

Abstract
Administration of a nitric oxide (NO) donor triggers migraine attacks, but the mechanisms by which this occurs are unknown. Reactive nitroxidative species, including NO and peroxynitrite (PN), have been implicated in nociceptive sensitization, and neutralizing PN is antinociceptive. We determined whether PN contributes to nociceptive responses in two distinct models of migraine headache. Female and male mice were subjected to 3 consecutive days of restraint stress or to dural stimulation with the proinflammatory cytokine interleukin-6. Following resolution of the initial poststimulus behavioral responses, animals were tested for hyperalgesic priming using a normally non-noxious dose of the NO donor sodium nitroprusside (SNP) or dural pH 7.0, respectively. We measured periorbital von Frey and grimace responses in both models and measured stress-induced changes in 3-nitrotyrosine (3-NT) expression (a marker for PN activity) and trigeminal ganglia (TGs) mitochondrial function. Additionally, we recorded the neuronal activity of TGs in response to the PN generator SIN-1 [5-amino-3-(4-morpholinyl)−1,2,3-oxadiazolium chloride]. We then tested the effects of the PN decomposition catalysts Fe(III)5,10,15,20-tetrakis(N-methylpyridinium-4-yl) porphyrin (FeTMPyP) and FeTPPS [Fe(III)5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrinato chloride], or the PN scavenger MnTBAP [Mn(III)tetrakis(4-benzoic acid)porphyrin] against these behavioral, molecular, and neuronal changes. Neutralizing PN attenuated stress-induced periorbital hypersensitivity and priming to SNP, with no effect on priming to dural pH 7.0. These compounds also prevented stress-induced increases in 3-NT expression in both the TGs and dura mater, and attenuated TG neuronal hyperexcitability caused by SIN-1. Surprisingly, FeTMPyP attenuated changes in TG mitochondrial function caused by SNP in stressed males only. Together, these data strongly implicate PN in migraine mechanisms and highlight the therapeutic potential of targeting PN.
SIGNIFICANCE STATEMENT Among the most reliable experimental triggers of migraine are nitric oxide donors. The mechanisms by which nitric oxide triggers attacks are unclear but may be because of reactive nitroxidative species such as peroxynitrite. Using mouse models of migraine headache, we show that peroxynitrite-modulating compounds attenuate behavioral, neuronal, and molecular changes caused by repeated stress and nitric oxide donors (two of the most common triggers of migraine in humans). Additionally, our results show a sex-specific regulation of mitochondrial function by peroxynitrite following stress, providing novel insight into the ways in which peroxynitrite may contribute to migraine-related mechanisms. Critically, our data underscore the potential in targeting peroxynitrite formation as a novel therapeutic for the treatment of migraine headache.
Keywords: dura mater, headache, migraine, nitric oxide, peroxynitrite, trigeminal ganglia
Introduction
Migraine is an extremely complex disorder that is the second leading cause of disability worldwide. While the pathology of migraine is poorly understood, the headache phase is thought to be the result of abnormal activation and sensitization of trigeminal sensory afferents innervating the meninges (Noseda and Burstein, 2013). Despite recent progress in therapeutics, the larger migraine population is still burdened by issues of low drug efficacy and high rates of headache relapse following treatment, underscoring the critical need for better treatments (Goadsby, 2013).
One of the most consistent pharmacological triggers of migraine is administration of a nitric oxide (NO) donor, after which migraine patients typically develop an attack within several hours of administration (Olesen et al., 1993). NO donors cause activation of hypothalamic circuits and trigger premonitory symptoms, both of which are also known to occur during the early stages of naturally occurring migraines (Afridi et al., 2004; Maniyar et al., 2014). Notably, these effects are not observed in healthy control subjects, implying that migraine patients are sensitized to mechanisms that are triggered by NO donors (Olesen and Ashina, 2015). However, given the relatively short half-life of NO (merely seconds), it is unclear how attacks can occur hours after NO has cleared the system. Additionally, NO plays a critical role in maintaining normal physiological processing, and, therefore, targeting NO as a therapeutic strategy has achieved mixed results (Lassen et al., 1997; Høivik et al., 2010; Pradhan et al., 2018).
NO reacts with superoxide (SO) to produce peroxynitrite (PN), a reactive nitroxidative species (RNOS) that promotes oxidative damage through the inactivation of essential enzymes, the inhibition of mitochondrial respiration, and upregulating mechanisms of neuroinflammation and nitrosative stress (Radi et al., 1994, 2002a; Virág et al., 2003). Numerous studies have implicated PN in the development and maintenance of painful states and have underscored its ability to promote spinal transmission, sensitize neurons, and disrupt homeostasis (Salvemini et al., 2011; Little et al., 2012). PN promotes dorsal root ganglia and dorsal horn hyperexcitability in models of neuropathic pain and a wide variety of PN-modulating compounds (PNMCs) have demonstrated efficacy in rodent models of neuropathic, inflammatory, and visceral pain (Doyle et al., 2012; Little et al., 2012; Slosky and Vanderah, 2015). Although it is still unclear whether PN formation contributes to migraine, clinical studies have generated support for a potential role. A meta-analysis of >1000 migraine patients found significantly decreased concentrations of SO dismutase (SOD; indicating increased SO levels) and increased levels of NO during migraine attacks, both of which are necessary for PN formation (Neri et al., 2015). Interestingly, administration of the NO synthase (NOS) precursor, l-arginine, led to increased levels of PN in the platelets of migraineurs during headache-free periods, an effect that is thought to be regulated by NO pathways (D’Andrea et al., 1994; Gallai et al., 1996; Taffi et al., 2005). Preclinically, a recent study found that the administration of a PNMC, but not a NOS inhibitor, prevented nociceptive responses caused by systemic injection of calcitonin gene-related peptide (CGRP), providing compelling evidence for a role of PN in migraine pathophysiology (Akerman et al., 2021). Furthermore, PN plays a critical role in mediating mitochondria dysfunction via inactivation of essential enzymes and proteins involved in cellular respiration and is believed to be the primary mechanism by which NO disrupts mitochondrial function (Radi et al., 1994, 2002b; Schweizer and Richter, 1996). In human migraine patients, changes in energy production are marked by impairments in enzymes that are critical for respiration. In preclinical migraine models, mitochondrial dysfunction is marked by increases in calcium release as well as impairments in mitochondrial biogenesis in the trigeminal ganglia (TGs), both of which can directly modulate neuronal excitability (Fried et al., 2014; Dong et al., 2017).
We recently demonstrated that rodents become sensitized to normally non-noxious doses of a NO donor following repeated restraint stress, an important observation given that stress is the number one reported trigger of migraine in humans (Peroutka, 2014; Avona et al., 2020). Additionally, our laboratory has previously shown that noninvasive dural stimulation with noxious compounds is able to induce hyperalgesic priming in mice (Burgos-Vega et al., 2019). Based on the above observations, we sought to explore whether PNMCs were efficacious in reducing nociceptive responses in these preclinical migraine models. Furthermore, we examined PN-related mechanisms by measuring the effects of PNMCs on the hyperexcitability of TGs as well as stress-induced 3-nitrotyrosine (3-NT) expression (a marker for PN activity) and mitochondrial bioenergetics.
Materials and Methods
Experimental animals.
Unless otherwise indicated, all behavioral experiments presented in this article used female and male ICR (CD-1) mice (age, 6–8 weeks; weight, ∼25–30 g), which were outbred and purchased from Envigo. All experiments were performed between the hours of 9:00 A.M. and 5:00 P.M. All mice were housed in groups of four animals per cage on a 12 h light/dark cycle and had access to food and water ad libitum. Upon arrival at the animal care facility, animals were allowed a minimum of 72 h to acclimate to their new environment before being handled for experiments. All procedures were conducted with prior approval of the Institutional Animal Care and Use Committee at the University of Texas at Dallas. Anesthetized mice were placed on a warm heating pad and allowed to recover before being placed in their respective home cages. Animal health was monitored for any adverse reactions to compounds administered and injection sites were examined for bleeding. Stressed mice were routinely checked for adverse events potentially because of the restraint tube. The health of stressed mice was also monitored daily by assessing changes in feeding behavior and/or significant changes in weight. Across all experiments, no adverse events were observed.
Drugs and compounds.
For dural injections, a human recombinant interleukin-6 (IL-6) protein (R&D Systems) stock solution (100 mg/ml) was prepared in sterile 0.1% BSA and diluted to 1 ng/ml in synthetic interstitial fluid (SIF) consisting of 135 mm NaCl, 5 mm KCl, 10 mm HEPES, 2 mm CaCl2, 10 mm glucose, and 1 mm MgCl2, at pH 7.4 and 310 mOsm. The NO donor sodium nitroprusside (SNP; Sigma-Aldrich) was prepared fresh in sterile PBS at the time of use and was kept away from light. To assess the role of PN, a 150 µl 30 mg/kg intraperitoneal injection of the PN scavenger Mn(III)tetrakis(4-benzoic acid)porphyrin (MnTBAP) or the PN decomposition catalyst (PNDC) Fe(III)5,10,15,20-tetrakis(N-methylpyridinium-4-yl) porphyrin (FeTMPyP) was administered following repeated stress or before hyperalgesic priming. This dose was based on previous studies showing that PNMCs administered intraperitoneally at this dose achieved efficacy in behavioral pain and trigeminal nociception assays (Akerman et al., 2002; Doyle et al., 2012). To test for the presence of hyperalgesic priming, mice were given a 150 µl intraperitoneal injection of 0.1 mg/kg SNP or a 5 µl dural injection of SIF pH 7.0 solution. For electrophysiology experiments, the PNDC Fe(III)5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrinato chloride (FeTPPS) was purchased from Cayman Chemicals, and a stock was made in deionized water at 5 mg/ml. A final working concentration of 100 or 10 μm was diluted in external solution. SIN-1 [5-amino-3-(4-morpholinyl)−1,2,3-oxadiazolium chloride] was purchased from Tocris Bioscience and was dissolved in external solution to a final concentration of 1 mm. SIN-1 was freshly prepared before patching each cell reported here. SNP was dissolved in the L15 culture medium and the external solution to a final concentration of 100 μm.
Mouse dural injections.
Mouse dural injections were performed as previously described (Burgos-Vega et al., 2019). Mice were anesthetized under isoflurane for <2 min with <2.5–3% isoflurane via a chamber and given a 5 µl injection via a modified internal cannula (Invivo1, part #8IC313ISPCXC, Internal Cannula; standard, 28 gauge, fit to 0.5 mm). The inner projection of the cannula was used to inject through the soft tissue at the intersection of the lambdoidal and sagittal sutures. Using a caliper, the length of the projection was adjusted to be from 0.6 to 0.7 mm based on animal weight (25–30 g) to avoid puncturing the dura mater. Control mice received a 5 µl dural injection of SIF, at pH 7.4 and 310 mOsm. Upon completion of injections, mice were placed back into their respective cups in the testing chamber for 1 h before testing.
Repeated restraint stress.
Mice were stressed as previously described (Avona et al., 2020). Mice were stressed between the hours of 10:00 A.M. and 12:00 P.M. for 2 h/d for 3 consecutive days. Mice were placed right side up in tail vein injection tubes (catalog #51338, Stoelting) with the nose through the provided breathing hole. The slotted tail piece was tightened so as to prevent the mouse from rotating in the tube, but loose enough to allow the animal to breathe. Mice were restrained at a level that allowed for adequate respiration, and care was taken to avoid any trauma caused by the restraint tube. Control mice were placed into a separate room and deprived of food and water for the same 2 h interval for 3 consecutive days. Animals subjected to stress were housed separately from control mice to avoid potential transfer of the stress phenotype.
Measuring mechanical hypersensitivity and grimace.
