

Abstract
Background
Myhre syndrome is a rare multisystem genetic disorder that is caused by de novo heterozygous gain‐of‐function variants in SMAD4. Patients with Myhre syndrome exhibit several phenotypes at different ages such as small size, autism, developmental delay, left‐sided heart defects, and hearing loss and often have a characteristic facial appearance. The early clinical diagnosis of Myhre syndrome remains a major challenge, particularly in the first year of life.
Methods
A Chinese male infant with syndactyly of fingers, hypertelorism, short palpebral fissures, and short philtrum was enrolled into the ENT department of the Chinese PLA General Hospital. Whole exome sequencing analysis was used to detect the disease‐causing variant. A literature review of Myhre syndrome was also performed.
Results
A recurrent de novo missense variant c.1498A > G p.I500V(p. Ile500Val) in SMAD4 was detected confirming the clinical diagnosis of Myhre syndrome at the age of 38 days. The infant appears to be the youngest reported case of Myhre syndrome. At 23‐month follow‐up, the affected infant has dysmorphic facial features, growth retardation, and previously undescribed complete syndactyly. Review the literatures noted several common features in Myhre syndrome patients including hearing loss (72.7%), characteristic facial features (26.0%–54.5%), finger and toe abnormalities (3.9%–48.1%), short stature (45.5%), and respiratory (30.0%) and cardiovascular problems (65.0%).
Conclusions
Clinicians should have a low threshold to perform genetic testing on patients with features suggesting Myhre syndrome even in the first year of life. Although some individuals with Myhre syndrome have normal hearing, early onset or progressive hearing loss usually occur in one or both ears in most patients, with remarkable phenotypic heterogeneity. Syndactyly may be minor such as typical 2–3 toe involvement, or more complicated as was observed in our patient.
Keywords: complete syndactyly, early diagnosis, Myhre syndrome, SMAD4
The affected infant we described is the youngest patient with Myhre syndrome, who was diagnosed at the age of 38 days.
The case we described is the first case of Myhre syndrome with complete syndactyly.
We reviewed literatures on Myhre syndrome focused on the hearing loss, characteristic facial feature, finger, and toe abnormalities.
1. INTRODUCTION
Myhre syndrome (MIM #139210) is a rare autosomal dominant multiple systematic disorder that was first described by Myhre et al. in two unrelated males in 1981 (Myhre et al., 1981). The clinical features of this disorder include poor growth, variable degrees of intellectual disability, distinctive facial dysmorphic features, brachydactyly, restricted joint mobility, hearing loss, stiff and thick skin, and cardiovascular and respiratory disorders (Caputo et al., 2012; Le Goff & Cormier‐Daire, 2012; Lin et al., 2016).
Gain‐of‐function pathogenic variants in SMAD4 (MIM #600993) are the only molecular etiology that is known to cause Myhre syndrome. Currently, four pathogenic variants of SMAD4 (Ile500Thr, Ile500Val, Ile500Met, and Arg496Cys) have been described, and almost occur de novo (sporadic) (Asakura et al., 2012; Caputo et al., 2012, 2014; Lin et al., 2020). Only one familial case of Myhre syndrome with SMAD4 c.1486C > T (p.Arg496Cys) has previously been reported (Meerschaut et al., 2019). Most patients with Myhre syndrome are diagnosed at school age with postnatal progression. The dysmorphic features associated with the syndrome may be observed in early childhood, but the final diagnosis is made after the clinical features are fully manifested during the late‐childhood period.
The early diagnosis of Myhre syndrome is challenging and relies on the detection of characteristic clinical features and confirmation by genetic analysis. Differential diagnosis includes other genetic syndromes such as acromicric dysplasia (MIM #102370) (Le Goff & Cormier‐Daire, 2012) and geleophysic dysplasia (MIM ## 231,050) (McInerney‐Leo et al., 2016). These syndromes are associated with growth retardation, facial dysmorphic features, finger deformity, stiff gait and cardiovascular involvement.
In this study, we report on a case of Myhre syndrome in a 23‐month‐old Chinese male infant with distinctive facial features, complete syndactyly of fingers and short stature. The diagnosis of Myhre syndrome was confirmed by the identification of one de novo missense variant c.1498A > G (p.Ile500Val) in SMAD4 at 38‐day and nearly 2 years of follow‐up. This case is remarkable for the early diagnosis and the complete syndactyly that have not been previously reported in individuals with Myhre syndrome.
2. METHODS
2.1. Clinical evaluation
A 12‐day‐old male was admitted to the ENT Department of the Chinese PLA General Hospital (Beijing, China). Physical examinations and medical history were collected on the patient through multiple interviews. An initial hearing screen was performed within 72 hours using transiently evoked otoacoustic emission (TEOAE) testing. Auditory brainstem response (ABR), auditory steady state response (ASSR), distortion product otoacoustic emission (DPOAE), and acoustic immittance were performed by the age of 23 months to evaluate hearing loss that was diagnosed according to the WHO 1997 criteria. The parents reported no history of stillbirth or miscarriage. Cardiac structure and function were evaluated by electrocardiography (ECG) and color Doppler echocardiography.
2.2. Whole exome sequencing and bioinformatics analysis
Whole exome sequencing (WES) genetic analyses was performed designed on parent‐offspring trios including two unaffected parents (I:1 and I:2) and one affected individual (II:1). Peripheral venous blood was collected from the proband and his parents. Genomic DNA was extracted using a QIAamp DNA blood mini‐kit (Qiagen, Germany) following standard protocols. Sequencing libraries were prepared and xGen Exome Research Panel probes (IDT) were used to capture the target sequences. WES of the captured sequences was performed on an Illumina NovaSeq 6000 by Euler Genomics (Beijing, China). Reads were mapped to the UCSC hg19 human reference genome. The reference sequence of SMAD4 used in alignment was NM_005359.6. Sequence variants were checked in the gnomAD population databases (http://gnomad‐old. broadinstitute.org/). Variant pathogenicity was interpreted according to the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015). The variants segregation analysis was confirmed by Sanger sequencing.
3. RESULTS
3.1. Clinical findings
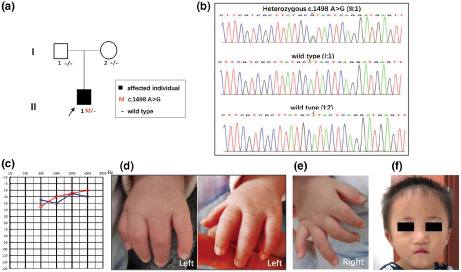
The affected male infant (II:1) and his unaffected parents from the family (I:1 and I:2) (Figure 1a) were clinically analyzed. This Chinese male infant (II:1) was born to unaffected non‐consanguineous parents (I:1 and I:2) by spontaneous vaginal delivery at 37 weeks +3 of gestation with a birthweight of 1.99 kg (<3rd centile). His mother suffered from gestational hypertension and gestational diabetes during pregnancy. The proband was 41 cm tall (<3rd centile) and had a head circumference of 29 cm (<3rd centile). The child had mild facial dysmorphic features including short palpebral fissures and a short philtrum (shortened distance between the nose and upper lip). Furthermore, we noted deformed 4 digits of the left hand (Figure 1d).
FIGURE 1.
