

SUMMARY
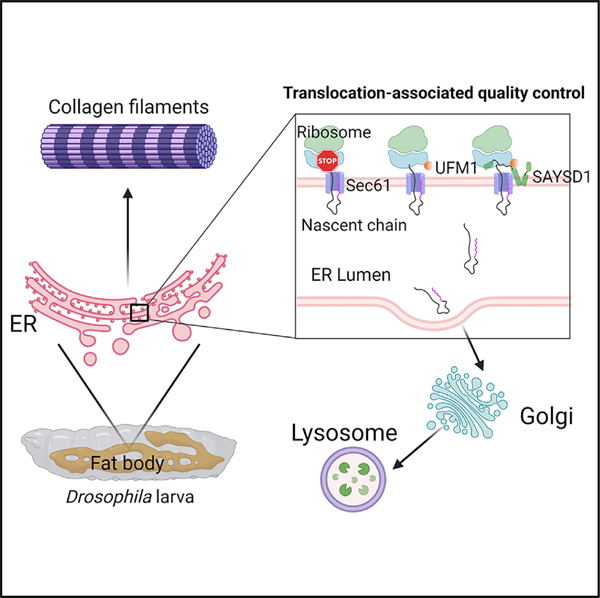
Translocon clogging at the endoplasmic reticulum (ER) as a result of translation stalling triggers ribosome UFMylation, activating translocation-associated quality control (TAQC) to degrade clogged substrates. How cells sense ribosome UFMylation to initiate TAQC is unclear. We conduct a genome-wide CRISPR-Cas9 screen to identify an uncharacterized membrane protein named SAYSD1 that facilitates TAQC. SAYSD1 associates with the Sec61 translocon and also recognizes both ribosome and UFM1 directly, engaging a stalled nascent chain to ensure its transport via the TRAPP complex to lysosomes for degradation. Like UFM1 deficiency, SAYSD1 depletion causes the accumulation of translocation-stalled proteins at the ER and triggers ER stress. Importantly, disrupting UFM1- and SAYSD1-dependent TAQC in Drosophila leads to intracellular accumulation of translocation-stalled collagens, defective collagen deposition, abnormal basement membranes, and reduced stress tolerance. Thus, SAYSD1 acts as a UFM1 sensor that collaborates with ribosome UFMylation at the site of clogged translocon, safeguarding ER homeostasis during animal development.
Graphical abstract
In brief
Wang et al. use a genetic screen to identify factors safeguarding protein translocation at the ER. They establish SAYSD1 as a Sec61-associated regulator sensing translocon clogging by binding to UFMylated ribosomes. SAYSD1 facilitates the clearance of translocation-stalled proteins to maintain ER protein homeostasis.
INTRODUCTION
In eukaryotes, proteins of the secretory pathway are mostly synthesized by endoplasmic reticulum (ER)-bound ribosomes and inserted into the ER lumen or integrated into the membrane co-translationally through the Sec61 translocon.1 This process is highly sensitive to perturbations by translation stalling or defects in protein modification, folding, and assembly, all of which can generate faulty polypeptides that clog the translocon.2,3 Failure to clear clogged translocons prevents nascent polypeptides from entering the ER, posing great harm to cells.2 While polypeptides stalled during post-translational translocation are cleared by a recently identified metalloprotease Ste24,4 not much is known about how cells cope with polypeptides stalled during co-translational protein translocation, the major protein translocation pathway at the ER.
UFM1 is a small protein structurally homologous to ubiquitin. Like ubiquitin, it can be conjugated to target proteins through UFMylation, a process mediated by an enzyme cascade involving UBA5 (E1), UFC1 (E2), and an ER-localized trimeric ligase complex (E3) composed of UFL1, DDRGK1 (also named UFBP1), and CDK5RAP3.5 The reaction is reversible as UFM1 can be cleaved off from substrates by two deUFMylating enzymes, UFSP1 and UFSP2.6 Genetic inactivation of UFM1 or UFMylating enzymes are known to cause ER stress7–9 and erythropoiesis defects,8,10 whereas mutations in UFM1 or genes encoding UFMylating enzymes have been linked to a variety of neurological disorders such as hypomyelinating leukodystrophy and epileptic encephalopathy.5,11–14
We and others recently established the ribosomal subunit RPL26 as the principal target of UFMylation.15,16 Importantly, we showed that RPL26 UFMylation occurs on the ER membrane and is specifically activated by co-translational translocation stalling.16 Ribosome UFMylation enables the export of stalled substrates from the ER for lysosomal degradation, suggesting a translocation-associated quality control (TAQC) mechanism that is distinct from the previously established ER-associated protein degradation (ERAD)17 or ribosome-associated quality control (RQC).18 Besides ribosomes, UFM1 may modify other proteins at lower levels to regulate other cellular processes such as ERphagy and DNA damage response.19–21
To dissect the mechanisms of TAQC, we conducted a genome-wide CRISPR-Cas9 knockout (KO) screen using a genetically engineered translocon-clogging reporter. The identified genes collectively reconstruct a ‘‘translocon-declogging’’ system centered around the UFMylation system, ribosome, the Sec61 translocon, and a translocon-associated membrane protein, SAYSD1. We show that SAYSD1 binds UFMylated ribosome in a bipartite manner to facilitate TAQC. Importantly, our study establishes collagens as endogenous TAQC substrates and a critical role for SAYSD1 and UFM1 in safeguarding collagen biogenesis.
RESULTS
Translocation-stalled nascent chains are eliminated by ER-to-lysosome transport
To study TAQC, we used a GFP-based reporter, ERGFP_K20 (Figure 1A), which contains a signal sequence (SS) for co-translational ER targeting, a N-glycosylation site (−CHO) followed by GFP. Importantly, ERGFP_K20 contains 20 consecutive lysine residues encoded by a polyadenine (poly-A) segment known to stall ribosomes during elongation.22,23 It also contains mCherry (mCh) downstream of the stalling sequence whose expression would indicate translation readthrough. Consistent with reported lysosomal degradation of ERGFP_K20,16 live-cell imaging using ERGFP_K20 stable cells showed that after treatment with the lysosome inhibitor bafilomycin A1 (Baf A1), ERGFP_K20 was first accumulated in a perinuclear region and then in lysosomes (Video S1). Immunostaining showed an extensive co-localization of perinuclear ERGFP_K20 with the Golgi protein GM130 (Figure 1B). This observation, together with the observation that ERGFP_K20 is sensitive to the Golgi-disrupting agent brefeldin A,16 suggests that trafficking through the Golgi is important for lysosomal degradation of ERGFP_K20.
Figure 1. ERGFP_K20 is transported to lysosome via the Golgi by the TRAPP complex.
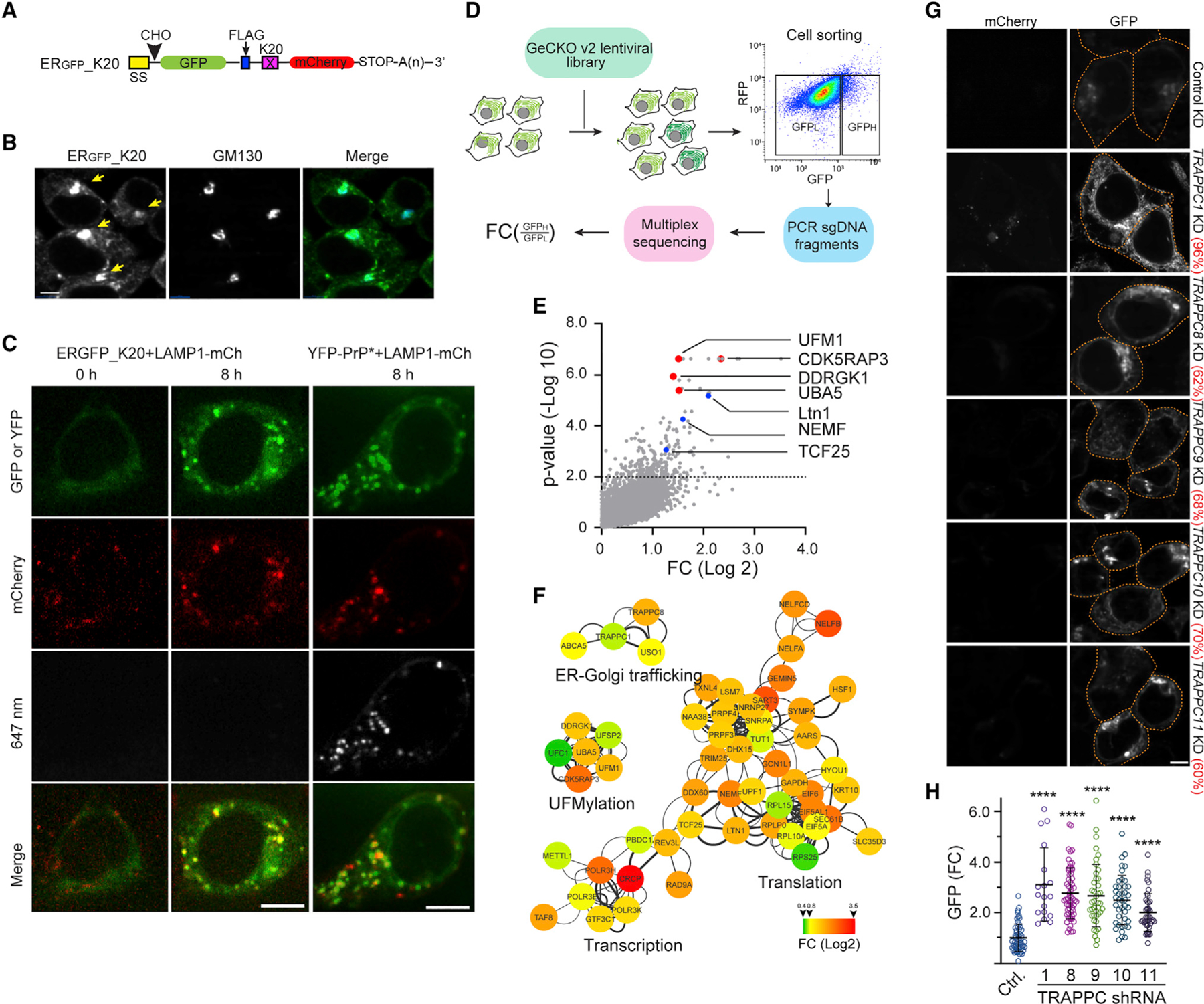
(A) A schematic diagram of the model TAQC substrate. SS, signal sequence; CHO, N-glycosylation site; X = K20.
(B) ERGFP_K20 is exported to lysosomes via the Golgi. ERGFP_K20-expressing 293T cells were treated with bafilomycin A1 (Baf A1) (200 nM), fixed, and stained by GM130 antibodies (blue). Arrows indicate Golgi-localized ERGFP_K20. Scale bar, 5 μm.
(C) Representative images from time-lapse videos show that ERGFP_K20 and YFP-PrP* are transported to lysosomes via distinct routes. ERGFP_K20 stable cells transfected with LAMP1-mCherry (mCh) were imaged at the indicated time points after treatment with Baf A1 and Alexa647-labeled GFP antibodies (left panels). The right panels show cells transfected with YFP-PrP* and LAMP1-mCh as a control. Scale bar, 5 μm.
(D) A schematic diagram of the CRISPR-Cas9 screen. LFC, log fold change.
(E) A scatterplot shows the distribution of genes positively enriched in ERGFP_K20 high cells. The dotted line indicates p = 0.01. Note that sgRNAs targeting the UFM1 pathway are enriched.
(F) A Cytoscape gene interaction map for sgRNAs positively enriched in ERGFP_K20-high cells. The color key indicates fold change (FC) in log 2.
(G and H) Knockdown of TRAPPC genes causes ERGFP_K20 accumulation in cells.
(G) Representative confocal images showing ERGFP_K20 stable cells transfected with the indicated shRNA constructs. The numbers indicate average knockdown efficiency determined by qRT-PCR (n = 3). Scale bar, 5 μm.
(H) The graphs show the quantification of GFP fluorescence in individual cells. Error bars indicate means ± SD; ****p < 0.0001 by one-way ANOVA with Dunnett’s multiple comparisons test. n = 3 independent experiments.
The perinuclear trafficking of ERGFP_K20 is reminiscent of rapid ER stress-induced export (RESET) of misfolded glycosylphosphatidylinositol (GPI)-anchored proteins.24 Because RESET substrates are first exported to the plasma membrane before entering the endolysosomal system,24 we tested whether ERGFP_K20 followed the same trafficking itinerary. We incubated ERGFP_K20 cells expressing LAMP1-mCh with Alexa647-labeled GFP antibody in the presence of Baf A1. If ERGFP-K20 was transiently exposed to the cell exterior, it should capture the GFP antibody, resulting in the co-internalization of ERGFP_K20 with the antibody. As a positive control, we used cells expressing the RESET substrate YFP-tagged PrP*, which was also recognized by the GFP antibody.24 As expected, YFP-PrP* cells accumulated GFP antibodies in LAMP1-positive lysosomes after incubation. By contrast, we failed to detect GFP antibodies in lysosomes of ERGFP-K20 cells under the same condition (Figures 1C and S1A–S1C), suggesting that unlike RESET, ERGFP_K20 is transported via the Golgi without going through the plasma membrane. Because pulse chase combined with ribosome fractionation showed that S35-methionine labeled, newly synthesized ERGFP-K20 was released from ribosomes similarly in wild-type (WT) and UFM1 KO cells (Figure S1D), and because ERGFP-K20 is entirely shielded by the ER membrane,16 we conclude that ERGFP-K20 is released into the ER lumen in a UFMylation-independent manner and then transported to lysosomes via the Golgi.
A genome-wide CRISPR-Cas9 screen identifies TAQC regulators
To identify regulators of TAQC, we performed a genome-wide CRISPR-Cas9 KO screen using 293T cells stably expressing ERGFP_K20. We transduced cells with a lentivirus-based CRISPR-Cas9 library, targeting 19,050 human genes each with 6 single guide RNAs (sgRNAs).25 Cells were separated into GFP-high (10%) and GFP-low (85%) populations by fluorescence-activated cell sorting (Figure 1D). Next-generation sequencing of PCR-amplified sgRNAs identified those enriched in the GFP-high populations from two independent repeats, which correspond to 234 significant gene hits (p < 0.01) (Figure 1E; Table S1). STRING-based protein network analysis highlighted 88 high-confidence hits with a combined false discovery rate (FDR) below 0.05 (see STAR Methods). Most genes in this group are functionally interlinked, forming one large and two smaller clusters (Figure 1F). One small cluster includes most UFMylation genes such as UFM1, UBA5, DDRGK1, CDK5RAP3, and UFSP2 (Figures 1E and 1F), which validated the genetic screen. The large cluster includes many genes involve in transcription and mRNA splicing. They might regulate ERGFP_K20 at the mRNA level and thus were excluded from further studies. This cluster also includes genes involved in translation and ER protein translocation (e.g., ribosome subunits RPL15, RPLP0, RPL10A, RPS25, EIF6, EIF5A, and Sec61β). These proteins might have a dual role: regulating protein synthesis while ensuring proper ER translocation via TAQC. Interestingly, the screen also identified three RQC factors: Ltn1, NEMF1/Rqc2, and TCF25/Rqc1. In the canonical RQC pathway, NEMF1/Rqc2 recruits Ltn1 to 60S ribosome following ribosome splitting, resulting in the ubiquitination of stalled substrates,26,27 whereas TCF25/Rqc1 recruits the Cdc48 ATPase to facilitate RQC.28 How these factors regulate ERGFP_K20 turnover remains to be elucidated.
