

Summary
Centromeres are essential for chromosome segregation in most animals and plants yet are among the most rapidly evolving genome elements. The mechanisms underlying this paradoxical phenomenon remain enigmatic. Here, we report that human centromeres innately harbor a striking enrichment of DNA breaks within functionally active centromere regions. Establishing a single-cell imaging strategy that enables comparative assessment of DNA breaks at repetitive regions, we show that centromeric DNA breaks are induced not only during active cellular proliferation but also de novo during quiescence. Markedly, centromere DNA breaks in quiescent cells are resolved enzymatically by the evolutionarily conserved RAD51 recombinase, which in turn safeguards the specification of functional centromeres. This study highlights the innate fragility of centromeres, which may have been co-opted over time to reinforce centromere specification whilst driving rapid evolution. The findings also provide insights into how fragile centromeres are likely to contribute to human disease.
Keywords: Centromeres, DNA damage, Homologous Recombination, RAD51, CENP-A
eTOC blurb
Centromeres are essential chromosomal regions that mediate the accurate inheritance of genetic information, but paradoxically centromere DNA sequences change rapidly. Saayman et al. reveal the frequent breakage of centromeric DNA even in resting cells, which are repaired in a manner that maintains centromere function while allowing DNA sequence change.
Graphical Abstract
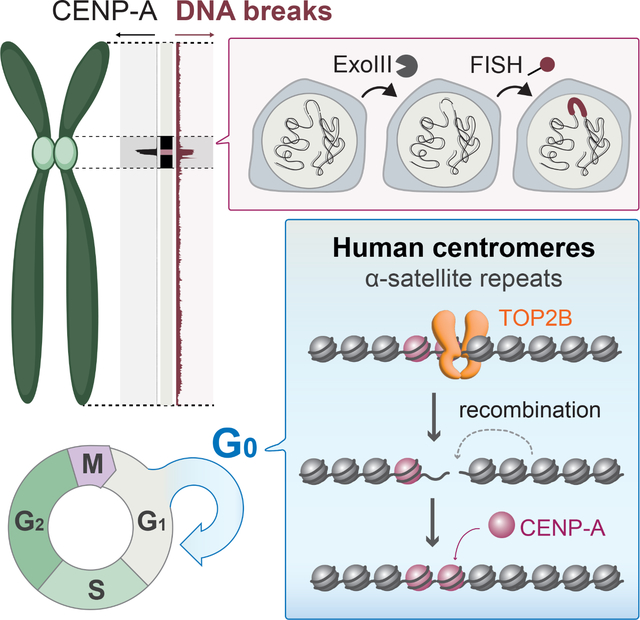
Introduction
Centromeres are essential regions of the eukaryotic genome, acting as structural platforms for kinetochore establishment and microtubule attachment during chromosome segregation. While a wide diversity of centromere structures has been reported, most plant and animal centromeres are composed of tandem repeat sequences known as satellite repeats, creating mega-base sized arrays. In humans, 171-bp monomeric ‘alpha-satellites’ are repeated in head-to-tail tandem fashion, which can then be further repeated to form higher-order repeats (HORs)1–3.
Although the repetitive structure of centromeres is highly conserved4, centromere positioning and functionality is principally defined epigenetically. Indeed, kinetochore establishment is specified by the presence of the centromere-specific histone variant CENP-A that can, to a large degree, be propagated in a sequence-independent manner5–8. CENP-A generally occupies a single HOR per chromosome, designating the functionally active centromere core9. In support of an epigenetic model for centromere definition, the only sequence-specific centromere-binding protein is CENP-B, which binds a 17-bp motif within certain alpha satellite repeats, but CENP-B does not appear to be essential in mammals10,11.
Curiously, centromeres have long been recognized as one the most rapidly evolving regions of the genome, with alpha satellite sequences substantially diverging between closely related species and even between chromosomes within species2,12–14. The centromere paradox describes the dichotomy between rapidly evolving centromere DNA sequences and highly conserved centromere functionality15, and has remained enigmatic for several decades.
Recent genomic evidence has provided some insight into the evolutionary dynamics of centromeres. The first completion of a human reference genome including repetitive regions has revealed a ‘layered’ centromere structure16,17. In this structure, functionally active centromere cores contain highly ordered and homogenous alpha satellites HORs, while inactive centromere peripheries are comprised of symmetric array layers with progressively increasing sequence divergence, structural rearrangements, and transposable element insertions. Based on these findings, the ‘expanding centromere’ model has been proposed, whereby new alpha satellite sequences periodically emerge, expanding and homogenizing rapidly within active centromere HORs, eventually displacing older arrays16,18.
The expanding centromere model can theoretically be explained by repeated homology-dependent unequal crossovers or gene conversion events driven by homologous recombination (HR)12,19. Indeed, although centromeres have been described as recombination ‘cold-spots’ during meiosis, it has become evident that sister-chromatid exchanges (SCEs), one of multiple HR repair products, occur frequently at centromeres in mitotically growing mammalian cells20,21. In addition, HR factors have recently been shown to be recruited to Cas9-induced DNA double-strand breaks (DSBs) at centromeres, revealing the capacity for centromeric HR in response to exogenously induced DNA damage22. Finally, previous reports have indicated a role of the evolutionally conserved Rad51 recombinase in facilitating non-crossover recombination between intra-chromosomal repeats flanking centromeres in S. pombe23. Mounting experimental evidence therefore supports a role for recombination at centromeres during mitotic proliferation. However, the question of why functionally active centromere cores specifically are so prone to recombination, and the pathways driving centromeric HR in unperturbed conditions, has remained unanswered.
Results
Spontaneous accumulation of DNA strand breaks within functionally active centromere cores
We first asked why recombination might occur more frequently within functionally active (i.e., CENP-A-bound) centromere regions than elsewhere in the genome. Recombination can be initiated by double-strand breaks (DSBs) or ssDNA gaps or nicks (collectively single-strand breaks, SSBs)24. We therefore wondered whether these DNA lesions were enriched within normally growing human centromeres in the absence of any exogenous perturbation.
To gain unbiased and direct evidence, we assessed genome-wide distributions of DNA breaks using publicly available next-generation sequencing (NGS)-based datasets. Although repetitive regions have typically been excluded from previous studies due to the lack of an appropriate reference sequence, the recent completion of a full human reference genome derived from the CHM13-hTERT cell line enables the alignment of short-read data to centromeric arrays (Figure S1A)17. Building on this development, we specifically focused on re-evaluating two NGS DNA break datasets in the chromosomally stable HCT116 cell line. First, GLOE-seq identifies free 3’hydroxy (OH) ends on DNA, thereby mapping both SSBs and DSBs25. Second, END-seq exploits the direct ligation of an adaptor to DSB ends, thereby selectively mapping DSBs (Figure S1B)26,27. The analysis was complemented by two publicly available CENP-A positioning datasets (CUT&RUN and ChIP-seq) to mark functionally active centromere loci28.
Our genome-wide analyses of DNA breaks, as detected by GLOE-seq, revealed a striking correlation with CENP-A positioning, as detected by both ChIP-seq and CUT&RUN, within centromeres (Figures 1A, S1C). DNA breaks were also enriched within genome repeats such as ribosomal DNA (rDNA) arrays on acrocentric chromosomes and telomeres (Figure 1A). Calculating the percent NGS reads relative to the percent size of all available repeat type annotations, we found that DNA breaks were enriched predominantly within satellite repeats and simple repeats (e.g., telomeres) (Figure 1B). CENP-A positioning, as detected by ChIP-seq, revealed more selective enrichment within satellite repeats compared to CUT&RUN, suggesting that ChIP-seq may detect stably incorporated CENP-A within centromeric HOR chromatin more accurately (Figures 1A, 1B, S1C). Within annotated subregions of centromeres, DNA breaks were specifically enriched within HOR-associated alpha satellites (hor), rDNA and other satellite repeats (e.g., hsat1, hsat2, censat) (Figure S2A). We further found that DNA breaks were enriched within functionally active core HORs to a greater extent than divergent HORs (dHOR) or individual alpha satellite monomers located towards the periphery of active centromeres (Figure 1C). Once again, this selective enrichment within core HORs was consistent with both CUT&RUN-based and ChIP-seq-based CENP-A positioning. Together, these data support the notion that intrinsic DNA breaks in asynchronous cells are enriched within the functionally active (i.e., CENP-A-bound) regions of human centromeres.
Figure 1 |. DNA strand breaks are enriched at human centromeres.
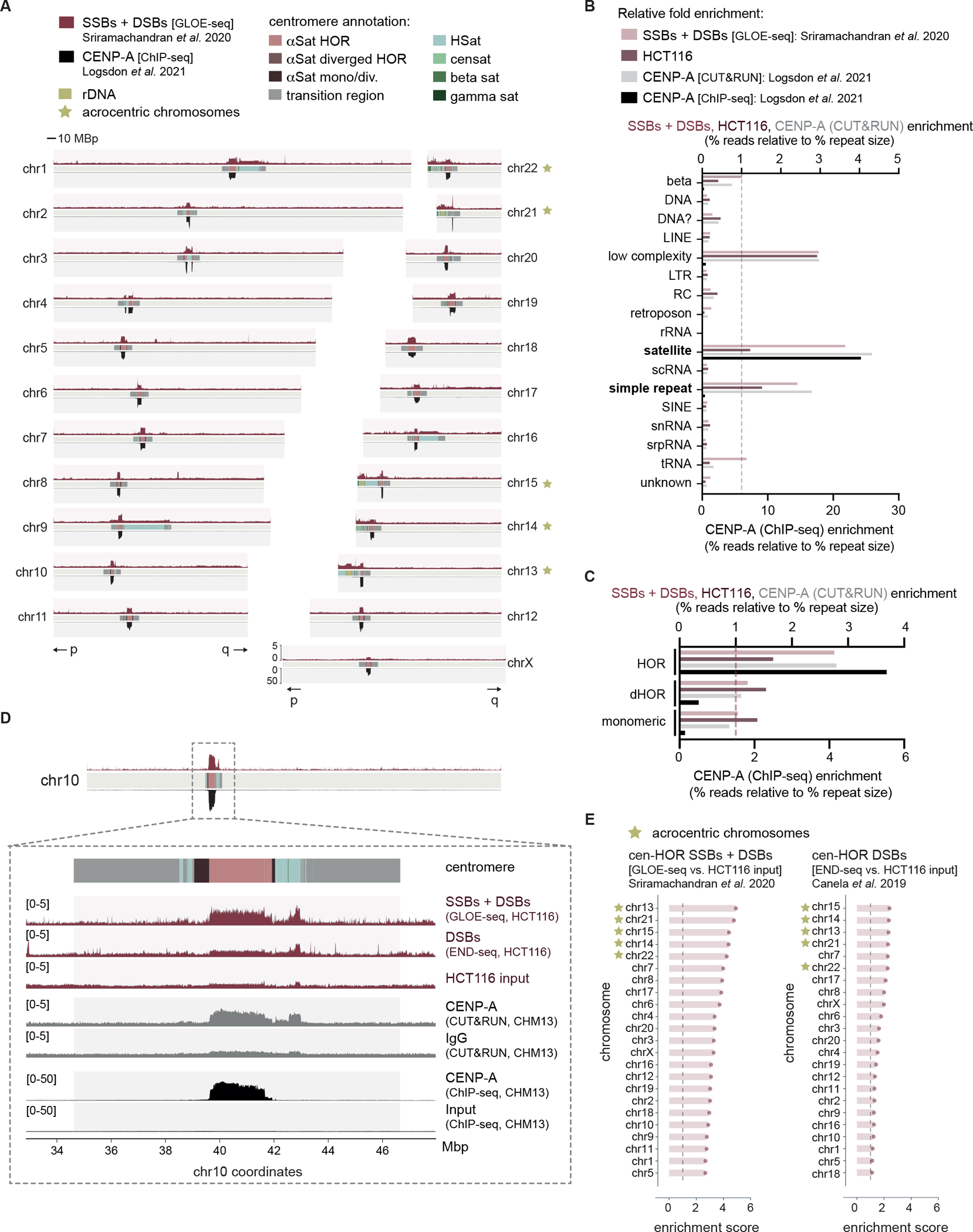
(A) Alignment of publicly available next generation sequencing data detecting DNA strand breaks (GLOE-seq25) above and CENP-A localization (ChIP-seq28) below across 22 autosome chromosomes and chromosome X of the human T2T-CHM13 reference assembly. Centromeric regions are composed of alpha satellites within higher order repeats (αSat HORs), alpha satellites within diverged HORs (αSat diverged HORs), monomeric / diverged alpha satellites (αSat mono/div.), human satellites (HSat), beta satellites (beta sat), gamma satellites (gamma sat), transition regions, other satellites (censat) and ribosomal DNA arrays (rDNA). p and q chromosome arms indicated below. (B) Fold enrichment of DNA breaks, HCT116 input and CENP-A (both CUT&RUN and ChIP-seq) reads across all annotated repeat types of the T2T-CHM13 reference genome. (C) Fold enrichment of DNA breaks, HCT116 input and CENP-A CUT&RUN reads over alpha satellites annotated as being either part of HORs, divergent HORs (dHOR) or monomeric. (D) Inset of chr10 centromere depicting total DNA breaks (GLOE-seq25) and DNA DSBs (END-seq27) with the corresponding input cell line (HCT116), as well as CENP-A localization (CUT&RUN above, ChIP-seq below) with the corresponding negative control (IgG above, ChIP-seq input below)28. (E) Enrichment scores of GLOE-seq or END-seq in HCT116 cells across human centromere HORs. Enrichment scores were calculated as the sums of mapped reads within centromeric HORs of each chromosome relative to that of the corresponding input control, after normalizing to read depth. Acrocentric chromosomes containing rDNA arrays are marked as indicated. See also Figures S1 and S2.
We similarly assessed NGS datasets specific to DSBs, as detected by END-seq27, to determine whether centromeric DNA breaks were single- or double-stranded in nature. Our analysis revealed less pronounced enrichment of END-seq reads across centromeres compared to GLOE-seq (Figure 1D). To quantify this trend, we calculated enrichment scores of reads across centromere HORs of each individual chromosome. After normalizing to read depth, the sums of mapped GLOE-seq or END-seq reads within each chromosome’s HOR were compared to an input sample to account for any cell line-specific copy number variations. From this, it became apparent that centromere HOR DNA breaks are composed of both SSBs and DSBs, although a subset of chromosomes (e.g., chr1, chr5, chr18) selectively harbor SSBs. (Figure 1E). Intriguingly, we also found that DNA breaks were particularly enriched on centromere HORs of acrocentric chromosomes.