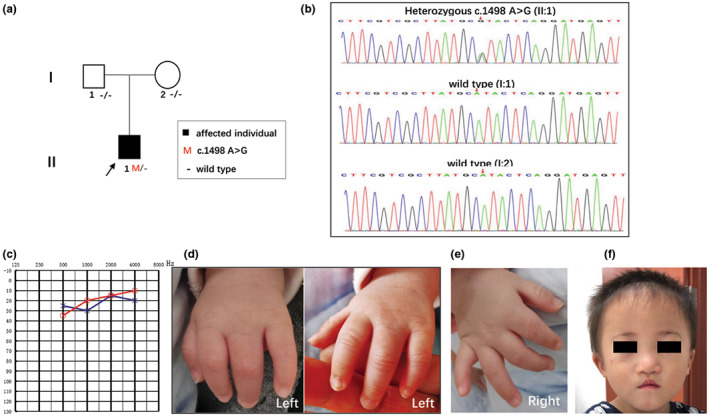
Pedigree, variant analysis and multi‐system anomalies of the patient (II:1). (a) The proband is indicated by an arrow. Subjects I:1, I:2 and II:1 were tested by WES. (b) The chromatogram shows SMAD4 heterozygous c.1498A > G mutated in patient II:1. (c) The audiogram showed normal hearing of II:1 (red, right ear; blue, left ear). (d) The left‐hand panel presents complete syndactyly of fingers 3–4. Complete syndactyly extends to the tip of the digits with the inclusion of the nail folds. (e) The right hand was normal. (f) A summary of facial features including short palpebral fissures, a short philtrum, a narrow mouth with a thin upper lip, an underdeveloped upper jaw, and a protruding lower jaw at 23 months.
WES is increasingly used in the clinical analysis of patients with rare disorders and was performed on the proband and his parents at 12 days after birth. At 38 days old, a definitive molecular diagnosis was reached, consistent with the clinical presentation (Table 1). In addition to the features in the newborn period, other later‐onset features associated with postnatal progression (e.g., hearing loss, growth and intellectual retardation) were sought. Physical examination at 23 months showed severe growth retardation with length 72 cm (<3rd centile), weight 8.5 kg (<3rd centile), and head circumference of 46 cm (<3rd centile). His psychomotor development was normal for age. Multiple facial dysmorphic features included short palpebral fissures, short philtrum, a narrow mouth with a thin upper lip, maxillary hypoplasia, and prognathism (Figure 1f). He walked independently on his tiptoes. According to the pediatrician, walking instability was related to knee flexion (moderate to severe) and popliteal ligament tightness. Complete syndactyly of fingers 3–4 with fusion of the bones in the left hand was identified by inspection and palpation. The boy's right testicle had descended into the scrotum and his left testicle was not. Although the patient did not pass the hearing screening, a systematic auditory functional evaluation showed normal hearing (Figure 1c); therefore, follow‐up is needed.
TABLE 1.
A summary of the common clinical features of subjects with a clinical and molecular diagnosis of Myhre syndrome and identified pathogenic variants
| Study No. | Youngest age of diagnosis | Variant of SMAD4 {counts} | Clinical features | Reference | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hearing loss | IUGR | Short stature | Facial features | Musculoskeletal dysplasia | Respiratory problem | Cardiovascular problem | Thick skin | Visual problem | Puberty/genital problem | |||||||||||||
| Short palpebral fissure | Midface hypoplasia | Short philtrum | Prognathism | Narrow mouth | Thin upper lip | Small ears | Brachydactyly | Clinodactyly | Muscular body build | Limited joint mobility | ||||||||||||
| 1 | 13yo | c.1500A > G {1} | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | – | – | 1 | – | 1 | 1 | 1 | 1 | 0 | Whiteford et al. (2001) |
| 2 | 28yo | c.1498A > G {5} | 8 | 0 | 0 | – | 11 | 11 | – | – | 6 | 5 | – | – | – | – | 3 | 8 | 9 | 6 | 5 | Le Goff et al. (2011) |
| c.1499 T > C {5} | ||||||||||||||||||||||
| c.1500A > G {1} | ||||||||||||||||||||||
| 3 | 28yo | c.1498A > G {5} | 8 | 5 | 1 | 8 | 8 | 8 | 8 | 6 | 6 | – | 8 | – | 8 | – | – | 6 | 6 | 4 | 1 | Caputo et al. (2012) |
| c.1499 T > C {3} | ||||||||||||||||||||||
| 4 | 12yo | c.1486C > T {1} | 2 | 1 | 1 | 2 | 0 | 1 | 1 | 2 | 1 | 2 | 2 | – | 2 | – | – | 1 | 2 | 2 | 0 | Al Ageeli et al. (2012) |
| c.1499 T > C {1} | ||||||||||||||||||||||
| 5 | 9yo | c.1499 T > C {1} | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | – | 1 | – | 1 | – | 1 | 0 | 1 | 1 | – | (Asakura et al., 2012) |
| 6 | – | c.1498A > G {1} | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 2 | 2 | 2 | – | – | 2 | 2 | 2 | 2 | 1 | 2 | Lindor et al. (2012) |
| c.1499 T > C {1} | ||||||||||||||||||||||
| 7 | 26yo | c.1486C > T {1} | 1 | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | Kenis et al. (2014) |
| 8 | 15yo | c.1486C > T {1} | 1 | – | – | – | – | – | 1 | – | – | – | 1 | – | – | 1 | – | – | – | – | – | Caputo et al. (2014) |
| 9 | 12yo |
c.1498A > G {1} |
1 | – | – | – | – | – | – | – | – | – | – | – | – | 1 | 1 | 1 | – | – | 1 | Piccolo et al. (2014) |
| 10 | 18yo | c.1499 T > C {1} | 1 | – | – | 1 | – | – | 1 | – | – | – | 1 | – | – | 1 | – | – | 1 | – | – | Ishibashi et al. (2014) |
| 11 | 2.5yo | c.1499 T > C {1} | 1 | – | 1 | – | – | 1 | 1 | – | – | – | 1 | 1 | – | – | 1 | 1 | – | – | – | Hawkes and Kini (2015) |
| 12 | 8yo | c.1498A > G {2} | 5 | 1 | – | – | 2 | – | 4 | 3 | 3 | – | 2 | 1 | – | 2 | 4 | 4 | 2 | 2 | – | Starr et al. (2015) |
| c.1499 T > C {3} | ||||||||||||||||||||||
| 13 | 6yo | c.1498A > G {3} | 3 | 1 | – | – | – | – | – | – | – | – | – | – | – | 4 | – | – | 2 | – | 4 | Lin et al. (2016) |
| c.1499 T > C {1} | ||||||||||||||||||||||
| 14 | 12yo | c.1499 T > C {1} | 1 | – | – | 1 | 1 | – | 1 | – | – | – | 1 | 1 | 1 | 1 | 1 | 1 | 1 | – | – | Garavelli et al. (2016) |
| 15 | 7yo | c.1498A > G {1} | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 0 | – | – | – | – | – | Bassett et al. (2016) |
| 16 | – | c.1498A > G {1} | – | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | – | 0 | 1 | 1 | 1 | 0 | Nomura et al. (2017) |
| 17 | 2yo | c.1498A > G {1} | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | – | 0 | 1 | 0 | 1 | 1 | 1 | 0 | Alagia et al. (2018) |
| 18 | 14yo | c.1498A > G {1} | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | – | 0 | – | 0 | 0 | 0 | 1 | 1 | Erdem et al. (2018) |
| 19 | – | c.1498A > G {1} | – | – | 1 | – | – | – | – | – | – | – | – | – | 1 | – | 1 | 1 | 1 | 1 | 0 | |
| 20 | 5mo | c.1498A > G {3} | 2 | 3 | 5 | 5 | 6 | 6 | 6 | 4 | 5 | – | 6 | – | 6 | – | 1 | 3 | 2 | 1 | 1 | Yu et al. (2019) |
| c.1499 T > C {3} | ||||||||||||||||||||||
| 21 | 33yo | c.1486C > T {2} | 2 | – | 2 | 2 | 2 | 1 | – | – | – | 1 | 2 | – | – | – | 1 | 1 | 1 | 2 | – | Lin et al. (2020) |
| 22 | 2mo | c.1486C > T {4} | 2 | – | 1 | 1 | – | – | – | 1 | – | 3 | – | 3 | – | 1 | 2 | 2 | 1 | 1 | – | Meerschaut et al. (2019) |
