

Abstract
Problem:
Preeclampsia (PE), new-onset hypertension during pregnancy, is associated with a pro-inflammatory state with activated T cells, cytolytic natural killer (NK) cells, dysregulated complement proteins, and B cells secreting agonistic autoantibodies to the angiotensin II type-1 receptor (AT1-AA). The reduced uterine perfusion pressure (RUPP) model of placental ischemia recapitulates these features of PE. Blocking CD40L-CD40 communication between T and B cells or B cell depletion with Rituximab prevents hypertension and AT1-AA production in RUPP rats. This suggests that T cell-dependent B cell activation contributes to the hypertension and AT1-AA associated with PE. B2 cells maturing into antibody producing plasma cells are the product of T cell-dependent B cell-interactions and B cell Activating Factor (BAFF) is an integral cytokine in the development of B2 cells specifically. Thus, we hypothesize that BAFF blockade will selectively deplete B2 cells, therefore reducing blood pressure, AT1-AA, activated NK Cells, and complement in the RUPP rat model of PE.
Method of Study:
Gestational Day (GD) 14 pregnant rats underwent the RUPP procedure, and a subset were treated with 1 mg/kg Anti-BAFF antibodies via jugular catheters. On GD19, blood pressure was measured, B cells and NK cells were measured by flow cytometry, AT1-AA was measured by cardiomyocyte bioassay, and complement activation was measured by ELISA.
Results:
Anti-BAFF therapy attenuated hypertension, AT1-AA, NK cell activation, and APRIL levels in RUPP rats without negatively impacting fetal outcomes.
Conclusions:
This study demonstrates that B2 cells contribute to hypertension, AT1-AA, and NK cell activation in response to placental ischemia during pregnancy.
Keywords: Preeclampsia, Inflammation, Autoantibodies, BAFF, Animal Model, Hypertension, Pregnancy
Introduction
Preeclampsia (PE) is new-onset hypertension during pregnancy and affects ~8% of pregnancies in the US [1]. PE is believed to stem from insufficient spiral artery remodeling, which results in placental ischemia and fetal growth restriction (FGR) [2]. Placental ischemia in animal models results in hypertension, FGR, endothelial dysfunction, renal dysfunction, and chronic inflammation, similar to what is seen in patients with PE [3, 4]. Currently, the only cure for PE is delivery of the fetal-placental unit, thereby increasing the risk for premature birth [5]. While the incidence of PE has continually increased [6], the treatment options for PE have not changed in nearly 50 years. Therefore, there is a dire need for new therapeutic options in treating women with PE [7].
The inflammatory profile in PE is characterized by activated CD4+ T cells shifted towards a Th1/Th17 phenotype, cytolytic natural killer (NK) cells, activation of the complement system, and activated B cells producing autoantibodies to the angiotensin II type-1 receptor (AT1-AA) in both humans and in animal models of placental ischemia [8–11]. AT1-AA is an IgG3 antibody that binds to the second extracellular loop of the AT1 receptor and activates downstream pathways similar to that of angiotensin II [12]. We have shown that AT1-AA contributes to multiple factors associated with PE including hypertension, Th1/Th17 cells, NK Cell activation, mitochondrial dysfunction, and endothelial dysfunction in the Reduced Uterine Perfusion Pressure (RUPP) model of placental ischemia [13–18]. Importantly AT1-AA has been found in serum of previously PE patients 8 years postpartum [19], and we believe it to play an important role in the development of cardiovascular disease in previously preeclamptic women.
We have demonstrated the importance of T cell-B cell communication as an important step in the pathophysiology of hypertension and AT1-AA production in the RUPP rat model of placental ischemia [20, 21]. T cell-B cell communication is a pivotal step in B cell activation and formation of immune memory [22], allowing for long-term production of antibodies after antigen exposure. We have shown that B cell depletion with Rituximab attenuates the hypertension associated with placental ischemia in the RUPP preclinical model of placental ischemia; therefore, implicating a role for B cells in PE pathophysiology [23].
B cells are divided into two subsets, B1 and B2, which develop independently of each other. B1 cells are innate-like B cells that develop from a fetal liver progenitor cell and spontaneously produce low-specificity Ig [24]. In clinical studies, B1 cells have been implicated in the pathophysiology of PE by producing AT1-AA [25], but B1 cells are not associated with long-term immune memory and may not be a source of postpartum AT1-AA [19]. B2 cells are classical B cells that produce high-specificity IgG after communication with CD4+ T cells and transform into memory B cells [24]. B2 cells stem from bone marrow progenitor cells and require stimulation by B cell Activating Factor (BAFF) as a secondary activation signal following T cell-B cell communication [26] in order to progress through B cell development and maturation. Following B2 cell activation, A Proliferation Inducing Ligand (APRIL) promotes Ig class switching, activated B cell survival, and survival of memory B cells [27–29]. B1 cells develop independent of BAFF [30]. Kowalczyk-Quintas et al. previously showed that infusion of mouse anti-BAFF antibodies reduced circulating and splenic B cells [31]. In addition, patients with systemic lupus erythematosus (SLE) that receive anti-BAFF therapy (Belimumab) have improved renal outcomes compared to standard SLE treatment alone [32, 33]. These data suggest that BAFF inhibition, such as anti-BAFF therapy with Belimumab, may be a therapeutic strategy to deplete developing B2 cells and reduce production of AT1-AA. This strategy would leave B1 cells viable to produce natural antibodies for the patient. We sought to investigate the effect of anti-BAFF therapy on B2 cell depletion and AT1-AA production, and hypertension in the RUPP model of placental ischemia in order to distinguish the importance of B2 cells vs B1 cells as producers of AT1-AA and to determine preclinical relevance of this avenue as a potential treatment for PE. We hypothesize that BAFF blockade with anti-BAFF antibodies would reduce circulating B cells and attenuate hypertension, FGR, AT1-AA and its downstream sequelae observed in the RUPP model of placental ischemia.
Methods
Animals
Twelve-week-old time-pregnant Sprague-Dawley rats (200–250g) were purchased from Envigo (Harlan Laboratories Inc., Indianapolis, IN, USA). Animals were housed in temperature-controlled rooms (23°C) with 12:12h light-dark cycles and free access to water and chow. The authors confirm that the ethical policies of the journal and the University of Mississippi Medical Center’s Institutional Animal Care and Use Committee were followed. All experiments were in accordance with the National Institute of Health’s “Guidelines for the Care and Use of Laboratory Animals.”
Reduced Uterine Perfusion Pressure (RUPP) Procedure
A group of control rats consisted of normal pregnant (NP) and reduced uterine perfusion pressure (RUPP) animals. On gestational day (GD) 14, animals underwent the RUPP procedure as previously described [34, 35] under 2% isoflurane anesthesia delivered by a vaporizer (Ohio Medical Products, Champagne, IL, USA). The RUPP model of placental ischemia has been shown to reduce uterine blood flow by ~54% [36] and recapitulate many of the characteristics seen in human PE. In brief, animals were placed under isoflurane anesthesia, and a midline incision was made. A restrictive silver clip (0.203 mm) was placed superior to the iliac bifurcation on the abdominal aorta. To reduce compensatory blood flow, another restrictive clip (0.100 mm) was placed at each bilateral uterine arcade at the ovarian end. All animals undergoing any surgical procedures were given carprofen (5 mg/kg) to aid with post-operative pain.
Infusion of Anti-BAFF
A protein basic local alignment search tool (BLAST) search was performed to compare mouse BAFF to rat BAFF through the NIH online search (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The BLAST search showed rat BAFF has ~85% sequence homology to mouse BAFF. For in vivo blockade of BAFF signaling, gestational day (GD) 13 pregnant rats underwent jugular catheterization under 2% isoflurane anesthesia delivered by a vaporizer (Ohio Medical Products, Champaign, IL, USA) and received carprofen (5mg/kg) to account for postoperative pain. On GD14, subsets of NP or RUPP rats were infused with 1 mg/kg of mouse Anti-BAFF antibodies (Adipogen Life Sciences, San Diego, CA, USA) via jugular catheters immediately following the RUPP procedure. The four groups of rats were normal pregnant (NP; n=9) rats, NP + Anti-BAFF (n=8), RUPP (n=12) and RUPP + anti-BAFF (n=11).
Mean Arterial Pressure Measurement
On GD 18, all groups underwent carotid catheterization for mean arterial pressure (MAP) measurement, as previously described [37]. On GD 19, animals were placed in restrainers and allowed one hour to acclimate to their surroundings. Following the acclimation period, MAP was measured continuously for 30 minutes (Cobe III Transducer CDX Sema) as previously described [37]. Average pup and placenta weight were determined, and each were reported as an n of 1. Blood and tissues were collected for analysis.
Determination of Placental and Circulating B cells
Circulating peripheral B cells were measured by flow cytometry, as previously described [38]. Briefly, whole blood was collected on GD 19, mixed with RPMI medium, and isolated by centrifugation over a Ficoll-Hypaque (Lymphoprep, Accurate Chemical & Scientific Corp., Westbury, NY) cushion according to manufacturer’s instructions. Placentas were collected from each animal and homogenized in nine mL of RPMI + 1mL of fetal bovine serum. Homogenized placentas were then filtered through a 30μm cell strainer. The homogenate was then layered over a Ficoll-Hypaque cushion (Lymphoprep, Accurate Chemical & Scientific Corp., Westbury, NY, USA) and spun at 300g to isolate placental leukocytes. Next, one million isolated peripheral blood mononuclear cells (PBMCs) were stained for 10 minutes at 4°C with antibodies against rat Anti-CD3 (Vioblue) (Miltenyi Biotec, San Diego, CA, USA), rat anti-CD68 (PE-Vio 770) (Myltenyi Biotec), and rat anti-CD45R (APC-Vio770) (Myltenyi Biotec). Cells were washed and analyzed using the MACSQuant analyzer 10 (Myltenyi Biotec) and quantified using FlowLogic Software (Innovai, Sydney, Australia) with fluorescence-minus-one (FMO) controls. B cells were considered CD3−CD68−CD45R+, B1 cells were considered CD3−CD68−CD45R+CD43+, B2 cells were considered CD3−CD68−CD45R+CD43−.