Such a striking enrichment of DNA breaks within centromere cores was unexpected. However, these analyses contained several potential drawbacks. First, we observed an under-enrichment of NGS PCR duplicates within alpha satellite repeats. As such, the enrichment of DNA breaks within centromere cores in HCT116 datasets was only apparent if PCR duplicate removal steps were included in the alignment strategy. As this phenomenon could theoretically result from the inherent difficulty in amplifying GC-rich satellite sequences, as previously reported in Drosophila29,30, we assessed the prevalence of this PCR bias across repeats throughout the genome. Surprisingly, this bias was unique to satellite repeats and selectively present in libraries with high levels of PCR duplication (Figure S2B, C, D). While including PCR duplicate removal steps were unavoidable due to the PCR bias found across the genome and the variability in PCR duplicate levels between NGS libraries, conducting PCR-free DNA break analyses has posed significant technical challenges. Hence, the possibility of a mapping artefact could not be conclusively excluded under the current state-of-the-art. Second, these publicly available NGS data were generated in separate studies, leading to potential pitfalls in their comparative assessment. Third, the precise sequence of centromeres, as well as exact positionings of CENP-A, likely varies between different cell lines. i.e., hTERT-CHM13 and HCT116. In a similar line, it is technically not possible to map exact locations of NGS reads within repetitive regions, as exemplified by CENP-A ChIP-seq and CUT&RUN (Figures 1A, 1B, S1C). While these short-read sequence analyses show CENP-A distribution to the entire regions of HOR or, in the case of CUT&RUN, HSat, recent studies using long-read sequence analysis revealed that CENP-A localization is limited to narrow regions within HORs16,31,32. These observations suggest that caution should be taken while interpreting short-read sequences within these regions. Fourth, NGS-based analyses, which detect averaged enrichments of DNA breaks in bulk cell populations, cannot detect cell-to-cell variation in centromere DNA breakage. These limitations collectively raised the need to verify the presence of DNA breaks within centromere cores by independent methods.
exo-FISH for the detection of centromere HOR DNA breaks
To investigate DNA breakage within centromere cores independently of NGS-based technologies, we developed a microscopy-based method to directly detect DNA breaks at defined repetitive loci of the genome in single cells. This method, exo-FISH, relies on in vitro end resection of undenatured DNA by Exonuclease III (ExoIII). ExoIII digests DNA from 3’ ends, using either a DSB or SSB as an initial substrate, thereby exposing single-strand DNA. The resulting single-stranded DNA is then hybridized with a fluorescently labeled complementary probe using fluorescent in-situ hybridization (FISH) (Figure 2A). Any increase in the number of DNA breaks would thereby facilitate more ExoIII digestion, exposing more ssDNA and producing higher FISH signal intensities. Importantly, exo-FISH includes a high-concentration RNase A treatment prior to ExoIII digestion to remove both free RNA and chromatin associated RNA, including RNA-DNA hybrids33,34, such that the potential hybridization of FISH probe to RNA and the RNase H1-like activity of ExoIII can be disregarded. Throughout these analyses, we used FISH probes complementary to the 17-bp CENP-B box to label centromere cores (cenFISH), probes complementary to telomeric repeats to label telomeres (telFISH) and a combined probe against human satellites 2 and 3 to label pericentromere-associated satellite repeats (HSatFISH) (Figure S3A). The hybridization patterns of the FISH probes matched their predicted localization, with large tracts of human satellites 2/3 on chromosomes 1, 9 and 16 visible when using HSatFISH, and chromosome ends with telFISH (Figure 2B).
Figure 2 |. Detection of spontaneous centromere HOR breaks with exo-FISH.
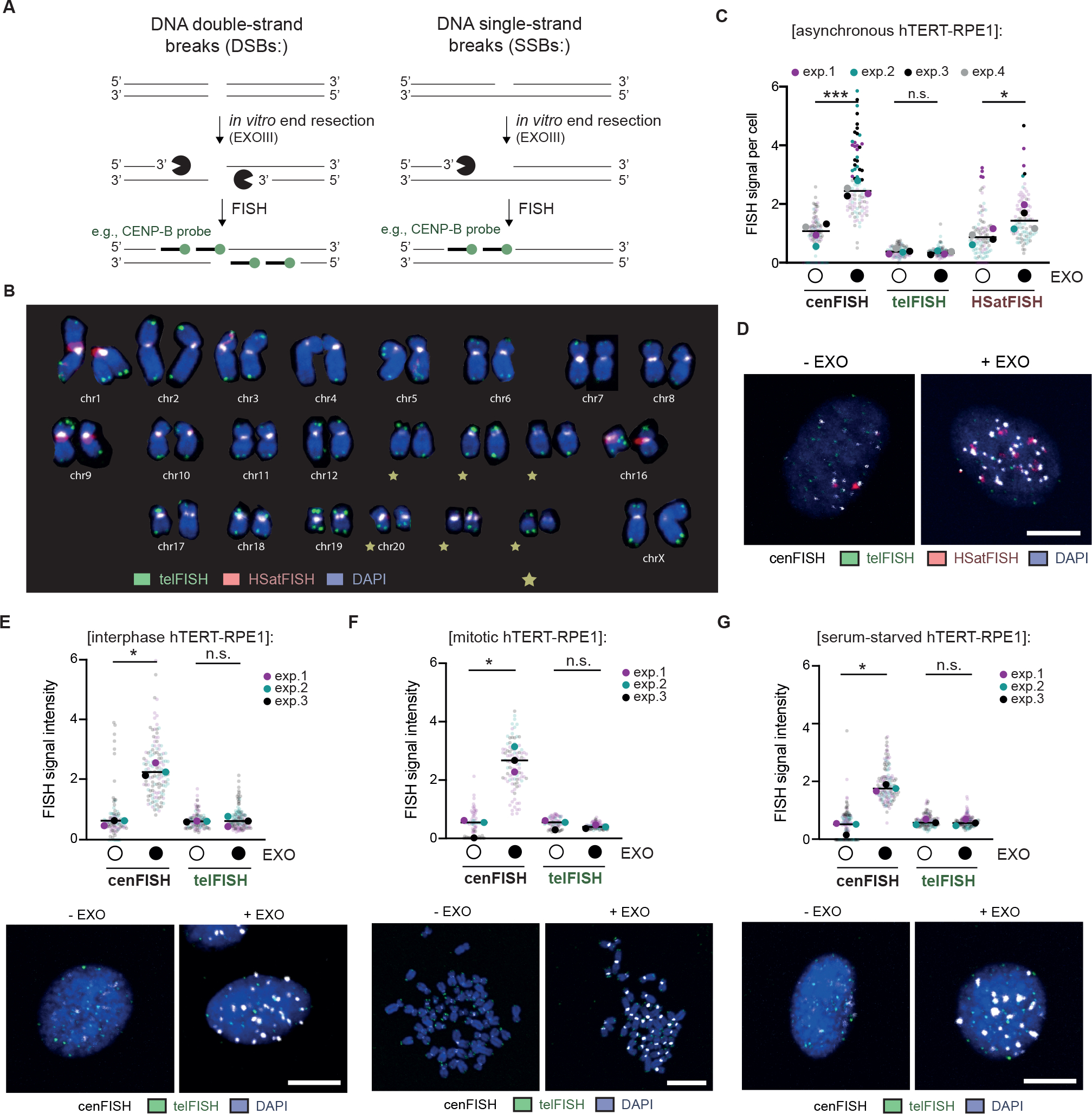
(A) Schematic of the detection of DNA breaks at repetitive elements using exo-FISH. (B) Karyogram depicting FISH probe hybridization patterns. FISH probes against HOR-associated centromere repeats (cenFISH, white), telomere repeats (telFISH, green) and human satellites 2 and 3 (HSatFISH, red) are depicted. (C, D) Quantification and representative images of exo-FISH applied to asynchronous hTERT-RPE1 cells with and without Exonuclease III (EXO) treatment. FISH signal intensity was calculated by first taking the sum of the fluorescence signal surrounding each cenFISH, telFISH or HSatFISH focus in a 14×14–20×20 pixel box, following a perimeter-estimated background subtraction. The median value for each cell is then calculated and plotted above, with each data point representing a cell median. (E) Representative images and quantification of exo-FISH in interphase hTERT-RPE1 cells. (F) Representative images and quantification of exo-FISH in mitotic hTERT-RPE1 cells. Cells were arrested in mitosis with a 3–5-hour STLC treatment and harvested by mitotic shake-off prior to spreading. (G) Representative images and quantification of exo-FISH in quiescent (i.e., serum-starved hTERT-RPE1) cells. Cells were serum-starved for ~120 hours before harvesting. Scale bar represents 10 μm. FISH signal intensity is X10,000 arbitrary units (A.U.). At least 30 cells were imaged per experimental condition. The medians of each experimental condition were used to perform a two-sided unpaired t-test (*p<0.05, **p<0.01). Filled and empty circles indicate presence and absence, respectively. See also Figure S3.
To demonstrate that exo-FISH can detect DNA breaks at human centromeres, we used hTERT-immortalized retinal pigment epithelial-1 (hTERT-RPE1) cells, a commonly used cell line with non-cancerous origin. These cells, arrested in mitosis, were fixed and DSBs or SSBs were subsequently introduced in vitro using the restriction enzyme BsmAI or the DNA nicking enzyme Nt.BsmAI, respectively. The 6-bp recognition sequences are present genome-wide including centromeres, with an average of one break per 895 bp within human centromeres. Without ExoIII treatment, neither BsmAI nor Nt.BsmAI pre-treatment had any effect on cenFISH signals. With ExoIII treatment, pre-treatment with either BsmAI or Nt.BsmAI increased cenFISH signal intensity (Figure S3B–E). The ExoIII-induced increase in signal intensity therefore confirmed that exo-FISH is able to detect both DSBs and SSBs at centromere HORs.
Applying exo-FISH to unperturbed hTERT-RPE1, we observed a consistent increase in cenFISH signals upon ExoIII treatment, confirming the presence of endogenous DNA breaks at centromere HORs (Figure 2C). This increase was not detected with the telFISH probe, suggesting that centromere-associated DNA breaks are more abundant than DNA breaks within telomeres. A smaller yet significant increase was also observed with the HSatFISH probe, correlating with the reduced enrichment of DNA breaks within HSat2 and HSat3 arrays, as suggested by GLOE-seq in HCT116 (Figure 1A, S2A). Importantly, the responsiveness of cenFISH probes to ExoIII treatment was equally apparent in HCT116 and HeLa demonstrating the universality of centromere HOR breaks across cell lines (Figure S3F).
DNA replication -dependent and -independent sources of centromere HOR DNA breaks
We next set out to investigate the nature of DNA breaks within centromere HORs in hTERT-RPE1. First, centromere HOR breaks were universally observed in both interphase and mitotically arrested hTERT-RPE1 (Figure 2E, F). Perturbation of normal DNA replication progression with low-dose replication stress (0.4 μM aphidicolin for 24 hours) also increased the ExoIII responsiveness of both cenFISH and telFISH (Figure S3G–J). These observations suggested that centromere HOR breaks are at least in part associated with DNA replication. To determine whether centromere HOR DNA breaks were totally dependent on active cellular proliferation, we next asked whether breaks persisted during quiescence, a temporary state in which cells reversibly halt cellular proliferation and associated processes. hTERT-RPE1 can be induced into quiescence within 24 hours of serum starvation, significantly reducing subsequent EdU incorporation as a marker of active DNA replication (Figure S4A, B). Surprisingly, centromere HOR breaks were detectable even after prolonged (~120 hours) serum starvation, raising the possibility that centromere HOR breaks can occur independently of active proliferation and DNA replication (Figure 2G).
Given the unexpected presence of centromere HOR DNA breaks in non-proliferating conditions, we sought to determine the kinetics of HOR DNA breaks in hTERT-RPE1 over the course of 7 days after serum starvation (Figure 3A). Centromere HOR breaks initially decreased over 3 days, followed by a subsequent increase after 5–7 days (Figure 3B, C). Cells had not resumed proliferation by this point, as the number of cells harvested after the full 7 days of serum starvation matched expected yields (Figure S4C). Together, these data suggest that centromere HOR DNA breaks are initially dependent on cellular proliferation, they can also be induced de novo during quiescence.
Figure 3 |. DNA replication -dependent and -independent sources of centromere HOR DNA breaks.
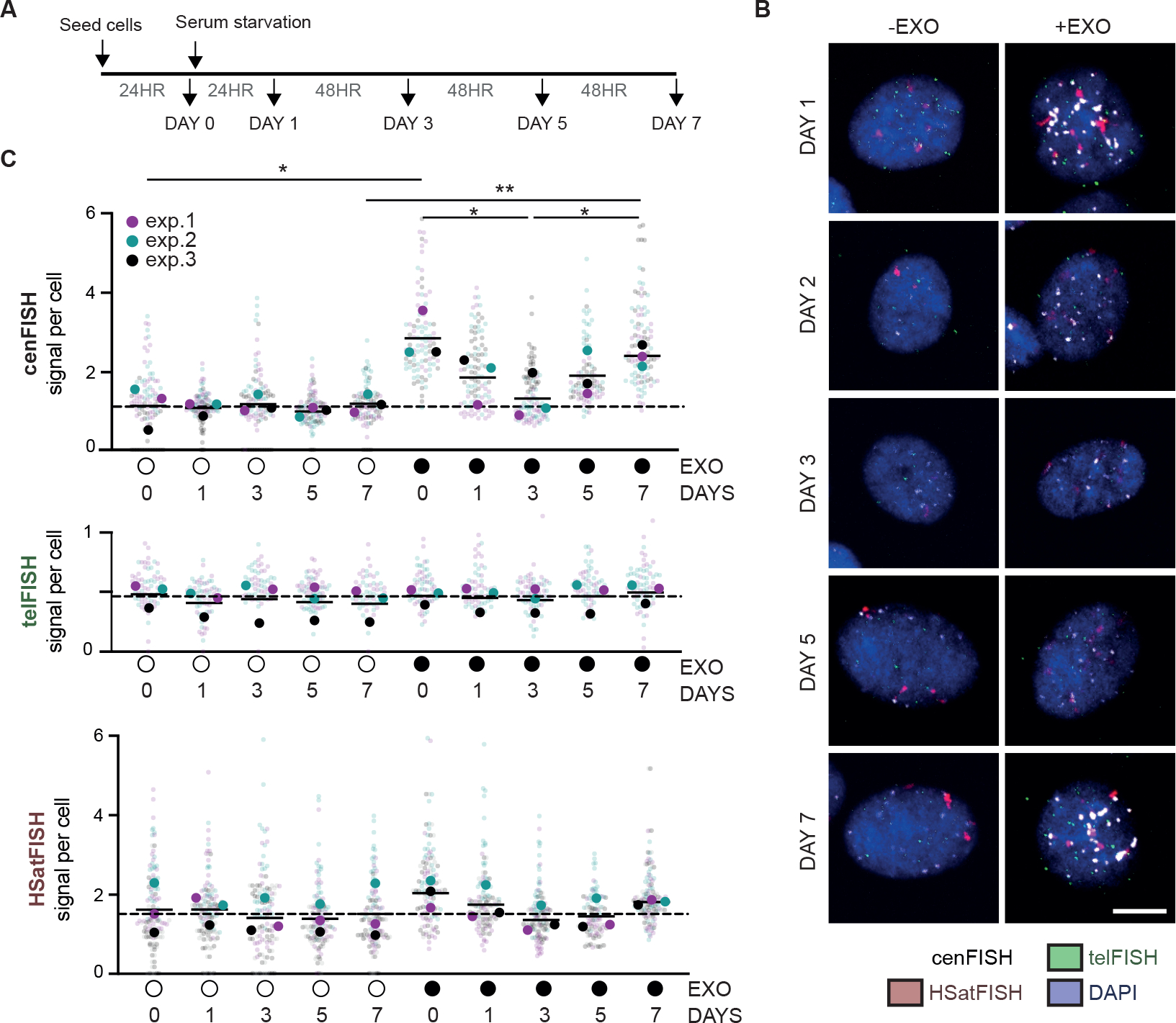
(A) Experimental schematic of time course following induction of quiescence through serum starvation in hTERT-RPE1 cells. (B, C) Quantification and representative images of exo-FISH performed at the indicated time points. Quantification was performed as in Figure 2. Scale bar represents 10 μm. FISH signal intensity is X10,000 arbitrary units (A.U.). At least 30 cells were imaged per experimental condition. The medians of each experimental condition were used to perform a two-sided unpaired t-test (*p<0.05, **p<0.01). Filled and empty circles indicate presence and absence, respectively. See also Figure S4.
