutzfeldt–Jakob disease
Rapid disease progression with a disease course <1 year
Corticobasal syndrome
CBS, the classic motor presentation of CBD as first described by Rebeiz and co-workers,4 accounts for approximately 50% of autopsy-confirmed cases (D. W. Dickson, unpublished observations from the CurePSP Brain Bank). Core features of CBS include levodopa-unresponsive parkinsonism, asymmetric akinesia, and rigidity, accompanied by other cortical and basal ganglia dysfunction, such as limb and oculomotor apraxia, cortical sensory deficits, dystonic posturing of a limb, myoclonus, and alien limb phenomenon.10,24 These cortical signs and symptoms are attributed to a predominant frontal and parietal involvement. Patients can present with either motor or cognitive complaints or with a combination of the two. In a combined series of 27 individuals with CBD confirmed by autopsy, the most frequent features at onset of the disease were limb clumsiness in 37% of the patients, followed by memory loss and behavioral changes in another 33%.19,25,26 A separate longitudinal study of 15 individuals with CBD reported an exclusively cognitive disturbance presentation in about half of the patients; another 20% had an exclusively motor abnormality presentation, and the remaining 30% had both motor and cognitive complaints.16 Owing to the remarkably low sensitivity and positive predictive value of these features for diagnosing CBD, studies of the neuropsychiatric features of patients with a clinical diagnosis of CBD should be interpreted with caution.
The accuracy of clinical diagnosis of CBD does, however, improve with disease progression. In a review of 27 autopsy-confirmed CBD by Litvan and co-workers, 22% of the patients had CBS as the initial diagnosis, increasing to 41% before death.26 Similarly, in a study by Kertesz and co-workers of 13 autopsy-confirmed cases of CBD, 35% had CBS as an intial diagnosis, increasing to 54% before death.27 Being able to predict CBD pathology as early in the disease course as possible is of great importance owing to the growing number of tau-directed therapies currently being developed.
The CBD mimickers
The CBD mimickers, which resemble CBS but have different underlying pathologies, include AD, PSP and frontotemporal lobar degeneration with TDP-43 pathology (FTLD–TDP, sporadic and familial), among others.9,28–32 Whitwell et al. reported that although a region of atrophy was common to all pathologies that result in CBS, CBS caused by FTLD-TDP was associated with notable frontotemporal gray matter atrophy; CBS caused by AD was associated with prominent temporoparietal atrophy; and in CBS caused by CBD or PSP gray matter atrophy was focused in the premotor cortex with additional involvement of superior posterior frontal cortex.33
Richardson syndrome
Richardson syndrome is the typical presentation of PSP,14,34 and is characterized by severe postural instability leading to falls soon after disease onset, vertical supranuclear gaze palsy, dysarthria, dysphagia, and frontal cognitive disturbances.35 The occurrence of PSP presenting as CBS has been attributed to either greater cortical tau pathology than is observed in the typical PSP presentation36 or an asymmetrical tau burden.37 A study published in 2010, which assessed differences in cortical atrophy in patients with CBS by use of MRI, showed that different patterns of atrophy are associated with specific histopathological abnormalities.33 As discussed below, CBD and PSP were originally believed to be distinct clinical and pathological entities, but as additional studies have been performed, their overlapping features have become increasingly apparent. When patients with CBD present with bilateral motor disturbances and early frontal dementia, they are frequently misdiagnosed as having PSP since both disorders involve levodopa-unresponsive parkinsonism and can share several other clinical features.38,39 Asymmetry of clinical signs and symptoms might have been overemphasized in the CBD literature, as several studies of autopsy-confirmed cases have reported the occurrence of symmetric motor disturbances.17,38,40,41
Patients with CBD can develop abnormal eye movements similar to those seen in patients with PSP. Decreased vertical saccadic velocities preceding vertical supranuclear gaze palsy are typically seen in patients with PSP, whereas increased saccadic latencies with preserved velocity are seen in patients with CBD, and the horizontal and vertical planes are similarly affected.42–44 In 2010, Ling et al. described clinical features of patients with CBD presenting with Richardson syndrome and confirmed that differences in these eye movement abnormalities are diagnostically helpful in differentiating CBD from PSP.17
Postural instability and falls occurring within the first year of symptom onset are indicative of PSP.45 By contrast, if patients with CBD do experience falls, they usually occur late in the disease course; when the patient has asymmetric motor disturbances, falls tend to incline towards the affected side of the body. Few studies have compared the neuropsychiatric profiles of patients with CBD and PSP. In a clinical series, Cummings and Litvan used the Neuropsychiatric Inventory to assess behavioral symptoms in patients with dementia and found that depression and irritability were notably more frequent in patients with CBD than in those with PSP. The latter, by contrast, exhibited an increased frequency of apathy.46
Patients with pathologically confirmed CBD and PSP show differing patterns of brain atrophy on MRI.47 When grouped according to their presenting feature (extrapyramidal versus cognitive impairment), patients with CBD who had an extrapyramidal presentation had greater cortical atrophy and less brainstem atrophy compared to patients with PSP who had an extrapyramidal presentation. Among patients who presented with cognitive impairment, those with CBD had substantially greater atrophy of cortical and subcortical gray matter, and less atrophy of subcortical white matter, than those who had PSP.47
Behavioral variant frontotemporal dementia
Patients who present with behavioral variant FTD (bvFTD) exhibit marked behavioral and personality changes, including apathy, disinhibition, perseveration, executive dysfunction, obsessive–compulsive behaviors and dramatically impaired insight.48 In approximately 50% of such patients, the underlying pathology is tau-positive (PSP, CBD or Pick disease). The other 50% have tau-negative FTLD with ubiquitin immunoreactive inclusions (FTLD-U), which in the majority of cases are TDP-43-positive.49,50 Although imaging signatures have been identified for specific pathologies, in one large study of patients with bvFTD no specific signature pattern of atrophy could distinguish tau-positive individuals from tau-negative patients.51 Josephs et al. found that bvFTD was the syndrome least predictive of underlying pathology.52 In spite of this finding, some studies have identified clinical and neuropsychiatric features that could help to distinguish tau-positive from tau-negative bvFTD. Hodges et al. found that patients with tau-positive bvFTD were substantially older at the time of diagnosis (mean 61.7 years) than the tau-negative cases (mean 53.7 years).49 Forman et al. found that the presence of both a behavioral disorder and aphasia at disease onset was more predictive of FTLD-U than were AD or other tauopathies.50 Grossman et al. found that tau-positive FTLD was associated with visual perceptual and spatial difficulties, whereas tau-negative FTLD was linked to language and executive dysfunction.53 In a cluster analysis, Hu et al. found that poor planning and judgment was associated with tau pathology, whereas impaired personal conduct and an absence of dysexecutive symptoms were predictive of FTLD-U pathology.31 These observations suggest that longitudinal evaluations of neuropsychological features can be useful in the antemortem differentiation between tau-positive and tau-negative pathology.