| 23 | 25yo | c.1486C > T {1} | – | – | 1 | 1 | 1 | – | 1 | 1 | 1 | 1 | 1 | – | 1 | – | – | – | 1 | 1 | – | Artemios et al. (2019) |
| 24 | 3yo | c.1498A > G {2} | – | 2 | 2 | 1 | 2 | 1 | – | 1 | – | – | 1 | – | – | – | 1 | 1 | – | – | 1 | H. Li et al. (2020) |
| 25 | 9.6yo | c.1498A > G {1} | 1 | – | – | 1 | – | 1 | – | 1 | 1 | – | 1 | – | 1 | – | – | 1 | 1 | 1 | – | Gursoy et al. (2020) |
| 26 | 25yo | c.1498A > G {1} | – | – | – | – | – | – | – | – | – | – | – | – | – | – | 1 | 1 | – | – | – | Alape et al. (2020) |
| 27 | 16yo | c.1498A > G {1} | 1 | – | 1 | – | – | – | – | 1 | – | – | – | 1 | – | – | – | 1 | – | 1 | 1 | Varenyiova et al. (2020) |
| 28 | 13yo | c.1499 T > C {1} | 1 | – | 1 | – | – | – | 1 | – | – | 1 | – | 1 | – | – | – | 1 | 1 | – | 1 | Jensen et al. (2020) |
| 29 | 55yo | c.1486C > T {1} | 1 | – | 1 | – | – | – | – | 1 | – | – | – | – | – | – | – | – | – | – | – | Kandhaya‐Pillai et al. (2021) |
| 30 | 58yo | c.1498A > G {1} | 1 | – | 1 | 1 | 1 | – | 1 | 1 | 1 | 1 | 1 | – | – | – | 1 | 1 | 1 | 1 | – | Di Cesare et al. (2021) |
| 31 | 28yo | c.1498A > G {1} | 1 | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | 1 | – | – | Inoue et al. (2021) |
| 32 | 2mo | c.1499 T > C {1} | 0 | 1 | 1 | 0 | – | – | – | – | – | 1 | 1 | – | 1 | – | 1 | 1 | 1 | – | – | Jeon et al. (2021) |
| 33 | 13yo | c.1498A > G {1} | 0 | – | 1 | 0 | – | – | 1 | – | – | – | – | 1 | – | – | – | 1 | – | – | 1 | Lim et al. (2021) |
| 34 | 32mo | c.1498A > G {1} | 1 | – | 1 | 0 | – | – | – | – | – | – | – | – | – | – | – | 1 | – | – | – | Li et al. (2021) |
| 35 | 13yo | c.1498A > G {1} | 1 | 1 | 1 | 0 | – | – | 1 | – | – | – | 1 | 1 | – | – | – | 1 | – | – | 1 | Kilci et al. (2022) |
| 36 | 5yo | c.1486C > T {1} | 4 | 2 | 3 | – | – | – | – | – | – | – | – | – | – | 5 | – | 5 | 3 | 3 | 3 | Cappuccio et al. (2022) |
| c.1498A > G {4} | ||||||||||||||||||||||
| 37 | 38do | c.1498A > G {1} | 0 | – | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 1 | This study |
| Proportion |
c.1498A > G 51.9% (40/77) |
72.7% (56/77) |
31.2% (24/77) |
45.5% (35/77) |
41.6% (32/77) |
54.5% (42/77) |
49.4% (38/77) |
42.9% (33/77) |
36.4% (28/77) |
41.6% (32/77) |
26.0% (20/77) |
48.1% (37/77) |
14.3% (11/77) |
31.2% (24/77) |
23.4% (18/77) |
30.0% (23/77) |
65.0% (50/77) |
54.5% (42/77) |
41.6% (32/77) |
31.2% (24/77) |
||
|
c.1499 T > C 29.9% (23/77) | ||||||||||||||||||||||
|
c.1486C > T 15.6% (12/77) | ||||||||||||||||||||||
|
c.1500A > G 2.6% (2/77) | ||||||||||||||||||||||
Note: 117 cases diagnosed cases of Myhre syndrome were identified. Table 1 has integrated the data from 77 cases with molecular confirmation, whilst the other 40 cases were not confirmed by molecular analysis. All of the listed cases were molecularly confirmed to be related to SMAD4 (NM_005359.6).
Abbreviations: do, days old; mo, months old; yo, years old; −, no information.
3.2. Genotyping
The median sequencing depth of the exon target regions exceeded 50× and the sequencing depth of 95% of the target regions exceeded 20×. The exon loss rate was <0.2%. The variants remaining after filtering were manually assessed based on the frequency/presence in the known SNP databases, any previous association with disease, predicted functional impact, any nucleotide/amino acid conservation, and the potential detrimental biochemistry of any amino acid substitution observed.
Using Sanger sequencing, three participating family members (one affected and two unaffected) were genotyped to identify the candidate variants. One heterozygous de novo variant c.1498A > G (p.Ile500Val) in SMAD4 (NM_005359.6) remained after the filtering process and validation by Sanger sequencing (Figure 1b) which was consistent with autosomal dominant inheritance. The SMAD4 c.1498A > G (p.Ile500Val) change occurs in an evolutionarily highly conserved region across different species in the MH1 domain resulting in one amino acid change from leucine to valine at position 500. This pathogenic variant causes the p.Ile500Val substitution and has been previously reported in multiple patients (Caputo et al., 2012; Di Cesare et al., 2021; Le Goff et al., 2011; Lin et al., 2016; Starr et al., 2015). According to the ACMG classification system, the c.1498A > G (p.Ile500Val) is classified as a pathogenic variant (PS1 + PS2 + PM2 + PP3).
3.3. Review of the literature
We identified 117 cases of Myhre syndrome, 77 (65.8%) of whom had clear molecular confirmations that were related to the SMAD4 gene, while 40 cases did not have confirmation. The common clinical features of the 77 cases with molecular confirmations (Table 1) include hearing loss (72.7%, 56/77), cardiovascular problem (65.0%, 50/77), midface hypoplasia (54.5%, 42/77), thick skin (54.5%, 42/77), a short philtrum (49.4%, 38/77), brachydactyly (48.1%, 37/77), prognathism (42.9%, 33/77), short stature (45.5%, 35/77), short palpebral fissures (41.6%, 32/77), a thin upper lip (41.6%, 32/77), visual problems (41.6%, 32/77), a narrow mouth (36.4%, 28/77), muscular body build (31.2%, 24/77), respiratory problems (30.0%, 23/77), intrauterine growth retardation (IUGR) (31.2%, 24/77), puberty/genital problems (31.2%, 24/77), small ears (26%, 20/77), limited joint mobility (23.4%, 18/77), and clinodactyly (14.3%, 11/77). Other clinical features which were not listed include poor articulation/delayed speech development 9.1% (7/77), neoplasia 6.5% (5/77), syndactyly (5.2%, 4/77), and polydactyly (3.9%, 3/77). Individual patients had facial paralysis (No.11), intellectual disability (No.14), obsessive–compulsive disorder, attention deficit hyperactivity disorders (No.18), kidney hypoplasia, vesicoureteral reflux, a high‐pitched voice, premature thinning of the hair, diabetes mellitus, osteoporosis, hypogonadism (No.29), choanal atresia, intellectual disability, schizophrenia and platyspondyly (No.31).
The 40 (34.2%, 40/117) patients who did not have clear molecular confirmations included 13 patients who were reported as having a pathogenic variant in SMAD4 “Ile500” without including the base variant. The remaining 27 (67.5%, 27/40) patients did not have any molecular information reported. 70% (28 of 40) of these patients suffered from hearing loss.