Determination of Placental and Circulating NK cell Populations
On GD 19, placentas and whole blood were collected from all animal groups and PBMCs were collected from whole blood and placentas, as described above. One million PBMCs were stained for 10 min at 4°C with rat anti-CD3 (Viogreen) (Myltenyi), rat NK cell activation structure (ANK61) (Abcam, Cambridge, MA) with FITC secondary (Abcam), and rat NK cell antibody (ANK44) (Abcam) with alexa405 secondary (Abcam). Cells were analyzed by the MACSQuant analyzer (Myltenyi) and quantified using FlowLogic Software (Inivai) using FMO controls. NK cells were considered CD3−Ank61+ cells, and activated NK cells were considered CD3−Ank61+ANK44+ cells [39].
Determination of Complement Components
Plasma, placental, and renal complement component C1q were measured using an enzyme-linked immunosorbent assay (ELISA) (Novus Biologicals, Littleton, CO). The sensitivity of the assay is 0.47 ng/mL, inter-assay variability <5.23 and intra-assay variability <4.82%. Plasma samples were diluted 1:80,000 and reported as mg/mL. ~100mg of placental or kidney tissue was homogenized in 900μL of ice-cold saline (pH = 7.4). Tissue homogenate was centrifuged for 5 minutes at 5,000g at 4°C, and the supernatant collected for use in the ELISA. Tissue homogenate supernatant from placentas and kidney was diluted 1:500 for analysis. Placental and renal C1q levels are reported as μg C1q/mg total protein.
Plasma complement component C3 was measured using an ELISA (Abcam) and used according to the manufacturer’s protocol. The sensitivity of the assay is 2.82 ng/mL, the inter-assay variability <10%, and the intra-assay variability <10%. The plasma samples were diluted 1:10,000 for analysis.
Plasma complement component C3a was measured using an ELISA kit (MyBioSource, San Diego, CA, USA) and used according to the manufacturer’s protocol. The assay is listed to have a sensitivity of 4.69 ng/mL and a coefficient of variation listed as <10%. Plasma samples were diluted 1:10 for analysis.
Plasma complement component C4 was measured using an ELISA kit (MyBioSource) according to the manufacturer’s protocol. The sensitivity of the assay is <3.12 ng/mL, the intra-assay variability CV<8%, and the inter-assay precision CV%<10%. Plasma samples were diluted 1:10,000.
Determination of Circulating AT1-AA
AT1-AA were collected from serum and analyzed using a modified version of the previously described cardiomyocyte assay [40]. Neonatal rat cardiomyocytes were isolated as previously described [41], and incubated with 2.5 μM Fluo-4 AM (Thermofisher, Waltham, MA, USA). Chronotropic events in response to AT1 receptor-mediated stimulation of cultured rat neonatal cardiomyocytes were quantified using the Biotek Cytation 5 cell imaging multimode reader (Agilent, Santa Clara, CA, USA). The change in beats per minute (Δbpm) was calculated and positively correlates to amount of AT1-AA in serum. Serum IgG fraction was incubated with the seven amino acid (7AA) sequence corresponding to the specific epitope binding region of the AT1-AA to confirm change in beats per minute were associated with AT1-AA stimulation. Data are reported as Δbpm.
Determination of Circulating BAFF, APRIL, and other cytokines
Circulating BAFF was determined using a commercially available ELISA (MyBioSource) and used according to the manufacturer’s protocol. The sensitivity of the ELISA is listed as <28pg/mL, intra-assay precision CV<10% and inter-assay precision CV<12%.
Circulating APRIL was determined using a commercially available ELISA (MyBioSource) according to the manufacturer’s protocol. The sensitivity of the ELISA is 0.1ng/mL, the inter-assay precision CV<10%, and intra-assay precision <10%.
Plasma samples were analyzed for Macrophage Inflammatory Protein (MIP)-1α, MIP-3α, and Monocyte Chemoattractant Protein (MCP)-1 using the Bio-plex Pro Rat Cytokine 23-Plex Immunoassay Kit (Bio-Rat, Hercules, CA, USA) according to the manufacturer’s protocol.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 9.2 software (GraphPad Software, San Diego, CA). Comparisons between groups were made using a two-way ANOVA with a Bonferroni post hoc test. Results were reported as means ± SEM and were considered significant when p<0.05.
Results
Anti-BAFF therapy Normalized Hypertension Associated with Placental Ischemia
RUPP rats had increased MAP (119 ± 2 mmHg,) compared to NP rats (99 ± 3mmHg, p<0.01) (Figure 1.a.). Anti-BAFF therapy did not change blood pressure in NP+Anti-BAFF (96 ± 3.19 mmHg) compared to control NP. Anti-BAFF therapy normalized blood pressure in RUPP+Anti-BAFF (100 ± 3 mmHg, p<0.01) compared to RUPP.
Figure 1.
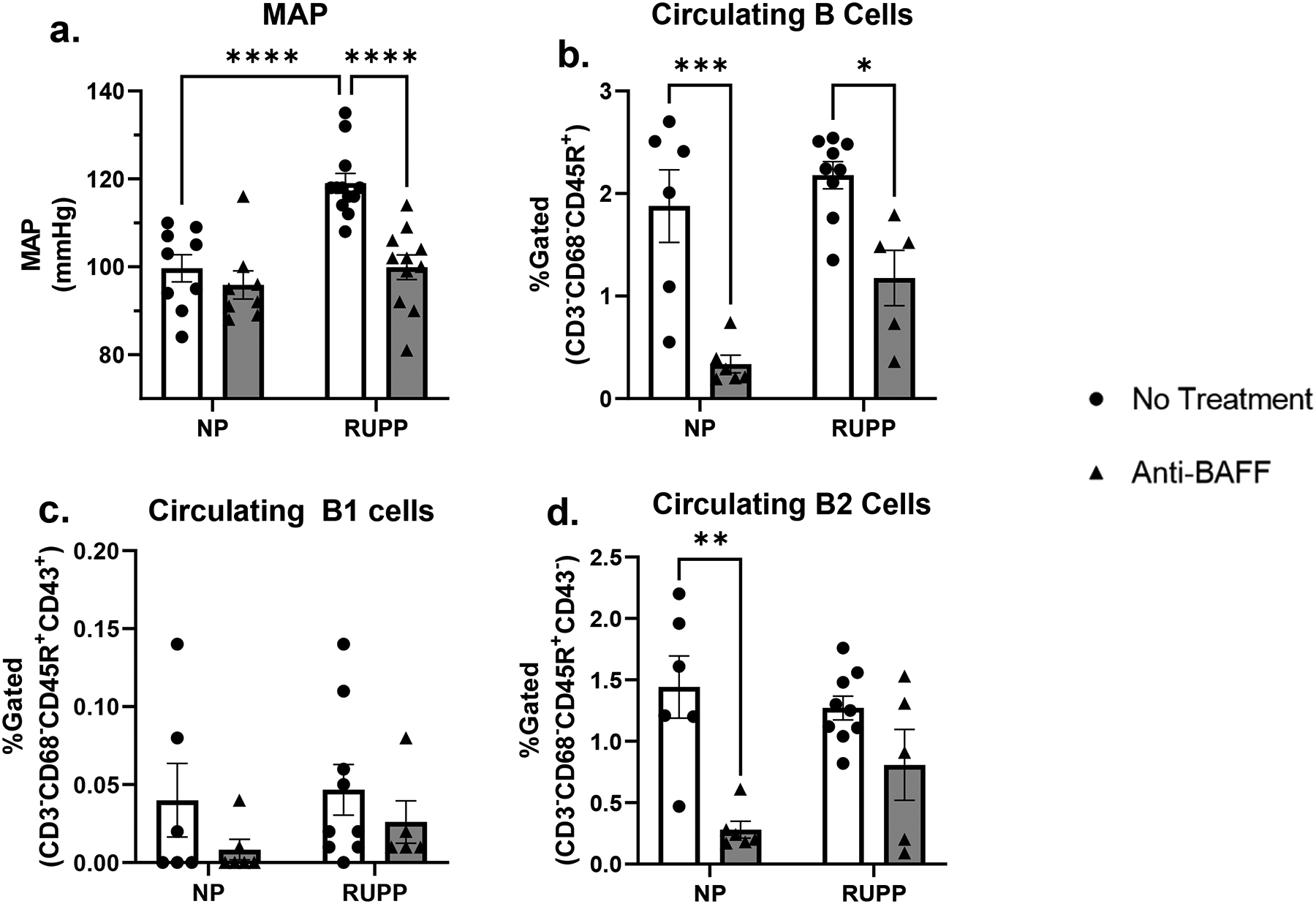
RUPP rats (n=12) had increased Mean Arterial Pressure (MAP) compared to NP (n=9, p<0.01. NP+Anti-BAFF (n=8) compared to NP, but RUPP+Anti-BAFF (n=11, p<0.01) had normalized MAP compared to RUPP. NP+Anti-BAFF had decreased circulating B cells (n=6) compared to untreated NP rats (n=6, <0.01) and RUPP+Anti-BAFF (n=5) had decreased circulating B cells compared to untreated RUPP rats (n=9, p<0.05). There was no difference in circulating B1 cells in NP (n=6) compared to RUPP (n=9). There was a trending decrease in circulating B1 cells between NP compared to NP+Anti-BAFF (n=6) and between RUPP compared to RUPP+Anti-BAFF (n=5). There was no change in circulating B2 cells between NP (n=6) compared to RUPP (n=9). There was a significant decrease in circulating B2 cells in NP+Anti-BAFF (n=6, p<0.05) compared to NP. There was a trending decrease in circulating B2 cells in RUPP+Anti-BAFF (n=5) compared to RUPP.