The nearly equal distribution of tau-positive and tau-negative underlying pathologies in bvFTD syndrome highlights the considerable overlap in clinical features of CBD, PSP and FTLD. Indeed, some neurologists argue in favor of grouping these three conditions under the single designation of FTD–Pick complex.20 Kertesz et al. have shown that bvFTD followed by the late development of progressive aphasia is usually caused by tau-negative FTLD.27 In another distinct syndrome progression, which is highly predictive of tau-positive pathology, the patient presents with a movement disorder (such as CBS) or the movement disorder develops after primary progressive aphasia. The differential diagnosis, however, is complicated because some patients with CBD develop only minimal CBS features late in the disease course, but present with notable features of FTD.38 The latter CBD phenotype is rarely mentioned in the literature.
Primary progressive aphasia
Primary progressive aphasia is a focal dementia disorder where the dominant deficit is progressive language impairment.54 A distinct form of primary progressive aphasia—termed progressive nonfluent aphasia—is most often associated with tau-positive pathology, including CBD, PSP and Pick disease.22,52,55 The other forms of primary progressive aphasia—semantic dementia and logopenic progressive aphasia—are more often associated with FTLD–TDP and AD pathology, respectively.56 The characteristics of progressive nonfluent aphasia include errors in language production with agrammatism and grammatical simplification.48 Speech production is typically strained or halting, with speech-sound errors occurring as a result of coexisting motor speech problems, referred to as apraxia of speech.
Clinical and pathological studies in patients with progressive nonfluent aphasia have highlighted the difficulty of obtaining an accurate antemortem diagnosis. As with the other clinical syndromes discussed above, several different pathologies have been reported to occur at variable frequencies in patients with progressive nonfluent aphasia. Some researchers have suggested that quantitative studies of neuropsychological features can be useful to predict the underlying pathology in patients with progressive language deterioration. Apraxia of speech, with or without progressive nonfluent aphasia, is often observed in patients with a tauopathy, but not in those with FTLD–TDP.22 Josephs et al. suggested that one could differentiate between patients with CBD and PSP since apraxia of speech without nonfluent aphasia was more frequent in the patients with PSP, whereas those with CBD tended to have both apraxia of speech and nonfluent aphasia. In three combined series of 93 pathologically confirmed individuals, 80% of patients with progressive nonfluent aphasia had tau-positive pathology.49,52,57
Posterior cortical atrophy syndrome
Patients with posterior cortical atrophy primarily exhibit disturbances in visual perception with features of Balint syndrome, Gerstmann syndrome, visual agnosia and alexia.58 The occipitoparietal cortices are predominantly affected. Posterior cortical atrophy is most often associated with AD or Creutzfeldt–Jakob disease,59 although posterior cortical atrophy syndrome (also known as Benson syndrome), has been described, albeit rarely, in patients with pathologically confirmed CBD.59–61 Two patients with CBD who had posterior cortical atrophy syndrome carried the brunt of the tau pathology in the posterior parietal cortex and Brodmann areas 17 and 18.60
Classifying the 4R tauopathies
When they were originally described, CBD and PSP were considered distinct clinical and pathological disorders,4,35 and they have remained distinct nosological entities over the years. However, a consensus exists that CBD and PSP could represent a disease spectrum, since they have overlapping clinical, pathological, genetic and biochemical features. Both disorders involve an accumulation of the abnormal 4R tau isoform in neurons and glial cells (oligodendroglia and astrocytes). They also share a similar genetic basis, with a higher frequency of the microtubule-associated protein tau (MAPT) H1 haplotype (and the H1c subhaplotype) than is found in healthy controls (Box 3). These characteristics of CBD and PSP support grouping them as the 4R tauopathies. This approach could prove useful in the design of tau-directed therapeutic clinical trials. Whether CBD and PSP share the same disease mechanism remains unknown; nevertheless, diagnostic accuracy is substantially improved when CBD and PSP are grouped as a single entity.
Box 3 |. Tau protein and corticobasal degeneration.
Microtubule-associated protein tau, encoded by the MAPT gene, is required for microtubule assembly and stability, and is highly expressed in neuronal axons.108–110 The phosphorylation status of tau regulates its function. In tauopathies, tau protein is aberrantly phosphorylated and has an abnormal conformation that favors aggregation and inclusion formation.13
The human MAPT gene is located on chromosome 17q21.3.111 Alternative splicing of exons 2, 3 and 10 produces six tau protein isoforms in the adult CNS.111,112 Moreover, genetic analyses of MAPT have revealed two major tau haplotypes, H1 and H2.113,114 Either the H1 haplotype or a specific H1 sub-haplotype (H1c) is associated with an increased risk of corticobasal degeneration or progressive supranuclear palsy.113,115–118
Patients with MAPT mutations can exhibit clinical and pathological features associated with corticobasal degeneration.119–121 Over 40 pathogenetic MAPT mutations have been identified in more than 100 families affected by frontotemporal dementia and parkinsonism linked to chromosome 17.122
Clinical presentations
In the archetypal presentations of CBD and PSP (CBS and Richardson syndrome, respectively), the two disorders have distinct clinical features; however, the differential diagnosis is often complicated and misdiagnosis is not infrequent, even at autopsy. Some studies suggest PSP has a slightly later age of onset than CBD, but few meaningful differences in any clinical or demographic feature have been demonstrated.
Neuropathological features
Neuropathological diagnostic criteria for CBD have been refined and validated on the basis of tau immunohistochemical analysis, with an emphasis on tau-positive neuropil threads in gray and white matter of the cortex, basal ganglia, diencephalon and rostral brainstem (Figure 1).62 In contrast to the original description by Rebeiz et al., these updated criteria place reduced emphasis on the ballooned neuron because this finding lacks diagnostic specificity.63
Figure 1 |.