4. DISCUSSION
The SMAD4 gene (MIM #600993) is a protein‐coding gene that is characterized by a highly conserved 230‐amino‐acid motif (323‐552aa) known as the MH2 domain. SMAD4 is required for transforming growth factor‐beta (TGF‐β) and bone morphogenic protein (BMP) signaling. SMAD4 participates in various cellular processes including cell proliferation, differentiation and apoptosis (Kandhaya‐Pillai et al., 2021; Massague & Wotton, 2000; Shi & Massague, 2003). Structural analysis of mutant SMAD4 suggests that a conformational change results in the perturbation of TGF‐β signaling and leads to enhanced levels of non‐ubiquitinated SMAD4 (Caputo et al., 2014).
Diseases associated with SMAD4 loss‐of‐function include Juvenile polyposis/hereditary hemorrhagic telangiectasia syndrome (MIM #175050), pancreatic cancer (MIM #260350), and Juvenile intestinal polyposis (MIM #174900). Until now, 159 pathogenic variants in SMAD4 have been reported according to the HGMD professional database including one pre‐initiation codon variation (c.‐127‐650C > T), one post‐termination codon variation (c.*1G > A) and 16 structural variations (Figure 2). Myhre syndrome is caused by the heterozygous gain‐of‐function pathogenic variant in SMAD4.
FIGURE 2.

Summary of the identified pathogenic variants in SMAD4 (NM_005359.6).
The most common clinical features of Myhre syndrome are the distinctive dysmorphic facial features (short palpebral fissures, maxillary hypoplasia, a small mouth with a thin upper lip and a short philtrum, prognathism), short stature, limited joint mobility (with particular difficulty in fist‐clenching and arm‐raising), thickened skin, and muscular pseudohypertrophy, which become recognizable in late childhood (Michot et al., 2014). Mild‐to‐moderate intellectual disability and developmental delay are common but not constant. Autistic‐like behavior has been reported (Lin et al., 2020). Cardiovascular abnormalities affect about 70% of patients and include restrictive cardiomyopathy and pericardial disease. In addition, congenital heart defects and tetralogy of Fallot have also been reported (Alagia et al., 2018; Cappuccio et al., 2022; Lin et al., 2016, 2020; Starr et al., 2015). Respiratory involvement consists of choanal stenosis and laryngotracheal proliferative fibrosis manifested as progressive tachypnea, exertional dyspnea, and respiratory distress. Early clinical diagnosis is important because of potentially fatal respiratory compromise (Alape et al., 2020; Kilci et al., 2022; McGowan et al., 2011; Oldenburg et al., 2015).
Gastrointestinal involvement includes pyloric stenosis, duodenal atresia and severe constipation (Lin et al., 2016; Oldenburg et al., 2015). Recurrent infections (particularly otitis media and pneumonia) are frequently reported (Erdem et al., 2018; Lin et al., 2020). Other uncommon features include syndactyly, cleft palate, cleft lip, hypermetropia, cryptorchidism and neoplasia (Asakura et al., 2012; Becerra‐Solano et al., 2008; Ishibashi et al., 2014; Lin et al., 2020). A recent study reported that Myhre syndrome is related to schizophrenia (Inoue et al., 2021).
In 2016, Lin et al. reported that hearing loss was frequently observed in 83% of 54 cases with Myhre syndrome and could be presented as sensorineural, conductive or mixed loss (Lin et al., 2016; Yu et al., 2019). We reviewed 56 Myhre syndrome cases with hearing loss, all of whom were molecularly confirmed with SMAD4 variants. The detailed information about the hearing phenotypes is summarized in Table 2. From our literature review, conductive hearing loss accounted for 26.8% (15 of 56) of cases whilst sensorineural and mixed hearing loss accounted for 10.8% and 17.9% of cases, respectively. Hearing loss may be early onset or progressive and our analysis showed that the age of onset ranged from 6 months to 58 years old. 30.4% of 56 cases suffered from bilateral hearing loss and 1.8% were proven to be unilateral. No detailed information concerning hearing loss was presented in the other 67.8% of cases.
TABLE 2.
The hearing phenotypes associated with Myhre syndrome caused by SMAD4 pathogenic variants
| Case No. | Variant (NM_005359.6) | Age of onset | Types | Bilateral/unilateral | Degree | Progressive | Frequency | Reference |
|---|---|---|---|---|---|---|---|---|
| 1 | c.1486C > T (p.Arg496Cys) | – | – | unilateral | mild | – | – | Al Ageeli et al. (2012) |
| 2 | c.1486C > T (p.Arg496Cys) | – | conductive | bilateral | – | – | – | Caputo et al. (2014) |
| 3 | c.1486C > T (p.Arg496Cys) | 26yo | mixed | bilateral | severe | progressive | – | Kenis et al. (2014) |
| 4 | c.1486C > T (p.Arg496Cys) | – | conductive | – | mild | – | – | Meerschaut et al. (2019) |
| 5 | c.1486C > T (p.Arg496Cys) | – | – | – | – | – | high | Meerschaut et al. (2019) |
| 6 | c.1486C > T (p.Arg496Cys) | – | sensorineural | – | – | – | – | Lin et al. (2020) |
| 7 | c.1486C > T (p.Arg496Cys) | – | mixed | – | – | progressive | – | Lin et al. (2020) |
| 8 | c.1486C > T (p.Arg496Cys) | 55yo | – | – | – | – | – | Kandhaya‐Pillai et al. (2021) |
| 9 | c.1486C > T (p.Arg496Cys) | – | – | – | – | – | – | Cappuccio et al. (2022) |
| 10 | c.1498A > G (p.Ile500Val) | – | sensorineural | – | – | – | – | Caputo et al. (2012) |
| 11 | c.1498A > G (p.Ile500Val) | – | conductive | – | – | – | – | Caputo et al. (2012) |
| 12 | c.1498A > G (p.Ile500Val) | – | mixed | – | – | – | – | Caputo et al. (2012) |
| 13 | c.1498A > G (p.Ile500Val) | – | mixed | – | – | – | – | Caputo et al. (2012) |
| 14 | c.1498A > G (p.Ile500Val) | – | mixed | – | – | – | – | Caputo et al. (2012) |
| 15 | c.1498A > G (p.Ile500Val) | – | – | – | – | – | – | Lindor et al. (2012) |
| 16 | c.1498A > G (p.Ile500Val) | – | – | – | severe | – | – | Le Goff et al. (2014) |
| 17 | c.1498A > G (p.Ile500Val) | – | – | – | severe | – | – | Le Goff et al. (2014) |
| 18 | c.1498A > G (p.Ile500Val) | – | – | – | – | – | – | Le Goff et al. (2014) |
| 19 | c.1498A > G (p.Ile500Val) | – | – | – | – | – | – | Le Goff et al. (2014) |
| 20 | c.1498A > G (p.Ile500Val) | – | – | – | severe | – | – | Le Goff et al. (2014) |
| 21 | c.1498A > G (p.Ile500Val) | – | sensorineural | bilateral | severe | – | – | Piccolo et al. (2014) |
| 22 | c.1498A > G (p.Ile500Val) | – | conductive | – | – | progressive | – | Starr et al. (2015) |
| 23 | c.1498A > G (p.Ile500Val) | 2yo | – | bilateral | – | – | – | Starr et al. (2015) |
| 24 | c.1498A > G (p.Ile500Val) | 4yo | conductive | bilateral | moderate | – | – | Bassett et al. (2016) |
| 25 | c.1498A > G (p.Ile500Val) | – | – | – | – | – | – | Lin et al. (2016) |
| 26 | c.1498A > G (p.Ile500Val) | – | – | – | – | – | – | Lin et al. (2016) |
| 27 | c.1498A > G (p.Ile500Val) | 14yo | mixed | – | – | – | – | Erdem et al. (2018) |
| 28 | c.1498A > G (p.Ile500Val) | 8yo | conductive | bilateral | – | – | high | Yu et al. (2019) |
| 29 | c.1498A > G (p.Ile500Val) | 9.6yo | mixed | bilateral | severe | – | – | Gursoy et al. (2020) |