To further evaluate centromere HOR DNA breaks in non-proliferating conditions, we sought to determine genome-wide distributions of DNA breaks in terminally differentiated post-mitotic cells. Recently, DSBs were mapped in post-mitotic neurons induced from pluripotent stem cells (iNeurons) using END-seq35. Our genome-wide quantification of END-seq reads across all human repeat types revealed enrichments of DNA DSBs within low-complexity repeats, satellite repeats and simple repeats (e.g., telomeres) (Figure S4D). In addition, DNA breaks were found to accumulate within centromere HORs (Figure S4E). Surprisingly, unlike cycling HCT116 cells (Figure 1E), centromere HOR DSBs in non-dividing cells were enriched uniformly across all chromosomes (Figure S4F). These observations collectively support the presence of spontaneous DNA DSBs within centromere HORs, even in non-dividing cells.
Topoisomerase IIβ induces spontaneous centromere HOR breaks
We next sought to identify sources of centromere HOR DNA breaks during quiescence. First, we assessed the impact of CENP-A in the generation of spontaneous DNA breaks, given the close spatial correlation between CENP-A and DNA break enrichment (Figure 1). The depletion of CENP-A protein levels after 96 hours RNAi treatment in asynchronous hTERT-RPE1 reduced CENP-A levels efficiently (Figure S5A). However, the depletion of CENP-A in serum-starved cells proved more challenging to detect by Western blot (Figure S5A), as previously reported36. This may be due to most CENP-A being stably incorporated in chromatin, with only a small proportion of CENP-A that is both centromeric and turning over during quiescence. Indeed, by assessing CENP-A levels within CENP-B-defined centromeres by immunofluorescence, we were able to detect a near total depletion in asynchronous cells (Figure S5B, C) and a partial depletion in serum-starved cells (Figure S5D, E), confirming that CENP-A was indeed lost from centromeres. Applying exo-FISH in these conditions, we found that there was no equivalent disruption in the ExoIII responsiveness of cenFISH probes after 96 hours CENP-A depletion in either condition (Figure S5F, G). Therefore, the innate DNA fragility of centromere HORs is unlikely related to the presence of CENP-A-containing nucleosomes.
We next tested the involvement of human topoisomerases. Topoisomerases are a family of enzymes, broadly categorized into type I and type II, that generate transient SSBs or DSBs, respectively, to facilitate transcription, DNA replication, chromatin remodeling and chromosome organization. In addition, topoisomerases have long-established links to centromere functionality, particularly during mitosis37–39. As such, topoisomerases served as likely candidates for the spontaneous DNA breaks observed within centromere HORs. Following 24 hours of serum starvation, hTERT-RPE1 were treated with siRNA targeting the five human nuclear topoisomerases (Figure 4A, B). Exo-FISH analysis 72 hours after siRNA treatment revealed that depletion of the type II topoisomerase IIβ (TOP2B), and partially the type I topoisomerase IIIα (TOP3A), nullified the ExoIII responsiveness of cenFISH probes, implying their involvement in generating de novo DNA breaks during quiescence (Figure 4C, D).
Figure 4 |. Topoisomerase IIβ induces centromere DNA breaks in quiescent cells.
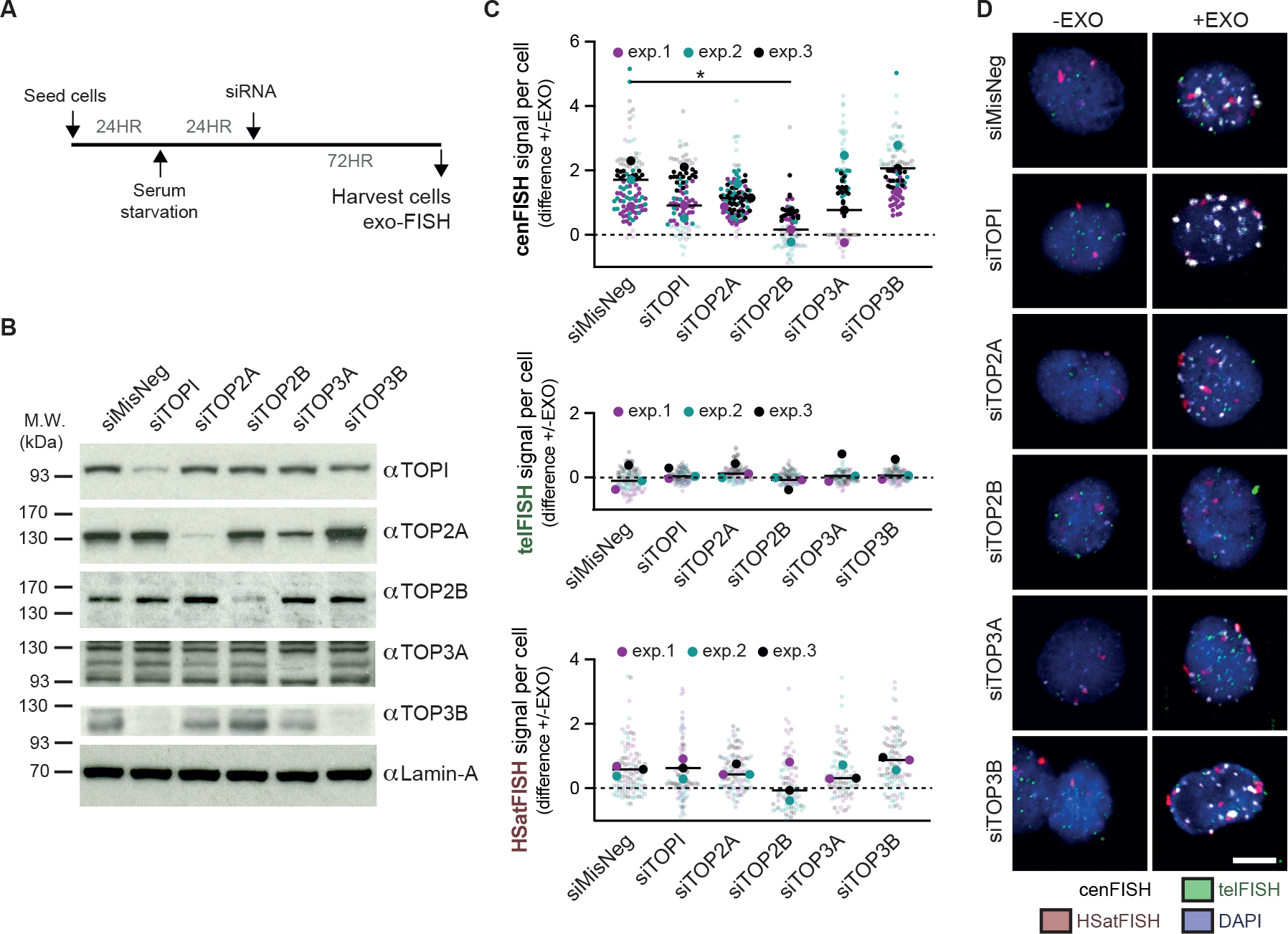
(A) Experimental schematic for the depletion of the five human nuclear topoisomerases in serum-starved hTERT-RPE1 cells. hTERT-RPE1 cells were serum-starved for 24 hours prior to RNAi treatment and harvested after another 72 hours. (B) Western blot verifying the depletion of topoisomerase I (TOPI), topoisomerase IIα (TOP2A), topoisomerase IIβ (TOP2B), topoisomerase IIIα (TOP3A) and topoisomerase IIIβ (TOP3B) 72 hours after RNAi treatment with 50 nM siRNA. Lamin-A levels were used as a loading control. (C, D) Quantification and representative images of exo-FISH following 72 hours depletion of human topoisomerases. Quantification was performed as in Figure 2. The difference in exo-FISH signals between +ExoIII and −ExoIII was then calculated and plotted. Scale bar represents 10 μm. FISH signal intensity is X10,000 arbitrary units (A.U.). At least 30 cells were imaged per experimental condition. The medians of each experimental condition were used to perform a two-sided unpaired t-test (*p<0.05, **p<0.01). See also Figure S5.
The recombinase RAD51 prevents the accumulation of centromere HOR DNA breaks
Having established that centromere HORs exhibit an unusually high degree of spontaneous DNA breakage in serum-starved cells, we next sought to determine whether these breaks could drive HR events. We reasoned that, if the spontaneous DNA breaks observed in centromere HORs serve as HR substrates, the impairment of HR would increase the level of centromere HOR DNA breaks.
The evolutionarily highly conserved RecA/RAD51 family of DNA recombinase plays an essential role both in homology search and strand exchange phases of HR40. In humans, RAD51 catalyzes DSB- and SSB-induced HR using double-stranded DNA as repair templates41. Hence, we focused on assessing the impact of RAD51 depletion on centromere HOR DNA breaks, particularly in non-dividing hTERT-RPE1 (Figure 5A). Few cells had incorporated EdU over 72 hours of siRNA exposure, confirming that cells were not actively replicating (Figure S6A). RAD51 depletion was verified with both western blotting (Figure 5B) and RT-qPCR (Figure S6B). In this condition, we indeed found that RAD51 depletion conferred an increase in DNA breaks at centromeres to a greater extent than telomeres or pericentromeric human satellite repeats (Figure 5C, D).
Figure 5 |. The recombinase RAD51 prevents the accumulation of centromere HOR DNA DSBs during quiescence.
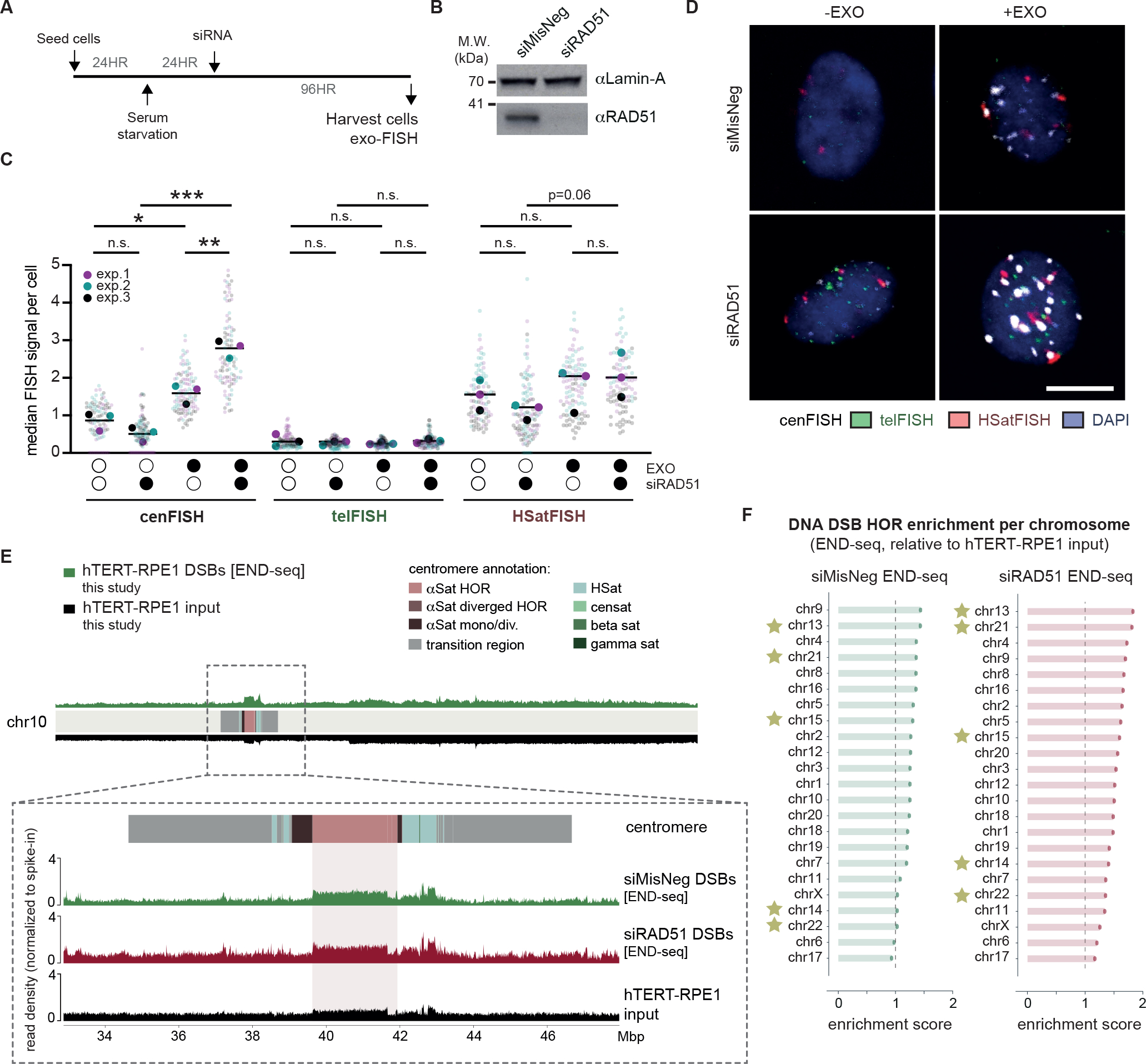
(A) Experimental schematic for the detection of centromeric breaks upon RAD51 depletion in quiescent hTERT-RPE1 cells. (B) Western blot confirming the depletion of RAD51 protein levels 72 hours after siRNA treatment. Lamin-A levels were used as a loading control. (C, D) Representative images and quantification of exo-FISH following RAD51 depletion. Quantification of FISH signals were performed as in Figure 2. Scale bar represents 10 μm. FISH signal intensity is X10,000 arbitrary units (A.U.). At least 30 cells were imaged per experimental condition. The averages of each experimental condition were used to perform a two-sided unpaired t-test (*p<0.05, **p<0.01). Filled and empty circles indicate presence and absence, respectively. (E) Inset of chr10 centromere depicting DNA DSBs (END-seq) following 96 hours depletion of RAD51 or the negative control in serum-starved hTERT-RPE1 cells. NGS alignment was performed with both PCR duplicate removal and spike-in normalisation. Baseline deviation of reads on the right arm of chr10 likely indicate some copy-number variation of hTERT-RPE1 relative to the T2T-CHM13 reference genome. (F) Enrichment END-seq scores across hTERT-RPE1 centromere HORs. NGS alignment was performed with both PCR duplicate removal and spike-in normalisation, and enrichment scores were quantified as in Figure 1. See also Figure S6.
