

Abstract

A library of pyridine-based 1,2,4-triazolo-tethered indole conjugates were designed, synthesized, and evaluated for anti-proliferative activity against a panel of six human cancer cell lines. All the synthesized conjugates (14a–q) were found to be effective against the HT-29 cell line. Particularly conjugates 14a, 14n, and 14q exhibited promising cytotoxicity, with IC50 values of 1 μM, 2.4 μM, and 3.6 μM, respectively, compared to the standard 5-fluorouracil (IC50 = 5.31 μM). Cell cycle arrest at the G0/G1 phase was observed with these compounds, the mitochondrial membrane potential was interrupted, and the total ROS production was enhanced. Western blot and immunofluorescence experiments illustrated that these compounds inhibit the expression of markers that are involved in β-catenin and PI3K pathways. Molecular dynamics simulations demonstrated that compound 14a has major hydrophobic interactions and few H-bonding interactions with both PI3K and tankyrase proteins.
Keywords: Pyridine-based 1,2,4-triazolo-tethered indole conjugates; β-catenin pathway; anti-proliferative; PI3K/tankyrase inhibitors; colon cancer
Colorectal cancer (CRC) is the third most lethal and fourth most frequently diagnosed cancer around the globe, according to GLOBOCAN 2020 data, and the prevalence of the disease is rising.1,2 The likelihood of having CRC is 4–5%, and factors like age, family history of chronic diseases, and lifestyle have been linked to a higher chance of developing the disease.3 Timely identification of the related gene mutations may aid to prevent progression of the disease. Several mutated genes, such as AXIN, Wnt (Wingless-related integration site), GSK3β (glycogen synthase kinase-3 beta), PI3K (phosphoinositide 3-kinase), etc., are mainly responsible for CRC.4
It is well established that mutations in the components of the Wnt/β-catenin pathway very often lead to aberrant activation of this pathway, thereby resulting in the overexpression of target genes that are responsible for cell proliferation, growth, apoptosis, metastasis, and cell-cycle control.5−7 In the cytoplasm, overexpressed β-catenin can form a destruction complex with a multitude of proteins, including APC (adenomatous polyposis coli), AXIN2, GSK3β, and CK1α. In the absence of Wnt, β-catenin is phosphorylated by GSK3β and undergoes proteasomal degradation, resulting in cell apoptosis.8 Meanwhile, in the presence of Wnt, the activated Frizzled receptor activates a Dishevelled protein that disrupts the destruction complex through TNKS, which interacts with the complex at the AXIN binding site.9 The disruption of the destruction complex results in the release of β-catenin into the nucleus, which leads to abnormal cell proliferation.10
On the other hand, the PI3K catalytic alpha polypeptide recurrently mutates and activates the downstream signaling molecule Akt. Activated Akt arbitrates cell survival, proliferation, and migration.11 After phosphorylation, the activated Akt inactivates other apoptogenic factors, including GSKβ, thereby inhibiting programmed cell death.12 The dual inhibition of PI3K/Akt and tankyrase 1/2 leads to programmed cell death through the stabilization of the β-catenin destruction complex via GSKβ.13
Several natural and synthetic compounds bearing scaffolds like indoles,14 1,2,4-triazoles,15 pyridines,16 and quinazolines17 (see Figure 1) have individually exhibited the inhibition of either Wnt/β-catenin (tankyrase) or PI3K pathways.18 When specific tankyrase 1/2 (8, G007-LK) and PI3K (2, BKM120) inhibitors have been applied in combination, the dual inhibition of both targets has been observed in CRC cell lines.19 Similarly, different combinations of TNKS (7, NVP-TNKS656) along with diverse PI3K inhibitors were found to block the Wnt/β-catenin pathway and revert the resistance to PI3K and AKT inhibition synergistically in CRC.20 Further, 7-O-succinylmacrolactin A (5, SMA) acted as a dual inhibitor for PI3K/Akt and TNKS pathways in vitro and in vivo.21 XAV-939 (compound 3), a small-molecule tankyrase inhibitor) was found to inhibit the disruption of the β-catenin destruction complex, thereby reducing the nuclear concentration of β-catenin and sensitizing the resistant cells specifically to PI3K and AKT inhibitors in CRC.22
Figure 1.

Some PI3K, tankyrase, and dual inhibitors under current clinical development.
Due to the lack of potential dual inhibitors (for both PI3K and tankyrase), researchers have been using combination therapies for the potential inhibition of PI3K and tankyrases in the treatment of CRC. Nowadays, there is an immense interest in the development of such dual inhibitors. In this regard, we attempted to develop some newer dual inhibitors of tankyrase and PI3K. Therefore, in view of the biological importance of 1,2,4-triazoles as tankyrase inhibitors, a virtual library of compounds was designed based on 1,2,4-triazoles tethered to pyridine as well as indole scaffolds. The rational design for the targeted molecules through the hybrid conjugation of the bioactive conjugate is depicted in Figure 2.
Figure 2.

Rational design for targeted bioactive conjugates.
In the present studies, based on 1,2,4-triazole-tethered pyridine derivatives and different bioactive indole scaffolds, more than 150 molecules were rationally designed by using the PubChem and ZINC15 databases (Table S1). The designed molecules that passed through the PAINS filter were subjected to virtual screening (see Figure 3) against the target proteins tankyrase (PDB ID: 4OA7) and PI3K (PDB ID: 3L54) to identify the potentiality of the aforementioned molecules in CRC. Initially, the docking procedure was confirmed by redocking the co-crystallized ligand at its reported binding sites and then computing the RMSD values of 0.53 Å and 0.54 Å compared with the X-ray structure. All the virtually designed processes and docking protocols are discussed in the materials and methods section in the Supporting Information (SI). More than 100 compounds were discarded based on very low docking scores (< –3.5 kcal/mol).23 Some of the compounds were dismissed because of non-compliance with Lipinski’s Rule of 5, and a few compounds were not considered because of a lack of synthetic feasibility. Finally, 17 compounds were selected based on the docking scores and the feasibility of synthesis. Among them, compounds 14a, 14n, and 14q have shown the best glide score in the active sites of tankyrase and PI3K proteins. Docking scores and MM-GBSA analysis of these compounds are illustrated in Table 1. 2D as well as 3D overlays of ligands 14a, 14n, and 14q with proteins 4OA7 and 3L54 are depicted in the SI, Figure S1.
Figure 3.

Structure-based virtual screening workflow.
