

Abstract
Background
μ‐opioid receptor agonists (MORAs) are indispensable for analgesia in bladder cancer (BC) patients, both during surgery and for chronic pain treatment. Whether MORAs affect BC progression and metastasis remains largely unknown. This study focused on the effects of MORAs on the formation of circulating tumor cells (CTCs) in BC and aimed to provide potential therapeutic targets, which would retain the pain‐relieving effects of MORAs in BC patients without sacrificing their long‐term prognosis.
Methods
Different preclinical models were used to identify the effects of MORAs on the progression of BC. A novel immunocapture microfluidic chip was utilized to analyze whether MORAs affected the number of CTCs in mouse models and clinical BC patients. Bioinformatic analyses, total transcriptome sequencing, and molecular biology methods were then used to investigate the underlying mechanisms in these models and in BC cell lines.
Results
Mouse models of hematogenous metastasis and in situ BC demonstrated that tumor metastasis was significantly increased after MORA treatment. A significant increase in the number of mesenchymal and/or epithelial CTCs was detected after MORA treatment in both the mouse models and clinical trial patients. Mechanistically, MORAs facilitated the formation of CTCs by activating the MOR/PI3K/AKT/Slug signaling pathway, hereby promoting the epithelial‐mesenchymal transition (EMT) of BC cells, as knockdown of MOR, Slug or blockade of PI3K inhibited the EMT process and CTC formation.
Conclusion
MORAs promoted BC metastasis by facilitating CTC formation. The EMT‐CTC axis could be targeted for preventive measures during MORA treatment to inhibit the associated tumor metastasis or recurrence in BC patients.
Keywords: μ‐opioid receptor agonist, AKT, bladder cancer, circulating tumor cell, epithelial‐mesenchymal transition, microfluidic chip, MOR, PI3K, Slug
Abbreviations
- BC
bladder cancer
- MOR
μ‐opioid receptor
- MORA
μ‐opioid receptor agonist
- MOR‐KD
μ‐opioid receptor knockdown
- EMT
epithelial‐mesenchymal transition
- CTC
circulating tumor cell
- MH
morphine hydrochloride
- NS
normal saline
- IVI
in vivo imaging
- IVIS
in vivo imaging system
- PBS
phosphate buffer
- SEM
scanning electron microscope
- HE
hematoxylin and eosin
- WB
Western blotting
- IHC
immunohistochemistry
- TF
transcription factor
- TCGA
the cancer genome atlas
- EpCAM
epithelial cell adhesion molecule
- CSV
cell surface vimentin
- RCT
randomized controlled trial
- GA
general anesthesia
- GA+E
general‐epidural anesthesia
- VAS
visual analogue scale
- SD
standard deviation
- GO
Gene Ontology
- GSEA
Gene Set Enrichment Analysis
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- KD
knockdown
- OV
overexpression
- OS
Overall survival
- RFS
Recurrence‐free survival
- Ctrl
Control
- E‐CTC
Epithelial circulating tumor cell
- M‐CTC
Mesenchymal circulating tumor cell
- GPCR
G protein‐coupled receptor
- SDI
Size Dictated Immunocapture
- RARC
Robot‐assisted laparoscopic radical cystectomy
- MB49‐Luc
luciferase‐labeled MB49
- RT‐qPCR
Real Time‐ quantitative Polymerase Chain Reaction
- shRNA
short hairpin RNA
- siRNA
small interfering RNA
- CONSORT
Consolidated Standards of Reporting Trials
1. BACKGROUND
Cancer remains a major human health concern worldwide. The latest data from the American Cancer Society show that the United States of America had 1,898,160 new cases and 608,570 cancer‐related deaths in 2020 [1, 2]. Bladder cancer (BC) is among the most prevalent cancers [3], with 573,278 new cases and 212,536 new deaths reported in 2020 worldwide, ranking 6th and 13th in yearly cancer incidence and mortality rates in males, respectively [3, 4, 5]. Data show that the five‐year survival rate for BC patients with distant metastasis is merely 6%, which is much lower than many other cancers [1].
The primary therapy for BC patients today is oncologic surgery [6]. Opioid agonists, especially μ‐opioid receptor agonists (MORAs), are indispensable for analgesia and sedation during and after surgery. As the most prevalent analgesics, MORAs are also commonly used for chronic pain treatment worldwide [7]. They are necessary for pain relief in terminal BC cases and are used in high doses long‐term in these patients [7]. In recent years, the possibility that MORAs impact tumor behavior and cancer patients’ prognosis has been a hot topic and introduces a new dimension [8, 9, 10]. Clinical studies have uncovered a possible causal link between the use of MORAs and reduced resistance to multiple cancers, including BC [11, 12, 13]. Population‐based case‐control studies and meta‐analyses reported that consumption of opium, whose major active ingredient is morphine, is significantly associated with an increased incidence of BC, making it an important risk factor [14]. Guerrero Orriach et al. [15] demonstrated that BC surgery patients receiving opioids and inhalational anesthetics exhibited a decrease in disease‐free survival compared to those receiving propofol and local anesthesia. However, these studies did not investigate whether these effects are caused by MORAs per se, highlighting the importance of studies that directly demonstrate the effects of MORAs on BC metastasis, especially in clinically relevant models. Moreover, the underlying mechanisms through which MORAs promote minimal residual disease to progress to clinical metastasis need to be elucidated.
With the advent of liquid biopsy, the novel finding of circulating tumor cells (CTCs), which are cells that have detached from the primary tumor, entering circulation and potentially initiating sites of metastasis, have recently been identified as a particularly promising indicator for predicting tumor recurrence and metastasis [16, 17]. It has been reported that CTC formation and survival are influenced and regulated by multiple factors, such as the epithelial‐mesenchymal transition (EMT) process, surgery and chemotherapy [18]. Among these factors, EMT plays a vital role not only in promoting the genesis of CTCs from tumor sites, but also in increasing the survival and metastatic capability of CTCs in circulation [18]. Although studies have shown that CTCs are independently associated with increased tumor metastasis and reduced overall survival (OS) [19], the extraordinary rarity and the lack of required sensitivity and selectivity to isolate multiple types of CTCs (such as epithelial CTCs, mesenchymal CTCs, double positive CTCs and CTC clusters) from a high background of blood cells, make their isolation challenging and limits clinical utility and popularization of CTC detection.
Recently, our team has developed a novel immunocapture microfluidic chip (Size Dictated Immunocapture Chip, SDI‐Chip) to detect CTCs in various cancers based on the principle of Deterministic Lateral Displacement [20, 21]. This SDI‐Chip selectively enhances the interaction of CTCs with an antibody‐coated micropillar array, resulting in efficient capture of CTCs while minimizing nonspecific interaction of blood cells. In addition to this selectively enhanced interaction, the micropillar array of the SDI‐Chip was optimized to enable extended duration of contact of CTCs with antibody‐coated micropillars, and spatial resolution of CTC capture based on antigen expression levels have also been optimized. Therefore, CTCs with different antigen expression levels can be efficiently captured and spatially resolved around micropillars [20, 22]. The order of magnitude is approximately 100 CTC counts/mL blood in patients with BC, colorectal cancer, and hepatocellular carcinoma [22, 23, 24], making the SDI‐Chip a unique and especially suitable technology for efficient, sensitive and spatially resolved capture and detection of CTCs.
Since MORAs are so widely used in cancer patients for pain control, exploring the effects of MORAs on cancer progression and metastasis is of wide‐ranging impact and value. It is especially critical to investigate whether MORAs affect the formation of CTCs, which create metastatic tumors and impact recurrence. Therefore, we performed a comprehensive study, using both in vitro and in vivo approaches, as well as a clinical randomized controlled trial to investigate the connections among MORAs, CTCs and BC metastasis.
2. MATERIALS AND METHODS
2.1. Animal models
All animal studies were approved by the Animal Care and Use Committee of the Renji Hospital, Shanghai Jiaotong University School of Medicine, and all procedures were performed based on the Principles of Laboratory Animal Care formulated by the National Institutes of Health, as well as the guidelines of the Administrative Committee of Experimental Animal Care and Use of Shanghai.
Adult C57BL/6J female mice (6‐8 weeks old, 18‐22 g) were used in all animal experiments and were provided by the Animal Research Center of the Shanghai Jiaotong University School of Medicine (Shanghai, China). All mice were housed in a specific pathogen‐free animal facility under controlled environmental conditions of 22 ± 2°C and a 12 h light‐dark cycle. Mice were fed a standard lab chow and sacrificed by pentobarbital sodium 30 mg/kg intraperitoneal injection.
For the hematogenous metastasis mouse model, 1 × 106 MB49‐Luc cells per mouse were injected into the bloodstream via the tail vein. Mice were then randomly divided into a morphine hydrochloride (MH)‐treated group or a normal saline (NS) control group (n = 11 per group). After cell injection, mice in the MH group received daily intraperitoneal MH injections (10 mg/kg, dissolved in NS) for 14 days, and the Control (Ctrl) group received the same daily volume of NS vehicle. On the 7th, 14th and 21st day, in vivo imaging (IVI) was performed, and the mice were sacrificed on Day 42 post‐cell injection. Tissues were collected for subsequent experiments.
For the in situ BC mouse model, a previously published protocol was used and improved upon [25]. To minimize the possibility of BC cells entering the bloodstream through injured vessels caused by hydrochloric acid, sodium hydroxide, or trypsin, we did not pre‐treat the mouse bladders, and 1 × 106 MB49 cells were injected directly into the bladder. Mice were then randomly divided into an MH‐treated or NS control group (n = 9 per group). Mice received identical volumes of MH or NS as in the hematogenous metastasis model above, only that dosing began on Day 8 post‐injection, after the establishment of orthotopic BC, and continued for 14 days until Day 21 post‐cell injection. Mice were sacrificed on Day 35 post‐cell injection, and their blood and tissues were collected.
For the subcutaneous tumor model, 5 × 105 MB49 cells suspended in 100 μL NS were injected subcutaneously into the right upper armpit of C57BL/6 mice. Mice were then randomly divided into an MH‐treated or NS control group (n = 5 per group) and treated from Day 8 post‐injection until Day 21 as above, receiving identical volumes as above.
To explore the effects of Slug and MOR on tumor growth and/or morphine‐associated CTC formation, separate groups of mice were subcutaneously injected in the right upper armpit with either MB49 cells, Slug‐knockdown (si‐Slug) cells or MOR‐ knockdown (MOR‐KD) MB49 cells, and each group was then randomized again into MH‐treated or NS‐treated groups (n = 6‐12 per group). Mice in the MH, si‐Slug + MH and MOR‐KD + MH groups received intraperitoneal MH injections (10 mg/kg) for 14 days from Day 8 post cell injection until Day 21. Mice were sacrificed on Day 21 or 22 for ethical reasons due to limitations in tumor size. Blood and tissue samples were collected as needed.