The TRAPP complex facilitates the ER-to-Golgi transport of ERGFP-K20
The high-confidence list includes several genes previously implicated in ER-to-Golgi trafficking. These include TRAPPC1, TRAPPC8, and USO1 (Figures 1F and S1E). TRAPPC1 and TRAPPC8 encode subunits of the conserved transport protein particle (TRAPP) complex that is associated with the Golgi,29 while USO1 is a Golgi-localized receptor regulating the intercisternal vesicle transport within the Golgi.30
To validate the role of the TRAPP complex in TAQC, we used small hairpin RNAs (shRNAs) to deplete several TRAPP subunits in ERGFP_K20 cells. Confocal microscopy showed that, compared with a scrambled non-targeting shRNA control, shRNAs targeting TRAPPC1, TRAPPC8, TRAPPC9, TRAPPC10, and TRAPPC11 all increased the green fluorescence of ERGFP_K20 cells (Figures 1G and 1H). Pulse-chase analysis confirmed that ERGFP_K20 was stabilized in TRAPPC1-depleted cells (Figures S1F and S1G). Because depletion of the TRAPP complex did not elevate the level of mCh in ERGFP_K20 cells (Figure 1G), we concluded that the TRAPP complex specifically promotes the degradation of translation-stalled ERGFP_K20. These results further corroborate the role of the Golgi system in TAQC.
SAYSD1 regulates UFM1-dependent degradation of ERGFP-K20
Among the high-confidence gene list, a gene named SAYSvFN domain-containing protein 1 (SAYSD1) encodes an uncharacterized membrane protein (Figure 2A). Like genes in the UFMylation pathway, SAYSD1 is conserved in metazoan but missing from fungi (Figure S2A). Using small interfering RNA (siRNA)-mediated gene silencing and CRISPR-mediated KO of SAYSD1, we confirmed that depletion of SAYSD1 significantly increased GFP, but not mCh, fluorescence in ERGFP_K20 cells (Figures 2B, 2C, and S2B). Because treating SAYSD1-depleted ERGFP_K20 cells with Baf A1 caused a much smaller increase in GFP fluorescence than similarly treated control cells (Figure S2C), the ERGFP_K20 accumulation phenotype in SAYSD1-depleted cells could be attributed to diminished lysosomal degradation. Consistent with this notion, pulse-chase analysis showed that knockdown of SAYSD1 indeed stabilized ERGFP_K20 (Figure S2D).
Figure 2. SAYSD1 promotes the degradation of ERGFP_K20.
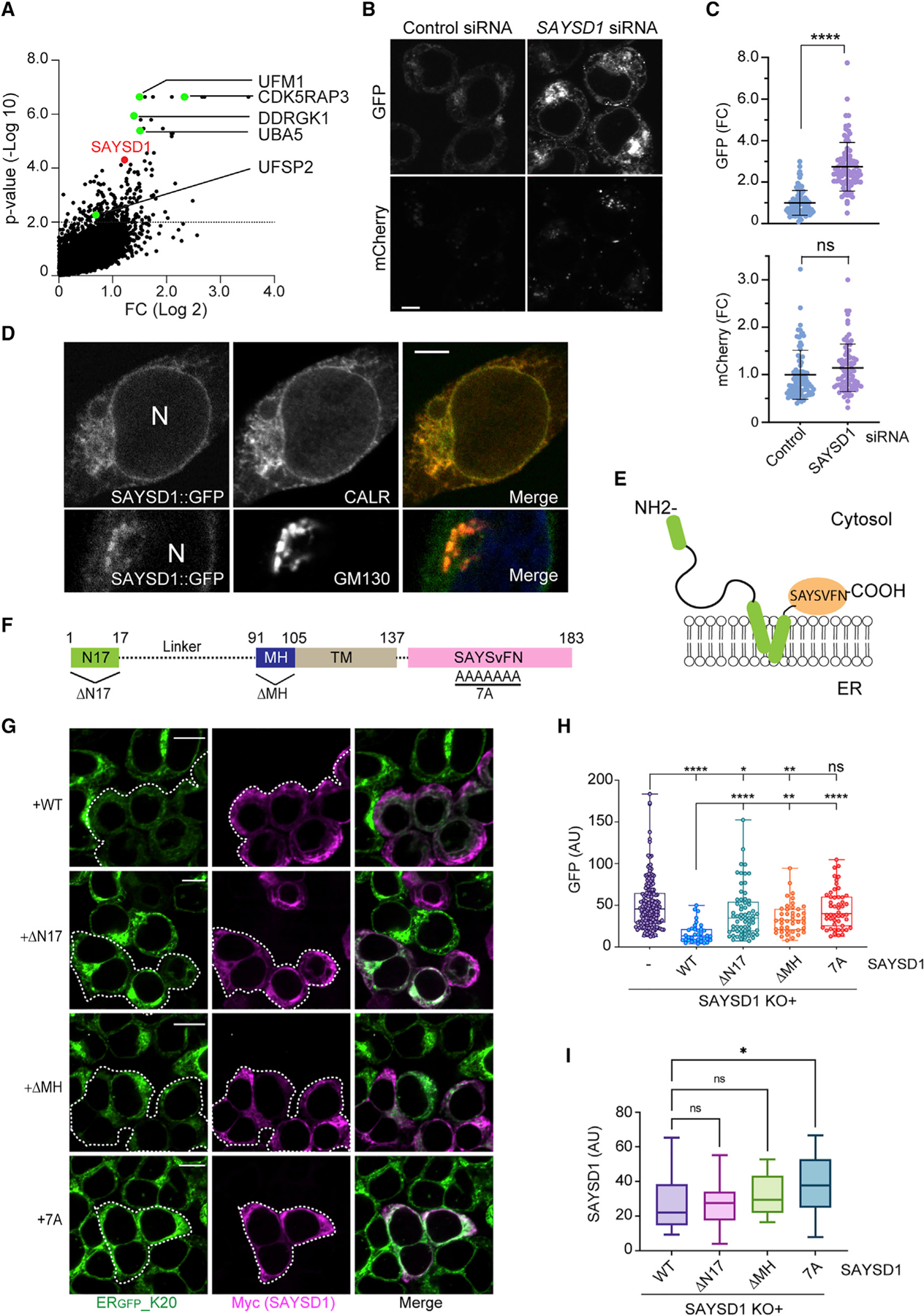
(A) sgRNAs targeting SAYSD1 are enriched in ERGFP_K20-high cells. The UFM1 pathway genes were highlighted in green.
(B and C) Knockdown of SAYSD1 increases the ERGFP_K20 green fluorescence without affecting the full-length readthrough product. (B) Shown are ERGFP_K20 stable 293T cells transfected with control or SAYSD1 siRNA for 48 h. Scale bar, 5 μm. The graphs in (C) show the quantification of the GFP and mC fluorescence in individual cells, respectively. Error bars indicate means ± SD, ****p < 0.0001 by unpaired Student’s t test; ns, not significant. n = 3 independent experiments.
(D) SAYSD1 is mainly localized to the ER. 293T cells expressing GFP-tagged SAYSD1 from the endogenous locus were fixed and stained with GM130 (bottom panels) or calreticulin (top panels) antibodies. N, nucleus. Scale bar, 5 μm.
(E) The predicted membrane topology of SAYSD1.
(F) The domain structure of SAYSD1 and the mutants used in the rescue study.
(G–I) Expression of Myc-tagged wild-type (WT) SAYSD1 but not mutants lacking either the SAYSVFN motif (7A), the N-terminal 17 residues (ΔN17), or the middle helical segment (ΔMH) in SAYSD1 knockout (KO) cells restores the degradation of ERGFP_K20. (G) Representative images from the experiments. Dashed lines indicate SAYSD1-positive cells. Scale bars, 10 μm. The graph in (H) shows the quantification of GFP fluorescence in individual cells. Error bars indicate means ± SD; ****p < 0.0001, **p < 0.01, *p < 0.05 by one-way ANVOA. ns, not significant. n = 4 independent experiments. (I) Quantification of SAYSD1 expression in (G).
As expected, neither UFM1 nor SAYSD1 KO affected the steady-state expression of ERGFP_K0, which lacks the ribosome-stalling sequence (Figure S2E). However, Baf A1 treatment led to more pronounced accumulation of ERGFP_K0 in UFM1 and SAYSD1 KO cells than in WT cells, suggesting that defects in UFM1- and SAYSD1-mediated TAQC may induce a compensatory mechanism to degrade damaged ER.
SAYSD1 KO did not affect the expression of either UFM1 or UFMylating enzymes such as UFC1, UFL1, or UFBP1/DDRGK1, nor did it reduce UFMylated ribosome (Figure S2F), suggesting that SAYSD1 may act downstream of ribosome UFMylation. Since UFM1 deficiency has been linked to ER stress induction, we treated cells with ER stress inducers thapsigargin and tunicamycin but found no effect on ERGFP_K20 (Figure S2G). Together, these results rule out the possibility that the TAQC defect in SAYSD1-depleted cells is caused by ER stress or lack of ribosome UFMylation.
We next determined the subcellular localization of SAYSD1 using cells bearing a GFP tag at the C terminus of endogenous SAYSD1 (Figure S2H). Confocal microscopy showed that SAYSD1 is present mostly in the ER, but a fraction was found in perinuclear puncta marked by the Golgi protein GM130 (Figure 2D), suggesting that it may cycle between these membrane compartments. By contrast, ERGFP_K20 was accumulated mostly in perinuclear puncta in SAYSD1-depleted cells, which were largely co-localized with the ER marker calreticulin (Figure S2I). These findings implicate SAYSD1 in the export of translation-stalled proteins from the ER in TAQC.
SAYSD1 is predicted to contain a kinked transmembrane domain (TMD) with both the N and C termini facing the cytosol (Figure 2E). A short helical segment precedes the TMD, which is followed by a highly conserved SAYSvFN-containing domain (SACD) (Figures 2F and S2A). While the accumulation of ERGFP_K20 in SAYSD1-depleted cells could be significantly reversed when WT SAYSD1 was re-expressed, expression of the SAYSD1-7A mutant (residues in the SAYSvFN motif mutated to alanine) or mutants lacking either the amino-terminal 17 residues (ΔN17) or the middle helical segment (ΔMH) (Figure 2F) failed to restore ERGFP_K20 to a level comparable to that in WT SAYSD1-expressing cells (Figures 2G–2I). This result suggests a critical role for these conserved motifs in TAQC.
UFM1-dependent association of SAYSD1 with a stalled nascent chain-ribosome complex
If SAYSD1 directly participates in TAQC, it may interact with the Sec61 translocon. Indeed, immunoprecipitation of Sec61β, a key component of the ER translocon, readily co-precipitated endogenous SAYSD1 but not abundant cytosolic proteins such as p97 (Figure 3A). The interaction of SAYSD1 with the translocon was further supported by co-sedimentation of endogenous SAYSD1 with Sec61β by sucrose gradient centrifugation (Figure S3A). Furthermore, reciprocal immunoprecipitation of GFP-tagged SAYSD1 from SAYSD1::GFP cells revealed that SAYSD1 not only interacted with the translocon but also the UFMylation ligase UFL1 (Figure S3B). Interestingly, ribosomes were not significantly co-precipitated with SAYSD1-GFP under these conditions (Figure S3B, lane 2 versus 1). However, when cells were exposed to anisomycin (ANS), a translation elongation inhibitor that causes ribosome UFMylation,16 ribosome could now be detected in complex with SAYSD1 (Figures S3B, lanes 3 and 4, and S3C). Upon ANS treatment, we also observed an increased association of SAYSD1 with Sec61β but not UFL1.
Figure 3. SAYSD1 preferentially engages translocation-stalled nascent chain-ribosome complexes in a UFMylation dependent manner.
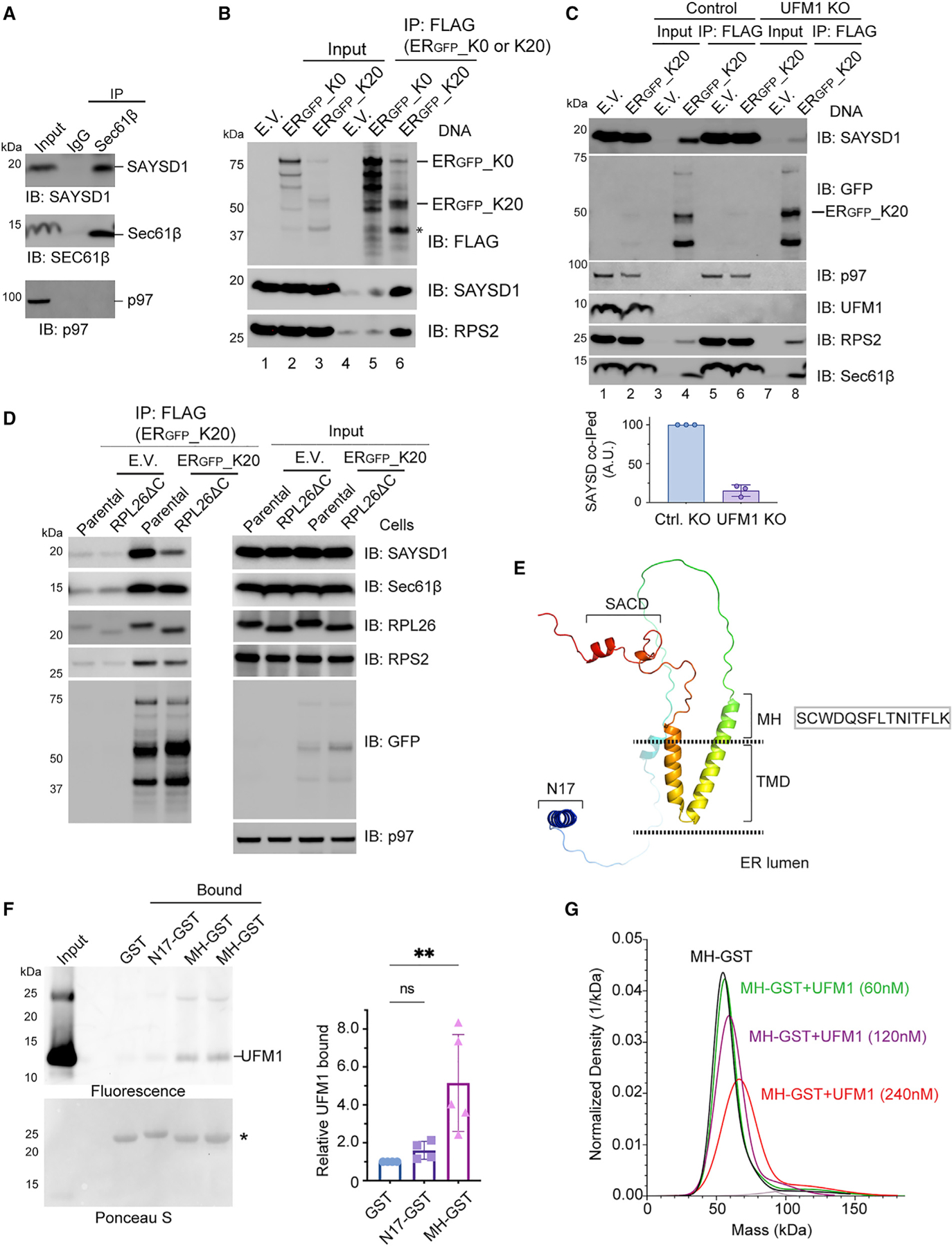
(A) Co-immunoprecipitation of endogenous SAYSD1 with the Sec61 translocon component Sec61β from 293T cells.
(B) SAYSD1 associates with ERGFP_K20 in a translation-stalling-dependent manner. 293T cells transfected with an empty vector (EV), ERGFP_K20, or ERGFP_K0 were subject to immunoprecipitation by FLAG beads. The asterisk indicates the stalled ERGFP_K20 species.
(C) The association of SAYSD1 with ERGFP_K20 is dependent on UFM1. Control or UFM1 CRISPR KO cells transfected as indicated were subject to FLAG pull-down and immunoblotting. The graph shows the quantification of three independent experiments.
(D) The association of SAYSD1 with ERGFP_K20 requires the RPL26 C tail. As in (C) except that cells that have the endogenous RPL26 C-terminal tail deleted by CRISPR-mediated homologous recombination were used.
(E) The predicted SAYSD1 structure by AlphaFold. SACD, SAYSvFN-containing domain; TMD, transmembrane domain.
