

Abstract
Large-scale whole-genome sequencing (WGS) studies have enabled analysis of noncoding rare variant (RV) associations with complex human diseases and traits. Variant set analysis is a powerful approach to study RV association. However, existing methods have limited ability in analyzing the noncoding genome. We propose a computationally efficient and robust noncoding RV association-detection framework, STAARpipeline, to automatically annotate a WGS study and perform flexible noncoding RV association analysis, including gene-centric analysis and fixed-window and dynamic-window-based non-gene-centric analysis by incorporating variant functional annotations. In gene-centric analysis, STAARpipeline uses STAAR to group noncoding variants based on functional categories of genes and incorporate multiple functional annotations. In non-gene-centric analysis, STAARpipeline uses SCANG-STAAR to incorporate dynamic window sizes and multiple functional annotations. We apply STAARpipeline to identify noncoding RV sets associated with four lipid traits in 21,015 discovery samples from the Trans-Omics for Precision Medicine (TOPMed) program and replicate several of them in additional 9,123 TOPMed samples. We also analyze five non-lipid TOPMed traits.
Introduction
Genome-wide association studies (GWASs) have successfully identified thousands of common genetic variants for complex diseases and traits; however, these common variants only explain a small fraction of heritability1. Recent studies suggest that the missing heritability of complex traits and diseases and causal variants may be accounted for in part by RVs (minor allele frequency (MAF) < 1%)2–4. Although whole-exome sequencing (WES) studies have identified exome-wide significant RV associations for complex diseases and traits5, 6, more than 98% of the genetic variants are located in the noncoding genome6. Many common variants identified by GWAS as being associated with phenotypes are located in noncoding regions7–9. Further, the ENCODE project shows that a significant fraction of noncoding regions are functionally active10, 11, indicating that rare noncoding regions may have an effect on diseases or traits.
An increasing number of whole-genome sequencing (WGS) association studies, such as the Genome Sequencing Program (GSP) of the National Human Genome Research Institute (NHGRI), the Trans-Omics for Precision Medicine (TOPMed) Program of the National Heart, Lung, and Blood Institute (NHLBI), and UK Biobank provide an opportunity to study the genetic contributions of noncoding RVs to complex traits and diseases. It is of substantial interest to use these rich WGS data to explore the role of noncoding RVs in the genetic underpinning of common human diseases.
Single-variant analyses are not appropriate for analysis of rare variants as they lack sufficient power12–14. To improve power, variant set tests have been proposed that assess the effects of sets of multiple RVs jointly. These include burden tests, SKAT, and most recently STAAR (variant-set test for association using annotation information), which incorporates multiple functional annotations for genetic variants to boost the power15–17. A key challenge of these approaches is the selection of RVs to form variant sets. Several methods have been proposed to create coding and noncoding variant sets for analysis of WGS/WES studies17–22. However, these methods have limited ability to define analysis units in the noncoding genome23. For example, for gene-centric analysis, STAAR uses two noncoding functional categories (masks) of regulatory regions: promoters and enhancers in GeneHancer24 overlaid with Cap Analysis of Gene Expression (CAGE) sites25, 26; for non-gene-centric analysis, STAAR uses fixed-size sliding windows to scan the genome.
As signal regions (variant-phenotype-association regions) are unknown in practice and their sizes vary across the genome, the fixed-size sliding window approach is likely to lead to power loss when the prespecified window sizes are too big or too small compared with the actual sizes of signal regions. Furthermore, it is often knowledge- and effort-intensive to functionally annotate variants in a WGS/WES study of interest using the existing resources. Limited tools exist for multi-faceted functional annotation and analytic integration of WGS/WES data for rare variant association tests (RVATs). Finally, there are few robust pipelines that perform scalable and comprehensive noncoding RV association analysis in large-scale WGS data with hundreds of millions of noncoding RVs across the genome. Much uncertainty remains on the best practices for performing computationally efficient RV analysis of large scale WGS studies.
To address these issues, we propose a computationally efficient and robust noncoding rare variant association-detection framework for WGS data. We make three new contributions toward automatically selecting interpretable and powerful variant sets for noncoding RV analysis. First, in gene-centric analysis, we propose additional strategies for grouping noncoding variants based on functional annotations, including untranslated regions, upstream regions, downstream regions, promoters, enhancers of protein-coding genes, and long noncoding RNA genes within STAAR. For promoters and enhancers, we offer additional options of overlaying promoters and GeneHancer-based enhancers with not only CAGE sites but also with DNase Hypersensitivity (DHS) sites10. Second, in non-gene-centric analysis, instead of using fixed-size sliding windows in STAAR, we propose SCANG-STAAR, a flexible data-adaptive window size RVAT method that extends the SCANG (scan the genome) method19 by incorporating multiple functional annotations through STAAR17, while accounting for both relatedness and population structure through the generalized linear mixed model (GLMM) framework27 for quantitative and dichotomous traits28, 29. Third, we develop STAARpipeline, a pipeline that (1) functionally annotates both noncoding and coding variants of a WGS study and builds an annotated genotype dataset using the multi-faceted functional annotation database FAVOR17, 30 (Functional Annotations of Variants - Online Resource), through FAVORannotator; and (2) performs RVATs using the proposed methods for both gene-centric analysis and non-gene-centric analysis.
We applied the proposed framework to detect noncoding RVs associated with four quantitative lipid traits: low-density lipoprotein cholesterol (LDL-C); high-density lipoprotein cholesterol (HDL-C); triglycerides (TG) and total cholesterol (TC) using 21,015 discovery samples and 9,123 replication samples from the NHLBI TOPMed Freeze 5 WGS data. We performed conditional analysis by conditioning on known lipids-associated variants and identified several novel replicated RVs sets associated with lipids. We also applied the proposed framework to identify RV associations in the noncoding genome for five additional non-lipid traits in TOPMed Freeze 5: C-reactive protein (CRP), estimated glomerular filtration rate (eGFR), fasting glucose (FG), fasting insulin (FI) and telomere length (TL).
Results
Overview of Noncoding RVATs
We propose a computationally efficient and robust noncoding RVAT framework for phenotype-genotype association analyses of whole-genome sequencing data, focusing on rare variant association analysis in the noncoding genome. This regression-based framework allows adjusting for covariates, population structure, and relatedness by fitting linear and logistic mixed models for quantitative and dichotomous traits28, 29. A central component of our approach is the development of strategies to aggregate noncoding rare variants using both flexible gene-centric and non-gene-centric approaches to empower RVATs. For the gene-centric approach, we group noncoding RVs for each gene using eight functional categories of regulatory regions provided by functional annotations and apply STAAR, which incorporates multiple in-silico variant functional annotation scores that prioritize functional variants using multi-dimensional variant biological functions17. For the non-gene-centric analysis, instead of using sliding windows with fixed sizes, we propose SCANG-STAAR, a procedure using dynamic windows with data-adaptive sizes and incorporating multi-dimensional functional annotations. We also perform analytical follow-up to dissect RV association signals independent of a given set of known variants via conditional analysis (Fig. 1).
Fig. 1 |. Workflow of STAARpipeline.
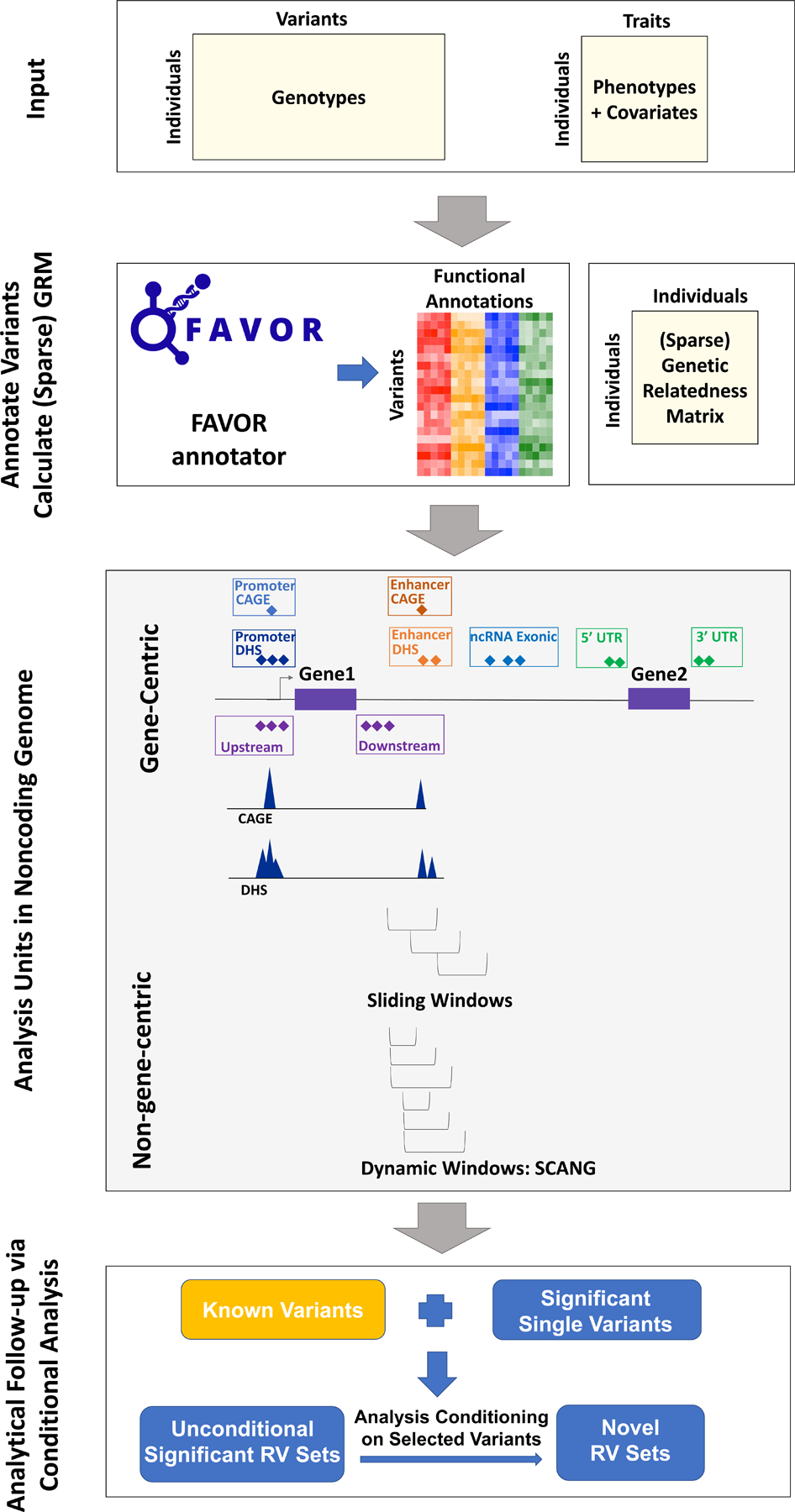
(a) Prepare the input data of STAARpipeline, including genotypes, phenotypes and covariates. (b) Annotate all variants in the genome using FAVORannotator through FAVOR database and calculate the (sparse) genetic relatedness matrix. (c) Define analysis units in the noncoding genome: eight functional categories of regulatory regions, sliding windows and dynamic windows using SCANG. (d) Obtain genome-wide significant associations and perform analytical follow-up via conditional analysis.
Gene-centric analysis of the noncoding genome
In gene-centric analysis of noncoding variants, we provide eight functional categories of regulatory regions to aggregate noncoding rare variants: (1) promoter RVs overlaid with CAGE sites, (2) promoter RVs overlaid with DHS sites, (3) enhancer RVs overlaid with CAGE sites, (4) enhancer RVs overlaid with DHS sites, (5) untranslated region (UTR) RVs, (6) upstream region RVs, (7) downstream region RVs and (8) noncoding RNA (ncRNA) RVs. The promoter RVs are defined as RVs in the +/− 3-kilobase (kb) window of transcription start sites with the overlap of CAGE sites or DHS sites. The enhancer RVs are defined as RVs in GeneHancer predicted regions with the overlap of CAGE sites or DHS sites10, 24–26. We define the UTR, upstream, downstream, and ncRNA RVs by GENCODE Variant Effect Predictor (VEP) categories31, 32. For the UTR mask, we include RVs in both 5' and 3' UTR regions. For the ncRNA mask, we include the exonic and splicing ncRNA RVs. We consider the protein-coding gene for the first seven categories provided by Ensembl33 and the ncRNA genes provided by GENCODE31, 32.
For each noncoding mask, we calculate its P value using the STAAR method that empowers RVATs by incorporating multiple variant functional annotation scores17. Functional annotations consist of diverse biological information of genomic elements. Incorporating this external biological information provided by functional annotations can increase the association analysis power34. For example, annotation principal components (aPCs) provide multi-dimensional summaries of variant annotations and capture the multi-faceted biological impact. The aPCs are calculated using the first principal component of the set of individual functional annotation scores measuring similar biological functionality17. We incorporate nine aPCs and three integrative scores (CADD35, LINSIGHT36, and FATHMM-XF37) as weights in constructing STAAR statistics17. We additionally incorporate a liver-tissue-specific aPC for lipids analysis. Details of these 13 functional annotations are given in Supplementary Table 1.
Specifically, we calculate the P value of each variant set using STAAR-O17, an omnibus test aggregating multiple annotation-weighted burden test15, SKAT16, and ACAT-V38 in the STAAR framework.
Non-gene-centric analysis using dynamic windows with SCANG-STAAR
We improve the STAAR-based fixed-size sliding window RVAT17, 18 by proposing a dynamic window based SCANG-STAAR method, which extends the SCANG19 procedure by incorporating multi-dimensional functional annotations to flexibly detect the locations and the sizes of signal windows across the genome. As the locations of regions associated with a disease or trait are often unknown in advance and their sizes may vary across the genome, the use of a pre-specified fixed-size sliding window for RVAT can lead to power loss, if the pre-specified window sizes do not align with the true locations of the signals.
Specifically, we extend the SCANG-SKAT (SCANG-S) procedure to SCANG-STAAR-S by calculating the STAAR-SKAT (STAAR-S) P value in each overlapping window by incorporating multiple variant functional annotations, instead of using just the MAF-weight-based SKAT P value. In SCANG-STAAR-S, we first calculate a threshold that controls the genome-wise type I error rate at a given 𝛼 level, based on the minimum value of the STAAR-S P value from all moving windows of different sizes in a range of windows (Online Methods). The procedure then selects the candidate significant windows whose set-based P value passes that threshold. When this results in multiple overlapping windows, we localize the detected significant window as the window whose P value is smaller than both the threshold and any window that overlaps with it. We then calculate the genome-wide P value of the detected windows by accounting for multiple comparisons of overlapping windows and controlling the corresponding genome-wise (family-wise) error rate (Online Methods).
Besides the SCANG-STAAR-S method, we also provide the SCANG-STAAR-B procedure, based on the STAAR-Burden P value. Compared with SCANG-STAAR-B, SCANG-STAAR-S has two advantages in detecting noncoding associations using dynamic windows in practice. First, the effects of causal variants in a neighborhood in the noncoding genome tend to be in different directions, especially in intergenic regions. Second, due to the different correlation structures of the two test statistics for overlapping windows, the genome-wide significance threshold of SCANG-STAAR-B is lower than that of SCANG-STAAR-S. For example, to control the genome-wise error rate at 0.05 level in our analysis of LDL-C, the P value thresholds for SCANG-STAAR-S and SCANG-STAAR-B are 3.80 × 10−9 and 2.31 × 10−10, respectively. We additionally provide the SCANG-STAAR-O procedure, which is based on an omnibus P value of SCANG-STAAR-S and SCANG-STAAR-B calculated by the ACAT method36. However, different from STAAR-O, we do not incorporate the ACAT-V test in the omnibus test, since the ACAT-V test is designed for sparse alternatives. Hence, it tends to detect the region with the smallest size that contains the most significant variant in the dynamic window procedure.