The distinct histological lesions of CBD and PSP visualized with tau immunohistochemistry. a | The astrocytic plaque is a hallmark of CBD. b | Tufted astrocytes are found in the superior frontal cortex of patients with PSP. c | Numerous neuropil threads in the internal capsule are found in patients with CBD; d | by contrast, those with PSP have sparse neuropil threads with oligodendroglial coiled bodies. e | Western blot of detergent-insoluble tau proteins (P3 fraction) from patients with CBD and PSP. The CBD and PSP samples both exhibit the characteristic major doublet of hyperphosphorylated full-length tau at 64 kDa and 68 kDa (asterisk). Cleaved tau fragments from a patient with CBD migrate as two bands at approximately 37 kDa (small arrow), whereas cleaved fragments from a patient with PSP migrate as a single band at 33 kDa (large arrow). Abbreviations: CBD, corticobasal degeneration; PSP, progressive supranuclear palsy.
The main difficulty in neuropathological diagnosis arises in patients with atypical CBD who have greater than usual brainstem and cerebellar pathology,40 and in patients with PSP who have greater than usual cortical pathology.36,64 In the majority of individuals, the distribution of tau pathology differs according to whether they have CBD or PSP: tau pathology in patients with CBD occurs predominantly in forebrain structures, whereas in those with PSP tau pathology occurs predominantly in hindbrain structures.39,65 However, exceptions to these general rules exist in patients with atypical presentations of both disorders. The strongest argument for separating these two diseases comprises their distinct pathological and biochemical features. For example, the astrocytic lesions associated with CBD and PSP are distinct and rarely (if ever) coexist.66 The pathognomonic astrocytic lesion in CBD is the astrocytic plaque,66,67 whereas tufted astrocytes are characteristic of PSP (Figure 1).39,68
Biochemical features
Although CBD and PSP are both 4R tauopathies, biochemical studies show differences in the tau protein profiles associated with the two disorders. In particular, the tau protein cleavage fragments differ in patients with CBD and PSP. The detergent-insoluble cleaved tau fragments from patients with CBD migrate as two bands (a doublet of around 37 kDa), whereas those from patients with PSP migrate as a single band of 33 kDa (Figure 1).69,70 These differences in proteolytic processing of tau might be related to the differences in glial lesions that occur in patients with CBD and PSP. Whether differences also exist between these biochemical profiles in various brain regions remains to be determined. Except for differences in the size of tau cleavage fragments, the biochemical properties of tau are similar in the brains of patients with CBD and PSP.
Mechanisms of tau-mediated disease
Under pathological conditions, tau becomes hyperphosphorylated, which reduces its binding affinity for microtubules. This change leads to a loss of proper microtubule functioning and/or a toxic gain of function, as dissociated tau has a greater propensity for multimerization than microtubule-bound tau. The cellular dysfunction that causes tau to become hyperphosphorylated in sporadic tauopathies is currently unknown; however, some evidence supports a role for microglia in sporadic tauopathies,71,72 and a molecular biology study published in 2010 described a mechanism through which microglial signaling enhances tau hyperphosphorylation in neurons in the setting of neuroinflammation.73 Further work is needed, however, to determine whether this mechanism, which provides a drug target upstream of tau hyperphosphorylation, will lead to the development of new strategies for the treatment of tauopathies.
Although an extensive amount of research has gone into elucidating the mechanism of tau-mediated neurodegeneration, many gaps remain in our current knowledge. Two studies have localized initial tau dysfunction to the synapse, which is the region where cell–cell communication occurs. Ittner et al. showed that tau has a role in targeting fyn kinase to postsynaptic compartments and modulates N-methyl-d-aspartate (NMDA) receptors, resulting in excitotoxic downstream signaling.74 Hoover et al. also showed that tau accumulates in dendritic spines, causing a disruption in synaptic function by reducing the levels of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and NMDA receptors.75 These studies have advanced our understanding of the pathophysiological processes underlying tauopathies that could provide new strategies for the development of tau-directed therapeutics.
Tau-directed therapeutics
Interest in the development of tau-directed therapeutics is growing. Several different approaches to reducing tau-mediated neurodegeneration focus on compensating for the loss of tau function using microtubule-stabilizing agents, reducing hyperphosphorylation of this protein through the inhibition of tau kinases, inhibiting the toxic aggregation of tau fibrils using agents that block protein–protein interactions, and enhancing intracellular tau degradation by targeting proteins involved in the ubiquitin–proteasome system.
Three compounds have progressed to human clinical testing: methylene blue, lithium chloride and an octapeptide (Asn–Ala–Pro–Val–Ser–Ile–Pro–Gln) known as NAP.76 Methylene blue was the first drug to demonstrate inhibition of tau aggregation and to alter the structure of paired-helical filaments that make up neurofibrillary tangles in the brains of patients with AD.77 Methylene blue has already been approved by the FDA for several conditions and is currently undergoing phase III clinical testing in patients with AD. Patients with tauopathy might also benefit from methylene blue treatment.
Lithium chloride is an inhibitor of glycogen synthase kinase 3β (GSK3β), one of several kinases that phosphorylate tau.78,79 GSK3β has been extensively studied as a therapeutic target in AD. Treatment with lithium chloride consistently results in an attenuation of tau hyperphosphorylation and pathology in animal models of tauopathies.80–84 Transgenic animals that harbor human MAPT mutations recapitulate key features of the neuropathology seen in human tauopathies, and these models are an excellent tool for preclinical drug discovery.85
Microtubule-stabilizing compounds, including the anticancer drug paclitaxel, showed improved microtubule density, accelerated axonal transport and increased motor function in a mouse model of tauopathy.86 These benefits were accompanied by intolerable adverse effects, however, because paclitaxel does not readily cross the blood–brain barrier and, therefore, high doses are required to observe any therapeutic effect. Efforts to explore other microtubule-stabilizing agents with improved blood–brain barrier permeability led to discovery of the neuroprotective octapeptide NAP, which has shown promise as a therapy for tauopathies by reducing the levels of hyperphosphorylated tau.87–89 Treatment with another microtubule-stabilizing compound, epothilone D, was reported in 2010 to increase microtubule density and reduce cognitive deficits in an animal model of tauopathy.90 Enhancing the cellular machinery responsible for degrading pathological tau species using inhibitors of heat-shock protein 90 reduces levels of hyperphosphorylated tau in vitro and in vivo.91,92 High-throughput drug library screens have been performed to identify novel molecules capable of inhibiting tau fibrillization. None of these molecules has yet been tested in animal models, but the technique shows promise for future identification of therapeutic compounds.93
Identification of biomarkers
Development of imaging and fluid biomarkers for CBD and related tauopathies is still in the early stages; however, such markers have the potential to improve diagnostic accuracy and specificity. Tau levels in cerebrospinal fluid (CSF) have been studied quite extensively in patients with AD and, since 1997, these levels have been monitored in clinical series of patients with PSP and CBD.94–97 The authors of some of these studies report an increase in total tau levels in patients with CBD compared with controls,95,98 whereas others found no notable difference in CSF tau levels in patients with CBD compared with both healthy controls and patients with PSP.97,99 Although these initial studies have assessed total tau and phosphorylated tau levels, the evidence suggests that evaluating levels of tau fragments and tau phosphorylation patterns could be useful for the differential diagnosis of neurodegenerative diseases and/or in clinical trials.96,100,101
Levels of other CSF proteins, including amyloid-β42, neurofilament light chain and neurofilament heavy chain, can differentiate patients with PSP from healthy control individuals or those with other parkinsonian disorders.97,102,103 Further studies are needed to explore the differences in expression of these and other proteins between individuals with CBD and PSP by means of unbiased proteomic methods in brain tissue and CSF.