Table 1. Docking Scores, Glide Energies, and Free Energy Calculations of Synthesized Compounds against Tankyrase.
| entry | compd | Xp G score (kcal/mol), 4OA7 | glide energy (kcal/mol) | MM-GBSA ΔGbind (kcal/mol) | interactions of tankyrase 1 in complex with IWR1 at the ligand binding site, 4OA7 |
|---|---|---|---|---|---|
| 1 | 14a | –9.747 | –55.234 | –57.19 | π–π: PHE 1188, HID 1201 |
| 2 | 14b | –6.457 | –38.711 | –55.04 | H-Bond: ASP 1198 |
| π–π: HIE 1201, TYR 1224 | |||||
| 3 | 14c | –8.889 | –53.135 | –53.64 | H-Bond: GLY 1185, ASP 1198 |
| π–π: HIE 1201, TYR 1213, TYR 1224 | |||||
| 4 | 14d | –7.033 | –38.204 | –37.48 | H-Bond: TYR 1203 |
| π–π: PHE 1188, HIE 1201 | |||||
| 5 | 14e | –8.097 | –41.346 | –44.59 | π -π: PHE 1188, TYR 1224 |
| 6 | 14f | –8.829 | –39.425 | –40.86 | H-Bond: ASP 1198 |
| π–π: HIE 1201, TYR 1224 | |||||
| 7 | 14g | –7.376 | –38.884 | –38.29 | π -π: PHE 1188 |
| 8 | 14h | –7.476 | –40.172 | –46.79 | H-Bond: TYR 1203 |
| π–π: HIS 1184, HIE 1201, TYR 1213, TYR 1224 | |||||
| 9 | 14i | –5.78 | –30.16 | –54.29 | π–π: HIE 1201, TYR 1203, TYR 1213 |
| 10 | 14j | –8.01 | –45.253 | –47.31 | π–π: TYR 1213, TYR 1224 |
| π–cation: LYS 890 | |||||
| 11 | 14k | –8.934 | –48.807 | –47.44 | H-Bond: GLY 1211 |
| π–π: PHE 1188, TYR 1224 | |||||
| 12 | 141 | –6.54 | –40.963 | –45.23 | π–π: PHE 1188, TYR 1224 |
| 13 | 14m | –9.105 | –35.82 | –44.64 | H-Bond: TYR 1203 |
| π–π: PHE 1188, HIE 1201 | |||||
| 14 | 14n | –9.747 | –55.234 | –57.19 | H-Bond: GLY 1196 |
| π–π: HIE 1201 | |||||
| 15 | 14o | –6.457 | –38.711 | –55.04 | H-Bond: GLY 1211 |
| π–π: PHE 1188, TYR 1224 | |||||
| 16 | 14p | –8.889 | –53.135 | –53.64 | π–π: PHE 1188 |
| 17 | 14q | –7.033 | –38.204 | –51.48 | H-Bond: GLY 1211 |
| π–π: PHE 1188 |
The protein 4OA7 showed interactions with key amino acids within the active site of tankyrase—both hydrophobic and hydrophilic contacts were seen between active conjugates and tankyrase protein. Conjugate 14a demonstrated π–π stacking with amino acid residues PHE 1188 and HID 1201 (Figure S1A). It exhibited a glide score of −9.747 and ΔGfree binding energy of −57.19 kcal/mol as calculated by prime MM-GBSA and MM-GBSA ΔGbind. Similarly, 14n interacted with the amino acid residue TYR 1213 via π–π stacking in chain A, as shown in Figure S1B, whereas 14q displayed H-bonding with GLY 1211 in addition to π–π stacking with amino acid residues PHE 1188 and TYR 1224 (Figure S1C). Conjugates 14n and 14q exhibited docking scores of −9.679 kcal/mol and −9.317 kcal/mol and free binding energies of −55.12 kcal/mol and −53.89 kcal/mol, respectively, as calculated by prime MM-GBSA and MM-GBSA ΔGbind (Table 1).
On the other hand, PDB ID 3L54, a PI3K (phosphoinositide 3-kinase) protein, and compound 14a exhibited interactions at amino acid residues TYR 867 and HIS 967 via π–π stacking, along with amino acid residues LYS 890 and ASP 950 through H-bonding with a bond length of 1.7 Å (Figure S1D). Similarly, conjugate 14n exhibited interactions with amino acid residue TYR 867 via π–π stacking in chain A, as shown in Figure S1E. It displayed a glide score of −6.59 kcal/mol and ΔGfree binding energy of −53.57 kcal/mol as calculated by prime MM-GBSA and revealed by MM-GBSA ΔGbind. Moreover, 14q displayed H-bonding with amino acid residues LYS 890 and ASP 950 and π–π stacking with TYR 867 and HIS 967 (Figure S1F). The 14n and 14q conjugates exhibited docking scores of −6.052 kcal/mol and −5.971 kcal/mol and free binding energies of −49.04 kcal/mol and −48.41 kcal/mol, respectively, as calculated by prime MM-GBSA and MM-GBSA ΔGbind (Table 2).
Table 2. Docking Scores, Glide Energies, and Free Energy Calculations of Synthesized Conjugates against PI3K.
| entry | compd | Xp G score (kcal/mol), 3L54 | glide energy (kcal/mol) | MM-GBSA ΔGbind (kcal/mol) | interactions of Pi3K gamma at the ligand binding site, 3L54 |
|---|---|---|---|---|---|
| 1 | 14a | –6.59 | –46.288 | –53.57 | H-Bond: LYS 890, ASP 950 |
| π–π: TYR 867, HIS 967 | |||||
| 2 | 14b | –5.065 | –45.715 | –47.66 | H-Bond: ASP 950 |
| π–π: TYR 867 | |||||
| π–Cation: LYS 890 | |||||
| 3 | 14c | –5.04 | –33.231 | –37.02 | H-Bond: ASP 950 |
| π–π: HIS 967 | |||||
| 4 | 14d | –5.918 | –41.972 | –42.92 | π–π: TYR 867 |
| Halogen Bond: SER 806 | |||||
| π–Cation: LYS 890 | |||||
| 5 | 14e | –3.732 | –40.986 | –52 | π–π: TRP 812 |
| 6 | 14f | –3.714 | –42.793 | –49.11 | H-Bond: ASP 950, VAL 882 |
| π–Cation: LYS 890 | |||||
| 7 | 14g | –5.039 | –33.229 | –43.89 | H-Bond: VAL 882, ASP 950 |
| π–π: HIS 967 | |||||
| 8 | 14h | –6.037 | –36.938 | –42.37 | H-Bond: VAL 882 |
| π–π: TYR 867 | |||||
| π–Cation: LYS 890 | |||||
| 9 | 14i | –4.103 | –43.35 | –49.64 | H-Bond: VAL 882, LYS 890 |
| 10 | 14j | –5.347 | –43.162 | –35.65 | π–π: TYR 867 |
| π–Cation: LYS 890 | |||||
| 11 | 14k | –5.376 | –32.939 | –47.04 | H-Bond: LYS 890, ASP 950 |
| π–π: TYR 867, HIS 967 | |||||
| 12 | 14l | –3.714 | –42.793 | –40.48 | H-Bond: ASP 950 |
| π–π: TYR 867 | |||||
| Halogen Bond: THR 887, LYS 890 | |||||
| π–Cation: LYS 890 | |||||
| 13 | 14m | –3.651 | –40.466 | –51.61 | H-Bond: VAL 882 |
| π–π: LYS 890 | |||||
| 14 | 14n | –6.052 | –45.653 | –49.04 | H-Bond: VAL 882 |
| π–π: LYS 890 | |||||
| 15 | 14o | –5.487 | –41.338 | –43.21 | H-Bond: VAL 882, LYS 890 |
| 16 | 14p | –5.673 | –46.366 | –42.7 | H-Bond: VAL 882 |
| π–π: TYR 867 | |||||
| 17 | 14q | –5.971 | –44.729 | –48.41 | H-Bond: LYS 890, ASP 950 |
| π–π: TYR 867, HIS 967 |
It is well known that most drug candidates fail in clinical trials because of their poor ADME profiles. Molecules need to possess a favorable pharmacokinetic profile and as well as biological activity to be deemed as potential therapeutic candidates. ADMET studies of our library compounds were performed by using the QikProp tool. Here, the “drug-likeness” metric was evaluated using Lipinski’s Rule of 5. All parameters of tested molecules were within the permitted limits. QikProp results suggested that the identified hits have drug-like physicochemical parameters which are illustrated in Table 3.