Mice were handled and conditioned for a single 5 min session, ∼24 h before habituation. Mice were habituated to paper cups (Choice 4 ounce paper cups: top diameter, 6.5 cm; bottom diameter, 4.5 cm; length, 72.5 cm) while in testing chambers for 2 h/d and for at least 2 d before measuring a baseline. Each mouse typically used their same assigned paper cup for the remainder of the experiment. Animals were given food while in testing chambers. Grimace measurements were recorded for each animal in 10 min increments using an Apple iPhone 11 Pro video camera and were analyzed as previously described (Langford et al., 2010; Avona et al., 2019). Analysis of five characterized pain behaviors (orbital tightening, nose bulging, cheek bulging, flattening of whiskers, and flattening of the ears) was scored on a scale from 0 to 2 (0 = not present, 1 = somewhat present, 2 = clearly present). Following grimace measurements, von Frey testing of the periorbital region of the face was used to measure mechanical hypersensitivity of the face as previously described (Burgos-Vega et al., 2019; Lackovic et al., 2021). Filament thresholds were determined using the Dixon “up-and-down” method. Testing in mice began with 0.07 g on the face and increased in weight to a maximum of 0.6 g on the face. The testing timelines for dural injection experiments and stress experiments were conducted as previously described (Burgos-Vega et al., 2019; Avona et al., 2020). In both experimental paradigms, once the mice returned to baseline, a subthreshold dose of compound was administered either onto the dura, at pH 7.0, or intraperitoneally (sodium nitroprusside) to test for hyperalgesic priming. Responses were defined as a mouse removing/swiping the filament away from its face on brief application of the filament. All animals were randomly allocated to experimental groups by drawing for groups. All experimenters were blinded to animal treatments.
Patch-clamp electrophysiology.
Animals were anesthetized with 5% isoflurane and killed by decapitation. TGs were dissected and placed in ice-cold HBSS (divalent free) and incubated at 37°C for 15 min in 20 U/ml papain (Worthington) followed by 15 min in 3 mg/ml collagenase type II (Worthington). After trituration through fire-polished Pasteur pipettes of progressively smaller opening sizes, cells were plated on plates (diameter, 3.5 cm; Corning) coated with poly-d-lysine and laminin (Sigma-Aldrich). Cells were allowed to adhere for several hours at room temperature in a humidified chamber and then nourished with Liebovitz L-15 medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum, 10 mm glucose, 10 mm phosphate buffer, and 50 U/ml penicillin/streptomycin. Whole-cell patch-clamp experiments were performed on isolated mouse TG neurons within 24 h of dissociation using a patch-clamp amplifier (MultiClamp 700B, Molecular Devices) and acquisition software (PClamp 9, Molecular Devices) at room temperature. Recordings were sampled at 20 kHz and filtered at 3 kHz (Digidata 1550B, Molecular Devices). Pipettes (outer diameter, 1.5 mm; inner diameter, 1.1 mm, BF150-110–10, Sutter Instruments) were pulled using a PC-100 puller (Narishige) and heat polished to 3–5 MΩ resistance using a microforge (model MF-83, Narishige). Series resistance was typically 7 MΩ and was compensated up to 60%. Data were analyzed using Clampfit 10 (Molecular Devices). All neurons included in the analysis had a resting membrane potential (RMP) more negative than −40 mV. The RMP was recorded 1–3 min after achieving whole-cell configuration. In current-clamp mode, cells were held at −60 mV, and action potentials (APs) were elicited by injecting slow ramp currents from 100 to 700 pA with Δ200 pA over 1 s to mimic slow depolarization. Basic membrane properties of AP threshold, AP amplitude, half-width, and rheobase were measured using a standard step protocol of 5 ms duration, depolarizing the cell in 10 pA increments until a single action potential was fired. A hyperpolarizing 25 pA pulse of 500 ms duration was applied to the cell in current-clamp mode to determine the input resistance. The pipette solution contained the following (in mm): 120 K-gluconate, 6 KCl, 4 ATP-Mg, 0.3 GTP-Na, 0.1 EGTA, 10 HEPES, and 10 phosphocreatine, at pH 7.4 (adjusted with N-methyl glucamine) and osmolarity at ∼290 mOsm. The external solution contained the following (in mm): 135 NaCl, 2 CaCl2, 1 MgCl2, 5 KCl, 10 glucose, and 2 phosphate buffer, at pH 7.4 (adjusted with N-methyl glucamine) and osmolarity adjusted to ∼315 mOsm with sucrose. Neurons were treated with MnTBAP (100 μm) mixed with the NO donor SIN-1 (1 mm) or FeTPPS (100 μm) mixed with SIN-1 for ∼20–30 min before recording.
Trigeminal mitochondrial bioenergetics.
At 24 h or 14 d following repeated stress, animals were deeply anesthetized with isoflurane and decapitated. TGs were dissected and collected into prechilled tubes of 1× HBSS and then centrifuged at 440 rpm for 30 s. Supernatants were removed and TGs were treated with Collagenase A (catalog #10103586001, Sigma-Aldrich) and incubated in a water bath at 37°C for 25 min. Cells were centrifuged at 440 rpm for 30 s, and supernatant was again removed. Cells were then treated with Collagenase D (catalog #1188866001, Sigma-Aldrich) and incubated at 37°C for another 20 min. Cells were centrifuged at 440 rpm for 2 min, and the pellet was resuspended in Enzyme T (trypsin inhibitor). Digested tissues were triturated ∼20 times using a 1 ml pipette tip and passed through a 70 μm nylon mesh filter followed by a wash with DMEM/F12 media. The resulting solution was centrifuged at 440 rpm for 4 min, and the pellet was resuspended in DMEM/F12 media (supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin). The number of cells was counted using a hemocytometer and trypan blue dye exclusion. For bioenergetic analysis, 60,000 cells were seeded directly into XFp Cell Culture Miniplates (catalog #103025–100, Agilent) coated with 1× poly-d-lysine (catalog #P6407, Sigma-Aldrich) and 1× laminin (catalog #L6274, Sigma-Aldrich). The following day, XF Assay Media (Seahorse Bioscience) supplemented with 2 mm glutamine, 5 mm glucose, 1 mm sodium pyruvate, and pH adjusted to 7.4 was warmed to 37°C in a 0% CO2 incubator. Culture media were replaced with Seahorse XF Calibrant Solution 1 h before running the Seahorse Mito Stress Test according to the manufacturer protocol (User Guide Kit 103010–100). From the oxygen consumption rates (OCRs) measured by the Seahorse apparatus, parameters of mitochondria function, including basal respiration, maximal respiration, spare reserve capacity, proton leak, ATP production, and coupling efficiency were calculated as described in the Seahorse XF Cell Mito Stress Test Report Generator manual in Seahorse Wave Software (Agilent).
3-Nitrotyrosine Western blotting.
At 24 h or 14 d following repeated stress, animals were deeply anesthetized with isoflurane and decapitated. TGs and dura mater were dissected and flash frozen on dry ice. Frozen tissues were homogenized in lysis buffer (50 mm Tris, pH 7.4; 150 nm NaCl; 1 mm EDTA, pH 8.0; and 1% Triton X-100) containing protease and phosphatase inhibitors (Sigma-Aldrich) using cell lysis tubes (catalog #P000918, Bertin Corp.), which were shaken for 60 s in a Minilys (Bertin Corp.). Tubes were then centrifuged at 14,000 rpm for 15 min, and supernatant was collected and transferred to prechilled tubes. Twenty micrograms of protein was loaded into each well of a 10% SDS-PAGE gel, and proteins were transferred to a 0.45 PVDF membrane (Millipore) at 30 V overnight at 4°C. The following day, membranes were blocked with 5% nonfat dry milk in 1× Tris buffer solution containing Tween 20 (TTBS) at room temperature for 1 h and then incubated with anti-3-nitrotyrosine antibody (catalog #ab61392, Abcam) overnight at 4°C. Membranes were then washed in 1× TTBS three times for 5 min each and then incubated with the corresponding secondary antibody at room temperature for 1 h. Subsequently, membranes were then washed in 1× TTBS six times for 5 min each, and signals were detected using Immobilon Western Chemiluminescence HRP Substrate (Millipore). Bands were visualized using a ChemiDoc Touch (Bio-Rad). Overexposed or saturated pixels were not used in the analysis. Analysis was performed using ImageJ version 2.1.0.
Experimental design and statistical analysis.
Female and male mice were used in almost all experiments to determine any sex differences. Behavioral data were analyzed for multiple comparisons at each time point via two-way ANOVA followed by Bonferroni’s post hoc analysis. F-values for these analyses are presented in Table 1. Significance was set at p < 0.05 for all statistical comparisons. Each experiment was independently replicated at least twice. All investigators were blinded to treatments during testing and scoring. Western blot and Seahorse data were also analyzed using a two-way ANOVA with Bonferroni’s correction. For Seahorse data comparisons within sex, a Welch’s two-tailed t test was used. Analysis of electrophysiology traces was performed using either Clampfit 10 or Axograph software. Comparisons between groups was performed using one-way or two-way ANOVA, as appropriate, followed by Dunnett’s test to assess changes in all experimental groups relative to the vehicle group. In experiments measuring ramp-induced excitability changes between a control group and a single treatment group, a repeated-measures ANOVA followed by Bonferroni’s post hoc analysis. For the comparison of drug effects on membrane properties between a control group and a drug group, either the paired or the unpaired t test was used, as appropriate. All data are represented in the Results and figures as the mean ± SEM. Significance was set at p < 0.05 for all statistical comparisons. All data analyses were performed using Prism version 9.2 for Mac OS X, and figures were composed in Prism or Inkscape (www.inkscape.org).
Table 1.
F-Values for between-factors two-way ANOVAs of behavioral data are presented for each figure
| Figure | Interaction | Row factor | Column factor |
|---|---|---|---|
| 1 B | F(27,540) = 10.07 | F(9,540) = 65.95 | F(3,540) = 136.1 |
| 1 C | F(27,370) = 8.502 | F(9,370) = 38.76 | F(3,370) = 91.74 |
| 1 D | F(27,280) = 10.52 | F(9,280) = 81.07 | F(3,280) = 117.4 |
| 1 E | F(27,280) = 17.65 | F(9,280) = 173.9 | F(3,280) = 167.9 |
| 2 B | F(30,396) = 3.329 | F(10,396) = 29.68 | F(3,396) = 63.92 |
| 2 C | F(30,330) = 4.342 | F(10,330) = 52.70 | F(3,330) = 107.2 |
| 2 D | F(30,220) = 2.900 | F(10,220) = 21.53 | F(3,220) = 51.06 |
| 2 E | F(30,220) = 2.331 | F(10,220) = 23.51 | F(3,220) = 84.19 |
| 3 B | F(27,200) = 7.308 | F(9,200) = 20.29 | F(3,200) = 139.3 |
| 3 C | F(27,200) = 12.74 | F(9,200) = 33.59 | F(3,200) = 139.0 |
| 4 B | F(36,260) = 4.852 | F(9,260) = 21.86 | F(4,260) = 92.94 |
| 4 C | F(36,260) = 8.091 | F(9,260) = 40.22 | F(4,260) = 61.78 |
| 4 D | F(36,350) = 6.744 | F(9,350) = 37.44 | F(4,350) = 113.7 |
| 4 E | F(36,350) = 12.98 | F(9,350) = 78.00 | F(4,350) = 103.0 |
| 5 B | F(27,120) = 5.911 | F(9,120) = 19.79 | F(3,120) = 134.1 |
| 5 C | F(27,120) = 5.806 | F(9,120) = 18.64 | F(3,120) = 47.11 |
In all cases, Bonferroni’s post hoc analysis was used for multiple comparisons.
Results
PN contributes to NO donor-induced hypersensitivity following repeated stress
Although the exact mechanism of how NO donors cause sensitization in migraine or other types of pain is still not understood, targeting the downstream formation the reactive nitro-oxidative molecule PN has demonstrated a role in mediating painful states (Radi et al., 2001; Little et al., 2012; Slosky and Vanderah, 2015; Akerman et al., 2021). Previously, our laboratory published that repeated restraint stress is capable of sensitizing mice to subthreshold, typically non-noxious doses of the NO donor SNP (Avona et al., 2020). Based on this, we tested whether administering a PN-modulating compound before NO donor administration was capable of preventing hyperalgesic priming following repeated stress. Following baseline measurements, female and male mice were subjected to repeated restraint stress and tested for grimace responses and von Frey thresholds. Upon returning to baseline thresholds, mice were administered 30 mg/kg PN scavenger MnTBAP, the decomposition catalyst FeTMPyP, or vehicle ∼30 min before intraperitoneal administration of 0.1 mg/kg SNP and again tested for facial hypersensitivity (Fig. 1A). Stress caused acute facial hypersensitivity and noxious grimace responses in both females (Fig. 1B,C) and males (Fig. 1D,E) that lasted for ∼14 d; however, stress-primed mice that received MnTBAP or FeTMPyP, but not vehicle, before SNP exhibited significantly reduced facial withdrawal thresholds, suggesting a role for PN formation in mediating NO donor-induced behavioral responses following stress. Grimace measurements were insignificant in the priming phase of these experiments, consistent with what we have previously reported (Avona et al., 2020).