(F) SAYSD1 binds the MH domain of UFM1 directly. The indicated that GST-tagged protein or GST immobilized on glutathione beads were incubated with Atto565-labeled UFM1. The precipitated proteins were SDS-PAGE fractionated and detected by a fluorescence scanner (top) or Ponceau S staining (bottom). The graph shows the quantification of 4 independent experiments. Error bars, means ± SD; **p < 0.01 by unpaired Student’s t test. n = 4.
(G) Mass photometry shows a direction interaction of MH-GST with UFM1.
To further probe the engagement of SAYSD1 with the stalled nascent chain-translocon-ribosome complex, we immunoprecipitated ERGFP-K20 or, as a control, ERGFP_K0, which lacks the ribosome-stalling sequence but otherwise is identical to ERGFP_K20.16 As expected, immunoblotting detected a strong interaction of ribosome (as indicated by the ribosome subunit RPS2) with ERGFP_K20 but not ERGFP_K0 or empty beads (Figure 3B, bottom panels, lane 6 versus 5 and 4). ERGFP_K20 immunoprecipitation also brought down endogenous SAYSD1 and Sec61β, whereas ERGFP_K0 only co-precipitated a negligible amount of SAYSD1 (Figures 3B, lane 6, and 3C, lane 4). Interestingly, the interaction of SAYSD1 with ERGFP_K20 was significantly reduced in UFM1 KO cells (Figure 3C, top panel, lane 4 versus 8) and in cells lacking the UFMylation site on RPL26 (RPL26 ΔC) (Figure 3D). By contrast, UFM1 depletion or KO of the RPL26 UFMylation site did not affect the interaction of ERGFP_K20 with either ribosome or Sec61β (Figures 3C and 3D, bottom panels). Collectively, these results suggest a dynamic interaction between SAYSD1 and the ER translocon that is regulated by ribosome UFMylation.
A middle helical (MH) segment in SAYSD1 binds UFM1 directly
The UFMylation-dependent association of SAYSD1 with ERGFP-K20 raised the possibility of SAYSD1 being a UFM1 ‘‘sensor.’’ To test this possibility, we expressed and purified GST-tagged proteins containing either the N17, the MH segment, or the C-terminal SACD (138–183) from E. coli (Figure S3D). We chose these regions because of sequence conservation and also because they are required for efficient ERGFP-K20 turnover (Figure 2). Additionally, AlphaFold predicts that these segments are folded independently in the cytosol (Figure 3E).31 We incubated purified proteins and a GST control with recombinant UFM1. Glutathione bead pull-down showed that only MH-GST consistently pulled down UFM1, whereas GST, N17-GST, or GST-SAYSD1137–183 failed to interact with UFM1 (Figures 3F and S3E).
We further confirmed the interaction between MH-GST and UFM1 using single-molecule mass photometry. We first examined the effect of UFM1 on the mass of MH-GST using N17-GST as a negative control. As expected, incubating UFM1 with N17-GST did not change the mass of the latter (Figure S3F). By contrast, incubating UFM1 with MH-GST resulted in a concentration-dependent increase in the mass of MH-GST (Figures 3G and S3F), suggesting the formation of a complex consisting of UFM1 and MH-GST. The specificity of the interaction was further supported by the observation that MH-GST did not bind ubiquitin under the same condition (Figure S3F). Collectively, these results demonstrate a direct and specific interaction between the MH segment of SAYSD1 and UFM1, which provides a molecular basis of UFM1-dependent engagement of SAYSD1 with the stalled nascent chain-ribosome complex.
A conserved SAYSD1 N-domain binds ribosome to promote TAQC
The N17 domain of SAYSD1 is predicted to form a helix with a conserved phenylalanine (F) followed by four positively charged residues (Figures 4A and 4B). Interestingly, the bacterial trigger factor uses a similar N-terminal ‘‘signature motif’’ to interact with ribosome (Figure 4A).32 Likewise, the ribosome binding site in the nascent polypeptide-associated complex (NAC) is formed by a stretch of positively charged residues (RRKxxKK) at the N terminus.33 These observations suggest that the N17 motif of SAYSD1 may also bind ribosome. To test this idea, we performed GST pull-down after incubating recombinant GST-N17 or GST (control) with a 293T cell extract. Immunoblotting showed that GST-N17, but not GST, readily co-precipitated with ribosomes (Figure 4C). Several additional lines of evidence further demonstrate N17 as a ribosome-binding domain that recognizes ribosome independent of UFMylation. First, although immunoblotting with UFM1 antibodies showed co-precipitation of N17 with UFM1-conjugated RPL26, immunoblotting with RPL26 antibodies also detect unmodified RPL26 in association with GST-N17 (Figure 4C, top panel). Second, the association of GST-N17 with ribosome was not affected by UFM1 depletion or by treating cells with ANS, which abolished and increased UFMylated ribosome, respectively (Figures S4A and S4B). Likewise, KO of the ribosome UFMylation site in RPL26 had no effect on this interaction (Figure S4C). Importantly, ribosome purified from rabbit reticulocyte lysate similarly interacted with GST-N17 (Figure 4D), suggesting that N17 interacts directly with ribosomes. This interaction is highly specific because GST-N17 did not pull down the abundant p97 ATPase or HSP90 (Figures 4C and 4E). Importantly, SAYSD1 variants carrying mutations on conserved residues (3A: L5A, F8A, R12A, or E7A_R14A) lost the ribosome-binding activity completely (Figure 4E). These results suggest N17 as a ribosome-binding site during TAQC.
Figure 4. SAYSD1 recognizes ribosome directly via an N-terminal segment.
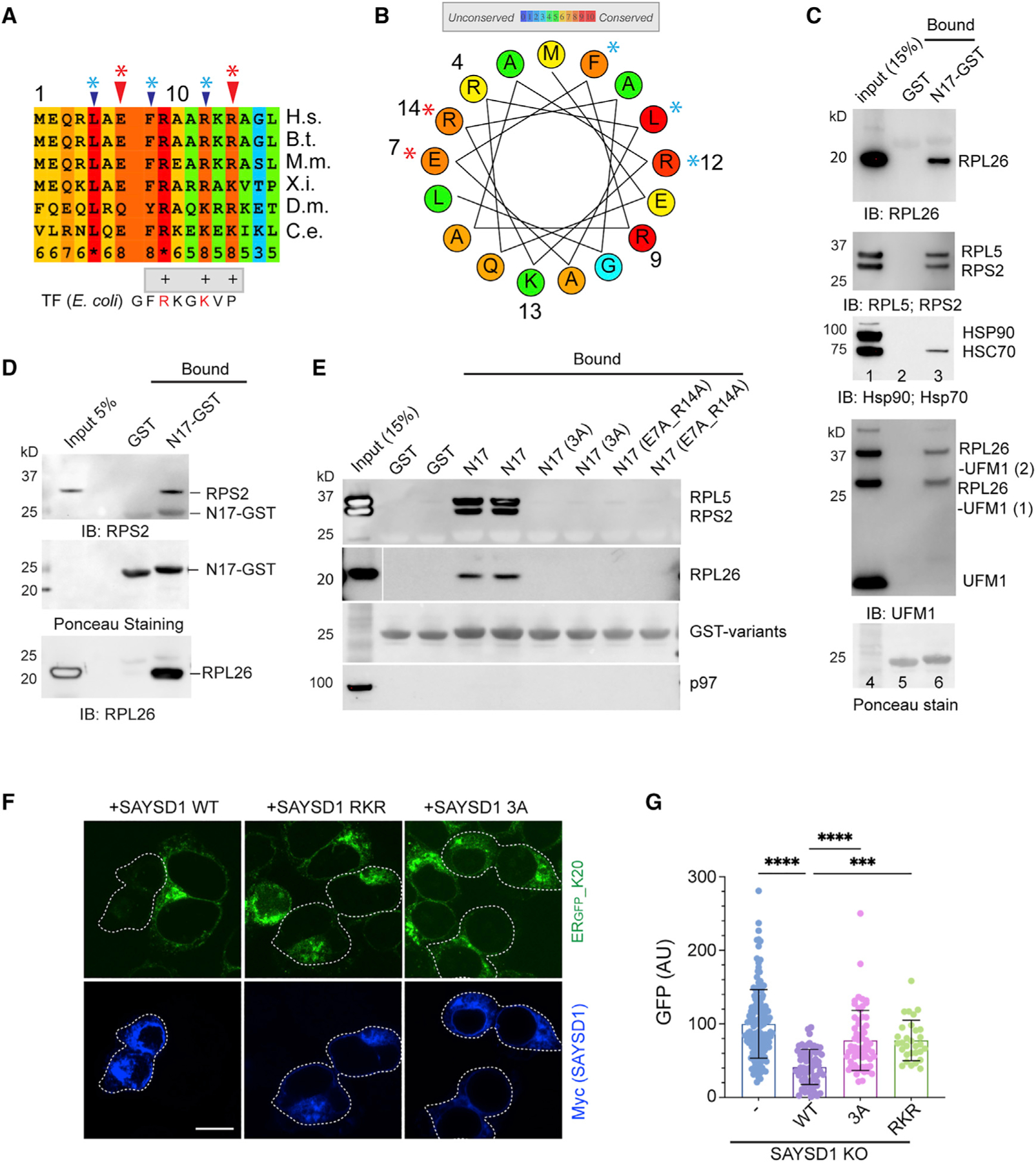
(A) Sequence alignment of the N17 domain of SAYSD1. The arrows indicate the two groups of conserved residues mutated in the study.
(B) A helix wheel view of the SAYSD1 N17 domain. Colors indicate the degree of conservation. Asterisks indicate residues mutated.
(C and D) GST-N17 interacts with ribosome directly. (C) GST-N17 or GST immobilized on glutathione beads were incubated with 293T cell extract. Precipitated proteins were analyzed by immunoblotting with the indicated antibodies. (D) As in (C) except that ribosome purified from rabbit reticulocyte lysate was used.
(E) The conserved residues in N17 are required for ribosome binding. The indicated GST-tagged N17 variants or GST was immobilized in duplicate and incubated with 293T cell lysate. The precipitated proteins were analyzed by immunoblotting using indicated antibodies.
(F and G) SAYSD1 ribosome binding is required for efficient turnover of ERGFP_K20. (F) SAYSD1 KO cells expressing ERGFP_K20 were transfected with the indicated DNA and imaged. SAYSD1-positive cells are highlighted by dashed lines. Scale bar, 10 μm. The graph in (G) shows the quantification of randomly imaged cells from two independent experiments. Error bars, means ± SD; ****p < 0.0001; ***p < 0.001 by unpaired Student’s t test.
To confirm the role of ribosome binding in TAQC, we expressed the SAYSD1 3A and another mutant that had three positively charged residues (RKR) mutated to alanine in ERGFP_K20-expressing SAYSD1 KO cells. Compared with WT SAYSD1, these mutants were significantly less effective in restoring the degradation of ERGFP_K20 (Figures 4F and 4G). Thus, ribosome binding by the N17 domain plays a critical role in TAQC.
UFM1 and SAYSD1 eliminate translation-stalled collagen I in mammalian cells
To elucidate the physiological function of TAQC, we investigated the role of UFM1 and SAYSD1 in collagen biogenesis. Collagens are the most abundant secretory proteins in metazoan.34 Most collagen molecules have a triple-helical domain (THD) that contains many repeated ‘‘PPG’’ motifs whose translation is known to cause ribosome pausing/stalling.35,36 This unique feature renders collagens a good candidate as an endogenous TAQC substrate.
To test whether the translation of the THD of collagens could cause translation arrest, we cloned the human collagen A1 (ColA1) THD-coding sequence between a GFP- and a mCh-coding sequence (Figure 5A). A viral 2A sequence was inserted both upstream and downstream of the THD sequence. This sequence causes ribosomes to re-initiate translation, resulting in two equally translated polypeptides from a single mRNA.37 In our design, the translation products should include GFP, Col1A1 THD, and mCh all in an equal amount if the Col1A1 THD sequence did not stall the ribosome. We used the ribosome-stalling poly-(A) sequence (K20) as a positive control and a sequence with no stalling motif (K0) as the negative reference. Flow cytometry analyses of cells expressing these reporters showed that the poly-A sequence reduced the mCh/GFP ratio by about 75% when compared with cells over-expressing GFP-K0, consistent with K20 being a strong stalling motif.23 The Col1A1 THD sequence reduced the mCh/GFP ratio by about 30% (Figure 5B), suggesting that it does cause translation arrest, albeit less efficiently than the poly-A sequence.
Figure 5. UFM1- and SAYSD1-dependent TAQC degrades translation-stalled Col1A1.
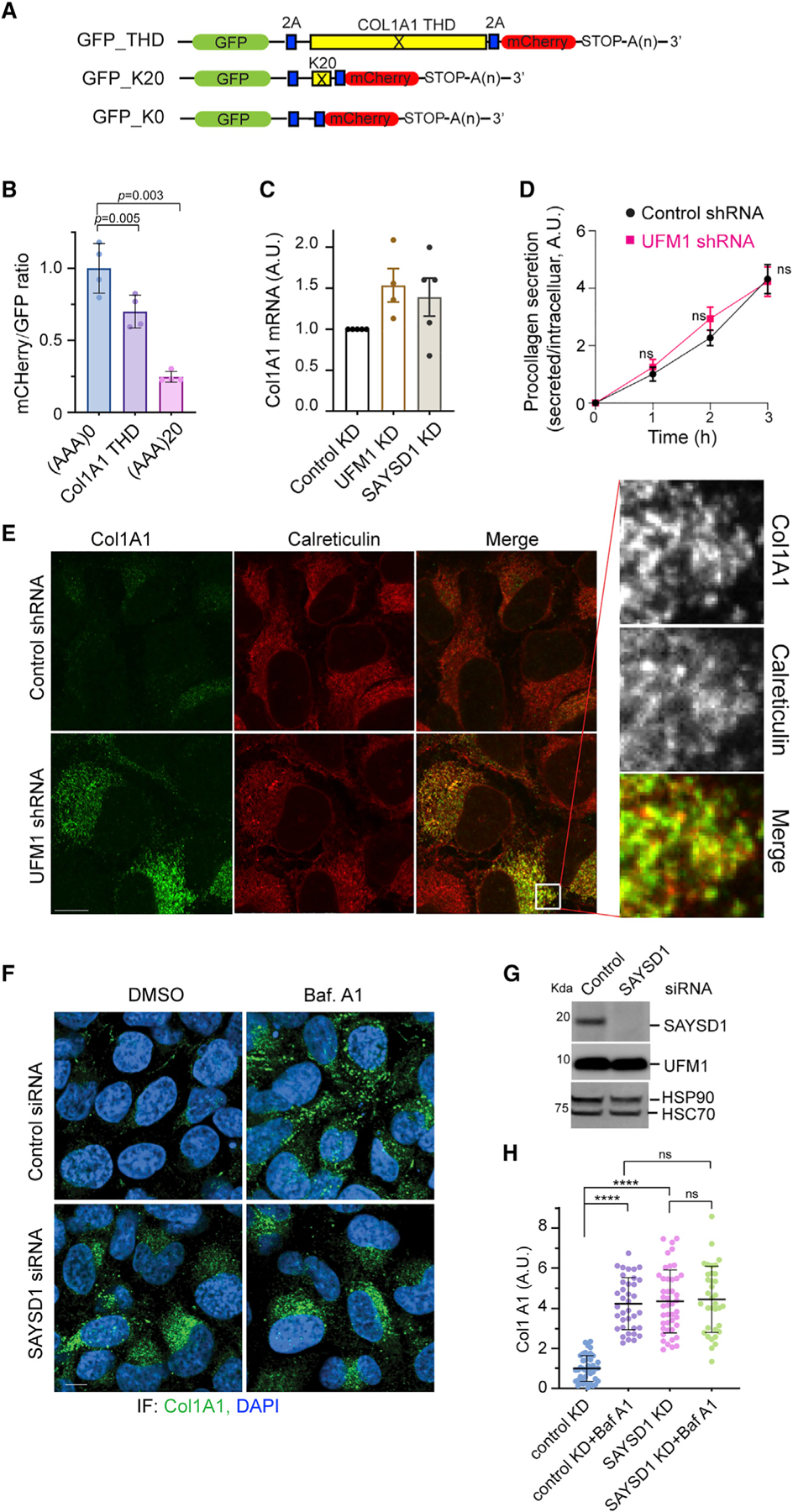
(A) A schematic view of the translation stalling reporters and the K0 control.