Distinctive patterns of atrophy on brain imaging have been observed in patients with autopsy-confirmed CBD and PSP.47 Patients with CBD have a characteristic pattern of posterior frontal atrophy regardless of the clinical syndrome, suggesting that this pattern of atrophy could prove to be a useful biomarker of CBD pathology (Figure 2). Differences in atrophy patterns elsewhere in the brain seem to reflect clinical differences: for example, marked prefrontal atrophy is observed in patients with CBD related to bvFTD, and substantial atrophy in posterior regions of the brain is observed in those with CBD related to posterior cortical atrophy. If CBD and PSP can be differentiated in living patients by use of inexpensive and relatively noninvasive methods, our knowledge of the epidemiology of these disorders will increase, which could ultimately lead to refined treatments.
Figure 2 |.
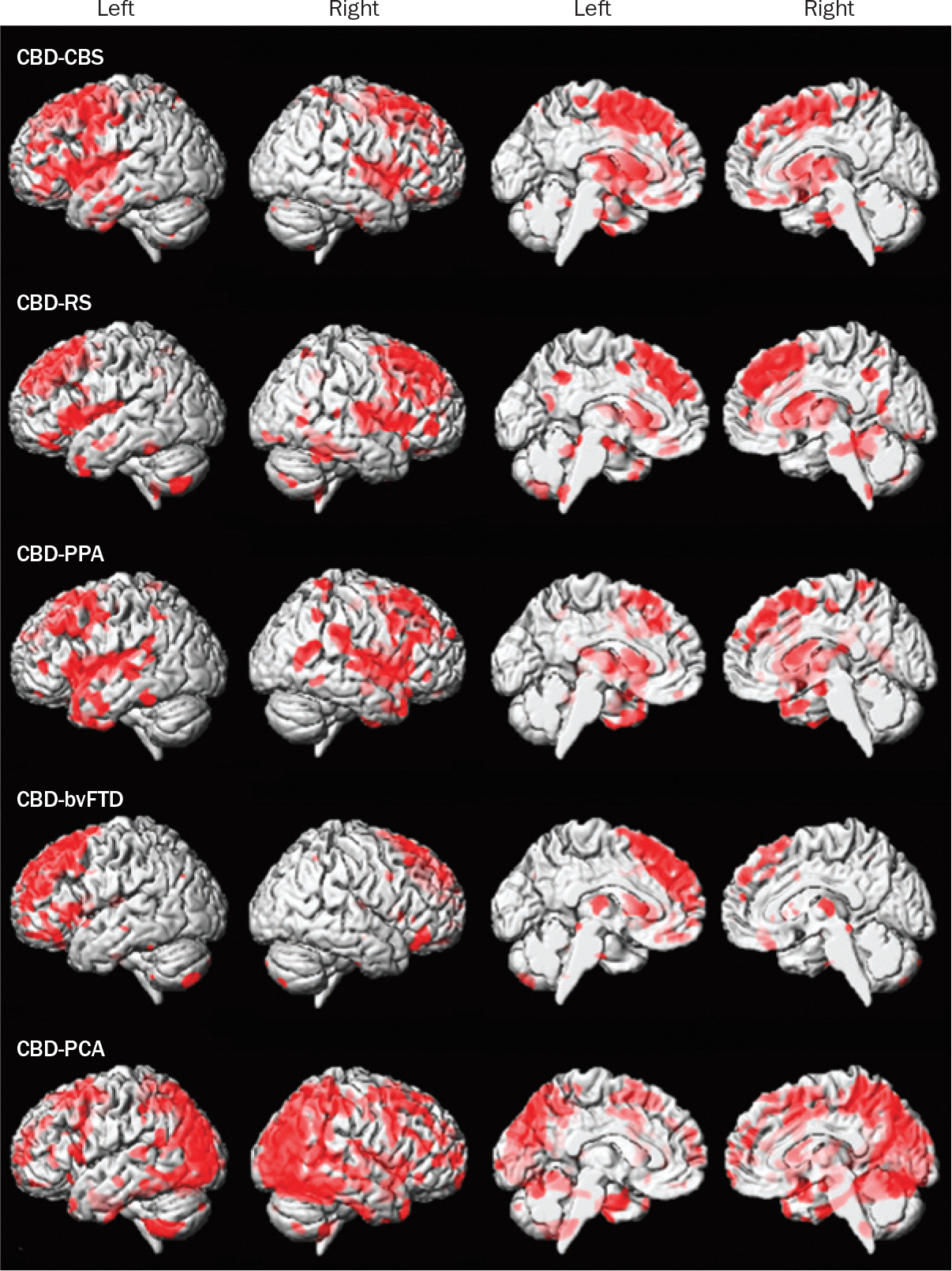
Patterns of gray matter loss in groups of patients with autopsy-confirmed CBD but different clinical syndromes. The results were generated using voxel-based morphometry and are shown on three-dimensional surface and medial renders of the brain. All clinical groups show gray matter loss as indicated by red coloration in the posterior lateral and medial frontal lobe, suggesting that this is a signature pattern of CBD pathology; however, patterns of gray matter loss that differ between groups extend into other regions of the brain. These differences are thought to cause the various clinical syndromes associated with CBD. Abbreviations: bvFTD, behavioral variant frontotemporal dementia; CBD, corticobasal degeneration; CBS, corticobasal syndrome; PCA, posterior cortical atrophy; PPA, primary progressive aphasia; RS, Richardson syndrome.