2.2. Cell culture
T24 and 5637 human BC cell lines, MB49 and luciferase‐labeled MB49 (MB49‐Luc) mouse BC cell lines were obtained from the FuHeng Cell Center (Shanghai, China) and authenticated by short tandem repeat tests before cell culture. All cell lines were cultured in high glucose Dulbecco's modified Eagle's medium (DMEM, Gibco, Jenks, Oklahoma, USA) supplemented with 10% fetal bovine serum (FBS, Gibco) and 50 U/mL penicillin and 50 μg/mL streptomycin (Yeasen, Shanghai, China) in a 37°C humidified incubator containing 5% CO2. An additional 5 μg/mL puromycin (Yeasen) was added into the medium for MB49‐Luc cells. When cells reached 80%‐90% confluency, they were detached with trypsin‐EDTA (0.25%; Yeasen) and subcultured or used for subsequent experiments.
2.3. Western blot analysis
Protein was isolated from cells and tumor tissues (weight = 0.1 g) using a RIPA buffer (Beyotime, Shanghai, China) containing 1% protease inhibitor cocktail (Sigma‐Aldrich, Merck KGaA, Darmstadt, Germany) and 1% phosphatase inhibitor (Beyotime). Protein concentrations were detected using the Bicinchoninic Acid Protein Assay kit (Vazyme, Nanjing, China). Samples (15 μg) were loaded onto 4%‐20% or 10% polyacrylamide gels and transferred to polyvinylidene fluoride membranes (EMD Millipore, Massachusetts, USA) using a semi‐dry Trans‐Blot Cell (Bio‐Rad Laboratories, California, USA). Membranes were blocked with QuickBlock™ Blocking Buffer (Beyotime) for 1 h at room temperature and incubated overnight at 4°C with the following primary antibodies: GAPDH (1:2000; Cat# 2118, Cell Signaling Technologies, Boston, MA, USA), β‐Tublin (1:2000; Cat# 2146, Cell Signaling Technologies), E‐cadherin (1:1000; Cat# 3195, Cell Signaling Technologies), N‐cadherin (1:1000; Cat# 13116, Cell Signaling Technologies), Vimentin (1:1000; Cat# 5741, Cell Signaling Technologies), Slug (1:1000; Cat# 9585, Cell Signaling Technologies), PI3K (1:1000; Cat# 4257, Cell Signaling Technologies), p‐PI3K (p85 [Tyr458]/p55 [Tyr199], 1:1000; Cat# 17366, Cell Signaling Technologies), AKT (1:1000; Cat# 4691, Cell Signaling Technologies), p‐AKT (Ser473, 1:1000; Cat# 9271, Cell Signaling Technologies), CK19 (1:1000; Cat# ab52625, Abcam, Boston, MA, USA) and MOR (1:1000; Cat# ab134054, Abcam,). After incubation with an HRP‐conjugated secondary antibody (1:3000; Cat# A0208, Beyotime) for 1 h at room temperature, bands were detected using super enhanced chemiluminescent reagents (Cat# 36208ES60, Yeasen) and exposed to x‐ray film (Bio‐Rad Laboratories, California, USA).
2.4. RNA isolation and Real Time‐ quantitative Polymerase Chain Reaction (RT‐qPCR) analysis
Total RNA was isolated with the EZ‐press RNA Purification Kit (EZ Bioscience, Roseville, MN, USA) according to the manufacturer's protocol. RNA (0.5 μg) was then reverse transcribed using the PrimeScriptTM RT‐PCR kit (Takara, Beijing, China). 2 × SYBR Green qPCR Master Mix (EZ Bioscience) was used for RT‐qPCR and reaction conditions were as follows: Pre‐denaturation at 95°C for 5 min, then 40 cycles of 95°C for 10 sec and 60°C for 30 sec. Ct values were calculated using the 2−ΔΔCt method. Primer sequences are listed in Supplementary Table S1.
2.5. Immunofluorescent staining
Cells were seeded into 12‐well plates covered with cell slides made of high‐quality glass (WHB‐12‐CS, WHB scientific, Shanghai, China) at a density of 2 × 104 cells/well and treated with morphine hydrochloride (MH) for 24 h. Next, cell slides were fixed with 4% paraformaldehyde (Servicebio, Wuhan, China) for 20 min and treated with 0.1% Triton X‐100 (P0096, Beyotime) for 10 min. Then, cell slides were blocked with 3% bovine serum albumin (Beyotime) in phosphate buffer (PBS; Corning, New York, USA) for 1 h at room temperature and incubated with primary antibody (MOR, 1:400, Cat# ab134054, Abcam; CK19, 1:400, Cat# ab52625, Abcam; N‐cadherin, 1:400, Cat# 13116, Cell Signaling Technologies; Vimentin, 1:400, Cat# 5741, Cell Signaling Technologies; Slug, 1:400, Cat# 9585, Cell Signaling Technologies) overnight at 4°C. Finally, slides were incubated for 1 h with Cy3‐conjugated secondary antibody (Beyotime), and then with DAPI (Vector, Newark, USA) for 10 min at room temperature. Representative fields were imaged with a laser confocal microscope (Model IX83, Olympus, Tokyo, Japan).
2.6. F‐actin cytoskeleton staining and scanning electron microscope (SEM) imaging
Cytoskeleton changes were detected by F‐actin staining. T24 cells (5 × 104 cells/well) were seeded into 12‐well plates covered with cell slides made of high‐quality glass (WHB scientific) and treated with MH for 24 h. After fixation with 4% paraformaldehyde, cell slides were stained with FITC‐conjugated phalloidin (Yeasen) for 30 min and DAPI (Vector) for 10 min at room temperature. For F‐actin staining, tumors were frozen in OCT (Servicebio) embedding medium, and 20 μm sections were cut sequentially and mounted on superfrost plus slides. Slices were permeabilized in 3% PBST and stained with FITC‐conjugated phalloidin for 30 min and DAPI for 10 min at room temperature. Images were acquired with a laser confocal microscope (Model IX83, Olympus).
SEM was performed with a Hitachi Regulus SU8100 field emission scope using an accelerating voltage of 3 kV. T24 cells were seeded in 6‐well plates (1 × 105 cells/well) and fixed with an electron microscope fixative (Servicebio) for 24 h at 4°C. Cell morphology was observed and representative pictures were acquired during SEM imaging.
2.7. Wound healing assay
Cells were seeded into 12‐well culture plates (1 × 105 cells/well). After reaching a confluent monolayer, a straight scratch was created with a 200 μL size pipet tip. Reference points were marked at the bottom of the cell culture plates. Cell debris was then washed away with phosphate buffer (Gibco), fresh high glucose DMEM (Gibco) was added to the wells, and healing was observed. Images of the scratch were captured under a phase‐contrast microscope at specific time points (0 h, 24 h, and 36 h). Three microscope images were taken of each well at 0 h, 24 h, and 36 h, respectively, and the distance of cell migration was measured for statistical analysis. Percent wound healing was calculated to reflect the migration activity of the sample: Percentage of wound healing = [(Blank area at 0 h) – (Blank area at 24 h)]/(Blank area at 0 h) × 100%.
2.8. Transwell migration and invasion assays
Cell migration and invasion ability were determined using Transwell chambers with an 8‐μm pore size (Corning) and Matrigel (BD Bioscience, Shanghai, China). Cells were seeded at a 1 × 105/mL density into the upper chambers and treated with MH or DMEM. For migration assays, cells (1 × 105/mL) and 200 μL DMEM without FBS were added to the upper chamber, and for invasion assays, the upper chambers were pre‐treated with 0.5 mg/L Matrigel for 30 min to form a membrane. 600 μL DMEM (with 10% FBS) was added to the lower chambers. Plates were incubated for 24 h and chambers were fixed with 4% paraformaldehyde for 20 min and stained with 0.5% crystal violet for 20 min. For both assays, 3 random fields per chamber were counted using the Image J 1.51 software (National Institutes of Health, Washington, USA) and averages were calculated to reflect the migration or invasion activity for each sample. All experiments were performed in triplicate. Digital images were acquired with an Olympus fluorescent microscope using its bright field function.
2.9. Live 2‐dimensional (2D) cell migration assay
The migration ability of T24 cells was monitored in real‐time and recorded using a laser confocal microscope. For live‐cell culture imaging, cells were seeded into 6‐cm dishes with glass bottoms for 12 h. Next, dishes were placed in a live cell culture system (Tokai Hit, Shizuoka, Japan) equipped with a humidified environmental chamber and maintained at 37°C with 5% CO2. Media were changed to a serum‐free medium containing either MH or saline before placing the dishes into the chamber. The temperature of the objective lens was also maintained at 37°C, and the airflow rate was 5 L/min. Three fields in different directions within each dish were chosen and imaged sequentially every 15 min for a total of 96 cycles, overall 24 h were recorded. Cells that remained within the frame throughout the observation period were selected for analysis. Adobe Premier software (version: CS4, Adobe Systems Incorporation, Beijing, China) was used to generate video files, and Image J was used to process and analyze the images.
2.10. RNAi transfection assay
Slug expression was knocked down by transfecting cells with small interfering RNA (Slug siRNA). 2 × 106 cells were plated in 6‐well plates and transfected at 80%‐90% confluency with 20 μmol/L Slug siRNA or non‐targeting control siRNA using the Lipo8000 transfection reagent (Beyotime) according to the manufacturer's instructions. After 24 h, transfection complexes were removed and cells were incubated with DMEM/10% FBS for an additional 48 h. Cells were then harvested and screened for siRNA knockdown by RT‐qPCR and Western blot. siRNA was synthesized by Sangon Biotech (Shanghai, China) and the sequences were as follows: si‐Slug‐sense 5’‐CAUUAGUGAUGAAGAGGAATT‐3’ and si‐Slug‐antisense 5’‐UUCCUCUUCAUCACUAAUGGG‐3’ for T24 cells; si‐Slug‐sense 5’‐CCUUGUGUCUGCAAGAUCUTT‐3’ and si‐Slug‐antisense 5’‐AGAUCUUGCA GACACAAGGTT‐3’ for MB49 cells. For the in vivo experiments, si‐Slug (10 nmol in 0.1 mL of NS per tumor nodule) was injected into tumor masses twice weekly starting on Day 8 post subcutaneous cancer cell injection (when tumor size reached approximately 5 × 5 × 5 mm3).