Analytical follow-up using conditional analysis
We perform follow-up conditional analysis to identify RV association signals that are independent of known single variant associations. We first select a list of known variants by including the previously identified trait-associated variants, for example, variants indexed in the GWAS Catalog39. We then perform stepwise selection to select the subset of independent variants from the known variants list to be used in the conditional analysis. We perform iterative conditional association analysis until the P values of all variants in the known variant list are larger than a cut-off (1 × 10−4, Online Methods). Instead of adjusting for all known trait-associated variants in the entire chromosome, we adjust for variants in an extended region of the specific variant, for example, a +/− 1-megabase (Mb) window beyond the variant of interest. Finally, we perform conditional analysis of each variant set by fitting the regression model adjusting for the selected known variants near the variant set (for example, in a +/− 1-Mb window).
STAARpipeline and computation cost
Our R package STAARpipeline performs scalable phenotype-genotype association analyses of functionally annotated WGS data using the developed RVAT methods. An additional package, STAARpipelineSummary summarizes the rare variant findings generated by STAARpipeline, including the results of both unconditional and conditional analysis and visualization of analysis results.
Specifically, to perform RVATs for a given WGS study, we first need to functionally annotate the variants and create variant sets. To achieve this, we use FAVORannotator, a workflow that annotates the variants of a given WGS study using the FAVOR database and generates annotated genotype files for use in STAARpipeline. Across the genome, STAARpipeline runs gene-centric noncoding and sliding window tests using STAAR and dynamic window analysis using SCANG-STAAR. STAARpipeline can also perform RV analysis of coding variants and single variant analysis of common and low-frequency variants (Discussion).
All analyses can be computed with attractive time and memory resources, even for large-scale WGS/WES datasets such as TOPMed, GSP and UK Biobank. We benchmarked STAARpipeline’s WGS association analysis of n=30,138 pooled related TOPMed lipids samples including both discovery and replication data in: 15 hours using 200 2.10 GHz computing cores with 11 Gb memory of gene-centric noncoding analysis; or 11 hours using 200 cores with 11 Gb memory of sliding window analysis; or 20 hours using 800 cores with 15 Gb memory of dynamic window analysis (including SCANG-STAAR-S, SCANG-STAAR-B and SCANG-STAAR-O). STAARpipelineSummary summarizes the results from STAARpipeline and provides analytical follow-up via conditional analysis. Summarizing the genome-wide TOPMed results took 24 hours using one core with 25 Gb memory.
Rare variant association analysis of lipid traits in the TOPMed WGS data
We applied STAARpipeline to identify RV-sets associated with four quantitative lipid traits (LDL-C, HDL-C, TG and TC) using TOPMed WGS data4, 17, 21. DNA samples were sequenced at the >30X target coverage4. The discovery phase consisted of six study cohorts with 21,015 samples sequenced in TOPMed Freeze 5. The replication phase consisted of eight remaining study cohorts with 9,123 samples in TOPMed Freeze 5 (Supplementary Note, Supplementary Table 2). Sample-level and variant-level quality control (QC) procedures were performed4, 21. Race/ethnicity was defined using a combination of self-reported race/ethnicity and study recruitment information40. The discovery cohorts consisted of 5,849 (27.8%) Black or African American, 12,313 (58.6%) White, 675 (3.2%) Asian American, 1,075 (5.1%) Hispanic/Latino American, and 1,103 (5.3%) Samoan participants. Among all samples in the discovery phase, 3,610 (17.2%) had first degree relatedness, 546 (2.6%) had second degree relatedness, and 472 (2.2%) had third degree relatedness (Supplementary Fig. 1). There were 215 million single-nucleotide variants (SNVs) observed in the discovery phase, and 205 million (94.9%) were rare variants (MAF < 1%). Among these 205 million rare variants, 202 million (98.8%) were noncoding variants defined by GENCODE VEP. Details of the study-specific demographics, summaries of lipid levels, and variant number distributions are given in Supplementary Tables 2–3 and Extended Data Fig. 1.
For each phenotype, we applied rank-based inverse normal transformation of the phenotype. We adjusted for age, age2, sex, race/ethnicity, study, and the first 10 ancestral PCs, and controlled for relatedness through heteroscedastic linear mixed models with sparse genetic relatedness matrices (GRMs) plus study-race/ethnicity-specific group-specific residual variance components (Online Methods). We accounted for the presence of medications of LDL-C and TC as before21. We tested for an association between lipid traits and RVs (MAF < 1%) in each variant set. In gene-centric analysis, we defined the eight analysis units as previously described: seven noncoding functional categories of protein-coding genes and one category for ncRNA genes. In non-gene-centric analysis, we performed a 2-kb sliding window analysis with 1-kb skip length, and a dynamic window analysis using SCANG-STAAR-S of all moving windows containing 40 to 300 variants19. In unconditional analysis we used Bonferroni-corrected genome-wide significance thresholds of 𝛼 = 0.05/(20,000 × 7) = 3.57 × 10−7 accounting for 7 different noncoding masks across protein-coding genes; 𝛼 = 0.05/20,000 = 2.50 × 10−6 accounting for ncRNA genes, and 𝛼 = 0.05/(2.66 × 106) = 1.88 × 10−8 accounting for 2.66 million 2-kb sliding windows across the genome. We controlled the genome-wise (family-wise) error rate for SCANG-STAAR-S dynamic window analysis at 𝛼 = 0.05 level19. We selected individual variants to be adjusted for in conditional analysis from the list of phenotype-associated common and low-frequency variants (MAF ≥ 1%) indexed in the GWAS Catalog39. Then we obtained the independent known variants using the algorithm described before in the analytical follow-up via conditional analysis section (Online Methods, Supplementary Table 4).
In gene-centric noncoding unconditional analysis of the discovery samples, STAARpipeline identified 43 genome-wide significant associations with at least one of the four lipid levels (Supplementary Table 5, Extended Data Figs. 2a–d, 3a–d, 4a–d, 5a–d). After conditioning on known lipid-associated variants, 14 out of the 43 associations remained significant at the Bonferroni-corrected level 𝛼 = 0.05/43 = 1.16 × 10−3 (Table 1). In the replication data, after adjusting for known lipid-associated variants, 4 of these 14 associations achieved significance at Bonferroni-corrected level 𝛼 = 0.05/14 = 3.57 × 10−3. These included enhancer DHS RVs in APOA1 and HDL-C, promoter CAGE RVs in APOE and TG, and enhancer CAGE or DHS RVs in APOE and TG. After further adjustment for known individual rare variants (minor allele count, MAC ≥ 20, Supplementary Table 6), none of the associations remained significant at the same significance level of 3.57 × 10−3 (Supplementary Table 7).
Table 1 |. Gene-centric noncoding analysis results of both unconditional analysis and analysis conditional on known common and low-frequency variants.
21,015 discovery samples and 9,123 replication samples from the NHLBI Trans-Omics for Precision Medicine (TOPMed) program are considered in the analysis. Results for the conditionally significant genes (unconditional STAAR-O P < 3.57 × 10−7 and conditional STAAR-O P < 1.16 × 10−3 for 7 different noncoding masks across protein-coding genes; unconditional STAAR-O P < 2.50 × 10−6 and conditional STAAR-O P < 1.16 × 10−3 for ncRNA genes) using discovery samples are presented in the table. The unconditional significant thresholds for both protein-coding genes of 7 different noncoding masks and ncRNA genes were defined by the multiple comparisons using the Bonferroni correction, that is, 0.05/(20,000 × 7) = 3.57 × 10−7 and 0.05/20,000 = 2.50 × 10−6. STAAR-O is a two-sided test. Chr (Chromosome); Category (Functional category); #SNV (Number of rare variants (MAF < 1%) of the particular functional category in the gene); STAAR-O (STAAR-O P value); HDL-C (High-density lipoprotein cholesterol); LDL-C (Low-density lipoprotein cholesterol); TG (Triglycerides); TC (Total cholesterol); Variants Adjusted (Adjusted variants in conditional analysis); n/a, no variant adjusted in the conditional analysis.
| Trait | Gene | Chr | Category | Discovery |
Replication |
Variants Adjusted | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| #SNV | STAAR-O (Unconditional) | STAAR-O (Conditional) | #SNV | STAAR-O (Unconditional) | STAAR-O (Conditional) | |||||
| HDL-C | APOA1 | 11 | enhancer_DHS | 1862 | 2.19E-07 | 7.67E-07 | 1005 | 1.50E-03 | 3.17E-03 | rs964184, rs12269901 |
|
| ||||||||||
| LDL-C | LDLR | 19 | upstream | 68 | 2.35E-17 | 4.24E-04 | 27 | 5.58E-01 | 6.31E-01 | rs12151108, rs688, rs6511720 |
| LDLR | 19 | promoter_CAGE | 131 | 1.88E-17 | 3.37E-04 | 56 | 2.51E-02 | 9.50E-02 | rs12151108, rs688, rs6511720 | |
| APOE | 19 | promoter_CAGE | 91 | 1.45E-11 | 4.88E-12 | 35 | 1.86E-01 | 4.36E-02 | rs7412, rs429358, rs35136575 | |
| LDLR | 19 | promoter_DHS | 257 | 4.03E-17 | 7.21E-04 | 113 | 5.74E-02 | 2.27E-01 | rs12151108, rs688, rs6511720 | |
| APOE | 19 | promoter_DHS | 162 | 9.81E-11 | 3.41E-12 | 64 | 7.45E-02 | 3.42E-02 | rs7412, rs429358, rs35136575 | |
| LDLR | 19 | enhancer_CAGE | 150 | 2.82E-17 | 5.01E-04 | 71 | 1.20E-02 | 4.05E-02 | rs12151108, rs688, rs6511720 | |
| APOE | 19 | enhancer_DHS | 239 | 9.84E-11 | 2.03E-11 | 112 | 2.55E-01 | 1.34E-01 | rs7412, rs429358, rs35136575 | |
| CTC-527H23.4 | 16 | ncRNA | 32 | 1.15E-06 | 1.15E-06 | 17 | 9.12E-01 | 9.12E-01 | n/a | |
|
| ||||||||||
| TG | APOE | 19 | promoter_CAGE | 92 | 4.45E-12 | 7.48E-06 | 36 | 9.45E-06 | 3.53E-05 | rs12721054, rs5112, rs429358 |
| APOA5 | 11 | promoter_DHS | 175 | 2.39E-08 | 3.47E-05 | 84 | 1.19E-04 | 8.78E-03 | rs964184, rs9804646, rs3135506, rs2266788 | |
| APOE | 19 | promoter_DHS | 163 | 1.80E-11 | 9.56E-06 | 65 | 2.96E-06 | 1.13E-05 | rs12721054, rs5112, rs429358 | |
| COL18A1 | 21 | enhancer_CAGE | 256 | 1.92E-07 | 1.92E-07 | 147 | 4.57E-02 | 4.57E-02 | n/a | |
| APOE | 19 | enhancer_DHS | 241 | 2.02E-11 | 8.44E-05 | 116 | 1.12E-05 | 4.15E-05 | rs12721054, rs5112, rs429358 | |
In unconditional analysis of the discovery samples, using the 2-kb sliding window procedure we identified 140 windows as genome-wide significant (Supplementary Table 8, Extended Data Figs. 2e–f, 3e–f, 4e–f, 5e–f). Among these 140 significant sliding windows, 14 were located in noncoding regions and, after conditioning on known lipid-associated variants, all remained significant at the Bonferroni-corrected level 𝛼 = 0.05/140 = 3.57 × 10−4 (Table 2). In replication data, 9 of the 14 associations were significant at the Bonferroni-corrected level 𝛼 = 0.05/14 = 3.57 × 10−3 after adjusting for known phenotype-specific variants. When we further adjusted these 9 associations for known individual variants (MAC ≥ 20), associations for two intronic sliding windows (PAFAH1B2 and TG) remained significant at the same level of 3.57 × 10−3 (Supplementary Table 9).
Table 2 |. 2-kb sliding window analysis results of unconditional analysis and analysis conditional on known common and low-frequency variants.
21,015 discovery samples and 9,123 replication samples from the NHLBI Trans-Omics for Precision Medicine (TOPMed) program are considered in the analysis. Results for the conditionally significant sliding windows (unconditional STAAR-O P < 1.88 × 10−8; conditional STAAR-O P < 3.57 × 10−4) using discovery samples are presented in the table. The unconditional significant threshold was defined by the multiple comparisons using the Bonferroni correction (0.05/(2.66 × 106) = 1.88 × 10−8). STAAR-O is a two-sided test. Chr (Chromosome); Start Location (Start location of the 2kb sliding window); End Location (End location of the 2-kb sliding window); #SNV (Number of rare variants (MAF < 1%) in the 2-kb sliding window; STAAR-O (STAAR-O P value); HDL-C (High-density lipoprotein cholesterol); LDL-C (Low-density lipoprotein cholesterol); TG (Triglycerides); TC (Total cholesterol); Variants Adjusted (Adjusted variants in conditional analysis); n/a, no variant adjusted in the conditional analysis. Physical positions of each window are on build hg38.
| Trait | Chr | Start Location | End Location | Gene | Discovery |
Replication |
Variants Adjusted | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| #SNV | STAAR-O (Unconditional) | STAAR-O (Conditional) | #SNV | STAAR-O (Unconditional) | STAAR-O (Conditional) | ||||||
| HDL-C | 8 | 57,071,644 | 57,073,643 | Intergenic (IMPAD1) | 111 | 1.79E-08 | 1.79E-08 | 53 | 8.38E-01 | 8.38E-01 | n/a |
| 11 | 116,802,930 | 116,804,929 | Intergenic (ZPR1) | 135 | 1.25E-08 | 4.31E-08 | 76 | 9.49E-05 | 2.02E-04 | rs964184, rs12269901 | |
| 11 | 117,146,930 | 117,148,929 | Intronic (PAFAH1B2) | 165 | 5.98E-09 | 8.28E-08 | 98 | 6.02E-04 | 1.12E-03 | rs964184, rs12269901 | |
| 11 | 117,147,930 | 117,149,929 | Intronic (PAFAH1B2) | 168 | 8.85E-09 | 1.22E-07 | 96 | 8.72E-04 | 1.64E-03 | rs964184, rs12269901 | |
| 16 | 56,760,029 | 56,762,028 | Intronic (NUP93) | 132 | 1.38E-08 | 9.65E-06 | 68 | 2.45E-01 | 1.15E-01 | rs247616, rs5883, rs7499892, rs17231520, rs5880 | |
| 16 | 56,761,029 | 56,763,028 | Intronic (NUP93) | 141 | 1.50E-08 | 1.09E-05 | 73 | 5.87E-01 | 2.26E-01 | rs247616, rs5883, rs7499892, rs17231520, rs5880 | |
|
| |||||||||||
| LDL-C | 1 | 55,333,498 | 55,335,497 | Intergenic (GOT2P1) | 171 | 6.66E-16 | 5.81E-07 | 95 | 1.27E-06 | 5.81E-07 | rs11591147, rs28362263, rs505151, rs12117661, rs472495 |
| 1 | 55,334,498 | 55,336,497 | Intergenic (GOT2P1) | 148 | 5.55E-16 | 5.49E-07 | 81 | 1.20E-06 | 5.49E-07 | rs11591147, rs28362263, rs505151, rs12117661, rs472495 | |
|
| |||||||||||
| TG | 11 | 117,146,930 | 117,148,929 | Intronic (PAFAH1B2) | 164 | 7.81E-19 | 4.13E-18 | 93 | 2.17E-17 | 5.66E-17 | rs964184, rs9804646, rs3135506, rs2266788 |
| 11 | 117,147,930 | 117,149,929 | Intronic (PAFAH1B2) | 165 | 1.15E-18 | 6.11E-18 | 94 | 3.47E-17 | 9.13E-17 | rs964184, rs9804646, rs3135506, rs2266788 | |
| 19 | 44,882,528 | 44,884,527 | Intronic (NECTIN2) | 145 | 1.06E-08 | 2.18E-07 | 88 | 2.71E-02 | 8.07E-01 | rs12721054, rs5112, rs429358 | |
|
| |||||||||||
| TC | 1 | 55,333,498 | 55,335,497 | Intergenic (GOT2P1) | 175 | 1.98E-13 | 3.83E-14 | 101 | 5.84E-07 | 1.88E-07 | rs11591147, rs28362263, rs505151, rs12117661, rs2495477 |
| 1 | 55,334,498 | 55,336,497 | Intergenic (GOT2P1) | 149 | 1.80E-13 | 3.49E-14 | 90 | 5.53E-07 | 1.78E-07 | rs11591147, rs28362263, rs505151, rs12117661, rs2495477 | |
| 19 | 44,894,528 | 44,896,527 | Intronic (TOMM40) | 180 | 2.73E-10 | 8.95E-08 | 97 | 2.68E-03 | 4.22E-01 | rs7412, rs429358, rs12721054 | |
We further compared the unconditional P values of different tests using the sliding window procedures41, including burden, SKAT and ACAT-V with only MAFs as the weights, and STAAR-O, which incorporates multiple variant functional annotations. Overall, by dynamically incorporating multiple functional annotations that captures different aspects of variant functions, STAAR-O detected more significant sliding windows, and showed consistently smaller P values for top sliding windows compared to existing RVATs without incorporating functional annotations (Supplementary Figs. 2–5). These results suggest that incorporating multiple functional annotations using the STAAR framework can boost the power for WGS RV association analysis.