Figure 1.

Peroxynitrite mediates NO donor-induced mechanical hypersensitivity in stress-primed mice. A, A schematic of the stress paradigm used is shown. Mice were subjected to repeated restraint stress or control conditions and tested for facial allodynia via von Frey assessment and mean grimace scores. Upon returning to baseline thresholds 14 d after stress, mice received a 30 mg/kg intraperitoneal injection of a PN scavenger (MnTBAP), a PN decomposition catalyst (FeTMPyP), or vehicle (PBS) 30 min before injection of the NO donor SNP (0.1 mg/kg, i.p.) and were again tested for facial allodynia. B, D, MnTBAP and FeTMPyP both significantly attenuated facial hypersensitivity caused by SNP in stress-primed female (B) and male (D) mice. C, E, No differences in grimace scoring were found in either sex (C, E). All control groups received vehicle before SNP. Two-way ANOVA followed by Bonferroni’s post hoc analysis revealed significant differences in the priming phase between stressed mice that received vehicle before SNP and stressed mice that received MnTBAP (denoted by *) or FeTMPyP (denoted by †) before SNP. n ≥ 6 for all groups in B and C; n = 8 for all groups in D and E. Data are represented as the mean ± SEM. Table 1, see for F-values. *†p < 0.05, **p < 0.01, ***†††p < 0.001, ****††††p < 0.0001.
PN does not contribute to dural pH 7.0 responses in IL-6-primed mice
Although modulating PN demonstrated efficacy in the SNP model where the production of PN is more likely to occur following the generation of NO, it was unclear what effect it would have against other stimuli. Thus, we asked whether PN plays a role in the development of responses to dural pH 7.0 in mice primed by dural IL-6. Our laboratory has previously shown that dural IL-6 is capable of inducing facial hypersensitivity in mice and sensitizing them to dural pH 7.0, which is typically non-noxious in healthy control mice (Burgos-Vega et al., 2019). Female and male mice were administered a 5 µl injection of IL-6 onto their dura mater and tested for facial hypersensitivity and grimacing. Upon resolution of acute behavioral responses, mice were given an intraperitoneal injection of 30 mg/kg MnTBAP, FeTMPyP, or vehicle followed by a 5 µl dural injection of pH 7.0 solution ∼30 min later (Fig. 2A). Although grimace responses were slightly altered in the early time points following dural pH 7.0, we found that neither MnTBAP or FeTMPyP was able to attenuate the facial hypersensitivity caused by dural pH 7.0 in either sex (Fig. 2B–E), suggesting that PN formation may not be critical to the development of pH 7.0 responses in IL-6-primed mice.
Figure 2.

Modulating PN does not attenuate facial priming to dural pH 7.0. A, Dural injections and behavioral testing timelines are presented. B–E, Female (B, C) and male (D, E) mice received a 5 µl dural injection of vehicle (SIF) or IL-6 (0.1 ng) to induce acute periorbital hypersensitivity and grimacing that lasted out to 72 h. After the pain resolved, mice were given a 30 mg/kg intraperitoneal injection of MnTBAP, FeTMPyP, or vehicle (SIF) 30 min before a second 5 µl dural injection of a SIF pH 7.0 solution to check for the presence of hyperalgesic priming. Mice that received a PN-modulating compound did not exhibit significant differences in nociceptive thresholds from those that received vehicle after IL-6; however, a two-way ANOVA with Bonferroni’s post hoc analysis of the priming phase revealed significantly lower grimace scores between the group that received MnTBAP (denoted by *) and the IL-6/vehicle group within the first 3 h following dural pH 7.0. All control mice received pH 7.0 solution in the priming phase. n ≥ 8 for all groups in A and B; n = 6 for all groups in D and E. Data are represented as the mean ± SEM. Table 1, see for F-values. *p < 0.05, **p < 0.01.
Temporal effects of modulating PN following repeated stress
After observing that modulation of PN was capable of preventing NO donor-induced hypersensitivity in stress-primed mice, we examined a potential role of PN in the acute stress response. As mentioned earlier, stress is the number one reported trigger of migraine in human patients. Based on this, we wanted to explore how PN formation plays a role in stress-induced acute facial hypersensitivity. Following the third day of stress, female mice were given a 30 mg/kg intraperitoneal injection of FeTMPyP or vehicle at ∼1, 24, 48, and 72 h following stress and were tested for facial allodynia 1 h following injections (Fig. 3A). This repeated dose regimen was found to significantly attenuate facial hypersensitivity (Fig. 3B), grimacing behaviors (Fig. 3C), and priming to SNP compared with stressed mice that received vehicle.
Figure 3.

Multiple dosing with a PNMC attenuates stress-induced hypersensitivity and priming to a NO donor. A, Stress paradigm and dosing regimen are shown. B, C, Following 3 d of repeated stress, female ICR mice were administered FeTMPyP (30 mg/kg, i.p.) or vehicle at 1, 24, 48, and 72 h poststress and tested for acute facial hypersensitivity (B) and grimacing (C). Upon returning to baseline thresholds, mice were checked for priming to low-dose SNP (0.1 mg/kg, i.p.). Stress-induced acute mechanical hypersensitivity and grimace responses in mice that received multiple injections of vehicle; however, these effects were attenuated by multiple injections of FeTMPyP, determined by a two-way ANOVA with Bonferroni’s post hoc analysis. *Significance between stressed mice that received FeTMPyP and those that received vehicle. All control groups received vehicle and were administered SNP before the priming phase (n = 6 for all groups). Data are represented as the mean ± SEM. Table 1, see for F-values. *p < 0.05, **p < 0.01, ****p < 0.0001.
Based on these results, we tested whether administering a single dose of a PNMC was enough to prevent stress-induced acute facial hypersensitivity. We gave female mice a single dose of FeTMPyP (30 mg/kg, i.p.) at 24 h following stress (∼1 h before our first behavioral time point; Fig. 4A) and checked for priming to SNP (0.1 mg/kg, i.p.). Contrary to the effects of multidosing FeTMPyP, we found no significant differences in acute facial hypersensitivity or grimacing between the treated group and the stress/vehicle group (Fig. 4B,C). Additionally, FeTMPyP at 24 h poststress did not prevent priming to low-dose SNP.
Figure 4.

Administration of a PNMC 24 h following repeated stress does not block facial allodynia. A, Stress paradigm and dosing regimen are shown. B, C, Following stress, female ICR mice exhibited robust facial hypersensitivity (B) and grimacing (C), and were primed to low-dose SNP (0.1 mg/kg, i.p.). A two-way ANOVA with Bonferroni’s post hoc analysis revealed no significant differences in acute hypersensitivity or priming in stressed mice that received a PNMC at 24 h following stress compared with stressed mice that received vehicle (n = 3–5 for all groups). Data are represented as the mean ± SEM. Table 1, see for F-values.
We next determined whether these effects were temporally dependent on when the compound was given following stress. We gave mice a single dose of FeTMPyP or MnTBAP (30 mg/kg, i.p.) 1 h after removing them from the restraint tubes and again tested them for acute facial hypersensitivity and grimace. Upon returning to baseline thresholds, we administered SNP (0.1 mg/kg, i.p.) to test for hyperalgesic priming (Fig. 5A). Strikingly, stressed female (Fig. 5B,C) mice that received a single PNMC dose at this time point exhibited significantly reduced facial withdrawal thresholds and grimace responses and did not prime to low-dose SNP compared with those that received vehicle. Because of the remarkable effect of single dosing at 1 h poststress, we additionally assessed these results in male mice (Fig. 5D,E) and found similar efficacy (i.e., no sex differences) in response to the PNMCs following stress. Together, these data suggest that PN contributes to the acute stress response, that the effects of PNMC administration on the acute stress response may be temporally dependent, and that a role for PN formation is critical in the hours immediately following stress exposure.
Figure 5.
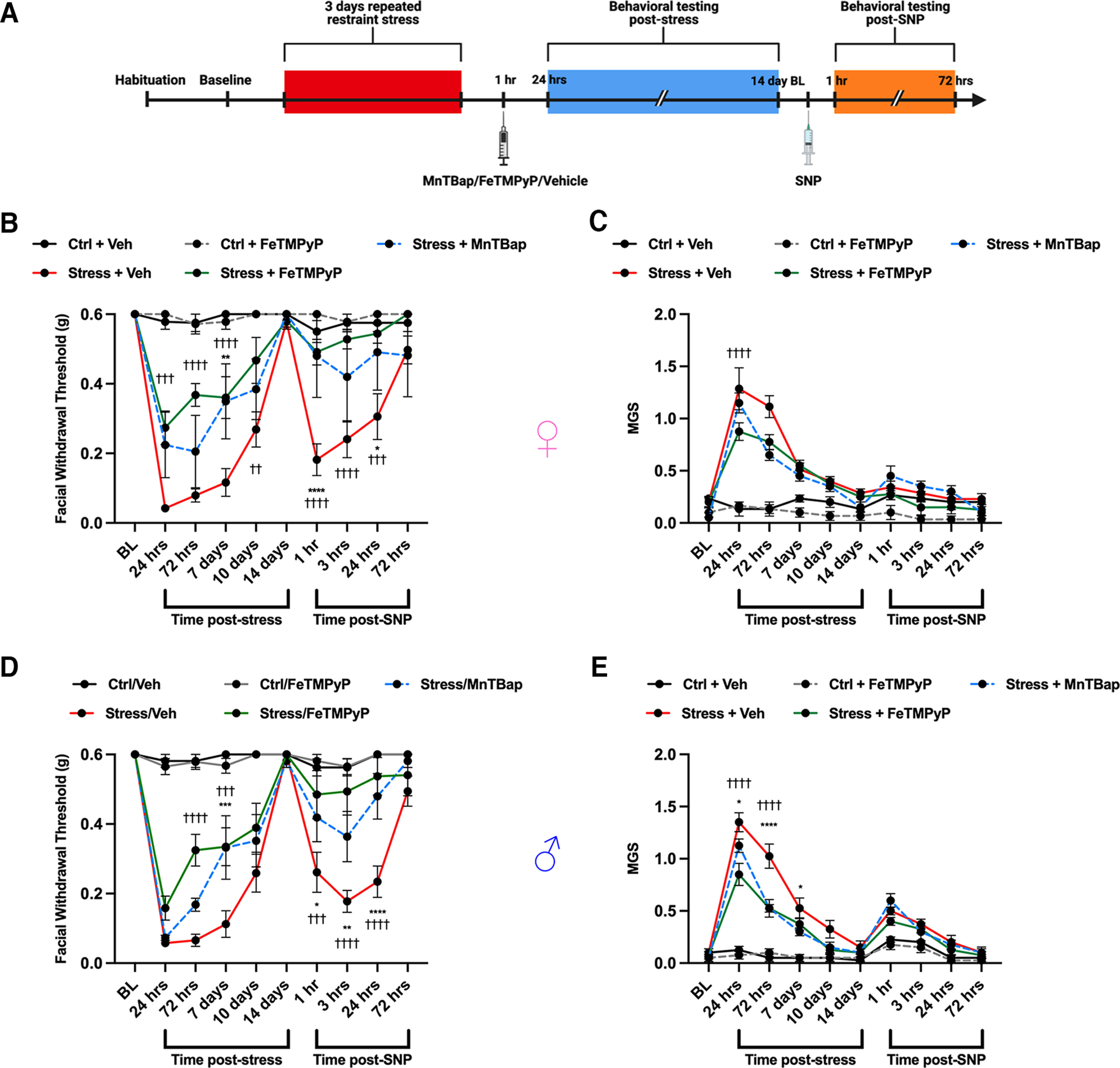
Administration of a PNMC at 1 h following stress results in attenuation of acute facial hypersensitivity and prevents priming to a NO donor. A, Following repeated stress, mice were administered FeTMPyP or MnTBAP (30 mg/kg, i.p.) 1 h poststress and tested for facial hypersensitivity, grimacing, and priming to low-dose SNP (0.1 mg/kg, i.p.). B–E, Compared with stressed mice that received vehicle, stressed mice that received a PNMC were found to have significant attenuation of acute allodynia and grimace scores, and did not prime to SNP in both females (B, C) and males (D, E). *Significance between stressed mice that received MnTBAP and those that received vehicle. †Significance between Stress/FeTMPyP and Stress/Vehicle groups. All control groups received vehicle and were administered SNP before the priming phase (n ≥ 4 in B and C; n = 8 in D and E). Data are represented as the mean ± SEM. Table 1, see for F-values. *†p < 0.05, **††p < 0.01, ***†††p < 0.001, ****††††p < 0.0001. Ctrl, Control.