2.11. Lentiviral vector transduction to generate stable MOR knockdown
Lentiviral plasmids containing a short hairpin RNA (shRNA) for the gene that encodes MOR (OPRM1) and a negative control were purchased from Zorin Biological Technology (Zorin Biological Technology, Shanghai, China). The sequence of shRNA‐MOR was: Forward 5’‐ GATCCGGATCCTCTCTTCTGCCATTG CTTCCTGTCAGACAATGGCAGAAGAGAGGATCCTTTTTG ‐3’ and Reverse 5’‐ AATTCAAAAAGGATCCTCTCTTCTGCCATTGTCTGACAGGAAG CAATGGCAGAAGAGAGGATCCG ‐3’. Before lentiviral vector transduction, 2 × 106 MB49 cells were seeded into 6‐well plates, and transduction was performed when cells were 30%‐50% confluent. Briefly, negative control or OPRM1‐Lentivirus (multiplicity of infection = 20 for both) and 8 μg/ml Polybrene (Beyotime) were added to MB49 cells together into fresh DMEM + 10% FBS. After incubation for 24 h, infected cells were cultured in a selection medium containing 3 μg/mL puromycin. Stably silenced OPRM1 MB49 cells (MOR‐KD MB49 cell) were then maintained in DMEM with 0.6 μg/mL puromycin.
2.12. Hematoxylin and eosin (HE), immunohistochemistry (IHC) staining and tissue fixation
Paraffin sections (5 μm) were rehydrated in graded ethanols and stained with an HE Staining Kit (Servicebio) according to the manufacturer's instructions. After permeabilization with xylene, sections were sealed with neutral resin and in situ tumors and metastases were detected under an Olympus fluorescence microscope.
Expression levels of epithelial‐mesenchymal transition (EMT)‐related markers, which included CK‐19 (1:400; Cat# ab52625, Abcam), E‐cadherin (1:200; Cat# 3195, Cell Signaling Technologies), N‐cadherin (1:400; Cat# 13116, Cell Signaling Technologies) and Vimentin (1:400; Cat# 5741, Cell Signaling Technologies), and expression levels of Slug (1:200; Cat# 9585, Cell Signaling Technologies) and p‐AKT (Ser473; 1:400; Cat# 9271, Cell Signaling Technologies) were analyzed by IHC staining. Paraffin sections of tumor tissues (5 μm) were incubated with primary antibodies overnight at 4°C. Next, sections were incubated with an anti‐rabbit‐HRP polymer (Beyotime) for 1 h, followed by incubation with DAB (Beyotime) for 20 min. Results were examined by two independent pathologists, who evaluated and scored the slides based on the intensity and extent of staining observed under a microscope. A final score between 0 and 12 was calculated by multiplying the positivity rate and intensity scores.
Liver, bladder and tumor samples were fixed with 4% paraformaldehyde. To observe lung metastases more clearly, all lung samples were first fixed in Bouin's fixative (Servicebio) for 24 h. Next, tissues were moved into 70% ethanol to dehydrate for 24 h. Then, metastases were verified both by visual observation during necropsy and HE staining.
2.13. IVI system (IVIS) imaging
Tumor metastases of the hematogenous metastasis mouse model were monitored using an IVIS Lumina III In Vivo Imaging System (PerkinElmer, Shanghai, China) and images were processed by the Living Image software (PerkinElmer). Before imaging, mice were injected intraperitoneally with 150 mg/kg firefly d‐Luciferin potassium salt (Yeasen). After 15 minutes, mice were anesthetized with 2% sevoflurane (Hengrui Medicine Co., Ltd., Lianyungang, China) for induction and 1% sevoflurane for maintenance during bioluminescence imaging. The measured temperature for the IVIS camera was ‐90°C, luminescent exposure time was 60 seconds, and imaging was performed in automatic mode.
2.14. RNA sequencing (RNA‐seq)
MB49 cells were seeded into 6‐well plates (2 × 105 cells/well) and cultured for 24 h. At 80% confluency, medium was replaced with serum‐free DMEM containing 10 μmol/L MH or normal saline. After 24 h, cells were lysed in Trizol Reagent (Invitrogen, California, USA) and total RNA was extracted for mRNA sequencing. Concentrations and RNA integrity were verified before library preparation. Library preparation and sequencing were performed by the Biomarker Technologies Corporation, Beijing, China. Sequencing was performed on an HiSeq2500 instrument (Illumina, California, USA) with 150 bp paired‐end reads, and all sequencing results were analyzed by two professional bioinformatics engineers.
2.15. Bioinformatic analysis
Correlations of the overall survival of BC patients and EMT‐transcription factor levels (EMT‐TFs) were analyzed using the Kaplan Meier plotter (http://kmplot.com/analysis/) [26]. Data from 404 BC patients from The Cancer Genome Atlas (TCGA) dataset were used to generate the Kaplan‐Meier survival curves. To explore potential interactions among Slug, AKT and EMT pathways, Biovista was used as a visual exploration tool (https://www.biovista.com/vizit/). The most relevant connections for genes or pathways shown are based on known datasets and published data. Moreover, Kyoto Encyclopedia of Genes and Genomes (KEGG), Gene Ontology (GO) enrichment analysis and Gene Set Enrichment Analysis (GSEA) were used for pathway enrichments.
2.16. CTC capture, detection, and analysis
CTC capture chips were produced and supplied by the Wisdom Healthy Biotech company and the Institute of Molecular Medicine, Renji Hospital, Jiaotong University School of Medicine, Shanghai. It is a novel, highly efficient and highly selective immunocapture microfluidic chip, which has been verified and improved upon for multiple applications [20]. More details can be found on their website http://www.yang‐lab.com/. To capture epithelial and mesenchymal CTCs, the verified markers epithelial cell adhesion molecule (EpCAM) and cell surface vimentin (CSV) were used, respectively [24].
Raw CTC capture chips were pre‐treated with 100 μL biotinylated antibody against EpCAM (10 μg/mL, Cat# BAF960, R&D system, Minneapolis, USA) and biotinylated antibody against CSV (10 μg/mL, Cat# H00007431‐M08, Abnova, Taiwan, China) for 1 h at room temperature. Then, 0.5‐1 mL of whole blood was injected slowly through the middle inlet with a syringe pump at a flow rate of 0.33 mL/h. The other two inlets were injected with PBS at the same flow rate. After injection, chips were washed with 0.1% PBST and incubated with 3% goat serum for 30 min at room temperature. Then, chips were incubated with 70 μL of fluorescently‐labeled secondary antibody mixtures (EpCAM‐FITC, 1:10, Cat# A15755, Thermo Fisher, Massachusetts, USA; CD45‐PE, 1:20, Cat# ab134202, Abcam; CSV‐APC, 1:200, Cat# H00007431‐MA08, Abnova; DAPI, 1:40, Cat# 62248, Thermo Fisher) for 1 h at room temperature. Finally, full images of the different channels (bright channel, FITC channel, PE channel, APC channel and DAPI channel) were imaged and merged. Captured CTCs were identified and counted by using the NIS‐Elements Viewer 4.50 software (Nikon, Tokyo, Japan). Cells with EpCAM+/CD45−/DAPI+ or CSV+/CD45−/DAPI+ fluorescence and displaying a clear cell type in bright field microscopy were identified as CTCs. The whole process of CTC capture and profiling is shown in Supplementary Figure S1.
2.17. Clinical randomized controlled trial (RCT) of the number of CTCs in patients undergoing BC surgery
The NCT04358718 clinical trial was conducted from June 2020 to January 2021 at the Renji Hospital, which is affiliated with the Shanghai Jiaotong University School of Medicine. This study was approved by the Renji Hospital Ethics Committee (KY2019‐192). Written informed consents were obtained from all patients or authorized family members. The trial complied with the Helsinki Declaration and the Consolidated Standards of Reporting Trials (CONSORT) statement.
Patients receiving elective robot‐assisted laparoscopic radical cystectomy (RARC) due to primary stage T2a or higher BC were eligible for the study and were randomly assigned via a computer‐generated random numbers table to one of the two groups: a general anesthesia (GA) group or a combined general‐epidural anesthesia (GA + E) group. Patients in the GA group received regular GA, which utilized MORAs (sufentanil and remifentanil) for pain relief during surgery and sufentanil for postoperative pain control. Patients in the GA + E group received a combined epidural‐GA, where both intra‐ and postoperative pain relief was provided by epidural ropivacaine. Only a very small amount of MORA was used during anesthesia induction in the GA + E group (see supplemental methods). Primary outcomes observed were the number of CTCs 3 days post‐surgery. Secondary outcomes observed were the number of CTCs immediately after surgery and 1 month post‐surgery, and the postoperative visual analogue scale (VAS) scores at 24 h and 48 h after surgery. Detailed study design and procedures are supplied in the Supplementary Materials.
2.18. Statistical analysis
Statistical analyses were conducted using the IBM SPSS Statistics 26.0 software (SPSS Inc., Armonk, New York, USA) and GraphPad Prism 7 software. Categorical variables were presented as frequency (n) or proportion (%), while continuous variables were expressed as mean ± standard deviation (SD) or median (25% interquartile range, 75% interquartile range). Differences between two groups were analyzed with independent samples/paired student's t‐tests. Differences between four groups were analyzed by repeated measures/block randomized one‐way analysis (or two‐way analysis as needed) of variance, followed by post‐hoc analysis (Tukey's test) as appropriate. If the data did not meet normal distribution standards, non‐parametric tests followed by a Kruskal‐Wallis test were utilized for comparisons. Categorical variables were compared using the χ2 test with the Yates correction or Fisher's exact test (when the total sample was < 40 or the expected frequency was < 1). Survival curves were created using Kaplan‐Meier survival analysis with log‐rank t‐test. Pearson correlation analysis and regression analysis models were used to explore the correlation between CTC counts and tumor size. All statistical tests were two‐sided with P values < 0.05 considered statistically significant.
3. RESULTS
3.1. MORAs promoted BC metastasis and facilitated CTC formation
Results of our hematogenous metastasis BC mouse model (Figure 1A) show that after 42 days, mice in the MH group developed more severe lung metastases than the control (Ctrl) group, as indicated by the appearance of lung tumor granules being joined together in the MH group, and lungs of the MH treated group exhibited significantly higher lung weights and larger areas of tumor tissue than the Ctrl group (Figure 1B). Results also show that continuous intraperitoneal morphine treatment in these mice promoted macroscopic metastases in other organs, all of which were confirmed by histopathological examination (Supplementary Figure S2A, Supplementary Table S2). IVI revealed that tissue metastasis occurred much earlier in mice from the MH group compared to the Ctrl group (Figure 1C‐D), accompanied by a significantly lower overall survival rate (P < 0.001) in MORA‐treated mice (Figure 1E).
FIGURE 1.