Table 3. In Silico ADME Profiles of Synthesized Conjugates.
| entry | compd | molecular weight | QPlogP o/w | HB donor | HB acceptor | SASA | % human absorption | Lipinski’s rule of 5 |
|---|---|---|---|---|---|---|---|---|
| 1 | 14a | 427.523 | 4.427 | 1 | 4.25 | 696.238 | 100 | 0 |
| 2 | 14b | 377.506 | 4.098 | 1 | 3.5 | 670.818 | 100 | 0 |
| 3 | 14c | 361.464 | 3.87 | 1 | 3.5 | 659.261 | 100 | 0 |
| 4 | 14d | 431.942 | 4.874 | 1 | 3.5 | 688.014 | 97.5 | 0 |
| 5 | 14e | 411.523 | 4.682 | 1 | 3.5 | 692.761 | 100 | 0 |
| 6 | 14f | 363.479 | 3.912 | 1 | 3.5 | 659.679 | 100 | 0 |
| 7 | 14g | 476.393 | 4.891 | 1 | 3.5 | 680.138 | 100 | 0 |
| 8 | 14h | 425.55 | 5.081 | 1 | 3.5 | 740.976 | 100 | 1 |
| 9 | 14i | 476.396 | 4.603 | 1 | 3.5 | 677.93 | 100 | 0 |
| 10 | 14j | 442.497 | 3.636 | 1 | 3.5 | 610.415 | 100 | 0 |
| 11 | 14k | 415.49.4 | 4.218 | 1 | 3.5 | 658.182 | 100 | 0 |
| 12 | 14l | 431.942 | 4.916 | 1 | 3.5 | 700.478 | 100 | 0 |
| 13 | 14m | 397.500 | 4.569 | 1 | 3.5 | 681.993 | 100 | 0 |
| 14 | 14n | 415.487 | 4.446 | 1 | 3.5 | 679.823 | 100 | 0 |
| 15 | 14o | 411.527 | 4.927 | 1 | 3.5 | 728.317 | 100 | 0 |
| 16 | 14p | 411.523 | 4.916 | 1 | 3.5 | 700.478 | 100 | 0 |
| 17 | 14q | 427.523 | 4.071 | 1 | 4.25 | 631.032 | 100 | 0 |
These pyridine-based 1,2,4-triazolo-tethered indole conjugates (14a–q) were synthesized by using a multi-step synthetic strategy starting from isonicotinohydrazide (9), as depicted in Scheme 1.
Scheme 1. Synthesis of Pyridine-Based 1,2,4-Triazolo-Tethered Indole Conjugates (14a–q).

Compound 9 was treated with various aromatic and aliphatic isothiocyanates (10a–q) to afford the corresponding thiosemicarbazides (11a–q). These thiosemicarbazides (11a–q) were cyclized by using an aqueous KOH to form 5-mercapto-1,2,4-triazoles (12a–q) in 90–95% yields. Finally, these 5-mercapto-1,2,4-triazoles (12a–q) were treated with 3-(2-bromoethyl)-1H-indole (13) in the presence of trimethylamine to achieve the desired conjugates (14a–q) with 75–90% yields. The library of 17 synthesized compounds is depicted in Table 4.
Table 4. Synthesized Pyridine-Based 1,2,4-Triazolo-Tethered Indole Conjugates (14a–q).

All the synthesized conjugates were investigated for their in vitro anti-proliferative activity against a panel of six human cancer cell lines, including HT-29, DLD1, CACO2 (colorectal adenocarcinoma), A549 (lung adenocarcinoma), A172 (glioblastoma, brain cancer), and TERA-1 (teratocarcinoma, testicular cancer) by using (3-4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.24,25 IC50 values of the active conjugates and a standard (5-FU)26 are depicted in Table 5.
Table 5. Anti-proliferative Activity of Synthesized Pyridine-Based 1,2,4-Triazolo-Tethered Indole Conjugates (14a–q) against Different Human Cancer Cell Lines.