3-Nitrotyrosine expression
PN plays a critical role in the nitration of proteins and essential enzymes, ultimately modifying their activity and leading to potential changes in neuronal excitability (Radi, 2004, 2013; Grace et al., 2016). Additionally, 3-NT is a well established marker of PN activity and has been shown to be upregulated in the blood platelets of human migraine patients between attacks (Van der Schueren et al., 2009; Bandookwala and Sengupta, 2020). Based on this, we measured 3-NT expression levels in the TGs and dura of mice that were subjected to repeated restraint stress. Approximately 24 h following repeated stress, an increase in the density of 3-NT expression identified at a nonspecific protein band (∼15–25 kDa) was observed in both the TGs and dura of stressed mice compared with controls, suggesting an increase in PN activity (Fig. 6). Furthermore, treatment with FeTMPyP (30 mg/kg, i.p.) at 1 h poststress was able to attenuate these increases. Similarly, 3-NT expression was increased in both tissues in stressed mice following administration of SNP (0.1 mg/kg, i.p.) ∼14 d poststress (Fig. 7), the time point at which mice return to baseline facial withdrawal thresholds. Interestingly, treatment with FeTMPyP 30 min before injection of SNP was able to prevent these increases in both sexes. Together, these observations suggest that repeated stress leads to increased PN activity and, subsequently, nitrotyrosination of proteins that may contribute to neuronal hyperexcitability.
Figure 6.
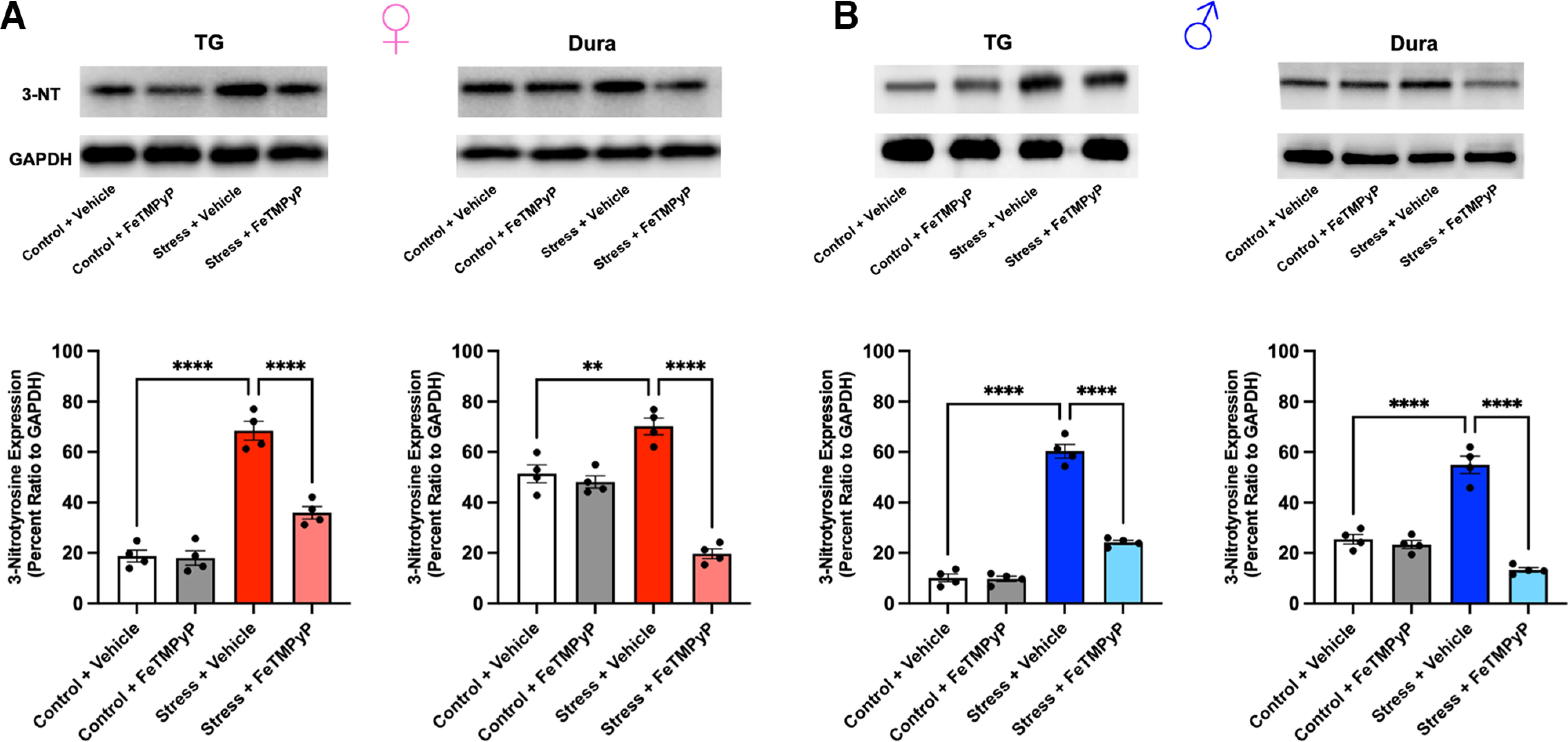
PN mediates stress-induced increases in 3-NT expression in the TGs and dura. A, B, Approximately 24 h following the final day of repeated stress, an increase in 3-NT expression, a marker for PN, was observed in the TGs and dura of female (A) and male (B) mice compared with controls, suggesting an increase in PN activity. These changes were prevented by the administration of FeTMPyP (30 mg/kg) at 1 h poststress, as indicated by a one-way ANOVA with post hoc Bonferroni’s correction (n = 4 independent replicates/group). Representative Western blots are shown. Data are represented as the mean ± SEM. **p < 0.01, ****p < 0.0001.
Figure 7.
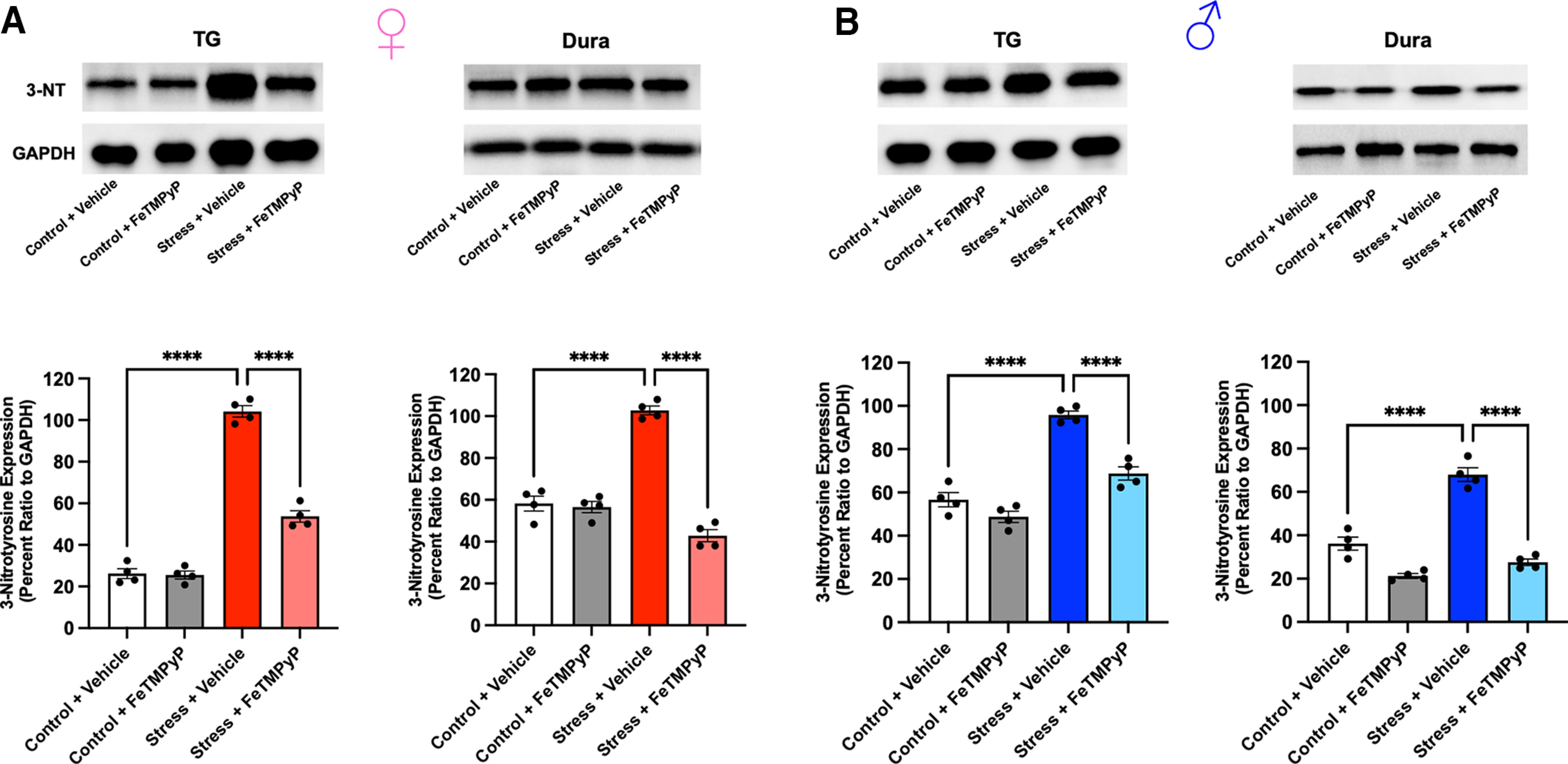
A, B, Administration of low-dose SNP (0.1 mg/kg, i.p.) ∼14 d after repeated stress (when mice typically return to baseline withdrawal thresholds) induces a robust increase in 3-NT expression in the TGs and dura of stressed female (A) and male (B) mice compared with controls. Notably, pretreatment with FeTMPyP (30 mg/kg, i.p.) 30 min before injection of SNP prevented this increase in 3-NT expression, as indicated by one-way ANOVA with Bonferroni’s post hoc correction (n = 4 independent replicates/group). Representative Western blots shown. Data are represented as the mean ± SEM. ****p < 0.0001.