In unconditional analysis of the discovery samples using the dynamic window procedure SCANG-STAAR-S, we identified 90 genome-wide significant associations (Supplementary Table 10). Among them, 10 were located in noncoding regions and remained significant at the Bonferroni-corrected level 𝛼 = 0.05/90 = 5.56 × 10−4 after conditioning on known lipid-associated variants (Table 3). In the replication data, after adjusting for known phenotype-specific variants, 7 were significant at the Bonferroni-corrected level 𝛼 = 0.05/10 = 5 × 10−3. After further adjustment for known individual rare variants (MAC ≥ 20), 3 associations remained significant, including RVs in an intronic region of PAFAH1B2 and TG, RVs in an intronic region of SIDT2 and TG, and RVs in an intronic region of CEP164 and TG (Supplementary Table 11).
Table 3 |. Dynamic window analysis results of unconditional analysis and analysis conditional on known common and low-frequency variants.
21,015 discovery samples and 9,123 replication samples from the NHLBI Trans-Omics for Precision Medicine (TOPMed) program are considered in the analysis. Results for the conditionally significant sliding windows (unconditional genome-wide error rate GW𝐸𝑅 < 0.05; conditional STAAR-S P < 5.56 × 10−4) using discovery samples are presented in the table. STAAR-S is a two-sided test. Chr (Chromosome); Start Location (Start location of the dynamic window); End Location (End location of the dynamic window); #SNV (Number of rare variants (MAF < 1%) in the dynamic window; GWER (genome-wide error rate); STAAR-S (STAAR-S P value); HDL-C (High-density lipoprotein cholesterol); LDL-C (Low-density lipoprotein cholesterol); TG (Triglycerides); TC (Total cholesterol); Variants Adjusted (Adjusted variants in conditional analysis). Physical positions of each window are on build hg38.
| Trait | Chr | Start Location | End Location | Gene | Discovery |
Replication |
Variants Adjusted | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| #SNV | GWER | STAAR-S (Unconditional) | STAAR-S (Conditional) | #SNV | STAAR-S (Unconditional) | STAAR-S (Conditional) | ||||||
| HDL-C | 11 | 116,866,780 | 116,867,288 | Intronic (SIK3) | 40 | 0.0295 | 2.24E-09 | 8.45E-09 | 19 | 2.22E-05 | 5.46E-05 | rs964184, rs12269901 |
| 11 | 116,928,564 | 116,929,045 | Intronic (SIK3) | 40 | 0.0025 | 1.50E-10 | 4.43E-10 | 18 | 7.81E-04 | 1.06E-03 | rs964184, rs12269901 | |
|
| ||||||||||||
| LDL-C | 1 | 55,335,150 | 55,335,701 | Intergenic (GOT2P1) | 40 | <0.0005 | 8.58E-18 | 7.49E-19 | 21 | 9.29E-07 | 4.80E-07 | rs11591147, rs28362263, rs505151, rs12117661, rs472495 |
| 19 | 11,319,992 | 11,320,870 | Intronic (TSPAN16) | 60 | 0.02 | 1.44E-09 | 3.16E-05 | 41 | 5.04E-01 | 5.10E-01 | rs12151108, rs688, rs6511720 | |
|
| ||||||||||||
| TG | 11 | 117,147,061 | 117,148,086 | Intronic (PAFAH1B2) | 80 | <0.0005 | 5.10E-16 | 8.55E-15 | 41 | 9.48E-19 | 3.44E-18 | rs964184, rs9804646, rs3135506, rs2266788 |
| 11 | 117,182,856 | 117,183,310 | Intronic (SIDT2) | 40 | <0.0005 | 3.96E-12 | 1.08E-11 | 15 | 3.77E-14 | 6.53E-14 | rs964184, rs9804646, rs3135506, rs2266788 | |
| 11 | 117,349,560 | 117,350,171 | Intronic (CEP164) | 50 | 0.013 | 1.08E-09 | 1.26E-09 | 29 | 4.12E-11 | 6.39E-11 | rs964184, rs9804646, rs3135506, rs2266788 | |
|
| ||||||||||||
| TC | 1 | 55,291,905 | 55,293,502 | Intergenic (GOT2P1) | 140 | 0.0055 | 3.17E-10 | 8.77E-05 | 68 | 4.76E-01 | 2.30E-01 | rs11591147, rs28362263, rs505151, rs12117661, rs2495477 |
| 1 | 55,335,119 | 55,335,584 | Intergenic (GOT2P1) | 40 | <0.0005 | 1.63E-15 | 4.44E-16 | 26 | 2.23E-07 | 7.03E-08 | rs11591147, rs28362263, rs505151, rs12117661, rs2495477 | |
| 19 | 11,319,627 | 11,320,925 | Intronic (TSPAN16) | 110 | <0.0005 | 2.95E-12 | 2.32E-05 | 75 | 3.40E-01 | 5.90E-01 | rs73015024, rs688, rs2278426, rs6511720 | |
Rare variant analysis of five non-lipid traits in the TOPMed WGS data
We further applied STAARpipeline to analyzing a broader spectrum of five phenotypes in the TOPMed Freeze 5 WGS data: CRP (𝑛 = 22,775)42, eGFR (𝑛 = 23,732)43, FG (𝑛 = 23,859)44, FI (𝑛 = 21,900)44 and TL (𝑛 = 39,742)45. Similar to the lipids analysis, for each phenotype, we performed gene-centric analysis, 2-kb sliding window analysis, and dynamic window analysis to detect RV associations in the noncoding genome (Online Methods).
In gene-centric noncoding unconditional analysis, STAARpipeline identified 6 genome-wide significant associations, and all 6 associations remained significant at the Bonferroni-corrected level 𝛼 = 0.05/6 = 8.33 × 10−3 after conditioning on known phenotype-specific variants (Supplementary Table 12, Supplementary Figs. 6a–d, 7a–d, 8a–d, 9a–d, 10a–d). After further adjustment for known individual rare variants using conditional analysis, although the strengths of 5 associations were reduced, all 6 associations remained significant at the same significance level of 𝛼 = 8.33 × 10−3 (Supplementary Table 12). In 2-kb sliding window unconditional analysis, we identified 19 genome-wide significant associations and 12 of them were in the noncoding genome (Supplementary Table 13, Supplementary Figs. 6e–f, 7e–f, 8e–f, 9e–f, 10e–f). After adjusting for known phenotype-specific variants, all the 12 associations remained significant at the Bonferroni-corrected level 𝛼 = 0.05/19 = 2.63 × 10−3, and 8 of 12 associations remained significant after further adjusting for known rare variants with MAC ≥ 20 (Supplementary Table 13).
In dynamic window unconditional analysis, we identified 17 genome-wide significant associations and 11 of them were in the noncoding genome (Supplementary Table 14). These 11 associations included 7 non-overlapping noncoding significant associations detected by the sliding window procedure, and 4 associations that were missed by the sliding window procedure. After adjusting for known phenotype-specific variants, all 11 associations remained significant at the Bonferroni-corrected level 𝛼 = 0.05/17 = 2.94 × 10−3, and 8 of 11 associations remained significant after further adjusting for known rare variants (Supplementary Table 14).
Simulation studies
We performed simulation studies to evaluate the type I error rate and power of SCANG-STAAR in a variety of configurations. We generated sequence data by simulating 100,000 chromosomes in a 10-Mb region using the calibration coalescent model (COSI)46 that mimics the linkage disequilibrium (LD) structure of samples from African Americans. The simulation studies used the 10-Mb sequence to mimic whole genome sequencing data and focused on rare variants (MAF < 1%). We considered the total sample sizes 𝑛 = 50,000 in all simulations. Quantitative and dichotomous phenotypes were generated by following the steps described in Data simulation (Online Methods).
Type I error simulations
For both quantitative and dichotomous traits, we performed 10,000 simulations using SCANG-STAAR-S, SCANG-STAAR-B and SCANG-STAAR-O to analyze a 10-Mb genome, and evaluated the empirical genome-wise (family-wise) type I error rates at nominal 𝛼 = 0.05 and 0.01 (Supplementary Table 15). The results show that all the three tests based on SCANG-STAAR provide a good control of the type I error rates for both continuous and dichotomous traits at the two 𝛼 levels.
Empirical power simulations
We then compared the empirical power of SCANG-STAAR with the existing methods, including the sliding window procedures using burden15, SKAT16, SKAT-O47 and STAAR17, and the dynamic window procedure using SCANG19 at genome-wise (family-wise) error rate 𝛼 = 0.01 level with 1,000 replicates. The genome-wise (family-wise) type I error rate was controlled using the empirical threshold for SCANG-STAAR and SCANG, and the Bonferroni correction for the sliding window procedures. We randomly selected two signal regions (variant-phenotype association regions) across the 10-Mb genome in each replicate. The lengths of the signal regions were randomly selected from lengths of 1 kb, 1.5 kb and 2 kb. We considered the proportions of causal variants is 15% on average among the signal regions, and the probability that variants are causal was allowed to be dependent on different sets of annotations through a logistic model, of which five were informative and the other five were non-informative. All the 10 annotations were used in SCANG-STAAR and STAAR. In order to evaluate power, we considered two criteria, causal variant detection rate and signal region detection rate19 (Online Methods).
For both quantitative and dichotomous traits, SCANG-STAAR had a higher power than the 1-kb and 2-kb sliding window procedure using burden, SKAT, SKAT-O and STAAR in terms of both causal variant detection rate and signal region detection rate across different proportions of effect size directions (Supplementary Figs. 11–16). Our simulation studies indicate that SCANG-STAAR improves power by flexibly detecting the locations and the sizes of signal regions. In addition, SCANG-STAAR had a higher power than SCANG for both causal variant detection rate and signal region detection rate (Supplementary Figs. 17–18). Our simulation studies indicate that SCANG-STAAR improves power by incorporating informative variant functional annotations.
Discussion
We developed a comprehensive association analysis framework for detecting noncoding rare variant set associations in large-scale WGS studies by defining a variety of noncoding variant sets and incorporating multi-faceted variant functional annotations. Our approach allows for analyzing both continuous and binary traits and accounts for both population structure and relatedness using generalized linear mixed models in gene-centric analysis and non-gene-centric analysis. It could further account for the stratification of recent population structure using the principal components calculated from RVs through the regression framework48. For gene-centric analysis, we proposed several strategies to define analysis units of RVs in the noncoding genome, including seven functional categories of regulatory regions for protein-coding genes, ncRNA genes, and performed RVATs of each noncoding mask using STAAR. For non-gene-centric analysis, to overcome the limitations of fixed-size sliding windows, we proposed SCANG-STAAR, a data-adaptive-size dynamic window scan procedure that incorporates multi-faceted functional annotations. We proposed STAARpipeline to perform RVATs using these methods for both noncoding and coding variants in unconditional analysis and conditional analyses, which provides an analytical follow-up to distinguish novel RV association signals independent of known variants.
STAARpipeline is a fast and resource-efficient tool for RV association analysis of WGS data that scales linearly on hundreds of thousands of samples.
STAARpipeline allows researchers to conveniently functionally annotate a WGS/WES study using the variant functional annotation database FAVOR and the FAVORannotator workflow. STAARpipeline optimizes computational feasibility of RV association analysis in two steps. First, STAARpipeline reduces the computation burden of fitting the null mixed model using the estimated sparse GRM17, 49. Second, STAARpipeline performs the RV association tests by taking advantage of sparse genotype dosages of RVs50.
We demonstrated the power gain of STAARpipeline over the existing approaches in the data analysis of 9 traits from TOPMed Freeze 5. First, STAARpipeline detected 49 significant associations in gene-centric noncoding analysis, and 35 associations (71.4%) were detected by the 6 newly proposed noncoding masks (Supplementary Tables 16). Second, the proposed dynamic window analysis procedure SCANG-STAAR detected 43 non-overlapped significant noncoding associations in the noncoding genome, which was 19.4% more than the existing 2-kb sliding window procedure that detected 36 non-overlapped significant noncoding associations (Supplementary Tables 17). In addition, SCANG-STAAR only missed one non-overlapped associations detected by 2-kb sliding window procedure (Supplementary Tables 8, 10, 13–14).
In the WGS RV analysis of lipid traits in TOPMed, we identified and replicated using our STAARpipeline several conditional associations in the noncoding genome, including RVs in an intronic region of PAFAH1B2 and TG, RVs in an intronic region of SIDT2 and TG, and RVs in an intronic region of CEP164 and TG, which were not detected by previous analysis of TOPMed Freeze 317, 21. Several coding rare variants in PAFAH1B2 have been previously detected associated with TG51, our findings detected additionally significant RV association in the noncoding region of PAFAH1B2. Two intronic common variants in SIDT2 have been reported associated with TG52, additional intronic rare variant association in SIDT2 was detected using STAARpipeline.
Since SCANG-STAAR considers many more overlapping windows than the sliding window procedure, the genome-wide significance threshold is smaller than that of the sliding window procedure. For example, to control the genome-wise error rate at 0.05 level in our analysis of LDL-C, the P value threshold of SCANG-STAAR-S was 3.80 × 10−9 while the Bonferroni-corrected threshold of the 2-kb sliding window procedure was 1.88 × 10−8. When the window size of the signal region is close to the sliding window size, the sliding window procedure may detect associations missed by the dynamic window procedure because of this gap of the P value thresholds. In STAARpipeline we pragmatically provide both procedures.