Conclusions
The complex and highly variable clinical presentation of CBD is the prevailing difficulty in identifying and accurately diagnosing this disorder in living patients. Efforts are currently being made to improve the accuracy of diagnosis using imaging and CSF biomarkers. Although considerable progress has been made in delineating the clinical picture of CBD, these studies are inherently limited because they can only be performed retrospectively owing to the necessity of obtaining an autopsy-confirmed diagnosis. The first completed genome-wide association study for PSP and CBD is a key endeavor that will help to elucidate the genetic basis of these 4R tauopathies.104 These genomic data will provide important insight into novel pathways and the fundamental biology involved in the pathogenesis of tauopathy, which might lead to new pharmacological strategies for the treatment of patients with this condition.
Key points.
Corticobasal degeneration (CBD) is associated with neuronal and glial tau pathology (including astrocytic plaques) in gray and white matter of the cortex, basal ganglia, diencephalon and rostral brainstem
The most common clinical presentation of CBD is asymmetric progressive rigidity and apraxia with limb dystonia and myoclonus, termed corticobasal syndrome
CBD pathology can be found in patients with Richardson syndrome (the most common clinical presentation of progressive supranuclear palsy), behavioral variant frontotemporal dementia and primary progressive aphasia
The underlying pathology in corticobasal syndrome can be Alzheimer disease, progressive supranuclear palsy, frontotemporal lobar degeneration or Pick disease; corticobasal syndrome is not, therefore, a specific phenotype of CBD
Given the remarkable clinical heterogeneity of CBD and poor accuracy of diagnosis in living patients, the findings of studies based on non-autopsy-confirmed cases should be interpreted with caution
Current efforts to improve the diagnostic accuracy of CBD include imaging and cerebrospinal fluid biomarkers, as well as genome-wide association studies
Review criteria.
We searched MEDLINE using the terms “corticobasal degeneration” and “cortical basal ganglionic degeneration”, and their variants, synonyms, and acronyms, for articles published between January 1988 and December 2010. We also examined the bibliographies of key articles to identify additional relevant studies. Research papers with pathologically confirmed diagnosis of corticobasal degeneration were specifically sought. Reports based entirely on clinical diagnoses were included only if they were the sole available literature on that particular topic.
Acknowledgments
K. A. Josephs is supported by NIH grants R01-DC10367 and R01-AG37491. J. L. Whitwell is supported by NIH grant R21-AG38736. D. W. Dickson is supported by the Mayo Foundation (Robert E. Jacoby Professorship for Alzheimer’s Research), Mangurian Foundation, CurePSP The Society for Progressive Supranuclear Palsy and NIH grants P50-AG16574, P50-NS72187 and P01-AG17216.
Footnotes
Competing interests
The authors declare no competing interests.
Contributor Information
Naomi Kouri, Department of Neuroscience Mayo Clinic, 4500 San Pablo Road, Jacksonville, FL 32224, USA.
Jennifer L. Whitwell, Department of Radiology, Mayo Clinic, 200 First Street S. W., Rochester, MN 55905, USA
Keith A. Josephs, Department of Neurology, Mayo Clinic, 200 First Street S. W., Rochester, MN 55905, USA
Rosa Rademakers, Department of Neuroscience Mayo Clinic, 4500 San Pablo Road, Jacksonville, FL 32224, USA.
Dennis W. Dickson, Department of Neuroscience Mayo Clinic, 4500 San Pablo Road, Jacksonville, FL 32224, USA
References
- 1.Litvan I et al. Accuracy of the clinical diagnosis of corticobasal degeneration: a clinicopathologic study. Neurology 48, 119–125 (1997). [DOI] [PubMed] [Google Scholar]
- 2.Togasaki DM & Tanner CM Epidemiologic aspects. Adv. Neurol. 82, 53–59 (2000). [PubMed] [Google Scholar]
- 3.Winter Y et al. Incidence of Parkinson’s disease and atypical parkinsonism: Russian population-based study. Mov. Disord. 25, 349–356 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Rebeiz JJ, Kolodny EH & Richardson EP Jr. Corticodentatonigral degeneration with neuronal achromasia. Arch. Neurol. 18, 20–33 (1968). [DOI] [PubMed] [Google Scholar]
- 5.Gibb WR, Luthert PJ & Marsden CD Corticobasal degeneration. Brain 112, 1171–1192 (1989). [DOI] [PubMed] [Google Scholar]
- 6.Bergeron C, Davis A & Lang AE Corticobasal ganglionic degeneration and progressive supranuclear palsy presenting with cognitive decline. Brain Pathol. 8, 355–365 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Watts RL, Mirra SS & Richarson EP Jr in Movement Disorders III: Blue Books of Practical Neurology Vol. 13 (eds Marsden CD & Fahn S) 282–299 (Butterworth–Heinemann, Oxford, 1994). [Google Scholar]
- 8.Riley DE & Lang AE Corticobasal ganglionic degeneration (CBGD): further observations in six additional cases. Neurology 38, 360 (1988). [Google Scholar]
- 9.Boeve BF et al. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology 53, 795–800 (1999). [DOI] [PubMed] [Google Scholar]
- 10.Riley DE et al. Cortical–basal ganglionic degeneration. Neurology 40, 1203–1212 (1990). [DOI] [PubMed] [Google Scholar]
- 11.Bak TH & Hodges JR Corticobasal degeneration: clinical aspects. Handb. Clin. Neurol. 89, 509–521 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Lang AE, Riley DE & Bergeron C in Neurodegenerative Diseases Ch. 49 (ed. Calne DB) 877–894 (W. B. Saunders, Philadelphia, 1994). [Google Scholar]
- 13.Grundke-Iqbal I et al. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 261, 6084–6089 (1986). [PubMed] [Google Scholar]
- 14.Williams DR et al. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSP-parkinsonism. Brain 128, 1247–1258 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Bergeron C, Pollanen MS, Weyer L, Black SE & Lang AE Unusual clinical presentations of cortical–basal ganglionic degeneration. Ann. Neurol. 40, 893–900 (1996). [DOI] [PubMed] [Google Scholar]
- 16.Murray R et al. Cognitive and motor assessment in autopsy-proven corticobasal degeneration. Neurology 68, 1274–1283 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Ling H et al. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain 133, 2045–2057 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Schneider JA, Watts RL, Gearing M, Brewer RP & Mirra SS Corticobasal degeneration: neuropathologic and clinical heterogeneity. Neurology 48, 959–969 (1997). [DOI] [PubMed] [Google Scholar]