Electrophysiological characterization of the effects of PN on TG neurons in vitro
Since cutaneous periorbital hypersensitivity and grimace could be mediated by actions of PN on TG neurons, we tested this question using TG cultures and direct exposure to NO donors in vitro. TG cultures were generated for patch-clamp recordings from adult male and female mice that were otherwise naive. All data were recorded from neurons having capacitances between 20 and 30 pF. APs were evoked by the application of a 1 s slow ramp current of four intensities (100 pA, 300 pA, 500 pA and 700 pA). APs that exceeded at least 40 mV were counted for each ramp injection. In initial experiments, TG cultures were treated with SNP (100 μm) for up to 1 h. However, we did not observe a significant increase in excitability compared with the vehicle group [vehicle, n = 5; number of APs: 100 pA, 1 ± 0.63; 300 pA, 5 ± 1.7; 500 pA, 7.4 ± 1.75; 700 pA, 9 ± 1.52; SNP (n = 8), 1.78 ± 0.43; 300 pA, 6.56 ± 0.96; 500 pA, 10.11 ± 1; 700 pA, 12.89 ± 0.89; main effect of treatment: F(1,12) = 2.33, p = 0.15]. Therefore, we tested an alternate NO donor compound, SIN-1, that leads to the direct formation of PN and measured changes in TG neuron excitability. Compared with the vehicle group, treatment of the TG neurons with SIN-1 (1 mm) increased the number of APs fired at all ramp intensities (Fig. 8A,B, top). Treatment of the neurons with MnTBAP (100 μm) mixed with SIN-1 (1 mm) prevented the increase in hyperexcitability. Next, we evaluated whether treatment with MnTBAP alone influenced the excitability of TG neurons. Compared with the vehicle group, MnTBAP did not alter the number of APs fired at the various ramp intensities (MnTBAP: number of APs: 100 pA, 0.62 ± 0.21; 300 pA, 4.84 ± 0.59; 500 pA, 8.07 ± 0.87; 700 pA, 11.23 ± 1.01; main effect of treatment: F(1,34) = 0.14, p = 0.71). Additionally, we tested the PN decomposition catalyst FeTPPS (100 μm) and found that, like MnTBAP, FeTPPS was also able to prevent the increase in the hyperexcitability caused by SIN-1. Treatment with SIN-1 also reduced the latency to the first AP at all ramp intensities, and this was prevented by coapplication of SIN-1 with MnTBAP or FeTPPS (Fig. 8B, bottom). Of the 20 cells recorded, 11 were from male and 9 from females. We found that there was no effect of sex in the number of APs elicited by SIN-1 (main effect of sex: F(1,18) = 0.4223, p = 0.52). Therefore, the data from male and female mice were pooled together for all treatment groups. We also tested whether a shorter application of SIN-1 would have the same effects on the excitability of TG neurons. In a separate set of experiments, baseline levels of AP firing in response to ramps were recorded and then SIN-1 (1 mm) was applied for 2 min, followed by a repeat of the ramp protocols. SIN-1 treatment for 2 min did not increase the excitability of TG neurons (n = 6 cells; capacitance, 25.83 ± 1.93 pF; baseline number of APs: 100 pA, 1.14 ± 0.4; 300 pA, 4.85 ± 1.33; 500 pA, 8.14 ± 1.71; 700 pA, 10.57 ± 1.78; SIN-1: 100 pA, 1.29 ± 0.52; 300 pA, 6.57 ± 1.48; 500 pA, 9.57 ± 1.88; 700 pA, 11.42 ± 1.88; main effect of treatment: F(1,5) = 4.495, p = 0.09).
Figure 8.
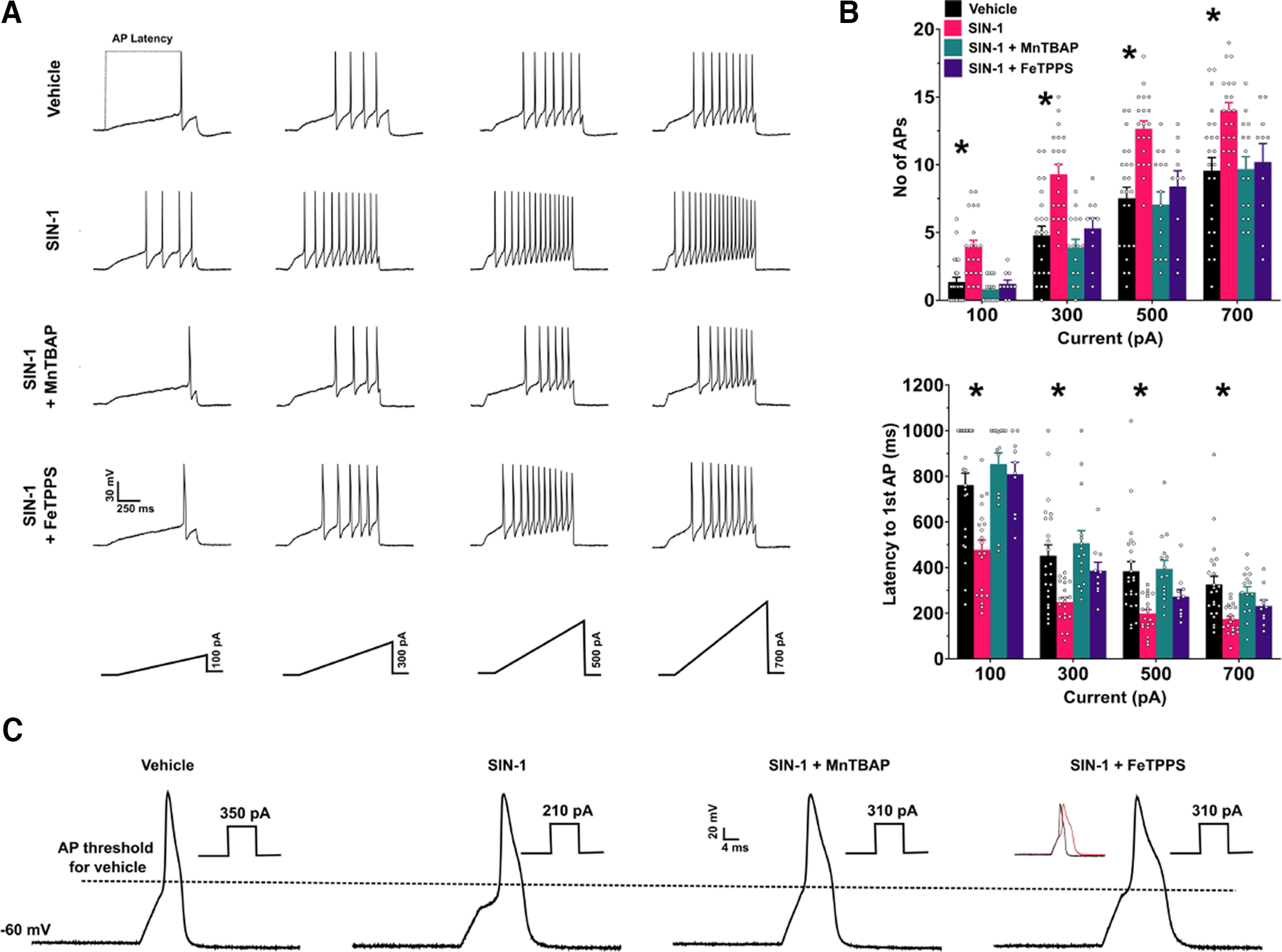
SIN-1 (1 mm) increases the excitability of TG neurons, and this hyperexcitability is prevented by the coapplication with SIN-1 of the peroxynitrite scavenger MnTBAP (100 μm) or the peroxynitrite decomposition catalyst FeTPPS (100 μm). A, Example traces of action potentials in the different treatment groups in response to the slow ramp injection of varying intensities. B, Top, Bar graphs representing the number of APs evoked by ramps. Main effect of treatment, F(3,64) = 11.01, p < 0.0001; *p < 0.05 for vehicle versus SIN-1 groups, post hoc Dunnett’s test. Bottom, Bar graphs representing the latency to the first AP at each ramp injection. Main effect of treatment, F(3,64) = 9.38, p < 0.0001, *p < 0.05 for vehicle versus SIN-1 groups, post hoc Dunnett’s test. Bottom, Bar graphs representing the latency to the first AP at each ramp injection. Main effect of treatment, F(3,64) = 9.38, p < 0.0001, *p < 0.05 for vehicle versus SIN-1 groups, post hoc Dunnett’s test. C, Raw traces of single APs evoked by a depolarizing step protocol. The dotted line is used to represent the lowering of threshold to fire an AP by SIN-1 and the prevention of this change in relation to vehicle treatment. The corresponding current rheobase used to evoke the APs is shown near each AP trace. Inset, Comparison of the AP half-width between vehicle group (black) and the SIN-1 + FeTPPS 100 μm group (red). Vehicle, n = 23 cells from 18 mice; SIN-1, n = 20 cells from 11 mice; SIN-1 + MnTBAP, n = 15 cells from 6 mice; SIN-1 + FeTPPS, n = 10 cells from 3 mice.
We next used a 5 ms step protocol to elicit a single AP to measure basic membrane properties (Table 2). We found that SIN-1 significantly reduced the AP threshold, increased the AP amplitude (measured from threshold to peak), and decreased the rheobase (Fig. 8C). Consistent with the data obtained with the ramp protocol, all of these changes were prevented by the cotreatment of SIN-1 with either MnTBAP or FeTPPS. However, we found that in the SIN-1 + FeTPPS group, the AP half-width was significantly broader compared with the vehicle group (Fig. 8C). In a small group of cells (n =3), FeTPPS (100 μm) alone did not change the number of APs in the ramp protocol compared with vehicle (FeTPPS: 100 pA, 1 ± 0.58; 300 pA, 5.33 ± 1.76; 500 pA, 8.33 ± 2.73; 700 pA, 10.67 ± 3.38) although the AP half-width was high in this group of cells (9.05 ± 2.78 ms).
Table 2.
AP Membrane properties of the different treatment groups
| Vehicle | SIN-1 | SIN-1 + MnTBAP | SIN-1 + FeTPPS | MnTBAP only | ANOVA | |
|---|---|---|---|---|---|---|
| Capacitance (pF) | 25.82 ± 0.94 | 25 ± 0.66 | 25.73 ± 1.13 | 26.78 ± 1.15 | 23.17 ± 1.15 |
F(4,73) = 1.387, p = 0.25 |
| RMP (mV) | −53.2 ± 0.96 | −50.1 ± 1.39 | −51.1 ± 1.72 | −52.6 ± 2.22 | −52.4 ± 1.41 |
F(4,73) = 0.9081, p = 0.46 |
| AP threshold (mV) | −18.09 ± 0.95 | −22.53 ± 0.63* | −19.12 ± 0.89 | −20.92 ± 1.03 | −17.69 ± 1.27 |
F(4,71) = 4.957, p = 0.0014; *p = 0.0012 |
| AP amplitude (mV) | 77.58 ± 1.24 | 82.31 ± 0.52* | 77.35 ± 1.1 | 80.38 ± 1.28 | 75.81 ± 1.52 |
F(4,71) = 5.241, p = 0.0009; *p = 0.005 |
| AP half-width (ms) | 3.19 ± 0.13 | 3.6 ± 0.1 | 3.17 ± 0.13 | 5.26 ± 0.38* | 3.38 ± 0.16 |
F(4,71) = 19.05, p < 0.0001; *p < 0.0001 |
| Rheobase (pA) | 301.36 ± 15.81 | 220 ± 9.85* | 288.57 ± 13.86 | 275.56 ± 11.68 | 275 ± 10.84 |
F(4,71) = 6.319, p = 0.0002; *p < 0.0001 |
| Input resistance (MΩ) | 1168.8 ± 85.1 | 1193 ± 69.57 | 1137.19 ± 137.8 | 1203.4 ± 59.58 | 1015.38 ± 116.05 |
F(4,68) = 0.4791, p = 0.751 |
One-way ANOVA followed by post hoc Dunnett's test. Data are represented as mean ± SEM. Asterisks denote a significant difference between the marked group and all other groups, with p-values being reported in the far right column for each row.
Effects of stress, sex, NO donors, and PNMCs on TG mitochondrial energetics
One of the most important consequences of nitroxidative stress is the disruption of proper mitochondrial function, ultimately culminating in impaired bioenergetics and homeostasis (Radi et al., 2001, 2002a). Based on this, we decided to explore whether stress induces mitochondrial dysfunction and whether PN plays a role mediating these effects. Following repeated restraint stress, we harvested and cultured mouse TGs from male and female ICR mice and allowed them to grow overnight. The next day, we processed these cultures using the Mito Stress Test Kit (Agilent) and measured OCRs using an Agilent Seahorse XFp apparatus. Approximately 24 h following day 3 of repeated restraint stress, basal respiration levels are significantly increased in stressed male mice, but not female mice, compared with controls; however, maximal respiration levels are significantly upregulated in both sexes, suggesting that female and male mice exhibit differences in respiration outputs shortly after repeated stress (Fig. 9). Conversely, at 14 d following repeated stress, when animals typically return to baseline nociceptive thresholds, we found significant increases in spare respiratory capacity (a measure of the ability of the cell to adapt to environmental stressors) and ATP production in female stressed mice, but not in male mice (Fig. 10). Additionally, comparisons within sex and between stressed and control conditions at both time points identified significant increases in maximal respiration, spare respiratory capacity, and ATP production in female stressed mice at 14 d poststress. In stressed male mice, we observed an increase in maximal respiration at both time points (Table 3). To parallel the behavior experiments with stress, we also measured OCRs in mice that were administered either vehicle or FeTMPyP 30 min before SNP at day 14 of stress. Interestingly, we found that maximal respiration, spare respiratory capacity, and nonmitochondrial respiration are all increased in male mice, but not female mice, following SNP, and, critically, this effect is attenuated by pretreatment with FeTMPyP (Fig. 11).