In addition to noncoding rare variants association analysis, STAARpipeline also provides single variant analysis for common and low-frequency variants and gene-centric analysis for coding rare variants. The single variant analysis in STAARpipeline provides individual P values of variants given a MAF or MAC cut-off, for example, MAC ≥ 20. The gene-centric coding analysis provides five functional categories to aggregate coding rare variants of each protein-coding gene: (1) putative loss of function (stop gain, stop loss and splice) RVs, (2) missense RVs, (3) disruptive missense RVs, (4) putative loss of function and disruptive missense RVs, and (5) synonymous RVs. The putative loss of function, missense, and synonymous RVs are defined by GENCODE VEP categories29,30. The disruptive variants are further defined by MetaSVM53, which measures the deleteriousness of missense mutations. As in the noncoding RV association analysis, single variant and gene-centric coding analyses also scale well in computation time and memory for large-scale WGS data. Using 30,138 related TOPMed samples these two analyses respectively took 3 hours and 5 hours for 100 cores with 6 Gb memory. Thus, STAARpipeline provides an efficient and comprehensive analysis tool for both coding and noncoding variant association discovery in large-scale sequencing studies.
With the emergence of large-scale WGS data, there is a pressing need to identify genetic components of complex traits in the noncoding genome. Here we introduce a powerful and scalable framework, STAARpipeline, for noncoding RV association detection across the genome. STAARpipeline provides several strategies to aggregate noncoding rare variants to empower RV association analysis in the noncoding region. We demonstrate the computational efficiency of STAARpipeline in application to the WGS association analysis of a range of traits up to ~40,000 TOPMed samples. The optimization approaches of STAARpipeline make it scalable for even larger data sets. Thus, our framework provides an essential solution for noncoding RV association detection in large-scale WGS data analysis and dissects the genetic contribution of noncoding rare variants to complex diseases.
Methods
Notations and model
Suppose there are n subjects with M total variants sequenced across the whole genome. For subject i, let 𝑌i denote a continuous or dichotomous trait with mean denote q covariates, such as age, gender, ancestral principal components; and denote the genotype information of the p genetic variants in a given variant set.
We consider the Generalized Linear Model for unrelated samples,
| (1) |
where g(μ) = μ for a continuous trait, g(μ) = logit(μ) for a dichotomous trait, α0 is an intercept, is a vector of regression coefficients for Xi, and is a vector of regression coefficients for Gi.
We consider the following Generalized Linear Mixed Model27, 28, 54 for related samples,
| (2) |
where the random effects bi account for remaining population structure unaccounted by ancestral principal components and relatedness. Let with variance components θ and a genetic relatedness matrix Φ17, 49. Our goal is testing the null hypothesis of whether the variant-set is associated with the phenotype, adjusting for covariates and relatedness, which corresponds to H0: β = 0, that is, β1 = β2 = ⋯ = βp = 0.
Variant set test using STAAR
The STAARpipeline calculates the variant set P value of each analysis unit using the STAAR method that incorporates multiple variant functional annotation scores17. Assume there are K annotations and , where Ajk is the kth annotation for the jth variant (k = 1, ⋯ , K; j = 1, ⋯ , p). For 𝑘 = 0, we assume . Assume wjl = Beta(MAFj; a1l, a2l), where (a11, a21) = (1,25), (a12, a22) = (1,1) and MAFj is the MAF of the jth variant (j = 1, ⋯ , p). The burden test statistic using kth variant functional annotation and lth beta density as the weight is given by
The SKAT test statistic using kth variant functional annotation and lth beta density as the weight is given by
(k = 0, ⋯ , K; l = 1,2). The ACAT-V test statistic using 𝑘th variant functional annotation and 𝑙th beta density as the weight is given by
where is the average of the weights among the extremely rare variants with MAC ≤ 10, and p′ is the number of variants with MAC > 10 in the variant set.
Let pBurden,l,k be the P value of QBurden,l,k, pSKAT,l,k be the P value of QSKAT,l,k, and pACAT–V,l,k be the P value of QACAT–V,l,k (k = 0, ⋯ , K; l = 1,2). We define STAAR-Burden (STAAR-B), STAAR-SKAT (STAAR-S), and STAAR-ACAT-V (STAAR-A) as , and the corresponding P value is calculated by , where . The STAAR-O test statistic is defined as
and the corresponding P value is calculated by
In gene-centric and sliding window analysis, we use the STAAR-O test for each analysis unit.
Dynamic window analysis using SCANG-STAAR
The STAARpipeline performs dynamic window analysis using the SCANG-STAAR procedure, which extends the dynamic window rare variant test procedure SCANG by incorporating multiple variant functional annotations using the STAAR method. Under the global null hypothesis, there is no variant associated with the phenotype across the genome. Under the alternative hypothesis, there exists at least one region associated with the phenotype. SCANG-STAAR procedure provides a valid test by using the minimum value of the P value of all candidate moving windows of different sizes
where p(I) is the P value of region I, |I| is the number of variants in a window I, and Lmin and Lmax are the smallest and largest number of variants in the searching windows, respectively. For SCANG-STAAR-S and SCANG-STAAR-B procedures, p(I) is the STAAR-S and STAAR-B P value of window I, respectively. For SCANG-STAAR-O, (𝐼) is the omnibus P value of STAAR-S and STAAR-B calculated by ACAT method38. Similar to the SCANG procedure, SCANG-STAAR controls the genome-wise type I error rate at a given α level by using the αth quantile of the empirical distribution of pmin as an empirical threshold ℎ(α, pmin, Lmin, Lmax)19. We reject the null hypothesis if the P value of any window is smaller than ℎ(α, pmin, Lmin, Lmax). If this results in only one window, the detected window is . If this results in multiple overlapping windows, we localize the signals as the window whose P value is smaller than both the threshold and the windows that overlap with it.
Conditional analysis
The STAARpipeline performs conditional analysis to identify RV association independent of known variants. We first select a list of known variants by including the trait-associated variants identified in literature, for example, variants indexed in GWAS Catalog39 or significant variants in large-scale GWAS. The significant variants detected in individual analysis using the same data could also be added into the known variants list to ensure the RV signals are not captured by the significant individual variants. We then use the following stepwise selection strategy to select a subset of independent variants representing the known variant list as the variants adjusted in the conditional analysis:
Calculate the individual P value of all variants in the known variants list and select the most significant variant.
For each step, calculate the P values of all the remaining variants conditional on the variant(s) that have already been selected. For each variant, we only condition on the selected variants within a specified region of that variant, such as the +/− 1-Mb window.
Select the variant with minimum conditional P value that is lower than the cutoff P value, for example, 1 × 10−4.
Repeat steps 2–3 until no variants can be selected.
Finally, we calculate the conditional P value of each significant RV analysis unit by adjusting for the selected variants residing in an extended region (for example, +/− 1-Mb window) of the analysis unit.
Statistical analysis of lipid traits in the TOPMed data
The TOPMed WGS data consist of ancestrally diverse and multi-ethnic related samples4. Race/ethnicity was defined using a combination of self-reported race/ethnicity and study recruitment information (Supplementary Note)40. The discovery cohorts consist of 5,849 (27.8%) Black or African American, 12,313 (58.6%) White, 675 (3.2%) Asian American, 1,075 (5.1%) Hispanic/Latino American and 1,103 (5.3%) Samoans. The replication cohorts consist of 2,265 (24.8%) Black or African American, 5,615 (61.5%) White, and 1,243 (13.6%) Hispanic/Latino American.
We applied STAARpipeline to identify RV sets associated with four quantitative lipid traits (LDL-C, HDL-C, TG and TC) using the TOPMed WGS data. LDL-C and TC were adjusted for the presence of medications as before21. Linear regression model adjusting for age, age2, sex was first fit for each study-race/ethnicity-specific group. In addition, for Old Order Amish, we also adjusted for APOB p.R3527Q in LDL-C and TC analyses and adjusted for APOC3 p.R19Ter in TG and HDL-C analyses21. The residuals were rank-based inverse normal transformed and rescaled by the standard deviation of the original phenotype within each group. We then fit a heteroscedastic linear mixed model (HLMM) for the rank normalized residuals, adjusting for 10 ancestral PCs, study-ethnicity group indicators, and a variance component for empirically derived kinship matrix plus separate group-specific residual variance components to account for population structure and relatedness. The output of HLMM was then used to perform following variant set analyses for rare variants (MAF < 1%) by scanning the genome, including gene-centric analysis using seven variant categories (promoter RVs overlaid with CAGE sites, promoter RVs overlaid with DHS sites, enhancer RVs overlaid with CAGE sites, enhancer RVs overlaid with DHS sites, UTR RVs, upstream RVs and downstream RVs) for each protein coded gene, ncRNA RVs, 2-kb sliding windows with 1-kb skip length, and dynamic windows with variants number between 40 and 300. The WGS RVAT analysis was performed using R packages STAAR (version 0.9.6), STAARpipeline (version 0.9.6) and STAARpipelineSummary (version 0.9.6).
Rare variant association analysis of CRP, eGFR, FG, FI, and TL in the TOPMed data
We applied STAARpipeline to identify RV sets associated with five non-lipid traits from the 14 cohorts in TOPMed Freeze 5, including CRP of 22,775 individuals, eGFR of 23,732 individuals, FG of 23,859 individuals, FI of 21,900 individuals and TL of 39,742 individuals (Supplementary Note). These five traits were defined the same as in the previous studies42–45. For CRP and FI, we additionally performed log-transformation of the trait in the analysis42, 44. For each trait, we first fit a linear regression model adjusting for age and sex for each study-race/ethnicity group, with additional adjustment of age2 for CRP, age2 and body mass index (BMI) for FG and FI, and sequencing center and 10 ancestral PCs for TL42–45. The residuals were transformed using the rank-based inverse normal transformation and rescaled by the standard deviation of the original phenotype within each study-race/ethnicity group. We then fit a heteroscedastic linear mixed model (HLMM) for the rank normalized residuals, adjusting for 10 ancestral PCs, study-ethnicity group indicators, and a variance component for empirically derived kinship matrix plus separate group-specific residual variance components to account for population structure and relatedness. We additionally adjusted for age, sex, and sequencing center for TL. The output of HLMM was then used in the RV association analysis of the STAARpipeline.
In gene-centric noncoding analysis, STAARpipeline identified 6 conditionally significant associations with at least one of the five traits compared with the previous analyses42–45. These included promoter CAGE or enhancer DHS RVs in associating CRP and CRP, ncRNA RVs in CTC-523E23.15 and FI, enhancer CAGE or DHS RVs in TINF2 and TL, enhancer DHS RVs in MRVI1 and TL (Supplementary Table 12).
In non-gene-centric noncoding analysis using 2-kb sliding windows, we identified 8 conditionally significant associations with at least one of the five traits. These included associations for 2 intergenic sliding windows near CRP and CRP, 2 intergenic sliding windows near AC073409.1 and TL, an intronic sliding window in TERT and TL, an intergenic sliding window near RNGTT and TL, and 2 intronic sliding windows in ZGPAT and TL (Supplementary Table 13). We also identified 8 conditionally significant associations in the noncoding genome with at least one of the five traits in dynamic window analysis. These included associations for 2 intergenic regions near CRP and CRP, an intergenic region near AC073409.1 and TL, an intronic region in MLIP and TL, an intronic region in TERT and TL, an intergenic region near RNGTT and TL, an intronic region in NOS1 and TL, and an intronic region in ZGPAT and TL (Supplementary Table 14). Note that the associations between RVs in the intronic region of MLIP or NOS1 with TL are missed by the 2-kb sliding window analysis.
Data simulation
Type I error rate simulations
We performed extensive simulation studies to show that the proposed SCANG-STAAR method controls the genome-wise (family-wise) type I error rate. We generated genotypes by simulating 100,000 sequencing chromosomes for a 10-Mb region that represent the whole genome. The data were generated to mimic the LD structure of an African American population by using the calibration coalescent model (COSI)46. We considered the total sample sizes n = 50,000 in all simulations. We generated continuous traits from a linear model
where X1i ∼ N(0,1), X2i ∼ Bernoulli(0.5), and ϵi ∼ N(0,1). We generated dichotomous traits from a logistic model
where X1i and X2i were defined the same as continuous traits and α0 was determined to set the prevalence to 1%. We used case-control sampling. In each simulation replicate, 10 annotations were generated as A1, … , A10 i.i.d. N(0,1) for each variant. For the size simulation, under the null, these annotations are not associated with the regression coefficients of the phenotype models. We applied SCANG-STAAR-S, SCANG-STAAR-B, SCANG-STAAR-O by incorporating the MAF and the 10 annotations as weights and repeated the procedure with 10,000 replicates to examine the genome-wise (family-wise) type I error rates for both continuous and dichotomous traits at α = 0.05 and 0.01 levels.
Empirical power simulations
Next, we carried out simulation studies to assess the power gain of SCANG-STAAR compared to the existing methods, including the sliding window procedures using burden15, SKAT16, SKAT-O47 and STAAR17, and the dynamic window procedure using SCANG19, with 1,000 replicates. In each simulation replicate, we randomly selected two signal regions (variant-phenotype association regions) across the 10-Mb genome for power simulations, where the length of the signal regions was randomly selected from 1 kb, 1.5 kb and 2 kb. For each signal region, causal variants were generated according to a logistic model
where five annotations {k1, ⋯ , k5} ⊂ {1, ⋯ ,10} were randomly sampled for each region. This assumes the probability of a variant being causal is a function of five randomly selected annotations. Note that we generated 10 functional annotations for each variant and used 5 to determine the probability of causal variants. For RVATs using SCANG-STAAR, we used all annotations, including 5 informative annotations and 5 non-informative annotations. For different regions, causality of variants was allowed to be dependent on different sets of annotations. We set for all annotations and δ0 = logit(0.015), resulting in 15% causal variants on average in signal regions.
We generated continuous traits from a linear model given by
where X1i, X2i, ϵi were defined as in the type I error rate simulations, G1j, … , Gsj were the genotypes of the s causal variants in the signal region, and β1, … , βs were the corresponding effect sizes of causal variants. Similarly, we generated dichotomous traits from a logistic model given by
where α0, X1i, X2i were defined as in the type I error rate simulations, G1j, … , Gsj were the genotypes of the s randomly selected causal variants in the signal region, and β1, … , βs were the corresponding log odds ratios (ORs) of the 𝑠 causal variants. We used case-control sampling. For both models, we set the effect sizes of causal variants as a decreasing function of MAFs,; for continuous trait, c0 = 0.10, and for dichotomous traits, c0 = 0.14, which gives an odds ratio of 2 for a variant with MAF 1 × 10−5. For each region, we varied the proportions of causal variant effect size directions by setting 50%, 80% or 100% variants with positive effects.
We applied SCANG-STAAR, SCANG, and 1-kb and 2-kb sliding window methods using burden, SKAT, SKAT-O and STAAR. We repeated the procedure with 1,000 replicates to examine power at genome-wise (family-wise) error rate α = 0.01 level. The genome-wise (family-wise) type I error rate was controlled using the empirical threshold for SCANG-STAAR and SCANG, and the Bonferroni correction for the sliding window procedures. For SCANG-STAAR and sliding window methods using STAAR, we incorporated all 10 annotations in the weighting scheme, including the 5 annotations that are associated with the variant being causal and the 5 annotations that are not.
To evaluate power, we considered two criteria, causal variant detection rate and signal region detection rate. The causal variant detection rate can be regarded as the power of causal variants detection. The signal region detection rate can be regarded as the power of signal regions detection. The causal variant detection rate is defined as
We define a causal variant as “detected” if it is in one of the detected signal regions. The signal region detection rate is defined as
We define the signal region as “detected” if it overlaps with one of the detected signal regions. Both the causal variant detection rate and the signal region detection rate can be regarded as measures of the test’s power.