| IC50 against the selected cell
lines |
|||||||
|---|---|---|---|---|---|---|---|
| entry | compd | CACO2 | DLD1 | A172 | A549 | TERA-1 | HT-29 |
| 1 | 14a | 3.9 ± 0.6 | 7 ± 1.3 | 9 ± 0.8 | 6.7 ± 1.2 | 4.3 ± 0.4 | 1.0 ± 0.4 |
| 2 | 14b | >30 | >30 | >30 | >30 | >30 | >30 |
| 3 | 14c | >30 | >30 | >30 | >30 | >30 | >30 |
| 4 | 14d | 25 ± 1.0 | 21.8 ± 1.2 | 23.3 ± 0.7 | 18.7 ± 1.6 | >30 | >30 |
| 5 | 14e | 17.8 ± 1.2 | 16.5 ± 0.8 | 20.3 ± 0.8 | 13.5 ± 1.6 | >30 | >30 |
| 6 | 14f | >30 | >30 | >30 | >30 | >30 | >30 |
| 7 | 14g | 10.5 ± 0.5 | 15.3 ± 1.2 | 14.7 ± 0.9 | 14.5 ± 0.3 | 24.6 ± 1.0 | 12.2 ± 1.6 |
| 8 | 14h | >30 | >30 | >30 | >30 | >30 | >30 |
| 9 | 14i | >30 | >30 | >30 | 4.0 ± 0.7 | >30 | >30 |
| 10 | 14] | >30 | >30 | 16.9 ± 1.1 | >30 | >30 | >30 |
| 11 | 14k | 10.3 ± 0.4 | 10.6 ± 1.1 | >30 | 8.09 ± 0.2 | >30 | 5.8 ± 1.2 |
| 12 | 14l | 13.5 ± 0.9 | 11 ± 1.5 | >30 | >30 | >30 | 7.3 ± 0.8 |
| 13 | 14m | >30 | 19 ± 0.3 | 26.8 ± 1.6 | >30 | >30 | >30 |
| 14 | 14n | 9.1 ± 0.8 | 7 ± 1.2 | 9.5 ± 1.4 | 5.3 ± 0.2 | 10.5 ± 0.5 | 2.4 ± 0.6 |
| 15 | 14o | >30 | >30 | >30 | >30 | >30 | >30 |
| 16 | 14p | >30 | 25.8 ± 1.6 | 8.1 ± 0.7 | 13.9 ± 1.2 | 10.7 ± 1.4 | >30 |
| 17 | 14q | 15.5 ± 1.1 | 9.3 ± 1.4 | 26.8 ± 1.6 | 25.1 ± 0.8 | 19.4 ± 0.5 | 3.6 ± 0.3 |
Among the tested conjugates, 14g, 14i, 14k, 14l, and 14p showed promising activity in different cell lines, whereas compounds 14d, 14e, 14j, and 14m exhibited moderate anti-proliferative activity against various cancer cell lines. 14a, 14n, and 14q were found to be very active in most of the cancer cell lines, with IC50 values ranging between 1 μM and 10.5 μM, as shown in Table 5. Among the cell lines tested, HT-29, a CRC cell line, was found to be more susceptible to this class of conjugates. Compound 14a was found to be the most active among the compounds tested against HT-29, with an IC50 value of 1.0 μM. Compounds 14a, 14n, and 14q were chosen for further biological studies.
Among the evaluated conjugates, 14a, 14n, and 14q showed promising anti-proliferative activity against the HT-29 cell line, with IC50 values of 1 μM, 2.4 μM, and 3.6 μM, respectively, compared to the standard drug 5-FU’s IC50 value of 5.31 μM. Using the HT-29 cancer cell line, the impact of these conjugates on the progression of the cell cycle was analyzed. Cell cycle analysis in different phases of the cell was performed using flow cytometry. The results in Figure 4 show that an increased cell population was found in the G0/G1 phase, wherein 14a, 14n, and 14q inhibit at 59.672%, 67.660%, and 42.657% at 1 μM, 2 μM, and 4 μM concentrations, respectively. These conjugates provoked the cell cycle arrest at the G0/G1 phase, thereby suggesting their ability to bind to the DNA and inhibit the replication in the HT-29 cancer cell line.
Figure 4.
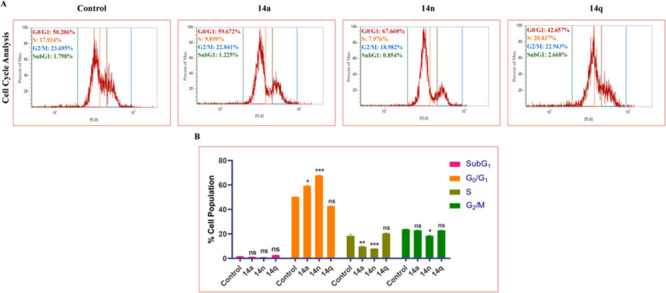
(A) Treatment with 14a (1 μM), 14n (2 μM), and 14q (4 μM) in HT-29 cells displayed significant G0/G1 phase arrest, as indicated by the increase in G0/G1 phase cell population. (B) Bar graphs depicting SubG1, G0/G1, S, and G2/M cell populations. Statistical significance was analyzed by two-way ANOVA followed by Tukey’s post hoc analysis, where *p < 0.05, **p < 0.01, and ***p < 0.001 represent control vs potent compounds 14a, 14n, and 14q in the treatment groups.
Mitochondria play a key role in regulating energy production and cancer progression. Therefore, the MMP was evaluated by flow cytometry using dye JC-1 in the HT-29 colon cancer cell line to determine whether these conjugates (14a, 14n, and 14q) were involved in causing damage to the integrity of the mitochondrial membrane. Cells were treated with concentrations of 1 μM, 2 μM, and 4 μM of each compound for 24 h. Increased monomer formation (see Figure 5) was critically observed in the HT-29 cells treated with the compounds, suggesting mitochondrial damage caused by the compounds. Overall, the results showed promising effects in the disruption of the MMP, thereby suggesting that the compounds actively act on the mitochondria and reduce the Δψm loss.
Figure 5.
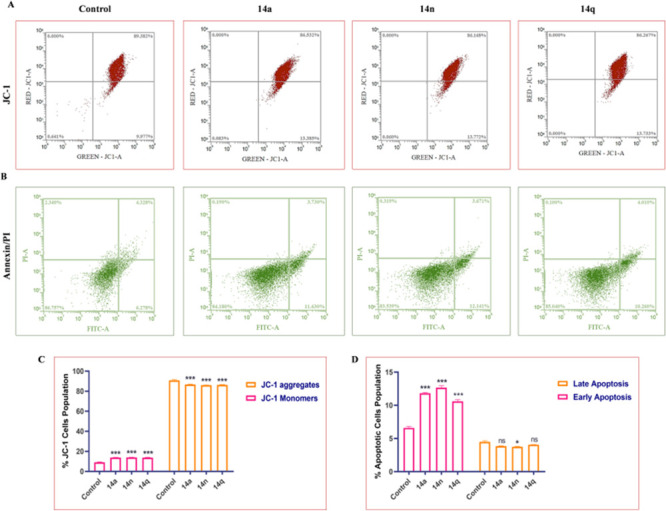
(A) Treatment of HT-29 cells with 14a (1 μM), 14n (2 μM), and 14q (4 μM) aggravates the formation of JC-1 aggregates and reduces the formation of monomers compared to the control. (B) Compounds induce apoptosis activation in HT-29 cells. Flow cytometry images depicting early apoptosis, late apoptosis, and necrotic population in HT-29 cells. (C) Flow cytometry bar graph depicting the % JC-1 cells’ population of HT-29 cells. (D) Bar graphs representing population of apoptotic cells as observed after Annexin/PI staining. Statistical significance was analyzed by two-way ANOVA followed by Tukey’s post hoc analysis, where *p < 0.05, **p < 0.01, and ***p < 0.001 represent control vs potent compounds 14a, 14n, and 14q in the treatment groups.