- 19.Grimes DA, Lang AE & Bergeron CB Dementia as the most common presentation of cortical-basal ganglionic degeneration. Neurology 53, 1969–1974 (1999). [DOI] [PubMed] [Google Scholar]
- 20.Kertesz A, Martinez-Lage P, Davidson W & Munoz DG The corticobasal degeneration syndrome overlaps progressive aphasia and frontotemporal dementia. Neurology 55, 1368–1375 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Gorno-Tempini ML, Murray RC, Rankin KP, Weiner MW & Miller BL Clinical, cognitive and anatomical evolution from nonfluent progressive aphasia to corticobasal syndrome: a case report. Neurocase 10, 426–436 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Josephs KA et al. Clinicopathological and imaging correlates of progressive aphasia and apraxia of speech. Brain 129, 1385–1398 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raggi A et al. The clinical overlap between the corticobasal degeneration syndrome and other diseases of the frontotemporal spectrum: three case reports. Behav. Neurol. 18, 159–164 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gibb WR, Luthert PJ & Marsden CD Clinical and pathological features of corticobasal degeneration. Adv. Neurol. 53, 51–54 (1990). [PubMed] [Google Scholar]
- 25.Wenning GK et al. Natural history and survival of 14 patients with corticobasal degeneration confirmed at postmortem examination. J. Neurol. Neurosurg. Psychiatry 64, 184–189 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Litvan I, Grimes DA & Lang AE Phenotypes and prognosis: clinicopathologic studies of corticobasal degeneration. Adv. Neurol. 82, 183–196 (2000). [PubMed] [Google Scholar]
- 27.Kertesz A, McMonagle P, Blair M, Davidson W & Munoz DG The evolution and pathology of frontotemporal dementia. Brain 128, 1996–2005 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Lang AE, Bergeron C, Pollanen MS & Ashby P Parietal Pick’s disease mimicking cortical–basal ganglionic degeneration. Neurology 44, 1436–1440 (1994). [DOI] [PubMed] [Google Scholar]
- 29.Grimes DA, Bergeron CB & Lang AE Motor neuron disease-inclusion dementia presenting as cortical–basal ganglionic degeneration. Mov. Disord. 14, 674–680 (1999). [DOI] [PubMed] [Google Scholar]
- 30.Horoupian DS & Wasserstein PH Alzheimer’s disease pathology in motor cortex in dementia with Lewy bodies clinically mimicking corticobasal degeneration. Acta Neuropathol. 98, 317–322 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Hu WT et al. Alzheimer’s disease and corticobasal degeneration presenting as corticobasal syndrome. Mov. Disord. 24, 1375–1379 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Benussi L et al. Progranulin Leu271LeufsX10 is one of the most common FTLD and CBS associated mutations worldwide. Neurobiol. Dis. 33, 379–385 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Whitwell JL et al. Imaging correlates of pathology in corticobasal syndrome. Neurology 75, 1879–1887 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williams DR, Lees AJ, Wherrett JR & Steele JCJ Clifford Richardson and 50 years of progressive supranuclear palsy. Neurology 70, 566–573 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Steele JC, Richardson JC & Olszewski J Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch. Neurol. 10, 333–359 (1964). [DOI] [PubMed] [Google Scholar]
- 36.Tsuboi Y et al. Increased tau burden in the cortices of progressive supranuclear palsy presenting with corticobasal syndrome. Mov. Disord. 20, 982–988 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Oide T et al. Progressive supranuclear palsy with asymmetric tau pathology presenting with unilateral limb dystonia. Acta Neuropathol. 104, 209–214 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Litvan I et al. Clinical features differentiating patients with postmortem confirmed progressive supranuclear palsy and corticobasal degeneration. J. Neurol. 246 (Suppl. 2), II1–II5 (1999). [DOI] [PubMed] [Google Scholar]
- 39.Dickson DW Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J. Neurol. 246 (Suppl. 2), II6–II15 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Shiozawa M et al. Corticobasal degeneration: an autopsy case clinically diagnosed as progressive supranuclear palsy. Clin. Neuropathol. 19, 192–199 (2000). [PubMed] [Google Scholar]
- 41.Hassan A et al. Symmetric corticobasal degeneration (S-CBD). Parkinsonism Relat. Disord. 16, 208–214 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vidailhet M et al. Eye movements in parkinsonian syndromes. Ann. Neurol. 35, 420–426 (1994). [DOI] [PubMed] [Google Scholar]
- 43.Rivaud-Péchoux S et al. Longitudinal ocular motor study in corticobasal degeneration and progressive supranuclear palsy. Neurology 54, 1029–1032 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Zadikoff C & Lang AE Apraxia in movement disorders. Brain 128, 1480–1497 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Houghton DJ & Litvan I Unraveling progressive supranuclear palsy: from the bedside back to the bench. Parkinsonism Relat. Disord. 13 (Suppl. 3), S341–S346 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Cummings JL & Litvan I Neuropsychiatric aspects of corticobasal degeneration. Adv. Neurol. 82, 147–152 (2000). [PubMed] [Google Scholar]
- 47.Josephs KA et al. Voxel-based morphometry in autopsy proven PSP and CBD. Neurobiol. Aging 29, 280–289 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neary D et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51, 1546–1554 (1998). [DOI] [PubMed] [Google Scholar]
- 49.Hodges JR et al. Clinicopathological correlates in frontotemporal dementia. Ann. Neurol. 56, 399–406 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Forman MS et al. Frontotemporal dementia: clinicopathological correlations. Ann. Neurol. 59, 952–962 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Whitwell JL et al. MRI correlates of protein deposition and disease severity in postmortem frontotemporal lobar degeneration. Neurodegener. Dis. 6, 106–117 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Josephs KA et al. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology 66, 41–48 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Grossman M et al. Longitudinal decline in autopsy-defined frontotemporal lobar degeneration. Neurology 70, 2036–2045 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mesulam MM Primary progressive aphasia. Ann. Neurol. 49, 425–432 (2001). [PubMed] [Google Scholar]