We also did a sensitivity analysis where all ten annotations were uninformative. Specifically, 15% of variants within each signal region were randomly chosen as causal variants without using the annotation information. SCANG-STAAR had a similar performance to SCANG, and had a higher power than the sliding window methods with fixed window sizes in terms of both causal variant detection rate and signal region detection rate (Supplementary Figs. 19–20). Our simulation results indicate that SCANG-STAAR is robust to the noninformative annotations and improves power by flexibly detecting the locations and the sizes of signal regions when functional annotations are informative.
Genome build
All genome coordinates are given in NCBI GRCh38/UCSC hg38.
Extended Data
Extended Data Fig. 1|. Rare variant (MAF < 0.01) distribution in the discovery phase using TOPMed cohorts (n=21,015).
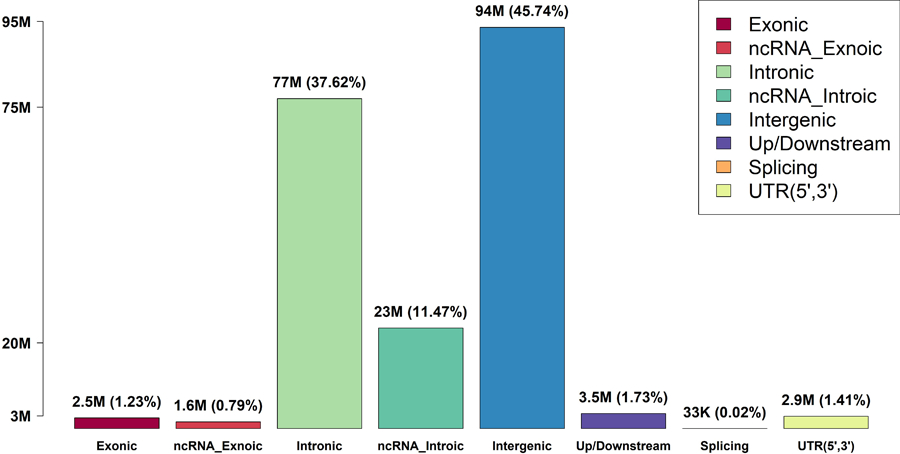
Variant categories are defined by GENCODE VEP categories.
Extended Data Fig. 2|. Manhattan plots and Q-Q plots for unconditional gene-centric noncoding analysis and sliding window analysis of high-density lipoprotein cholesterol (HDL-C) in the discovery phase (n=21,015).
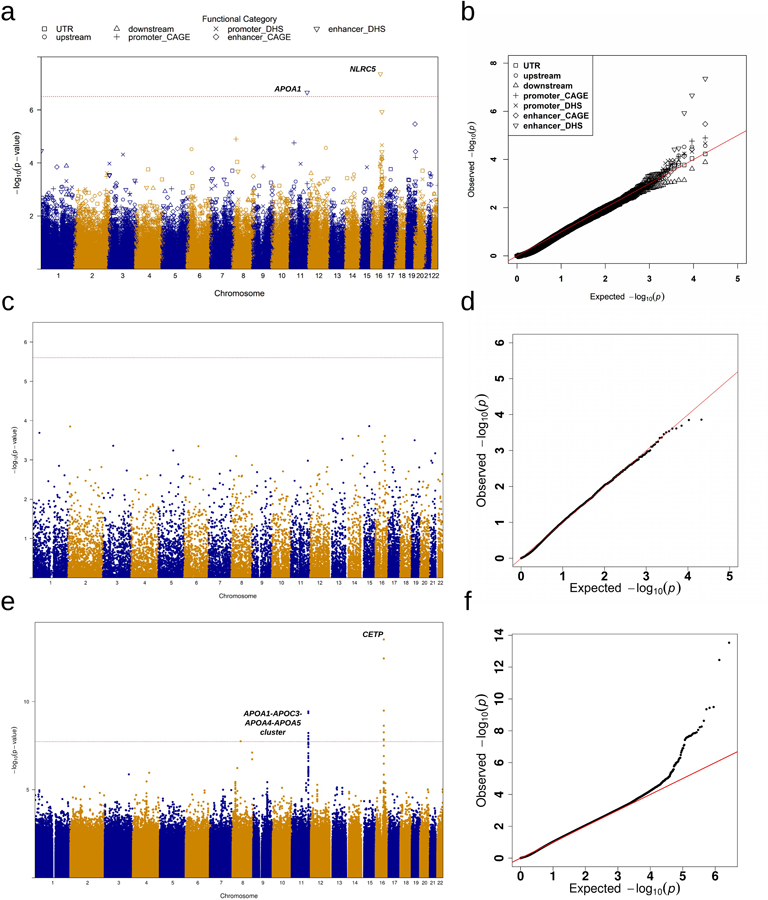
a, Manhattan plots for unconditional gene-centric noncoding analysis of protein-coding gene. The horizontal line indicates a genome-wide STAAR-O P value threshold of . The significant threshold is defined by multiple comparisons using the Bonferroni correction . Different symbols represent the STAAR-O P value of the protein-coding gene using different functional categories (upstream, downstream, UTR, promoter_CAGE, promoter_DHS, enhancer_CAGE, enhancer_DHS). Promoter_CAGE and promoter_DHS are the promoters with overlap of Cap Analysis of Gene Expression (CAGE) sites and DNase hypersensitivity (DHS) sites for a given gene, respectively. Enhancer_CAGE and enhancer_DHS are the enhancers in GeneHancer predicted regions with the overlap of CAGE sites and DHS sites for a given gene, respectively. b, Quantile-quantile plots for unconditional gene-centric noncoding analysis of protein-coding gene. Different symbols represent the STAAR-O P-value of the gene using different functional categories (upstream, downstream, UTR, promoter_CAGE, promoter_DHS, enhancer_CAGE, enhancer_DHS). c, Manhattan plots for unconditional gene-centric noncoding analysis of ncRNA gene. The horizontal line indicates a genome-wide STAAR-O P value threshold of . The significant threshold is defined by multiple comparisons using the Bonferroni correction . d, Quantile-quantile plots for unconditional gene-centric noncoding analysis of ncRNA gene. e, Manhattan plot for 2-kb sliding windows. The horizontal line indicates a genome-wide P value threshold of . The significant threshold is defined by multiple comparisons using the Bonferroni correction . f, Quantile-quantile plot for 2-kb sliding windows. In panels, a, c and e, the chromosome number are indicated by the colors of dots. In all panels, STAAR-O is a two-sided test.
Extended Data Fig. 3|. Manhattan plots and Q-Q plots for unconditional gene-centric noncoding analysis and sliding window analysis of low-density lipoprotein cholesterol (LDL-C) in the discovery phase (n=21,015).
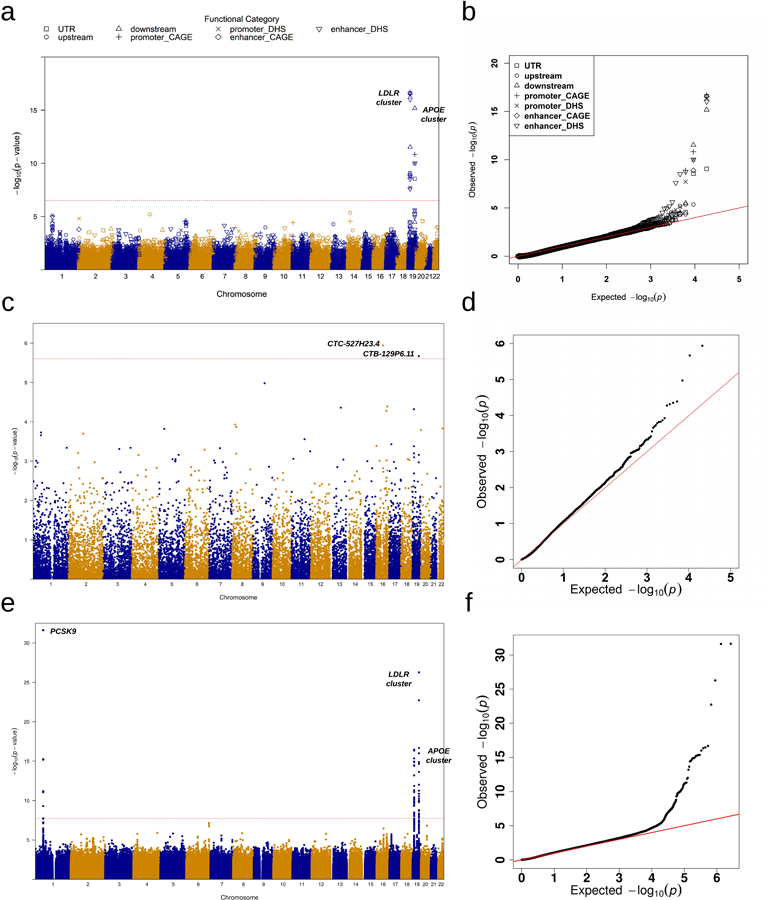
a, Manhattan plots for unconditional gene-centric noncoding analysis of protein-coding gene. The horizontal line indicates a genome-wide STAAR-O P-value threshold of . The significant threshold is defined by multiple comparisons using the Bonferroni correction . Different symbols represent the STAAR-O P-value of the protein-coding gene using different functional categories (upstream, downstream, UTR, promoter_CAGE, promoter_DHS, enhancer_CAGE, enhancer_DHS). Promoter_CAGE and promoter_DHS are the promoters with overlap of Cap Analysis of Gene Expression (CAGE) sites and DNase hypersensitivity (DHS) sites for a given gene, respectively. Enhancer_CAGE and enhancer_DHS are the enhancers in GeneHancer predicted regions with the overlap of CAGE sites and DHS sites for a given gene, respectively. b, Quantile-quantile plots for unconditional gene-centric noncoding analysis of protein-coding gene. Different symbols represent the STAAR-O P-value of the gene using different functional categories (upstream, downstream, UTR, promoter_CAGE, promoter_DHS, enhancer_CAGE, enhancer_DHS). c, Manhattan plots for unconditional gene-centric noncoding analysis of ncRNA gene. The horizontal line indicates a genome-wide STAAR-O P-value threshold of . The significant threshold is defined by multiple comparisons using the Bonferroni correction . d, Quantile-quantile plots for unconditional gene-centric noncoding analysis of ncRNA gene. e, Manhattan plot for 2-kb sliding windows. The horizontal line indicates a genome-wide P-value threshold of . The significant threshold is defined by multiple comparisons using the Bonferroni correction . f, Quantile-quantile plot for 2-kb sliding windows. In panels, a, c and e, the chromosome number are indicated by the colors of dots. In all panels, STAAR-O is a two-sided test.
Extended Data Fig. 4|. Manhattan plots and Q-Q plots for unconditional gene-centric noncoding analysis and sliding window analysis of triglycerides (TG) in the discovery phase (n=21,015).
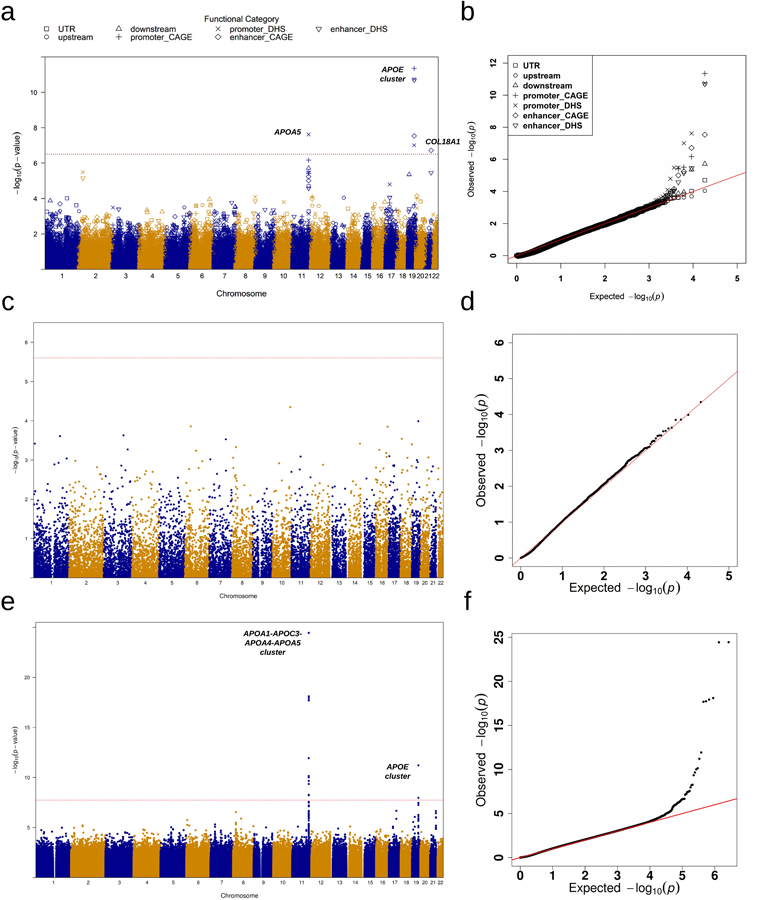
a, Manhattan plots for unconditional gene-centric noncoding analysis of protein-coding gene. The horizontal line indicates a genome-wide STAAR-O P-value threshold of . The significant threshold is defined by multiple comparisons using the Bonferroni correction . Different symbols represent the STAAR-O P-value of the protein-coding gene using different functional categories (upstream, downstream, UTR, promoter_CAGE, promoter_DHS, enhancer_CAGE, enhancer_DHS). Promoter_CAGE and promoter_DHS are the promoters with overlap of Cap Analysis of Gene Expression (CAGE) sites and DNase hypersensitivity (DHS) sites for a given gene, respectively. Enhancer_CAGE and enhancer_DHS are the enhancers in GeneHancer predicted regions with the overlap of CAGE sites and DHS sites for a given gene, respectively. b, Quantile-quantile plots for unconditional gene-centric noncoding analysis of protein-coding gene. Different symbols represent the STAAR-O P-value of the gene using different functional categories (upstream, downstream, UTR, promoter_CAGE, promoter_DHS, enhancer_CAGE, enhancer_DHS). c, Manhattan plots for unconditional gene-centric noncoding analysis of ncRNA gene. The horizontal line indicates a genome-wide STAAR-O P-value threshold of . The significant threshold is defined by multiple comparisons using the Bonferroni correction . d, Quantile-quantile plots for unconditional gene-centric noncoding analysis of ncRNA gene. e, Manhattan plot for 2-kb sliding windows. The horizontal line indicates a genome-wide P-value threshold of . The significant threshold is defined by multiple comparisons using the Bonferroni correction . f, Quantile-quantile plot for 2-kb sliding windows. In panels, a, c and e, the chromosome number are indicated by the colors of dots. In all panels, STAAR-O is a two-sided test.
Extended Data Fig. 5|. Manhattan plots and Q-Q plots for unconditional gene-centric noncoding analysis and sliding window analysis of total cholesterol (TC) in the discovery phase (n=21,015).