To further dissect RAD51 activity during quiescence, we performed END-seq to detect DNA DSBs genome-wide, once again in serum-starved hTERT-RPE1. A spike-in was included to quantitively compare the total number of DNA breaks between samples accurately, and an input sample for hTERT-RPE1 was included to control for any copy-number variations of hTERT-RPE1 relative to the T2T-CHM13 reference genome. DNA DSBs were then detected as any enrichment of END-seq reads relative to the hTERT-RPE1 input. RAD51 depletion resulted in an increase of DNA DSBs within centromere HORs across all centromeres (Figure 5E, F). Importantly, these trends persisted regardless of whether PCR duplicate removal was included during NGS read alignment (Figure S6C, D). To compare DSB enrichments within HORs relative to the rest of the genome, we compared read enrichments within HORs to read enrichments across whole chromosomes excluding HORs. We observed a significant increase of DNA DSBs within HORs, and a further increase upon RAD51 depletion, once again regardless of PCR duplicate removal (Figure S6E). Finally, in agreement with the exo-FISH findings, RAD51 depletion had no impact on the levels of DNA breaks at telomeres (Figure S6F). However, END-seq analysis also revealed a role for RAD51 in protecting against DNA breaks in human satellite repeats, particularly of HSat1 arrays on acrocentric chromosomes (Figure S6F).
RAD51 strand-exchange activity required for the protection of centromere HORs
We hypothesized that the enhanced END-seq signal observed within centromere HORs in RAD51-depleted cells reflects DSBs that are left unrepaired by RAD51-mediated recombination. However, it was also conceivable that RAD51 protects against de novo DSB formation. To fully understand the mode of RAD51 function, we exploited a series of RAD51 separation-of-function variants, once again in serum-starved hTERT-RPE1 (Figure 6A). First, RAD51-K133R is a variant of RAD51 that binds ATP and is defective in ATP hydrolysis, thereby able to stably bind DNA and perform strand exchange, but unable to disassemble RAD51 filaments42. Second, RAD51-II3A contains three amino acid substitutions (R130A, R303A, K313A) within its second low affinity DNA binding interface (site II), which mediates homology searches over intact dsDNA during strand invasion. This variant retains its high-affinity ssDNA binding interface (site I), hence binds ssDNA with an affinity equivalent to WT RAD51, and is thereby able to protect ssDNA but unable to catalyze strand exchange43. Finally, the Fanconi anemia associated RAD51-T131P displays DNA-independent ATPase activity, and hence is unable to bind DNA on its own in any capacity, unless a similar level of WT RAD51 is present. It may exhibit reduced strand exchange activity, although its strand exchange activity requires the presence of significant amount of WT RAD51 (i.e., one fifth of T131P RAD51)44. Using these variants, we sought to determine whether RAD51 strand-exchange activity is required to prevent the accumulation of DNA breaks within centromere HORs.
Figure 6 |. RAD51 strand-exchange activity is required for the protection of centromere HORs in quiescent cells.
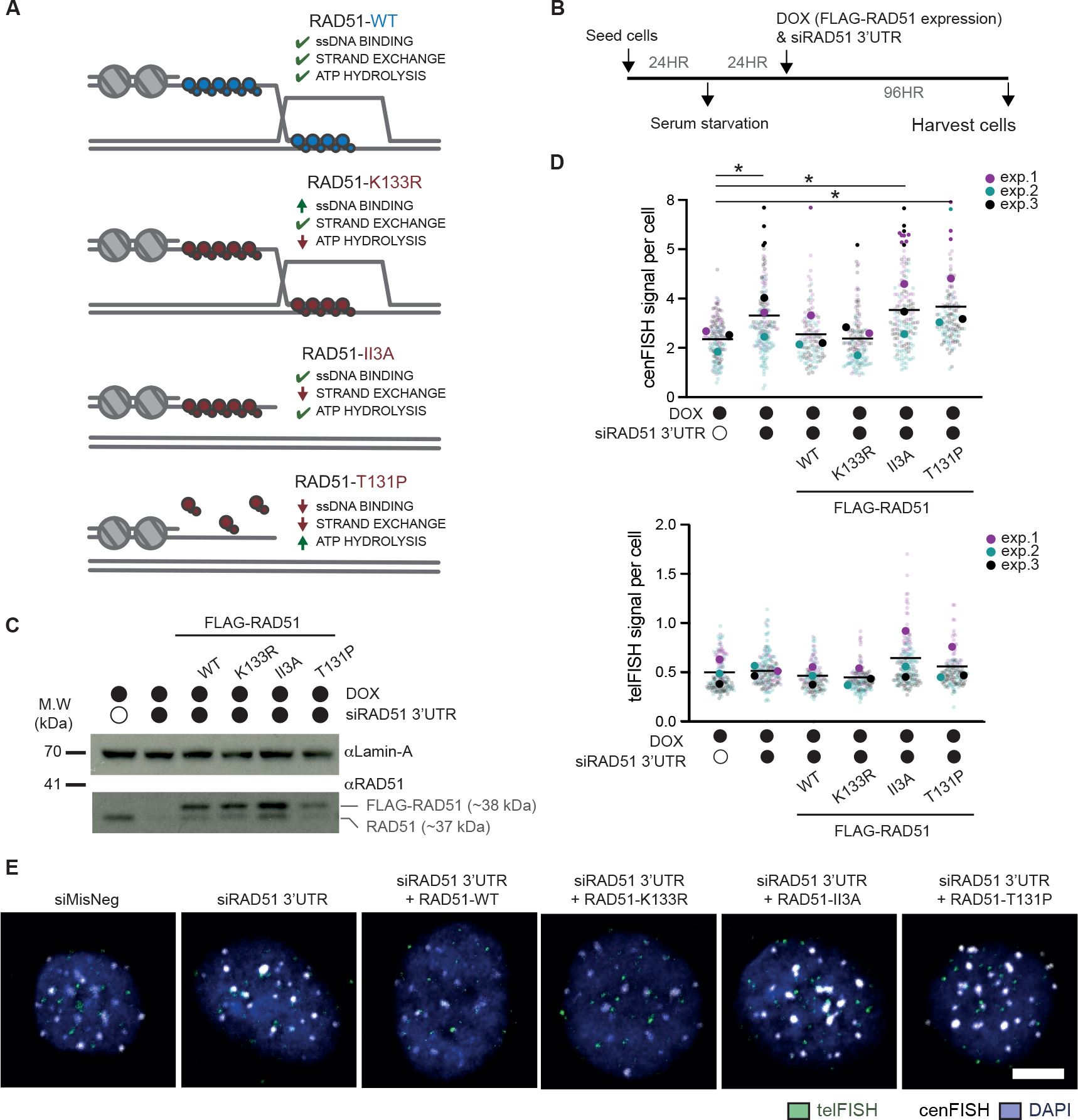
(A) Schematic of RAD51 separation-of-function variant functionalities. (B) Experimental schematic for the detection of centromeric breaks upon endogenous RAD51 depletion and expression of FLAG fusion of RAD51 variants in quiescent hTERT-RPE1 cells. (C) Western blot confirming the depletion of endogenous RAD51 (end. RAD51) 48 hours after siRNA treatment, and expression of separation-of-function variants upon doxycycline (DOX) treatment. Lamin-A levels were used as a loading control. (D, E) Quantification and representative images of exo-FISH following depletion of endogenous RAD51 and re-expression of the RAD51 separation-of-function variants, 96 hours after siRNA treatment in serum-starved hTERT-RPE1 cells. Quantification of FISH signals were performed as in Figure 2. Scale bar represents 10 μm. FISH signal intensity is X10,000 arbitrary units (A.U.). At least 30 cells were imaged per experimental condition. The averages of each experimental condition were used to perform a two-sided unpaired t-test (*p<0.05, **p<0.01). Filled and empty circles indicate presence and absence, respectively. See also Figure S7.
Cells were induced into quiescence and endogenous RAD51 was subsequently depleted using siRNA targeting the 3’UTR. We then asked whether re-expression of the FLAG-RAD51 variants could rescue the centromeric DNA damage phenotype (Figure 6B). In this condition, we observed an appearance of a faint band at the size of endogenous RAD51 (Figure 6C). This secondary band likely reflects the cleavage of FLAG-epitope from FLAG-RAD51 fusions, as it was equally apparent even when FLAG-RAD51 variants were induced 48 hours after siRNA treatment (Figure S7A). In support of this notion, all cell lines, except those complemented with wild-type (WT) FLAG-RAD51, exhibited reduced survival upon endogenous RAD51 depletion, confirming the functional impairment of those expressing FLAG-RAD51 variants (Figure S7B). The variants also exhibited expected phenotypes in RAD51 foci formation as a proxy of their ability to form stable RAD51 filaments. Re-expression of RAD51-WT, RAD51-K133R and RAD51-II3A variants, but not RAD51-T131P, were able to form RAD51 foci (Figure S7C, D).
We then used exo-FISH to determine the level of centromere HOR breaks in these serum starved cells (Figure 6D, E). While depletion of endogenous RAD51 increased centromere HOR breakage, expression of exogenous RAD51-WT or RAD51-K133R was able to rescue this phenotype to a similar degree. However, expression of RAD51-II3A or RAD51-T131P did not reduce the levels of centromere HOR breaks. These data highlight the importance of RAD51 strand exchange activity, which is selectively present in RAD51-WT and RAD51-K133R, at human centromere HORs. Conversely, the ability of RAD51 to bind single-stranded DNA, which is present in the RAD51-II3A variant, appears inconsequential to the overall levels of DNA breaks at centromere HORs. These data support the notion that both the homology-pairing and strand-exchange activities of RAD51 are critical for limiting the accumulation of centromere HOR DNA breaks.
RAD51 maintains CENP-A identity in serum-starved hTERT-RPE1 cells
The observation that the recombination activity of RAD51 suppresses spontaneous centromere HOR breaks in serum-starved cells provides critical experimental evidence for the proposed role of HR in centromere rearrangements and homogenization within functionally active centromere cores. However, it remains unclear why serum-starved cells tolerate such high levels of genome instability at centromeres. We therefore speculated that there may be some selective advantage at the cost of frequent breakage and recombination occurring within human centromere HORs.
One intriguing possibility was that frequent centromere HOR breaks, and subsequent recombination, could reinforce the specification (i.e., CENP-A occupancy) of centromeres. This hypothesis was founded on previous observations that CENP-A can be recruited to DSBs and, as part of the ‘expanding centromere model’, CENP-A occupancy often colocalizes with the source of array expansion and homogenization within the most recently emerged HOR arrays16,22,45,46. The spatial colocalization between CENP-A and array rearrangements suggests that CENP-A chromatin occupancy and centromeric recombination may be intimately linked.
We therefore sought to determine whether these data support a role for DNA breaks in driving CENP-A occupancy. First, we found that the levels of DNA breaks correlated with CENP-A chromatin occupancy across centromeres in asynchronous HCT116 (Figures 1 and S8A, B), supporting previous reports of CENP-A recruitment to DNA breaks. Importantly, long-term depletion of CENP-A in both asynchronous and serum-starved hTERT-RPE1 cells did not influence the levels of spontaneous DNA breaks within centromere HORs (Figure S5). Hence, this correlation was not a function of CENP-A occupancy itself. Second, we noticed a peculiar enrichment of spontaneous DNA breaks within the q arm of chromosome 8, as detected by GLOE-seq in HCT116 cells (e.g., Figure 1A). This DNA breakage hotspot was located between 8q21.1 and 8q21.3, a variable number tandem repeat identified as a site of recurrent neocentromere formation28,47. Mapping CENP-A ChIP-seq datasets from an 8q21 neocentromere cell line MS4221, we found a direct overlap with this break site (Figure S8C).
We next asked the potential involvement of HR in this relationship between DNA fragility and CENP-A occupancy. Indeed, in all conditions where spontaneous DNA breaks were detected (i.e., asynchronous HCT116 and both asynchronous and serum-starved hTERT-RPE1), we observed the striking reduction of CENP-A levels within CENP-B-defined centromeres in the absence of RAD51 (Figure 7A–F). Importantly, we detected no change in the levels of CENP-B in any condition, which acts as an internal control for fluorescence. Given that RAD51 limits centromeric DNA damage in serum-starved hTERT-RPE1 cells (Figure 5), we turned to serum-starved hTERT-RPE1 to further validate the relationship between RAD51 and CENP-A levels. RAD51 depletion did not significantly impact the total levels of CENP-A, as detected by western blotting (Figure 7E), nor CENP-A subcellular localization (Figure S8D). Regardless, as genome-wide DNA damage can lead to the mislocalization of CENP-A45,46, we considered a possibility that non-centromeric DNA damage induced upon RAD51 depletion might result in CENP-A mislocalization and a loss of CENP-A at core centromeres. However, quantification of the number of CENP-A foci co-localizing with CENP-B foci (i.e., foci centers within 5 pixels) reveal no obvious impact of RAD51 depletion on CENP-A mislocalization outside of centromere cores in serum-starved hTERT-RPE1 (Figure S8E). In addition, we observe no strong induction of γH2A.X, a marker for DNA damage, upon RAD51 depletion in serum-starved hTERT-RPE1 cells (Figure S8F). These data support a model wherein DNA breaks and subsequent recombination promote CENP-A levels within centromere HORs, distinct from previous proposals that DNA damage is sufficient for CENP-A deposition22,45.
Figure 7 |. Loss of centromere identity in the absence of RAD51.