Anti-BAFF therapy Reduced Circulating B cells but Did Not Reduce Placental B cells
Anti-BAFF decreased circulating B cells (0.34 ± 0.09% gated) in NP rats compared to control NP rats (1.89 ± 0.35% gated p<0.01) (Figure 1.b.). RUPP+Anti-BAFF had decreased circulating B cells (p<0.05) compared to RUPP rats (1.18 ± 0.27% gated vs 2.18 ± 0.13% gated). There was no significant difference between circulating B1 cells between NP (0.04 ± 0.024% gated) compared to RUPP (0.047 ± 0.016% gated) (Figure 1.c.). There were trending decreases in circulating B1 cells in NP+Anti-BAFF (0.008 ± 0.007% gated) and RUPP+Anti-BAFF (0.026 ± 0.014% gated) compared to control NP and RUPP groups. There was no change in circulating B2 cells between NP (1.442 ± 0.254% gated) and RUPP (1.721 ± 0.097% gated) (Figure 1.d). NP+Anti-BAFF had significantly lower circulating B2 cells (0.280 ± 0.068% gated, p<0.05) compared to NP. There was a trending decrease in circulating B2 cells in RUPP+Anti-BAFF (0.808 ± 0.289% gated) compared to control RUPP.
There was no change between total placental B cells between NP (1.567 ± 0.362% gated) compared to RUPP (1.460 ± 0.280% gated) or compared to NP+Anti-BAFF (1.203 ± 0.358% gated) (S3). There was also no change in total placental B cells between RUPP and RUPP+Anti-BAFF (0.968 ± 0.276% gated). There were no changes in placental B1 cells between any of the groups: NP (0.082 ± 0.034% gated), NP+Anti-BAFF (0.100 ± 0.061% gated), RUPP (0.112 ± 0.015% gated), RUPP+Anti-BAFF (0.088 ± 0.018% gated). There were no changes in placental B2 cells between any of the groups: NP (1.148 ± 0.356% gated), NP+Anti-BAFF (0.485 ± 0.208% gated), RUPP (1.480 ± 0.361% gated), RUPP+Anti-BAFF (1.078 ± 0.466% gated).
Anti-BAFF therapy did not worsen fetal weight, placental weight, or fetal viability during pregnancy
RUPP rats had reduced pup weight (1.97 ± 0.08 g, n=12, p<0.05) compared to NP pups (2.21 ± 0.09 g, n=8) (Figure 2.a). NP+Anti-BAFF had no change in pup weight (2.33 ± 0.05, n=8) compared to NP. RUPP+Anti-BAFF had reduced pup weight (1.82 ± 0.10 g, n=9, p<0.05) compared to NP+Anti-BAFF. RUPP+Anti-BAFF had no change in pup weight compared to RUPP, indicating pup weight was not worse with anti-BAFF therapy. Placenta weight was reduced in RUPP rats (0.46 ± 0.02, p<0.01) compared to NP (0.59 ± 0.03,) (Figure 2.b). Anti-BAFF therapy did not change placenta weight in NP+Anti-BAFF rats (0.55 ± 0.31, n=8) compared to NP. There was no change in placenta weight between NP+Anti-BAFF and RUPP+Anti-BAFF (0.54 ± 0.2, n=9). There was no change in placenta weight between RUPP+Anti-BAFF and RUPPs. Fetal viability was reduced in RUPP rats (47 ± 8 %, n=13, p<0.01) compared to NP (97 ± 2 %, n=9) (Figure 2.c.). NP+Anti-BAFF had no change in fetal viability (99 ± 1 %, n=8) compared to NP. RUPP+Anti-BAFF had reduced fetal viability (43 ± 10 %, n=11, p<0.01) compared to NP+Anti-BAFF which was not worsened compared to RUPP.
Figure 2.
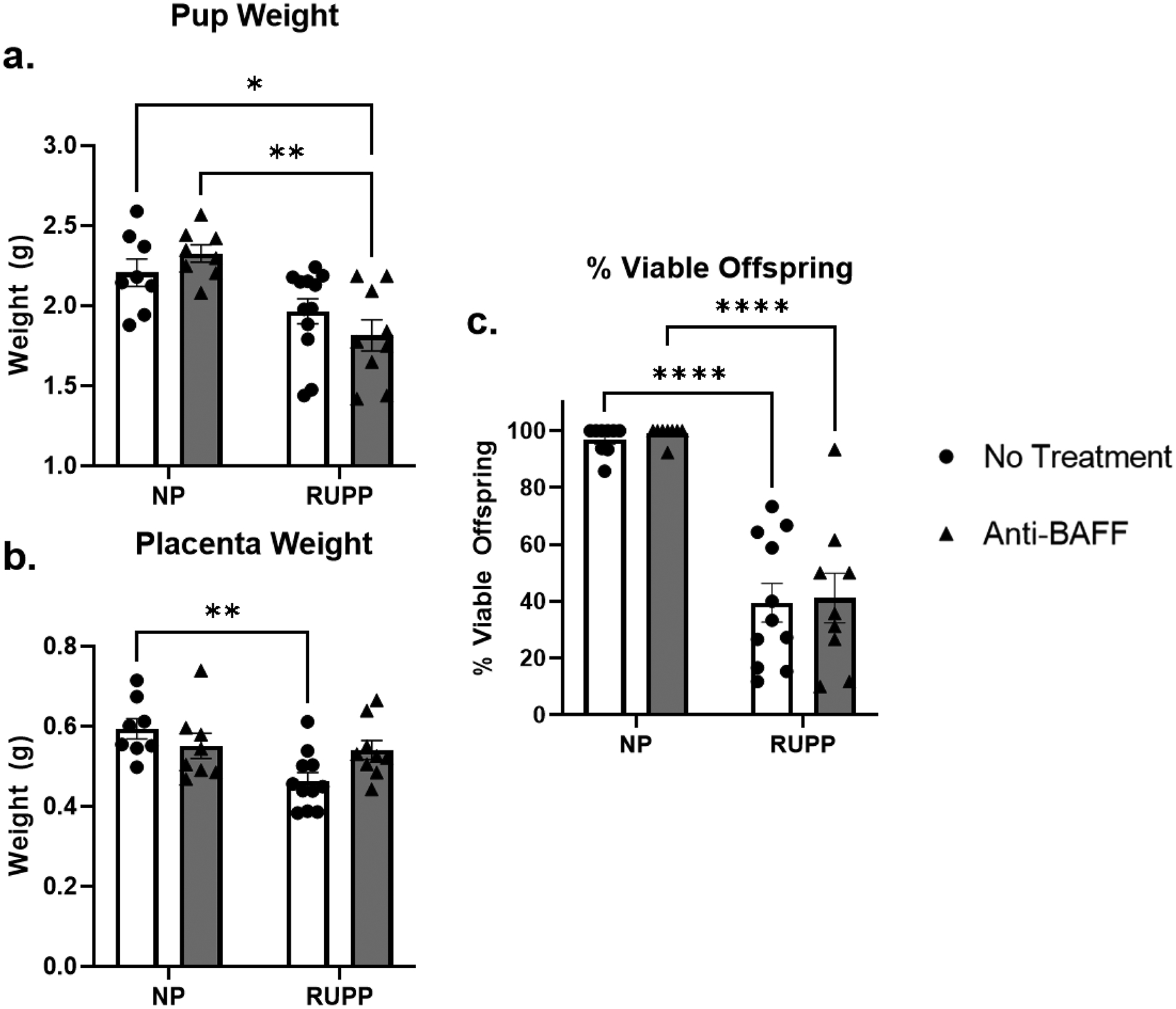
NP+Anti-BAFF (n=8) had no change in pup weight compared to NP (n=8). RUPP rats had smaller pups (n=12, p<0.05) compared to NP. RUPP+Anti-BAFF (n=9, p<0.01) had smaller pups compared to NP+Anti-BAFF, but there was no change between RUPP and RUPP+Anti-BAFF. There was no change in placenta weight between NP (n=8) and NP+Anti-BAFF (n=8). RUPPs (n=11) had smaller placentas compared to NP, but RUPP+Anti-BAFF (n=9) did not have smaller pups than NP+Anti-BAFF. There was no change in offspring viability between NP (n=8) and NP+Anti-BAFF (n=8). RUPP rats (n=11) had decreased fetal viability compared to NP and RUPP+Anti-BAFF (n=9) had decreased fetal viability compared to NP+Anti-BAFF. There was no change in fetal viability between RUPP and RUPP+Anti-BAFF.
Anti-BAFF therapy suppressed AT1-AA in response to RUPP
As previously shown, RUPP had increased AT1-AA (16±2 ΔBPM, n=6) compared to NP (2±1 ΔBPM, n=6, p<0.001) (Figure 3). There was no change in AT1-AA between NP+Anti-BAFF (8±2 ΔBPM, n=5) and NP. RUPP+Anti-BAFF had significantly lower AT1-AA (7±2 ΔBPM, n=7, p<0.01) compared to RUPP.
Figure 3.