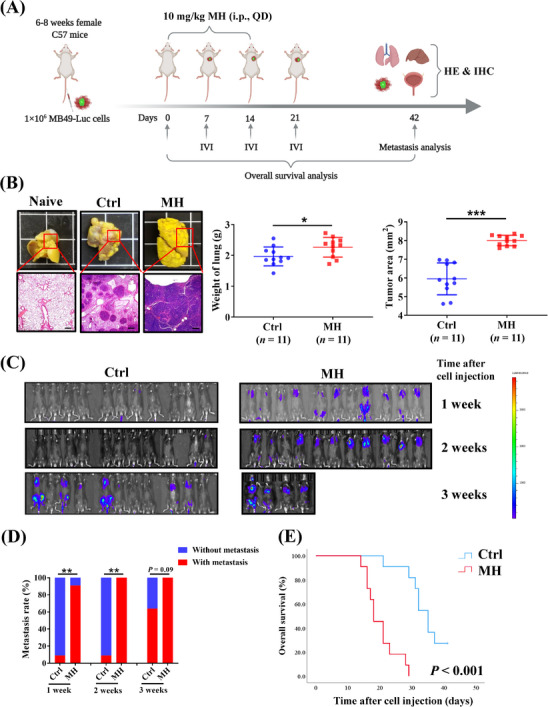
MORA treatment promotes bladder cancer cell metastasis in a hematogenous metastasis mouse model, (A) Study diagram: adult C57BL/6J female mice were injected with luciferase‐expressing MB49‐luc cells into the vein tail (Day 0). 10 mg/kg MH or normal saline were injected intraperitoneally daily for 14 days (Days 1‐14), (B) Lung metastases were more extensive in the MH treated group (scale bar = 100 μm), (C) IVI indicated that tumor metastases occurred visibly earlier in mice from the MH group than the Ctrl group, (D) Comparison of cumulative metastasis rates at indicated time points, (E) Survival analysis of the two groups showed a significantly lower overall survival rate in MH‐treated mice. A‐E: n = 11 per group, mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001, Abbreviations: MORA, μ‐opioid receptor agonist; MH, morphine hydrochloride; Ctrl, normal saline controls; QD, once a day; HE, hematoxylin and eosin; IHC, immunohistochemistry; IVI, in vivo live imaging; SD, standard deviation.
Next, the effects of a MORA on tumor metastasis were further investigated in an in situ BC mouse model (Figure 2A, Supplementary Figure S2B). As shown in Supplementary Figure S2C, significantly more bladder tumors formed in the MH group compared to the Ctrl group (7 [77.8%] vs. 3 [33.3%]). Concordantly, the average weights and volumes of bladders were significantly higher in mice from the MH group than in those from the Ctrl group (Supplementary Figure S2C). Moreover, mice from the MH group developed more numerous macroscopic lung metastases that were also larger in size compared to those from the Ctrl group (Supplementary Figure S2D).
FIGURE 2.
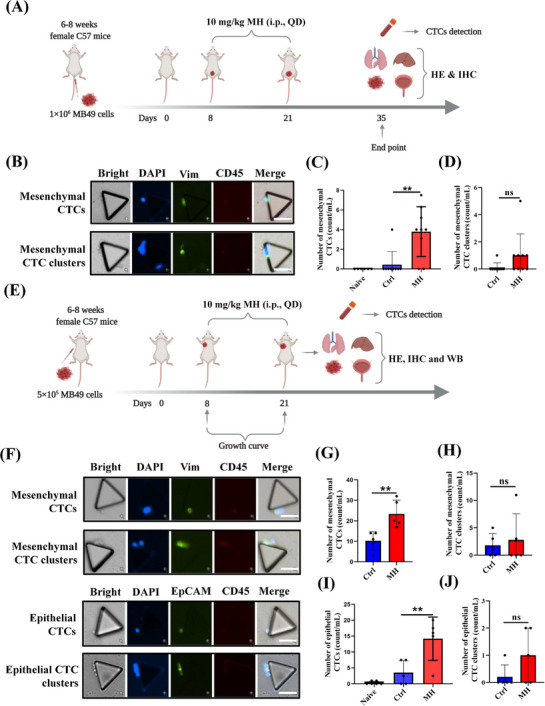
MORA treatment increases generation of mesenchymal and epithelial CTCs in murine BC models, (A) Study diagram: MB49 cells were injected into mouse bladders to establish BC in situ BC. 10 mg/kg of MH or normal saline was injected intraperitoneally daily for 14 days (Days 8‐21), (B‐D) Representative images of mesenchymal CTCs and CTC clusters detected by microfluidic chip in mice with in situ BC (B). The number of mesenchymal CTCs (C) and mesenchymal CTC clusters (D) were increased in the MH group. The former reached statistical significance, the latter showed a trend (B‐D: n = 6 for naïve, n = 9 each in the Ctrl and MH groups, mean ± SD, scale bar = 50 μm), (E) Study diagram: MB49 cells were injected subcutaneously into the right upper “arm” pit of mice to establish a subcutaneous BC model. 10 mg/kg of MH or normal saline were injected intraperitoneally daily for 14 days (Days 8‐21), (F‐J) Representative images of mesenchymal CTCs and CTC clusters, and epithelial CTCs and CTC clusters, detected by microfluidic chip in mice with subcutaneous BC (F). Quantification of mesenchymal CTCs (G), mesenchymal CTC clusters (H), epithelial CTCs (I), and epithelial CTC clusters (J) in mice with subcutaneous BC. (F‐J: n = 6 for naïve, n = 5 each in the Ctrl and MH groups, mean ± SD, scale bar = 50 μm). **P < 0.01; ns, not significant. naïve, no tumor cell injection and no vehicle treatment, Abbreviations: MORA, μ‐opioid receptor agonist; CTC, circulating tumor cell; BC, bladder cancer; MH, morphine hydrochloride; Ctrl, normal saline controls; QD, once a day; HE, hematoxylin and eosin; IHC, immunohistochemistry; WB, western blot; SD, standard deviation.
It is now widely accepted that CTCs play an important role in tumor metastasis and recurrence [16]. CTCs can be classified into two subgroups: epithelial (E‐CTC) and mesenchymal (M‐CTC); and some CTCs co‐express both epithelial and mesenchymal markers and can be classified as both E‐CTC and M‐CTC. Recent studies indicate that M‐CTCs are associated with a stronger metastatic potential compared to E‐CTCs [27]. Therefore, we next analyzed the effects of MH treatment on M‐CTC formation in tumors of the in situ BC mouse model by using the SDI‐Chip previously reported by our group [20]. The number of M‐CTCs were significantly higher in the MH group than in the Ctrl group, and M‐CTC clusters were also increased in the MH group compared to those in the Ctrl group, although it did not reach statistical significance (Figure 2B‐D). We also generated a subcutaneous BC mouse model (Figure 2E, Supplementary Figure S2E), and validated that the number of M‐CTCs was significantly higher in the MH group, and the M‐CTC clusters displayed an increase in the trend in the MH group but remained statistically similar between the two groups (Figure 2F‐H). Moreover, morphine treatment also significantly increased the number of E‐CTCs,but not E‐CTC clusters, in this model (Figure 2F, I and J). These findings suggest that MORA treatment facilitated CTC formation, which could be a reason why MORA treatment accelerates BC metastasis.
3.2. MORA treatment promoted cell deformation, migration, and EMT
We first confirmed that MOR was expressed in both the MB49 and T24 bladder carcinoma cell lines (Supplementary Figure S3A‐B). To explore how MORAs facilitate CTC formation, we then cultured MB49 cells with MH and performed an RNA‐seq (Figure 3A‐D, Supplementary Figure S3C‐D). GO enrichment analysis revealed that cell adhesion, developmental process, cell locomotion and cell junction pathways were significantly enriched after MH treatment (Figures 3A‐B, Supplementary Figure S3D). GSEA also demonstrated an enrichment of the biological processes of EMT, cell morphogenesis, and cell adhesion and migration‐related signaling pathways (Figure 3C‐D, Supplementary Figure S3E). Since it is well‐known that EMT contributes to the generation of CTCs, we hypothesized that the MORA‐induced EMT process facilitated CTC formation and therefore promoted tumor metastasis.
FIGURE 3.
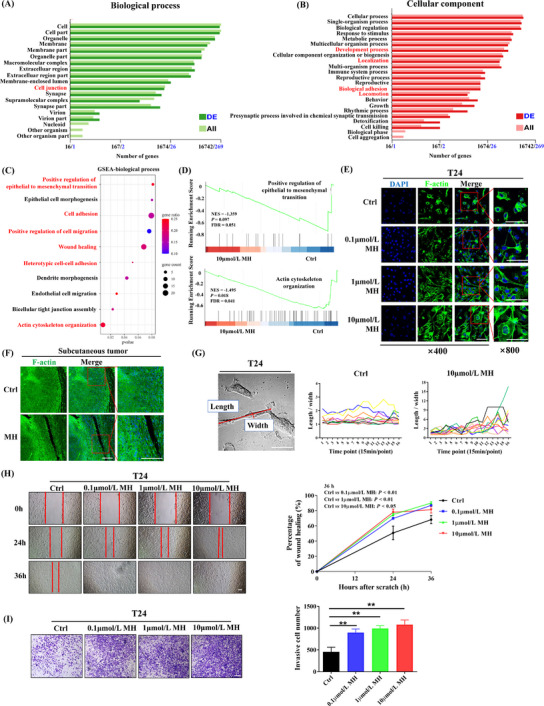
MORA treatment promotes EMT in BC cells, (A‐B) Map of significantly enriched biological processes (A) and cellular component pathway (B) enrichment analyses of RNA‐seq data from MH‐treated or Ctrl MB49 BC cells. The black numbers along the x‐axis are the coordinates of total genes, and the blue numbers are the coordinates of differentially expressed genes, (C) GSEA in MB49 cells showed that EMT, cell morphogenesis, cell adhesion and cell migration‐related signaling pathways were significantly enriched after MH treatment. (A‐C, n = 3 per group), (D) GSEA showing a positive association between epithelial to mesenchymal transition and actin cytoskeleton organization, (E) F‐actin staining in T24 BC cells. Cytoskeleton transformation into a mesenchymal phenotype was observed after MH treatment (E, n = 3 per group; left three panels scale bar = 100 μm), (F) F‐actin staining in subcutaneous tumors showed an elongated phenotype in the MH group and a signet‐ring phenotype in the Ctrl group (F, n = 3 per group; left three panels scale bar = 100 μm), (G) T24 live‐cell imaging verified that cells underwent a remarkable phenotypic change to an elongated shape after MH treatment (G, n = 10 per group, scale bar = 50 μm). Time points on the x‐axis represent data collected for each 15‐minute interval, (H‐I) Migrative (H) and invasive abilities (I) of T24 cells were both significantly increased during MH treatment (H‐I, n = 3 per group; mean ± SD, scale bar = 100 μm). **P<0.01, Abbreviations: MORA, μ‐opioid receptor agonist; EMT, epithelial‐mesenchymal transition; BC, bladder cancer; Ctrl, control; GSEA, gene set enrichment analysis; MH, morphine hydrochloride; SD, standard deviation; DE, differential expression.