Figure 9.

Mitochondrial respiration is increased in the TGs of male and female mice at 24 h following repeated restraint stress. A, B, Mitochondrial OCRs were measured for females (A) and males (B) from which several metabolic parameters were calculated. C, D, Overall, male mice exhibited an increase in basal respiration (C) levels while both sexes were found to have increased levels of maximal respiration (D), indicating that stress increases respiration rates in TG mitochondria (n = 3 replicate runs). Data are represented as the mean ± SEM. *p < 0.05.
Figure 10.

At day 14 following repeated stress (when mice typically return to baseline withdrawal thresholds), spare respiratory capacity and ATP production are increased in female mice. A, B, Mitochondrial OCRs were measured for females (A) and males (B) from which several metabolic parameters were calculated. C, D, Female mice exhibited increased mitochondrial spare respiratory capacity (C) and ATP production (D) in their TGs, an effect that was not observed in male mice, suggesting a potential sex difference in the long-term effects of stress on mitochondrial function (n = 3 replicate runs). Data are represented as the mean ± SEM. ****p < 0.0001.
Table 3.
Comparison of stress-induced TG mitochondrial activity within sex
| Metabolic parameters (pmol/min/100,000 cells) |
||||
|---|---|---|---|---|
| 24 h |
14 d |
|||
| Control | Stress | Control | Stress | |
| Female | ||||
| Basal respiration | 7.21 ± 1.50 | 12.56 ± 1.41 | 12.68 ± 0.79 | 15.15 ± 2.06 |
| Maximal respiration | 16.14 ± 2.99 | 34.38 ± 5.91 | 27.59 ± 5.59 | 52.57 ± 6.50* |
| Spare reserve capacity | 8.93 ± 2.89 | 21.82 ± 6.27 | 11.58 ± 2.25 | 34.08 ± 1.64** |
| Non-mitochondrial respiration |
8.14 ± 0.88 | 9.77 ± 2.52 | 3.03 ± 0.90 | 5.74 ± 1.20 |
| Proton leak | 3.22 ± 0.84 | 3.82 ± 0.42 | 4.63 ± 0.82 | 4.98 ± 0.49 |
| ATP production | 3.99 ± 0.66 | 8.74 ± 1.64 | 8.05 ± 1.07 | 23.50 ± 1.24*** |
| Coupling efficiency | 57.67 ± 1.25 | 68.75 ± 5.86 | 64.03 ± 4.76 | 69.70 ± 8.34 |
| Male | ||||
| Basal respiration | 6.22 ± 0.19 | 13.78 ± 2.25 | 11.43 ± 0.27 | 11.25 ± 1.45 |
| Maximal respiration | 21.69 ± 3.05 | 45.29 ± 3.05** | 23.59 ± 0.93 | 27.14 ± 0.39* |
| Spare reserve capacity | 15.47 ± 3.06 | 31.51 ± 1.24* | 12.36 ± 0.84 | 15.62 ± 1.78 |
| Non-mitochondrial respiration |
4.26 ± 0.46 | 4.96 ± 2.09 | 4.08 ± 0.46 | 6.42 ± 1.50 |
| Proton leak | 2.85 ± 0.17 | 6.63 ± 2.13 | 0.73 ± 0.18 | 1.79 ± 0.36 |
| ATP production | 3.37 ± 0.31 | 7.15 ± 2.46 | 10.84 ± 0.05 | 12.27 ± 0.42 |
| Coupling efficiency | 54.65 ± 4.11 | 48.69 ± 14.91 | 93.75 ± 1.55 | 91.81 ± 0.40 |
Seahorse metabolic data are presented for female and male mice at 24 h and 14 d poststress. A two-tailed Welch’s t test found a significant increase in maximal respiration, spare respiratory capacity, and ATP production in stressed female mice compared with controls at 14 d poststress. Similarly, an increase in maximal respiration and spare respiratory capacity was identified in stressed male mice at 24 h poststress followed by an increase in maximal respiration between conditions at 14 d poststress. Data are represented as the mean ± SEM.
*p < 0.05, **p < 0.01, ***p < 0.001.
Figure 11.
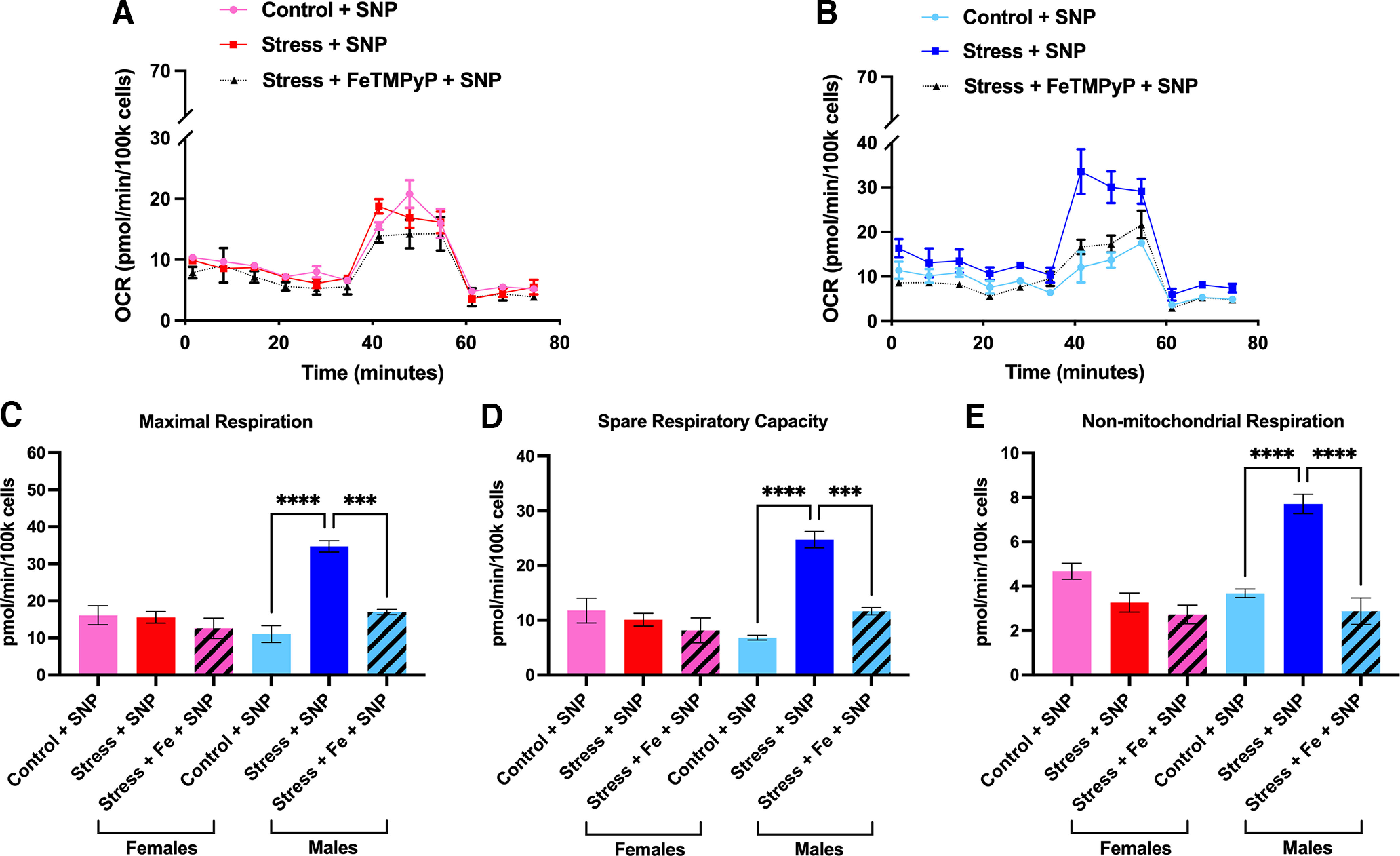
Administration of low-dose SNP induces robust changes in mitochondrial function in the TGs of male, but not female mice, an effect that is attenuated by pretreatment with FeTMPyP. Fourteen days following repeated stress, mice were administered either FeTMPyP (30 mg/kg, i.p.) or vehicle and were primed with SNP (0.1 mg/kg, i.p.). A, B, Mitochondrial OCRs were measured for females (A) and males (B) from which several metabolic parameters were calculated. C–E, Interestingly, stress-primed male mice exhibited robust increases in maximal respiration (C), spare respiratory capacity (D), and nonmitochondrial respiration (E) in response to SNP. Notably, these changes were attenuated by pretreatment with FeTMPyP. No changes were observed in female mice (n = 3 replicate runs). Data are represented as the mean ± SEM. **p < 0.01, ***p < 0.001, ***p < 0.0001.
Discussion
Here, we demonstrate a critical role for PN in contributing to long-lasting facial hypersensitivity and priming to a low-dose NO donor following repeated restraint stress, a preclinical model of migraine headache with important clinical implications given the extreme sensitivity of migraine patients to both stress and NO-producing agents (Peroutka, 2014). Administering FeTMPyP or MnTBAP before administration of low-dose SNP in stress-primed mice demonstrated efficacy in reducing nociceptive responses, suggesting that PN mediates at least some of the effects of NO donor-induced behavioral responses. Critically, our data also implicate endogenous PN formation in the mechanisms responsible for the onset of acute facial hypersensitivity following stress. Stress has been shown to induce NOS expression in the dura, to stimulate production of RNOS, and to promote hyperalgesic priming to NO donors in preclinical migraine models (Olivenza et al., 2000; Costa et al., 2005; Zinck et al., 2006; Avona et al., 2020). Repeated daily dosing with FeTMPyP beginning 1 h poststress and lasting out to 72 h was able to attenuate acute facial hypersensitivity in mice, an effect that was also achieved with just a single dose given 1 h poststress. To our surprise, this efficacy was not achieved when administered as a single dose 24 h poststress, suggesting that PN temporally mediates the development of acute hypersensitivity. Indeed, future experiments should test the efficacy of other multidosing strategies to confirm these time-dependent effects. Critically, these findings support previous studies that implicate PN in nociceptive processing, but also suggest a critical role for PN formation in the mechanisms responsible for the onset of acute hypersensitivity (Little et al., 2012; Akerman et al., 2021). Additionally, the observation that targeted PN degradation partially attenuates stress-induced facial hypersensitivity in both the acute and priming phase indicates that PN contributes to sustained hyperalgesia, which is likely caused by sensitization of the trigeminal sensory system, given the enhanced susceptibility to normally non-noxious stimuli in both preclinical migraine models as well as in human migraine patients (Goadsby et al., 2017).
Stimulation of the dura mater with proinflammatory mediators sensitizes meningeal afferents to pH 7.0 (Burgos-Vega et al., 2019), an effect that is mediated by acid-sensing ion channels (ASICs), which have been implicated in migraine pathophysiology (Yan et al., 2011; Holland et al., 2012; Dussor, 2015; Karsan et al., 2018). Of particular relevance, NO has been shown to modulate ASICs in DRG neurons, an effect that is reversed on treatment with oxidative reducing agents, but not inhibitors of cGMP, suggesting a role for RNOS in the modulation of ASICS during inflammation (Cadiou et al., 2007). Additionally, inhibiting ASIC3 prevents durovascular and NO-mediated trigeminal pain in rats (Holton et al., 2020). Based on this, we tested the efficacy of PNMCs in preventing facial hypersensitivity to dural pH 7.0 in mice primed with dural IL-6 and discovered that neither PNMC was successful in reducing nociceptive responses (Cadiou et al., 2007). Although these data suggest that PN does not contribute to the nociceptive response to pH 7.0 following IL-6, it may still be involved in other mechanisms of trigeminal nociception.