(B) The translation of the Col1A1 triple-helical domain (THD) causes ribosome stalling. 293T cells were transfected with the indicated reporters. The ratio of mC versus GFP was determined by flow cytometry. Error bars, means ± SD; p values are determined by one-way ANOVA followed by Dunnett’s multiple comparisons test; n = 4 independent experiments.
(C) UFM1 or SAYSD1 knockdown does not significantly increase Col1A1 mRNA as determined by qRT-PCR. n = 3 independent experiments. Error bars, means ± SD.
(D) UFM1 depletion does not affect Col1A1 secretion. Conditioned medium from control or UFM1-depleted U2OS cells were analyzed by ELISA. Error bars indicate means ± SD. ns, not significant by unpaired Student’s t test, n = 3 independent experiments.
(E) UFM1 depletion stabilizes Col1A1 in the ER. Control and UFM1 knockdown U2OS cells were stained by Col1A1 (green) and calreticulin (red) antibodies. Scale bar, 10 μm. Right panels show an enlarged view of the boxed area.
(F–H) Baf A1 treatment does not further enhance SAYSD1-depletion-induced Col1A1 accumulation.
(F) Control and SAYSD1 knockdown U2OS cells were treated with DMSO as a control or with Baf A1 (200 nM, 5 h) and stained by Col1A1 antibodies (green) or DAPI (blue). Scale bar, 10 μm. (G) A fraction of the siRNA-treated cells in (F) were analyzed by immunoblotting to confirm the knockdown of SAYSD1. (H) Quantification of the Col1A1 levels in (F). Error bars, means ± SD; ****p < 0.0001, by one-way ANOVA with Dunnett’s multiple comparisons test. n = 3 independent experiments.
We next analyzed the level of endogenous Col1A1 in control and UFM1 knockdown human osteosarcoma U2OS cells, which naturally produce and secrete collagen 1.38 Immunostaining showed that UFM1 knockdown increased the Col1A1 protein level by ~5-fold (Figures S5A and S5B), which was at least partially regulated at a post-transcriptional level because qRT-PCR revealed only a small increase in the Col1A1 mRNA level by UFM1 depletion (Figure 5C). The accumulation of Col1A1 was not caused by lack of protein secretion because ELISA analysis of conditioned medium detected no difference in secreted Col1A1 between control and UFM1 knockdown cells (Figure 5D). Similar to UFM1 depletion, Baf A1 treatment also caused Col1A1 to accumulate; combining Baf A1 treatment with UFM1 knockdown did not lead to further Col1A1 accumulation compared with Baf A1 treatment or UFM1 depletion alone (Figures S5B and S5C). These results suggest that Col1A1 accumulation in UFM1-depleted cells is mainly caused by reduced lysosomal degradation. Importantly, re-expressing UFM1 in UFM1 knockdown cells drastically reduced Col1A1 levels (Figures S5D and S5E), demonstrating a specific causal link between UFM1 depletion and Col1A1 stabilization.
Dual-color fluorescence confocal analyses showed that Col1A1 was mainly co-localized with the ER protein calreticulin in UFM1 knockdown cells (Figure 5E). By contrast, in Baf A1-treated cells, Col1A1 accumulated in vesicle-like puncta, which were mostly free of calreticulin (Figure S5F). These results are consistent with the established role of UFM1 in TAQC, suggesting that UFMylation also facilitates the export of stalled Col1A1 for lysosomal degradation.
Like in UFM1-depleted cells, knockdown of SAYSD1 also caused a significant accumulation of Col1A1 protein (Figures 5F–5H) but no significant increase in its mRNA (Figure 5C). Baf A1 treatment did not further enhance Col1A1 accumulation in SAYSD1 knockdown cells (Figures 5F–5H). Altogether, these results indicate that UFM1- and SAYSD1-mediated TAQC constitutively targets a fraction of newly synthesized Col1A1 for lysosomal degradation (see discussion).
UFMylation and SAYSD1 promote collagen biogenesis in flies
To study the in vivo function of TAQC, we first performed immunostaining using a UFM1-specific antibody to determine the expression of UFM1 in fat body (FB) and salivary gland (SG) of Drosophila third-instar larvae. These tissues are dedicated to protein secretion with UFM1 readily detectable by confocal microscopy (Figure S6A). Interestingly, we observed low UFM1 staining in immature SG but high UFM1 staining in mature, secretion-competent SG bearing a sizable lumen (Figures S6Aiii versus S6Aii and S6Ai). The observation suggests a conserved role for UFMylation in ER protein biogenesis in flies.
To further study TAQC in flies, we generated transgenic flies carrying the ERGFP_K20 reporter downstream of a UAS regulatory element. ERGFP_K20 flies were crossed to a GMR-Gal4 enhancer line, which specifically expressed the UAS-binding transcription factor Gal4 in photoreceptor cells of larval eye discs. This resulted in the expression of ERGFP_K20 in a similar pattern as Gal4. Similar as in mammalian cells, we detected weak GFP and no mCh in eye discs of GMR > ERGFP_K20 flies (Figures 6Ai and 6B), suggesting that translation stalling by the poly-A segment also occurs in flies. Knockdown of UFM1 significantly increased the GFP signal with no impact on mCh, as did depletion of TRAPPC11 (Figures 6Aii and 6Aiii versus 6Ai and 6B). Similar results were obtained when we used the Cg-Gal4 line, in which Gal4 expression is driven by an FB-specific promoter of the COL4 gene (Viking) (Figures S6B and S6C). Collectively, these results suggest that UFM1-dependent TAQC is conserved in Drosophila.
Figure 6. UFMylation and SAYSD1 regulate TAQC and ER protein biogenesis in Drosophila.
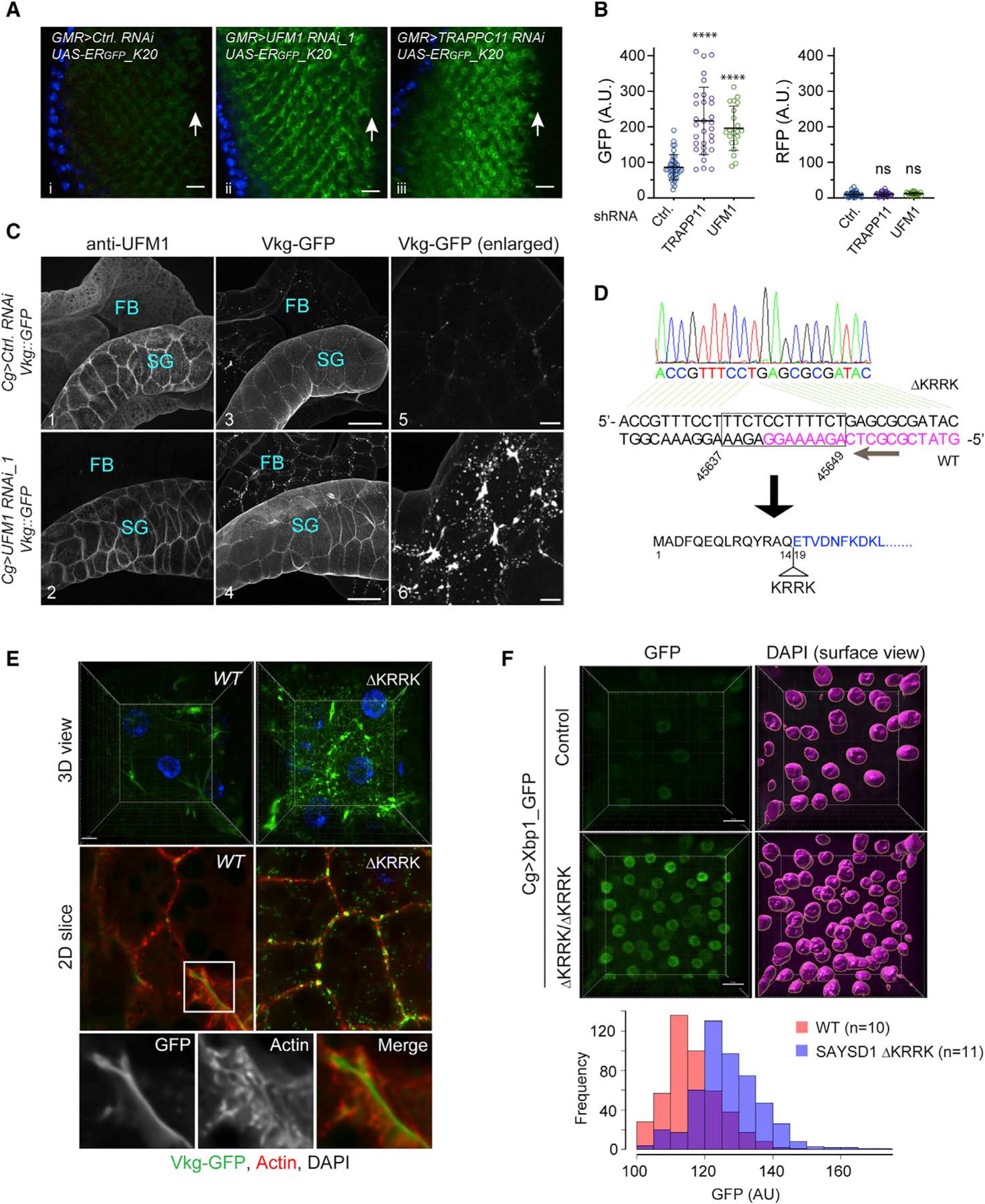
(A) Knockdown of UFM1 or TRAPPC11 in photoreceptor cells caused accumulation of ERGFP_K20. Scale bars, 10 μm.
(B) Quantification of the fluorescence intensity in individual eye discs as shown in (A). Error bars indicate means ± SD. ****p < 0.001 by one-way ANOVA with Dunnett’s multiple comparisons test. ns, not significant. n = 4 independent experiments.
(C) Fat body (FB) specific knockdown of UFM1 causes Viking-GFP (Vkg-GFP) to accumulate in FBs. Salivary gland (SG)-associated FBs from larvae of the indicated genotypes were stained with UFM1 antibodies (panels 1 and 2). Shown are maximum projected views of the confocal sections. Panels 5 and 6 show an enlarged view of Vkg-GFP in FBs. Scale bars, panels 1–4, 100 μm; panels 5 and 6, 20 μm.
(D) DNA sequencing to validate the genotype of SAYSD1 CRISPR flies. DNA from homozygous SAYSD1 CRISPR second-instar larvae was used to amplify SAYSD1 genomic locus, which was then sequenced. Magenta indicates the sgRNA sequence.
(E) As in (C) except that FBs from second-instar larvae of WT and SAYSD1 homozygous ΔKRRK mutants were stained with an actin dye (red) were imaged. All flies also bear the Vkg:GFP reporter. Scale bar, 10 μm.
(F) ER stress is induced in SAYSD1 ΔKRRK homozygous flies. FBs from control or SAYSD1 ΔKRRK second-instar larvae bearing an ER stress reporter (Xbp1-GFP) were stained with DAPI and imaged by 3D confocal microscopy. 3D surfaces were rendered by Imaris for individual nucleus, which were used to quantify the GFP signal. The graph shows the quantification results from two independent experiments. Scale bar, 15 μm.
To test whether TAQC regulates collagen biogenesis in flies, we knocked down UFM1 in COL4-producing FBs in larvae that had a GFP-encoding sequence inserted in the endogenous Viking locus (Viking-GFP).39 Drosophila COL4 consists of two polypeptide chains, COL4A1 and COL4A2 (also named Viking), which are a major component of the basement membrane.40 COL4 is secreted from FB and then incorporated into the basement membranes of imaginal discs and guts to maintain tissue homeostasis.41 To rule out potential off-target effects, we used two UFM1 shRNA lines. As a negative control, flies carrying mCh shRNA were used. Immunostaining with UFM1 antibodies confirmed that UFM1 was specifically depletedin FBsof Cg> UFM1 RNAi flies, but its level in nearby tissues such as SG remained unchanged (Figure 6C2 versus 6C1). Confocal microscopy detected only a weak Viking-GFP signal in FBs in control larvae (Figure 6C3 and 6C5), suggesting that newly synthesized COL4, if not secreted, is degraded rapidly. By contrast, we observed many Viking-GFP-containing puncta in UFM1-depleted FBs (Figures 6C4 and 6C6 versus 6C3 and 6C5; Figure S6D). This phenotype could not be attributed to a secretion defect because the Viking-GFP signal in the basement membranes around SG and guts (see below) was comparable between control larvae and larvae with FB-specific UFM1 depletion (Figure 6C). Thus, a significant fraction of COL4 appears to be downregulated in a UFM1-dependent manner during Drosophila development.
To validate the role of SAYSD1 in collagen biogenesis in flies, we used CRISPR-Cas9 to generate SAYSD1 loss-of-function mutant flies (see STAR Methods). An initial screening of more than 30 individual CRISPR lines identified a line that harbors a 12-nt deletion in the SAYSD1 locus. The resulting allele encodes a protein missing four charged residues (ΔKRRK) in the ribosome-binding N17 domain (Figure 6D). Although homozygous ΔKRRK flies died before the third-instar larval stage, Viking-GFP accumulation was observed by 3D confocal microscopy in FBs of second-instar SAYSD1ΔKRRK mutant larvae. In WT FBs, Viking-GFP was detected as a weak, smooth signal in spaces between FB cells, suggesting that it is mostly secreted. In SAYSD1ΔKRRK homozygous FBs, Viking-GFP was detected as bright green puncta both in and outside the cell (Figure 6E). Thus, like UFM1, SAYSD1 also contributes to TAQC of COL4 in flies.
To further evaluate the role of SAYSD1 in ER homeostasis regulation, we crossed Cg > Xbp1-GFP flies to either control or SAYSD1 ΔKRRK flies. The Xbp1-GFP reporter contains an unconventional intron from Xbp1. Only when this intron is spliced out during ER stress will the downstream GFP be translated in frame with Xbp1. Thus, the level of GFP expression reflects the level of ER stress. As anticipated, FB cells from SAYSD1 ΔKRRK larvae have a much higher GFP expression compared with control WT FBs (Figure 6F). Because ER stress upregulation was reported in UFM1 KO cells,7–9 these results further corroborate the notion that UFM1 and SAYSD1 function together to promote ER protein homeostasis.
TAQC controls the quality of secreted collagen in flies
Since deficiency in TAQC does not reduce collagen secretion but rather leads to reduced lysosomal degradation, we postulated that translation-stalled Viking-GFP, if not efficiently degraded, may escape from UFM1- or SAYSD1-deficient FBs. Incorporation of such defective collagens into the basement membranes might disrupt the basement membrane integrity. To test this idea, we examined the Viking-GFP-containing collagen fibrils on the gut of third-instar larvae by confocal microscopy, focusing on the proventriculus and middle midgut, where Viking-GFP forms stereotyped patterns at this developmental stage (Figure S7A). On the proventriculus of control larvae, Viking-GFP forms six evenly spaced rings, but in larvae that had UFM1 depleted from FBs, most proventriculus only have 4 Viking-GFP-positive rings, which were often irregularly spaced (Figure 7A1–7A3). On the middle midgut, WT larvae have long parallel and evenly spaced longitudinal collagen fibrils (LCFs), which are intersected perpendicularly by regularly spaced circular collagen fibrils (CCFs) (Figures 7A4, 7A7, and S7A). By contrast, in larvae with FB-specific depletion of UFM1, LCFs are often disrupted or formed intersections, while CCFs are curled, irregularly spaced, or missing in some areas (Figure 7A5, 7A6, 7A8, and 7A9). These phenotypes were even more prominent in the gut of adult Cg-Gal4; Viking-GFP/UAS-UFM1 RNAi flies (Figures S7B and S7C). As a result, the visceral muscle cells, which are normally attached to Viking-GFP-containing COL4 fibrils, are no longer positioned properly (Figure S7B).