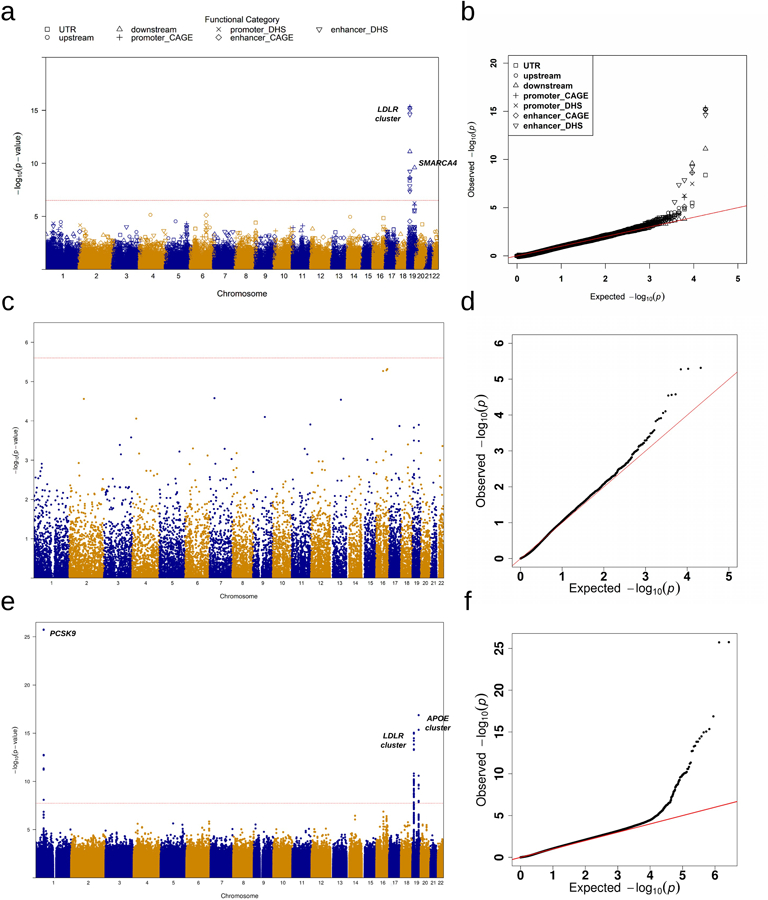
a, Manhattan plots for unconditional gene-centric noncoding analysis of protein-coding gene. The horizontal line indicates a genome-wide STAAR-O P-value threshold of . The significant threshold is defined by multiple comparisons using the Bonferroni correction . Different symbols represent the STAAR-O P-value of the protein-coding gene using different functional categories (upstream, downstream, UTR, promoter_CAGE, promoter_DHS, enhancer_CAGE, enhancer_DHS). Promoter_CAGE and promoter_DHS are the promoters with overlap of Cap Analysis of Gene Expression (CAGE) sites and DNase hypersensitivity (DHS) sites for a given gene, respectively. Enhancer_CAGE and enhancer_DHS are the enhancers in GeneHancer predicted regions with the overlap of CAGE sites and DHS sites for a given gene, respectively. b, Quantile-quantile plots for unconditional gene-centric noncoding analysis of protein-coding gene. Different symbols represent the STAAR-O P-value of the gene using different functional categories (upstream, downstream, UTR, promoter_CAGE, promoter_DHS, enhancer_CAGE, enhancer_DHS). c, Manhattan plots for unconditional gene-centric noncoding analysis of ncRNA gene. The horizontal line indicates a genome-wide STAAR-O P-value threshold of . The significant threshold is defined by multiple comparisons using the Bonferroni correction . d, Quantile-quantile plots for unconditional gene-centric noncoding analysis of ncRNA gene. e, Manhattan plot for 2-kb sliding windows. The horizontal line indicates a genome-wide P-value threshold of . The significant threshold is defined by multiple comparisons using the Bonferroni correction . f, Quantile-quantile plot for 2-kb sliding windows. In panels, a, c and e, the chromosome number are indicated by the colors of dots. In all panels, STAAR-O is a two-sided test.
Supplementary Material
Acknowledgments
This work was supported by grants R35-CA197449, U19-CA203654, R01-HL113338, and U01-HG009088 (X. Lin), R01-HL142711 and R01-HL127564 (P.N. and G.M.P), R35-HL135824 (C.J.W.), 75N92020D00001, HHSN268201500003I, N01-HC-95159, 75N92020D00005, N01-HC-95160, 75N92020D00002, N01-HC-95161, 75N92020D00003, N01-HC-95162, 75N92020D00006, N01-HC-95163, 75N92020D00004, N01-HC-95164, 75N92020D00007, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079, UL1-TR-001420, UL1-TR001881, DK063491, R01-HL071051, R01-HL071205, R01-HL071250, R01-HL071251, R01-HL071258, R01-HL071259, and UL1-RR033176 (J.I.R. and X.G.), U01-HL72518, HL087698, HL49762, HL59684, HL58625, HL071025, HL112064, NR0224103, and M01-RR000052 (to the Johns Hopkins General Clinical Research Center), NO1-HC-25195, HHSN268201500001I, 75N92019D00031, and R01-HL092577-06S1 (R.S.V. and L.A.C.), the Evans Medical Foundation and the Jay and Louis Coffman Endowment from the Department of Medicine, Boston University School of Medicine (R.S.V.), HHSN268201800001I and U01-HL137162 (K.M.R., M.P.C.), R01-HL133040 (D.E.W.), R35-HL135818, R01-HL113338, and HL436801 (S.R.), KL2TR002490 (L.M.R.), R01-HL92301, R01-HL67348, R01-NS058700, R01-AR48797, and R01-AG058921 (N.D.P. and D.W.B.), R01-DK071891 (N.D.P., B.I.F., and D.W.B.), M01-RR07122 and F32-HL085989 (to the General Clinical Research Center of the Wake Forest University School of Medicine), the American Diabetes Association, P60-AG10484 (to the Claude Pepper Older Americans Independence Center of Wake Forest University Health Sciences), U01-HL137181 (J.R.O.), R01-HL141944 (R.A.M.), R.A.M. receives support as the Sarah Miller Coulson Scholar in the Johns Hopkins Center for Innovative Medicine, HHSN268201600018C, HHSN268201600001C, HHSN268201600002C, HHSN268201600003C, and HHSN268201600004C (C.L.K.), R01-HL113323, U01-DK085524, R01-HL045522, R01-MH078143, R01-MH078111, and R01-MH083824 (H.H.H.G., R.D., J.E.C., and J.B.), R01-DK117445 and R01-MD012765 (N.F. and B.M.L.), U01-DK078616, UM1-DK0786 and R01-DK078616 (J.B.M.), 18CDA34110116 from American Heart Association (P.S.d.V.), HHSN268201800010I, HHSN268201800011I, HHSN268201800012I, HHSN268201800013I, HHSN268201800014I, and HHSN268201800015I (A.C.), R01-HL153805, R03-HL154284 (B.E.C.), HHSN268201700001I, HHSN268201700002I, HHSN268201700003I, HHSN268201700005I, and HHSN268201700004I (E.B.), U01-HL072524, R01-HL104135-04S1, U01-HL054472, U01-HL054473, U01-HL054495, U01-HL054509, and R01-HL055673-18S1 (D.K.A.). Molecular data for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung and Blood Institute (NHLBI). Core support including centralized genomic read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Core support including phenotype harmonization, data management, sample-identity QC, and general program coordination were provided by the TOPMed Data Coordinating Center (R01HL-120393; U01HL-120393; contract HHSN268201800001I). We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed. We gratefully acknowledge the support from The Samoan Obesity, Lifestyle and Genetic Adaptations Study (OLaGA) Group. The full study specific acknowledgements are detailed in Supplementary Note.
NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium
Namiko Abe55, Gonçalo Abecasis56, Francois Aguet57, Christine Albert58, Laura Almasy59, Alvaro Alonso60, Seth Ament61, Peter Anderson62, Pramod Anugu63, Deborah Applebaum-Bowden64, Kristin Ardlie57, Dan Arking65, Allison Ashley-Koch66, Stella Aslibekyan67, Tim Assimes68, Dimitrios Avramopoulos65, Najib Ayas69, Adithya Balasubramanian70, John Barnard71, Kathleen Barnes72, R. Graham Barr73, Emily Barron-Casella65, Lucas Barwick74, Terri Beaty65, Gerald Beck75, Diane Becker76, Lewis Becker65, Rebecca Beer77, Amber Beitelshees61, Emelia Benjamin78, Takis Benos79, Marcos Bezerra80, Nathan Blue81, Russell Bowler82, Ulrich Broeckel83, Jai Broome62, Deborah Brown84, Karen Bunting55, Esteban Burchard85, Carlos Bustamante86, Erin Buth87, Jonathan Cardwell88, Vincent Carey89, Julie Carrier90, April Carson91, Cara Carty92, Richard Casaburi93, Juan P. Casas Romero94, James Casella65, Peter Castaldi95, Mark Chaffin57, Christy Chang61, Yi-Cheng Chang96, Daniel Chasman97, Sameer Chavan88, Bo-Juen Chen55, Wei-Min Chen98, Yii-Der Ida Chen99, Michael Cho89, Seung Hoan Choi57, Lee-Ming Chuang100, Mina Chung101, Ren-Hua Chung102, Clary Clish103, Suzy Comhair104, Elaine Cornell105, Carolyn Crandall93, James Crapo106, Jeffrey Curtis107, Brian Custer108, Coleen Damcott61, Dawood Darbar109, Sean David110, Colleen Davis62, Michelle Daya88, Mariza de Andrade111, Lisa de las Fuentes112, Michael DeBaun113, Ranjan Deka114, Dawn DeMeo89, Scott Devine61, Huyen Dinh70, Harsha Doddapaneni115, Qing Duan116, Shannon Dugan-Perez70, Jon Peter Durda105, Susan K. Dutcher117, Charles Eaton118, Lynette Ekunwe63, Adel El Boueiz119, Patrick Ellinor120, Leslie Emery62, Serpil Erzurum71, Charles Farber98, Jesse Farek70, Tasha Fingerlin121, Matthew Flickinger56, Myriam Fornage122, Chris Frazar62, Mao Fu61, Stephanie M. Fullerton62, Lucinda Fulton123, Stacey Gabriel57, Weiniu Gan77, Shanshan Gao88, Yan Gao63, Margery Gass124, Heather Geiger125, Bruce Gelb126, Mark Geraci127, Soren Germer55, Robert Gerszten128, Auyon Ghosh89, Richard Gibbs70, Chris Gignoux68, Mark Gladwin79, David Glahn129, Stephanie Gogarten62, Da-Wei Gong61, Sharon Graw130, Kathryn J. Gray131, Daniel Grine88, Colin Gross56, C. Charles Gu123, Yue Guan61, Namrata Gupta57, Michael Hall132, Yi Han70, Patrick Hanly133, Daniel Harris134, Nicola L. Hawley135, Jiang He136, Ben Heavner87, Susan Heckbert137, Ryan Hernandez85, David Herrington138, Craig Hersh139, Bertha Hidalgo67, James Hixson122, Brian Hobbs89, John Hokanson88, Elliott Hong61, Karin Hoth140, Chao (Agnes) Hsiung141, Jianhong Hu70, Yi-Jen Hung142, Haley Huston143, Chii Min Hwu144, Marguerite Ryan Irvin67, Rebecca Jackson145, Deepti Jain62, Cashell Jaquish146, Jill Johnsen147, Andrew Johnson77, Craig Johnson62, Rich Johnston60, Kimberly Jones65, Hyun Min Kang148, Robert Kaplan149, Sharon Kardia56, Shannon Kelly150, Eimear Kenny126, Michael Kessler61, Alyna Khan62, Ziad Khan70, Wonji Kim151, John Kimoff152, Greg Kinney153, Barbara Konkle154, Holly Kramer155, Christoph Lange156, Ethan Lange88, Cathy Laurie62, Cecelia Laurie62, Meryl LeBoff89, Jiwon Lee89, Sandra Lee70, Wen-Jane Lee144, Jonathon LeFaive56, David Levine62, Dan Levy77, Joshua Lewis61, Xiaohui Li99, Yun Li116, Henry Lin99, Honghuang Lin157, Simin Liu158, Yongmei Liu159, Yu Liu160, Ruth J.F. Loos161, Steven Lubitz120, Kathryn Lunetta162, James Luo77, Ulysses Magalang163, Michael Mahaney164, Barry Make65, JoAnn Manson89, Melissa Marton125, Susan Mathai88, Susanne May87, Patrick McArdle61, Merry-Lynn McDonald165, Sean McFarland151, Daniel McGoldrick166, Caitlin McHugh87, Becky McNeil167, Hao Mei63, Vipin Menon70, Luisa Mestroni130, Ginger Metcalf70, Deborah A. Meyers168, Emmanuel Mignot169, Julie Mikulla77, Nancy Min63, Mollie Minear170, Ryan L. Minster79, Matt Moll95, Zeineen Momin70, Courtney Montgomery171, Donna Muzny70, Josyf C. Mychaleckyj98, Girish Nadkarni126, Rakhi Naik65, Sergei Nekhai172, Sarah C. Nelson87, Bonnie Neltner88, Caitlin Nessner70, Deborah Nickerson173, Osuji Nkechinyere70, Kari North116, Tim O'Connor61, Heather Ochs-Balcom174, Geoffrey Okwuonu70, Allan Pack175, David T. Paik176, James Pankow177, George Papanicolaou77, Cora Parker178, Juan Manuel Peralta179, Marco Perez68, James Perry61, Ulrike Peters180, Lawrence S Phillips60, Jacob Pleiness56, Toni Pollin61, Wendy Post181, Julia Powers Becker182, Meher Preethi Boorgula88, Michael Preuss126, Pankaj Qasba77, Dandi Qiao89, Zhaohui Qin60, Nicholas Rafaels183, Mahitha Rajendran70, D.C. Rao123, Laura Rasmussen-Torvik184, Aakrosh Ratan98, Robert Reed61, Catherine Reeves185, Elizabeth Regan106, Rebecca Robillard186, Nicolas Robine125, Dan Roden187, Carolina Roselli57, Ingo Ruczinski65, Alexi Runnels125, Pamela Russell88, Sarah Ruuska143, Kathleen Ryan61, Ester Cerdeira Sabino188, Danish Saleheen189, Shabnam Salimi190, Sejal Salvi70, Steven Salzberg65, Kevin Sandow191, Vijay G. Sankaran192, Jireh Santibanez70, Karen Schwander123, David Schwartz88, Frank Sciurba79, Christine Seidman193, Jonathan Seidman194, Frédéric Sériès195, Vivien Sheehan196, Stephanie L. Sherman197, Amol Shetty61, Aniket Shetty88, Wayne Hui-Heng Sheu144, M. Benjamin Shoemaker198, Brian Silver199, Edwin Silverman89, Robert Skomro200, Albert Vernon Smith201, Josh Smith62, Nicholas Smith137, Tanja Smith55, Sylvia Smoller149, Beverly Snively202, Michael Snyder68, Tamar Sofer89, Nona Sotoodehnia62, Adrienne M. Stilp62, Garrett Storm203, Elizabeth Streeten61, Jessica Lasky Su204, Yun Ju Sung123, Jody Sylvia89, Adam Szpiro62, Daniel Taliun56, Hua Tang205, Margaret Taub65, Matthew Taylor130, Simeon Taylor61, Marilyn Telen66, Timothy A. Thornton62, Machiko Threlkeld206, Lesley Tinker207, David Tirschwell62, Sarah Tishkoff208, Hemant Tiwari209, Catherine Tong210, Russell Tracy211, Michael Tsai177, Dhananjay Vaidya65, David Van Den Berg212, Peter VandeHaar56, Scott Vrieze177, Tarik Walker88, Robert Wallace140, Avram Walts88, Fei Fei Wang62, Heming Wang213, Jiongming Wang214, Karol Watson93, Jennifer Watt70, Joshua Weinstock148, Bruce Weir62, Scott T. Weiss215, Lu-Chen Weng120, Jennifer Wessel216, Kayleen Williams87, L. Keoki Williams217, Carla Wilson89, Lara Winterkorn125, Quenna Wong62, Joseph Wu176, Huichun Xu61, Ivana Yang88, Ketian Yu56, Seyedeh Maryam Zekavat57, Yingze Zhang218, Snow Xueyan Zhao106, Xiaofeng Zhu219, Elad Ziv220, Michael Zody55, Sebastian Zoellner56
55 - New York Genome Center, New York, New York, 10013, US; 56 - University of Michigan, Ann Arbor, Michigan, 48109, US; 57 - Broad Institute, Cambridge, Massachusetts, 2142, US; 58 - Cedars Sinai, Boston, Massachusetts, 2114, US; 59 - Children's Hospital of Philadelphia, University of Pennsylvania, Philadelphia, Pennsylvania, 19104, US; 60 - Emory University, Atlanta, Georgia, 30322, US; 61 - University of Maryland, Baltimore, Maryland, 21201, US; 62 - University of Washington, Seattle, Washington, 98195, US; 63 - University of Mississippi Medical Center, Jackson, Mississippi, 39216, US; 64 - National Institutes of Health, Bethesda, Maryland, 20892, US; 65 - Johns Hopkins University, Baltimore, Maryland, 21218, US; 66 - Duke University, Durham, North Carolina, 27708, US; 67 - University of Alabama, Birmingham, Alabama, 35487, US; 68 - Stanford University, Stanford, California, 94305, US; 69 - Providence Health Care, Medicine, Vancouver, CA; 70 - Baylor College of Medicine Human Genome Sequencing Center, Houston, Texas, 77030, US; 71 - Cleveland Clinic, Cleveland, Ohio, 44195, US; 72 - Tempus, University of Colorado Anschutz Medical Campus, Aurora, Colorado, 80045, US; 73 - Columbia University, New York, New York, 10032, US; 74 - The Emmes Corporation, LTRC, Rockville, Maryland, 20850, US; 75 - Cleveland Clinic, Quantitative Health Sciences, Cleveland, Ohio, 44195, US; 76 - Johns Hopkins University, Medicine, Baltimore, Maryland, 21218, US; 77 - National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, Maryland, 20892, US; 78 - Boston University, Massachusetts General Hospital, Boston University School of Medicine, Boston, Massachusetts, 2114, US; 79 - University of Pittsburgh, Pittsburgh, Pennsylvania, 15260, US; 80 - Fundação de Hematologia e Hemoterapia de Pernambuco - Hemope, Recife, 52011-000, BR; 81 - University of Utah, Obstetrics and Gynecology, Salt Lake City, Utah, 84132, US; 82 - National Jewish Health, National Jewish Health, Denver, Colorado, 80206, US; 83 - Medical College of Wisconsin, Pediatrics, Milwaukee, Wisconsin, 53226, US; 84 - University of Texas Health at Houston, Pediatrics, Houston, Texas, 77030, US; 85 - University of California, San Francisco, San Francisco, California, 94143, US; 86 - Stanford