Targeting apoptosis is one of the most promising non-surgical therapeutic approaches to combat cancer. Defects in apoptosis are crucial for cancer cells’ survival and proliferation. Loss of apoptosis leads to tumor progression, angiogenesis stimulation, and cell proliferation deregulation. Conjugates 14a, 14n, and 14q significantly increased the early and late apoptotic populations in HT-29 cells. The percentage of early and late apoptotic cell populations after treatment with these conjugates at doses of 1 μM, 2 μM, and 4 μM, respectively, for 24 h increased significantly compared to the control group.
Generation of reactive oxygen species (ROS) by cancer chemotherapeutics plays a key role in treating a variety of cancers. Targeted inhibition of the antioxidant responses and increasing ROS production is a major mechanism involved in treating cancer. It has been demonstrated that accumulated ROS disrupts redox homeostasis, severely harming cancer cells. In HT-29 colon cancer cells, compounds 14a, 14n, and 14q increased the production of ROS (see Figure 6), which ultimately resulted in cell death. These conjugates also increased the mitochondrial ROS and superoxide levels, as well as total ROS generation, showing an effect on the most prominent and effective methods for the treatment of cancer.
Figure 6.
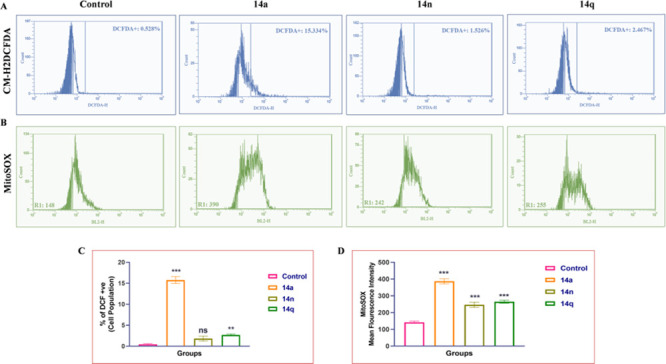
HT-29 cells were treated with compounds 14a (1 μM), 14n (2 μM), and 14q (4 μM), causing increased production of ROS. (A) Flow cytometric analysis graphs showing the percentage of ROS produced by HT-29 cells. (B) Intra-mitochondrial superoxide production was significantly enhanced in HT-29 cells after treatment with conjugates 14a (1 μM), 14n (2 μM), and 14q (4 μM), as measured by a mitochondrial-localizing MitoSOX Red fluorescent probe. (C) Bar representation for mean fluorescence intensity of the conjugates 14a, 14n, and 14q. (D) Representative flow cytometry histograms of MitoSOX Red fluorescence in HT-29 cells (10,000 cells). One-way ANOVA was used to analyze statistical significance, followed by Tukey’s post hoc analysis, where *p < 0.05, **p < 0.01, and ***p < 0.001 represent control vs potent compounds 14a, 14n, and 14q in the treatment groups.
A variety of studies have reported the effects of β-catenin and TAB-182 inhibition in cancer treatment. Cancer cells were found to have a higher expression of β-catenin and TAB-182, which are major markers responsible for tumor initiation and progression. The expression levels of these proteins in HT-29 colon cancer cells (see Figure 7) were found to be significantly reduced after treatment with 14a, 14n, and 14q at doses of 1 μM, 2 μM, and 4 μM, respectively. Further, this treatment inhibits cancer cell progression by reducing the nuclear translocation of β-catenin in HT-29 cells in a dose-dependent manner.
Figure 7.
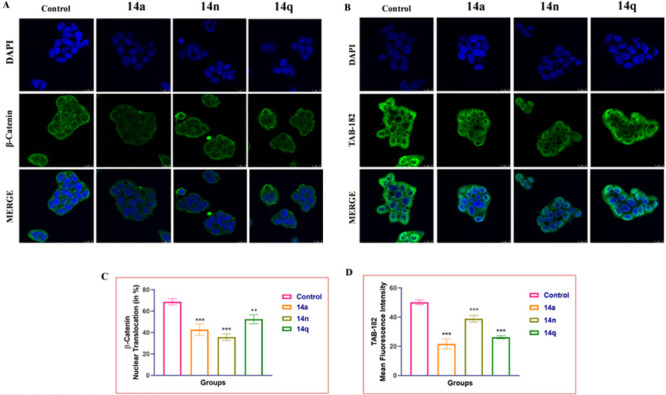
Immunostaining analysis of (A) β-catenin and (B) TAB-182 in HT-29 cells. Graph represent the expression levels of TAB-182 and β-catenin levels (C) and (D). Images (63× and 2×) were acquired using a confocal microscope. One-way ANOVA was used to analyze statistical significance, followed by Tukey’s post hoc analysis, where *p < 0.05, **p < 0.01, and ***p < 0.001 represent control vs potent conjugates 14a, 14n, and 14q in the treatment groups.
High levels of inflammatory markers such as NF-κB are expressed in cancer cells, while drugs targeted at reducing cancer progression inhibit the nuclear translocation of NF-κB. Treatment with the conjugates 14a, 14n, and 14q was found to significantly lower the expression of PI3K-P85 and other inflammatory markers like nuclear translocation and NF-κB expression levels (see Figure 8). According to the findings, these conjugates are effective in treating colon cancer and have been shown to reduce the expression of markers that promote the growth and proliferation of cancer cells. Similar to this, the actin filaments in the HT-29 cells showed obvious damage when stained with F-Actin and Phalloidin Red, further demonstrating the conjugates’ cytotoxic activity.
Figure 8.