Figure 7. TAQC safeguards collagen biogenesis in Drosophila.
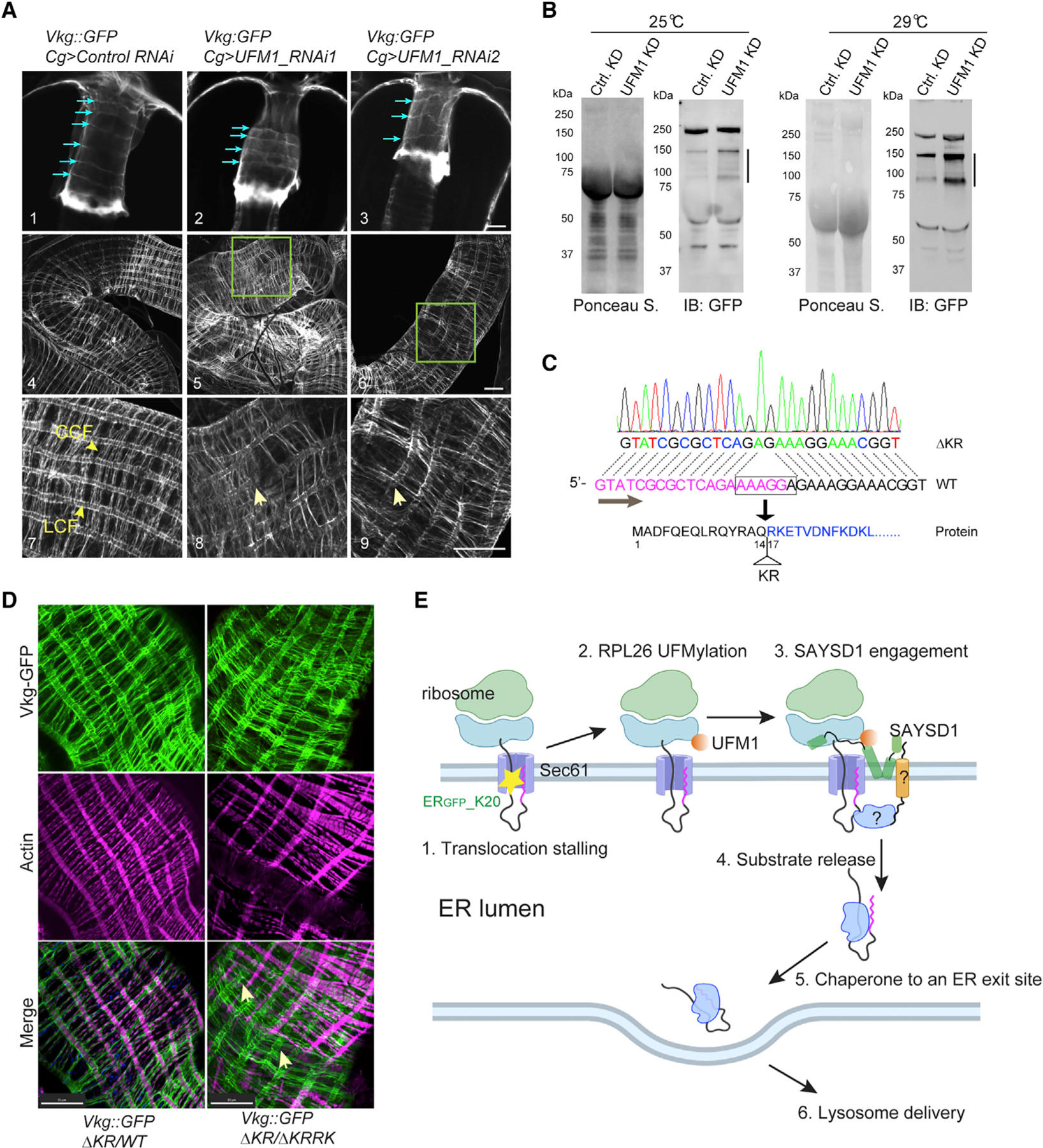
(A) Abnormal Viking deposition by UFM1-depleted FB in fly larvae. Shown are representative confocal images of Vkg-GFP on proventriculus (panels 1–3) or middle midgut (panels 4–9) from third-instar larvae of the indicated genotypes. Blue arrows in panels 1–3 indicate collagen fibrils around the proventriculus. Arrows in panels 8 and 9 indicate disrupted LCFs. Panels 7–9 are enlarged views. Scale bars, 20 μm.
(B) Defective Vkg-GFP accumulation in hemolymph in larvae with FB-specific UFM1 depletion. The lines indicate truncated Vkg-GFP species.
(C) Identification of a second SAYSD1 allele deleting two charged residues in the N17 domain.
(D) Abnormal Viking-containing basement membranes on middle midgut of fly larvae bearing the indicated mutant SAYSD1 alleles. The guts from third-instar larvae were stained by phalloidin to label actin (magenta). Arrows indicate Vkg-GFP-containing fibrils that are detached from muscle cells. Scale bars, 50 μm.
(E) A model of ribosome UFMylation sensing by SAYSD1 in TAQC. Created by Biorender.com.
To further prove that defective collagens are indeed secreted from UFM1-deficient FBs, we used immunoblotting to examine the secreted Viking-GFP in hemolymph collected from either control or FB specific UFM1 knockdown larvae. Consistent with our hypothesis, we detected several truncated Viking-GFP species that were elevated by FB-specific UFM1 knockdown (KD) (Figure 7B). By contrast, the secretion of full-length Viking was largely unaffected.
To examine the role of SAYSD1 in basement membrane quality control, we screened additional CRISPR lines to identify a mutant that has only two positively charged residues (ΔKR) deleted from the ribosome-binding N17 domain (Figure 7C). Flies bearing homozygous DKR alleles or trans-heterozygous ΔKR and ΔKRRK alleles are viable, probably because the smaller deletion had a less severe impact on the SAYSD1 function. Nonetheless, trans-heterozygous SAYSD1 third-instar larvae (ΔKR/ΔKRRK) have pronounced defects in collagen quality control and basement membrane assembly. These include Viking-GFP accumulation in FB cells and irregularly positioned Viking-GFP fibrils with many detached from the underlying muscle cells along the middle midgut (Figure 7D). Altogether, these results establish a critical role for UFM1- and SYASD1-mediated TAQC in the elimination of defective collagen molecules, which maintains the structural integrity of the basement membrane in Drosophila.
Intriguingly, despite the abnormal basement membrane morphology, flies with FB-specific UFM1 KD developed normally with no discernable phenotype upon eclosion. FB-specific UFM1 KD also did not affect the fly lifespan under normal growth conditions. However, when raised at an elevated temperature (29°C), flies with FB-specific UFM1 KD had a shortened lifespan (Figures S7D versus S7E). These findings suggest that basement membrane integrity is not essential for fly development but confers stress resistance in adult flies.
DISCUSSION
In this study, we screened for genes involved in TAQC-mediated elimination of polypeptides stalled during co-translational protein translocation. Our study re-discovers the UFMylation pathway, underscoring the significance of this post-translational modification in TAQC. Additionally, we uncover SAYSD1 as a functional partner for the UFMylation system in this process. SAYSD1 encodes a membrane protein, which associates with the Sec61 translocon in a stalling-regulated manner. It contains a highly conserved TMD, which may be the site of Sec61 binding. This TMD is predicted to form a kinked configuration, exposing a conserved N-terminal segment (N17), an MH before the TMD, and the highly conserved SACD downstream of the TMD all to the cytosol. We demonstrate that N17 is a ribosome-binding domain and that the MH segment forms a UFM1-binding site. Since the UFMylation site on RPL26 is close to the Sec61 translocon,42 the bipartite mode of ribosome recognition would allow N17 and MH to jointly sense UFMylated ribosomes on the clogged Sec61 translocon (Figure 7E). Although the N17 domain of SAYSD1 can readily interact with ribosome in cell extracts, we did not detect significant association of endogenous SAYSD1 with ribosome in the absence of translocation stalling. Thus, N17 might be kept in an inactivated form until ribosome UFMylation upon translocation stalling.
Our findings suggest that in TAQC, translocation-stalled polypeptides are released into the ER lumen and then transported to lysosomes via the Golgi. Our screen identifies the TRAPP complex as a facilitator of TAQC. The TRAPP complex is thought to tether ER-derived vesicles to the Golgi apparatus in the canonical ER-to-Golgi trafficking pathway.43 However, our screen does not recover any COPII components. This may be due to genetic redundancy or unsaturated screen design. Intriguingly, the release of TAQC substrates from UFMylated ribosomes is independent of ribosome UFMylation. Nevertheless, their delivery to lysosomes still requires both UFM1 and SAYSD1. Thus, ribosome UFMylation might elicit an ‘‘outside-in’’ signal, which allows the ‘‘sorting’’ of faulty substrates away from properly folded proteins that are destined to the cell surface or cell exterior (Figure 7E). SAYSD1 may interact with a membrane protein with a luminal domain, which in turn recruits a downstream chaperone to escort TAQC substrates to the correct destination. Alternatively, the stalled translocon may adopt a special conformation to engage a luminal protein.
Previous studies suggested that a significant fraction (10%–20%) of the newly synthesized collagens are rapidly degraded via numerous quality control mechanisms including ERAD and ERphagy.44,45 Our study shows that translocation pausing due to the enrichment of the stalling-prone PPX motifs in collagens is another source of defective collagens endogenously produced in animals. Our data suggest that the delicate mechanism of collagen biogenesis evolved by nature creates an unavoidable translation hardship, necessitating the UFM1- and SAYSD1-dependent TAQC to safeguard collagen biogenesis. This notion would explain the absence of the UFMylation system and SAYSD1 in yeast, which has no orthologous collagen genes. Intriguingly, UFM1 was recently identified as a positive regulator of ERphagy by a CRISPR-Cas9-based genetic screen, and an ERphagy receptor FAM134 was reported to play a role in collagen quality control at the ER in cultured cells.20,38,46 These observations raise the possibility that UFMylation-regulated ER cargoes may use both the canonical ER-to-Golgi pathway and ERphagy to reach lysosomes depending on whether an ERphagy receptor is present on the cargo-bearing vesicles. Given the previously established links between collagen and CNS disorders,47,48 out study hints at the possibility that defective basement membrane assembly and/or abnormal collagen deposition may contribute to neuronal dysfunctions associated with genetic lesions in the UFMylation system.
Limitations of the study
We acknowledge that our study has several limitations. First, while we identified a region in SAYSD1 for UFM1 binding, we cannot exclude the involvement of other SAYSD1 sequences or yet-to-be identified SAYSD1-binding proteins in this recognition. For tested TAQC reporter, KO of SAYSD1 only results in partial stabilization of the substrate, suggesting that additional mechanisms may also contribute to TAQC. In this regard, it is intriguing that our screen also identifies several canonical RQC factors as potential TAQC regulators. These proteins may collaborate with or operate in parallel to the reported lysosomal degradation mechanism; both possibilities were hinted at by previous studies.49,50 Additionally, the reported lysosomal degradation mechanism is demonstrated mostly with one model substrate. It is thus possible that other translation-stalled membrane substrates may use different mechanisms (e.g., ERAD or ERphagy) for degradation after being released into the ER lumen, particularly if they bear additional folding defects or other unstable structural signatures. Along the same line, whether translation stalled-collagens use the exact same ER-to-Golgi route as ERGFP_K20 for trafficking to lysosomes awaits further characterization.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Yihong Ye (yihongy@mail.nih.gov).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
All sequencing data generated from the CRISPR screen in this paper have been deposited to GEO under accession codes GEO: GSE222104.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
The human 293T, 293FT, U2OS cell lines were purchased from ATCC. ER_GFPK20 cells were made using 293T as the parental cells, which were transfected with ER_GFPK20. GFP positive cells were selected by FACS. CRISPR knockout cells were made on either 293T or ER_GFPK20 stable cell background. These cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM, Corning) containing 10% fetal bovine serum (FBS) and antibiotics (penicillin/streptomycin, 10 U/mL) at 37°C in a 5% CO2 humidified atmosphere.
Fly lines
Fly lines were ordered from the Bloomington Drosophila Stock Center. UAS-ERGFP_K20 transgenic flies were generated using a strain (R8622) carrying an attB integration site at 68A4 by Rainbow Transgenic. SAYSD1 CRISPR mutant flies were generated using a transgenic line (82,685) expressing a SAYSD1 targeting sgRNA and CAS9. Flies were maintained on standard food in a 25°C incubator. The genetic backgrounds of the flies used are:
36,796: y[1] sc[*] v[1] sev[21]; P{y[+t7.7] v[+t1.8] = TRiP.GL00536}attP2/TM3, Sb[1]
39,054: y[1] sc[*] v[1] sev[21]; P{y[+t7.7] v[+t1.8] = TRiP.HMS01974}attP40.
64,038: y[1] v[1]; P{y[+t7.7] v[+t1.8] = TRiP.HMJ22898}attP40.
35,785: y[1] sc[*] v[1] sev[21]; P{y[+t7.7] v[+t1.8] = VALIUM20-mCherry}attP2.
82,685: y[1] v[1]; P{y[+t7.7] v[+t1.8] = TKO.GS05018}attP40.
METHOD DETAILS
Cell culture and transfections
To facilitate procollagen biogenesis in U2OS cells, 50 μg/mL L-ascorbic acid was added in the culture medium. Plasmid transfection was performed using the TransIT-293 reagent (Mirus) for 293T and 293FT cells or Lipofectamine 2000 (Invitrogen) for U2OS cells following the manufacturer’s instructions. Lipofectamine RNAiMAX (Invitrogen) was used for siRNA transfections according to the manufacturer’s protocol.
Plasmids, shRNAs and antibodies
The ERGFP_K20 construct were generated as previously reported. To generate lentiviral expression plasmids, the ERGFP_K20 sequence were cloned into pLenti-CMV-GFP hygro (656-4) vector (Addgene #17446).
The human SAYSD1 cDNA was PCR-amplified from a human SAYSD1 cDNA clone (Origene) and inserted into the pLenti-cmv-GFP hygro (656-4) vector (Addgene #17446) to generate either N-terminally HA-tagged or C-terminally Myc-FLAG-tagged wildtype SAYSD1 (SAYSD1 WT) and N-terminal 17 residue-deleted SAYSD1 (Δ1-17 SAYSD1). The conserved ‘‘SAYSVFN’’ motif was mutated to ‘‘AAAAAAA’’ motif in the HA-SAYSD1 plasmid to generate the HA-SAYSD1 ‘‘7A’’ mutant. To generate a bacterial vector expressing human UFM1, the codon-optimized gene fragment encoding human UFM1 was inserted into the pET28a vector between NdeI and BamHI sites.
All shRNA lentiviral plasmids were generated based on the pLKO.1 puro vector (Addgene #8453) followed by the protocol in Addgene. The target sequences for shRNAs are listed below.
| Target gene | shRNA sequence |
|---|---|
| UFM1 | GTGTTGGAAGTTGTTAATATC |
| TRAPPC1 | GTCTCAGGAGAATTCATGTCT |
| TRAPPC8 | GTCAATGGTATACGAATATTA |
| TRAPPC9 | GGCCACACAAAATGAAAAGCT |
| TRAPPC10 | CCTGTGCTGGAGATCAGAATT |
| TRAPPC11 | CATGAAAGAATGTCAGACCAT |
To generate GST-fusion proteins, the corresponding DNA fragments encoding different domains of SAYSD1 were either inserted into the pET41a vector to construct N-terminal GST fusion proteins or used to replace Mff(1–61) in the pET28-Mff(1–61)-PP-GST vector (Addgene #73042) to generate C-terminal GST-fusion proteins.
To generate transgenic flies expressing ERGFP_K20, the pUAST-attb-ERGFP_K20 plasmid was generated by inserting the ERGFP_K20 sequence into the EcoRI and XbaI sites of pUAST-attb vector.