University, Biomedical Data Science, Stanford, California, 94305, US; 87 - University of Washington, Biostatistics, Seattle, Washington, 98195, US; 88 - University of Colorado at Denver, Denver, Colorado, 80204, US; 89 - Brigham & Women's Hospital, Boston, Massachusetts, 2115, US; 90 - University of Montreal, US; 91 - University of Mississippi, Medicine, Jackson, Mississippi, 39213, US; 92 - Washington State University, Pullman, Washington, 99164, US; 93 - University of California, Los Angeles, Los Angeles, California, 90095, US; 94 - Brigham & Women's Hospital, US; 95 - Brigham & Women's Hospital, Medicine, Boston, Massachusetts, 2115, US; 96 - National Taiwan University, Taipei, 10617, TW; 97 - Brigham & Women's Hospital, Division of Preventive Medicine, Boston, Massachusetts, 2215, US; 98 - University of Virginia, Charlottesville, Virginia, 22903, US; 99 - Lundquist Institute, Torrance, California, 90502, US; 100 - National Taiwan University, National Taiwan University Hospital, Taipei, 10617, TW; 101 - Cleveland Clinic, Cleveland Clinic, Cleveland, Ohio, 44195, US; 102 - National Health Research Institute Taiwan, Miaoli County, 350, TW; 103 - Broad Institute, Metabolomics Platform, Cambridge, Massachusetts, 2142, US; 104 - Cleveland Clinic, Immunity and Immunology, Cleveland, Ohio, 44195, US; 105 - University of Vermont, Burlington, Vermont, 5405, US; 106 - National Jewish Health, Denver, Colorado, 80206, US; 107 - University of Michigan, Internal Medicine, Ann Arbor, Michigan, 48109, US; 108 - Vitalant Research Institute, San Francisco, California, 94118, US; 109 - University of Illinois at Chicago, Chicago, Illinois, 60607, US; 110 - University of Chicago, Chicago, Illinois, 60637, US; 111 - Mayo Clinic, Health Quantitative Sciences Research, Rochester, Minnesota, 55905, US; 112 - Washington University in St Louis, Department of Medicine, Cardiovascular Division, St. Louis, Missouri, 63110, US; 113 - Vanderbilt University, Nashville, Tennessee, 37235, US; 114 - University of Cincinnati, Cincinnati, Ohio, 45220, US; 115 - Baylor College of Medicine Human Genome Sequencing Center, Houston, Texas, 77030; 116 - University of North Carolina, Chapel Hill, North Carolina, 27599, US; 117 - Washington University in St Louis, Genetics, St Louis, Missouri, 63110, US; 118 - Brown University, Providence, Rhode Island, 2912, US; 119 - Harvard University, Channing Division of Network Medicine, Cambridge, Massachusetts, 2138, US; 120 - Massachusetts General Hospital, Boston, Massachusetts, 2114, US; 121 - National Jewish Health, Center for Genes, Environment and Health, Denver, Colorado, 80206, US; 122 - University of Texas Health at Houston, Houston, Texas, 77225, US; 123 - Washington University in St Louis, St Louis, Missouri, 63130, US; 124 - Fred Hutchinson Cancer Research Center, Seattle, Washington, 98109, US; 125 - New York Genome Center, New York City, New York, 10013, US; 126 - Icahn School of Medicine at Mount Sinai, New York, New York, 10029, US; 127 - University of Pittsburgh, Pittsburgh, Pennsylvania, US; 128 - Beth Israel Deaconess Medical Center, Boston, Massachusetts, 2215, US; 129 - Boston Children's Hospital, Harvard Medical School, Department of Psychiatry, Boston, Massachusetts, 2115, US; 130 - University of Colorado Anschutz Medical Campus, Aurora, Colorado, 80045, US; 131 - Mass General Brigham, Obstetrics and Gynecology, Boston, Massachusetts, 2115, US; 132 - University of Mississippi, Cardiology, Jackson, Mississippi, 39216, US; 133 - University of Calgary, Medicine, Calgary, CA; 134 - University of Maryland, Genetics, Philadelphia, Pennsylvania, 19104, US; 135 - Yale University, Department of Chronic Disease Epidemiology, New Haven, Connecticut, 6520, US; 136 - Tulane University, New Orleans, Louisiana, 70118, US; 137 - University of Washington, Epidemiology, Seattle, Washington, 98195, US; 138 - Wake Forest Baptist Health, Winston-Salem, North Carolina, 27157, US; 139 - Brigham & Women's Hospital, Channing Division of Network Medicine, Boston, Massachusetts, 2115, US; 140 - University of Iowa, Iowa City, Iowa, 52242, US; 141 - National Health Research Institute Taiwan, Institute of Population Health Sciences, NHRI, Miaoli County, 350, TW; 142 - Tri-Service General Hospital National Defense Medical Center, TW; 143 - Blood Works Northwest, Seattle, Washington, 98104, US; 144 - Taichung Veterans General Hospital Taiwan, Taichung City, 407, TW; 145 - Oklahoma State University Medical Center, Internal Medicine, DIvision of Endocrinology, Diabetes and Metabolism, Columbus, Ohio, 43210, US; 146 - National Heart, Lung, and Blood Institute, National Institutes of Health, NHLBI, Bethesda, Maryland, 20892, US; 147 - Blood Works Northwest, Research Institute, Seattle, Washington, 98104, US; 148 - University of Michigan, Biostatistics, Ann Arbor, Michigan, 48109, US; 149 - Albert Einstein College of Medicine, New York, New York, 10461, US; 150 - University of California, San Francisco, San Francisco, California, 94118, US; 151 - Harvard University, Cambridge, Massachusetts, 2138, US; 152 - McGill University, Montréal, QC H3A 0G4, CA; 153 - University of Colorado at Denver, Epidemiology, Aurora, Colorado, 80045, US; 154 - Blood Works Northwest, Medicine, Seattle, Washington, 98104, US; 155 - Loyola University, Public Health Sciences, Maywood, Illinois, 60153, US; 156 - Harvard School of Public Health, Biostats, Boston, Massachusetts, 2115, US; 157 - Boston University, University of Massachusetts Chan Medical School, Worcester, Massachusetts, 1655, US; 158 - Brown University, Epidemiology and Medicine, Providence, Rhode Island, 2912, US; 159 - Duke University, Cardiology, Durham, North Carolina, 27708, US; 160 - Stanford University, Cardiovascular Institute, Stanford, California, 94305, US; 161 - Icahn School of Medicine at Mount Sinai, The Charles Bronfman Institute for Personalized Medicine, New York, New York, 10029, US; 162 - Boston University, Boston, Massachusetts, 2215, US; 163 - Ohio State University, Division of Pulmonary, Critical Care and Sleep Medicine, Columbus, Ohio, 43210, US; 164 - University of Texas Rio Grande Valley School of Medicine, Brownsville, Texas, 78520, US; 165 - University of Alabama, University of Alabama at Birmingham, Birmingham, Alabama, 35487, US; 166 – University of Washington, Genome Sciences, Seattle, Washington, 98195, US; 167 - RTI International, US; 168 - University of Arizona, Tucson, Arizona, 85721, US; 169 - Stanford University, Center For Sleep Sciences and Medicine, Palo Alto, California, 94304, US; 170 - National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, Maryland, 20892, US; 171 - Oklahoma Medical Research Foundation, Genes and Human Disease, Oklahoma City, Oklahoma, 73104, US; 172 - Howard University, Washington, District of Columbia, 20059, US; 173 - University of Washington, Department of Genome Sciences, Seattle, Washington, 98195, US; 174 - University at Buffalo, Buffalo, New York, 14260, US; 175 - University of Pennsylvania, Division of Sleep Medicine/Department of Medicine, Philadelphia, Pennsylvania, 19104-3403, US; 176 - Stanford University, Stanford Cardiovascular Institute, Stanford, California, 94305, US; 177 - University of Minnesota, Minneapolis, Minnesota, 55455, US; 178 - RTI International, Biostatistics and Epidemiology Division, Research Triangle Park, North Carolina, 27709-2194, US; 179 - University of Texas Rio Grande Valley School of Medicine, Edinburg, Texas, 78539, US; 180 - Fred Hutchinson Cancer Research Center, Fred Hutch and UW, Seattle, Washington, 98109, US; 181 - Johns Hopkins University, Cardiology/Medicine, Baltimore, Maryland, 21218, US; 182 - University of Colorado at Denver, Medicine, Denver, Colorado, 80204, US; 183 - University of Colorado at Denver, CCPM, Denver, Colorado, 80045, US; 184 - Northwestern University, Chicago, Illinois, 60208, US; 185 - New York Genome Center, New York Genome Center, New York City, New York, 10013, US; 186 - University of Ottawa, Sleep Research Unit, University of Ottawa Institute for Mental Health Research, Ottawa, ON K1Z 7K4, CA; 187 - Vanderbilt University, Medicine, Pharmacology, Biomedicla Informatics, Nashville, Tennessee, 37235, US; 188 - Universidade de Sao Paulo, Faculdade de Medicina, Sao Paulo, 1310000, BR; 189 - Columbia University, New York, New York, 10027, US; 190 - University of Maryland, Pathology, Seattle, Washington, 98195, US; 191 - Lundquist Institute, TGPS, Torrance, California, 90502, US; 192 - Harvard University, Division of Hematology/Oncology, Boston, Massachusetts, 2115, US; 193 - Harvard Medical School, Genetics, Boston, Massachusetts, 2115, US; 194 - Harvard Medical School, Boston, Massachusetts, 2115, US; 195 - Université Laval, Quebec City, G1V 0A6, CA; 196 - Emory University, Pediatrics, Atlanta, Georgia, 30307, US; 197 - Emory University, Human Genetics, Atlanta, Georgia, 30322, US; 198 - Vanderbilt University, Medicine/Cardiology, Nashville, Tennessee, 37235, US; 199 - UMass Memorial Medical Center, Worcester, Massachusetts, 1655, US; 200 - University of Saskatchewan, Saskatoon, SK S7N 5C9, CA; 201 - University of Michigan; 202 - Wake Forest Baptist Health, Biostatistical Sciences, Winston-Salem, North Carolina, 27157, US; 203 - University of Colorado at Denver, Genomic Cardiology, Aurora, Colorado, 80045, US; 204 - Brigham & Women's Hospital, Channing Department of Medicine, Boston, Massachusetts, 2115, US; 205 - Stanford University, Genetics, Stanford, California, 94305, US; 206 - University of Washington, University of Washington, Department of Genome Sciences, Seattle, Washington, 98195, US; 207 - Fred Hutchinson Cancer Research Center, Cancer Prevention Division of Public Health Sciences, Seattle, Washington, 98109, US; 208 - University of Pennsylvania, Genetics, Philadelphia, Pennsylvania, 19104, US; 209 - University of Alabama, Biostatistics, Birmingham, Alabama, 35487, US; 210 - University of Washington, Department of Biostatistics, Seattle, Washington, 98195, US; 211 - University of Vermont, Pathology & Laboratory Medicine, Burlington, Vermont, 5405, US; 212 - University of Southern California, USC Methylation Characterization Center, University of Southern California, California, 90033, US; 213 - Brigham & Women's Hospital, Mass General Brigham, Boston, Massachusetts, 2115, US; 214 - University of Michigan, US; 215 - Brigham & Women's Hospital, Channing Division of Network Medicine, Department of Medicine, Boston, Massachusetts, 2115, US; 216 - Indiana University, Epidemiology, Indianapolis, Indiana, 46202, US; 217 - Henry Ford Health System, Detroit, Michigan, 48202, US; 218 - University of Pittsburgh, Medicine, Pittsburgh, Pennsylvania, 15260, US; 219 - Case Western Reserve University, Department of Population and Quantitative Health Sciences, Cleveland, Ohio, 44106, US; 220 - University of California, San Francisco, US
TOPMed Lipids Working Group
Gonçalo Abecasis56, Donna K. Arnett10, Stella Aslibekyan67, Tim Assimes68, Elizabeth Atkinson57, Christie Ballantyne70, Wei Bao140, Amber Beitelshees61, Romit Bhattacharya57, Larry Bielak12, Joshua Bis13, Corneliu Bodea120, Eric Boerwinkle7,16, Donald W. Bowden17, Jennifer Brody13, Brian Cade4,18,19, Sarah Calvo57, Jenna Carlson79, I-Shou Chang102, Yii-Der Ida Chen99, So Mi Cho57, Seung Hoan Choi57, Ren-Hua Chung102, Adolfo Correa21, L. Adrienne Cupples22,23, Coleen Damcott61, Paul de Vries7, Ana F. Diallo221, Ron Do126, Jacqueline Dron57, Amanda Elliott57, Hilary Finucane57, Caitlin Floyd222, Mao Fu61, Andrea Ganna57, Dawei Gong61, Sarah Graham107, Mary Haas57, Bernhard Haring223, Jiang He136, Scott Heemann222, Blanca Himes224, James Hixson122, Marguerite Ryan Irvin67, Gail Jarvik62, Jicai Jiang61, Roby Joehanes194, Paule Valery Joseph64, Goo Jun122, Rita Kalyani27, Masahiro Kanai57, Sharon Kardia56, Sekar Kathiresan225, Amit Khera57, Sumeet Khetarpal57, Derek Klarin57, Charles Kooperberg28, Satoshi Koyama57, Brian Kral29, Leslie Lange30, Cathy Laurie62, Rozenn Lemaitre13, Zilin Li1,2, Xihao Li1, Changwei Li136, Xihong Lin1,4,53, Yingchang Lu113, Michael Mahaney164, Ani Manichaikul32, Lisa Martin32, Rasika Mathias27, Ravi Mathur167, Stephen McGarvey118, John McLenithan61, Julie Mikulla77, Amy Miller222, Braxton D. Mitchell36,37, May E. Montasser38, Vamsi Mootha57, Andrew Moran73, Alanna C. Morrison7, Tetsushi Nakao57, Pradeep Natarajan3,4,5, Kari North116, Jeff O’Connell36, Christopher O’Donnell77, Nicholette Palmer17, Kaavya Paruchuri57, Aniruddh Patel57, Gina Peloso22,23, James Perry61, Ulrike Peters180, Mary Pettinger124, Patricia Peyser12, James Pirruccello120, Toni Pollin61, Michael Preuss126, Bruce Psaty13,40,41, Susan Redline18,19,43, Robert Reed61, Alex Reiner28,40, Stephen Rich30, Samantha Rosenthal127, Jerome Rotter26, Margaret Sunitha Selvaraj3,4,5, Wayne Hui-Heng Sheu144, Jennifer Smith12,45, Tamar Sofer89, Adrienne M. Stilp62, Shamil R. Sunyaev94, Ida Surakka56, Carole Sztalryd61, Hua Tang205, Kent D. Taylor26, Mark Trinder57, Michael Tsai177, Md Mesbah Uddin57, Sarah Urbut57, Eric Van Buren1, Marie Verbanck126, Ann Von Holle116, Heming Wang213, Yuxuan Wang22, Kerri Wiggins62, John Wilkins184, Cristen Willer50,51,52, James Wilson49, Brooke Wolford56, Huichun Xu61, Lisa Yanek27, Zhi Yu57, Norann Zaghloul61, Seyedeh Maryam Zekavat57, Jingwen Zhang1, Ying Zhou124
221 - Virginia Commonwealth University, Richmond, Virginia, US; 222 - Westat, Atlanta, Georgia, US; 223 - Saarland University Medical Center, Homburg, Germany; 224 - Department of Biostatistics, Epidemiology and Informatics, University of Pennsylvania, Philadelphia, Pennsylvania, US; 225 - Verve Therapeutics, Cambridge, Massachusetts, US
Footnotes
A full list of consortium members appears at the Supplementary Note.