To confirm that hypothesis, we first microscopically examined morphological cell changes in MH‐treated T24 human bladder carcinoma cells and in the subcutaneous tumor tissues. F‐actin staining revealed cytoskeleton transformation of T24 cells to the characteristic spindly morphology of the mesenchymal phenotype after treatment with MH for 24 h, which became more pronounced with increased MH concentration (Figure 3E). Scanning electron microscope (SEM) imaging confirmed these cell morphology changes (Supplementary Figure S3F). Concordantly, F‐actin staining of the subcutaneous tumors showed an elongated phenotype in the MH‐treated group and a signet‐ring phenotype in tumors from the Ctrl group (Figure 3F). Live‐cell imaging of T24 cells under a laser confocal microscope further verified that the cells displayed remarkable and frequent phenotypic changes to an elongated shape after MH treatment (Figure 3G, Supplementary Figure S3G, and Supplementary Video S1‐2). To observe cell activity, we then evaluated cell migration and invasion ability. Exposure to MH resulted in significantly enhanced migration and invasion abilities (Figure 3H‐I).
Next, we measured changes in cell surface biomarkers to monitor EMT in vitro and in tumors. As shown in Supplementary Figure S4A‐D, MH treatment resulted in a significant decrease in protein expression of the epithelial markers E‐cadherin and CK19 and a significant increase in the mesenchymal markers N‐cadherin and Vimentin in three different BC cell lines (T24, MB49 and 5637). Analysis of the subcutaneous tumors and the in situ bladder tumors revealed a similar decrease in epithelial and increase in mesenchymal markers, verified with both Western blot analysis and IHC staining (Supplementary Figure S4E‐G). Together, these data provide convincing evidence, both in vitro and in vivo, that MORA treatment induced the EMT process in bladder cancer cells.
3.3. MORA facilitated EMT by upregulating the EMT‐transcription factor (TF) Slug
Previous studies have suggested that EMT‐TFs are key factors in mediating EMT in cancer [18]. Using the GO‐molecular function and KEGG pathway enrichment analysis, we uncovered that TF activity was significantly changed after MH treatment, and TF dysregulation contributed to the MH‐related biological events in MB49 cells (Supplementary Figure S5A‐B). Therefore, we next evaluated the mRNA expression levels of eight crucial EMT‐TFs in T24 and MB49 cells. RT‐qPCR analysis revealed that SLUG was the only TF that was significantly increased after MH treatment in both cell lines (Figure 4A‐B). We verified that Slug protein levels were also increased in both MH‐treated cell lines as well as in subcutaneous tumors of MH‐treated mice (Figure 4C‐D, Supplementary Figure S5C‐E).
FIGURE 4.
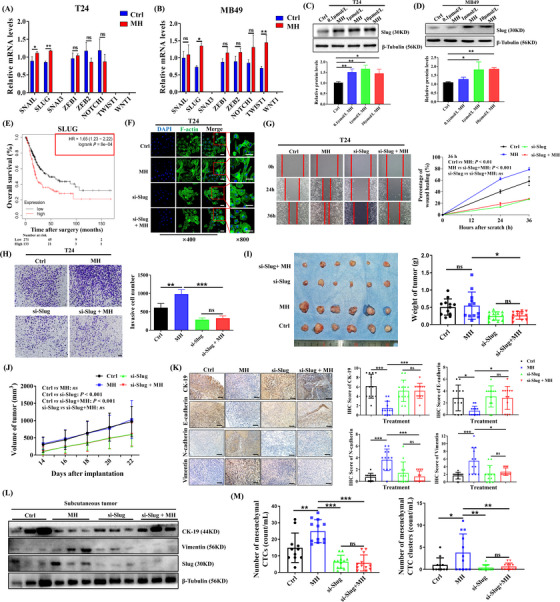
MORA promotes EMT in BC cells by upregulating the EMT‐TF Slug, (A‐B) mRNA expression patterns of eight EMT‐TFs in MH‐treated T24 cells (A) and MB49 cells (B). (A‐B, n = 3 per group; mean ± SD). Expression levels of SNAI3, TWIST1 and WNT1 were below level of detection in T24 cells. Expression levels of SNAI3 and WNT1 were below level of detection in MB49 cells, (C‐D) Western blots showing a significant upregulation of Slug protein expression in T24 (C) and MB49 cells (D) after MH treatment (C‐D, n = 3 per group; mean ± SD), (E) Kaplan‐Meier analysis from the TCGA‐BC dataset, examining the correlation between SLUG expression levels and overall BC patient survival, showing that higher SLUG levels were significantly associated with shorter overall patient survival, (F‐H) Slug knockdown in T24 cells significantly reversed MH‐induced morphological changes (F), increases in migrative (G), and increases in invasive abilities (H). (F‐H, n = 3 per group; mean ± SD, scale bar = 100 μm in left three panels of F and in G and H), (I‐J) Tumor growth was significantly inhibited in Slug‐deficient MB49 cell tumors, with or without MH treatment, (K) IHC staining showing inhibition of the morphine‐induced EMT process in Slug knockdown tumors (scale bar = 100 μm), (L) Western Blots of subcutaneous BC tumors showing reversal of MH‐induced changes of EMT markers when Slug was knocked down, (M) The mesenchymal CTC and CTC cluster increase seen in the MH‐treated group was significantly reduced in the absence of Slug. (n = 12 per group for I‐L, and n = 11‐12 per group for M) All data was shown as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001; ns, not significant, Abbreviations: MORA, μ‐opioid receptor agonist; EMT, epithelial‐mesenchymal transition; BC, bladder cancer; TF, transcription factor; MH, morphine hydrochloride; Ctrl, control; SD, standard deviation; IHC, immunohistochemistry; CTC, circulating tumor cell.
Furthermore, we then generated Kaplan‐Meier survival analyses from 404 BC patients of TCGA dataset. This dataset showed that among the 8 critical EMT‐TFs, SLUG was the only TF whose expression levels inversely correlated with patients’ long‐term survival (Figure 4E, Supplementary Figure S5F), leading us to the hypothesis that MORA treatment upregulated Slug, resulting in BC tumor progression.
To confirm this hypothesis in vitro, Slug was knocked down in T24 and MB49 cells with siRNA (Supplementary Figure S6A). As shown in Figure 4F‐H, Slug silencing remarkably reversed the morphological changes of these cells and reversed the increases in cell migration and invasion ability induced by MH treatment in T24 cells. We validated that expression levels of the mesenchymal markers, N‐cadherin and Vimentin, were significantly inhibited in Slug‐deficient cells (Supplementary Figure S6B). Then we injected mice subcutaneously with Slug‐knockdown MB49 cells to monitor tumor development in vivo and to observe the formation of CTCs. Tumor growth was significantly inhibited without Slug, even when treated with MH (Figure 4I‐J). Moreover, morphine‐induced downregulation of the epithelial markers CK19 and E‐cadherin and upregulation of the mesenchymal markers N‐cadherin and Vimentin were abolished in tumors formed by Slug‐deficient cells (Figure 4K‐L). Concordantly, the increase in the number of mesenchymal CTCs and CTC clusters seen in the MH group was significantly reduced in the absence of Slug, indicating that Slug plays a key role in MORA‐facilitated CTC formation (Figure 4M).
3.4. MORA upregulated Slug expression by activating the PI3K/AKT signaling pathway in a MOR‐dependent manner
Previous studies have shown that the PI3K/AKT signaling pathway regulates Slug expression in cancer and is involved in the EMT process [18, 28]. Using a visual exploration tool, we confirmed that Slug connected tightly with the AKT signaling pathway (Figure 5A). Activation of the AKT signaling pathway by MH was then analyzed both in vitro and in vivo. In vitro, T24 cells showed significant increases in phosphorylated PI3K (p‐PI3K)/PI3K and p‐AKT/AKT ratios when exposed directly to MH (Figure 5B). In vivo, the MH‐treated groups of all three tumor tissues (the in situ bladder tumors, the subcutaneous tumors and the metastatic lung tumors from the hematogenous model) showed increased expression of p‐AKT compared to the control groups (Ctrl; Figure 5C). In addition, when adding the PI3K inhibitor LY294002 to the T24 cell medium, MH‐induced activation of PI3K and AKT was significantly inhibited and not different from the control. The upregulation of Slug and the morphological changes and enhancement of cell migration and invasion ability that was observed previously with MH treatment were all abolished when PI3K inhibitor was added, indicating that PI3K signaling acted upstream of Slug and the EMT process (Figure 5D‐G, Supplementary Figure S6C‐D).
FIGURE 5.
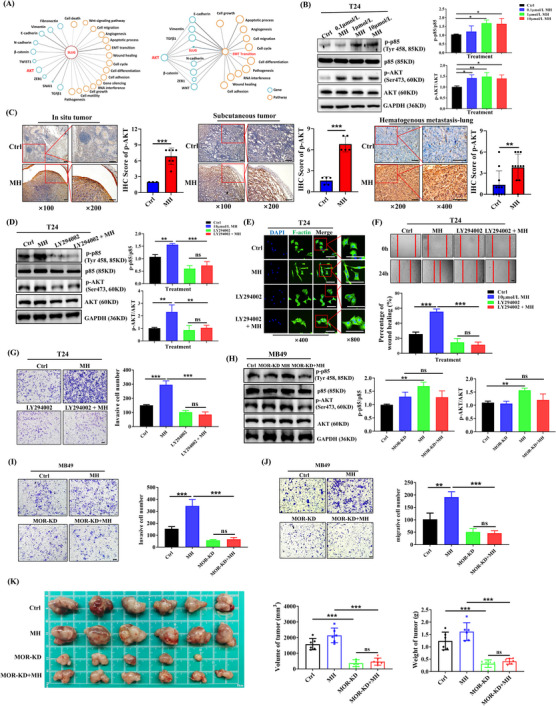
Activation of the MOR/PI3K/AKT pathway contributes to the EMT process induced by a MORA, (A) Correlations among Slug, AKT and EMT. By using the visiable tool Biovista, genes (blue circle) or pathways (orange circle) that connect tightly with each other are shown. Comprehensive relationships suggested that Slug connected tightly with the AKT signaling pathway and EMT‐related pathways, (B) Western blot showing activation of the PI3K/AKT pathway with MH treatment for 24 h in T24 cells (B, n = 3 per group; mean ± SD), (C) IHC staining of in situ bladder tumors, subcutaneous BC tumors and lung metastases showing significant upregulation of p‐AKT in the MH treated groups (C, n = 3‐11; left panels scale bar = 100 μm), (D‐G) PI3K inhibitor LY294002 significantly inhibited the MH‐induced activation of PI3K and AKT (D), transition to the mesenchymal phenotype (E), increase in cell migration (F), and increase in invasion ability (G) of T24 cells, (H) MOR‐KD MB49 cells did not show activation of PI3K/AKT signaling with MH treatment, (I‐J) MH‐induced increased cell migration and invasion were absent in MOR‐KD MB49 cells, (K) Tumor growth was significantly inhibited in MOR‐KD MB49 cell tumors, with or without MH treatment. (n = 3 per group in D‐J and n = 6 per group in K, mean ± SD, scale bar = 100 μm in left three panels of E, and in F, G, I and J) *P < 0.05, **P < 0.01, ***P < 0.001; ns, not significant, Abbreviations: MORA, μ‐opioid receptor agonist; EMT, epithelial‐mesenchymal transition; MH, morphine hydrochloride; Ctrl, control; BC, bladder cancer; IHC, immunohistochemistry; KD, knockdown; SD, standard deviation.