All plasmids were verified via Sanger sequencing and prepared using the Qiagen midi-prep kit before use. Antibodies are listed in the resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Calreticulin | ThermoFisher | Cat# PA3-900; RRID: AB_325990 |
| c-Myc (9E10) | Santa Cruz | Cat# SC-40; RRID: AB_627268 |
| FLAG (M2) | Sigma | Cat# F-1804; RRID: AB_262044 |
| GFP | Santa Cruz | Cat# SC-9996; RRID: AB_627695 |
| GFP | Developmental Studies Hybridoma Bank | Cat# DSHB-GFP-4C9; RRID: AB_2617422 |
| GFP | ThermoFisher | Cat# A31852; RRID: AB_162553 |
| GM130 | Abcam | Cat# Ab169276; RRID: AB_2894838 |
| GRP78 (BiP) | R&D Systems | Cat# MAB4846; RRID: AB_2233235 |
| HA | Sigma | Cat# H-3663; RRID: AB_262051 |
| HSC70 | Santa Cruz | Cat# SC-7298; RRID: AB_627761 |
| HSP90 | Santa Cruz | Cat# SC-13119; RRID: AB_675659 |
| mCherry | Abcam | Cat# Ab167453; RRID: AB_2571870 |
| PDI (P4HB) | Abcam | Cat# Ab2792; RRID: AB_303304 |
| Procollagen I | Developmental Studies Hybridoma Bank | Cat# SP1.D8; RRID: AB_528438 |
| RPL5 | Abcam | Cat# Ab157099 |
| RPS2 | Bethyl | Cat# A303-794A; RRID: AB_11218192 |
| SAYSD1 | Abcam | Cat# Ab189918 |
| Sec61β | Tom Rapoport lab | N/A |
| UBE1 | Sigma | Cat# E–3152; RRID: AB_259297 |
| UFL1 | Bethyl | Cat# A303-456A; RRID: AB_10951658 |
| UFM1 | Abcam | Cat# Ab109305; RRID: AB_10864675 |
| VCP | Fitzgerald | Cat# 10R-P104A; RRID: AB_1287614 |
| Goat anti-Mouse IgG (AlexaFluor®680-conjugated) | ThermoFisher | Cat# A21058; RRID: AB_2535724 |
| Goat anti-Mouse IgG (HRP-conjugated) | Sigma | Cat# A4416; RRID: AB_258167 |
| Goat anti-Rabbit IgG (HRP-conjugated) | Sigma | Cat# A6154; RRID: AB_2766784 |
| Goat anti-Rabbit IgG (IRDye800-Conjugated) | Rockland | Cat# 611-132-003; RRID: AB_220151 |
| Goat anti-Rabbit IgG (AlexaFluor®568-conjugated) | ThermoFisher | Cat# A11011; RRID: AB_143157 |
| Goat anti-Mouse IgG (AlexaFluor®405-conjugated) | ThermoFisher | Cat# A48255; RRID: AB_2890536 |
| Goat anti-Mouse IgG (AlexaFluor®488-conjugated) | ThermoFisher | Cat# A32723; RRID: AB_2633275 |
|
| ||
| Bacterial and virus strains | ||
| TOP10 | ThermoFisher | C404010 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
| UFM1 | R&D Systems, Inc. | UL-522 |
| Ubiquitin | Sigma | U5507 |
| Acti-stain 555 phalloidin | Cytoskeleton, Inc. | PHDH1 |
| Anisomycin | Sigma | A9789 |
| L-Ascorbic acid | Sigma | L5960 |
| Bafilomycin A1 | LC Laboratories | B-1080 |
| cycloheximide | Sigma | C7698 |
|
| ||
| Critical commercial assays | ||
| Human Pro-Collagen I alpha 1 ELISA Kit | Abcam | Ab210966 |
|
| ||
| Deposited data | ||
| Raw and processed data | This paper | GEO: GSE222104 |
|
| ||
| Experimental models: Cell lines | ||
| 293T | ATCC | CRL-3216 |
| U2OS | ATCC | HTB-96 |
|
| ||
| Experimental models: Organisms/strains | ||
| UFM1-RNAi_1 | Bloomington Drosophila Stock Center | 36,796 |
| UFM1-RNAi_2 | Bloomington Drosophila Stock Center | 39,054 |
| gry (TRAPPC11)-RNAi | Bloomington Drosophila Stock Center | 64,038 |
| mCherry-RNAi | Bloomington Drosophila Stock Center | 35,785 |
| CG13663 sgRNA | Bloomington Drosophila Stock Center | 82,685 |
|
| ||
| Oligonucleotides | ||
| Silencer Select Negative Control No. 2 siRNA | Life Technologies | 4,390,847 |
| Silencer Select Pre-designed siRNA against UFM1 | Life Technologies | s28345 |
| Silencer Select Pre-designed siRNA against SAYSD1 | Life Technologies | s31482 |
| Silencer Select Pre-designed siRNA against SAYSD1 | Life Technologies | s31483 |
|
| ||
| Recombinant DNA | ||
| pLenti-CMV-GFP | Addgene | 17,446 |
| pLKO.1 puro | Addgene | 8453 |
| pET28-Mff(1–61)-PP-GST | Addgene | 73,042 |
| GeckoV2 sgRNA libraries | Addgene | 1,000,000,048 |
|
| ||
| Software and algorithms | ||
| Adobe Illustrator | Adobe | N/A |
| Adobe Photoshop | Adobe | N/A |
| Cutadapt | Open source | N/A |
| Cytoscape | Open source | N/A |
| FASTQC | Open source | N/A |
| GraphPad Prism | Dotmatics | N/A |
| ImageLab | BioRad | N/A |
| ImageJ | Open source | N/A |
| NIS Elements | Nikon | N/A |
| Zen | Zeiss | N/A |
Lentivirus production and transduction
For the generation of human GeCKOv2 lentiviral pooled libraries, two 15-cm dishes of 293FT cells were seeded at 40% confluence. In the next day, 1 h prior to transfection, medium was replaced with 13 mL pre-warmed Opti-MEM medium (Life Technologies). For each dish, 6.8 μg pCMV-VSV-G, 10.1 μg psPAX2 (Addgene), 13 μg pLentiCRISPRv2-sgRNA plasmids pool and 135 μL PLUS reagent (Invitrogen) were added to 4 mL Opti-MEM as mixture A, which is then mixed with mixture B containing 68 μL lipofectamine 2000 and 4 mL Opti-MEM. The complete mixture was incubated for 20 min at RT before being added to cells. After 6 h, the medium was changed to 25 mL D10 medium (DMEM medium with 10% FBS and 1% BSA) with penicillin/streptomycin (10 U/mL) for virus production. After 60 h of incubation, virus-containing medium from two culture dishes were combined and centrifuged at 3,000 rpm at 4°C for 10 min and the supernatant was filtered through a 0.45 μm low protein-binding membrane (Steriflip HV/PVDF, Millipore). To concentrate lentivirus, the cleared supernatant was ultracentrifuged at 24,000 rpm for 2 h at 4°C using the JA25.50 rotor (Beckman). Virus was resuspended overnight in 180 μL D10 medium at 4°C. Virus was aliquoted, flash-frozen in liquid nitrogen and stored at −80°C.
For the shRNA lentivirus production, 293FT cells were seeded in a 6-well plate at 40% confluence. After 24h, cells in each well were transfected with 0.4 μg pCMV-VSV-G, 0.6 μg psPAX2 and 0.8 μg lentiviral shRNA plasmids using the TransIT-293 reagent (Mirus). After 6 h, the medium was replaced with fresh growth media and continued incubating for 60 h. To collect the lentivirus, the medium was filtered through a 0.45 μm PVDF membrane and used without further concentration.
Genome-wide CRISPR/Cas9 knockout screen
The GeckoV2 library was purchased from Addgene (1,000,000,048) and amplified according to the Addgene’s protocol and the online protocol from Feng Zhang’s lab.25 The complexity of the sgRNA library was verified by high-throughput sequencing by the NIDDK Genomic Core. Lentiviruses containing the sgRNA library and Cas9 were generated and used for transduction via spinfection. Briefly, 60 million ERGFP_K20 stable 293T cells supplemented with 8 μg/mL polybrene (Sigma) were seeded in two 12-well plates at a density of 3 million cells per well. Concentrated GeCKO v2 lentivirus was added to each well at a multiplicity of infection (MOI) at 0.3. Cells were then spun at 1000 g at room temperature for 2h followed by incubation at 37°C in a humidified incubator for 1h. After the medium was removed, fresh growth medium was added and incubated cells for 48 h before the start of selection for lentiviral integration using puromycin (0.3 μg/mL). The transduced cells were subcultured in medium supplemented with puromycin every 2 days for a total of 8 days and 80 million cells were maintained for each passage. After puromycin treatment, cells were recovered in a medium lacking puromycin for 24h before cell sorting. In total, 60 million cells were sorted into GFP high (1% of total cells) or GFP low (80% of total cells) cell populations by a FACS AriaII cell sorter. Genomic DNA was extracted from each cell population using a QIAGEN Blood Maxi kit (for GFP low cells) or QIAGEN Blood Midi kit (for GFP high cells) according to the manufacturer’s instructions. sgRNAs sequences were amplified from genomic DNA samples using the Herculase II Fusion DNA Polymerase (Agilent Technologies) in two PCR steps as follows. In the first PCR, the genomic region containing sgRNAs were amplified from 130 μg total DNA using the following primers: Forward: 5′-AATGGACTATCATATGCTTACCGTAACTTGAAAGTATTTCG-3′; Reverse: 5′-CTTTAGTTTGTATGTCTGTTGCTATTATGTCTACTATTCTTTCC-3′. In total, 13 PCR reactions were performed in parallel, with 10 μg genomic DNA in each reaction using Herculase II Fusion DNA Polymerase (Agilent) for 18 cycles and the resulting PCR products were combined. In the second step PCR, 5 μL of first PCR product was used in a 100 μL reaction volume and 24 PCR cycles was used. The primers used for the second PCR include stagger sequences of variable lengths and a 6bp barcode for multiplexing of different biological samples. The second step PCR primers were used as the following: Forward: 5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT (1-9bp variable length sequence)-(6bp barcode)tcttgtggaaaggacgaaacaccg-3’; Reverse: 5′-CAAGCAGAAGACGGCATACGAGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTtctactattctttcccctgcactgt-3′. The final PCR products were gel extracted, quantified, and sequenced using a NovaSeq sequencer (Illumina) at the NHLBI DNA Sequencing and Genomics Core. sgRNA sequences were obtained per sample by extracting 20 bps followed by the index sequence of ‘‘TTGTGGAAAGGAC GAAACACCG’’ on the de-multiplexed FASTQ files from Illumina’s NGS sequencer using the Cutadapt software, version 2.8 (https://doi.org/10.14806/ej.17.1.200). FASTQC, version 0.11.9 was used to assess the sequencing quality (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). MAGeCK, version 0.5.9, was used to quantify and to identify differentially expressed sgRNAs.51 MAGeCK count command was run on the merged two-half libraries of A and B to quantify. Differentially expressed sgRNAs with statistical significance were determined by running the MAGeCK test command in paired mode. Genes were ranked based on the number of unique sgRNA enriched in the GFP high population versus the GFP low population. Data was derived from two biological repeats.
Immunofluorescence microscopy and live-cell imaging
293T or COS7 cells were co-transfected with LAMP1-mCherry and either ERGFP_K20 or YFP-PrP* (C179A) using Lipofectamine 3000. Medium was changed after 16h and cells were seeded onto a glass coverslip. For 293T cells only, coverslips were pre-coated for 3 h with 0.01% poly-L-lysine solution to promote adherence to the coverslip. Forty-eight hours after transfection, cells were incubated with 0.4 mg/mL Alexa Fluor 647-conjugated anti-GFP antibody and 250 nM Bafilomycin A1, and imaged every 30 min for 15 h or more, as indicated. To detect antibody uptake of Alexa Fluor 647-conjugated anti-GFP antibody, image acquisition in the 647 nm channel from ERGFP_K20-expressing or YFP-PrPC179-expressing cells were identical in every way, including using identical excitation, emission, gain and contrast parameters. The imaging parameters were set to detect ERGFP_K20 or YFP-PrP* at steady-state (t = 0). While imaging ERGFP_K20 or YFP-PrP* in Bafilomycin A1 treated cells, ERGFP_K20 or YFP-PrP* quickly saturated the pixels due to the rapid rate of accumulation in lysosomes and limitations in the dynamic range of our camera.
Images and time lapse series were collected with Nikon inverted spinning disk confocal microscope equipped with Yokogawa CSU-X1 Spinning Disk, EMCCD camera, laser launch including 488 nm (to image GFP), 561 nm (to image mCherry) and 647 nm (to image Alexa Fluor 647), focus drift correction through the Perfect Focus System, 60x Plan Apo 1.40 NA oil/0.13mm WD, and controlled by the NIS-Elements software. For live cell imaging experiments, cells seeded in # 1.5 coverslip bottom-dishes and incubated in complete culture medium (described above) at 37°C with 5% CO2 within a Tokai incubator on the microscope stage. Imaging experiments were performed at 60–80% confluency 48 h after transient transfection.
For fixed cells imaging, cells were fixed in 4% paraformaldehyde at room temperature for 12 min followed by washing with PBS for two times. The cells were permeabilized in PBS containing 0.1% Triton X-100 and 10% FBS at room temperature for 45 min. Fixed cells were then incubated with respective primary antibodies at indicated concentration at 4°C overnight, followed by incubation with a secondary antibody at room temperature for 1h. DAPI was used to stain nuclei. The cells were then imaged either using the Zeiss LSM 780 confocal microscope or the Nikon CSU-W1 SoRa microscope.
Immunoblotting
To perform protein immunoblotting, proteins were separated in NuPAGE (4%–12%) Bis-Tris gels (Invitrogen) and transferred onto nitrocellulose membranes (BioRad). Target protein was detected by specific primary antibodies followed by secondary horseradish peroxidase (HRP)-conjugated antibodies or fluorescently labeled secondary antibodies (Invitrogen). Enhanced chemiluminescence (ECL) method was used to develop protein signals immunoblotted with HRP-conjugated secondary antibodies. Immunoblotting signal was detected using the BioRad ChemiDoc Imaging System. The intensity of the detected protein bands was quantified by the BioRad Image Lab software.
Generation of SAYSD1 CRISPR knockout cells
To generate SAYSD1 knockout cells, a pLentiCRISPRv2 plasmid containing a SAYSD1-targeting sgRNA sequence (CTCAGCTAACCGCTGTTCCA) was generated and packed into lentivirus. 293T cells transduced by the lentivirus were selected with puromycin for 7 days and the surviving cells were single-cell sorted into 96-well plate. Clones were screened by immunoblotting using the anti-SAYSD1 antibody. Knockout clones were pooled and used for the experiments.
Procollagen I secretion
U2OS cells were seeded at 1.5 million cells per well in a 6-well plate supplemented with 50 μg/mL L-ascorbate acid. Twenty hours later, cells were washed once and incubated in 1.2 mL culture media supplemented with 50 μg/mL L-ascorbate acid and 50 μg/mL cycloheximide (CHX). The medium was collected at 0, 1, 2 and 3 h time points, centrifuged at 2000 g for 5 min to remove any cells or cell debris. To prepare cell lysate, cells were washed once with ice-cold PBS and lysed in 90 μL NP40 lysis buffer and incubated for 15 min on ice, followed by centrifugation at 15.6 kg for 10 min. The medium and lysate were diluted 10 or 50 times, respectively, and the procollagen I level in medium and cell lysate was measured by the human Procollagen I alpha 1 ELISA Kit from Abcam.
To assess the effect of Baf A1 treatment on procollagen I secretion, U2OS cells were treated with DMSO or 250 nM Baf A1 for 5 h and the procollagen I level in medium and cell lysate were measured as above-mentioned.