Indeed, the production of RNOS in response to noxious stimulation or injury has been shown to contribute to the hyperexcitability of primary sensory neurons. For example, NO and PN are capable of directly increasing the excitability of neurons via proinflammatory, proapoptotic, and nitroxidative stress pathways (Salvemini et al., 2011; Doyle et al., 2012; Little et al., 2012; Liu et al., 2013; Grace et al., 2016). Additionally, PN directly or indirectly acts as a neuromodulator for mechanisms of neuroinflammatory signaling, mitochondrial dysfunction, protein tyrosine nitration, and lipid peroxidation, all of which can contribute to neuronal hyperexcitability and, ultimately, peripheral sensitization (Radi et al., 2002a; Grace et al., 2016). A role for PN in TG nociceptor sensitization was recently demonstrated, in which treatment with FeTPPS was able to attenuate neuronal hyperexcitability caused by systemic injection of CGRP in rats (Goadsby et al., 2017; Akerman et al., 2021). In further support of this, we show that treatment with either MnTBAP or FeTPPS prevents NO donor-induced hyperexcitability in cultured TG neurons, implicating PN in this process. We also demonstrate that repeated stress increases 3-NT expression in the TGs and dura of mice and that low-dose SNP further increases this expression in stress-primed mice, indicating a general increase in PN activity as well as the modulation of protein activity that may underscore neuronal hyperexcitability. Importantly, targeting PN formation prevents these changes. Tyrosine nitration by PN is known to modulate the activity of several critical proteins, including MnSOD, the essential antioxidant enzyme that detoxifies free radical species, as well as numerous excitatory channels, leading to enhanced neurotransmission, neurotoxicity, and hyperexcitability (Trotti et al., 1996; Zanelli et al., 2002; Radi, 2004; Görg et al., 2005; Little et al., 2012; Janes et al., 2013; Muscoli et al., 2013). Together, these studies strongly implicate PN in mechanisms underlying peripheral sensitization.
Our data also suggest that repeated stress induces several changes in mouse mitochondrial function that may contribute to TG neuronal hyperexcitability. These changes are marked by increases in TG mitochondrial respiration levels 24 h following repeated stress and, in stressed female mice specifically, are sustained by increases in spare respiratory capacity and ATP production out to at least 2 weeks. Although an important output of mitochondrial respiration is the production of ATP, abnormal production can lead to abnormal Ca2+ signaling and, ultimately, to neuronal hyperexcitability (Boyman et al., 2020; Agalave et al., 2021). Based on this, it is possible that alterations in TG mitochondrial activity may contribute to peripheral sensitization in this model.
Regulation of mitochondrial function is a highly dynamic process and, in response to cellular stress, mitochondria adapt their energy production to meet the metabolic demands of the cell. Spare respiratory capacity characterizes the mitochondrial capacity to meet this demand and, thus, can be used to gauge the relative metabolic fitness (and health) of the cell (Nicholls, 2009). Although we do not see any significant changes in this parameter by 24 h poststress, there is a robust increase in female spare respiratory capacity and ATP production at 14 d poststress, an effect not observed in males, suggesting that female TG mitochondria have a higher capacity to adapt to stress than male TG mitochondria. Because female mice exhibit this level of metabolic flexibility at 14 d poststress, this may potentially explain why the administration of SNP at this time point does not cause a substantial change in respiration output in female TG mitochondria. In other words, stressed female TG mitochondria may not require as much energy to respond to the cellular stress induced by the administration of SNP, whereas stressed male TG mitochondria, which do not appear to be as metabolically flexible at this time point, are generating significant amounts of respiration to meet the energy requirements necessary to initiate an appropriate cellular response. This sex difference in mitochondrial function may be driven by evolutionary selection, considering that the majority of mitochondrial DNA is inherited from the mother. Additionally, several studies have demonstrated that female mitochondria tend to be more resilient to cellular stress than male mitochondria, and this is likely influenced by hormones (Justo et al., 2005; Klinge, 2008; Rutkai et al., 2015; Ventura-Clapier et al., 2017). Interestingly, treatment with FeTMPyP before SNP was able to attenuate these male-specific effects, providing metabolic evidence that PN at least partially mediates NO donor-induced mitochondrial dysfunction. Indeed, these cellular metabolic differences could potentially explain some of the sex differences observed in the development of hypersensitivity to various stimuli (Avona et al., 2019). Because we did not observe sex differences in stress-induced mechanical hypersensitivity, we believe these differences in metabolic fitness may reflect a sex-dependent role for PN in the regulation of mechanisms that underlie stress and NO donor-induced hypersensitivity (i.e., stress-induced PN production may lead to similar phenotypic outputs using distinct cellular mechanisms). Future studies should aim to elucidate the extent to which PN-mediated mitochondrial dysfunction is involved in migraine-related mechanisms, including determining potential contributions to TG neuronal excitability.
In summary, the above data highlight a novel role for PN in contributing to the development and maintenance of hypersensitivity in a preclinical model of migraine headache and further implicate PN as a therapeutically attractive target for this disease. To our knowledge, our study is the first to demonstrate that PN contributes to stress-induced increases in mechanical hypersensitivity, protein tyrosine nitration, neuronal excitability, and mitochondrial activity, providing a basic framework for how PN may be involved in mechanisms underlying migraine. One caveat to our study involves the ability of FeTMPyP to scavenge SO in addition to PN, making it difficult to differentiate between their activity; however, MnTBAP and FeTPPS, which are more specific to PN decomposition, were both found to attenuate nociceptive responses or neuronal hyperexcitability, respectively, providing a strong rationale for PN being the primary RNOS behind these effects. It is also important to acknowledge the other actions of NO and how they may relate to migraine. For example, stimulation of the NO receptor soluble guanylyl cyclase has been shown to contribute to migraine-related pain in mice, suggesting an alternative mechanism of action for NO in trigeminal nociception (Ben Aissa et al., 2018). Thus, future therapeutic development in this area may be driven by dual-action compounds with effects on more than one pathway downstream of NO production. Regardless, given the important roles of NO and SO in maintaining normal physiological function, targeting the overproduction of PN presents a unique approach to resolving nitro-oxidative stress and other consequences of RNOS formation that may play a critical role in migraine pathophysiology. Currently, more pharmacologically efficacious PNMCs are being developed and phase II trials have begun for chemotherapy-induced peripheral neuropathy as well as surgical, osteoarthritic, and diabetic pain; the data shown here suggest that these compounds should be tested as potential novel migraine treatments (Doyle et al., 2021).
Footnotes
This work was supported by National Institutes of Health Grants NS-104200 and NS-104990. We thank Nikhil Gogineni, Brian Nguyen, and Meghna Suresh for technical assistance on this project.
The authors declare no competing financial interests.
References
- Afridi SK, Kaube H, Goadsby PJ (2004) Glyceryl trinitrate triggers premonitory symptoms in migraineurs. Pain 110:675–680. 10.1016/j.pain.2004.05.007 [DOI] [PubMed] [Google Scholar]
- Agalave NM, Mody PH, Szabo-Pardi TA, Jeong HS, Burton MD (2021) Neuroimmune consequences of eIF4E phosphorylation on chemotherapy-induced peripheral neuropathy. Front Immunol 12:642420. 10.3389/fimmu.2021.642420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerman S, Williamson DJ, Kaube H, Goadsby PJ (2002) Nitric oxide synthase inhibitors can antagonize neurogenic and calcitonin gene-related peptide induced dilation of dural meningeal vessels. Br J Pharmacol 137:62–68. 10.1038/sj.bjp.0704842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerman S, Salvemini D, Romero-Reyes M (2021) Targeting reactive nitroxidative species in preclinical models of migraine. Cephalalgia 41:1187–1200. 10.1177/03331024211017884 [DOI] [PubMed] [Google Scholar]
- Avona A, Burgos-Vega C, Burton MD, Akopian AN, Price TJ, Dussor G (2019) Dural calcitonin gene-related peptide produces female-specific responses in rodent migraine models. J Neurosci 39:4323–4331. 10.1523/JNEUROSCI.0364-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avona A, Mason BN, Lackovic J, Wajahat N, Motina M, Quigley L, Burgos-Vega C, Loomis CM, Garcia-Martinez LF, Akopian AN, Price TJ, Dussor G (2020) Repetitive stress in mice causes migraine-like behaviors and calcitonin gene-related peptide-dependent hyperalgesic priming to a migraine trigger. Pain 161:2539–2550. 10.1097/j.pain.0000000000001953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandookwala M, Sengupta P (2020) 3-Nitrotyrosine: a versatile oxidative stress biomarker for major neurodegenerative diseases. Int J Neurosci 130:1047–1062. 10.1080/00207454.2020.1713776 [DOI] [PubMed] [Google Scholar]
- Ben Aissa M, et al., (2018) Soluble guanylyl cyclase is a critical regulator of migraine-associated pain. Cephalalgia 38:1471–1484. 10.1177/0333102417737778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyman L, Karbowski M, Lederer WJ (2020) Regulation of mitochondrial ATP production: ca(2+) signaling and quality control. Trends Mol Med 26:21–39. 10.1016/j.molmed.2019.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos-Vega CC, Quigley LD, Trevisan Dos Santos G, Yan F, Asiedu M, Jacobs B, Motina M, Safdar N, Yousuf H, Avona A, Price TJ, Dussor G (2019) Non-invasive dural stimulation in mice: a novel preclinical model of migraine. Cephalalgia 39:123–134. 10.1177/0333102418779557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadiou H, Studer M, Jones NG, Smith ES, Ballard A, McMahon SB, McNaughton PA (2007) Modulation of acid-sensing ion channel activity by nitric oxide. J Neurosci 27:13251–13260. 10.1523/JNEUROSCI.2135-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A, Smeraldi A, Tassorelli C, Greco R, Nappi G (2005) Effects of acute and chronic restraint stress on nitroglycerin-induced hyperalgesia in rats. Neurosci Lett 383:7–11. 10.1016/j.neulet.2005.03.026 [DOI] [PubMed] [Google Scholar]
- D’Andrea G, Cananzi AR, Perini F, Alecci M, Zamberlan F, Hasselmark L, Welch KM (1994) Decreased collagen-induced platelet aggregation and increased platelet arginine levels in migraine: a possible link with the NO pathway. Cephalalgia 14:352–356. 10.1046/j.1468-2982.1994.1405352.x [DOI] [PubMed] [Google Scholar]
- Dong X, Guan X, Chen K, Jin S, Wang C, Yan L, Shi Z, Zhang X, Chen L, Wan Q (2017) Abnormal mitochondrial dynamics and impaired mitochondrial biogenesis in trigeminal ganglion neurons in a rat model of migraine. Neurosci Lett 636:127–133. 10.1016/j.neulet.2016.10.054 [DOI] [PubMed] [Google Scholar]