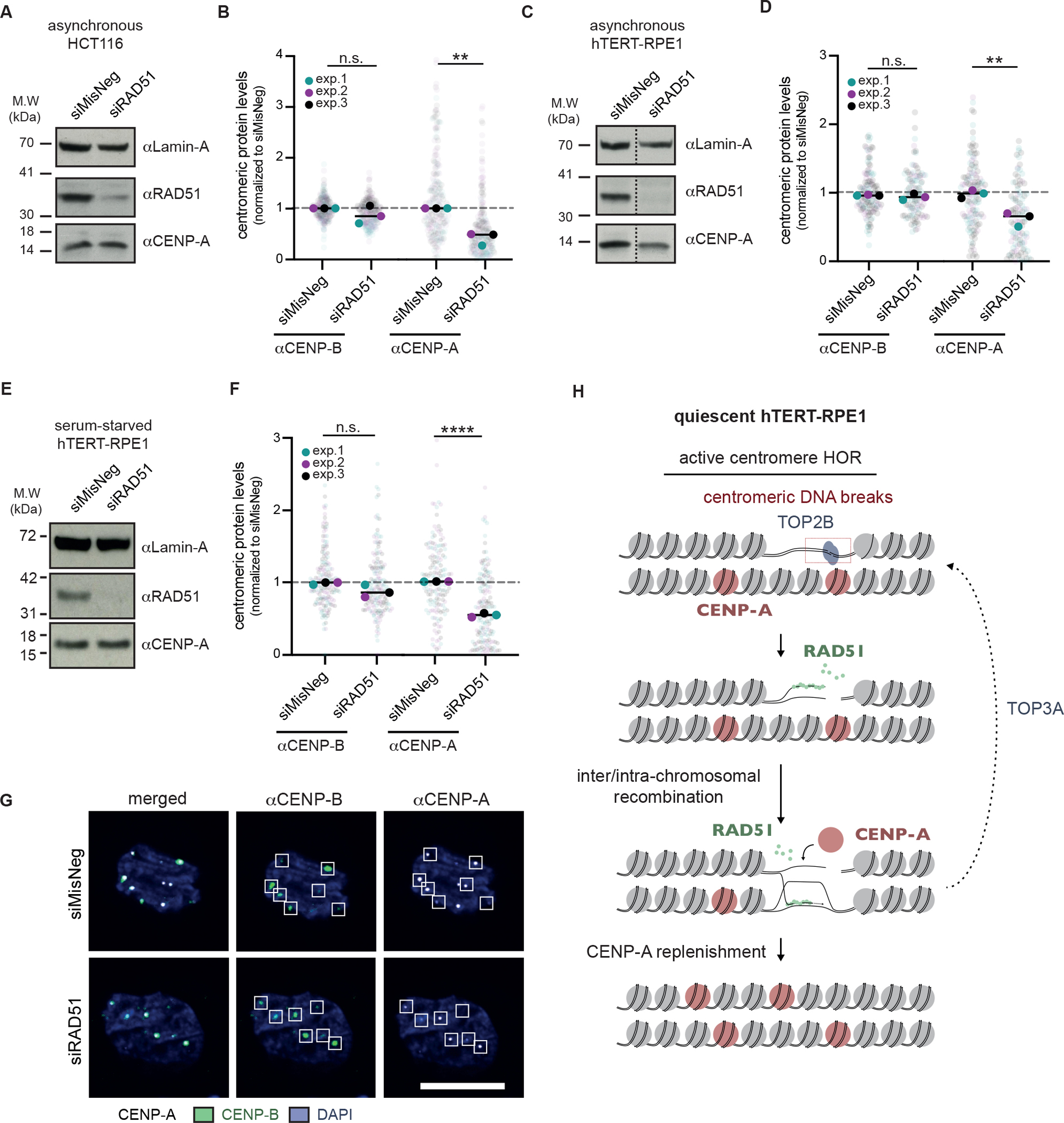
(A) Western blot confirming the depletion of RAD51 in asynchronous HCT116 cells, 72 hours after siRNA transfection. For all Western blots, Lamin-A levels were used as a loading control. (B) Quantification of CENP-A and CENP-B levels in asynchronous HCT116 cells, 96 hours after siRNA-mediated depletion of RAD51. CENP-B foci were used to define centromere loci, and quantification was performed as in Figure S5. (C) Western blot confirming the depletion of RAD51 in asynchronous hTERT-RPE1 cells, 96 hours after siRNA transfection. Dashed line indicates where the blot was cropped. (D) As in (B) but for asynchronous hTERT-RPE1 cells. (E) Western blot confirming the depletion of RAD51 in serum-starved hTERT-RPE1 cells. Cells were serum-starved for 24 hours prior to siRNA treatment and harvested 72 hours later. (F) As in (B) but for serum-starved hTERT-RPE1 cells. (G) Representative images of (F). At least 30 cells were imaged per experimental condition, and the medians of each experimental condition were used to perform a two-sided unpaired t-test (*p<0.05, **p<0.01). (H) Model for the role of RAD51-mediated recombination at spontaneous centromere HOR DNA breaks. See also Figure S8.
Discussion
Since 1976, active recombination processes at centromeres have been assumed in models of the rapid evolution of centromeric satellite arrays, proposed to originate within functionally active centromere cores12,18,19. In this work, we address two outstanding questions in the field. First, why are functionally active centromere cores specifically prone to recombination and rapid evolution? Second, does canonical RAD51-mediated recombination act at centromere cores, and if so, does it have a physiologically important function?
We tackled these questions by (1) assessing the genome-wide distribution of DNA breaks, as detected by NGS, against the latest complete human genome assembly, and (2) developing and applying the versatile exo-FISH technique to assess the frequency of DNA breaks within repetitive regions of the genome in single cells. These complementary approaches revealed that CENP-A-bound centromere HORs harbor a remarkable degree of DNA breaks even in non-cancerous and non-dividing human cells. An enrichment of DNA breaks at centromeres has been previously reported in S. cerevisiae, which, despite having point (i.e., non-repetitive) centromeres, are also rapidly evolving25,48. This highlights an evolutionary conservation of centromeric DNA fragility that appears to prevail even within non-repetitive DNA.
Spontaneous DNA breaks within centromere HORs are initially dependent on active DNA replication, but also induced de novo during cellular quiescence (Figure 3, Figure S3G–J). Replication-dependent and mitotic DNA breaks observed in asynchronous cells may reflect ssDNA gaps or Okazaki fragments associated with ongoing DNA replication and mitotic chromosome de-catenation, respectively. However, we speculate that de novo DNA breaks in quiescent cells are predominantly small nicks or double stranded breaks, as supported by our identification of topoisomerase IIβ as a source of centromere HOR DNA breaks during quiescence (Figure 4). Unlike the more extensively characterized topoisomerase IIα, topoisomerase IIβ is expressed in most adult tissues and the predominant topoisomerase II isoform expressed in quiescence49. The ubiquitous expression of topoisomerase IIβ conceivably underlies the universality of centromere HOR DNA breaks detected by both NGS and exo-FISH. Intriguingly, type II topoisomerases act as homodimers, where each monomer transiently introduces proximal nicks50. The dynamic nature of topoisomerase IIβ-induced DNA breaks, either as DNA nicks or DSBs, may explain why canonical DNA DSB response pathways (e.g., γH2A.X) have not been previously observed within centromere cores.
The observation of DNA break accumulation within centromere cores during quiescence is particularly interesting in light of the fact that mammalian oocytes will remain suspended in a prolonged state of quiescence prior to fertilization. Hence, it is tempting to speculate that centromere DNA breaks induced during quiescence may drive satellite array rearrangements that are subsequently passed through the germline (Figure S8G). In line with this notion, we further found that RAD51 suppresses centromere HOR DNA DSBs in quiescent hTERT-RPE1 cells, and its strand exchange activity is required for this functionality (Figure 5, 6). We therefore propose that DSBs or small nicks induced by topoisomerase IIβ within centromere HORs are targeted for RAD51-mediated recombination (Figure 7H).
Following recombination between centromere repeats, recombination intermediates may be resolved in either crossover or non-crossover modes, which would drive satellite arrays expansions/contractions and homogenization, respectively. We observed a partial impact of topoisomerase IIIα (Figure 4), which is widely described to act with BLM helicase to mediate non-crossover resolution of HR-intermediates, in the induction of DNA breaks in quiescent hTERT-RPE1 cells. Therefore, it seems that at least some of centromeric recombination intermediates are resolved in a non-crossover mode, which may assist the maintenance of centromere stability. An outstanding question remains as to the mechanisms in place driving crossover and non-crossover modes of intermediate resolution and their impact for centromere structure and stability.
Importantly, this work also provides mechanistic insight into the functional importance of DNA breaks within centromere HORs, as innate DNA fragility may be implicated with the epigenetic signaling of centromere identity. Previous studies have reported a relationship between exogenously induced DNA breaks and CENP-A recruitment22,45,46. In line with these studies, we found a correlation between centromeric DNA break enrichment and CENP-A occupancy within endogenous centromeres as well as spontaneous DNA breakage within neocentromere hotspots (Figure S8A, C). In quiescent cells, CENP-A loading also occurs with similar kinetics to DNA break induction, with accelerated CENP-A loading between 4 and 7 days after serum-starvation in hTERT-RPE1 cells36, corresponding to the onset of de novo DNA breaks in our system (Figure 3). Notably, active repair of centromeric DNA breaks appears to be important for the relationship between DNA breaks and CENP-A occupancy, as we observed the universal loss of centromeric CENP-A upon RAD51 depletion in both cycling and quiescent hTERT-RPE1 and HCT116 cells (Figure 7). We therefore propose that both DNA break incidence and active repair within centromere cores are important for the epigenetic signaling of centromere identity. It remains unclear what specifies CENP-A loading to RAD51-repaired breaks within centromere HORs, as opposed to other break-prone regions that are similarly repaired by RAD51 (e.g., HSat1 arrays). One possibility is that a combination of local transcription and chromatin accessibility within centromere HORs further specifies CENP-A loading, but further work will be required to clarify this question.
Beyond their implications in centromere evolution and signaling, our findings shed fresh light on genome instability originating at centromeres. Centromeres are frequently the origin of chromosome breaks across several tumor types. One intriguing possibility is that spontaneous DNA breakage may in fact drive frequent centromeric breakage, genome rearrangements and the onset of cancer. In this context, DNA breaks at centromeres may serve as a double-edged sword, reinforcing the identity of centromeres at the cost of potential genome instability.
Limitations of the study
The complete human genome assembly, used in this study, builds on long-read sequencing of the hTERT-CHM13 cell line, the genome of which is uniformly homozygous for one set of alleles and therefore effectively haploid. However, centromere DNA sequences are shown to be highly variable between cell lines16. Further, mapping short-read sequences to highly repetitive regions poses significant challenges, especially when NGS library preparation requires PCR amplification. Conversely, exo-FISH provides a versatile method to detect DNA breaks at repetitive regions of the genome on a single-cell level. Regardless, evidence from this study has highlighted some limitations in its application. First, increased exo-FISH signals detected upon endogenous RAD51 depletion were rescued in cells expressing RAD51-K133R, which catalyzes strand invasion and stabilizes the resultant structure (i.e., displacement loops, D-loops), but is unable to complete HR repair (Figure 6). In addition, telomere ends, which are considered naturally-occurring DNA breaks but are protected by telomere loops (T-loops), were also not responsive to Exonuclease III treatment using exo-FISH (e.g., Figure 2). These observations suggest that Exonuclease III only uses free DNA ends as substrates for resection, but not DNA ends protected by structures such as D-loops and T-loops. This property can, however, be exploited to distinguish free DNA ends and protected DNA ends. Second, exo-FISH relies on the enzymatic activities of Exonuclease III and is thereby limited to specific initial substrates. These include both SSBs and DSBs and, under the conditions in which chromatin-associated RNA molecules are preserved (i.e., untreated with RNase A), RNA-DNA hybrids through its RNaseH-like activity. In future studies, this shortcoming could be resolved by modifying exo-FISH to use alternative exonucleases probing different kinds of DNA lesions, for example using truncated Exonuclease VIII to specifically detect DSBs. Finally, exo-FISH provides information limited to specific regions of interest (ROI) and involves in vitro ExoIII-mediated resection from 3’ DNA ends, which, in theory, could start from those located at the vicinity of the ROI, proceeding into the ROI. As such, the true degree of enrichment of DNA strand breaks at centromere HOR regions and their precise locations remain unclear. Development of long-read sequencing-based methods to detect DNA damage sites, along with further releases of full genome sequences in various cell lines, such as hTERT-RPE1 and HCT116, will be instrumental to more precisely map DNA breaks within centromeres and centromere HORs.
STAR Methods
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Prof. Fumiko Esashi ( fumiko.esashi@path.ox.ac.uk ).
Materials availability
Plasmids and cell lines generated in this study are available upon request.
Data and code availability
The sequencing data generated in this study has been deposited to NCBI under BioProject ID PRJNA885500. This paper also analyses existing, publicly available data. These accession numbers for the datasets are listed in the Table S1. The source data generated and/or analysed in this study are included or referred to in the manuscript, or available in Mendeley Data with the identifier [doi:10.17632/65jt7xwr2p.1].
This paper does not contain original code.
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Cell lines
hTERT-immortalized retinal pigmented epithelial cells (hTERT-RPE1, RRID:CVCL_4388) were cultured in standard growth conditions (5% CO2, 37°C) in complete media (1:1 Dulbecco Modified Eagle Medium / Nutrient Mixture F12 supplemented with 10% fetal bovine serum (FBS) (F9665, Merck), 1% penicillin-streptomycin (15140122, Life Technologies) and 0.123% sodium bicarbonate (S8761, Sigma-Aldrich)). hTERT-RPE1 Flp-In T-REx were grown under similar conditions, including 100 μg/ml Zeocin (ant-zn-1, InvivoGen) and 10 μg/ml Blasticidin (ant-bl-1, InvivoGen). hTERT-RPE1 Flp-In T-REx cells expressing RAD51 separation-of-function variants were generated by transfecting cells with pDEST_FRT_TO plasmids containing FLAG, FLAG-RAD51-WT, FLAG-RAD51-K133R, FLAG-RAD51-T131P or FLAG-RAD51-II3A. Selection was done in 500 μg/ml G418 (G8168, Sigma-Aldrich), and maintained in 200 μg/ml G418 and 10 μg/ml Blasticidin. To induce variant expression, cells were incubated with complete media containing 1 μg/ml doxycycline (D9891, Sigma Aldrich) for at least 24 hours. HCT116 cells were cultured in standard growing conditions (5% CO2, 37°C) in McCoy’s 5A medium (26600080, Gibco) supplemented with 10% FBS and 1% penicillin-streptomycin. HeLa cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with high glucose (D6429, Sigma Aldrich) with 10% FBS and 1% penicillin-streptomycin.
Method details
Short-read sequencing analysis
Short-read sequencing data (Table S1) were trimmed using Trimmomatic (RRID:SCR_011848, v. 0.39)51 and read quality was verified using FASTQC (RRID:SCR_014583, v.0.11.5). Reads were then aligned to the CHM13v1.0 assembly 17 using BWA-ALN (RRID:SCR_010910)52 with high stringency (no mismatches allowed, -n 0). BWA-SAMSE and SAMtools (RRID:SCR_002105, v1.8)53 were used to convert aligned reads into BAM format. Where indicated, PCR duplicates were removed using SAMtools markdup -r. Using deepTools (RRID:SCR_016366, v. 3.5.1)54, read scores across chromosomal coordinates were generated after normalizing to RPKM for comparisons between datasets. To calculate enrichment scores across centromeres of each chromosome, the sums of RPKM-normalized read scores over CHM13v1.0 centromeric coordinates17 (available on https://github.com/marbl/CHM13) were compared to that of the relevant negative control or input sequence. To analyze spike-in reads, NGS reads were trimmed and aligned to the mouse reference genome (mm10) using Trimommatic, BWA-ALN, BWA-SAMSE and SAMtools, with the same parameters described above (no mismatches allowed, -n 0). Where indicated, PCR duplicates were removed using SAMtools markdup -r. Reads were then counted within the Zinc-finger nuclease spike-in locus (mm10 chr6:41,551,380–41,558,579) and normalised to siMisNeg END-seq to generate normalisation scaling factors for each sample, which were then applied genome-wide to END-seq datasets. For correlation analyses between GLOE-seq and CENP-A datasets, enrichment scores were calculated within HOR arrays rather than full centromeres using the same approach described above. To determine statistical significance, linear regression analyses were performed using GraphPAD Prism 8 for Mac OSX (RRID:SCR_002798, v.8.4.3) (www.graphpad.com). Python (RRID:SCR_008394, v.3.8.8)55, Matplotlib (RRID:SCR_008624, v.3.4.2)56, NumPy (RRID:SCR_008633, v.1.19.1)57, pandas (RRID:SCR_018214, v1.2.3)58 and pyGenomeTracks (v3.6)59,60 were all used for data processing and visualization.