Quantitative reverse transcription PCR (qRT-PCR)
Total cellular RNA was extracted using the TriPure reagent (Roche) and purified using the RNeasy MinElute Cleanup Kit (Promega) following standard protocols. RNA concentration was measured by a Nanodrop UV spectrophotometer, and 500 ng RNA was used to prepare cDNA library using the BioRad iScript Reverse Transcription Supermix. The qPCR reactions were carried out using the BioRad SsoAdvanced SYBR Green Supermix kit on a BioRad CFX96 machine. Data was analyzed using BioRad CFX manager 3.0 software. Ribosomal 18S RNA was used as a reference gene for quantification of gene expression. The following primers were used for qPCR: COPB1: forward, 5′-TTAGCTTAAAAAATGATCTAG-3′; reverse, 5′-GAAAACGAAGAGTAGATCCTC-3′; KDELR2: forward, 5′-CTGCTGAAGATCTGGAAGAC-3′; reverse, 5′-AGAGGATCTCAAGAGGAGAG-3′; RAB1B: forward, 5′-CCTGCTCCTGCGGTTTGCTG-3′; reverse, 5′- TCTTGTTGCCCACCAGGAGC-3′; TMED3: forward, 5′-AGTTCTCCCTGGATTACCAG-3′; reverse, 5′-CCTCATGGATGGTCACGCAG-3′; TRAPPC1: forward, 5′- GAAGCAAGCAGGGATTCCCAA-3′; reverse, 5′- TAGATGTGGTGCAGCACATCT-3′; TRAPPC8: forward, 5′- AGCAGGAGATTATGACCTTAA-3′; reverse, 5′- GTATTTAAGTGTATTTGGTAT-3′; TRAPPC9: forward, 5′- GAGCTTCACAGAGGAAGTGAA-3′; reverse, 5′- AGTGGTGCCTGTAGCGGATGT-3′; TRAPPC10: forward, 5′- CTTCCAAGAGAACCAATGGAA-3′; reverse, 5′-CTTCAGAACATTCTGCCACTT-3′; TRAPPC11: forward, 5′- CCAAATGTAGACCCAAGAGAA-3′; reverse, 5′- GAACCACTGCAACTTTTGTGT-3′; COL1A1, forward, 5′- GATTCCCTGGACCTAAAGGTGC-3′; reverse, 5′-AGCCTCTCCATCTTTGCCAGCA-3′.
Radiolabeling pulse-chase assay
Radiolabeling pulse-chase assay was performed as previously described.16 To measure the turnover of ERGFP_K20, 4.0 million 293T cells stably expressing ERGFP_K20 or in some cases cells transfected with ERGFP_K20 were starved in 2 mL starvation medium (DMEM medium lacking cysteine and methionine, supplemented with 10% FBS) for 30 min at 37°C, followed by labeling with 4 mCi [35S]-methionine/cysteine in 300 μL starvation medium for 16 min at 37°C. After labeling, cells were resuspended in 300 μL starvation medium supplemented with 2.5 mM methionine and cysteine. An aliquot of cells (65 μL) was immediately taken out and mixed with 500 μL RIPA buffer with 1 mM DTT and protease inhibitors. The remaining cells were incubated at 37°C for 20, 40, and 60 min. Equal amounts of cells (65 μL) were taken at each time points and lysed in RIPA buffer. After centrifugation of cell lysate at 140,00× g for 5 min, the cleared cell extracts were subjected to immunoprecipitation using anti-GFP antibody and protein A beads. The beads were then washed twice with the NET buffer with 0.1% SDS and bound proteins were eluted in 36 μL Laemmli sample buffer by boiling at 95°C for 5 min. Eluted proteins were analyzed by the SDS-PAGE using Criterion TGX precast gels (10%) followed by autoradiography analyses.
Ribosome fractionation
293T cells were seed into two wells at 0.5 million per well. Two wells were used for each siRNA transfection. siRNA transfections were performed twice on the following consecutive two days, with 60 pmol scrambled or UFM1 siRNA for each transfection. At 48 h posttransfection, cells from two wells were combined and seeded into a 10-cm dish. One day later, cells were then transfected with 5 μg plasmid encoding ERGFP_K20. The cells were then harvested 24 h later and used for radiolabeling pulse-chase as described above. Cells were chased for 0, 5, 10 and 20 min and 2 million cells were lysed at each time point in 200 μL polysome lysis buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 15 mM MgCl2, 1 mM DTT, 1% Triton X-100, 200 U/mL SUPERase In RNase inhibitor, 20 U/mL Turbo DNase and complete EDTA-free protease inhibitor cocktail) and incubated on ice for 10 min followed by centrifugation at 14,000 g for 5 min. Two hundred microliter supernatant was loaded onto a sucrose cushion (0.8 M sucrose, 1 mM DTT, 200 U/mL SUPERase In RNase inhibitor and complete EDTA-free protease inhibitor cocktail) and centrifuged at 69,000 rpm using a TLA100.4 rotor for 2.5 h at 4°C. After the ribosome-free supernatant was removed, the ribosome pellet was washed once with water and dissolved in 500 μL RIPA buffer. After centrifugation at 14,000 g for 5 min, the cleared ribosome solutions and the ribosome-free fractions were used for immunoprecipitation using anti-FLAG beads. Samples were resolved using SDS-PAGE and analyzed by autoradiograph.
To examine the association of SAYSD1 with Sec61β and ribosomes, 293T cells plated in a 10 cm dish were transfected with 5 μg ERGFP_K20 plasmid. 72 h post transfection, cells were lysed by 1% CHAPS in buffer LC (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 15 mM MgCl2, 1 mM DTT) that also contains 100 μg/mL cycloheximide, 200 U/mL SUPERase In RNase inhibitor, 20 U/mL Turbo DNase and a complete EDTA-free protease inhibitor cocktail. Cleared cell lysate (12,000 g 10min) was layered on top of a 10–50% sucrose gradient in the buffer LC containing 0.1% CHAPS. The sample was centrifuged at 44,000 rpm for 3 h in an SW60 rotor. 500 μL fractions were collected for immunoblotting analysis.
Immunoprecipitation
To detect interaction of endogenous SAYSD1 with the translocon and ribosomes. Four million of SAYSD1::GFP 293T cells or parental cells were seed in 10-cm dishes. One day later, cells were treated with 200 nM anisomycin (ANS) or DMSO for 1 or 2 h and lysed in 600 μl of digitonin lysis buffer (25 mM HEPES, pH 7.3, 250 mM KOAc, 10 mM MgCl2, 5 mM NaOAc and 0.5 mM EGTA, 2% digitonin, 1 mM DTT and complete protease inhibitors) at 4°C for 15 min. Sucrose was added to 250 mM and cell lysate was centrifuged at 15.6 kg for 5 min. For immunoprecipitation, 0.5 mL cleared cell lysate was incubated with 30 μL slurry of pre-washed GFP-Trap nanobeads (ChromoTek) for 1.5 h at 4°C. After incubation, beads were washed with digitonin wash buffer (25 mM HEPES, pH 7.3, 115 mM KOAc, 10 mM Mg KOAc, 5 mM NaOAc, 0.2% digitonin, and 0.5 mM EGTA) followed by boiling in 50 μL Laemmli sample buffer at 95°C for 10 min before SDS-PAGE and immunoblotting analyses. To immunoprecipitate the translocon, 293T cells were lysed in the digitonin lysis buffer and cleared supernatant was incubated with 10 μL pre-immune or Sec61β serum for 1 h at 4°C. Protein A beads were used to precipitate antibody protein complexes.
To detect interaction of TAQC substrates with SAYSD1, 293T cells were seeded in a 6-well plate at 0.5 million per well and grown for 24 h followed by transfection of 1.5 μg ERGFP_K20-expressing plasmid. For control experiments, cells were transfected with ER_K0 or pcDNA3 empty vectors. After 48 h, cells were washed with ice-cold PBS and lysed in 0.5 mL CHAPS lysis buffer (1% CHAPS, 50 mM HEPES pH7.3, 100 mM NaCl, 1 mM DTT and complete protease inhibitors) for 15 min at 4°C. The cell extracts were then centrifuged at 15.6 kg for 10 min at 4°C. To immunoprecipitate ERGFP_k20, the cleared supernatant was incubated with 30 μL pre-equilibrated anti-FLAG M2 beads at 4°C for 1 h using a head-over-tail rotator. For GST-N17 pulldown of ribosome, 2 x 106 cells were lysed in 0.4 mL CHAPS lysis buffer. The cleared supernatant was adjusted to include 1mM EGTA and 2.5mM MgCl2 and then incubated with glutathione beads prebound with GST-tagged recombinant proteins for 30 min at 4°C. After incubation, beads were washed twice with CHAPS wash buffer (0.02% CHAPS, 50 mM HEPES pH7.3, 100 mM NaCl, 1 mM DTT) and bound proteins were eluted by boiling in 50 μL Laemmli sample buffer at 95°C for 10 min. Samples were analyzed by SDS-PAGE and immunoblotting using respective antibodies as indicated.
To detect the interaction of GST-SAYSD1 with UFM1, GST-tagged proteins (2 μg) and His6-UFM1 or Atto565-UFM1 (16 μg) were mixed in 200 μL UFM1 binding buffer (25 mM of Tris-HCl, pH 7.4, 150 mM of NaCl, 2 mM of MgCl2, 2 mM of KCl, and 0.05% of NP40). After incubation at 4°C for 1 h, the mixture was transferred into a new microtube containing 25 μL (bed volume) GST beads equilibrated with the UFM1 binding buffer and further incubated for another hour. The beads were spun down at 1,000 g 1min and washed twice with the UFM1 binding buffer. The proteins on beads were eluted with 50 μL of Laemmli sample buffer and assayed by SDS-PAGE followed by immunoblotting or fluorescence scanning.
Endogenous tagging
GFP tagging of endogenous SAYSD1 was carried out using a CRISPR/Cas12-Assisted PCR Tagging system as described (http://www.pcr-tagging.com).52 Briefly, the PCR cassette was amplified from pMaCTag-05 plasmid by the AccuPrime Pfx DNA Polymerase with the primers, M1_SAYSD1 and M2_SAYSD1, listed below. The PCR product was gel purified with QIAGEN Gel Extraction Kit. 293T cells were transiently transfected with 1 μg of the PCR cassette and 1 μg of pcDNA3.1-hAsCpf1/TYCV/pY210 (gift from Feng Zhang, Addgene plasmid # 89351) using TransIT293 (Mirus) according to the manufacturing protocol and GFP positive cells were sorted two weeks later with Flow Cytometry.
M1_SAYSD1: 5′-GTGTTCAATCCAGGCTGTGAAGCCATCCAGGGCACCCTGACTGCAGAGCAGTTGGAGCGCGAGTTACAGTTGAGACCCCTGGCAGGGAGATCAGGTGGAGGAGGTAGTG-3′
M2_SAYSD1: 5′-CCAATGGTGAGGAAGACCACATCAGAGGTTAGCTGCATGACAGCACAGCTGGGTCAAAAAAGGGTCCTATCTCCCTGCCAGATCTACAAGAGTAGAAATTAGCTAGCTGCATCGGTACC-3′
Recombinant protein purification and mass photometry
Plasmids encoding the N-terminally His6-tagged UFM1 were transformed into BL21(DE3) cells (Agilent) and the transformed cells were cultured in LB medium supplemented with kanamycin 50 μg/mL and chloramphenicol 25 μg/mL. To induce protein expression, 0.5 mM IPTG were added when the optical density at 600 nm of the bacterial culture reached 0.9, and the proteins are expressed for 22 h at 19°C. For protein purification, bacterial pellet was lysed in buffer R (20 mM Tris pH 8.0, 500 mM KCl, 10% glycerol) supplemented with the EDTA-free complete protease inhibitor cocktail by sonication and centrifuged for 45 min at 15,000 rpm using a JA15.25 rotor. The cleared supernatant was incubated with Ni-NTA agarose (Qiagen) for 1 h at 4°C. After washing, the His6-UFM1 protein was eluted in buffer R supplemented with 50-300 mM imidazole. Elution fractions were analyzed using SDS-PAGE followed by Coomassie blue staining. The proteins are pooled, concentrated and dialyzed in a protein buffer (50 mM Tris, pH 7.4, 50 mM NaCl). GST-fusion proteins were expressed following the abovementioned procedure and purified using the Glutathione Sepharose 4B resin using the standard protocol. Proteins were then dialyzed against assay buffer. Protein concentration was determined by the Bradford assay.
The isolation of ribosomes from rabbit reticulocyte lysate (RRL) was performed according to a published protocol.53 Briefly, in a fresh ultracentrifuge tube, 250 μL RRL was laid on 1 mL of a cushion buffer (50 mM Tris-HCl, pH7.5, 5mM MgCl2, 25 mM KCl, 2 M Sucrose, filter-sterilized through a 0.22 mm filter). The sample was placed in a TLA120.2 rotor, and centrifuged at 4°C at 48,000 rpm, for 20 h in Beckman OptimaTM MAX ultracentrifuger. After centrifuge, the supernatant was carefully removed, and the ribosome-containing pellet was resuspended in 250 μL PBS buffer.
Mass photometry was performed on an OneMP instrument (Refeyn, UK) at room temperature following the published protocol.54 Briefly, microscope coverslips (24 × 50 mm, Fisher Scientific) were rinsed consecutively in isopropanol and H2O and blow dry in a stream of clean nitrogen. 30nM GST or MH-GST were mixed with the indicated concentrations of UFM1 in a phosphate saline buffer. After applying 10 μL solution on the slide, the mass distribution plot was obtained with the software provided by the instrument manufacturer (Refeyn, UK).
Fly experiments
To examine COL4 biogenesis, Viking-GFP was recombined to the same chromosome as Cg-Gal4 in female germlines. Progenies were screened based on eye color. Cg-Gal4, Viking-GFP females were crossed to w1118/y; UAS-UFM1 shRNA/UAS-mCherry or UAS-Control shRNA/UAS-mCherry. Third-instar larvae without UAS-mCherry were picked for dissection, fixing, and confocal imaging. For phalloidin staining, tissues were fixed in 4% formaldehyde in phosphate buffer saline (PBS) at room temperature for 15 min, permeabilized in PBS with 0.1% Saponin and 10% fetal bovine serum and stained by Acti-stain 555 Phalloidin at 50 nM for 30 min at room temperature. Tissue dissection and immunostaining were performed as described previously.55 Specifically, third-instar larvae were rinsed with PBS and then dissected in the same buffer. Tissues were fixed in PBS containing 4% paraformaldehyde at room temperature for 20 min and then rinsed 3 times with PBS. Tissues were incubated with a staining solution (PBS with 0.1% Saponin and 10% fetal bovine serum) for 10 min and then stained with primary antibodies in the same buffer at 4°C overnight. After three rinses with PBS, tissues were stained with secondary antibodies in the staining solution at room temperature for 1 h. Tissues were then washed three times with PBS and mounted in Vectashield mounting medium with DAPI followed by confocal microscopy.
To generate SAYSD1 CRISPR KO flies, female flies expressing sgRNA ubiquitously for Cas9-mediated mutagenesis of CG13663 (FBgn0039291) (#82685) were crossed to flies expressing Cas9 in the male germline cells with the Nos promoter.56 Male F1 progenies bearing both the sgRNA-expressing chromosome and the Nos-Cas9 chromosome were then crossed to female flies bearing double balancers for the second and third chromosome (x/y, w−; Sp/Cyo; TM2/TM6B). Individual F2 male progenies with white eyes were then crossed to flies with the double balancers to establish candidate KO lines. Genomic DNA from homozygous SAYSD1 mutant was extracted and used to amplify the SAYSD1 genomic DNA and for sequencing validation.
QUANTIFICATION AND STATISTICAL ANALYSES
Immunoblotting plots were digitally scanned by a ChemDoc Imaging System. Band intensity was measured by Image Lab (BioRad). Imaging data were processed by either Zen 2.3 SP1 (Zeiss) or by Nikon Elements (Nikon). Fluorescence intensity was measured by Imaged J or by Imaris in the case of 3D imaging. Unless specified, the n values in the figure legend indicate the number of independent biological replicates. Statistically analyses were performed using GraphPad Prism 9. For two group comparison, unpaired Student’s t-test was used. For multi-group comparison, we used one-way ANOVA. For comparing life span in flies, Gehan-Breslow-Wilcoxon test was used. Error bars, shown as mean ± SD and p values were calculated by GraphPad Prism 9. A value of p < 0.05 was considered as statistically significant. Graphs were prepared with GraphPad Prism 7.