| 30 | c.1498A > G (p.Ile500Val) | 9yo | – | – | – | – | – | Varenyiova et al. (2020) |
| 31 | c.1498A > G (p.Ile500Val) | 58yo | conductive | bilateral | – | progressive | – | Di Cesare et al. (2021) |
| 32 | c.1498A > G (p.Ile500Val) | 4yo | – | – | – | – | – | Inoue et al. (2021) |
| 33 | c.1498A > G (p.Ile500Val) | 1yo | – | – | – | – | – | Li et al. (2021) |
| 34 | c.1498A > G (p.Ile500Val) | 12yo | conductive | bilateral | – | – | – | Kilci et al. (2022) |
| 35 | c.1498A > G (p.Ile500Val) | 12yo | conductive | – | – | – | – | Cappuccio et al. (2022) |
| 36 | c.1498A > G (p.Ile500Val) | – | sensorineural | bilateral | mild | – | – | Cappuccio et al. (2022) |
| 37 | c.1498A > G (p.Ile500Val) | – | conductive | bilateral | moderate | – | – | Cappuccio et al. (2022) |
| 38 | c.1499 T > C (p.Ile500Thr) | – | – | bilateral | severe | – | – | Al Ageeli et al. (2012) |
| 39 | c.1499 T > C (p.Ile500Thr) | 9yo | mixed | – | severe | – | – | Asakura et al. (2012) |
| 40 | c.1499 T > C (p.Ile500Thr) | – | mixed | – | – | – | – | Caputo et al. (2012) |
| 41 | c.1499 T > C (p.Ile500Thr) | – | conductive | – | – | – | – | Caputo et al. (2012) |
| 42 | c.1499 T > C (p.Ile500Thr) | – | sensorineural | – | – | – | – | Caputo et al. (2012) |
| 43 | c.1499 T > C (p.Ile500Thr) | – | – | – | – | – | – | Lindor et al. (2012) |
| 44 | c.1499 T > C (p.Ile500Thr) | 21mo | – | – | – | – | – | Ishibashi et al. (2014) |
| 45 | c.1499 T > C (p.Ile500Thr) | – | – | – | severe | – | – | Le Goff et al. (2014) |
| 46 | c.1499 T > C (p.Ile500Thr) | – | – | – | severe | – | – | Le Goff et al. (2014) |
| 47 | c.1499 T > C (p.Ile500Thr) | – | – | – | severe | – | – | Le Goff et al. (2014) |
| 48 | c.1499 T > C (p.Ile500Thr) | – | – | – | – | – | – | Hawkes and Kini (2015) |
| 49 | c.1499 T > C (p.Ile500Thr) | 29yo | conductive | – | – | progressive | – | Starr et al. (2015) |
| 50 | c.1499 T > C (p.Ile500Thr) | – | sensorineural | bilateral | – | – | – | Starr et al. (2015) |
| 51 | c.1499 T > C (p.Ile500Thr) | 4yo | mixed (left); conductive (right) | bilateral | – | – | – | Starr et al. (2015) |
| 52 | c.1499 T > C (p.Ile500Thr) | – | – | – | – | – | – | Lin et al. (2016) |
| 53 | c.1499 T > C (p.Ile500Thr) | childhood | conductive | bilateral | – | – | – | Garavelli et al. (2016) |
| 54 | c.1499 T > C (p.Ile500Thr) | – | – | bilateral | – | – | – | Yu et al. (2019) |
| 55 | c.1499 T > C (p.Ile500Thr) | – | conductive | – | – | – | – | Jensen et al. (2020) |
| 56 | c.1500A > G (p.Ile500Met) | infancy | conductive | bilateral | severe | progressive | – | Whiteford et al. (2001) |
| Proportion | c.1486C > T, 16.1% (9/56) | ≤1yo, 1.8% (1/56) |
Conductive 26.8% (15/56) |
Bilateral 30.4% (17/56) |
Mild 5.4% (3/56) |
Progressive 10.7% (6/56) |
High frequency 3.6% (2/56) |
|
| c.1498A > G, 50% (28/56) | >1yo, 30.4% (17/56) |
Sensorineural 10.8% (6/56) |
Unilateral 1.8% (1/56) |
Moderate 3.6% (2/56) |
||||
| c.1499 T > C, 32.1% (18/56) |
Mixed 17.9% (10/56) |
Severe 21.4% (12/56) |
||||||
| c.1500A > G, 1.8% (1/56) |
Note: Previous reports described 84 cases of Myhre syndrome involving hearing loss. Table 2 integrated data from 56 cases with hearing phenotypes and molecular confirmation, whilst the other 28 cases that were not confirmed by molecular analysis were not listed. All of the listed 56 cases were molecularly confirmed to be related to the SMAD4 gene (NM_005359.6).
Abbreviations: do, days old; mo, months old; yo, years old; –, no information.
We failed to analyze differences in the age of onset, the side of hearing loss, the degree of severity and the affected frequency between the groups of different pathogenic variants because of the limited number of cases with complete hearing phenotype information. In addition to the 56 patients with molecular confirmation, we reviewed an additional 11 patients who were clinically diagnosed. All of the patients were reported between 1983 and 2011 and are not listed in Table 2. Amongst the hearing phenotypes in this group of patients, bilateral hearing loss accounted for 72.7% (8 of 11) cases, whilst unilateral hearing loss accounted for 9.1% (1 of 11). Conductive hearing loss accounted for 45.5% (5 of 11) of cases, and sensorineural and mixed hearing loss accounted for 27.3% and 36.4% of cases, respectively. Also, 17 Myhre syndrome patients reported hearing loss but lacked details of the hearing phenotype and molecular confirmation (not listed in Table 2). Hearing loss was predominantly bilateral, late‐onset, severe, progressive and conductive. It should be noted that, in many reports, details about hearing phenotypes of patients with Myhre syndrome were absent. One possible explanation is that the hearing loss is mild and covered by other severe manifestations.
However, certain mechanisms of etiopathogenesis of auditory impairment cannot be deduced. In 2021, Tiziana Di Cesare et al. reported on a 58‐year‐old patient who was diagnosed with Myhre Syndrome and had thickened tympanic membranes, mastoid sclerosis and otospngiosis. The patient underwent cochlear implantation on the right side and is the first report of this treatment in Myhre Syndrome. Despite the failure to evoke postoperative electrical potentials via medium‐frequency electrodes, the patient benefited from an improved quality of life. Cochlear implantation is an effective and well‐tolerated auditory rehabilitation strategy that can achieve surprisingly good results in speech discrimination despite possible nerve involvement (Di Cesare et al., 2021).
Some neonates diagnosed with Myhre Syndrome pass newborn hearing screens but then show delay‐onset and progressive hearing loss (Di Cesare et al., 2021). In this study, the proband did not pass the newborn hearing screening. However, his hearing was within the normal range at 23 months. According to a study of newborn hearing screening of 1,585,892 neonates performed in Beijing, 1.016% of neonates failed the newborn hearing screening yet 82.13% of these cases progressed to have normal hearing (Dai et al., 2019). However, the possibility of delayed hearing loss in the proband was not excluded. To avoid learning and behavioral problems caused by hearing loss, we recommend that patients are regularly monitored and undertake frequent hearing tests. This approach will enable prompt interventions to avoid delayed language development.
Syndactyly occurs occasionally in patients with Myhre syndrome. However, it is the most common feature in congenital hand deformities with an incidence of around 1 in 2000 live births (Jordan et al., 2012). Syndactyly may occur as an isolated finding or may be a symptom of a genetic syndrome. Syndactyly is an established shared feature of more than 300 genetic syndromes (Malik, 2012), ranging in severity from complete “mitten‐like” syndactyly in Apert syndrome to the minor two 2–3 syndactyly in Smith‐Lemli‐Opitz syndrome.