Replication stress induction and serum starvation time course
For induction of replication stress, cells were seeded at ~200K per 6-well plated and treated for 24 hours with 0.4 μM aphidicolin (APH) (sc-201535, Santa Cruz) dissolved in DMSO. Cells were then harvested at the indicated time points and processed for flow cytometry or exo-FISH. When performing the serum starvation time course, technical variations of exo-FISH sample preparation was minimized by harvest samples at the same point. To this end, hTERT-RPE1 cells were seeded in serial dilutions and the media was changed to serum-starved media (0.1% FBS (F9665, Merck), 1% penicillin-streptomycin (15140122, Life Technologies) and 0.123% sodium bicarbonate (S8761, Sigma-Aldrich)) at the indicated time points. Cells were split if they approached confluence before serum starvation.
Plasmids
pDEST-FLAG_FRT_TO plasmids expressing FLAG, RAD51-WT, RAD51-K133R and RAD51-T131P were previously generated61. The hygromycin resistance cassette was switched for a neomycin resistance cassette to be used in the hTERT-RPE1 Flp-In T-REx cell line. RAD51-II3A point mutations (R130A, R303A, K131A) were introduced using site-directed PCR mutagenesis using primers listed in Table S2. All RAD51 variant sequence mutations were confirmed by DNA sequencing.
RNAi depletion
Lipofectamine RNAiMAX (13778075, ThermoFisher) was used for transfecting siRNA (Table S3) according to manufacturer’s recommendation. A final concentration of 20 nM siRNA was used for siRAD51, siCENP-A or the universal negative control siMisNeg (SIC001, Merck). For topoisomerase depletions, a final concentration of 50 nM siRNA was used (including the siMisNeg negative control). siRNA targeting topoisomerases were Silencer Select RNAi (4390824, ThermoFisher Scientific) targeting TOP1 (siRNA ID s14305), TOP2A (siRNA ID s14308), TOP2B (siRNA ID s106), TOP3A (siRNA ID s14311) and TOP3B (siRNA ID s17099). Cells were further incubated for 24 hours before changing media to fresh complete media and harvesting at the indicated times for analysis.
Western blotting & chromatin fractionation
To verify protein depletion by western blot, cell extracts were prepared using NETN150 buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM EDTA, 0.5% NP-40, 10 mM Benzamidine HCl, 10 mM NaF, 1 mM sodium glycerophosphate, 1 mM dithiothreitol, 25U/ml Benzonase nuclease supplemented with protease inhibit cocktail (P2714, Sigma-Aldrich)). Following separation by SDS-PAGE, standard western blotting procedures were used for the detection of RAD51 (1:2000, ab176458, Abcam), CENP-A (1:1000, GTX13939, Abcam) and Lamin-A (1:20 000, L1293, Sigma-Aldrich) (Table S4).
For chromatin fractionation, hTERT-RPE1 cells were seeded in 10-cm dishes. Following 24 hours of serum starvations, cells were treated with siRNA targeting RAD51. Media was changed 24 hours after siRNA treatment and incubated for a further 72 hours prior to harvesting. Cells were harvested by trypsinization, washed once in ice-cold PBS, and resuspended in PBS. Whole-cell extracts were prepared by taking an aliquot of this cell suspension, centrifuging, and resuspending in NETN150 buffer (100 μl per 10 mg cells). Following 30 minutes incubation, extracts were obtained by centrifuging 30 min (16,000 g) and collecting the supernatant. For fractionation, the remainder of the cell suspension was centrifuged and washed with ice-cold sucrose buffer (10 mM Tris-HCl pH 7.5, 20 nM KCl, 250 nM sucrose, 2.5 mM MgCl2, 10 mM benzamidine hydrochloride, 1 mM Na3VO4 supplemented with protease inhibitor cocktail) (100 μl ice-cold sucrose buffer per 10 mg cells). 0.2% Triton X-100 was added, and suspensions were vortexed three times, 10 seconds each. Nuclei were then pelleted by centrifugation (5 min, 500 g). Supernatants collected at this point were used as cytoplasmic fraction and left on ice. Nuclei pellets were then washed with 1X volume sucrose buffer, resuspended in 1X volume NETN150 and incubated on ice for 30 minutes. All fractions were then spun for 30 min (16,000 g) and supernatants were collected as nuclear fractions. All samples were prepared for Western blotting by adding 4X sample buffer (NP0007, Invitrogen) with 100 mM DTT, heating at 70°C for 10 minutes and loading ~10 μg protein per lane. Following separation by SDS-PAGE, standard western blotting procedures were used for the detection of RAD51 (1:2000, 7946, homemade), CENP-A (1:1000, GTX13939, Abcam), Lamin-A (1:20,000, L1293, Sigma-Aldrich), Tubulin (1:2000, 3873, Cell Signalling Technologies), and Histone H3 (1:2000 A300–823A-T, Bethyl Laboratories) (Table S4).
Immunofluorescence (IF)
Cells were harvested at the indicated times, counted and seeded at ~50K cells per 12-mm coverslip pre-treated with poly-L-lysine. Cells were left at 4°C for 10 minutes until firmly attached to coverslips and fixed in 4% PFA (28906, Pierce) in PBS at room temperature for 10 minutes. Samples were then quenched in 0.1M Tris-HCl pH 7.5 for 5 minutes, washed in PBS and stored for up to 2 weeks in PBS containing 0.04% NaN3 at 4°C. Slides were permeabilized in PBS-TX (PBS, 0.1% (v/v) Triton X-100) for 10 minutes, incubated in IF blocking buffer (PBS, 2% (v/v) FBS, 2% (w/v) BSA, 0.1% (v/v) Triton X-100, 0.04% (w/v) NaN3) for 30 minutes at 37°C, and incubated with primary antibodies (Table S4) diluted in IF blocking buffer for 1 hour at 37°C, both in a humidified chamber. Slides were then washed three times with PBS-TX, incubated with secondary antibodies (Table S5) diluted in IF blocking buffer for 1 hour at 37°C and washed again thrice in PBS-TX before mounting using ProLong mounting media containing DAPI (P10144, ThermoFisher Scientific). Slides were imaged with the Olympus FV1000 Fluoview Laser Scanning Microscope with Becker and Hickel FLIM system and FV10-ASW software (RRID:SCR_014215, v4.2), and cell aggregates were avoided when imaging. To ensure unbiased imaging and quantification, cells were selected by DAPI staining only and quantification was automated. Image processing and quantification are described below.
Exo-FISH
Cells were harvested, counted and swollen in 0.56% KCl at 20K cells / ml for 20 minutes, fixed in 3:1 methanol:acetic acid for 20 minutes and spread onto glass slides (12392138, Thermo Fisher Scientific). Since the density of cells can influence FISH signal intensity, a consistent number of cells (~20–50K) were spread homogenously on each slide. Slides were dried overnight at room temperature in the dark. Slides were then rehydrated in PBS for 5 minutes at room temperature, treated with 0.5 mg/ml RNaseA (R6513, Sigma-Aldrich) for 10 minutes at 37°C, and then incubated with 50–200 mU/μl Exonuclease III (M1811, Promega) for 30 minutes at 37°C in a humidified chamber. For experiments where HSatFISH was included (Figure 2C, 2D, 3B, 3C, 4C, 4D, 5C, 5D), a 100 μg/ml pepsin treatment in 0.1M HCl (37°C, 10 min) followed by a 10 minutes wash in PBS was included between RNaseA and Exonuclease III treatments. Slides were then serially dehydrated with 70%, 95% and 100% EtOH, 5 minutes each, and air-dried.
For FISH probe hybridization, dried slides were treated with 200 nM CENPBR-Cy3 (F3009, PNABio), 200 nM TelC-Cy5 (F1003, PNABio) and (optionally) 200 nM HSat2/3-FISH (custom made) diluted in hybridization solution (10 mM Tris-HCl pH 7.2, 70% formamide, 0.5% blocking solution (blocking solution: 10% blocking reagent (w/v, 11096176001, Roche) dissolved in maleic acid buffer (100 mM maleic acid, 150 mM NaCl, pH 7.5))) for 90–120 minutes at room temperature. FISH probe sequences are listed in Table S6. Slides were washed for 15 minutes in hybridization wash buffer #1 (10 mM Tris-HCl pH 7.2, 70% formamide, 0.1% BSA), followed by three times washes, 5 minutes each, in hybridization wash buffer #2 (0.1M Tris-HCl pH 7.2, 0.15 M NaCl, 0.08% Tween-20). Finally, slides were once again serially dehydrated in 70%, 95% and 100% EtOH, 5 minutes each, before air-drying and mounting on ProLong mounting media containing DAPI (P10144, ThermoFisher Scientific).
Slides were imaged with the Olympus FV1000 Fluoview Laser Scanning Microscope with Becker and Hickel FLIM system and FV10-ASW software (RRID:SCR_014215, v4.2), and cell aggregates were avoided when imaging using the DAPI channel. To ensure unbiased imaging and quantification, cells were selected by DAPI staining only and quantification was automated. Image processing and quantification are described below.
To isolate mitotic cells, hTERT-RPE1 cells were incubated with 5 μM S-trityl-L-cysteine (164739, Sigma-Aldrich) for 3–6 hours and collected by mitotic shake-off. For the validation of exo-FISH using restriction enzymes in vitro, cells were treated with 25 mU/μl (total 5U) BsmAI (R0529S, NEB) or Nt.BsmAI (R0121S, NEB) in 1X CutSmart Buffer (NEB) for 1 hour at 37°C in a humidified chamber, between the RNaseA digestion and the ExoIII treatment. The negative control was treated with 1X CutSmart Buffer alone.
END-seq
hTERT RPE1 cells were grown in DMEM-F12 media supplemented with 0.123% sodium bicarbonate (Sigma-Aldrich), 10% FBS (Sigma-Aldrich), and 1% pen-strep (Gibco). Upon reaching confluency, media was switched to serum-starvation media (0.1% FBS). One day later, cells were treated with siRNA targeting RAD51 (siRAD51), as described above. Control cells were treated with non-targeting siRNA. After 24 hours, media was replaced with fresh serum-starvation media. Cells were grown for three more days with media being replaced with fresh serum-starvation media every 24 hours. Following this, live cells were harvested and processed by END-seq as previously described26. Briefly, harvested cells were embedded in 1% low melting agarose plugs, which were subsequently treated with Proteinase K (50°C for 1 hour, followed by 37°C for 7 hours) and RNaseA (37 °C for 1 hour). DNA ends within plugs were then blunted using Exonuclease T and A-tailed using Klenow fragment. Biotinylated proximal hairpin adaptors with 3’ T overhangs and Illumina p5 primers were then used to selectively biotinylate A-tailed DNA breaks. Subsequently, agarose plugs were melted and sheared by sonication to produce DNA fragments (150–200 bp). Biotinylated DNA fragments were then captured with streptavidin beads. Captured DNA fragments were end-repaired using T4 DNA polymerase, Klenow fragment, T4 polynucleotide kinase and once again A-tailed using Klenow fragment. Distal hairpin adaptors containing 3’ T overhangs were then ligated to captured DNA fragments to introduce p7 Illumina primers, using the NEB Quick ligation kit. Finally, hairpins were digested using USER enzyme (NEB), and libraries were PCR amplified prior to Illumina sequencing. END-seq spike-ins were conducted using Abelson-transformed Lig4−/− mouse pre-B cells, carrying a doxycycline-inducible zinc-finger nuclease (ZFN) which can efficiently introduce a single DSB near the T-cell receptor β-chain (TCRβ) gene enhancer 62. Specifically, the mouse pre-B cells were arrested in G1 by treatment with 3 μM imatinib for 48 hours when cells were at a density of 1 × 106 cells/mL. To induce expression of the ZFN targeting the TCRβ spike-in locus (mm10 chr6:41,551,380–41,558,579), cells were treated with doxycycline at 1 μg/mL for 24 hours. Cells were then frozen and stored at −80°C until agarose plug preparation. During agarose plug preparation, spike-in mouse pre-B cells were added to the indicated RPE-1 cells so that 10% of all cells in each sample were spike-in cells, before proceeding with making agarose plugs as previously described26.
exo-FISH & IF quantification & statistical analysis
For exo-FISH: cenFISH, HSatFISH or telFISH foci were automatically detected using Fiji (RRID:SCR_002285, v. 2.0.0-rc-69/1.52p)63 based on an arbitrarily determined threshold (~200–400 arbitrary units). A 10×10–20×20 pixel box was generated around each focus and saved, after which the medial signal across all foci of a given cell was calculated to create a ‘median focus’ per cell. The median value of the perimeter readings was then used to estimate the background signal and this was subtracted from the median focus. Representative median foci were plotted using seaborn (v.0.11.1)64. For beeswarm plotting in Prism (RRID:SCR_002798, v.8.4.3) (www.graphpad.com), the sum of fluorescence signal within the background-subtracted median focus was calculated to encapsulate both focus intensity as well as size. For IF, the same approach as described above was used except CENP-B foci were automatically detected and used to define the 10×10–20×20 pixel box. The background-subtracted signal intensity of CENP-B as well as the secondary signal (e.g., CENP-A) was then calculated within the CENP-B-defined box. The proportion of CENP-A foci colocalizing with CENP-B foci was determined by calculating the proportion of CENP-A foci, as determined by a standardized threshold, being within 5 pixels of CENP-B foci for each cell. For total nuclear γH2A.X quantification, the total γH2A.X signal was calculated for each cell and divided by the cell size to determine mean signal.
For all analyses, student’s two-sided unpaired t-tests were used to compare the medians/averages of three biological replicates between experimental conditions using GraphPAD Prism 8 for Mac OSX (RRID:SCR_002798, v.8.4.3) (www.graphpad.com). p value ≤ 0.05 = *; ≤ 0.01 = **; ≤ 0.001 = ***; ≤ 0.0001 = ****. Since the sample size (n=3) for each statistical test was too small to perform normality tests, parametric tests were chosen. No adjustments for multiple comparisons were made since any data with multiple comparisons only contained positive controls as additional comparisons. Automated imaging analysis scripts available upon request. Python (RRID:SCR_008394, v.3.8.8)55, Matplotlib (RRID:SCR_008624, v.3.4.2)56, NumPy (RRID:SCR_008633, v.1.19.1)57, pandas (RRID:SCR_018214, v1.2.3)58 and seaborn (v.0.11.1)64 were all used for data processing and visualization.