Studies have verified that G protein‐coupled receptor (GPCR) and the PI3K/AKT signaling pathway are connected [29]. Therefore, we constructed a MOR‐KD MB49 cell line (Supplementary Figure S6E) and investigated whether the effects of a MORA occurred in a MOR‐dependent manner. Compared to wild‐type MB49 cells, MOR‐KD MB49 cells did not show an activation of PI3K/AKT signaling with MH treatment (Figure 5H). The MH‐induced mesenchymal phenotype and increase in cell activity were also absent in these cells (Figure 5I‐J, Supplementary Figure S6F), showing that the MOR plays a fundamental role in MORA‐induced PI3K/AKT activation and EMT. The role of MOR on tumor growth and activation of the AKT signaling and EMT progress were also confirmed in vivo by generating subcutaneous tumors with MOR‐KD MB49 cells (Figure 5K, Supplementary Figure S6G).
3.5. Perioperative opioid treatment promoted CTC formation in BC patients undergoing surgery
Following our preclinical findings, we conducted a clinical study at the Shanghai Renji Hospital to evaluate whether perioperative opioid treatment affected the number of CTCs postoperatively. Between June 2020 and January 2021, 44 patients receiving elective RARC surgery due to primary BC were enrolled and randomly assigned to a GA group receiving peri‐ and postoperative MORA treatment (n = 22) or a combined general‐epidural anesthesia (GA + E) group receiving only a very small amount of MORA during anesthesia induction (n = 22) (Supplementary Figure S7). Comparisons of patients’ baseline characteristics, perioperative variables and postoperative complications did not show significant differences between the two groups, except for the MORA (sufentanil and remifentanil) treatment doses (Supplementary Table S3, Supplementary Table S4). Postoperative VAS scores were similarly low in both groups, indicating that both anesthesia methods provided effective pain relief in these patients (Supplementary Table S4). Blood samples were collected from patients of both groups at indicated time points (pre‐surgery, immediately after surgery, 3rd day post‐surgery, and 1‐month post‐surgery) for CTC detection using the immunocapture microfluidic chip. Utilizing EpCAM and cell surface vimentin (CSV) antibodies, 6 types of CTCs/ CTC clusters were identified, including single epithelial CTCs, single mesenchymal CTCs, double‐positive CTCs, single epithelial CTC clusters, single mesenchymal CTC clusters, and double‐positive CTC clusters, all of which were present in very low numbers in healthy controls (Supplementary Figure S8A‐B). No pre‐surgery differences in total epithelial CTCs (single epithelial CTCs + double‐positive CTCs + single epithelial CTC clusters + double‐positive CTC clusters), total mesenchymal CTCs (single mesenchymal CTCs + double‐positive CTCs + single mesenchymal CTC clusters + double‐positive CTC clusters), total double‐positive CTCs (double‐positive CTCs + double‐positive CTC clusters) and total CTCs (single epithelial CTCs + single mesenchymal CTCs + double‐positive CTCs + single epithelial CTC clusters + single mesenchymal CTC clusters + double‐positive CTC clusters), were observed between the two groups (Figure 6A‐D). All cell numbers, except the number of total double‐positive CTCs, remained similar between the two groups immediately after surgery; however, on the 3rd day post‐surgery, all cell numbers were significantly lower in the GA + E group compared to the GA group, and this effect was still observed at the 1 month after surgery time point (Figure 6A‐D). Notably, 3 days post‐surgery, the number of total CTC clusters (single epithelial CTC clusters + single mesenchymal CTC clusters + double‐positive CTC clusters), which are reported to possess significantly higher metastasis‐promoting capability than individual CTCs [30], were significantly lower in the GA + E group than in the GA group, and still showed a lower trend 1 month after surgery, although that difference did not reach statistical significance (Figure 6E). When comparing fold changes of the numbers of CTCs at the three postoperative time points to their respective values before surgery, the CTC decline trends in the GA + E group was significantly more pronounced than those in the GA group in most cases (Figure 6F).
FIGURE 6.
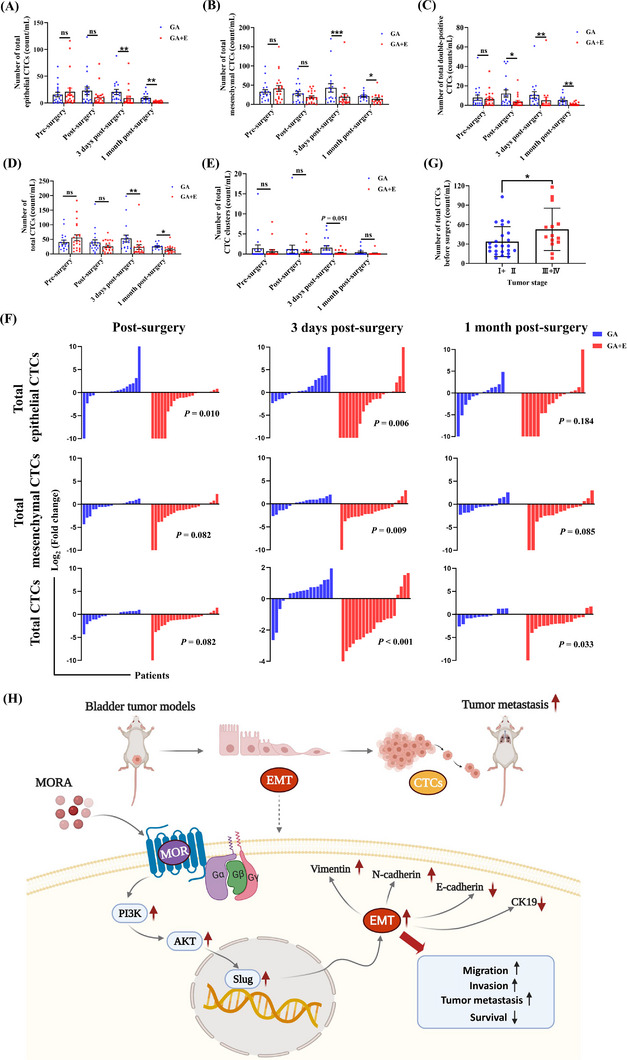
Perioperative opioid treatment promotes CTC formation in BC patients receiving robot‐assisted laparoscopic radical cystectomy, (A‐E) The numbers of total epithelial CTCs (A), total mesenchymal CTCs (B), total double‐positive CTCs (C), total CTCs (D), and total CTC clusters (E) in BC patients receiving surgery under GA or a combined GA + E, measured pre‐surgery, immediately after surgery, 3rd day post‐surgery, and 1 month post‐surgery, respectively. Patients in the GA + E group received only a very small amount of MORA during anesthesia induction, whereas patients in the GA group received continuous perioperative doses of MORAs, (F) Fold changes of the different types of CTCs at indicated time points. When CTC count = 0 pre‐surgery but > 0 at the other three time points, the Log2(fold change) was defined as 10. Similarly, when CTC count > 0 pre‐surgery but = 0 at the other three time points, the Log2(fold change) was defined as ‐10. Change trends of patients whose post‐surgery blood samples were missing are not shown. The decline trends of CTCs in the GA + E group were significantly more pronounced than those in the GA group in most cases, (G) Before surgery, patients with higher tumor stages (III and IV) had significantly more total CTCs compared to those diagnosed with lower tumor stages (I and II), (n = 13‐22 in A‐G, mean ± SD) *P < 0.05, **P < 0.01, ***P < 0.001; ns, not significant, (H) Schematic model showing the mechanisms how a MORA facilitates the formation of CTCs leading to increases in tumor metastases in BC, Abbreviations: CTC, circulating tumor cell; BC, bladder cancer; GA, general anesthesia; E, epidural anesthesia; MORA, μ‐opioid receptor agonist; MOR, μ‐opioid receptor; EMT, epithelial‐mesenchymal transition.
Even though there were fluctuations in the number of CTC counts at the time and immediately after surgery, as well as on the 3rd day post‐surgery, all CTC counts were lower 1 month post‐surgery than their corresponding preoperative values in both groups (Figure 6A‐E), verifying the effectiveness of surgery in removing tumor mass and thereby reducing CTC formation. We also confirmed that more CTCs were detected in patients diagnosed with higher tumor stages (III + IV) than in those with lower tumor stages (I + II) (Figure 6G). However, in contrast to previous studies [31], we found no correlation between CTC numbers and tumor size in our patients (Supplementary Figure S8C), which was also seen in our animal models (Supplementary Figure S8D), indicating more complex mechanisms for CTC formation.
4. DISCUSSION
This current study demonstrated that morphine, a representative of MORAs, promoted BC metastasis and facilitated CTC formation in vitro, in vivo and in a human clinical trial. Investigations into the underlying mechanisms revealed that MORA treatment significantly increased the EMT process via activating the PI3K/AKT/Slug signaling pathway in a MOR‐dependent manner, as knockdown of MOR, Slug or blockade of PI3K inhibited the EMT process and CTC formation (Figure 6H). Taking advantage of a novel, highly efficient and selective immunocapture microfluidic chip, the present study also greatly expands our current knowledge of the complex impact of MORAs on cancer progression by exploring their influence on CTC formation, and presents a rationale to target the EMT‐CTC axis to prevent the tumor‐promoting effects of MORAs, so that sufficient pain relief can be provided in BC patients without sacrificing their long‐term prognosis.
Opioids, especially MORAs, are the most effective analgesics to date and are widely used all over the world [32]. Concomitantly, the opioid crisis has led to serious social issues such as addiction, violent crimes, drug abuse and huge economic losses, and has threatened global health [12, 32]. In cancer patients, MORAs are indispensable to pain control. Besides the above‐mentioned social problems, recent discoveries have brought attention to whether they affect cancer progression, and previous clinical and basic studies have uncovered close connections between MORA use and tumor metastasis, recurrence and malignant biological behaviors of cancer cells [28, 33]. However, some cellular studies have challenged this view by showing the potential benefits of MORAs in cancer prognosis. For example, Tegeder et al. [34] showed that morphine reduces growth and promotes apoptosis of breast cancer cells through the activation of p53. Zhang et al. [35] demonstrated that morphine, at clinical concentrations, suppresses the malignant properties of hepatocellular carcinoma cells. Several recently published clinical trials showed no differences in long‐term prognosis between opioid‐sparing anesthetic techniques and routine techniques in patients undergoing surgery due to malignant tumors [36, 37]. This discrepancy may be ascribed to differences in tumor types, animal models used, anesthetic techniques, and exposure patterns to opioids, indicating the necessity for more research in this field. In this present study, we demonstrated that MORA treatment significantly promoted BC formation and metastasis in several in vivo mouse models, which included a hematogenous metastasis model, an in situ BC model and a subcutaneous BC model. These findings strongly indicated a carcinogenic effect of chronic exposure to MORAs in BC individuals, demonstrating that caution is warranted when using MORAs long‐term for pain control in BC patients.