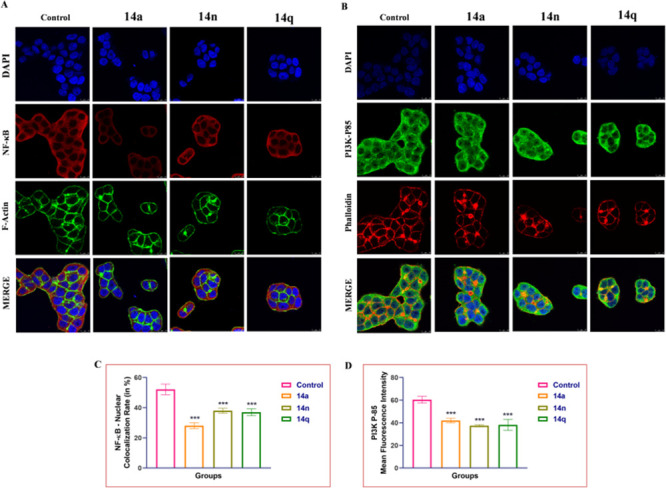
Representative images depicting immunofluorescence analysis of (A) NF-κB along with F-Actin in HT-29 cells. Similarly, HT-29 cells were stained with PI3K-P85 and Phalloidin Red. Treatment with different 14a, 14n, and 14q derivatives has prominently reduced the expression of NF-κB in the nucleus and PI3K P-85 levels in HT-29 colon cancer cells. Images (63× and 2×) were acquired using a confocal microscope. One-way ANOVA was used to determine statistical significance, followed by Tukey’s post hoc analysis, where *p < 0.05, **p < 0.01, and ***p < 0.001 represent control vs potent compounds 14a, 14n, and 14q in treatment groups.
The effects of 14a, 14n, and 14q on the expression levels of APC, AXIN-1, AXIN-2, p-Pan-akt, Pan-akt, β-actin, TAB-182, β-catenin, p-PI3K p85, PI3K p85, p-GSK3β, and GSK3β were determined by Western blotting. Phosphorylation of the PI3K p85 molecule, TAB-182, β-catenin, and GSK3β was significantly inhibited at 1 μM, 2 μM, and 4 μM concentrations, respectively, of compounds 14a, 14n, and 14q compared with the control. However, no change was observed in the expression levels of the p-PI3K p85, PI3K p85 molecule, and TAB-182. Furthermore, it was found that these compounds lowered the expression levels of the cell proliferation marker β-catenin pathway and PI3K/Akt pathway, as shown in Figure 9. Inhibition of the expression levels of APC, AXIN-1, AXIN-2, β-actin, TAB-182, p-GSK3β, GSK3β, and β-catenin illustrates the interruption of the Wnt pathway by this class of compounds, while inhibition of the expression levels of p-PI3K p85, PI3K p85, Pan-Akt, and Akt illustrates the interruption of the PI3K pathway. Therefore, it is evident that conjugates 14a, 14n, and 14q are inhibiting the key markers of both the β-catenin and PI3K pathways in CRC.
Figure 9.

Effects of 14a (1 μM), 14n (2 μM), and 14q (4 μM) on major proteins related to cell proliferation. Western blot analysis detected the expression levels APC, AXIN-1, AXIN-2, p-Pan-akt, Pan-akt, β-actin, TAB-182, β-catenin, p-PI3K p85, PI3K p85, p-GSK3β, and GSK3β proteins.
Structure–activity relationships (SARs) have been drawn based on (i) the nature of the N-substitution (aromatic/aliphatic) on the 1,2,4-triazolyl ring and (ii) the nature as well as the position of the substituent (electron-donating/withdrawing) that is present on the aromatic ring, as displayed in Figure 10. Between the aromatic and aliphatic N-substituted 1,2,4-triazolo compounds, N-aryl-substituted compounds have revealed a better activity profile. In the case of N-aryl conjugates, the nature and the position of the substituent on the aromatic ring have a remarkable influence on the activity. Among these N-aryl conjugates, 4-methoxy- (14a), 2-florophenyl- (14n), and 3-methoxy-substituted (14q) compounds are found to be the most active ones. Among the halogenated compounds, F > Br > Cl is the order in which the substituent has the greatest impact on biological activity. Thus, conjugates 14a, 14n, and 14q showed excellent activity, with IC50 values of 1.0 μM, 2.4 μM, and 3.6 μM, respectively, against the HT-29 cancer cell line. Conjugate 14i, having a 2-bromophenyl substitution, also exhibited good activity, with an IC50 of 4.0 μM against the A549 cancer cell line. Moreover, the 4-fluorophenyl-substituted conjugate (14k) demonstrated better activity against most of the cell lines, and the remaining other aromatic derivatives demonstrated moderate activity.
Figure 10.

SAR for synthesized compounds against the anti-proliferative activity.
Similarly, among the N-aliphatic-containing conjugates, the nature of the substituent greatly influences the biological activity. The N-benzyl-substituted conjugate (14p) demonstrated better activity in comparison to other substituents such as N-butyl, N-propyl, and N-allyl. Among all the conjugates, it has been observed that compounds 14a, 14n, and 14q exhibited the most promising anti-proliferative activity, and they were taken up for detailed biological studies.
The docking complexes of 14a with the proteins (4OA7 and 3L54) were subjected to molecular dynamics (MD) studies employing Schrödinger’s software (Schrödinger’s, LLC, New York) over a time scale of 10 ns. The possible key interactions between ligand 14a and these proteins were examined in this study.
During the simulation, a frame was recorded for every 10 ps and saved into a trajectory. Overall, around 1000 frames were generated in the simulation process. The RMSD (Root Mean Square Deviation) between the ligand and protein was calculated by aligning the structures generated during the MD simulation in the trajectory with the initial frame. The RMSD between the 4OA7–14a complex made it quite evident that the complex was stable throughout the whole simulation period, but a minor variation was observed at 2 ns. Later, the complex got stabilized toward the end of the simulation at 1.4 Å (Figure S2A). Similarly, Figure S2B displays the RMSD between the 3L54–14a complex, wherein a minor variation was observed at 2–6 ns and stabilized was noted at 1.1 Å. The conformational changes along the protein side chain were analyzed by employing the RMSF (Root Mean Square Fluctuation) (Figure S2C,D). Flexibility within the ranges of 0.6–3.6 Å and 0.6–4.2 Å can be interpreted from the RMSF data for the proteins 4OA7 and 3L54, respectively.
The interaction of 14a with proteins 4OA7 and 3L54 was also examined throughout the simulation study, and an analysis report is illustrated in Figure S2E,F. It was observed that the ligand interacted with the target proteins in both a hydrophilic and a hydrophobic fashion. Conjugate 14a showed strong interactions with TYR 1201 and GLY 1185 of 4OA7 and TYR 867, VAL 882, and HIS 967 of 3L54, respectively. Moreover, the MD simulation studies illustrated that the RMSD value of 14a and the protein complexes did not exceed 2.0 Å. The RMSF plot fluctuations of the amino acid residues were less than 5.0 Å, thereby representing the stability of the protein conformation. The binding energy profiles of the protein–ligand complexes, ascribed from various components like RMSD, rGyr (Radius of Gyration), MolSA (Molecular Surface Area), intraHB (Intramolecular Hydrogen Bonds), PSA (Polar Surface Area), and SASA (Solvent Accessible Surface Area), are depicted in Figure S3A,B.