- 55.Knibb JA, Xuereb JH, Patterson K & Hodges JR Clinical and pathological characterization of progressive aphasia. Ann. Neurol. 59, 156–165 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Grossman M Primary progressive aphasia: clinicopathological correlations. Nat. Rev. Neurol. 6, 88–97 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Josephs KA et al. Frontotemporal lobar degeneration and ubiquitin immunohistochemistry. Neuropathol. Appl. Neurobiol. 30, 369–373 (2004). [DOI] [PubMed] [Google Scholar]
- 58.Benson DF, Davis RJ & Snyder BD Posterior cortical atrophy. Arch. Neurol. 45, 789–793 (1988). [DOI] [PubMed] [Google Scholar]
- 59.Renner JA et al. Progressive posterior cortical dysfunction: a clinicopathologic series. Neurology 63, 1175–1180 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Tang-Wai DF et al. Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology 63, 1168–1174 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Jellinger KA et al. Four-repeat tauopathy clinically presenting as posterior cortical atrophy: atypical corticobasal degeneration? Acta Neuropathol. 121, 267–277 (2011). [DOI] [PubMed] [Google Scholar]
- 62.Dickson DW et al. Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J. Neuropathol. Exp. Neurol. 61, 935–946 (2002). [DOI] [PubMed] [Google Scholar]
- 63.Fujino Y, Delucia MW, Davies P & Dickson DW Ballooned neurones in the limbic lobe are associated with Alzheimer type pathology and lack diagnostic specificity. Neuropathol. Appl. Neurobiol. 30, 676–682 (2004). [DOI] [PubMed] [Google Scholar]
- 64.Josephs KA et al. Atypical progressive supranuclear palsy with corticospinal tract degeneration. J. Neuropathol. Exp. Neurol. 65, 396–405 (2006). [DOI] [PubMed] [Google Scholar]
- 65.Dickson DW in The Neuropathology of Dementia Ch. 11 (eds Esiri MM et al. ) 227–256 (Cambridge University Press, Cambridge, 2004). [Google Scholar]
- 66.Komori T et al. Astrocytic plaques and tufts of abnormal fibers do not coexist in corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. 96, 401–408 (1998). [DOI] [PubMed] [Google Scholar]
- 67.Feany MB & Dickson DW Widespread cytoskeletal pathology characterizes corticobasal degeneration. Am. J. Pathol. 146, 1388–1396 (1995). [PMC free article] [PubMed] [Google Scholar]
- 68.Yamada T, McGeer PL & McGeer EG Appearance of paired nucleated, tau-positive glia in patients with progressive supranuclear palsy brain tissue. Neurosci. Lett. 135, 99–102 (1992). [DOI] [PubMed] [Google Scholar]
- 69.Arai T et al. Identification of amino-terminally cleaved tau fragments that distinguish progressive supranuclear palsy from corticobasal degeneration. Ann. Neurol. 55, 72–79 (2004). [DOI] [PubMed] [Google Scholar]
- 70.Arai T et al. Intracellular processing of aggregated tau differs between corticobasal degeneration and progressive supranuclear palsy. Neuroreport 12, 935–938 (2001). [DOI] [PubMed] [Google Scholar]
- 71.Ishizawa K & Dickson DW Microglial activation parallels system degeneration in progressive supranuclear palsy and corticobasal degeneration. J. Neuropathol. Exp. Neurol. 60, 647–657 (2001). [DOI] [PubMed] [Google Scholar]
- 72.Gerhard A et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in progressive supranuclear palsy. Mov. Disord. 21, 89–93 (2006). [DOI] [PubMed] [Google Scholar]
- 73.Bhaskar K et al. Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68, 19–31 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ittner LM et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimers’s disease mouse models. Cell 142, 387–397 (2010). [DOI] [PubMed] [Google Scholar]
- 75.Hoover BR et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brunden KR et al. Tau-directed drug discovery for Alzheimer’s disease and related tauopathies: a focus on tau assembly inhibitors. Exp. Neurol. 223, 304–310 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wischik CM, Edwards PC, Lai RY, Roth M & Harrington CR Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc. Natl Acad. Sci. USA 93, 11213–11218 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gong CX, Grundke-Iqbal I & Iqbal K Targeting tau protein in Alzheimer’s disease. Drugs Aging 27, 351–365 (2010). [DOI] [PubMed] [Google Scholar]
- 79.Mandelkow EM et al. Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 314, 315–321 (1992). [DOI] [PubMed] [Google Scholar]
- 80.Pérez M, Hernandez F, Lim F, Diaz-Nido J & Avila J Chronic lithium treatment decreases mutant tau protein aggregation in a transgenic mouse model. J. Alzheimers Dis. 5, 301–308 (2003). [DOI] [PubMed] [Google Scholar]
- 81.Noble W et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl Acad. Sci. USA 102, 6990–6995 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nakashima H et al. Chronic lithium treatment decreases tau lesions by promoting ubiquitination in a mouse model of tauopathies. Acta Neuropathol. 110, 547–556 (2005). [DOI] [PubMed] [Google Scholar]
- 83.Engel T, Goñi-Oliver P, Lucas JJ, Avila J & Hernández F Chronic lithium administration to FTDP-17 tau and GSK-3β overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J. Neurochem. 99, 1445–1455 (2006). [DOI] [PubMed] [Google Scholar]
- 84.Caccamo A, Oddo S, Tran LX & LaFerla FM Lithium reduces tau phosphorylation but not Aβ or working memory deficits in a transgenic model with both plaques and tangles. Am. J. Pathol. 170, 1669–1675 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee VM, Kenyon TK & Trojanowski JQ Transgenic animal models of tauopathies. Biochim. Biophys. Acta 1739, 251–259 (2005). [DOI] [PubMed] [Google Scholar]
- 86.Zhang B et al. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc. Natl Acad. Sci. USA 102, 227–231 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gozes I & Divinski I The femtomolar-acting NAP interacts with microtubules: novel aspects of astrocyte protection. J. Alzheimers Dis. 6, S37–S41 (2004). [DOI] [PubMed] [Google Scholar]
- 88.Matsuoka Y et al. Intranasal NAP administration reduces accumulation of amyloid peptide and tau hyperphosphorylation in a transgenic mouse model of Alzheimer’s disease at early pathological stage. J. Mol. Neurosci. 31, 165–170 (2007). [DOI] [PubMed] [Google Scholar]
- 89.Matsuoka Y et al. A neuronal microtubule-interacting agent, NAPVSIPQ, reduces tau pathology and enhances cognitive function in a mouse model of Alzheimer’s disease. J. Pharmacol. Exp. Ther. 325, 146–153 (2008). [DOI] [PubMed] [Google Scholar]