We noted syndactyly in 10.3% (12/117) patients with Myhre syndrome. Syndactyly was more common in the toes compared to the fingers and only one patient reported the soft tissue fusion in the fingers which was classified as incomplete syndactyly. The patient in our study showed complete syndactyly of the fingers, although radiographs were not available. In this case, surgical intervention was not suggested, and the patient's functional needs should be considered. Also, incomplete syndactyly of the fingers or toes is common in previously observed cases of Myhre syndrome. The ability to make generalized conclusions of Myhre syndrome related to complete syndactyly of the fingers based on this study is limited and coincidental occurrence could not be ruled out.
Recent reports focused on the progressive nature of the clinical manifestations of Myhre syndrome. The early clinical diagnosis of Myhre syndrome in infancy is challenging because the typical clinical dysmorphic features are mild and may be overlooked. The affected infant in this study was born with short palpebral fissures, a small mouth with a thin upper lip, a short philtrum and complete syndactyly of fingers 3–4 on the left hand which can be nonspecific. Early genetic testing can help diagnosed Myhre syndrome.
5. CONCLUSIONS
This Chinese male infant with Myhre syndrome had facial features and complete syndactyly of the fingers, and was successfully diagnosed at 38 days by WES which showed a recurrent pathogenic variant in SMAD4, c.1498A > G (p.Ile500Val) in the patient. Generally, patients with Myhre syndrome are characterized by hearing loss and other cardiac disorders, which underline the necessity of definite diagnosis by genetic testing along with subsequent early prevention of other associated defects. We recommended regular hearing follow‐up for patients with Myhre syndrome. The potential life threatening cardiovascular and respiratory manifestations of Myhre syndrome require regular examinations. Surgical intervention should be evaluated with extreme caution and with as little invasion as possible due to the tendency to develop fibrosis which may cause significant morbidity and mortality (Yu et al., 2019). Consequently, it is important to confirm the clinical diagnosis of Myhre syndrome with molecular analysis.
AUTHOR CONTRIBUTIONS
Y.Y.Y., X.G., and J.C.X. conceived of the study and participated in its design and draft the manuscript. K.Y. and W.Q.W participated in the next generation sequencing and literature review. X.W. and L.M.H. participated in the data analysis and results discussion. M.Y.H., J.Y.Y., M.L. and D.Y.K. participated in the collection of clinical data and blood samples. All authors read and approved the final manuscript.
FUNDING INFORMATION
This study was supported by grants from the National Natural Science Foundation of China (81873704, 82271177) to Yong‐Yi Yuan, the National Natural Science Foundation of China (82171158) to Xue Gao, the National Natural Science Foundation of China (81900953) to Ming‐Yu Han, and the Natural Science Foundation of Hainan Province (819MS110) to Ming‐Yu Han.
CONFLICT OF INTEREST
The authors declare that there is no competing interest in this research. The authors have no financial relationships relevant to the article to disclose.
ETHICAL COMPLIANCE
The study was approved by the PLA General Hospital Ethics Committees. Written informed consent was obtained from the parents of the minor subject (23‐month‐old) for participation in the clinical and genetic research study, and publication of their clinical data.
ACKNOWLEDGMENTS
The authors would like to thank all the family members for their participation and cooperation in this study, all the reviewers who participated in the review, and MJEditor (www.mjeditor.com) for providing English editing services during the preparation of this manuscript.
Yang, K. , Wang, X. , Wang, W.‐Q. , Han, M.‐Y. , Hu, L.‐M. , Kang, D.‐Y. , Yang, J.‐Y. , Liu, M. , Gao, X. , Yuan, Y.‐Y. , & Xu, J.‐C. (2023). A newborn male with Myhre syndrome, hearing loss, and complete syndactyly of fingers 3–4. Molecular Genetics & Genomic Medicine, 11, e2103. 10.1002/mgg3.2103
Kun Yang, Xi Wang, and Wei‐Qian Wang are listed as co‐first authors.
Contributor Information
Xue Gao, Email: mixueer0110@126.com.
Yong‐Yi Yuan, Email: yyymzh@163.com.
Jin‐Cao Xu, Email: xujincao@126.com.
DATA AVAILABILITY STATEMENT
The patient's phenotype and the detected variants have been submitted to the ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and the accession number is SCV002072466.
REFERENCES
- al Ageeli, E. , Mignot, C. , Afenjar, A. , Whalen, S. , Dorison, N. , Mayer, M. , Esteva, B. , Dubern, B. , Momtchilova, M. , le Gargasson, J. F. , Bursztyn, J. , & Héron, D. (2012). Retinal involvement in two unrelated patients with Myhre syndrome. European Journal of Medical Genetics, 55(10), 541–547. 10.1016/j.ejmg.2012.05.006 [DOI] [PubMed] [Google Scholar]
- Alagia, M. , Cappuccio, G. , Pinelli, M. , Torella, A. , Brunetti‐Pierri, R. , Simonelli, F. , Limongelli, G. , Oppido, G. , Nigro, V. , Brunetti‐Pierri, N. , & TUDP . (2018). A child with Myhre syndrome presenting with corectopia and tetralogy of Fallot. American Journal of Medical Genetics. Part A, 176(2), 426–430. 10.1002/ajmg.a.38560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alape, D. , Singh, R. , Folch, E. , Fernandez Bussy, S. , Agnew, A. , & Majid, A. (2020). Life‐threatening multilevel airway stenosis due to Myhre syndrome. American Journal of Respiratory and Critical Care Medicine, 201(6), 731–732. 10.1164/rccm.201905-0922IM [DOI] [PubMed] [Google Scholar]
- Artemios, P. , Areti, S. , Katerina, P. , Helen, F. , Eirini, T. , & Charalambos, P. (2019). Autism Spectrum disorder and psychiatric comorbidity in a patient with Myhre syndrome. Journal of Autism and Developmental Disorders, 49(7), 3031–3035. 10.1007/s10803-019-04015-y [DOI] [PubMed] [Google Scholar]
- Asakura, Y. , Muroya, K. , Sato, T. , Kurosawa, K. , Nishimura, G. , & Adachi, M. (2012). First case of a Japanese girl with Myhre syndrome due to a heterozygous SMAD4 mutation. American Journal of Medical Genetics. Part A, 158A(8), 1982–1986. 10.1002/ajmg.a.35440 [DOI] [PubMed] [Google Scholar]
- Bassett, J. K. , Douzgou, S. , & Kerr, B. (2016). Severe constipation in a patient with Myhre syndrome: A case report. Clinical Dysmorphology, 25(2), 54–57. 10.1097/MCD.0000000000000109 [DOI] [PubMed] [Google Scholar]