Flow cytometry
To measure EdU incorporation, cells were incubated in 10 μM EdU in complete media and harvested at the indicated times by trypsinization. Cell pellets were then washed twice in PBS and fixed in 70% EtOH overnight at 4°C. On the following day, fixed cells were washed twice in PBS, followed by a resuspension in PBSTri-BSA (PBS, 0.1% Triton X-100, 1% BSA) and incubation on ice for 15 minutes. Cells were then washed thrice in PBST-BSA (PBS, 0.1% Tween 20, 1% BSA). The Click-iT reaction was used to label any incorporated EdU with a 1 hour incubation in Click-iT reaction buffer (PBS pH 7.2, 2 mM CuSO4, 10 mM sodium ascorbate (A4034, Sigma-Aldrich), 10 μM Alexa Fluor Azide (A20012, ThermoFisher Scientific)) at room temperature. Any remaining Click-iT solution was washed out with three washes in PBST-BSA, and cells were finally resuspended in DAPI staining solution (PBS, 0.1% BSA, 100 μl RNaseA and 2 μg/ml DAPI) prior to flow cytometry. For DNA content analysis upon replication stress treatment, cells were harvested as above, washed twice in PBST-BSA and incubated with DAPI staining solution as above. All flow cytometry samples were analyzed on the Cytoflex LX using CytExpert (RRID:SCR_017217, v.2.3.0.84) and data processed using FlowJo (RRID:SCR_008520, v10.6.2) (https://www.flowjo.com/). Single cells were selectively analyzed by SSC-A, FSC-A and FSC-W using standard gating strategies (as described in Figure S4A).
RT-qPCR
RNA was extracted from mitotic or quiescent populations using TRI reagent solution (AM9738, Thermo Fisher Scientific) according to manufacturer’s recommendations. Contaminating genomic DNA was removed by two successive rounds of DNase treatment using the TURBO DNA-free kit (AM1907, Invitrogen). RNA was then reverse-transcribed to cDNA using a high-capacity cDNA reverse transcription kit (4368814, Applied Biosystems), including a non-reverse transcribed (RT-) sample to detect any signal contributions from contaminating genomic DNA. The equivalent of 100 ng cDNA input was then used for the detection of RNA using primers listed in Table S7 with SensiFAST SYBR No-ROX kit (BIO-98005, Meridian Bioscience) on the Rotorgene Q Real-Time PCR System (Qiagen) using Q-Rex software (RRID:SCR_015740). Arbitrary thresholds were chosen to determine Ct, and relative RNA levels were calculated as 2−ΔCt(treatment-control).
Cell survival analysis by WST-1
To determine cell survival in the hTERT-RPE1 RAD51 variant lines, cells were seeded at 1K per well in 96-well plate in technical triplicate, with or without 1 μg/ml doxycycline where indicated. The following day, endogenous RAD51 was depleted using the protocol described above, and cells received fresh media with or without 1 μg/ml doxycycline the following day. Fresh media was given every second day. Another 96 hours later, cells were incubated with complete media containing 10% WST-1 (5015944001, Roche) for 1 hour at 37°C before plate absorbance reading at 450 and 650 nm. Cell survival was calculated based on 450 nm readings subtracted from the background (650 nm) and converted to a percentage of the negative control.
Supplementary Material
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-RAD51 | Abcam | Cat#ab176458; RRID: AB_2665405 |
| Rabbit anti-RAD51 (7946) | homemade | N/A |
| Rabbit anti-Lamin-A | Sigma-Aldrich | Cat#L1293; RRID: AB_532254 |
| Rabbit anti-CENP-B | Bethyl Laboratories | Cat#IHC-00064; RRID: AB_669682 |
| Mouse anti-CENP-A | Abcam | Cat #ab13939; RRID: AB_300766 |
| Mouse anti-CENP-A | GeneTex | Cat#GTC13939; RRID: AB_369391 |
| Rabbit anti-TOP1 | Abcam | Cat#ab109374; RRID: AB_10861978 |
| Rabbit anti-TOP2A | Cell Signaling Technology | Cat#12286; RRID: AB_2797871 |
| Rabbit anti-TOP2B | GeneTex | Cat#GTX102640-GTX-25ul; RRID: AB_11169314 |
| Rabbit anti-TOP3A | Proteintech | Cat#14525-1-AP; RRID: AB_2205881 |
| Rabbit anti-TOP3B | Biorbyt | Cat#ORB127293 |
| Mouse anti-Tubulin | Cell Signaling Technology | Cat#3873; RRID: AB_1904178 |
| Rabbit anti-Histone H3 | Bethyl Laboratories | Cat#A300-823A-T; RRID: AB_2118462 |
| Goat anti-rabbit Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A11070; RRID: AB_142134 |
| Goat anti-rabbit Alexa Fluor 555 | Thermo Fisher Scientific | Cat#A21430; RRID: AB_1500773 |
| Goat anti-rabbit Alexa 647 | Thermo Fisher Scientific | Cat#A27040; RRID: AB_2536101 |
| Goat anti-mouse Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A11017; RRID: AB_143160 |
| Goat anti-mouse Alexa Fluor 555 | Thermo Fisher Scientific | Cat#A21425; RRID: AB_1500751 |
| Goat anti-mouse Alexa 647 | Thermo Fisher Scientific | Cat#A21237; RRID: AB_1500743 |
| Goat anti-mouse HRP-conjugated | Agilent | Cat#P0447; RRID: AB_2617137 |
| Goat anti-rabbit HRP-conjugated | Agilent | Cat#P0448; RRID: AB_2617138 |
| Chemicals, peptides, and recombinant proteins | ||
| Exonuclease III | Promega | Cat#M1811 |
| Aphidicolin | Santa Cruz Biotechnology | Cat#sc-201535 |
| Lipofectamine RNAiMAX | Thermo Fisher Scientific | Cat#13778075 |
| FISH blocking solution | Roche | Cat#11096176001 |
| BsmAI | NEB | Cat# R0529S |
| Nt.BsmAI | NEB | Cat#R0121S |
| Critical commercial assays | ||
| TURBO DNA-free kit | Invitrogen | Cat#AM1907 |
| cDNA reverse transcription kit | Applied Biosystems | Cat#4368814 |
| Deposited data | ||
| Raw and analysed data | This paper | Mendeley Data DOI: 10.17632/65jt7xwr2p.1 |
| Raw sequencing data | This paper | PRJNA885500 |
| T2T-CHM13 reference genome | GitHub | https://github.com/marbl/CHM13 |
| Experimental models: Cell lines | ||
| hTERT-RPE1 | ATCC | RRID:CVCL4388 |
| hTERT-RPE1 Flp-In T-REx | Jonathon Pine | CancerTools.org:Cat#153242 |
| HCT116 | ATCC | RRID:CVCL0291 |
| HeLa | ATCC | RRID:CVCL0030 |
| Oligonucleotides | ||
| Universal negative control siMisNeg | Merck | Cat#SIC001 |
| siRAD51 #1 (5’ GACUGCCAGGAUAAAGCUU 3’) |
Custom order: Integrated DNA Technologies (IDT) | N/A |
| siRAD51 #2 (5’ GUGCUGCAGCCUAAUGAGA 3’) |
Custom order: Integrated DNA Technologies (IDT) | N/A |
| siRAD51 3’UTR #1 (5’ GACUGCCAGGAUAAAGCUU 3’) |
Custom order: Integrated DNA Technologies (IDT) | N/A |
| siRAD51 3’UTR #2 (5’ GUGCUGCAGCCUAAUGAGA 3’) |
Custom order: Integrated DNA Technologies (IDT) | N/A |
| siCENP-A (5’ GGACUCUCCAGAGCCAUGAUU 3’) |
Custom order: Integrated DNA Technologies (IDT) | N/A |
| Silencer Select RNAi targeting TOP1 | Thermo Fisher Scientific | Cat#4390824; siRNA ID s14305 |
| Silencer Select RNAi targeting TOP2A | Thermo Fisher Scientific | Cat#4390824; siRNA ID s14308 |
| Silencer Select RNAi targeting TOP2B | Thermo Fisher Scientific | Cat#4390824; siRNA ID s106 |
| Silencer Select RNAi targeting TOP3A | Thermo Fisher Scientific | Cat#4390824; siRNA ID s14311 |
| Silencer Select RNAi targeting TOP3B | Thermo Fisher Scientific | Cat#4390824; siRNA ID s17099 |
| II3A mutagenesis primers (R130A_F: 5’ AGAAATGTTTGGAGAATTCGCAACTGGGAAGACCCAGATC 3’) | Custom order: Integrated DNA Technologies (IDT) | N/A |
| II3A mutagenesis primers (R130A_R: 5’ GATCTGGGTCTTCCCAGTTGCGAATTCTCCAAACATTTCT 3’) | Custom order: Integrated DNA Technologies (IDT) | N/A |
| II3A mutagenesis primers (R303A_F: 5’ AACAACCAGATTGTATCTGGCGAAAGGAAGAGGGGAAACC 3’) | Custom order: Integrated DNA Technologies (IDT) | N/A |
| II3A mutagenesis primers (R303A_R: 5’ GGTTTCCCCTCTTCCTTTCGCCAGATACAATCTGGTTGTT 3’) | Custom order: Integrated DNA Technologies (IDT) | N/A |
| II3A mutagenesis primers (K131A_F: 5’ GGGGAAACCAGAATCTGCGCAATCTACGACTCTCCCTG 3’) | Custom order: Integrated DNA Technologies (IDT) | N/A |
| II3A mutagenesis primers (K131A_R: 5’ CAGGGAGAGTCGTAGATTGCGCAGATTCTGGTTTCCCC 3’) | Custom order: Integrated DNA Technologies (IDT) | N/A |
| RT-qPCR primer against RAD51 F: TCTCTGGCAGTGATGTCCTGGA R: TAAAGGGCGGTGGCACTGTCTA |
Custom order: Integrated DNA Technologies (IDT) | N/A |
| RT-qPCR primer against GAPDH F: CTGTTGCTGTAGCCAAATTCGT R: ACCCACTCCTCCACCTTTGAC |
Custom order: Integrated DNA Technologies (IDT) | N/A |
| Software and algorithms | ||
| Trimmomatic v0.39 | Bolger et al.51 | RRID:SCR_011848 |
| FASTQC v0.11.5 | Babraham Bioinformatics | RRID:SCR_014583 |
| BWA-ALN | Li52 | RRID:SCR_010910 |
| SAMtools v1.8 | Li et al.53 | RRID:SCR_002105 |
| deepTools v3.5.1 | Ramírez et al.54 | RRID:SCR_016366 |
| GraphPAD Prism v8.4.3 | N/A | RRID:SCR_002798 |
| Python v3.8.8 | Van Rossum and Drake Jr55 | RRID:SCR_008394 |
| Matplotplib v3.4.2 | Hunter56 | RRID:SCR_008624 |
| NumPy v1.19.1 | Harris et al.57 | RRID:SCR_008633 |
| Pandas v1.2.3 | McKinney58 | RRID:SCR_018214 |
| pyGenomeTracks v3.6 | Ramírez et al.59 Lopez-Delisle et al.60 |
N/A |
| Fiji v2.0.0 | Schindelin et al.63 | RRID:SCR_002285 |
| Seaborn v0.11.1 | Waskom64 | - |
| Olympus FV1000 Software FV10-ASW v4.2 | Olympus | RRID:SCR_014215 |
| Cytoflex LX CytExpert v2.3.0.84 | Cytoflex | RRID:SCR_017217 |
| FlowJo v10.6.2 | FlowJo | RRID:SCR_008520 |
| Q-Rex software | QIAGEN | RRID:SCR_015740 |
| Other | ||
| FISH probe against centromere HORs (Cy3) 5’ ATTCGTTGGAAACGGGA 3’ |
PNABio | Cat#F3009 |
| FISH probe against telomeres (Cy5) 5’ CCCTAACCCTAACCCTAA 3’ |
PNABio | Cat#F1003 |
| FISH probe against HSat2 (A488) 5’ TCGAGTCCATTCGATGAT 3’ |
Custom order from PNABio | N/A |
| FISH probe against HSat3 (A488) 5’ TCCACTCGGGTTGATT 3’ |
Custom order from PNABio | N/A |
| Publicly available NGS dataset: HCT116 GLOE-seq (Sriramachandran et al. 2020) | NCBI Sequence Read Archive | SRR9676440 |
| Publicly available NGS dataset: HCT116 END-seq (Canela et al. 2019) | NCBI Sequence Read Archive | SRR8870099 |
| Publicly available NGS dataset: HCT116 input (ENCODE) | NCBI Sequence Read Archive | SRR577511 |
| Publicly available NGS dataset: CHM13 CUT&RUN CENP-A (Logsdon et al. 2021) | NCBI Sequence Read Archive | SRR15395852 |
| Publicly available NGS dataset: CHM13 CUT&RUN IgG (Logsdon et al. 2021) | NCBI Sequence Read Archive | SRR15395848 |
| Publicly available NGS dataset: CHM13 ChIP-seq CENP-A (Logsdon et al. 2021) | NCBI Sequence Read Archive | SRR13278683 |
| Publicly available NGS dataset: CHM13 ChIP-seq input (Logsdon et al. 2021) | NCBI Sequence Read Archive | SRR13278681 |
| Publicly available NGS dataset: iNeuron END-seq (Wu et al. 2021) | NCBI Sequence Read Archive | SRR13764826 |
| Publicly available NGS dataset: iNeuron input (Wu et al. 2021) | NCBI Sequence Read Archive | SRR13764817 |
| Publicly available NGS dataset: MS4221 CENP-A ChIP-seq (Hasson et al. 2013) | NCBI Sequence Read Archive | SRR8870099 |
| Publicly available NGS dataset: MS4221 input ChIP-seq (Hasson et al. 2013) | NCBI Sequence Read Archive | SRR577511 |
Highlights.
DNA strand breaks are enriched within active centromere cores in human cells.
Centromere DNA breaks are newly induced in quiescent cells by Topoisomerase IIβ.
RAD51 enzymatic activity limits DNA strand breaks in quiescent cells.
RAD51 maintains centromere specification through CENP-A occupancy.