The mechanisms of the carcinogenic effects of MORAs have not yet been well elucidated. Several possible mechanisms have been proposed, including (1) inhibition of the immune system, particularly inhibiting the function of immune cells such as natural killer cells, effector T cells, lymphocytes, dendritic cells and B cells [38], (2) upregulation of stromal angiogenesis [28], and (3) direct effects on cancer cells, i.e., increasing the proliferative, migrative and invasive abilities of cancer cells [39, 40, 41]. However, the exact mechanism of how MORAs promote cancer recurrence and metastasis remain unclear.
CTCs are cancer cells that shed from the tumor nidus and enter the circulatory system, thereby having the potential to form new distant metastases, and they are key components in determining cancer prognosis [16, 42]. In a clinical setting, both CTC enumeration and molecular analyses have been proven valuable as predictive biomarkers for cancer progression and provide new means to monitor the efficiency of individual treatments [43]. Whether MORAs affect CTC formation, however, has not been elucidated. The influence of anesthetics on CTCs is starting to attract researchers’ attention. A recent clinical trial report's new finding on the effects of sevoflurane versus propofol in regard to CTC counts over time in breast cancer surgery patients, and according to the ClinicalTrials.gov website, two clinical trials are currently ongoing, investigating the impact of opioids on CTCs (NCT03700411 and NCT03700541). However, the number of trials in this field is still few, which we suspect are being limited by technological challenges due to the rarity of CTCs compared to blood cells and an inability to isolate them in a viable state [44].
For the present study, we utilized a novel, highly efficient and selective immunocapture microfluidic chip [21, 24, 45], which successfully captured CTCs in several BC mouse models. There are several advantages to our SDI chips when comparing them to other popular CTC detection systems. First, a higher efficiency for CTC capture, requiring only 0.5∼1 mL of blood for our microfluidic chip, whereas most of the other CTCs detection methods need 5∼10 mL of blood [46, 47]. Moreover, dozens of CTCs per mL can be detected with this chip in cancer patients, compared to only several CTCs per ml when using other methods [22, 47]. Second, the ability to identify different kinds of CTCs, six kinds of CTC types, including E‐CTCs, M‐CTCs and CTC clusters, can be detected at the same time with our microfluidic chip, which is unachievable by other CTCs detection systems such as Cellsearch or Cellcollector devices [47, 48].
Using this novel immunocapture microfluidic chip, our experiments showed that MORA treatment significantly increased the number of both M‐CTCs and E‐CTCs. Moreover, when comparing CTC numbers in patients receiving primary BC surgery using routine intraoperative GA and postoperative MORA analgesia to an alternative, almost opioid‐free intraoperative anesthesia and postoperative analgesia technique, we discovered that perioperative opioid‐sparing approaches were associated with a reduced number of CTCs postoperatively in BC patients, especially in the number of M‐CTCs and total CTC clusters, both of which reportedly have a higher capacity to form metastases when compared to other CTC subpopulations [24]. Notably, most subgroups of CTCs remained similar in number immediately after surgery and were higher on the 3rd day post‐surgery compared to their pre‐surgery values. This is reasonable because surgical tumor resection can cause CTCs to be released into the bloodstream [49, 50]. The number of CTCs then declines with time since surgery removed the source of CTCs, and only those with long survival rates can be detected 1 month post‐surgery. Therefore, we speculate that the lower number of CTCs in the opioid‐sparing group on the 3rd day after surgery reflected the fact that almost no opioids were used that would promote additional CTC formation. The fact that this difference was sustained for up to 1 month after surgery, revealed the possibility that opioids can increase CTC survival. Together, our findings provided compelling evidence that MORAs promoted the formation of CTCs and demonstrated a novel mechanism, increasing our understanding of the carcinogenic effects of MORAs.
EMT has been a focal point in the field of oncology not only due to its importance in tumor progression and survival but also its critical role in promoting CTC formation, survival and metastasis to distant organs [16, 18, 42, 51‐53]. Previous studies have shown that cancer cells that undergo EMT more easily invade and survive in the bloodstream [42, 51]. Our RNA‐seq analysis in MB49 cells demonstrated enrichment of cells exhibiting EMT upon MORA treatment. In this context, we then investigated whether a MORA could promote CTC formation by increasing the EMT of BC cells. Our hypothesis was confirmed by multiple experiments. By taking advantage of a live‐cell imaging system, we provided live evidence visualizing the changes in cellular phenotype and activity after MORA treatment. Further investigation into the upstream signaling pathways revealed that inhibiting PI3K/AKT and knocking down Slug both inhibited the EMT process and CTC formation induced by morphine treatment. Strikingly, with our μ‐opioid receptor knockdown BC cell line MOR‐KD MB49, we showed that MOR was required for MORA to exert these effects. These results point towards targeting the MOR/PI3K/AKT/Slug signaling pathway to prevent the promoting effects of MORAs on CTC formation.
The current study has certain limitations. Firstly, we did not perform a molecular analysis of the captured CTCs, due to technological restrictions. With recent development of whole‐genome single‐cell sequencing of CTCs by our research group [54], we will be able to identify MORA‐induced changes in gene expression signatures in CTCs in the future, and biomarker analyses in CTCs could later be used to stratify patients with unfavorable prognoses due to MORA treatment prior to therapy initiation. Secondly, the clinical trial of this current study focused on the number of CTCs instead of patients’ long‐term survival. A large‐scale, multicenter clinical trial utilizing recurrence‐free survival (RFS) or overall survival (OS) as primary outcomes in BC surgery patients is needed to further clarify the relationship among MORA use, CTC formation and patient prognosis.
5. CONCLUSIONS
The current study represents a major advance in the understanding of the effects of MORAs on bladder cancer metastasis. This work uncovered that MORAs facilitated CTC formation, which is due, at least in part, to the activation of the MOR/PI3K/AKT/Slug signaling pathway. These findings indicated that MORAs should be used judiciously in BC patients during the perioperative period or for chronic cancer pain control. Molecules targeting peripheral MORs or the PI3K/AKT/Slug pathway are suggested as adjuvants when MORAs are indispensable in BC patients.
DECLARATIONS
Ethics approval and consent to participate
The study protocol was approved by the ethics committee of Renji Hospital (permit number: KY2019‐192). Tissue samples were obtained with written informed consent from each patient. The animal study was carried out in compliance with the guidance suggestions of the Animal Care Committee of the Renji Hospital (permit number: RJ‐2017‐10‐10).
CONSENT FOR PUBLICATION
Not applicable.
AUTHOR CONTRIBUTIONS
Xiaoqiang Wang, Song Zhang, Di Jin, Jiamei Luo carried out the experiments and analyzed the data. Jie Tian, Weifeng Yu, Chaoyong Yang, and Ming Cao jointly conceived and supervised the project and drafted the manuscript with input from all authors. Yumiao Shi, Yiqi Zhang, Lingling Wu, Yanling Song, Diansan Su, Zhiying Pan, and Haige Chen provided advice and resource to the studies and carried out data collection. All authors read and approved the final manuscript.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
Supporting information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors sincerely appreciate Dr. Hexin Yan (Renji Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China), Dr. Yu Gan (Shanghai Cancer Institute, Renji Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China), Dr. Zhaoli Zhou (Shanghai University of Medicine & Health Sciences, Shanghai, China), and Dr. Yingfu Jiao (Renji Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China) for providing valuable suggestions and generous support. We also thank Isabel Lambertz (Research Specialist, Texas A&M University, College Station, Texas, USA) for English language editing.
Wang X, Zhang S, Jin D, Luo J, Shi Y, Zhang Y, et al. μ‐opioid receptor agonist facilitates circulating tumor cell formation in bladder cancer via the MOR/AKT/Slug pathway: a comprehensive study including randomized controlled trial. Cancer Communications. 2023;43:365–386. 10.1002/cac2.12408
Xiaoqiang Wang, Song Zhang and Di Jin contributed equally to this work.
Contributor Information
Ming Cao, Email: Chowming2007@gmail.com.
Chaoyong Yang, Email: cyyang@xmu.edu.cn.
Weifeng Yu, Email: ywf808@yeah.net.