The present investigation has employed rational design and structure-based virtual screening on the target proteins (3L54 and 4OA7) to identify the potential ligands targeting both tankyrase and PI3K. Based on the virtual screening, a focused library of 17 pyridine-based 1,2,4-triazolo-linked indole conjugates were selected from the ZINC15 and PubChem databases, and these were prepared by using a multi-step synthetic strategy. All the synthesized conjugates were evaluated for their anti-proliferative activity. Among the compounds tested, conjugates 14a, 14n, and 14q illustrated promising anti-proliferative activity, with IC50 values of 1 μM, 2.4 μM, and 4.6 μM, respectively, against colorectal adenocarcinoma cell lines (HT-29). These conjugates induced cell cycle arrest at the G0/G1 phase and also enhanced cellular ROS production with increased superoxide levels. They demonstrated prominent effects in disrupting the mitochondrial membrane potential of the cancer cells. They also significantly increased the early as well as late apoptotic cell population, as evidenced by the annexin/PI staining.
Immunofluorescence assays in the subsequent experiments revealed some crucial changes in the expression levels of proteins that are responsible for cancer cell proliferation. Furthermore, 14a, 14n, and 14q significantly lowered the expression levels of TAB-182 and PI3K-P85 and also inhibited the nuclear translocation of NF-κB as well as β-catenin. Western blot results showed that conjugates 14a, 14n, and 14q inhibit the expression levels of APC, AXIN-1, AXIN-2, p-Pan-Akt, Pan-Akt, β-actin, TAB-182, β-catenin, p-PI3K p85, PI3K p85, p-GSK3β, and GSK3β proteins, which is important as these markers are involved in the PI3K and β-catenin pathways in CRC. MM-GBSA calculations were carried out to identify the strength of interactions and their patterns. The combination of MD simulation and experimental methods was useful for understanding the relationship between the protein’s structure and its functions. Based on the docking results, immunocytochemistry, and Western blot assays, it appears that conjugates 14a, 14n, and 14q are suitable ligands for targeting tankyrase as well as PI3K in CRC. Therefore, this class of compounds could serve as an excellent template for drug discovery and development for the dual inhibition of tankyrase and PI3K.
Acknowledgments
This research was funded by the Department of Science and Technology (DST) of the Government of India, grant number DST/IMRCD/BRICS/PilotCall2/CCT/2018-G, the Russian Foundation for Basic Research, grant number 18-515-80028 and the National Research Foundation (NRF) of South Africa, grant number 116014, under the BRICS STI cooperation program, grant number BRICS2017-236. Jamia Hamdard is thanked for providing the necessary laboratory facilities.
Glossary
Abbreviations
- CRC
colorectal cancer
- MMP
mitochondrial membrane potential
- ROS
reactive oxygen species
- IC50
half-maximal inhibitory concentration
- PI3K
phosphoinositide 3-kinase
- SAR
structure–activity relationship
- TNKS
tankyrase
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00475.
Experimental procedure for the synthesis of compounds with their analytical data, assay procedure, 1H NMR, and 13C NMR (PDF)
Author Contributions
‡ P.A.Y. and S.R.P. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Sung H.; Ferlay J.; Siegel R. L.; Laversanne M.; Soerjomataram I.; Jemal A.; Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71 (3), 209–249. 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- Sawicki T.; Ruszkowska M.; Danielewicz A.; Niedźwiedzka E.; Arłukowicz T.; Przybyłowicz K. E. A Review of Colorectal Cancer in Terms of Epidemiology, Risk Factors, Development, Symptoms and Diagnosis. Cancers (Basel) 2021, 13 (9), 2025. 10.3390/cancers13092025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladabaum U.; Dominitz J. A.; Kahi C.; Schoen R. E. Strategies for Colorectal Cancer Screening. Gastroenterology 2020, 158 (2), 418–432. 10.1053/j.gastro.2019.06.043. [DOI] [PubMed] [Google Scholar]
- Armaghany T.; Wilson J. D.; Chu Q.; Mills G. Genetic Alterations in Colorectal Cancer. Gastrointest. Cancer Res. 2012, 5 (1), 19–27. [PMC free article] [PubMed] [Google Scholar]
- Yang P.; Zhu Y.; Zheng Q.; Meng S.; Wu Y.; Shuai W.; Sun Q.; Wang G. Recent Advances of β-Catenin Small Molecule Inhibitors for Cancer Therapy: Current Development and Future Perspectives. Eur. J. Med. Chem. 2022, 243, 114789. 10.1016/j.ejmech.2022.114789. [DOI] [PubMed] [Google Scholar]
- Lee E.; Salic A.; Krüger R.; Heinrich R.; Kirschner M. W. The Roles of APC and Axin Derived from Experimental and Theoretical Analysis of the Wnt Pathway. PLoS Biol. 2003, 1 (1), e10. 10.1371/journal.pbio.0000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandurangan A. K. Potential Targets for Prevention of Colorectal Cancer: A Focus on PI3K/Akt/MTOR and Wnt Pathways. Asian Pacific J. Cancer Prev. 2013, 14 (4), 2201–2205. 10.7314/APJCP.2013.14.4.2201. [DOI] [PubMed] [Google Scholar]
- Ma L.; Wang X.; Jia T.; Wei W.; Chua M. S.; So S. Tankyrase Inhibitors Attenuate WNT/ß-Catenin Signaling and Inhibit Growth of Hepatocellular Carcinoma Cells. Oncotarget 2015, 6 (28), 25390–25401. 10.18632/oncotarget.4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. C.; Kim O. H.; Lee S. K.; Kim S. J. IWR-1 Inhibits Epithelial-Mesenchymal Transition of Colorectal Cancer Cells through Suppressing Wnt/β-Catenin Signaling as Well as Survivin Expression. Oncotarget 2015, 6 (29), 27146–27159. 10.18632/oncotarget.4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C.; Chen Y. G. Dishevelled: The Hub of Wnt Signaling. Cell. Signal. 2010, 22 (5), 717–727. 10.1016/j.cellsig.2009.11.021. [DOI] [PubMed] [Google Scholar]