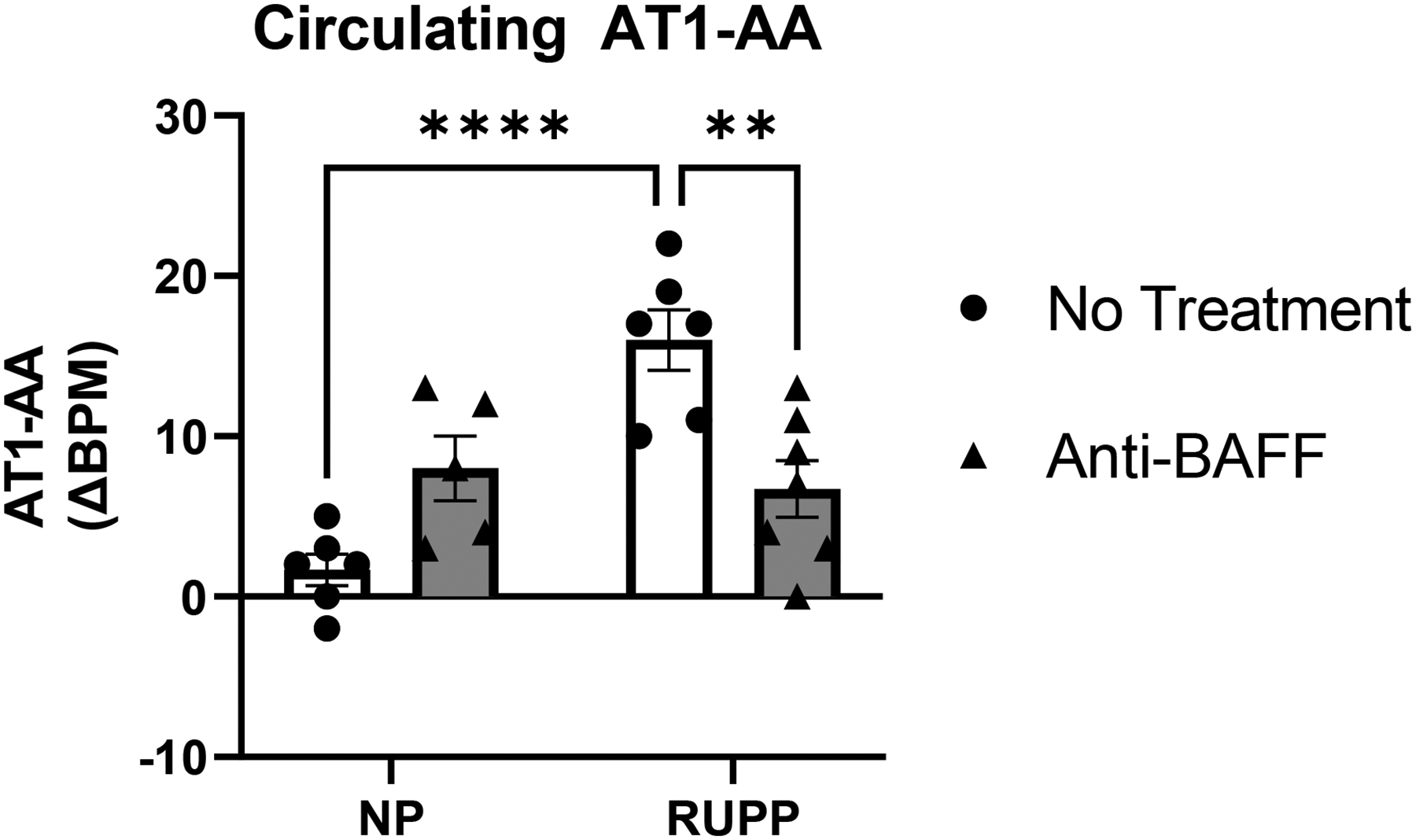
There was no change in circulating AT1-AA between NP (n=6) and NP+Anti-BAFF (n=5). As previously described, RUPPs had significantly increased AT1-AA (n=6, p<0.001) compared to NP. RUPP+Anti-BAFF (n=6, p<0.01) had significantly decreased AT1-AA compared to RUPP.
Anti-BAFF therapy prevented NK cell activation in response to RUPP
RUPP rats had increased circulating cytolytic NK cells (16.50 ± 2.98 %gated, n=7, p<0.05) compared to NP (7.34 ± 3.06 %gated, n=6) (Figure 4.a.). NP+Anti-BAFF had no change in total circulating NK cells (39.10 ± 7.29 %gated, n=7) or cytolytic circulating NK cells (1.34 ± 0.83 %gated, n=5) compared to NP (S 4. a.). RUPP+Anti-BAFF had increased total circulating NK cells (77.08 ± 4.13 %gated, n=6, p<0.05), but no change in circulating cytolytic NK cells (3.88 ± 0.99 %gated, n=8) compared to NP+Anti-BAFF. There was no change in total circulating NK cells between RUPP and RUPP+Anti-BAFF, however RUPP+Anti-BAFF had reduced circulating cytolytic NK cells (p<0.05) compared to RUPP.
Figure 4.
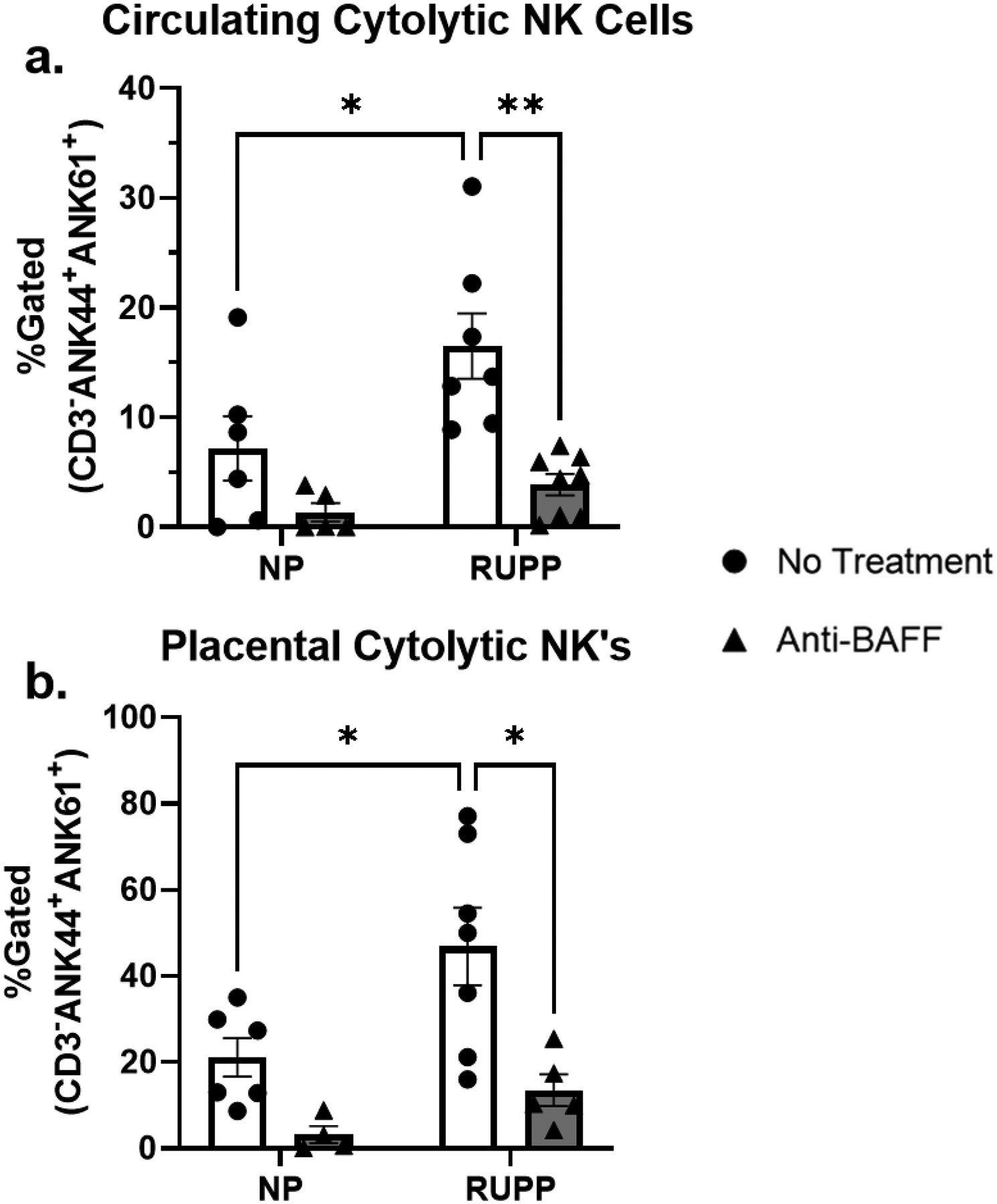
There was no change in circulating cytolytic NK cells between NP (n=6) and NP+Anti-BAFF (n=5), but RUPP rats (n=7, p<0.05) had increased circulating cytolytic NK cells compared to NP. RUPP+Anti-BAFF (n=8, p<0.01) had decreased circulating cytolytic NK cells compared to RUPP. There was no change in placental cytolytic NK cells between NP (n=6) and NP+Anti-BAFF (n=4), but RUPP rats (n=7, p<0.05) had increased placental cytolytic NK cells compared to NP. RUPP+Anti-BAFF (n=5, p<0.05) compared to RUPP.
There were no changes in total placental NK cells between NP (45.76 ± 14.43 %gated, n=6) and RUPP (33.08 ± 12.03 %gated, n=6) (S 4. b.), but RUPP rats had increased placental cytolytic NK cells (46.87 ± 8.99 %gated, n=7, p<0.05) compared to NP (21.18 ± 4.47 %gated, n=6) (Figure 4. b.). NP+Anti-BAFF had no changes in either total placental NK cells (13.67 ± 1.42 %gated, n=4) or cytolytic placental NK cells (3.2 ± 2.01, n=4) compared to NP. RUPP+Anti-BAFF had no changes in either total placental NK cells (46.12 ± 9.27 %gated, n=5) or cytolytic placental NK cells (13.55 ± 3.65 %gated, n=5) compared to NP+Anti-BAFF. RUPP+Anti-BAFF had no change in total placental NK cells compared to RUPP, yet RUPP+Anti-BAFF had reduced placental cytolytic NK cells (p<0.05) compared to RUPP.
Anti-BAFF Antibodies have no Effect on Circulating, Placental, or Renal C1q
There were no changes in plasma C1q across any of the groups: NP (2.95 ± 0.06 mg/mL, n=6), NP+Anti-BAFF (2.86 ± 0.28 mg/mL, n=6), RUPP (3.25 ± 0.18 mg/mL, n=6), RUPP+Anti-BAFF (3.16 ± 0.24 mg/mL, n=5) (S 5. a.). There were no changes in placental C1q across any of the groups: NP (2.83 ± 0.35 ug/mg protein, n=5), NP+Anti-BAFF (2.57 ± 0.47 ug/mg protein, n=5), RUPP (2.90 ± 0.36 ug/mg protein, n=5), or RUPP+Anti-BAFF (2.26 ± 0.48 ug/mg protein, n=5) (S 5.b.). There were no changes in renal-deposited C1q across any of the groups: NP (1.42 ± 0.39 ug/mg protein, n=5), NP+Anti-BAFF (1.02 ± 0.12 ug/mg protein, n=5), RUPP (1.16 ± 0.28 ug/mg protein, n=5), or RUPP+Anti-BAFF (1.08 ± 0.12 ug/mg protein, n=4) (S 5. c.).