Jie Tian, Email: vaseline2001@hotmail.com.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33. [DOI] [PubMed] [Google Scholar]
- 2. Qiu H, Cao S, Xu R. Cancer incidence, mortality, and burden in China: a time‐trend analysis and comparison with the United States and United Kingdom based on the global epidemiological data released in 2020. Cancer Commun (Lond). 2021;41(10):1037–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–49. [DOI] [PubMed] [Google Scholar]
- 4. Antoni S, Ferlay J, Soerjomataram I, Znaor A, Jemal A, Bray F. Bladder Cancer Incidence and Mortality: A Global Overview and Recent Trends. Eur Urol. 2017;71(1):96–108. [DOI] [PubMed] [Google Scholar]
- 5. Cumberbatch MGK, Jubber I, Black PC, Esperto F, Figueroa JD, Kamat AM, et al. Epidemiology of Bladder Cancer: A Systematic Review and Contemporary Update of Risk Factors in 2018. Eur Urol. 2018;74(6):784–95. [DOI] [PubMed] [Google Scholar]
- 6. Waks AG, Winer EP. Breast Cancer Treatment: A Review. JAMA. 2019;321(3):288–300. [DOI] [PubMed] [Google Scholar]
- 7. Kolodny A, Courtwright DT, Hwang CS, Kreiner P, Eadie JL, Clark TW, et al. The prescription opioid and heroin crisis: a public health approach to an epidemic of addiction. Annu Rev Public Health. 2015;36:559–74. [DOI] [PubMed] [Google Scholar]
- 8. Perry NJS, Buggy D, Ma D. Can Anesthesia Influence Cancer Outcomes After Surgery? JAMA Surg. 2019;154(4):279–80. [DOI] [PubMed] [Google Scholar]
- 9. Brinkman D, Wang JH, Redmond HP. Morphine as a treatment of cancer‐induced pain‐is it safe? A review of in vivo studies and mechanisms. Naunyn Schmiedebergs Arch Pharmacol. 2018;391(11):1169–78. [DOI] [PubMed] [Google Scholar]
- 10. Ramirez MF, Gorur A, Cata JP. Opioids and cancer prognosis: A summary of the clinical evidence. Neurosci Lett. 2021;746:135661. [DOI] [PubMed] [Google Scholar]
- 11. Akbari M, Naghibzadeh‐Tahami A, Khanjani N, Baneshi M, Kamali E, Hesampour M, et al. Opium as a Risk Factor for Bladder Cancer: A Population‐based Case‐control Study in Iran. Arch Iran Med. 2015;18(9):567–71. [PubMed] [Google Scholar]
- 12. Bidary MZ, Sahranavard M, Rezayat AA, Omranzadeh A, Hoseiny SH, Kabirian A, et al. Opium as a carcinogen: A systematic review and meta‐analysis. EClinicalMedicine. 2021;33:100768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kamangar F, Shakeri R, Malekzadeh R, Islami F. Opium use: an emerging risk factor for cancer? Lancet Oncol. 2014;15(2):e69–e77. [DOI] [PubMed] [Google Scholar]
- 14. Afshari M, Janbabaei G, Bahrami MA, Moosazadeh M. Opium and bladder cancer: A systematic review and meta‐analysis of the odds ratios for opium use and the risk of bladder cancer. PloS one. 2017;12(6):e0178527–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guerrero Orriach JL, Raigon Ponferrada A, Malo Manso A, Herrera Imbroda B, Escalona Belmonte JJ, Ramirez Aliaga M, et al. Anesthesia in Combination with Propofol Increases Disease‐Free Survival in Bladder Cancer Patients Who Undergo Radical Tumor Cystectomy as Compared to Inhalational Anesthetics and Opiate‐Based Analgesia. Oncology. 2020;98(3):161–7. [DOI] [PubMed] [Google Scholar]
- 16. Rodrigues P, Vanharanta S. Circulating Tumor Cells: Come Together, Right Now, Over Metastasis. Cancer Discov. 2019;9(1):22–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang HM, Li HX. Tumor heterogeneity and the potential role of liquid biopsy in bladder cancer. Cancer Commun (Lond). 2021;41(2):91–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer. 2018;18(2):128–34. [DOI] [PubMed] [Google Scholar]
- 19. Yang L, Yan X, Chen J, Zhan Q, Hua Y, Xu S, et al. Hexokinase 2 discerns a novel circulating tumor cell population associated with poor prognosis in lung cancer patients. Proc Natl Acad Sci U S A. 2021;118(11):e2012228118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Song Y, Shi Y, Huang M, Wang W, Wang Y, Cheng J, et al. Bioinspired Engineering of a Multivalent Aptamer‐Functionalized Nanointerface to Enhance the Capture and Release of Circulating Tumor Cells. Angew Chem Int Ed Engl. 2019;58(8):2236–40. [DOI] [PubMed] [Google Scholar]
- 21. Wu L, Ding H, Qu X, Shi X, Yang J, Huang M, et al. Fluidic Multivalent Membrane Nanointerface Enables Synergetic Enrichment of Circulating Tumor Cells with High Efficiency and Viability. J Am Chem Soc. 2020;142(10):4800–6. [DOI] [PubMed] [Google Scholar]
- 22. Ahmed MG, Abate MF, Song Y, Zhu Z, Yan F, Xu Y, et al. Isolation, Detection, and Antigen‐Based Profiling of Circulating Tumor Cells Using a Size‐Dictated Immunocapture Chip. Angew Chem Int Ed Engl. 2017;56(36):10681–5. [DOI] [PubMed] [Google Scholar]
- 23. Zhu L, Lin H, Wan S, Chen X, Wu L, Zhu Z, et al. Efficient Isolation and Phenotypic Profiling of Circulating Hepatocellular Carcinoma Cells via a Combinatorial‐Antibody‐Functionalized Microfluidic Synergetic‐Chip. Anal Chem. 2020;92(22):15229–35. [DOI] [PubMed] [Google Scholar]
- 24. Zheng Y, Zhang J, Huang M, Wang T, Qu X, Wu L, et al. Selection of Aptamers Against Vimentin for Isolation and Release of Circulating Tumor Cells Undergoing Epithelial Mesenchymal Transition. Anal Chem. 2020;92(7):5178–84. [DOI] [PubMed] [Google Scholar]
- 25. Xiong Q, Liu A, Ren Q, Xue Y, Yu X, Ying Y, et al. Cuprous oxide nanoparticles trigger reactive oxygen species‐induced apoptosis through activation of erk‐dependent autophagy in bladder cancer. Cell Death Dis. 2020;11(5):366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nagy Á, Munkácsy G, Győrffy B. Pancancer survival analysis of cancer hallmark genes. Sci Rep. 2021;11(1):6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Poruk KE, Valero V 3rd, Fau, Saunders T, Saunders T Fau, Blackford AL, Blackford Al Fau, Griffin JF, Griffin Jf Fau, Poling J, Poling J Fau, et al. Circulating Tumor Cell Phenotype Predicts Recurrence and Survival in Pancreatic Adenocarcinoma. Ann Surg. 2016;264(6):1073–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lennon FE, Mirzapoiazova T, Mambetsariev B, Poroyko VA, Salgia R, Moss J, et al. The Mu opioid receptor promotes opioid and growth factor‐induced proliferation, migration and Epithelial Mesenchymal Transition (EMT) in human lung cancer. PloS one. 2014;9(3):e91577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fritsch R, de Krijger I, Fritsch K, George R, Reason B, Kumar MS, et al. RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3‐kinase isoforms. Cell. 2013;153(5):1050–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158(5):1110–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yin W, Han YM, Li ZL, Huang ZX, Huang L, Zhong XG. Clinical significance of perioperative EMT‐CTC in rectal cancer patients receiving open/laparoscopic surgery. Neoplasma. 2020;67(5):1131–38. [DOI] [PubMed] [Google Scholar]
- 32. Hodder SL, Feinberg J, Strathdee SA, Shoptaw S, Altice FL, Ortenzio L, et al. The opioid crisis and HIV in the USA: deadly synergies. Lancet. 2021;397(10279):1139–50. [DOI] [PubMed] [Google Scholar]
- 33. Chen DT, Pan JH, Chen YH, Xing W, Yan Y, Yuan YF, et al. The mu‐opioid receptor is a molecular marker for poor prognosis in hepatocellular carcinoma and represents a potential therapeutic target. Br J Anaesth. 2019;122(6):e157–e67. [DOI] [PubMed] [Google Scholar]
- 34. Tegeder I, Grösch S, Schmidtko A, Häussler A, Schmidt H, Niederberger E, et al. G protein‐independent G1 cell cycle block and apoptosis with morphine in adenocarcinoma cells: involvement of p53 phosphorylation. Cancer Res. 2003;63(8):1846–52. [PubMed] [Google Scholar]
- 35. Zhang HW, Wang F, Zhou YQ, Xu SP, Yu SY, Zhang ZG. Morphine Suppresses Liver Cancer Cell Tumor Properties In Vitro and In Vivo. Front Oncol. 2021;11:666446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sessler DI, Pei L, Huang Y, Fleischmann E, Marhofer P, Kurz A, et al. Recurrence of breast cancer after regional or general anaesthesia: a randomised controlled trial. Lancet. 2019;394(10211):1807–15. [DOI] [PubMed] [Google Scholar]
- 37. Falk W, Magnuson A, Eintrei C, Henningsson R, Myrelid P, Matthiessen P, et al. Comparison between epidural and intravenous analgesia effects on disease‐free survival after colorectal cancer surgery: a randomised multicentre controlled trial. Br J Anaesth. 2021;127(1):65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boland JW, Pockley AG. Influence of opioids on immune function in patients with cancer pain: from bench to bedside. Br J Pharmacol. 2018;175(14):2726–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Harper P, Hald O, Lwaleed BA, Kyyaly A, Johnston D, Cooper AJ, et al. The impact of morphine treatment on bladder cancer cell proliferation and apoptosis: in vitro studies. Exp oncol. 2018;40(3):190–3. [PubMed] [Google Scholar]
- 40. Singleton PA, Mirzapoiazova T, Hasina R, Salgia R, Moss J. Increased μ‐opioid receptor expression in metastatic lung cancer. Br J Anaesth. 2014;113 Suppl 1(Suppl 1):i103–i8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Niu DG, Peng F, Zhang W, Guan Z, Zhao HD, Li JL, et al. Morphine promotes cancer stem cell properties, contributing to chemoresistance in breast cancer. Oncotarget. 2015;6(6):3963–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tayoun T, Faugeroux V, Oulhen M, Aberlenc A, Pawlikowska P, Farace F. CTC‐Derived Models: A Window into the Seeding Capacity of Circulating Tumor Cells (CTCs). Cells. 2019;8(10):1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Castro‐Giner F, Aceto N. Tracking cancer progression: from circulating tumor cells to metastasis. Genome Med. 2020;12(1):31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rushton AJ, Nteliopoulos G, Shaw JA, Coombes RC. A Review of Circulating Tumour Cell Enrichment Technologies. Cancers (Basel). 2021;13(5):970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang J, Lin B, Wu L, Huang M, Li X, Zhang H, et al. DNA Nanolithography Enables a Highly Ordered Recognition Interface in a Microfluidic Chip for the Efficient Capture and Release of Circulating Tumor Cells. Angew Chem Int Ed Engl. 2020;59(33):14115–9. [DOI] [PubMed] [Google Scholar]
- 46. Gkountela S, Castro‐Giner F, Szczerba BM, Vetter M, Landin J, Scherrer R, et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell. 2019;176(1‐2):98–112.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Riethdorf S, O'Flaherty L, Hille C, Pantel K. Clinical applications of the CellSearch platform in cancer patients. Adv Drug Deliv Rev. 2018;125:102–21. [DOI] [PubMed] [Google Scholar]
- 48. He Y, Shi J, Shi G, Xu X, Liu Q, Liu C, et al. Using the New CellCollector to Capture Circulating Tumor Cells from Blood in Different Groups of Pulmonary Disease: A Cohort Study. Sci Rep. 2017;7(1):9542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Weitz J, Kienle P, Lacroix J, Willeke F, Benner A, Lehnert T, et al. Dissemination of tumor cells in patients undergoing surgery for colorectal cancer. Clin Cancer Res. 1998;4(2):343–8. [PubMed] [Google Scholar]
- 50. Martin OA, Anderson RL, Narayan K, MacManus MP. Does the mobilization of circulating tumour cells during cancer therapy cause metastasis? Nat Rev Clin Oncol. 2017;14(1):32–44. [DOI] [PubMed] [Google Scholar]
- 51. Qi L‐N, Xiang B‐D, Wu F‐X, Ye J‐Z, Zhong J‐H, Wang Y‐Y, et al. Circulating Tumor Cells Undergoing EMT Provide a Metric for Diagnosis and Prognosis of Patients with Hepatocellular Carcinoma. Cancer Res. 2018;78(16):4731–44. [DOI] [PubMed] [Google Scholar]
- 52. Gorin MA, Verdone JE, van der Toom E, Bivalacqua TJ, Allaf ME, Pienta KJ. Circulating tumour cells as biomarkers of prostate, bladder, and kidney cancer. Nat Rev Urol. 2017;14(2):90–7. [DOI] [PubMed] [Google Scholar]
- 53. Nieto MA, Huang RY‐J, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166(1):21–45. [DOI] [PubMed] [Google Scholar]
- 54. Ruan Q, Ruan W, Lin X, Wang Y, Zou F, Zhou L, et al. Digital‐WGS: Automated, highly efficient whole‐genome sequencing of single cells by digital microfluidics. Sci Adv. 2020;6(50):eabd6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.