Competing interests
S.M.G. is now an employee of Regeneron Genetics Center. J.B.M. is an Academic Associate for Quest Diagnostics R&D. For B.D.M.: The Amish Research Program receives partial support from Regeneron Pharmaceuticals. M.E.M. reports grant from Regeneron Pharmaceutical unrelated to the present work. B.M.P. serves on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson. L.M.R. is a consultant for the TOPMed Admistrative Coordinating Center (through Westat). For S.R.: Jazz Pharma, Eli Lilly, Apnimed, unrelated to the present work. The spouse of C.J.W. works at Regeneron Pharmaceuticals. P.N. reports investigator-initiated grants from Amgen, Apple, AstraZeneca, Boston Scientific, and Novartis, personal fees from Apple, AstraZeneca, Blackstone Life Sciences, Foresite Labs, Novartis, Roche / Genentech, is a co-founder of TenSixteen Bio, is a shareholder of geneXwell and TenSixteen Bio, and spousal employment at Vertex, all unrelated to the present work. X. Lin is a consultant of AbbVie Pharmaceuticals and Verily Life Sciences. The remaining authors declare no competing interests.
Code availability
STAARpipeline is implemented as an open-source R package available at https://github.com/xihaoli/STAARpipeline56 and https://content.sph.harvard.edu/xlin/software.html. STAARpipelineSummary is implemented as an open-source R package available at https://github.com/xihaoli/STAARpipelineSummary57 and https://content.sph.harvard.edu/xlin/software.html. The scripts used to generate the results have been archived on Zenodo using https://doi.org/10.5281/zenodo.687140858.
Data analysis was performed in R (3.6.1). STAAR v0.9.6, STAARpipeline v0.9.6, and STAARpipelineSummary v0.9.6 were used in simulation and real data analysis, seqMeta v1.6.7 was used in simulation. Wget v1.14 was used for downloading the annotation data. FAVORannotator v1.0.0 (https://github.com/xihaoli/STAARpipeline-Tutorial) was used for functionally annotate the whole-genome data.
Data availability
This paper used the TOPMed Freeze 5 Whole Genome Sequencing data and phenotype data of lipids, C-reactive protein, estimated glomerular filtration rate, fasting glucose, fasting insulin and telomere length. The genotype and phenotype data are both available in dbGAP. The TOPMed data were from the following fourteen studies, where the accession numbers are provided in parenthesis: Framingham Heart Study (phs000974.v1.p1), Old Order Amish (phs000956.v1.p1), Jackson Heart Study (phs000964.v1.p1), Multi-Ethnic Study of Atherosclerosis (phs001416.v1.p1), Genome-wide Association Study of Adiposity in Samoans (phs000972) and Women’s Health Initiative (phs001237), Atherosclerosis Risk in Communities Study (phs001211), Cleveland Family Study (phs000954), Cardiovascular Health Study (phs001368), Diabetes Heart Study (phs001412), Genetic Study of Atherosclerosis Risk (phs001218), Genetic Epidemiology Network of Arteriopathy (phs001345), Genetics of Lipid Lowering Drugs and Diet Network (phs001359) and San Antonio Family Heart Study (phs001215).
The functional annotation data are publicly available and were downloaded from the following links: GRCh38 CADD v1.4 (https://cadd.gs.washington.edu/download), ANNOVAR dbNSFP v3.3a (https://annovar.openbioinformatics.org/en/latest/user-guide/download), LINSIGHT (https://github.com/CshlSiepelLab/LINSIGHT), FATHMM-XF (http://fathmm.biocompute.org.uk/fathmm-xf), CAGE (https://fantom.gsc.riken.jp/5/data), GeneHancer (https://www.genecards.org), and Umap/Bismap (https://bismap.hoffmanlab.org). In addition, recombination rate and nucleotide diversity were obtained from Gazal et al55. The tissue-specific functional annotations were downloaded from ENCODE (https://www.encodeproject.org/report/?type=Experiment). The assembled functional annotation data from these sources are available at http://favor.genohub.org.
References
- 1.Manolio TA et al. Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wainschtein P et al. Assessing the contribution of rare variants to complex trait heritability from whole-genome sequence data. Nature Genetics 54, 263–273 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hernandez RD et al. Ultrarare variants drive substantial cis heritability of human gene expression. Nature genetics 51, 1349–1355 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taliun D et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flannick J et al. Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature 570, 71–76 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Hout CV et al. Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature 586, 749–756 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang F & Lupski JR Non-coding genetic variants in human disease. Human molecular genetics 24, R102–R110 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khurana E et al. Role of non-coding sequence variants in cancer. Nature Reviews Genetics 17, 93–108 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Lee PH et al. Principles and methods of in-silico prioritization of non-coding regulatory variants. Human genetics 137, 15–30 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moore JE et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bansal V, Libiger O, Torkamani A & Schork NJ Statistical analysis strategies for association studies involving rare variants. Nature Reviews Genetics 11, 773 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee S, Abecasis GR, Boehnke M & Lin X Rare-variant association analysis: study designs and statistical tests. The American Journal of Human Genetics 95, 5–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kiezun A et al. Exome sequencing and the genetic basis of complex traits. Nature genetics 44, 623 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li B & Leal SM Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. The American Journal of Human Genetics 83, 311–321 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu MC et al. Rare-variant association testing for sequencing data with the sequence kernel association test. The American Journal of Human Genetics 89, 82–93 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X et al. Dynamic incorporation of multiple in silico functional annotations empowers rare variant association analysis of large whole-genome sequencing studies at scale. Nature genetics 52, 969–983 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morrison AC et al. Practical approaches for whole-genome sequence analysis of heart-and blood-related traits. The American Journal of Human Genetics 100, 205–215 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Z et al. Dynamic scan procedure for detecting rare-variant association regions in whole-genome sequencing studies. The American Journal of Human Genetics 104, 802–814 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He Z, Xu B, Buxbaum J & Ionita-Laza I A genome-wide scan statistic framework for whole-genome sequence data analysis. Nature communications 10, 1–11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Natarajan P et al. Deep-coverage whole genome sequences and blood lipids among 16,324 individuals. Nature communications 9, 1–12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Z, Liu Y & Lin X Simultaneous Detection of Signal Regions Using Quadratic Scan Statistics With Applications to Whole Genome Association Studies. Journal of the American Statistical Association 117, 823–834 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bocher O & Génin E Rare variant association testing in the non-coding genome. Human Genetics 139, 1345–1362 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Fishilevich S et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database 2017. (2017). [DOI] [PMC free article] [PubMed]
- 25.Fantom Consortium A promoter-level mammalian expression atlas. Nature 507, 462 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andersson R et al. An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Breslow NE & Clayton DG Approximate inference in generalized linear mixed models. Journal of the American statistical Association 88, 9–25 (1993). [Google Scholar]
- 28.Chen H et al. Control for population structure and relatedness for binary traits in genetic association studies via logistic mixed models. The American Journal of Human Genetics 98, 653–666 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen H et al. Efficient variant set mixed model association tests for continuous and binary traits in large-scale whole-genome sequencing studies. The American Journal of Human Genetics (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou H, Arapoglou T, Li X, Li Z & Lin X, Edn. V1 (Harvard Dataverse, 2022).
- 31.Harrow J et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome research 22, 1760–1774 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frankish A et al. GENCODE reference annotation for the human and mouse genomes. Nucleic acids research 47, D766–D773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kinsella RJ et al. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database 2011. (2011). [DOI] [PMC free article] [PubMed]
- 34.Povysil G et al. Rare-variant collapsing analyses for complex traits: guidelines and applications. Nature Reviews Genetics 20, 747–759 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Kircher M et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nature genetics 46, 310 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang Y-F, Gulko B & Siepel A Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nature genetics 49, 618–624 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rogers MF et al. FATHMM-XF: accurate prediction of pathogenic point mutations via extended features. Bioinformatics 34, 511–513 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Y et al. ACAT: a fast and powerful p value combination method for rare-variant analysis in sequencing studies. The American Journal of Human Genetics 104, 410–421 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buniello A et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic acids research 47, D1005–D1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stilp AM et al. A System for Phenotype Harmonization in the National Heart, Lung, and Blood Institute Trans-Omics for Precision Medicine (TOPMed) Program. American Journal of Epidemiology (2021). [DOI] [PMC free article] [PubMed]
- 41.Moutsianas L et al. The power of gene-based rare variant methods to detect disease-associated variation and test hypotheses about complex disease. PLoS genetics 11, e1005165 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raffield LM et al. Allelic heterogeneity at the CRP locus identified by whole-genome sequencing in multi-ancestry cohorts. The American Journal of Human Genetics 106, 112–120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin BM et al. Whole genome sequence analyses of eGFR in 23,732 people representing multiple ancestries in the NHLBI trans-omics for precision medicine (TOPMed) consortium. EBioMedicine 63, 103157 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DiCorpo D et al. Whole Genome Sequence Association Analysis of Fasting Glucose and Fasting Insulin Levels in Diverse Cohorts from the NHLBI TOPMed Program. medRxiv, 2020.2012. 2031.20234310 (2021). [DOI] [PMC free article] [PubMed]
- 45.Taub MA et al. Genetic determinants of telomere length from 109,122 ancestrally diverse whole-genome sequences in TOPMed. Cell Genomics 2, 100084 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schaffner SF et al. Calibrating a coalescent simulation of human genome sequence variation. Genome research 15, 1576–1583 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee S, Wu MC & Lin X Optimal tests for rare variant effects in sequencing association studies. Biostatistics 13, 762–775 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zaidi AA & Mathieson I Demographic history mediates the effect of stratification on polygenic scores. Elife 9, e61548 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gogarten SM et al. Genetic association testing using the GENESIS R/Bioconductor package. Bioinformatics 35, 5346–5348 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zheng X & Davis JW SAIGEgds—an efficient statistical tool for large-scale PheWAS with mixed models. Bioinformatics (2020). [DOI] [PubMed]
- 51.Peloso GM et al. Association of low-frequency and rare coding-sequence variants with blood lipids and coronary heart disease in 56,000 whites and blacks. The American Journal of Human Genetics 94, 223–232 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moon S, Lee Y, Won S & Lee J Multiple genotype–phenotype association study reveals intronic variant pair on SIDT2 associated with metabolic syndrome in a Korean population. Human genomics 12, 1–10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dong C et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Human molecular genetics 24, 2125–2137 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references
- 54.Chen H et al. Efficient variant set mixed model association tests for continuous and binary traits in large-scale whole-genome sequencing studies. The American Journal of Human Genetics 104, 260–274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gazal S et al. Linkage disequilibrium–dependent architecture of human complex traits shows action of negative selection. Nature Genetics 49, 1421–1427 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li X & Li Z xihaoli/STAARpipeline: STAARpipeline_v0.9.6 Version 0.9.6 10.5281/zenodo.6871504 (2022). [DOI]
- 57.Li X & Li Z xihaoli/STAARpipelineSummary: STAARpipelineSummary_v0.9.6 Version 0.9.6 10.5281/zenodo.6871524 (2022). [DOI]
- 58.Li X & Li Z xihaoli/STAARpipeline-Tutorial: v0.9.6 Version 0.9.6 10.5281/zenodo.6871408 (2022). [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper used the TOPMed Freeze 5 Whole Genome Sequencing data and phenotype data of lipids, C-reactive protein, estimated glomerular filtration rate, fasting glucose, fasting insulin and telomere length. The genotype and phenotype data are both available in dbGAP. The TOPMed data were from the following fourteen studies, where the accession numbers are provided in parenthesis: Framingham Heart Study (phs000974.v1.p1), Old Order Amish (phs000956.v1.p1), Jackson Heart Study (phs000964.v1.p1), Multi-Ethnic Study of Atherosclerosis (phs001416.v1.p1), Genome-wide Association Study of Adiposity in Samoans (phs000972) and Women’s Health Initiative (phs001237), Atherosclerosis Risk in Communities Study (phs001211), Cleveland Family Study (phs000954), Cardiovascular Health Study (phs001368), Diabetes Heart Study (phs001412), Genetic Study of Atherosclerosis Risk (phs001218), Genetic Epidemiology Network of Arteriopathy (phs001345), Genetics of Lipid Lowering Drugs and Diet Network (phs001359) and San Antonio Family Heart Study (phs001215).
The functional annotation data are publicly available and were downloaded from the following links: GRCh38 CADD v1.4 (https://cadd.gs.washington.edu/download), ANNOVAR dbNSFP v3.3a (https://annovar.openbioinformatics.org/en/latest/user-guide/download), LINSIGHT (https://github.com/CshlSiepelLab/LINSIGHT), FATHMM-XF (http://fathmm.biocompute.org.uk/fathmm-xf), CAGE (https://fantom.gsc.riken.jp/5/data), GeneHancer (https://www.genecards.org), and Umap/Bismap (https://bismap.hoffmanlab.org). In addition, recombination rate and nucleotide diversity were obtained from Gazal et al55. The tissue-specific functional annotations were downloaded from ENCODE (https://www.encodeproject.org/report/?type=Experiment). The assembled functional annotation data from these sources are available at http://favor.genohub.org.