Anti-BAFF did not Prevent Complement C3 Activation in Response to RUPP
There was no difference in complement component C3 across any of the groups: NP (913 ± 142 ug/mL, n=5), NP+Anti-BAFF (1223 ± 181 ug/mL, n=5), RUPP (1179 ± 186 ug/mL, n=4), RUPP+Anti-BAFF (1279 ± 131 ug/mL, n=6) (Figure 5.a.). Circulating complement component C3a was increased in RUPP (188 ± 11 ng/mL, n=5, p<0.05) compared to NP (128 ± 16 ng/mL, n=5) (Figure 5.b.). There was no change in C3a between NP and NP+Anti-BAFF (119 ± 17 ng/mL, n=5). C3a was increased in RUPP+Anti-BAFF (195 ± 10 ng/mL, n=6, p<0.01) compared to NP+Anti-BAFF. There were no changes in C3a between RUPP and RUPP+Anti-BAFF groups.
Figure 5.
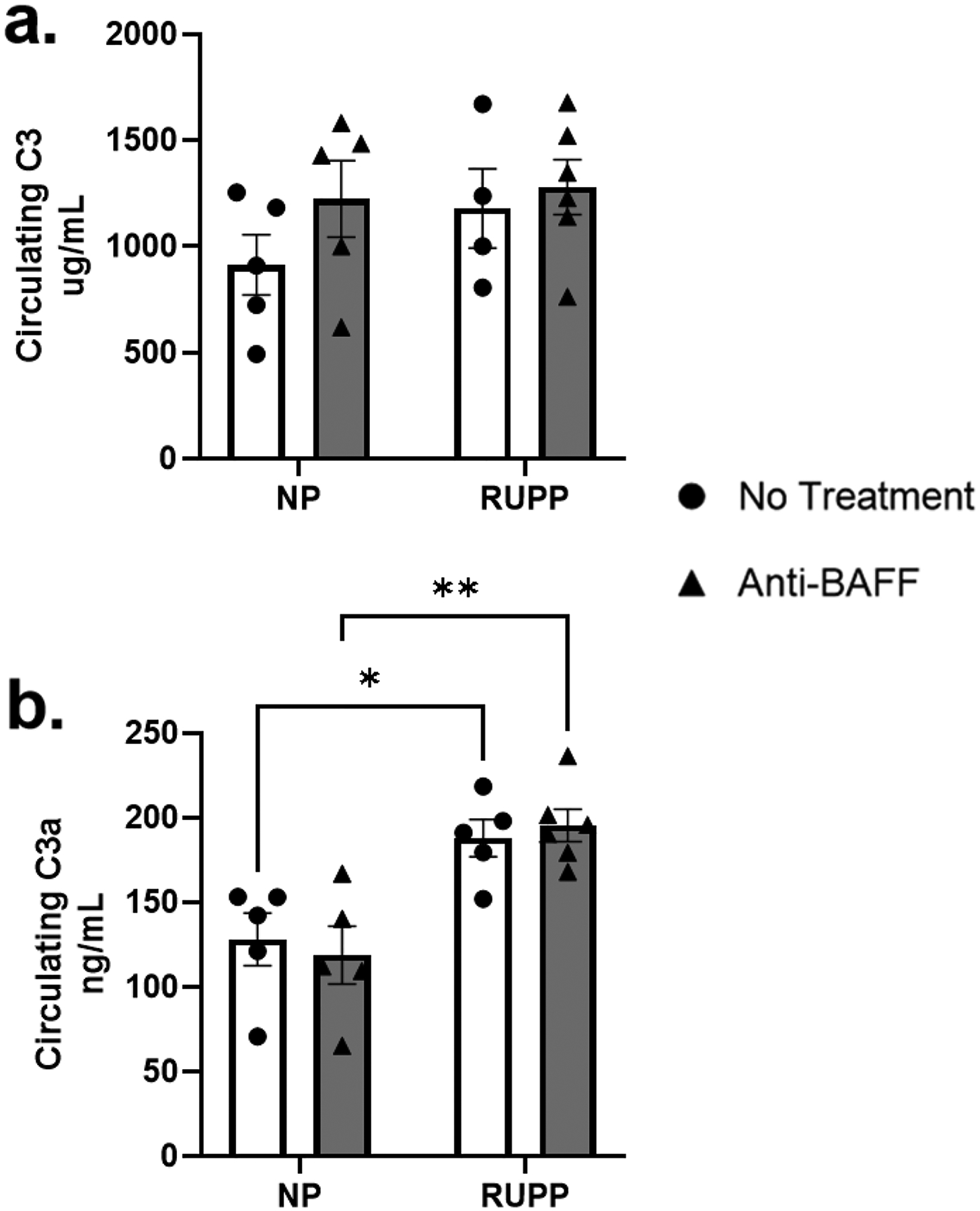
There was no change in circulating complement C3 between NP (n=5), NP+Anti-BAFF (n=5), RUPP (n=4), or RUPP+Anti-BAFF (n=6). There was no change in circulating C3a between NP (n=5) compared to NP+Anti-BAFF (n=5), but RUPP (n=5, p<0.05) had increased C3a compared to NP. RUPP+Anti-BAFF (n=6, p<0.01) had increased C3a compared to NP+Anti-BAFF, but there was no difference between RUPP and RUPP+Anti-BAFF.
Anti-BAFF Antibodies have no Effect on Circulating Complement C4
There were no changes in circulating Complement C4 across any of the groups: NP (1377 ± 116 ug/mL, n=4), NP+Anti-BAFF (1082 ± 115 ug/mL, n=4), RUPP (1539 ± 173 ug/mL, n=5), RUPP+Anti-BAFF (1212 ± 122 ug/mL, n=5) (S 5. d.).
Anti-BAFF Therapy Reduced Circulating APRIL in Response to RUPP
Although lowered in the anti-BAFF treated groups, these differences did not reach statistical significance in circulating BAFF across any of the groups: plasma BAFF was (738 ± 67 pg/mL, n=7) in NP, (499 ± 77 pg/mL, n=7) in NP+Anti-BAFF, (733 ± 112 pg/mL, n=7) in RUPP, and (598 ± 77 pg/mL, n=6) in RUPP+Anti-BAFF (Figure 6.a.).
Figure 6.
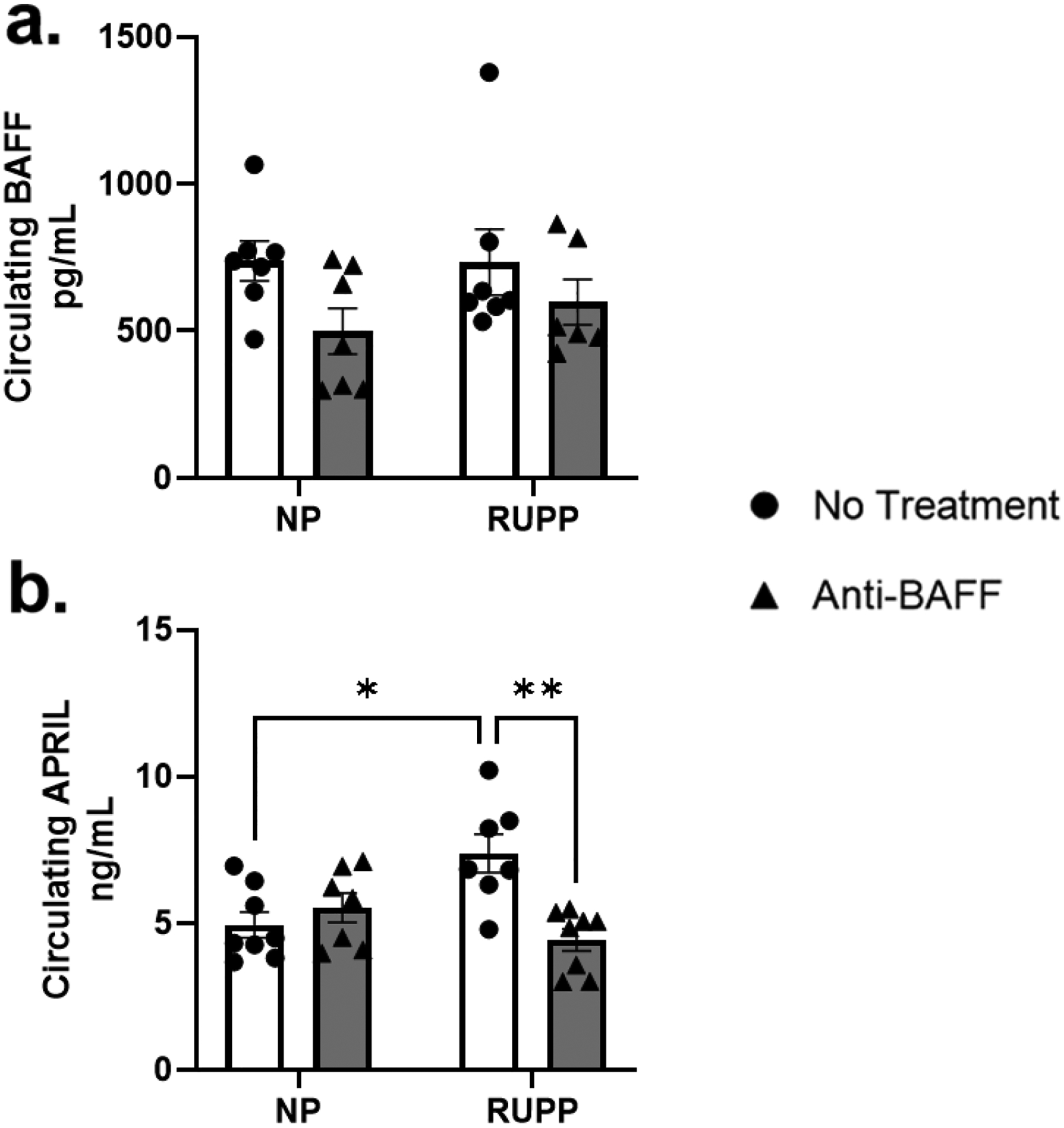
Circulating BAFF trended to decrease between NP (n=7) and NP+Anti-BAFF (n=7) but it did not reach significance. There was no change in BAFF between NP and RUPP (n=7), there was also no change in BAFF between RUPP and RUPP+Anti-BAFF (n=6). There was no change in circulating APRIL between NP (n=8) and NP+Anti-BAFF (n=7), but RUPPs (n=7, p<0.05) had increased APRIL compared to NP. Increased APRIL in the RUPP was attenuated in RUPP+Anti-BAFF (n=8, p<0.01).