- 90.Brunden KR et al. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J. Neurosci. 30, 13861–13866 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Luo W et al. Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proc. Natl Acad. Sci. USA 104, 9511–9516 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dickey CA et al. The high-affinity HSP90–CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Invest. 117, 648–658 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pickhardt M et al. Anthraquinones inhibit tau aggregation and dissolve Alzheimer’s paired helical filaments in vitro and in cells. J. Biol. Chem. 280, 3628–3635 (2005). [DOI] [PubMed] [Google Scholar]
- 94.Urakami K et al. A comparison of tau protein in cerebrospinal fluid between corticobasal degeneration and progressive supranuclear palsy. Neurosci. Lett. 259, 127–129 (1999). [DOI] [PubMed] [Google Scholar]
- 95.Urakami K et al. Diagnostic significance of tau protein in cerebrospinal fluid from patients with corticobasal degeneration or progressive supranuclear palsy. J. Neurol. Sci. 183, 95–98 (2001). [DOI] [PubMed] [Google Scholar]
- 96.Borroni B et al. Pattern of tau forms in CSF is altered in progressive supranuclear palsy. Neurobiol. Aging 30, 34–40 (2009). [DOI] [PubMed] [Google Scholar]
- 97.Noguchi M et al. Decreased β-amyloid peptide42 in cerebrospinal fluid of patients with progressive supranuclear palsy and corticobasal degeneration. J. Neurol. Sci. 237, 61–65 (2005). [DOI] [PubMed] [Google Scholar]
- 98.Mitani K et al. Increased CSF tau protein in corticobasal degeneration. J. Neurol. 245, 44–46 (1998). [DOI] [PubMed] [Google Scholar]
- 99.Arai H et al. Cerebrospinal fluid tau levels in neurodegenerative diseases with distinct tau-related pathology. Biochem. Biophys. Res. Commun. 236, 262–264 (1997). [DOI] [PubMed] [Google Scholar]
- 100.Portelius E et al. Characterization of tau in cerebrospinal fluid using mass spectrometry. J. Proteome Res. 7, 2114–2120 (2008). [DOI] [PubMed] [Google Scholar]
- 101.Guillozet-Bongaarts AL et al. Phosphorylation and cleavage of tau in non-AD tauopathies. Acta Neuropathol. 113, 513–520 (2007). [DOI] [PubMed] [Google Scholar]
- 102.Holmberg B, Rosengren L, Karlsson JE & Johnels B Increased cerebrospinal fluid levels of neurofilament protein in progressive supranuclear palsy and multiple-system atrophy compared with Parkinson’s disease. Mov. Disord. 13, 70–77 (1998). [DOI] [PubMed] [Google Scholar]
- 103.Brettschneider J et al. Neurofilament heavy-chain NfHSMI35 in cerebrospinal fluid supports the differential diagnosis of parkinsonian syndromes. Mov. Disord. 21, 2224–2227 (2006). [DOI] [PubMed] [Google Scholar]
- 104.Müller U GWAS in PSP: results at disease-associated loci other than MAPT. Proc. CurePSP 2010 International Research Symposium (San Diego, CA, November 18, 2010). [Google Scholar]
- 105.Sergeant N, Wattez A & Delacourte A Neurofibrillary degeneration in progressive supranuclear palsy and corticobasal degeneration: tau pathologies with exclusively “exon 10” isoforms. J. Neurochem. 72, 1243–1249 (1999). [DOI] [PubMed] [Google Scholar]
- 106.Delacourte A, Sergeant N, Wattez A, Gauvreau D & Robitaille Y Vulnerable neuronal subsets in Alzheimer’s and Pick’s disease are distinguished by their tau isoform distribution and phosphorylation. Ann. Neurol. 43, 193–204 (1998). [DOI] [PubMed] [Google Scholar]
- 107.Hutton M Missense and splice site mutations in tau associated with FTDP-17: multiple pathogenic mechanisms. Neurology 56, S21–S25 (2001). [DOI] [PubMed] [Google Scholar]
- 108.Binder LI, Frankfurter A & Rebhun LI The distribution of tau in the mammalian central nervous system. J. Cell. Biol. 101, 1371–1378 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.LoPresti P, Szuchet S, Papasozomenos SC, Zinkowski RP & Binder LI Functional implications for the microtubule-associated protein tau: localization in oligodendrocytes. Proc. Natl Acad. Sci. USA 92, 10369–10373 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Witman GB, Cleveland DW, Weingarten MD & Kirschner MW Tubulin requires tau for growth onto microtubule initiating sites. Proc. Natl Acad. Sci. USA 73, 4070–4074 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Andreadis A, Brown WM & Kosik KS Structure and novel exons of the human tau gene. Biochemistry 31, 10626–10633 (1992). [DOI] [PubMed] [Google Scholar]
- 112.Goedert M, Spillantini MG, Jakes R, Rutherford D & Crowther RA Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3, 519–526 (1989). [DOI] [PubMed] [Google Scholar]
- 113.Baker M et al. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum. Mol. Genet. 8, 711–715 (1999). [DOI] [PubMed] [Google Scholar]
- 114.Stefansson H et al. A common inversion under selection in Europeans. Nat. Genet. 37, 129–137 (2005). [DOI] [PubMed] [Google Scholar]
- 115.Conrad C et al. Genetic evidence for the involvement of tau in progressive supranuclear palsy. Ann. Neurol. 41, 277–281 (1997). [DOI] [PubMed] [Google Scholar]
- 116.Houlden H et al. Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology 56, 1702–1706 (2001). [DOI] [PubMed] [Google Scholar]
- 117.Rademakers R et al. High-density SNP haplotyping suggests altered regulation of tau gene expression in progressive supranuclear palsy. Hum. Mol. Genet. 14, 3281–3292 (2005). [DOI] [PubMed] [Google Scholar]
- 118.Pittman AM et al. Linkage disequilibrium fine mapping and haplotype association analysis of the tau gene in progressive supranuclear palsy and corticobasal degeneration. J. Med. Genet. 42, 837–846 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bugiani O et al. Frontotemporal dementia and corticobasal degeneration in a family with a P301S mutation in tau. J. Neuropathol. Exp. Neurol. 58, 667–677 (1999). [DOI] [PubMed] [Google Scholar]
- 120.Spillantini MG et al. A novel tau mutation (N296N) in familial dementia with swollen achromatic neurons and corticobasal inclusion bodies. Ann. Neurol. 48, 939–943 (2000). [DOI] [PubMed] [Google Scholar]
- 121.Rossi G et al. The G389R mutation in the MAPT gene presenting as sporadic corticobasal syndrome. Mov. Disord. 23, 892–895 (2008). [DOI] [PubMed] [Google Scholar]
- 122.Rademakers R, Cruts M & van Broeckhoven C The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum. Mutat. 24, 277–295 (2004). [DOI] [PubMed] [Google Scholar]