Supplementary Material
Highlights.
A CRISPR screen identifies genes involved in translocation-associated quality control
SAYSD1 associates with the Sec61 translocon to safeguard ER protein translocation
SAYSD1 senses a clogged translocon by recognizing UFMylated ribosome
Ribosome UFMylation and SAYSD1 control the quality of secreted collagens
ACKNOWLEDGMENTS
We thank J. Skeath (Washington University) for Viking-GFP flies; H. Ryoo (New York University) for the Xbp1-GFP fly strain; the NHLBI Genomic, Flow Cytometry, Biophysics, and NIDDK Advanced Light Microscopy & Image Analysis Cores (ALMIAC) for services; and N. Guydosh (NIDDK) and J. Yewdell (NIAID) for critical reading of the manuscript. This research was supported by an intramural research program of the NIDDK (Y.Y.) and of NINDS (Q.Y.) at the National Institutes of Health and by NIH/NIGMS R01 GM134327 to P.-S.K.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112028.
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
REFERENCES
- 1.Rapoport TA, Li L, and Park E (2017). Structural and mechanistic insights into protein translocation. Annu. Rev. Cell Dev. Biol 33, 369–390. 10.1146/annurev-cellbio-100616-060439. [DOI] [PubMed] [Google Scholar]
- 2.Wang L, and Ye Y (2020). Clearing traffic jams during protein translocation across membranes. Front. Cell Dev. Biol 8, 610689. 10.3389/fcell.2020.610689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Phillips BP, and Miller EA (2020). Ribosome-associated quality control of membrane proteins at the endoplasmic reticulum. J. Cell Sci 133, jcs251983. 10.1242/jcs.251983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ast T, Michaelis S, and Schuldiner M (2016). The protease Ste24 clears clogged translocons. Cell 164, 103–114. 10.1016/j.cell.2015.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerakis Y, Quintero M, Li H, and Hetz C (2019). The UFMylation system in proteostasis and beyond. Trends Cell Biol 29, 974–986. 10.1016/j.tcb.2019.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang SH, Kim GR, Seong M, Baek SH, Seol JH, Bang OS, Ovaa H, Tatsumi K, Komatsu M, Tanaka K, and Chung CH (2007). Two novel ubiquitin-fold modifier 1 (Ufm1)-specific proteases, UfSP1 and UfSP2. J. Biol. Chem 282, 5256–5262. 10.1074/jbc.M610590200. [DOI] [PubMed] [Google Scholar]
- 7.Lemaire K, Moura RF, Granvik M, Igoillo-Esteve M, Hohmeier HE, Hendrickx N, Newgard CB, Waelkens E, Cnop M, and Schuit F (2011). Ubiquitin fold modifier 1 (UFM1) and its target UFBP1 protect pancreatic beta cells from ER stress-induced apoptosis. PLoS One 6, e18517. 10.1371/journal.pone.0018517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai Y, Pi W, Sivaprakasam S, Zhu X, Zhang M, Chen J, Makala L, Lu C, Wu J, Teng Y, et al. (2015). UFBP1, a key component of the Ufm1 conjugation system, is essential for ufmylation-mediated regulation of erythroid development. PLoS Genet 11, e1005643. 10.1371/journal.pgen.1005643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Zhang M, Wu J, Lei G, and Li H (2012). Transcriptional regulation of the Ufm1 conjugation system in response to disturbance of the endoplasmic reticulum homeostasis and inhibition of vesicle trafficking. PLoS One 7, e48587. 10.1371/journal.pone.0048587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tatsumi K, Yamamoto-Mukai H, Shimizu R, Waguri S, Sou YS, Sakamoto A, Taya C, Shitara H, Hara T, Chung CH, et al. (2011). The Ufm1-activating enzyme Uba5 is indispensable for erythroid differentiation in mice. Nat. Commun 2, 181. 10.1038/ncomms1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nahorski MS, Maddirevula S, Ishimura R, Alsahli S, Brady AF, Begemann A, Mizushima T, Guzmán-Vega FJ, Obata M, Ichimura Y, et al. (2018). Biallelic UFM1 and UFC1 mutations expand the essential role of ufmylation in brain development. Brain 141, 1934–1945. 10.1093/brain/awy135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muona M, Ishimura R, Laari A, Ichimura Y, Linnankivi T, Keski-Filppula R, Herva R, Rantala H, Paetau A, Pöyhönen M, et al. (2016). Biallelic variants in UBA5 link dysfunctional UFM1 ubiquitin-like modifier pathway to severe infantile-onset encephalopathy. Am. J. Hum. Genet 99, 683–694. 10.1016/j.ajhg.2016.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Colin E, Daniel J, Ziegler A, Wakim J, Scrivo A, Haack TB, Khiati S, Denommé AS, Amati-Bonneau P, Charif M, et al. (2016). Biallelic variants in UBA5 reveal that disruption of the UFM1 cascade can result in early-onset encephalopathy. Am. J. Hum. Genet 99, 695–703. 10.1016/j.ajhg.2016.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duan R, Shi Y, Yu L, Zhang G, Li J, Lin Y, Guo J, Wang J, Shen L, Jiang H, et al. (2016). UBA5 mutations cause a new form of autosomal recessive cerebellar ataxia. PLoS One 11, e0149039. 10.1371/journal.pone.0149039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walczak CP, Leto DE, Zhang L, Riepe C, Muller RY, DaRosa PA, Ingolia NT, Elias JE, and Kopito RR (2019). Ribosomal protein RPL26 is the principal target of UFMylation. Proc. Natl. Acad. Sci. USA 116, 1299–1308. 10.1073/pnas.1816202116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L, Xu Y, Rogers H, Saidi L, Noguchi CT, Li H, Yewdell JW, Guydosh NR, and Ye Y (2020). UFMylation of RPL26 links translocation-associated quality control to endoplasmic reticulum protein homeostasis. Cell Res 30, 5–20. 10.1038/s41422-019-0236-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun Z, and Brodsky JL (2019). Protein quality control in the secretory pathway. J. Cell Biol 218, 3171–3187. 10.1083/jcb.201906047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joazeiro CAP (2017). Ribosomal stalling during translation: providing substrates for ribosome-associated protein quality control. Annu. Rev. Cell Dev. Biol 33, 343–368. 10.1146/annurev-cellbio-111315-125249. [DOI] [PubMed] [Google Scholar]
- 19.Stephani M, Picchianti L, and Dagdas Y (2021). C53 is a cross-kingdom conserved reticulophagy receptor that bridges the gap between-selective autophagy and ribosome stalling at the endoplasmic reticulum. Autophagy 17, 586–587. 10.1080/15548627.2020.1846304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang JR, Lingeman E, Luong T, Ahmed S, Muhar M, Nguyen T, Olzmann JA, and Corn JE (2020). A genome-wide ER-phagy screen highlights key roles of mitochondrial metabolism and ER-resident UFMylation. Cell 180, 1160–1177.e20. 10.1016/j.cell.2020.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qin B, Yu J, Nowsheen S, Wang M, Tu X, Liu T, Li H, Wang L, and Lou Z (2019). UFL1 promotes histone H4 ufmylation and ATM activation. Nat. Commun 10, 1242. 10.1038/s41467-019-09175-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ito-Harashima S, Kuroha K, Tatematsu T, and Inada T (2007). Translation of the poly(A) tail plays crucial roles in nonstop mRNA surveillance via translation repression and protein destabilization by proteasome in yeast. Genes Dev 21, 519–524. 10.1101/gad.1490207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Juszkiewicz S, and Hegde RS (2017). Initiation of quality control during poly(A) translation requires site-specific ribosome ubiquitination. Mol. Cell 65, 743–750.e4. 10.1016/j.molcel.2016.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Satpute-Krishnan P, Ajinkya M, Bhat S, Itakura E, Hegde RS, and Lippincott-Schwartz J (2014). ER stress-induced clearance of misfolded GPI-anchored proteins via the secretory pathway. Cell 158, 522–533. 10.1016/j.cell.2014.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bengtson MH, and Joazeiro CAP (2010). Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature 467, 470–473. 10.1038/nature09371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shao S, Brown A, Santhanam B, and Hegde RS (2015). Structure and assembly pathway of the ribosome quality control complex. Mol. Cell 57, 433–444. 10.1016/j.molcel.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Defenouillère Q, Yao Y, Mouaikel J, Namane A, Galopier A, Decourty L, Doyen A, Malabat C, Saveanu C, Jacquier A, and Fromont-Racine M (2013). Cdc48-associated complex bound to 60S particles is required for the clearance of aberrant translation products. Proc. Natl. Acad. Sci. USA 110, 5046–5051. 10.1073/pnas.1221724110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sacher M, Jiang Y, Barrowman J, Scarpa A, Burston J, Zhang L, Schieltz D, Yates JR 3rd, Abeliovich H, and Ferro-Novick S (1998). TRAPP, a highly conserved novel complex on the cis-Golgi that mediates vesicle docking and fusion. EMBO J 17, 2494–2503. 10.1093/emboj/17.9.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sapperstein SK, Walter DM, Grosvenor AR, Heuser JE, and Waters MG (1995). p115 is a general vesicular transport factor related to the yeast endoplasmic reticulum to Golgi transport factor Uso1p. Proc. Natl. Acad. Sci. USA 92, 522–526. 10.1073/pnas.92.2.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kramer G, Rauch T, Rist W, Vorderwülbecke S, Patzelt H, Schulze-Specking A, Ban N, Deuerling E, and Bukau B (2002). L23 protein functions as a chaperone docking site on the ribosome. Nature 419, 171–174. 10.1038/nature01047. [DOI] [PubMed] [Google Scholar]
- 33.Wegrzyn RD, Hofmann D, Merz F, Nikolay R, Rauch T, Graf C, and Deuerling E (2006). A conserved motif is prerequisite for the interaction of NAC with ribosomal protein L23 and nascent chains. J. Biol. Chem 281, 2847–2857. 10.1074/jbc.M511420200. [DOI] [PubMed] [Google Scholar]
- 34.Gelse K, Pöschl E, and Aigner T (2003). Collagens–structure, function, and biosynthesis. Adv. Drug Deliv. Rev 55, 1531–1546. 10.1016/j.addr.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 35.Peil L, Starosta AL, Lassak J, Atkinson GC, Virumäe K, Spitzer M, Tenson T, Jung K, Remme J, and Wilson DN (2013). Distinct XPPX sequence motifs induce ribosome stalling, which is rescued by the translation elongation factor EF-P. Proc. Natl. Acad. Sci. USA 110, 15265–15270. 10.1073/pnas.1310642110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huter P, Arenz S, Bock LV, Graf M, Frister JO, Heuer A, Peil L, Starosta AL, Wohlgemuth I, Peske F, et al. (2017). Structural basis for polyproline-mediated ribosome stalling and rescue by the translation elongation factor EF-P. Mol. Cell 68, 515–527.e6. 10.1016/j.molcel.2017.10.014. [DOI] [PubMed] [Google Scholar]
- 37.Ryan MD, King AM, and Thomas GP (1991). Cleavage of foot-and-mouth disease virus polyprotein is mediated by residues located within a 19 amino acid sequence. J. Gen. Virol 72, 2727–2732. 10.1099/0022-1317-72-11-2727. [DOI] [PubMed] [Google Scholar]
- 38.Forrester A, De Leonibus C, Grumati P, Fasana E, Piemontese M, Staiano L, Fregno I, Raimondi A, Marazza A, Bruno G, et al. (2019). A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex. EMBO J 38, e99847. 10.15252/embj.201899847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morin X, Daneman R, Zavortink M, and Chia W (2001). A protein trap strategy to detect GFP-tagged proteins expressed from their endogenous loci in Drosophila. Proc. Natl. Acad. Sci. USA 98, 15050–15055. 10.1073/pnas.261408198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yurchenco PD, and Furthmayr H (1984). Self-assembly of basement membrane collagen. Biochemistry 23, 1839–1850. 10.1021/bi00303a040. [DOI] [PubMed] [Google Scholar]
- 41.Pastor-Pareja JC, and Xu T (2011). Shaping cells and organs in Drosophila by opposing roles of fat body-secreted Collagen IV and perlecan. Dev. Cell 21, 245–256. 10.1016/j.devcel.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Voorhees RM, Fernández IS, Scheres SHW, and Hegde RS (2014). Structure of the mammalian ribosome-Sec61 complex to 3.4 A resolution. Cell 157, 1632–1643. 10.1016/j.cell.2014.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barrowman J, Bhandari D, Reinisch K, and Ferro-Novick S (2010). TRAPP complexes in membrane traffic: convergence through a common Rab. Nat. Rev. Mol. Cell Biol 11, 759–763. 10.1038/nrm2999. [DOI] [PubMed] [Google Scholar]
- 44.Bienkowski RS (1989). Intracellular degradation of newly synthesized collagen. Revis. Biol. Celular 21, 423–443. [PubMed] [Google Scholar]
- 45.Ito S, and Nagata K (2021). Quality control of procollagen in cells. Annu. Rev. Biochem 90, 631–658. 10.1146/annurev-biochem-013118-111603. [DOI] [PubMed] [Google Scholar]
- 46.Reggio A, Buonomo V, Berkane R, Bhaskara RM, Tellechea M, Peluso I, Polishchuk E, Di Lorenzo G, Cirillo C, Esposito M, et al. (2021). Role of FAM134 paralogues in endoplasmic reticulum remodeling, ERphagy, and Collagen quality control. EMBO Rep 22, e52289. 10.15252/embr.202052289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gregorio I, Braghetta P, Bonaldo P, and Cescon M (2018). Collagen VI in healthy and diseased nervous system. Dis. Model. Mech 11, dmm032946. 10.1242/dmm.032946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Palacios-Macapagal D, Cann J, Cresswell G, Zerrouki K, Dacosta K, Wang J, Connor J, and Davidso TS (2021). Chronic CNS pathology is associated with abnormal collagen deposition and fibrotic-like changes. Preprint at bioRxiv 10.1101/2021.07.06.451298. [DOI] [Google Scholar]
- 49.Arakawa S, Yunoki K, Izawa T, Tamura Y, Nishikawa SI, and Endo T (2016). Quality control of nonstop membrane proteins at the ER membrane and in the cytosol. Sci. Rep 6, 30795. 10.1038/srep30795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.von der Malsburg K, Shao S, and Hegde RS (2015). The ribosome quality control pathway can access nascent polypeptides stalled at the Sec61 translocon. Mol. Biol. Cell 26, 2168–2180. 10.1091/mbc.E15-01-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554. 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fueller J, Herbst K, Meurer M, Gubicza K, Kurtulmus B, Knopf JD, Kirrmaier D, Buchmuller BC, Pereira G, Lemberg MK, and Knop M (2020). CRISPR-Cas12a-assisted PCR tagging of mammalian genes. J. Cell Biol 219, e201910210. 10.1083/jcb.201910210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rivera MC, Maguire B, and Lake JA (2015). Purification of polysomes. Cold Spring Harb. Protoc 2015, 303–305. 10.1101/pdb.prot081364. [DOI] [PubMed] [Google Scholar]
- 54.Wu D, and Piszczek G (2020). Measuring the affinity of protein-protein interactions on a single-molecule level by mass photometry. Anal. Biochem 592, 113575. 10.1016/j.ab.2020.113575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ye Y, and Fortini ME (1998). Characterization of Drosophila presenilin and its colocalization with notch during development. Mech. Dev 79, 199–211. 10.1016/s0925-4773(98)00169-5. [DOI] [PubMed] [Google Scholar]
- 56.Port F, Chen HM, Lee T, and Bullock SL (2014). Optimized CRISPR/Cas tools for efficient germline and somatic genome engineering in Drosophila. Proc. Natl. Acad. Sci. USA 111, E2967–E2976. 10.1073/pnas.1405500111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data generated from the CRISPR screen in this paper have been deposited to GEO under accession codes GEO: GSE222104.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.