Circulating APRIL was increased in RUPP (7.39 ± 0.7 ng/mL, n=7, p<0.05) compared to NP (4.95 ± 0.4 ng/mL, n=8), was unchanged in NP+Anti-BAFF (5.53 ± 0.5 ng/mL, n=7) vs NP (Figure 6.b.), and was normalized in RUPP+Anti-BAFF (4.43 ± 0.4 ng/mL, n=8, p<0.01) compared to RUPP.
Circulating Cytokine Responses to RUPP and Anti-BAFF Antibodies
There was no change in plasma MIP-1α between NP (3.94 ± 1.17pg/mL, n=8) and NP+Anti-BAFF (4.05 ± 0.65pg/mL, n=7) (S 6. a.). RUPP There was a significant increase in plasma MIP-1α between RUPP (20.19 ± 7.03pg/mL, n=9, p<0.05) and NP. RUPP+Anti-BAFF had significantly lower plasma MIP-1α (2.24 ± 0.84pg/mL, n=9, p<0.05) compared to RUPP.
There was no difference in plasma MIP-3α between NP (4.59 ± 0.47pg/mL, n=8) and NP+Anti-BAFF (5.08 ± 1.14pg/mL, n=8) (S 6. b.). There was a significant increase in plasma MIP-3α in RUPPs (12.12 ± 2.42, n=8, p<0.05) compared to NP. There was significantly higher plasma MIP-3α in RUPP+Anti-BAFF (13.42 ± 2.73pg/mL, n=7, p<0.05) compared to NP+Anti-BAFF. There was no change in plasma MIP-3α between RUPP and RUPP+Anti-BAFF.
There was no change in plasma MCP-1 between NP (76.38 ± 13.00pg/mL, n=8) and NP+Anti-BAFF (110.04 ± 20.29pg/mL, n=8) (S 6. c.). There was a significant increase in plasma MCP-1 in RUPPs (359.28 ± 105.92pg/mL, n=9, p<0.05) compared to NP. There was also a trending decrease in plasma MCP-1 in RUPP+Anti-BAFF (223.04 ± 34.42pg/mL, n=9) compared to RUPP.
Discussion
Our lab previously showed that B cell depletion with the anti-CD20 antibody Rituximab attenuates hypertension in the RUPP model [23]. However, Rituximab depletes all B cells including developing B cells, mature B cells, and newly activated B cells; and CD20 is down regulated when cells reach the plasma cell differentiation stage [42, 43]. In this study we also measured total placental B cells, placental B1 cells, and placental B2 cells and saw no changes in response to RUPP or Anti-BAFF antibody infusion (S 3. a, b, c.). It has been previously reported that a subset of CD5+ B1 cells (B1a) cells are increased in the placentas of women with PE [25]. In this study we did not differentiate into B1a and B1b B cells which may contribute to the differences between our study and previously published work. Moreover, Jensen et al. investigated B1a cells in human placentas while we measured B1 cells in rat placentas drawing another important distinction between these two studies. However, here we demonstrate that anti-BAFF antibodies can deplete circulating B cells, and BAFF depletion can be used to normalize the hypertension, NK cells, and AT1-AA without harming fetal development and survival in the RUPP model of placental ischemia.
In contrast to prior studies in nonpregnant mice [31], at GD19 there was no significant decrease in circulating BAFF in the anti-BAFF treated groups, which could be due to our use of a different dose and dosing schedule of anti-BAFF in this study compared to the cited literature. In our study we used single infusion of Ant-BAFF at a 1 mg/kg dose; whereas in the cited study, repeated administration of 2 mg/kg anti-BAFF antibodies were used every two weeks. Moreover, this observation could be due to the ELISA measuring both bound and unbound circulating BAFF. Importantly, our study shows that a single dose of anti-BAFF antibodies reduces circulating B cells and prevents hypertension in response to placental ischemia. This suggests that administering anti-BAFF at the time of placental ischemia prevents B cell activation, which then prevents AT1-AA and hypertension in RUPP rats. We also demonstrated that the sister cytokine to BAFF, APRIL, is increased in response to placental ischemia and treatment with anti-BAFF antibodies reduced APRIL in placental ischemic rats. To our knowledge, this is the first study to show a relationship between BAFF, APRIL, and autoantibody production in an animal model of PE. APRIL is produced by myeloid lineage cells and promotes B cell class-switching, survival of activated B cells long-term, and plasma cell survival [29]. Unlike BAFF, APRIL supports both B1 and B2 cells post activation in humans [29]. In a mouse model of kidney rejection, depleting APRIL and BAFF together reduced autoantibody production and lessened B cell mediated renal rejection [44]. In the RUPP model, increased APRIL could be downstream of B cell activation in response to placental ischemia and myeloid cells supporting the autoreactive B cells; however, in anti-BAFF treated RUPPs, reduced B cell activation could prevent the need for increased APRIL production. However, more studies are necessary to better understand relationship between BAFF/APRIL, AT1-AA, and hypertension in response to placental ischemia.
Human anti-BAFF antibodies (Belimumab) have been approved for treatment of SLE and has been shown to deplete B cells while preventing transformation of B cells into Ig-producing plasma cells [32]. Importantly, treatment with Belimumab reduced production of autoreactive antibodies associated with SLE [45], thereby demonstrating a potential role for BAFF as a therapeutic target to prevent autoantibody production. Moreover, toxicological studies using Belimumab in cynomolgus monkeys showed B cell depletion without altering CD4+ T cell levels or treatment-related infections. Considering the off-target effects that Rituximab has shown in patients and animal models of disease, these data further support BAFF as a potential therapeutic to inhibit B cell involvement in disease [46]. Kowalczyk-Quintas et al. showed that depletion of BAFF in mice reduced circulating B cells in one week without depleting T cells [31]. Rituximab is noted for having off-target effects that may suppress other immune cell types and functions [47]; therefore, studies indicating that Belimumab is more specific for B cell suppression/depletion is important from a therapeutic approach [48, 49]. Treatment with anti-BAFF antibodies could be beneficial in selectively depleting newly activated B2 cells while sparing existing B1 cells and maintaining natural antibodies in the patients undergoing therapy. Contrary to our previous studies [38], there was no change in B cells between NP and RUPP rats, which is likely due to the large variation in data collected in the B cell number in the NP control group.
In this study we showed that RUPP rats had increase plasma Macrophage Inflammatory Protein (MIP)-1α, MIP-3α, and Monocyte Chemoattractant Protein (MCP)-1, while treatment with Anti-BAFF attenuated the increase in MIP-1α. These three cytokines were first noted for their interactions with macrophages and monocytes, but were later discovered to be produced by many cell types including endothelial cells and all hematopoietic cells [50]. MIP-1α, MIP-3α, and MCP-1 can all recruit macrophages, monocytes, dendritic cells, or Th17s. In this study, increases in MIP-1α, MIP-3α, and MCP-1 indicate recruitment of immune cells, including monocytes and macrophages, into tissues, but our study did not differentiate which tissues had changes in immune cells outside of B cells and natural killer cells as shown in the results. Nevertheless, our data indicate that while there is increased signaling for inflammatory cell invasion in the RUPP, Anti-BAFF attenuates MIP-1α which could suggest an interaction between macrophage activation and B cell secretion of AT1-AA.
Here, we show that anti-BAFF treatment reduced circulating AT1-AA, as well as activated NK cells in the circulation and placentas in response to placental ischemia. In normal pregnancies, uterine NK cells help in placentation and vascular remodeling [51, 52], but dysregulation of uterine NK cells has been associated with multiple pathologies of pregnancy in humans including PE [53–55]. Activated NK cells can induce production of anti-angiogenic factors, resulting in decreased placental vascularization and contributing to FGR [56–58]. While AT1-AA can activate the AT1 receptor [59], they can also target cells for destruction through antibody dependent cellular cytotoxicity (ADCC), which involves activated NK cells. In ADCC, an antigen is bound by an antibody’s binding region, while the constant region of the antibody can be recognized by NK cells, which induce apoptosis of the targeted cell [60]. ADCC activates NK cells, but we have shown that blockade of AT1-AA in the RUPP reduced activation of NK cells, suggesting a link between AT1-AA and NK cell activation in response to placental ischemia [61]. As NK cell activation has been linked with the pathogenesis of PE, targeting B cells or AT1-AA could potentially ameliorate both AT1-AA induced pathologies and NK cell activation [56, 62]. Our data suggests that by depleting B cells, we can reduce AT1-AA, which leads to decreased NK cell activation in response to placental ischemia and improves fetal weight in some studies [63, 64].