- Fernandes M. S.; Sanches J. M.; Seruca R.. Targeting the PI3K Signalling as a Therapeutic Strategy in Colorectal Cancer. Targeted Therapy of Colorectal Cancer Subtypes; Advances in Experimental Medicine and Biology 1110; Springer International Publishing: Cham, 2018; p 35. 10.1007/978-3-030-02771-1_4 [DOI] [PubMed] [Google Scholar]
- Rubinfeld B.; Albert I.; Porfiri E.; Fiol C.; Munemitsu S.; Polakis P. Binding of GSK3beta to the APC-Beta-Catenin Complex and Regulation of Complex Assembly. Science 1996, 272 (5264), 1023–1026. 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- Yu M.; Yang Y.; Sykes M.; Wang S. Small-Molecule Inhibitors of Tankyrases as Prospective Therapeutics for Cancer. J. Med. Chem. 2022, 65 (7), 5244–5273. 10.1021/acs.jmedchem.1c02139. [DOI] [PubMed] [Google Scholar]
- Ahmad A.; Biersack B.; Li Y.; Kong D.; Bao B.; Schobert R.; Padhye S. B.; Sarkar F. H. Targeted Regulation of PI3K/Akt/MTOR/NF-KB Signaling by Indole Compounds and Their Derivatives: Mechanistic Details and Biological Implications for Cancer Therapy. Anticancer. Agents Med. Chem. 2013, 13 (7), 1002–1013. 10.2174/18715206113139990078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haikarainen T.; Waaler J.; Ignatev A.; Nkizinkiko Y.; Venkannagari H.; Obaji E.; Krauss S.; Lehtiö L. Development and Structural Analysis of Adenosine Site Binding Tankyrase Inhibitors. Bioorg. Med. Chem. Lett. 2016, 26 (2), 328–333. 10.1016/j.bmcl.2015.12.018. [DOI] [PubMed] [Google Scholar]
- Waaler J.; Machon O.; Von Kries J. P.; Wilson S. R.; Lundenes E.; Wedlich D.; Gradl D.; Paulsen J. E.; Machonova O.; Dembinski J. L.; Dinh H.; Krauss S. Novel Synthetic Antagonists of Canonical Wnt Signaling Inhibit Colorectal Cancer Cell Growth. Cancer Res. 2011, 71 (1), 197–205. 10.1158/0008-5472.CAN-10-1282. [DOI] [PubMed] [Google Scholar]
- Zheng S.; Liu J.; Wu Y.; Huang T. L.; Wang G. Small-Molecule Inhibitors of Wnt Signaling Pathway: Towards Novel Anticancer Therapeutics. Future Med. Chem. 2015, 7 (18), 2485–2505. 10.4155/fmc.15.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A.; Riyaz S.; Srivastava A.; Rahim A. Tankyrase Inhibitors as Therapeutic Targets for Cancer. Curr. Top. Med. Chem. 2014, 14 (17), 1967–1976. 10.2174/1568026614666140929115831. [DOI] [PubMed] [Google Scholar]
- Solberg N. T.; Waaler J.; Lund K.; Mygland L.; Olsen P. A.; Krauss S. TANKYRASE Inhibition Enhances the Antiproliferative Effect of PI3K and EGFR Inhibition, Mutually Affecting β-CATENIN and AKT Signaling in Colorectal Cancer. Mol. Cancer Res. 2018, 16 (3), 543–553. 10.1158/1541-7786.MCR-17-0362. [DOI] [PubMed] [Google Scholar]
- Arqués O.; Chicote I.; Puig I.; Tenbaum S. P.; Argilés G.; Dienstmann R.; Fernández N.; Caratù G.; Matito J.; Silberschmidt D.; Rodon J.; Landolfi S.; Prat A.; Espín E.; Charco R.; Nuciforo P.; Vivancos A.; Shao W.; Tabernero J.; Palmer H. G. Tankyrase Inhibition Blocks Wnt/b-Catenin Pathway and Reverts Resistance to PI3K and AKT Inhibitors in the Treatment of Colorectal Cancer. Clin. Cancer Res. 2016, 22 (3), 644–656. 10.1158/1078-0432.CCR-14-3081. [DOI] [PubMed] [Google Scholar]
- Regmi S. C.; Park S. Y.; Kim S. J.; Banskota S.; Shah S.; Kim D. H.; Kim J. A. The Anti-Tumor Activity of Succinyl Macrolactin a Is Mediated through the β-Catenin Destruction Complex via the Suppression of Tankyrase and PI3K/Akt. PLoS One 2015, 10 (11), e0141753. 10.1371/journal.pone.0141753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenbaum S. P; Ordonez-Moran P.; Puig I.; Chicote I.; Arques O.; Landolfi S.; Fernandez Y.; Herance J. R.; Gispert J. D; Mendizabal L.; Aguilar S.; Cajal S. R. y; Schwartz S.; Vivancos A.; Espin E.; Rojas S.; Baselga J.; Tabernero J.; Munoz A.; Palmer H. G β-Catenin Confers Resistance to PI3K and AKT Inhibitors and Subverts FOXO3a to Promote Metastasis in Colon Cancer. Nat. Med. 2012, 18 (6), 892–901. 10.1038/nm.2772. [DOI] [PubMed] [Google Scholar]
- Yakkala P. A.; Panda S. R.; Shafi S.; Naidu V. G. M.; Yar M. S.; Ubanako P. N.; Adeyemi S. A.; Kumar P.; Choonara Y. E.; Radchenko E. V.; Palyulin V. A.; Kamal A. Synthesis and Cytotoxic Activity of 1,2,4-Triazolo-Linked Bis-Indolyl Conjugates as Dual Inhibitors of Tankyrase and PI3K. Molecules 2022, 27 (21), 7642. 10.3390/molecules27217642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namballa H. K.; Anchi P.; Lakshmi Manasa K.; Soni J. P.; Godugu C.; Shankaraiah N.; Kamal A. Bioorganic Chemistry β -Carboline Tethered Cinnamoyl 2-Aminobenzamides as Class I Selective HDAC Inhibitors : Design, Synthesis, Biological Activities and Modelling Studies. Bioorg. Chem. 2021, 117 (August), 105461. 10.1016/j.bioorg.2021.105461. [DOI] [PubMed] [Google Scholar]
- Sathish M.; Chetan Dushantrao S.; Nekkanti S.; Tokala R.; Thatikonda S.; Tangella Y.; Srinivas G.; Cherukommu S.; Hari Krishna N.; Shankaraiah N.; Nagesh N.; Kamal A. Synthesis of DNA Interactive C3-Trans-Cinnamide Linked β-Carboline Conjugates as Potential Cytotoxic and DNA Topoisomerase I Inhibitors. Bioorg. Med. Chem. 2018, 26 (17), 4916–4929. 10.1016/j.bmc.2018.08.031. [DOI] [PubMed] [Google Scholar]
- Wang N.; Yang L.; Dai J.; Wu Y.; Zhang R.; Jia X.; Liu C. 5-FU Inhibits Migration and Invasion of CRC Cells through PI3K/AKT Pathway Regulated by MARCH1. Cell Biol. Int. 2021, 45 (2), 368–381. 10.1002/cbin.11493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.