- Doyle T, Chen Z, Muscoli C, Bryant L, Esposito E, Cuzzocrea S, Dagostino C, Ryerse J, Rausaria S, Kamadulski A, Neumann WL, Salvemini D (2012) Targeting the overproduction of peroxynitrite for the prevention and reversal of paclitaxel-induced neuropathic pain. J Neurosci 32:6149–6160. 10.1523/JNEUROSCI.6343-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle TM, Braden K, Harada CM, Mufti F, Schafer RM, Salvemini D (2021) Novel non-opioid based therapeutics for chronic neuropathic pain. Mo Med 118:327–333. [PMC free article] [PubMed] [Google Scholar]
- Dussor G (2015) ASICs as therapeutic targets for migraine. Neuropharmacology 94:64–71. 10.1016/j.neuropharm.2014.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried NT, Moffat C, Seifert EL, Oshinsky ML (2014) Functional mitochondrial analysis in acute brain sections from adult rats reveals mitochondrial dysfunction in a rat model of migraine. Am J Physiol Cell Physiol 307:C1017–1030. 10.1152/ajpcell.00332.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallai V, Floridi A, Mazzotta G, Codini M, Tognoloni M, Vulcano MR, Sartori M, Russo S, Alberti A, Michele F, Sarchielli P (1996) L-arginine/nitric oxide pathway activation in platelets of migraine patients with and without aura. Acta Neurol Scand 94:151–160. 10.1111/j.1600-0404.1996.tb07046.x [DOI] [PubMed] [Google Scholar]
- Goadsby PJ (2013) Therapeutic prospects for migraine: can paradise be regained? Ann Neurol 74:423–434. 10.1002/ana.23996 [DOI] [PubMed] [Google Scholar]
- Goadsby PJ, Holland PR, Martins-Oliveira M, Hoffmann J, Schankin C, Akerman S (2017) Pathophysiology of migraine: a disorder of sensory processing. Physiol Rev 97:553–622. 10.1152/physrev.00034.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görg B, Wettstein M, Metzger S, Schliess F, Häussinger D (2005) Lipopolysaccharide-induced tyrosine nitration and inactivation of hepatic glutamine synthetase in the rat. Hepatology 41:1065–1073. 10.1002/hep.20662 [DOI] [PubMed] [Google Scholar]
- Grace PM, Gaudet AD, Staikopoulos V, Maier SF, Hutchinson MR, Salvemini D, Watkins LR (2016) Nitroxidative signaling mechanisms in pathological pain. Trends Neurosci 39:862–879. 10.1016/j.tins.2016.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Høivik HO, Laurijssens BE, Harnisch LO, Twomey CK, Dixon RM, Kirkham AJ, Williams PM, Wentz AL, Lunnon MW (2010) Lack of efficacy of the selective iNOS inhibitor GW274150 in prophylaxis of migraine headache. Cephalalgia 30:1458–1467. 10.1177/0333102410370875 [DOI] [PubMed] [Google Scholar]
- Holland PR, Akerman S, Andreou AP, Karsan N, Wemmie JA, Goadsby PJ (2012) Acid-sensing ion channel 1: a novel therapeutic target for migraine with aura. Ann Neurol 72:559–563. 10.1002/ana.23653 [DOI] [PubMed] [Google Scholar]
- Holton CM, Strother LC, Dripps I, Pradhan AA, Goadsby PJ, Holland PR (2020) Acid-sensing ion channel 3 blockade inhibits durovascular and nitric oxide-mediated trigeminal pain. Br J Pharmacol 177:2478–2486. 10.1111/bph.14990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes K, Doyle T, Bryant L, Esposito E, Cuzzocrea S, Ryerse J, Bennett GJ, Salvemini D (2013) Bioenergetic deficits in peripheral nerve sensory axons during chemotherapy-induced neuropathic pain resulting from peroxynitrite-mediated post-translational nitration of mitochondrial superoxide dismutase. Pain 154:2432–2440. 10.1016/j.pain.2013.07.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justo R, Boada J, Frontera M, Oliver J, Bermúdez J, Gianotti M (2005) Gender dimorphism in rat liver mitochondrial oxidative metabolism and biogenesis. Am J Physiol Cell Physiol 289:C372–C378. 10.1152/ajpcell.00035.2005 [DOI] [PubMed] [Google Scholar]
- Karsan N, Gonzales EB, Dussor G (2018) Targeted acid-sensing ion channel therapies for migraine. Neurotherapeutics 15:402–414. 10.1007/s13311-018-0619-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinge CM (2008) Estrogenic control of mitochondrial function and biogenesis. J Cell Biochem 105:1342–1351. 10.1002/jcb.21936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackovic J, Price TJ, Dussor G (2021) De novo protein synthesis is necessary for priming in preclinical models of migraine. Cephalalgia 41:237–246. 10.1177/0333102420970514 [DOI] [PubMed] [Google Scholar]
- Langford DJ, Bailey AL, Chanda ML, Clarke SE, Drummond TE, Echols S, Glick S, Ingrao J, Klassen-Ross T, Lacroix-Fralish ML, Matsumiya L, Sorge RE, Sotocinal SG, Tabaka JM, Wong D, van den Maagdenberg AMJM, Ferrari MD, Craig KD, Mogil JS (2010) Coding of facial expressions of pain in the laboratory mouse. Nat Methods 7:447–449. 10.1038/nmeth.1455 [DOI] [PubMed] [Google Scholar]
- Lassen LH, Ashina M, Christiansen I, Ulrich V, Olesen J (1997) Nitric oxide synthase inhibition in migraine. Lancet 349:401–402. 10.1016/s0140-6736(97)80021-9 [DOI] [PubMed] [Google Scholar]
- Little JW, Doyle T, Salvemini D (2012) Reactive nitroxidative species and nociceptive processing: determining the roles for nitric oxide, superoxide, and peroxynitrite in pain. Amino Acids 42:75–94. 10.1007/s00726-010-0633-0 [DOI] [PubMed] [Google Scholar]
- Liu D, Shan Y, Valluru L, Bao F (2013) Mn (III) tetrakis (4-benzoic acid) porphyrin scavenges reactive species, reduces oxidative stress, and improves functional recovery after experimental spinal cord injury in rats: comparison with methylprednisolone. BMC Neurosci 14:23. 10.1186/1471-2202-14-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniyar FH, Sprenger T, Monteith T, Schankin C, Goadsby PJ (2014) Brain activations in the premonitory phase of nitroglycerin-triggered migraine attacks. Brain 137:232–241. 10.1093/brain/awt320 [DOI] [PubMed] [Google Scholar]
- Muscoli C, Dagostino C, Ilari S, Lauro F, Gliozzi M, Bardhi E, Palma E, Mollace V, Salvemini D (2013) Posttranslational nitration of tyrosine residues modulates glutamate transmission and contributes to N-methyl-D-aspartate-mediated thermal hyperalgesia. Mediators Inflamm 2013:950947. 10.1155/2013/950947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri M, Frustaci A, Milic M, Valdiglesias V, Fini M, Bonassi S, Barbanti P (2015) A meta-analysis of biomarkers related to oxidative stress and nitric oxide pathway in migraine. Cephalalgia 35:931–937. 10.1177/0333102414564888 [DOI] [PubMed] [Google Scholar]
- Nicholls DG (2009) Spare respiratory capacity, oxidative stress and excitotoxicity. Biochem Soc Trans 37:1385–1388. 10.1042/BST0371385 [DOI] [PubMed] [Google Scholar]
- Noseda R, Burstein R (2013) Migraine pathophysiology: anatomy of the trigeminovascular pathway and associated neurological symptoms, cortical spreading depression, sensitization and modulation of pain. Pain 154:S44–S53. 10.1016/j.pain.2013.07.021 [DOI] [PubMed] [Google Scholar]
- Olesen J, Ashina M (2015) Can nitric oxide induce migraine in normal individuals? Cephalalgia 35:1125–1129. 10.1177/0333102414566201 [DOI] [PubMed] [Google Scholar]
- Olesen J, Iversen HK, Thomsen LL (1993) Nitric oxide supersensitivity: a possible molecular mechanism of migraine pain. Neuroreport 4:1027–1030. 10.1097/00001756-199308000-00008 [DOI] [PubMed] [Google Scholar]
- Olivenza R, Moro MA, Lizasoain I, Lorenzo P, Fernández AP, Rodrigo J, Boscá L, Leza JC (2000) Chronic stress induces the expression of inducible nitric oxide synthase in rat brain cortex. J Neurochem 74:785–791. 10.1046/j.1471-4159.2000.740785.x [DOI] [PubMed] [Google Scholar]
- Peroutka SJ (2014) What turns on a migraine? A systematic review of migraine precipitating factors. Curr Pain Headache Rep 18:454. 10.1007/s11916-014-0454-z [DOI] [PubMed] [Google Scholar]
- Pradhan AA, Bertels Z, Akerman S (2018) Targeted nitric oxide synthase inhibitors for migraine. Neurotherapeutics 15:391–401. 10.1007/s13311-018-0614-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radi R (2004) Nitric oxide, oxidants, and protein tyrosine nitration. Proc Natl Acad Sci U S A 101:4003–4008. 10.1073/pnas.0307446101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radi R (2013) Protein tyrosine nitration: biochemical mechanisms and structural basis of functional effects. Acc Chem Res 46:550–559. 10.1021/ar300234c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radi R, Rodriguez M, Castro L, Telleri R (1994) Inhibition of mitochondrial electron transport by peroxynitrite. Arch Biochem Biophys 308:89–95. 10.1006/abbi.1994.1013 [DOI] [PubMed] [Google Scholar]
- Radi R, Peluffo G, Alvarez MN, Naviliat M, Cayota A (2001) Unraveling peroxynitrite formation in biological systems. Free Radic Biol Med 30:463–488. 10.1016/s0891-5849(00)00373-7 [DOI] [PubMed] [Google Scholar]
- Radi R, Cassina A, Hodara R (2002a) Nitric oxide and peroxynitrite interactions with mitochondria. Biol Chem 383:401–409. 10.1515/BC.2002.044 [DOI] [PubMed] [Google Scholar]
- Radi R, Cassina A, Hodara R, Quijano C, Castro L (2002b) Peroxynitrite reactions and formation in mitochondria. Free Radic Biol Med 33:1451–1464. 10.1016/s0891-5849(02)01111-5 [DOI] [PubMed] [Google Scholar]
- Rutkai I, Dutta S, Katakam PV, Busija DW (2015) Dynamics of enhanced mitochondrial respiration in female compared with male rat cerebral arteries. Am J Physiol Heart Circ Physiol 309:H1490–H1500. 10.1152/ajpheart.00231.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvemini D, Little JW, Doyle T, Neumann WL (2011) Roles of reactive oxygen and nitrogen species in pain. Free Radic Biol Med 51:951–966. 10.1016/j.freeradbiomed.2011.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer M, Richter C (1996) Peroxynitrite stimulates the pyridine nucleotide-linked Ca2+ release from intact rat liver mitochondria. Biochemistry 35:4524–4528. 10.1021/bi952708+ [DOI] [PubMed] [Google Scholar]
- Slosky LM, Vanderah TW (2015) Therapeutic potential of peroxynitrite decomposition catalysts: a patent review. Expert Opin Ther Pat 25:443–466. 10.1517/13543776.2014.1000862 [DOI] [PubMed] [Google Scholar]
- Taffi R, Vignini A, Lanciotti C, Luconi R, Nanetti L, Mazzanti L, Provinciali L, Silvestrini M, Bartolini M (2005) Platelet membrane fluidity and peroxynitrite levels in migraine patients during headache-free periods. Cephalalgia 25:353–358. 10.1111/j.1468-2982.2004.00863.x [DOI] [PubMed] [Google Scholar]
- Trotti D, Rossi D, Gjesdal O, Levy LM, Racagni G, Danbolt NC, Volterra A (1996) Peroxynitrite inhibits glutamate transporter subtypes. J Biol Chem 271:5976–5979. 10.1074/jbc.271.11.5976 [DOI] [PubMed] [Google Scholar]
- Van der Schueren BJ, Lunnon MW, Laurijssens BE, Guillard F, Palmer J, Van Hecken A, Depré M, Vanmolkot FH, de Hoon JN (2009) Does the unfavorable pharmacokinetic and pharmacodynamic profile of the iNOS inhibitor GW273629 lead to inefficacy in acute migraine? J Clin Pharmacol 49:281–290. 10.1177/0091270008329548 [DOI] [PubMed] [Google Scholar]
- Ventura-Clapier R, Moulin M, Piquereau J, Lemaire C, Mericskay M, Veksler V, Garnier A (2017) Mitochondria: a central target for sex differences in pathologies. Clin Sci (Lond) 131:803–822. 10.1042/CS20160485 [DOI] [PubMed] [Google Scholar]
- Virág L, Szabó E, Gergely P, Szabó C (2003) Peroxynitrite-induced cytotoxicity: mechanism and opportunities for intervention. Toxicol Lett 140-141:113–124. 10.1016/s0378-4274(02)00508-8 [DOI] [PubMed] [Google Scholar]
- Yan J, Edelmayer RM, Wei X, De Felice M, Porreca F, Dussor G (2011) Dural afferents express acid-sensing ion channels: a role for decreased meningeal pH in migraine headache. Pain 152:106–113. 10.1016/j.pain.2010.09.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanelli SA, Ashraf QM, Mishra OP (2002) Nitration is a mechanism of regulation of the NMDA receptor function during hypoxia. Neuroscience 112:869–877. 10.1016/s0306-4522(02)00141-0 [DOI] [PubMed] [Google Scholar]
- Zinck T, Illum R, Jansen-Olesen I (2006) Increased expression of endothelial and neuronal nitric oxide synthase in dura and pia mater after air stress. Cephalalgia 26:14–25. 10.1111/j.1468-2982.2005.00978.x [DOI] [PubMed] [Google Scholar]