An additional mechanism of antibody-mediated cell death is activation of the complement system [65]. The complement system is part of the innate immune system that aids in cell turnover, cell destruction, and inflammation [66]. The complement system is important in uterine remodeling during normal pregnancy [67]. Dysregulation of the complement system is associated with PE and other pregnancy complications [11, 65, 68]. The stimulus for the classical complement cascade is an antibody binding its antigen, which starts a cascade of proteolytic cleavages resulting in the formation of the membrane attack complex and induction of cell death [11]. The initiating factor of the classical complement cascade (C1q) and a marker of classical complement activation (C4a) are reduced in circulation in PE compared to NP, and this decrease has been attributed to complement deposition and activation in the kidney [68]. Another study later found that C1q and C4a are increased in glomeruli of women with PE, suggesting a role for classical complement activation in PE [69]. In addition, placentas of women with PE have increased activation marker C4d compared to NP, but showed no difference in deposited C1q [70]. AT1-AA has been implicated as a potential activator of classical complement and could contribute to cell death and tissue damage in PE via this mechanism [65, 71]. In this study, we found no significant changes in complement component C1q in the circulation, placenta, or kidney, nor did we see changes in circulating complement C4 associated with the RUPP model or in response to anti-BAFF administration. Although data regarding C1q is not prominently investigated using animals, Singh et al. showed that C1q deficient mice have dysregulation of trophoblast development and appear to develop a PE-like syndrome characterized by hypertension, increased soluble VEGF-1 receptor (sflt-1), and albuminuria [72].
In contrast to C1 and C4, our study showed increased circulating complement C3a in response to RUPP which was not reduced in the RUPP with anti-BAFF antibodies. All complement system pathways converge at the cleavage of C3 to C3a and C3b; therefore, C3a is a common marker of complement system activation [73]. Chronically elevated C3a assists in maintaining a pro-inflammatory response, as in rheumatoid arthritis [74]. This increase in C3a could cause a feed-forward mechanism in PE, further promoting inflammatory cytokine production and immune cell activation. Some clinical studies have reported no changes in circulating C3a in women with PE [68, 75], but others have shown that elevated C3a may be more closely associated with early onset PE [65, 67, 76, 77]. Together this suggests that complement dysregulation and activation is involved in the pathology of PE; however, because our model is one of late onset PE, we may not be able to detect changes in C3a with an intervention. Overall, our data are congruent with other studies demonstrating that the complement system is activated in the RUPP model, but that complement activation may not be involved in the hypertension or NK cell activation seen in the RUPP. It is also important to mention that studies in animal models do not perfectly recapitulate every characteristic of human PE; therefore, some aspects of this study may not translate to the clinic. However, this study implicates B2 cells as important contributors to the pathophysiology of hypertension in the RUPP model.
Collectively, our study showed that targeting BAFF in the RUPP model of placental ischemia normalizes hypertension, reduces AT1-AA, and reduces NK cell activation but does not prevent complement activation. This could be due to the therapy being an antibody itself, which may not allow for a decrease in the activation of complement, but additional studies are needed to verify this. Nevertheless, targeting BAFF could be a potential therapeutic option in the treatment of PE. Future studies exploring BAFF depletion in other preclinical models of PE will further determine the viability of this therapeutic approach. Importantly, this study is the first to show that reducing APRIL in response to placental ischemia is associated with improved blood pressure, immune activation, and AT1-AA. Future studies could determine whether the changes in circulating APRIL occur in other models of PE and investigate if blockade of APRIL has similar affects in response to placental ischemia as seen with BAFF blockade.
Supplementary Material
Acknowledgments
This study was supported by NIH grants RO1HD067541 (BL) and P20GM121334 (BL, LMA) and 1U54GM115428 (LMA), American heart association (AHA) early career award 19CDA34670055 (LMA).
Footnotes
Ethics Statement
The authors confirm that the ethical policies of the journal and the University of Mississippi Medical Center’s Institutional Animal Care and Use Committee were followed. All experiments were in accordance with the National Institute of Health’s “Guidelines for the Care and Use of Laboratory Animals.”
Disclosures
The authors declare no conflicts of interest.
References
- 1.Rana S, et al. , Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circ Res, 2019. 124(7): p. 1094–1112. [DOI] [PubMed] [Google Scholar]
- 2.Staff AC, et al. , Failure of physiological transformation and spiral artery atherosis: their roles in preeclampsia. Am J Obstet Gynecol, 2022. 226(2S): p. S895–S906. [DOI] [PubMed] [Google Scholar]
- 3.Staff AC, Dechend R, and Redman CW, Review: Preeclampsia, acute atherosis of the spiral arteries and future cardiovascular disease: two new hypotheses. Placenta, 2013. 34 Suppl: p. S73–8. [DOI] [PubMed] [Google Scholar]
- 4.Zhang P, Decidual Vasculopathy in Preeclampsia and Spiral Artery Remodeling Revisited: Shallow Invasion versus Failure of Involution. AJP Rep, 2018. 8(4): p. e241–e246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies EL, Bell JS, and Bhattacharya S, Preeclampsia and preterm delivery: A population-based case-control study. Hypertens Pregnancy, 2016. 35(4): p. 510–519. [DOI] [PubMed] [Google Scholar]
- 6.Jeyabalan A, Epidemiology of preeclampsia: impact of obesity. Nutr Rev, 2013. 71 Suppl 1: p. S18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sahlman H, et al. , Maternal use of drugs and preeclampsia. Br J Clin Pharmacol, 2019. 85(12): p. 2848–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Udenze I, et al. , The role of cytokines as inflammatory mediators in preeclampsia. Pan Afr Med J, 2015. 20: p. 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harmon AC, et al. , The role of inflammation in the pathology of preeclampsia. Clin Sci (Lond), 2016. 130(6): p. 409–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cornelius DC, et al. , Inflammatory mediators: a causal link to hypertension during preeclampsia. Br J Pharmacol, 2019. 176(12): p. 1914–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burwick RM and Feinberg BB, Complement activation and regulation in preeclampsia and hemolysis, elevated liver enzymes, and low platelet count syndrome. Am J Obstet Gynecol, 2022. 226(2S): p. S1059–S1070. [DOI] [PubMed] [Google Scholar]
- 12.Dechend R, et al. , AT1 receptor agonistic antibodies, hypertension, and preeclampsia. Semin Nephrol, 2004. 24(6): p. 571–9. [DOI] [PubMed] [Google Scholar]
- 13.LaMarca B, et al. , Hypertension in response to autoantibodies to the angiotensin II type I receptor (AT1-AA) in pregnant rats: role of endothelin-1. Hypertension, 2009. 54(4): p. 905–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herse F and LaMarca B, Angiotensin II type 1 receptor autoantibody (AT1-AA)-mediated pregnancy hypertension. Am J Reprod Immunol, 2013. 69(4): p. 413–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.LaMarca B, et al. , Identifying immune mechanisms mediating the hypertension during preeclampsia. Am J Physiol Regul Integr Comp Physiol, 2016. 311(1): p. R1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaka R, et al. , Characterization of Mitochondrial Bioenergetics in Preeclampsia. J Clin Med, 2021. 10(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wallace K, et al. , CD4+ T-helper cells stimulated in response to placental ischemia mediate hypertension during pregnancy. Hypertension, 2011. 57(5): p. 949–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cornelius DC, et al. , Reduced uterine perfusion pressure T-helper 17 cells cause pathophysiology associated with preeclampsia during pregnancy. Am J Physiol Regul Integr Comp Physiol, 2016. 311(6): p. R1192–R1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rieber-Mohn AB, et al. , Auto-antibodies against the angiotensin II type I receptor in women with uteroplacental acute atherosis and preeclampsia at delivery and several years postpartum. J Reprod Immunol, 2018. 128: p. 23–29. [DOI] [PubMed] [Google Scholar]
- 20.Cornelius DC, et al. , Blockade of CD40 ligand for intercellular communication reduces hypertension, placental oxidative stress, and AT1-AA in response to adoptive transfer of CD4+ T lymphocytes from RUPP rats. Am J Physiol Regul Integr Comp Physiol, 2015. 309(10): p. R1243–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reeve KE, et al. , Placental CD4+ T cells from preeclamptic patients cause autoantibodies to the angiotensin II type I receptor and hypertension in a pregnant rat model of preeclampsia. Exploration of Medicine, 2022. 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petersone L, et al. , T Cell/B Cell Collaboration and Autoimmunity: An Intimate Relationship. Front Immunol, 2018. 9: p. 1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.LaMarca B, et al. , Hypertension in response to placental ischemia during pregnancy: role of B lymphocytes. Hypertension, 2011. 57(4): p. 865–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tarlinton DM, McLean M, and Nossal GJ, B1 and B2 cells differ in their potential to switch immunoglobulin isotype. Eur J Immunol, 1995. 25(12): p. 3388–93. [DOI] [PubMed] [Google Scholar]
- 25.Jensen F, et al. , CD19+CD5+ cells as indicators of preeclampsia. Hypertension, 2012. 59(4): p. 861–8. [DOI] [PubMed] [Google Scholar]
- 26.Schneider P, The role of APRIL and BAFF in lymphocyte activation. Curr Opin Immunol, 2005. 17(3): p. 282–9. [DOI] [PubMed] [Google Scholar]
- 27.Khodadadi L, et al. , The Maintenance of Memory Plasma Cells. Front Immunol, 2019. 10: p. 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Belnoue E, et al. , APRIL is critical for plasmablast survival in the bone marrow and poorly expressed by early-life bone marrow stromal cells. Blood, 2008. 111(5): p. 2755–64. [DOI] [PubMed] [Google Scholar]