- Becerra‐Solano, L. E. , Díaz‐Rodriguez, M. , Nastasi‐Catanese, J. A. , Toscano‐Flores, J. J. , Bañuelos‐Robles, O. , Figuera, L. E. , Matute, E. , & de Lourdes Ramírez‐Dueñas, M. (2008). The fifth female patient with Myhre syndrome: Further delineation. Clinical Dysmorphology, 17(2), 113–117. 10.1097/MCD.0b013e3282f52828 [DOI] [PubMed] [Google Scholar]
- Cappuccio, G. , Brunetti‐Pierri, N. , Clift, P. , Learn, C. , Dykes, J. C. , Mercer, C. L. , Callewaert, B. , Meerschaut, I. , Spinelli, A. M. , Bruno, I. , Gillespie, M. J. , Dorfman, A. T. , Grimberg, A. , Lindsay, M. E. , & Lin, A. E. (2022). Expanded cardiovascular phenotype of Myhre syndrome includes tetralogy of Fallot suggesting a role for SMAD4 in human neural crest defects. American Journal of Medical Genetics. Part A, 188(5), 1384–1395. 10.1002/ajmg.a.62645 [DOI] [PubMed] [Google Scholar]
- Caputo, V. , Bocchinfuso, G. , Castori, M. , Traversa, A. , Pizzuti, A. , Stella, L. , Grammatico, P. , & Tartaglia, M. (2014). Novel SMAD4 mutation causing Myhre syndrome. American Journal of Medical Genetics. Part A, 164A(7), 1835–1840. 10.1002/ajmg.a.36544 [DOI] [PubMed] [Google Scholar]
- Caputo, V. , Cianetti, L. , Niceta, M. , Carta, C. , Ciolfi, A. , Bocchinfuso, G. , Carrani, E. , Dentici, M. L. , Biamino, E. , Belligni, E. , Garavelli, L. , Boccone, L. , Melis, D. , Andria, G. , Gelb, B. D. , Stella, L. , Silengo, M. , Dallapiccola, B. , & Tartaglia, M. (2012). A restricted spectrum of mutations in the SMAD4 tumor‐suppressor gene underlies Myhre syndrome. American Journal of Human Genetics, 90(1), 161–169. 10.1016/j.ajhg.2011.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, P. , Huang, L. H. , Wang, G. J. , Gao, X. , Qu, C. Y. , Chen, X. W. , Ma, F. R. , Zhang, J. , Xing, W. L. , Xi, S. Y. , Ma, B. R. , Pan, Y. , Cheng, X.‐H. , Duan, H. , Yuan, Y. Y. , Zhao, Y.‐Y. , Zhao, L.‐P. , Chang, L. , Gao, R.‐Z. , & Han, D. M. (2019). Concurrent hearing and genetic screening of 180,469 neonates with follow‐up in Beijing, China. American Journal of Human Genetics, 105(4), 803–812. 10.1016/j.ajhg.2019.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cesare, T. , Rossi, G. , Girotto, G. , & Di Nardo, W. (2021). Benefit of cochlear implantation in a patient with Myhre syndrome. BML Case Reports, 14(8), e243164. 10.1136/bcr-2021-243164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdem, H. B. , Sahin, I. , & Tatar, A. (2018). Myhre syndrome with novel findings: Bilateral congenital cortical cataract, bilateral papilledema, accessory nipple, and adenoid hypertrophy. Clinical Dysmorphology, 27(1), 12–14. 10.1097/MCD.0000000000000188 [DOI] [PubMed] [Google Scholar]
- Garavelli, L. , Maini, I. , Baccilieri, F. , Ivanovski, I. , Pollazzon, M. , Rosato, S. , Iughetti, L. , Unger, S. , Superti‐Furga, A. , & Tartaglia, M. (2016). Natural history and life‐threatening complications in Myhre syndrome and review of the literature. European Journal of Pediatrics, 175(10), 1307–1315. 10.1007/s00431-016-2761-3 [DOI] [PubMed] [Google Scholar]
- Gursoy, S. , Hazan, F. , Ozturk, T. , & Ates, H. (2020). Novel ocular and inner ear anomalies in a patient with Myhre syndrome. Molecular Syndromology, 10(6), 339–343. 10.1159/000504829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes, L. , & Kini, U. (2015). Myhre syndrome with facial paralysis and branch pulmonary stenosis. Clinical Dysmorphology, 24(2), 84–85. 10.1097/MCD.0000000000000068 [DOI] [PubMed] [Google Scholar]
- Inoue, K. , Eiro, T. , Semoto, M. , Roppongi, T. , Nomoto, M. , Takahashi, Y. , & Hishimoto, A. (2021). First case of Myhre syndrome with schizophrenia. Clinical Dysmorphology, 30(4), 207–208. 10.1097/MCD.0000000000000386 [DOI] [PubMed] [Google Scholar]
- Ishibashi, N. , Sasaki, Y. , & Asakura, Y. (2014). Myhre syndrome: A rare craniofacial disorder. Cranio, 32(4), 300–306. 10.1179/0886963413Z.00000000024 [DOI] [PubMed] [Google Scholar]
- Jensen, B. , James, R. , Hong, Y. , Omoyinmi, E. , Pilkington, C. , Sebire, N. J. , Howell, K. J. , Brogan, P. A. , & Eleftheriou, D. (2020). A case of Myhre syndrome mimicking juvenile scleroderma. Pediatric Rheumatology Online Journal, 18(1), 72. 10.1186/s12969-020-00466-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon, M. J. , Kim, M. J. , Kim, J. H. , Park, J. S. , Yim, J. , Kim, M. , Kwon, S. K. , Lee, S. , Ko, J. M. , Chae, J. H. , & Suh, D. I. (2021). Multilevel airway stenosis being bypassed by a customized tracheostomy tube in an infant with Myhre syndrome. Pediatric Allergy, Immunology and Pulmonology, 34(2), 83–87. 10.1089/ped.2021.0029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan, D. , Hindocha, S. , Dhital, M. , Saleh, M. , & Khan, W. (2012). The epidemiology, genetics and future management of syndactyly. The Open Orthopaedics Journal, 6, 14–27. 10.2174/1874325001206010014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandhaya‐Pillai, R. , Hou, D. , Zhang, J. , Yang, X. , Compoginis, G. , Mori, T. , Tchkonia, T. , Martin, G. M. , Hisama, F. M. , Kirkland, J. L. , & Oshima, J. (2021). SMAD4 mutations and cross‐talk between TGF‐beta/IFNgamma signaling accelerate rates of DNA damage and cellular senescence, resulting in a segmental progeroid syndrome‐the Myhre syndrome. Geroscience, 43(3), 1481–1496. 10.1007/s11357-020-00318-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenis, C. , Verstreken, M. , Gieraerts, K. , De Foer, B. , Van der Aa, N. , Offeciers, E. F. , & Casselman, J. W. (2014). Bilateral otospongiosis and a unilateral vestibular schwannoma in a patient with Myhre syndrome. Otology & Neurotology, 35(9), e253–e255. 10.1097/MAO.0000000000000314 [DOI] [PubMed] [Google Scholar]
- Kilci, F. , Hurmuzlu‐Kozler, S. , Jones, J. , Dogan, K. , Cerrah Gunes, M. , & Cizmecioglu‐Jones, F. M. (2022). Myhre syndrome associated with hyperinsulinism and impaired glucose tolerance: A novel finding. Clinical Dysmorphology, 31(1), 42–44. 10.1097/MCD.0000000000000396 [DOI] [PubMed] [Google Scholar]
- Le Goff, C. , & Cormier‐Daire, V. (2012). From tall to short: The role of TGFbeta signaling in growth and its disorders. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics, 160C(3), 145–153. 10.1002/ajmg.c.31337 [DOI] [PubMed] [Google Scholar]
- Le Goff, C. , Michot, C. , & Cormier‐Daire, V. (2014). Myhre syndrome. Clinical Genetics, 85(6), 503–513. 10.1111/cge.12365 [DOI] [PubMed] [Google Scholar]
- le Goff, C. , Mahaut, C. , Abhyankar, A. , le Goff, W. , Serre, V. , Afenjar, A. , Destrée, A. , di Rocco, M. , Héron, D. , Jacquemont, S. , Marlin, S. , Simon, M. , Tolmie, J. , Verloes, A. , Casanova, J. L. , Munnich, A. , & Cormier‐Daire, V. (2011). Mutations at a single codon in mad homology 2 domain of SMAD4 cause Myhre syndrome. Nature Genetics, 44(1), 85–88. 10.1038/ng.1016 [DOI] [PubMed] [Google Scholar]