Acknowledgements
We thank Prof Jonathon Pines for the kind gift of the hTERT-RPE1 Flp-In T-REx cell line; Profs Lars Jansen, Daniele Fachinetti, Nicholas Proudfoot and Jordan Raff for the insightful discussion; Dr Nicola Zilio in Prof Helle Ulrich’s laboratory and Dr Wei Wu in the A.N. laboratory for their guidance on sequencing analysis; and Brian Leung and Jacob Wall in the F.E. laboratory for their assistance in sample preparations. This work was supported by the Medical Research Council (MR/W017601, to F.E.). F.E. was supported by the Wellcome Trust Senior Research Fellowships in Basic Biomedical Science (101009/Z/13/Z) and is thankful for supports from the Edward Penley Abraham Research Fund. X.S. was funded by the Oxford Cancer Centre Cancer Research UK D.Phil studentship (CRUK-OC-DPhil17-XS). E.G. is a recipient of the Medical Sciences Graduate School studentship, funded by the Medical Research Council (18/19_MSD_2111222). A.N and W.J.N. are supported by the Intramural Research Program of the NIH funded in part with Federal funds from the NCI under contract HHSN261201500003I.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wu JC, and Manuelidis L (1980). Sequence definition and organization of a human repeated DNA. J Mol Biol 142, 363–386. 10.1016/0022-2836(80)90277-6. [DOI] [PubMed] [Google Scholar]
- 2.Willard HF (1985). Chromosome-specific organization of human alpha satellite DNA. Am J Hum Genet 37, 524–532. [PMC free article] [PubMed] [Google Scholar]
- 3.Willard HF, and Waye JS (1987). Chromosome-specific subsets of human alpha satellite DNA: analysis of sequence divergence within and between chromosomal subsets and evidence for an ancestral pentameric repeat. J Mol Evol. 25, 207–214. [DOI] [PubMed] [Google Scholar]
- 4.Melters DP, Bradnam KR, Young HA, Telis N, May MR, Ruby JG, Sebra R, Peluso P, Eid J, Rank D, et al. (2013). Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biology 14, R10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marshall OJ, Chueh AC, Wong LH, and Choo KH (2008). Neocentromeres: new insights into centromere structure, disease development, and karyotype evolution. Am J Hum Genet 82, 261–282. 10.1016/j.ajhg.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hori T, Shang WH, Takeuchi K, and Fukagawa T (2013). The CCAN recruits CENP-A to the centromere and forms the structural core for kinetochore assembly. J Cell Biol 200, 45–60. 10.1083/jcb.201210106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mendiburo MJ, Padeken J, Fülöp S, Schepers A, and Heun P (2011). Drosophila CENH3 is sufficient for centromere formation. Science 334, 686–690. [DOI] [PubMed] [Google Scholar]
- 8.Murillo-Pineda M, Valente LP, Dumont M, Mata JF, Fachinetti D, and Jansen LET (2021). Induction of spontaneous human neocentromere formation and long-term maturation. J Cell Biol 220. 10.1083/jcb.202007210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McNulty SM, and Sullivan BA (2018). Alpha satellite DNA biology: finding function in the recesses of the genome. Chromosome Res 26, 115–138. 10.1007/s10577-018-9582-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masumoto H, Masukata H, Muro Y, Nozaki N, and Okazaki T (1989). A human centromere antigen (CENP-B) interacts with a short specific sequence in alphoid DNA, a human centromeric satellite. J Cell Biol 109, 1963–1973. 10.1083/jcb.109.5.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hudson DF, Fowler KJ, Earle E, Saffery R, Kalitsis P, Trowell H, Hill J, Wreford NG, de Kretser DM, Cancilla MR, et al. (1998). Centromere protein B null mice are mitotically and meiotically normal but have lower body and testis weights. J Cell Biol 141, 309–319. 10.1083/jcb.141.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balzano E, and Giunta S (2020). Centromeres under Pressure: Evolutionary Innovation in Conflict with Conserved Function. Genes (Basel) 11, 912. 10.3390/genes11080912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Talbert PB, Kasinathan S, and Henikoff S (2018). Simple and Complex Centromeric Satellites in Drosophila Sibling Species. Genetics 208, 977–990. 10.1534/genetics.117.300620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alexandrov I, Kazakov A, Tumeneva I, Shepelev V, and Yurov Y (2001). Alpha-satellite DNA of primates: old and new families. Chromosoma 110, 253–266. 10.1007/s004120100146. [DOI] [PubMed] [Google Scholar]
- 15.Henikoff S, Ahmad K, and Malik HS (2001). The centromere paradox: stable inheritance with rapidly evolving DNA. Science 293, 1098–1102. 10.1126/science.1062939. [DOI] [PubMed] [Google Scholar]
- 16.Altemose N, Logsdon GA, Bzikadze AV, Sidhwani P, Langley SA, Caldas GV, Hoyt SJ, Uralsky L, Ryabov FD, Shew CJ, et al. (2022). Complete genomic and epigenetic maps of human centromeres. Science 376, eabl4178. 10.1126/science.abl4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, Vollger MR, Altemose N, Uralsky L, Gershman A, et al. (2022). The complete sequence of a human genome. Science 376, 44–53. 10.1126/science.abj6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miga KH, and Alexandrov IA (2021). Variation and Evolution of Human Centromeres: A Field Guide and Perspective. Annu Rev Genet 55, 583–602. 10.1146/annurev-genet-071719-020519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith GP (1976). Evolution of repeated DNA sequences by unequal crossover. Science 191, 528–535. 10.1126/science.1251186. [DOI] [PubMed] [Google Scholar]
- 20.Giunta S, and Funabiki H (2017). Integrity of the human centromere DNA repeats is protected by CENP-A, CENP-C, and CENP-T. Proc Natl Acad Sci U S A 114, 1928–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaco I, Canela A, Vera E, and Blasco MA (2008). Centromere mitotic recombination in mammalian cells. J Cell Biol 181, 885–892. 10.1083/jcb.200803042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yilmaz D, Furst A, Meaburn K, Lezaja A, Wen Y, Altmeyer M, Reina-San-Martin B, and Soutoglou E (2021). Activation of homologous recombination in G1 preserves centromeric integrity. Nature 600, 748–753. 10.1038/s41586-021-04200-z. [DOI] [PubMed] [Google Scholar]
- 23.Onaka AT, Toyofuku N, Inoue T, Okita AK, Sagawa M, Su J, Shitanda T, Matsuyama R, Zafar F, Takahashi TS, et al. (2016). Rad51 and Rad54 promote noncrossover recombination between centromere repeats on the same chromatid to prevent isochromosome formation. Nucleic Acids Res 44, 10744–10757. 10.1093/nar/gkw874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vriend LE, and Krawczyk PM (2017). Nick-initiated homologous recombination: Protecting the genome, one strand at a time. DNA Repair (Amsterdam) 50, 1–13. [DOI] [PubMed] [Google Scholar]
- 25.Sriramachandran AM, Petrosino G, Mendez-Lago M, Schafer AJ, Batista-Nascimento LS, Zilio N, and Ulrich HD (2020). Genome-wide Nucleotide-Resolution Mapping of DNA Replication Patterns, Single-Strand Breaks, and Lesions by GLOE-Seq. Molecular Cell 78, 975–985 e977. 10.1016/j.molcel.2020.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong N, John S, Nussenzweig A, and Canela A (2021). END-seq: An Unbiased, High-Resolution, and Genome-Wide Approach to Map DNA Double-Strand Breaks and Resection in Human Cells. Methods Mol Biol 2153, 9–31. 10.1007/978-1-0716-0644-5_2. [DOI] [PubMed] [Google Scholar]
- 27.Canela A, Maman Y, Huang SN, Wutz G, Tang W, Zagnoli-Vieira G, Callen E, Wong N, Day A, Peters JM, et al. (2019). Topoisomerase II-Induced Chromosome Breakage and Translocation Is Determined by Chromosome Architecture and Transcriptional Activity. Molecular Cell 75, 252–266 e258. 10.1016/j.molcel.2019.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Logsdon GA, Vollger MR, Hsieh P, Mao Y, Liskovykh MA, Koren S, Nurk S, Mercuri L, Dishuck PC, Rhie A, et al. (2021). The structure, function and evolution of a complete human chromosome 8. Nature 593, 101–107. 10.1038/s41586-021-03420-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei KH, Lower SE, Caldas IV, Sless TJS, Barbash DA, and Clark AG (2018). Variable Rates of Simple Satellite Gains across the Drosophila Phylogeny. Mol Biol Evol 35, 925–941. 10.1093/molbev/msy005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benjamini Y, and Speed TP (2012). Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res 40, e72. 10.1093/nar/gks001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gershman A, Sauria MEG, Guitart X, Vollger MR, Hook PW, Hoyt SJ, Jain M, Shumate A, Razaghi R, Koren S, et al. (2022). Epigenetic patterns in a complete human genome. Science 376, eabj5089. 10.1126/science.abj5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altemose N, Maslan A, Smith OK, Sundararajan K, Brown RR, Mishra R, Detweiler AM, Neff N, Miga KH, Straight AF, and Streets A (2022). DiMeLo-seq: a long-read, single-molecule method for mapping protein-DNA interactions genome wide. Nat Methods 19, 711–723. 10.1038/s41592-022-01475-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Halasz L, Karanyi Z, Boros-Olah B, Kuik-Rozsa T, Sipos E, Nagy E, Mosolygo LA, Mazlo A, Rajnavolgyi E, Halmos G, and Szekvolgyi L (2017). RNA-DNA hybrid (R-loop) immunoprecipitation mapping: an analytical workflow to evaluate inherent biases. Genome Res 27, 1063–1073. 10.1101/gr.219394.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cristini A, Groh M, Kristiansen MS, and Gromak N (2018). RNA/DNA Hybrid Interactome Identifies DXH9 as a Molecular Player in Transcriptional Termination and R-Loop-Associated DNA Damage. Cell Rep 23, 1891–1905. 10.1016/j.celrep.2018.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu W, Hill SE, Nathan WJ, Paiano J, Callen E, Wang D, Shinoda K, van Wietmarschen N, Colon-Mercado JM, Zong D, et al. (2021). Neuronal enhancers are hotspots for DNA single-strand break repair. Nature 593, 440–444. 10.1038/s41586-021-03468-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Swartz SZ, McKay LS, Su KC, Bury L, Padeganeh A, Maddox PS, Knouse KA, and Cheeseman IM (2019). Quiescent Cells Actively Replenish CENP-A Nucleosomes to Maintain Centromere Identity and Proliferative Potential. Dev Cell 51, 35–48 e37. 10.1016/j.devcel.2019.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Porter AC, and Farr CJ (2004). Topoisomerase II: untangling its contribution at the centromere. Chromosome Res 12, 569–583. 10.1023/B:CHRO.0000036608.91085.d1. [DOI] [PubMed] [Google Scholar]
- 38.Norman-Axelsson U, Durand-Dubief M, Prasad P, and Ekwall K (2013). DNA topoisomerase III localizes to centromeres and affects centromeric CENP-A levels in fission yeast. PLoS Genet 9, e1003371. 10.1371/journal.pgen.1003371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bizard AH, Allemand JF, Hassenkam T, Paramasivam M, Sarlos K, Singh MI, and Hickson ID (2019). PICH and TOP3A cooperate to induce positive DNA supercoiling. Nat Struct Mol Biol 26, 267–274. 10.1038/s41594-019-0201-6. [DOI] [PubMed] [Google Scholar]
- 40.Baumann P, Benson FE, and West SC (1996). Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell 87, 757–766. 10.1016/s0092-8674(00)81394-x. [DOI] [PubMed] [Google Scholar]
- 41.Davis L, and Maizels N (2014). Homology-directed repair of DNA nicks via pathways distinct from canonical double-strand break repair. Proc Natl Acad Sci U S A 111, E924–932. 10.1073/pnas.1400236111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chi P, Van Komen S, Sehorn MG, Sigurdsson S, and Sung P (2006). Roles of ATP binding and ATP hydrolysis in human Rad51 recombinase function. DNA Repair (Amst) 5, 381–391. 10.1016/j.dnarep.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 43.Mason JM, Chan Y-L, Weichselbaum RW, and Bishop DK (2019). Non-enzymatic roles of human RAD51 at stalled replication forks. Nature Communications 10, 4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang AT, Kim T, Wagner JE, Conti BA, Lach FP, Huang AL, Molina H, Sanborn EM, Zierhut H, Cornes BK, et al. (2015). A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination. Molecular Cell 59, 478–490. 10.1016/j.molcel.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zeitlin SG, Baker NM, Chapados BR, Soutoglou E, Wang JY, Berns MW, and Cleveland DW (2009). Double-strand DNA breaks recruit the centromeric histone CENP-A. Proc Natl Acad Sci U S A 106, 15762–15767. 10.1073/pnas.0908233106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hedouin S, Grillo G, Ivkovic I, Velasco G, and Francastel C (2017). CENP-A chromatin disassembly in stressed and senescent murine cells. Sci Rep 7, 42520. 10.1038/srep42520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hasson D, Alonso A, Cheung F, Tepperberg JH, Papenhausen PR, Engelen JJ, and Warburton PE (2011). Formation of novel CENP-A domains on tandem repetitive DNA and across chromosome breakpoints on human chromosome 8q21 neocentromeres. Chromosoma 120, 621–632. 10.1007/s00412-011-0337-6. [DOI] [PubMed] [Google Scholar]
- 48.Bensasson D (2011). Evidence for a high mutation rate at rapidly evolving yeast centromeres. BMC Evol Biol 11, 211. 10.1186/1471-2148-11-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vavrova A, and Simunek T (2012). DNA topoisomerase IIbeta: a player in regulation of gene expression and cell differentiation. Int J Biochem Cell Biol 44, 834–837. 10.1016/j.biocel.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 50.Vos SM, Tretter EM, Schmidt BH, and Berger JM (2011). All tangled up: how cells direct, manage and exploit topoisomerase function. Nat Rev Mol Cell Biol 12, 827–841. 10.1038/nrm3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li H (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv 1303. [Google Scholar]
- 53.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van Rossum G, and Drake FL Jr (1995). Python reference manual. Centrum voor Wiskunde en Informatica; Amsterdam. [Google Scholar]
- 56.Hunter JD (2007). Matplotlib: A 2D Graphics Environment. Computing in Science & Engineering 9, 90–95. [Google Scholar]
- 57.Harris CR, Millman KJ, van der Walt SJ, Gommers R, Virtanen P, Cournapeau D, Wieser E, Taylor J, Berg S, Smith NJ, et al. (2020). Array programming with NumPy. Nature 585, 357–362. 10.1038/s41586-020-2649-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McKinney W (2010). Data Structures for Statistical Computing in Python. Proceedings of the 9th Python in Science Conference. [Google Scholar]
- 59.Ramirez F, Bhardwaj V, Arrigoni L, Lam KC, Gruning BA, Villaveces J, Habermann B, Akhtar A, and Manke T (2018). High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat Commun 9, 189. 10.1038/s41467-017-02525-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lopez-Delisle L, Rabbani L, Wolff J, Bhardwaj V, Backofen R, Gruning B, Ramirez F, and Manke T (2021). pyGenomeTracks: reproducible plots for multivariate genomic datasets. Bioinformatics 37, 422–423. 10.1093/bioinformatics/btaa692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wassing IE, Graham E, Saayman X, Rampazzo L, Ralf C, Bassett A, and Esashi F (2021). The RAD51 recombinase protects mitotic chromatin in human cells. Nat Commun 12, 5380. 10.1038/s41467-021-25643-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Canela A, Sridharan S, Sciascia N, Tubbs A, Meltzer P, Sleckman BP, and Nussenzweig A (2016). DNA Breaks and End Resection Measured Genome-wide by End Sequencing. Molecular Cell 63, 898–911. 10.1016/j.molcel.2016.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Waskom ML (2021). seaborn: statistical data visualization. Journal of Open Source Software 6, 3021. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data generated in this study has been deposited to NCBI under BioProject ID PRJNA885500. This paper also analyses existing, publicly available data. These accession numbers for the datasets are listed in the Table S1. The source data generated and/or analysed in this study are included or referred to in the manuscript, or available in Mendeley Data with the identifier [doi:10.17632/65jt7xwr2p.1].
This paper does not contain original code.
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.