- 29.Kawakami T, et al. , Abundant a proliferation-inducing ligand (APRIL)-producing macrophages contribute to plasma cell accumulation in immunoglobulin G4-related disease. Nephrol Dial Transplant, 2019. 34(6): p. 960–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mongini PK, et al. , APRIL and BAFF promote increased viability of replicating human B2 cells via mechanism involving cyclooxygenase 2. J Immunol, 2006. 176(11): p. 6736–51. [DOI] [PubMed] [Google Scholar]
- 31.Kowalczyk-Quintas C, et al. , Antibodies That Block or Activate Mouse B Cell Activating Factor of the Tumor Necrosis Factor (TNF) Family (BAFF), Respectively, Induce B Cell Depletion or B Cell Hyperplasia. J Biol Chem, 2016. 291(38): p. 19826–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blair HA and Duggan ST, Belimumab: A Review in Systemic Lupus Erythematosus. Drugs, 2018. 78(3): p. 355–366. [DOI] [PubMed] [Google Scholar]
- 33.Furie R, et al. , Two-Year, Randomized, Controlled Trial of Belimumab in Lupus Nephritis. N Engl J Med, 2020. 383(12): p. 1117–1128. [DOI] [PubMed] [Google Scholar]
- 34.Granger JP, et al. , Reduced uterine perfusion pressure (RUPP) model for studying cardiovascular-renal dysfunction in response to placental ischemia. Methods Mol Med, 2006. 122: p. 383–92. [DOI] [PubMed] [Google Scholar]
- 35.Harmon A, et al. , IL-10 supplementation increases Tregs and decreases hypertension in the RUPP rat model of preeclampsia. Hypertens Pregnancy, 2015. 34(3): p. 291–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sholook MM, et al. , Systemic hemodynamic and regional blood flow changes in response to chronic reductions in uterine perfusion pressure in pregnant rats. Am J Physiol Heart Circ Physiol, 2007. 293(4): p. H2080–4. [DOI] [PubMed] [Google Scholar]
- 37.Deer E, et al. , CD4+ T cells cause renal and placental mitochondrial oxidative stress as mechanisms of hypertension in response to placental ischemia. Am J Physiol Renal Physiol, 2021. 320(1): p. F47–F54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cottrell JN, et al. , Interleukin-4 supplementation improves the pathophysiology of hypertension in response to placental ischemia in RUPP rats. Am J Physiol Regul Integr Comp Physiol, 2019. 316(2): p. R165–R171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Travis OK, et al. , Interleukin-17 signaling mediates cytolytic natural killer cell activation in response to placental ischemia. Am J Physiol Regul Integr Comp Physiol, 2020. 318(6): p. R1036–R1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harmon AC, et al. , Placental CD4(+) T cells isolated from preeclamptic women cause preeclampsia-like symptoms in pregnant nude-athymic rats. Pregnancy Hypertens, 2019. 15: p. 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Booz GW and Baker KM, Role of type 1 and type 2 angiotensin receptors in angiotensin II-induced cardiomyocyte hypertrophy. Hypertension, 1996. 28(4): p. 635–40. [DOI] [PubMed] [Google Scholar]
- 42.Memon AB, et al. , Long-term safety of rituximab induced peripheral B-cell depletion in autoimmune neurological diseases. PLoS One, 2018. 13(1): p. e0190425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crickx E, et al. , Anti-CD20-mediated B-cell depletion in autoimmune diseases: successes, failures and future perspectives. Kidney Int, 2020. 97(5): p. 885–893. [DOI] [PubMed] [Google Scholar]
- 44.Bath NM, et al. , Autoantibody production significantly decreased with APRIL/BLyS blockade in murine chronic rejection kidney transplant model. PLoS One, 2019. 14(10): p. e0223889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Navarra SV, et al. , Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet, 2011. 377(9767): p. 721–31. [DOI] [PubMed] [Google Scholar]
- 46.Halpern WG, et al. , Chronic administration of belimumab, a BLyS antagonist, decreases tissue and peripheral blood B-lymphocyte populations in cynomolgus monkeys: pharmacokinetic, pharmacodynamic, and toxicologic effects. Toxicol Sci, 2006. 91(2): p. 586–99. [DOI] [PubMed] [Google Scholar]
- 47.Luo C, et al. , Efficacy and safety of new anti-CD20 monoclonal antibodies versus rituximab for induction therapy of CD20(+) B-cell non-Hodgkin lymphomas: a systematic review and meta-analysis. Sci Rep, 2021. 11(1): p. 3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shipa M, et al. , Effectiveness of Belimumab After Rituximab in Systemic Lupus Erythematosus : A Randomized Controlled Trial. Ann Intern Med, 2021. 174(12): p. 1647–1657. [DOI] [PubMed] [Google Scholar]
- 49.Wise LM and Stohl W, Belimumab and Rituximab in Systemic Lupus Erythematosus: A Tale of Two B Cell-Targeting Agents. Front Med (Lausanne), 2020. 7: p. 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Menten P, Wuyts A, and Van Damme J, Macrophage inflammatory protein-1. Cytokine Growth Factor Rev, 2002. 13(6): p. 455–81. [DOI] [PubMed] [Google Scholar]
- 51.Fraser R, et al. , Decidual natural killer cells regulate vessel stability: implications for impaired spiral artery remodelling. J Reprod Immunol, 2015. 110: p. 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robson A, et al. , Uterine natural killer cells initiate spiral artery remodeling in human pregnancy. FASEB J, 2012. 26(12): p. 4876–85. [DOI] [PubMed] [Google Scholar]
- 53.Murphy SP, et al. , Uterine NK cells mediate inflammation-induced fetal demise in IL-10-null mice. J Immunol, 2005. 175(6): p. 4084–90. [DOI] [PubMed] [Google Scholar]
- 54.Aneman I, et al. , Mechanisms of Key Innate Immune Cells in Early- and Late-Onset Preeclampsia. Front Immunol, 2020. 11: p. 1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Giezeman-Smits KM, et al. , Novel monoclonal antibodies against membrane structures that are preferentially expressed on IL-2-activated rat NK cells. J Leukoc Biol, 1998. 63(2): p. 209–15. [DOI] [PubMed] [Google Scholar]
- 56.Travis OK, et al. , Adoptive transfer of placental ischemia-stimulated natural killer cells causes a preeclampsia-like phenotype in pregnant rats. Am J Reprod Immunol, 2021. 85(6): p. e13386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang HL, et al. , Decidual stromal cells promote the differentiation of CD56(bright) CD16(−) NK cells by secreting IL-24 in early pregnancy. Am J Reprod Immunol, 2019. 81(6): p. e13110. [DOI] [PubMed] [Google Scholar]
- 58.Fukui A, et al. , Changes of NK cells in preeclampsia. Am J Reprod Immunol, 2012. 67(4): p. 278–86. [DOI] [PubMed] [Google Scholar]
- 59.Dechend R, et al. , AT1 receptor agonistic antibodies from preeclamptic patients stimulate NADPH oxidase. Circulation, 2003. 107(12): p. 1632–9. [DOI] [PubMed] [Google Scholar]
- 60.Lo Nigro C, et al. , NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: biological evidence and clinical perspectives. Ann Transl Med, 2019. 7(5): p. 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cunningham MW Jr., et al. , Renal natural killer cell activation and mitochondrial oxidative stress; new mechanisms in AT1-AA mediated hypertensive pregnancy. Pregnancy Hypertens, 2019. 15: p. 72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.El-Azzamy H, et al. , Dysregulated uterine natural killer cells and vascular remodeling in women with recurrent pregnancy losses. Am J Reprod Immunol, 2018. 80(4): p. e13024. [DOI] [PubMed] [Google Scholar]
- 63.Cottrell JN, et al. , Progesterone-induced blocking factor improves blood pressure, inflammation, and pup weight in response to reduced uterine perfusion pressure (RUPP). Am J Physiol Regul Integr Comp Physiol, 2021. 320(5): p. R719–R727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Elfarra JT, et al. , 17-Hydroxyprogesterone caproate improves T cells and NK cells in response to placental ischemia; new mechanisms of action for an old drug. Pregnancy Hypertens, 2020. 19: p. 226–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pierik E, et al. , Dysregulation of Complement Activation and Placental Dysfunction: A Potential Target to Treat Preeclampsia? Front Immunol, 2019. 10: p. 3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sarma JV and Ward PA, The complement system. Cell Tissue Res, 2011. 343(1): p. 227–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Derzsy Z, et al. , Activation of the complement system in normal pregnancy and preeclampsia. Mol Immunol, 2010. 47(7–8): p. 1500–6. [DOI] [PubMed] [Google Scholar]
- 68.Agostinis C, et al. , Complement component C1q as potential diagnostic but not predictive marker of preeclampsia. Am J Reprod Immunol, 2016. 76(6): p. 475–481. [DOI] [PubMed] [Google Scholar]
- 69.Penning M, et al. , Classical Complement Pathway Activation in the Kidneys of Women With Preeclampsia. Hypertension, 2015. 66(1): p. 117–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Buurma A, et al. , Preeclampsia is characterized by placental complement dysregulation. Hypertension, 2012. 60(5): p. 1332–7. [DOI] [PubMed] [Google Scholar]
- 71.Wang W, et al. , Autoantibody-mediated complement C3a receptor activation contributes to the pathogenesis of preeclampsia. Hypertension, 2012. 60(3): p. 712–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Singh J, Ahmed A, and Girardi G, Role of complement component C1q in the onset of preeclampsia in mice. Hypertension, 2011. 58(4): p. 716–24. [DOI] [PubMed] [Google Scholar]
- 73.Campo A, et al. , [Complement activation products (C3a and C5b-9) as markers of activity of dermatomyositis. Comparison with usual biochemical parameters]. Actas Dermosifiliogr, 2007. 98(6): p. 403–14. [PubMed] [Google Scholar]
- 74.Coulthard LG and Woodruff TM, Is the complement activation product C3a a proinflammatory molecule? Re-evaluating the evidence and the myth. J Immunol, 2015. 194(8): p. 3542–8. [DOI] [PubMed] [Google Scholar]
- 75.Lynch AM, et al. , The interrelationship of complement-activation fragments and angiogenesis-related factors in early pregnancy and their association with pre-eclampsia. BJOG, 2010. 117(4): p. 456–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Halmos A, et al. , Circulating ficolin-2 and ficolin-3 in normal pregnancy and pre-eclampsia. Clin Exp Immunol, 2012. 169(1): p. 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Boij R, et al. , Biomarkers of coagulation, inflammation, and angiogenesis are independently associated with preeclampsia. Am J Reprod Immunol, 2012. 68(3): p. 258–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.