- Li, H. , Cheng, B. , Hu, X. , Li, C. , Su, J. , Zhang, S. , Li, L. , Li, M. , Yang, K. , He, S. , Chen, S. , Wang, H. , Liu, G. , & Shen, Y. (2020). The first two Chinese Myhre syndrome patients with the recurrent SMAD4 pathogenic variants: Functional consequences and clinical diversity. Clinica Chimica Acta, 500, 128–134. 10.1016/j.cca.2019.10.006 [DOI] [PubMed] [Google Scholar]
- Li, J. , Zhu, T. , Yang, S. , Yang, F. , Wu, J. , & Xiong, F. (2021). Myhre syndrome misdiagnosed as Marfan syndrome: An educational presentation. Brazilian Journal of Cardiovascular Surgery, 36(5), 700–702. 10.21470/1678-9741-2020-0592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, D. , Kim, J. H. , & Lee, J. (2021). Myhre syndrome: The first case in Korea. Annals of Pediatric Endocrinology & Metabolism, 26(3), 210–214. 10.6065/apem.2040214.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, A. E. , Alali, A. , Starr, L. J. , Shah, N. , Beavis, A. , Pereira, E. M. , Lindsay, M. E. , & Klugman, S. (2020). Gain‐of‐function pathogenic variants in SMAD4 are associated with neoplasia in Myhre syndrome. American Journal of Medical Genetics. Part A, 182(2), 328–337. 10.1002/ajmg.a.61430 [DOI] [PubMed] [Google Scholar]
- Lin, A. E. , Michot, C. , Cormier‐Daire, V. , L'Ecuyer, T. J. , Matherne, G. P. , Barnes, B. H. , Humberson, J. B. , Edmondson, A. C. , Zackai, E. , O'Connor, M. J. , Kaplan, J. D. , Ebeid, M. R. , Krier, J. , Krieg, E. , Ghoshhajra, B. , & Lindsay, M. E. (2016). Gain‐of‐function mutations in SMAD4 cause a distinctive repertoire of cardiovascular phenotypes in patients with Myhre syndrome. American Journal of Medical Genetics. Part A, 170(10), 2617–2631. 10.1002/ajmg.a.37739 [DOI] [PubMed] [Google Scholar]
- Lindor, N. M. , Gunawardena, S. R. , & Thibodeau, S. N. (2012). Mutations of SMAD4 account for both LAPS and Myhre syndromes. American Journal of Medical Genetics. Part A, 158A(6), 1520–1521. 10.1002/ajmg.a.35374 [DOI] [PubMed] [Google Scholar]
- Malik, S. (2012). Syndactyly: Phenotypes, genetics and current classification. European Journal of Human Genetics, 20(8), 817–824. 10.1038/ejhg.2012.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague, J. , & Wotton, D. (2000). Transcriptional control by the TGF‐beta/Smad signaling system. The EMBO Journal, 19(8), 1745–1754. 10.1093/emboj/19.8.1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan, R. , Gulati, R. , McHenry, P. , Cooke, A. , Butler, S. , Keng, W. T. , Murday, V. , Whiteford, M. , Dikkers, F. G. , Sikkema‐Raddatz, B. , van Essen, T. , & Tolmie, J. (2011). Clinical features and respiratory complications in Myhre syndrome. European Journal of Medical Genetics, 54(6), e553–e559. 10.1016/j.ejmg.2011.07.001 [DOI] [PubMed] [Google Scholar]
- McInerney‐Leo, A. M. , le Goff, C. , Leo, P. J. , Kenna, T. J. , Keith, P. , Harris, J. E. , Steer, R. , Bole‐Feysot, C. , Nitschke, P. , Kielty, C. , Brown, M. A. , Zankl, A. , Duncan, E. L. , & Cormier‐Daire, V. (2016). Mutations in LTBP3 cause acromicric dysplasia and geleophysic dysplasia. Journal of Medical Genetics, 53(7), 457–464. 10.1136/jmedgenet-2015-103647 [DOI] [PubMed] [Google Scholar]
- Meerschaut, I. , Beyens, A. , Steyaert, W. , de Rycke, R. , Bonte, K. , de Backer, T. , Janssens, S. , Panzer, J. , Plasschaert, F. , de Wolf, D. , & Callewaert, B. (2019). Myhre syndrome: A first familial recurrence and broadening of the phenotypic spectrum. American Journal of Medical Genetics. Part A, 179(12), 2494–2499. 10.1002/ajmg.a.61377 [DOI] [PubMed] [Google Scholar]
- Michot, C. , le Goff, C. , Mahaut, C. , Afenjar, A. , Brooks, A. S. , Campeau, P. M. , Destree, A. , di Rocco, M. , Donnai, D. , Hennekam, R. , Heron, D. , Jacquemont, S. , Kannu, P. , Lin, A. E. , Manouvrier‐Hanu, S. , Mansour, S. , Marlin, S. , McGowan, R. , Murphy, H. , … Cormier‐Daire, V. (2014). Myhre and LAPS syndromes: Clinical and molecular review of 32 patients. European Journal of Human Genetics, 22(11), 1272–1277. 10.1038/ejhg.2013.288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myhre, S. A. , Ruvalcaba, R. H. , & Graham, C. B. (1981). A new growth deficiency syndrome. Clinical Genetics, 20(1), 1–5. 10.1111/j.1399-0004.1981.tb01798.x [DOI] [PubMed] [Google Scholar]
- Nomura, R. , Miyai, K. , Nishimura, G. , Kashimada, K. , & Morio, T. (2017). Myhre syndrome: Age‐dependent progressive phenotype. Pediatrics International, 59(11), 1205–1206. 10.1111/ped.13413 [DOI] [PubMed] [Google Scholar]
- Oldenburg, M. S. , Frisch, C. D. , Lindor, N. M. , Edell, E. S. , Kasperbauer, J. L. , & O'Brien, E. K. (2015). Myhre‐LAPs syndrome and intubation related airway stenosis: Keys to diagnosis and critical therapeutic interventions. American Journal of Otolaryngology, 36(5), 636–641. 10.1016/j.amjoto.2015.04.005 [DOI] [PubMed] [Google Scholar]
- Piccolo, P. , Mithbaokar, P. , Sabatino, V. , Tolmie, J. , Melis, D. , Schiaffino, M. C. , Filocamo, M. , Andria, G. , & Brunetti‐Pierri, N. (2014). SMAD4 mutations causing Myhre syndrome result in disorganization of extracellular matrix improved by losartan. European Journal of Human Genetics, 22(8), 988–994. 10.1038/ejhg.2013.283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, Y. , & Massague, J. (2003). Mechanisms of TGF‐beta signaling from cell membrane to the nucleus. Cell, 113(6), 685–700. 10.1016/s0092-8674(03)00432-x [DOI] [PubMed] [Google Scholar]
- Starr, L. J. , Grange, D. K. , Delaney, J. W. , Yetman, A. T. , Hammel, J. M. , Sanmann, J. N. , Perry, D. A. , Schaefer, G. B. , & Olney, A. H. (2015). Myhre syndrome: Clinical features and restrictive cardiopulmonary complications. American Journal of Medical Genetics. Part A, 167A(12), 2893–2901. 10.1002/ajmg.a.37273 [DOI] [PubMed] [Google Scholar]
- Varenyiova, Z. , Hrckova, G. , Ilencikova, D. , & Podracka, L. (2020). Myhre syndrome associated with Dunbar syndrome and urinary tract abnormalities: A case report. Frontiers in Pediatrics, 8, 72. 10.3389/fped.2020.00072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteford, M. L. , Doig, W. B. , Raine, P. A. , Hollman, A. S. , & Tolmie, J. L. (2001). A new case of Myhre syndrome. Clinical Dysmorphology, 10(2), 135–140. 10.1097/00019605-200104000-00011 [DOI] [PubMed] [Google Scholar]
- Yu, K. P. , Luk, H. M. , Chung, B. H. , & Lo, I. F. (2019). Myhre syndrome: A report of six Chinese patients and literature review. Clinical Dysmorphology, 28(3), 145–150. 10.1097/MCD.0000000000000271 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The patient's phenotype and the detected variants have been submitted to the ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and the accession number is SCV002072466.